User login
Hepcidin levels vary according to cause of anemia
showing anemia
Measuring levels of the hormone hepcidin can help us distinguish anemia caused by iron deficiency from anemia caused by other conditions, a new study suggests.
Investigators say the findings, published in Science Translational Medicine, could help public officials make more informed decisions about distributing iron supplements.
Giving iron supplements to individuals who don’t need them can promote or exacerbate malaria and other infections.
With this research, the investigators linked low levels of hepcidin to iron-deficiency anemia in preschool children living in Africa.
Sant-Rayn Pasricha, PhD, of the University of Oxford in the UK, and his colleagues tested hepcidin levels in 1313 samples taken in 2001 and 2008 from children in The Gambia and Tanzania, respectively.
The team also looked at retrospective data from 25 Gambian children with either postmalarial or nonmalarial anemia.
The mean hepcidin level was significantly lower in children with iron-deficiency anemia than in children with anemia due to inflammation/infection (1.8 ng/mL and 21.7 ng/mL, respectively; P<0.0001).
To expand upon this finding, the investigators modeled the potential impact of screening children for iron supplementation needs based on hepcidin levels rather than anemia status.
If a screen was based on the presence of anemia (hemoglobin <11 g/dL), 77% of iron-deficient children would have received iron supplements, but so would 73% of children with Plasmodium falciparum parasitemia and all of the children with anemia due to inflammation.
On the other hand, if a screen was based on a hepcidin cutoff of <5.5 ng/mL, iron supplements would be given to 77% of children with iron deficiency, 80% of children with iron-deficiency anemia, 20% of children with P falciparum infection, and 14% of children with anemia due to inflammation.
The investigators noted that a lower hepcidin cutoff would have reduced the proportion of individuals with infection and/or inflammation receiving iron, but it would have increased the risk that more children with iron-deficiency anemia would not receive iron. And of course, the reverse is true of a higher hepcidin cutoff.
Nevertheless, the team believes these results are promising. They are now conducting clinical trials to test whether hepcidin levels can be used as a marker to guide iron supplementation decisions. ![]()
showing anemia
Measuring levels of the hormone hepcidin can help us distinguish anemia caused by iron deficiency from anemia caused by other conditions, a new study suggests.
Investigators say the findings, published in Science Translational Medicine, could help public officials make more informed decisions about distributing iron supplements.
Giving iron supplements to individuals who don’t need them can promote or exacerbate malaria and other infections.
With this research, the investigators linked low levels of hepcidin to iron-deficiency anemia in preschool children living in Africa.
Sant-Rayn Pasricha, PhD, of the University of Oxford in the UK, and his colleagues tested hepcidin levels in 1313 samples taken in 2001 and 2008 from children in The Gambia and Tanzania, respectively.
The team also looked at retrospective data from 25 Gambian children with either postmalarial or nonmalarial anemia.
The mean hepcidin level was significantly lower in children with iron-deficiency anemia than in children with anemia due to inflammation/infection (1.8 ng/mL and 21.7 ng/mL, respectively; P<0.0001).
To expand upon this finding, the investigators modeled the potential impact of screening children for iron supplementation needs based on hepcidin levels rather than anemia status.
If a screen was based on the presence of anemia (hemoglobin <11 g/dL), 77% of iron-deficient children would have received iron supplements, but so would 73% of children with Plasmodium falciparum parasitemia and all of the children with anemia due to inflammation.
On the other hand, if a screen was based on a hepcidin cutoff of <5.5 ng/mL, iron supplements would be given to 77% of children with iron deficiency, 80% of children with iron-deficiency anemia, 20% of children with P falciparum infection, and 14% of children with anemia due to inflammation.
The investigators noted that a lower hepcidin cutoff would have reduced the proportion of individuals with infection and/or inflammation receiving iron, but it would have increased the risk that more children with iron-deficiency anemia would not receive iron. And of course, the reverse is true of a higher hepcidin cutoff.
Nevertheless, the team believes these results are promising. They are now conducting clinical trials to test whether hepcidin levels can be used as a marker to guide iron supplementation decisions. ![]()
showing anemia
Measuring levels of the hormone hepcidin can help us distinguish anemia caused by iron deficiency from anemia caused by other conditions, a new study suggests.
Investigators say the findings, published in Science Translational Medicine, could help public officials make more informed decisions about distributing iron supplements.
Giving iron supplements to individuals who don’t need them can promote or exacerbate malaria and other infections.
With this research, the investigators linked low levels of hepcidin to iron-deficiency anemia in preschool children living in Africa.
Sant-Rayn Pasricha, PhD, of the University of Oxford in the UK, and his colleagues tested hepcidin levels in 1313 samples taken in 2001 and 2008 from children in The Gambia and Tanzania, respectively.
The team also looked at retrospective data from 25 Gambian children with either postmalarial or nonmalarial anemia.
The mean hepcidin level was significantly lower in children with iron-deficiency anemia than in children with anemia due to inflammation/infection (1.8 ng/mL and 21.7 ng/mL, respectively; P<0.0001).
To expand upon this finding, the investigators modeled the potential impact of screening children for iron supplementation needs based on hepcidin levels rather than anemia status.
If a screen was based on the presence of anemia (hemoglobin <11 g/dL), 77% of iron-deficient children would have received iron supplements, but so would 73% of children with Plasmodium falciparum parasitemia and all of the children with anemia due to inflammation.
On the other hand, if a screen was based on a hepcidin cutoff of <5.5 ng/mL, iron supplements would be given to 77% of children with iron deficiency, 80% of children with iron-deficiency anemia, 20% of children with P falciparum infection, and 14% of children with anemia due to inflammation.
The investigators noted that a lower hepcidin cutoff would have reduced the proportion of individuals with infection and/or inflammation receiving iron, but it would have increased the risk that more children with iron-deficiency anemia would not receive iron. And of course, the reverse is true of a higher hepcidin cutoff.
Nevertheless, the team believes these results are promising. They are now conducting clinical trials to test whether hepcidin levels can be used as a marker to guide iron supplementation decisions. ![]()
Eculizumab gets full FDA approval for aHUS
Credit: Globovision
The US Food and Drug Administration (FDA) has granted full approval for eculizumab (Soliris) to treat adult and pediatric patients with atypical
hemolytic uremic syndrome (aHUS).
The drug received accelerated approval for this indication in 2011.
Now, eculizumab has received full FDA approval based on the fulfillment of post-marketing requirements, including the submission of data from 2 additional prospective trials of eculizumab in patients with aHUS.
The revised eculizumab label now includes results with 2 years of ongoing treatment in aHUS patients and data on the use of eculizumab prior to supportive care with plasma or plasma exchange in prospective clinical trials.
The drug’s label also includes a boxed warning informing readers that life-threatening and fatal meningococcal infections have occurred in patients treated with eculizumab.
About aHUS and eculizumab
aHUS is a chronic, life-threatening disease in which a genetic deficiency in one or more complement regulatory genes causes chronic, uncontrolled complement activation. This results in complement-mediated thrombotic microangiopathy (TMA), the formation of blood clots in small blood vessels throughout the body.
Permanent, uncontrolled complement activation in aHUS causes a life-long risk for TMA, which leads to sudden and life-threatening damage to the kidney, brain, heart, and other vital organs, as well as premature death. Complement-mediated TMA also causes thrombocytopenia and hemolysis.
Eculizumab is a first-in-class terminal complement inhibitor indicated to inhibit complement-mediated TMA. The drug’s effectiveness in aHUS is based on its effects on TMA and renal function.
Eculizumab received accelerated FDA approval to treat aHUS in September 2011. The FDA granted this approval based on the results of 2 trials that suggested the drug was likely to provide a clinical benefit.
To achieve traditional FDA approval, the drug’s developer, Alexion Pharmaceuticals, was required to submit additional data that confirm the drug provides a clinical benefit.
To that end, the Clinical Studies section (Section 14.2) of the revised eculizumab prescribing information now contains results from 4 prospective, single-arm studies in patients with aHUS.
This includes updated data from the first 2 trials, as well as data from 2 new trials, 1 in pediatric patients with aHUS and the other in adolescents and adults with aHUS.
For details on these trials, see the full prescribing information. ![]()
Credit: Globovision
The US Food and Drug Administration (FDA) has granted full approval for eculizumab (Soliris) to treat adult and pediatric patients with atypical
hemolytic uremic syndrome (aHUS).
The drug received accelerated approval for this indication in 2011.
Now, eculizumab has received full FDA approval based on the fulfillment of post-marketing requirements, including the submission of data from 2 additional prospective trials of eculizumab in patients with aHUS.
The revised eculizumab label now includes results with 2 years of ongoing treatment in aHUS patients and data on the use of eculizumab prior to supportive care with plasma or plasma exchange in prospective clinical trials.
The drug’s label also includes a boxed warning informing readers that life-threatening and fatal meningococcal infections have occurred in patients treated with eculizumab.
About aHUS and eculizumab
aHUS is a chronic, life-threatening disease in which a genetic deficiency in one or more complement regulatory genes causes chronic, uncontrolled complement activation. This results in complement-mediated thrombotic microangiopathy (TMA), the formation of blood clots in small blood vessels throughout the body.
Permanent, uncontrolled complement activation in aHUS causes a life-long risk for TMA, which leads to sudden and life-threatening damage to the kidney, brain, heart, and other vital organs, as well as premature death. Complement-mediated TMA also causes thrombocytopenia and hemolysis.
Eculizumab is a first-in-class terminal complement inhibitor indicated to inhibit complement-mediated TMA. The drug’s effectiveness in aHUS is based on its effects on TMA and renal function.
Eculizumab received accelerated FDA approval to treat aHUS in September 2011. The FDA granted this approval based on the results of 2 trials that suggested the drug was likely to provide a clinical benefit.
To achieve traditional FDA approval, the drug’s developer, Alexion Pharmaceuticals, was required to submit additional data that confirm the drug provides a clinical benefit.
To that end, the Clinical Studies section (Section 14.2) of the revised eculizumab prescribing information now contains results from 4 prospective, single-arm studies in patients with aHUS.
This includes updated data from the first 2 trials, as well as data from 2 new trials, 1 in pediatric patients with aHUS and the other in adolescents and adults with aHUS.
For details on these trials, see the full prescribing information. ![]()
Credit: Globovision
The US Food and Drug Administration (FDA) has granted full approval for eculizumab (Soliris) to treat adult and pediatric patients with atypical
hemolytic uremic syndrome (aHUS).
The drug received accelerated approval for this indication in 2011.
Now, eculizumab has received full FDA approval based on the fulfillment of post-marketing requirements, including the submission of data from 2 additional prospective trials of eculizumab in patients with aHUS.
The revised eculizumab label now includes results with 2 years of ongoing treatment in aHUS patients and data on the use of eculizumab prior to supportive care with plasma or plasma exchange in prospective clinical trials.
The drug’s label also includes a boxed warning informing readers that life-threatening and fatal meningococcal infections have occurred in patients treated with eculizumab.
About aHUS and eculizumab
aHUS is a chronic, life-threatening disease in which a genetic deficiency in one or more complement regulatory genes causes chronic, uncontrolled complement activation. This results in complement-mediated thrombotic microangiopathy (TMA), the formation of blood clots in small blood vessels throughout the body.
Permanent, uncontrolled complement activation in aHUS causes a life-long risk for TMA, which leads to sudden and life-threatening damage to the kidney, brain, heart, and other vital organs, as well as premature death. Complement-mediated TMA also causes thrombocytopenia and hemolysis.
Eculizumab is a first-in-class terminal complement inhibitor indicated to inhibit complement-mediated TMA. The drug’s effectiveness in aHUS is based on its effects on TMA and renal function.
Eculizumab received accelerated FDA approval to treat aHUS in September 2011. The FDA granted this approval based on the results of 2 trials that suggested the drug was likely to provide a clinical benefit.
To achieve traditional FDA approval, the drug’s developer, Alexion Pharmaceuticals, was required to submit additional data that confirm the drug provides a clinical benefit.
To that end, the Clinical Studies section (Section 14.2) of the revised eculizumab prescribing information now contains results from 4 prospective, single-arm studies in patients with aHUS.
This includes updated data from the first 2 trials, as well as data from 2 new trials, 1 in pediatric patients with aHUS and the other in adolescents and adults with aHUS.
For details on these trials, see the full prescribing information. ![]()
Finding could aid treatment of Fanconi anemia
Credit: Tom Ellenberger
Understanding the interaction between 2 genes may be the key to better treatment of Fanconi anemia, according to a paper published in Cell Cycle.
Researchers investigated the relationship between FANCD2 and DNA2, 2 genes known to play roles in DNA repair.
A defective version of FANCD2 can result in Fanconi anemia. And although DNA2 has not been associated with a Fanconi anemia family
yet, genetic studies have implicated DNA2 in the Fanconi anemia DNA repair pathway.
With the current study, the researchers found that deleting either FANCD2 or DNA2 alone makes cells susceptible to DNA damage. But the deletion of both genes enables DNA repair.
“A key implication of this finding is the potential to manipulate DNA2 to improve the survival of FANCD2-deficient cells, and hopefully, by extension, the survival of [Fanconi anemia] patients,” said study author Kenneth Karanja, PhD, a former postdoctoral scholar at the California Institute of Technology in Pasadena.
To uncover the relationship between the genes, Dr Karanja and his colleagues applied DNA-damaging substances—formaldehyde and cisplatin—to 3 types of cells: those lacking FANCD2, those lacking DNA2, and cells lacking both genes.
The groups of cells in which only 1 of the 2 genes had been deleted quickly succumbed to the substance-induced DNA damage. However, the cells lacking both FANCD2 and DNA2 were able to repair the DNA damage and survive.
So the researchers concluded that depletion of DNA2 in FANCD2-deficient cells reverses the cells’ sensitivity to DNA-damaging substances. And this finding may have implications for Fanconi anemia treatment.
“DNA2 is a well-studied gene, and this recent discovery could potentially become the basis for ameliorating the symptoms of this incurable disorder,” said study author Judith Campbell, PhD, of the California Institute of Technology.
“Since much is known about the mechanism of action of DNA2, it is an attractive target for future drug treatments—like small-molecule inhibitors that could reduce [a Fanconi anemia] patient’s cancer predisposition—as well as a possible gene therapy for aiding a patient’s blood cell development.” ![]()
Credit: Tom Ellenberger
Understanding the interaction between 2 genes may be the key to better treatment of Fanconi anemia, according to a paper published in Cell Cycle.
Researchers investigated the relationship between FANCD2 and DNA2, 2 genes known to play roles in DNA repair.
A defective version of FANCD2 can result in Fanconi anemia. And although DNA2 has not been associated with a Fanconi anemia family
yet, genetic studies have implicated DNA2 in the Fanconi anemia DNA repair pathway.
With the current study, the researchers found that deleting either FANCD2 or DNA2 alone makes cells susceptible to DNA damage. But the deletion of both genes enables DNA repair.
“A key implication of this finding is the potential to manipulate DNA2 to improve the survival of FANCD2-deficient cells, and hopefully, by extension, the survival of [Fanconi anemia] patients,” said study author Kenneth Karanja, PhD, a former postdoctoral scholar at the California Institute of Technology in Pasadena.
To uncover the relationship between the genes, Dr Karanja and his colleagues applied DNA-damaging substances—formaldehyde and cisplatin—to 3 types of cells: those lacking FANCD2, those lacking DNA2, and cells lacking both genes.
The groups of cells in which only 1 of the 2 genes had been deleted quickly succumbed to the substance-induced DNA damage. However, the cells lacking both FANCD2 and DNA2 were able to repair the DNA damage and survive.
So the researchers concluded that depletion of DNA2 in FANCD2-deficient cells reverses the cells’ sensitivity to DNA-damaging substances. And this finding may have implications for Fanconi anemia treatment.
“DNA2 is a well-studied gene, and this recent discovery could potentially become the basis for ameliorating the symptoms of this incurable disorder,” said study author Judith Campbell, PhD, of the California Institute of Technology.
“Since much is known about the mechanism of action of DNA2, it is an attractive target for future drug treatments—like small-molecule inhibitors that could reduce [a Fanconi anemia] patient’s cancer predisposition—as well as a possible gene therapy for aiding a patient’s blood cell development.” ![]()
Credit: Tom Ellenberger
Understanding the interaction between 2 genes may be the key to better treatment of Fanconi anemia, according to a paper published in Cell Cycle.
Researchers investigated the relationship between FANCD2 and DNA2, 2 genes known to play roles in DNA repair.
A defective version of FANCD2 can result in Fanconi anemia. And although DNA2 has not been associated with a Fanconi anemia family
yet, genetic studies have implicated DNA2 in the Fanconi anemia DNA repair pathway.
With the current study, the researchers found that deleting either FANCD2 or DNA2 alone makes cells susceptible to DNA damage. But the deletion of both genes enables DNA repair.
“A key implication of this finding is the potential to manipulate DNA2 to improve the survival of FANCD2-deficient cells, and hopefully, by extension, the survival of [Fanconi anemia] patients,” said study author Kenneth Karanja, PhD, a former postdoctoral scholar at the California Institute of Technology in Pasadena.
To uncover the relationship between the genes, Dr Karanja and his colleagues applied DNA-damaging substances—formaldehyde and cisplatin—to 3 types of cells: those lacking FANCD2, those lacking DNA2, and cells lacking both genes.
The groups of cells in which only 1 of the 2 genes had been deleted quickly succumbed to the substance-induced DNA damage. However, the cells lacking both FANCD2 and DNA2 were able to repair the DNA damage and survive.
So the researchers concluded that depletion of DNA2 in FANCD2-deficient cells reverses the cells’ sensitivity to DNA-damaging substances. And this finding may have implications for Fanconi anemia treatment.
“DNA2 is a well-studied gene, and this recent discovery could potentially become the basis for ameliorating the symptoms of this incurable disorder,” said study author Judith Campbell, PhD, of the California Institute of Technology.
“Since much is known about the mechanism of action of DNA2, it is an attractive target for future drug treatments—like small-molecule inhibitors that could reduce [a Fanconi anemia] patient’s cancer predisposition—as well as a possible gene therapy for aiding a patient’s blood cell development.” ![]()
Mutations implicated in hematologic disorders
Credit: Jeremy L. Grisham
An analysis of more than 30,000 individuals has revealed several genetic mutations that appear to play roles in hematologic disorders.
Investigators discovered variants that showed correlations with platelet counts, white blood cell (WBC) counts, hemoglobin concentration, and hematocrit levels.
The group believes these findings could have implications for a range of conditions, including cytopenias, myeloproliferative neoplasms, and stroke.
Guillaume Lettre, PhD, of Université de Montréal and the Montreal Heart Institute in Canada, and his colleagues recounted their discoveries in a letter to Nature Genetics.
The investigators analyzed hemoglobin concentration, hematocrit levels, WBC counts, and platelet counts in 31,340 individuals genotyped on an exome array.
This revealed several missense variants in CXCR2 that were associated with a decreased WBC count. And in a resequencing study, the team identified a CXCR2 frameshift mutation that was associated with congenital neutropenia.
The group also discovered several missense and splice-site variants in genes known to regulate hematopoiesis—TFR2, HBB, TUBB1, SH2B3, and EPO.
A TFR2 mutation (rs139178017) was independently associated with higher hematocrit levels and hemoglobin concentration.
An HBB variant (rs33971440) and an EPO variant (rs62483572), on the other hand, were associated with lower hematocrit levels and hemoglobin concentrations. Further analyses confirmed that having these mutations increased a person’s risk of anemia, with odds ratios of 36.1 and 1.7, respectively.
A TUBB1 missense variant (rs41303899) was associated with decreased platelet count, while 2 missense variants of SH2B3 (rs148636776 and rs72650673) were associated with increased platelet counts.
Lastly, a mutation in JAK2 (rs77375493) was associated with increases in platelets, WBCs, hemoglobin, and hematocrit. And further analyses suggested that individuals with this variant had early stage myeloproliferative neoplasms.
“[T]hese donors also had a higher risk of having a stroke during their lifetime,” said study author Jean-Claude Tardif, MD, of Université de Montréal and the Montreal Heart Institute.
He and his colleagues believe these findings are encouraging, as they provide additional insight into hematologic disorders. But the results also suggest the experimental approach used in this study can be applied to other diseases as well. ![]()
Credit: Jeremy L. Grisham
An analysis of more than 30,000 individuals has revealed several genetic mutations that appear to play roles in hematologic disorders.
Investigators discovered variants that showed correlations with platelet counts, white blood cell (WBC) counts, hemoglobin concentration, and hematocrit levels.
The group believes these findings could have implications for a range of conditions, including cytopenias, myeloproliferative neoplasms, and stroke.
Guillaume Lettre, PhD, of Université de Montréal and the Montreal Heart Institute in Canada, and his colleagues recounted their discoveries in a letter to Nature Genetics.
The investigators analyzed hemoglobin concentration, hematocrit levels, WBC counts, and platelet counts in 31,340 individuals genotyped on an exome array.
This revealed several missense variants in CXCR2 that were associated with a decreased WBC count. And in a resequencing study, the team identified a CXCR2 frameshift mutation that was associated with congenital neutropenia.
The group also discovered several missense and splice-site variants in genes known to regulate hematopoiesis—TFR2, HBB, TUBB1, SH2B3, and EPO.
A TFR2 mutation (rs139178017) was independently associated with higher hematocrit levels and hemoglobin concentration.
An HBB variant (rs33971440) and an EPO variant (rs62483572), on the other hand, were associated with lower hematocrit levels and hemoglobin concentrations. Further analyses confirmed that having these mutations increased a person’s risk of anemia, with odds ratios of 36.1 and 1.7, respectively.
A TUBB1 missense variant (rs41303899) was associated with decreased platelet count, while 2 missense variants of SH2B3 (rs148636776 and rs72650673) were associated with increased platelet counts.
Lastly, a mutation in JAK2 (rs77375493) was associated with increases in platelets, WBCs, hemoglobin, and hematocrit. And further analyses suggested that individuals with this variant had early stage myeloproliferative neoplasms.
“[T]hese donors also had a higher risk of having a stroke during their lifetime,” said study author Jean-Claude Tardif, MD, of Université de Montréal and the Montreal Heart Institute.
He and his colleagues believe these findings are encouraging, as they provide additional insight into hematologic disorders. But the results also suggest the experimental approach used in this study can be applied to other diseases as well. ![]()
Credit: Jeremy L. Grisham
An analysis of more than 30,000 individuals has revealed several genetic mutations that appear to play roles in hematologic disorders.
Investigators discovered variants that showed correlations with platelet counts, white blood cell (WBC) counts, hemoglobin concentration, and hematocrit levels.
The group believes these findings could have implications for a range of conditions, including cytopenias, myeloproliferative neoplasms, and stroke.
Guillaume Lettre, PhD, of Université de Montréal and the Montreal Heart Institute in Canada, and his colleagues recounted their discoveries in a letter to Nature Genetics.
The investigators analyzed hemoglobin concentration, hematocrit levels, WBC counts, and platelet counts in 31,340 individuals genotyped on an exome array.
This revealed several missense variants in CXCR2 that were associated with a decreased WBC count. And in a resequencing study, the team identified a CXCR2 frameshift mutation that was associated with congenital neutropenia.
The group also discovered several missense and splice-site variants in genes known to regulate hematopoiesis—TFR2, HBB, TUBB1, SH2B3, and EPO.
A TFR2 mutation (rs139178017) was independently associated with higher hematocrit levels and hemoglobin concentration.
An HBB variant (rs33971440) and an EPO variant (rs62483572), on the other hand, were associated with lower hematocrit levels and hemoglobin concentrations. Further analyses confirmed that having these mutations increased a person’s risk of anemia, with odds ratios of 36.1 and 1.7, respectively.
A TUBB1 missense variant (rs41303899) was associated with decreased platelet count, while 2 missense variants of SH2B3 (rs148636776 and rs72650673) were associated with increased platelet counts.
Lastly, a mutation in JAK2 (rs77375493) was associated with increases in platelets, WBCs, hemoglobin, and hematocrit. And further analyses suggested that individuals with this variant had early stage myeloproliferative neoplasms.
“[T]hese donors also had a higher risk of having a stroke during their lifetime,” said study author Jean-Claude Tardif, MD, of Université de Montréal and the Montreal Heart Institute.
He and his colleagues believe these findings are encouraging, as they provide additional insight into hematologic disorders. But the results also suggest the experimental approach used in this study can be applied to other diseases as well. ![]()
Study links serum phosphorous levels and anemia risk

Graham Colm
LAS VEGAS—New research suggests a link between serum phosphorous levels and anemia in patients without chronic kidney disease (CKD).
Previous studies have shown that elevations in serum phosphorous are associated with anemia in patients with end-stage renal disease, but whether the link exists in patients without CKD has been unclear.
Now, results of a large study indicate that patients without CKD who have elevated serum phosphorus also have an increased risk of anemia.
John J. Sim, MD, of the Kaiser Permanente Los Angeles Medical Center, and his colleagues presented these findings at the National Kidney Foundation’s 2014 Spring Clinical Meetings (abstract 1708).
The researchers evaluated 32,907 patients with documented serum phosphorus levels, hemoglobin values, and estimated glomerular filtration rates of 60 mL/min or greater. Anemia was defined as having a hemoglobin level below 11 g/dL.
The mean age was 52 years, and 62% of patients were female. The majority of patients were classified as white, 26% as Hispanic, 15% as black, and 7% as Asian.
Serum phosphorus levels ranged from 1.9 mg/dL to 5.7 mg/dL. And 13% of subjects met the criteria for anemia.
Multivariable analysis revealed that each 0.5 mg/dL increase in serum phosphorus level was associated with a 7% increase in the risk of anemia.
The researchers also divided subjects into quartiles according to serum phosphorous levels and calculated the odds ratios (ORs) for anemia.
For the 3.1 mg/dL to 3.5 mg/dL quartile, the OR was 0.85. For the 3.5 mg/dL to 3.9 mg/dL quartile, the OR was 0.90. And for the 3.9 mg/dL to 5.7 mg/dL quartile, the OR was 1.05.
The researchers noted that the link was “more pronounced” in men, but the results suggest that elevated serum phosphorous levels are associated with anemia in non-CKD patients of both sexes. ![]()
Graham Colm
LAS VEGAS—New research suggests a link between serum phosphorous levels and anemia in patients without chronic kidney disease (CKD).
Previous studies have shown that elevations in serum phosphorous are associated with anemia in patients with end-stage renal disease, but whether the link exists in patients without CKD has been unclear.
Now, results of a large study indicate that patients without CKD who have elevated serum phosphorus also have an increased risk of anemia.
John J. Sim, MD, of the Kaiser Permanente Los Angeles Medical Center, and his colleagues presented these findings at the National Kidney Foundation’s 2014 Spring Clinical Meetings (abstract 1708).
The researchers evaluated 32,907 patients with documented serum phosphorus levels, hemoglobin values, and estimated glomerular filtration rates of 60 mL/min or greater. Anemia was defined as having a hemoglobin level below 11 g/dL.
The mean age was 52 years, and 62% of patients were female. The majority of patients were classified as white, 26% as Hispanic, 15% as black, and 7% as Asian.
Serum phosphorus levels ranged from 1.9 mg/dL to 5.7 mg/dL. And 13% of subjects met the criteria for anemia.
Multivariable analysis revealed that each 0.5 mg/dL increase in serum phosphorus level was associated with a 7% increase in the risk of anemia.
The researchers also divided subjects into quartiles according to serum phosphorous levels and calculated the odds ratios (ORs) for anemia.
For the 3.1 mg/dL to 3.5 mg/dL quartile, the OR was 0.85. For the 3.5 mg/dL to 3.9 mg/dL quartile, the OR was 0.90. And for the 3.9 mg/dL to 5.7 mg/dL quartile, the OR was 1.05.
The researchers noted that the link was “more pronounced” in men, but the results suggest that elevated serum phosphorous levels are associated with anemia in non-CKD patients of both sexes. ![]()
Graham Colm
LAS VEGAS—New research suggests a link between serum phosphorous levels and anemia in patients without chronic kidney disease (CKD).
Previous studies have shown that elevations in serum phosphorous are associated with anemia in patients with end-stage renal disease, but whether the link exists in patients without CKD has been unclear.
Now, results of a large study indicate that patients without CKD who have elevated serum phosphorus also have an increased risk of anemia.
John J. Sim, MD, of the Kaiser Permanente Los Angeles Medical Center, and his colleagues presented these findings at the National Kidney Foundation’s 2014 Spring Clinical Meetings (abstract 1708).
The researchers evaluated 32,907 patients with documented serum phosphorus levels, hemoglobin values, and estimated glomerular filtration rates of 60 mL/min or greater. Anemia was defined as having a hemoglobin level below 11 g/dL.
The mean age was 52 years, and 62% of patients were female. The majority of patients were classified as white, 26% as Hispanic, 15% as black, and 7% as Asian.
Serum phosphorus levels ranged from 1.9 mg/dL to 5.7 mg/dL. And 13% of subjects met the criteria for anemia.
Multivariable analysis revealed that each 0.5 mg/dL increase in serum phosphorus level was associated with a 7% increase in the risk of anemia.
The researchers also divided subjects into quartiles according to serum phosphorous levels and calculated the odds ratios (ORs) for anemia.
For the 3.1 mg/dL to 3.5 mg/dL quartile, the OR was 0.85. For the 3.5 mg/dL to 3.9 mg/dL quartile, the OR was 0.90. And for the 3.9 mg/dL to 5.7 mg/dL quartile, the OR was 1.05.
The researchers noted that the link was “more pronounced” in men, but the results suggest that elevated serum phosphorous levels are associated with anemia in non-CKD patients of both sexes. ![]()
Groups investigate malaria complications in children
Credit: Peter H. Seeberger
Two studies published in PLOS Pathogens provide new insight into the malaria-related complications that can occur in children.
One study revealed how the immune system manages to prevent malaria fever in children infected with Plasmodium falciparum.
And with the other study, researchers identified proteins that can help them distinguish children with complicated malaria syndromes from those with uncomplicated malaria.
Analyzing immune response
In the first study, Peter Crompton, MD, of the US National Institute of Allergy and Infectious Diseases in Rockville, Maryland, and his colleagues analyzed immune cells from healthy children before the malaria season and from the same children after their first bout of malaria fever during the ensuing malaria season.
The researchers exposed both sets of immune cells to parasite-infected red blood cells and found that their responses were different.
When confronted with parasites before the malaria season, the children’s immune cells produced large amounts of molecules that promote inflammation—such as IL-1b, IL-6, and IL-8—which results in fever and other malaria symptoms.
But after a malaria fever episode, the immune cells responded by producing more anti-inflammatory molecules—such as IL-10 and TGF-b—and showed evidence of an enhanced ability to recognize and destroy parasites.
The ability of the immune cells to mount this response—somewhat effective in controlling the parasites but avoiding systemic inflammation and fever—seems to depend on the continued exposure to parasites through bites of infected mosquitoes.
When the researchers took blood again from the same children after the subsequent dry season (when there are few or no new infections) and exposed the immune cells to parasite-infected red blood cells, the anti-inflammatory response had returned to baseline, leaving children susceptible again to malaria-induced inflammation and fever.
The researchers said these findings shed new light on the notion of premunition, an immune response that protects against illness and high numbers of parasites in the blood without completely eliminating the infection.
They suggested that it evolved as an appropriate immune response to at least partially protect young children from potentially life-threatening inflammation and unchecked parasite replication before they acquire antibodies that protect against the onset of malaria symptoms.
Proteins provide answers
In the second study, Peter Nilsson, PhD, of SciLifeLab in Stockholm, Sweden, and his colleagues used a systematic proteomics approach to distinguish children who develop malaria-related complications from those who do not.
The researchers compared proteins in the blood of uninfected children with proteins in malaria-infected children. And they compared proteins in children with severe malaria syndromes to proteins in uncomplicated cases.
The team analyzed 1015 proteins in blood samples from more than 719 children. They divided the samples into “discovery” and “verification” sets, and only associations found in both sets were reported.
The researchers identified 41 proteins that distinguished malaria patients from uninfected children from the same community. Most of these were components of the inflammatory response.
Thirteen proteins helped the team distinguish uncomplicated malaria from severe malaria syndromes. They identified proteins specific to the 2 most deadly complicated malaria syndromes in children—severe malarial anemia and cerebral malaria.
Markers of oxidative stress were related to severe malarial anemia. And markers of endothelial activation, platelet adhesion, and muscular damage were identified in children with cerebral malaria.
The researchers said their study could aid the discovery of distinct mechanisms in the human response to malaria infection between the 2 most fatal syndromes of childhood malaria. ![]()
Credit: Peter H. Seeberger
Two studies published in PLOS Pathogens provide new insight into the malaria-related complications that can occur in children.
One study revealed how the immune system manages to prevent malaria fever in children infected with Plasmodium falciparum.
And with the other study, researchers identified proteins that can help them distinguish children with complicated malaria syndromes from those with uncomplicated malaria.
Analyzing immune response
In the first study, Peter Crompton, MD, of the US National Institute of Allergy and Infectious Diseases in Rockville, Maryland, and his colleagues analyzed immune cells from healthy children before the malaria season and from the same children after their first bout of malaria fever during the ensuing malaria season.
The researchers exposed both sets of immune cells to parasite-infected red blood cells and found that their responses were different.
When confronted with parasites before the malaria season, the children’s immune cells produced large amounts of molecules that promote inflammation—such as IL-1b, IL-6, and IL-8—which results in fever and other malaria symptoms.
But after a malaria fever episode, the immune cells responded by producing more anti-inflammatory molecules—such as IL-10 and TGF-b—and showed evidence of an enhanced ability to recognize and destroy parasites.
The ability of the immune cells to mount this response—somewhat effective in controlling the parasites but avoiding systemic inflammation and fever—seems to depend on the continued exposure to parasites through bites of infected mosquitoes.
When the researchers took blood again from the same children after the subsequent dry season (when there are few or no new infections) and exposed the immune cells to parasite-infected red blood cells, the anti-inflammatory response had returned to baseline, leaving children susceptible again to malaria-induced inflammation and fever.
The researchers said these findings shed new light on the notion of premunition, an immune response that protects against illness and high numbers of parasites in the blood without completely eliminating the infection.
They suggested that it evolved as an appropriate immune response to at least partially protect young children from potentially life-threatening inflammation and unchecked parasite replication before they acquire antibodies that protect against the onset of malaria symptoms.
Proteins provide answers
In the second study, Peter Nilsson, PhD, of SciLifeLab in Stockholm, Sweden, and his colleagues used a systematic proteomics approach to distinguish children who develop malaria-related complications from those who do not.
The researchers compared proteins in the blood of uninfected children with proteins in malaria-infected children. And they compared proteins in children with severe malaria syndromes to proteins in uncomplicated cases.
The team analyzed 1015 proteins in blood samples from more than 719 children. They divided the samples into “discovery” and “verification” sets, and only associations found in both sets were reported.
The researchers identified 41 proteins that distinguished malaria patients from uninfected children from the same community. Most of these were components of the inflammatory response.
Thirteen proteins helped the team distinguish uncomplicated malaria from severe malaria syndromes. They identified proteins specific to the 2 most deadly complicated malaria syndromes in children—severe malarial anemia and cerebral malaria.
Markers of oxidative stress were related to severe malarial anemia. And markers of endothelial activation, platelet adhesion, and muscular damage were identified in children with cerebral malaria.
The researchers said their study could aid the discovery of distinct mechanisms in the human response to malaria infection between the 2 most fatal syndromes of childhood malaria. ![]()
Credit: Peter H. Seeberger
Two studies published in PLOS Pathogens provide new insight into the malaria-related complications that can occur in children.
One study revealed how the immune system manages to prevent malaria fever in children infected with Plasmodium falciparum.
And with the other study, researchers identified proteins that can help them distinguish children with complicated malaria syndromes from those with uncomplicated malaria.
Analyzing immune response
In the first study, Peter Crompton, MD, of the US National Institute of Allergy and Infectious Diseases in Rockville, Maryland, and his colleagues analyzed immune cells from healthy children before the malaria season and from the same children after their first bout of malaria fever during the ensuing malaria season.
The researchers exposed both sets of immune cells to parasite-infected red blood cells and found that their responses were different.
When confronted with parasites before the malaria season, the children’s immune cells produced large amounts of molecules that promote inflammation—such as IL-1b, IL-6, and IL-8—which results in fever and other malaria symptoms.
But after a malaria fever episode, the immune cells responded by producing more anti-inflammatory molecules—such as IL-10 and TGF-b—and showed evidence of an enhanced ability to recognize and destroy parasites.
The ability of the immune cells to mount this response—somewhat effective in controlling the parasites but avoiding systemic inflammation and fever—seems to depend on the continued exposure to parasites through bites of infected mosquitoes.
When the researchers took blood again from the same children after the subsequent dry season (when there are few or no new infections) and exposed the immune cells to parasite-infected red blood cells, the anti-inflammatory response had returned to baseline, leaving children susceptible again to malaria-induced inflammation and fever.
The researchers said these findings shed new light on the notion of premunition, an immune response that protects against illness and high numbers of parasites in the blood without completely eliminating the infection.
They suggested that it evolved as an appropriate immune response to at least partially protect young children from potentially life-threatening inflammation and unchecked parasite replication before they acquire antibodies that protect against the onset of malaria symptoms.
Proteins provide answers
In the second study, Peter Nilsson, PhD, of SciLifeLab in Stockholm, Sweden, and his colleagues used a systematic proteomics approach to distinguish children who develop malaria-related complications from those who do not.
The researchers compared proteins in the blood of uninfected children with proteins in malaria-infected children. And they compared proteins in children with severe malaria syndromes to proteins in uncomplicated cases.
The team analyzed 1015 proteins in blood samples from more than 719 children. They divided the samples into “discovery” and “verification” sets, and only associations found in both sets were reported.
The researchers identified 41 proteins that distinguished malaria patients from uninfected children from the same community. Most of these were components of the inflammatory response.
Thirteen proteins helped the team distinguish uncomplicated malaria from severe malaria syndromes. They identified proteins specific to the 2 most deadly complicated malaria syndromes in children—severe malarial anemia and cerebral malaria.
Markers of oxidative stress were related to severe malarial anemia. And markers of endothelial activation, platelet adhesion, and muscular damage were identified in children with cerebral malaria.
The researchers said their study could aid the discovery of distinct mechanisms in the human response to malaria infection between the 2 most fatal syndromes of childhood malaria. ![]()
Agent can reduce ESR in SCD, study suggests
Credit: Graham Colm
MIAMI—Results of a small study suggest an experimental agent can decrease the erythrocyte sedimentation rate (ESR) in patients with sickle cell disease (SCD).
Previous research has shown the ESR is elevated in SCD patients during vaso-occlusive crisis.
In the current study, the experimental agent MST-188 decreased elevated ESRs by 50% in blood from SCD patients.
According to researchers, this reflects reduced red blood cell (RBC) aggregation and suggests improved microvascular blood flow.
“The data from this study are consistent with observations in prior studies that MST-188 decreases blood viscosity and RBC aggregation and improves microvascular blood flow, and supportive of the potential for MST-188 to shorten the duration of sickle cell crisis,” said Martin Emanuele, PhD, of Mast Therapeutics, the company developing MST-188.
Dr Emanuele presented the study data at the recent 8th Annual Sickle Cell Disease Research & Educational Symposium.
MST-188 is a non-ionic, linear block copolymer composed of a central chain of hydrophobic polyoxypropylene and 2 flanking chains of hydrophylic polyoxyethylene. In previous studies, the agent has shown hemorheologic properties that result in improved microvascular blood flow.
For the current study, the researchers compared MST-188 to dextrans, evaluating their effects on the ESR in blood collected from SCD patients and healthy controls. Dextrans are branched polysaccharides of 10-70 kDa that have been used as antithrombotic agents and plasma expanders.
The researchers analyzed EDTA-anticoagulated whole blood collected from 8 healthy individuals and 11 SCD patients. The team treated samples with MST-188; dextran 10K, 18K , 40K, and 70K at various concentrations; or saline control.
At baseline, ESRs for SCD patients were significantly higher than for the healthy subjects. The mean ESRs were 26.4 ± 7.1 mm/hr and 14.6 ± 2.1 mm/hr, respectively.
However, adding MST-188 to the SCD patient samples decreased the mean ESR to 14.1 ± 4.6 mm/hr (Δ47%). On the other hand, comparable concentrations of dextrans showed little or no effect on the ESR in SCD samples.
The researchers said MST-188 may reduce the ESR by inhibiting acute-phase-reactant-induced RBC aggregates, and this may result from the effect of MST-188 on RBC membranes or cell-protein interactions.
Regardless of the exact mechanism, the team said lowering the ESR reflects reduced RBC aggregation and suggests improved microvascular blood flow, which indicates that MST-188 may be able to shorten the duration of vaso-occlusive crisis.
“It is widely understood that multiple biological processes contribute to vaso-occlusion and that an effective solution requires a broad, multi-modal approach rather than a single targeted therapy,” Dr Emanuele said.
“In addition to the effects on RBC aggregation, our data suggest that MST-188 addresses cell adhesion and platelet activation, reduces hemolysis, lowers blood viscosity, and limits reperfusion injury following restoration of blood flow.”
He and his colleagues at Mast Therapeutics are planning additional studies of MST-188 in SCD. The agent is currently under investigation in a phase 3 trial. ![]()
Credit: Graham Colm
MIAMI—Results of a small study suggest an experimental agent can decrease the erythrocyte sedimentation rate (ESR) in patients with sickle cell disease (SCD).
Previous research has shown the ESR is elevated in SCD patients during vaso-occlusive crisis.
In the current study, the experimental agent MST-188 decreased elevated ESRs by 50% in blood from SCD patients.
According to researchers, this reflects reduced red blood cell (RBC) aggregation and suggests improved microvascular blood flow.
“The data from this study are consistent with observations in prior studies that MST-188 decreases blood viscosity and RBC aggregation and improves microvascular blood flow, and supportive of the potential for MST-188 to shorten the duration of sickle cell crisis,” said Martin Emanuele, PhD, of Mast Therapeutics, the company developing MST-188.
Dr Emanuele presented the study data at the recent 8th Annual Sickle Cell Disease Research & Educational Symposium.
MST-188 is a non-ionic, linear block copolymer composed of a central chain of hydrophobic polyoxypropylene and 2 flanking chains of hydrophylic polyoxyethylene. In previous studies, the agent has shown hemorheologic properties that result in improved microvascular blood flow.
For the current study, the researchers compared MST-188 to dextrans, evaluating their effects on the ESR in blood collected from SCD patients and healthy controls. Dextrans are branched polysaccharides of 10-70 kDa that have been used as antithrombotic agents and plasma expanders.
The researchers analyzed EDTA-anticoagulated whole blood collected from 8 healthy individuals and 11 SCD patients. The team treated samples with MST-188; dextran 10K, 18K , 40K, and 70K at various concentrations; or saline control.
At baseline, ESRs for SCD patients were significantly higher than for the healthy subjects. The mean ESRs were 26.4 ± 7.1 mm/hr and 14.6 ± 2.1 mm/hr, respectively.
However, adding MST-188 to the SCD patient samples decreased the mean ESR to 14.1 ± 4.6 mm/hr (Δ47%). On the other hand, comparable concentrations of dextrans showed little or no effect on the ESR in SCD samples.
The researchers said MST-188 may reduce the ESR by inhibiting acute-phase-reactant-induced RBC aggregates, and this may result from the effect of MST-188 on RBC membranes or cell-protein interactions.
Regardless of the exact mechanism, the team said lowering the ESR reflects reduced RBC aggregation and suggests improved microvascular blood flow, which indicates that MST-188 may be able to shorten the duration of vaso-occlusive crisis.
“It is widely understood that multiple biological processes contribute to vaso-occlusion and that an effective solution requires a broad, multi-modal approach rather than a single targeted therapy,” Dr Emanuele said.
“In addition to the effects on RBC aggregation, our data suggest that MST-188 addresses cell adhesion and platelet activation, reduces hemolysis, lowers blood viscosity, and limits reperfusion injury following restoration of blood flow.”
He and his colleagues at Mast Therapeutics are planning additional studies of MST-188 in SCD. The agent is currently under investigation in a phase 3 trial. ![]()
Credit: Graham Colm
MIAMI—Results of a small study suggest an experimental agent can decrease the erythrocyte sedimentation rate (ESR) in patients with sickle cell disease (SCD).
Previous research has shown the ESR is elevated in SCD patients during vaso-occlusive crisis.
In the current study, the experimental agent MST-188 decreased elevated ESRs by 50% in blood from SCD patients.
According to researchers, this reflects reduced red blood cell (RBC) aggregation and suggests improved microvascular blood flow.
“The data from this study are consistent with observations in prior studies that MST-188 decreases blood viscosity and RBC aggregation and improves microvascular blood flow, and supportive of the potential for MST-188 to shorten the duration of sickle cell crisis,” said Martin Emanuele, PhD, of Mast Therapeutics, the company developing MST-188.
Dr Emanuele presented the study data at the recent 8th Annual Sickle Cell Disease Research & Educational Symposium.
MST-188 is a non-ionic, linear block copolymer composed of a central chain of hydrophobic polyoxypropylene and 2 flanking chains of hydrophylic polyoxyethylene. In previous studies, the agent has shown hemorheologic properties that result in improved microvascular blood flow.
For the current study, the researchers compared MST-188 to dextrans, evaluating their effects on the ESR in blood collected from SCD patients and healthy controls. Dextrans are branched polysaccharides of 10-70 kDa that have been used as antithrombotic agents and plasma expanders.
The researchers analyzed EDTA-anticoagulated whole blood collected from 8 healthy individuals and 11 SCD patients. The team treated samples with MST-188; dextran 10K, 18K , 40K, and 70K at various concentrations; or saline control.
At baseline, ESRs for SCD patients were significantly higher than for the healthy subjects. The mean ESRs were 26.4 ± 7.1 mm/hr and 14.6 ± 2.1 mm/hr, respectively.
However, adding MST-188 to the SCD patient samples decreased the mean ESR to 14.1 ± 4.6 mm/hr (Δ47%). On the other hand, comparable concentrations of dextrans showed little or no effect on the ESR in SCD samples.
The researchers said MST-188 may reduce the ESR by inhibiting acute-phase-reactant-induced RBC aggregates, and this may result from the effect of MST-188 on RBC membranes or cell-protein interactions.
Regardless of the exact mechanism, the team said lowering the ESR reflects reduced RBC aggregation and suggests improved microvascular blood flow, which indicates that MST-188 may be able to shorten the duration of vaso-occlusive crisis.
“It is widely understood that multiple biological processes contribute to vaso-occlusion and that an effective solution requires a broad, multi-modal approach rather than a single targeted therapy,” Dr Emanuele said.
“In addition to the effects on RBC aggregation, our data suggest that MST-188 addresses cell adhesion and platelet activation, reduces hemolysis, lowers blood viscosity, and limits reperfusion injury following restoration of blood flow.”
He and his colleagues at Mast Therapeutics are planning additional studies of MST-188 in SCD. The agent is currently under investigation in a phase 3 trial.
Findings could increase use of delayed cord clamping
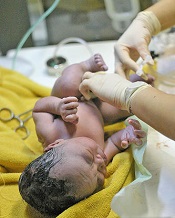
Credit: Meutia Chaerani
and Indradi Soemardjan
A baby’s position prior to delayed umbilical cord clamping does not affect the volume of placental blood transferred, according to a study published in The Lancet.
Researchers found that placing a baby on the mother’s chest or abdomen before clamping does not decrease the amount of blood transferred when compared to holding the child in the recommended introitus position.
As the chest/abdomen position is more desirable, the researchers believe this discovery could help increase the use of delayed cord clamping, which has been shown to reduce the risk of iron deficiency in infancy.
Current recommendations for delayed cord clamping are based on studies conducted 35 years ago. They suggest that, for effective placental transfusion to occur, a baby must be held at the level of the placenta—the introitus position.
The researchers noted that this position can be uncomfortable for the person holding the baby and interferes with immediate contact between the mother and child. These issues could be contributing to low compliance with delayed cord clamping, ultimately resulting in higher-than-necessary levels of iron deficiency in babies.
So the team decided to examine whether the transfer of blood in delayed cord clamping procedures is affected by the position in which the baby is held immediately after birth.
They conducted the study in 3 university-affiliated hospitals in Argentina, evaluating 197 babies who were held in the introitus position and 194 babies who were immediately placed on the mother’s abdomen or chest.
By measuring the babies’ weights at the point of birth and immediately after the delayed cord clamping procedure, the researchers were able to measure the volume of blood that had transferred from the placenta to the child.
They found no statistically significant difference between the 2 groups in the volume of blood transferred. The mean weight change was 56 g for babies in the introitus group and 53 g for babies in the abdomen/chest group (P=0.45).
“Our study suggests that when umbilical cord clamping is delayed for 2 minutes, holding the baby on the mother’s chest or abdomen is no worse than the currently recommended practice of holding the baby below this level,” said study author Nestor Vain, MD, of the Foundation for Maternal and Child Health (FUNDASAMIN) in Buenos Aires, Argentina.
“Because of the potential of enhanced bonding between mother and baby, increased success of breastfeeding, and the compliance with the procedure, holding the infant by the mother immediately after birth should be strongly recommended.”
Writing in a related comment article, Tonse Raju, MD, of the National Institute of Child Health and Human Development in Bethesda, Maryland, noted that introducing delayed cord clamping into practice has not been easy, and logistical issues might be partly responsible.
“Intuitively, to keep the newborn baby’s position below the level of the placenta in situ should maximize the volume of placental transfusion,” Dr Raju wrote. “However, trying to hold on to a wet, vigorously crying, and wriggling infant at the perineum for 2 minutes, in gloved hands, is awkward and can be risky.”
“[This study] should bring a sigh of relief from those trying to incorporate delayed umbilical cord clamping into practice. The results are convincing and show that gravity did not have an effect on volume of placental transfusion.”
Credit: Meutia Chaerani
and Indradi Soemardjan
A baby’s position prior to delayed umbilical cord clamping does not affect the volume of placental blood transferred, according to a study published in The Lancet.
Researchers found that placing a baby on the mother’s chest or abdomen before clamping does not decrease the amount of blood transferred when compared to holding the child in the recommended introitus position.
As the chest/abdomen position is more desirable, the researchers believe this discovery could help increase the use of delayed cord clamping, which has been shown to reduce the risk of iron deficiency in infancy.
Current recommendations for delayed cord clamping are based on studies conducted 35 years ago. They suggest that, for effective placental transfusion to occur, a baby must be held at the level of the placenta—the introitus position.
The researchers noted that this position can be uncomfortable for the person holding the baby and interferes with immediate contact between the mother and child. These issues could be contributing to low compliance with delayed cord clamping, ultimately resulting in higher-than-necessary levels of iron deficiency in babies.
So the team decided to examine whether the transfer of blood in delayed cord clamping procedures is affected by the position in which the baby is held immediately after birth.
They conducted the study in 3 university-affiliated hospitals in Argentina, evaluating 197 babies who were held in the introitus position and 194 babies who were immediately placed on the mother’s abdomen or chest.
By measuring the babies’ weights at the point of birth and immediately after the delayed cord clamping procedure, the researchers were able to measure the volume of blood that had transferred from the placenta to the child.
They found no statistically significant difference between the 2 groups in the volume of blood transferred. The mean weight change was 56 g for babies in the introitus group and 53 g for babies in the abdomen/chest group (P=0.45).
“Our study suggests that when umbilical cord clamping is delayed for 2 minutes, holding the baby on the mother’s chest or abdomen is no worse than the currently recommended practice of holding the baby below this level,” said study author Nestor Vain, MD, of the Foundation for Maternal and Child Health (FUNDASAMIN) in Buenos Aires, Argentina.
“Because of the potential of enhanced bonding between mother and baby, increased success of breastfeeding, and the compliance with the procedure, holding the infant by the mother immediately after birth should be strongly recommended.”
Writing in a related comment article, Tonse Raju, MD, of the National Institute of Child Health and Human Development in Bethesda, Maryland, noted that introducing delayed cord clamping into practice has not been easy, and logistical issues might be partly responsible.
“Intuitively, to keep the newborn baby’s position below the level of the placenta in situ should maximize the volume of placental transfusion,” Dr Raju wrote. “However, trying to hold on to a wet, vigorously crying, and wriggling infant at the perineum for 2 minutes, in gloved hands, is awkward and can be risky.”
“[This study] should bring a sigh of relief from those trying to incorporate delayed umbilical cord clamping into practice. The results are convincing and show that gravity did not have an effect on volume of placental transfusion.”
Credit: Meutia Chaerani
and Indradi Soemardjan
A baby’s position prior to delayed umbilical cord clamping does not affect the volume of placental blood transferred, according to a study published in The Lancet.
Researchers found that placing a baby on the mother’s chest or abdomen before clamping does not decrease the amount of blood transferred when compared to holding the child in the recommended introitus position.
As the chest/abdomen position is more desirable, the researchers believe this discovery could help increase the use of delayed cord clamping, which has been shown to reduce the risk of iron deficiency in infancy.
Current recommendations for delayed cord clamping are based on studies conducted 35 years ago. They suggest that, for effective placental transfusion to occur, a baby must be held at the level of the placenta—the introitus position.
The researchers noted that this position can be uncomfortable for the person holding the baby and interferes with immediate contact between the mother and child. These issues could be contributing to low compliance with delayed cord clamping, ultimately resulting in higher-than-necessary levels of iron deficiency in babies.
So the team decided to examine whether the transfer of blood in delayed cord clamping procedures is affected by the position in which the baby is held immediately after birth.
They conducted the study in 3 university-affiliated hospitals in Argentina, evaluating 197 babies who were held in the introitus position and 194 babies who were immediately placed on the mother’s abdomen or chest.
By measuring the babies’ weights at the point of birth and immediately after the delayed cord clamping procedure, the researchers were able to measure the volume of blood that had transferred from the placenta to the child.
They found no statistically significant difference between the 2 groups in the volume of blood transferred. The mean weight change was 56 g for babies in the introitus group and 53 g for babies in the abdomen/chest group (P=0.45).
“Our study suggests that when umbilical cord clamping is delayed for 2 minutes, holding the baby on the mother’s chest or abdomen is no worse than the currently recommended practice of holding the baby below this level,” said study author Nestor Vain, MD, of the Foundation for Maternal and Child Health (FUNDASAMIN) in Buenos Aires, Argentina.
“Because of the potential of enhanced bonding between mother and baby, increased success of breastfeeding, and the compliance with the procedure, holding the infant by the mother immediately after birth should be strongly recommended.”
Writing in a related comment article, Tonse Raju, MD, of the National Institute of Child Health and Human Development in Bethesda, Maryland, noted that introducing delayed cord clamping into practice has not been easy, and logistical issues might be partly responsible.
“Intuitively, to keep the newborn baby’s position below the level of the placenta in situ should maximize the volume of placental transfusion,” Dr Raju wrote. “However, trying to hold on to a wet, vigorously crying, and wriggling infant at the perineum for 2 minutes, in gloved hands, is awkward and can be risky.”
“[This study] should bring a sigh of relief from those trying to incorporate delayed umbilical cord clamping into practice. The results are convincing and show that gravity did not have an effect on volume of placental transfusion.”
NICE supports use of ESAs in cancer patients

Anemic cancer patients in the UK may soon have 6 erythropoiesis-stimulating agents (ESAs) available through the National Health Service.
The National Institute for Health and Care Excellence (NICE) has drafted a guidance recommending 5 epoetins—2 alfas, 1 beta, 1 theta, and 1 zeta—and 1 darbepoetin to treat anemia in adult cancer patients receiving chemotherapy.
NICE has started a consultation on the guidance, and it will be open for comment until May 9.
To inform NICE on the clinical effectiveness of the 6 ESAs, an independent appraisal committee looked at results from several clinical trials and reviewed the existing guidance.
The committee concluded that the ESAs were effective at increasing hemoglobin levels and managing anemia when compared to current standards of care. The drugs reduced the need for blood transfusions in patients receiving cancer treatment.
Furthermore, analyses suggested the ESAs are cost-effective. However, the draft guidance recommends that healthcare professionals offer patients the appropriate ESA with the lowest cost.
The guidance recommends the ESAs for their approved indications.
Epoetin alfa (marketed as Eprex and Binocrit) and epoetin zeta (marketed as Retacrit) are approved in the UK to treat anemia and reduce transfusion requirements in adult patients receiving chemotherapy for solid tumors, malignant lymphoma, or multiple myeloma. Patients must be at risk of transfusion, as assessed by their general status (eg, cardiovascular status, pre-existing anemia at the start of chemotherapy).
Eprex, Binocrit, and Retacrit are available in pre-filled syringes at net prices of £5.53, £5.09, and £5.66 per 1000 units, respectively.
Epoetin beta (marketed as NeoRecormon) and Epoetin theta (marketed as Eporatio) are approved to treat symptomatic anemia in adult patients with non-myeloid malignancies who are receiving chemotherapy.
NeoRecormo is available in a pre-filled syringe at a net price of £3.51 per 500 units, and Eporatio is available in a pre-filled syringe at a net price of £5.99 per 1000 units.
Darbepoetin alfa (marketed as Aranesp) is approved to treat symptomatic anemia in adult patients with non-myeloid malignancies who are receiving chemotherapy. It is recommended for use at hemoglobin concentrations of 100 g/l or lower, with target values up to 120 g/l. Aranesp is available in a pre-filled syringe at a net price of £14.68 per 10 micrograms.
Costs exclude tax and may vary in different settings. For more information, see the draft guidance.
Anemic cancer patients in the UK may soon have 6 erythropoiesis-stimulating agents (ESAs) available through the National Health Service.
The National Institute for Health and Care Excellence (NICE) has drafted a guidance recommending 5 epoetins—2 alfas, 1 beta, 1 theta, and 1 zeta—and 1 darbepoetin to treat anemia in adult cancer patients receiving chemotherapy.
NICE has started a consultation on the guidance, and it will be open for comment until May 9.
To inform NICE on the clinical effectiveness of the 6 ESAs, an independent appraisal committee looked at results from several clinical trials and reviewed the existing guidance.
The committee concluded that the ESAs were effective at increasing hemoglobin levels and managing anemia when compared to current standards of care. The drugs reduced the need for blood transfusions in patients receiving cancer treatment.
Furthermore, analyses suggested the ESAs are cost-effective. However, the draft guidance recommends that healthcare professionals offer patients the appropriate ESA with the lowest cost.
The guidance recommends the ESAs for their approved indications.
Epoetin alfa (marketed as Eprex and Binocrit) and epoetin zeta (marketed as Retacrit) are approved in the UK to treat anemia and reduce transfusion requirements in adult patients receiving chemotherapy for solid tumors, malignant lymphoma, or multiple myeloma. Patients must be at risk of transfusion, as assessed by their general status (eg, cardiovascular status, pre-existing anemia at the start of chemotherapy).
Eprex, Binocrit, and Retacrit are available in pre-filled syringes at net prices of £5.53, £5.09, and £5.66 per 1000 units, respectively.
Epoetin beta (marketed as NeoRecormon) and Epoetin theta (marketed as Eporatio) are approved to treat symptomatic anemia in adult patients with non-myeloid malignancies who are receiving chemotherapy.
NeoRecormo is available in a pre-filled syringe at a net price of £3.51 per 500 units, and Eporatio is available in a pre-filled syringe at a net price of £5.99 per 1000 units.
Darbepoetin alfa (marketed as Aranesp) is approved to treat symptomatic anemia in adult patients with non-myeloid malignancies who are receiving chemotherapy. It is recommended for use at hemoglobin concentrations of 100 g/l or lower, with target values up to 120 g/l. Aranesp is available in a pre-filled syringe at a net price of £14.68 per 10 micrograms.
Costs exclude tax and may vary in different settings. For more information, see the draft guidance.
Anemic cancer patients in the UK may soon have 6 erythropoiesis-stimulating agents (ESAs) available through the National Health Service.
The National Institute for Health and Care Excellence (NICE) has drafted a guidance recommending 5 epoetins—2 alfas, 1 beta, 1 theta, and 1 zeta—and 1 darbepoetin to treat anemia in adult cancer patients receiving chemotherapy.
NICE has started a consultation on the guidance, and it will be open for comment until May 9.
To inform NICE on the clinical effectiveness of the 6 ESAs, an independent appraisal committee looked at results from several clinical trials and reviewed the existing guidance.
The committee concluded that the ESAs were effective at increasing hemoglobin levels and managing anemia when compared to current standards of care. The drugs reduced the need for blood transfusions in patients receiving cancer treatment.
Furthermore, analyses suggested the ESAs are cost-effective. However, the draft guidance recommends that healthcare professionals offer patients the appropriate ESA with the lowest cost.
The guidance recommends the ESAs for their approved indications.
Epoetin alfa (marketed as Eprex and Binocrit) and epoetin zeta (marketed as Retacrit) are approved in the UK to treat anemia and reduce transfusion requirements in adult patients receiving chemotherapy for solid tumors, malignant lymphoma, or multiple myeloma. Patients must be at risk of transfusion, as assessed by their general status (eg, cardiovascular status, pre-existing anemia at the start of chemotherapy).
Eprex, Binocrit, and Retacrit are available in pre-filled syringes at net prices of £5.53, £5.09, and £5.66 per 1000 units, respectively.
Epoetin beta (marketed as NeoRecormon) and Epoetin theta (marketed as Eporatio) are approved to treat symptomatic anemia in adult patients with non-myeloid malignancies who are receiving chemotherapy.
NeoRecormo is available in a pre-filled syringe at a net price of £3.51 per 500 units, and Eporatio is available in a pre-filled syringe at a net price of £5.99 per 1000 units.
Darbepoetin alfa (marketed as Aranesp) is approved to treat symptomatic anemia in adult patients with non-myeloid malignancies who are receiving chemotherapy. It is recommended for use at hemoglobin concentrations of 100 g/l or lower, with target values up to 120 g/l. Aranesp is available in a pre-filled syringe at a net price of £14.68 per 10 micrograms.
Costs exclude tax and may vary in different settings. For more information, see the draft guidance.
NICE standard aims to improve care of SCD patients

and a normal one
Betty Pace
The UK’s National Institute for Health and Care Excellence (NICE) has published a new quality standard to improve care for patients with acute painful episodes resulting from sickle cell disease (SCD).
NICE quality standards include statements that describe high-priority areas for improvement in a defined care or service area.
The current standard builds upon the 2012 NICE clinical guideline for the management of acute painful sickle cell episodes.
The standard states that SCD patients who present at the hospital with a pain episode should have a thorough assessment and receive appropriate pain relief within 30 minutes.
They should then be assessed regularly until satisfactory pain relief has been achieved, with careful monitoring of adverse events in those who are taking strong opioids.
Patients should be assessed for symptoms of acute chest syndrome, such as chest pain, fever, and abnormal respiratory signs.
Healthcare professionals must have access to locally agreed protocols on treatment and management, as well as support from specialist centers.
Healthcare professionals should also provide SCD patients with clear written information to encourage involvement in their continuing care.
“We know that the management of this condition is variable across the country, and there is a need to address patient concerns, such as unacceptable delays in receiving pain relief,” said Gillian Leng, Deputy Chief Executive and Director of Health and Social Care at NICE.
“This new standard will drive up the quality of care people with sickle cell receive, so that they can be confident they will be comfortable during their stay in hospital.”
NICE quality standards are not requirements or targets, but the health and social care system is obliged to consider them in planning and delivering services.
and a normal one
Betty Pace
The UK’s National Institute for Health and Care Excellence (NICE) has published a new quality standard to improve care for patients with acute painful episodes resulting from sickle cell disease (SCD).
NICE quality standards include statements that describe high-priority areas for improvement in a defined care or service area.
The current standard builds upon the 2012 NICE clinical guideline for the management of acute painful sickle cell episodes.
The standard states that SCD patients who present at the hospital with a pain episode should have a thorough assessment and receive appropriate pain relief within 30 minutes.
They should then be assessed regularly until satisfactory pain relief has been achieved, with careful monitoring of adverse events in those who are taking strong opioids.
Patients should be assessed for symptoms of acute chest syndrome, such as chest pain, fever, and abnormal respiratory signs.
Healthcare professionals must have access to locally agreed protocols on treatment and management, as well as support from specialist centers.
Healthcare professionals should also provide SCD patients with clear written information to encourage involvement in their continuing care.
“We know that the management of this condition is variable across the country, and there is a need to address patient concerns, such as unacceptable delays in receiving pain relief,” said Gillian Leng, Deputy Chief Executive and Director of Health and Social Care at NICE.
“This new standard will drive up the quality of care people with sickle cell receive, so that they can be confident they will be comfortable during their stay in hospital.”
NICE quality standards are not requirements or targets, but the health and social care system is obliged to consider them in planning and delivering services.
and a normal one
Betty Pace
The UK’s National Institute for Health and Care Excellence (NICE) has published a new quality standard to improve care for patients with acute painful episodes resulting from sickle cell disease (SCD).
NICE quality standards include statements that describe high-priority areas for improvement in a defined care or service area.
The current standard builds upon the 2012 NICE clinical guideline for the management of acute painful sickle cell episodes.
The standard states that SCD patients who present at the hospital with a pain episode should have a thorough assessment and receive appropriate pain relief within 30 minutes.
They should then be assessed regularly until satisfactory pain relief has been achieved, with careful monitoring of adverse events in those who are taking strong opioids.
Patients should be assessed for symptoms of acute chest syndrome, such as chest pain, fever, and abnormal respiratory signs.
Healthcare professionals must have access to locally agreed protocols on treatment and management, as well as support from specialist centers.
Healthcare professionals should also provide SCD patients with clear written information to encourage involvement in their continuing care.
“We know that the management of this condition is variable across the country, and there is a need to address patient concerns, such as unacceptable delays in receiving pain relief,” said Gillian Leng, Deputy Chief Executive and Director of Health and Social Care at NICE.
“This new standard will drive up the quality of care people with sickle cell receive, so that they can be confident they will be comfortable during their stay in hospital.”
NICE quality standards are not requirements or targets, but the health and social care system is obliged to consider them in planning and delivering services.