User login
Multiple Painless Whitish Papules on the Vulva and Perianal Region
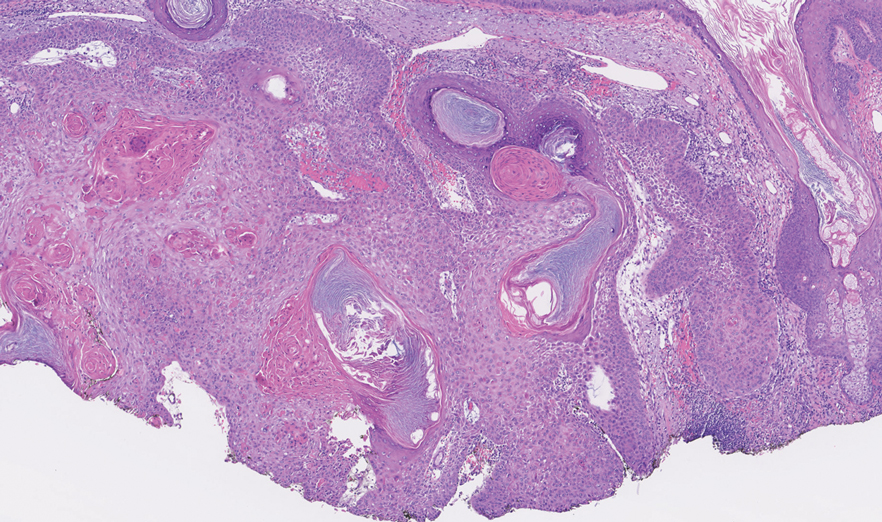
THE DIAGNOSIS: Papular Acantholytic Dyskeratosis
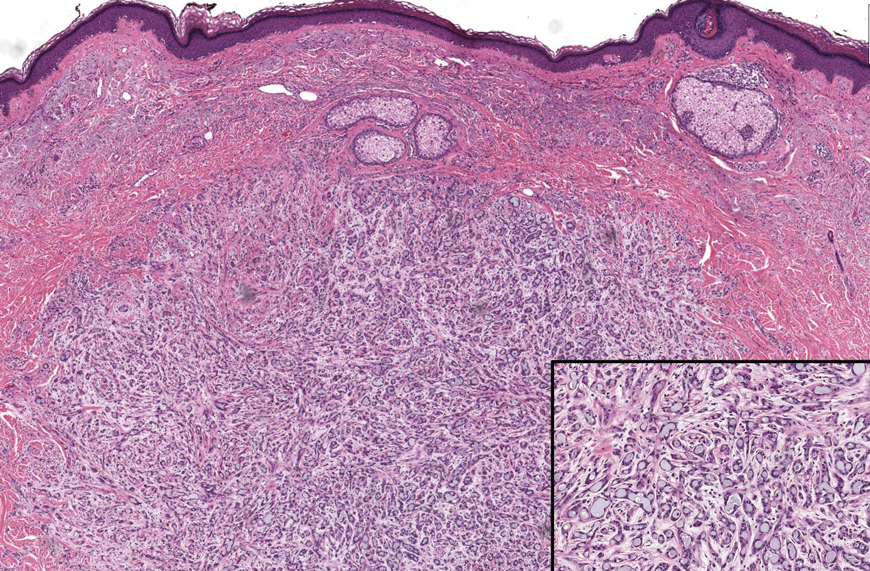
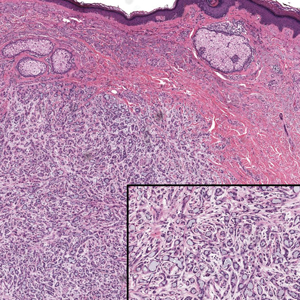
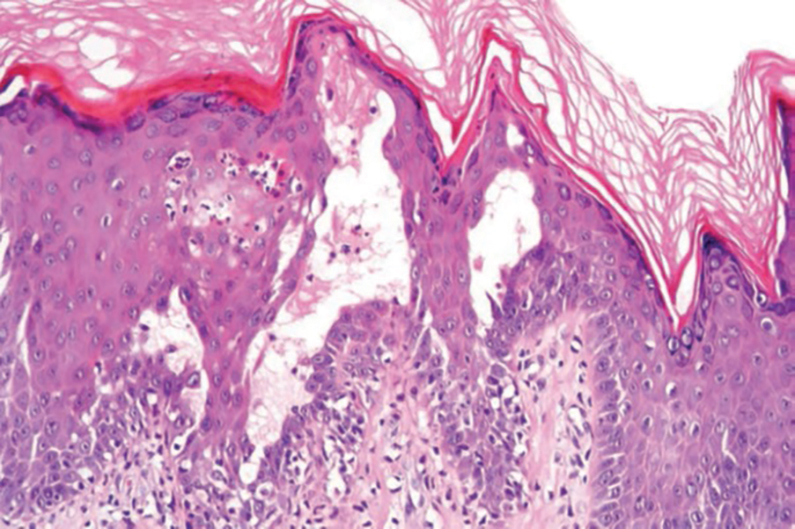
Histopathology of the lesion in our patient revealed hyperkeratosis, parakeratosis, dyskeratosis, and acantholysis of keratinocytes. The dermis showed variable chronic inflammatory cells. Corps ronds and grains in the acantholytic layer of the epidermis were identified. Hair follicles were not affected by acantholysis. Anti–desmoglein 1 and anti–desmoglein 3 serum antibodies were negative. Based on the combined clinical and histologic findings, the patient was diagnosed with papular acantholytic dyskeratosis (PAD) of the genitocrural area.
Although its typical histopathologic pattern mimics both Hailey-Hailey disease and Darier disease, PAD is a rare unique clinicopathologic entity recognized by dermatopathologists. It usually occurs in middle-aged women with no family history of similar conditions. The multiple localized, flesh-colored to whitish papules of PAD tend to coalesce into plaques in the anogenital and genitocrural regions. Plaques usually are asymptomatic but may be pruritic. Histopathologically, PAD will demonstrate hyperkeratosis, dyskeratosis, and acantholysis. Corps ronds and grains will be present in the acantholytic layer of the epidermis.1,2
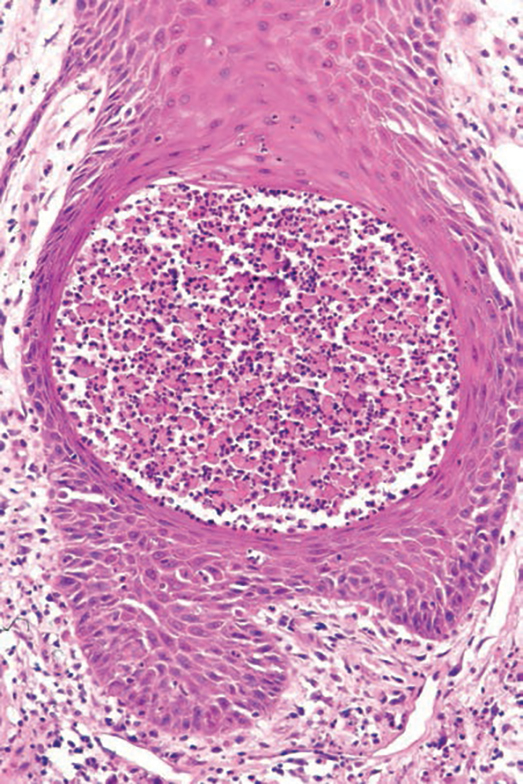
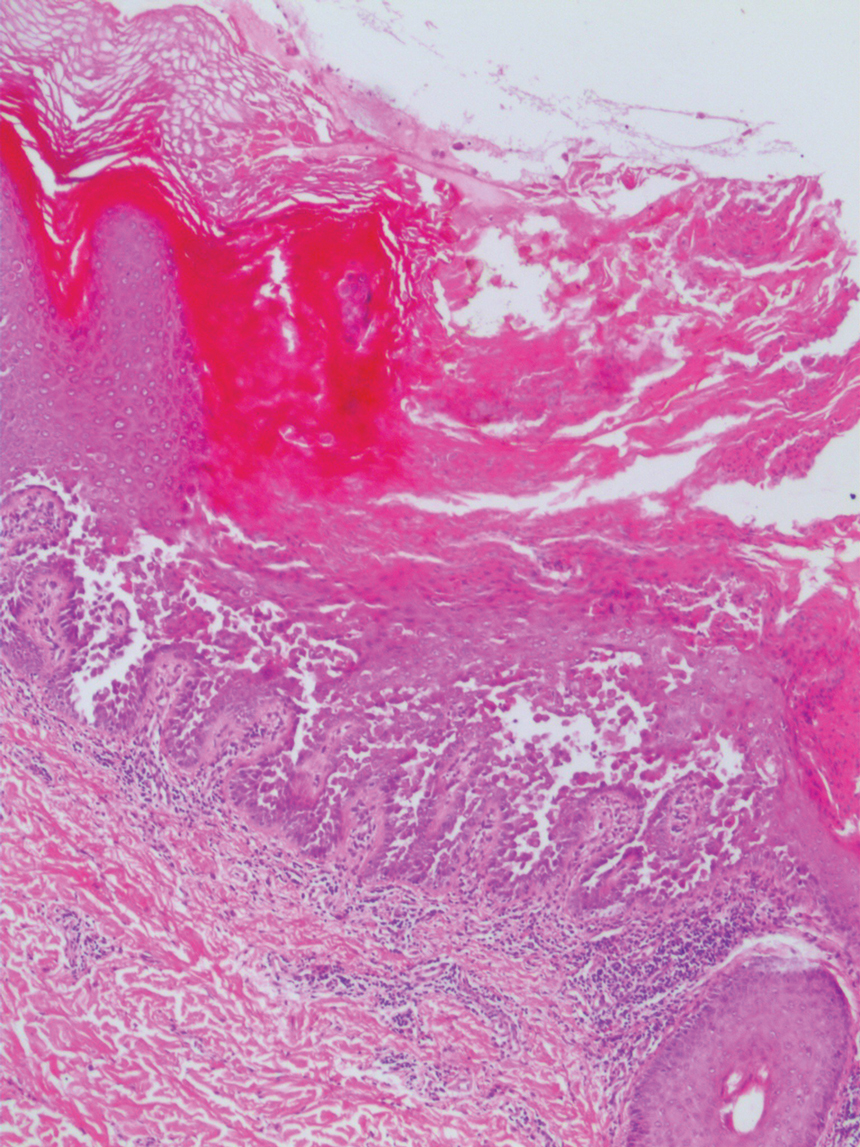
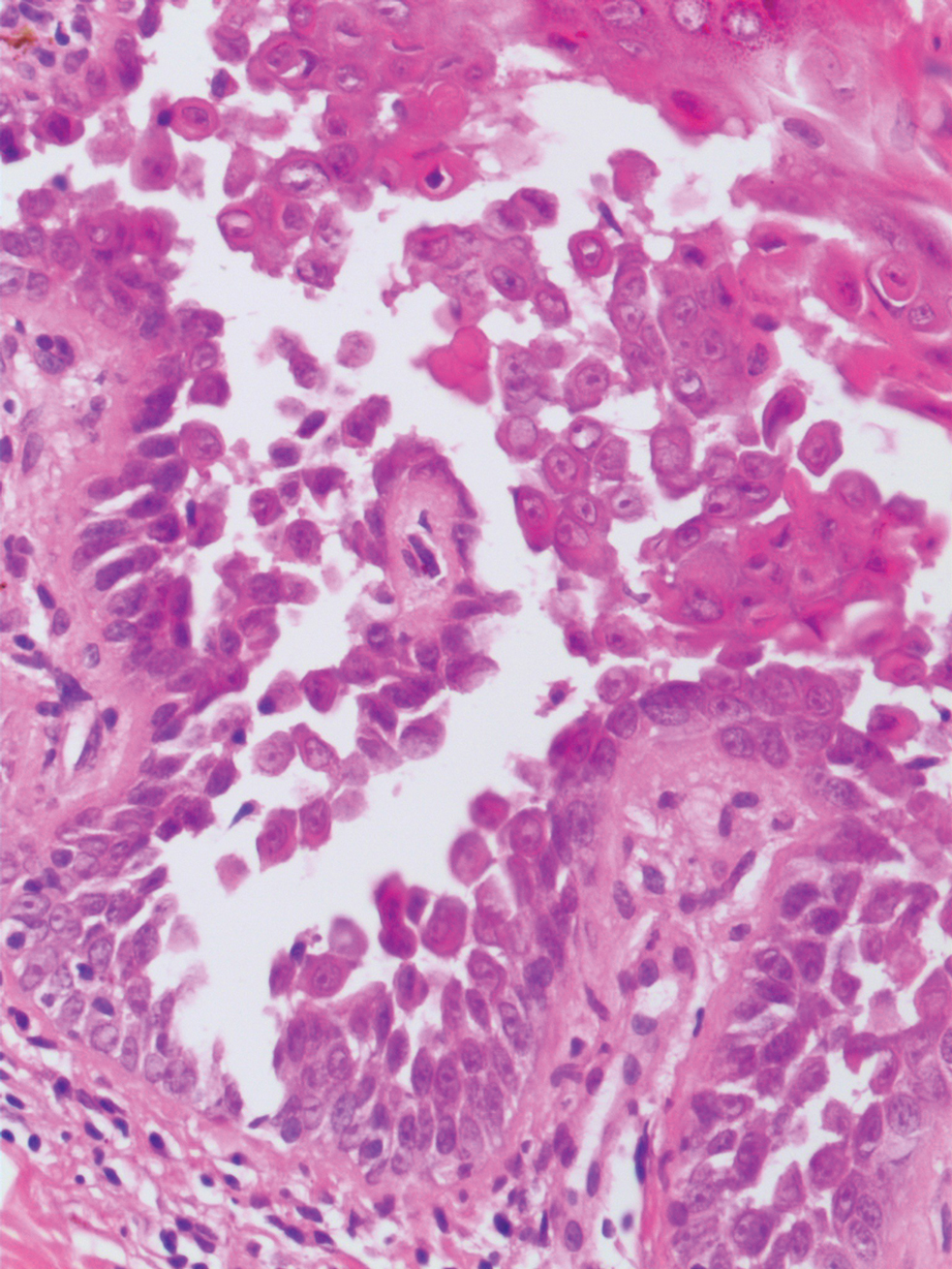
The differential diagnosis for PAD includes pemphigus vegetans, Hailey-Hailey disease, Darier disease, and Grover disease. Patients usually develop pemphigus vegetans at an older age (typically 50–70 years).3 Histopathologically, it is characterized by pseudoepitheliomatous hyperplasia with an eosinophilic microabscess as well as acantholysis that involves the follicular epithelium (Figure 1),4 which were not seen in our patient. Direct immunofluorescence will show the intercellular pattern of the pemphigus group, and antidesmoglein antibodies can be detected by enzyme-linked immunosorbent assay.4,5
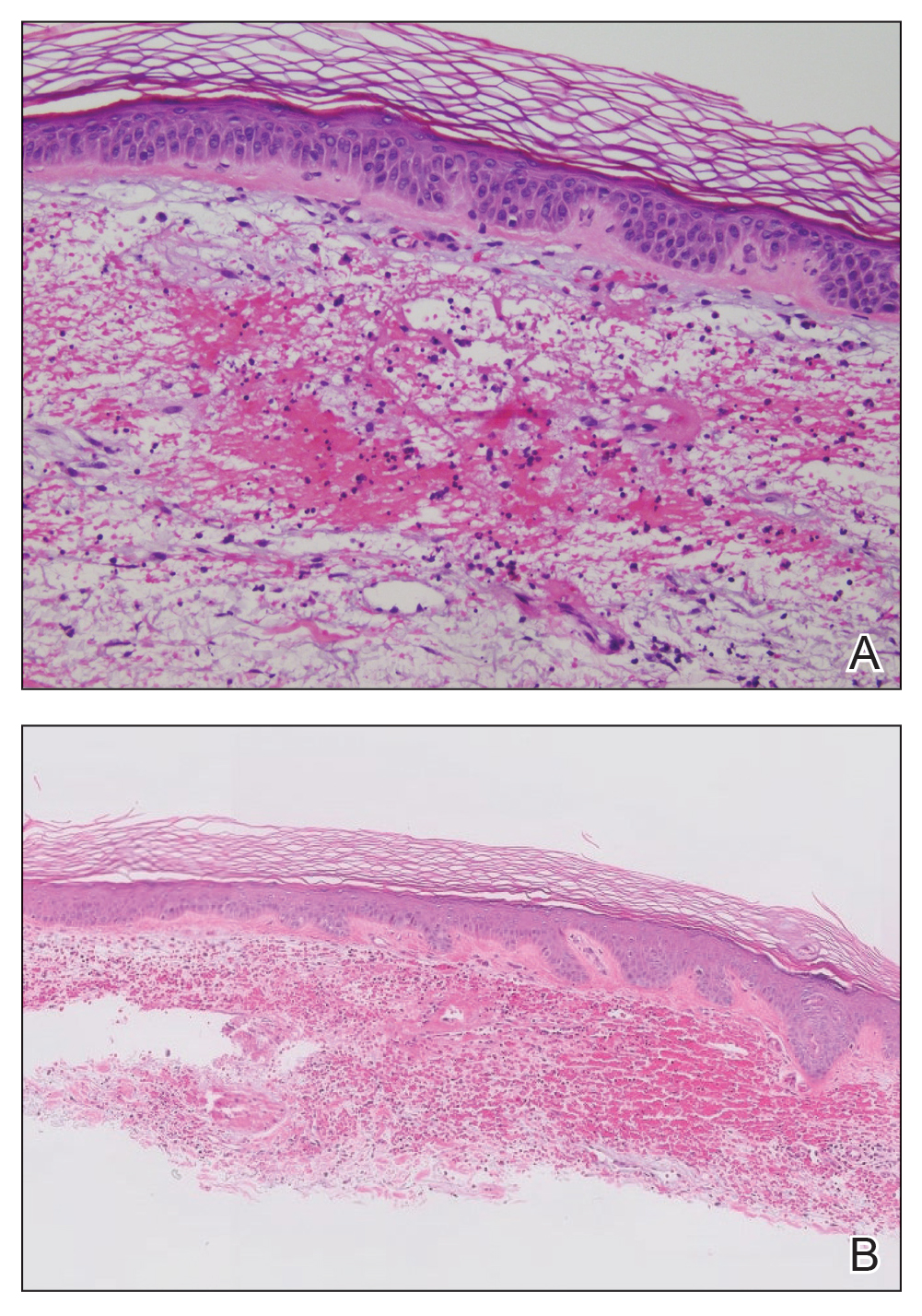
Hailey-Hailey disease (also known as benign familial pemphigus) typically manifests as itchy malodorous vesicles and erosions, especially in intertriginous areas. The most commonly affected sites are the groin, neck, under the breasts, and between the buttocks. In one study, two-thirds of affected patients reported a relevant family history.4 Histopathology will show minimal dyskeratosis and suprabasilar acantholysis with loss of intercellular bridges, classically described as resembling a dilapidated brick wall (Figure 2).4,5 There is no notable follicular involvement with acantholysis.4
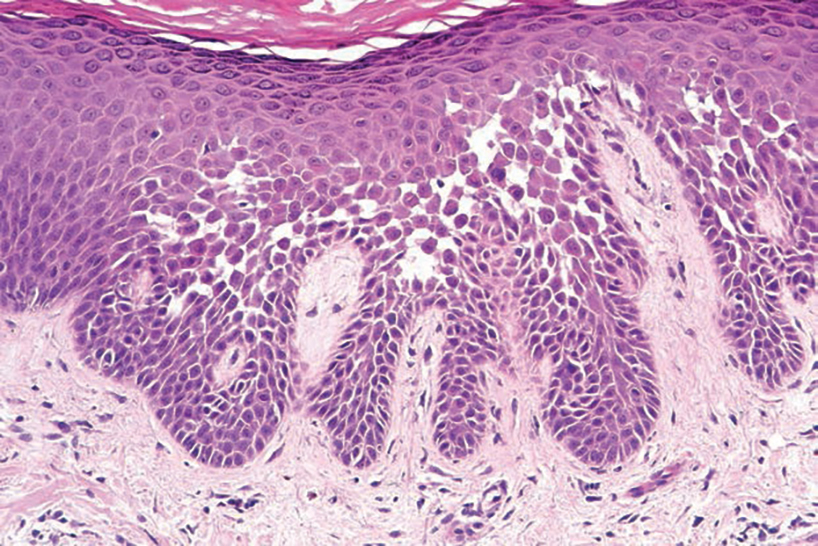
characteristic dilapidated brick wall appearance (H&E, original
magnification ×40).
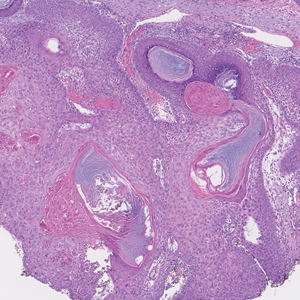
Darier disease (also known as keratosis follicularis) typically is inherited in an autosomal-dominant pattern.4 It is found on the seborrheic areas such as the scalp, forehead, nasolabial folds, and upper chest. Characteristic features include distal notching of the nails, mucosal lesions, and palmoplantar papules. Histopathology will reveal acantholysis, dyskeratosis, suprabasilar acantholysis, and corps ronds and grains.4 Acantholysis in Darier disease can be in discrete foci and/or widespread (Figure 3).4 Darier disease demonstrates more dyskeratosis than Hailey-Hailey disease.4,5

Grover disease (also referred to as transient acantholytic dermatosis) is observed predominantly in individuals who are middle-aged or older, though occurrence in children has been rarely reported.4 It affects the trunk, neck, and proximal limbs but spares the genital area. Histopathology may reveal acantholysis (similar to Hailey-Hailey disease or pemphigus vulgaris), dyskeratosis (resembling Darier disease), spongiosis, parakeratosis, and a superficial perivascular lymphocytic infiltrate with eosinophils.4 A histologic clue to the diagnosis is small lesion size (1–3 mm). Usually, only 1 or 2 small discrete lesions that span a few rete ridges are noted (Figure 4).4 Grover disease can cause follicular or acrosyringeal involvement.4
- Al-Muriesh M, Abdul-Fattah B, Wang X, et al. Papular acantholytic dyskeratosis of the anogenital and genitocrural area: case series and review of the literature. J Cutan Pathol. 2016;43:749-758. doi:10.1111/cup.12736
- Harrell J, Nielson C, Beers P, et al. Eruption on the vulva and groin. JAAD Case Reports. 2019;6:6-8. doi:10.1016/j.jdcr.2019.11.003
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls [Internet]. Updated June 26, 2023. Accessed September 18, 2024. https://www.ncbi.nlm.nih.gov/books/NBK545229
- Acantholytic disorders. In: Calonje E, Brenn T, Lazar A, et al, eds. McKee’s Pathology of the Skin: With Clinical Correlations. Elsevier/ Saunders; 2012:171-200.
- Mohr MR, Erdag G, Shada AL, et al. Two patients with Hailey- Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211215. doi:10.1001/archdermatol.2010.445
THE DIAGNOSIS: Papular Acantholytic Dyskeratosis
Histopathology of the lesion in our patient revealed hyperkeratosis, parakeratosis, dyskeratosis, and acantholysis of keratinocytes. The dermis showed variable chronic inflammatory cells. Corps ronds and grains in the acantholytic layer of the epidermis were identified. Hair follicles were not affected by acantholysis. Anti–desmoglein 1 and anti–desmoglein 3 serum antibodies were negative. Based on the combined clinical and histologic findings, the patient was diagnosed with papular acantholytic dyskeratosis (PAD) of the genitocrural area.
Although its typical histopathologic pattern mimics both Hailey-Hailey disease and Darier disease, PAD is a rare unique clinicopathologic entity recognized by dermatopathologists. It usually occurs in middle-aged women with no family history of similar conditions. The multiple localized, flesh-colored to whitish papules of PAD tend to coalesce into plaques in the anogenital and genitocrural regions. Plaques usually are asymptomatic but may be pruritic. Histopathologically, PAD will demonstrate hyperkeratosis, dyskeratosis, and acantholysis. Corps ronds and grains will be present in the acantholytic layer of the epidermis.1,2
The differential diagnosis for PAD includes pemphigus vegetans, Hailey-Hailey disease, Darier disease, and Grover disease. Patients usually develop pemphigus vegetans at an older age (typically 50–70 years).3 Histopathologically, it is characterized by pseudoepitheliomatous hyperplasia with an eosinophilic microabscess as well as acantholysis that involves the follicular epithelium (Figure 1),4 which were not seen in our patient. Direct immunofluorescence will show the intercellular pattern of the pemphigus group, and antidesmoglein antibodies can be detected by enzyme-linked immunosorbent assay.4,5
Hailey-Hailey disease (also known as benign familial pemphigus) typically manifests as itchy malodorous vesicles and erosions, especially in intertriginous areas. The most commonly affected sites are the groin, neck, under the breasts, and between the buttocks. In one study, two-thirds of affected patients reported a relevant family history.4 Histopathology will show minimal dyskeratosis and suprabasilar acantholysis with loss of intercellular bridges, classically described as resembling a dilapidated brick wall (Figure 2).4,5 There is no notable follicular involvement with acantholysis.4
characteristic dilapidated brick wall appearance (H&E, original
magnification ×40).
Darier disease (also known as keratosis follicularis) typically is inherited in an autosomal-dominant pattern.4 It is found on the seborrheic areas such as the scalp, forehead, nasolabial folds, and upper chest. Characteristic features include distal notching of the nails, mucosal lesions, and palmoplantar papules. Histopathology will reveal acantholysis, dyskeratosis, suprabasilar acantholysis, and corps ronds and grains.4 Acantholysis in Darier disease can be in discrete foci and/or widespread (Figure 3).4 Darier disease demonstrates more dyskeratosis than Hailey-Hailey disease.4,5
Grover disease (also referred to as transient acantholytic dermatosis) is observed predominantly in individuals who are middle-aged or older, though occurrence in children has been rarely reported.4 It affects the trunk, neck, and proximal limbs but spares the genital area. Histopathology may reveal acantholysis (similar to Hailey-Hailey disease or pemphigus vulgaris), dyskeratosis (resembling Darier disease), spongiosis, parakeratosis, and a superficial perivascular lymphocytic infiltrate with eosinophils.4 A histologic clue to the diagnosis is small lesion size (1–3 mm). Usually, only 1 or 2 small discrete lesions that span a few rete ridges are noted (Figure 4).4 Grover disease can cause follicular or acrosyringeal involvement.4
THE DIAGNOSIS: Papular Acantholytic Dyskeratosis
Histopathology of the lesion in our patient revealed hyperkeratosis, parakeratosis, dyskeratosis, and acantholysis of keratinocytes. The dermis showed variable chronic inflammatory cells. Corps ronds and grains in the acantholytic layer of the epidermis were identified. Hair follicles were not affected by acantholysis. Anti–desmoglein 1 and anti–desmoglein 3 serum antibodies were negative. Based on the combined clinical and histologic findings, the patient was diagnosed with papular acantholytic dyskeratosis (PAD) of the genitocrural area.
Although its typical histopathologic pattern mimics both Hailey-Hailey disease and Darier disease, PAD is a rare unique clinicopathologic entity recognized by dermatopathologists. It usually occurs in middle-aged women with no family history of similar conditions. The multiple localized, flesh-colored to whitish papules of PAD tend to coalesce into plaques in the anogenital and genitocrural regions. Plaques usually are asymptomatic but may be pruritic. Histopathologically, PAD will demonstrate hyperkeratosis, dyskeratosis, and acantholysis. Corps ronds and grains will be present in the acantholytic layer of the epidermis.1,2
The differential diagnosis for PAD includes pemphigus vegetans, Hailey-Hailey disease, Darier disease, and Grover disease. Patients usually develop pemphigus vegetans at an older age (typically 50–70 years).3 Histopathologically, it is characterized by pseudoepitheliomatous hyperplasia with an eosinophilic microabscess as well as acantholysis that involves the follicular epithelium (Figure 1),4 which were not seen in our patient. Direct immunofluorescence will show the intercellular pattern of the pemphigus group, and antidesmoglein antibodies can be detected by enzyme-linked immunosorbent assay.4,5
Hailey-Hailey disease (also known as benign familial pemphigus) typically manifests as itchy malodorous vesicles and erosions, especially in intertriginous areas. The most commonly affected sites are the groin, neck, under the breasts, and between the buttocks. In one study, two-thirds of affected patients reported a relevant family history.4 Histopathology will show minimal dyskeratosis and suprabasilar acantholysis with loss of intercellular bridges, classically described as resembling a dilapidated brick wall (Figure 2).4,5 There is no notable follicular involvement with acantholysis.4
characteristic dilapidated brick wall appearance (H&E, original
magnification ×40).
Darier disease (also known as keratosis follicularis) typically is inherited in an autosomal-dominant pattern.4 It is found on the seborrheic areas such as the scalp, forehead, nasolabial folds, and upper chest. Characteristic features include distal notching of the nails, mucosal lesions, and palmoplantar papules. Histopathology will reveal acantholysis, dyskeratosis, suprabasilar acantholysis, and corps ronds and grains.4 Acantholysis in Darier disease can be in discrete foci and/or widespread (Figure 3).4 Darier disease demonstrates more dyskeratosis than Hailey-Hailey disease.4,5
Grover disease (also referred to as transient acantholytic dermatosis) is observed predominantly in individuals who are middle-aged or older, though occurrence in children has been rarely reported.4 It affects the trunk, neck, and proximal limbs but spares the genital area. Histopathology may reveal acantholysis (similar to Hailey-Hailey disease or pemphigus vulgaris), dyskeratosis (resembling Darier disease), spongiosis, parakeratosis, and a superficial perivascular lymphocytic infiltrate with eosinophils.4 A histologic clue to the diagnosis is small lesion size (1–3 mm). Usually, only 1 or 2 small discrete lesions that span a few rete ridges are noted (Figure 4).4 Grover disease can cause follicular or acrosyringeal involvement.4
- Al-Muriesh M, Abdul-Fattah B, Wang X, et al. Papular acantholytic dyskeratosis of the anogenital and genitocrural area: case series and review of the literature. J Cutan Pathol. 2016;43:749-758. doi:10.1111/cup.12736
- Harrell J, Nielson C, Beers P, et al. Eruption on the vulva and groin. JAAD Case Reports. 2019;6:6-8. doi:10.1016/j.jdcr.2019.11.003
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls [Internet]. Updated June 26, 2023. Accessed September 18, 2024. https://www.ncbi.nlm.nih.gov/books/NBK545229
- Acantholytic disorders. In: Calonje E, Brenn T, Lazar A, et al, eds. McKee’s Pathology of the Skin: With Clinical Correlations. Elsevier/ Saunders; 2012:171-200.
- Mohr MR, Erdag G, Shada AL, et al. Two patients with Hailey- Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211215. doi:10.1001/archdermatol.2010.445
- Al-Muriesh M, Abdul-Fattah B, Wang X, et al. Papular acantholytic dyskeratosis of the anogenital and genitocrural area: case series and review of the literature. J Cutan Pathol. 2016;43:749-758. doi:10.1111/cup.12736
- Harrell J, Nielson C, Beers P, et al. Eruption on the vulva and groin. JAAD Case Reports. 2019;6:6-8. doi:10.1016/j.jdcr.2019.11.003
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls [Internet]. Updated June 26, 2023. Accessed September 18, 2024. https://www.ncbi.nlm.nih.gov/books/NBK545229
- Acantholytic disorders. In: Calonje E, Brenn T, Lazar A, et al, eds. McKee’s Pathology of the Skin: With Clinical Correlations. Elsevier/ Saunders; 2012:171-200.
- Mohr MR, Erdag G, Shada AL, et al. Two patients with Hailey- Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211215. doi:10.1001/archdermatol.2010.445
A 21-year-old woman presented with a chronic eruption in the anogenital region of 4 years’ duration. Clinical examination revealed numerous painless, mildly itchy, malodorous, whitish papules on an erythematous base that were distributed on the vulva and perianal region. There were no erosions, and no other areas were involved. Routine laboratory tests were within reference range. The patient had no sexual partner and no family history of similar lesions. A skin biopsy was performed.
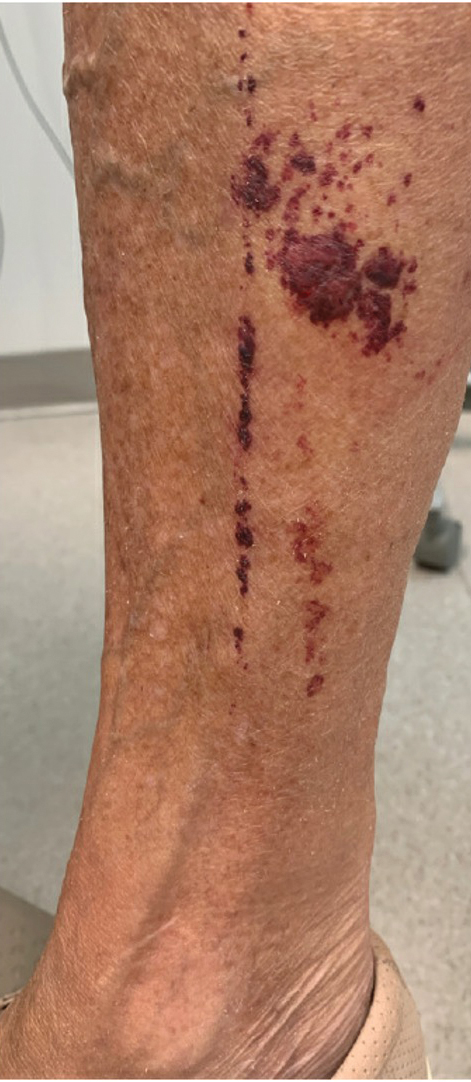
Purpuric Lesions on the Leg
THE DIAGNOSIS: Dengue Hemorrhagic Fever
The retiform purpura observed in our patient was suggestive of a vasculitic, thrombotic, or embolic etiology. Dengue IgM serologic testing performed based on her extensive travel history and recent return from a dengue-endemic area was positive, indicating acute infection. A clinical diagnosis of dengue hemorrhagic fever (DHF) was made based on the hemorrhagic appearance of the lesion. Histopathology revealed leukocytoclastic vasculitis (Figure). Anti–double-stranded DNA, antideoxyribonuclease, C3 and C4, CH50 (total hemolytic complement), antineutrophil cytoplasmic antibodies, HIV, and hepatitis B virus tests were normal. Direct immunofluorescence was negative.
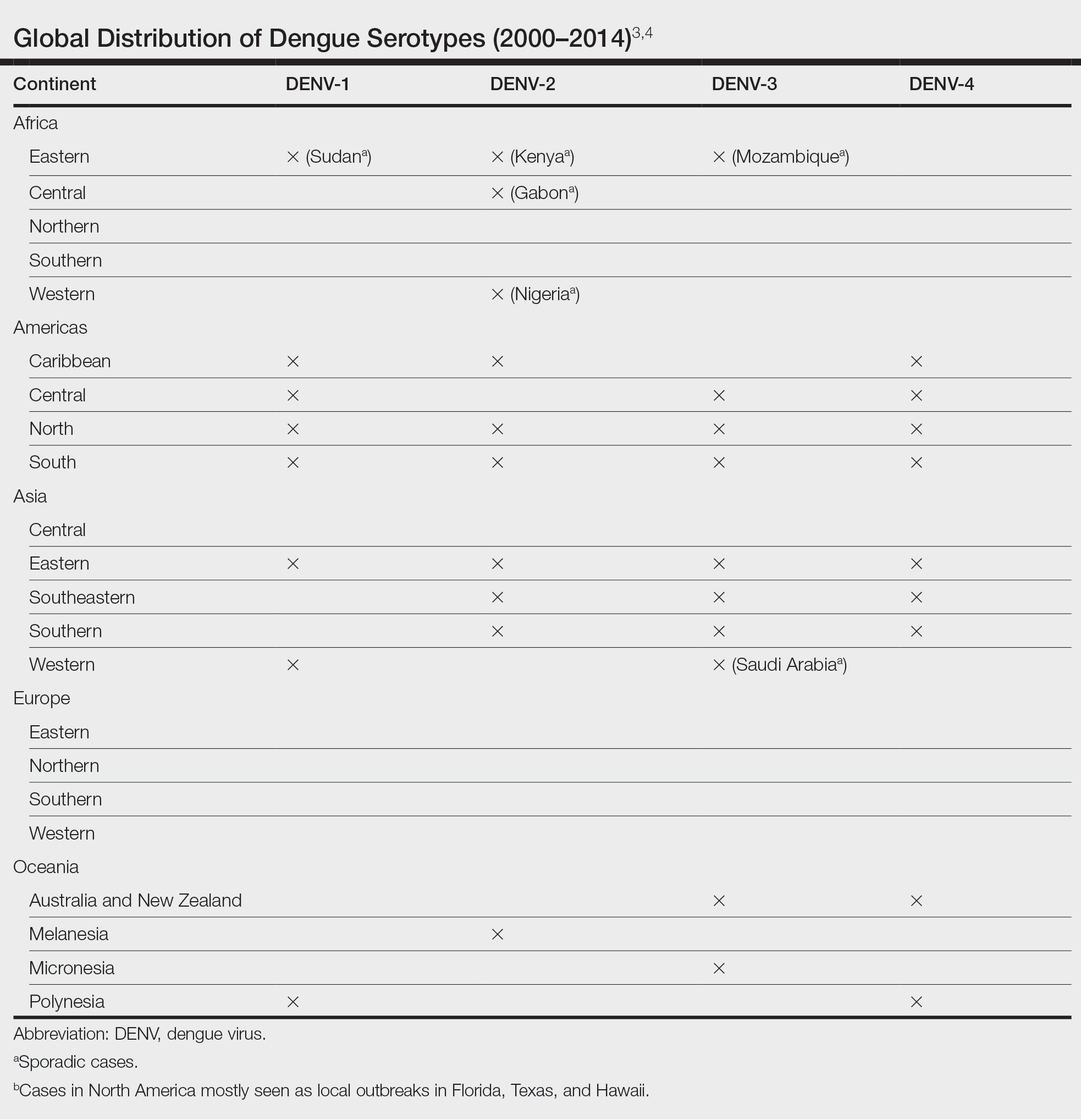
Dengue virus is a single-stranded RNA virus transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is one of the most prevalent arthropod-borne viruses affecting humans today.1,2 Infection with the dengue virus generally is seen in travelers visiting tropical regions of Africa, Mexico, South America, South and Central Asia, Southeast Asia, and the Caribbean.1 The Table shows the global distribution of dengue serotypes from 2000 to 2014.3,4 There are 4 serotypes of the dengue virus: DENV-1 to DENV-4. Infection with 1 strain elicits longlasting immunity to that strain, but subsequent infection with another strain can result in severe DHF due to antibody cross-reaction.1
Dengue virus infection ranges from mildly symptomatic to a spectrum of increasingly severe conditions that comprise dengue fever (DF) and DHF, as well as dengue shock syndrome and brain stem hemorrhage, which may be fatal.2,5 Dengue fever manifests as severe myalgia, fever, headache (usually retro-orbital), arthralgia, erythema, and rubelliform exanthema.6 The frequency of skin eruptions in patients with DF varies with the virus strain and outbreaks.7 The lesions initially develop with the onset of fever and manifest as flushing or erythematous mottling of the face, neck, and chest areas.1,7 The morbilliform eruption develops 2 to 6 days after the onset of the fever, beginning on the trunk and spreading to the face and extremities.1,7 The rash may become confluent with characteristic sparing of small round areas of normal skin described as white islands in a sea of red.2 Verrucous papules on the ears also have been described and may resemble those seen in Cowden syndrome. In patients with prior infection with a different strain of the virus, hemorrhagic lesions may develop, including characteristic retiform purpura, a positive tourniquet test, and the appearance of petechiae on the lower legs. Pruritus and desquamation, especially on the palms and soles, may follow the termination of the eruption.7
The differential diagnosis of DF includes measles, rubella, enteroviruses, and influenza. Chikungunya and West Nile viruses in Asia and Africa and the O’nyong-nyong virus in Africa are also arboviruses that cause a clinical picture similar to DF but not DHF. Other diagnostic considerations include phases of scarlet fever, typhoid, malaria, leptospirosis, hepatitis A, and trypanosomal and rickettsial diseases.7 The differential diagnosis of DHF includes antineutrophil cytoplasmic antibody–associated vasculitis, rheumatoid vasculitis, and bacterial septic vasculitis.
Acute clinical diagnosis of DF can be challenging because of the nonspecific symptoms that can be seen in almost every infectious disease. Clinical presentation assessment should be confirmed with laboratory testing.6 Dengue virus infection usually is confirmed by the identification of viral genomic RNA, antigens, or the antibodies it elicits. Enzyme-linked immunosorbent assay–based serologic tests are cost-effective and easy to perform.5 IgM antibodies usually show cross-reactivity with platelets, but the antibody levels are not positively correlated with the severity of DF.8 Primary infection with the dengue virus is characterized by the elevation of specific IgM levels that usually occurs 3 to 5 days after symptom onset and persists during the postfebrile stage (up to 30 to 60 days). In secondary infections, the IgM levels usually rise more slowly and reach a lower level than in primary infections.9 For both primary and secondary infections, testing IgM levels after the febrile stage may be helpful with the laboratory diagnosis.
Currently, there is no antiviral drug available for dengue. Treatment of dengue infection is symptomatic and supportive.2
Dengue hemorrhagic fever is indicated by a rising hematocrit (≥20%) and a falling platelet count (>100,000/mm3) accompanying clinical signs of hemorrhage. Treatment includes intravenous fluid replacement and careful clinical monitoring of hematocrit levels, platelet count, vitals, urine output, and other signs of shock.5 For patients with a history of dengue infection, travel to areas with other serotypes is not recommended.
If any travel to a high-risk area is planned, countryspecific travel recommendations and warnings should be reviewed from the Centers for Disease Control and Prevention’s website (https://wwwnc.cdc.gov/travel/notices/level1/dengue-global). Use of an Environmental Protection Agency–registered insect repellent to avoid mosquito bites and acetaminophen for managing symptoms is advised. During travel, staying in places with window and door screens and using a bed net during sleep are suggested. Long-sleeved shirts and long pants also are preferred. Travelers should see a health care provider if they have symptoms of dengue.10
African tick bite fever (ATBF) is caused by Rickettsia africae transmitted by Amblyomma ticks. Skin findings in ATBF include erythematous, firm, tender papules with central eschars consistent with the feeding patterns of ticks.11 Histopathology of ATBF usually includes fibrinoid necrosis of vessels in the dermis with a perivascular inflammatory infiltrate and coagulation necrosis of the surrounding dermis consistent with eschar formation.12 The lack of an eschar weighs against this diagnosis.
African trypanosomiasis (also known as sleeping sickness) is caused by protozoa transmitted by the tsetse fly. A chancrelike, circumscribed, rubbery, indurated red or violaceous nodule measuring 2 to 5 cm in diameter often develops as the earliest cutaneous sign of the disease.13 Nonspecific histopathologic findings, such as infiltration of lymphocytes and macrophages and proliferation of endothelial cells and fibroblasts, may be observed.14 Extravascular parasites have been noted in skin biopsies.15 In later stages, skin lesions called trypanids may be observed as macular, papular, annular, targetoid, purpuric, and erythematous lesions, and histopathologic findings consistent with vasculitis also may be seen.13
Chikungunya virus infection is an acute-onset, mosquito-borne viral disease. Skin manifestations may start with nonspecific, generalized, morbilliform, maculopapular rashes coinciding with fever, which also may be seen initially with DHF. Skin hyperpigmentation, mostly centrofacial and involving the nose (chik sign); purpuric and ecchymotic lesions over the trunk and flexors of limbs in adults, often surmounted by subepidermal bullae and lesions resembling toxic epidermal necrolysis; and nonhealing ulcers in the genital and groin areas are common skin manifestations of chikungunya infection.16 Intraepithelial splitting with acantholysis and perivascular lymphohistiocytic infiltration may be observed in the histopathology of blistering lesions, which are not consistent with DHF.17
Zika virus infection is caused by an arbovirus within the Flaviviridae family, which also includes the dengue virus. Initial mucocutaneous findings of the Zika virus include nonspecific diffuse maculopapular eruptions. The eruption generally spares the palms and soles; however, various manifestations including involvement of the palms and soles have been reported.18 The morbilliform eruption begins on the face and extends to the trunk and extremities. Mild hemorrhagic manifestations, including petechiae and bleeding gums, may be observed. Distinguishing between dengue and Zika virus infection relies on the severity of symptoms and laboratory tests, including polymerase chain reaction or IgM antibody testing.19 The other conditions listed do not produce hemorrhagic fever.
- Pincus LB, Grossman ME, Fox LP. The exanthem of dengue fever: clinical features of two US tourists traveling abroad. J Am Acad Dermatol. 2008;58:308-316. doi:10.1016/j.jaad.2007.08.042
- Radakovic-Fijan S, Graninger W, Müller C, et al. Dengue hemorrhagic fever in a British travel guide. J Am Acad Dermatol. 2002;46:430-433. doi:10.1067/mjd.2002.111904
- Yamashita A, Sakamoto T, Sekizuka T, et al. DGV: dengue genographic viewer. Front Microbiol. 2016;7:875. doi:10.3389/fmicb.2016.00875
- Centers for Disease and Prevention. Dengue in the US states and territories. Updated October 7, 2020. Accessed September 30, 2024. https://www.cdc.gov/dengue/data-research/facts-stats/?CDC_AAref_Val=https://www.cdc.gov/dengue/areaswithrisk/in-the-us.html
- Khetarpal N, Khanna I. Dengue fever: causes, complications, and vaccine strategies. J Immunol Res. 2016;2016:6803098. doi:10.1155/2016/6803098
- Muller DA, Depelsenaire AC, Young PR. Clinical and laboratory diagnosis of dengue virus infection. J Infect Dis. 2017;215(suppl 2):S89-S95. doi:10.1093/infdis/jiw649
- Waterman SH, Gubler DJ. Dengue fever. Clin Dermatol. 1989;7:117-122. doi:10.1016/0738-081x(89)90034-5
- Lin CF, Lei HY, Liu CC, et al. Generation of IgM anti-platelet autoantibody in dengue patients. J Med Virol. 2001;63:143-149. doi:10.1002/1096- 9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L
- Tripathi NK, Shrivastava A, Dash PK, et al. Detection of dengue virus. Methods Mol Biol. 2011;665:51-64. doi:10.1007/978-1-60761-817-1_4
- Centers for Disease Control and Prevention. Plan for travel. Accessed September 30, 2024. https://wwwnc.cdc.gov/travel
- Mack I, Ritz N. African tick-bite fever. N Engl J Med. 2019;380:960. doi:10.1056/NEJMicm1810093
- Lepidi H, Fournier PE, Raoult D. Histologic features and immunodetection of African tick-bite fever eschar. Emerg Infect Dis. 2006;12:1332- 1337. doi:10.3201/eid1209.051540
- McGovern TW, Williams W, Fitzpatrick JE, et al. Cutaneous manifestations of African trypanosomiasis. Arch Dermatol. 1995;131:1178-1182.
- Kristensson K, Bentivoglio M. Pathology of African trypanosomiasis. In: Dumas M, Bouteille B, Buguet A, eds. Progress in Human African Trypanosomiasis, Sleeping Sickness. Springer; 1999:157-181.
- Capewell P, Cren-Travaillé C, Marchesi F, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5:e17716. doi:10.7554/eLife.17716
- Singal A. Chikungunya and skin: current perspective. Indian Dermatol Online J. 2017;8:307-309. doi:10.4103/idoj.IDOJ_93_17
- Robin S, Ramful D, Zettor J, et al. Severe bullous skin lesions associated with chikungunya virus infection in small infants. Eur J Pediatr. 2009;169:67-72. doi:10.1007/s00431-009-0986-0
- Hussain A, Ali F, Latiwesh OB, et al. A comprehensive review of the manifestations and pathogenesis of Zika virus in neonates and adults. Cureus. 2018;10:E3290. doi:10.7759/cureus.3290
- Farahnik B, Beroukhim K, Blattner CM, et al. Cutaneous manifestations of the Zika virus. J Am Acad Dermatol. 2016;74:1286-1287. doi:10.1016/j.jaad.2016.02.1232
THE DIAGNOSIS: Dengue Hemorrhagic Fever
The retiform purpura observed in our patient was suggestive of a vasculitic, thrombotic, or embolic etiology. Dengue IgM serologic testing performed based on her extensive travel history and recent return from a dengue-endemic area was positive, indicating acute infection. A clinical diagnosis of dengue hemorrhagic fever (DHF) was made based on the hemorrhagic appearance of the lesion. Histopathology revealed leukocytoclastic vasculitis (Figure). Anti–double-stranded DNA, antideoxyribonuclease, C3 and C4, CH50 (total hemolytic complement), antineutrophil cytoplasmic antibodies, HIV, and hepatitis B virus tests were normal. Direct immunofluorescence was negative.
Dengue virus is a single-stranded RNA virus transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is one of the most prevalent arthropod-borne viruses affecting humans today.1,2 Infection with the dengue virus generally is seen in travelers visiting tropical regions of Africa, Mexico, South America, South and Central Asia, Southeast Asia, and the Caribbean.1 The Table shows the global distribution of dengue serotypes from 2000 to 2014.3,4 There are 4 serotypes of the dengue virus: DENV-1 to DENV-4. Infection with 1 strain elicits longlasting immunity to that strain, but subsequent infection with another strain can result in severe DHF due to antibody cross-reaction.1
Dengue virus infection ranges from mildly symptomatic to a spectrum of increasingly severe conditions that comprise dengue fever (DF) and DHF, as well as dengue shock syndrome and brain stem hemorrhage, which may be fatal.2,5 Dengue fever manifests as severe myalgia, fever, headache (usually retro-orbital), arthralgia, erythema, and rubelliform exanthema.6 The frequency of skin eruptions in patients with DF varies with the virus strain and outbreaks.7 The lesions initially develop with the onset of fever and manifest as flushing or erythematous mottling of the face, neck, and chest areas.1,7 The morbilliform eruption develops 2 to 6 days after the onset of the fever, beginning on the trunk and spreading to the face and extremities.1,7 The rash may become confluent with characteristic sparing of small round areas of normal skin described as white islands in a sea of red.2 Verrucous papules on the ears also have been described and may resemble those seen in Cowden syndrome. In patients with prior infection with a different strain of the virus, hemorrhagic lesions may develop, including characteristic retiform purpura, a positive tourniquet test, and the appearance of petechiae on the lower legs. Pruritus and desquamation, especially on the palms and soles, may follow the termination of the eruption.7
The differential diagnosis of DF includes measles, rubella, enteroviruses, and influenza. Chikungunya and West Nile viruses in Asia and Africa and the O’nyong-nyong virus in Africa are also arboviruses that cause a clinical picture similar to DF but not DHF. Other diagnostic considerations include phases of scarlet fever, typhoid, malaria, leptospirosis, hepatitis A, and trypanosomal and rickettsial diseases.7 The differential diagnosis of DHF includes antineutrophil cytoplasmic antibody–associated vasculitis, rheumatoid vasculitis, and bacterial septic vasculitis.
Acute clinical diagnosis of DF can be challenging because of the nonspecific symptoms that can be seen in almost every infectious disease. Clinical presentation assessment should be confirmed with laboratory testing.6 Dengue virus infection usually is confirmed by the identification of viral genomic RNA, antigens, or the antibodies it elicits. Enzyme-linked immunosorbent assay–based serologic tests are cost-effective and easy to perform.5 IgM antibodies usually show cross-reactivity with platelets, but the antibody levels are not positively correlated with the severity of DF.8 Primary infection with the dengue virus is characterized by the elevation of specific IgM levels that usually occurs 3 to 5 days after symptom onset and persists during the postfebrile stage (up to 30 to 60 days). In secondary infections, the IgM levels usually rise more slowly and reach a lower level than in primary infections.9 For both primary and secondary infections, testing IgM levels after the febrile stage may be helpful with the laboratory diagnosis.
Currently, there is no antiviral drug available for dengue. Treatment of dengue infection is symptomatic and supportive.2
Dengue hemorrhagic fever is indicated by a rising hematocrit (≥20%) and a falling platelet count (>100,000/mm3) accompanying clinical signs of hemorrhage. Treatment includes intravenous fluid replacement and careful clinical monitoring of hematocrit levels, platelet count, vitals, urine output, and other signs of shock.5 For patients with a history of dengue infection, travel to areas with other serotypes is not recommended.
If any travel to a high-risk area is planned, countryspecific travel recommendations and warnings should be reviewed from the Centers for Disease Control and Prevention’s website (https://wwwnc.cdc.gov/travel/notices/level1/dengue-global). Use of an Environmental Protection Agency–registered insect repellent to avoid mosquito bites and acetaminophen for managing symptoms is advised. During travel, staying in places with window and door screens and using a bed net during sleep are suggested. Long-sleeved shirts and long pants also are preferred. Travelers should see a health care provider if they have symptoms of dengue.10
African tick bite fever (ATBF) is caused by Rickettsia africae transmitted by Amblyomma ticks. Skin findings in ATBF include erythematous, firm, tender papules with central eschars consistent with the feeding patterns of ticks.11 Histopathology of ATBF usually includes fibrinoid necrosis of vessels in the dermis with a perivascular inflammatory infiltrate and coagulation necrosis of the surrounding dermis consistent with eschar formation.12 The lack of an eschar weighs against this diagnosis.
African trypanosomiasis (also known as sleeping sickness) is caused by protozoa transmitted by the tsetse fly. A chancrelike, circumscribed, rubbery, indurated red or violaceous nodule measuring 2 to 5 cm in diameter often develops as the earliest cutaneous sign of the disease.13 Nonspecific histopathologic findings, such as infiltration of lymphocytes and macrophages and proliferation of endothelial cells and fibroblasts, may be observed.14 Extravascular parasites have been noted in skin biopsies.15 In later stages, skin lesions called trypanids may be observed as macular, papular, annular, targetoid, purpuric, and erythematous lesions, and histopathologic findings consistent with vasculitis also may be seen.13
Chikungunya virus infection is an acute-onset, mosquito-borne viral disease. Skin manifestations may start with nonspecific, generalized, morbilliform, maculopapular rashes coinciding with fever, which also may be seen initially with DHF. Skin hyperpigmentation, mostly centrofacial and involving the nose (chik sign); purpuric and ecchymotic lesions over the trunk and flexors of limbs in adults, often surmounted by subepidermal bullae and lesions resembling toxic epidermal necrolysis; and nonhealing ulcers in the genital and groin areas are common skin manifestations of chikungunya infection.16 Intraepithelial splitting with acantholysis and perivascular lymphohistiocytic infiltration may be observed in the histopathology of blistering lesions, which are not consistent with DHF.17
Zika virus infection is caused by an arbovirus within the Flaviviridae family, which also includes the dengue virus. Initial mucocutaneous findings of the Zika virus include nonspecific diffuse maculopapular eruptions. The eruption generally spares the palms and soles; however, various manifestations including involvement of the palms and soles have been reported.18 The morbilliform eruption begins on the face and extends to the trunk and extremities. Mild hemorrhagic manifestations, including petechiae and bleeding gums, may be observed. Distinguishing between dengue and Zika virus infection relies on the severity of symptoms and laboratory tests, including polymerase chain reaction or IgM antibody testing.19 The other conditions listed do not produce hemorrhagic fever.
THE DIAGNOSIS: Dengue Hemorrhagic Fever
The retiform purpura observed in our patient was suggestive of a vasculitic, thrombotic, or embolic etiology. Dengue IgM serologic testing performed based on her extensive travel history and recent return from a dengue-endemic area was positive, indicating acute infection. A clinical diagnosis of dengue hemorrhagic fever (DHF) was made based on the hemorrhagic appearance of the lesion. Histopathology revealed leukocytoclastic vasculitis (Figure). Anti–double-stranded DNA, antideoxyribonuclease, C3 and C4, CH50 (total hemolytic complement), antineutrophil cytoplasmic antibodies, HIV, and hepatitis B virus tests were normal. Direct immunofluorescence was negative.
Dengue virus is a single-stranded RNA virus transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is one of the most prevalent arthropod-borne viruses affecting humans today.1,2 Infection with the dengue virus generally is seen in travelers visiting tropical regions of Africa, Mexico, South America, South and Central Asia, Southeast Asia, and the Caribbean.1 The Table shows the global distribution of dengue serotypes from 2000 to 2014.3,4 There are 4 serotypes of the dengue virus: DENV-1 to DENV-4. Infection with 1 strain elicits longlasting immunity to that strain, but subsequent infection with another strain can result in severe DHF due to antibody cross-reaction.1
Dengue virus infection ranges from mildly symptomatic to a spectrum of increasingly severe conditions that comprise dengue fever (DF) and DHF, as well as dengue shock syndrome and brain stem hemorrhage, which may be fatal.2,5 Dengue fever manifests as severe myalgia, fever, headache (usually retro-orbital), arthralgia, erythema, and rubelliform exanthema.6 The frequency of skin eruptions in patients with DF varies with the virus strain and outbreaks.7 The lesions initially develop with the onset of fever and manifest as flushing or erythematous mottling of the face, neck, and chest areas.1,7 The morbilliform eruption develops 2 to 6 days after the onset of the fever, beginning on the trunk and spreading to the face and extremities.1,7 The rash may become confluent with characteristic sparing of small round areas of normal skin described as white islands in a sea of red.2 Verrucous papules on the ears also have been described and may resemble those seen in Cowden syndrome. In patients with prior infection with a different strain of the virus, hemorrhagic lesions may develop, including characteristic retiform purpura, a positive tourniquet test, and the appearance of petechiae on the lower legs. Pruritus and desquamation, especially on the palms and soles, may follow the termination of the eruption.7
The differential diagnosis of DF includes measles, rubella, enteroviruses, and influenza. Chikungunya and West Nile viruses in Asia and Africa and the O’nyong-nyong virus in Africa are also arboviruses that cause a clinical picture similar to DF but not DHF. Other diagnostic considerations include phases of scarlet fever, typhoid, malaria, leptospirosis, hepatitis A, and trypanosomal and rickettsial diseases.7 The differential diagnosis of DHF includes antineutrophil cytoplasmic antibody–associated vasculitis, rheumatoid vasculitis, and bacterial septic vasculitis.
Acute clinical diagnosis of DF can be challenging because of the nonspecific symptoms that can be seen in almost every infectious disease. Clinical presentation assessment should be confirmed with laboratory testing.6 Dengue virus infection usually is confirmed by the identification of viral genomic RNA, antigens, or the antibodies it elicits. Enzyme-linked immunosorbent assay–based serologic tests are cost-effective and easy to perform.5 IgM antibodies usually show cross-reactivity with platelets, but the antibody levels are not positively correlated with the severity of DF.8 Primary infection with the dengue virus is characterized by the elevation of specific IgM levels that usually occurs 3 to 5 days after symptom onset and persists during the postfebrile stage (up to 30 to 60 days). In secondary infections, the IgM levels usually rise more slowly and reach a lower level than in primary infections.9 For both primary and secondary infections, testing IgM levels after the febrile stage may be helpful with the laboratory diagnosis.
Currently, there is no antiviral drug available for dengue. Treatment of dengue infection is symptomatic and supportive.2
Dengue hemorrhagic fever is indicated by a rising hematocrit (≥20%) and a falling platelet count (>100,000/mm3) accompanying clinical signs of hemorrhage. Treatment includes intravenous fluid replacement and careful clinical monitoring of hematocrit levels, platelet count, vitals, urine output, and other signs of shock.5 For patients with a history of dengue infection, travel to areas with other serotypes is not recommended.
If any travel to a high-risk area is planned, countryspecific travel recommendations and warnings should be reviewed from the Centers for Disease Control and Prevention’s website (https://wwwnc.cdc.gov/travel/notices/level1/dengue-global). Use of an Environmental Protection Agency–registered insect repellent to avoid mosquito bites and acetaminophen for managing symptoms is advised. During travel, staying in places with window and door screens and using a bed net during sleep are suggested. Long-sleeved shirts and long pants also are preferred. Travelers should see a health care provider if they have symptoms of dengue.10
African tick bite fever (ATBF) is caused by Rickettsia africae transmitted by Amblyomma ticks. Skin findings in ATBF include erythematous, firm, tender papules with central eschars consistent with the feeding patterns of ticks.11 Histopathology of ATBF usually includes fibrinoid necrosis of vessels in the dermis with a perivascular inflammatory infiltrate and coagulation necrosis of the surrounding dermis consistent with eschar formation.12 The lack of an eschar weighs against this diagnosis.
African trypanosomiasis (also known as sleeping sickness) is caused by protozoa transmitted by the tsetse fly. A chancrelike, circumscribed, rubbery, indurated red or violaceous nodule measuring 2 to 5 cm in diameter often develops as the earliest cutaneous sign of the disease.13 Nonspecific histopathologic findings, such as infiltration of lymphocytes and macrophages and proliferation of endothelial cells and fibroblasts, may be observed.14 Extravascular parasites have been noted in skin biopsies.15 In later stages, skin lesions called trypanids may be observed as macular, papular, annular, targetoid, purpuric, and erythematous lesions, and histopathologic findings consistent with vasculitis also may be seen.13
Chikungunya virus infection is an acute-onset, mosquito-borne viral disease. Skin manifestations may start with nonspecific, generalized, morbilliform, maculopapular rashes coinciding with fever, which also may be seen initially with DHF. Skin hyperpigmentation, mostly centrofacial and involving the nose (chik sign); purpuric and ecchymotic lesions over the trunk and flexors of limbs in adults, often surmounted by subepidermal bullae and lesions resembling toxic epidermal necrolysis; and nonhealing ulcers in the genital and groin areas are common skin manifestations of chikungunya infection.16 Intraepithelial splitting with acantholysis and perivascular lymphohistiocytic infiltration may be observed in the histopathology of blistering lesions, which are not consistent with DHF.17
Zika virus infection is caused by an arbovirus within the Flaviviridae family, which also includes the dengue virus. Initial mucocutaneous findings of the Zika virus include nonspecific diffuse maculopapular eruptions. The eruption generally spares the palms and soles; however, various manifestations including involvement of the palms and soles have been reported.18 The morbilliform eruption begins on the face and extends to the trunk and extremities. Mild hemorrhagic manifestations, including petechiae and bleeding gums, may be observed. Distinguishing between dengue and Zika virus infection relies on the severity of symptoms and laboratory tests, including polymerase chain reaction or IgM antibody testing.19 The other conditions listed do not produce hemorrhagic fever.
- Pincus LB, Grossman ME, Fox LP. The exanthem of dengue fever: clinical features of two US tourists traveling abroad. J Am Acad Dermatol. 2008;58:308-316. doi:10.1016/j.jaad.2007.08.042
- Radakovic-Fijan S, Graninger W, Müller C, et al. Dengue hemorrhagic fever in a British travel guide. J Am Acad Dermatol. 2002;46:430-433. doi:10.1067/mjd.2002.111904
- Yamashita A, Sakamoto T, Sekizuka T, et al. DGV: dengue genographic viewer. Front Microbiol. 2016;7:875. doi:10.3389/fmicb.2016.00875
- Centers for Disease and Prevention. Dengue in the US states and territories. Updated October 7, 2020. Accessed September 30, 2024. https://www.cdc.gov/dengue/data-research/facts-stats/?CDC_AAref_Val=https://www.cdc.gov/dengue/areaswithrisk/in-the-us.html
- Khetarpal N, Khanna I. Dengue fever: causes, complications, and vaccine strategies. J Immunol Res. 2016;2016:6803098. doi:10.1155/2016/6803098
- Muller DA, Depelsenaire AC, Young PR. Clinical and laboratory diagnosis of dengue virus infection. J Infect Dis. 2017;215(suppl 2):S89-S95. doi:10.1093/infdis/jiw649
- Waterman SH, Gubler DJ. Dengue fever. Clin Dermatol. 1989;7:117-122. doi:10.1016/0738-081x(89)90034-5
- Lin CF, Lei HY, Liu CC, et al. Generation of IgM anti-platelet autoantibody in dengue patients. J Med Virol. 2001;63:143-149. doi:10.1002/1096- 9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L
- Tripathi NK, Shrivastava A, Dash PK, et al. Detection of dengue virus. Methods Mol Biol. 2011;665:51-64. doi:10.1007/978-1-60761-817-1_4
- Centers for Disease Control and Prevention. Plan for travel. Accessed September 30, 2024. https://wwwnc.cdc.gov/travel
- Mack I, Ritz N. African tick-bite fever. N Engl J Med. 2019;380:960. doi:10.1056/NEJMicm1810093
- Lepidi H, Fournier PE, Raoult D. Histologic features and immunodetection of African tick-bite fever eschar. Emerg Infect Dis. 2006;12:1332- 1337. doi:10.3201/eid1209.051540
- McGovern TW, Williams W, Fitzpatrick JE, et al. Cutaneous manifestations of African trypanosomiasis. Arch Dermatol. 1995;131:1178-1182.
- Kristensson K, Bentivoglio M. Pathology of African trypanosomiasis. In: Dumas M, Bouteille B, Buguet A, eds. Progress in Human African Trypanosomiasis, Sleeping Sickness. Springer; 1999:157-181.
- Capewell P, Cren-Travaillé C, Marchesi F, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5:e17716. doi:10.7554/eLife.17716
- Singal A. Chikungunya and skin: current perspective. Indian Dermatol Online J. 2017;8:307-309. doi:10.4103/idoj.IDOJ_93_17
- Robin S, Ramful D, Zettor J, et al. Severe bullous skin lesions associated with chikungunya virus infection in small infants. Eur J Pediatr. 2009;169:67-72. doi:10.1007/s00431-009-0986-0
- Hussain A, Ali F, Latiwesh OB, et al. A comprehensive review of the manifestations and pathogenesis of Zika virus in neonates and adults. Cureus. 2018;10:E3290. doi:10.7759/cureus.3290
- Farahnik B, Beroukhim K, Blattner CM, et al. Cutaneous manifestations of the Zika virus. J Am Acad Dermatol. 2016;74:1286-1287. doi:10.1016/j.jaad.2016.02.1232
- Pincus LB, Grossman ME, Fox LP. The exanthem of dengue fever: clinical features of two US tourists traveling abroad. J Am Acad Dermatol. 2008;58:308-316. doi:10.1016/j.jaad.2007.08.042
- Radakovic-Fijan S, Graninger W, Müller C, et al. Dengue hemorrhagic fever in a British travel guide. J Am Acad Dermatol. 2002;46:430-433. doi:10.1067/mjd.2002.111904
- Yamashita A, Sakamoto T, Sekizuka T, et al. DGV: dengue genographic viewer. Front Microbiol. 2016;7:875. doi:10.3389/fmicb.2016.00875
- Centers for Disease and Prevention. Dengue in the US states and territories. Updated October 7, 2020. Accessed September 30, 2024. https://www.cdc.gov/dengue/data-research/facts-stats/?CDC_AAref_Val=https://www.cdc.gov/dengue/areaswithrisk/in-the-us.html
- Khetarpal N, Khanna I. Dengue fever: causes, complications, and vaccine strategies. J Immunol Res. 2016;2016:6803098. doi:10.1155/2016/6803098
- Muller DA, Depelsenaire AC, Young PR. Clinical and laboratory diagnosis of dengue virus infection. J Infect Dis. 2017;215(suppl 2):S89-S95. doi:10.1093/infdis/jiw649
- Waterman SH, Gubler DJ. Dengue fever. Clin Dermatol. 1989;7:117-122. doi:10.1016/0738-081x(89)90034-5
- Lin CF, Lei HY, Liu CC, et al. Generation of IgM anti-platelet autoantibody in dengue patients. J Med Virol. 2001;63:143-149. doi:10.1002/1096- 9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L
- Tripathi NK, Shrivastava A, Dash PK, et al. Detection of dengue virus. Methods Mol Biol. 2011;665:51-64. doi:10.1007/978-1-60761-817-1_4
- Centers for Disease Control and Prevention. Plan for travel. Accessed September 30, 2024. https://wwwnc.cdc.gov/travel
- Mack I, Ritz N. African tick-bite fever. N Engl J Med. 2019;380:960. doi:10.1056/NEJMicm1810093
- Lepidi H, Fournier PE, Raoult D. Histologic features and immunodetection of African tick-bite fever eschar. Emerg Infect Dis. 2006;12:1332- 1337. doi:10.3201/eid1209.051540
- McGovern TW, Williams W, Fitzpatrick JE, et al. Cutaneous manifestations of African trypanosomiasis. Arch Dermatol. 1995;131:1178-1182.
- Kristensson K, Bentivoglio M. Pathology of African trypanosomiasis. In: Dumas M, Bouteille B, Buguet A, eds. Progress in Human African Trypanosomiasis, Sleeping Sickness. Springer; 1999:157-181.
- Capewell P, Cren-Travaillé C, Marchesi F, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5:e17716. doi:10.7554/eLife.17716
- Singal A. Chikungunya and skin: current perspective. Indian Dermatol Online J. 2017;8:307-309. doi:10.4103/idoj.IDOJ_93_17
- Robin S, Ramful D, Zettor J, et al. Severe bullous skin lesions associated with chikungunya virus infection in small infants. Eur J Pediatr. 2009;169:67-72. doi:10.1007/s00431-009-0986-0
- Hussain A, Ali F, Latiwesh OB, et al. A comprehensive review of the manifestations and pathogenesis of Zika virus in neonates and adults. Cureus. 2018;10:E3290. doi:10.7759/cureus.3290
- Farahnik B, Beroukhim K, Blattner CM, et al. Cutaneous manifestations of the Zika virus. J Am Acad Dermatol. 2016;74:1286-1287. doi:10.1016/j.jaad.2016.02.1232
A 74-year-old woman who frequently traveled abroad presented to the dermatology department with retiform purpura of the lower leg along with gastrointestinal cramps, fatigue, and myalgia. The patient reported that the symptoms had started 10 days after returning from a recent trip to Africa.
Inspection of Deep Tumor Margins for Accurate Cutaneous Squamous Cell Carcinoma Staging
To the Editor:
Histopathologic analysis of debulk specimens in Mohs micrographic surgery (MMS) may augment identification of high-risk factors in cutaneous squamous cell carcinoma (cSCC), which may warrant tumor upstaging.1 Intratumor location has not been studied when looking at these high-risk factors. Herein, we report 4 cSCCs initially categorized as well differentiated that were reclassified as moderate to poorly differentiated on analysis of debulk specimens obtained via shave removal.
An 80-year-old man (patient 1) presented with a tender 2-cm erythematous plaque with dried hemorrhagic crusting on the frontal scalp. He had a history of nonmelanoma skin cancers. A biopsy revealed a well-differentiated cSCC, which was upgraded from a T2a tumor to T2b during MMS due to galea involvement. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells at the deep margin (Figure 1A). Given T2b staging, baseline imaging and radiation therapy were recommended.
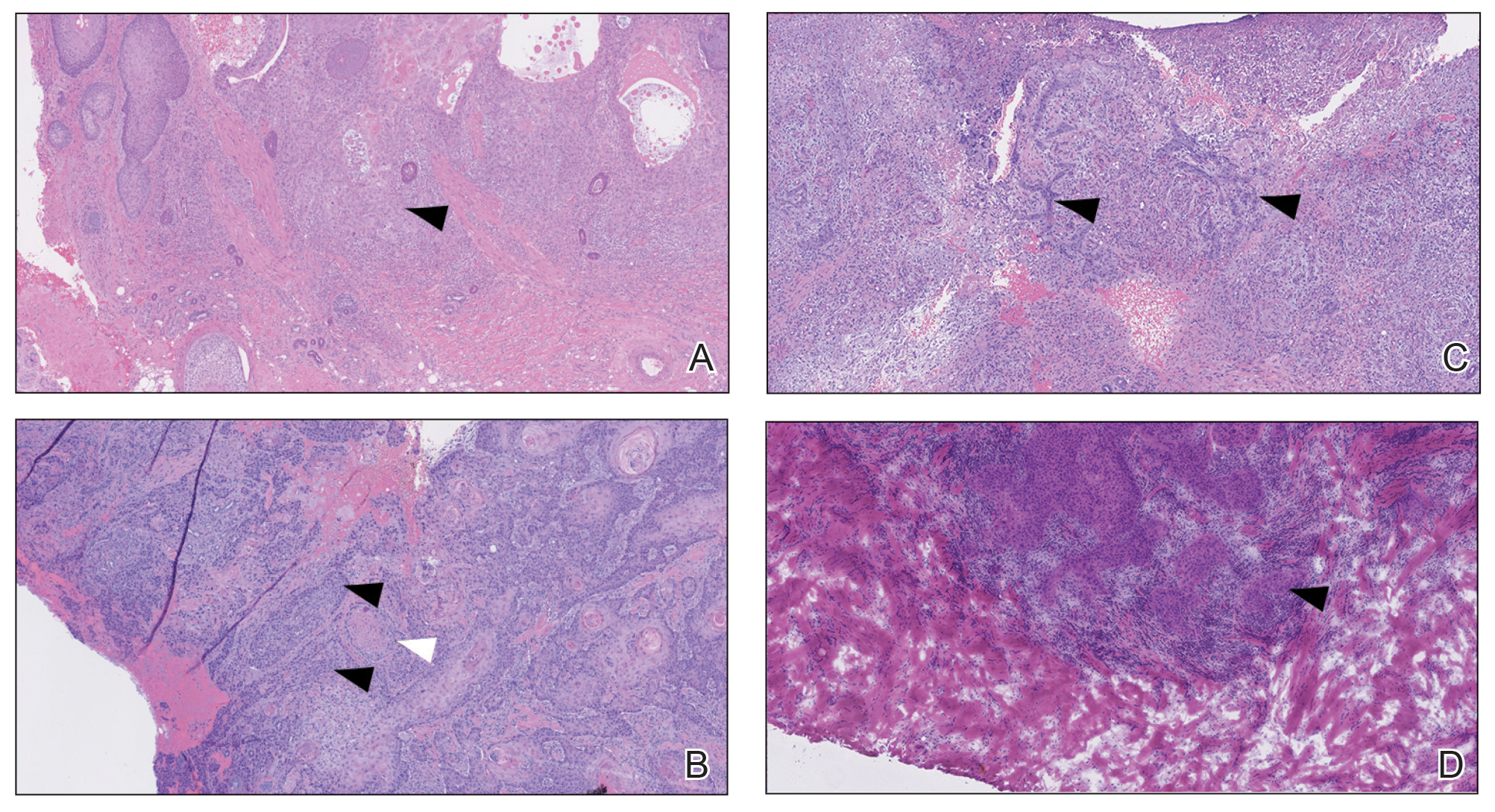
A 75-year-old man (patient 2) presented with a 2-cm erythematous plaque on the left vertex scalp with hemorrhagic crusting, yellow scale, and purulent drainage. He had a history of cSCCs. A biopsy revealed well-differentiated invasive cSCC, which was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Examination of the second Mohs stage revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, infiltration beyond the subcutaneous fat, and perineural invasion (Figure 1B). Given T2b staging, baseline imaging and radiation therapy were recommended.
An 86-year-old woman (patient 3) presented with a tender 2.4-cm plum-colored nodule on the right lower leg. She had a history of basal cell carcinoma. A biopsy revealed a well-differentiated invasive cSCC staged at T2a. Debulk analysis revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, though the staging remained the same (Figure 1C).
An 82-year-old man (patient 4) presented with a 2.7-cm ulcerated nodule with adjacent scaling on the vertex scalp. He had no history of skin cancer. A biopsy revealed a well-differentiated cSCC (Figure 2) that was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells with single-cell extension at the deep margin in the galea (Figure 1D). Given T2b staging, baseline imaging and radiation therapy were recommended.
Tumor differentiation is a factor included in the Brigham and Women’s Hospital staging system, and intratumor variability can be clinically relevant for tumor staging.1 Specifically, cSCCs may exhibit intratumor heterogeneity in which predominantly well-differentiated tumors contain focal areas of poorer differentiation.2 This intratumor heterogeneity complicates estimation of tumor risk, as a well-differentiated tumor on biopsy may exhibit poor differentiation at a deeper margin. Our cases highlight that the cells at the deeper margin indeed can show poorer differentiation or other higher-risk tumor features. Thus, the most clinically relevant cells for tumor staging and prognostication may not be visible on initial biopsy, underscoring the utility of close examination of the deep layer of the debulk specimen and Mohs layer for comprehensive staging.
Genetic studies have attempted to identify gene expression patterns in cSCCs that predispose to invasion.3 Three of the top 6 genes in this “invasion signature gene set” were matrix metalloproteases; additionally, IL-24 messenger RNA was upregulated in both the cSCC invasion front and in situ cSCCs. IL-24 has been shown to upregulate the expression of matrix metalloprotease 7 in vitro, suggesting that it may influence tumor progression.3 Although gene expression was not included in this series, the identification of genetic variability in the most poorly differentiated cells residing in the deep margins is of great interest and may reveal mutations contributing to irregular cell morphology and cSCC invasiveness.
Prior studies have indicated that a proportion of cSCCs are histopathologically upgraded from the initial biopsy during MMS due to evidence of perineural invasion, bony invasion, or lesser differentiation noted during MMS stages or debulk analysis.1,4 However, the majority of Mohs surgeons report immediately discarding debulk specimens without further evaluation.5 Herein, we highlight 4 cSCC cases in which the deep margins of the debulk specimen contained the most dedifferentiated cells. Our findings emphasize the importance of thoroughly examining deep tumor margins for complete staging yet also highlight that identifying cells at these margins may not change patient management when high-risk criteria are already met.
- McIlwee BE, Abidi NY, Ravi M, et al. Utility of debulk specimens during Mohs micrographic surgery for cutaneous squamous cell carcinoma. Dermatol Surg. 2021;47:599-604.
- Ramón y Cajal S, Sesé M, Capdevila C, et al. Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med. 2020;98:161-177.
- Mitsui H, Suárez-Fariñas M, Gulati N, et al. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J Invest Dermatol. 2014;134:1418-1427.
- Chung E, Hoang S, McEvoy AM, et al. Histopathologic upgrading of cutaneous squamous cell carcinomas during Mohs micrographic surgery: a retrospective cohort study. J Am Acad Dermatol. 2021;85:923-930.
- Alniemi DT, Swanson AM, Lasarev M, et al. Tumor debulking trends for keratinocyte carcinomas among Mohs surgeons. Dermatol Surg. 2021;47:1660-1661.
To the Editor:
Histopathologic analysis of debulk specimens in Mohs micrographic surgery (MMS) may augment identification of high-risk factors in cutaneous squamous cell carcinoma (cSCC), which may warrant tumor upstaging.1 Intratumor location has not been studied when looking at these high-risk factors. Herein, we report 4 cSCCs initially categorized as well differentiated that were reclassified as moderate to poorly differentiated on analysis of debulk specimens obtained via shave removal.
An 80-year-old man (patient 1) presented with a tender 2-cm erythematous plaque with dried hemorrhagic crusting on the frontal scalp. He had a history of nonmelanoma skin cancers. A biopsy revealed a well-differentiated cSCC, which was upgraded from a T2a tumor to T2b during MMS due to galea involvement. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells at the deep margin (Figure 1A). Given T2b staging, baseline imaging and radiation therapy were recommended.
A 75-year-old man (patient 2) presented with a 2-cm erythematous plaque on the left vertex scalp with hemorrhagic crusting, yellow scale, and purulent drainage. He had a history of cSCCs. A biopsy revealed well-differentiated invasive cSCC, which was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Examination of the second Mohs stage revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, infiltration beyond the subcutaneous fat, and perineural invasion (Figure 1B). Given T2b staging, baseline imaging and radiation therapy were recommended.
An 86-year-old woman (patient 3) presented with a tender 2.4-cm plum-colored nodule on the right lower leg. She had a history of basal cell carcinoma. A biopsy revealed a well-differentiated invasive cSCC staged at T2a. Debulk analysis revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, though the staging remained the same (Figure 1C).
An 82-year-old man (patient 4) presented with a 2.7-cm ulcerated nodule with adjacent scaling on the vertex scalp. He had no history of skin cancer. A biopsy revealed a well-differentiated cSCC (Figure 2) that was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells with single-cell extension at the deep margin in the galea (Figure 1D). Given T2b staging, baseline imaging and radiation therapy were recommended.
Tumor differentiation is a factor included in the Brigham and Women’s Hospital staging system, and intratumor variability can be clinically relevant for tumor staging.1 Specifically, cSCCs may exhibit intratumor heterogeneity in which predominantly well-differentiated tumors contain focal areas of poorer differentiation.2 This intratumor heterogeneity complicates estimation of tumor risk, as a well-differentiated tumor on biopsy may exhibit poor differentiation at a deeper margin. Our cases highlight that the cells at the deeper margin indeed can show poorer differentiation or other higher-risk tumor features. Thus, the most clinically relevant cells for tumor staging and prognostication may not be visible on initial biopsy, underscoring the utility of close examination of the deep layer of the debulk specimen and Mohs layer for comprehensive staging.
Genetic studies have attempted to identify gene expression patterns in cSCCs that predispose to invasion.3 Three of the top 6 genes in this “invasion signature gene set” were matrix metalloproteases; additionally, IL-24 messenger RNA was upregulated in both the cSCC invasion front and in situ cSCCs. IL-24 has been shown to upregulate the expression of matrix metalloprotease 7 in vitro, suggesting that it may influence tumor progression.3 Although gene expression was not included in this series, the identification of genetic variability in the most poorly differentiated cells residing in the deep margins is of great interest and may reveal mutations contributing to irregular cell morphology and cSCC invasiveness.
Prior studies have indicated that a proportion of cSCCs are histopathologically upgraded from the initial biopsy during MMS due to evidence of perineural invasion, bony invasion, or lesser differentiation noted during MMS stages or debulk analysis.1,4 However, the majority of Mohs surgeons report immediately discarding debulk specimens without further evaluation.5 Herein, we highlight 4 cSCC cases in which the deep margins of the debulk specimen contained the most dedifferentiated cells. Our findings emphasize the importance of thoroughly examining deep tumor margins for complete staging yet also highlight that identifying cells at these margins may not change patient management when high-risk criteria are already met.
To the Editor:
Histopathologic analysis of debulk specimens in Mohs micrographic surgery (MMS) may augment identification of high-risk factors in cutaneous squamous cell carcinoma (cSCC), which may warrant tumor upstaging.1 Intratumor location has not been studied when looking at these high-risk factors. Herein, we report 4 cSCCs initially categorized as well differentiated that were reclassified as moderate to poorly differentiated on analysis of debulk specimens obtained via shave removal.
An 80-year-old man (patient 1) presented with a tender 2-cm erythematous plaque with dried hemorrhagic crusting on the frontal scalp. He had a history of nonmelanoma skin cancers. A biopsy revealed a well-differentiated cSCC, which was upgraded from a T2a tumor to T2b during MMS due to galea involvement. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells at the deep margin (Figure 1A). Given T2b staging, baseline imaging and radiation therapy were recommended.
A 75-year-old man (patient 2) presented with a 2-cm erythematous plaque on the left vertex scalp with hemorrhagic crusting, yellow scale, and purulent drainage. He had a history of cSCCs. A biopsy revealed well-differentiated invasive cSCC, which was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Examination of the second Mohs stage revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, infiltration beyond the subcutaneous fat, and perineural invasion (Figure 1B). Given T2b staging, baseline imaging and radiation therapy were recommended.
An 86-year-old woman (patient 3) presented with a tender 2.4-cm plum-colored nodule on the right lower leg. She had a history of basal cell carcinoma. A biopsy revealed a well-differentiated invasive cSCC staged at T2a. Debulk analysis revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, though the staging remained the same (Figure 1C).
An 82-year-old man (patient 4) presented with a 2.7-cm ulcerated nodule with adjacent scaling on the vertex scalp. He had no history of skin cancer. A biopsy revealed a well-differentiated cSCC (Figure 2) that was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells with single-cell extension at the deep margin in the galea (Figure 1D). Given T2b staging, baseline imaging and radiation therapy were recommended.
Tumor differentiation is a factor included in the Brigham and Women’s Hospital staging system, and intratumor variability can be clinically relevant for tumor staging.1 Specifically, cSCCs may exhibit intratumor heterogeneity in which predominantly well-differentiated tumors contain focal areas of poorer differentiation.2 This intratumor heterogeneity complicates estimation of tumor risk, as a well-differentiated tumor on biopsy may exhibit poor differentiation at a deeper margin. Our cases highlight that the cells at the deeper margin indeed can show poorer differentiation or other higher-risk tumor features. Thus, the most clinically relevant cells for tumor staging and prognostication may not be visible on initial biopsy, underscoring the utility of close examination of the deep layer of the debulk specimen and Mohs layer for comprehensive staging.
Genetic studies have attempted to identify gene expression patterns in cSCCs that predispose to invasion.3 Three of the top 6 genes in this “invasion signature gene set” were matrix metalloproteases; additionally, IL-24 messenger RNA was upregulated in both the cSCC invasion front and in situ cSCCs. IL-24 has been shown to upregulate the expression of matrix metalloprotease 7 in vitro, suggesting that it may influence tumor progression.3 Although gene expression was not included in this series, the identification of genetic variability in the most poorly differentiated cells residing in the deep margins is of great interest and may reveal mutations contributing to irregular cell morphology and cSCC invasiveness.
Prior studies have indicated that a proportion of cSCCs are histopathologically upgraded from the initial biopsy during MMS due to evidence of perineural invasion, bony invasion, or lesser differentiation noted during MMS stages or debulk analysis.1,4 However, the majority of Mohs surgeons report immediately discarding debulk specimens without further evaluation.5 Herein, we highlight 4 cSCC cases in which the deep margins of the debulk specimen contained the most dedifferentiated cells. Our findings emphasize the importance of thoroughly examining deep tumor margins for complete staging yet also highlight that identifying cells at these margins may not change patient management when high-risk criteria are already met.
- McIlwee BE, Abidi NY, Ravi M, et al. Utility of debulk specimens during Mohs micrographic surgery for cutaneous squamous cell carcinoma. Dermatol Surg. 2021;47:599-604.
- Ramón y Cajal S, Sesé M, Capdevila C, et al. Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med. 2020;98:161-177.
- Mitsui H, Suárez-Fariñas M, Gulati N, et al. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J Invest Dermatol. 2014;134:1418-1427.
- Chung E, Hoang S, McEvoy AM, et al. Histopathologic upgrading of cutaneous squamous cell carcinomas during Mohs micrographic surgery: a retrospective cohort study. J Am Acad Dermatol. 2021;85:923-930.
- Alniemi DT, Swanson AM, Lasarev M, et al. Tumor debulking trends for keratinocyte carcinomas among Mohs surgeons. Dermatol Surg. 2021;47:1660-1661.
- McIlwee BE, Abidi NY, Ravi M, et al. Utility of debulk specimens during Mohs micrographic surgery for cutaneous squamous cell carcinoma. Dermatol Surg. 2021;47:599-604.
- Ramón y Cajal S, Sesé M, Capdevila C, et al. Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med. 2020;98:161-177.
- Mitsui H, Suárez-Fariñas M, Gulati N, et al. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J Invest Dermatol. 2014;134:1418-1427.
- Chung E, Hoang S, McEvoy AM, et al. Histopathologic upgrading of cutaneous squamous cell carcinomas during Mohs micrographic surgery: a retrospective cohort study. J Am Acad Dermatol. 2021;85:923-930.
- Alniemi DT, Swanson AM, Lasarev M, et al. Tumor debulking trends for keratinocyte carcinomas among Mohs surgeons. Dermatol Surg. 2021;47:1660-1661.
Practice Points
- A proportion of cutaneous squamous cell carcinomas are upgraded from the initial biopsy during Mohs micrographic surgery due to evidence of perineural invasion, bony invasion, or lesser differentiation noted on Mohs stages or debulk analysis.
- Thorough inspection of the deep tumor margins may be required for accurate tumor staging and evaluation of metastatic risk. Cells at the deep margin of the tumor may demonstrate poorer differentiation and/or other higher-risk tumor features than those closer to the surface.
- Tumor staging may be incomplete until the deep margins are assessed to find the most dysplastic and likely clinically relevant cells, which may be missed without evaluation of the debulked tumor.
Transient Eruption of Verrucous Keratoses During Encorafenib Therapy: Adverse Event or Paraneoplastic Phenomenon?
To the Editor:
Mutations of the BRAF protein kinase gene are implicated in a variety of malignancies.1 BRAF mutations in malignancies cause the mitogen-activated protein kinase (MAPK) pathway to become constitutively active, which results in unchecked cellular proliferation,2,3 making the BRAF mutation an attractive target for inhibition with pharmacologic agents to potentially halt cancer growth.4 Vemurafenib—the first selective BRAF inhibitor used in clinical practice—initially was approved by the US Food and Drug Administration in 2011. The approval of dabrafenib followed in 2013 and most recently encorafenib in 2018.5
Although targeted treatment of BRAF-mutated malignancies with BRAF inhibitors has become common, it often is associated with cutaneous adverse events (AEs), such as rash, pruritus, photosensitivity, actinic keratosis, and verrucous keratosis. Some reports demonstrate these events in up to 95% of patients undergoing BRAF inhibitor treatment.6 In several cases the eruption of verrucous keratoses is among the most common cutaneous AEs seen among patients receiving BRAF inhibitor treatment.5-7
In general, lesions can appear days to months after therapy is initiated and may resolve after switching to dual therapy with a MEK inhibitor or with complete cessation of BRAF inhibitor therapy.5,7,8 One case of spontaneous resolution of vemurafenib-associated panniculitis during ongoing BRAF inhibitor therapy has been reported9; however, spontaneous resolution of cutaneous AEs is uncommon. Herein, we describe verrucous keratoses in a patient undergoing treatment with encorafenib that resolved spontaneously despite ongoing BRAF inhibitor therapy.
A 61-year-old woman presented to the emergency department with pain in the right lower quadrant. Computed tomography (CT) of the abdomen and pelvis revealed a large ovarian mass. Subsequent bloodwork revealed elevated carcinoembryonic antigen levels. The patient underwent a hysterectomy, bilateral salpingo-oophorectomy, omentectomy, right hemicolectomy with ileotransverse side-to-side anastomosis, right pelvic lymph node reduction, and complete cytoreduction. Histopathology revealed an adenocarcinoma of the cecum with tumor invasion into the visceral peritoneum and metastases to the left ovary, fallopian tube, and omentum. A BRAF V600E mutation was detected.
Two months after the initial presentation, the patient started her first cycle of chemotherapy with a combination of folinic acid, fluorouracil, and oxaliplatin. She completed 11 cycles of this regimen, then was switched to capecitabine and oxaliplatin for an additional 2 cycles due to insurance concerns. At the end of treatment, there was no evidence of disease on CT, thus the patient was followed with observation. However, she presented 10 months later to the emergency department with abdominal pain, and CT revealed new lesions in the liver that were concerning for potential metastases. She started oral encorafenib 300 mg/d and intravenous cetuximab 500 mg weekly; after 1 week, encorafenib was reduced to 150 mg/d due to nausea and loss of appetite. Within 2 weeks of starting treatment, the patient reported the relatively abrupt appearance of more than 50 small papules across the shoulders and back (Figure 1A). She was referred to dermatology, and shave biopsies of 2 lesions—one from the left anterior thigh, the other from the right posterior shoulder—revealed verrucous keratosis pathology (Figure 2). At this time, encorafenib was increased again to 300 mg/d as the patient had been tolerating the reduced dose. She continued to report the appearance of new lesions for the next 3 months, after which the lesions were stable for approximately 2 months. By 2.5 months after initiation of therapy, the patient had undergone CT demonstrating resolution of the liver lesions. At 5 months of therapy, the patient reported a stable to slightly reduced number of skin lesions but had begun to experience worsening joint pain, and the dosage of encorafenib was reduced to 225 mg/d. At 7 months of therapy, the dosage was further reduced to 150 mg/d due to persistent arthralgia. A follow-up examination at 10 months of therapy showed improvement in the number and size of the verrucous keratoses, and near resolution was seen by 14 months after the initial onset of the lesions (Figure 1B). At 20 months after initial onset, only 1 remaining verrucous keratosis was identified on physical examination and biopsy. The patient had continued a regimen of encorafenib 150 mg/d and weekly intravenous 500 mg cetuximab up to this point. Over the entire time period that the patient was seen, up to 12 lesions located in high-friction areas had become irritated and were treated with cryotherapy, but this contributed only minorly to the patient’s overall presentation.
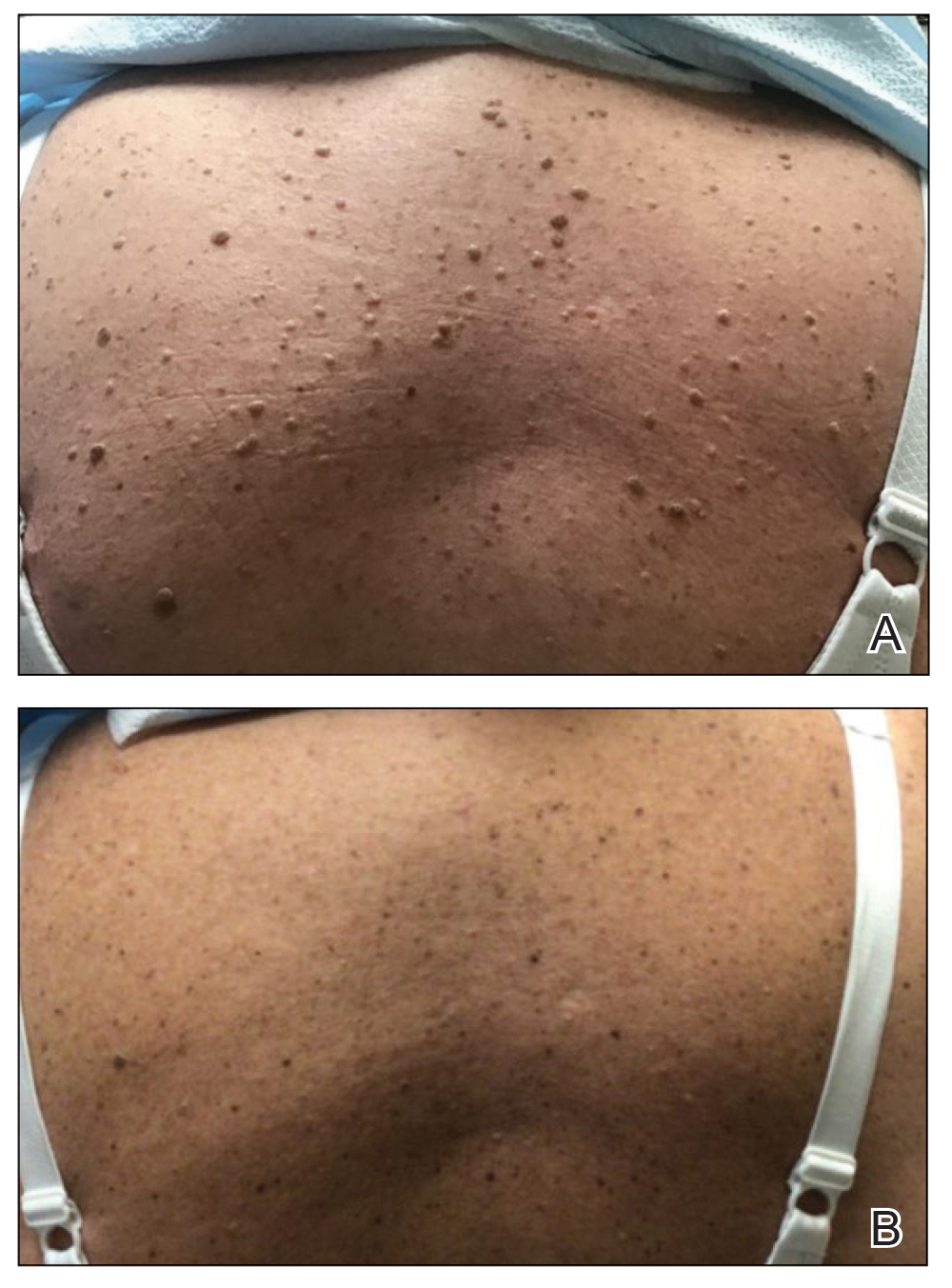
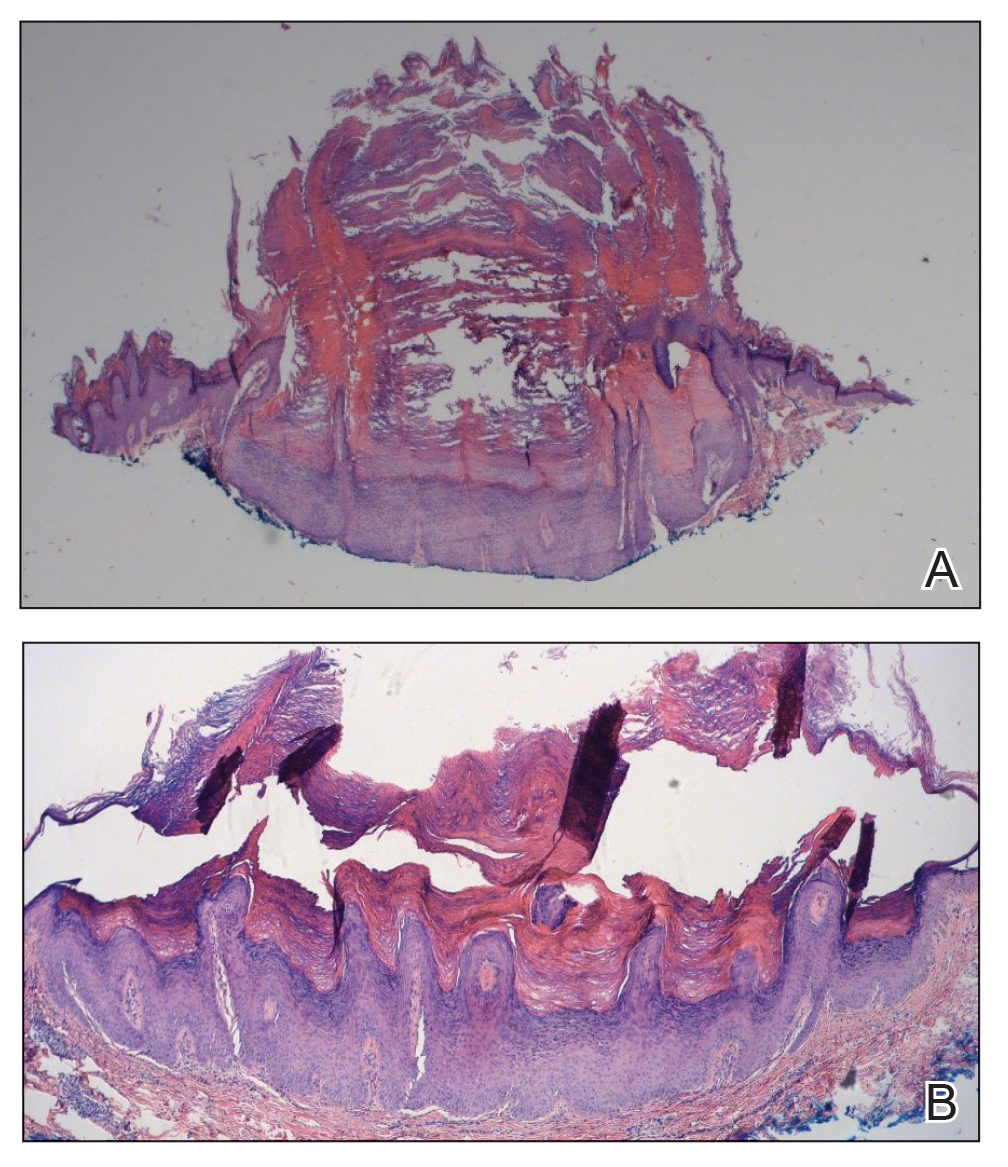
Verrucous keratosis is a known cutaneous AE of BRAF inhibitor treatment with vemurafenib and dabrafenib, with fewer cases attributed to encorafenib.5,6 Within the oncologic setting, the eruption of verrucous papules as a paraneoplastic phenomenon is heavily debated in the literature and is known as the Leser-Trélat sign. This phenomenon is commonly associated with adenocarcinomas of the gastrointestinal tract, as seen in our patient.10 Based on Curth’s postulates—the criteria used to evaluate the relationship between an internal malignancy and a cutaneous disorder—this was unlikely in our patient. The criteria, which do not all need to be met to suggest a paraneoplastic phenomenon, include concurrent onset of the malignancy and the dermatosis, parallel course, association of a specific dermatosis with a specific malignancy, statistical significance of the association, and the presence of a genetic basis for the association.11 Several features favored a drug-related cutaneous eruption vs a paraneoplastic phenomenon: (1) the malignancy was identified months before the cutaneous eruptions manifested; (2) the cutaneous lesions appeared once treatment had already been initiated; and (3) the cutaneous lesions persisted long after the malignancy was no longer identifiable on CT. Indeed, eruption of the papules temporally coincided closely with the initiation of BRAF inhibitor therapy, arguing for correlation.
As a suspected BRAF inhibitor–associated cutaneous AE, the eruption of verrucous keratoses in our patient is remarkable for its spontaneous resolution despite ongoing therapy. It is speculated that keratinocytic proliferation while on BRAF inhibitor therapy may be caused by a paradoxical increase in signaling through CRAF, another Raf isoform that plays a role in the induction of terminal differentiation of keratinocytes, with a subsequent increase in MAPK signaling.12-14 Self-resolution of this cycle despite continuing BRAF inhibitor therapy suggests the possible involvement of balancing and/or alternative mechanistic pathways that may be related to the immune system. Although verrucous keratoses are considered benign proliferations and do not necessarily require any specific treatment or reduction in BRAF inhibitor dosage, they may be treated with cryotherapy, electrocautery, shave removal, or excision,15 which often is done if the lesions become inflamed and cause pain. Additionally, some patients may feel distress from the appearance of the lesions and desire treatment for this reason. Understanding that verrucous keratoses can be a transient cutaneous AE rather than a persistent one may be useful to clinicians as they manage AEs during BRAF inhibitor therapy.
- Pakneshan S, Salajegheh A, Smith RA, Lam AK. Clinicopathological relevance of BRAF mutations in human cancer. Pathology. 2013;45:346-356. doi:10.1097/PAT.0b013e328360b61d
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am. 2009;23:529-545. doi:10.1016/j.hoc.2009.04.001
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246. doi:10.1200/JCO.2010.32.4327
- Ji Z, Flaherty KT, Tsao H. Targeting the RAS pathway in melanoma. Trends Mol Med. 2012;18:27-35. doi:10.1016/j.molmed.2011.08.001
- Gouda MA, Subbiah V. Precision oncology for BRAF-mutant cancers with BRAF and MEK inhibitors: from melanoma to tissue-agnostic therapy. ESMO Open. 2023;8:100788. doi:10.1016/j.esmoop.2023.100788
- Gençler B, Gönül M. Cutaneous side effects of BRAF inhibitors in advanced melanoma: review of the literature. Dermatol Res Pract. 2016;2016:5361569. doi:10.1155/2016/5361569.
- Chu EY, Wanat KA, Miller CJ, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol. 2012;67:1265-1272. doi:10.1016/j.jaad.2012.04.008
- Naqash AR, File DM, Ziemer CM, et al. Cutaneous adverse reactions in B-RAF positive metastatic melanoma following sequential treatment with B-RAF/MEK inhibitors and immune checkpoint blockade or vice versa. a single-institutional case-series. J Immunother Cancer. 2019;7:4. doi:10.1186/s40425-018-0475-y
- Maldonado-Seral C, Berros-Fombella JP, Vivanco-Allende B, et al. Vemurafenib-associated neutrophilic panniculitis: an emergent adverse effect of variable severity. Dermatol Online J. 2013;19:16. doi:10.5070/d370x41670
- Mirali S, Mufti A, Lansang RP, et al. Eruptive seborrheic keratoses are associated with a co-occurring malignancy in the majority of reported cases: a systematic review. J Cutan Med Surg. 2022;26:57-62. doi:10.1177/12034754211035124
- Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin. 2009;59:73-98. doi:10.3322/caac.20005
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435. doi:10.1038/nature08833
- Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209-221. doi:10.1016/j.cell.2009.12.040
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signaling in cells with wild-type BRAF. Nature. 2010;464:427-430. doi:10.1038/nature08902
- Hayat MA. Brain Metastases from Primary Tumors, Volume 3: Epidemiology, Biology, and Therapy of Melanoma and Other Cancers. Academic Press; 2016.
To the Editor:
Mutations of the BRAF protein kinase gene are implicated in a variety of malignancies.1 BRAF mutations in malignancies cause the mitogen-activated protein kinase (MAPK) pathway to become constitutively active, which results in unchecked cellular proliferation,2,3 making the BRAF mutation an attractive target for inhibition with pharmacologic agents to potentially halt cancer growth.4 Vemurafenib—the first selective BRAF inhibitor used in clinical practice—initially was approved by the US Food and Drug Administration in 2011. The approval of dabrafenib followed in 2013 and most recently encorafenib in 2018.5
Although targeted treatment of BRAF-mutated malignancies with BRAF inhibitors has become common, it often is associated with cutaneous adverse events (AEs), such as rash, pruritus, photosensitivity, actinic keratosis, and verrucous keratosis. Some reports demonstrate these events in up to 95% of patients undergoing BRAF inhibitor treatment.6 In several cases the eruption of verrucous keratoses is among the most common cutaneous AEs seen among patients receiving BRAF inhibitor treatment.5-7
In general, lesions can appear days to months after therapy is initiated and may resolve after switching to dual therapy with a MEK inhibitor or with complete cessation of BRAF inhibitor therapy.5,7,8 One case of spontaneous resolution of vemurafenib-associated panniculitis during ongoing BRAF inhibitor therapy has been reported9; however, spontaneous resolution of cutaneous AEs is uncommon. Herein, we describe verrucous keratoses in a patient undergoing treatment with encorafenib that resolved spontaneously despite ongoing BRAF inhibitor therapy.
A 61-year-old woman presented to the emergency department with pain in the right lower quadrant. Computed tomography (CT) of the abdomen and pelvis revealed a large ovarian mass. Subsequent bloodwork revealed elevated carcinoembryonic antigen levels. The patient underwent a hysterectomy, bilateral salpingo-oophorectomy, omentectomy, right hemicolectomy with ileotransverse side-to-side anastomosis, right pelvic lymph node reduction, and complete cytoreduction. Histopathology revealed an adenocarcinoma of the cecum with tumor invasion into the visceral peritoneum and metastases to the left ovary, fallopian tube, and omentum. A BRAF V600E mutation was detected.
Two months after the initial presentation, the patient started her first cycle of chemotherapy with a combination of folinic acid, fluorouracil, and oxaliplatin. She completed 11 cycles of this regimen, then was switched to capecitabine and oxaliplatin for an additional 2 cycles due to insurance concerns. At the end of treatment, there was no evidence of disease on CT, thus the patient was followed with observation. However, she presented 10 months later to the emergency department with abdominal pain, and CT revealed new lesions in the liver that were concerning for potential metastases. She started oral encorafenib 300 mg/d and intravenous cetuximab 500 mg weekly; after 1 week, encorafenib was reduced to 150 mg/d due to nausea and loss of appetite. Within 2 weeks of starting treatment, the patient reported the relatively abrupt appearance of more than 50 small papules across the shoulders and back (Figure 1A). She was referred to dermatology, and shave biopsies of 2 lesions—one from the left anterior thigh, the other from the right posterior shoulder—revealed verrucous keratosis pathology (Figure 2). At this time, encorafenib was increased again to 300 mg/d as the patient had been tolerating the reduced dose. She continued to report the appearance of new lesions for the next 3 months, after which the lesions were stable for approximately 2 months. By 2.5 months after initiation of therapy, the patient had undergone CT demonstrating resolution of the liver lesions. At 5 months of therapy, the patient reported a stable to slightly reduced number of skin lesions but had begun to experience worsening joint pain, and the dosage of encorafenib was reduced to 225 mg/d. At 7 months of therapy, the dosage was further reduced to 150 mg/d due to persistent arthralgia. A follow-up examination at 10 months of therapy showed improvement in the number and size of the verrucous keratoses, and near resolution was seen by 14 months after the initial onset of the lesions (Figure 1B). At 20 months after initial onset, only 1 remaining verrucous keratosis was identified on physical examination and biopsy. The patient had continued a regimen of encorafenib 150 mg/d and weekly intravenous 500 mg cetuximab up to this point. Over the entire time period that the patient was seen, up to 12 lesions located in high-friction areas had become irritated and were treated with cryotherapy, but this contributed only minorly to the patient’s overall presentation.
Verrucous keratosis is a known cutaneous AE of BRAF inhibitor treatment with vemurafenib and dabrafenib, with fewer cases attributed to encorafenib.5,6 Within the oncologic setting, the eruption of verrucous papules as a paraneoplastic phenomenon is heavily debated in the literature and is known as the Leser-Trélat sign. This phenomenon is commonly associated with adenocarcinomas of the gastrointestinal tract, as seen in our patient.10 Based on Curth’s postulates—the criteria used to evaluate the relationship between an internal malignancy and a cutaneous disorder—this was unlikely in our patient. The criteria, which do not all need to be met to suggest a paraneoplastic phenomenon, include concurrent onset of the malignancy and the dermatosis, parallel course, association of a specific dermatosis with a specific malignancy, statistical significance of the association, and the presence of a genetic basis for the association.11 Several features favored a drug-related cutaneous eruption vs a paraneoplastic phenomenon: (1) the malignancy was identified months before the cutaneous eruptions manifested; (2) the cutaneous lesions appeared once treatment had already been initiated; and (3) the cutaneous lesions persisted long after the malignancy was no longer identifiable on CT. Indeed, eruption of the papules temporally coincided closely with the initiation of BRAF inhibitor therapy, arguing for correlation.
As a suspected BRAF inhibitor–associated cutaneous AE, the eruption of verrucous keratoses in our patient is remarkable for its spontaneous resolution despite ongoing therapy. It is speculated that keratinocytic proliferation while on BRAF inhibitor therapy may be caused by a paradoxical increase in signaling through CRAF, another Raf isoform that plays a role in the induction of terminal differentiation of keratinocytes, with a subsequent increase in MAPK signaling.12-14 Self-resolution of this cycle despite continuing BRAF inhibitor therapy suggests the possible involvement of balancing and/or alternative mechanistic pathways that may be related to the immune system. Although verrucous keratoses are considered benign proliferations and do not necessarily require any specific treatment or reduction in BRAF inhibitor dosage, they may be treated with cryotherapy, electrocautery, shave removal, or excision,15 which often is done if the lesions become inflamed and cause pain. Additionally, some patients may feel distress from the appearance of the lesions and desire treatment for this reason. Understanding that verrucous keratoses can be a transient cutaneous AE rather than a persistent one may be useful to clinicians as they manage AEs during BRAF inhibitor therapy.
To the Editor:
Mutations of the BRAF protein kinase gene are implicated in a variety of malignancies.1 BRAF mutations in malignancies cause the mitogen-activated protein kinase (MAPK) pathway to become constitutively active, which results in unchecked cellular proliferation,2,3 making the BRAF mutation an attractive target for inhibition with pharmacologic agents to potentially halt cancer growth.4 Vemurafenib—the first selective BRAF inhibitor used in clinical practice—initially was approved by the US Food and Drug Administration in 2011. The approval of dabrafenib followed in 2013 and most recently encorafenib in 2018.5
Although targeted treatment of BRAF-mutated malignancies with BRAF inhibitors has become common, it often is associated with cutaneous adverse events (AEs), such as rash, pruritus, photosensitivity, actinic keratosis, and verrucous keratosis. Some reports demonstrate these events in up to 95% of patients undergoing BRAF inhibitor treatment.6 In several cases the eruption of verrucous keratoses is among the most common cutaneous AEs seen among patients receiving BRAF inhibitor treatment.5-7
In general, lesions can appear days to months after therapy is initiated and may resolve after switching to dual therapy with a MEK inhibitor or with complete cessation of BRAF inhibitor therapy.5,7,8 One case of spontaneous resolution of vemurafenib-associated panniculitis during ongoing BRAF inhibitor therapy has been reported9; however, spontaneous resolution of cutaneous AEs is uncommon. Herein, we describe verrucous keratoses in a patient undergoing treatment with encorafenib that resolved spontaneously despite ongoing BRAF inhibitor therapy.
A 61-year-old woman presented to the emergency department with pain in the right lower quadrant. Computed tomography (CT) of the abdomen and pelvis revealed a large ovarian mass. Subsequent bloodwork revealed elevated carcinoembryonic antigen levels. The patient underwent a hysterectomy, bilateral salpingo-oophorectomy, omentectomy, right hemicolectomy with ileotransverse side-to-side anastomosis, right pelvic lymph node reduction, and complete cytoreduction. Histopathology revealed an adenocarcinoma of the cecum with tumor invasion into the visceral peritoneum and metastases to the left ovary, fallopian tube, and omentum. A BRAF V600E mutation was detected.
Two months after the initial presentation, the patient started her first cycle of chemotherapy with a combination of folinic acid, fluorouracil, and oxaliplatin. She completed 11 cycles of this regimen, then was switched to capecitabine and oxaliplatin for an additional 2 cycles due to insurance concerns. At the end of treatment, there was no evidence of disease on CT, thus the patient was followed with observation. However, she presented 10 months later to the emergency department with abdominal pain, and CT revealed new lesions in the liver that were concerning for potential metastases. She started oral encorafenib 300 mg/d and intravenous cetuximab 500 mg weekly; after 1 week, encorafenib was reduced to 150 mg/d due to nausea and loss of appetite. Within 2 weeks of starting treatment, the patient reported the relatively abrupt appearance of more than 50 small papules across the shoulders and back (Figure 1A). She was referred to dermatology, and shave biopsies of 2 lesions—one from the left anterior thigh, the other from the right posterior shoulder—revealed verrucous keratosis pathology (Figure 2). At this time, encorafenib was increased again to 300 mg/d as the patient had been tolerating the reduced dose. She continued to report the appearance of new lesions for the next 3 months, after which the lesions were stable for approximately 2 months. By 2.5 months after initiation of therapy, the patient had undergone CT demonstrating resolution of the liver lesions. At 5 months of therapy, the patient reported a stable to slightly reduced number of skin lesions but had begun to experience worsening joint pain, and the dosage of encorafenib was reduced to 225 mg/d. At 7 months of therapy, the dosage was further reduced to 150 mg/d due to persistent arthralgia. A follow-up examination at 10 months of therapy showed improvement in the number and size of the verrucous keratoses, and near resolution was seen by 14 months after the initial onset of the lesions (Figure 1B). At 20 months after initial onset, only 1 remaining verrucous keratosis was identified on physical examination and biopsy. The patient had continued a regimen of encorafenib 150 mg/d and weekly intravenous 500 mg cetuximab up to this point. Over the entire time period that the patient was seen, up to 12 lesions located in high-friction areas had become irritated and were treated with cryotherapy, but this contributed only minorly to the patient’s overall presentation.
Verrucous keratosis is a known cutaneous AE of BRAF inhibitor treatment with vemurafenib and dabrafenib, with fewer cases attributed to encorafenib.5,6 Within the oncologic setting, the eruption of verrucous papules as a paraneoplastic phenomenon is heavily debated in the literature and is known as the Leser-Trélat sign. This phenomenon is commonly associated with adenocarcinomas of the gastrointestinal tract, as seen in our patient.10 Based on Curth’s postulates—the criteria used to evaluate the relationship between an internal malignancy and a cutaneous disorder—this was unlikely in our patient. The criteria, which do not all need to be met to suggest a paraneoplastic phenomenon, include concurrent onset of the malignancy and the dermatosis, parallel course, association of a specific dermatosis with a specific malignancy, statistical significance of the association, and the presence of a genetic basis for the association.11 Several features favored a drug-related cutaneous eruption vs a paraneoplastic phenomenon: (1) the malignancy was identified months before the cutaneous eruptions manifested; (2) the cutaneous lesions appeared once treatment had already been initiated; and (3) the cutaneous lesions persisted long after the malignancy was no longer identifiable on CT. Indeed, eruption of the papules temporally coincided closely with the initiation of BRAF inhibitor therapy, arguing for correlation.
As a suspected BRAF inhibitor–associated cutaneous AE, the eruption of verrucous keratoses in our patient is remarkable for its spontaneous resolution despite ongoing therapy. It is speculated that keratinocytic proliferation while on BRAF inhibitor therapy may be caused by a paradoxical increase in signaling through CRAF, another Raf isoform that plays a role in the induction of terminal differentiation of keratinocytes, with a subsequent increase in MAPK signaling.12-14 Self-resolution of this cycle despite continuing BRAF inhibitor therapy suggests the possible involvement of balancing and/or alternative mechanistic pathways that may be related to the immune system. Although verrucous keratoses are considered benign proliferations and do not necessarily require any specific treatment or reduction in BRAF inhibitor dosage, they may be treated with cryotherapy, electrocautery, shave removal, or excision,15 which often is done if the lesions become inflamed and cause pain. Additionally, some patients may feel distress from the appearance of the lesions and desire treatment for this reason. Understanding that verrucous keratoses can be a transient cutaneous AE rather than a persistent one may be useful to clinicians as they manage AEs during BRAF inhibitor therapy.
- Pakneshan S, Salajegheh A, Smith RA, Lam AK. Clinicopathological relevance of BRAF mutations in human cancer. Pathology. 2013;45:346-356. doi:10.1097/PAT.0b013e328360b61d
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am. 2009;23:529-545. doi:10.1016/j.hoc.2009.04.001
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246. doi:10.1200/JCO.2010.32.4327
- Ji Z, Flaherty KT, Tsao H. Targeting the RAS pathway in melanoma. Trends Mol Med. 2012;18:27-35. doi:10.1016/j.molmed.2011.08.001
- Gouda MA, Subbiah V. Precision oncology for BRAF-mutant cancers with BRAF and MEK inhibitors: from melanoma to tissue-agnostic therapy. ESMO Open. 2023;8:100788. doi:10.1016/j.esmoop.2023.100788
- Gençler B, Gönül M. Cutaneous side effects of BRAF inhibitors in advanced melanoma: review of the literature. Dermatol Res Pract. 2016;2016:5361569. doi:10.1155/2016/5361569.
- Chu EY, Wanat KA, Miller CJ, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol. 2012;67:1265-1272. doi:10.1016/j.jaad.2012.04.008
- Naqash AR, File DM, Ziemer CM, et al. Cutaneous adverse reactions in B-RAF positive metastatic melanoma following sequential treatment with B-RAF/MEK inhibitors and immune checkpoint blockade or vice versa. a single-institutional case-series. J Immunother Cancer. 2019;7:4. doi:10.1186/s40425-018-0475-y
- Maldonado-Seral C, Berros-Fombella JP, Vivanco-Allende B, et al. Vemurafenib-associated neutrophilic panniculitis: an emergent adverse effect of variable severity. Dermatol Online J. 2013;19:16. doi:10.5070/d370x41670
- Mirali S, Mufti A, Lansang RP, et al. Eruptive seborrheic keratoses are associated with a co-occurring malignancy in the majority of reported cases: a systematic review. J Cutan Med Surg. 2022;26:57-62. doi:10.1177/12034754211035124
- Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin. 2009;59:73-98. doi:10.3322/caac.20005
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435. doi:10.1038/nature08833
- Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209-221. doi:10.1016/j.cell.2009.12.040
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signaling in cells with wild-type BRAF. Nature. 2010;464:427-430. doi:10.1038/nature08902
- Hayat MA. Brain Metastases from Primary Tumors, Volume 3: Epidemiology, Biology, and Therapy of Melanoma and Other Cancers. Academic Press; 2016.
- Pakneshan S, Salajegheh A, Smith RA, Lam AK. Clinicopathological relevance of BRAF mutations in human cancer. Pathology. 2013;45:346-356. doi:10.1097/PAT.0b013e328360b61d
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am. 2009;23:529-545. doi:10.1016/j.hoc.2009.04.001
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246. doi:10.1200/JCO.2010.32.4327
- Ji Z, Flaherty KT, Tsao H. Targeting the RAS pathway in melanoma. Trends Mol Med. 2012;18:27-35. doi:10.1016/j.molmed.2011.08.001
- Gouda MA, Subbiah V. Precision oncology for BRAF-mutant cancers with BRAF and MEK inhibitors: from melanoma to tissue-agnostic therapy. ESMO Open. 2023;8:100788. doi:10.1016/j.esmoop.2023.100788
- Gençler B, Gönül M. Cutaneous side effects of BRAF inhibitors in advanced melanoma: review of the literature. Dermatol Res Pract. 2016;2016:5361569. doi:10.1155/2016/5361569.
- Chu EY, Wanat KA, Miller CJ, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol. 2012;67:1265-1272. doi:10.1016/j.jaad.2012.04.008
- Naqash AR, File DM, Ziemer CM, et al. Cutaneous adverse reactions in B-RAF positive metastatic melanoma following sequential treatment with B-RAF/MEK inhibitors and immune checkpoint blockade or vice versa. a single-institutional case-series. J Immunother Cancer. 2019;7:4. doi:10.1186/s40425-018-0475-y
- Maldonado-Seral C, Berros-Fombella JP, Vivanco-Allende B, et al. Vemurafenib-associated neutrophilic panniculitis: an emergent adverse effect of variable severity. Dermatol Online J. 2013;19:16. doi:10.5070/d370x41670
- Mirali S, Mufti A, Lansang RP, et al. Eruptive seborrheic keratoses are associated with a co-occurring malignancy in the majority of reported cases: a systematic review. J Cutan Med Surg. 2022;26:57-62. doi:10.1177/12034754211035124
- Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin. 2009;59:73-98. doi:10.3322/caac.20005
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435. doi:10.1038/nature08833
- Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209-221. doi:10.1016/j.cell.2009.12.040
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signaling in cells with wild-type BRAF. Nature. 2010;464:427-430. doi:10.1038/nature08902
- Hayat MA. Brain Metastases from Primary Tumors, Volume 3: Epidemiology, Biology, and Therapy of Melanoma and Other Cancers. Academic Press; 2016.
Practice Points
- Verrucous keratoses are common cutaneous adverse events (AEs) associated with BRAF inhibitor therapy.
- Verrucous papules may be a paraneoplastic phenomenon and can be differentiated from a treatment-related AE based on the timing and progression in relation to tumor burden.
- Although treatment of particularly bothersome lesions with cryotherapy may be warranted, verrucous papules secondary to BRAF inhibitor therapy may resolve spontaneously.
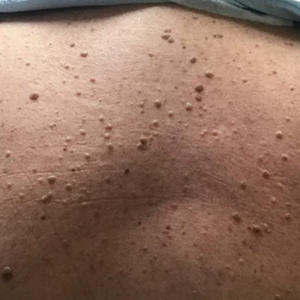
Rare Case of Photodistributed Hyperpigmentation Linked to Kratom Consumption
To the Editor:
Kratom (Mitragyna speciosa) is an evergreen tree native to Southeast Asia.1 Its leaves contain psychoactive compounds including mitragynine and 7-hydroxymitragynine, which exert dose-dependent effects on the central nervous system through opioid and monoaminergic receptors.2,3 At low doses (1–5 g), kratom elicits mild stimulant effects such as increased sociability, alertness, and talkativeness. At high doses (5–15 g), kratom has depressant effects that can provide relief from pain and opioid-withdrawal symptoms.3
Traditionally, kratom has been used in Southeast Asia for recreational and ceremonial purposes, to ease opioid-withdrawal symptoms, and to reduce fatigue from manual labor.4 In the 21st century, availability of kratom expanded to Europe, Australia, and the United States, largely facilitated by widespread dissemination of deceitful marketing and unregulated sales on the internet.1 Although large-scale epidemiologic studies evaluating kratom’s prevalence are scarce, available evidence indicates rising worldwide usage, with a notable increase in kratom-related poison center calls between 2011 and 2017 in the United States.5 In July 2023, kratom made headlines due to the death of a woman in Florida following use of the substance.6
A cross-sectional study revealed that in the United States, kratom typically is used by White individuals for self-treatment of anxiety, depression, pain, and opioid withdrawal.7 However, the potential for severe adverse effects and dependence on kratom can outweigh the benefits.6,8 Reported adverse effects of kratom include tachycardia, hypercholesteremia, liver injury, hallucinations, respiratory depression, seizure, coma, and death.9,10 We present a case of kratom-induced photodistributed hyperpigmentation.
A 63-year-old man presented to the dermatology clinic with diffuse tender, pruritic, hyperpigmented skin lesions that developed over the course of 1 year. The lesions were distributed on sun-exposed areas, including the face, neck, and forearms (Figure 1). The patient reported no other major symptoms, and his health was otherwise unremarkable. He had a medical history of psoriasiform and spongiotic dermatitis consistent with eczema, psoriasis, hypercholesteremia, and hyperlipidemia. The patient was not taking any medications at the time of presentation. He had a family history of plaque psoriasis in his father. Five years prior to the current presentation, the patient was treated with adalimumab for steroid-resistant psoriasis; however, despite initial improvement, he experienced recurrence of scaly erythematous plaques and had discontinued adalimumab the year prior to presentation.
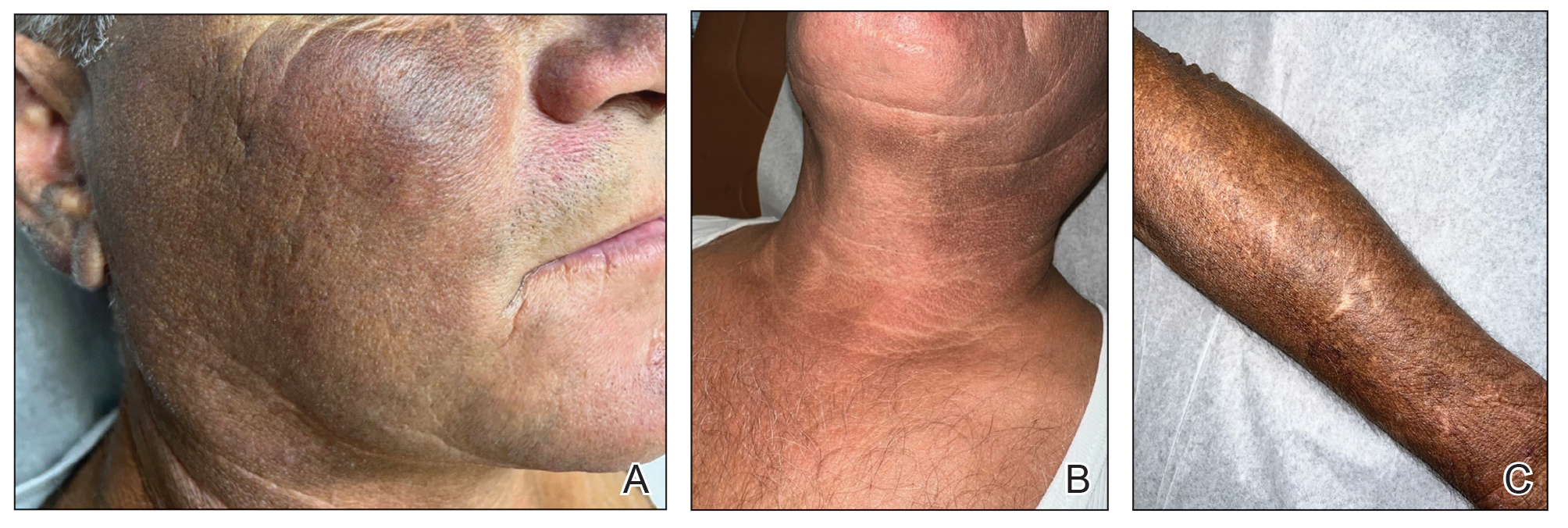
When adalimumab was discontinued, the patient sought alternative treatment for the skin symptoms and began self-administering kratom in an attempt to alleviate associated physical discomfort. He ingested approximately 3 bottles of liquid kratom per day, with each bottle containing 180 mg of mitragynine and less than 8 mg of 7-hydroxymitragynine. Although not scientifically proven, kratom has been colloquially advertised to improve psoriasis.11 The patient reported no other medication use or allergies.
Shave biopsies of hyperpigmented lesions on the right side of the neck, ear, and forearm were performed. Histopathology revealed a sparse superficial, perivascular, lymphocytic infiltrate accompanied by a prominent number of melanophages in the superficial dermis (Figure 2). Special stains further confirmed that the pigment was melanin; the specimens stained positive with Fontana-Masson stain (Figure 3) and negative with an iron stain (Figure 4).
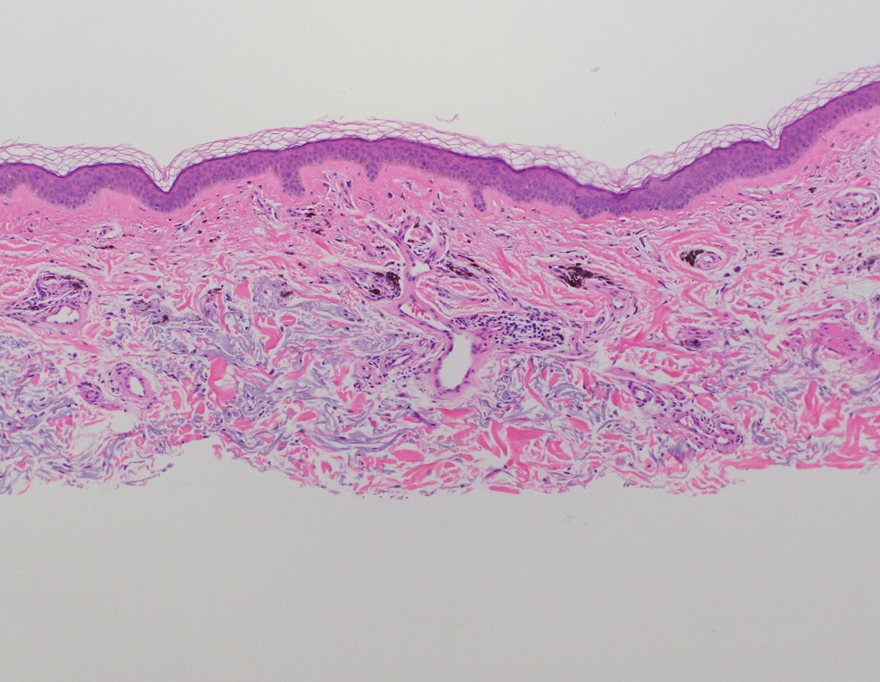
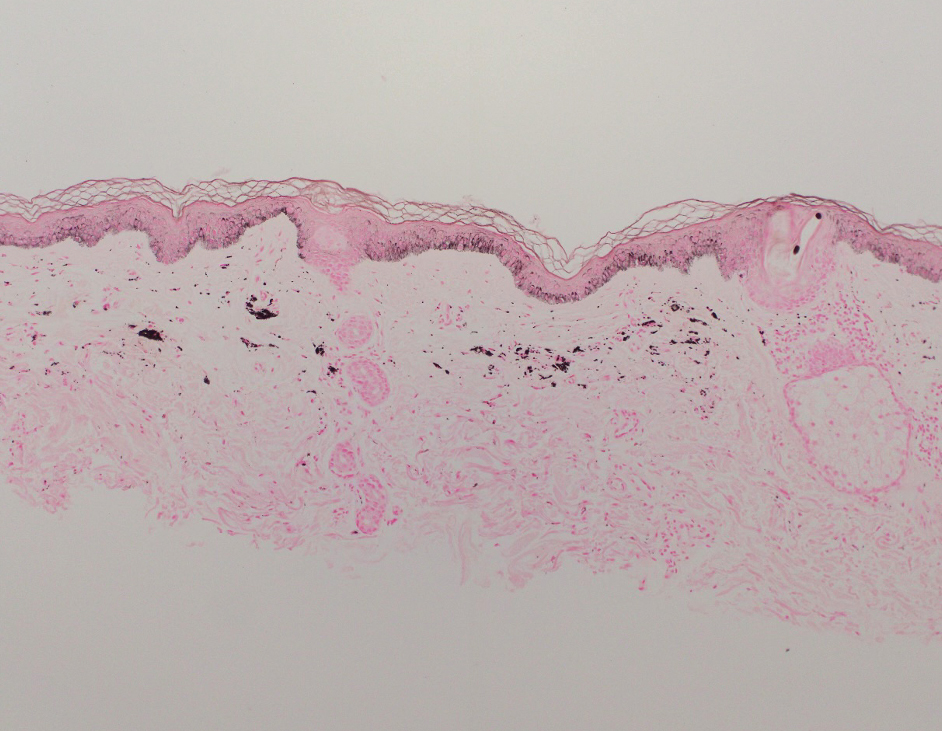
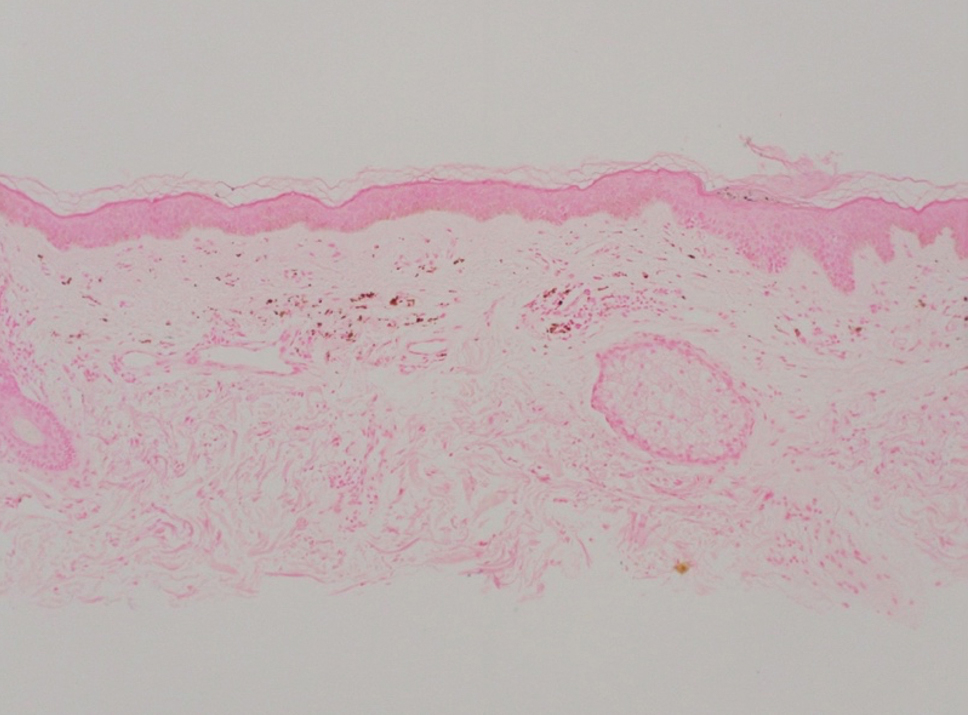
Adalimumab-induced hyperpigmentation was considered. A prior case of adalimumab-induced hyperpigmentation manifested on the face. Histopathology was consistent with a superficial, perivascular, lymphocytic infiltrate with melanophages in the dermis; however, hyperpigmentation was absent in the periorbital area, and affected areas faded 4 months after discontinuation of adalimumab.12 Our patient presented with hyperpigmentation 1 year after adalimumab cessation, and the hyperpigmented areas included the periorbital region. Because of the distinct temporal and clinical features, adalimumab-induced hyperpigmentation was eliminated from the differential diagnosis.
Based on the photodistributed pattern of hyperpigmentation, histopathology, and the temporal relationship between hyperpigmentation onset and kratom usage, a diagnosis of kratom-induced photodistributed hyperpigmentation was made. The patient was advised to discontinue kratom use and use sun protection to prevent further photodamage. The patient subsequently was lost to follow-up.
Kratom alkaloids bind all 3 opioid receptors—μOP, δOP, and κOPs—in a G-protein–biased manner with 7-hydroxymitragynine, the most pharmacologically active alkaloid, exhibiting a higher affinity for μ-opioid receptors.13,14 In human epidermal melanocytes, binding between μ-opioid receptors and β-endorphin, an endogenous opioid, is associated with increased melanin production. This melanogenesis has been linked to hyperpigmentation.15 Given the similarity between kratom alkaloids and β-endorphin in opioid-receptor binding, it is possible that kratom-induced hyperpigmentation may occur through a similar mechanism involving μ-opioid receptors and melanogenesis in epidermal melanocytes. Moreover, some researchers have theorized that sun exposure may result in free radical formation of certain drugs or their metabolites. These free radicals then can interact with cellular DNA, triggering the release of pigmentary mediators and resulting in hyperpigmentation.16 This theory may explain the photodistributed pattern of kratom-induced hyperpigmentation. Further studies are needed to understand the mechanism behind this adverse reaction and its implications for patient treatment.
Literature on kratom-induced hyperpigmentation is limited. Powell et al17 reported a similar case of kratom-induced photodistributed hyperpigmentation—a White man had taken kratom to reduce opioid use and subsequently developed hyperpigmented patches on the arms and face. Moreover, anonymous Reddit users have shared anecdotal reports of hyperpigmentation following kratom use.18
Physicians should be aware of hyperpigmentation as a potential adverse reaction of kratom use as its prevalence increases globally. Further research is warranted to elucidate the mechanism behind this adverse reaction and identify risk factors.
- Prozialeck WC, Avery BA, Boyer EW, et al. Kratom policy: the challenge of balancing therapeutic potential with public safety. Int J Drug Policy. 2019;70:70-77. doi:10.1016/j.drugpo.2019.05.003
- Bergen-Cico D, MacClurg K. Kratom (Mitragyna speciosa) use, addiction potential, and legal status. In: Preedy VR, ed. Neuropathology of Drug Addictions and Substance Misuse. 2016:903-911. doi:10.1016/B978-0-12-800634-4.00089-5
- Warner ML, Kaufman NC, Grundmann O. The pharmacology and toxicology of kratom: from traditional herb to drug of abuse. Int J Legal Med. 2016;130:127-138. doi:10.1007/s00414-015-1279-y
- Transnational Institute. Kratom in Thailand: decriminalisation and community control? May 3, 2011. Accessed August 23, 2024. https://www.tni.org/en/publication/kratom-in-thailand-decriminalisation-and-community-control
- Eastlack SC, Cornett EM, Kaye AD. Kratom—pharmacology, clinical implications, and outlook: a comprehensive review. Pain Ther. 2020;9:55-69. doi:10.1007/s40122-020-00151-x
- Reyes R. Family of Florida mom who died from herbal substance kratom wins $11M suit. New York Post. July 30, 2023. Updated July 31, 2023. Accessed August 23, 2024. https://nypost.com/2023/07/30/family-of-florida-mom-who-died-from-herbal-substance-kratom-wins-11m-suit/
- Garcia-Romeu A, Cox DJ, Smith KE, et al. Kratom (Mitragyna speciosa): user demographics, use patterns, and implications for the opioid epidemic. Drug Alcohol Depend. 2020;208:107849. doi:10.1016/j.drugalcdep.2020.107849
- Mayo Clinic. Kratom: unsafe and ineffective. Accessed August 23, 2024. https://www.mayoclinic.org/healthy-lifestyle/consumer-health/in-depth/kratom/art-20402171
- Sethi R, Hoang N, Ravishankar DA, et al. Kratom (Mitragyna speciosa): friend or foe? Prim Care Companion CNS Disord. 2020;22:19nr02507.
- Eggleston W, Stoppacher R, Suen K, et al. Kratom use and toxicities in the United States. Pharmacother J Hum Pharmacol Drug Ther. 2019;39:775-777. doi:10.1002/phar.2280
- Qrius. 6 benefits of kratom you should know for healthy skin. March 21, 2023. Accessed August 23, 2024. https://qrius.com/6-benefits-of-kratom-you-should-know-for-healthy-skin/
- Blomberg M, Zachariae COC, Grønhøj F. Hyperpigmentation of the face following adalimumab treatment. Acta Derm Venereol. 2009;89:546-547. doi:10.2340/00015555-0697
- Matsumoto K, Hatori Y, Murayama T, et al. Involvement of μ-opioid receptors in antinociception and inhibition of gastrointestinal transit induced by 7-hydroxymitragynine, isolated from Thai herbal medicine Mitragyna speciosa. Eur J Pharmacol. 2006;549:63-70. doi:10.1016/j.ejphar.2006.08.013
- Jentsch MJ, Pippin MM. Kratom. In: StatPearls. StatPearls Publishing; 2023.
- Bigliardi PL, Tobin DJ, Gaveriaux-Ruff C, et al. Opioids and the skin—where do we stand? Exp Dermatol. 2009;18:424-430.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10. doi:10.5070/D33c97j4z5
- Powell LR, Ryser TJ, Morey GE, et al. Kratom as a novel cause of photodistributed hyperpigmentation. JAAD Case Rep. 2022;28:145-148. doi:10.1016/j.jdcr.2022.07.033
- Haccoon. Skin discoloring? Reddit. June 30, 2019. Accessed August 23, 2024. https://www.reddit.com/r/quittingkratom/comments/c7b1cm/skin_discoloring/
To the Editor:
Kratom (Mitragyna speciosa) is an evergreen tree native to Southeast Asia.1 Its leaves contain psychoactive compounds including mitragynine and 7-hydroxymitragynine, which exert dose-dependent effects on the central nervous system through opioid and monoaminergic receptors.2,3 At low doses (1–5 g), kratom elicits mild stimulant effects such as increased sociability, alertness, and talkativeness. At high doses (5–15 g), kratom has depressant effects that can provide relief from pain and opioid-withdrawal symptoms.3
Traditionally, kratom has been used in Southeast Asia for recreational and ceremonial purposes, to ease opioid-withdrawal symptoms, and to reduce fatigue from manual labor.4 In the 21st century, availability of kratom expanded to Europe, Australia, and the United States, largely facilitated by widespread dissemination of deceitful marketing and unregulated sales on the internet.1 Although large-scale epidemiologic studies evaluating kratom’s prevalence are scarce, available evidence indicates rising worldwide usage, with a notable increase in kratom-related poison center calls between 2011 and 2017 in the United States.5 In July 2023, kratom made headlines due to the death of a woman in Florida following use of the substance.6
A cross-sectional study revealed that in the United States, kratom typically is used by White individuals for self-treatment of anxiety, depression, pain, and opioid withdrawal.7 However, the potential for severe adverse effects and dependence on kratom can outweigh the benefits.6,8 Reported adverse effects of kratom include tachycardia, hypercholesteremia, liver injury, hallucinations, respiratory depression, seizure, coma, and death.9,10 We present a case of kratom-induced photodistributed hyperpigmentation.
A 63-year-old man presented to the dermatology clinic with diffuse tender, pruritic, hyperpigmented skin lesions that developed over the course of 1 year. The lesions were distributed on sun-exposed areas, including the face, neck, and forearms (Figure 1). The patient reported no other major symptoms, and his health was otherwise unremarkable. He had a medical history of psoriasiform and spongiotic dermatitis consistent with eczema, psoriasis, hypercholesteremia, and hyperlipidemia. The patient was not taking any medications at the time of presentation. He had a family history of plaque psoriasis in his father. Five years prior to the current presentation, the patient was treated with adalimumab for steroid-resistant psoriasis; however, despite initial improvement, he experienced recurrence of scaly erythematous plaques and had discontinued adalimumab the year prior to presentation.
When adalimumab was discontinued, the patient sought alternative treatment for the skin symptoms and began self-administering kratom in an attempt to alleviate associated physical discomfort. He ingested approximately 3 bottles of liquid kratom per day, with each bottle containing 180 mg of mitragynine and less than 8 mg of 7-hydroxymitragynine. Although not scientifically proven, kratom has been colloquially advertised to improve psoriasis.11 The patient reported no other medication use or allergies.
Shave biopsies of hyperpigmented lesions on the right side of the neck, ear, and forearm were performed. Histopathology revealed a sparse superficial, perivascular, lymphocytic infiltrate accompanied by a prominent number of melanophages in the superficial dermis (Figure 2). Special stains further confirmed that the pigment was melanin; the specimens stained positive with Fontana-Masson stain (Figure 3) and negative with an iron stain (Figure 4).
Adalimumab-induced hyperpigmentation was considered. A prior case of adalimumab-induced hyperpigmentation manifested on the face. Histopathology was consistent with a superficial, perivascular, lymphocytic infiltrate with melanophages in the dermis; however, hyperpigmentation was absent in the periorbital area, and affected areas faded 4 months after discontinuation of adalimumab.12 Our patient presented with hyperpigmentation 1 year after adalimumab cessation, and the hyperpigmented areas included the periorbital region. Because of the distinct temporal and clinical features, adalimumab-induced hyperpigmentation was eliminated from the differential diagnosis.
Based on the photodistributed pattern of hyperpigmentation, histopathology, and the temporal relationship between hyperpigmentation onset and kratom usage, a diagnosis of kratom-induced photodistributed hyperpigmentation was made. The patient was advised to discontinue kratom use and use sun protection to prevent further photodamage. The patient subsequently was lost to follow-up.
Kratom alkaloids bind all 3 opioid receptors—μOP, δOP, and κOPs—in a G-protein–biased manner with 7-hydroxymitragynine, the most pharmacologically active alkaloid, exhibiting a higher affinity for μ-opioid receptors.13,14 In human epidermal melanocytes, binding between μ-opioid receptors and β-endorphin, an endogenous opioid, is associated with increased melanin production. This melanogenesis has been linked to hyperpigmentation.15 Given the similarity between kratom alkaloids and β-endorphin in opioid-receptor binding, it is possible that kratom-induced hyperpigmentation may occur through a similar mechanism involving μ-opioid receptors and melanogenesis in epidermal melanocytes. Moreover, some researchers have theorized that sun exposure may result in free radical formation of certain drugs or their metabolites. These free radicals then can interact with cellular DNA, triggering the release of pigmentary mediators and resulting in hyperpigmentation.16 This theory may explain the photodistributed pattern of kratom-induced hyperpigmentation. Further studies are needed to understand the mechanism behind this adverse reaction and its implications for patient treatment.
Literature on kratom-induced hyperpigmentation is limited. Powell et al17 reported a similar case of kratom-induced photodistributed hyperpigmentation—a White man had taken kratom to reduce opioid use and subsequently developed hyperpigmented patches on the arms and face. Moreover, anonymous Reddit users have shared anecdotal reports of hyperpigmentation following kratom use.18
Physicians should be aware of hyperpigmentation as a potential adverse reaction of kratom use as its prevalence increases globally. Further research is warranted to elucidate the mechanism behind this adverse reaction and identify risk factors.
To the Editor:
Kratom (Mitragyna speciosa) is an evergreen tree native to Southeast Asia.1 Its leaves contain psychoactive compounds including mitragynine and 7-hydroxymitragynine, which exert dose-dependent effects on the central nervous system through opioid and monoaminergic receptors.2,3 At low doses (1–5 g), kratom elicits mild stimulant effects such as increased sociability, alertness, and talkativeness. At high doses (5–15 g), kratom has depressant effects that can provide relief from pain and opioid-withdrawal symptoms.3
Traditionally, kratom has been used in Southeast Asia for recreational and ceremonial purposes, to ease opioid-withdrawal symptoms, and to reduce fatigue from manual labor.4 In the 21st century, availability of kratom expanded to Europe, Australia, and the United States, largely facilitated by widespread dissemination of deceitful marketing and unregulated sales on the internet.1 Although large-scale epidemiologic studies evaluating kratom’s prevalence are scarce, available evidence indicates rising worldwide usage, with a notable increase in kratom-related poison center calls between 2011 and 2017 in the United States.5 In July 2023, kratom made headlines due to the death of a woman in Florida following use of the substance.6
A cross-sectional study revealed that in the United States, kratom typically is used by White individuals for self-treatment of anxiety, depression, pain, and opioid withdrawal.7 However, the potential for severe adverse effects and dependence on kratom can outweigh the benefits.6,8 Reported adverse effects of kratom include tachycardia, hypercholesteremia, liver injury, hallucinations, respiratory depression, seizure, coma, and death.9,10 We present a case of kratom-induced photodistributed hyperpigmentation.
A 63-year-old man presented to the dermatology clinic with diffuse tender, pruritic, hyperpigmented skin lesions that developed over the course of 1 year. The lesions were distributed on sun-exposed areas, including the face, neck, and forearms (Figure 1). The patient reported no other major symptoms, and his health was otherwise unremarkable. He had a medical history of psoriasiform and spongiotic dermatitis consistent with eczema, psoriasis, hypercholesteremia, and hyperlipidemia. The patient was not taking any medications at the time of presentation. He had a family history of plaque psoriasis in his father. Five years prior to the current presentation, the patient was treated with adalimumab for steroid-resistant psoriasis; however, despite initial improvement, he experienced recurrence of scaly erythematous plaques and had discontinued adalimumab the year prior to presentation.
When adalimumab was discontinued, the patient sought alternative treatment for the skin symptoms and began self-administering kratom in an attempt to alleviate associated physical discomfort. He ingested approximately 3 bottles of liquid kratom per day, with each bottle containing 180 mg of mitragynine and less than 8 mg of 7-hydroxymitragynine. Although not scientifically proven, kratom has been colloquially advertised to improve psoriasis.11 The patient reported no other medication use or allergies.
Shave biopsies of hyperpigmented lesions on the right side of the neck, ear, and forearm were performed. Histopathology revealed a sparse superficial, perivascular, lymphocytic infiltrate accompanied by a prominent number of melanophages in the superficial dermis (Figure 2). Special stains further confirmed that the pigment was melanin; the specimens stained positive with Fontana-Masson stain (Figure 3) and negative with an iron stain (Figure 4).
Adalimumab-induced hyperpigmentation was considered. A prior case of adalimumab-induced hyperpigmentation manifested on the face. Histopathology was consistent with a superficial, perivascular, lymphocytic infiltrate with melanophages in the dermis; however, hyperpigmentation was absent in the periorbital area, and affected areas faded 4 months after discontinuation of adalimumab.12 Our patient presented with hyperpigmentation 1 year after adalimumab cessation, and the hyperpigmented areas included the periorbital region. Because of the distinct temporal and clinical features, adalimumab-induced hyperpigmentation was eliminated from the differential diagnosis.
Based on the photodistributed pattern of hyperpigmentation, histopathology, and the temporal relationship between hyperpigmentation onset and kratom usage, a diagnosis of kratom-induced photodistributed hyperpigmentation was made. The patient was advised to discontinue kratom use and use sun protection to prevent further photodamage. The patient subsequently was lost to follow-up.
Kratom alkaloids bind all 3 opioid receptors—μOP, δOP, and κOPs—in a G-protein–biased manner with 7-hydroxymitragynine, the most pharmacologically active alkaloid, exhibiting a higher affinity for μ-opioid receptors.13,14 In human epidermal melanocytes, binding between μ-opioid receptors and β-endorphin, an endogenous opioid, is associated with increased melanin production. This melanogenesis has been linked to hyperpigmentation.15 Given the similarity between kratom alkaloids and β-endorphin in opioid-receptor binding, it is possible that kratom-induced hyperpigmentation may occur through a similar mechanism involving μ-opioid receptors and melanogenesis in epidermal melanocytes. Moreover, some researchers have theorized that sun exposure may result in free radical formation of certain drugs or their metabolites. These free radicals then can interact with cellular DNA, triggering the release of pigmentary mediators and resulting in hyperpigmentation.16 This theory may explain the photodistributed pattern of kratom-induced hyperpigmentation. Further studies are needed to understand the mechanism behind this adverse reaction and its implications for patient treatment.
Literature on kratom-induced hyperpigmentation is limited. Powell et al17 reported a similar case of kratom-induced photodistributed hyperpigmentation—a White man had taken kratom to reduce opioid use and subsequently developed hyperpigmented patches on the arms and face. Moreover, anonymous Reddit users have shared anecdotal reports of hyperpigmentation following kratom use.18
Physicians should be aware of hyperpigmentation as a potential adverse reaction of kratom use as its prevalence increases globally. Further research is warranted to elucidate the mechanism behind this adverse reaction and identify risk factors.
- Prozialeck WC, Avery BA, Boyer EW, et al. Kratom policy: the challenge of balancing therapeutic potential with public safety. Int J Drug Policy. 2019;70:70-77. doi:10.1016/j.drugpo.2019.05.003
- Bergen-Cico D, MacClurg K. Kratom (Mitragyna speciosa) use, addiction potential, and legal status. In: Preedy VR, ed. Neuropathology of Drug Addictions and Substance Misuse. 2016:903-911. doi:10.1016/B978-0-12-800634-4.00089-5
- Warner ML, Kaufman NC, Grundmann O. The pharmacology and toxicology of kratom: from traditional herb to drug of abuse. Int J Legal Med. 2016;130:127-138. doi:10.1007/s00414-015-1279-y
- Transnational Institute. Kratom in Thailand: decriminalisation and community control? May 3, 2011. Accessed August 23, 2024. https://www.tni.org/en/publication/kratom-in-thailand-decriminalisation-and-community-control
- Eastlack SC, Cornett EM, Kaye AD. Kratom—pharmacology, clinical implications, and outlook: a comprehensive review. Pain Ther. 2020;9:55-69. doi:10.1007/s40122-020-00151-x
- Reyes R. Family of Florida mom who died from herbal substance kratom wins $11M suit. New York Post. July 30, 2023. Updated July 31, 2023. Accessed August 23, 2024. https://nypost.com/2023/07/30/family-of-florida-mom-who-died-from-herbal-substance-kratom-wins-11m-suit/
- Garcia-Romeu A, Cox DJ, Smith KE, et al. Kratom (Mitragyna speciosa): user demographics, use patterns, and implications for the opioid epidemic. Drug Alcohol Depend. 2020;208:107849. doi:10.1016/j.drugalcdep.2020.107849
- Mayo Clinic. Kratom: unsafe and ineffective. Accessed August 23, 2024. https://www.mayoclinic.org/healthy-lifestyle/consumer-health/in-depth/kratom/art-20402171
- Sethi R, Hoang N, Ravishankar DA, et al. Kratom (Mitragyna speciosa): friend or foe? Prim Care Companion CNS Disord. 2020;22:19nr02507.
- Eggleston W, Stoppacher R, Suen K, et al. Kratom use and toxicities in the United States. Pharmacother J Hum Pharmacol Drug Ther. 2019;39:775-777. doi:10.1002/phar.2280
- Qrius. 6 benefits of kratom you should know for healthy skin. March 21, 2023. Accessed August 23, 2024. https://qrius.com/6-benefits-of-kratom-you-should-know-for-healthy-skin/
- Blomberg M, Zachariae COC, Grønhøj F. Hyperpigmentation of the face following adalimumab treatment. Acta Derm Venereol. 2009;89:546-547. doi:10.2340/00015555-0697
- Matsumoto K, Hatori Y, Murayama T, et al. Involvement of μ-opioid receptors in antinociception and inhibition of gastrointestinal transit induced by 7-hydroxymitragynine, isolated from Thai herbal medicine Mitragyna speciosa. Eur J Pharmacol. 2006;549:63-70. doi:10.1016/j.ejphar.2006.08.013
- Jentsch MJ, Pippin MM. Kratom. In: StatPearls. StatPearls Publishing; 2023.
- Bigliardi PL, Tobin DJ, Gaveriaux-Ruff C, et al. Opioids and the skin—where do we stand? Exp Dermatol. 2009;18:424-430.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10. doi:10.5070/D33c97j4z5
- Powell LR, Ryser TJ, Morey GE, et al. Kratom as a novel cause of photodistributed hyperpigmentation. JAAD Case Rep. 2022;28:145-148. doi:10.1016/j.jdcr.2022.07.033
- Haccoon. Skin discoloring? Reddit. June 30, 2019. Accessed August 23, 2024. https://www.reddit.com/r/quittingkratom/comments/c7b1cm/skin_discoloring/
- Prozialeck WC, Avery BA, Boyer EW, et al. Kratom policy: the challenge of balancing therapeutic potential with public safety. Int J Drug Policy. 2019;70:70-77. doi:10.1016/j.drugpo.2019.05.003
- Bergen-Cico D, MacClurg K. Kratom (Mitragyna speciosa) use, addiction potential, and legal status. In: Preedy VR, ed. Neuropathology of Drug Addictions and Substance Misuse. 2016:903-911. doi:10.1016/B978-0-12-800634-4.00089-5
- Warner ML, Kaufman NC, Grundmann O. The pharmacology and toxicology of kratom: from traditional herb to drug of abuse. Int J Legal Med. 2016;130:127-138. doi:10.1007/s00414-015-1279-y
- Transnational Institute. Kratom in Thailand: decriminalisation and community control? May 3, 2011. Accessed August 23, 2024. https://www.tni.org/en/publication/kratom-in-thailand-decriminalisation-and-community-control
- Eastlack SC, Cornett EM, Kaye AD. Kratom—pharmacology, clinical implications, and outlook: a comprehensive review. Pain Ther. 2020;9:55-69. doi:10.1007/s40122-020-00151-x
- Reyes R. Family of Florida mom who died from herbal substance kratom wins $11M suit. New York Post. July 30, 2023. Updated July 31, 2023. Accessed August 23, 2024. https://nypost.com/2023/07/30/family-of-florida-mom-who-died-from-herbal-substance-kratom-wins-11m-suit/
- Garcia-Romeu A, Cox DJ, Smith KE, et al. Kratom (Mitragyna speciosa): user demographics, use patterns, and implications for the opioid epidemic. Drug Alcohol Depend. 2020;208:107849. doi:10.1016/j.drugalcdep.2020.107849
- Mayo Clinic. Kratom: unsafe and ineffective. Accessed August 23, 2024. https://www.mayoclinic.org/healthy-lifestyle/consumer-health/in-depth/kratom/art-20402171
- Sethi R, Hoang N, Ravishankar DA, et al. Kratom (Mitragyna speciosa): friend or foe? Prim Care Companion CNS Disord. 2020;22:19nr02507.
- Eggleston W, Stoppacher R, Suen K, et al. Kratom use and toxicities in the United States. Pharmacother J Hum Pharmacol Drug Ther. 2019;39:775-777. doi:10.1002/phar.2280
- Qrius. 6 benefits of kratom you should know for healthy skin. March 21, 2023. Accessed August 23, 2024. https://qrius.com/6-benefits-of-kratom-you-should-know-for-healthy-skin/
- Blomberg M, Zachariae COC, Grønhøj F. Hyperpigmentation of the face following adalimumab treatment. Acta Derm Venereol. 2009;89:546-547. doi:10.2340/00015555-0697
- Matsumoto K, Hatori Y, Murayama T, et al. Involvement of μ-opioid receptors in antinociception and inhibition of gastrointestinal transit induced by 7-hydroxymitragynine, isolated from Thai herbal medicine Mitragyna speciosa. Eur J Pharmacol. 2006;549:63-70. doi:10.1016/j.ejphar.2006.08.013
- Jentsch MJ, Pippin MM. Kratom. In: StatPearls. StatPearls Publishing; 2023.
- Bigliardi PL, Tobin DJ, Gaveriaux-Ruff C, et al. Opioids and the skin—where do we stand? Exp Dermatol. 2009;18:424-430.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10. doi:10.5070/D33c97j4z5
- Powell LR, Ryser TJ, Morey GE, et al. Kratom as a novel cause of photodistributed hyperpigmentation. JAAD Case Rep. 2022;28:145-148. doi:10.1016/j.jdcr.2022.07.033
- Haccoon. Skin discoloring? Reddit. June 30, 2019. Accessed August 23, 2024. https://www.reddit.com/r/quittingkratom/comments/c7b1cm/skin_discoloring/
Practice Points
- Clinicians should be aware of photodistributed hyperpigmentation as a potential adverse effect of kratom usage.
- Kratom-induced photodistributed hyperpigmentation should be suspected in patients with hyperpigmented lesions in sun-exposed areas of the skin following kratom use. A biopsy of lesions should be obtained to confirm the diagnosis.
- Cessation of kratom should be recommended.
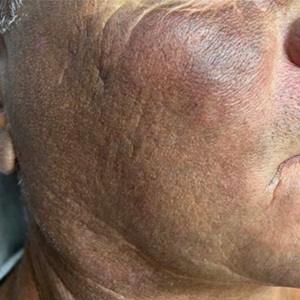
Acute Tender Papules on the Arms and Legs
The Diagnosis: Erythema Nodosum Leprosum
Erythema nodosum leprosum (ENL) is a type 2 reaction sometimes seen in patients infected with Mycobacterium leprae—primarily those with lepromatous or borderline lepromatous subtypes. Clinically, ENL manifests with abrupt onset of tender erythematous papules with associated fevers and general malaise. Studies have demonstrated a complex immune system reaction in ENL, but the detailed pathophysiology is not fully understood.1 Biopsies conducted within 24 hours of lesion formation are most elucidating. Foamy histiocytes admixed with neutrophils are seen in the subcutis, often causing a lobular panniculitis (quiz image).2 Neutrophils rarely are seen in other types of leprosy and thus are a useful diagnostic clue for ENL. Vasculitis of small- to medium-sized vessels can be seen but is not a necessary diagnostic criterion. Fite staining will highlight many acid-fast bacilli within the histiocytes (Figure 1).
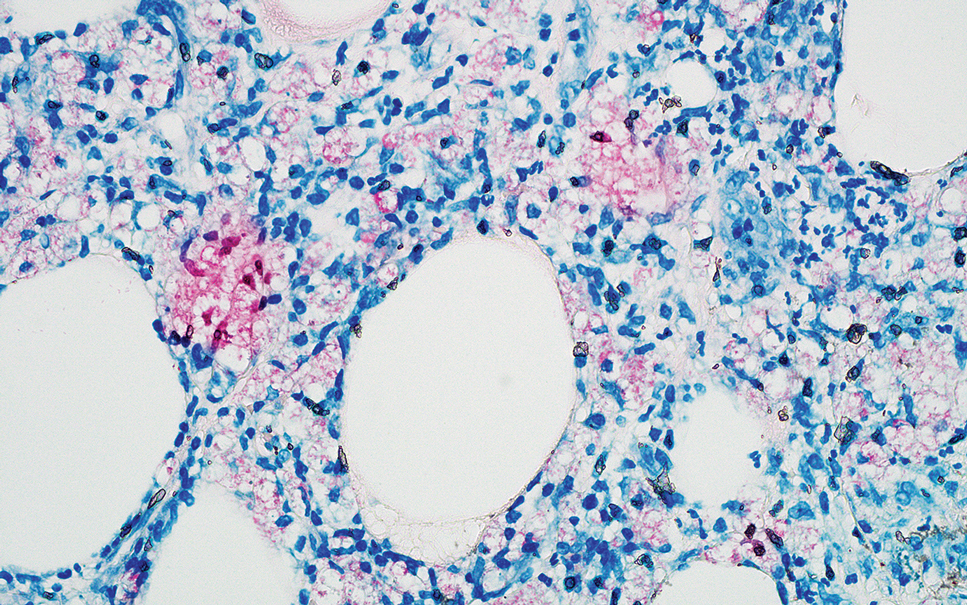
Erythema nodosum leprosum is treated with a combination of immunosuppressants such as prednisone and thalidomide. Our patient was taking triple-antibiotic therapy—dapsone, rifampin, and clofazimine—for lepromatous leprosy when the erythematous papules developed on the arms and legs. After a skin biopsy confirmed the diagnosis of ENL, he was started on prednisone 20 mg daily with plans for close follow-up. Unfortunately, the patient was subsequently lost to follow-up.
Acute febrile neutrophilic dermatosis (also known as Sweet syndrome) is an acute inflammatory disease characterized by abrupt onset of painful erythematous papules, plaques, or nodules on the skin. It often is seen in association with preceding infections (especially those in the upper respiratory or gastrointestinal tracts), hematologic malignancies, inflammatory bowel disease, or exposure to certain classes of medications (eg, granulocyte colony-stimulating factor, tyrosine kinase inhibitors, various antibiotics).3 Histologically, acute febrile neutrophilic dermatosis is characterized by dense neutrophilic infiltrates, often with notable dermal edema (Figure 2).4 Many cases also show leukocytoclastic vasculitis; however, foamy histiocytes are not a notable component of the inflammatory infiltrate, though a histiocytoid form of acute febrile neutrophilic dermatosis has been described.5 Infections must be rigorously ruled out prior to diagnosing a patient with acute febrile neutrophilic dermatosis, making it a diagnosis of exclusion.
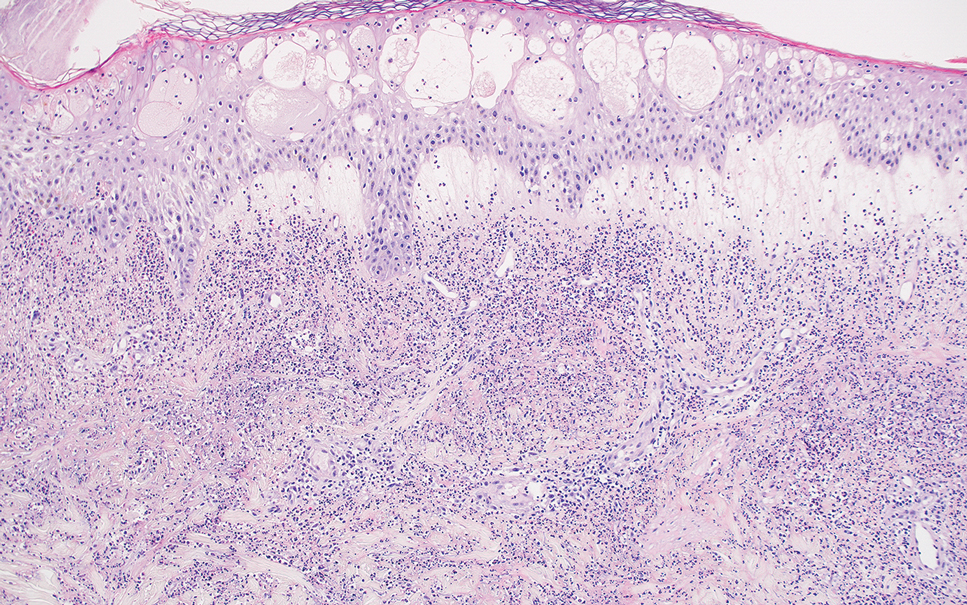
Cutaneous coccidioidomycosis is an infection caused by the dimorphic fungi Coccidioides immitis or Coccidioides posadasii. Cutaneous disease is rare but can occur from direct inoculation or dissemination from pulmonary disease in immunocompetent or immunocompromised patients. Papules, pustules, or plaques are seen clinically. Histologically, cutaneous coccidioidomycosis shows spherules that vary from 10 to 100 μm and are filled with multiple smaller endospores (Figure 3).6 Pseudoepitheliomatous hyperplasia with dense suppurative and granulomatous infiltrates also is seen.
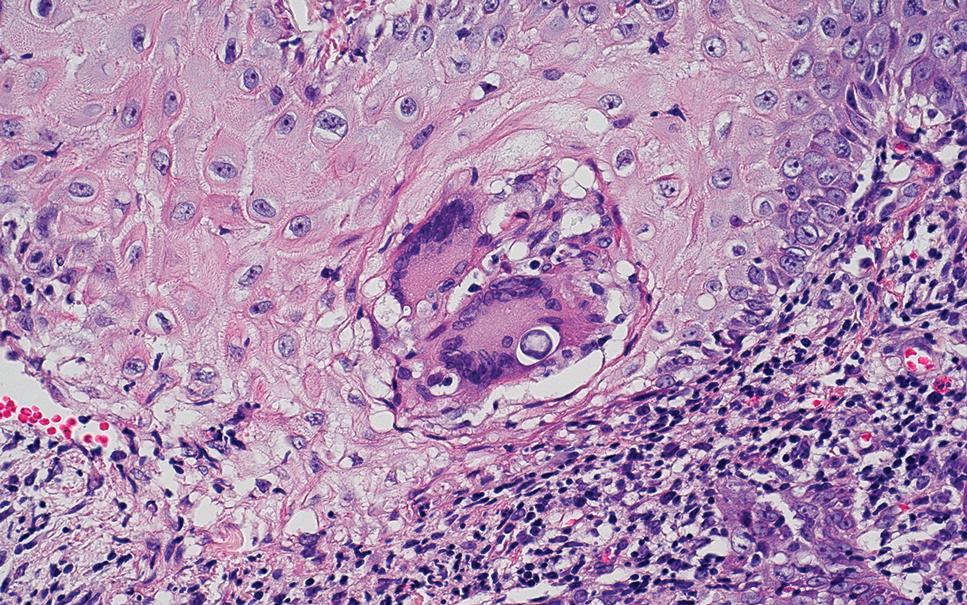
Erythema induratum is characterized by tender nodules on the lower extremities and has a substantial female predominance. Many cases are associated with Mycobacterium tuberculosis infection. The bacteria are not seen directly in the skin but are instead detectable through DNA polymerase chain reaction testing or investigation of other organ systems.7,8 Histologically, lesions show a lobular panniculitis with a mixed infiltrate. Vasculitis is seen in approximately 90% of erythema induratum cases vs approximately 25% of classic ENL cases (Figure 4),2,9 which has led some to use the term nodular vasculitis to describe this disease entity. Nodular vasculitis is considered by others to be a distinct disease entity in which there are clinical and histologic features similar to erythema induratum but no evidence of M tuberculosis infection.9
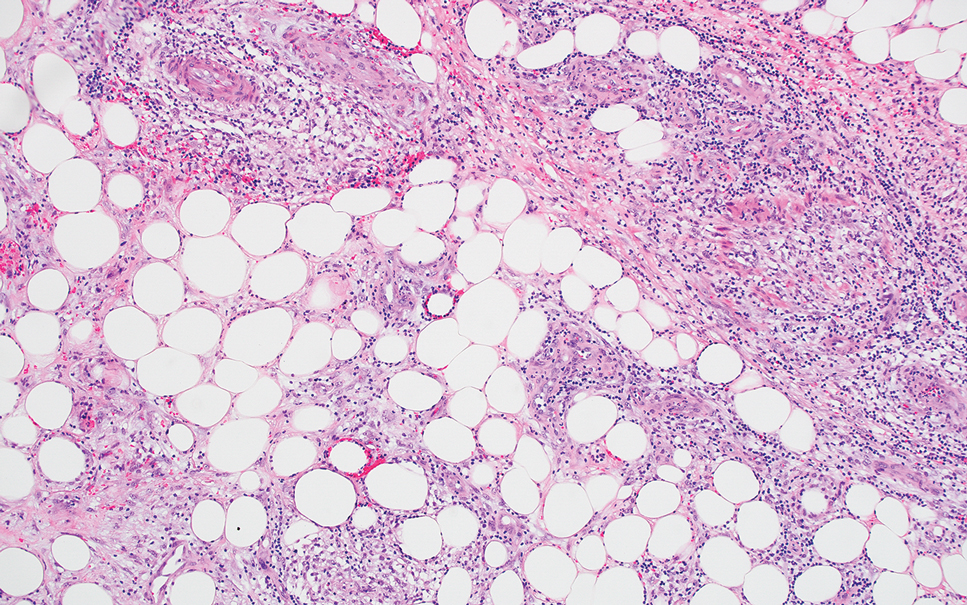
Polyarteritis nodosa is a vasculitis that affects medium- sized vessels of various organ systems. The presenting signs and symptoms vary based on the affected organ systems. Palpable to retiform purpura, livedo racemosa, subcutaneous nodules, or ulcers are seen when the skin is involved. The histologic hallmark is necrotizing vasculitis of medium-sized arterioles (Figure 5), although leukocytoclastic vasculitis of small-caliber vessels also can be seen in biopsies of affected skin.10 The vascular changes are said to be segmental, with uninvolved segments interspersed with involved segments. Antineutrophil cytoplasmic antibody (ANCA)– associated vasculitis also must be considered when one sees leukocytoclastic vasculitis of small-caliber vessels in the skin, as it can be distinguished most readily by detecting circulating antibodies specific for myeloperoxidase (MPO-ANCA) or proteinase 3 (PR3-ANCA).
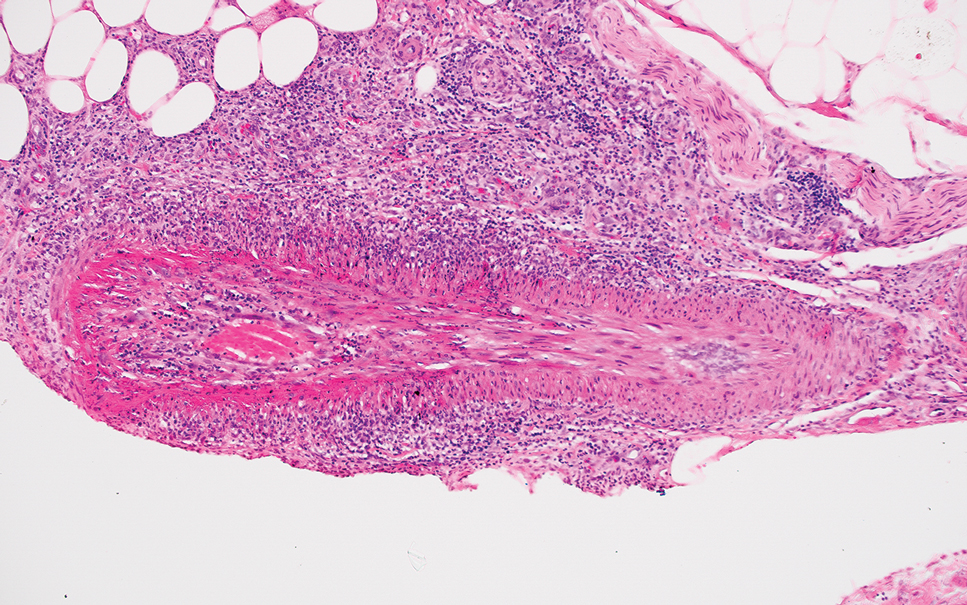
- Polycarpou A, Walker SL, Lockwood DNJ. A systematic review of immunological studies of erythema nodosum leprosum. Front Immunol. 2017;8:233. doi:10.3389/fimmu.2017.00233
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45. doi:10.1016/j.clindermatol.2014.10.003
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:1-28. doi:10.1186/1750-1172-2-34
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133. doi:10.1097/01.dad.0000249887.59810.76
- Wilson TC, Stone MS, Swick BL. Histiocytoid Sweet syndrome with haloed myeloid cells masquerading as a cryptococcal infection. Am J Dermatopathology. 2014;36:264-269. doi:10.1097/DAD.0b013e31828b811b
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin: a clinicopathological study of 20 cases of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356. doi:10.1097/00000372-199508000-00008
- Boonchai W, Suthipinittharm P, Mahaisavariya P. Panniculitis in tuberculosis: a clinicopathologic study of nodular panniculitis associated with tuberculosis. Int J Dermatol. 1998;37:361-363. doi:10.1046/j.1365-4362.1998.00299.x
- Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851. doi:10.1016/j.jaad.2008.07.030
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathological analysis and a review of the published work. J Dermatol. 2010;37:85-93. doi:10.1111/j.1346-8138.2009.00752.x
The Diagnosis: Erythema Nodosum Leprosum
Erythema nodosum leprosum (ENL) is a type 2 reaction sometimes seen in patients infected with Mycobacterium leprae—primarily those with lepromatous or borderline lepromatous subtypes. Clinically, ENL manifests with abrupt onset of tender erythematous papules with associated fevers and general malaise. Studies have demonstrated a complex immune system reaction in ENL, but the detailed pathophysiology is not fully understood.1 Biopsies conducted within 24 hours of lesion formation are most elucidating. Foamy histiocytes admixed with neutrophils are seen in the subcutis, often causing a lobular panniculitis (quiz image).2 Neutrophils rarely are seen in other types of leprosy and thus are a useful diagnostic clue for ENL. Vasculitis of small- to medium-sized vessels can be seen but is not a necessary diagnostic criterion. Fite staining will highlight many acid-fast bacilli within the histiocytes (Figure 1).
Erythema nodosum leprosum is treated with a combination of immunosuppressants such as prednisone and thalidomide. Our patient was taking triple-antibiotic therapy—dapsone, rifampin, and clofazimine—for lepromatous leprosy when the erythematous papules developed on the arms and legs. After a skin biopsy confirmed the diagnosis of ENL, he was started on prednisone 20 mg daily with plans for close follow-up. Unfortunately, the patient was subsequently lost to follow-up.
Acute febrile neutrophilic dermatosis (also known as Sweet syndrome) is an acute inflammatory disease characterized by abrupt onset of painful erythematous papules, plaques, or nodules on the skin. It often is seen in association with preceding infections (especially those in the upper respiratory or gastrointestinal tracts), hematologic malignancies, inflammatory bowel disease, or exposure to certain classes of medications (eg, granulocyte colony-stimulating factor, tyrosine kinase inhibitors, various antibiotics).3 Histologically, acute febrile neutrophilic dermatosis is characterized by dense neutrophilic infiltrates, often with notable dermal edema (Figure 2).4 Many cases also show leukocytoclastic vasculitis; however, foamy histiocytes are not a notable component of the inflammatory infiltrate, though a histiocytoid form of acute febrile neutrophilic dermatosis has been described.5 Infections must be rigorously ruled out prior to diagnosing a patient with acute febrile neutrophilic dermatosis, making it a diagnosis of exclusion.
Cutaneous coccidioidomycosis is an infection caused by the dimorphic fungi Coccidioides immitis or Coccidioides posadasii. Cutaneous disease is rare but can occur from direct inoculation or dissemination from pulmonary disease in immunocompetent or immunocompromised patients. Papules, pustules, or plaques are seen clinically. Histologically, cutaneous coccidioidomycosis shows spherules that vary from 10 to 100 μm and are filled with multiple smaller endospores (Figure 3).6 Pseudoepitheliomatous hyperplasia with dense suppurative and granulomatous infiltrates also is seen.
Erythema induratum is characterized by tender nodules on the lower extremities and has a substantial female predominance. Many cases are associated with Mycobacterium tuberculosis infection. The bacteria are not seen directly in the skin but are instead detectable through DNA polymerase chain reaction testing or investigation of other organ systems.7,8 Histologically, lesions show a lobular panniculitis with a mixed infiltrate. Vasculitis is seen in approximately 90% of erythema induratum cases vs approximately 25% of classic ENL cases (Figure 4),2,9 which has led some to use the term nodular vasculitis to describe this disease entity. Nodular vasculitis is considered by others to be a distinct disease entity in which there are clinical and histologic features similar to erythema induratum but no evidence of M tuberculosis infection.9
Polyarteritis nodosa is a vasculitis that affects medium- sized vessels of various organ systems. The presenting signs and symptoms vary based on the affected organ systems. Palpable to retiform purpura, livedo racemosa, subcutaneous nodules, or ulcers are seen when the skin is involved. The histologic hallmark is necrotizing vasculitis of medium-sized arterioles (Figure 5), although leukocytoclastic vasculitis of small-caliber vessels also can be seen in biopsies of affected skin.10 The vascular changes are said to be segmental, with uninvolved segments interspersed with involved segments. Antineutrophil cytoplasmic antibody (ANCA)– associated vasculitis also must be considered when one sees leukocytoclastic vasculitis of small-caliber vessels in the skin, as it can be distinguished most readily by detecting circulating antibodies specific for myeloperoxidase (MPO-ANCA) or proteinase 3 (PR3-ANCA).
The Diagnosis: Erythema Nodosum Leprosum
Erythema nodosum leprosum (ENL) is a type 2 reaction sometimes seen in patients infected with Mycobacterium leprae—primarily those with lepromatous or borderline lepromatous subtypes. Clinically, ENL manifests with abrupt onset of tender erythematous papules with associated fevers and general malaise. Studies have demonstrated a complex immune system reaction in ENL, but the detailed pathophysiology is not fully understood.1 Biopsies conducted within 24 hours of lesion formation are most elucidating. Foamy histiocytes admixed with neutrophils are seen in the subcutis, often causing a lobular panniculitis (quiz image).2 Neutrophils rarely are seen in other types of leprosy and thus are a useful diagnostic clue for ENL. Vasculitis of small- to medium-sized vessels can be seen but is not a necessary diagnostic criterion. Fite staining will highlight many acid-fast bacilli within the histiocytes (Figure 1).
Erythema nodosum leprosum is treated with a combination of immunosuppressants such as prednisone and thalidomide. Our patient was taking triple-antibiotic therapy—dapsone, rifampin, and clofazimine—for lepromatous leprosy when the erythematous papules developed on the arms and legs. After a skin biopsy confirmed the diagnosis of ENL, he was started on prednisone 20 mg daily with plans for close follow-up. Unfortunately, the patient was subsequently lost to follow-up.
Acute febrile neutrophilic dermatosis (also known as Sweet syndrome) is an acute inflammatory disease characterized by abrupt onset of painful erythematous papules, plaques, or nodules on the skin. It often is seen in association with preceding infections (especially those in the upper respiratory or gastrointestinal tracts), hematologic malignancies, inflammatory bowel disease, or exposure to certain classes of medications (eg, granulocyte colony-stimulating factor, tyrosine kinase inhibitors, various antibiotics).3 Histologically, acute febrile neutrophilic dermatosis is characterized by dense neutrophilic infiltrates, often with notable dermal edema (Figure 2).4 Many cases also show leukocytoclastic vasculitis; however, foamy histiocytes are not a notable component of the inflammatory infiltrate, though a histiocytoid form of acute febrile neutrophilic dermatosis has been described.5 Infections must be rigorously ruled out prior to diagnosing a patient with acute febrile neutrophilic dermatosis, making it a diagnosis of exclusion.
Cutaneous coccidioidomycosis is an infection caused by the dimorphic fungi Coccidioides immitis or Coccidioides posadasii. Cutaneous disease is rare but can occur from direct inoculation or dissemination from pulmonary disease in immunocompetent or immunocompromised patients. Papules, pustules, or plaques are seen clinically. Histologically, cutaneous coccidioidomycosis shows spherules that vary from 10 to 100 μm and are filled with multiple smaller endospores (Figure 3).6 Pseudoepitheliomatous hyperplasia with dense suppurative and granulomatous infiltrates also is seen.
Erythema induratum is characterized by tender nodules on the lower extremities and has a substantial female predominance. Many cases are associated with Mycobacterium tuberculosis infection. The bacteria are not seen directly in the skin but are instead detectable through DNA polymerase chain reaction testing or investigation of other organ systems.7,8 Histologically, lesions show a lobular panniculitis with a mixed infiltrate. Vasculitis is seen in approximately 90% of erythema induratum cases vs approximately 25% of classic ENL cases (Figure 4),2,9 which has led some to use the term nodular vasculitis to describe this disease entity. Nodular vasculitis is considered by others to be a distinct disease entity in which there are clinical and histologic features similar to erythema induratum but no evidence of M tuberculosis infection.9
Polyarteritis nodosa is a vasculitis that affects medium- sized vessels of various organ systems. The presenting signs and symptoms vary based on the affected organ systems. Palpable to retiform purpura, livedo racemosa, subcutaneous nodules, or ulcers are seen when the skin is involved. The histologic hallmark is necrotizing vasculitis of medium-sized arterioles (Figure 5), although leukocytoclastic vasculitis of small-caliber vessels also can be seen in biopsies of affected skin.10 The vascular changes are said to be segmental, with uninvolved segments interspersed with involved segments. Antineutrophil cytoplasmic antibody (ANCA)– associated vasculitis also must be considered when one sees leukocytoclastic vasculitis of small-caliber vessels in the skin, as it can be distinguished most readily by detecting circulating antibodies specific for myeloperoxidase (MPO-ANCA) or proteinase 3 (PR3-ANCA).
- Polycarpou A, Walker SL, Lockwood DNJ. A systematic review of immunological studies of erythema nodosum leprosum. Front Immunol. 2017;8:233. doi:10.3389/fimmu.2017.00233
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45. doi:10.1016/j.clindermatol.2014.10.003
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:1-28. doi:10.1186/1750-1172-2-34
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133. doi:10.1097/01.dad.0000249887.59810.76
- Wilson TC, Stone MS, Swick BL. Histiocytoid Sweet syndrome with haloed myeloid cells masquerading as a cryptococcal infection. Am J Dermatopathology. 2014;36:264-269. doi:10.1097/DAD.0b013e31828b811b
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin: a clinicopathological study of 20 cases of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356. doi:10.1097/00000372-199508000-00008
- Boonchai W, Suthipinittharm P, Mahaisavariya P. Panniculitis in tuberculosis: a clinicopathologic study of nodular panniculitis associated with tuberculosis. Int J Dermatol. 1998;37:361-363. doi:10.1046/j.1365-4362.1998.00299.x
- Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851. doi:10.1016/j.jaad.2008.07.030
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathological analysis and a review of the published work. J Dermatol. 2010;37:85-93. doi:10.1111/j.1346-8138.2009.00752.x
- Polycarpou A, Walker SL, Lockwood DNJ. A systematic review of immunological studies of erythema nodosum leprosum. Front Immunol. 2017;8:233. doi:10.3389/fimmu.2017.00233
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45. doi:10.1016/j.clindermatol.2014.10.003
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:1-28. doi:10.1186/1750-1172-2-34
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133. doi:10.1097/01.dad.0000249887.59810.76
- Wilson TC, Stone MS, Swick BL. Histiocytoid Sweet syndrome with haloed myeloid cells masquerading as a cryptococcal infection. Am J Dermatopathology. 2014;36:264-269. doi:10.1097/DAD.0b013e31828b811b
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin: a clinicopathological study of 20 cases of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356. doi:10.1097/00000372-199508000-00008
- Boonchai W, Suthipinittharm P, Mahaisavariya P. Panniculitis in tuberculosis: a clinicopathologic study of nodular panniculitis associated with tuberculosis. Int J Dermatol. 1998;37:361-363. doi:10.1046/j.1365-4362.1998.00299.x
- Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851. doi:10.1016/j.jaad.2008.07.030
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathological analysis and a review of the published work. J Dermatol. 2010;37:85-93. doi:10.1111/j.1346-8138.2009.00752.x
A 66-year-old man presented with new tender erythematous papules scattered over the arms and legs. A biopsy of a lesion on the left thigh was performed.
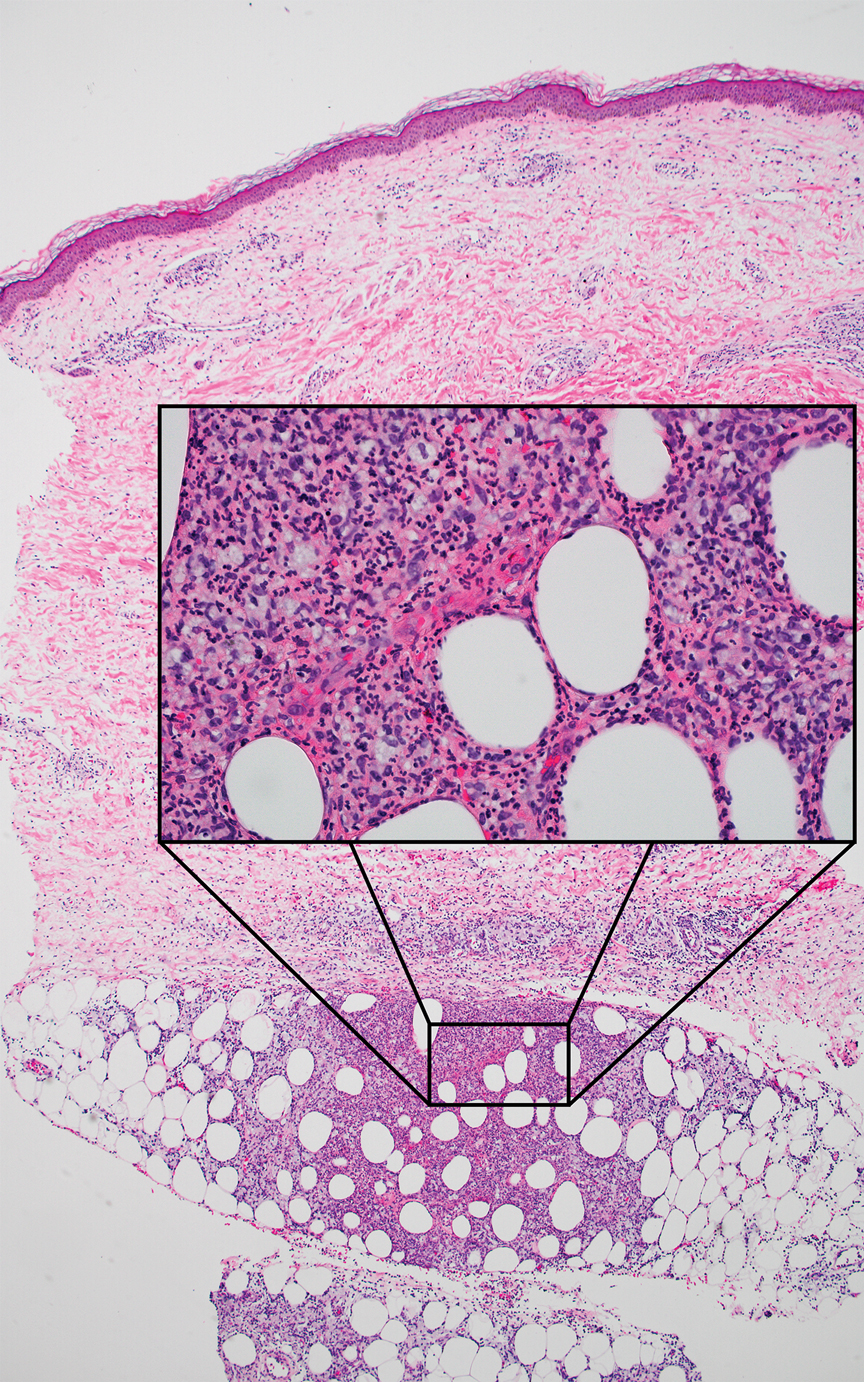
Necrotic Papules in a Pediatric Patient
The Diagnosis: Pityriasis Lichenoides et Varioliformis Acuta
Sectioned punch biopsies were performed on the patient’s right arm. Histopathology showed acanthosis and parakeratosis in the epidermis, with vacuolar degeneration and dyskeratosis in the basal layer. Dermal changes included extravasated red blood cells in the papillary dermis as well as perivascular lymphocytic infiltrates in both the papillary and reticular dermis (Figure). Direct immunofluorescence of a perilesional biopsy using anti–human IgG, IgM, IgA, C3, and fibrin conjugates showed no findings of immune deposition. Biopsy results were consistent with pityriasis lichenoides et varioliformis acuta (PLEVA), and the patient was treated with a 5-day course of oral azithromycin, triamcinolone ointment 0.1% twice daily, and phototherapy with narrowband UVB 3 times weekly. Rapid improvement was noted at 2-month follow-up.
Pityriasis lichenoides et varioliformis acuta is a form of pityriasis lichenoides, a group of inflammatory dermatoses that are characterized clinically by successive crops of morphologically diverse lesions. Epidemiologic studies have shown a slight male predominance. It primarily affects children and young adults, with peak ages of 8 and 32 years in pediatric and adult populations, respectively.1
The pathogenesis of PLEVA remains unclear. An abnormal immune response to Toxoplasma, Epstein-Barr virus, HIV, and other pathogens has been suggested based on serologic evidence of concurrent disease activity with the onset of lesions as well as cutaneous improvement in some patients after treatment of the infection.1 A T-cell lymphoproliferative etiology also has been considered based on histopathologic similarities between PLEVA and lymphomatoid papulosis (LyP) as well as findings of clonality in T-cell receptor gene rearrangement in many patients.1,2 Some clinicians consider LyP and PLEVA as separate entities on one disease spectrum.
Eruptions of PLEVA tend to favor the trunk and proximal extremities. Lesions may begin as macules measuring 2 to 3 mm in diameter that quickly evolve into papules with fine scale that remains attached centrally. Ulcerations with hemorrhagic crusts also may be noted as the lesions progress in stage. The rash may persist for weeks to years, and overlapping crops of macules and papules at varying stages of development may be seen in the same patient.1
Histopathologic findings of PLEVA include spongiosis, dyskeratosis, parakeratosis, and focal keratinocyte necrosis within the epidermis, as well as vacuolar degeneration of the basal layer. Lymphocyte and erythrocyte extravasation may extend into the epidermis. Dermal findings may include edema and wedge-shaped perivascular lymphocytic infiltrates extending into the reticular dermis.1

Important differential diagnoses to consider include LyP, mycosis fungoides (MF), pemphigus foliaceus, and varicella. Lymphomatoid papulosis is a benign CD30+ lymphoproliferative disorder that is characterized by an indolent course of recurrent, often self-resolving papules that occur most frequently on the trunk, arms, and legs of older patients. There are several histologic subtypes of LyP, but the most common (type A) may manifest with wedge-shaped perivascular lymphocytic infiltrates in the dermis, similar to PLEVA. T-cell receptor gene rearrangement studies characteristically reveal clonality in LyP, and clonality has been reported in PLEVA. However, LyP demonstrates a higher cytologic grade and lacks the characteristic parakeratotic scale and superficial dermal microhemorrhage of PLEVA.3
Mycosis fungoides is a malignant lymphoproliferative disorder that is characterized by an indolent clinical course of persistent patches, plaques, or tumors of various sizes that often manifest in non–sun-exposed areas of the skin. Early stages of MF are difficult to detect histologically, but biopsies may show atypical lymphocytes with hyperchromatic, irregularly contoured nuclei arranged along the basal layer of the epidermis. Epidermal aggregates of atypical lymphocytes (also known as Pautrier microabscesses) are considered highly specific for MF. T-cell receptor and immunopathologic studies also are important adjuncts in the diagnosis of MF.4
Pemphigus foliaceus is an autoimmune blistering disease caused by antibodies directed against desmoglein 1, which is found in the granular layer of the epidermis. It manifests with a subtle onset of scattered crusted lesions in the seborrheic areas, such as the scalp, face, chest, and upper back. Histopathologic findings of early blisters may include acantholysis and dyskeratosis in the stratum granulosum as well as vacuolization of the granular layer. The blisters may coalesce into superficial bullae containing fibrin and neutrophils. Immunofluorescence studies that demonstrate intraepidermal C3 and IgG deposition are key to the diagnosis of pemphigus.5
Varicella (also known as chickenpox) manifests with crops of vesicles on an erythematous base in a centripetal distribution favoring the trunk and proximal extremities. It often is preceded by prodromal fever, malaise, and myalgia. Histopathologic evaluation of varicella is uncommon but may reveal acantholysis, multinucleation, and nuclear margination of keratinocytes. Viral culture or nucleic acid amplification testing of lesions can be used to verify the diagnosis.6
Most cases of PLEVA resolve without intervention.7 Treatment is directed at speeding recovery, providing symptomatic relief, and limiting permanent sequelae. Topical steroids often are used to alleviate inflammation and pruritus. Systemic antibiotics such as doxycycline, minocycline, and erythromycin have been used for their anti-inflammatory properties. Phototherapy of various wavelengths, including broadband and narrowband UVB as well as psoralen plus UVA, have led to improvements in affected patients. Refractory disease may warrant consideration of therapy with methotrexate, acitretin, dapsone, or cyclosporine.7
There have been rare reports of PLEVA evolving into its potentially lethal variant, febrile ulceronecrotic Mucha-Habermann disease, which is differentiated by the presence of systemic manifestations, including high fever, sore throat, diarrhea, central nervous system symptoms, abdominal pain, interstitial pneumonitis, splenomegaly, arthritis, sepsis, megaloblastic anemia, or conjunctival ulcers. The orogenital mucosa may be affected. Cutaneous lesions may rapidly progress to large, generalized, coalescent ulcers with necrotic crusts and vasculitic features on biopsy.8 Malignant transformation of PLEVA into LyP or MF rarely may occur and warrants continued follow-up of unresolved lesions.9
- Bowers S, Warshaw EM. Pityriasis lichenoides and its subtypes. J Am Acad Dermatol. 2006;55:557-572. doi:10.1016/j.jaad.2005.07.058
- Teklehaimanot F, Gade A, Rubenstein R. Pityriasis lichenoides et varioliformis acuta (PLEVA). In: StatPearls. StatPearls Publishing; 2023.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73. doi:10.1111/jdv.15931
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Lepe K, Yarrarapu SNS, Zito PM. Pemphigus foliaceus. In: StatPearls. StatPearls Publishing; 2023.
- Ayoade F, Kumar S. Varicella zoster (chickenpox). In: StatPearls. StatPearls Publishing; 2023.
- Bellinato F, Maurelli M, Gisondi P, et al. A systematic review of treatments for pityriasis lichenoides. J Eur Acad Dermatol Venereol. 2019;33:2039-2049. doi:10.1111/jdv.15813
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738. doi:10.1111/ijd.13195
- Thomson KF, Whittaker SJ, Russell-Jones R, et al. Childhood cutaneous T-cell lymphoma in association with pityriasis lichenoides chronica. Br J Dermatol. 1999;141:1136-1152. doi:10.1046/j.1365-2133.1999.03232.x
The Diagnosis: Pityriasis Lichenoides et Varioliformis Acuta
Sectioned punch biopsies were performed on the patient’s right arm. Histopathology showed acanthosis and parakeratosis in the epidermis, with vacuolar degeneration and dyskeratosis in the basal layer. Dermal changes included extravasated red blood cells in the papillary dermis as well as perivascular lymphocytic infiltrates in both the papillary and reticular dermis (Figure). Direct immunofluorescence of a perilesional biopsy using anti–human IgG, IgM, IgA, C3, and fibrin conjugates showed no findings of immune deposition. Biopsy results were consistent with pityriasis lichenoides et varioliformis acuta (PLEVA), and the patient was treated with a 5-day course of oral azithromycin, triamcinolone ointment 0.1% twice daily, and phototherapy with narrowband UVB 3 times weekly. Rapid improvement was noted at 2-month follow-up.
Pityriasis lichenoides et varioliformis acuta is a form of pityriasis lichenoides, a group of inflammatory dermatoses that are characterized clinically by successive crops of morphologically diverse lesions. Epidemiologic studies have shown a slight male predominance. It primarily affects children and young adults, with peak ages of 8 and 32 years in pediatric and adult populations, respectively.1
The pathogenesis of PLEVA remains unclear. An abnormal immune response to Toxoplasma, Epstein-Barr virus, HIV, and other pathogens has been suggested based on serologic evidence of concurrent disease activity with the onset of lesions as well as cutaneous improvement in some patients after treatment of the infection.1 A T-cell lymphoproliferative etiology also has been considered based on histopathologic similarities between PLEVA and lymphomatoid papulosis (LyP) as well as findings of clonality in T-cell receptor gene rearrangement in many patients.1,2 Some clinicians consider LyP and PLEVA as separate entities on one disease spectrum.
Eruptions of PLEVA tend to favor the trunk and proximal extremities. Lesions may begin as macules measuring 2 to 3 mm in diameter that quickly evolve into papules with fine scale that remains attached centrally. Ulcerations with hemorrhagic crusts also may be noted as the lesions progress in stage. The rash may persist for weeks to years, and overlapping crops of macules and papules at varying stages of development may be seen in the same patient.1
Histopathologic findings of PLEVA include spongiosis, dyskeratosis, parakeratosis, and focal keratinocyte necrosis within the epidermis, as well as vacuolar degeneration of the basal layer. Lymphocyte and erythrocyte extravasation may extend into the epidermis. Dermal findings may include edema and wedge-shaped perivascular lymphocytic infiltrates extending into the reticular dermis.1
Important differential diagnoses to consider include LyP, mycosis fungoides (MF), pemphigus foliaceus, and varicella. Lymphomatoid papulosis is a benign CD30+ lymphoproliferative disorder that is characterized by an indolent course of recurrent, often self-resolving papules that occur most frequently on the trunk, arms, and legs of older patients. There are several histologic subtypes of LyP, but the most common (type A) may manifest with wedge-shaped perivascular lymphocytic infiltrates in the dermis, similar to PLEVA. T-cell receptor gene rearrangement studies characteristically reveal clonality in LyP, and clonality has been reported in PLEVA. However, LyP demonstrates a higher cytologic grade and lacks the characteristic parakeratotic scale and superficial dermal microhemorrhage of PLEVA.3
Mycosis fungoides is a malignant lymphoproliferative disorder that is characterized by an indolent clinical course of persistent patches, plaques, or tumors of various sizes that often manifest in non–sun-exposed areas of the skin. Early stages of MF are difficult to detect histologically, but biopsies may show atypical lymphocytes with hyperchromatic, irregularly contoured nuclei arranged along the basal layer of the epidermis. Epidermal aggregates of atypical lymphocytes (also known as Pautrier microabscesses) are considered highly specific for MF. T-cell receptor and immunopathologic studies also are important adjuncts in the diagnosis of MF.4
Pemphigus foliaceus is an autoimmune blistering disease caused by antibodies directed against desmoglein 1, which is found in the granular layer of the epidermis. It manifests with a subtle onset of scattered crusted lesions in the seborrheic areas, such as the scalp, face, chest, and upper back. Histopathologic findings of early blisters may include acantholysis and dyskeratosis in the stratum granulosum as well as vacuolization of the granular layer. The blisters may coalesce into superficial bullae containing fibrin and neutrophils. Immunofluorescence studies that demonstrate intraepidermal C3 and IgG deposition are key to the diagnosis of pemphigus.5
Varicella (also known as chickenpox) manifests with crops of vesicles on an erythematous base in a centripetal distribution favoring the trunk and proximal extremities. It often is preceded by prodromal fever, malaise, and myalgia. Histopathologic evaluation of varicella is uncommon but may reveal acantholysis, multinucleation, and nuclear margination of keratinocytes. Viral culture or nucleic acid amplification testing of lesions can be used to verify the diagnosis.6
Most cases of PLEVA resolve without intervention.7 Treatment is directed at speeding recovery, providing symptomatic relief, and limiting permanent sequelae. Topical steroids often are used to alleviate inflammation and pruritus. Systemic antibiotics such as doxycycline, minocycline, and erythromycin have been used for their anti-inflammatory properties. Phototherapy of various wavelengths, including broadband and narrowband UVB as well as psoralen plus UVA, have led to improvements in affected patients. Refractory disease may warrant consideration of therapy with methotrexate, acitretin, dapsone, or cyclosporine.7
There have been rare reports of PLEVA evolving into its potentially lethal variant, febrile ulceronecrotic Mucha-Habermann disease, which is differentiated by the presence of systemic manifestations, including high fever, sore throat, diarrhea, central nervous system symptoms, abdominal pain, interstitial pneumonitis, splenomegaly, arthritis, sepsis, megaloblastic anemia, or conjunctival ulcers. The orogenital mucosa may be affected. Cutaneous lesions may rapidly progress to large, generalized, coalescent ulcers with necrotic crusts and vasculitic features on biopsy.8 Malignant transformation of PLEVA into LyP or MF rarely may occur and warrants continued follow-up of unresolved lesions.9
The Diagnosis: Pityriasis Lichenoides et Varioliformis Acuta
Sectioned punch biopsies were performed on the patient’s right arm. Histopathology showed acanthosis and parakeratosis in the epidermis, with vacuolar degeneration and dyskeratosis in the basal layer. Dermal changes included extravasated red blood cells in the papillary dermis as well as perivascular lymphocytic infiltrates in both the papillary and reticular dermis (Figure). Direct immunofluorescence of a perilesional biopsy using anti–human IgG, IgM, IgA, C3, and fibrin conjugates showed no findings of immune deposition. Biopsy results were consistent with pityriasis lichenoides et varioliformis acuta (PLEVA), and the patient was treated with a 5-day course of oral azithromycin, triamcinolone ointment 0.1% twice daily, and phototherapy with narrowband UVB 3 times weekly. Rapid improvement was noted at 2-month follow-up.
Pityriasis lichenoides et varioliformis acuta is a form of pityriasis lichenoides, a group of inflammatory dermatoses that are characterized clinically by successive crops of morphologically diverse lesions. Epidemiologic studies have shown a slight male predominance. It primarily affects children and young adults, with peak ages of 8 and 32 years in pediatric and adult populations, respectively.1
The pathogenesis of PLEVA remains unclear. An abnormal immune response to Toxoplasma, Epstein-Barr virus, HIV, and other pathogens has been suggested based on serologic evidence of concurrent disease activity with the onset of lesions as well as cutaneous improvement in some patients after treatment of the infection.1 A T-cell lymphoproliferative etiology also has been considered based on histopathologic similarities between PLEVA and lymphomatoid papulosis (LyP) as well as findings of clonality in T-cell receptor gene rearrangement in many patients.1,2 Some clinicians consider LyP and PLEVA as separate entities on one disease spectrum.
Eruptions of PLEVA tend to favor the trunk and proximal extremities. Lesions may begin as macules measuring 2 to 3 mm in diameter that quickly evolve into papules with fine scale that remains attached centrally. Ulcerations with hemorrhagic crusts also may be noted as the lesions progress in stage. The rash may persist for weeks to years, and overlapping crops of macules and papules at varying stages of development may be seen in the same patient.1
Histopathologic findings of PLEVA include spongiosis, dyskeratosis, parakeratosis, and focal keratinocyte necrosis within the epidermis, as well as vacuolar degeneration of the basal layer. Lymphocyte and erythrocyte extravasation may extend into the epidermis. Dermal findings may include edema and wedge-shaped perivascular lymphocytic infiltrates extending into the reticular dermis.1
Important differential diagnoses to consider include LyP, mycosis fungoides (MF), pemphigus foliaceus, and varicella. Lymphomatoid papulosis is a benign CD30+ lymphoproliferative disorder that is characterized by an indolent course of recurrent, often self-resolving papules that occur most frequently on the trunk, arms, and legs of older patients. There are several histologic subtypes of LyP, but the most common (type A) may manifest with wedge-shaped perivascular lymphocytic infiltrates in the dermis, similar to PLEVA. T-cell receptor gene rearrangement studies characteristically reveal clonality in LyP, and clonality has been reported in PLEVA. However, LyP demonstrates a higher cytologic grade and lacks the characteristic parakeratotic scale and superficial dermal microhemorrhage of PLEVA.3
Mycosis fungoides is a malignant lymphoproliferative disorder that is characterized by an indolent clinical course of persistent patches, plaques, or tumors of various sizes that often manifest in non–sun-exposed areas of the skin. Early stages of MF are difficult to detect histologically, but biopsies may show atypical lymphocytes with hyperchromatic, irregularly contoured nuclei arranged along the basal layer of the epidermis. Epidermal aggregates of atypical lymphocytes (also known as Pautrier microabscesses) are considered highly specific for MF. T-cell receptor and immunopathologic studies also are important adjuncts in the diagnosis of MF.4
Pemphigus foliaceus is an autoimmune blistering disease caused by antibodies directed against desmoglein 1, which is found in the granular layer of the epidermis. It manifests with a subtle onset of scattered crusted lesions in the seborrheic areas, such as the scalp, face, chest, and upper back. Histopathologic findings of early blisters may include acantholysis and dyskeratosis in the stratum granulosum as well as vacuolization of the granular layer. The blisters may coalesce into superficial bullae containing fibrin and neutrophils. Immunofluorescence studies that demonstrate intraepidermal C3 and IgG deposition are key to the diagnosis of pemphigus.5
Varicella (also known as chickenpox) manifests with crops of vesicles on an erythematous base in a centripetal distribution favoring the trunk and proximal extremities. It often is preceded by prodromal fever, malaise, and myalgia. Histopathologic evaluation of varicella is uncommon but may reveal acantholysis, multinucleation, and nuclear margination of keratinocytes. Viral culture or nucleic acid amplification testing of lesions can be used to verify the diagnosis.6
Most cases of PLEVA resolve without intervention.7 Treatment is directed at speeding recovery, providing symptomatic relief, and limiting permanent sequelae. Topical steroids often are used to alleviate inflammation and pruritus. Systemic antibiotics such as doxycycline, minocycline, and erythromycin have been used for their anti-inflammatory properties. Phototherapy of various wavelengths, including broadband and narrowband UVB as well as psoralen plus UVA, have led to improvements in affected patients. Refractory disease may warrant consideration of therapy with methotrexate, acitretin, dapsone, or cyclosporine.7
There have been rare reports of PLEVA evolving into its potentially lethal variant, febrile ulceronecrotic Mucha-Habermann disease, which is differentiated by the presence of systemic manifestations, including high fever, sore throat, diarrhea, central nervous system symptoms, abdominal pain, interstitial pneumonitis, splenomegaly, arthritis, sepsis, megaloblastic anemia, or conjunctival ulcers. The orogenital mucosa may be affected. Cutaneous lesions may rapidly progress to large, generalized, coalescent ulcers with necrotic crusts and vasculitic features on biopsy.8 Malignant transformation of PLEVA into LyP or MF rarely may occur and warrants continued follow-up of unresolved lesions.9
- Bowers S, Warshaw EM. Pityriasis lichenoides and its subtypes. J Am Acad Dermatol. 2006;55:557-572. doi:10.1016/j.jaad.2005.07.058
- Teklehaimanot F, Gade A, Rubenstein R. Pityriasis lichenoides et varioliformis acuta (PLEVA). In: StatPearls. StatPearls Publishing; 2023.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73. doi:10.1111/jdv.15931
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Lepe K, Yarrarapu SNS, Zito PM. Pemphigus foliaceus. In: StatPearls. StatPearls Publishing; 2023.
- Ayoade F, Kumar S. Varicella zoster (chickenpox). In: StatPearls. StatPearls Publishing; 2023.
- Bellinato F, Maurelli M, Gisondi P, et al. A systematic review of treatments for pityriasis lichenoides. J Eur Acad Dermatol Venereol. 2019;33:2039-2049. doi:10.1111/jdv.15813
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738. doi:10.1111/ijd.13195
- Thomson KF, Whittaker SJ, Russell-Jones R, et al. Childhood cutaneous T-cell lymphoma in association with pityriasis lichenoides chronica. Br J Dermatol. 1999;141:1136-1152. doi:10.1046/j.1365-2133.1999.03232.x
- Bowers S, Warshaw EM. Pityriasis lichenoides and its subtypes. J Am Acad Dermatol. 2006;55:557-572. doi:10.1016/j.jaad.2005.07.058
- Teklehaimanot F, Gade A, Rubenstein R. Pityriasis lichenoides et varioliformis acuta (PLEVA). In: StatPearls. StatPearls Publishing; 2023.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73. doi:10.1111/jdv.15931
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Lepe K, Yarrarapu SNS, Zito PM. Pemphigus foliaceus. In: StatPearls. StatPearls Publishing; 2023.
- Ayoade F, Kumar S. Varicella zoster (chickenpox). In: StatPearls. StatPearls Publishing; 2023.
- Bellinato F, Maurelli M, Gisondi P, et al. A systematic review of treatments for pityriasis lichenoides. J Eur Acad Dermatol Venereol. 2019;33:2039-2049. doi:10.1111/jdv.15813
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738. doi:10.1111/ijd.13195
- Thomson KF, Whittaker SJ, Russell-Jones R, et al. Childhood cutaneous T-cell lymphoma in association with pityriasis lichenoides chronica. Br J Dermatol. 1999;141:1136-1152. doi:10.1046/j.1365-2133.1999.03232.x
A 7-year-old boy was referred to the dermatology clinic for evaluation of a diffuse pruritic rash of 3 months’ duration. The rash began as scant erythematous papules on the face, and crops of similar lesions later erupted on the trunk, arms, and legs. He was treated previously by a pediatrician for scabies with topical permethrin followed by 2 doses of oral ivermectin 200 μg/kg without improvement. Physical examination revealed innumerable erythematous macules and papules with centrally adherent scaling distributed on the trunk, arms, and legs, as well as scant necrotic papules with a hemorrhagic crust and a peripheral rim of scale.


Scarring Head Wound
The Diagnosis: Brunsting-Perry Cicatricial Pemphigoid
Physical examination and histopathology are paramount in diagnosing Brunsting-Perry cicatricial pemphigoid (BPCP). In our patient, histopathology showed subepidermal blistering with a mixed superficial dermal inflammatory cell infiltrate. Direct immunofluorescence was positive for linear IgG and C3 antibodies along the basement membrane. The scarring erosions on the scalp combined with the autoantibody findings on direct immunofluorescence were consistent with BPCP. He was started on dapsone 100 mg daily and demonstrated complete resolution of symptoms after 10 months, with the exception of persistent scarring hair loss (Figure).

Brunsting-Perry cicatricial pemphigoid is a rare dermatologic condition. It was first defined in 1957 when Brunsting and Perry1 examined 7 patients with cicatricial pemphigoid that predominantly affected the head and neck region, with occasional mucous membrane involvement but no mucosal scarring. Characteristically, BPCP manifests as scarring herpetiform plaques with varied blisters, erosions, crusts, and scarring.1 It primarily affects middle-aged men.2
Historically, BPCP has been considered a variant of cicatricial pemphigoid (now known as mucous membrane pemphigoid), bullous pemphigoid, or epidermolysis bullosa acquisita.3 The antigen target has not been established clearly; however, autoantibodies against laminin 332, collagen VII, and BP180 and BP230 have been proposed.2,4,5 Jacoby et al6 described BPCP on a spectrum with bullous pemphigoid and cicatricial pemphigoid, with primarily circulating autoantibodies on one end and tissue-fixed autoantibodies on the other.
The differential for BPCP also includes anti-p200 pemphigoid and anti–laminin 332 pemphigoid. Anti-p200 pemphigoid also is known as bullous pemphigoid with antibodies against the 200-kDa protein.7 It may clinically manifest similar to bullous pemphigoid and other subepidermal autoimmune blistering diseases; thus, immunopathologic differentiation can be helpful. Anti–laminin 332 pemphigoid (also known as anti–laminin gamma-1 pemphigoid) is characterized by autoantibodies targeting the laminin 332 protein in the basement membrane zone, resulting in blistering and erosions.8 Similar to BPCP and epidermolysis bullosa aquisita, anti–laminin 332 pemphigoid may affect cephalic regions and mucous membrane surfaces, resulting in scarring and cicatricial changes. Anti–laminin 332 pemphigoid also has been associated with internal malignancy.8 The use of the salt-split skin technique can be utilized to differentiate these entities based on their autoantibody-binding patterns in relation to the lamina densa.
Treatment options for mild BPCP include potent topical or intralesional steroids and dapsone, while more severe cases may require systemic therapy with rituximab, azathioprine, mycophenolate mofetil, or cyclophosphamide.4
This case highlights the importance of histopathologic examination of skin lesions with an unusual history or clinical presentation. Dermatologists should consider BPCP when presented with erosions, ulcerations, or blisters of the head and neck in middle-aged male patients.
- Brunsting LA, Perry HO. Benign pemphigoid? a report of seven cases with chronic, scarring, herpetiform plaques about the head and neck. AMA Arch Derm. 1957;75:489-501. doi:10.1001 /archderm.1957.01550160015002
- Jedlickova H, Neidermeier A, Zgažarová S, et al. Brunsting-Perry pemphigoid of the scalp with antibodies against laminin 332. Dermatology. 2011;222:193-195. doi:10.1159/000322842
- Eichhoff G. Brunsting-Perry pemphigoid as differential diagnosis of nonmelanoma skin cancer. Cureus. 2019;11:E5400. doi:10.7759/cureus.5400
- Asfour L, Chong H, Mee J, et al. Epidermolysis bullosa acquisita (Brunsting-Perry pemphigoid variant) localized to the face and diagnosed with antigen identification using skin deficient in type VII collagen. Am J Dermatopathol. 2017;39:e90-e96. doi:10.1097 /DAD.0000000000000829
- Zhou S, Zou Y, Pan M. Brunsting-Perry pemphigoid transitioning from previous bullous pemphigoid. JAAD Case Rep. 2020;6:192-194. doi:10.1016/j.jdcr.2019.12.018
- Jacoby WD Jr, Bartholome CW, Ramchand SC, et al. Cicatricial pemphigoid (Brunsting-Perry type). case report and immunofluorescence findings. Arch Dermatol. 1978;114:779-781. doi:10.1001/archderm.1978.01640170079018
- Kridin K, Ahmed AR. Anti-p200 pemphigoid: a systematic review. Front Immunol. 2019;10:2466. doi:10.3389/fimmu.2019.02466
- Shi L, Li X, Qian H. Anti-laminin 332-type mucous membrane pemphigoid. Biomolecules. 2022;12:1461. doi:10.3390/biom12101461
The Diagnosis: Brunsting-Perry Cicatricial Pemphigoid
Physical examination and histopathology are paramount in diagnosing Brunsting-Perry cicatricial pemphigoid (BPCP). In our patient, histopathology showed subepidermal blistering with a mixed superficial dermal inflammatory cell infiltrate. Direct immunofluorescence was positive for linear IgG and C3 antibodies along the basement membrane. The scarring erosions on the scalp combined with the autoantibody findings on direct immunofluorescence were consistent with BPCP. He was started on dapsone 100 mg daily and demonstrated complete resolution of symptoms after 10 months, with the exception of persistent scarring hair loss (Figure).
Brunsting-Perry cicatricial pemphigoid is a rare dermatologic condition. It was first defined in 1957 when Brunsting and Perry1 examined 7 patients with cicatricial pemphigoid that predominantly affected the head and neck region, with occasional mucous membrane involvement but no mucosal scarring. Characteristically, BPCP manifests as scarring herpetiform plaques with varied blisters, erosions, crusts, and scarring.1 It primarily affects middle-aged men.2
Historically, BPCP has been considered a variant of cicatricial pemphigoid (now known as mucous membrane pemphigoid), bullous pemphigoid, or epidermolysis bullosa acquisita.3 The antigen target has not been established clearly; however, autoantibodies against laminin 332, collagen VII, and BP180 and BP230 have been proposed.2,4,5 Jacoby et al6 described BPCP on a spectrum with bullous pemphigoid and cicatricial pemphigoid, with primarily circulating autoantibodies on one end and tissue-fixed autoantibodies on the other.
The differential for BPCP also includes anti-p200 pemphigoid and anti–laminin 332 pemphigoid. Anti-p200 pemphigoid also is known as bullous pemphigoid with antibodies against the 200-kDa protein.7 It may clinically manifest similar to bullous pemphigoid and other subepidermal autoimmune blistering diseases; thus, immunopathologic differentiation can be helpful. Anti–laminin 332 pemphigoid (also known as anti–laminin gamma-1 pemphigoid) is characterized by autoantibodies targeting the laminin 332 protein in the basement membrane zone, resulting in blistering and erosions.8 Similar to BPCP and epidermolysis bullosa aquisita, anti–laminin 332 pemphigoid may affect cephalic regions and mucous membrane surfaces, resulting in scarring and cicatricial changes. Anti–laminin 332 pemphigoid also has been associated with internal malignancy.8 The use of the salt-split skin technique can be utilized to differentiate these entities based on their autoantibody-binding patterns in relation to the lamina densa.
Treatment options for mild BPCP include potent topical or intralesional steroids and dapsone, while more severe cases may require systemic therapy with rituximab, azathioprine, mycophenolate mofetil, or cyclophosphamide.4
This case highlights the importance of histopathologic examination of skin lesions with an unusual history or clinical presentation. Dermatologists should consider BPCP when presented with erosions, ulcerations, or blisters of the head and neck in middle-aged male patients.
The Diagnosis: Brunsting-Perry Cicatricial Pemphigoid
Physical examination and histopathology are paramount in diagnosing Brunsting-Perry cicatricial pemphigoid (BPCP). In our patient, histopathology showed subepidermal blistering with a mixed superficial dermal inflammatory cell infiltrate. Direct immunofluorescence was positive for linear IgG and C3 antibodies along the basement membrane. The scarring erosions on the scalp combined with the autoantibody findings on direct immunofluorescence were consistent with BPCP. He was started on dapsone 100 mg daily and demonstrated complete resolution of symptoms after 10 months, with the exception of persistent scarring hair loss (Figure).
Brunsting-Perry cicatricial pemphigoid is a rare dermatologic condition. It was first defined in 1957 when Brunsting and Perry1 examined 7 patients with cicatricial pemphigoid that predominantly affected the head and neck region, with occasional mucous membrane involvement but no mucosal scarring. Characteristically, BPCP manifests as scarring herpetiform plaques with varied blisters, erosions, crusts, and scarring.1 It primarily affects middle-aged men.2
Historically, BPCP has been considered a variant of cicatricial pemphigoid (now known as mucous membrane pemphigoid), bullous pemphigoid, or epidermolysis bullosa acquisita.3 The antigen target has not been established clearly; however, autoantibodies against laminin 332, collagen VII, and BP180 and BP230 have been proposed.2,4,5 Jacoby et al6 described BPCP on a spectrum with bullous pemphigoid and cicatricial pemphigoid, with primarily circulating autoantibodies on one end and tissue-fixed autoantibodies on the other.
The differential for BPCP also includes anti-p200 pemphigoid and anti–laminin 332 pemphigoid. Anti-p200 pemphigoid also is known as bullous pemphigoid with antibodies against the 200-kDa protein.7 It may clinically manifest similar to bullous pemphigoid and other subepidermal autoimmune blistering diseases; thus, immunopathologic differentiation can be helpful. Anti–laminin 332 pemphigoid (also known as anti–laminin gamma-1 pemphigoid) is characterized by autoantibodies targeting the laminin 332 protein in the basement membrane zone, resulting in blistering and erosions.8 Similar to BPCP and epidermolysis bullosa aquisita, anti–laminin 332 pemphigoid may affect cephalic regions and mucous membrane surfaces, resulting in scarring and cicatricial changes. Anti–laminin 332 pemphigoid also has been associated with internal malignancy.8 The use of the salt-split skin technique can be utilized to differentiate these entities based on their autoantibody-binding patterns in relation to the lamina densa.
Treatment options for mild BPCP include potent topical or intralesional steroids and dapsone, while more severe cases may require systemic therapy with rituximab, azathioprine, mycophenolate mofetil, or cyclophosphamide.4
This case highlights the importance of histopathologic examination of skin lesions with an unusual history or clinical presentation. Dermatologists should consider BPCP when presented with erosions, ulcerations, or blisters of the head and neck in middle-aged male patients.
- Brunsting LA, Perry HO. Benign pemphigoid? a report of seven cases with chronic, scarring, herpetiform plaques about the head and neck. AMA Arch Derm. 1957;75:489-501. doi:10.1001 /archderm.1957.01550160015002
- Jedlickova H, Neidermeier A, Zgažarová S, et al. Brunsting-Perry pemphigoid of the scalp with antibodies against laminin 332. Dermatology. 2011;222:193-195. doi:10.1159/000322842
- Eichhoff G. Brunsting-Perry pemphigoid as differential diagnosis of nonmelanoma skin cancer. Cureus. 2019;11:E5400. doi:10.7759/cureus.5400
- Asfour L, Chong H, Mee J, et al. Epidermolysis bullosa acquisita (Brunsting-Perry pemphigoid variant) localized to the face and diagnosed with antigen identification using skin deficient in type VII collagen. Am J Dermatopathol. 2017;39:e90-e96. doi:10.1097 /DAD.0000000000000829
- Zhou S, Zou Y, Pan M. Brunsting-Perry pemphigoid transitioning from previous bullous pemphigoid. JAAD Case Rep. 2020;6:192-194. doi:10.1016/j.jdcr.2019.12.018
- Jacoby WD Jr, Bartholome CW, Ramchand SC, et al. Cicatricial pemphigoid (Brunsting-Perry type). case report and immunofluorescence findings. Arch Dermatol. 1978;114:779-781. doi:10.1001/archderm.1978.01640170079018
- Kridin K, Ahmed AR. Anti-p200 pemphigoid: a systematic review. Front Immunol. 2019;10:2466. doi:10.3389/fimmu.2019.02466
- Shi L, Li X, Qian H. Anti-laminin 332-type mucous membrane pemphigoid. Biomolecules. 2022;12:1461. doi:10.3390/biom12101461
- Brunsting LA, Perry HO. Benign pemphigoid? a report of seven cases with chronic, scarring, herpetiform plaques about the head and neck. AMA Arch Derm. 1957;75:489-501. doi:10.1001 /archderm.1957.01550160015002
- Jedlickova H, Neidermeier A, Zgažarová S, et al. Brunsting-Perry pemphigoid of the scalp with antibodies against laminin 332. Dermatology. 2011;222:193-195. doi:10.1159/000322842
- Eichhoff G. Brunsting-Perry pemphigoid as differential diagnosis of nonmelanoma skin cancer. Cureus. 2019;11:E5400. doi:10.7759/cureus.5400
- Asfour L, Chong H, Mee J, et al. Epidermolysis bullosa acquisita (Brunsting-Perry pemphigoid variant) localized to the face and diagnosed with antigen identification using skin deficient in type VII collagen. Am J Dermatopathol. 2017;39:e90-e96. doi:10.1097 /DAD.0000000000000829
- Zhou S, Zou Y, Pan M. Brunsting-Perry pemphigoid transitioning from previous bullous pemphigoid. JAAD Case Rep. 2020;6:192-194. doi:10.1016/j.jdcr.2019.12.018
- Jacoby WD Jr, Bartholome CW, Ramchand SC, et al. Cicatricial pemphigoid (Brunsting-Perry type). case report and immunofluorescence findings. Arch Dermatol. 1978;114:779-781. doi:10.1001/archderm.1978.01640170079018
- Kridin K, Ahmed AR. Anti-p200 pemphigoid: a systematic review. Front Immunol. 2019;10:2466. doi:10.3389/fimmu.2019.02466
- Shi L, Li X, Qian H. Anti-laminin 332-type mucous membrane pemphigoid. Biomolecules. 2022;12:1461. doi:10.3390/biom12101461
A 60-year-old man presented to a dermatology clinic with a wound on the scalp that had persisted for 11 months. The lesion started as a small erosion that eventually progressed to involve the entire parietal scalp. He had a history of type 2 diabetes mellitus, hypertension, and Graves disease. Physical examination demonstrated a large scar over the vertex scalp with central erosion, overlying crust, peripheral scalp atrophy, hypopigmentation at the periphery, and exaggerated superficial vasculature. Some oral erosions also were observed. A review of systems was negative for any constitutional symptoms. A month prior, the patient had been started on dapsone 50 mg with a prednisone taper by an outside dermatologist and noticed some improvement.


Painful Plaque on the Forearm
The Diagnosis: Mycobacterium marinum Infection
A repeat excisional biopsy showed suppurative granulomatous dermatitis with negative stains for infectious organisms; however, tissue culture grew Mycobacterium marinum. The patient had a history of exposure to fish tanks, which are a potential habitat for nontuberculous mycobacteria. These bacteria can enter the body through a minor laceration or cut in the skin, which was likely due to her occupation and pet care activities.1 Her fish tank exposure combined with the cutaneous findings of a long-standing indurated plaque with proximal nodular lymphangitis made M marinum infection the most likely diagnosis.2
Due to the limited specificity and sensitivity of patient symptoms, histologic staining, and direct microscopy, the gold standard for diagnosing acid-fast bacilli is tissue culture. 3 Tissue polymerase chain reaction testing is most useful in identifying the species of mycobacteria when histologic stains identify acid-fast bacilli but repeated tissue cultures are negative.4 With M marinum, a high clinical suspicion is needed to acquire a positive tissue culture because it needs to be grown for several weeks and at a temperature of 30 °C.5 Therefore, the physician should inform the laboratory if there is any suspicion for M marinum to increase the likelihood of obtaining a positive culture.
The differential diagnosis for M marinum infection includes other skin diseases that can cause nodular lymphangitis (also known as sporotrichoid spread) such as sporotrichosis, leishmaniasis, and certain bacterial and fungal infections. Although cat scratch disease, which is caused by Bartonella henselae, can appear similar to M marinum on histopathology, it clinically manifests with a single papulovesicular lesion at the site of inoculation that then forms a central eschar and resolves within a few weeks. Cat scratch disease typically causes painful lymphadenopathy, but it does not cause nodular lymphangitis or sporotrichoid spread.6 Sporotrichosis can have a similar clinical and histologic manifestation to M marinum infection, but the patient history typically includes exposure to Sporothrix schenckii through gardening or other contact with thorns, plants, or soil.2 Cutaneous sarcoidosis can have a similar clinical appearance to M marinum infection, but nodular lymphangitis does not occur and histopathology would demonstrate noncaseating epithelioid cell granulomas.7 Lastly, although vegetative pyoderma gangrenosum can have some of the same histologic findings as M marinum, it typically also demonstrates sinus tract formation, which was not present in our case. Additionally, vegetative pyoderma gangrenosum manifests with a verrucous and pustular plaque that would not have lymphocutaneous spread.8
Treatment of cutaneous M marinum infection is guided by antibiotic susceptibility testing. One regimen is clarithromycin (500 mg twice daily9) plus ethambutol. 10 Treatment often entails a multidrug combination due to the high rates of antibiotic resistance. Other antibiotics that potentially can be used include rifampin, trimethoprim-sulfamethoxazole, minocycline, and quinolones. The treatment duration typically is more than 3 months, and therapy is continued for 4 to 6 weeks after the skin lesions resolve.11 Excision of the lesion is reserved for patients with M marinum infection that fails to respond to antibiotic therapy.5
- Wayne LG, Sramek HA. Agents of newly recognized or infrequently encountered mycobacterial diseases. Clin Microbiol Rev. 1992;5:1-25. doi:10.1128/CMR.5.1.1
- Tobin EH, Jih WW. Sporotrichoid lymphocutaneous infections: etiology, diagnosis and therapy. Am Fam Physician. 2001;63:326-332.
- van Ingen J. Diagnosis of nontuberculous mycobacterial infections. Semin Respir Crit Care Med. 2013;34:103-109. doi:10.1055/s-0033-1333569
- Williamson H, Phillips R, Sarfo S, et al. Genetic diversity of PCR-positive, culture-negative and culture-positive Mycobacterium ulcerans isolated from Buruli ulcer patients in Ghana. PLoS One. 2014;9:E88007. doi:10.1371/journal.pone.0088007
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Baranowski K, Huang B. Cat scratch disease. StatPearls [Internet]. Updated June 12, 2023. Accessed July 15, 2024. https://www.ncbi.nlm .nih.gov/books/NBK482139/
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416. doi:10.1016/j.det.2015.03.006
- Borg Grech S, Vella Baldacchino A, Corso R, et al. Superficial granulomatous pyoderma successfully treated with intravenous immunoglobulin. Eur J Case Rep Intern Med. 2021;8:002656. doi:10.12890/2021_002656
- Krooks J, Weatherall A, Markowitz S. Complete resolution of Mycobacterium marinum infection with clarithromycin and ethambutol: a case report and a review of the literature. J Clin Aesthet Dermatol. 2018;11:48-51.
- Medel-Plaza M., Esteban J. Current treatment options for Mycobacterium marinum cutaneous infections. Expert Opin Pharmacother. 2023;24:1113-1123. doi:10.1080/14656566.2023.2211258
- Tirado-Sánchez A, Bonifaz A. Nodular lymphangitis (sporotrichoid lymphocutaneous infections): clues to differential diagnosis. J Fungi (Basel). 2018;4:56. doi:10.3390/jof4020056
The Diagnosis: Mycobacterium marinum Infection
A repeat excisional biopsy showed suppurative granulomatous dermatitis with negative stains for infectious organisms; however, tissue culture grew Mycobacterium marinum. The patient had a history of exposure to fish tanks, which are a potential habitat for nontuberculous mycobacteria. These bacteria can enter the body through a minor laceration or cut in the skin, which was likely due to her occupation and pet care activities.1 Her fish tank exposure combined with the cutaneous findings of a long-standing indurated plaque with proximal nodular lymphangitis made M marinum infection the most likely diagnosis.2
Due to the limited specificity and sensitivity of patient symptoms, histologic staining, and direct microscopy, the gold standard for diagnosing acid-fast bacilli is tissue culture. 3 Tissue polymerase chain reaction testing is most useful in identifying the species of mycobacteria when histologic stains identify acid-fast bacilli but repeated tissue cultures are negative.4 With M marinum, a high clinical suspicion is needed to acquire a positive tissue culture because it needs to be grown for several weeks and at a temperature of 30 °C.5 Therefore, the physician should inform the laboratory if there is any suspicion for M marinum to increase the likelihood of obtaining a positive culture.
The differential diagnosis for M marinum infection includes other skin diseases that can cause nodular lymphangitis (also known as sporotrichoid spread) such as sporotrichosis, leishmaniasis, and certain bacterial and fungal infections. Although cat scratch disease, which is caused by Bartonella henselae, can appear similar to M marinum on histopathology, it clinically manifests with a single papulovesicular lesion at the site of inoculation that then forms a central eschar and resolves within a few weeks. Cat scratch disease typically causes painful lymphadenopathy, but it does not cause nodular lymphangitis or sporotrichoid spread.6 Sporotrichosis can have a similar clinical and histologic manifestation to M marinum infection, but the patient history typically includes exposure to Sporothrix schenckii through gardening or other contact with thorns, plants, or soil.2 Cutaneous sarcoidosis can have a similar clinical appearance to M marinum infection, but nodular lymphangitis does not occur and histopathology would demonstrate noncaseating epithelioid cell granulomas.7 Lastly, although vegetative pyoderma gangrenosum can have some of the same histologic findings as M marinum, it typically also demonstrates sinus tract formation, which was not present in our case. Additionally, vegetative pyoderma gangrenosum manifests with a verrucous and pustular plaque that would not have lymphocutaneous spread.8
Treatment of cutaneous M marinum infection is guided by antibiotic susceptibility testing. One regimen is clarithromycin (500 mg twice daily9) plus ethambutol. 10 Treatment often entails a multidrug combination due to the high rates of antibiotic resistance. Other antibiotics that potentially can be used include rifampin, trimethoprim-sulfamethoxazole, minocycline, and quinolones. The treatment duration typically is more than 3 months, and therapy is continued for 4 to 6 weeks after the skin lesions resolve.11 Excision of the lesion is reserved for patients with M marinum infection that fails to respond to antibiotic therapy.5
The Diagnosis: Mycobacterium marinum Infection
A repeat excisional biopsy showed suppurative granulomatous dermatitis with negative stains for infectious organisms; however, tissue culture grew Mycobacterium marinum. The patient had a history of exposure to fish tanks, which are a potential habitat for nontuberculous mycobacteria. These bacteria can enter the body through a minor laceration or cut in the skin, which was likely due to her occupation and pet care activities.1 Her fish tank exposure combined with the cutaneous findings of a long-standing indurated plaque with proximal nodular lymphangitis made M marinum infection the most likely diagnosis.2
Due to the limited specificity and sensitivity of patient symptoms, histologic staining, and direct microscopy, the gold standard for diagnosing acid-fast bacilli is tissue culture. 3 Tissue polymerase chain reaction testing is most useful in identifying the species of mycobacteria when histologic stains identify acid-fast bacilli but repeated tissue cultures are negative.4 With M marinum, a high clinical suspicion is needed to acquire a positive tissue culture because it needs to be grown for several weeks and at a temperature of 30 °C.5 Therefore, the physician should inform the laboratory if there is any suspicion for M marinum to increase the likelihood of obtaining a positive culture.
The differential diagnosis for M marinum infection includes other skin diseases that can cause nodular lymphangitis (also known as sporotrichoid spread) such as sporotrichosis, leishmaniasis, and certain bacterial and fungal infections. Although cat scratch disease, which is caused by Bartonella henselae, can appear similar to M marinum on histopathology, it clinically manifests with a single papulovesicular lesion at the site of inoculation that then forms a central eschar and resolves within a few weeks. Cat scratch disease typically causes painful lymphadenopathy, but it does not cause nodular lymphangitis or sporotrichoid spread.6 Sporotrichosis can have a similar clinical and histologic manifestation to M marinum infection, but the patient history typically includes exposure to Sporothrix schenckii through gardening or other contact with thorns, plants, or soil.2 Cutaneous sarcoidosis can have a similar clinical appearance to M marinum infection, but nodular lymphangitis does not occur and histopathology would demonstrate noncaseating epithelioid cell granulomas.7 Lastly, although vegetative pyoderma gangrenosum can have some of the same histologic findings as M marinum, it typically also demonstrates sinus tract formation, which was not present in our case. Additionally, vegetative pyoderma gangrenosum manifests with a verrucous and pustular plaque that would not have lymphocutaneous spread.8
Treatment of cutaneous M marinum infection is guided by antibiotic susceptibility testing. One regimen is clarithromycin (500 mg twice daily9) plus ethambutol. 10 Treatment often entails a multidrug combination due to the high rates of antibiotic resistance. Other antibiotics that potentially can be used include rifampin, trimethoprim-sulfamethoxazole, minocycline, and quinolones. The treatment duration typically is more than 3 months, and therapy is continued for 4 to 6 weeks after the skin lesions resolve.11 Excision of the lesion is reserved for patients with M marinum infection that fails to respond to antibiotic therapy.5
- Wayne LG, Sramek HA. Agents of newly recognized or infrequently encountered mycobacterial diseases. Clin Microbiol Rev. 1992;5:1-25. doi:10.1128/CMR.5.1.1
- Tobin EH, Jih WW. Sporotrichoid lymphocutaneous infections: etiology, diagnosis and therapy. Am Fam Physician. 2001;63:326-332.
- van Ingen J. Diagnosis of nontuberculous mycobacterial infections. Semin Respir Crit Care Med. 2013;34:103-109. doi:10.1055/s-0033-1333569
- Williamson H, Phillips R, Sarfo S, et al. Genetic diversity of PCR-positive, culture-negative and culture-positive Mycobacterium ulcerans isolated from Buruli ulcer patients in Ghana. PLoS One. 2014;9:E88007. doi:10.1371/journal.pone.0088007
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Baranowski K, Huang B. Cat scratch disease. StatPearls [Internet]. Updated June 12, 2023. Accessed July 15, 2024. https://www.ncbi.nlm .nih.gov/books/NBK482139/
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416. doi:10.1016/j.det.2015.03.006
- Borg Grech S, Vella Baldacchino A, Corso R, et al. Superficial granulomatous pyoderma successfully treated with intravenous immunoglobulin. Eur J Case Rep Intern Med. 2021;8:002656. doi:10.12890/2021_002656
- Krooks J, Weatherall A, Markowitz S. Complete resolution of Mycobacterium marinum infection with clarithromycin and ethambutol: a case report and a review of the literature. J Clin Aesthet Dermatol. 2018;11:48-51.
- Medel-Plaza M., Esteban J. Current treatment options for Mycobacterium marinum cutaneous infections. Expert Opin Pharmacother. 2023;24:1113-1123. doi:10.1080/14656566.2023.2211258
- Tirado-Sánchez A, Bonifaz A. Nodular lymphangitis (sporotrichoid lymphocutaneous infections): clues to differential diagnosis. J Fungi (Basel). 2018;4:56. doi:10.3390/jof4020056
- Wayne LG, Sramek HA. Agents of newly recognized or infrequently encountered mycobacterial diseases. Clin Microbiol Rev. 1992;5:1-25. doi:10.1128/CMR.5.1.1
- Tobin EH, Jih WW. Sporotrichoid lymphocutaneous infections: etiology, diagnosis and therapy. Am Fam Physician. 2001;63:326-332.
- van Ingen J. Diagnosis of nontuberculous mycobacterial infections. Semin Respir Crit Care Med. 2013;34:103-109. doi:10.1055/s-0033-1333569
- Williamson H, Phillips R, Sarfo S, et al. Genetic diversity of PCR-positive, culture-negative and culture-positive Mycobacterium ulcerans isolated from Buruli ulcer patients in Ghana. PLoS One. 2014;9:E88007. doi:10.1371/journal.pone.0088007
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Baranowski K, Huang B. Cat scratch disease. StatPearls [Internet]. Updated June 12, 2023. Accessed July 15, 2024. https://www.ncbi.nlm .nih.gov/books/NBK482139/
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416. doi:10.1016/j.det.2015.03.006
- Borg Grech S, Vella Baldacchino A, Corso R, et al. Superficial granulomatous pyoderma successfully treated with intravenous immunoglobulin. Eur J Case Rep Intern Med. 2021;8:002656. doi:10.12890/2021_002656
- Krooks J, Weatherall A, Markowitz S. Complete resolution of Mycobacterium marinum infection with clarithromycin and ethambutol: a case report and a review of the literature. J Clin Aesthet Dermatol. 2018;11:48-51.
- Medel-Plaza M., Esteban J. Current treatment options for Mycobacterium marinum cutaneous infections. Expert Opin Pharmacother. 2023;24:1113-1123. doi:10.1080/14656566.2023.2211258
- Tirado-Sánchez A, Bonifaz A. Nodular lymphangitis (sporotrichoid lymphocutaneous infections): clues to differential diagnosis. J Fungi (Basel). 2018;4:56. doi:10.3390/jof4020056
A 30-year-old woman presented to the dermatology clinic with lesions on the right forearm of 2 years’ duration. Her medical history was unremarkable. She reported working as a chef and caring for multiple pets in her home, including 3 cats, 6 fish tanks, 3 dogs, and 3 lizards. Physical examination revealed a painful, indurated, red-violaceous plaque on the right forearm with satellite pink nodules that had been slowly migrating proximally up the forearm. An outside excisional biopsy performed 1 year prior had shown suppurative granulomatous dermatitis with negative stains for infectious organisms and negative tissue cultures. At that time, the patient was diagnosed with ruptured folliculitis; however, a subsequent lack of clinical improvement prompted her to seek a second opinion at our clinic.
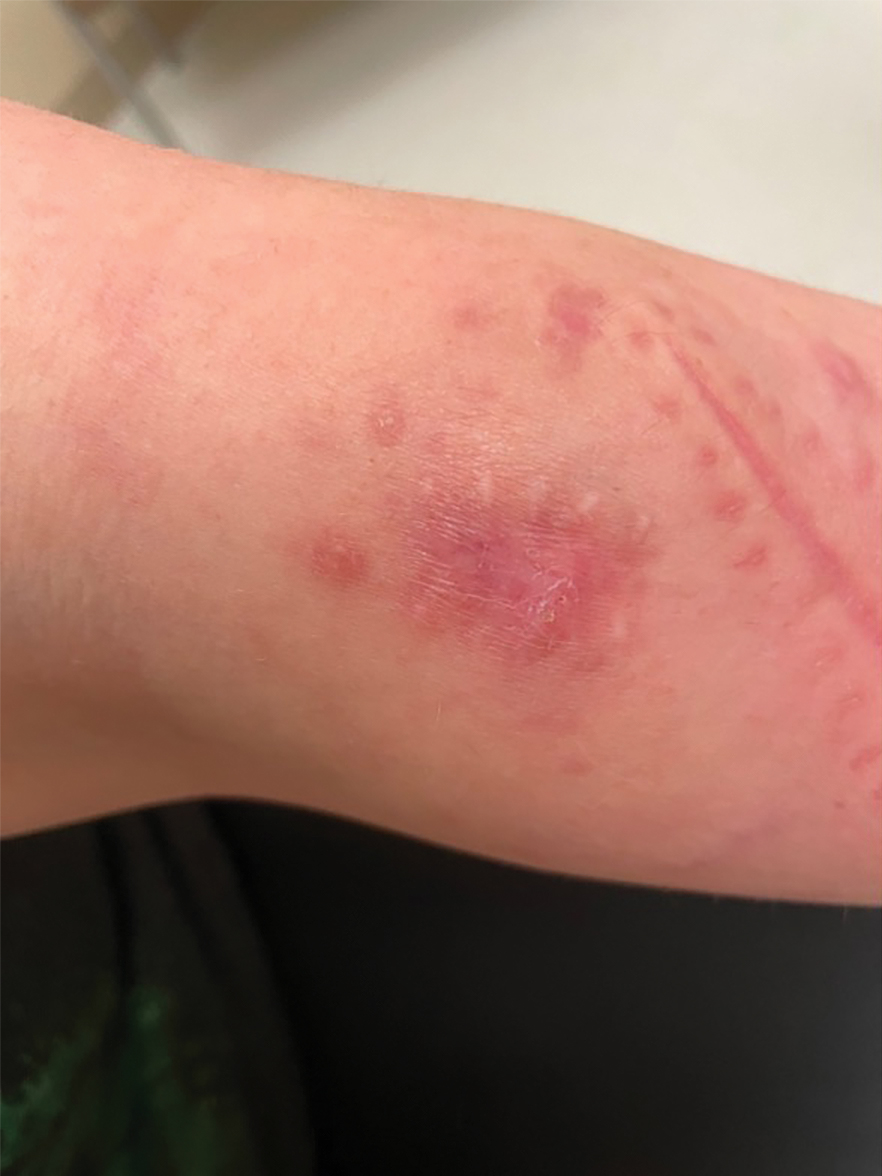
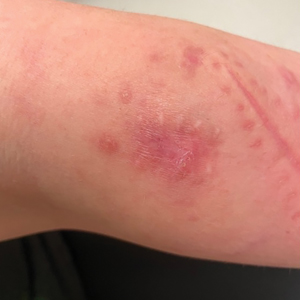
Slowly Enlarging Nodule on the Neck
The Diagnosis: Microsecretory Adenocarcinoma
Microscopically, the tumor was relatively well circumscribed but had irregular borders. It consisted of microcysts and tubules lined by flattened to plump eosinophilic cells with mildly enlarged nuclei and intraluminal basophilic secretions. Peripheral lymphocytic aggregates also were seen in the mid and deep reticular dermis. Tumor necrosis, lymphovascular invasion, and notable mitotic activity were absent. Immunohistochemistry was diffusely positive for cytokeratin (CK) 7 and CK5/6. Occasional tumor cells showed variable expression of alpha smooth muscle actin, S-100 protein, and p40 and p63 antibodies. Immunohistochemistry was negative for CK20; GATA binding protein 3; MYB proto-oncogene, transcription factor; and insulinoma-associated protein 1. A dual-color, break-apart fluorescence in situ hybridization probe identified a rearrangement of the SS18 (SYT) gene locus on chromosome 18. The nodule was excised with clear surgical margins, and the patient had no evidence of recurrent disease or metastasis at 2-year follow-up.
In recent years, there has been a growing recognition of the pivotal role played by gene fusions in driving oncogenesis, encompassing a diverse range of benign and malignant cutaneous neoplasms. These investigations have shed light on previously unknown mechanisms and pathways contributing to the pathogenesis of these neoplastic conditions, offering invaluable insights into their underlying biology. As a result, our ability to classify and diagnose these cutaneous tumors has improved. A notable example of how our current understanding has evolved is the discovery of the new cutaneous adnexal tumor microsecretory adenocarcinoma (MSA). Initially described by Bishop et al1 in 2019 as predominantly occurring in the intraoral minor salivary glands, rare instances of primary cutaneous MSA involving the head and neck regions also have been reported.2 Microsecretory adenocarcinoma represents an important addition to the group of fusion-driven tumors with both salivary gland and cutaneous adnexal analogues, characterized by a MEF2C::SS18 gene fusion. This entity is now recognized as a group of cutaneous adnexal tumors with distinct gene fusions, including both relatively recently discovered entities (eg, secretory carcinoma with NTRK fusions) and previously known entities with newly identified gene fusions (eg, poroid neoplasms with NUTM1, YAP1, or WWTR1 fusions; hidradenomatous neoplasms with CRTC1::MAML2 fusions; and adenoid cystic carcinoma with MYB, MYBL1, and/or NFIB rearrangements).3
Microsecretory adenocarcinoma exhibits a high degree of morphologic consistency, characterized by a microcystic-predominant growth pattern, uniform intercalated ductlike tumor cells with attenuated eosinophilic to clear cytoplasm, monotonous oval hyperchromatic nuclei with indistinct nucleoli, abundant basophilic luminal secretions, and a variably cellular fibromyxoid stroma. It also shows rounded borders with subtle infiltrative growth. Occasionally, pseudoepitheliomatous hyperplasia, tumor-associated lymphoid proliferation, or metaplastic bone formation may accompany MSA. Perineural invasion is rare, necrosis is absent, and mitotic rates generally are low, contributing to its distinctive histopathologic features that aid in accurate diagnosis and differentiation from other entities. Immunohistochemistry reveals diffuse positivity for CK7 and patchy to diffuse expression of S-100 in tumor cells as well as variable expression of p40 and p63. Highly specific SS18 gene translocations at chromosome 18q are useful for diagnosing MSA when found alongside its characteristic appearance, and SS18 break-apart fluorescence in situ hybridization can serve reliably as an accurate diagnostic method (Figure 1).4 Our case illustrates how molecular analysis assists in distinguishing MSA from other cutaneous adnexal tumors, exemplifying the power of our evolving understanding in refining diagnostic accuracy and guiding targeted therapies in clinical practice.
The differential diagnosis of MSA includes tubular adenoma, secretory carcinoma, cribriform tumor (previously carcinoma), and metastatic adenocarcinoma. Tubular adenoma is a rare benign neoplasm that predominantly affects females and can manifest at any age in adulthood. It typically manifests as a slow-growing, occasionally pedunculated nodule, often measuring less than 2 cm. Although it most commonly manifests on the scalp, tubular adenoma also may arise in diverse sites such as the face, axillae, lower extremities, or genitalia.
Notably, scalp lesions often are associated with nevus sebaceus of Jadassohn or syringocystadenoma papilliferum. Microscopically, tubular adenoma is well circumscribed within the dermis and may extend into the subcutis in some cases. Its distinctive appearance consists of variably sized tubules lined by a double or multilayered cuboidal to columnar epithelium, frequently displaying apocrine decapitation secretion (Figure 2). Cystic changes and intraluminal papillae devoid of true fibrovascular cores frequently are observed. Immunohistochemically, luminal epithelial cells express epithelial membrane antigen and carcinoembryonic antigen, while the myoepithelial layer expresses smooth muscle markers, p40, and S-100 protein. BRAF V600E mutation can be detected using immunohistochemistry, with excellent sensitivity and specificity using the anti-BRAF V600E antibody (clone VE1).5 Distinguishing tubular adenoma from MSA is achievable by observing its larger, more variable tubules, along with the consistent presence of a peripheral myoepithelial layer.
Secretory carcinoma is recognized as a low-grade gene fusion–driven carcinoma that primarily arises in salivary glands (both major and minor), with occasional occurrences in the breast and extremely rare instances in other locations such as the skin, thyroid gland, and lung.6 Although the axilla is the most common cutaneous site, diverse locations such as the neck, eyelids, extremities, and nipples also have been documented. Secretory carcinoma affects individuals across a wide age range (13–71 years).6 The hallmark tumors exhibit densely packed, sievelike microcystic glands and tubular spaces filled with abundant eosinophilic intraluminal secretions (Figure 3). Additionally, morphologic variants, such as predominantly papillary, papillary-cystic, macrocystic, solid, partially mucinous, and mixed-pattern neoplasms, have been described. Secretory carcinoma shares certain features with MSA; however, it is distinguished by the presence of pronounced eosinophilic secretions, plump and vacuolated cytoplasm, and a less conspicuous fibromyxoid stroma. Immunohistochemistry reveals tumor cells that are positive for CK7, SOX-10, S-100, mammaglobin, MUC4, and variably GATA-3. Genetically, secretory carcinoma exhibits distinct characteristics, commonly showing the ETV6::NTRK3 fusion, detectable through molecular techniques or pan-TRK immunohistochemistry, while RET fusions and other rare variants are less frequent.7
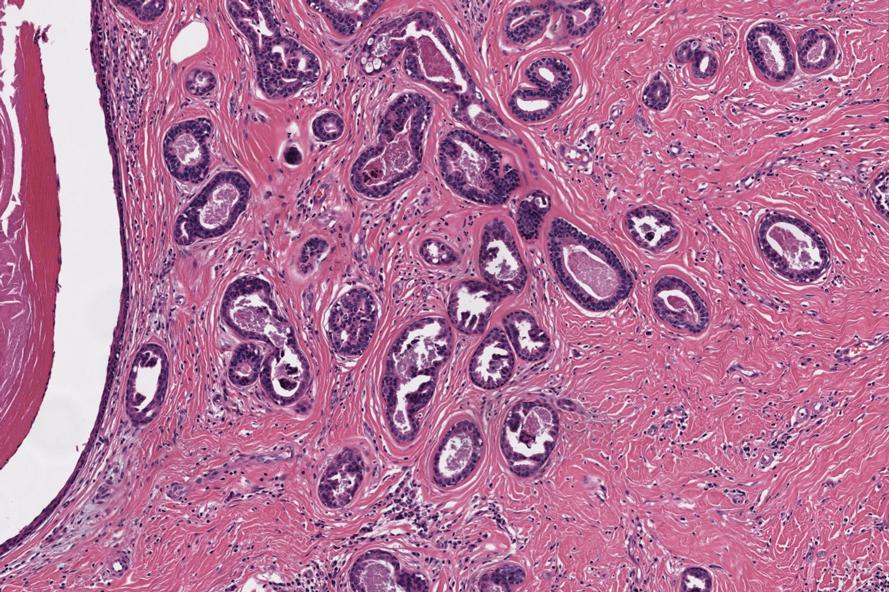
In 1998, Requena et al8 introduced the concept of primary cutaneous cribriform carcinoma. Despite initially being classified as a carcinoma, the malignant potential of this tumor remains uncertain. Consequently, the term cribriform tumor now has become the preferred terminology for denoting this rare entity.9 Primary cutaneous cribriform tumors are observed more commonly in women and typically affect individuals aged 20 to 55 years (mean, 44 years). Predominant locations include the upper and lower extremities, especially the thighs, knees, and legs, with additional cases occurring on the head and trunk. Microscopically, cribriform tumor is characterized by a partially circumscribed, unencapsulated dermal nodule composed of round or oval nuclei displaying hyperchromatism and mild pleomorphism. The defining aspect of its morphology revolves around interspersed small round cavities that give rise to the hallmark cribriform pattern (Figure 4). Although MSA occasionally may exhibit a cribriform architectural pattern, it typically lacks the distinctive feature of thin, threadlike, intraluminal bridging strands observed in cribriform tumors. Similarly, luminal cells within the cribriform tumor express CK7 and exhibit variable S-100 expression. It is recognized as an indolent neoplasm with uncertain malignant potential.
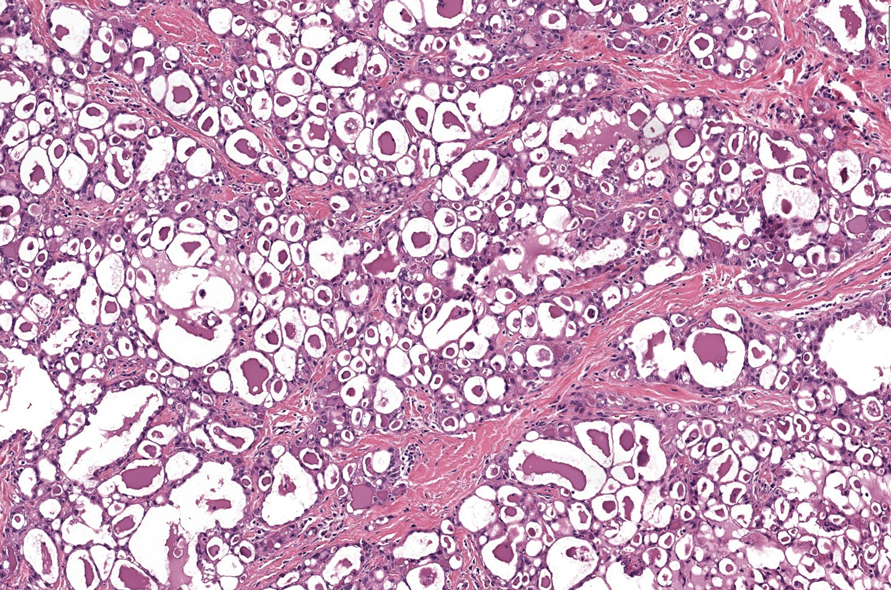
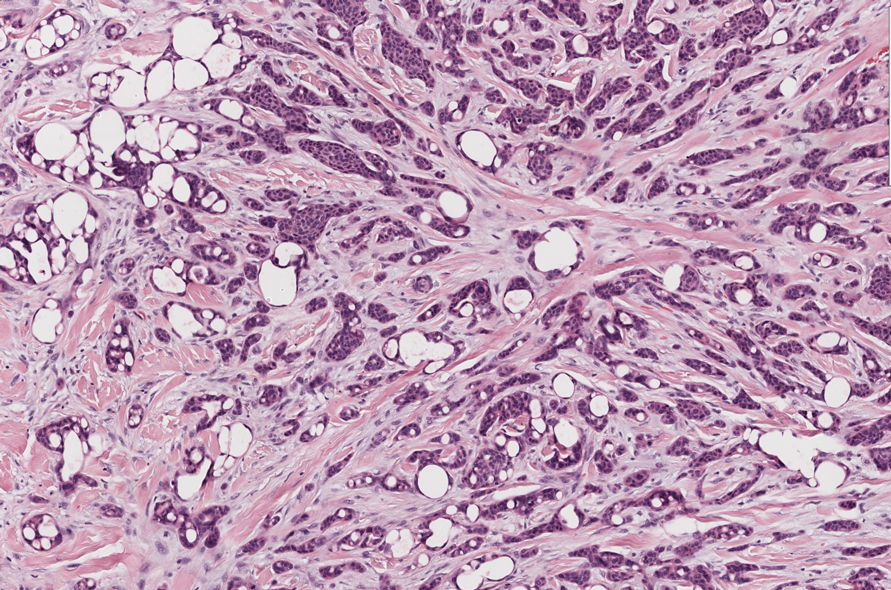
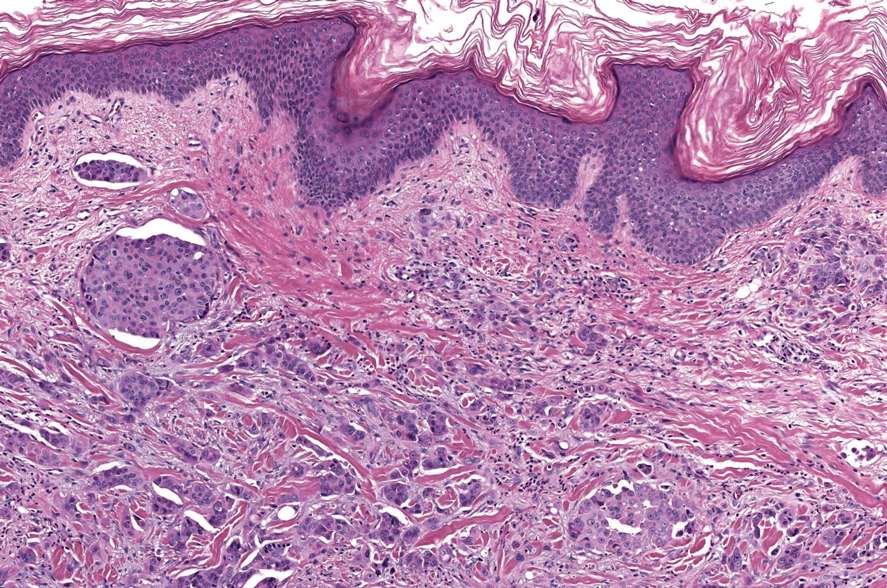
The histopathologic features of metastatic carcinomas can overlap with those of primary cutaneous tumors, particularly adnexal neoplasms.10 However, several key features can aid in the differentiation of cutaneous metastases, including a dermal-based growth pattern with or without subcutaneous involvement, the presence of multiple lesions, and the occurrence of lymphovascular invasion (Figure 5). Conversely, features that suggest a primary cutaneous adnexal neoplasm include the presence of superimposed in situ disease, carcinoma developing within a benign adnexal neoplasm, and notable stromal and/or vascular hyalinization within benign-appearing areas. In some cases, it can be difficult to determine the primary site of origin of a metastatic carcinoma to the skin based on morphologic features alone. In these cases, immunohistochemistry can be helpful. The most cost-effective and time-efficient approach to accurate diagnosis is to obtain a comprehensive clinical history. If there is a known history of cancer, a small panel of organ-specific immunohistochemical studies can be performed to confirm the diagnosis. If there is no known history, an algorithmic approach can be used to identify the primary site of origin. In all circumstances, it cannot be stressed enough that acquiring a thorough clinical history before conducting any diagnostic examinations is paramount.
- Bishop JA, Weinreb I, Swanson D, et al. Microsecretory adenocarcinoma: a novel salivary gland tumor characterized by a recurrent MEF2C-SS18 fusion. Am J Surg Pathol. 2019;43:1023-1032.
- Bishop JA, Williams EA, McLean AC, et al. Microsecretory adenocarcinoma of the skin harboring recurrent SS18 fusions: a cutaneous analog to a newly described salivary gland tumor. J Cutan Pathol. 2023;50:134-139.
- Macagno N, Sohier Pierre, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers (Basel). 2022;14:476.
- Bishop JA, Koduru P, Veremis BM, et al. SS18 break-apart fluorescence in situ hybridization is a practical and effective method for diagnosing microsecretory adenocarcinoma of salivary glands. Head Neck Pathol. 2021;15:723-726.
- Liau JY, Tsai JH, Huang WC, et al. BRAF and KRAS mutations in tubular apocrine adenoma and papillary eccrine adenoma of the skin. Hum Pathol. 2018;73:59-65.
- Chang MD, Arthur AK, Garcia JJ, et al. ETV6 rearrangement in a case of mammary analogue secretory carcinoma of the skin. J Cutan Pathol. 2016;43:1045-1049.
- Skalova A, Baneckova M, Thompson LDR, et al. Expanding the molecular spectrum of secretory carcinoma of salivary glands with a novel VIM-RET fusion. Am J Surg Pathol. 2020;44:1295-1307.
- Requena L, Kiryu H, Ackerman AB. Neoplasms With Apocrine Differentiation. Lippencott-Raven; 1998.
- Kazakov DV, Llamas-Velasco M, Fernandez-Flores A, et al. Cribriform tumour (previously carcinoma). In: WHO Classification of Tumours: Skin Tumours. 5th ed. International Agency for Research on Cancer; 2024.
- Habaermehl G, Ko J. Cutaneous metastases: a review and diagnostic approach to tumors of unknown origin. Arch Pathol Lab Med. 2019;143:943-957.
The Diagnosis: Microsecretory Adenocarcinoma
Microscopically, the tumor was relatively well circumscribed but had irregular borders. It consisted of microcysts and tubules lined by flattened to plump eosinophilic cells with mildly enlarged nuclei and intraluminal basophilic secretions. Peripheral lymphocytic aggregates also were seen in the mid and deep reticular dermis. Tumor necrosis, lymphovascular invasion, and notable mitotic activity were absent. Immunohistochemistry was diffusely positive for cytokeratin (CK) 7 and CK5/6. Occasional tumor cells showed variable expression of alpha smooth muscle actin, S-100 protein, and p40 and p63 antibodies. Immunohistochemistry was negative for CK20; GATA binding protein 3; MYB proto-oncogene, transcription factor; and insulinoma-associated protein 1. A dual-color, break-apart fluorescence in situ hybridization probe identified a rearrangement of the SS18 (SYT) gene locus on chromosome 18. The nodule was excised with clear surgical margins, and the patient had no evidence of recurrent disease or metastasis at 2-year follow-up.
In recent years, there has been a growing recognition of the pivotal role played by gene fusions in driving oncogenesis, encompassing a diverse range of benign and malignant cutaneous neoplasms. These investigations have shed light on previously unknown mechanisms and pathways contributing to the pathogenesis of these neoplastic conditions, offering invaluable insights into their underlying biology. As a result, our ability to classify and diagnose these cutaneous tumors has improved. A notable example of how our current understanding has evolved is the discovery of the new cutaneous adnexal tumor microsecretory adenocarcinoma (MSA). Initially described by Bishop et al1 in 2019 as predominantly occurring in the intraoral minor salivary glands, rare instances of primary cutaneous MSA involving the head and neck regions also have been reported.2 Microsecretory adenocarcinoma represents an important addition to the group of fusion-driven tumors with both salivary gland and cutaneous adnexal analogues, characterized by a MEF2C::SS18 gene fusion. This entity is now recognized as a group of cutaneous adnexal tumors with distinct gene fusions, including both relatively recently discovered entities (eg, secretory carcinoma with NTRK fusions) and previously known entities with newly identified gene fusions (eg, poroid neoplasms with NUTM1, YAP1, or WWTR1 fusions; hidradenomatous neoplasms with CRTC1::MAML2 fusions; and adenoid cystic carcinoma with MYB, MYBL1, and/or NFIB rearrangements).3
Microsecretory adenocarcinoma exhibits a high degree of morphologic consistency, characterized by a microcystic-predominant growth pattern, uniform intercalated ductlike tumor cells with attenuated eosinophilic to clear cytoplasm, monotonous oval hyperchromatic nuclei with indistinct nucleoli, abundant basophilic luminal secretions, and a variably cellular fibromyxoid stroma. It also shows rounded borders with subtle infiltrative growth. Occasionally, pseudoepitheliomatous hyperplasia, tumor-associated lymphoid proliferation, or metaplastic bone formation may accompany MSA. Perineural invasion is rare, necrosis is absent, and mitotic rates generally are low, contributing to its distinctive histopathologic features that aid in accurate diagnosis and differentiation from other entities. Immunohistochemistry reveals diffuse positivity for CK7 and patchy to diffuse expression of S-100 in tumor cells as well as variable expression of p40 and p63. Highly specific SS18 gene translocations at chromosome 18q are useful for diagnosing MSA when found alongside its characteristic appearance, and SS18 break-apart fluorescence in situ hybridization can serve reliably as an accurate diagnostic method (Figure 1).4 Our case illustrates how molecular analysis assists in distinguishing MSA from other cutaneous adnexal tumors, exemplifying the power of our evolving understanding in refining diagnostic accuracy and guiding targeted therapies in clinical practice.
The differential diagnosis of MSA includes tubular adenoma, secretory carcinoma, cribriform tumor (previously carcinoma), and metastatic adenocarcinoma. Tubular adenoma is a rare benign neoplasm that predominantly affects females and can manifest at any age in adulthood. It typically manifests as a slow-growing, occasionally pedunculated nodule, often measuring less than 2 cm. Although it most commonly manifests on the scalp, tubular adenoma also may arise in diverse sites such as the face, axillae, lower extremities, or genitalia.
Notably, scalp lesions often are associated with nevus sebaceus of Jadassohn or syringocystadenoma papilliferum. Microscopically, tubular adenoma is well circumscribed within the dermis and may extend into the subcutis in some cases. Its distinctive appearance consists of variably sized tubules lined by a double or multilayered cuboidal to columnar epithelium, frequently displaying apocrine decapitation secretion (Figure 2). Cystic changes and intraluminal papillae devoid of true fibrovascular cores frequently are observed. Immunohistochemically, luminal epithelial cells express epithelial membrane antigen and carcinoembryonic antigen, while the myoepithelial layer expresses smooth muscle markers, p40, and S-100 protein. BRAF V600E mutation can be detected using immunohistochemistry, with excellent sensitivity and specificity using the anti-BRAF V600E antibody (clone VE1).5 Distinguishing tubular adenoma from MSA is achievable by observing its larger, more variable tubules, along with the consistent presence of a peripheral myoepithelial layer.
Secretory carcinoma is recognized as a low-grade gene fusion–driven carcinoma that primarily arises in salivary glands (both major and minor), with occasional occurrences in the breast and extremely rare instances in other locations such as the skin, thyroid gland, and lung.6 Although the axilla is the most common cutaneous site, diverse locations such as the neck, eyelids, extremities, and nipples also have been documented. Secretory carcinoma affects individuals across a wide age range (13–71 years).6 The hallmark tumors exhibit densely packed, sievelike microcystic glands and tubular spaces filled with abundant eosinophilic intraluminal secretions (Figure 3). Additionally, morphologic variants, such as predominantly papillary, papillary-cystic, macrocystic, solid, partially mucinous, and mixed-pattern neoplasms, have been described. Secretory carcinoma shares certain features with MSA; however, it is distinguished by the presence of pronounced eosinophilic secretions, plump and vacuolated cytoplasm, and a less conspicuous fibromyxoid stroma. Immunohistochemistry reveals tumor cells that are positive for CK7, SOX-10, S-100, mammaglobin, MUC4, and variably GATA-3. Genetically, secretory carcinoma exhibits distinct characteristics, commonly showing the ETV6::NTRK3 fusion, detectable through molecular techniques or pan-TRK immunohistochemistry, while RET fusions and other rare variants are less frequent.7
In 1998, Requena et al8 introduced the concept of primary cutaneous cribriform carcinoma. Despite initially being classified as a carcinoma, the malignant potential of this tumor remains uncertain. Consequently, the term cribriform tumor now has become the preferred terminology for denoting this rare entity.9 Primary cutaneous cribriform tumors are observed more commonly in women and typically affect individuals aged 20 to 55 years (mean, 44 years). Predominant locations include the upper and lower extremities, especially the thighs, knees, and legs, with additional cases occurring on the head and trunk. Microscopically, cribriform tumor is characterized by a partially circumscribed, unencapsulated dermal nodule composed of round or oval nuclei displaying hyperchromatism and mild pleomorphism. The defining aspect of its morphology revolves around interspersed small round cavities that give rise to the hallmark cribriform pattern (Figure 4). Although MSA occasionally may exhibit a cribriform architectural pattern, it typically lacks the distinctive feature of thin, threadlike, intraluminal bridging strands observed in cribriform tumors. Similarly, luminal cells within the cribriform tumor express CK7 and exhibit variable S-100 expression. It is recognized as an indolent neoplasm with uncertain malignant potential.
The histopathologic features of metastatic carcinomas can overlap with those of primary cutaneous tumors, particularly adnexal neoplasms.10 However, several key features can aid in the differentiation of cutaneous metastases, including a dermal-based growth pattern with or without subcutaneous involvement, the presence of multiple lesions, and the occurrence of lymphovascular invasion (Figure 5). Conversely, features that suggest a primary cutaneous adnexal neoplasm include the presence of superimposed in situ disease, carcinoma developing within a benign adnexal neoplasm, and notable stromal and/or vascular hyalinization within benign-appearing areas. In some cases, it can be difficult to determine the primary site of origin of a metastatic carcinoma to the skin based on morphologic features alone. In these cases, immunohistochemistry can be helpful. The most cost-effective and time-efficient approach to accurate diagnosis is to obtain a comprehensive clinical history. If there is a known history of cancer, a small panel of organ-specific immunohistochemical studies can be performed to confirm the diagnosis. If there is no known history, an algorithmic approach can be used to identify the primary site of origin. In all circumstances, it cannot be stressed enough that acquiring a thorough clinical history before conducting any diagnostic examinations is paramount.
The Diagnosis: Microsecretory Adenocarcinoma
Microscopically, the tumor was relatively well circumscribed but had irregular borders. It consisted of microcysts and tubules lined by flattened to plump eosinophilic cells with mildly enlarged nuclei and intraluminal basophilic secretions. Peripheral lymphocytic aggregates also were seen in the mid and deep reticular dermis. Tumor necrosis, lymphovascular invasion, and notable mitotic activity were absent. Immunohistochemistry was diffusely positive for cytokeratin (CK) 7 and CK5/6. Occasional tumor cells showed variable expression of alpha smooth muscle actin, S-100 protein, and p40 and p63 antibodies. Immunohistochemistry was negative for CK20; GATA binding protein 3; MYB proto-oncogene, transcription factor; and insulinoma-associated protein 1. A dual-color, break-apart fluorescence in situ hybridization probe identified a rearrangement of the SS18 (SYT) gene locus on chromosome 18. The nodule was excised with clear surgical margins, and the patient had no evidence of recurrent disease or metastasis at 2-year follow-up.
In recent years, there has been a growing recognition of the pivotal role played by gene fusions in driving oncogenesis, encompassing a diverse range of benign and malignant cutaneous neoplasms. These investigations have shed light on previously unknown mechanisms and pathways contributing to the pathogenesis of these neoplastic conditions, offering invaluable insights into their underlying biology. As a result, our ability to classify and diagnose these cutaneous tumors has improved. A notable example of how our current understanding has evolved is the discovery of the new cutaneous adnexal tumor microsecretory adenocarcinoma (MSA). Initially described by Bishop et al1 in 2019 as predominantly occurring in the intraoral minor salivary glands, rare instances of primary cutaneous MSA involving the head and neck regions also have been reported.2 Microsecretory adenocarcinoma represents an important addition to the group of fusion-driven tumors with both salivary gland and cutaneous adnexal analogues, characterized by a MEF2C::SS18 gene fusion. This entity is now recognized as a group of cutaneous adnexal tumors with distinct gene fusions, including both relatively recently discovered entities (eg, secretory carcinoma with NTRK fusions) and previously known entities with newly identified gene fusions (eg, poroid neoplasms with NUTM1, YAP1, or WWTR1 fusions; hidradenomatous neoplasms with CRTC1::MAML2 fusions; and adenoid cystic carcinoma with MYB, MYBL1, and/or NFIB rearrangements).3
Microsecretory adenocarcinoma exhibits a high degree of morphologic consistency, characterized by a microcystic-predominant growth pattern, uniform intercalated ductlike tumor cells with attenuated eosinophilic to clear cytoplasm, monotonous oval hyperchromatic nuclei with indistinct nucleoli, abundant basophilic luminal secretions, and a variably cellular fibromyxoid stroma. It also shows rounded borders with subtle infiltrative growth. Occasionally, pseudoepitheliomatous hyperplasia, tumor-associated lymphoid proliferation, or metaplastic bone formation may accompany MSA. Perineural invasion is rare, necrosis is absent, and mitotic rates generally are low, contributing to its distinctive histopathologic features that aid in accurate diagnosis and differentiation from other entities. Immunohistochemistry reveals diffuse positivity for CK7 and patchy to diffuse expression of S-100 in tumor cells as well as variable expression of p40 and p63. Highly specific SS18 gene translocations at chromosome 18q are useful for diagnosing MSA when found alongside its characteristic appearance, and SS18 break-apart fluorescence in situ hybridization can serve reliably as an accurate diagnostic method (Figure 1).4 Our case illustrates how molecular analysis assists in distinguishing MSA from other cutaneous adnexal tumors, exemplifying the power of our evolving understanding in refining diagnostic accuracy and guiding targeted therapies in clinical practice.
The differential diagnosis of MSA includes tubular adenoma, secretory carcinoma, cribriform tumor (previously carcinoma), and metastatic adenocarcinoma. Tubular adenoma is a rare benign neoplasm that predominantly affects females and can manifest at any age in adulthood. It typically manifests as a slow-growing, occasionally pedunculated nodule, often measuring less than 2 cm. Although it most commonly manifests on the scalp, tubular adenoma also may arise in diverse sites such as the face, axillae, lower extremities, or genitalia.
Notably, scalp lesions often are associated with nevus sebaceus of Jadassohn or syringocystadenoma papilliferum. Microscopically, tubular adenoma is well circumscribed within the dermis and may extend into the subcutis in some cases. Its distinctive appearance consists of variably sized tubules lined by a double or multilayered cuboidal to columnar epithelium, frequently displaying apocrine decapitation secretion (Figure 2). Cystic changes and intraluminal papillae devoid of true fibrovascular cores frequently are observed. Immunohistochemically, luminal epithelial cells express epithelial membrane antigen and carcinoembryonic antigen, while the myoepithelial layer expresses smooth muscle markers, p40, and S-100 protein. BRAF V600E mutation can be detected using immunohistochemistry, with excellent sensitivity and specificity using the anti-BRAF V600E antibody (clone VE1).5 Distinguishing tubular adenoma from MSA is achievable by observing its larger, more variable tubules, along with the consistent presence of a peripheral myoepithelial layer.
Secretory carcinoma is recognized as a low-grade gene fusion–driven carcinoma that primarily arises in salivary glands (both major and minor), with occasional occurrences in the breast and extremely rare instances in other locations such as the skin, thyroid gland, and lung.6 Although the axilla is the most common cutaneous site, diverse locations such as the neck, eyelids, extremities, and nipples also have been documented. Secretory carcinoma affects individuals across a wide age range (13–71 years).6 The hallmark tumors exhibit densely packed, sievelike microcystic glands and tubular spaces filled with abundant eosinophilic intraluminal secretions (Figure 3). Additionally, morphologic variants, such as predominantly papillary, papillary-cystic, macrocystic, solid, partially mucinous, and mixed-pattern neoplasms, have been described. Secretory carcinoma shares certain features with MSA; however, it is distinguished by the presence of pronounced eosinophilic secretions, plump and vacuolated cytoplasm, and a less conspicuous fibromyxoid stroma. Immunohistochemistry reveals tumor cells that are positive for CK7, SOX-10, S-100, mammaglobin, MUC4, and variably GATA-3. Genetically, secretory carcinoma exhibits distinct characteristics, commonly showing the ETV6::NTRK3 fusion, detectable through molecular techniques or pan-TRK immunohistochemistry, while RET fusions and other rare variants are less frequent.7
In 1998, Requena et al8 introduced the concept of primary cutaneous cribriform carcinoma. Despite initially being classified as a carcinoma, the malignant potential of this tumor remains uncertain. Consequently, the term cribriform tumor now has become the preferred terminology for denoting this rare entity.9 Primary cutaneous cribriform tumors are observed more commonly in women and typically affect individuals aged 20 to 55 years (mean, 44 years). Predominant locations include the upper and lower extremities, especially the thighs, knees, and legs, with additional cases occurring on the head and trunk. Microscopically, cribriform tumor is characterized by a partially circumscribed, unencapsulated dermal nodule composed of round or oval nuclei displaying hyperchromatism and mild pleomorphism. The defining aspect of its morphology revolves around interspersed small round cavities that give rise to the hallmark cribriform pattern (Figure 4). Although MSA occasionally may exhibit a cribriform architectural pattern, it typically lacks the distinctive feature of thin, threadlike, intraluminal bridging strands observed in cribriform tumors. Similarly, luminal cells within the cribriform tumor express CK7 and exhibit variable S-100 expression. It is recognized as an indolent neoplasm with uncertain malignant potential.
The histopathologic features of metastatic carcinomas can overlap with those of primary cutaneous tumors, particularly adnexal neoplasms.10 However, several key features can aid in the differentiation of cutaneous metastases, including a dermal-based growth pattern with or without subcutaneous involvement, the presence of multiple lesions, and the occurrence of lymphovascular invasion (Figure 5). Conversely, features that suggest a primary cutaneous adnexal neoplasm include the presence of superimposed in situ disease, carcinoma developing within a benign adnexal neoplasm, and notable stromal and/or vascular hyalinization within benign-appearing areas. In some cases, it can be difficult to determine the primary site of origin of a metastatic carcinoma to the skin based on morphologic features alone. In these cases, immunohistochemistry can be helpful. The most cost-effective and time-efficient approach to accurate diagnosis is to obtain a comprehensive clinical history. If there is a known history of cancer, a small panel of organ-specific immunohistochemical studies can be performed to confirm the diagnosis. If there is no known history, an algorithmic approach can be used to identify the primary site of origin. In all circumstances, it cannot be stressed enough that acquiring a thorough clinical history before conducting any diagnostic examinations is paramount.
- Bishop JA, Weinreb I, Swanson D, et al. Microsecretory adenocarcinoma: a novel salivary gland tumor characterized by a recurrent MEF2C-SS18 fusion. Am J Surg Pathol. 2019;43:1023-1032.
- Bishop JA, Williams EA, McLean AC, et al. Microsecretory adenocarcinoma of the skin harboring recurrent SS18 fusions: a cutaneous analog to a newly described salivary gland tumor. J Cutan Pathol. 2023;50:134-139.
- Macagno N, Sohier Pierre, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers (Basel). 2022;14:476.
- Bishop JA, Koduru P, Veremis BM, et al. SS18 break-apart fluorescence in situ hybridization is a practical and effective method for diagnosing microsecretory adenocarcinoma of salivary glands. Head Neck Pathol. 2021;15:723-726.
- Liau JY, Tsai JH, Huang WC, et al. BRAF and KRAS mutations in tubular apocrine adenoma and papillary eccrine adenoma of the skin. Hum Pathol. 2018;73:59-65.
- Chang MD, Arthur AK, Garcia JJ, et al. ETV6 rearrangement in a case of mammary analogue secretory carcinoma of the skin. J Cutan Pathol. 2016;43:1045-1049.
- Skalova A, Baneckova M, Thompson LDR, et al. Expanding the molecular spectrum of secretory carcinoma of salivary glands with a novel VIM-RET fusion. Am J Surg Pathol. 2020;44:1295-1307.
- Requena L, Kiryu H, Ackerman AB. Neoplasms With Apocrine Differentiation. Lippencott-Raven; 1998.
- Kazakov DV, Llamas-Velasco M, Fernandez-Flores A, et al. Cribriform tumour (previously carcinoma). In: WHO Classification of Tumours: Skin Tumours. 5th ed. International Agency for Research on Cancer; 2024.
- Habaermehl G, Ko J. Cutaneous metastases: a review and diagnostic approach to tumors of unknown origin. Arch Pathol Lab Med. 2019;143:943-957.
- Bishop JA, Weinreb I, Swanson D, et al. Microsecretory adenocarcinoma: a novel salivary gland tumor characterized by a recurrent MEF2C-SS18 fusion. Am J Surg Pathol. 2019;43:1023-1032.
- Bishop JA, Williams EA, McLean AC, et al. Microsecretory adenocarcinoma of the skin harboring recurrent SS18 fusions: a cutaneous analog to a newly described salivary gland tumor. J Cutan Pathol. 2023;50:134-139.
- Macagno N, Sohier Pierre, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers (Basel). 2022;14:476.
- Bishop JA, Koduru P, Veremis BM, et al. SS18 break-apart fluorescence in situ hybridization is a practical and effective method for diagnosing microsecretory adenocarcinoma of salivary glands. Head Neck Pathol. 2021;15:723-726.
- Liau JY, Tsai JH, Huang WC, et al. BRAF and KRAS mutations in tubular apocrine adenoma and papillary eccrine adenoma of the skin. Hum Pathol. 2018;73:59-65.
- Chang MD, Arthur AK, Garcia JJ, et al. ETV6 rearrangement in a case of mammary analogue secretory carcinoma of the skin. J Cutan Pathol. 2016;43:1045-1049.
- Skalova A, Baneckova M, Thompson LDR, et al. Expanding the molecular spectrum of secretory carcinoma of salivary glands with a novel VIM-RET fusion. Am J Surg Pathol. 2020;44:1295-1307.
- Requena L, Kiryu H, Ackerman AB. Neoplasms With Apocrine Differentiation. Lippencott-Raven; 1998.
- Kazakov DV, Llamas-Velasco M, Fernandez-Flores A, et al. Cribriform tumour (previously carcinoma). In: WHO Classification of Tumours: Skin Tumours. 5th ed. International Agency for Research on Cancer; 2024.
- Habaermehl G, Ko J. Cutaneous metastases: a review and diagnostic approach to tumors of unknown origin. Arch Pathol Lab Med. 2019;143:943-957.
A 74-year-old man presented with an asymptomatic nodule on the left neck measuring approximately 2 cm. An excisional biopsy was obtained for histopathologic evaluation.