User login
Why genetic screening isn’t preventing SCD

and a normal one
Credit: Betty Pace
There may be a simple reason why genetic screening has failed to fulfill the promise of preventing sickle cell disease (SCD).
According to an article published in JAMA, it’s a lack of communication.
We’ve long had the technical capacity to screen individuals for the sickle cell trait (SCT). Yet few individuals of child-bearing age who were born in the US actually know their SCT status.
So they aren’t aware that they might pass SCT or SCD down to their children.
And this may boil down to a lack of communication among healthcare professionals, patients, and family members.
“[P]arents are routinely notified by NBS [newborn screening] programs if their child has SCD, but only 37% are notified if their child has SCT,” said author Barry Zuckerman, MD, of Boston Medical Center in Massachusetts.
Even if parents do receive SCT screening results, we don’t know whether they understand the implications or share them with their child. And counseling or referrals to genetic counsellors are not provided in a standard fashion.
Furthermore, although NBS programs notify primary care physicians of screening results at the time of birth, results may not be readily available during routine clinic visits, and patients may not have the same physician throughout their childhood.
The lack of knowledge regarding SCT status represents a missed opportunity to provide appropriate health and prenatal counseling and testing, according to Dr Zuckerman and his colleagues.
They said that timely knowledge of genetic vulnerability and genetic counseling are necessary for informed decision-making with regard to reproduction. It is important to increase the number of adolescents and young adults who know their SCT status to decrease the number of individuals inheriting SCD.
To increase awareness of SCT status and facilitate informed decision-making about reproductive options, we must do 2 things, according to the authors.
First, the results of positive screens for SCT must be communicated to primary care clinicians, recorded in the patient’s medical record as part of a problem list, and shared with parents and the individual.
And second, we must provide effective communication and information through genetic counseling on reproductive options for those with SCT.
The authors also stressed that schools and community organizations have potentially important roles in communicating the importance of SCT status to adolescents and young adults. And by working together, the healthcare system, schools, and community organizations may be able to improve SCT knowledge and awareness. ![]()
and a normal one
Credit: Betty Pace
There may be a simple reason why genetic screening has failed to fulfill the promise of preventing sickle cell disease (SCD).
According to an article published in JAMA, it’s a lack of communication.
We’ve long had the technical capacity to screen individuals for the sickle cell trait (SCT). Yet few individuals of child-bearing age who were born in the US actually know their SCT status.
So they aren’t aware that they might pass SCT or SCD down to their children.
And this may boil down to a lack of communication among healthcare professionals, patients, and family members.
“[P]arents are routinely notified by NBS [newborn screening] programs if their child has SCD, but only 37% are notified if their child has SCT,” said author Barry Zuckerman, MD, of Boston Medical Center in Massachusetts.
Even if parents do receive SCT screening results, we don’t know whether they understand the implications or share them with their child. And counseling or referrals to genetic counsellors are not provided in a standard fashion.
Furthermore, although NBS programs notify primary care physicians of screening results at the time of birth, results may not be readily available during routine clinic visits, and patients may not have the same physician throughout their childhood.
The lack of knowledge regarding SCT status represents a missed opportunity to provide appropriate health and prenatal counseling and testing, according to Dr Zuckerman and his colleagues.
They said that timely knowledge of genetic vulnerability and genetic counseling are necessary for informed decision-making with regard to reproduction. It is important to increase the number of adolescents and young adults who know their SCT status to decrease the number of individuals inheriting SCD.
To increase awareness of SCT status and facilitate informed decision-making about reproductive options, we must do 2 things, according to the authors.
First, the results of positive screens for SCT must be communicated to primary care clinicians, recorded in the patient’s medical record as part of a problem list, and shared with parents and the individual.
And second, we must provide effective communication and information through genetic counseling on reproductive options for those with SCT.
The authors also stressed that schools and community organizations have potentially important roles in communicating the importance of SCT status to adolescents and young adults. And by working together, the healthcare system, schools, and community organizations may be able to improve SCT knowledge and awareness. ![]()
and a normal one
Credit: Betty Pace
There may be a simple reason why genetic screening has failed to fulfill the promise of preventing sickle cell disease (SCD).
According to an article published in JAMA, it’s a lack of communication.
We’ve long had the technical capacity to screen individuals for the sickle cell trait (SCT). Yet few individuals of child-bearing age who were born in the US actually know their SCT status.
So they aren’t aware that they might pass SCT or SCD down to their children.
And this may boil down to a lack of communication among healthcare professionals, patients, and family members.
“[P]arents are routinely notified by NBS [newborn screening] programs if their child has SCD, but only 37% are notified if their child has SCT,” said author Barry Zuckerman, MD, of Boston Medical Center in Massachusetts.
Even if parents do receive SCT screening results, we don’t know whether they understand the implications or share them with their child. And counseling or referrals to genetic counsellors are not provided in a standard fashion.
Furthermore, although NBS programs notify primary care physicians of screening results at the time of birth, results may not be readily available during routine clinic visits, and patients may not have the same physician throughout their childhood.
The lack of knowledge regarding SCT status represents a missed opportunity to provide appropriate health and prenatal counseling and testing, according to Dr Zuckerman and his colleagues.
They said that timely knowledge of genetic vulnerability and genetic counseling are necessary for informed decision-making with regard to reproduction. It is important to increase the number of adolescents and young adults who know their SCT status to decrease the number of individuals inheriting SCD.
To increase awareness of SCT status and facilitate informed decision-making about reproductive options, we must do 2 things, according to the authors.
First, the results of positive screens for SCT must be communicated to primary care clinicians, recorded in the patient’s medical record as part of a problem list, and shared with parents and the individual.
And second, we must provide effective communication and information through genetic counseling on reproductive options for those with SCT.
The authors also stressed that schools and community organizations have potentially important roles in communicating the importance of SCT status to adolescents and young adults. And by working together, the healthcare system, schools, and community organizations may be able to improve SCT knowledge and awareness. ![]()
New approach for treating PNH

Investigators have identified a novel strategy for treating paroxysmal nocturnal hemoglobinuria (PNH), according to a paper published in Blood.
In patients with PNH, defective expression of regulatory proteins on the surface of red blood cells leaves the cells vulnerable to attack by the complement immune system.
This can lead to hemolysis, which results in severe anemia and contributes to a high risk of thrombosis.
Eculizumab is the only approved therapeutic for PNH. The drug reduces hemolysis and can provide patients with relief from blood transfusions.
However, eculizumab is costly (currently more than $400,000 per year per patient), and one third of PNH patients who receive eculizumab continue to require blood transfusions to manage their anemia.
Investigators previously discovered that this non-response is due to fragments of complement C3 proteins on the surface of red blood cells, which are eventually attacked by immune cells.
Therefore, John Lambris, PhD, of the University of Pennsylvania, and his colleagues hypothesized that using small molecules to inhibit the complement cascade at the level of C3 proteins might be an effective strategy for treating PNH.
The team thought this method would prevent both hemolysis and immune cell recognition, and it might be more cost-effective than the current antibody-based treatment.
So they investigated the effect of a C3 inhibitor called Cp40 and its long-acting form, PEG-Cp40, on self-attack and resulting hemolysis using human PNH cells. Both compounds effectively inhibited hemolysis and efficiently prevented deposition of C3 fragments on PNH red blood cells.
In non-human primates, a single injection of PEG-Cp40 had an elimination half-life of more than 5 days. However, the investigators found evidence to suggest the drug may affect plasma levels of C3.
“We think these 2 compounds are excellent and potentially cost-effective candidates for further clinical investigation,” Dr Lambris said.
He hopes the compounds will be tested in clinical trials by 2015. Dr Lambris and his colleague, Daniel Ricklin, PhD, are the inventors of patents and patent applications owned by the University of Pennsylvania that describe the use of complement inhibitors for therapeutic purposes.
And Dr Lambris is a founder and equity holder of Amyndas Pharmaceuticals, which has exclusively licensed the Cp40 and PEG-Cp40 technologies from the university and is developing complement inhibitors for clinical applications. ![]()
Investigators have identified a novel strategy for treating paroxysmal nocturnal hemoglobinuria (PNH), according to a paper published in Blood.
In patients with PNH, defective expression of regulatory proteins on the surface of red blood cells leaves the cells vulnerable to attack by the complement immune system.
This can lead to hemolysis, which results in severe anemia and contributes to a high risk of thrombosis.
Eculizumab is the only approved therapeutic for PNH. The drug reduces hemolysis and can provide patients with relief from blood transfusions.
However, eculizumab is costly (currently more than $400,000 per year per patient), and one third of PNH patients who receive eculizumab continue to require blood transfusions to manage their anemia.
Investigators previously discovered that this non-response is due to fragments of complement C3 proteins on the surface of red blood cells, which are eventually attacked by immune cells.
Therefore, John Lambris, PhD, of the University of Pennsylvania, and his colleagues hypothesized that using small molecules to inhibit the complement cascade at the level of C3 proteins might be an effective strategy for treating PNH.
The team thought this method would prevent both hemolysis and immune cell recognition, and it might be more cost-effective than the current antibody-based treatment.
So they investigated the effect of a C3 inhibitor called Cp40 and its long-acting form, PEG-Cp40, on self-attack and resulting hemolysis using human PNH cells. Both compounds effectively inhibited hemolysis and efficiently prevented deposition of C3 fragments on PNH red blood cells.
In non-human primates, a single injection of PEG-Cp40 had an elimination half-life of more than 5 days. However, the investigators found evidence to suggest the drug may affect plasma levels of C3.
“We think these 2 compounds are excellent and potentially cost-effective candidates for further clinical investigation,” Dr Lambris said.
He hopes the compounds will be tested in clinical trials by 2015. Dr Lambris and his colleague, Daniel Ricklin, PhD, are the inventors of patents and patent applications owned by the University of Pennsylvania that describe the use of complement inhibitors for therapeutic purposes.
And Dr Lambris is a founder and equity holder of Amyndas Pharmaceuticals, which has exclusively licensed the Cp40 and PEG-Cp40 technologies from the university and is developing complement inhibitors for clinical applications. ![]()
Investigators have identified a novel strategy for treating paroxysmal nocturnal hemoglobinuria (PNH), according to a paper published in Blood.
In patients with PNH, defective expression of regulatory proteins on the surface of red blood cells leaves the cells vulnerable to attack by the complement immune system.
This can lead to hemolysis, which results in severe anemia and contributes to a high risk of thrombosis.
Eculizumab is the only approved therapeutic for PNH. The drug reduces hemolysis and can provide patients with relief from blood transfusions.
However, eculizumab is costly (currently more than $400,000 per year per patient), and one third of PNH patients who receive eculizumab continue to require blood transfusions to manage their anemia.
Investigators previously discovered that this non-response is due to fragments of complement C3 proteins on the surface of red blood cells, which are eventually attacked by immune cells.
Therefore, John Lambris, PhD, of the University of Pennsylvania, and his colleagues hypothesized that using small molecules to inhibit the complement cascade at the level of C3 proteins might be an effective strategy for treating PNH.
The team thought this method would prevent both hemolysis and immune cell recognition, and it might be more cost-effective than the current antibody-based treatment.
So they investigated the effect of a C3 inhibitor called Cp40 and its long-acting form, PEG-Cp40, on self-attack and resulting hemolysis using human PNH cells. Both compounds effectively inhibited hemolysis and efficiently prevented deposition of C3 fragments on PNH red blood cells.
In non-human primates, a single injection of PEG-Cp40 had an elimination half-life of more than 5 days. However, the investigators found evidence to suggest the drug may affect plasma levels of C3.
“We think these 2 compounds are excellent and potentially cost-effective candidates for further clinical investigation,” Dr Lambris said.
He hopes the compounds will be tested in clinical trials by 2015. Dr Lambris and his colleague, Daniel Ricklin, PhD, are the inventors of patents and patent applications owned by the University of Pennsylvania that describe the use of complement inhibitors for therapeutic purposes.
And Dr Lambris is a founder and equity holder of Amyndas Pharmaceuticals, which has exclusively licensed the Cp40 and PEG-Cp40 technologies from the university and is developing complement inhibitors for clinical applications. ![]()
Defect causes bone marrow failure, group finds
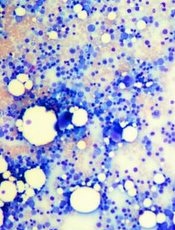
Credit: Daniel E. Sabath
Researchers say they’ve discovered a distinct bone-marrow-failure syndrome and the genetic defect that causes it.
In its natural form, the gene ERCC6L2 plays a role in DNA repair and mitochondrial function.
But investigators found evidence to suggest that mutations in ERCC6L2, and the subsequent DNA damage, were the underlying cause of tri-lineage bone marrow failure in a pair of patients with neurological dysfunction.
“New DNA sequencing technology has enabled us to identify and define a new gene defect which causes a particular type of bone marrow failure,” said Inderjeet Dokal, MD, of Queen Mary University of London in the UK.
“Clinicians treating patients with bone marrow failure should now include analysis for this gene in their investigation.”
Dr Dokal and his colleagues described this research in The American Journal of Human Genetics.
The team performed exome sequencing in 3 patients with genetically uncharacterized, tri-lineage bone marrow failure.
The patients came from consanguineous families (their parents were first-degree cousins), they had developmental delays characterized by learning disabilities, and 2 of the patients had microcephaly.
The sequencing did not uncover variations in any of the known genes associated with bone marrow failure. And the researchers could not find any obvious disease-causing variants in 1 of the patients.
However, the other 2 patients shared homozygous truncating mutations in ERCC6L2—c.1963C>T (p.Arg655*) and c.1236_1239delAACA (p.Thr413Cysfs*2). The c.1963C>T variant had already been identified, but, to the researchers’ knowledge, the other variant had not.
Additional experiments suggested that these mutations affect the subcellular localization and stability of ERCC6L2.
The investigators then speculated that ERCC6L2 plays a role in the DNA-damage response. To test that theory, they mimicked the truncating mutations by knocking down ERCC6L2 expression in human A549 cells.
This significantly reduced cell viability when the cells were exposed to the DNA-damaging agents mitomycin C and irofulven.
To further confirm their theory, the researchers looked at another marker of DNA damage. Previous research had suggested that Snf2 protein complexes are involved in the recruitment of γH2AX, a phosphorylated form of histone 2A, to sites of DNA damage.
So the team performed immunostaining with a γH2AX-specific antibody. And they found that ERCC6L2-knockdown cells displayed H2AX phosphorylation, an effect that increased upon genotoxic stress (treatment with irofulven).
These results indicate that ERCC6L2 plays a role in the DNA-damage-response pathway, and knockdown of this gene sensitizes cells to genotoxic agents.
Additional experiments showed that ERCC6L2 translocated to the mitochondria and the nucleus in response to DNA damage. And ERCC6L2 knockdown induced intracellular reactive oxygen species (ROS).
But introducing the ROS scavenger N-acetyl cysteine diminished the cytotoxicity induced by irofulven, and it halted ERCCGL2 traffic to the mitochondria and nucleus.
The investigators said these results point to a distinct bone-marrow-failure syndrome resulting from mutations in ERCC6L2.
“This is a promising finding which we hope, one day, could lead to finding an effective treatment for this type of gene defect,” Dr Dokal said. “Now [that] we know this research technique works, we plan to carry out further studies to shed more light on the genetic basis of many other cases of bone marrow failure.” ![]()
Credit: Daniel E. Sabath
Researchers say they’ve discovered a distinct bone-marrow-failure syndrome and the genetic defect that causes it.
In its natural form, the gene ERCC6L2 plays a role in DNA repair and mitochondrial function.
But investigators found evidence to suggest that mutations in ERCC6L2, and the subsequent DNA damage, were the underlying cause of tri-lineage bone marrow failure in a pair of patients with neurological dysfunction.
“New DNA sequencing technology has enabled us to identify and define a new gene defect which causes a particular type of bone marrow failure,” said Inderjeet Dokal, MD, of Queen Mary University of London in the UK.
“Clinicians treating patients with bone marrow failure should now include analysis for this gene in their investigation.”
Dr Dokal and his colleagues described this research in The American Journal of Human Genetics.
The team performed exome sequencing in 3 patients with genetically uncharacterized, tri-lineage bone marrow failure.
The patients came from consanguineous families (their parents were first-degree cousins), they had developmental delays characterized by learning disabilities, and 2 of the patients had microcephaly.
The sequencing did not uncover variations in any of the known genes associated with bone marrow failure. And the researchers could not find any obvious disease-causing variants in 1 of the patients.
However, the other 2 patients shared homozygous truncating mutations in ERCC6L2—c.1963C>T (p.Arg655*) and c.1236_1239delAACA (p.Thr413Cysfs*2). The c.1963C>T variant had already been identified, but, to the researchers’ knowledge, the other variant had not.
Additional experiments suggested that these mutations affect the subcellular localization and stability of ERCC6L2.
The investigators then speculated that ERCC6L2 plays a role in the DNA-damage response. To test that theory, they mimicked the truncating mutations by knocking down ERCC6L2 expression in human A549 cells.
This significantly reduced cell viability when the cells were exposed to the DNA-damaging agents mitomycin C and irofulven.
To further confirm their theory, the researchers looked at another marker of DNA damage. Previous research had suggested that Snf2 protein complexes are involved in the recruitment of γH2AX, a phosphorylated form of histone 2A, to sites of DNA damage.
So the team performed immunostaining with a γH2AX-specific antibody. And they found that ERCC6L2-knockdown cells displayed H2AX phosphorylation, an effect that increased upon genotoxic stress (treatment with irofulven).
These results indicate that ERCC6L2 plays a role in the DNA-damage-response pathway, and knockdown of this gene sensitizes cells to genotoxic agents.
Additional experiments showed that ERCC6L2 translocated to the mitochondria and the nucleus in response to DNA damage. And ERCC6L2 knockdown induced intracellular reactive oxygen species (ROS).
But introducing the ROS scavenger N-acetyl cysteine diminished the cytotoxicity induced by irofulven, and it halted ERCCGL2 traffic to the mitochondria and nucleus.
The investigators said these results point to a distinct bone-marrow-failure syndrome resulting from mutations in ERCC6L2.
“This is a promising finding which we hope, one day, could lead to finding an effective treatment for this type of gene defect,” Dr Dokal said. “Now [that] we know this research technique works, we plan to carry out further studies to shed more light on the genetic basis of many other cases of bone marrow failure.” ![]()
Credit: Daniel E. Sabath
Researchers say they’ve discovered a distinct bone-marrow-failure syndrome and the genetic defect that causes it.
In its natural form, the gene ERCC6L2 plays a role in DNA repair and mitochondrial function.
But investigators found evidence to suggest that mutations in ERCC6L2, and the subsequent DNA damage, were the underlying cause of tri-lineage bone marrow failure in a pair of patients with neurological dysfunction.
“New DNA sequencing technology has enabled us to identify and define a new gene defect which causes a particular type of bone marrow failure,” said Inderjeet Dokal, MD, of Queen Mary University of London in the UK.
“Clinicians treating patients with bone marrow failure should now include analysis for this gene in their investigation.”
Dr Dokal and his colleagues described this research in The American Journal of Human Genetics.
The team performed exome sequencing in 3 patients with genetically uncharacterized, tri-lineage bone marrow failure.
The patients came from consanguineous families (their parents were first-degree cousins), they had developmental delays characterized by learning disabilities, and 2 of the patients had microcephaly.
The sequencing did not uncover variations in any of the known genes associated with bone marrow failure. And the researchers could not find any obvious disease-causing variants in 1 of the patients.
However, the other 2 patients shared homozygous truncating mutations in ERCC6L2—c.1963C>T (p.Arg655*) and c.1236_1239delAACA (p.Thr413Cysfs*2). The c.1963C>T variant had already been identified, but, to the researchers’ knowledge, the other variant had not.
Additional experiments suggested that these mutations affect the subcellular localization and stability of ERCC6L2.
The investigators then speculated that ERCC6L2 plays a role in the DNA-damage response. To test that theory, they mimicked the truncating mutations by knocking down ERCC6L2 expression in human A549 cells.
This significantly reduced cell viability when the cells were exposed to the DNA-damaging agents mitomycin C and irofulven.
To further confirm their theory, the researchers looked at another marker of DNA damage. Previous research had suggested that Snf2 protein complexes are involved in the recruitment of γH2AX, a phosphorylated form of histone 2A, to sites of DNA damage.
So the team performed immunostaining with a γH2AX-specific antibody. And they found that ERCC6L2-knockdown cells displayed H2AX phosphorylation, an effect that increased upon genotoxic stress (treatment with irofulven).
These results indicate that ERCC6L2 plays a role in the DNA-damage-response pathway, and knockdown of this gene sensitizes cells to genotoxic agents.
Additional experiments showed that ERCC6L2 translocated to the mitochondria and the nucleus in response to DNA damage. And ERCC6L2 knockdown induced intracellular reactive oxygen species (ROS).
But introducing the ROS scavenger N-acetyl cysteine diminished the cytotoxicity induced by irofulven, and it halted ERCCGL2 traffic to the mitochondria and nucleus.
The investigators said these results point to a distinct bone-marrow-failure syndrome resulting from mutations in ERCC6L2.
“This is a promising finding which we hope, one day, could lead to finding an effective treatment for this type of gene defect,” Dr Dokal said. “Now [that] we know this research technique works, we plan to carry out further studies to shed more light on the genetic basis of many other cases of bone marrow failure.” ![]()
Drug gets breakthrough designation for SAA
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the thrombopoietin receptor agonist eltrombopag (Promacta/Revolade) to treat patients with severe aplastic anemia (SAA) who have had an insufficient response to immunosuppressive therapy (IST).
Eltrombopag is not approved in this setting anywhere in the world, and there are no approved therapies for SAA patients who are unresponsive to initial IST.
Of those patients who are unresponsive to initial IST, approximately 40% die from infection or bleeding within 5 years of their diagnosis.
Breakthrough therapy designation is the newest of the FDA’s programs aimed at accelerating the development and review of drugs for serious or life-threatening conditions. A drug receives the designation when preliminary clinical evidence suggests it may offer substantial improvement over available therapies on at least one clinically significant endpoint.
Eltrombopag was granted breakthrough designation based on results from an open-label, phase 2 study in 43 heavily pretreated SAA patients with an insufficient response to IST. Updated results of this trial were published in December (Desmond et al, Blood 2013).
Patients received varying doses of eltrombopag to improve blood counts. At 3 to 4 months of follow-up, the overall response rate was 40% (17/43), which included 1 tri-lineage and 5 bi-lineage responses.
Most of the 17 patients who remained on eltrombopag in an extension study continued to show improvement, but 3 lost their response. Ultimately, 7 patients had significant increases in neutrophil, red cell, and platelet counts.
Five patients who experienced robust near-normalization of blood counts discontinued the drug and maintained stable counts a median of 13 months after discontinuation (range, 1-15).
Eight patients, including 2 who responded, developed new cytogenetic abnormalities while on eltrombopag. But none have evolved to acute myeloid leukemia to date.
The only dose-limiting toxicity was reversible transaminitis. Two patients had reversible transaminitis related to treatment, and both required dose interruption.
There were no thrombotic events while patients were on treatment, but 1 responding patient developed deep-vein thrombosis 14 months after treatment discontinuation. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the thrombopoietin receptor agonist eltrombopag (Promacta/Revolade) to treat patients with severe aplastic anemia (SAA) who have had an insufficient response to immunosuppressive therapy (IST).
Eltrombopag is not approved in this setting anywhere in the world, and there are no approved therapies for SAA patients who are unresponsive to initial IST.
Of those patients who are unresponsive to initial IST, approximately 40% die from infection or bleeding within 5 years of their diagnosis.
Breakthrough therapy designation is the newest of the FDA’s programs aimed at accelerating the development and review of drugs for serious or life-threatening conditions. A drug receives the designation when preliminary clinical evidence suggests it may offer substantial improvement over available therapies on at least one clinically significant endpoint.
Eltrombopag was granted breakthrough designation based on results from an open-label, phase 2 study in 43 heavily pretreated SAA patients with an insufficient response to IST. Updated results of this trial were published in December (Desmond et al, Blood 2013).
Patients received varying doses of eltrombopag to improve blood counts. At 3 to 4 months of follow-up, the overall response rate was 40% (17/43), which included 1 tri-lineage and 5 bi-lineage responses.
Most of the 17 patients who remained on eltrombopag in an extension study continued to show improvement, but 3 lost their response. Ultimately, 7 patients had significant increases in neutrophil, red cell, and platelet counts.
Five patients who experienced robust near-normalization of blood counts discontinued the drug and maintained stable counts a median of 13 months after discontinuation (range, 1-15).
Eight patients, including 2 who responded, developed new cytogenetic abnormalities while on eltrombopag. But none have evolved to acute myeloid leukemia to date.
The only dose-limiting toxicity was reversible transaminitis. Two patients had reversible transaminitis related to treatment, and both required dose interruption.
There were no thrombotic events while patients were on treatment, but 1 responding patient developed deep-vein thrombosis 14 months after treatment discontinuation. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the thrombopoietin receptor agonist eltrombopag (Promacta/Revolade) to treat patients with severe aplastic anemia (SAA) who have had an insufficient response to immunosuppressive therapy (IST).
Eltrombopag is not approved in this setting anywhere in the world, and there are no approved therapies for SAA patients who are unresponsive to initial IST.
Of those patients who are unresponsive to initial IST, approximately 40% die from infection or bleeding within 5 years of their diagnosis.
Breakthrough therapy designation is the newest of the FDA’s programs aimed at accelerating the development and review of drugs for serious or life-threatening conditions. A drug receives the designation when preliminary clinical evidence suggests it may offer substantial improvement over available therapies on at least one clinically significant endpoint.
Eltrombopag was granted breakthrough designation based on results from an open-label, phase 2 study in 43 heavily pretreated SAA patients with an insufficient response to IST. Updated results of this trial were published in December (Desmond et al, Blood 2013).
Patients received varying doses of eltrombopag to improve blood counts. At 3 to 4 months of follow-up, the overall response rate was 40% (17/43), which included 1 tri-lineage and 5 bi-lineage responses.
Most of the 17 patients who remained on eltrombopag in an extension study continued to show improvement, but 3 lost their response. Ultimately, 7 patients had significant increases in neutrophil, red cell, and platelet counts.
Five patients who experienced robust near-normalization of blood counts discontinued the drug and maintained stable counts a median of 13 months after discontinuation (range, 1-15).
Eight patients, including 2 who responded, developed new cytogenetic abnormalities while on eltrombopag. But none have evolved to acute myeloid leukemia to date.
The only dose-limiting toxicity was reversible transaminitis. Two patients had reversible transaminitis related to treatment, and both required dose interruption.
There were no thrombotic events while patients were on treatment, but 1 responding patient developed deep-vein thrombosis 14 months after treatment discontinuation. ![]()
Inhibitor strengthens RBCs in PNH
Credit: NHLBI
The apoptosis inhibitor aurin tricarboxylic acid (ATA) is active against paroxysmal nocturnal hemoglobinemia (PNH), according to research published in PLOS ONE.
PNH is a rare condition in which red blood cells (RBCs) become vulnerable to attacks by the complement immune system and subsequently rupture.
This can lead to complications such as anemia, kidney disease, and fatal thromboses.
PNH results from a lack of 2 proteins that protect RBCs from destruction: decay-accelerating factor (CD55), an inhibitor of alternative pathway C3 convertase, and protectin (CD59), an inhibitor of membrane attack complex (MAC) formation.
Because previous studies suggested that ATA selectively blocks complement activation at the C3 convertase stage and MAC formation at the C9 insertion stage, researchers thought ATA might prove effective against PNH.
First, they compared RBCs from 5 patients with PNH (who were on long-term treatment with eculizumab) to RBCs from healthy individuals.
Despite the eculizumab, the PNH patients’ RBCs were twice as vulnerable to complement-induced lysis as the healthy subjects’ RBCs. And western blot revealed both C3 and C5 convertases on the membranes of patients’ RBCs.
However, when the researchers added ATA to patients’ blood samples, the RBCs were protected from complement attack. In fact, the drug restored the RBCs’ resistance to the same level as normal RBCs.
“Our study suggests that ATA could offer more complete protection as an oral treatment for PNH, while eliminating the need for infusions,” said study author Patrick McGeer, MD, PhD, of the University of British Columbia in Vancouver, Canada.
“PNH is a disease that may happen to anyone through a chance mutation, and, if nature were to design a perfect fix for this mutation, it would be ATA.”
Dr McGeer added that many diseases are caused or worsened by an overactive complement immune system. So his group’s findings could have implications for conditions such as Alzheimer’s disease, Parkinson’s disease, macular degeneration, amyotrophic lateral sclerosis, multiple sclerosis, and rheumatoid arthritis.
He and his colleagues are now proceeding with further testing, and Dr McGeer expects ATA could be available in clinics within a year. ![]()
Credit: NHLBI
The apoptosis inhibitor aurin tricarboxylic acid (ATA) is active against paroxysmal nocturnal hemoglobinemia (PNH), according to research published in PLOS ONE.
PNH is a rare condition in which red blood cells (RBCs) become vulnerable to attacks by the complement immune system and subsequently rupture.
This can lead to complications such as anemia, kidney disease, and fatal thromboses.
PNH results from a lack of 2 proteins that protect RBCs from destruction: decay-accelerating factor (CD55), an inhibitor of alternative pathway C3 convertase, and protectin (CD59), an inhibitor of membrane attack complex (MAC) formation.
Because previous studies suggested that ATA selectively blocks complement activation at the C3 convertase stage and MAC formation at the C9 insertion stage, researchers thought ATA might prove effective against PNH.
First, they compared RBCs from 5 patients with PNH (who were on long-term treatment with eculizumab) to RBCs from healthy individuals.
Despite the eculizumab, the PNH patients’ RBCs were twice as vulnerable to complement-induced lysis as the healthy subjects’ RBCs. And western blot revealed both C3 and C5 convertases on the membranes of patients’ RBCs.
However, when the researchers added ATA to patients’ blood samples, the RBCs were protected from complement attack. In fact, the drug restored the RBCs’ resistance to the same level as normal RBCs.
“Our study suggests that ATA could offer more complete protection as an oral treatment for PNH, while eliminating the need for infusions,” said study author Patrick McGeer, MD, PhD, of the University of British Columbia in Vancouver, Canada.
“PNH is a disease that may happen to anyone through a chance mutation, and, if nature were to design a perfect fix for this mutation, it would be ATA.”
Dr McGeer added that many diseases are caused or worsened by an overactive complement immune system. So his group’s findings could have implications for conditions such as Alzheimer’s disease, Parkinson’s disease, macular degeneration, amyotrophic lateral sclerosis, multiple sclerosis, and rheumatoid arthritis.
He and his colleagues are now proceeding with further testing, and Dr McGeer expects ATA could be available in clinics within a year. ![]()
Credit: NHLBI
The apoptosis inhibitor aurin tricarboxylic acid (ATA) is active against paroxysmal nocturnal hemoglobinemia (PNH), according to research published in PLOS ONE.
PNH is a rare condition in which red blood cells (RBCs) become vulnerable to attacks by the complement immune system and subsequently rupture.
This can lead to complications such as anemia, kidney disease, and fatal thromboses.
PNH results from a lack of 2 proteins that protect RBCs from destruction: decay-accelerating factor (CD55), an inhibitor of alternative pathway C3 convertase, and protectin (CD59), an inhibitor of membrane attack complex (MAC) formation.
Because previous studies suggested that ATA selectively blocks complement activation at the C3 convertase stage and MAC formation at the C9 insertion stage, researchers thought ATA might prove effective against PNH.
First, they compared RBCs from 5 patients with PNH (who were on long-term treatment with eculizumab) to RBCs from healthy individuals.
Despite the eculizumab, the PNH patients’ RBCs were twice as vulnerable to complement-induced lysis as the healthy subjects’ RBCs. And western blot revealed both C3 and C5 convertases on the membranes of patients’ RBCs.
However, when the researchers added ATA to patients’ blood samples, the RBCs were protected from complement attack. In fact, the drug restored the RBCs’ resistance to the same level as normal RBCs.
“Our study suggests that ATA could offer more complete protection as an oral treatment for PNH, while eliminating the need for infusions,” said study author Patrick McGeer, MD, PhD, of the University of British Columbia in Vancouver, Canada.
“PNH is a disease that may happen to anyone through a chance mutation, and, if nature were to design a perfect fix for this mutation, it would be ATA.”
Dr McGeer added that many diseases are caused or worsened by an overactive complement immune system. So his group’s findings could have implications for conditions such as Alzheimer’s disease, Parkinson’s disease, macular degeneration, amyotrophic lateral sclerosis, multiple sclerosis, and rheumatoid arthritis.
He and his colleagues are now proceeding with further testing, and Dr McGeer expects ATA could be available in clinics within a year. ![]()
Sickle cell trait affects need for ESAs
Researchers may have discovered why African Americans on dialysis sometimes require higher doses of erythropoietin-stimulating agents (ESAs) than dialysis patients of other ethnicities.
The team found that sickle cell trait was more common in African Americans on dialysis than in the general African American population.
And patients with sickle cell trait required higher ESA doses than other African American dialysis patients to reach the same hemoglobin level.
The researchers reported these findings in the Journal of the American Society of Nephrology.
The team noted that kidney abnormalities have been reported in some individuals with sickle cell trait. And studies have shown that African Americans with kidney failure require higher doses of ESAs to treat anemia during dialysis. So researchers wondered if there was a correlation.
To find out, Vimal Derebail, MD, of the University of North Carolina at Chapel Hill, and his colleagues examined laboratory and clinical data concerning 5319 adult African American hemodialysis patients.
But the researchers looked at the presence of hemoglobin C trait as well as sickle cell trait.
In the entire study cohort, 542 (10.2%) patients had sickle cell trait, and 129 (2.4%) had hemoglobin C trait. There were no other hemoglobinopathy traits present.
Sickle cell trait was more common among dialysis patients than the general African American population—10.2% and 6.5%-8.7%, respectively (P<0.05).
Among the 5002 patients receiving ESAs, 10.3% had sickle cell trait and 2.4% had hemoglobin C trait.
The patients with hemoglobinopathy traits received higher median ESA doses than patients with normal traits—4737.4 units/treatment and 4364.1 units/treatment, respectively (P=0.02).
Having either hemoglobinopathy trait was associated with a 13.2% increase in ESA dose (P=0.001). And patients with either trait had a 30% increased risk of falling into the highest quartile of ESA dosing.
There was no significant difference in the dose increase according to trait type (P=0.10).
The researchers therefore said these findings suggest the presence of hemoglobinopathy traits, particularly sickle cell trait, may explain why greater ESA doses are administered to African American dialysis patients relative to Caucasian patients.
“While we don’t know whether there are any adverse consequences to this higher dose of medication yet, further policies and decisions regarding management of anemia in dialysis patients should take into account these findings,” Dr Derebail said.
He added that future research should also explore whether sickle cell trait is more common in dialysis patients because it contributes to kidney disease. ![]()
Researchers may have discovered why African Americans on dialysis sometimes require higher doses of erythropoietin-stimulating agents (ESAs) than dialysis patients of other ethnicities.
The team found that sickle cell trait was more common in African Americans on dialysis than in the general African American population.
And patients with sickle cell trait required higher ESA doses than other African American dialysis patients to reach the same hemoglobin level.
The researchers reported these findings in the Journal of the American Society of Nephrology.
The team noted that kidney abnormalities have been reported in some individuals with sickle cell trait. And studies have shown that African Americans with kidney failure require higher doses of ESAs to treat anemia during dialysis. So researchers wondered if there was a correlation.
To find out, Vimal Derebail, MD, of the University of North Carolina at Chapel Hill, and his colleagues examined laboratory and clinical data concerning 5319 adult African American hemodialysis patients.
But the researchers looked at the presence of hemoglobin C trait as well as sickle cell trait.
In the entire study cohort, 542 (10.2%) patients had sickle cell trait, and 129 (2.4%) had hemoglobin C trait. There were no other hemoglobinopathy traits present.
Sickle cell trait was more common among dialysis patients than the general African American population—10.2% and 6.5%-8.7%, respectively (P<0.05).
Among the 5002 patients receiving ESAs, 10.3% had sickle cell trait and 2.4% had hemoglobin C trait.
The patients with hemoglobinopathy traits received higher median ESA doses than patients with normal traits—4737.4 units/treatment and 4364.1 units/treatment, respectively (P=0.02).
Having either hemoglobinopathy trait was associated with a 13.2% increase in ESA dose (P=0.001). And patients with either trait had a 30% increased risk of falling into the highest quartile of ESA dosing.
There was no significant difference in the dose increase according to trait type (P=0.10).
The researchers therefore said these findings suggest the presence of hemoglobinopathy traits, particularly sickle cell trait, may explain why greater ESA doses are administered to African American dialysis patients relative to Caucasian patients.
“While we don’t know whether there are any adverse consequences to this higher dose of medication yet, further policies and decisions regarding management of anemia in dialysis patients should take into account these findings,” Dr Derebail said.
He added that future research should also explore whether sickle cell trait is more common in dialysis patients because it contributes to kidney disease. ![]()
Researchers may have discovered why African Americans on dialysis sometimes require higher doses of erythropoietin-stimulating agents (ESAs) than dialysis patients of other ethnicities.
The team found that sickle cell trait was more common in African Americans on dialysis than in the general African American population.
And patients with sickle cell trait required higher ESA doses than other African American dialysis patients to reach the same hemoglobin level.
The researchers reported these findings in the Journal of the American Society of Nephrology.
The team noted that kidney abnormalities have been reported in some individuals with sickle cell trait. And studies have shown that African Americans with kidney failure require higher doses of ESAs to treat anemia during dialysis. So researchers wondered if there was a correlation.
To find out, Vimal Derebail, MD, of the University of North Carolina at Chapel Hill, and his colleagues examined laboratory and clinical data concerning 5319 adult African American hemodialysis patients.
But the researchers looked at the presence of hemoglobin C trait as well as sickle cell trait.
In the entire study cohort, 542 (10.2%) patients had sickle cell trait, and 129 (2.4%) had hemoglobin C trait. There were no other hemoglobinopathy traits present.
Sickle cell trait was more common among dialysis patients than the general African American population—10.2% and 6.5%-8.7%, respectively (P<0.05).
Among the 5002 patients receiving ESAs, 10.3% had sickle cell trait and 2.4% had hemoglobin C trait.
The patients with hemoglobinopathy traits received higher median ESA doses than patients with normal traits—4737.4 units/treatment and 4364.1 units/treatment, respectively (P=0.02).
Having either hemoglobinopathy trait was associated with a 13.2% increase in ESA dose (P=0.001). And patients with either trait had a 30% increased risk of falling into the highest quartile of ESA dosing.
There was no significant difference in the dose increase according to trait type (P=0.10).
The researchers therefore said these findings suggest the presence of hemoglobinopathy traits, particularly sickle cell trait, may explain why greater ESA doses are administered to African American dialysis patients relative to Caucasian patients.
“While we don’t know whether there are any adverse consequences to this higher dose of medication yet, further policies and decisions regarding management of anemia in dialysis patients should take into account these findings,” Dr Derebail said.
He added that future research should also explore whether sickle cell trait is more common in dialysis patients because it contributes to kidney disease. ![]()
FDA doesn’t hold drug trials to same standards

Credit: FDA
A new study suggests the US Food and Drug Administration (FDA) does not hold drug trials to the same set of standards.
The research revealed substantial differences in trials used to support drugs approved between 2005 and 2012.
Some drugs were approved based on results from multiple studies, while other approvals were based on data from a single trial.
Furthermore, trials varied greatly with regard to size, length of study period, type of comparator, and metrics of efficacy.
These results appear in the current issue of JAMA.
“Based on our analysis, some drugs are approved on the basis of large, high-quality clinical trials, while others are approved based on results of smaller trials,” said senior study author Joseph Ross, MD, of the Yale School of Medicine in New Haven, Connecticut.
“There was a lack of uniformity in the level of evidence the FDA used. We also found that only 40% of drug approvals involved a clinical trial that compared a new drug to existing treatment offerings. This is an important step for determining whether the new drug is a better option than existing, older drugs.”
Dr Ross and his colleagues evaluated the strength of clinical trial evidence supporting FDA approval decisions by characterizing key features of efficacy trials, such as size, duration, and endpoints.
The researchers used publicly available FDA documents to identify 188 drugs approved between 2005 and 2012 for 206 indications on the basis of 448 pivotal efficacy trials.
The team identified trials for 201 of the indications. Four drugs (including 1 used for 2 different indications) were approved without a pivotal efficacy trial.
So among the 201 indications, the median number of trials reviewed per indication was 2 (interquartile range [IQR], 1-2.5). Seventy-four indications (36.8%) were approved on the basis of a single trial, 77 (38.3%) on data from 2 trials, and 50 (24.9%) on data from 3 or more trials.
Most trials were randomized (89.3%) and double-blinded (79.5%). The median duration of a trial was 14.0 weeks (IQR, 6.0-26.0 weeks), and 113 trials (25.2%) lasted 6 months or longer.
The median number of total subjects enrolled in a trial was 446 (IQR, 205-678), and the median number of patients in the intervention arm of a study was 271 (IQR, 133-426).
More than half of trials (55.1%) used a placebo for comparison, 31.9% used an active comparator (such as another drug), and 12.9% had no comparator.
The primary endpoint was a surrogate outcome in 48.9% of trials, a clinical outcome for 29%, and a clinical scale for 22.1%.
These results suggest the quality of clinical trial evidence the FDA uses to make approval decisions varies widely across indications, the researchers said.
Study author Nicholas S. Downing, a student at the Yale School of Medicine, noted that survey data suggest patients expect drugs approved by the FDA to be both safe and effective.
“Based on our study of the data, we can’t be certain that this expectation is necessarily justified,” he said, “given the quantity and quality of the variability we saw in the drug approval process.” ![]()
Credit: FDA
A new study suggests the US Food and Drug Administration (FDA) does not hold drug trials to the same set of standards.
The research revealed substantial differences in trials used to support drugs approved between 2005 and 2012.
Some drugs were approved based on results from multiple studies, while other approvals were based on data from a single trial.
Furthermore, trials varied greatly with regard to size, length of study period, type of comparator, and metrics of efficacy.
These results appear in the current issue of JAMA.
“Based on our analysis, some drugs are approved on the basis of large, high-quality clinical trials, while others are approved based on results of smaller trials,” said senior study author Joseph Ross, MD, of the Yale School of Medicine in New Haven, Connecticut.
“There was a lack of uniformity in the level of evidence the FDA used. We also found that only 40% of drug approvals involved a clinical trial that compared a new drug to existing treatment offerings. This is an important step for determining whether the new drug is a better option than existing, older drugs.”
Dr Ross and his colleagues evaluated the strength of clinical trial evidence supporting FDA approval decisions by characterizing key features of efficacy trials, such as size, duration, and endpoints.
The researchers used publicly available FDA documents to identify 188 drugs approved between 2005 and 2012 for 206 indications on the basis of 448 pivotal efficacy trials.
The team identified trials for 201 of the indications. Four drugs (including 1 used for 2 different indications) were approved without a pivotal efficacy trial.
So among the 201 indications, the median number of trials reviewed per indication was 2 (interquartile range [IQR], 1-2.5). Seventy-four indications (36.8%) were approved on the basis of a single trial, 77 (38.3%) on data from 2 trials, and 50 (24.9%) on data from 3 or more trials.
Most trials were randomized (89.3%) and double-blinded (79.5%). The median duration of a trial was 14.0 weeks (IQR, 6.0-26.0 weeks), and 113 trials (25.2%) lasted 6 months or longer.
The median number of total subjects enrolled in a trial was 446 (IQR, 205-678), and the median number of patients in the intervention arm of a study was 271 (IQR, 133-426).
More than half of trials (55.1%) used a placebo for comparison, 31.9% used an active comparator (such as another drug), and 12.9% had no comparator.
The primary endpoint was a surrogate outcome in 48.9% of trials, a clinical outcome for 29%, and a clinical scale for 22.1%.
These results suggest the quality of clinical trial evidence the FDA uses to make approval decisions varies widely across indications, the researchers said.
Study author Nicholas S. Downing, a student at the Yale School of Medicine, noted that survey data suggest patients expect drugs approved by the FDA to be both safe and effective.
“Based on our study of the data, we can’t be certain that this expectation is necessarily justified,” he said, “given the quantity and quality of the variability we saw in the drug approval process.” ![]()
Credit: FDA
A new study suggests the US Food and Drug Administration (FDA) does not hold drug trials to the same set of standards.
The research revealed substantial differences in trials used to support drugs approved between 2005 and 2012.
Some drugs were approved based on results from multiple studies, while other approvals were based on data from a single trial.
Furthermore, trials varied greatly with regard to size, length of study period, type of comparator, and metrics of efficacy.
These results appear in the current issue of JAMA.
“Based on our analysis, some drugs are approved on the basis of large, high-quality clinical trials, while others are approved based on results of smaller trials,” said senior study author Joseph Ross, MD, of the Yale School of Medicine in New Haven, Connecticut.
“There was a lack of uniformity in the level of evidence the FDA used. We also found that only 40% of drug approvals involved a clinical trial that compared a new drug to existing treatment offerings. This is an important step for determining whether the new drug is a better option than existing, older drugs.”
Dr Ross and his colleagues evaluated the strength of clinical trial evidence supporting FDA approval decisions by characterizing key features of efficacy trials, such as size, duration, and endpoints.
The researchers used publicly available FDA documents to identify 188 drugs approved between 2005 and 2012 for 206 indications on the basis of 448 pivotal efficacy trials.
The team identified trials for 201 of the indications. Four drugs (including 1 used for 2 different indications) were approved without a pivotal efficacy trial.
So among the 201 indications, the median number of trials reviewed per indication was 2 (interquartile range [IQR], 1-2.5). Seventy-four indications (36.8%) were approved on the basis of a single trial, 77 (38.3%) on data from 2 trials, and 50 (24.9%) on data from 3 or more trials.
Most trials were randomized (89.3%) and double-blinded (79.5%). The median duration of a trial was 14.0 weeks (IQR, 6.0-26.0 weeks), and 113 trials (25.2%) lasted 6 months or longer.
The median number of total subjects enrolled in a trial was 446 (IQR, 205-678), and the median number of patients in the intervention arm of a study was 271 (IQR, 133-426).
More than half of trials (55.1%) used a placebo for comparison, 31.9% used an active comparator (such as another drug), and 12.9% had no comparator.
The primary endpoint was a surrogate outcome in 48.9% of trials, a clinical outcome for 29%, and a clinical scale for 22.1%.
These results suggest the quality of clinical trial evidence the FDA uses to make approval decisions varies widely across indications, the researchers said.
Study author Nicholas S. Downing, a student at the Yale School of Medicine, noted that survey data suggest patients expect drugs approved by the FDA to be both safe and effective.
“Based on our study of the data, we can’t be certain that this expectation is necessarily justified,” he said, “given the quantity and quality of the variability we saw in the drug approval process.”
Sickle cell crises curtailed with experimental cellular adhesion inhibitor
NEW ORLEANS – An experimental cellular adhesion inhibitor was successful at reducing the severity and duration of vaso-occlusive crises in patients with sickle cell disease.
In a phase II trial of 76 patients with sickle cell disease, patients randomized to receive the pan-selectin inhibitor GMI 1070 early in their hospitalization for a vaso-occlusive crisis (VOC) had shorter lengths of stay and needed significantly lower cumulative doses of narcotics for pain control than did patients randomized to placebo, reported Dr. Marilyn J. Telen, chief of the hematology division at Duke University, Durham, N.C.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
"We see somewhere between 75,000 and 90,000 admissions [annually] for acute painful vaso-occlusive crisis among this patient population. Indeed, these crises are the most common and essentially the archetypal presentation of sickle cell disease. Nevertheless, up till this time, treatment for these crises or VOC in sickle cell disease, remain only supportive, focusing largely on using narcotics for symptom relief, and then other measures, some of which are used to counteract the ill effects of narcotics," said Dr. Telen at the annual meeting of the American Society of Hematology.
GMI 1070 (being developed by GlycoMimetics, in partnership with Pfizer) is a synthetic molecule designed to inhibit the glycoprotein cellular-adhesion molecules involved in inflammation. In previous studies, the drug has been shown to be safe, and in a mouse model of VOC, was successful at restoring blood flow, Dr. Telen said. The drug has received both orphan drug and fast-track status from the Food and Drug Administration, according to GlycoMimetics.
Dr. Telen and her colleagues enrolled 76 patients aged 12-51 years with sickle cell disease and randomized them to receive a loading dose of GMI 1070 delivered intravenously (43 patients), followed by up to 14 subsequent doses delivered every 12 hours, or placebo (33 patients), with other treatment left to the discretion of the participating institutions. After an interim pharmacokinetic analysis showed that the drug did not reach target nadir levels, the dose was doubled.
All 76 patients reached the primary endpoint of VOC resolution, defined as a composite of decreased pain, termination of the need for intravenous opioids, patient and physician agreement on the ability to discharge the patient, and actual hospital discharge.
A total of 58 patients continued on the assigned drug until they either reached the primary endpoint criteria or received the maximum number of doses allowed. The remaining 18 patients discontinued the drug either for adverse events, no improvement by day 5 on the assigned drug, or other reasons.
In an analysis pooling all patients assigned to GMI 1070, including those who started out on the lower dose, there was a consistent reduction over placebo in the mean time to resolution of VOC: 103 hours vs. 144 hours for patients treated with placebo. This difference was not statistically significant, however.
A Kaplan-Meier analysis showed a median time to resolution of 69.6 hours for GMI 1070, compared with 139 hours with placebo, a difference that was not significant.
There was an 83% reduction in the secondary endpoint of cumulative opioid analgesics administered during hospitalization, a difference that was statistically significant (P =.010). There was also a reduction by 84 hours in the median time to discharge, and by 55 hours in the mean time to discharge, among patients treated with the active drug, compared with those on placebo. These differences, while large, were not significant, Dr. Telen said.
She noted that although most of the endpoints in this study failed to reach statistical significance, the separation of the curves between the placebo- and GMI 1070–treated patients began early, usually within 2 days of the start of treatment.
Total adverse event rates, including serious events and those deemed to be treatment related, were comparable between the two study arms for all subgroups.
Dr. Telen noted that because the population of patients enrolled was more clinically diverse than the available literature would suggest, the study was underpowered to detect differences, given the size of the sample. She predicted that given the size of the effects seen, statistical significance would emerge in a larger study.
GlycoMimetics is currently working with Pfizer to develop a phase III trial of GM 1070 for this indication.
The study was supported by GlycoMimetics. Dr. Telen is a consultant to the company, and several coauthors are employees of the company.
NEW ORLEANS – An experimental cellular adhesion inhibitor was successful at reducing the severity and duration of vaso-occlusive crises in patients with sickle cell disease.
In a phase II trial of 76 patients with sickle cell disease, patients randomized to receive the pan-selectin inhibitor GMI 1070 early in their hospitalization for a vaso-occlusive crisis (VOC) had shorter lengths of stay and needed significantly lower cumulative doses of narcotics for pain control than did patients randomized to placebo, reported Dr. Marilyn J. Telen, chief of the hematology division at Duke University, Durham, N.C.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
"We see somewhere between 75,000 and 90,000 admissions [annually] for acute painful vaso-occlusive crisis among this patient population. Indeed, these crises are the most common and essentially the archetypal presentation of sickle cell disease. Nevertheless, up till this time, treatment for these crises or VOC in sickle cell disease, remain only supportive, focusing largely on using narcotics for symptom relief, and then other measures, some of which are used to counteract the ill effects of narcotics," said Dr. Telen at the annual meeting of the American Society of Hematology.
GMI 1070 (being developed by GlycoMimetics, in partnership with Pfizer) is a synthetic molecule designed to inhibit the glycoprotein cellular-adhesion molecules involved in inflammation. In previous studies, the drug has been shown to be safe, and in a mouse model of VOC, was successful at restoring blood flow, Dr. Telen said. The drug has received both orphan drug and fast-track status from the Food and Drug Administration, according to GlycoMimetics.
Dr. Telen and her colleagues enrolled 76 patients aged 12-51 years with sickle cell disease and randomized them to receive a loading dose of GMI 1070 delivered intravenously (43 patients), followed by up to 14 subsequent doses delivered every 12 hours, or placebo (33 patients), with other treatment left to the discretion of the participating institutions. After an interim pharmacokinetic analysis showed that the drug did not reach target nadir levels, the dose was doubled.
All 76 patients reached the primary endpoint of VOC resolution, defined as a composite of decreased pain, termination of the need for intravenous opioids, patient and physician agreement on the ability to discharge the patient, and actual hospital discharge.
A total of 58 patients continued on the assigned drug until they either reached the primary endpoint criteria or received the maximum number of doses allowed. The remaining 18 patients discontinued the drug either for adverse events, no improvement by day 5 on the assigned drug, or other reasons.
In an analysis pooling all patients assigned to GMI 1070, including those who started out on the lower dose, there was a consistent reduction over placebo in the mean time to resolution of VOC: 103 hours vs. 144 hours for patients treated with placebo. This difference was not statistically significant, however.
A Kaplan-Meier analysis showed a median time to resolution of 69.6 hours for GMI 1070, compared with 139 hours with placebo, a difference that was not significant.
There was an 83% reduction in the secondary endpoint of cumulative opioid analgesics administered during hospitalization, a difference that was statistically significant (P =.010). There was also a reduction by 84 hours in the median time to discharge, and by 55 hours in the mean time to discharge, among patients treated with the active drug, compared with those on placebo. These differences, while large, were not significant, Dr. Telen said.
She noted that although most of the endpoints in this study failed to reach statistical significance, the separation of the curves between the placebo- and GMI 1070–treated patients began early, usually within 2 days of the start of treatment.
Total adverse event rates, including serious events and those deemed to be treatment related, were comparable between the two study arms for all subgroups.
Dr. Telen noted that because the population of patients enrolled was more clinically diverse than the available literature would suggest, the study was underpowered to detect differences, given the size of the sample. She predicted that given the size of the effects seen, statistical significance would emerge in a larger study.
GlycoMimetics is currently working with Pfizer to develop a phase III trial of GM 1070 for this indication.
The study was supported by GlycoMimetics. Dr. Telen is a consultant to the company, and several coauthors are employees of the company.
NEW ORLEANS – An experimental cellular adhesion inhibitor was successful at reducing the severity and duration of vaso-occlusive crises in patients with sickle cell disease.
In a phase II trial of 76 patients with sickle cell disease, patients randomized to receive the pan-selectin inhibitor GMI 1070 early in their hospitalization for a vaso-occlusive crisis (VOC) had shorter lengths of stay and needed significantly lower cumulative doses of narcotics for pain control than did patients randomized to placebo, reported Dr. Marilyn J. Telen, chief of the hematology division at Duke University, Durham, N.C.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
"We see somewhere between 75,000 and 90,000 admissions [annually] for acute painful vaso-occlusive crisis among this patient population. Indeed, these crises are the most common and essentially the archetypal presentation of sickle cell disease. Nevertheless, up till this time, treatment for these crises or VOC in sickle cell disease, remain only supportive, focusing largely on using narcotics for symptom relief, and then other measures, some of which are used to counteract the ill effects of narcotics," said Dr. Telen at the annual meeting of the American Society of Hematology.
GMI 1070 (being developed by GlycoMimetics, in partnership with Pfizer) is a synthetic molecule designed to inhibit the glycoprotein cellular-adhesion molecules involved in inflammation. In previous studies, the drug has been shown to be safe, and in a mouse model of VOC, was successful at restoring blood flow, Dr. Telen said. The drug has received both orphan drug and fast-track status from the Food and Drug Administration, according to GlycoMimetics.
Dr. Telen and her colleagues enrolled 76 patients aged 12-51 years with sickle cell disease and randomized them to receive a loading dose of GMI 1070 delivered intravenously (43 patients), followed by up to 14 subsequent doses delivered every 12 hours, or placebo (33 patients), with other treatment left to the discretion of the participating institutions. After an interim pharmacokinetic analysis showed that the drug did not reach target nadir levels, the dose was doubled.
All 76 patients reached the primary endpoint of VOC resolution, defined as a composite of decreased pain, termination of the need for intravenous opioids, patient and physician agreement on the ability to discharge the patient, and actual hospital discharge.
A total of 58 patients continued on the assigned drug until they either reached the primary endpoint criteria or received the maximum number of doses allowed. The remaining 18 patients discontinued the drug either for adverse events, no improvement by day 5 on the assigned drug, or other reasons.
In an analysis pooling all patients assigned to GMI 1070, including those who started out on the lower dose, there was a consistent reduction over placebo in the mean time to resolution of VOC: 103 hours vs. 144 hours for patients treated with placebo. This difference was not statistically significant, however.
A Kaplan-Meier analysis showed a median time to resolution of 69.6 hours for GMI 1070, compared with 139 hours with placebo, a difference that was not significant.
There was an 83% reduction in the secondary endpoint of cumulative opioid analgesics administered during hospitalization, a difference that was statistically significant (P =.010). There was also a reduction by 84 hours in the median time to discharge, and by 55 hours in the mean time to discharge, among patients treated with the active drug, compared with those on placebo. These differences, while large, were not significant, Dr. Telen said.
She noted that although most of the endpoints in this study failed to reach statistical significance, the separation of the curves between the placebo- and GMI 1070–treated patients began early, usually within 2 days of the start of treatment.
Total adverse event rates, including serious events and those deemed to be treatment related, were comparable between the two study arms for all subgroups.
Dr. Telen noted that because the population of patients enrolled was more clinically diverse than the available literature would suggest, the study was underpowered to detect differences, given the size of the sample. She predicted that given the size of the effects seen, statistical significance would emerge in a larger study.
GlycoMimetics is currently working with Pfizer to develop a phase III trial of GM 1070 for this indication.
The study was supported by GlycoMimetics. Dr. Telen is a consultant to the company, and several coauthors are employees of the company.
AT ASH 2013
Major finding: The mean time to resolution of vaso-occlusive crisis in patients with sickle cell disease was 103 hours for patients treated with GMI 1070 vs. 144 for those treated with placebo.
Data source: A randomized, double blind multicenter study of 76 patients aged 12-51.
Disclosures: The study was supported by GlycoMimetics. Dr. Telen is a consultant to the company, and several coauthors are employees of the company.
Drug gets orphan designation for MDS
The US Food and Drug Administration (FDA) has granted orphan designation to an investigational drug for the treatment of myelodysplastic syndromes (MDS).
The drug, CPI-613, targets metabolic changes that are thought to occur in many cancer cells.
It has demonstrated activity and tolerability in a phase 1 trial of patients with advanced, relapsed/refractory hematologic malignancies.
CPI-613 previously received orphan designation for acute myeloid leukemia (AML) and pancreatic carcinoma.
Orphan designation is granted for drugs intended to treat diseases that affect fewer than 200,000 individuals in the US. This designation gives the makers of CPI-613, Cornerstone Pharmaceuticals, 7 years of US marketing exclusivity once the drug is approved.
The designation also allows the company to apply for government funding to defray trial costs, tax credits for clinical research expenses, and a potential waiver of the FDA’s application user fee.
CPI-613: Mechanism and phase 1 results
CPI-613 induces cancer-specific inhibition of the mitochondrial enzymes pyruvate dehydrogenase (PDH) and alpha ketoglutarate dehydrogenase (KGDH).
Disrupting the function of PDH and KGDH disrupts tumor mitochondrial metabolism. As a result, tumor cells are starved of energy and biosynthetic intermediates, which leads to cell death.
Researchers evaluated CPI-613 in a phase 1 study of patients with advanced, relapsed/refractory hematologic malignancies.
The team, led by Timothy S. Pardee, MD, of Wake Forest Baptist Medical Center in Winston-Salem, North Carolina, presented the results at the 2013 ASCO Annual Meeting as abstract 2516. (Information in the abstract differs slightly from that presented at the meeting.)
The trial was designed to determine the maximum tolerated dose, safety, and anticancer activity of CPI-613 as a single agent.
Twenty-one evaluable patients received CPI-613 on days 1 and 4 for 3 weeks every 28 days. Ten patients received more than 1 cycle of therapy.
The starting dose was 420 mg/m2. Treatment could be continued if the patient experienced clinical benefit. Doses were escalated to a final dose of 3780 mg/m2.
CPI-613 was generally well-tolerated when infused over 2 hours. Patients did not experience worsening cytopenias at any dose level. However, 1-hour infusions led to grade 3 renal failure in 2 patients.
At a dose of 3780 mg/m2, 1 patient had prolonged grade 3 nausea, and 1 patient had grade 3 renal failure. Six patients received a 2-hour infusion of 2940 mg/m2 without dose-limiting toxicities, so the researchers considered this the maximum tolerated dose.
Of the 21 patients, 9 achieved a response of stable disease or better. One MDS patient achieved a complete remission and maintained it over 23 cycles. One AML patient achieved a morphologic leukemia-free state.
A Burkitt lymphoma patient and a cutaneous T-cell lymphoma patient maintained partial responses over 16 and 15 cycles, respectively. Two multiple myeloma patients, 2 MDS patients, and 1 AML patient had stable disease.
“We are very encouraged by the tolerability and signals of activity seen in several patients in this phase 1 study for whom there is no available therapy shown to provide clinical benefit,” Dr Pardee said.
“We look forward to further evaluating CPI-613 in the early relapsed/refractory AML patient setting when administered in combination with a standard chemotherapeutic regimen, as well as in early relapsed or refractory MDS patients, with the hope of improving the outcomes and the quality of life for these patients through the combined use of this mechanistically novel agent.”
The AML study is a phase 1 trial investigating CPI-613 in combination with high-dose cytarabine and mitoxantrone, and the MDS study is a phase 2 trial investigating single-agent CPI-613.
The US Food and Drug Administration (FDA) has granted orphan designation to an investigational drug for the treatment of myelodysplastic syndromes (MDS).
The drug, CPI-613, targets metabolic changes that are thought to occur in many cancer cells.
It has demonstrated activity and tolerability in a phase 1 trial of patients with advanced, relapsed/refractory hematologic malignancies.
CPI-613 previously received orphan designation for acute myeloid leukemia (AML) and pancreatic carcinoma.
Orphan designation is granted for drugs intended to treat diseases that affect fewer than 200,000 individuals in the US. This designation gives the makers of CPI-613, Cornerstone Pharmaceuticals, 7 years of US marketing exclusivity once the drug is approved.
The designation also allows the company to apply for government funding to defray trial costs, tax credits for clinical research expenses, and a potential waiver of the FDA’s application user fee.
CPI-613: Mechanism and phase 1 results
CPI-613 induces cancer-specific inhibition of the mitochondrial enzymes pyruvate dehydrogenase (PDH) and alpha ketoglutarate dehydrogenase (KGDH).
Disrupting the function of PDH and KGDH disrupts tumor mitochondrial metabolism. As a result, tumor cells are starved of energy and biosynthetic intermediates, which leads to cell death.
Researchers evaluated CPI-613 in a phase 1 study of patients with advanced, relapsed/refractory hematologic malignancies.
The team, led by Timothy S. Pardee, MD, of Wake Forest Baptist Medical Center in Winston-Salem, North Carolina, presented the results at the 2013 ASCO Annual Meeting as abstract 2516. (Information in the abstract differs slightly from that presented at the meeting.)
The trial was designed to determine the maximum tolerated dose, safety, and anticancer activity of CPI-613 as a single agent.
Twenty-one evaluable patients received CPI-613 on days 1 and 4 for 3 weeks every 28 days. Ten patients received more than 1 cycle of therapy.
The starting dose was 420 mg/m2. Treatment could be continued if the patient experienced clinical benefit. Doses were escalated to a final dose of 3780 mg/m2.
CPI-613 was generally well-tolerated when infused over 2 hours. Patients did not experience worsening cytopenias at any dose level. However, 1-hour infusions led to grade 3 renal failure in 2 patients.
At a dose of 3780 mg/m2, 1 patient had prolonged grade 3 nausea, and 1 patient had grade 3 renal failure. Six patients received a 2-hour infusion of 2940 mg/m2 without dose-limiting toxicities, so the researchers considered this the maximum tolerated dose.
Of the 21 patients, 9 achieved a response of stable disease or better. One MDS patient achieved a complete remission and maintained it over 23 cycles. One AML patient achieved a morphologic leukemia-free state.
A Burkitt lymphoma patient and a cutaneous T-cell lymphoma patient maintained partial responses over 16 and 15 cycles, respectively. Two multiple myeloma patients, 2 MDS patients, and 1 AML patient had stable disease.
“We are very encouraged by the tolerability and signals of activity seen in several patients in this phase 1 study for whom there is no available therapy shown to provide clinical benefit,” Dr Pardee said.
“We look forward to further evaluating CPI-613 in the early relapsed/refractory AML patient setting when administered in combination with a standard chemotherapeutic regimen, as well as in early relapsed or refractory MDS patients, with the hope of improving the outcomes and the quality of life for these patients through the combined use of this mechanistically novel agent.”
The AML study is a phase 1 trial investigating CPI-613 in combination with high-dose cytarabine and mitoxantrone, and the MDS study is a phase 2 trial investigating single-agent CPI-613.
The US Food and Drug Administration (FDA) has granted orphan designation to an investigational drug for the treatment of myelodysplastic syndromes (MDS).
The drug, CPI-613, targets metabolic changes that are thought to occur in many cancer cells.
It has demonstrated activity and tolerability in a phase 1 trial of patients with advanced, relapsed/refractory hematologic malignancies.
CPI-613 previously received orphan designation for acute myeloid leukemia (AML) and pancreatic carcinoma.
Orphan designation is granted for drugs intended to treat diseases that affect fewer than 200,000 individuals in the US. This designation gives the makers of CPI-613, Cornerstone Pharmaceuticals, 7 years of US marketing exclusivity once the drug is approved.
The designation also allows the company to apply for government funding to defray trial costs, tax credits for clinical research expenses, and a potential waiver of the FDA’s application user fee.
CPI-613: Mechanism and phase 1 results
CPI-613 induces cancer-specific inhibition of the mitochondrial enzymes pyruvate dehydrogenase (PDH) and alpha ketoglutarate dehydrogenase (KGDH).
Disrupting the function of PDH and KGDH disrupts tumor mitochondrial metabolism. As a result, tumor cells are starved of energy and biosynthetic intermediates, which leads to cell death.
Researchers evaluated CPI-613 in a phase 1 study of patients with advanced, relapsed/refractory hematologic malignancies.
The team, led by Timothy S. Pardee, MD, of Wake Forest Baptist Medical Center in Winston-Salem, North Carolina, presented the results at the 2013 ASCO Annual Meeting as abstract 2516. (Information in the abstract differs slightly from that presented at the meeting.)
The trial was designed to determine the maximum tolerated dose, safety, and anticancer activity of CPI-613 as a single agent.
Twenty-one evaluable patients received CPI-613 on days 1 and 4 for 3 weeks every 28 days. Ten patients received more than 1 cycle of therapy.
The starting dose was 420 mg/m2. Treatment could be continued if the patient experienced clinical benefit. Doses were escalated to a final dose of 3780 mg/m2.
CPI-613 was generally well-tolerated when infused over 2 hours. Patients did not experience worsening cytopenias at any dose level. However, 1-hour infusions led to grade 3 renal failure in 2 patients.
At a dose of 3780 mg/m2, 1 patient had prolonged grade 3 nausea, and 1 patient had grade 3 renal failure. Six patients received a 2-hour infusion of 2940 mg/m2 without dose-limiting toxicities, so the researchers considered this the maximum tolerated dose.
Of the 21 patients, 9 achieved a response of stable disease or better. One MDS patient achieved a complete remission and maintained it over 23 cycles. One AML patient achieved a morphologic leukemia-free state.
A Burkitt lymphoma patient and a cutaneous T-cell lymphoma patient maintained partial responses over 16 and 15 cycles, respectively. Two multiple myeloma patients, 2 MDS patients, and 1 AML patient had stable disease.
“We are very encouraged by the tolerability and signals of activity seen in several patients in this phase 1 study for whom there is no available therapy shown to provide clinical benefit,” Dr Pardee said.
“We look forward to further evaluating CPI-613 in the early relapsed/refractory AML patient setting when administered in combination with a standard chemotherapeutic regimen, as well as in early relapsed or refractory MDS patients, with the hope of improving the outcomes and the quality of life for these patients through the combined use of this mechanistically novel agent.”
The AML study is a phase 1 trial investigating CPI-613 in combination with high-dose cytarabine and mitoxantrone, and the MDS study is a phase 2 trial investigating single-agent CPI-613.
Autoimmune Hemolytic Anemia
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons. AIHA is mediated by antibodies, and in the majority of cases immunglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. This manual reviews the most common types of AIHA, with emphasis on diagnosis and treatment.
To read the full article in PDF:
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons. AIHA is mediated by antibodies, and in the majority of cases immunglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. This manual reviews the most common types of AIHA, with emphasis on diagnosis and treatment.
To read the full article in PDF:
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons. AIHA is mediated by antibodies, and in the majority of cases immunglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. This manual reviews the most common types of AIHA, with emphasis on diagnosis and treatment.
To read the full article in PDF: