User login
Hep B vaccine response varied among youth with inflammatory, autoimmune disorders

“Hepatitis B is a common viral infection with 2 billion people worldwide having evidence of prior or current infection, and it can present as an acute or chronic infection,” or with chronic sequelae, including cirrhosis and hepatocellular carcinoma, Alexandra Ritter said during the annual meeting of the Society for Pediatric Dermatology. A three-dose vaccination series is recommended beginning at birth, and in 2016, the Centers for Disease Control and Prevention reported that 90.5% of U.S. children aged 19-35 months had completed the series.
While the vaccine series provides protection in healthy individuals more than 95% of the time, a decreased response has been noted in specific pediatric populations, including those with inflammatory and autoimmune diseases. “This is important to note and investigate further because a decreased vaccine response increases the risk for this high-risk population, and the use of boosters is currently debated,” said Ms. Ritter, who is a fourth-year student at the Medical University of South Carolina, Charleston.
To determine the percent of pediatric patients with inflammatory or autoimmune disease who lack evidence of immunity following the hepatitis B vaccine series, Ms. Ritter and colleagues Abigail Truitt and pediatric dermatologist Lara Wine Lee, MD, PhD, of MUSC, retrospectively reviewed the charts of 160 patients between the ages of 6 months and 21 years, who were diagnosed with an autoimmune or autoinflammatory disease, or inflammatory bowel disease (IBD), and had documented evidence of vaccination and serologic testing prior to the start of immunosuppressive therapy.
Of the 160 patients, 100 (63%) had IBD, 34 (21%) had an autoimmune disease, 26 (16%) had an autoinflammatory disease, 89 (56%) were female, and their mean age was 15 years.
The researchers observed variation in the testing ordered between the three patient groups. Specifically, 88.2% of autoimmune patients had hepatitis B surface antigen (HBsAg) testing, compared with 96.15% of patients with an autoinflammatory disease and 67% of patients with IBD, while 76.47% of patients with an autoimmune disease had hepatitis B core antibody (anti-HBc) testing, compared with 88.46% of patients with an autoinflammatory disease and 31% of patients with IBD.
In addition, 82.35% of patients with an autoimmune disease had HBsAg testing, compared with 100% of patients with an autoinflammatory disease and 94% of patients with IBD.
Of the 148 patients who had HBsAg testing ordered and completed prior to starting an immunosuppressive drug, there was no statistically significant difference in the percent of patients showing evidence of an immune response to the hepatitis B vaccine (32.14% among patients with an autoimmune disease, 34.62% among patients with an autoinflammatory disease, and 31.91% among patients with IBD). Combined, 67.57% of tested negative for the hepatitis B surface antibody.
“Our study showed that the majority of these patients did not show serologic evidence of immunity despite being fully vaccinated,” Ms. Ritter said. “There was also variation in the testing ordered and a more standardized approach is needed in this high-risk population.” She acknowledged certain limitations of the study, including its retrospective design and lack of a control group.
“This brings us to our next question of whether this indicates a failure of the vaccine, or the way immunity is tested,” she continued. “The CDC and the European Consensus Group on Hepatitis B Immunity recommend a cutoff of greater than 10 mIU/mL. Those that achieve immunity are protected for up to 20 years due to immune memory, even if their antibody levels later drop. There have been rare cases of immunocompetent individuals having evidence of transient asymptomatic infections when antibody levels drop. The chronic disease has only been documented in infants born to positive mothers. In hemodialysis patients, however, clinically significant infections have been documented when antibody levels drop.”
The CDC only recommends postvaccination testing to infants born to positive mothers, health care workers at high risk, hemodialysis patients, people with HIV and other immunocompromised people, and needle-sharing partners of chronically infected people. This is completed 1-2 months following the third vaccine dose, and those with antibody levels less than 10 mIU/mL should be revaccinated. “As some groups do not respond to the vaccine series, alternative dosing and the intradermal vaccine have been studied and shown to be effective in certain groups,” she said.
When it comes to monitoring immunocompromised individuals and giving booster shots, however, there are conflicting recommendations. The CDC recommends yearly testing and booster shots when levels drop below 10 mIU/mL only in hemodialysis patients, while the European Consensus Group recommends testing every 6-12 months for immunocompromised individuals and boosters when their levels drop below 10 mIU/mL.
“The CDC has not yet determined if other immunocompromised individuals should receive a booster, with more research required, but studies have shown it to be effective,” Ms. Ritter said. In a similar study looking at evidence of immunity in children with connective tissue disease who were on immunosuppressive treatment, 50% had no evidence of protective antibodies, compared with 96% in the control group. “In that study, a booster shot was given, and protective antibody concentrations were found at follow-up,” she said.
The researchers reported having no financial disclosures.
[email protected]
“Hepatitis B is a common viral infection with 2 billion people worldwide having evidence of prior or current infection, and it can present as an acute or chronic infection,” or with chronic sequelae, including cirrhosis and hepatocellular carcinoma, Alexandra Ritter said during the annual meeting of the Society for Pediatric Dermatology. A three-dose vaccination series is recommended beginning at birth, and in 2016, the Centers for Disease Control and Prevention reported that 90.5% of U.S. children aged 19-35 months had completed the series.
While the vaccine series provides protection in healthy individuals more than 95% of the time, a decreased response has been noted in specific pediatric populations, including those with inflammatory and autoimmune diseases. “This is important to note and investigate further because a decreased vaccine response increases the risk for this high-risk population, and the use of boosters is currently debated,” said Ms. Ritter, who is a fourth-year student at the Medical University of South Carolina, Charleston.
To determine the percent of pediatric patients with inflammatory or autoimmune disease who lack evidence of immunity following the hepatitis B vaccine series, Ms. Ritter and colleagues Abigail Truitt and pediatric dermatologist Lara Wine Lee, MD, PhD, of MUSC, retrospectively reviewed the charts of 160 patients between the ages of 6 months and 21 years, who were diagnosed with an autoimmune or autoinflammatory disease, or inflammatory bowel disease (IBD), and had documented evidence of vaccination and serologic testing prior to the start of immunosuppressive therapy.
Of the 160 patients, 100 (63%) had IBD, 34 (21%) had an autoimmune disease, 26 (16%) had an autoinflammatory disease, 89 (56%) were female, and their mean age was 15 years.
The researchers observed variation in the testing ordered between the three patient groups. Specifically, 88.2% of autoimmune patients had hepatitis B surface antigen (HBsAg) testing, compared with 96.15% of patients with an autoinflammatory disease and 67% of patients with IBD, while 76.47% of patients with an autoimmune disease had hepatitis B core antibody (anti-HBc) testing, compared with 88.46% of patients with an autoinflammatory disease and 31% of patients with IBD.
In addition, 82.35% of patients with an autoimmune disease had HBsAg testing, compared with 100% of patients with an autoinflammatory disease and 94% of patients with IBD.
Of the 148 patients who had HBsAg testing ordered and completed prior to starting an immunosuppressive drug, there was no statistically significant difference in the percent of patients showing evidence of an immune response to the hepatitis B vaccine (32.14% among patients with an autoimmune disease, 34.62% among patients with an autoinflammatory disease, and 31.91% among patients with IBD). Combined, 67.57% of tested negative for the hepatitis B surface antibody.
“Our study showed that the majority of these patients did not show serologic evidence of immunity despite being fully vaccinated,” Ms. Ritter said. “There was also variation in the testing ordered and a more standardized approach is needed in this high-risk population.” She acknowledged certain limitations of the study, including its retrospective design and lack of a control group.
“This brings us to our next question of whether this indicates a failure of the vaccine, or the way immunity is tested,” she continued. “The CDC and the European Consensus Group on Hepatitis B Immunity recommend a cutoff of greater than 10 mIU/mL. Those that achieve immunity are protected for up to 20 years due to immune memory, even if their antibody levels later drop. There have been rare cases of immunocompetent individuals having evidence of transient asymptomatic infections when antibody levels drop. The chronic disease has only been documented in infants born to positive mothers. In hemodialysis patients, however, clinically significant infections have been documented when antibody levels drop.”
The CDC only recommends postvaccination testing to infants born to positive mothers, health care workers at high risk, hemodialysis patients, people with HIV and other immunocompromised people, and needle-sharing partners of chronically infected people. This is completed 1-2 months following the third vaccine dose, and those with antibody levels less than 10 mIU/mL should be revaccinated. “As some groups do not respond to the vaccine series, alternative dosing and the intradermal vaccine have been studied and shown to be effective in certain groups,” she said.
When it comes to monitoring immunocompromised individuals and giving booster shots, however, there are conflicting recommendations. The CDC recommends yearly testing and booster shots when levels drop below 10 mIU/mL only in hemodialysis patients, while the European Consensus Group recommends testing every 6-12 months for immunocompromised individuals and boosters when their levels drop below 10 mIU/mL.
“The CDC has not yet determined if other immunocompromised individuals should receive a booster, with more research required, but studies have shown it to be effective,” Ms. Ritter said. In a similar study looking at evidence of immunity in children with connective tissue disease who were on immunosuppressive treatment, 50% had no evidence of protective antibodies, compared with 96% in the control group. “In that study, a booster shot was given, and protective antibody concentrations were found at follow-up,” she said.
The researchers reported having no financial disclosures.
[email protected]
“Hepatitis B is a common viral infection with 2 billion people worldwide having evidence of prior or current infection, and it can present as an acute or chronic infection,” or with chronic sequelae, including cirrhosis and hepatocellular carcinoma, Alexandra Ritter said during the annual meeting of the Society for Pediatric Dermatology. A three-dose vaccination series is recommended beginning at birth, and in 2016, the Centers for Disease Control and Prevention reported that 90.5% of U.S. children aged 19-35 months had completed the series.
While the vaccine series provides protection in healthy individuals more than 95% of the time, a decreased response has been noted in specific pediatric populations, including those with inflammatory and autoimmune diseases. “This is important to note and investigate further because a decreased vaccine response increases the risk for this high-risk population, and the use of boosters is currently debated,” said Ms. Ritter, who is a fourth-year student at the Medical University of South Carolina, Charleston.
To determine the percent of pediatric patients with inflammatory or autoimmune disease who lack evidence of immunity following the hepatitis B vaccine series, Ms. Ritter and colleagues Abigail Truitt and pediatric dermatologist Lara Wine Lee, MD, PhD, of MUSC, retrospectively reviewed the charts of 160 patients between the ages of 6 months and 21 years, who were diagnosed with an autoimmune or autoinflammatory disease, or inflammatory bowel disease (IBD), and had documented evidence of vaccination and serologic testing prior to the start of immunosuppressive therapy.
Of the 160 patients, 100 (63%) had IBD, 34 (21%) had an autoimmune disease, 26 (16%) had an autoinflammatory disease, 89 (56%) were female, and their mean age was 15 years.
The researchers observed variation in the testing ordered between the three patient groups. Specifically, 88.2% of autoimmune patients had hepatitis B surface antigen (HBsAg) testing, compared with 96.15% of patients with an autoinflammatory disease and 67% of patients with IBD, while 76.47% of patients with an autoimmune disease had hepatitis B core antibody (anti-HBc) testing, compared with 88.46% of patients with an autoinflammatory disease and 31% of patients with IBD.
In addition, 82.35% of patients with an autoimmune disease had HBsAg testing, compared with 100% of patients with an autoinflammatory disease and 94% of patients with IBD.
Of the 148 patients who had HBsAg testing ordered and completed prior to starting an immunosuppressive drug, there was no statistically significant difference in the percent of patients showing evidence of an immune response to the hepatitis B vaccine (32.14% among patients with an autoimmune disease, 34.62% among patients with an autoinflammatory disease, and 31.91% among patients with IBD). Combined, 67.57% of tested negative for the hepatitis B surface antibody.
“Our study showed that the majority of these patients did not show serologic evidence of immunity despite being fully vaccinated,” Ms. Ritter said. “There was also variation in the testing ordered and a more standardized approach is needed in this high-risk population.” She acknowledged certain limitations of the study, including its retrospective design and lack of a control group.
“This brings us to our next question of whether this indicates a failure of the vaccine, or the way immunity is tested,” she continued. “The CDC and the European Consensus Group on Hepatitis B Immunity recommend a cutoff of greater than 10 mIU/mL. Those that achieve immunity are protected for up to 20 years due to immune memory, even if their antibody levels later drop. There have been rare cases of immunocompetent individuals having evidence of transient asymptomatic infections when antibody levels drop. The chronic disease has only been documented in infants born to positive mothers. In hemodialysis patients, however, clinically significant infections have been documented when antibody levels drop.”
The CDC only recommends postvaccination testing to infants born to positive mothers, health care workers at high risk, hemodialysis patients, people with HIV and other immunocompromised people, and needle-sharing partners of chronically infected people. This is completed 1-2 months following the third vaccine dose, and those with antibody levels less than 10 mIU/mL should be revaccinated. “As some groups do not respond to the vaccine series, alternative dosing and the intradermal vaccine have been studied and shown to be effective in certain groups,” she said.
When it comes to monitoring immunocompromised individuals and giving booster shots, however, there are conflicting recommendations. The CDC recommends yearly testing and booster shots when levels drop below 10 mIU/mL only in hemodialysis patients, while the European Consensus Group recommends testing every 6-12 months for immunocompromised individuals and boosters when their levels drop below 10 mIU/mL.
“The CDC has not yet determined if other immunocompromised individuals should receive a booster, with more research required, but studies have shown it to be effective,” Ms. Ritter said. In a similar study looking at evidence of immunity in children with connective tissue disease who were on immunosuppressive treatment, 50% had no evidence of protective antibodies, compared with 96% in the control group. “In that study, a booster shot was given, and protective antibody concentrations were found at follow-up,” she said.
The researchers reported having no financial disclosures.
[email protected]
FROM SPD 2021
CAR T-cell therapy drives refractory SLE into remission
Chimeric antigen receptor T-cell (CAR T) therapy, a life-extending treatment for patients with advanced B-cell malignancies and multiple myeloma, has now been shown to be effective for treating refractory systemic lupus erythematosus (SLE) in at least one patient.

A 20-year-old woman with severe, refractory SLE, active lupus nephritis, pericarditis, and other serious symptoms had both serologic and clinical remission follow the infusion of a CAR T cell product directed against the B-cell surface antigen CD19, reported Georg Schett, MD, and colleagues from the German Center for Immunotherapy at Friedrich Alexander University Erlangen-Nuremberg in Erlangen, Germany.
“Given the role of B cells in a variety of severe autoimmune diseases, CAR T-cell therapy that targets B-cell antigens may have wider application,” they wrote in a letter to the editor of The New England Journal of Medicine.
Dr. Schett said in an email response to an interview request that the patient has remained healthy and asymptomatic without further treatment after 6 months of follow-up.
“The key question will be whether B cells return and whether these B cells will carry on to make antibodies against double-stranded DNA,” he said. “We think that the loss of B cells could be sustained given that CAR T cells are still present in the patient. The main question will be how long CAR T cells will be there and how long they deplete the B cells.”
Not just for cancer anymore
CAR T therapy involves harvesting autologous T cells and transducing them with a lentiviral vector to recognize CD19 or other B-cell surface antigens. The transduced cells are then expanded and reinfused into the patient following a lymphodepletion regimen.
There are currently five CAR T constructs approved by the Food and Drug Administration for the treatment of diffuse large B-cell lymphoma and other B-lineage lymphomas, acute lymphoblastic leukemia, multiple myeloma, and other hematologic malignancies.
For this patient, however, Dr. Schett and colleagues created their own CAR T construct rather than adapting an off-the-shelf product.
The use of this groundbreaking therapy to treat an autoimmune condition is novel, the investigators noted: “This technological breakthrough, together with recent convincing data on the role of B cells in disease pathogenesis derived from preclinical lupus models, provides a rationale for the use of CAR T-cell therapies in patients with SLE,” they wrote.
One such preclinical study was reported in Science Translational Medicine in 2019 by Marko Z. Radic, PhD, of the University of Tennessee Health Science Center in Memphis, and colleagues.

Those investigators generated CD19-targeted CAR T constructs and demonstrated that in mouse models of lupus, CD8-positive T cells from two different lupus strains could be successfully transfected, and that transfer of the CD19-targeting CAR T cells ablated both autoantibodies and CD19-positive cells.
“In both models, survival was remarkably extended, and target organs were spared. These exciting results could pave the way for using CD19-targeted T cells to treat patients with lupus,” they wrote.
Now, that prediction has come to fruition.
“It’s brilliant that the first case report has now been accomplished. I am fully convinced that this method will rid therapy refractory patients of their symptoms,” Dr. Radic said in an interview.
Anti-CD20 failures
B-cell depletion with the anti-CD20 monoclonal antibody rituximab has been shown to be an effective therapeutic strategy for patients with rheumatoid arthritis and multiple sclerosis, but was ineffective in two separate clinical trials for SLE.
“Incomplete B-cell depletion of tissue-resident B cells, or the transient nature of the treatment, may have contributed to the failure of the initial rituximab trials to attain satisfactory outcomes,” Dr. Radic and coauthors wrote.
In patients with severe lupus, autoreactive B cells may lurk in lymphatic organs and/or inflamed tissues. Alternatively, CD20-negative plasma cells, which are unaffected by rituximab, could also be a source of SLE autoantibodies, Dr. Schett and coinvestigators said.
Case details
As noted before, the 20-year-old patient described by Dr. Schett and colleagues presented with World Health Organization class IIIA active lupus nephritis, indicating focal proliferative disease. In addition, she also had nephritic syndrome, pericarditis, pleurisy, rash, and arthritis, and had a history of Libman-Sacks endocarditis.
Her disease was refractory to treatment with all the usual suspects, including hydroxychloroquine, high-dose glucocorticoids, cyclophosphamide, mycophenolate mofetil, tacrolimus, rituximab, and belimumab, another B-cell targeted agent.
The T cell collection, transduction, expansion, and infusion were all successfully performed. By day 9 following infusion, CAR T cells comprised nearly one-third of her total circulating T cells, and then began to decrease, but remained detectable in circulation for the ensuing 7 weeks.
Levels of anti–double-stranded DNA decreased from above 5,000 U/mL to 4 U/mL within 5 weeks, and her complement levels (C3 and C4) normalized.
“These signs of serologic remission were paralleled by clinical remission with proteinuria decreasing from above 2,000 mg of protein per gram of creatinine to less than 250 mg of protein per gram of creatinine,” the investigators wrote.
The patient’s SLE Disease Activity Index score with SELENA (Safety of Estrogens in Lupus National Assessment) modification dropped from 16 at baseline to 0 at follow-up.
The patient did not experience any of the adverse events that are commonly seen in patients treated with CAR T therapy, such as the cytokine release syndrome, neurotoxic adverse events, or prolonged cytopenias.
Unanswered questions
Dr. Radic said that it was unclear from the brief case report whether Dr. Schett and colleagues considered including a “kill switch” in their CAR T construct, which could be activated in the case of serious toxicities.
In addition, their use of both CD4-positive T cells in addition to CD8-positive cells in their construct raises some concern, because in patients with SLE there is evidence that CD4-positive helper T cells can be autoreactive, he noted.
The work by Dr. Schett and colleagues was supported by grants from the German government, European Union, and the Innovative Medicines Initiative. Dr. Schett reported having no conflicts of interest to disclose. Dr. Radic is listed as inventor on a patent for anti-CD19 CAR T cells in lupus.
Chimeric antigen receptor T-cell (CAR T) therapy, a life-extending treatment for patients with advanced B-cell malignancies and multiple myeloma, has now been shown to be effective for treating refractory systemic lupus erythematosus (SLE) in at least one patient.
A 20-year-old woman with severe, refractory SLE, active lupus nephritis, pericarditis, and other serious symptoms had both serologic and clinical remission follow the infusion of a CAR T cell product directed against the B-cell surface antigen CD19, reported Georg Schett, MD, and colleagues from the German Center for Immunotherapy at Friedrich Alexander University Erlangen-Nuremberg in Erlangen, Germany.
“Given the role of B cells in a variety of severe autoimmune diseases, CAR T-cell therapy that targets B-cell antigens may have wider application,” they wrote in a letter to the editor of The New England Journal of Medicine.
Dr. Schett said in an email response to an interview request that the patient has remained healthy and asymptomatic without further treatment after 6 months of follow-up.
“The key question will be whether B cells return and whether these B cells will carry on to make antibodies against double-stranded DNA,” he said. “We think that the loss of B cells could be sustained given that CAR T cells are still present in the patient. The main question will be how long CAR T cells will be there and how long they deplete the B cells.”
Not just for cancer anymore
CAR T therapy involves harvesting autologous T cells and transducing them with a lentiviral vector to recognize CD19 or other B-cell surface antigens. The transduced cells are then expanded and reinfused into the patient following a lymphodepletion regimen.
There are currently five CAR T constructs approved by the Food and Drug Administration for the treatment of diffuse large B-cell lymphoma and other B-lineage lymphomas, acute lymphoblastic leukemia, multiple myeloma, and other hematologic malignancies.
For this patient, however, Dr. Schett and colleagues created their own CAR T construct rather than adapting an off-the-shelf product.
The use of this groundbreaking therapy to treat an autoimmune condition is novel, the investigators noted: “This technological breakthrough, together with recent convincing data on the role of B cells in disease pathogenesis derived from preclinical lupus models, provides a rationale for the use of CAR T-cell therapies in patients with SLE,” they wrote.
One such preclinical study was reported in Science Translational Medicine in 2019 by Marko Z. Radic, PhD, of the University of Tennessee Health Science Center in Memphis, and colleagues.
Those investigators generated CD19-targeted CAR T constructs and demonstrated that in mouse models of lupus, CD8-positive T cells from two different lupus strains could be successfully transfected, and that transfer of the CD19-targeting CAR T cells ablated both autoantibodies and CD19-positive cells.
“In both models, survival was remarkably extended, and target organs were spared. These exciting results could pave the way for using CD19-targeted T cells to treat patients with lupus,” they wrote.
Now, that prediction has come to fruition.
“It’s brilliant that the first case report has now been accomplished. I am fully convinced that this method will rid therapy refractory patients of their symptoms,” Dr. Radic said in an interview.
Anti-CD20 failures
B-cell depletion with the anti-CD20 monoclonal antibody rituximab has been shown to be an effective therapeutic strategy for patients with rheumatoid arthritis and multiple sclerosis, but was ineffective in two separate clinical trials for SLE.
“Incomplete B-cell depletion of tissue-resident B cells, or the transient nature of the treatment, may have contributed to the failure of the initial rituximab trials to attain satisfactory outcomes,” Dr. Radic and coauthors wrote.
In patients with severe lupus, autoreactive B cells may lurk in lymphatic organs and/or inflamed tissues. Alternatively, CD20-negative plasma cells, which are unaffected by rituximab, could also be a source of SLE autoantibodies, Dr. Schett and coinvestigators said.
Case details
As noted before, the 20-year-old patient described by Dr. Schett and colleagues presented with World Health Organization class IIIA active lupus nephritis, indicating focal proliferative disease. In addition, she also had nephritic syndrome, pericarditis, pleurisy, rash, and arthritis, and had a history of Libman-Sacks endocarditis.
Her disease was refractory to treatment with all the usual suspects, including hydroxychloroquine, high-dose glucocorticoids, cyclophosphamide, mycophenolate mofetil, tacrolimus, rituximab, and belimumab, another B-cell targeted agent.
The T cell collection, transduction, expansion, and infusion were all successfully performed. By day 9 following infusion, CAR T cells comprised nearly one-third of her total circulating T cells, and then began to decrease, but remained detectable in circulation for the ensuing 7 weeks.
Levels of anti–double-stranded DNA decreased from above 5,000 U/mL to 4 U/mL within 5 weeks, and her complement levels (C3 and C4) normalized.
“These signs of serologic remission were paralleled by clinical remission with proteinuria decreasing from above 2,000 mg of protein per gram of creatinine to less than 250 mg of protein per gram of creatinine,” the investigators wrote.
The patient’s SLE Disease Activity Index score with SELENA (Safety of Estrogens in Lupus National Assessment) modification dropped from 16 at baseline to 0 at follow-up.
The patient did not experience any of the adverse events that are commonly seen in patients treated with CAR T therapy, such as the cytokine release syndrome, neurotoxic adverse events, or prolonged cytopenias.
Unanswered questions
Dr. Radic said that it was unclear from the brief case report whether Dr. Schett and colleagues considered including a “kill switch” in their CAR T construct, which could be activated in the case of serious toxicities.
In addition, their use of both CD4-positive T cells in addition to CD8-positive cells in their construct raises some concern, because in patients with SLE there is evidence that CD4-positive helper T cells can be autoreactive, he noted.
The work by Dr. Schett and colleagues was supported by grants from the German government, European Union, and the Innovative Medicines Initiative. Dr. Schett reported having no conflicts of interest to disclose. Dr. Radic is listed as inventor on a patent for anti-CD19 CAR T cells in lupus.
Chimeric antigen receptor T-cell (CAR T) therapy, a life-extending treatment for patients with advanced B-cell malignancies and multiple myeloma, has now been shown to be effective for treating refractory systemic lupus erythematosus (SLE) in at least one patient.
A 20-year-old woman with severe, refractory SLE, active lupus nephritis, pericarditis, and other serious symptoms had both serologic and clinical remission follow the infusion of a CAR T cell product directed against the B-cell surface antigen CD19, reported Georg Schett, MD, and colleagues from the German Center for Immunotherapy at Friedrich Alexander University Erlangen-Nuremberg in Erlangen, Germany.
“Given the role of B cells in a variety of severe autoimmune diseases, CAR T-cell therapy that targets B-cell antigens may have wider application,” they wrote in a letter to the editor of The New England Journal of Medicine.
Dr. Schett said in an email response to an interview request that the patient has remained healthy and asymptomatic without further treatment after 6 months of follow-up.
“The key question will be whether B cells return and whether these B cells will carry on to make antibodies against double-stranded DNA,” he said. “We think that the loss of B cells could be sustained given that CAR T cells are still present in the patient. The main question will be how long CAR T cells will be there and how long they deplete the B cells.”
Not just for cancer anymore
CAR T therapy involves harvesting autologous T cells and transducing them with a lentiviral vector to recognize CD19 or other B-cell surface antigens. The transduced cells are then expanded and reinfused into the patient following a lymphodepletion regimen.
There are currently five CAR T constructs approved by the Food and Drug Administration for the treatment of diffuse large B-cell lymphoma and other B-lineage lymphomas, acute lymphoblastic leukemia, multiple myeloma, and other hematologic malignancies.
For this patient, however, Dr. Schett and colleagues created their own CAR T construct rather than adapting an off-the-shelf product.
The use of this groundbreaking therapy to treat an autoimmune condition is novel, the investigators noted: “This technological breakthrough, together with recent convincing data on the role of B cells in disease pathogenesis derived from preclinical lupus models, provides a rationale for the use of CAR T-cell therapies in patients with SLE,” they wrote.
One such preclinical study was reported in Science Translational Medicine in 2019 by Marko Z. Radic, PhD, of the University of Tennessee Health Science Center in Memphis, and colleagues.
Those investigators generated CD19-targeted CAR T constructs and demonstrated that in mouse models of lupus, CD8-positive T cells from two different lupus strains could be successfully transfected, and that transfer of the CD19-targeting CAR T cells ablated both autoantibodies and CD19-positive cells.
“In both models, survival was remarkably extended, and target organs were spared. These exciting results could pave the way for using CD19-targeted T cells to treat patients with lupus,” they wrote.
Now, that prediction has come to fruition.
“It’s brilliant that the first case report has now been accomplished. I am fully convinced that this method will rid therapy refractory patients of their symptoms,” Dr. Radic said in an interview.
Anti-CD20 failures
B-cell depletion with the anti-CD20 monoclonal antibody rituximab has been shown to be an effective therapeutic strategy for patients with rheumatoid arthritis and multiple sclerosis, but was ineffective in two separate clinical trials for SLE.
“Incomplete B-cell depletion of tissue-resident B cells, or the transient nature of the treatment, may have contributed to the failure of the initial rituximab trials to attain satisfactory outcomes,” Dr. Radic and coauthors wrote.
In patients with severe lupus, autoreactive B cells may lurk in lymphatic organs and/or inflamed tissues. Alternatively, CD20-negative plasma cells, which are unaffected by rituximab, could also be a source of SLE autoantibodies, Dr. Schett and coinvestigators said.
Case details
As noted before, the 20-year-old patient described by Dr. Schett and colleagues presented with World Health Organization class IIIA active lupus nephritis, indicating focal proliferative disease. In addition, she also had nephritic syndrome, pericarditis, pleurisy, rash, and arthritis, and had a history of Libman-Sacks endocarditis.
Her disease was refractory to treatment with all the usual suspects, including hydroxychloroquine, high-dose glucocorticoids, cyclophosphamide, mycophenolate mofetil, tacrolimus, rituximab, and belimumab, another B-cell targeted agent.
The T cell collection, transduction, expansion, and infusion were all successfully performed. By day 9 following infusion, CAR T cells comprised nearly one-third of her total circulating T cells, and then began to decrease, but remained detectable in circulation for the ensuing 7 weeks.
Levels of anti–double-stranded DNA decreased from above 5,000 U/mL to 4 U/mL within 5 weeks, and her complement levels (C3 and C4) normalized.
“These signs of serologic remission were paralleled by clinical remission with proteinuria decreasing from above 2,000 mg of protein per gram of creatinine to less than 250 mg of protein per gram of creatinine,” the investigators wrote.
The patient’s SLE Disease Activity Index score with SELENA (Safety of Estrogens in Lupus National Assessment) modification dropped from 16 at baseline to 0 at follow-up.
The patient did not experience any of the adverse events that are commonly seen in patients treated with CAR T therapy, such as the cytokine release syndrome, neurotoxic adverse events, or prolonged cytopenias.
Unanswered questions
Dr. Radic said that it was unclear from the brief case report whether Dr. Schett and colleagues considered including a “kill switch” in their CAR T construct, which could be activated in the case of serious toxicities.
In addition, their use of both CD4-positive T cells in addition to CD8-positive cells in their construct raises some concern, because in patients with SLE there is evidence that CD4-positive helper T cells can be autoreactive, he noted.
The work by Dr. Schett and colleagues was supported by grants from the German government, European Union, and the Innovative Medicines Initiative. Dr. Schett reported having no conflicts of interest to disclose. Dr. Radic is listed as inventor on a patent for anti-CD19 CAR T cells in lupus.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
FDA approves anifrolumab (Saphnelo) as first new lupus treatment in more than 10 years
Anifrolumab, an inhibitor of type 1 interferons, received approval from the Food and Drug Administration for the treatment of adults with moderate to severe systemic lupus erythematosus (SLE) who are receiving standard therapy, according to a statement released Aug. 2 from its manufacturer, AstraZeneca.

Anifrolumab will be marketed as Saphnelo. It is a fully human monoclonal antibody against subunit 1 of the type 1 interferon receptor, and its approval represents the only new treatment approved for patients with SLE in a decade. The recommended dosage is 300 mg as an intravenous infusion over a 30-minute period every 4 weeks, according to its prescribing information, and it will be sold in a single-dose vial containing 300 mg/2 mL (150 mg/mL).
Increased type I interferon (IFN) signaling is associated with increased disease activity in patients with SLE, and the option of a type I IFN receptor antagonist may allow physicians to treat patients with fewer corticosteroids, according to the statement.
The approval was based on data from three trials. The TULIP (Treatment of Uncontrolled Lupus via the Interferon Pathway) phase 3 research included two randomized, double-blind, placebo-controlled studies, TULIP-1 and TULIP-2. The TULIP trials each enrolled seropositive patients with moderate to severe active disease despite standard-of-care therapy (SOC), which included oral corticosteroids, antimalarials, and immunosuppressants (methotrexate, azathioprine, or mycophenolate mofetil). All patients met American College of Rheumatology criteria and had an SLE Disease Activity Index (SLEDAI)-2K of 6 or greater, as well as British Isles Lupus Assessment Group (BILAG) index scoring showing one or more organ systems with grade A involvement or two or more with grade B. Both trials required stable SOC therapy throughout the study except for mandatory attempts at oral corticosteroid tapering for patients who were receiving 10 mg/day or more of prednisone or its equivalent at study entry.
TULIP-1 failed to meet its primary endpoint of SLE Responder Index (SRI) at 52 weeks, but investigators determined after the trial that some patients taking anifrolumab had been inappropriately labeled as nonresponders because the trial automatically required any patient who used a restricted drug, including NSAIDs, to be classified as a nonresponder even if they used the medication for something unrelated to SLE. When these rules were amended in a post hoc analysis, differences between the groups treated with anifrolumab and placebo widened in secondary endpoints for oral corticosteroid dose reduction, Cutaneous Lupus Erythematosus Disease Activity Severity Index response, and BILAG-Based Composite Lupus Assessment (BICLA) response.
The TULIP-2 trial included 362 patients who received a fixed dose of 300 mg anifrolumab or a placebo intravenously every 4 weeks for 48 weeks. In this study, anifrolumab patients showed significant improvement in disease activity on the BICLA scale, compared with placebo patients. The BICLA response was 47.8% in patients taking anifrolumab and 31.5% in placebo-treated patients (P = .001).
In the MUSE phase 2 trial, 305 adults with SLE were randomized to a fixed-dose intravenous infusion of 300 mg or 1,000 mg of anifrolumab or a placebo every 4 weeks, plus SOC, for 48 weeks. Patients in this study showed significant improvement on either dose, compared with placebo.
The results from the MUSE trial were published online in Arthritis & Rheumatology Nov. 7, 2016, followed by the TULIP-1 trial in The Lancet Rheumatology Nov. 11, 2019, and the TULIP-2 trial in the New England Journal of Medicine Jan. 16, 2020.
The most common treatment-related adverse events in all three studies were nasopharyngitis, upper respiratory tract infection, bronchitis, infusion-related reactions, herpes zoster, and cough. Infusion-related reactions in the trials were similar in anifrolumab and placebo patients, and included headache, nausea, vomiting, fatigue, and dizziness.
Anifrolumab has not been evaluated in patients with severe active lupus nephritis or severe active central nervous system lupus and is not recommended for these patients, according to the statement.
AstraZeneca said in its statement that anifrolumab is also under regulatory review in Japan and the European Union, and it continues to evaluate anifrolumab in patients with SLE in a long-term extension phase 3 trial and a phase 3 trial assessing subcutaneous delivery. The company said it “is exploring the potential of Saphnelo in a variety of diseases where type I IFN plays a key role, including lupus nephritis, cutaneous lupus erythematosus, and myositis.”
Anifrolumab, an inhibitor of type 1 interferons, received approval from the Food and Drug Administration for the treatment of adults with moderate to severe systemic lupus erythematosus (SLE) who are receiving standard therapy, according to a statement released Aug. 2 from its manufacturer, AstraZeneca.
Anifrolumab will be marketed as Saphnelo. It is a fully human monoclonal antibody against subunit 1 of the type 1 interferon receptor, and its approval represents the only new treatment approved for patients with SLE in a decade. The recommended dosage is 300 mg as an intravenous infusion over a 30-minute period every 4 weeks, according to its prescribing information, and it will be sold in a single-dose vial containing 300 mg/2 mL (150 mg/mL).
Increased type I interferon (IFN) signaling is associated with increased disease activity in patients with SLE, and the option of a type I IFN receptor antagonist may allow physicians to treat patients with fewer corticosteroids, according to the statement.
The approval was based on data from three trials. The TULIP (Treatment of Uncontrolled Lupus via the Interferon Pathway) phase 3 research included two randomized, double-blind, placebo-controlled studies, TULIP-1 and TULIP-2. The TULIP trials each enrolled seropositive patients with moderate to severe active disease despite standard-of-care therapy (SOC), which included oral corticosteroids, antimalarials, and immunosuppressants (methotrexate, azathioprine, or mycophenolate mofetil). All patients met American College of Rheumatology criteria and had an SLE Disease Activity Index (SLEDAI)-2K of 6 or greater, as well as British Isles Lupus Assessment Group (BILAG) index scoring showing one or more organ systems with grade A involvement or two or more with grade B. Both trials required stable SOC therapy throughout the study except for mandatory attempts at oral corticosteroid tapering for patients who were receiving 10 mg/day or more of prednisone or its equivalent at study entry.
TULIP-1 failed to meet its primary endpoint of SLE Responder Index (SRI) at 52 weeks, but investigators determined after the trial that some patients taking anifrolumab had been inappropriately labeled as nonresponders because the trial automatically required any patient who used a restricted drug, including NSAIDs, to be classified as a nonresponder even if they used the medication for something unrelated to SLE. When these rules were amended in a post hoc analysis, differences between the groups treated with anifrolumab and placebo widened in secondary endpoints for oral corticosteroid dose reduction, Cutaneous Lupus Erythematosus Disease Activity Severity Index response, and BILAG-Based Composite Lupus Assessment (BICLA) response.
The TULIP-2 trial included 362 patients who received a fixed dose of 300 mg anifrolumab or a placebo intravenously every 4 weeks for 48 weeks. In this study, anifrolumab patients showed significant improvement in disease activity on the BICLA scale, compared with placebo patients. The BICLA response was 47.8% in patients taking anifrolumab and 31.5% in placebo-treated patients (P = .001).
In the MUSE phase 2 trial, 305 adults with SLE were randomized to a fixed-dose intravenous infusion of 300 mg or 1,000 mg of anifrolumab or a placebo every 4 weeks, plus SOC, for 48 weeks. Patients in this study showed significant improvement on either dose, compared with placebo.
The results from the MUSE trial were published online in Arthritis & Rheumatology Nov. 7, 2016, followed by the TULIP-1 trial in The Lancet Rheumatology Nov. 11, 2019, and the TULIP-2 trial in the New England Journal of Medicine Jan. 16, 2020.
The most common treatment-related adverse events in all three studies were nasopharyngitis, upper respiratory tract infection, bronchitis, infusion-related reactions, herpes zoster, and cough. Infusion-related reactions in the trials were similar in anifrolumab and placebo patients, and included headache, nausea, vomiting, fatigue, and dizziness.
Anifrolumab has not been evaluated in patients with severe active lupus nephritis or severe active central nervous system lupus and is not recommended for these patients, according to the statement.
AstraZeneca said in its statement that anifrolumab is also under regulatory review in Japan and the European Union, and it continues to evaluate anifrolumab in patients with SLE in a long-term extension phase 3 trial and a phase 3 trial assessing subcutaneous delivery. The company said it “is exploring the potential of Saphnelo in a variety of diseases where type I IFN plays a key role, including lupus nephritis, cutaneous lupus erythematosus, and myositis.”
Anifrolumab, an inhibitor of type 1 interferons, received approval from the Food and Drug Administration for the treatment of adults with moderate to severe systemic lupus erythematosus (SLE) who are receiving standard therapy, according to a statement released Aug. 2 from its manufacturer, AstraZeneca.
Anifrolumab will be marketed as Saphnelo. It is a fully human monoclonal antibody against subunit 1 of the type 1 interferon receptor, and its approval represents the only new treatment approved for patients with SLE in a decade. The recommended dosage is 300 mg as an intravenous infusion over a 30-minute period every 4 weeks, according to its prescribing information, and it will be sold in a single-dose vial containing 300 mg/2 mL (150 mg/mL).
Increased type I interferon (IFN) signaling is associated with increased disease activity in patients with SLE, and the option of a type I IFN receptor antagonist may allow physicians to treat patients with fewer corticosteroids, according to the statement.
The approval was based on data from three trials. The TULIP (Treatment of Uncontrolled Lupus via the Interferon Pathway) phase 3 research included two randomized, double-blind, placebo-controlled studies, TULIP-1 and TULIP-2. The TULIP trials each enrolled seropositive patients with moderate to severe active disease despite standard-of-care therapy (SOC), which included oral corticosteroids, antimalarials, and immunosuppressants (methotrexate, azathioprine, or mycophenolate mofetil). All patients met American College of Rheumatology criteria and had an SLE Disease Activity Index (SLEDAI)-2K of 6 or greater, as well as British Isles Lupus Assessment Group (BILAG) index scoring showing one or more organ systems with grade A involvement or two or more with grade B. Both trials required stable SOC therapy throughout the study except for mandatory attempts at oral corticosteroid tapering for patients who were receiving 10 mg/day or more of prednisone or its equivalent at study entry.
TULIP-1 failed to meet its primary endpoint of SLE Responder Index (SRI) at 52 weeks, but investigators determined after the trial that some patients taking anifrolumab had been inappropriately labeled as nonresponders because the trial automatically required any patient who used a restricted drug, including NSAIDs, to be classified as a nonresponder even if they used the medication for something unrelated to SLE. When these rules were amended in a post hoc analysis, differences between the groups treated with anifrolumab and placebo widened in secondary endpoints for oral corticosteroid dose reduction, Cutaneous Lupus Erythematosus Disease Activity Severity Index response, and BILAG-Based Composite Lupus Assessment (BICLA) response.
The TULIP-2 trial included 362 patients who received a fixed dose of 300 mg anifrolumab or a placebo intravenously every 4 weeks for 48 weeks. In this study, anifrolumab patients showed significant improvement in disease activity on the BICLA scale, compared with placebo patients. The BICLA response was 47.8% in patients taking anifrolumab and 31.5% in placebo-treated patients (P = .001).
In the MUSE phase 2 trial, 305 adults with SLE were randomized to a fixed-dose intravenous infusion of 300 mg or 1,000 mg of anifrolumab or a placebo every 4 weeks, plus SOC, for 48 weeks. Patients in this study showed significant improvement on either dose, compared with placebo.
The results from the MUSE trial were published online in Arthritis & Rheumatology Nov. 7, 2016, followed by the TULIP-1 trial in The Lancet Rheumatology Nov. 11, 2019, and the TULIP-2 trial in the New England Journal of Medicine Jan. 16, 2020.
The most common treatment-related adverse events in all three studies were nasopharyngitis, upper respiratory tract infection, bronchitis, infusion-related reactions, herpes zoster, and cough. Infusion-related reactions in the trials were similar in anifrolumab and placebo patients, and included headache, nausea, vomiting, fatigue, and dizziness.
Anifrolumab has not been evaluated in patients with severe active lupus nephritis or severe active central nervous system lupus and is not recommended for these patients, according to the statement.
AstraZeneca said in its statement that anifrolumab is also under regulatory review in Japan and the European Union, and it continues to evaluate anifrolumab in patients with SLE in a long-term extension phase 3 trial and a phase 3 trial assessing subcutaneous delivery. The company said it “is exploring the potential of Saphnelo in a variety of diseases where type I IFN plays a key role, including lupus nephritis, cutaneous lupus erythematosus, and myositis.”
COVID-19 vaccination does not increase risk of flare in patients with lupus
COVID-19 vaccinations appear to be well tolerated in patients with systemic lupus erythematosus (SLE) and come with a low risk of flare, according to the results of a global, web-based survey.

“Disseminating these reassuring data might prove crucial to increasing vaccine coverage in patients with SLE,” wrote lead author Renaud Felten, MD, of Strasbourg (France) University Hospital. Their results were published as a comment in Lancet Rheumatology.
To assess vaccine tolerability among lupus patients, the cross-sectional Tolerance and Consequences of Vaccination Against COVID-19 in Lupus Patients (VACOLUP) study analyzed a 43-question survey of 696 participants with a self-reported, medically confirmed diagnosis of SLE from 30 countries between March 22, 2021, and May 17, 2021. The cohort was 96% women, and their median age was 42 (interquartile range, 34-51). Nearly 36% of respondents were from Italy, 27% were from Chile, 13% were from France, and just under 9% were Americans. All participants received at least one dose of COVID-19 vaccine, and 49% received a second dose. The most common vaccines were Pfizer-BioNTech (57%), Sinovac (22%), AstraZeneca (10%), and Moderna (8%).
Only 21 participants (3%) reported a medically confirmed SLE flare after a median of 3 days (IQR, 0-29) post COVID vaccination, with most experiencing musculoskeletal symptoms (90%) and fatigue (86%). Of the 21 cases, 15 reported a subsequent change in SLE treatment and 4 were admitted to the hospital. A previous flare that occurred within a year before vaccination was associated with an increased risk of flare post vaccination (relative risk, 5.52; 95% confidence interval, 2.17-14.03; P < .0001).
Side effects – including swelling, soreness, fever, chills, fatigue, joint and muscle pain, nausea, and headache – were reported in 45% of participants (n = 316) after their first dose and in 53% of the 343 participants who received a second dose. There was no notable difference in the likelihood of side effects across gender and age or in patients who received mRNA vaccines, compared with vaccines with other modes of action. Patients who reported side effects after the first dose were more likely to also report them after the second, compared with those who reported none (109 [81%] of 135 vs. 72 [35%] of 205; RR, 2.30; 95% CI, 1.88-2.82; P < .0001).
In the majority of cases (2,232 of 2,683), the side effects were of minor or moderate intensity and did not affect the participants’ ability to perform daily tasks. The study found no significant association between side effects and a SLE flare and SLE medications or previous SLE disease manifestations.

When asked to comment on the study, Amit Saxena, MD, of the Lupus Center at New York University Langone Health, said: “What we are seeing is pretty mild to moderate in terms of follow-up side effects or lupus-related activity. Several studies have shown this amongst our autoimmune rheumatology cohort, as well as what I’ve seen clinically in my own patients. More than anything else, numbers are the most important, and this is a large study.”
He acknowledged the benefits of going directly to patients to gauge their responses and reactions, giving them the opportunity to share concerns that physicians may not think about.
“As rheumatologists, we tend to focus on certain things that might not necessarily be what the patients themselves focus on,” he said. “I think the fact that this questionnaire dealt with a lot of what people complain about – fatigue, sore arm, things that we know are part of getting the vaccine – they aren’t necessarily things we capture with tools that screen for lupus flares, for example.”
More than anything, Dr. Saxena commended the study’s timeliness. “Patients are constantly asking us about the vaccine, and there’s so much misinformation,” he said. “People say, ‘Because I have lupus, I was told not to get vaccinated.’ I don’t know where they get that information from; we are telling everyone to get it, especially our lupus patients.”
The authors recognized their study’s main limitation as the self-reported and subjective nature of the survey, which they attempted to mitigate by asking for medically confirmed flares only. They noted, however, that the short median time between vaccination and flare onset could be caused by patients confusing expected side effects for something more serious, meaning the 3% figure “could be an overestimation of the actual flare rate.”
“Vaccination is recommended for patients with rheumatic and musculoskeletal diseases according to the American College of Rheumatology,” they added, “irrespective of disease activity and severity.”
Several authors reported potential conflicts of interest, including receiving consultancy fees and grants from Pfizer, GlaxoSmithKline, AbbVie, and Janssen, all unrelated to the study.
COVID-19 vaccinations appear to be well tolerated in patients with systemic lupus erythematosus (SLE) and come with a low risk of flare, according to the results of a global, web-based survey.
“Disseminating these reassuring data might prove crucial to increasing vaccine coverage in patients with SLE,” wrote lead author Renaud Felten, MD, of Strasbourg (France) University Hospital. Their results were published as a comment in Lancet Rheumatology.
To assess vaccine tolerability among lupus patients, the cross-sectional Tolerance and Consequences of Vaccination Against COVID-19 in Lupus Patients (VACOLUP) study analyzed a 43-question survey of 696 participants with a self-reported, medically confirmed diagnosis of SLE from 30 countries between March 22, 2021, and May 17, 2021. The cohort was 96% women, and their median age was 42 (interquartile range, 34-51). Nearly 36% of respondents were from Italy, 27% were from Chile, 13% were from France, and just under 9% were Americans. All participants received at least one dose of COVID-19 vaccine, and 49% received a second dose. The most common vaccines were Pfizer-BioNTech (57%), Sinovac (22%), AstraZeneca (10%), and Moderna (8%).
Only 21 participants (3%) reported a medically confirmed SLE flare after a median of 3 days (IQR, 0-29) post COVID vaccination, with most experiencing musculoskeletal symptoms (90%) and fatigue (86%). Of the 21 cases, 15 reported a subsequent change in SLE treatment and 4 were admitted to the hospital. A previous flare that occurred within a year before vaccination was associated with an increased risk of flare post vaccination (relative risk, 5.52; 95% confidence interval, 2.17-14.03; P < .0001).
Side effects – including swelling, soreness, fever, chills, fatigue, joint and muscle pain, nausea, and headache – were reported in 45% of participants (n = 316) after their first dose and in 53% of the 343 participants who received a second dose. There was no notable difference in the likelihood of side effects across gender and age or in patients who received mRNA vaccines, compared with vaccines with other modes of action. Patients who reported side effects after the first dose were more likely to also report them after the second, compared with those who reported none (109 [81%] of 135 vs. 72 [35%] of 205; RR, 2.30; 95% CI, 1.88-2.82; P < .0001).
In the majority of cases (2,232 of 2,683), the side effects were of minor or moderate intensity and did not affect the participants’ ability to perform daily tasks. The study found no significant association between side effects and a SLE flare and SLE medications or previous SLE disease manifestations.
When asked to comment on the study, Amit Saxena, MD, of the Lupus Center at New York University Langone Health, said: “What we are seeing is pretty mild to moderate in terms of follow-up side effects or lupus-related activity. Several studies have shown this amongst our autoimmune rheumatology cohort, as well as what I’ve seen clinically in my own patients. More than anything else, numbers are the most important, and this is a large study.”
He acknowledged the benefits of going directly to patients to gauge their responses and reactions, giving them the opportunity to share concerns that physicians may not think about.
“As rheumatologists, we tend to focus on certain things that might not necessarily be what the patients themselves focus on,” he said. “I think the fact that this questionnaire dealt with a lot of what people complain about – fatigue, sore arm, things that we know are part of getting the vaccine – they aren’t necessarily things we capture with tools that screen for lupus flares, for example.”
More than anything, Dr. Saxena commended the study’s timeliness. “Patients are constantly asking us about the vaccine, and there’s so much misinformation,” he said. “People say, ‘Because I have lupus, I was told not to get vaccinated.’ I don’t know where they get that information from; we are telling everyone to get it, especially our lupus patients.”
The authors recognized their study’s main limitation as the self-reported and subjective nature of the survey, which they attempted to mitigate by asking for medically confirmed flares only. They noted, however, that the short median time between vaccination and flare onset could be caused by patients confusing expected side effects for something more serious, meaning the 3% figure “could be an overestimation of the actual flare rate.”
“Vaccination is recommended for patients with rheumatic and musculoskeletal diseases according to the American College of Rheumatology,” they added, “irrespective of disease activity and severity.”
Several authors reported potential conflicts of interest, including receiving consultancy fees and grants from Pfizer, GlaxoSmithKline, AbbVie, and Janssen, all unrelated to the study.
COVID-19 vaccinations appear to be well tolerated in patients with systemic lupus erythematosus (SLE) and come with a low risk of flare, according to the results of a global, web-based survey.
“Disseminating these reassuring data might prove crucial to increasing vaccine coverage in patients with SLE,” wrote lead author Renaud Felten, MD, of Strasbourg (France) University Hospital. Their results were published as a comment in Lancet Rheumatology.
To assess vaccine tolerability among lupus patients, the cross-sectional Tolerance and Consequences of Vaccination Against COVID-19 in Lupus Patients (VACOLUP) study analyzed a 43-question survey of 696 participants with a self-reported, medically confirmed diagnosis of SLE from 30 countries between March 22, 2021, and May 17, 2021. The cohort was 96% women, and their median age was 42 (interquartile range, 34-51). Nearly 36% of respondents were from Italy, 27% were from Chile, 13% were from France, and just under 9% were Americans. All participants received at least one dose of COVID-19 vaccine, and 49% received a second dose. The most common vaccines were Pfizer-BioNTech (57%), Sinovac (22%), AstraZeneca (10%), and Moderna (8%).
Only 21 participants (3%) reported a medically confirmed SLE flare after a median of 3 days (IQR, 0-29) post COVID vaccination, with most experiencing musculoskeletal symptoms (90%) and fatigue (86%). Of the 21 cases, 15 reported a subsequent change in SLE treatment and 4 were admitted to the hospital. A previous flare that occurred within a year before vaccination was associated with an increased risk of flare post vaccination (relative risk, 5.52; 95% confidence interval, 2.17-14.03; P < .0001).
Side effects – including swelling, soreness, fever, chills, fatigue, joint and muscle pain, nausea, and headache – were reported in 45% of participants (n = 316) after their first dose and in 53% of the 343 participants who received a second dose. There was no notable difference in the likelihood of side effects across gender and age or in patients who received mRNA vaccines, compared with vaccines with other modes of action. Patients who reported side effects after the first dose were more likely to also report them after the second, compared with those who reported none (109 [81%] of 135 vs. 72 [35%] of 205; RR, 2.30; 95% CI, 1.88-2.82; P < .0001).
In the majority of cases (2,232 of 2,683), the side effects were of minor or moderate intensity and did not affect the participants’ ability to perform daily tasks. The study found no significant association between side effects and a SLE flare and SLE medications or previous SLE disease manifestations.
When asked to comment on the study, Amit Saxena, MD, of the Lupus Center at New York University Langone Health, said: “What we are seeing is pretty mild to moderate in terms of follow-up side effects or lupus-related activity. Several studies have shown this amongst our autoimmune rheumatology cohort, as well as what I’ve seen clinically in my own patients. More than anything else, numbers are the most important, and this is a large study.”
He acknowledged the benefits of going directly to patients to gauge their responses and reactions, giving them the opportunity to share concerns that physicians may not think about.
“As rheumatologists, we tend to focus on certain things that might not necessarily be what the patients themselves focus on,” he said. “I think the fact that this questionnaire dealt with a lot of what people complain about – fatigue, sore arm, things that we know are part of getting the vaccine – they aren’t necessarily things we capture with tools that screen for lupus flares, for example.”
More than anything, Dr. Saxena commended the study’s timeliness. “Patients are constantly asking us about the vaccine, and there’s so much misinformation,” he said. “People say, ‘Because I have lupus, I was told not to get vaccinated.’ I don’t know where they get that information from; we are telling everyone to get it, especially our lupus patients.”
The authors recognized their study’s main limitation as the self-reported and subjective nature of the survey, which they attempted to mitigate by asking for medically confirmed flares only. They noted, however, that the short median time between vaccination and flare onset could be caused by patients confusing expected side effects for something more serious, meaning the 3% figure “could be an overestimation of the actual flare rate.”
“Vaccination is recommended for patients with rheumatic and musculoskeletal diseases according to the American College of Rheumatology,” they added, “irrespective of disease activity and severity.”
Several authors reported potential conflicts of interest, including receiving consultancy fees and grants from Pfizer, GlaxoSmithKline, AbbVie, and Janssen, all unrelated to the study.
FROM THE LANCET RHEUMATOLOGY
Autoinflammatory diseases ‘not so rare after all,’ expert says
Not long ago, after all.

“Patients with autoinflammatory diseases are all around us, but many go several years without a diagnosis,” Dr. Dissanayake, a rheumatologist at the Autoinflammatory Disease Clinic at the Hospital for Sick Children, Toronto, said during the annual meeting of the Society for Pediatric Dermatology. “The median time to diagnosis has been estimated to be between 2.5 and 5 years. You can imagine that this type of delay can lead to significant issues, not only with quality of life but also morbidity due to unchecked inflammation that can cause organ damage, and in the most severe cases, can result in an early death.”
Effective treatment options such as biologic medications, however, can prevent these negative sequelae if the disease is recognized early. “Dermatologists are in a unique position because they will often be the first specialist to see these patients and therefore make the diagnosis early on and really alter the lives of these patients,” he said.
While it’s common to classify autoinflammatory diseases by presenting features, such as age of onset, associated symptoms, family history/ethnicity, and triggers/alleviating factors for episodes, Dr. Dissanayake prefers to classify them into one of three groups based on pathophysiology, the first being inflammasomopathies. “When activated, an inflammasome is responsible for processing cytokines from the [interleukin]-1 family from the pro form to the active form,” he explained. As a result, if there is dysregulation and overactivity of the inflammasome, there is excessive production of cytokines like IL-1 beta and IL-18 driving the disease.
Clinical characteristics include fevers and organ involvement, notably abdominal pain, nonvasculitic rashes, uveitis, arthritis, elevated white blood cell count/neutrophils, and highly elevated inflammatory markers. Potential treatments include IL-1 blockers.
The second category of autoinflammatory diseases are the interferonopathies, which are caused by overactivity of the antiviral side of the innate immune system. “For example, if you have overactivity of a sensor for a nucleic acid in your cytosol, the cell misinterprets this as a viral infection and will turn on type 1 interferon production,” said Dr. Dissanayake, who is also an assistant professor of pediatrics at the University of Toronto. “As a result, if you have dysregulation of these pathways, you will get excessive type 1 interferon that contributes to your disease manifestations.” Clinical characteristics include fevers and organ involvement, notably vasculitic rashes, interstitial lung disease, and intracranial calcifications. Inflammatory markers may not be as elevated, and autoantibodies may be present. Janus kinase inhibitors are a potential treatment, he said.
The third category of autoinflammatory diseases are the NF-kappaBopathies, which are caused by overactivity of the NF-kappaB signaling pathway. Clinical characteristics can include fevers with organ involvement that can be highly variable but may include mucocutaneous lesions or granulomatous disease as potential clues. Treatment options depend on the pathway that is involved but tumor necrosis factor blockers often play a role because of the importance of NF-KB in this signaling pathway.
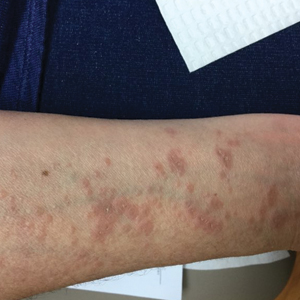
From a skin perspective, most of the rashes Dr. Dissanayake and colleagues see in the rheumatology clinic consist of nonspecific dermohypodermatitis: macules, papules, patches, or plaques. The most common monogenic autoinflammatory disease is Familial Mediterranean Fever syndrome, which “commonly presents as an erysipelas-like rash of the lower extremities, typically below the knee, often over the malleolus,” he said.
Other monogenic autoinflammatory diseases with similar rashes include TNF receptor–associated periodic syndrome, Hyper-IgD syndrome, and systemic juvenile idiopathic arthritis.
Other patients present with urticarial rashes, most commonly cryopyrin-associated periodic syndrome (CAPS). “This is a neutrophilic urticaria, so it tends not to be pruritic and can actually sometimes be tender,” he said. “It also tends not to be as transient as your typical urticaria.” Urticarial rashes can also appear with NLRP12-associated autoinflammatory syndrome (familial cold autoinflammatory syndrome–2), PLCgamma2-associated antibody deficiency and immune dysregulation, and Schnitzler syndrome (monoclonal IgM gammopathy).
Patients can also present with pyogenic or pustular lesions, which can appear with pyoderma gangrenosum–related diseases, such as pyogenic arthritis, pyoderma gangrenosum, arthritis (PAPA) syndrome; pyrin-associated inflammation with neutrophilic dermatosis; deficiency of the IL-1 receptor antagonist; deficiency of IL-36 receptor antagonist; and Majeed syndrome, a mutation in the LPIN2 gene.
The mucocutaneous system can also be affected in autoinflammatory diseases, often presenting with symptoms such as periodic fever, aphthous stomatitis, and pharyngitis. Cervical adenitis syndrome is the most common autoinflammatory disease in childhood and can present with aphthous stomatitis, he said, while Behcet’s disease typically presents with oral and genital ulcers. “More recently, monogenic forms of Behcet’s disease have been described, with haploinsufficiency of A20 and RelA, which are both part of the NF-KB pathway,” he said.
Finally, the presence of vasculitic lesions often suggest interferonopathies such as STING-associated vasculopathy in infancy, proteasome-associated autoinflammatory syndrome and deficiency of adenosine deaminase 2.
Dr. Dissanayake noted that dermatologists should suspect an autoimmune disease if a patient has recurrent fevers, evidence of systemic inflammation on blood work, and if multiple organ systems are involved, especially the lungs, gut, joints, CNS system, and eyes. “Many of these patients have episodic and stereotypical attacks,” he said.
“One of the tools we use in the autoinflammatory clinic is to have patients and families keep a symptom diary where they track the dates of the various symptoms. We can review this during their appointment and try to come up with a diagnosis based on the pattern,” he said.
Since many of these diseases are due to a single gene defect, if there’s any evidence to suggest a monogenic cause, consider an autoinflammatory disease, he added. “If there’s a family history, if there’s consanguinity, or if there’s early age of onset – these may all lead you to think about monogenic autoinflammatory disease.”
During a question-and-answer session, a meeting attendee asked what type of workup he recommends when an autoinflammatory syndrome is suspected. “It partially depends on what organ systems you suspect to be involved,” Dr. Dissanayake said. “As a routine baseline, typically what we would check is CBC and differential, [erythrocyte sedimentation rate] and [C-reactive protein], and we screen for liver transaminases and creatinine to check for liver and kidney issues. A serum albumin will also tell you if the patient is hypoalbuminemic, that there’s been some chronic inflammation and they’re starting to leak the protein out. It’s good to check blood work during the flare and off the flare, to get a sense of the persistence of that inflammation.”
Dr. Dissanayake disclosed that he has received research finding from Gilead Sciences and speaker fees from Novartis.
*This story was updated on 9/20/2021.
Not long ago, after all.
“Patients with autoinflammatory diseases are all around us, but many go several years without a diagnosis,” Dr. Dissanayake, a rheumatologist at the Autoinflammatory Disease Clinic at the Hospital for Sick Children, Toronto, said during the annual meeting of the Society for Pediatric Dermatology. “The median time to diagnosis has been estimated to be between 2.5 and 5 years. You can imagine that this type of delay can lead to significant issues, not only with quality of life but also morbidity due to unchecked inflammation that can cause organ damage, and in the most severe cases, can result in an early death.”
Effective treatment options such as biologic medications, however, can prevent these negative sequelae if the disease is recognized early. “Dermatologists are in a unique position because they will often be the first specialist to see these patients and therefore make the diagnosis early on and really alter the lives of these patients,” he said.
While it’s common to classify autoinflammatory diseases by presenting features, such as age of onset, associated symptoms, family history/ethnicity, and triggers/alleviating factors for episodes, Dr. Dissanayake prefers to classify them into one of three groups based on pathophysiology, the first being inflammasomopathies. “When activated, an inflammasome is responsible for processing cytokines from the [interleukin]-1 family from the pro form to the active form,” he explained. As a result, if there is dysregulation and overactivity of the inflammasome, there is excessive production of cytokines like IL-1 beta and IL-18 driving the disease.
Clinical characteristics include fevers and organ involvement, notably abdominal pain, nonvasculitic rashes, uveitis, arthritis, elevated white blood cell count/neutrophils, and highly elevated inflammatory markers. Potential treatments include IL-1 blockers.
The second category of autoinflammatory diseases are the interferonopathies, which are caused by overactivity of the antiviral side of the innate immune system. “For example, if you have overactivity of a sensor for a nucleic acid in your cytosol, the cell misinterprets this as a viral infection and will turn on type 1 interferon production,” said Dr. Dissanayake, who is also an assistant professor of pediatrics at the University of Toronto. “As a result, if you have dysregulation of these pathways, you will get excessive type 1 interferon that contributes to your disease manifestations.” Clinical characteristics include fevers and organ involvement, notably vasculitic rashes, interstitial lung disease, and intracranial calcifications. Inflammatory markers may not be as elevated, and autoantibodies may be present. Janus kinase inhibitors are a potential treatment, he said.
The third category of autoinflammatory diseases are the NF-kappaBopathies, which are caused by overactivity of the NF-kappaB signaling pathway. Clinical characteristics can include fevers with organ involvement that can be highly variable but may include mucocutaneous lesions or granulomatous disease as potential clues. Treatment options depend on the pathway that is involved but tumor necrosis factor blockers often play a role because of the importance of NF-KB in this signaling pathway.
From a skin perspective, most of the rashes Dr. Dissanayake and colleagues see in the rheumatology clinic consist of nonspecific dermohypodermatitis: macules, papules, patches, or plaques. The most common monogenic autoinflammatory disease is Familial Mediterranean Fever syndrome, which “commonly presents as an erysipelas-like rash of the lower extremities, typically below the knee, often over the malleolus,” he said.
Other monogenic autoinflammatory diseases with similar rashes include TNF receptor–associated periodic syndrome, Hyper-IgD syndrome, and systemic juvenile idiopathic arthritis.
Other patients present with urticarial rashes, most commonly cryopyrin-associated periodic syndrome (CAPS). “This is a neutrophilic urticaria, so it tends not to be pruritic and can actually sometimes be tender,” he said. “It also tends not to be as transient as your typical urticaria.” Urticarial rashes can also appear with NLRP12-associated autoinflammatory syndrome (familial cold autoinflammatory syndrome–2), PLCgamma2-associated antibody deficiency and immune dysregulation, and Schnitzler syndrome (monoclonal IgM gammopathy).
Patients can also present with pyogenic or pustular lesions, which can appear with pyoderma gangrenosum–related diseases, such as pyogenic arthritis, pyoderma gangrenosum, arthritis (PAPA) syndrome; pyrin-associated inflammation with neutrophilic dermatosis; deficiency of the IL-1 receptor antagonist; deficiency of IL-36 receptor antagonist; and Majeed syndrome, a mutation in the LPIN2 gene.
The mucocutaneous system can also be affected in autoinflammatory diseases, often presenting with symptoms such as periodic fever, aphthous stomatitis, and pharyngitis. Cervical adenitis syndrome is the most common autoinflammatory disease in childhood and can present with aphthous stomatitis, he said, while Behcet’s disease typically presents with oral and genital ulcers. “More recently, monogenic forms of Behcet’s disease have been described, with haploinsufficiency of A20 and RelA, which are both part of the NF-KB pathway,” he said.
Finally, the presence of vasculitic lesions often suggest interferonopathies such as STING-associated vasculopathy in infancy, proteasome-associated autoinflammatory syndrome and deficiency of adenosine deaminase 2.
Dr. Dissanayake noted that dermatologists should suspect an autoimmune disease if a patient has recurrent fevers, evidence of systemic inflammation on blood work, and if multiple organ systems are involved, especially the lungs, gut, joints, CNS system, and eyes. “Many of these patients have episodic and stereotypical attacks,” he said.
“One of the tools we use in the autoinflammatory clinic is to have patients and families keep a symptom diary where they track the dates of the various symptoms. We can review this during their appointment and try to come up with a diagnosis based on the pattern,” he said.
Since many of these diseases are due to a single gene defect, if there’s any evidence to suggest a monogenic cause, consider an autoinflammatory disease, he added. “If there’s a family history, if there’s consanguinity, or if there’s early age of onset – these may all lead you to think about monogenic autoinflammatory disease.”
During a question-and-answer session, a meeting attendee asked what type of workup he recommends when an autoinflammatory syndrome is suspected. “It partially depends on what organ systems you suspect to be involved,” Dr. Dissanayake said. “As a routine baseline, typically what we would check is CBC and differential, [erythrocyte sedimentation rate] and [C-reactive protein], and we screen for liver transaminases and creatinine to check for liver and kidney issues. A serum albumin will also tell you if the patient is hypoalbuminemic, that there’s been some chronic inflammation and they’re starting to leak the protein out. It’s good to check blood work during the flare and off the flare, to get a sense of the persistence of that inflammation.”
Dr. Dissanayake disclosed that he has received research finding from Gilead Sciences and speaker fees from Novartis.
*This story was updated on 9/20/2021.
Not long ago, after all.
“Patients with autoinflammatory diseases are all around us, but many go several years without a diagnosis,” Dr. Dissanayake, a rheumatologist at the Autoinflammatory Disease Clinic at the Hospital for Sick Children, Toronto, said during the annual meeting of the Society for Pediatric Dermatology. “The median time to diagnosis has been estimated to be between 2.5 and 5 years. You can imagine that this type of delay can lead to significant issues, not only with quality of life but also morbidity due to unchecked inflammation that can cause organ damage, and in the most severe cases, can result in an early death.”
Effective treatment options such as biologic medications, however, can prevent these negative sequelae if the disease is recognized early. “Dermatologists are in a unique position because they will often be the first specialist to see these patients and therefore make the diagnosis early on and really alter the lives of these patients,” he said.
While it’s common to classify autoinflammatory diseases by presenting features, such as age of onset, associated symptoms, family history/ethnicity, and triggers/alleviating factors for episodes, Dr. Dissanayake prefers to classify them into one of three groups based on pathophysiology, the first being inflammasomopathies. “When activated, an inflammasome is responsible for processing cytokines from the [interleukin]-1 family from the pro form to the active form,” he explained. As a result, if there is dysregulation and overactivity of the inflammasome, there is excessive production of cytokines like IL-1 beta and IL-18 driving the disease.
Clinical characteristics include fevers and organ involvement, notably abdominal pain, nonvasculitic rashes, uveitis, arthritis, elevated white blood cell count/neutrophils, and highly elevated inflammatory markers. Potential treatments include IL-1 blockers.
The second category of autoinflammatory diseases are the interferonopathies, which are caused by overactivity of the antiviral side of the innate immune system. “For example, if you have overactivity of a sensor for a nucleic acid in your cytosol, the cell misinterprets this as a viral infection and will turn on type 1 interferon production,” said Dr. Dissanayake, who is also an assistant professor of pediatrics at the University of Toronto. “As a result, if you have dysregulation of these pathways, you will get excessive type 1 interferon that contributes to your disease manifestations.” Clinical characteristics include fevers and organ involvement, notably vasculitic rashes, interstitial lung disease, and intracranial calcifications. Inflammatory markers may not be as elevated, and autoantibodies may be present. Janus kinase inhibitors are a potential treatment, he said.
The third category of autoinflammatory diseases are the NF-kappaBopathies, which are caused by overactivity of the NF-kappaB signaling pathway. Clinical characteristics can include fevers with organ involvement that can be highly variable but may include mucocutaneous lesions or granulomatous disease as potential clues. Treatment options depend on the pathway that is involved but tumor necrosis factor blockers often play a role because of the importance of NF-KB in this signaling pathway.
From a skin perspective, most of the rashes Dr. Dissanayake and colleagues see in the rheumatology clinic consist of nonspecific dermohypodermatitis: macules, papules, patches, or plaques. The most common monogenic autoinflammatory disease is Familial Mediterranean Fever syndrome, which “commonly presents as an erysipelas-like rash of the lower extremities, typically below the knee, often over the malleolus,” he said.
Other monogenic autoinflammatory diseases with similar rashes include TNF receptor–associated periodic syndrome, Hyper-IgD syndrome, and systemic juvenile idiopathic arthritis.
Other patients present with urticarial rashes, most commonly cryopyrin-associated periodic syndrome (CAPS). “This is a neutrophilic urticaria, so it tends not to be pruritic and can actually sometimes be tender,” he said. “It also tends not to be as transient as your typical urticaria.” Urticarial rashes can also appear with NLRP12-associated autoinflammatory syndrome (familial cold autoinflammatory syndrome–2), PLCgamma2-associated antibody deficiency and immune dysregulation, and Schnitzler syndrome (monoclonal IgM gammopathy).
Patients can also present with pyogenic or pustular lesions, which can appear with pyoderma gangrenosum–related diseases, such as pyogenic arthritis, pyoderma gangrenosum, arthritis (PAPA) syndrome; pyrin-associated inflammation with neutrophilic dermatosis; deficiency of the IL-1 receptor antagonist; deficiency of IL-36 receptor antagonist; and Majeed syndrome, a mutation in the LPIN2 gene.
The mucocutaneous system can also be affected in autoinflammatory diseases, often presenting with symptoms such as periodic fever, aphthous stomatitis, and pharyngitis. Cervical adenitis syndrome is the most common autoinflammatory disease in childhood and can present with aphthous stomatitis, he said, while Behcet’s disease typically presents with oral and genital ulcers. “More recently, monogenic forms of Behcet’s disease have been described, with haploinsufficiency of A20 and RelA, which are both part of the NF-KB pathway,” he said.
Finally, the presence of vasculitic lesions often suggest interferonopathies such as STING-associated vasculopathy in infancy, proteasome-associated autoinflammatory syndrome and deficiency of adenosine deaminase 2.
Dr. Dissanayake noted that dermatologists should suspect an autoimmune disease if a patient has recurrent fevers, evidence of systemic inflammation on blood work, and if multiple organ systems are involved, especially the lungs, gut, joints, CNS system, and eyes. “Many of these patients have episodic and stereotypical attacks,” he said.
“One of the tools we use in the autoinflammatory clinic is to have patients and families keep a symptom diary where they track the dates of the various symptoms. We can review this during their appointment and try to come up with a diagnosis based on the pattern,” he said.
Since many of these diseases are due to a single gene defect, if there’s any evidence to suggest a monogenic cause, consider an autoinflammatory disease, he added. “If there’s a family history, if there’s consanguinity, or if there’s early age of onset – these may all lead you to think about monogenic autoinflammatory disease.”
During a question-and-answer session, a meeting attendee asked what type of workup he recommends when an autoinflammatory syndrome is suspected. “It partially depends on what organ systems you suspect to be involved,” Dr. Dissanayake said. “As a routine baseline, typically what we would check is CBC and differential, [erythrocyte sedimentation rate] and [C-reactive protein], and we screen for liver transaminases and creatinine to check for liver and kidney issues. A serum albumin will also tell you if the patient is hypoalbuminemic, that there’s been some chronic inflammation and they’re starting to leak the protein out. It’s good to check blood work during the flare and off the flare, to get a sense of the persistence of that inflammation.”
Dr. Dissanayake disclosed that he has received research finding from Gilead Sciences and speaker fees from Novartis.
*This story was updated on 9/20/2021.
FROM SPD 2021
FDA approves intravenous immunoglobulin for dermatomyositis
, according to a statement from manufacturer Octapharma USA.

Dermatomyositis is a rare, idiopathic autoimmune disorder that affects approximately 10 out of every million people in the United States, mainly adults in their late 40s to early 60s, according to the company, but children aged 5-15 years can be affected. The disease is characterized by skin rashes, chronic muscle inflammation, progressive muscle weakness, and risk for mortality that is three times higher than for the general population.
There are no previously approved treatments for dermatomyositis prior to Octagam 10%, which also is indicated for chronic immune thrombocytopenic purpura in adults.
The approval for dermatomyositis was based on the results of a phase 3 randomized, double-blind, placebo-controlled clinical trial (the ProDERM trial) that included 95 adult patients at 36 sites worldwide, with 17 sites in the United States. In the trial, 78.7% of patients with dermatomyositis who were randomized to receive 2 g/kg of Octagam 10% every 4 weeks showed response at 16 weeks, compared with 43.8% of patients who received placebo. Response was based on the 2016 American College of Rheumatology/European Alliance of Associations for Rheumatology myositis response criteria. Placebo patients who switched to intravenous immunoglobulin (IVIG) during a trial extension had response rates at week 40 similar to the original patients at week 16.
“The study gives clinicians much more confidence in the efficacy and safety of intravenous immunoglobulin and provides valuable information about what type of patient is best suited for the treatment,” Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh and a member of the ProDERM study Steering Committee, said in the Octapharma statement.
Safety and tolerability were similar to profiles seen with other IVIG medications, according to the statement. The medication does carry a boxed warning from its chronic ITP approval, cautioning about the potential for thrombosis, renal dysfunction, and acute renal failure.
The most common adverse reactions reported by dermatomyositis patients in the ProDERM trial were headache, fever, nausea, vomiting, increased blood pressure, chills, musculoskeletal pain, increased heart rate, dyspnea, and reactions at the infusion sites.
Read the full prescribing information here.
, according to a statement from manufacturer Octapharma USA.
Dermatomyositis is a rare, idiopathic autoimmune disorder that affects approximately 10 out of every million people in the United States, mainly adults in their late 40s to early 60s, according to the company, but children aged 5-15 years can be affected. The disease is characterized by skin rashes, chronic muscle inflammation, progressive muscle weakness, and risk for mortality that is three times higher than for the general population.
There are no previously approved treatments for dermatomyositis prior to Octagam 10%, which also is indicated for chronic immune thrombocytopenic purpura in adults.
The approval for dermatomyositis was based on the results of a phase 3 randomized, double-blind, placebo-controlled clinical trial (the ProDERM trial) that included 95 adult patients at 36 sites worldwide, with 17 sites in the United States. In the trial, 78.7% of patients with dermatomyositis who were randomized to receive 2 g/kg of Octagam 10% every 4 weeks showed response at 16 weeks, compared with 43.8% of patients who received placebo. Response was based on the 2016 American College of Rheumatology/European Alliance of Associations for Rheumatology myositis response criteria. Placebo patients who switched to intravenous immunoglobulin (IVIG) during a trial extension had response rates at week 40 similar to the original patients at week 16.
“The study gives clinicians much more confidence in the efficacy and safety of intravenous immunoglobulin and provides valuable information about what type of patient is best suited for the treatment,” Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh and a member of the ProDERM study Steering Committee, said in the Octapharma statement.
Safety and tolerability were similar to profiles seen with other IVIG medications, according to the statement. The medication does carry a boxed warning from its chronic ITP approval, cautioning about the potential for thrombosis, renal dysfunction, and acute renal failure.
The most common adverse reactions reported by dermatomyositis patients in the ProDERM trial were headache, fever, nausea, vomiting, increased blood pressure, chills, musculoskeletal pain, increased heart rate, dyspnea, and reactions at the infusion sites.
Read the full prescribing information here.
, according to a statement from manufacturer Octapharma USA.
Dermatomyositis is a rare, idiopathic autoimmune disorder that affects approximately 10 out of every million people in the United States, mainly adults in their late 40s to early 60s, according to the company, but children aged 5-15 years can be affected. The disease is characterized by skin rashes, chronic muscle inflammation, progressive muscle weakness, and risk for mortality that is three times higher than for the general population.
There are no previously approved treatments for dermatomyositis prior to Octagam 10%, which also is indicated for chronic immune thrombocytopenic purpura in adults.
The approval for dermatomyositis was based on the results of a phase 3 randomized, double-blind, placebo-controlled clinical trial (the ProDERM trial) that included 95 adult patients at 36 sites worldwide, with 17 sites in the United States. In the trial, 78.7% of patients with dermatomyositis who were randomized to receive 2 g/kg of Octagam 10% every 4 weeks showed response at 16 weeks, compared with 43.8% of patients who received placebo. Response was based on the 2016 American College of Rheumatology/European Alliance of Associations for Rheumatology myositis response criteria. Placebo patients who switched to intravenous immunoglobulin (IVIG) during a trial extension had response rates at week 40 similar to the original patients at week 16.
“The study gives clinicians much more confidence in the efficacy and safety of intravenous immunoglobulin and provides valuable information about what type of patient is best suited for the treatment,” Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh and a member of the ProDERM study Steering Committee, said in the Octapharma statement.
Safety and tolerability were similar to profiles seen with other IVIG medications, according to the statement. The medication does carry a boxed warning from its chronic ITP approval, cautioning about the potential for thrombosis, renal dysfunction, and acute renal failure.
The most common adverse reactions reported by dermatomyositis patients in the ProDERM trial were headache, fever, nausea, vomiting, increased blood pressure, chills, musculoskeletal pain, increased heart rate, dyspnea, and reactions at the infusion sites.
Read the full prescribing information here.
Neuropsychiatric event etiology in lupus helps define predictors, outcomes
Different kinds of neuropsychiatric (NP) events in patients with systemic lupus erythematosus (SLE) have substantial variability in their occurrence, resolution, and recurrence over time, as well as in their predictors, according to new research from a large, prospective, international, inception cohort study.
Because “multiple NP events due to different causes may present concurrently in individual patients, the findings emphasize the importance of recognizing attribution of NP events as a determinant of clinical outcome,” John G. Hanly, MD, of Queen Elizabeth II Health Sciences Centre and Dalhousie University, Halifax, N.S., and colleagues wrote in Arthritis & Rheumatology.
In a previous study of the same group of 1,827 patients with SLE, NP events occurred in about half and approximately one-third of these events were deemed disease related. They also “occurred most frequently around the diagnosis of SLE and had a significant negative impact on health-related quality of life,” the researchers wrote.
Researchers involved with the Systemic Lupus International Collaborating Clinics recruited the 1,827 adults with SLE over an 11-year period during 1999-2011 from a total of 31 sites in Europe, Asia, and North America. The average age of the patients at study enrollment was 35 years, 89% were women, and 49% were White. The mean disease duration was 5.6 months, and 70% of patients were taking corticosteroids at enrollment.
Over an average follow-up period of 7.6 years, 955 patients (52.3%) experienced a single neuropsychiatric event, and 493 (27.0%) experienced two or more events; the total number of unique NP events was 1,910. Most of these unique events (92%) involved the central nervous system, and 8.4% involved the peripheral nervous system.
The researchers used multistate models to attribute NP events to SLE based on factors that included the temporal onset of NP events in relation to SLE diagnosis, concurrent non-SLE factors, and NP events that are common in healthy controls. The four states in the multistate models were no NP events, no current NP event but a history of at least one event, new or ongoing NP events, and death. The results included a multivariate analysis of a model involving 492 observed transitions into new or ongoing NP events.
In the multivariate analysis, factors positively associated with SLE-attributed NP events included male sex (hazard ratio, 1.35; P = .028), concurrent non-SLE NP events excluding headache (HR, 1.83; P < .001), active SLE based on the Systemic Lupus Erythematosus Disease Activity Index 2000 (HR, 1.19; P = .012), and corticosteroid use (HR, 1.59; P = .008). The researchers also found that SLE-attributed NP events were negatively associated with Asian race/ethnicity, postsecondary education, and use of immunosuppressive drugs.
Another multivariate analysis found that non-SLE NP events were positively associated with only concurrent SLE-attributed NP events excluding headache (HR, 2.31; P < .001), but negative associations were seen with non-U.S. African race/ethnicity and Asian race/ethnicity.
The researchers found that SLE-attributed NP events had higher rates of resolution, compared with non-SLE NP events, with the exception of headache, which had similar resolution for both event groups.
“Resolution of SLE events was more likely in patients with Asian race/ethnicity and those with Central/Focal nervous system disease with no effect seen for age at diagnosis,” the researchers noted. “For non-SLE NP events, African race/ethnicity at non-U.S. sites and younger age at diagnosis was associated with a better outcome.”
The study findings were limited by several factors including the predominantly White patient population and the clustering of NP events into limited categories, which may have reduced the identification of more specific associations, the researchers noted. Also, the assessment of NP event outcomes did not include patient perceptions, and the relatively short follow-up period does not allow for assessment of later NP events such as cerebrovascular disease. However, “despite these limitations the current study provides valuable data on the presentation, outcome and predictors of NP disease in SLE patients enrolled in a long-term, international, disease inception cohort,” the researchers concluded.
The study received no outside funding. Dr. Hanly was supported by a grant from the Canadian Institutes of Health Research but had no financial conflicts to disclose. Several coauthors received grant support from various institutions, but not from industry, and had no financial conflicts to disclose.
Different kinds of neuropsychiatric (NP) events in patients with systemic lupus erythematosus (SLE) have substantial variability in their occurrence, resolution, and recurrence over time, as well as in their predictors, according to new research from a large, prospective, international, inception cohort study.
Because “multiple NP events due to different causes may present concurrently in individual patients, the findings emphasize the importance of recognizing attribution of NP events as a determinant of clinical outcome,” John G. Hanly, MD, of Queen Elizabeth II Health Sciences Centre and Dalhousie University, Halifax, N.S., and colleagues wrote in Arthritis & Rheumatology.
In a previous study of the same group of 1,827 patients with SLE, NP events occurred in about half and approximately one-third of these events were deemed disease related. They also “occurred most frequently around the diagnosis of SLE and had a significant negative impact on health-related quality of life,” the researchers wrote.
Researchers involved with the Systemic Lupus International Collaborating Clinics recruited the 1,827 adults with SLE over an 11-year period during 1999-2011 from a total of 31 sites in Europe, Asia, and North America. The average age of the patients at study enrollment was 35 years, 89% were women, and 49% were White. The mean disease duration was 5.6 months, and 70% of patients were taking corticosteroids at enrollment.
Over an average follow-up period of 7.6 years, 955 patients (52.3%) experienced a single neuropsychiatric event, and 493 (27.0%) experienced two or more events; the total number of unique NP events was 1,910. Most of these unique events (92%) involved the central nervous system, and 8.4% involved the peripheral nervous system.
The researchers used multistate models to attribute NP events to SLE based on factors that included the temporal onset of NP events in relation to SLE diagnosis, concurrent non-SLE factors, and NP events that are common in healthy controls. The four states in the multistate models were no NP events, no current NP event but a history of at least one event, new or ongoing NP events, and death. The results included a multivariate analysis of a model involving 492 observed transitions into new or ongoing NP events.
In the multivariate analysis, factors positively associated with SLE-attributed NP events included male sex (hazard ratio, 1.35; P = .028), concurrent non-SLE NP events excluding headache (HR, 1.83; P < .001), active SLE based on the Systemic Lupus Erythematosus Disease Activity Index 2000 (HR, 1.19; P = .012), and corticosteroid use (HR, 1.59; P = .008). The researchers also found that SLE-attributed NP events were negatively associated with Asian race/ethnicity, postsecondary education, and use of immunosuppressive drugs.
Another multivariate analysis found that non-SLE NP events were positively associated with only concurrent SLE-attributed NP events excluding headache (HR, 2.31; P < .001), but negative associations were seen with non-U.S. African race/ethnicity and Asian race/ethnicity.
The researchers found that SLE-attributed NP events had higher rates of resolution, compared with non-SLE NP events, with the exception of headache, which had similar resolution for both event groups.
“Resolution of SLE events was more likely in patients with Asian race/ethnicity and those with Central/Focal nervous system disease with no effect seen for age at diagnosis,” the researchers noted. “For non-SLE NP events, African race/ethnicity at non-U.S. sites and younger age at diagnosis was associated with a better outcome.”
The study findings were limited by several factors including the predominantly White patient population and the clustering of NP events into limited categories, which may have reduced the identification of more specific associations, the researchers noted. Also, the assessment of NP event outcomes did not include patient perceptions, and the relatively short follow-up period does not allow for assessment of later NP events such as cerebrovascular disease. However, “despite these limitations the current study provides valuable data on the presentation, outcome and predictors of NP disease in SLE patients enrolled in a long-term, international, disease inception cohort,” the researchers concluded.
The study received no outside funding. Dr. Hanly was supported by a grant from the Canadian Institutes of Health Research but had no financial conflicts to disclose. Several coauthors received grant support from various institutions, but not from industry, and had no financial conflicts to disclose.
Different kinds of neuropsychiatric (NP) events in patients with systemic lupus erythematosus (SLE) have substantial variability in their occurrence, resolution, and recurrence over time, as well as in their predictors, according to new research from a large, prospective, international, inception cohort study.
Because “multiple NP events due to different causes may present concurrently in individual patients, the findings emphasize the importance of recognizing attribution of NP events as a determinant of clinical outcome,” John G. Hanly, MD, of Queen Elizabeth II Health Sciences Centre and Dalhousie University, Halifax, N.S., and colleagues wrote in Arthritis & Rheumatology.
In a previous study of the same group of 1,827 patients with SLE, NP events occurred in about half and approximately one-third of these events were deemed disease related. They also “occurred most frequently around the diagnosis of SLE and had a significant negative impact on health-related quality of life,” the researchers wrote.
Researchers involved with the Systemic Lupus International Collaborating Clinics recruited the 1,827 adults with SLE over an 11-year period during 1999-2011 from a total of 31 sites in Europe, Asia, and North America. The average age of the patients at study enrollment was 35 years, 89% were women, and 49% were White. The mean disease duration was 5.6 months, and 70% of patients were taking corticosteroids at enrollment.
Over an average follow-up period of 7.6 years, 955 patients (52.3%) experienced a single neuropsychiatric event, and 493 (27.0%) experienced two or more events; the total number of unique NP events was 1,910. Most of these unique events (92%) involved the central nervous system, and 8.4% involved the peripheral nervous system.
The researchers used multistate models to attribute NP events to SLE based on factors that included the temporal onset of NP events in relation to SLE diagnosis, concurrent non-SLE factors, and NP events that are common in healthy controls. The four states in the multistate models were no NP events, no current NP event but a history of at least one event, new or ongoing NP events, and death. The results included a multivariate analysis of a model involving 492 observed transitions into new or ongoing NP events.
In the multivariate analysis, factors positively associated with SLE-attributed NP events included male sex (hazard ratio, 1.35; P = .028), concurrent non-SLE NP events excluding headache (HR, 1.83; P < .001), active SLE based on the Systemic Lupus Erythematosus Disease Activity Index 2000 (HR, 1.19; P = .012), and corticosteroid use (HR, 1.59; P = .008). The researchers also found that SLE-attributed NP events were negatively associated with Asian race/ethnicity, postsecondary education, and use of immunosuppressive drugs.
Another multivariate analysis found that non-SLE NP events were positively associated with only concurrent SLE-attributed NP events excluding headache (HR, 2.31; P < .001), but negative associations were seen with non-U.S. African race/ethnicity and Asian race/ethnicity.
The researchers found that SLE-attributed NP events had higher rates of resolution, compared with non-SLE NP events, with the exception of headache, which had similar resolution for both event groups.
“Resolution of SLE events was more likely in patients with Asian race/ethnicity and those with Central/Focal nervous system disease with no effect seen for age at diagnosis,” the researchers noted. “For non-SLE NP events, African race/ethnicity at non-U.S. sites and younger age at diagnosis was associated with a better outcome.”
The study findings were limited by several factors including the predominantly White patient population and the clustering of NP events into limited categories, which may have reduced the identification of more specific associations, the researchers noted. Also, the assessment of NP event outcomes did not include patient perceptions, and the relatively short follow-up period does not allow for assessment of later NP events such as cerebrovascular disease. However, “despite these limitations the current study provides valuable data on the presentation, outcome and predictors of NP disease in SLE patients enrolled in a long-term, international, disease inception cohort,” the researchers concluded.
The study received no outside funding. Dr. Hanly was supported by a grant from the Canadian Institutes of Health Research but had no financial conflicts to disclose. Several coauthors received grant support from various institutions, but not from industry, and had no financial conflicts to disclose.
FROM ARTHRITIS & RHEUMATOLOGY
Lupus images fall short on diverse examples
Lupus images in medical resource materials underrepresent patients with skin of color, based on data from a review of more than 1,400 images published between 2014 and 2019 in materials from a university’s online medical library.
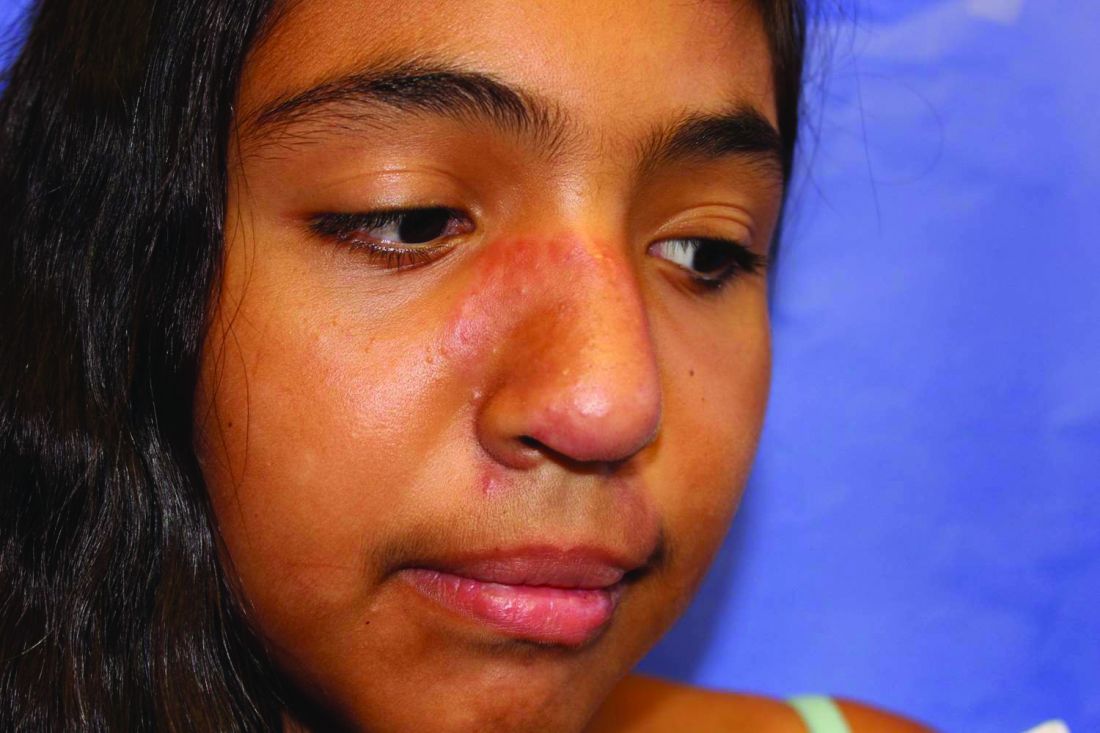
Patients with skin of color who develop lupus tend to present earlier and with more severe cases, and often experience worse outcomes, compared with other populations, wrote Amaad Rana, MD, of Washington University, St. Louis, and colleagues. Medical resources in general have historically underrepresented patients of color, and the researchers reviewed lupus materials for a similar publication bias.
In a study published in Arthritis Care & Research, the investigators identified 1,417 images in rheumatology, dermatology, and internal medicine resources, including 119 medical textbooks, 15 medical journals, 2 online image libraries, and the online image collections of Google and UpToDate. An additional 24 images came from skin of color atlases.
Excluding the skin of color atlases, 56.4% of the images represented light skin, 35.1% showed medium skin, and 8.5% showed dark skin. Overall, publishers were more than twice as likely to portray light skin tones and were significantly less likely to portray dark skin tones (odds ratios, 2.59 and 0.19, respectively), compared with an equal representation of skin tones; however, the difference was not significant for portrayal of medium skin tones (OR, 1.08).
By specialty, dermatology was more inclusive of skin of color images than rheumatology or internal medicine, although the internal medicine sample size was too small for comparable analysis, the researchers noted. Dermatology textbooks were 2.42 times more likely and rheumatology textbooks were 4.87 times more likely to depict light skin tones than an equal representation of light, medium, and dark skin tones.
The researchers rated the skin color in the images using the New Immigrant Survey Skin Color Scale and categorized the images as representing light (NISSCS scores, 1-2), medium (NISSCS scores, 3-5), or dark skin (NISSCS scores, 6-10). Medical journals had the most images of dark skin, excluding skin of color atlases. In a comparison of specialties, dermatology materials included the most images of medium and darker skin tones.
The underrepresentation of skin of color patients can contribute to a limited knowledge of lupus presentation that could lead to disparate health outcomes, the researchers noted.
The study findings were limited by several factors, including the review of only the online textbooks and journals available through the medical library of a single university, the researchers noted. In addition, definitions of light, medium, and dark skin tones were variable among studies, and the researchers did not distinguish among lupus pathologies.
“Further research is needed to quantitatively assess the influence these materials have on healthcare providers’ ability to care for patients with lupus and SOC, and new material and strategies will be required to correct this disparity and promote equitable representation,” the researchers emphasized. “Ultimately, this will arm practitioners with the resources to competently treat patients with any skin color and work towards reducing disparities in health outcomes.”
The study received no outside funding. The researchers had no financial conflicts to disclose.
Lupus images in medical resource materials underrepresent patients with skin of color, based on data from a review of more than 1,400 images published between 2014 and 2019 in materials from a university’s online medical library.
Patients with skin of color who develop lupus tend to present earlier and with more severe cases, and often experience worse outcomes, compared with other populations, wrote Amaad Rana, MD, of Washington University, St. Louis, and colleagues. Medical resources in general have historically underrepresented patients of color, and the researchers reviewed lupus materials for a similar publication bias.
In a study published in Arthritis Care & Research, the investigators identified 1,417 images in rheumatology, dermatology, and internal medicine resources, including 119 medical textbooks, 15 medical journals, 2 online image libraries, and the online image collections of Google and UpToDate. An additional 24 images came from skin of color atlases.
Excluding the skin of color atlases, 56.4% of the images represented light skin, 35.1% showed medium skin, and 8.5% showed dark skin. Overall, publishers were more than twice as likely to portray light skin tones and were significantly less likely to portray dark skin tones (odds ratios, 2.59 and 0.19, respectively), compared with an equal representation of skin tones; however, the difference was not significant for portrayal of medium skin tones (OR, 1.08).
By specialty, dermatology was more inclusive of skin of color images than rheumatology or internal medicine, although the internal medicine sample size was too small for comparable analysis, the researchers noted. Dermatology textbooks were 2.42 times more likely and rheumatology textbooks were 4.87 times more likely to depict light skin tones than an equal representation of light, medium, and dark skin tones.
The researchers rated the skin color in the images using the New Immigrant Survey Skin Color Scale and categorized the images as representing light (NISSCS scores, 1-2), medium (NISSCS scores, 3-5), or dark skin (NISSCS scores, 6-10). Medical journals had the most images of dark skin, excluding skin of color atlases. In a comparison of specialties, dermatology materials included the most images of medium and darker skin tones.
The underrepresentation of skin of color patients can contribute to a limited knowledge of lupus presentation that could lead to disparate health outcomes, the researchers noted.
The study findings were limited by several factors, including the review of only the online textbooks and journals available through the medical library of a single university, the researchers noted. In addition, definitions of light, medium, and dark skin tones were variable among studies, and the researchers did not distinguish among lupus pathologies.
“Further research is needed to quantitatively assess the influence these materials have on healthcare providers’ ability to care for patients with lupus and SOC, and new material and strategies will be required to correct this disparity and promote equitable representation,” the researchers emphasized. “Ultimately, this will arm practitioners with the resources to competently treat patients with any skin color and work towards reducing disparities in health outcomes.”
The study received no outside funding. The researchers had no financial conflicts to disclose.
Lupus images in medical resource materials underrepresent patients with skin of color, based on data from a review of more than 1,400 images published between 2014 and 2019 in materials from a university’s online medical library.
Patients with skin of color who develop lupus tend to present earlier and with more severe cases, and often experience worse outcomes, compared with other populations, wrote Amaad Rana, MD, of Washington University, St. Louis, and colleagues. Medical resources in general have historically underrepresented patients of color, and the researchers reviewed lupus materials for a similar publication bias.
In a study published in Arthritis Care & Research, the investigators identified 1,417 images in rheumatology, dermatology, and internal medicine resources, including 119 medical textbooks, 15 medical journals, 2 online image libraries, and the online image collections of Google and UpToDate. An additional 24 images came from skin of color atlases.
Excluding the skin of color atlases, 56.4% of the images represented light skin, 35.1% showed medium skin, and 8.5% showed dark skin. Overall, publishers were more than twice as likely to portray light skin tones and were significantly less likely to portray dark skin tones (odds ratios, 2.59 and 0.19, respectively), compared with an equal representation of skin tones; however, the difference was not significant for portrayal of medium skin tones (OR, 1.08).
By specialty, dermatology was more inclusive of skin of color images than rheumatology or internal medicine, although the internal medicine sample size was too small for comparable analysis, the researchers noted. Dermatology textbooks were 2.42 times more likely and rheumatology textbooks were 4.87 times more likely to depict light skin tones than an equal representation of light, medium, and dark skin tones.
The researchers rated the skin color in the images using the New Immigrant Survey Skin Color Scale and categorized the images as representing light (NISSCS scores, 1-2), medium (NISSCS scores, 3-5), or dark skin (NISSCS scores, 6-10). Medical journals had the most images of dark skin, excluding skin of color atlases. In a comparison of specialties, dermatology materials included the most images of medium and darker skin tones.
The underrepresentation of skin of color patients can contribute to a limited knowledge of lupus presentation that could lead to disparate health outcomes, the researchers noted.
The study findings were limited by several factors, including the review of only the online textbooks and journals available through the medical library of a single university, the researchers noted. In addition, definitions of light, medium, and dark skin tones were variable among studies, and the researchers did not distinguish among lupus pathologies.
“Further research is needed to quantitatively assess the influence these materials have on healthcare providers’ ability to care for patients with lupus and SOC, and new material and strategies will be required to correct this disparity and promote equitable representation,” the researchers emphasized. “Ultimately, this will arm practitioners with the resources to competently treat patients with any skin color and work towards reducing disparities in health outcomes.”
The study received no outside funding. The researchers had no financial conflicts to disclose.
FROM ARTHRITIS CARE & RESEARCH
New biomarkers may predict interstitial lung disease progression in patients with systemic sclerosis
Quantitative assessment of the extent of interstitial lung disease in patients with systemic sclerosis and levels of certain proteins in bronchoalveolar lavage samples have potential for predicting mortality and disease progression, according to two analyses of data from the Scleroderma Lung Study I and II.

The analyses, presented at the annual European Congress of Rheumatology, aim to improve current prognostic abilities in patients with systemic sclerosis–interstitial lung disease (SSc-ILD). Although forced vital capacity is commonly used as a biomarker for survival in many SSc-ILD trials, other factors can affect FVC, such as respiratory muscle weakness and skin fibrosis. Further, FVC correlates poorly with patient-reported outcomes, explained first author Elizabeth Volkmann, MD, director of the scleroderma program at the University of California, Los Angeles, and the founder and codirector of the UCLA connective tissue disease–related interstitial lung disease program.
Dr. Volkmann presented two studies that investigated the potential of radiographic and protein biomarkers for predicting mortality and identifying patients at risk for ILD progression. The biomarkers may also help to identify patients who would benefit most from immunosuppressive therapy.
The first study found that tracking the quantitative extent of ILD (QILD) over time with high-resolution CT (HRCT) predicted poorer outcomes and could therefore act as a surrogate endpoint for mortality among patients with SSc-ILD. The other study identified associations between specific proteins from bronchoalveolar lavage (BAL) and the likelihood of ILD progression, although some associations were treatment dependent.

Jacob M. van Laar, MD, PhD, professor of rheumatology at the University Medical Center Utrecht (the Netherlands), who was not involved in the study, found the results intriguing and noted the importance of further validation in research before these biomarkers are considered for clinical use.
“It would be wonderful if we can tailor therapy based on BAL biomarkers in the future, as clinicians often struggle to decide on selection, timing, and duration of immunosuppressive treatment,” Dr. van Laar told this news organization. “This has become even more relevant with the introduction of new drugs such as nintedanib.”
Extent of ILD progression as a surrogate for mortality
Scleroderma Lung Study I involved 158 patients with SSc-ILD who were randomly assigned to receive either cyclophosphamide or placebo for 12 months. Scleroderma Lung Study II included 142 patients with SSc-ILD who were randomly assigned to receive either mycophenolate for 24 months or cyclophosphamide for 12 months followed by placebo for 12 months.
The researchers calculated QILD in the whole lung at baseline, at 12 months in the first trial, and at 24 months in the second trial. However, only 82 participants from the first trial and 90 participants from the second trial underwent HRCT. Demographic and disease characteristics were similar between the two groups on follow-up scans.
Follow-up continued for 12 years for patients in the first trial and 8 years in the second. The researchers compared survival rates between the 41% of participants from the first study and 31% of participants from the second study who had poorer QILD scores (at least a 2% increase) with the participants who had stable or improved scores (less than 2% increase).
Participants from both trials had significantly poorer long-term survival if their QILD scores had increased by at least 2% at follow-up (P = .01 for I; P = .019 for II). The association was no longer significant after adjustment for baseline FVC, age, and modified Rodnan skin score in the first trial (hazard ratio, 1.98; P = .089), but it remained significant for participants of the second trial (HR, 3.86; P = .014).
“Data from two independent trial cohorts demonstrated that radiographic progression of SSc-ILD at 1 and 2 years is associated with worse long-term survival,” Dr. Volkmann told attendees.
However, FVC did not significantly predict risk of mortality in either trial.
“To me, the most striking finding from the first study was that change in QILD performed better as a predictor of survival than change in FVC,” Dr. van Laar said in an interview. “This indicates QILD is fit for purpose and worth including in future clinical trials.”
Limitations of the study included lack of HRCT for all participants in the trials and the difference in timing (1 year and 2 years) of HRCT assessment between the two trials. The greater hazard ratio for worsened QILD in the second trial may suggest that assessment at 2 years provides more reliable data as a biomarker, Dr. Volkmann said.
“QILD may represent a better proxy for how a patient feels, functions, and survives than FVC,” she said.
Treatment-dependent biomarkers for worsening lung fibrosis
In the second study, the researchers looked for any associations between changes in the radiographic extent of SSc-ILD and 68 proteins from BAL.
“Being able to risk-stratify patients with interstitial lung disease at the time of diagnosis and predict which patients are likely to have a stable versus progressive disease course is critical for making important treatment decisions for these patients,” Dr. Volkmann told attendees.
The second study she presented involved Scleroderma Lung Study I. Of the 158 participants, 144 underwent a bronchoscopy, yielding BAL protein samples from 103 participants. The researchers determined the extent of radiographic fibrosis in the whole lung with quantitative imaging analysis of HRCT of the chest at baseline and 12 months.
Although the researchers identified several statistically significant associations between certain proteins and changes in radiographic fibrosis, “baseline protein levels were differentially associated with the course of ILD based on treatment status,” she told attendees.
For example, increased levels of the following proteins were linked to poor radiographic fibrosis scores for patients who received placebo:
- Granulocyte-macrophage colony-stimulating factor
- Interleukin-1
- Monocyte chemoattractant protein–3
- Chemokine ligand–5
- Transforming growth factor–beta
- Hepatocyte growth factor
- Stem cell factor
- IL-4
- TGF-alpha
Yet increases in these proteins predicted improvement in radiographic fibrosis in patients who had taken cyclophosphamide.
Independently of treatment, the researchers also identified an association between higher levels of fractalkine and poorer radiographic fibrosis scores and between higher IL-7 levels and improved radiographic fibrosis scores.
After adjusting for treatment arm and baseline severity of ILD, significant associations remained between change in radiographic fibrosis score and IL-1, MCP-3, surfactant protein C, IL-7 and CCL-5 levels.
“Biomarker discovery is really central to our ability to risk stratify patients with SSc-ILD,” Dr. Volkmann told attendees. “Understanding how biomarkers predict outcomes in treated and untreated patients may improve personalized medicine to patients with SSc-ILD and could also reveal novel treatment targets.”
Dr. van Laar said in an interview that this study’s biggest strength lay in its large sample size and in the comprehensiveness of the biomarkers studied.
“The findings are interesting from a research perspective and potentially relevant for clinical practice, but the utility of measuring biomarkers in BAL should be further studied for predictive value on clinical endpoints,” Dr. van Laar said. “BAL is an invasive procedure [that] is not routinely done.”
The research was funded by the National Institutes of Health. Dr. Volkmann has consulted for Boehringer Ingelheim and received grant funding from Corbus, Forbius, and Kadmon. Dr. van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
Quantitative assessment of the extent of interstitial lung disease in patients with systemic sclerosis and levels of certain proteins in bronchoalveolar lavage samples have potential for predicting mortality and disease progression, according to two analyses of data from the Scleroderma Lung Study I and II.
The analyses, presented at the annual European Congress of Rheumatology, aim to improve current prognostic abilities in patients with systemic sclerosis–interstitial lung disease (SSc-ILD). Although forced vital capacity is commonly used as a biomarker for survival in many SSc-ILD trials, other factors can affect FVC, such as respiratory muscle weakness and skin fibrosis. Further, FVC correlates poorly with patient-reported outcomes, explained first author Elizabeth Volkmann, MD, director of the scleroderma program at the University of California, Los Angeles, and the founder and codirector of the UCLA connective tissue disease–related interstitial lung disease program.
Dr. Volkmann presented two studies that investigated the potential of radiographic and protein biomarkers for predicting mortality and identifying patients at risk for ILD progression. The biomarkers may also help to identify patients who would benefit most from immunosuppressive therapy.
The first study found that tracking the quantitative extent of ILD (QILD) over time with high-resolution CT (HRCT) predicted poorer outcomes and could therefore act as a surrogate endpoint for mortality among patients with SSc-ILD. The other study identified associations between specific proteins from bronchoalveolar lavage (BAL) and the likelihood of ILD progression, although some associations were treatment dependent.
Jacob M. van Laar, MD, PhD, professor of rheumatology at the University Medical Center Utrecht (the Netherlands), who was not involved in the study, found the results intriguing and noted the importance of further validation in research before these biomarkers are considered for clinical use.
“It would be wonderful if we can tailor therapy based on BAL biomarkers in the future, as clinicians often struggle to decide on selection, timing, and duration of immunosuppressive treatment,” Dr. van Laar told this news organization. “This has become even more relevant with the introduction of new drugs such as nintedanib.”
Extent of ILD progression as a surrogate for mortality
Scleroderma Lung Study I involved 158 patients with SSc-ILD who were randomly assigned to receive either cyclophosphamide or placebo for 12 months. Scleroderma Lung Study II included 142 patients with SSc-ILD who were randomly assigned to receive either mycophenolate for 24 months or cyclophosphamide for 12 months followed by placebo for 12 months.
The researchers calculated QILD in the whole lung at baseline, at 12 months in the first trial, and at 24 months in the second trial. However, only 82 participants from the first trial and 90 participants from the second trial underwent HRCT. Demographic and disease characteristics were similar between the two groups on follow-up scans.
Follow-up continued for 12 years for patients in the first trial and 8 years in the second. The researchers compared survival rates between the 41% of participants from the first study and 31% of participants from the second study who had poorer QILD scores (at least a 2% increase) with the participants who had stable or improved scores (less than 2% increase).
Participants from both trials had significantly poorer long-term survival if their QILD scores had increased by at least 2% at follow-up (P = .01 for I; P = .019 for II). The association was no longer significant after adjustment for baseline FVC, age, and modified Rodnan skin score in the first trial (hazard ratio, 1.98; P = .089), but it remained significant for participants of the second trial (HR, 3.86; P = .014).
“Data from two independent trial cohorts demonstrated that radiographic progression of SSc-ILD at 1 and 2 years is associated with worse long-term survival,” Dr. Volkmann told attendees.
However, FVC did not significantly predict risk of mortality in either trial.
“To me, the most striking finding from the first study was that change in QILD performed better as a predictor of survival than change in FVC,” Dr. van Laar said in an interview. “This indicates QILD is fit for purpose and worth including in future clinical trials.”
Limitations of the study included lack of HRCT for all participants in the trials and the difference in timing (1 year and 2 years) of HRCT assessment between the two trials. The greater hazard ratio for worsened QILD in the second trial may suggest that assessment at 2 years provides more reliable data as a biomarker, Dr. Volkmann said.
“QILD may represent a better proxy for how a patient feels, functions, and survives than FVC,” she said.
Treatment-dependent biomarkers for worsening lung fibrosis
In the second study, the researchers looked for any associations between changes in the radiographic extent of SSc-ILD and 68 proteins from BAL.
“Being able to risk-stratify patients with interstitial lung disease at the time of diagnosis and predict which patients are likely to have a stable versus progressive disease course is critical for making important treatment decisions for these patients,” Dr. Volkmann told attendees.
The second study she presented involved Scleroderma Lung Study I. Of the 158 participants, 144 underwent a bronchoscopy, yielding BAL protein samples from 103 participants. The researchers determined the extent of radiographic fibrosis in the whole lung with quantitative imaging analysis of HRCT of the chest at baseline and 12 months.
Although the researchers identified several statistically significant associations between certain proteins and changes in radiographic fibrosis, “baseline protein levels were differentially associated with the course of ILD based on treatment status,” she told attendees.
For example, increased levels of the following proteins were linked to poor radiographic fibrosis scores for patients who received placebo:
- Granulocyte-macrophage colony-stimulating factor
- Interleukin-1
- Monocyte chemoattractant protein–3
- Chemokine ligand–5
- Transforming growth factor–beta
- Hepatocyte growth factor
- Stem cell factor
- IL-4
- TGF-alpha
Yet increases in these proteins predicted improvement in radiographic fibrosis in patients who had taken cyclophosphamide.
Independently of treatment, the researchers also identified an association between higher levels of fractalkine and poorer radiographic fibrosis scores and between higher IL-7 levels and improved radiographic fibrosis scores.
After adjusting for treatment arm and baseline severity of ILD, significant associations remained between change in radiographic fibrosis score and IL-1, MCP-3, surfactant protein C, IL-7 and CCL-5 levels.
“Biomarker discovery is really central to our ability to risk stratify patients with SSc-ILD,” Dr. Volkmann told attendees. “Understanding how biomarkers predict outcomes in treated and untreated patients may improve personalized medicine to patients with SSc-ILD and could also reveal novel treatment targets.”
Dr. van Laar said in an interview that this study’s biggest strength lay in its large sample size and in the comprehensiveness of the biomarkers studied.
“The findings are interesting from a research perspective and potentially relevant for clinical practice, but the utility of measuring biomarkers in BAL should be further studied for predictive value on clinical endpoints,” Dr. van Laar said. “BAL is an invasive procedure [that] is not routinely done.”
The research was funded by the National Institutes of Health. Dr. Volkmann has consulted for Boehringer Ingelheim and received grant funding from Corbus, Forbius, and Kadmon. Dr. van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
Quantitative assessment of the extent of interstitial lung disease in patients with systemic sclerosis and levels of certain proteins in bronchoalveolar lavage samples have potential for predicting mortality and disease progression, according to two analyses of data from the Scleroderma Lung Study I and II.
The analyses, presented at the annual European Congress of Rheumatology, aim to improve current prognostic abilities in patients with systemic sclerosis–interstitial lung disease (SSc-ILD). Although forced vital capacity is commonly used as a biomarker for survival in many SSc-ILD trials, other factors can affect FVC, such as respiratory muscle weakness and skin fibrosis. Further, FVC correlates poorly with patient-reported outcomes, explained first author Elizabeth Volkmann, MD, director of the scleroderma program at the University of California, Los Angeles, and the founder and codirector of the UCLA connective tissue disease–related interstitial lung disease program.
Dr. Volkmann presented two studies that investigated the potential of radiographic and protein biomarkers for predicting mortality and identifying patients at risk for ILD progression. The biomarkers may also help to identify patients who would benefit most from immunosuppressive therapy.
The first study found that tracking the quantitative extent of ILD (QILD) over time with high-resolution CT (HRCT) predicted poorer outcomes and could therefore act as a surrogate endpoint for mortality among patients with SSc-ILD. The other study identified associations between specific proteins from bronchoalveolar lavage (BAL) and the likelihood of ILD progression, although some associations were treatment dependent.
Jacob M. van Laar, MD, PhD, professor of rheumatology at the University Medical Center Utrecht (the Netherlands), who was not involved in the study, found the results intriguing and noted the importance of further validation in research before these biomarkers are considered for clinical use.
“It would be wonderful if we can tailor therapy based on BAL biomarkers in the future, as clinicians often struggle to decide on selection, timing, and duration of immunosuppressive treatment,” Dr. van Laar told this news organization. “This has become even more relevant with the introduction of new drugs such as nintedanib.”
Extent of ILD progression as a surrogate for mortality
Scleroderma Lung Study I involved 158 patients with SSc-ILD who were randomly assigned to receive either cyclophosphamide or placebo for 12 months. Scleroderma Lung Study II included 142 patients with SSc-ILD who were randomly assigned to receive either mycophenolate for 24 months or cyclophosphamide for 12 months followed by placebo for 12 months.
The researchers calculated QILD in the whole lung at baseline, at 12 months in the first trial, and at 24 months in the second trial. However, only 82 participants from the first trial and 90 participants from the second trial underwent HRCT. Demographic and disease characteristics were similar between the two groups on follow-up scans.
Follow-up continued for 12 years for patients in the first trial and 8 years in the second. The researchers compared survival rates between the 41% of participants from the first study and 31% of participants from the second study who had poorer QILD scores (at least a 2% increase) with the participants who had stable or improved scores (less than 2% increase).
Participants from both trials had significantly poorer long-term survival if their QILD scores had increased by at least 2% at follow-up (P = .01 for I; P = .019 for II). The association was no longer significant after adjustment for baseline FVC, age, and modified Rodnan skin score in the first trial (hazard ratio, 1.98; P = .089), but it remained significant for participants of the second trial (HR, 3.86; P = .014).
“Data from two independent trial cohorts demonstrated that radiographic progression of SSc-ILD at 1 and 2 years is associated with worse long-term survival,” Dr. Volkmann told attendees.
However, FVC did not significantly predict risk of mortality in either trial.
“To me, the most striking finding from the first study was that change in QILD performed better as a predictor of survival than change in FVC,” Dr. van Laar said in an interview. “This indicates QILD is fit for purpose and worth including in future clinical trials.”
Limitations of the study included lack of HRCT for all participants in the trials and the difference in timing (1 year and 2 years) of HRCT assessment between the two trials. The greater hazard ratio for worsened QILD in the second trial may suggest that assessment at 2 years provides more reliable data as a biomarker, Dr. Volkmann said.
“QILD may represent a better proxy for how a patient feels, functions, and survives than FVC,” she said.
Treatment-dependent biomarkers for worsening lung fibrosis
In the second study, the researchers looked for any associations between changes in the radiographic extent of SSc-ILD and 68 proteins from BAL.
“Being able to risk-stratify patients with interstitial lung disease at the time of diagnosis and predict which patients are likely to have a stable versus progressive disease course is critical for making important treatment decisions for these patients,” Dr. Volkmann told attendees.
The second study she presented involved Scleroderma Lung Study I. Of the 158 participants, 144 underwent a bronchoscopy, yielding BAL protein samples from 103 participants. The researchers determined the extent of radiographic fibrosis in the whole lung with quantitative imaging analysis of HRCT of the chest at baseline and 12 months.
Although the researchers identified several statistically significant associations between certain proteins and changes in radiographic fibrosis, “baseline protein levels were differentially associated with the course of ILD based on treatment status,” she told attendees.
For example, increased levels of the following proteins were linked to poor radiographic fibrosis scores for patients who received placebo:
- Granulocyte-macrophage colony-stimulating factor
- Interleukin-1
- Monocyte chemoattractant protein–3
- Chemokine ligand–5
- Transforming growth factor–beta
- Hepatocyte growth factor
- Stem cell factor
- IL-4
- TGF-alpha
Yet increases in these proteins predicted improvement in radiographic fibrosis in patients who had taken cyclophosphamide.
Independently of treatment, the researchers also identified an association between higher levels of fractalkine and poorer radiographic fibrosis scores and between higher IL-7 levels and improved radiographic fibrosis scores.
After adjusting for treatment arm and baseline severity of ILD, significant associations remained between change in radiographic fibrosis score and IL-1, MCP-3, surfactant protein C, IL-7 and CCL-5 levels.
“Biomarker discovery is really central to our ability to risk stratify patients with SSc-ILD,” Dr. Volkmann told attendees. “Understanding how biomarkers predict outcomes in treated and untreated patients may improve personalized medicine to patients with SSc-ILD and could also reveal novel treatment targets.”
Dr. van Laar said in an interview that this study’s biggest strength lay in its large sample size and in the comprehensiveness of the biomarkers studied.
“The findings are interesting from a research perspective and potentially relevant for clinical practice, but the utility of measuring biomarkers in BAL should be further studied for predictive value on clinical endpoints,” Dr. van Laar said. “BAL is an invasive procedure [that] is not routinely done.”
The research was funded by the National Institutes of Health. Dr. Volkmann has consulted for Boehringer Ingelheim and received grant funding from Corbus, Forbius, and Kadmon. Dr. van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
Nivolumab-Induced Granuloma Annulare
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
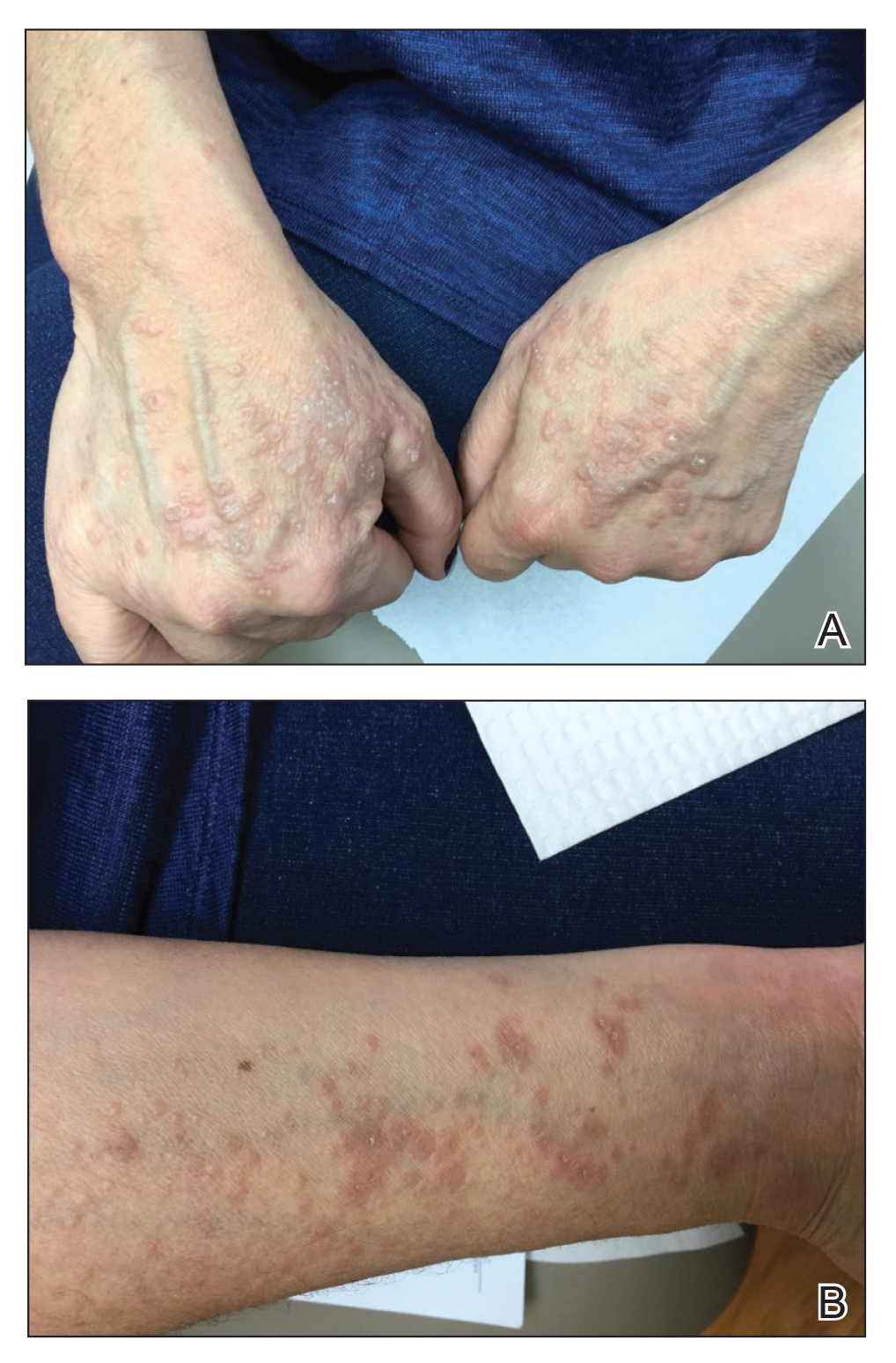
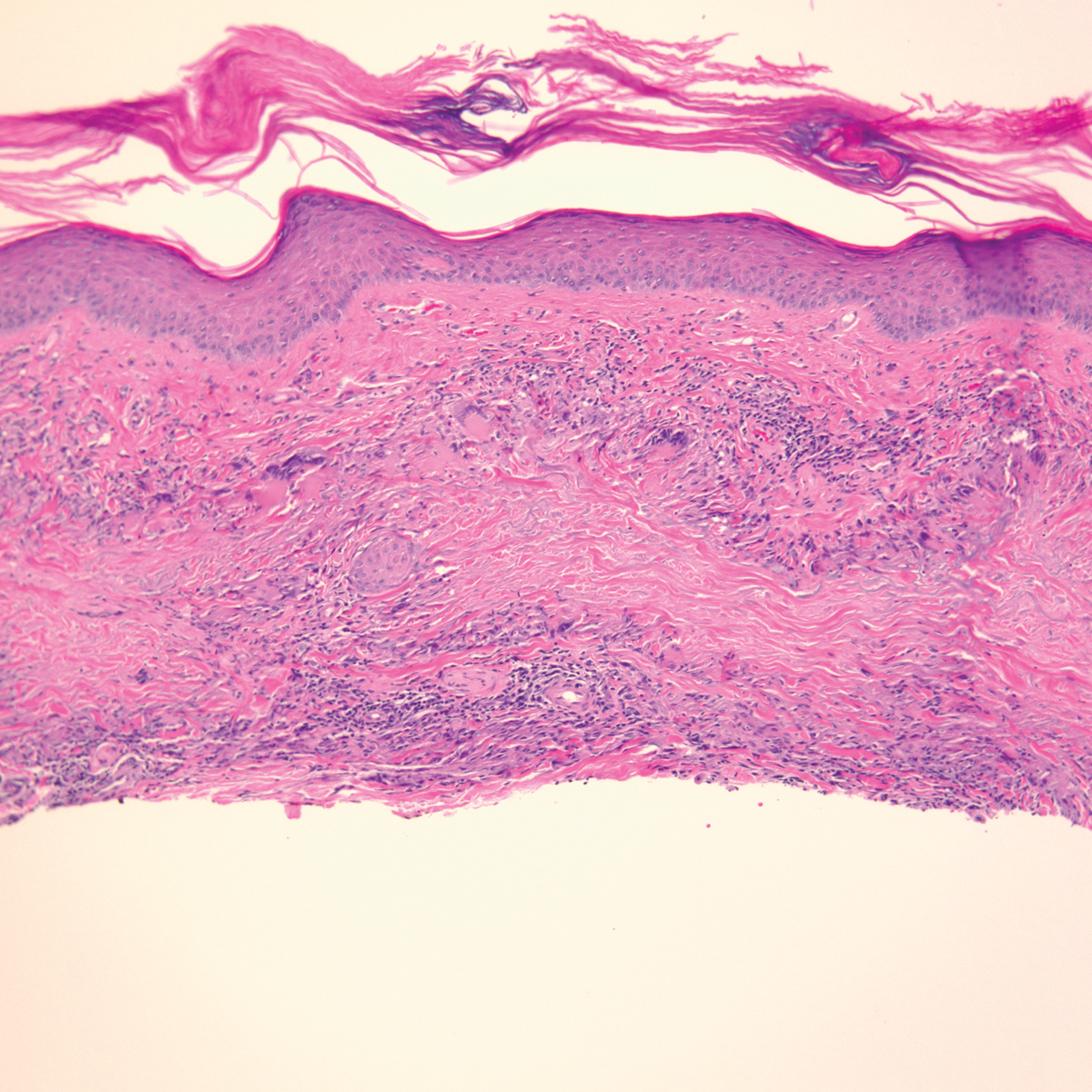
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
Practice Points
- Immune-related adverse events (irAEs) frequently occur in patients on immunotherapy, with the skin representing the most common site of involvement.
- Although rare, granulomatous reactions such as granuloma annulare increasingly are recognized as potential irAEs.
- Clinicians should be aware of this novel association to accurately diagnose and effectively treat adverse reactions in patients receiving immunotherapy.