User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Is A Patient Getting Under Your Skin? A Dermatologist Shares Tips for Coping
SAN DIEGO — In his role as chief medical officer for Ascension Medical Group–Texas, which employs about 1,000 physicians across every medical specialty,
At the annual meeting of the American Academy of Dermatology, Dr. Reichenberg, professor of dermatology at the University of Texas at Austin, shared several tips for managing such difficult patients:
Look for ‘red flags’ that raise concerns. This may include patients’ unrealistic expectations for a cure, “which could be because of their cultural or educational background,” he said. Difficult patients also may view physicians as enemies.

“They may quote legal jargon or threaten consequences if there is a bad outcome,” he explained. “They may say, ‘I’m a great reviewer on Yelp and I look forward to giving you a great Yelp review when we finish today.’ They may also have previously sued physicians, or they may tell you that their last physician was horrible.”
Shift into robot mode. In other words, don’t stray from your practice’s protocol by offering special treatment to difficult patients. For example, if a difficult patient shows up 15 minutes late and the office has a policy that patients should be rescheduled if they arrive 10 minutes late, “do not break that policy no matter what, because that’s your protocol,” he advised. “You also do not promise anything you don’t know or that nobody could know. If a difficult patient asks, ‘what is the statistical chance that I’ll get better with this treatment,’ you either say, ‘studies have shown that this is the exact percentage,’ or ‘I don’t know. We’re going to do our best.’”
Set expectations at the outset. “If I walk into the room and the nurse has been in there for 25 minutes doing the intake and I know it’s going to be a long visit, I’ll start by saying, ‘I have 8 minutes to see you today,’ ” Dr. Reichenberg said. “ ‘Whatever we don’t finish today we’ll have to do during a follow-up visit, so let’s please prioritize what we need to do.’ ” Sometimes he sets his smartphone alarm to 8 minutes and when the timer goes off, he’ll say, “I’m so sorry, but I have to go.” For talkative patients, he continued, “I’ll ask, ‘is it okay if I interrupt you if I have a clarifying question?’ That gives you permission to interrupt.”
Blame a third “party” or policy. When patients express anger, find an “enemy” that you can be angry at together. “You might say something like, ‘I’m as frustrated as you are; I can’t believe how broken our health care system is that I have only 8 minutes with you today,’ ” he advised. “Show that you’re on the same side as them.” You could also blame a policy by saying something like, “I’m sorry; I can’t do that for you. My practice has strict rules about that. I’m as frustrated as you are.”
Practice self-regulation. Here, the goal is to delay the time between being triggered by the patient who gets under your skin and your response to that person, such as saying you received “a page or an important text before you walk out of the exam room,” he said. This principle also applies to messages that unreasonable individuals send by e-mail or through messages on their patient portal. “Probably the biggest mistakes I’ve seen from physicians is when they get really angry and they write an angry portal message or e-mail and send it out,” Dr. Reichenberg said. “If I feel triggered, I wait to respond. I’ll sometimes forward [the response] it to my nurse and request that person to send it out the next morning, so the reply reads, ‘Dr. Reichenberg said…’ That gives me the chance to calm down. It also gives the patient a chance to calm down.”
Never worry alone. When struggling to communicate effectively with a difficult patient, he recommends seeking input from a trusted physician colleague. “Better yet, pick up the phone and call the patient’s primary care doctor or another specialist who takes care of that person, and talk about it,” he said. “Figure out if this is your problem or the patient’s problem. They may offer advice on how to handle that person.”
Know when the conflict is untenable. Sometimes it’s best to resign from providing care to difficult patients. “I might write or say something like, ‘I resign from your care. I do not have any expertise to help you with your problem,’ ” Dr. Reichenberg said. “Or, ‘I don’t know that I have the infrastructure to handle the kind of problems you have. I’m not sure we’re the best fit.’ I would suggest that you not give every single detail about why you’re firing them, because the patients could write a step-by-step response, arguing against that.” If you decide to terminate the relationship with a patient, make sure that he or she is not in an acute phase of their illness. “You do not want to get sued for patient abandonment,” he said. “Know your state laws. In general, you’re going to give them a statement of intent to terminate — usually in 30 days — but you have to agree to treat them emergently.” Dr. Reichenberg also provides them with a referral source so they can find a new physician and waives the fee for sending medical records to the new provider. “Also, though it’s not required, I’ll include a statement about the consequences of not receiving care, if I think that they’re [neglecting] their own care,” he said.
Dr. Reichenberg reported having no financial disclosures.
SAN DIEGO — In his role as chief medical officer for Ascension Medical Group–Texas, which employs about 1,000 physicians across every medical specialty,
At the annual meeting of the American Academy of Dermatology, Dr. Reichenberg, professor of dermatology at the University of Texas at Austin, shared several tips for managing such difficult patients:
Look for ‘red flags’ that raise concerns. This may include patients’ unrealistic expectations for a cure, “which could be because of their cultural or educational background,” he said. Difficult patients also may view physicians as enemies.
“They may quote legal jargon or threaten consequences if there is a bad outcome,” he explained. “They may say, ‘I’m a great reviewer on Yelp and I look forward to giving you a great Yelp review when we finish today.’ They may also have previously sued physicians, or they may tell you that their last physician was horrible.”
Shift into robot mode. In other words, don’t stray from your practice’s protocol by offering special treatment to difficult patients. For example, if a difficult patient shows up 15 minutes late and the office has a policy that patients should be rescheduled if they arrive 10 minutes late, “do not break that policy no matter what, because that’s your protocol,” he advised. “You also do not promise anything you don’t know or that nobody could know. If a difficult patient asks, ‘what is the statistical chance that I’ll get better with this treatment,’ you either say, ‘studies have shown that this is the exact percentage,’ or ‘I don’t know. We’re going to do our best.’”
Set expectations at the outset. “If I walk into the room and the nurse has been in there for 25 minutes doing the intake and I know it’s going to be a long visit, I’ll start by saying, ‘I have 8 minutes to see you today,’ ” Dr. Reichenberg said. “ ‘Whatever we don’t finish today we’ll have to do during a follow-up visit, so let’s please prioritize what we need to do.’ ” Sometimes he sets his smartphone alarm to 8 minutes and when the timer goes off, he’ll say, “I’m so sorry, but I have to go.” For talkative patients, he continued, “I’ll ask, ‘is it okay if I interrupt you if I have a clarifying question?’ That gives you permission to interrupt.”
Blame a third “party” or policy. When patients express anger, find an “enemy” that you can be angry at together. “You might say something like, ‘I’m as frustrated as you are; I can’t believe how broken our health care system is that I have only 8 minutes with you today,’ ” he advised. “Show that you’re on the same side as them.” You could also blame a policy by saying something like, “I’m sorry; I can’t do that for you. My practice has strict rules about that. I’m as frustrated as you are.”
Practice self-regulation. Here, the goal is to delay the time between being triggered by the patient who gets under your skin and your response to that person, such as saying you received “a page or an important text before you walk out of the exam room,” he said. This principle also applies to messages that unreasonable individuals send by e-mail or through messages on their patient portal. “Probably the biggest mistakes I’ve seen from physicians is when they get really angry and they write an angry portal message or e-mail and send it out,” Dr. Reichenberg said. “If I feel triggered, I wait to respond. I’ll sometimes forward [the response] it to my nurse and request that person to send it out the next morning, so the reply reads, ‘Dr. Reichenberg said…’ That gives me the chance to calm down. It also gives the patient a chance to calm down.”
Never worry alone. When struggling to communicate effectively with a difficult patient, he recommends seeking input from a trusted physician colleague. “Better yet, pick up the phone and call the patient’s primary care doctor or another specialist who takes care of that person, and talk about it,” he said. “Figure out if this is your problem or the patient’s problem. They may offer advice on how to handle that person.”
Know when the conflict is untenable. Sometimes it’s best to resign from providing care to difficult patients. “I might write or say something like, ‘I resign from your care. I do not have any expertise to help you with your problem,’ ” Dr. Reichenberg said. “Or, ‘I don’t know that I have the infrastructure to handle the kind of problems you have. I’m not sure we’re the best fit.’ I would suggest that you not give every single detail about why you’re firing them, because the patients could write a step-by-step response, arguing against that.” If you decide to terminate the relationship with a patient, make sure that he or she is not in an acute phase of their illness. “You do not want to get sued for patient abandonment,” he said. “Know your state laws. In general, you’re going to give them a statement of intent to terminate — usually in 30 days — but you have to agree to treat them emergently.” Dr. Reichenberg also provides them with a referral source so they can find a new physician and waives the fee for sending medical records to the new provider. “Also, though it’s not required, I’ll include a statement about the consequences of not receiving care, if I think that they’re [neglecting] their own care,” he said.
Dr. Reichenberg reported having no financial disclosures.
SAN DIEGO — In his role as chief medical officer for Ascension Medical Group–Texas, which employs about 1,000 physicians across every medical specialty,
At the annual meeting of the American Academy of Dermatology, Dr. Reichenberg, professor of dermatology at the University of Texas at Austin, shared several tips for managing such difficult patients:
Look for ‘red flags’ that raise concerns. This may include patients’ unrealistic expectations for a cure, “which could be because of their cultural or educational background,” he said. Difficult patients also may view physicians as enemies.
“They may quote legal jargon or threaten consequences if there is a bad outcome,” he explained. “They may say, ‘I’m a great reviewer on Yelp and I look forward to giving you a great Yelp review when we finish today.’ They may also have previously sued physicians, or they may tell you that their last physician was horrible.”
Shift into robot mode. In other words, don’t stray from your practice’s protocol by offering special treatment to difficult patients. For example, if a difficult patient shows up 15 minutes late and the office has a policy that patients should be rescheduled if they arrive 10 minutes late, “do not break that policy no matter what, because that’s your protocol,” he advised. “You also do not promise anything you don’t know or that nobody could know. If a difficult patient asks, ‘what is the statistical chance that I’ll get better with this treatment,’ you either say, ‘studies have shown that this is the exact percentage,’ or ‘I don’t know. We’re going to do our best.’”
Set expectations at the outset. “If I walk into the room and the nurse has been in there for 25 minutes doing the intake and I know it’s going to be a long visit, I’ll start by saying, ‘I have 8 minutes to see you today,’ ” Dr. Reichenberg said. “ ‘Whatever we don’t finish today we’ll have to do during a follow-up visit, so let’s please prioritize what we need to do.’ ” Sometimes he sets his smartphone alarm to 8 minutes and when the timer goes off, he’ll say, “I’m so sorry, but I have to go.” For talkative patients, he continued, “I’ll ask, ‘is it okay if I interrupt you if I have a clarifying question?’ That gives you permission to interrupt.”
Blame a third “party” or policy. When patients express anger, find an “enemy” that you can be angry at together. “You might say something like, ‘I’m as frustrated as you are; I can’t believe how broken our health care system is that I have only 8 minutes with you today,’ ” he advised. “Show that you’re on the same side as them.” You could also blame a policy by saying something like, “I’m sorry; I can’t do that for you. My practice has strict rules about that. I’m as frustrated as you are.”
Practice self-regulation. Here, the goal is to delay the time between being triggered by the patient who gets under your skin and your response to that person, such as saying you received “a page or an important text before you walk out of the exam room,” he said. This principle also applies to messages that unreasonable individuals send by e-mail or through messages on their patient portal. “Probably the biggest mistakes I’ve seen from physicians is when they get really angry and they write an angry portal message or e-mail and send it out,” Dr. Reichenberg said. “If I feel triggered, I wait to respond. I’ll sometimes forward [the response] it to my nurse and request that person to send it out the next morning, so the reply reads, ‘Dr. Reichenberg said…’ That gives me the chance to calm down. It also gives the patient a chance to calm down.”
Never worry alone. When struggling to communicate effectively with a difficult patient, he recommends seeking input from a trusted physician colleague. “Better yet, pick up the phone and call the patient’s primary care doctor or another specialist who takes care of that person, and talk about it,” he said. “Figure out if this is your problem or the patient’s problem. They may offer advice on how to handle that person.”
Know when the conflict is untenable. Sometimes it’s best to resign from providing care to difficult patients. “I might write or say something like, ‘I resign from your care. I do not have any expertise to help you with your problem,’ ” Dr. Reichenberg said. “Or, ‘I don’t know that I have the infrastructure to handle the kind of problems you have. I’m not sure we’re the best fit.’ I would suggest that you not give every single detail about why you’re firing them, because the patients could write a step-by-step response, arguing against that.” If you decide to terminate the relationship with a patient, make sure that he or she is not in an acute phase of their illness. “You do not want to get sued for patient abandonment,” he said. “Know your state laws. In general, you’re going to give them a statement of intent to terminate — usually in 30 days — but you have to agree to treat them emergently.” Dr. Reichenberg also provides them with a referral source so they can find a new physician and waives the fee for sending medical records to the new provider. “Also, though it’s not required, I’ll include a statement about the consequences of not receiving care, if I think that they’re [neglecting] their own care,” he said.
Dr. Reichenberg reported having no financial disclosures.
FROM AAD 2024
Expert Highlights Emerging Trends in Neuromodulators
SAN DIEGO — .
“This technique is more popular in Asia than it is here in the US,” Dr. Green, who practices dermatology in Coral Gables, Florida, said at the annual meeting of the American Academy of Dermatology. As opposed to intramuscular injections, “it’s an intradermal delivery, so you use numbing cream prior, and you’re injecting botulinum toxin A nearly parallel to the skin surface with the bevel of the needle up,” he said. “You want to use a precise product. It’s uncomfortable delivering volume so superficially due to the tissue distention, so I also use a massager. I inject approximately 0.05 mL to 0.1 mL per point. This does really work.”

This mode of delivery was evaluated in a prospective, double-blind, split-face study in South Korea, which enrolled 18 volunteers who received an intradermal injection of botulinum toxin A into one cheek and normal saline into the contralateral side as a control. Participants were between 30 and 54 years of age and were seen at the clinic 2, 4, 8, and 12 weeks after the injection. At each visit, investigators took photographs, used a facial analyzer to evaluate the pores and wrinkles of the infraorbital area, and used a Sebumeter to evaluate sebum secretions from both cheeks. Improvement or aggravation in skin texture was evaluated by both volunteers and clinicians on a numeric scale from –4 (severe aggravation) to +4 (marked improvement) at each visit, and following photographic review, the wrinkle score of the nasolabial fold was graded on a 5-point scale.
The researchers observed no significant effects on the wrinkles of the infraorbital area and on sebum secretion. However, on the side where botulinum toxin A was injected, there were significant improvements in the wrinkles of the nasolabial fold and skin texture, they reported. The effects on nasolabial fold wrinkles lasted 12 weeks, effects on skin texture lasted 8 weeks, and improvement in pore size was only observed at week 2, they wrote. One serious adverse event occurred: a case of facial palsy after the injection of 30 units of botulinum toxin A in one cheek. However, injection of 20 units of botulinum toxin A in one cheek was not associated with any adverse events.
“The duration of these treatments is yet to be determined, but I think this is definitely going to gain popularity in the US,” said Dr. Green, clinical assistant professor of dermatology at the University of Miami Department of Dermatology and Cutaneous Surgery.
Recently Approved Neurotoxin
He also discussed letibotulinumtoxinA-wlbg (Letybo), an injectable neurotoxin long used in South Korea, which the US Food and Drug Administration (FDA) approved for the temporary improvement in the appearance of moderate to severe glabellar (frown) lines in adults on March 4, 2024. Approval was based on positive results from three phase 3 trials of letibotulinumtoxinA-wlbg that enrolled more than 1,000 individuals in the United States and Europe.
“This is the sixth approved neurotoxin in the US,” Dr. Green said. “It is derived from the CBFC26 strain of Clostridium botulinum, and it’s a purified 900 kDa type A toxin complex with human serum albumin and sodium chloride as its excipients.” It comes in a 50-unit or 100-unit vial and requires refrigeration. “To me, the most fascinating thing about this product is that it has been the number-one selling botulinum toxin on the South Korea market for the last 5 years,” he said. “But what do we know about its characteristics?”
In a non-inferiority trial, Chinese researchers enrolled 500 patients with moderate to severe glabellar wrinkles to investigate the efficacy and safety of letibotulinumtoxinA-wlbg and onabotulinumtoxinA. Participants were randomized 3:1 to receive 20 U of letibotulinumtoxinA-wlbg or onabotulinumtoxinA and then observed them for 16 weeks. The primary endpoint was noninferiority in the proportion of study participants who received a score of 0 or 1 for glabellar wrinkles on a four-point photographic evaluation scale, as assessed by an evaluator at maximum frown at 4 weeks.
At week 4, 88.49% of participants in the letibotulinumtoxinA-wlbg arm achieved a score of 0 or 1 for glabellar wrinkles, compared with 87.39% of those in the onabotulinumtoxinA arm (P = .7469). No significant differences were observed for secondary efficacy or safety endpoints between the two treatments. “It will be interesting to see how this product does when it’s available to us,” Dr. Green said.
Another potential newcomer is ready-to-use liquid botulinum neurotoxin. RelabotulinumtoxinA is a complex, protein-free, ready-to-use liquid botulinum toxin A designed to avoid the traditional requirement to reconstitute it from powder, according to Galderma. It features a saline phosphate buffer solution, so it contains no human or animal-derived excipients, Dr. Green pointed out, and it eliminates the variability, errors, and risks associated with reconstitution.
“There was a report in the neurology literature of botulinum toxin being reconstituted with sterile water for cervical dystonia,” he noted. “When this was injected, it was excruciatingly painful, because it created an osmotic gradient within the muscle. So, if we can take a step away from human error, that would be a good thing.”
To date, Dr. Green said, four phase 3 trials of relabotulinumtoxinA involving more than 1,900 patients have been conducted in the United States and Canada evaluating its use for glabellar frown lines and lateral canthal lines, “and the data is impressive,” he said. This product is still investigational, said Dr. Green, who has not had experience injecting it in the clinical trial program.
The idea of a rapid onset botulinum toxin is also emerging. TrenibotulinumtoxinE, which is being developed by Allergan, “is similar to a type A neurotoxin,” Dr. Green said. “It inhibits neuromuscular transmission via presynaptic vesicular protein synaptosomal-associated protein (SNAP)-25 but at a different cleavage site. It has a faster onset — within one day — but a shorter duration — 3-4 weeks.”
In a dose escalation study of its use for glabellar frown lines, 80% of participants achieved a two-grade investigator-rated improvement in glabellar frown line severity at maximum frown at the highest dose. The maximum clinical effect of trenibotulinumtoxinE was seen within 24 hours and lasted between 14 and 30 days.
“The question is, if it is approved by the FDA, where would this product fit in our practices?” Dr. Green asked. “The effect is gone in 3 weeks as opposed to 4 months,” so this may be an option to recommend for someone who is reticent to try neurotoxins, he said, “or a patient who comes to you on a Friday and says, ‘I have a gala tomorrow night.’ ”
Dr. Green disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for many pharmaceutical companies, including Allergan and Galderma.
SAN DIEGO — .
“This technique is more popular in Asia than it is here in the US,” Dr. Green, who practices dermatology in Coral Gables, Florida, said at the annual meeting of the American Academy of Dermatology. As opposed to intramuscular injections, “it’s an intradermal delivery, so you use numbing cream prior, and you’re injecting botulinum toxin A nearly parallel to the skin surface with the bevel of the needle up,” he said. “You want to use a precise product. It’s uncomfortable delivering volume so superficially due to the tissue distention, so I also use a massager. I inject approximately 0.05 mL to 0.1 mL per point. This does really work.”
This mode of delivery was evaluated in a prospective, double-blind, split-face study in South Korea, which enrolled 18 volunteers who received an intradermal injection of botulinum toxin A into one cheek and normal saline into the contralateral side as a control. Participants were between 30 and 54 years of age and were seen at the clinic 2, 4, 8, and 12 weeks after the injection. At each visit, investigators took photographs, used a facial analyzer to evaluate the pores and wrinkles of the infraorbital area, and used a Sebumeter to evaluate sebum secretions from both cheeks. Improvement or aggravation in skin texture was evaluated by both volunteers and clinicians on a numeric scale from –4 (severe aggravation) to +4 (marked improvement) at each visit, and following photographic review, the wrinkle score of the nasolabial fold was graded on a 5-point scale.
The researchers observed no significant effects on the wrinkles of the infraorbital area and on sebum secretion. However, on the side where botulinum toxin A was injected, there were significant improvements in the wrinkles of the nasolabial fold and skin texture, they reported. The effects on nasolabial fold wrinkles lasted 12 weeks, effects on skin texture lasted 8 weeks, and improvement in pore size was only observed at week 2, they wrote. One serious adverse event occurred: a case of facial palsy after the injection of 30 units of botulinum toxin A in one cheek. However, injection of 20 units of botulinum toxin A in one cheek was not associated with any adverse events.
“The duration of these treatments is yet to be determined, but I think this is definitely going to gain popularity in the US,” said Dr. Green, clinical assistant professor of dermatology at the University of Miami Department of Dermatology and Cutaneous Surgery.
Recently Approved Neurotoxin
He also discussed letibotulinumtoxinA-wlbg (Letybo), an injectable neurotoxin long used in South Korea, which the US Food and Drug Administration (FDA) approved for the temporary improvement in the appearance of moderate to severe glabellar (frown) lines in adults on March 4, 2024. Approval was based on positive results from three phase 3 trials of letibotulinumtoxinA-wlbg that enrolled more than 1,000 individuals in the United States and Europe.
“This is the sixth approved neurotoxin in the US,” Dr. Green said. “It is derived from the CBFC26 strain of Clostridium botulinum, and it’s a purified 900 kDa type A toxin complex with human serum albumin and sodium chloride as its excipients.” It comes in a 50-unit or 100-unit vial and requires refrigeration. “To me, the most fascinating thing about this product is that it has been the number-one selling botulinum toxin on the South Korea market for the last 5 years,” he said. “But what do we know about its characteristics?”
In a non-inferiority trial, Chinese researchers enrolled 500 patients with moderate to severe glabellar wrinkles to investigate the efficacy and safety of letibotulinumtoxinA-wlbg and onabotulinumtoxinA. Participants were randomized 3:1 to receive 20 U of letibotulinumtoxinA-wlbg or onabotulinumtoxinA and then observed them for 16 weeks. The primary endpoint was noninferiority in the proportion of study participants who received a score of 0 or 1 for glabellar wrinkles on a four-point photographic evaluation scale, as assessed by an evaluator at maximum frown at 4 weeks.
At week 4, 88.49% of participants in the letibotulinumtoxinA-wlbg arm achieved a score of 0 or 1 for glabellar wrinkles, compared with 87.39% of those in the onabotulinumtoxinA arm (P = .7469). No significant differences were observed for secondary efficacy or safety endpoints between the two treatments. “It will be interesting to see how this product does when it’s available to us,” Dr. Green said.
Another potential newcomer is ready-to-use liquid botulinum neurotoxin. RelabotulinumtoxinA is a complex, protein-free, ready-to-use liquid botulinum toxin A designed to avoid the traditional requirement to reconstitute it from powder, according to Galderma. It features a saline phosphate buffer solution, so it contains no human or animal-derived excipients, Dr. Green pointed out, and it eliminates the variability, errors, and risks associated with reconstitution.
“There was a report in the neurology literature of botulinum toxin being reconstituted with sterile water for cervical dystonia,” he noted. “When this was injected, it was excruciatingly painful, because it created an osmotic gradient within the muscle. So, if we can take a step away from human error, that would be a good thing.”
To date, Dr. Green said, four phase 3 trials of relabotulinumtoxinA involving more than 1,900 patients have been conducted in the United States and Canada evaluating its use for glabellar frown lines and lateral canthal lines, “and the data is impressive,” he said. This product is still investigational, said Dr. Green, who has not had experience injecting it in the clinical trial program.
The idea of a rapid onset botulinum toxin is also emerging. TrenibotulinumtoxinE, which is being developed by Allergan, “is similar to a type A neurotoxin,” Dr. Green said. “It inhibits neuromuscular transmission via presynaptic vesicular protein synaptosomal-associated protein (SNAP)-25 but at a different cleavage site. It has a faster onset — within one day — but a shorter duration — 3-4 weeks.”
In a dose escalation study of its use for glabellar frown lines, 80% of participants achieved a two-grade investigator-rated improvement in glabellar frown line severity at maximum frown at the highest dose. The maximum clinical effect of trenibotulinumtoxinE was seen within 24 hours and lasted between 14 and 30 days.
“The question is, if it is approved by the FDA, where would this product fit in our practices?” Dr. Green asked. “The effect is gone in 3 weeks as opposed to 4 months,” so this may be an option to recommend for someone who is reticent to try neurotoxins, he said, “or a patient who comes to you on a Friday and says, ‘I have a gala tomorrow night.’ ”
Dr. Green disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for many pharmaceutical companies, including Allergan and Galderma.
SAN DIEGO — .
“This technique is more popular in Asia than it is here in the US,” Dr. Green, who practices dermatology in Coral Gables, Florida, said at the annual meeting of the American Academy of Dermatology. As opposed to intramuscular injections, “it’s an intradermal delivery, so you use numbing cream prior, and you’re injecting botulinum toxin A nearly parallel to the skin surface with the bevel of the needle up,” he said. “You want to use a precise product. It’s uncomfortable delivering volume so superficially due to the tissue distention, so I also use a massager. I inject approximately 0.05 mL to 0.1 mL per point. This does really work.”
This mode of delivery was evaluated in a prospective, double-blind, split-face study in South Korea, which enrolled 18 volunteers who received an intradermal injection of botulinum toxin A into one cheek and normal saline into the contralateral side as a control. Participants were between 30 and 54 years of age and were seen at the clinic 2, 4, 8, and 12 weeks after the injection. At each visit, investigators took photographs, used a facial analyzer to evaluate the pores and wrinkles of the infraorbital area, and used a Sebumeter to evaluate sebum secretions from both cheeks. Improvement or aggravation in skin texture was evaluated by both volunteers and clinicians on a numeric scale from –4 (severe aggravation) to +4 (marked improvement) at each visit, and following photographic review, the wrinkle score of the nasolabial fold was graded on a 5-point scale.
The researchers observed no significant effects on the wrinkles of the infraorbital area and on sebum secretion. However, on the side where botulinum toxin A was injected, there were significant improvements in the wrinkles of the nasolabial fold and skin texture, they reported. The effects on nasolabial fold wrinkles lasted 12 weeks, effects on skin texture lasted 8 weeks, and improvement in pore size was only observed at week 2, they wrote. One serious adverse event occurred: a case of facial palsy after the injection of 30 units of botulinum toxin A in one cheek. However, injection of 20 units of botulinum toxin A in one cheek was not associated with any adverse events.
“The duration of these treatments is yet to be determined, but I think this is definitely going to gain popularity in the US,” said Dr. Green, clinical assistant professor of dermatology at the University of Miami Department of Dermatology and Cutaneous Surgery.
Recently Approved Neurotoxin
He also discussed letibotulinumtoxinA-wlbg (Letybo), an injectable neurotoxin long used in South Korea, which the US Food and Drug Administration (FDA) approved for the temporary improvement in the appearance of moderate to severe glabellar (frown) lines in adults on March 4, 2024. Approval was based on positive results from three phase 3 trials of letibotulinumtoxinA-wlbg that enrolled more than 1,000 individuals in the United States and Europe.
“This is the sixth approved neurotoxin in the US,” Dr. Green said. “It is derived from the CBFC26 strain of Clostridium botulinum, and it’s a purified 900 kDa type A toxin complex with human serum albumin and sodium chloride as its excipients.” It comes in a 50-unit or 100-unit vial and requires refrigeration. “To me, the most fascinating thing about this product is that it has been the number-one selling botulinum toxin on the South Korea market for the last 5 years,” he said. “But what do we know about its characteristics?”
In a non-inferiority trial, Chinese researchers enrolled 500 patients with moderate to severe glabellar wrinkles to investigate the efficacy and safety of letibotulinumtoxinA-wlbg and onabotulinumtoxinA. Participants were randomized 3:1 to receive 20 U of letibotulinumtoxinA-wlbg or onabotulinumtoxinA and then observed them for 16 weeks. The primary endpoint was noninferiority in the proportion of study participants who received a score of 0 or 1 for glabellar wrinkles on a four-point photographic evaluation scale, as assessed by an evaluator at maximum frown at 4 weeks.
At week 4, 88.49% of participants in the letibotulinumtoxinA-wlbg arm achieved a score of 0 or 1 for glabellar wrinkles, compared with 87.39% of those in the onabotulinumtoxinA arm (P = .7469). No significant differences were observed for secondary efficacy or safety endpoints between the two treatments. “It will be interesting to see how this product does when it’s available to us,” Dr. Green said.
Another potential newcomer is ready-to-use liquid botulinum neurotoxin. RelabotulinumtoxinA is a complex, protein-free, ready-to-use liquid botulinum toxin A designed to avoid the traditional requirement to reconstitute it from powder, according to Galderma. It features a saline phosphate buffer solution, so it contains no human or animal-derived excipients, Dr. Green pointed out, and it eliminates the variability, errors, and risks associated with reconstitution.
“There was a report in the neurology literature of botulinum toxin being reconstituted with sterile water for cervical dystonia,” he noted. “When this was injected, it was excruciatingly painful, because it created an osmotic gradient within the muscle. So, if we can take a step away from human error, that would be a good thing.”
To date, Dr. Green said, four phase 3 trials of relabotulinumtoxinA involving more than 1,900 patients have been conducted in the United States and Canada evaluating its use for glabellar frown lines and lateral canthal lines, “and the data is impressive,” he said. This product is still investigational, said Dr. Green, who has not had experience injecting it in the clinical trial program.
The idea of a rapid onset botulinum toxin is also emerging. TrenibotulinumtoxinE, which is being developed by Allergan, “is similar to a type A neurotoxin,” Dr. Green said. “It inhibits neuromuscular transmission via presynaptic vesicular protein synaptosomal-associated protein (SNAP)-25 but at a different cleavage site. It has a faster onset — within one day — but a shorter duration — 3-4 weeks.”
In a dose escalation study of its use for glabellar frown lines, 80% of participants achieved a two-grade investigator-rated improvement in glabellar frown line severity at maximum frown at the highest dose. The maximum clinical effect of trenibotulinumtoxinE was seen within 24 hours and lasted between 14 and 30 days.
“The question is, if it is approved by the FDA, where would this product fit in our practices?” Dr. Green asked. “The effect is gone in 3 weeks as opposed to 4 months,” so this may be an option to recommend for someone who is reticent to try neurotoxins, he said, “or a patient who comes to you on a Friday and says, ‘I have a gala tomorrow night.’ ”
Dr. Green disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for many pharmaceutical companies, including Allergan and Galderma.
FROM AAD 2024
Analysis Finds Low Malignancy Rate in Pediatric Longitudinal Melanonychia
TOPLINE:
METHODOLOGY:
- LM — a pigmented band in the nail plate caused by increased melanin deposition — occurs in children and adults, resulting from melanocytic activation or proliferation in response to infection, systemic disease, medication, trauma, and other factors.
- Clinical features of LM in children mimic red-flag signs of subungual melanoma in adults although rarely is subungual melanoma.
- A biopsy can confirm the diagnosis, but other considerations include the scar, cost and stress of a procedure, and possibly pain or deformity.
- The researchers conducted a systematic review and meta-analysis of the prevalence of clinical and dermoscopic features in 1391 pediatric patients with LM (diagnosed at a mean age of 5-13 years) from 24 studies published between 1996 and 2023.
TAKEAWAY:
- Of 731 lesions in which a diagnosis was provided, benign nail matrix nevus accounted for 86% of cases.
- Only eight cases of subungual melanoma in situ were diagnosed, with no cases of invasive melanoma identified.
- Most lesions occurred on the fingernails (76%), particularly in the first digits (45%), and the most frequent clinical features included dark-colored bands (70%), multicolored bands (48%), broad bandwidth (41%), and pseudo-Hutchinson sign (41%).
- During a median follow-up of 1-5.5 years, 30% of lesions continued to evolve with changes in width or color, while 23% remained stable and 20% underwent spontaneous regression.
IN PRACTICE:
“In the pivotal clinical decision of whether to biopsy a child with longitudinal melanonychia, perhaps with features that would require a prompt biopsy in an adult, this study provides data to support the option of clinical monitoring,” the authors wrote.
SOURCE:
The meta-analysis, led by Serena Yun-Chen Tsai, MD, in the Department of Dermatology, Massachusetts General Hospital, Boston, Massachusetts, was published online in Pediatric Dermatology.
LIMITATIONS:
Most studies were conducted in Asia, and data stratified by skin type were limited. Inconsistent reporting and missing critical features could affect data quality. Also, certain features displayed high heterogeneity.
DISCLOSURES:
This meta-analysis was supported by the Pediatric Dermatology Research Alliance Career Bridge Research Grant. One co-author disclosed relationships with UpToDate (author, reviewer), Skin Analytics (consultant), and DermTech (research materials).
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- LM — a pigmented band in the nail plate caused by increased melanin deposition — occurs in children and adults, resulting from melanocytic activation or proliferation in response to infection, systemic disease, medication, trauma, and other factors.
- Clinical features of LM in children mimic red-flag signs of subungual melanoma in adults although rarely is subungual melanoma.
- A biopsy can confirm the diagnosis, but other considerations include the scar, cost and stress of a procedure, and possibly pain or deformity.
- The researchers conducted a systematic review and meta-analysis of the prevalence of clinical and dermoscopic features in 1391 pediatric patients with LM (diagnosed at a mean age of 5-13 years) from 24 studies published between 1996 and 2023.
TAKEAWAY:
- Of 731 lesions in which a diagnosis was provided, benign nail matrix nevus accounted for 86% of cases.
- Only eight cases of subungual melanoma in situ were diagnosed, with no cases of invasive melanoma identified.
- Most lesions occurred on the fingernails (76%), particularly in the first digits (45%), and the most frequent clinical features included dark-colored bands (70%), multicolored bands (48%), broad bandwidth (41%), and pseudo-Hutchinson sign (41%).
- During a median follow-up of 1-5.5 years, 30% of lesions continued to evolve with changes in width or color, while 23% remained stable and 20% underwent spontaneous regression.
IN PRACTICE:
“In the pivotal clinical decision of whether to biopsy a child with longitudinal melanonychia, perhaps with features that would require a prompt biopsy in an adult, this study provides data to support the option of clinical monitoring,” the authors wrote.
SOURCE:
The meta-analysis, led by Serena Yun-Chen Tsai, MD, in the Department of Dermatology, Massachusetts General Hospital, Boston, Massachusetts, was published online in Pediatric Dermatology.
LIMITATIONS:
Most studies were conducted in Asia, and data stratified by skin type were limited. Inconsistent reporting and missing critical features could affect data quality. Also, certain features displayed high heterogeneity.
DISCLOSURES:
This meta-analysis was supported by the Pediatric Dermatology Research Alliance Career Bridge Research Grant. One co-author disclosed relationships with UpToDate (author, reviewer), Skin Analytics (consultant), and DermTech (research materials).
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- LM — a pigmented band in the nail plate caused by increased melanin deposition — occurs in children and adults, resulting from melanocytic activation or proliferation in response to infection, systemic disease, medication, trauma, and other factors.
- Clinical features of LM in children mimic red-flag signs of subungual melanoma in adults although rarely is subungual melanoma.
- A biopsy can confirm the diagnosis, but other considerations include the scar, cost and stress of a procedure, and possibly pain or deformity.
- The researchers conducted a systematic review and meta-analysis of the prevalence of clinical and dermoscopic features in 1391 pediatric patients with LM (diagnosed at a mean age of 5-13 years) from 24 studies published between 1996 and 2023.
TAKEAWAY:
- Of 731 lesions in which a diagnosis was provided, benign nail matrix nevus accounted for 86% of cases.
- Only eight cases of subungual melanoma in situ were diagnosed, with no cases of invasive melanoma identified.
- Most lesions occurred on the fingernails (76%), particularly in the first digits (45%), and the most frequent clinical features included dark-colored bands (70%), multicolored bands (48%), broad bandwidth (41%), and pseudo-Hutchinson sign (41%).
- During a median follow-up of 1-5.5 years, 30% of lesions continued to evolve with changes in width or color, while 23% remained stable and 20% underwent spontaneous regression.
IN PRACTICE:
“In the pivotal clinical decision of whether to biopsy a child with longitudinal melanonychia, perhaps with features that would require a prompt biopsy in an adult, this study provides data to support the option of clinical monitoring,” the authors wrote.
SOURCE:
The meta-analysis, led by Serena Yun-Chen Tsai, MD, in the Department of Dermatology, Massachusetts General Hospital, Boston, Massachusetts, was published online in Pediatric Dermatology.
LIMITATIONS:
Most studies were conducted in Asia, and data stratified by skin type were limited. Inconsistent reporting and missing critical features could affect data quality. Also, certain features displayed high heterogeneity.
DISCLOSURES:
This meta-analysis was supported by the Pediatric Dermatology Research Alliance Career Bridge Research Grant. One co-author disclosed relationships with UpToDate (author, reviewer), Skin Analytics (consultant), and DermTech (research materials).
A version of this article appeared on Medscape.com.
Tooth Enamel Disorder Is a Feature of Kindler EB
TOPLINE:
METHODOLOGY:
- KEB or Kindler syndrome, a genetic skin-blistering disease associated with pathogenic variants in FERMT1, is the rarest type of EB. Early detection and preventive measures can minimize complications, such as gum disease and other oral health issues, that have been reported in patients with KEB.
- Amelogenesis imperfecta is a group of rare genetic developmental conditions characterized by tooth enamel defects and can be associated with hypersensitivity and eruption disturbances in teeth, as well as periodontal conditions.
- Researchers conducted a longitudinal study on 36 patients with KEB (age, 2 weeks to 70 years; 42% female) from two clinics in Germany and Chile from 2003 to 2023, with follow-up times of 1-24 years.
- The primary outcomes were presence of orofacial features, including amelogenesis imperfecta, intraoral wounds, and periodontal disease, and oral squamous cell carcinoma.
TAKEAWAY:
- All 11 patients with information on enamel structure in their records had pitted enamel anomalies (pitted amelogenesis imperfecta), with variable severity.
- Of patients whose enamel could not be analyzed, three had all teeth crowned in their 20s, suggesting enamel defects, and two had all teeth extracted in their teens or 20s, indicating severe periodontal disease.
- The most common orofacial features were periodontal disease (27 of 36 patients), intraoral lesions (16 of 22 patients), angular cheilitis (24 of 33 patients), and cheilitis (22 of 34 patients), gingival overgrowth (17 of 26 patients), microstomia (14 of 25 patients), and vestibular obliteration (8 of 16 patients).
- Oral squamous cell carcinoma was diagnosed at the site of chronic lip lesions in two patients, with lethal outcomes.
IN PRACTICE:
These findings highlight the extent and severity of oral manifestations in KEB, the authors concluded, adding that “oral care is mandatory” in patients with KEB.
SOURCE:
This report, led by Susanne Krämer, DDS, MSc, of Medical Faculty and Medical Center, University of Freiburg, Freiburg im Breisgau, Germany, was published online in JAMA Dermatology.
LIMITATIONS:
The small sample size and the retrospective nature of the study could limit its generalizability.
DISCLOSURES:
The authors did not disclose any source of funding. The authors declared no conflicts of interest.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- KEB or Kindler syndrome, a genetic skin-blistering disease associated with pathogenic variants in FERMT1, is the rarest type of EB. Early detection and preventive measures can minimize complications, such as gum disease and other oral health issues, that have been reported in patients with KEB.
- Amelogenesis imperfecta is a group of rare genetic developmental conditions characterized by tooth enamel defects and can be associated with hypersensitivity and eruption disturbances in teeth, as well as periodontal conditions.
- Researchers conducted a longitudinal study on 36 patients with KEB (age, 2 weeks to 70 years; 42% female) from two clinics in Germany and Chile from 2003 to 2023, with follow-up times of 1-24 years.
- The primary outcomes were presence of orofacial features, including amelogenesis imperfecta, intraoral wounds, and periodontal disease, and oral squamous cell carcinoma.
TAKEAWAY:
- All 11 patients with information on enamel structure in their records had pitted enamel anomalies (pitted amelogenesis imperfecta), with variable severity.
- Of patients whose enamel could not be analyzed, three had all teeth crowned in their 20s, suggesting enamel defects, and two had all teeth extracted in their teens or 20s, indicating severe periodontal disease.
- The most common orofacial features were periodontal disease (27 of 36 patients), intraoral lesions (16 of 22 patients), angular cheilitis (24 of 33 patients), and cheilitis (22 of 34 patients), gingival overgrowth (17 of 26 patients), microstomia (14 of 25 patients), and vestibular obliteration (8 of 16 patients).
- Oral squamous cell carcinoma was diagnosed at the site of chronic lip lesions in two patients, with lethal outcomes.
IN PRACTICE:
These findings highlight the extent and severity of oral manifestations in KEB, the authors concluded, adding that “oral care is mandatory” in patients with KEB.
SOURCE:
This report, led by Susanne Krämer, DDS, MSc, of Medical Faculty and Medical Center, University of Freiburg, Freiburg im Breisgau, Germany, was published online in JAMA Dermatology.
LIMITATIONS:
The small sample size and the retrospective nature of the study could limit its generalizability.
DISCLOSURES:
The authors did not disclose any source of funding. The authors declared no conflicts of interest.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- KEB or Kindler syndrome, a genetic skin-blistering disease associated with pathogenic variants in FERMT1, is the rarest type of EB. Early detection and preventive measures can minimize complications, such as gum disease and other oral health issues, that have been reported in patients with KEB.
- Amelogenesis imperfecta is a group of rare genetic developmental conditions characterized by tooth enamel defects and can be associated with hypersensitivity and eruption disturbances in teeth, as well as periodontal conditions.
- Researchers conducted a longitudinal study on 36 patients with KEB (age, 2 weeks to 70 years; 42% female) from two clinics in Germany and Chile from 2003 to 2023, with follow-up times of 1-24 years.
- The primary outcomes were presence of orofacial features, including amelogenesis imperfecta, intraoral wounds, and periodontal disease, and oral squamous cell carcinoma.
TAKEAWAY:
- All 11 patients with information on enamel structure in their records had pitted enamel anomalies (pitted amelogenesis imperfecta), with variable severity.
- Of patients whose enamel could not be analyzed, three had all teeth crowned in their 20s, suggesting enamel defects, and two had all teeth extracted in their teens or 20s, indicating severe periodontal disease.
- The most common orofacial features were periodontal disease (27 of 36 patients), intraoral lesions (16 of 22 patients), angular cheilitis (24 of 33 patients), and cheilitis (22 of 34 patients), gingival overgrowth (17 of 26 patients), microstomia (14 of 25 patients), and vestibular obliteration (8 of 16 patients).
- Oral squamous cell carcinoma was diagnosed at the site of chronic lip lesions in two patients, with lethal outcomes.
IN PRACTICE:
These findings highlight the extent and severity of oral manifestations in KEB, the authors concluded, adding that “oral care is mandatory” in patients with KEB.
SOURCE:
This report, led by Susanne Krämer, DDS, MSc, of Medical Faculty and Medical Center, University of Freiburg, Freiburg im Breisgau, Germany, was published online in JAMA Dermatology.
LIMITATIONS:
The small sample size and the retrospective nature of the study could limit its generalizability.
DISCLOSURES:
The authors did not disclose any source of funding. The authors declared no conflicts of interest.
A version of this article appeared on Medscape.com.
JAK Inhibitors for Vitiligo: Response Continues Over Time
SAN DIEGO — according to presentations at a late-breaking session at the annual meeting of the American Academy of Dermatology (AAD).
In one, the addition of narrow-band ultraviolet-B (NB-UVB) light therapy to ritlecitinib appears more effective than ritlecitinib alone. In the other study, the effectiveness of upadacitinib appears to improve over time.
Based on the ritlecitinib data, “if you have phototherapy in your office, it might be good to couple it with ritlecitinib for vitiligo patients,” said Emma Guttman-Yassky, MD, PhD, chair of the Department of Dermatology, Icahn School of Medicine at Mount Sinai, New York City, who presented the findings.
However, because of the relatively small numbers in the extension study, Dr. Guttman-Yassky characterized the evidence as preliminary and in need of further investigation.
For vitiligo, the only approved JAK inhibitor is ruxolitinib, 1.5%, in a cream formulation. In June, ritlecitinib (Litfulo) was approved by the Food and Drug Administration (FDA) for alopecia areata. Phototherapy, which has been used for decades in the treatment of vitiligo, has an established efficacy and safety profile as a stand-alone vitiligo treatment. Upadacitinib has numerous indications for inflammatory diseases, such as rheumatoid arthritis, and was granted FDA approval for atopic dermatitis in 2022.
NB-UVB Arm Added in Ritlecitinib Extension
The ritlecitinib study population was drawn from patients with non-segmental vitiligo who initially participated in a 24-week dose-ranging period of a phase 2b trial published last year. In that study, 364 patients were randomized to doses of once-daily ritlecitinib ranging from 10 to 50 mg with or without a 4-week loading regimen. Higher doses were generally associated with greater efficacy on the primary endpoint of facial vitiligo area scoring index (F-VASI) but not with a greater risk for adverse events.
In the 24-week extension study, 187 patients received a 4-week loading regimen of 200-mg ritlecitinib daily followed by 50 mg of daily ritlecitinib for the remaining 20 weeks. Another 43 patients were randomized to one of two arms: The same 4-week loading regimen of 200-mg ritlecitinib daily followed by 50 mg of daily ritlecitinib or to 50-mg daily ritlecitinib without a loading dose but combined with NB-UVB delivered twice per week.
Important to interpretation of results, there was an additional twist. Patients in the randomized arm who had < 10% improvement in the total vitiligo area severity index (T-VASI) at week 12 of the extension were discontinued from the study.
The endpoints considered when comparing ritlecitinib with or without NB-UVB at the end of the extension study were F-VASI, T-VASI, patient global impression of change, and adverse events. Responses were assessed on the basis of both observed and last observation carried forward (LOCF).
Of the 43 people, who were randomized in the extension study, nine (21%) had < 10% improvement in T-VASI and were therefore discontinued from the study.
At the end of 24 weeks, both groups had a substantial response to their assigned therapy, but the addition of NB-UVB increased rates of response, although not always at a level of statistical significance, according to Dr. Guttman-Yassky.
For the percent improvement in F-VASI, specifically, the increase did not reach significance on the basis of LOCF (57.9% vs 51.5%; P = .158) but was highly significant on the basis of observed responses (69.6% vs 55.1%; P = .009). For T-VASI, differences for adjunctive NB-UVB over monotherapy did not reach significance for either observed or LOCF responses, but it was significant for observed responses in a patient global impression of change.
Small Numbers Limit Strength of Ritlecitinib, NB-UVB Evidence
However, Dr. Guttman-Yassky said it is important “to pay attention to the sample sizes” when noting the lack of significance.
The combination appeared safe, and there were no side effects associated with the addition of twice-weekly NB-UVB to ritlecitinib.
She acknowledged that the design of this analysis was “complicated” and that the number of randomized patients was small. She suggested the findings support the potential for benefit from the combination of a JAK inhibitor and NB-UVB, both of which have shown efficacy as monotherapy in previous studies. She indicated that a trial of this combination is reasonable while awaiting a more definitive study.
One of the questions that might be posed in a larger study is the timing of NB-UVB, such as whether it is best reserved for those with inadequate early response to a JAK inhibitor or if optimal results are achieved when a JAK inhibitor and NB-UVB are initiated simultaneously.
Upadacitinib Monotherapy Results
One rationale for initiating therapy with the combination of a JAK inhibitor and NB-UVB is the potential for a more rapid response, but extended results from a second phase 2b study with a different oral JAK inhibitor, upadacitinib, suggested responses on JAK inhibitor monotherapy improve steadily over time.
“The overall efficacy continued to improve without reaching a plateau at 1 year,” reported Thierry Passeron, MD, PhD, professor and chair, Department of Dermatology, Université Côte d’Azur, Nice, France. He spoke at the same AAD late-breaking session as Dr. Guttman-Yassky.
The 24-week dose-ranging data from the upadacitinib trial were previously reported at the 2023 annual meeting of the European Association of Dermatology and Venereology. In the placebo-controlled portion, which randomized 185 patients with extensive non-segmental vitiligo to 6 mg, 11 mg, or 22 mg, the two higher doses were significantly more effective than placebo.
In the extension, patients in the placebo group were randomized to 11 mg or 22 mg, while those in the higher dose groups remained on their assigned therapies.
F-VASI Almost Doubled in Extension Trial
From week 24 to week 52, there was nearly a doubling of the percent F-VASI reduction, climbing from 32% to 60.8% in the 11-mg group and from 38.7% to 64.9% in the 22-mg group, Dr. Passeron said. Placebo groups who were switched to active therapy at 24 weeks rapidly approached the rates of F-VASI response of those initiated on upadacitinib.
The percent reductions in T-VASI, although lower, followed the same pattern. For the 11-mg group, the reduction climbed from 16% at 24 weeks to 44.7% at 52 weeks. For the 22-mg group, the reduction climbed from 22.9% to 44.4%. Patients who were switched from placebo to 11 mg or to 22 mg also experienced improvements in T-VASI up to 52 weeks, although the level of improvement was lower than that in patients initially randomized to the higher doses of upadacitinib.
There were “no new safety signals” for upadacitinib, which is FDA-approved for multiple indications, according to Dr. Passeron. He said acne-like lesions were the most bothersome adverse event, and cases of herpes zoster were “rare.”
A version of these data was published in a British Journal of Dermatology supplement just prior to the AAD meeting.
Phase 3 vitiligo trials are planned for both ritlecitinib and upadacitinib.
Dr. Guttman-Yassky reported financial relationships with approximately 45 pharmaceutical companies, including Pfizer, which makes ritlecitinib and provided funding for the study she discussed. Dr. Passeron reported financial relationships with approximately 40 pharmaceutical companies, including AbbVie, which makes upadacitinib and provided funding for the study he discussed.
A version of this article appeared on Medscape.com.
SAN DIEGO — according to presentations at a late-breaking session at the annual meeting of the American Academy of Dermatology (AAD).
In one, the addition of narrow-band ultraviolet-B (NB-UVB) light therapy to ritlecitinib appears more effective than ritlecitinib alone. In the other study, the effectiveness of upadacitinib appears to improve over time.
Based on the ritlecitinib data, “if you have phototherapy in your office, it might be good to couple it with ritlecitinib for vitiligo patients,” said Emma Guttman-Yassky, MD, PhD, chair of the Department of Dermatology, Icahn School of Medicine at Mount Sinai, New York City, who presented the findings.
However, because of the relatively small numbers in the extension study, Dr. Guttman-Yassky characterized the evidence as preliminary and in need of further investigation.
For vitiligo, the only approved JAK inhibitor is ruxolitinib, 1.5%, in a cream formulation. In June, ritlecitinib (Litfulo) was approved by the Food and Drug Administration (FDA) for alopecia areata. Phototherapy, which has been used for decades in the treatment of vitiligo, has an established efficacy and safety profile as a stand-alone vitiligo treatment. Upadacitinib has numerous indications for inflammatory diseases, such as rheumatoid arthritis, and was granted FDA approval for atopic dermatitis in 2022.
NB-UVB Arm Added in Ritlecitinib Extension
The ritlecitinib study population was drawn from patients with non-segmental vitiligo who initially participated in a 24-week dose-ranging period of a phase 2b trial published last year. In that study, 364 patients were randomized to doses of once-daily ritlecitinib ranging from 10 to 50 mg with or without a 4-week loading regimen. Higher doses were generally associated with greater efficacy on the primary endpoint of facial vitiligo area scoring index (F-VASI) but not with a greater risk for adverse events.
In the 24-week extension study, 187 patients received a 4-week loading regimen of 200-mg ritlecitinib daily followed by 50 mg of daily ritlecitinib for the remaining 20 weeks. Another 43 patients were randomized to one of two arms: The same 4-week loading regimen of 200-mg ritlecitinib daily followed by 50 mg of daily ritlecitinib or to 50-mg daily ritlecitinib without a loading dose but combined with NB-UVB delivered twice per week.
Important to interpretation of results, there was an additional twist. Patients in the randomized arm who had < 10% improvement in the total vitiligo area severity index (T-VASI) at week 12 of the extension were discontinued from the study.
The endpoints considered when comparing ritlecitinib with or without NB-UVB at the end of the extension study were F-VASI, T-VASI, patient global impression of change, and adverse events. Responses were assessed on the basis of both observed and last observation carried forward (LOCF).
Of the 43 people, who were randomized in the extension study, nine (21%) had < 10% improvement in T-VASI and were therefore discontinued from the study.
At the end of 24 weeks, both groups had a substantial response to their assigned therapy, but the addition of NB-UVB increased rates of response, although not always at a level of statistical significance, according to Dr. Guttman-Yassky.
For the percent improvement in F-VASI, specifically, the increase did not reach significance on the basis of LOCF (57.9% vs 51.5%; P = .158) but was highly significant on the basis of observed responses (69.6% vs 55.1%; P = .009). For T-VASI, differences for adjunctive NB-UVB over monotherapy did not reach significance for either observed or LOCF responses, but it was significant for observed responses in a patient global impression of change.
Small Numbers Limit Strength of Ritlecitinib, NB-UVB Evidence
However, Dr. Guttman-Yassky said it is important “to pay attention to the sample sizes” when noting the lack of significance.
The combination appeared safe, and there were no side effects associated with the addition of twice-weekly NB-UVB to ritlecitinib.
She acknowledged that the design of this analysis was “complicated” and that the number of randomized patients was small. She suggested the findings support the potential for benefit from the combination of a JAK inhibitor and NB-UVB, both of which have shown efficacy as monotherapy in previous studies. She indicated that a trial of this combination is reasonable while awaiting a more definitive study.
One of the questions that might be posed in a larger study is the timing of NB-UVB, such as whether it is best reserved for those with inadequate early response to a JAK inhibitor or if optimal results are achieved when a JAK inhibitor and NB-UVB are initiated simultaneously.
Upadacitinib Monotherapy Results
One rationale for initiating therapy with the combination of a JAK inhibitor and NB-UVB is the potential for a more rapid response, but extended results from a second phase 2b study with a different oral JAK inhibitor, upadacitinib, suggested responses on JAK inhibitor monotherapy improve steadily over time.
“The overall efficacy continued to improve without reaching a plateau at 1 year,” reported Thierry Passeron, MD, PhD, professor and chair, Department of Dermatology, Université Côte d’Azur, Nice, France. He spoke at the same AAD late-breaking session as Dr. Guttman-Yassky.
The 24-week dose-ranging data from the upadacitinib trial were previously reported at the 2023 annual meeting of the European Association of Dermatology and Venereology. In the placebo-controlled portion, which randomized 185 patients with extensive non-segmental vitiligo to 6 mg, 11 mg, or 22 mg, the two higher doses were significantly more effective than placebo.
In the extension, patients in the placebo group were randomized to 11 mg or 22 mg, while those in the higher dose groups remained on their assigned therapies.
F-VASI Almost Doubled in Extension Trial
From week 24 to week 52, there was nearly a doubling of the percent F-VASI reduction, climbing from 32% to 60.8% in the 11-mg group and from 38.7% to 64.9% in the 22-mg group, Dr. Passeron said. Placebo groups who were switched to active therapy at 24 weeks rapidly approached the rates of F-VASI response of those initiated on upadacitinib.
The percent reductions in T-VASI, although lower, followed the same pattern. For the 11-mg group, the reduction climbed from 16% at 24 weeks to 44.7% at 52 weeks. For the 22-mg group, the reduction climbed from 22.9% to 44.4%. Patients who were switched from placebo to 11 mg or to 22 mg also experienced improvements in T-VASI up to 52 weeks, although the level of improvement was lower than that in patients initially randomized to the higher doses of upadacitinib.
There were “no new safety signals” for upadacitinib, which is FDA-approved for multiple indications, according to Dr. Passeron. He said acne-like lesions were the most bothersome adverse event, and cases of herpes zoster were “rare.”
A version of these data was published in a British Journal of Dermatology supplement just prior to the AAD meeting.
Phase 3 vitiligo trials are planned for both ritlecitinib and upadacitinib.
Dr. Guttman-Yassky reported financial relationships with approximately 45 pharmaceutical companies, including Pfizer, which makes ritlecitinib and provided funding for the study she discussed. Dr. Passeron reported financial relationships with approximately 40 pharmaceutical companies, including AbbVie, which makes upadacitinib and provided funding for the study he discussed.
A version of this article appeared on Medscape.com.
SAN DIEGO — according to presentations at a late-breaking session at the annual meeting of the American Academy of Dermatology (AAD).
In one, the addition of narrow-band ultraviolet-B (NB-UVB) light therapy to ritlecitinib appears more effective than ritlecitinib alone. In the other study, the effectiveness of upadacitinib appears to improve over time.
Based on the ritlecitinib data, “if you have phototherapy in your office, it might be good to couple it with ritlecitinib for vitiligo patients,” said Emma Guttman-Yassky, MD, PhD, chair of the Department of Dermatology, Icahn School of Medicine at Mount Sinai, New York City, who presented the findings.
However, because of the relatively small numbers in the extension study, Dr. Guttman-Yassky characterized the evidence as preliminary and in need of further investigation.
For vitiligo, the only approved JAK inhibitor is ruxolitinib, 1.5%, in a cream formulation. In June, ritlecitinib (Litfulo) was approved by the Food and Drug Administration (FDA) for alopecia areata. Phototherapy, which has been used for decades in the treatment of vitiligo, has an established efficacy and safety profile as a stand-alone vitiligo treatment. Upadacitinib has numerous indications for inflammatory diseases, such as rheumatoid arthritis, and was granted FDA approval for atopic dermatitis in 2022.
NB-UVB Arm Added in Ritlecitinib Extension
The ritlecitinib study population was drawn from patients with non-segmental vitiligo who initially participated in a 24-week dose-ranging period of a phase 2b trial published last year. In that study, 364 patients were randomized to doses of once-daily ritlecitinib ranging from 10 to 50 mg with or without a 4-week loading regimen. Higher doses were generally associated with greater efficacy on the primary endpoint of facial vitiligo area scoring index (F-VASI) but not with a greater risk for adverse events.
In the 24-week extension study, 187 patients received a 4-week loading regimen of 200-mg ritlecitinib daily followed by 50 mg of daily ritlecitinib for the remaining 20 weeks. Another 43 patients were randomized to one of two arms: The same 4-week loading regimen of 200-mg ritlecitinib daily followed by 50 mg of daily ritlecitinib or to 50-mg daily ritlecitinib without a loading dose but combined with NB-UVB delivered twice per week.
Important to interpretation of results, there was an additional twist. Patients in the randomized arm who had < 10% improvement in the total vitiligo area severity index (T-VASI) at week 12 of the extension were discontinued from the study.
The endpoints considered when comparing ritlecitinib with or without NB-UVB at the end of the extension study were F-VASI, T-VASI, patient global impression of change, and adverse events. Responses were assessed on the basis of both observed and last observation carried forward (LOCF).
Of the 43 people, who were randomized in the extension study, nine (21%) had < 10% improvement in T-VASI and were therefore discontinued from the study.
At the end of 24 weeks, both groups had a substantial response to their assigned therapy, but the addition of NB-UVB increased rates of response, although not always at a level of statistical significance, according to Dr. Guttman-Yassky.
For the percent improvement in F-VASI, specifically, the increase did not reach significance on the basis of LOCF (57.9% vs 51.5%; P = .158) but was highly significant on the basis of observed responses (69.6% vs 55.1%; P = .009). For T-VASI, differences for adjunctive NB-UVB over monotherapy did not reach significance for either observed or LOCF responses, but it was significant for observed responses in a patient global impression of change.
Small Numbers Limit Strength of Ritlecitinib, NB-UVB Evidence
However, Dr. Guttman-Yassky said it is important “to pay attention to the sample sizes” when noting the lack of significance.
The combination appeared safe, and there were no side effects associated with the addition of twice-weekly NB-UVB to ritlecitinib.
She acknowledged that the design of this analysis was “complicated” and that the number of randomized patients was small. She suggested the findings support the potential for benefit from the combination of a JAK inhibitor and NB-UVB, both of which have shown efficacy as monotherapy in previous studies. She indicated that a trial of this combination is reasonable while awaiting a more definitive study.
One of the questions that might be posed in a larger study is the timing of NB-UVB, such as whether it is best reserved for those with inadequate early response to a JAK inhibitor or if optimal results are achieved when a JAK inhibitor and NB-UVB are initiated simultaneously.
Upadacitinib Monotherapy Results
One rationale for initiating therapy with the combination of a JAK inhibitor and NB-UVB is the potential for a more rapid response, but extended results from a second phase 2b study with a different oral JAK inhibitor, upadacitinib, suggested responses on JAK inhibitor monotherapy improve steadily over time.
“The overall efficacy continued to improve without reaching a plateau at 1 year,” reported Thierry Passeron, MD, PhD, professor and chair, Department of Dermatology, Université Côte d’Azur, Nice, France. He spoke at the same AAD late-breaking session as Dr. Guttman-Yassky.
The 24-week dose-ranging data from the upadacitinib trial were previously reported at the 2023 annual meeting of the European Association of Dermatology and Venereology. In the placebo-controlled portion, which randomized 185 patients with extensive non-segmental vitiligo to 6 mg, 11 mg, or 22 mg, the two higher doses were significantly more effective than placebo.
In the extension, patients in the placebo group were randomized to 11 mg or 22 mg, while those in the higher dose groups remained on their assigned therapies.
F-VASI Almost Doubled in Extension Trial
From week 24 to week 52, there was nearly a doubling of the percent F-VASI reduction, climbing from 32% to 60.8% in the 11-mg group and from 38.7% to 64.9% in the 22-mg group, Dr. Passeron said. Placebo groups who were switched to active therapy at 24 weeks rapidly approached the rates of F-VASI response of those initiated on upadacitinib.
The percent reductions in T-VASI, although lower, followed the same pattern. For the 11-mg group, the reduction climbed from 16% at 24 weeks to 44.7% at 52 weeks. For the 22-mg group, the reduction climbed from 22.9% to 44.4%. Patients who were switched from placebo to 11 mg or to 22 mg also experienced improvements in T-VASI up to 52 weeks, although the level of improvement was lower than that in patients initially randomized to the higher doses of upadacitinib.
There were “no new safety signals” for upadacitinib, which is FDA-approved for multiple indications, according to Dr. Passeron. He said acne-like lesions were the most bothersome adverse event, and cases of herpes zoster were “rare.”
A version of these data was published in a British Journal of Dermatology supplement just prior to the AAD meeting.
Phase 3 vitiligo trials are planned for both ritlecitinib and upadacitinib.
Dr. Guttman-Yassky reported financial relationships with approximately 45 pharmaceutical companies, including Pfizer, which makes ritlecitinib and provided funding for the study she discussed. Dr. Passeron reported financial relationships with approximately 40 pharmaceutical companies, including AbbVie, which makes upadacitinib and provided funding for the study he discussed.
A version of this article appeared on Medscape.com.
FROM AAD 2024
Tender Dermal Nodule on the Temple
The Diagnosis: Lymphoepithelioma-like Carcinoma
Lymphoepithelioma-like carcinoma (LELC) is a rare, poorly differentiated, primary cutaneous neoplasm that occurs on sun-exposed skin, particularly on the head and neck of elderly individuals. It often manifests as an asymptomatic, slow-growing, flesh-colored or erythematous dermal nodule, though ulceration and tenderness have been reported.1 Histopathologically, these neoplasms often are poorly circumscribed and can infiltrate surrounding subcutaneous and soft tissue. As a biphasic tumor, LELC is characterized by islands, nests, or trabeculae of epithelioid cells within the mid dermis surrounded by a dense lymphocytic infiltrate with plasma cells (Figure 1).1 The epithelial component rarely communicates with the overlying epidermis and is composed of atypical polygonal cells with eosinophilic cytoplasm, vesicular nuclei, prominent nucleoli, and frequent mitosis.2 These epithelial nests can be highlighted by pancytokeratin AE1/AE3 or other epithelial differentiation markers (eg, CAM 5.2, CK5/6, epithelial membrane antigen, high-molecular-weight cytokeratin), while the surrounding lymphocytic infiltrate consists of an admixture of T cells and B cells. Lymphoepithelioma-like carcinomas also can demonstrate sebaceous, eccrine, or follicular differentiations.3 The epithelial nests of LELC also are positive for p63 and epithelial membrane antigen.2
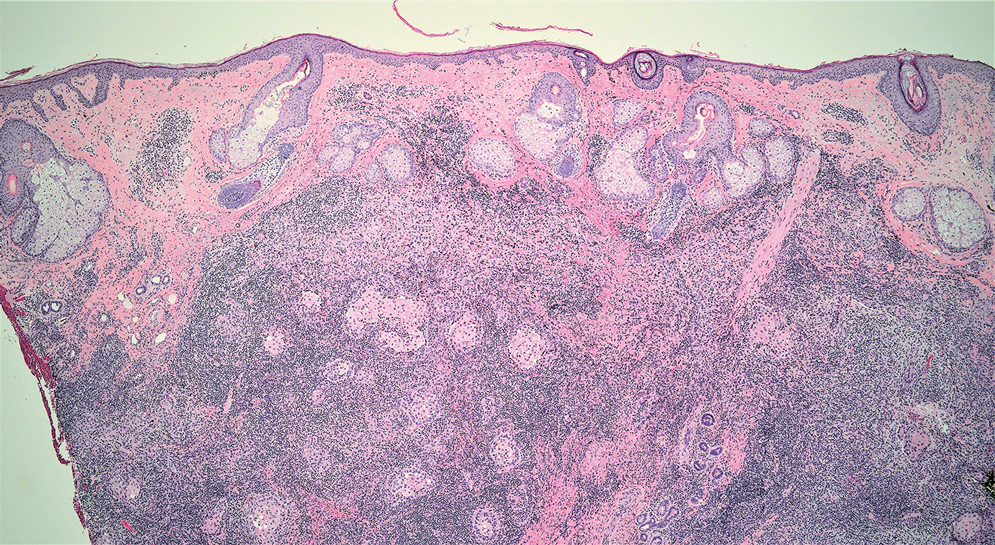
The usual treatment of LELC is wide local excision or Mohs micrographic surgery.1 Despite the poorly differentiated morphology of the tumor, LELC has a generally good prognosis with low metastatic potential and few reports of local recurrence after incomplete excision.3 Patients who are not candidates for surgery as well as recalcitrant cases are managed with radiotherapy.1
Cutaneous lymphadenoma (CL) is a benign adnexal neoplasm that manifests as a small, solitary, fleshcolored nodule usually in the head and neck region.4 Histologically, CL consists of well-circumscribed epithelial nests within the dermis that are peripherally outlined by palisading basaloid cells and filled with clear to eosinophilic epithelioid cells (Figure 2).5 The fibrotic tumor stroma often is infiltrated by numerous intralobular dendritic cells and lymphocytes that occasionally can be arranged in germinal center–like nodules.4 The lymphoepithelial nature of CL can be challenging to distinguish morphologically from LELC, and immunohistochemistry stains may be required. In CL, both the basaloid and epithelioid cells stain positive for pancytokeratin AE1/ AE3, but the peripheral palisaded basaloid cells also stain positive for BerEP4. Additionally, the fibrotic stroma can be highlighted by CD34 and the intralobular dendritic cells by S-100.4
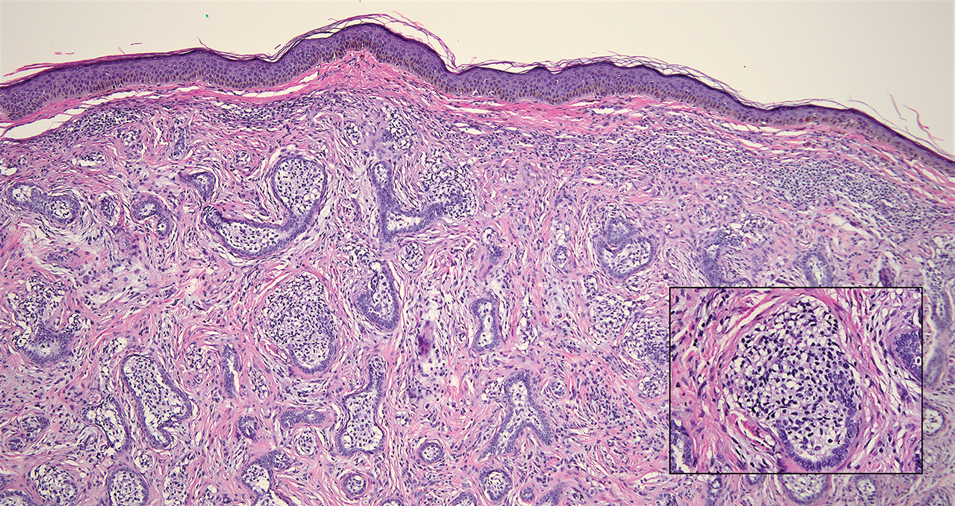
Nasopharyngeal carcinoma (NPC), formerly known as lymphoepithelioma, refers to carcinoma arising within the epithelium of the nasopharynx.6 Endemic to China, NPC manifests as an enlarging nasopharyngeal mass, causing clinical symptoms such as nasal obstruction and epistaxis.7 Histologically, nonkeratinizing NPC exhibits a biphasic morphology consisting of epithelioid neoplastic cells and background lymphocytic infiltrates (Figure 3). The epithelial component consists of round to oval neoplastic cells with amphophilic to eosinophilic cytoplasm, vesicular nuclei, and prominent nucleoli.6 Nasopharyngeal carcinoma is associated strongly with the Epstein-Barr virus while LELC is not; thus, Epstein- Barr encoding region in situ hybridization can reliably distinguish these entities. Metastatic NPC is rare but has been reported; therefore, it is highly recommended to perform an otolaryngologic examination in addition to testing for Epstein-Barr virus reactivity as part of a complete evaluation.8
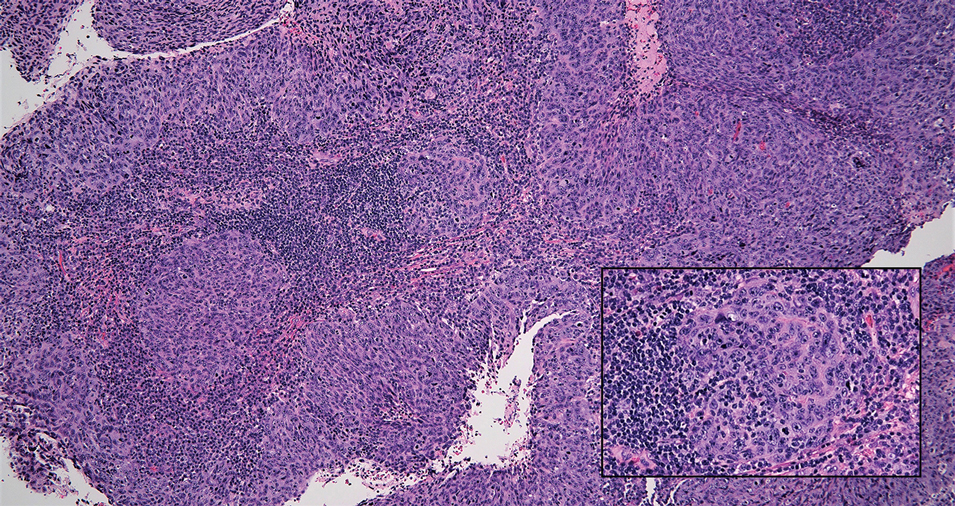
Cutaneous squamous cell carcinoma (SCC) is a common epidermal malignancy with multiple subtypes and variable morphology. The clinical presentation of SCC is similar to LELC—an enlarging hyperkeratotic papule or nodule on sun-exposed skin that often is ulcerated and tender.9 Histologically, poorly differentiated nonkeratinizing SCC can form nests and trabeculae of epithelioid cells that are stained by epithelial differentiation markers, resembling the epithelioid nests of LELC. Distinguishing between LELC and poorly differentiated SCC with robust inflammatory infiltrate can be challenging (Figure 4). In fact, some experts support LELC as an SCC variant rather than a separate entity.9 However, in contrast to LELC, the dermal nests of SCC usually maintain an epidermal connection and often are associated with an overlying area of SCC in situ or welldifferentiated SCC.3
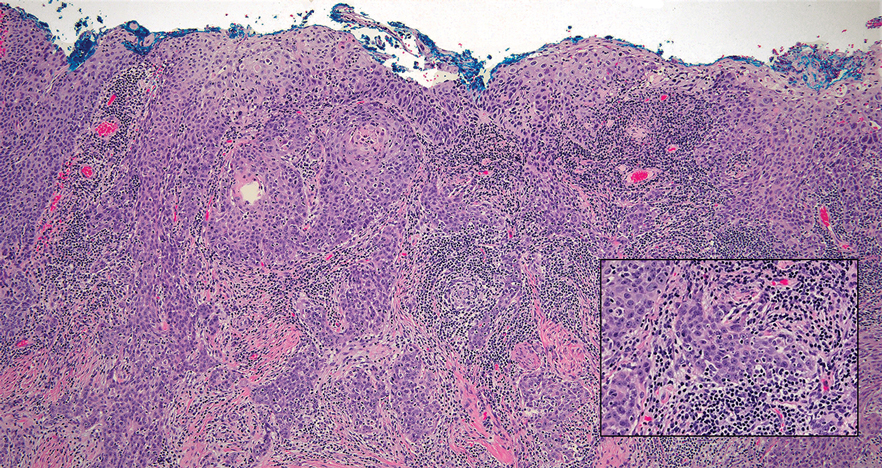
Mycosis fungoides (MF) is a primary cutaneous T-cell lymphoma. It is the most common type of cutaneous lymphoma, accounting for almost 50% of all reported cases.10 Classic MF has an indolent course and progresses through several clinical stages. Patches and plaques characterize early stages; lymphadenopathy indicates progression to later stages in which erythroderma may develop with coalescence of patches, plaques, and tumors; and MF present in blood or lymph nodes characterizes the late stage. Each stage of MF is different histologically—from a superficial lichenoid infiltrate with exocytosis of malignant T cells in the patch stage, to more robust epidermotropism and dermal infiltrate in the plaque stage, and finally a dense dermal infiltrate in the late stage.11 The rare syringotropic variant of MF clinically manifests as solitary or multiple erythematous lesions, often with overlying alopecia. Syringotropic MF uniquely exhibits folliculotropism and syringotropism along with syringometaplasia on histologic evaluation (Figure 5).12 The syringometaplasia can be difficult to distinguish from the epithelial nests of LELC, particularly with the lymphocytic background. Immunohistochemical panels for T-cell markers can highlight aberrant T cells in syringotropic MF through their usual loss of CD5 and CD7, in comparison to normal T cells in LELC.11 An elevated CD4:CD8 ratio of 4:1 and molecular analysis for T-cell receptor gene clonal rearrangements also can support the diagnosis of MF.12
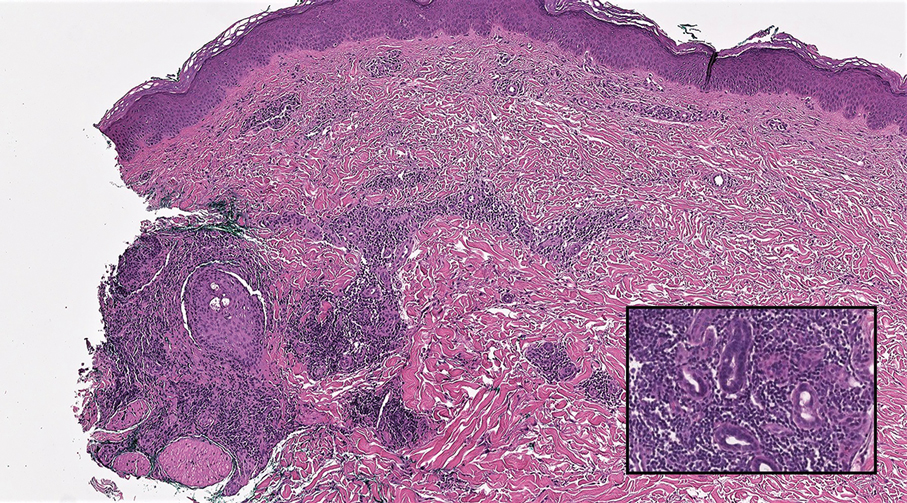
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Fisher JC, White RM, Hurd DS. Lymphoepithelioma-like carcinoma of the skin: a case of one patient presenting with two primary cutaneous neoplasms. J Am Osteopath Coll Dermatol. 2015;33:40-41.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Yu R, Salama S, Alowami S. Cutaneous lymphadenoma: a rare case and brief review of a diagnostic pitfall. Rare Tumors. 2014;6:5358.
- Monteagudo C, Fúnez R, Sánchez-Sendra B, et al. Cutaneous lymphadenoma is a distinct trichoblastoma-like lymphoepithelial tumor with diffuse androgen receptor immunoreactivity, Notch1 ligand in Reed-Sternberg-like Cells, and common EGFR somatic mutations. Am J Surg Pathol. 2021;45:1382-1390.
- Stelow EB, Wenig BM. Update from the 4th edition of the World Health Organization classification of head and neck tumours: nasopharynx. Head Neck Pathol. 2017;11:16-22.
- Almomani MH, Zulfiqar H, Nagalli S. Nasopharyngeal carcinoma (NPC, lymphoepithelioma). StatPearls Publishing; 2022.
- Lassen CB, Lock-Andersen J. Lymphoepithelioma-like carcinoma of the skin: a case with perineural invasion. Plast Reconstr Surg Glob Open. 2014;2:E252.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer Staging Guidelines, Prognostic Factors, and Histopathologic Variants. Adv Anat Pathol. 2017;24:171-194.
- Pileri A, Facchetti F, Rütten A, et al. Syringotropic mycosis fungoides: a rare variant of the disease with peculiar clinicopathologic features. Am J Surg Pathol. 2011;35:100-109.
- Ryu HJ, Kim SI, Jang HO, et al. Evaluation of the International Society for Cutaneous Lymphoma Algorithm for the Diagnosis of Early Mycosis Fungoides [published October 15, 2021]. Cells. 2021;10:2758. doi:10.3390/cells10102758
- Lehmer LM, Amber KT, de Feraudy SM. Syringotropic mycosis fungoides: a rare form of cutaneous T-cell lymphoma enabling a histopathologic “sigh of relief.” Am J Dermatopathol. 2017;39:920-923.
The Diagnosis: Lymphoepithelioma-like Carcinoma
Lymphoepithelioma-like carcinoma (LELC) is a rare, poorly differentiated, primary cutaneous neoplasm that occurs on sun-exposed skin, particularly on the head and neck of elderly individuals. It often manifests as an asymptomatic, slow-growing, flesh-colored or erythematous dermal nodule, though ulceration and tenderness have been reported.1 Histopathologically, these neoplasms often are poorly circumscribed and can infiltrate surrounding subcutaneous and soft tissue. As a biphasic tumor, LELC is characterized by islands, nests, or trabeculae of epithelioid cells within the mid dermis surrounded by a dense lymphocytic infiltrate with plasma cells (Figure 1).1 The epithelial component rarely communicates with the overlying epidermis and is composed of atypical polygonal cells with eosinophilic cytoplasm, vesicular nuclei, prominent nucleoli, and frequent mitosis.2 These epithelial nests can be highlighted by pancytokeratin AE1/AE3 or other epithelial differentiation markers (eg, CAM 5.2, CK5/6, epithelial membrane antigen, high-molecular-weight cytokeratin), while the surrounding lymphocytic infiltrate consists of an admixture of T cells and B cells. Lymphoepithelioma-like carcinomas also can demonstrate sebaceous, eccrine, or follicular differentiations.3 The epithelial nests of LELC also are positive for p63 and epithelial membrane antigen.2
The usual treatment of LELC is wide local excision or Mohs micrographic surgery.1 Despite the poorly differentiated morphology of the tumor, LELC has a generally good prognosis with low metastatic potential and few reports of local recurrence after incomplete excision.3 Patients who are not candidates for surgery as well as recalcitrant cases are managed with radiotherapy.1
Cutaneous lymphadenoma (CL) is a benign adnexal neoplasm that manifests as a small, solitary, fleshcolored nodule usually in the head and neck region.4 Histologically, CL consists of well-circumscribed epithelial nests within the dermis that are peripherally outlined by palisading basaloid cells and filled with clear to eosinophilic epithelioid cells (Figure 2).5 The fibrotic tumor stroma often is infiltrated by numerous intralobular dendritic cells and lymphocytes that occasionally can be arranged in germinal center–like nodules.4 The lymphoepithelial nature of CL can be challenging to distinguish morphologically from LELC, and immunohistochemistry stains may be required. In CL, both the basaloid and epithelioid cells stain positive for pancytokeratin AE1/ AE3, but the peripheral palisaded basaloid cells also stain positive for BerEP4. Additionally, the fibrotic stroma can be highlighted by CD34 and the intralobular dendritic cells by S-100.4
Nasopharyngeal carcinoma (NPC), formerly known as lymphoepithelioma, refers to carcinoma arising within the epithelium of the nasopharynx.6 Endemic to China, NPC manifests as an enlarging nasopharyngeal mass, causing clinical symptoms such as nasal obstruction and epistaxis.7 Histologically, nonkeratinizing NPC exhibits a biphasic morphology consisting of epithelioid neoplastic cells and background lymphocytic infiltrates (Figure 3). The epithelial component consists of round to oval neoplastic cells with amphophilic to eosinophilic cytoplasm, vesicular nuclei, and prominent nucleoli.6 Nasopharyngeal carcinoma is associated strongly with the Epstein-Barr virus while LELC is not; thus, Epstein- Barr encoding region in situ hybridization can reliably distinguish these entities. Metastatic NPC is rare but has been reported; therefore, it is highly recommended to perform an otolaryngologic examination in addition to testing for Epstein-Barr virus reactivity as part of a complete evaluation.8
Cutaneous squamous cell carcinoma (SCC) is a common epidermal malignancy with multiple subtypes and variable morphology. The clinical presentation of SCC is similar to LELC—an enlarging hyperkeratotic papule or nodule on sun-exposed skin that often is ulcerated and tender.9 Histologically, poorly differentiated nonkeratinizing SCC can form nests and trabeculae of epithelioid cells that are stained by epithelial differentiation markers, resembling the epithelioid nests of LELC. Distinguishing between LELC and poorly differentiated SCC with robust inflammatory infiltrate can be challenging (Figure 4). In fact, some experts support LELC as an SCC variant rather than a separate entity.9 However, in contrast to LELC, the dermal nests of SCC usually maintain an epidermal connection and often are associated with an overlying area of SCC in situ or welldifferentiated SCC.3
Mycosis fungoides (MF) is a primary cutaneous T-cell lymphoma. It is the most common type of cutaneous lymphoma, accounting for almost 50% of all reported cases.10 Classic MF has an indolent course and progresses through several clinical stages. Patches and plaques characterize early stages; lymphadenopathy indicates progression to later stages in which erythroderma may develop with coalescence of patches, plaques, and tumors; and MF present in blood or lymph nodes characterizes the late stage. Each stage of MF is different histologically—from a superficial lichenoid infiltrate with exocytosis of malignant T cells in the patch stage, to more robust epidermotropism and dermal infiltrate in the plaque stage, and finally a dense dermal infiltrate in the late stage.11 The rare syringotropic variant of MF clinically manifests as solitary or multiple erythematous lesions, often with overlying alopecia. Syringotropic MF uniquely exhibits folliculotropism and syringotropism along with syringometaplasia on histologic evaluation (Figure 5).12 The syringometaplasia can be difficult to distinguish from the epithelial nests of LELC, particularly with the lymphocytic background. Immunohistochemical panels for T-cell markers can highlight aberrant T cells in syringotropic MF through their usual loss of CD5 and CD7, in comparison to normal T cells in LELC.11 An elevated CD4:CD8 ratio of 4:1 and molecular analysis for T-cell receptor gene clonal rearrangements also can support the diagnosis of MF.12
The Diagnosis: Lymphoepithelioma-like Carcinoma
Lymphoepithelioma-like carcinoma (LELC) is a rare, poorly differentiated, primary cutaneous neoplasm that occurs on sun-exposed skin, particularly on the head and neck of elderly individuals. It often manifests as an asymptomatic, slow-growing, flesh-colored or erythematous dermal nodule, though ulceration and tenderness have been reported.1 Histopathologically, these neoplasms often are poorly circumscribed and can infiltrate surrounding subcutaneous and soft tissue. As a biphasic tumor, LELC is characterized by islands, nests, or trabeculae of epithelioid cells within the mid dermis surrounded by a dense lymphocytic infiltrate with plasma cells (Figure 1).1 The epithelial component rarely communicates with the overlying epidermis and is composed of atypical polygonal cells with eosinophilic cytoplasm, vesicular nuclei, prominent nucleoli, and frequent mitosis.2 These epithelial nests can be highlighted by pancytokeratin AE1/AE3 or other epithelial differentiation markers (eg, CAM 5.2, CK5/6, epithelial membrane antigen, high-molecular-weight cytokeratin), while the surrounding lymphocytic infiltrate consists of an admixture of T cells and B cells. Lymphoepithelioma-like carcinomas also can demonstrate sebaceous, eccrine, or follicular differentiations.3 The epithelial nests of LELC also are positive for p63 and epithelial membrane antigen.2
The usual treatment of LELC is wide local excision or Mohs micrographic surgery.1 Despite the poorly differentiated morphology of the tumor, LELC has a generally good prognosis with low metastatic potential and few reports of local recurrence after incomplete excision.3 Patients who are not candidates for surgery as well as recalcitrant cases are managed with radiotherapy.1
Cutaneous lymphadenoma (CL) is a benign adnexal neoplasm that manifests as a small, solitary, fleshcolored nodule usually in the head and neck region.4 Histologically, CL consists of well-circumscribed epithelial nests within the dermis that are peripherally outlined by palisading basaloid cells and filled with clear to eosinophilic epithelioid cells (Figure 2).5 The fibrotic tumor stroma often is infiltrated by numerous intralobular dendritic cells and lymphocytes that occasionally can be arranged in germinal center–like nodules.4 The lymphoepithelial nature of CL can be challenging to distinguish morphologically from LELC, and immunohistochemistry stains may be required. In CL, both the basaloid and epithelioid cells stain positive for pancytokeratin AE1/ AE3, but the peripheral palisaded basaloid cells also stain positive for BerEP4. Additionally, the fibrotic stroma can be highlighted by CD34 and the intralobular dendritic cells by S-100.4
Nasopharyngeal carcinoma (NPC), formerly known as lymphoepithelioma, refers to carcinoma arising within the epithelium of the nasopharynx.6 Endemic to China, NPC manifests as an enlarging nasopharyngeal mass, causing clinical symptoms such as nasal obstruction and epistaxis.7 Histologically, nonkeratinizing NPC exhibits a biphasic morphology consisting of epithelioid neoplastic cells and background lymphocytic infiltrates (Figure 3). The epithelial component consists of round to oval neoplastic cells with amphophilic to eosinophilic cytoplasm, vesicular nuclei, and prominent nucleoli.6 Nasopharyngeal carcinoma is associated strongly with the Epstein-Barr virus while LELC is not; thus, Epstein- Barr encoding region in situ hybridization can reliably distinguish these entities. Metastatic NPC is rare but has been reported; therefore, it is highly recommended to perform an otolaryngologic examination in addition to testing for Epstein-Barr virus reactivity as part of a complete evaluation.8
Cutaneous squamous cell carcinoma (SCC) is a common epidermal malignancy with multiple subtypes and variable morphology. The clinical presentation of SCC is similar to LELC—an enlarging hyperkeratotic papule or nodule on sun-exposed skin that often is ulcerated and tender.9 Histologically, poorly differentiated nonkeratinizing SCC can form nests and trabeculae of epithelioid cells that are stained by epithelial differentiation markers, resembling the epithelioid nests of LELC. Distinguishing between LELC and poorly differentiated SCC with robust inflammatory infiltrate can be challenging (Figure 4). In fact, some experts support LELC as an SCC variant rather than a separate entity.9 However, in contrast to LELC, the dermal nests of SCC usually maintain an epidermal connection and often are associated with an overlying area of SCC in situ or welldifferentiated SCC.3
Mycosis fungoides (MF) is a primary cutaneous T-cell lymphoma. It is the most common type of cutaneous lymphoma, accounting for almost 50% of all reported cases.10 Classic MF has an indolent course and progresses through several clinical stages. Patches and plaques characterize early stages; lymphadenopathy indicates progression to later stages in which erythroderma may develop with coalescence of patches, plaques, and tumors; and MF present in blood or lymph nodes characterizes the late stage. Each stage of MF is different histologically—from a superficial lichenoid infiltrate with exocytosis of malignant T cells in the patch stage, to more robust epidermotropism and dermal infiltrate in the plaque stage, and finally a dense dermal infiltrate in the late stage.11 The rare syringotropic variant of MF clinically manifests as solitary or multiple erythematous lesions, often with overlying alopecia. Syringotropic MF uniquely exhibits folliculotropism and syringotropism along with syringometaplasia on histologic evaluation (Figure 5).12 The syringometaplasia can be difficult to distinguish from the epithelial nests of LELC, particularly with the lymphocytic background. Immunohistochemical panels for T-cell markers can highlight aberrant T cells in syringotropic MF through their usual loss of CD5 and CD7, in comparison to normal T cells in LELC.11 An elevated CD4:CD8 ratio of 4:1 and molecular analysis for T-cell receptor gene clonal rearrangements also can support the diagnosis of MF.12
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Fisher JC, White RM, Hurd DS. Lymphoepithelioma-like carcinoma of the skin: a case of one patient presenting with two primary cutaneous neoplasms. J Am Osteopath Coll Dermatol. 2015;33:40-41.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Yu R, Salama S, Alowami S. Cutaneous lymphadenoma: a rare case and brief review of a diagnostic pitfall. Rare Tumors. 2014;6:5358.
- Monteagudo C, Fúnez R, Sánchez-Sendra B, et al. Cutaneous lymphadenoma is a distinct trichoblastoma-like lymphoepithelial tumor with diffuse androgen receptor immunoreactivity, Notch1 ligand in Reed-Sternberg-like Cells, and common EGFR somatic mutations. Am J Surg Pathol. 2021;45:1382-1390.
- Stelow EB, Wenig BM. Update from the 4th edition of the World Health Organization classification of head and neck tumours: nasopharynx. Head Neck Pathol. 2017;11:16-22.
- Almomani MH, Zulfiqar H, Nagalli S. Nasopharyngeal carcinoma (NPC, lymphoepithelioma). StatPearls Publishing; 2022.
- Lassen CB, Lock-Andersen J. Lymphoepithelioma-like carcinoma of the skin: a case with perineural invasion. Plast Reconstr Surg Glob Open. 2014;2:E252.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer Staging Guidelines, Prognostic Factors, and Histopathologic Variants. Adv Anat Pathol. 2017;24:171-194.
- Pileri A, Facchetti F, Rütten A, et al. Syringotropic mycosis fungoides: a rare variant of the disease with peculiar clinicopathologic features. Am J Surg Pathol. 2011;35:100-109.
- Ryu HJ, Kim SI, Jang HO, et al. Evaluation of the International Society for Cutaneous Lymphoma Algorithm for the Diagnosis of Early Mycosis Fungoides [published October 15, 2021]. Cells. 2021;10:2758. doi:10.3390/cells10102758
- Lehmer LM, Amber KT, de Feraudy SM. Syringotropic mycosis fungoides: a rare form of cutaneous T-cell lymphoma enabling a histopathologic “sigh of relief.” Am J Dermatopathol. 2017;39:920-923.
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Fisher JC, White RM, Hurd DS. Lymphoepithelioma-like carcinoma of the skin: a case of one patient presenting with two primary cutaneous neoplasms. J Am Osteopath Coll Dermatol. 2015;33:40-41.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Yu R, Salama S, Alowami S. Cutaneous lymphadenoma: a rare case and brief review of a diagnostic pitfall. Rare Tumors. 2014;6:5358.
- Monteagudo C, Fúnez R, Sánchez-Sendra B, et al. Cutaneous lymphadenoma is a distinct trichoblastoma-like lymphoepithelial tumor with diffuse androgen receptor immunoreactivity, Notch1 ligand in Reed-Sternberg-like Cells, and common EGFR somatic mutations. Am J Surg Pathol. 2021;45:1382-1390.
- Stelow EB, Wenig BM. Update from the 4th edition of the World Health Organization classification of head and neck tumours: nasopharynx. Head Neck Pathol. 2017;11:16-22.
- Almomani MH, Zulfiqar H, Nagalli S. Nasopharyngeal carcinoma (NPC, lymphoepithelioma). StatPearls Publishing; 2022.
- Lassen CB, Lock-Andersen J. Lymphoepithelioma-like carcinoma of the skin: a case with perineural invasion. Plast Reconstr Surg Glob Open. 2014;2:E252.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer Staging Guidelines, Prognostic Factors, and Histopathologic Variants. Adv Anat Pathol. 2017;24:171-194.
- Pileri A, Facchetti F, Rütten A, et al. Syringotropic mycosis fungoides: a rare variant of the disease with peculiar clinicopathologic features. Am J Surg Pathol. 2011;35:100-109.
- Ryu HJ, Kim SI, Jang HO, et al. Evaluation of the International Society for Cutaneous Lymphoma Algorithm for the Diagnosis of Early Mycosis Fungoides [published October 15, 2021]. Cells. 2021;10:2758. doi:10.3390/cells10102758
- Lehmer LM, Amber KT, de Feraudy SM. Syringotropic mycosis fungoides: a rare form of cutaneous T-cell lymphoma enabling a histopathologic “sigh of relief.” Am J Dermatopathol. 2017;39:920-923.
A 77-year-old man presented with a 1.2-cm dermal nodule on the left temple of 1 year’s duration. The lesion had become tender and darker in color. An excision was performed and submitted for histologic examination. Additional immunohistochemistry staining for Epstein-Barr virus was negative.
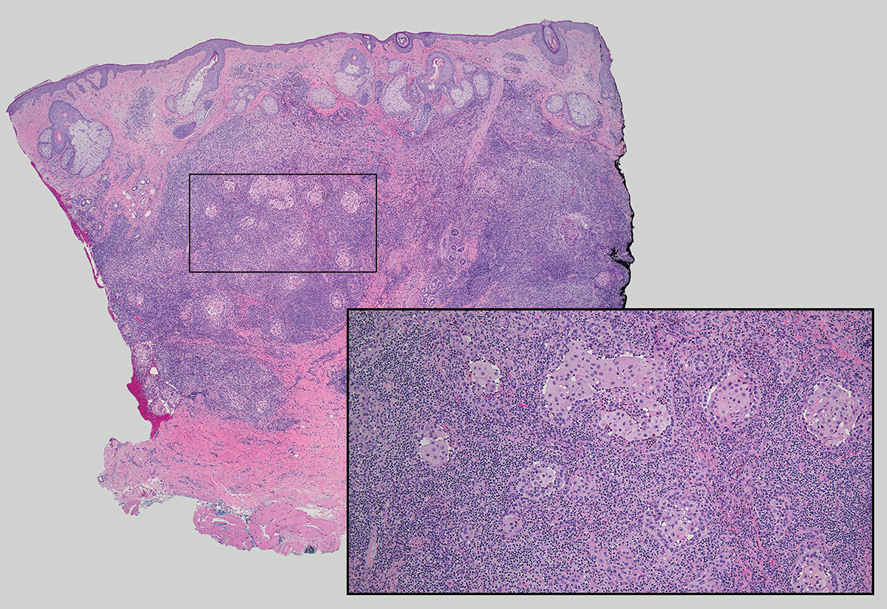
Botanical Briefs: Fig Phytophotodermatitis (Ficus carica)
Plant Parts and Nomenclature
Ficus carica (common fig) is a deciduous shrub or small tree with smooth gray bark that can grow up to 10 m in height (Figure 1). It is characterized by many spreading branches, but the trunk rarely grows beyond a diameter of 7 in. Its hairy leaves are coarse on the upper side and soft underneath with 3 to 7 deep lobes that can extend up to 25 cm in length or width; the leaves grow individually, alternating along the sides of the branches. Fig trees often can be seen adorning yards, gardens, and parks, especially in tropical and subtropical climates. Ficus carica should not be confused with Ficus benjamina (weeping fig), a common ornamental tree that also is used to provide shade in hot climates, though both can cause phototoxic skin eruptions.
.")
The common fig tree originated in the Mediterranean and western Asia1 and has been cultivated by humans since the second and third millennia

Ficus carica is a member of the Moraceae family (derived from the Latin name for the mulberry tree), which includes 53 genera and approximately 1400 species, of which about 850 belong to the genus Ficus (the Latin name for a fig tree). The term carica likely comes from the Latin word carricare (to load) to describe a tree loaded with figs. Family members include trees, shrubs, lianas, and herbs that usually contain laticifers with a milky latex.
Traditional Uses
For centuries, components of the fig tree have been used in herbal teas and pastes to treat ailments ranging from sore throats to diarrhea, though there is no evidence to support their efficacy.4 Ancient Indians and Egyptians used plants such as the common fig tree containing furocoumarins to induce hyperpigmentation in vitiligo.5
Phototoxic Components
The leaves and sap of the common fig tree contain psoralens, which are members of the furocoumarin group of chemical compounds and are the source of its phototoxicity. The fruit does not contain psoralens.6-9 The tree also produces proteolytic enzymes such as protease, amylase, ficin, triterpenoids, and lipodiastase that enhance its phototoxic effects.8 Exposure to UV light between 320 and 400 nm following contact with these phototoxic components triggers a reaction in the skin over the course of 1 to 3 days.5 The psoralens bind in epidermal cells, cross-link the DNA, and cause cell-membrane destruction, leading to edema and necrosis.10 The delay in symptoms may be attributed to the time needed to synthesize acute-phase reaction proteins such as tumor necrosis factor α and IL-1.11 In spring and summer months, an increased concentration of psoralens in the leaves and sap contribute to an increased incidence of phytophotodermatitis.9 Humidity and sweat also increase the percutaneous absorption of psoralens.12,13
Allergens
Fig trees produce a latex protein that can cause cross-reactive hypersensitivity reactions in those allergic to F benjamina latex and rubber latex.6 The latex proteins in fig trees can act as airborne respiratory allergens. Ingestion of figs can produce anaphylactic reactions in those sensitized to rubber latex and F benjamina latex.7 Other plant families associated with phototoxic reactions include Rutaceae (lemon, lime, bitter orange), Apiaceae (formerly Umbelliferae)(carrot, parsnip, parsley, dill, celery, hogweed), and Fabaceae (prairie turnip).
Cutaneous Manifestations
Most cases of fig phytophotodermatitis begin with burning, pain, and/or itching within hours of sunlight exposure in areas of the skin that encountered components of the fig tree, often in a linear pattern. The affected areas become erythematous and edematous with formation of bullae and unilocular vesicles over the course of 1 to 3 days.12,14,15 Lesions may extend beyond the region of contact with the fig tree as they spread across the skin due to sweat or friction, and pain may linger even after the lesions resolve.12,13,16 Adults who handle fig trees (eg, pruning) are susceptible to phototoxic reactions, especially those using chain saws or other mechanisms that result in spray exposure, as the photosensitizing sap permeates the wood and bark of the entire tree.17 Similarly, children who handle fig leaves or sap during outdoor play can develop bullous eruptions. Severe cases have resulted in hospital admission after prolonged exposure.16 Additionally, irritant dermatitis may arise from contact with the trichomes or “hairs” on various parts of the plant.

Patients who use natural remedies containing components of the fig tree without the supervision of a medical provider put themselves at risk for unsafe or unwanted adverse effects, such as phytophotodermatitis.12,15,16,18 An entire family presented with burns after they applied fig leaf extract to the skin prior to tanning outside in the sun.19 A 42-year-old woman acquired a severe burn covering 81% of the body surface after topically applying fig leaf tea to the skin as a tanning agent.20 A subset of patients ingesting or applying fig tree components for conditions such as vitiligo, dermatitis, onychomycosis, and motor retardation developed similar cutaneous reactions.13,14,21,22 Lesions resembling finger marks can raise concerns for potential abuse or neglect in children.22
The differential diagnosis for fig phytophotodermatitis includes sunburn, chemical burns, drug-related photosensitivity, infectious lesions (eg, herpes simplex, bullous impetigo, Lyme disease, superficial lymphangitis), connective tissue disease (eg, systemic lupus erythematosus), contact dermatitis, and nonaccidental trauma.12,15,18 Compared to sunburn, phytophotodermatitis tends to increase in severity over days following exposure and heals with dramatic hyperpigmentation, which also prompts visits to dermatology.12
Treatment
Treatment of fig phytophotodermatitis chiefly is symptomatic, including analgesia, appropriate wound care, and infection prophylaxis. Topical and systemic corticosteroids may aid in the resolution of moderate to severe reactions.15,23,24 Even severe injuries over small areas or mild injuries to a high percentage of the total body surface area may require treatment in a burn unit. Patients should be encouraged to use mineral-based sunscreens on the affected areas to reduce the risk for hyperpigmentation. Individuals who regularly handle fig trees should use contact barriers including gloves and protective clothing (eg, long-sleeved shirts, long pants).
- Ikegami H, Nogata H, Hirashima K, et al. Analysis of genetic diversity among European and Asian fig varieties (Ficus carica L.) using ISSR, RAPD, and SSR markers. Genetic Resources and Crop Evolution. 2009;56:201-209.
- Zohary D, Spiegel-Roy P. Beginnings of fruit growing in the Old World. Science. 1975;187:319-327.
- Young R. Young’s Analytical Concordance. Thomas Nelson; 1982.
- Duke JA. Handbook of Medicinal Herbs. CRC Press; 2002.
- Pathak MA, Fitzpatrick TB. Bioassay of natural and synthetic furocoumarins (psoralens). J Invest Dermatol. 1959;32:509-518.
- Focke M, Hemmer W, Wöhrl S, et al. Cross-reactivity between Ficus benjamina latex and fig fruit in patients with clinical fig allergy. Clin Exp Allergy. 2003;33:971-977.
- Hemmer W, Focke M, Götz M, et al. Sensitization to Ficus benjamina: relationship to natural rubber latex allergy and identification of foods implicated in the Ficus-fruit syndrome. Clin Exp Allergy. 2004;34:1251-1258.
- Bonamonte D, Foti C, Lionetti N, et al. Photoallergic contact dermatitis to 8-methoxypsoralen in Ficus carica. Contact Dermatitis. 2010;62:343-348.
- Zaynoun ST, Aftimos BG, Abi Ali L, et al. Ficus carica; isolation and quantification of the photoactive components. Contact Dermatitis. 1984;11:21-25.
- Tessman JW, Isaacs ST, Hearst JE. Photochemistry of the furan-side 8-methoxypsoralen-thymidine monoadduct inside the DNA helix. conversion to diadduct and to pyrone-side monoadduct. Biochemistry. 1985;24:1669-1676.
- Geary P. Burns related to the use of psoralens as a tanning agent. Burns. 1996;22:636-637.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:E238745.
- Ozdamar E, Ozbek S, Akin S. An unusual cause of burn injury: fig leaf decoction used as a remedy for a dermatitis of unknown etiology. J Burn Care Rehabil. 2003;24:229-233; discussion 228.
- Berakha GJ, Lefkovits G. Psoralen phototherapy and phototoxicity. Ann Plast Surg. 1985;14:458-461.
- Papazoglou A, Mantadakis E. Fig tree leaves phytophotodermatitis. J Pediatr. 2021;239:244-245.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Rouaiguia-Bouakkaz S, Amira-Guebailia H, Rivière C, et al. Identification and quantification of furanocoumarins in stem bark and wood of eight Algerian varieties of Ficus carica by RP-HPLC-DAD and RP-HPLC-DAD-MS. Nat Prod Commun. 2013;8:485-486.
- Oliveira AA, Morais J, Pires O, et al. Fig tree induced phytophotodermatitis. BMJ Case Rep. 2020;13:E233392.
- Bassioukas K, Stergiopoulou C, Hatzis J. Erythrodermic phytophotodermatitis after application of aqueous fig-leaf extract as an artificial suntan promoter and sunbathing. Contact Dermatitis. 2004;51:94-95.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Son JH, Jin H, You HS, et al. Five cases of phytophotodermatitis caused by fig leaves and relevant literature review. Ann Dermatol. 2017;29:86-90.
- Abali AE, Aka M, Aydogan C, et al. Burns or phytophotodermatitis, abuse or neglect: confusing aspects of skin lesions caused by the superstitious use of fig leaves. J Burn Care Res. 2012;33:E309-E312.
- Picard C, Morice C, Moreau A, et al. Phytophotodermatitis in children: a difficult diagnosis mimicking other dermatitis. 2017;5:1-3.
- Enjolras O, Soupre V, Picard A. Uncommon benign infantile vascular tumors. Adv Dermatol. 2008;24:105-124.
Plant Parts and Nomenclature
Ficus carica (common fig) is a deciduous shrub or small tree with smooth gray bark that can grow up to 10 m in height (Figure 1). It is characterized by many spreading branches, but the trunk rarely grows beyond a diameter of 7 in. Its hairy leaves are coarse on the upper side and soft underneath with 3 to 7 deep lobes that can extend up to 25 cm in length or width; the leaves grow individually, alternating along the sides of the branches. Fig trees often can be seen adorning yards, gardens, and parks, especially in tropical and subtropical climates. Ficus carica should not be confused with Ficus benjamina (weeping fig), a common ornamental tree that also is used to provide shade in hot climates, though both can cause phototoxic skin eruptions.
The common fig tree originated in the Mediterranean and western Asia1 and has been cultivated by humans since the second and third millennia
Ficus carica is a member of the Moraceae family (derived from the Latin name for the mulberry tree), which includes 53 genera and approximately 1400 species, of which about 850 belong to the genus Ficus (the Latin name for a fig tree). The term carica likely comes from the Latin word carricare (to load) to describe a tree loaded with figs. Family members include trees, shrubs, lianas, and herbs that usually contain laticifers with a milky latex.
Traditional Uses
For centuries, components of the fig tree have been used in herbal teas and pastes to treat ailments ranging from sore throats to diarrhea, though there is no evidence to support their efficacy.4 Ancient Indians and Egyptians used plants such as the common fig tree containing furocoumarins to induce hyperpigmentation in vitiligo.5
Phototoxic Components
The leaves and sap of the common fig tree contain psoralens, which are members of the furocoumarin group of chemical compounds and are the source of its phototoxicity. The fruit does not contain psoralens.6-9 The tree also produces proteolytic enzymes such as protease, amylase, ficin, triterpenoids, and lipodiastase that enhance its phototoxic effects.8 Exposure to UV light between 320 and 400 nm following contact with these phototoxic components triggers a reaction in the skin over the course of 1 to 3 days.5 The psoralens bind in epidermal cells, cross-link the DNA, and cause cell-membrane destruction, leading to edema and necrosis.10 The delay in symptoms may be attributed to the time needed to synthesize acute-phase reaction proteins such as tumor necrosis factor α and IL-1.11 In spring and summer months, an increased concentration of psoralens in the leaves and sap contribute to an increased incidence of phytophotodermatitis.9 Humidity and sweat also increase the percutaneous absorption of psoralens.12,13
Allergens
Fig trees produce a latex protein that can cause cross-reactive hypersensitivity reactions in those allergic to F benjamina latex and rubber latex.6 The latex proteins in fig trees can act as airborne respiratory allergens. Ingestion of figs can produce anaphylactic reactions in those sensitized to rubber latex and F benjamina latex.7 Other plant families associated with phototoxic reactions include Rutaceae (lemon, lime, bitter orange), Apiaceae (formerly Umbelliferae)(carrot, parsnip, parsley, dill, celery, hogweed), and Fabaceae (prairie turnip).
Cutaneous Manifestations
Most cases of fig phytophotodermatitis begin with burning, pain, and/or itching within hours of sunlight exposure in areas of the skin that encountered components of the fig tree, often in a linear pattern. The affected areas become erythematous and edematous with formation of bullae and unilocular vesicles over the course of 1 to 3 days.12,14,15 Lesions may extend beyond the region of contact with the fig tree as they spread across the skin due to sweat or friction, and pain may linger even after the lesions resolve.12,13,16 Adults who handle fig trees (eg, pruning) are susceptible to phototoxic reactions, especially those using chain saws or other mechanisms that result in spray exposure, as the photosensitizing sap permeates the wood and bark of the entire tree.17 Similarly, children who handle fig leaves or sap during outdoor play can develop bullous eruptions. Severe cases have resulted in hospital admission after prolonged exposure.16 Additionally, irritant dermatitis may arise from contact with the trichomes or “hairs” on various parts of the plant.
Patients who use natural remedies containing components of the fig tree without the supervision of a medical provider put themselves at risk for unsafe or unwanted adverse effects, such as phytophotodermatitis.12,15,16,18 An entire family presented with burns after they applied fig leaf extract to the skin prior to tanning outside in the sun.19 A 42-year-old woman acquired a severe burn covering 81% of the body surface after topically applying fig leaf tea to the skin as a tanning agent.20 A subset of patients ingesting or applying fig tree components for conditions such as vitiligo, dermatitis, onychomycosis, and motor retardation developed similar cutaneous reactions.13,14,21,22 Lesions resembling finger marks can raise concerns for potential abuse or neglect in children.22
The differential diagnosis for fig phytophotodermatitis includes sunburn, chemical burns, drug-related photosensitivity, infectious lesions (eg, herpes simplex, bullous impetigo, Lyme disease, superficial lymphangitis), connective tissue disease (eg, systemic lupus erythematosus), contact dermatitis, and nonaccidental trauma.12,15,18 Compared to sunburn, phytophotodermatitis tends to increase in severity over days following exposure and heals with dramatic hyperpigmentation, which also prompts visits to dermatology.12
Treatment
Treatment of fig phytophotodermatitis chiefly is symptomatic, including analgesia, appropriate wound care, and infection prophylaxis. Topical and systemic corticosteroids may aid in the resolution of moderate to severe reactions.15,23,24 Even severe injuries over small areas or mild injuries to a high percentage of the total body surface area may require treatment in a burn unit. Patients should be encouraged to use mineral-based sunscreens on the affected areas to reduce the risk for hyperpigmentation. Individuals who regularly handle fig trees should use contact barriers including gloves and protective clothing (eg, long-sleeved shirts, long pants).
Plant Parts and Nomenclature
Ficus carica (common fig) is a deciduous shrub or small tree with smooth gray bark that can grow up to 10 m in height (Figure 1). It is characterized by many spreading branches, but the trunk rarely grows beyond a diameter of 7 in. Its hairy leaves are coarse on the upper side and soft underneath with 3 to 7 deep lobes that can extend up to 25 cm in length or width; the leaves grow individually, alternating along the sides of the branches. Fig trees often can be seen adorning yards, gardens, and parks, especially in tropical and subtropical climates. Ficus carica should not be confused with Ficus benjamina (weeping fig), a common ornamental tree that also is used to provide shade in hot climates, though both can cause phototoxic skin eruptions.
The common fig tree originated in the Mediterranean and western Asia1 and has been cultivated by humans since the second and third millennia
Ficus carica is a member of the Moraceae family (derived from the Latin name for the mulberry tree), which includes 53 genera and approximately 1400 species, of which about 850 belong to the genus Ficus (the Latin name for a fig tree). The term carica likely comes from the Latin word carricare (to load) to describe a tree loaded with figs. Family members include trees, shrubs, lianas, and herbs that usually contain laticifers with a milky latex.
Traditional Uses
For centuries, components of the fig tree have been used in herbal teas and pastes to treat ailments ranging from sore throats to diarrhea, though there is no evidence to support their efficacy.4 Ancient Indians and Egyptians used plants such as the common fig tree containing furocoumarins to induce hyperpigmentation in vitiligo.5
Phototoxic Components
The leaves and sap of the common fig tree contain psoralens, which are members of the furocoumarin group of chemical compounds and are the source of its phototoxicity. The fruit does not contain psoralens.6-9 The tree also produces proteolytic enzymes such as protease, amylase, ficin, triterpenoids, and lipodiastase that enhance its phototoxic effects.8 Exposure to UV light between 320 and 400 nm following contact with these phototoxic components triggers a reaction in the skin over the course of 1 to 3 days.5 The psoralens bind in epidermal cells, cross-link the DNA, and cause cell-membrane destruction, leading to edema and necrosis.10 The delay in symptoms may be attributed to the time needed to synthesize acute-phase reaction proteins such as tumor necrosis factor α and IL-1.11 In spring and summer months, an increased concentration of psoralens in the leaves and sap contribute to an increased incidence of phytophotodermatitis.9 Humidity and sweat also increase the percutaneous absorption of psoralens.12,13
Allergens
Fig trees produce a latex protein that can cause cross-reactive hypersensitivity reactions in those allergic to F benjamina latex and rubber latex.6 The latex proteins in fig trees can act as airborne respiratory allergens. Ingestion of figs can produce anaphylactic reactions in those sensitized to rubber latex and F benjamina latex.7 Other plant families associated with phototoxic reactions include Rutaceae (lemon, lime, bitter orange), Apiaceae (formerly Umbelliferae)(carrot, parsnip, parsley, dill, celery, hogweed), and Fabaceae (prairie turnip).
Cutaneous Manifestations
Most cases of fig phytophotodermatitis begin with burning, pain, and/or itching within hours of sunlight exposure in areas of the skin that encountered components of the fig tree, often in a linear pattern. The affected areas become erythematous and edematous with formation of bullae and unilocular vesicles over the course of 1 to 3 days.12,14,15 Lesions may extend beyond the region of contact with the fig tree as they spread across the skin due to sweat or friction, and pain may linger even after the lesions resolve.12,13,16 Adults who handle fig trees (eg, pruning) are susceptible to phototoxic reactions, especially those using chain saws or other mechanisms that result in spray exposure, as the photosensitizing sap permeates the wood and bark of the entire tree.17 Similarly, children who handle fig leaves or sap during outdoor play can develop bullous eruptions. Severe cases have resulted in hospital admission after prolonged exposure.16 Additionally, irritant dermatitis may arise from contact with the trichomes or “hairs” on various parts of the plant.
Patients who use natural remedies containing components of the fig tree without the supervision of a medical provider put themselves at risk for unsafe or unwanted adverse effects, such as phytophotodermatitis.12,15,16,18 An entire family presented with burns after they applied fig leaf extract to the skin prior to tanning outside in the sun.19 A 42-year-old woman acquired a severe burn covering 81% of the body surface after topically applying fig leaf tea to the skin as a tanning agent.20 A subset of patients ingesting or applying fig tree components for conditions such as vitiligo, dermatitis, onychomycosis, and motor retardation developed similar cutaneous reactions.13,14,21,22 Lesions resembling finger marks can raise concerns for potential abuse or neglect in children.22
The differential diagnosis for fig phytophotodermatitis includes sunburn, chemical burns, drug-related photosensitivity, infectious lesions (eg, herpes simplex, bullous impetigo, Lyme disease, superficial lymphangitis), connective tissue disease (eg, systemic lupus erythematosus), contact dermatitis, and nonaccidental trauma.12,15,18 Compared to sunburn, phytophotodermatitis tends to increase in severity over days following exposure and heals with dramatic hyperpigmentation, which also prompts visits to dermatology.12
Treatment
Treatment of fig phytophotodermatitis chiefly is symptomatic, including analgesia, appropriate wound care, and infection prophylaxis. Topical and systemic corticosteroids may aid in the resolution of moderate to severe reactions.15,23,24 Even severe injuries over small areas or mild injuries to a high percentage of the total body surface area may require treatment in a burn unit. Patients should be encouraged to use mineral-based sunscreens on the affected areas to reduce the risk for hyperpigmentation. Individuals who regularly handle fig trees should use contact barriers including gloves and protective clothing (eg, long-sleeved shirts, long pants).
- Ikegami H, Nogata H, Hirashima K, et al. Analysis of genetic diversity among European and Asian fig varieties (Ficus carica L.) using ISSR, RAPD, and SSR markers. Genetic Resources and Crop Evolution. 2009;56:201-209.
- Zohary D, Spiegel-Roy P. Beginnings of fruit growing in the Old World. Science. 1975;187:319-327.
- Young R. Young’s Analytical Concordance. Thomas Nelson; 1982.
- Duke JA. Handbook of Medicinal Herbs. CRC Press; 2002.
- Pathak MA, Fitzpatrick TB. Bioassay of natural and synthetic furocoumarins (psoralens). J Invest Dermatol. 1959;32:509-518.
- Focke M, Hemmer W, Wöhrl S, et al. Cross-reactivity between Ficus benjamina latex and fig fruit in patients with clinical fig allergy. Clin Exp Allergy. 2003;33:971-977.
- Hemmer W, Focke M, Götz M, et al. Sensitization to Ficus benjamina: relationship to natural rubber latex allergy and identification of foods implicated in the Ficus-fruit syndrome. Clin Exp Allergy. 2004;34:1251-1258.
- Bonamonte D, Foti C, Lionetti N, et al. Photoallergic contact dermatitis to 8-methoxypsoralen in Ficus carica. Contact Dermatitis. 2010;62:343-348.
- Zaynoun ST, Aftimos BG, Abi Ali L, et al. Ficus carica; isolation and quantification of the photoactive components. Contact Dermatitis. 1984;11:21-25.
- Tessman JW, Isaacs ST, Hearst JE. Photochemistry of the furan-side 8-methoxypsoralen-thymidine monoadduct inside the DNA helix. conversion to diadduct and to pyrone-side monoadduct. Biochemistry. 1985;24:1669-1676.
- Geary P. Burns related to the use of psoralens as a tanning agent. Burns. 1996;22:636-637.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:E238745.
- Ozdamar E, Ozbek S, Akin S. An unusual cause of burn injury: fig leaf decoction used as a remedy for a dermatitis of unknown etiology. J Burn Care Rehabil. 2003;24:229-233; discussion 228.
- Berakha GJ, Lefkovits G. Psoralen phototherapy and phototoxicity. Ann Plast Surg. 1985;14:458-461.
- Papazoglou A, Mantadakis E. Fig tree leaves phytophotodermatitis. J Pediatr. 2021;239:244-245.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Rouaiguia-Bouakkaz S, Amira-Guebailia H, Rivière C, et al. Identification and quantification of furanocoumarins in stem bark and wood of eight Algerian varieties of Ficus carica by RP-HPLC-DAD and RP-HPLC-DAD-MS. Nat Prod Commun. 2013;8:485-486.
- Oliveira AA, Morais J, Pires O, et al. Fig tree induced phytophotodermatitis. BMJ Case Rep. 2020;13:E233392.
- Bassioukas K, Stergiopoulou C, Hatzis J. Erythrodermic phytophotodermatitis after application of aqueous fig-leaf extract as an artificial suntan promoter and sunbathing. Contact Dermatitis. 2004;51:94-95.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Son JH, Jin H, You HS, et al. Five cases of phytophotodermatitis caused by fig leaves and relevant literature review. Ann Dermatol. 2017;29:86-90.
- Abali AE, Aka M, Aydogan C, et al. Burns or phytophotodermatitis, abuse or neglect: confusing aspects of skin lesions caused by the superstitious use of fig leaves. J Burn Care Res. 2012;33:E309-E312.
- Picard C, Morice C, Moreau A, et al. Phytophotodermatitis in children: a difficult diagnosis mimicking other dermatitis. 2017;5:1-3.
- Enjolras O, Soupre V, Picard A. Uncommon benign infantile vascular tumors. Adv Dermatol. 2008;24:105-124.
- Ikegami H, Nogata H, Hirashima K, et al. Analysis of genetic diversity among European and Asian fig varieties (Ficus carica L.) using ISSR, RAPD, and SSR markers. Genetic Resources and Crop Evolution. 2009;56:201-209.
- Zohary D, Spiegel-Roy P. Beginnings of fruit growing in the Old World. Science. 1975;187:319-327.
- Young R. Young’s Analytical Concordance. Thomas Nelson; 1982.
- Duke JA. Handbook of Medicinal Herbs. CRC Press; 2002.
- Pathak MA, Fitzpatrick TB. Bioassay of natural and synthetic furocoumarins (psoralens). J Invest Dermatol. 1959;32:509-518.
- Focke M, Hemmer W, Wöhrl S, et al. Cross-reactivity between Ficus benjamina latex and fig fruit in patients with clinical fig allergy. Clin Exp Allergy. 2003;33:971-977.
- Hemmer W, Focke M, Götz M, et al. Sensitization to Ficus benjamina: relationship to natural rubber latex allergy and identification of foods implicated in the Ficus-fruit syndrome. Clin Exp Allergy. 2004;34:1251-1258.
- Bonamonte D, Foti C, Lionetti N, et al. Photoallergic contact dermatitis to 8-methoxypsoralen in Ficus carica. Contact Dermatitis. 2010;62:343-348.
- Zaynoun ST, Aftimos BG, Abi Ali L, et al. Ficus carica; isolation and quantification of the photoactive components. Contact Dermatitis. 1984;11:21-25.
- Tessman JW, Isaacs ST, Hearst JE. Photochemistry of the furan-side 8-methoxypsoralen-thymidine monoadduct inside the DNA helix. conversion to diadduct and to pyrone-side monoadduct. Biochemistry. 1985;24:1669-1676.
- Geary P. Burns related to the use of psoralens as a tanning agent. Burns. 1996;22:636-637.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:E238745.
- Ozdamar E, Ozbek S, Akin S. An unusual cause of burn injury: fig leaf decoction used as a remedy for a dermatitis of unknown etiology. J Burn Care Rehabil. 2003;24:229-233; discussion 228.
- Berakha GJ, Lefkovits G. Psoralen phototherapy and phototoxicity. Ann Plast Surg. 1985;14:458-461.
- Papazoglou A, Mantadakis E. Fig tree leaves phytophotodermatitis. J Pediatr. 2021;239:244-245.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Rouaiguia-Bouakkaz S, Amira-Guebailia H, Rivière C, et al. Identification and quantification of furanocoumarins in stem bark and wood of eight Algerian varieties of Ficus carica by RP-HPLC-DAD and RP-HPLC-DAD-MS. Nat Prod Commun. 2013;8:485-486.
- Oliveira AA, Morais J, Pires O, et al. Fig tree induced phytophotodermatitis. BMJ Case Rep. 2020;13:E233392.
- Bassioukas K, Stergiopoulou C, Hatzis J. Erythrodermic phytophotodermatitis after application of aqueous fig-leaf extract as an artificial suntan promoter and sunbathing. Contact Dermatitis. 2004;51:94-95.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Son JH, Jin H, You HS, et al. Five cases of phytophotodermatitis caused by fig leaves and relevant literature review. Ann Dermatol. 2017;29:86-90.
- Abali AE, Aka M, Aydogan C, et al. Burns or phytophotodermatitis, abuse or neglect: confusing aspects of skin lesions caused by the superstitious use of fig leaves. J Burn Care Res. 2012;33:E309-E312.
- Picard C, Morice C, Moreau A, et al. Phytophotodermatitis in children: a difficult diagnosis mimicking other dermatitis. 2017;5:1-3.
- Enjolras O, Soupre V, Picard A. Uncommon benign infantile vascular tumors. Adv Dermatol. 2008;24:105-124.
Practice Points
- Exposure to the components of the common fig tree (Ficus carica) can induce phytophotodermatitis.
- Notable postinflammatory hyperpigmentation typically occurs in the healing stage of fig phytophotodermatitis.

Micronutrient Deficiencies in Patients With Inflammatory Bowel Disease
In 2023, ESPEN (the European Society for Clinical Nutrition and Metabolism) published consensus recommendations highlighting the importance of regular monitoring and treatment of nutrient deficiencies in patients with inflammatory bowel disease (IBD) for improved prognosis, mortality, and quality of life.1 Suboptimal nutrition in patients with IBD predominantly results from inflammation of the gastrointestinal (GI) tract leading to malabsorption; however, medications commonly used to manage IBD also can contribute to malnutrition.2,3 Additionally, patients may develop nausea and food avoidance due to medication or the disease itself, leading to nutritional withdrawal and eventual deficiency.4 Even with the development of diets focused on balancing nutritional needs and decreasing inflammation,5 offsetting this aversion to food can be difficult to overcome.2
Cutaneous manifestations of IBD are multifaceted and can be secondary to the disease, reactive to or associated with IBD, or effects from nutritional deficiencies. The most common vitamin and nutrient deficiencies in patients with IBD include iron; zinc; calcium; vitamin D; and vitamins B6 (pyridoxine), B9 (folic acid), and B12.6 Malnutrition may manifest with cutaneous disease, and dermatologists can be the first to identify and assess for nutritional deficiencies. In this article, we review the mechanisms of these micronutrient depletions in the context of IBD, their subsequent dermatologic manifestations (Table), and treatment and monitoring guidelines for each deficiency.

Iron
A systematic review conducted from 2007 to 2012 in European patients with IBD (N=2192) found the overall prevalence of anemia in this population to be 24% (95% CI, 18%-31%), with 57% of patients with anemia experiencing iron deficiency.7 Anemia is observed more commonly in patients hospitalized with IBD and is common in patients with both Crohn disease and ulcerative colitis.8
Pathophysiology—Iron is critically important in oxygen transportation throughout the body as a major component of hemoglobin. Physiologically, the low pH of the duodenum and proximal jejunum allows divalent metal transporter 1 to transfer dietary Fe3+ into enterocytes, where it is reduced to the transportable Fe2+.9,10 Distribution of Fe2+ ions from enterocytes relies on ferroportin, an iron-transporting protein, which is heavily regulated by the protein hepcidin.11 Hepcidin, a known acute phase reactant, will increase in the setting of active IBD, causing a depletion of ferroportin and an inability of the body to utilize the stored iron in enterocytes.12 This poor utilization of iron stores combined with blood loss caused by inflammation in the GI tract is the proposed primary mechanism of iron-deficiency anemia observed in patients with IBD.13
Cutaneous Manifestations—From a dermatologic perspective, iron-deficiency anemia can manifest with a wide range of symptoms including glossitis, koilonychia, xerosis and/or pruritus, and brittle hair or hair loss.14,15 Although the underlying pathophysiology of these cutaneous manifestations is not fully understood, there are several theories assessing the mechanisms behind the skin findings of iron deficiency.
Atrophic glossitis has been observed in many patients with iron deficiency and is thought to manifest due to low iron concentrations in the blood, thereby decreasing oxygen delivery to the papillae of the dorsal tongue with resultant atrophy.16,17 Similarly, decreased oxygen delivery to the nail bed capillaries may cause deformities in the nail called koilonychia (or “spoon nails”).18 Iron is a key co-factor in collagen lysyl hydroxylase that promotes collagen binding; iron deficiency may lead to disruptions in the epidermal barrier that can cause pruritus and xerosis.19 An observational study of 200 healthy patients with a primary concern of pruritus found a correlation between low serum ferritin and a higher degree of pruritus (r=−0.768; P<.00001).20
Evidence for iron’s role in hair growth comes from a mouse model study with a mutation in the serine protease TMPRSS6—a protein that regulates hepcidin and iron absorption—which caused an increase in hepcidin production and subsequent systemic iron deficiency. Mice at 4 weeks of age were devoid of all body hair but had substantial regrowth after initiation of a 2-week iron-rich diet, which suggests a connection between iron repletion and hair growth in mice with iron deficiency.21 Additionally, a meta-analysis analyzing the comorbidities of patients with alopecia areata found them to have higher odds (odds ratio [OR]=2.78; 95% CI, 1.23-6.29) of iron-deficiency anemia but no association with IBD (OR=1.48; 95% CI, 0.32-6.82).22
Diagnosis and Monitoring—The American Gastroenterological Association recommends a complete blood cell count (CBC), serum ferritin, transferrin saturation (TfS), and C-reactive protein (CRP) as standard evaluations for iron deficiency in patients with IBD. Patients with active IBD should be screened every 3 months,and patients with inactive disease should be screened every 6 to 12 months.23
Although ferritin and TfS often are used as markers for iron status in healthy individuals, they are positive and negative acute phase reactants, respectively. Using them to assess iron status in patients with IBD may inaccurately represent iron status in the setting of inflammation from the disease.24 The European Crohn’s and Colitis Organisation (ECCO) produced guidelines to define iron deficiency as a TfS less than 20% or a ferritin level less than 30 µg/L in patients without evidence of active IBD and a ferritin level less than 100 µg/L for patients with active inflammation.25
A 2020 multicenter observational study of 202 patients with diagnosed IBD found that the ECCO guideline of ferritin less than 30 µg/L had an area under the receiver operating characteristic (AUROC) curve of 0.69, a sensitivity of 0.43, and a specificity of 0.95 in their population.26 In a sensitivity analysis stratifying patients by CRP level (<10 or ≥10 mg/L), the authors found that for patients with ulcerative colitis and a CRP less than 10 mg/L, a cut-off value of ferritin less than 65 µg/L (AUROC=0.78) had a sensitivity of 0.78 and specificity of 0.76, and a TfS value of less than 16% (AUROC=0.88) had a sensitivity of 0.79 and a specificity of 0.9. In patients with a CRP of 10 mg/L or greater, a cut-off value of ferritin 80 µg/L (AUROC=0.76) had a sensitivity of 0.75 and a specificity of 0.82, and a TfS value of less than 11% (AUROC=0.69) had a sensitivity of 0.79 and a specificity of 0.88. There were no ferritin cut-off values associated with good diagnostic performance (defined as both sensitivity and specificity >0.70) for iron deficiency in patients with Crohn disease.26
The authors recommended using an alternative iron measurement such as soluble transferrin receptor (sTfR)/log ferritin ratio (TfR-F) that is not influenced by active inflammation and has a good correlation with ferritin values (TfR-F: r=0.66; P<.001).26 However, both sTfR and TfR-F have high costs and intermethod variability as well as differences in their reference ranges depending on which laboratory performs the analysis, limiting the accessibility and practicality of easily obtaining these tests.27 Although there may be inaccuracies for standard ferritin or TfS under ECCO guidelines, proposed alternatives have their own limitations, which may make ferritin and TfS the most reasonable evaluations of iron status as long as disease activity status at the time of testing is taken into consideration.
Treatment—Treatment of underlying iron deficiency in patients with IBD requires reversing the cause of the deficiency and supplementing iron. In patients with IBD, the options to supplement iron may be limited by active disease, making oral intake less effective. Oral iron supplementation also is associated with notable GI adverse effects that may be exacerbated in patients with IBD. A systematic review of 43 randomized controlled trials (RCTs) evaluating GI adverse effects (eg, nausea, abdominal pain, diarrhea, constipation, and black or tarry stools) of oral ferrous sulfate compared with placebo or intravenous (IV) iron supplementation in healthy nonanemic individuals found a significant increase in GI adverse effects with oral supplementation (placebo: OR=2.32; P<.0001; IV: OR=3.05; P<.0001).28
Therefore, IV iron repletion may be necessary in patients with IBD and may require numerous infusions depending on the formulation of iron. In an RCT conducted in 2011, patients with iron-deficiency anemia with quiescent or mild to moderate IBD were treated with either IV iron sulfate or ferric carboxymaltose.29 With a primary end point of hemoglobin response greater than 2 g/dL, the authors found that 150 of 240 patients responded to ferric carboxymaltose vs 118 of 235 treated with iron sulfate (P=.004). The dosing for ferric carboxymaltose was 1 to 3 infusions of 500 to 1000 mg of iron and for iron sulfate up to 11 infusions of 200 mg of iron.29
Zinc
A systematic review of zinc deficiency in patients with IBD identified 7 studies including 2413 patients and revealed those with Crohn disease had a higher prevalence of zinc deficiency compared with patients with ulcerative colitis (54% vs 41%).30
Pathophysiology—Zinc serves as a catalytic cofactor for enzymatic activity within proteins and immune cells.31 The homeostasis of zinc is tightly regulated within the brush border of the small intestine by zinc transporters ZIP4 and ZIP1 from the lumen of enterocytes into the bloodstream.32 Inflammation in the small intestine due to Crohn disease can result in zinc malabsorption.
Ranaldi et al33 exposed intestinal cells and zinc-depleted intestinal cells to tumor necrosis factor α media to simulate an inflammatory environment. They measured transepithelial electrical resistance as a surrogate for transmembrane permeability and found that zinc-depleted cells had a statistically significantly higher transepithelial electrical resistance percentage (60% reduction after 4 hours; P<1.10–6) when exposed to tumor necrosis factor α signaling compared with normal intestinal cells. They concluded that zinc deficiency can increase intestinal permeability in the presence of inflammation, creating a cycle of further nutrient malabsorption and inflammation exacerbating IBD symptoms.33
Cutaneous Manifestations—After absorption in the small intestine, approximately 5% of zinc resides in the skin, with the highest concentration in the stratum spinosum.34 A cell study found that keratinocytes in zinc-deficient environments had higher rates of apoptosis compared with cells in normal media. The authors proposed that this higher rate of apoptosis and the resulting inflammation could be a mechanism for developing the desquamative or eczematous scaly plaques that are common cutaneous manifestations of zinc deficiency.35
Other cutaneous findings may include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.36 The histopathology of these skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.37
Diagnosis and Monitoring—Assessing serum zinc levels is challenging, as they may decrease during states of inflammation.38 A mouse model study showed a 3.1-fold increase (P<.001) in ZIP14 expression in wild-type mice compared with an IL-6 -/- knock-down model after IL-6 exposure. The authors concluded that the upregulation of ZIP14 in the liver due to inflammatory cytokine upregulation decreases zinc availability in serum.39 Additionally, serum zinc can overestimate the level of deficiency in IBD because approximately 75% of serum zinc is bound to albumin, which decreases in the setting of inflammation.40-42
Alternatively, alkaline phosphatase (AP), a zinc-dependent metalloenzyme, may be a better evaluator of zinc status during periods of inflammation. A study in rats evaluated zinc through serum zinc levels and AP levels after a period of induced stress to mimic a short-term inflammatory state.43 The researchers found that total body stores of zinc were unaffected throughout the experiment; only serum zinc declined throughout the experiment duration while AP did not. Because approximately 75% of serum zinc is bound to serum albumin,42 the researchers concluded the induced inflammatory state depleted serum albumin and redistributed zinc to the liver, causing the observed serum zinc changes, while total body zinc levels and AP were largely unaffected in comparison.43 Comorbid conditions such as liver or bone disease can increase AP levels, which limits the utility of AP as a surrogate for zinc in patients with comorbidities.44 However, even in the context of active IBD, serum zinc still is currently considered the best biomarker to evaluate zinc status.45
Treatment—The recommended dose for zinc supplementation is 20 to 40 mg daily with higher doses (>50 mg/d) for patients with malabsorptive syndromes such as IBD.46 It can be administered orally or parenterally. Although rare, zinc replacement therapy may be associated with diarrhea, nausea, vomiting, mild headaches, and fatigue.46 Additional considerations should be taken when repleting other micronutrients with zinc, as calcium and folate can inhibit zinc reabsorption, while zinc itself can inhibit iron and copper reabsorption.47
Vitamin D and Calcium
Low vitamin D levels (<50 nmol/L) and hypocalcemia (<8.8 mg/dL) are common in patients with IBD.48,49
Pathophysiology—Vitamin D levels are maintained via 2 mechanisms. The first mechanism is through the skin, as keratinocytes produce 7-dehydrocholesterol after exposure to UV light, which is converted into previtamin D3 and then thermally isomerizes into vitamin D3. This vitamin D3 is then transported to the liver on vitamin D–binding protein.50 The second mechanism is through oral vitamin D3 that is absorbed through vitamin D receptors in intestinal epithelium and transported to the liver, where it is hydroxylated into 25-hydroxyvitamin D (25[OH]D), then to the kidneys for hydroxylation to 1,25(OH)2D for redistribution throughout the body.50 This activated form of vitamin D regulates calcium absorption in the intestine, and optimal vitamin D levels are necessary to absorb calcium efficiently.51 Inflammation from IBD within the small intestine can downregulate vitamin D receptors, causing malabsorption and decreased serum vitamin D.52
Vitamin D signaling also is vital to maintaining the tight junctions and adherens junctions of the intestinal epithelium. Weakening the permeability of the epithelium further exacerbates malabsorption and subsequent vitamin D deficiency.52 A meta-analysis of 27 studies including 8316 patients with IBD showed low vitamin D levels were associated with increased odds of disease activity (OR=1.53; 95% CI, 1.32-1.77), mucosal inflammation (OR=1.25; 95% CI, 1.06-1.47), and future clinical relapse (OR=1.23; 95% CI, 1.03-1.47) in patients with Crohn disease. The authors concluded that low levels of vitamin D could be used as a potential biomarker of inflammatory status in Crohn disease.53
Vitamin D and calcium are further implicated in maintaining skeletal health,47 while vitamin D specifically helps maintain intestinal homeostasis54 and immune system modulation in the skin.55
Cutaneous Manifestations—Vitamin D is thought to play crucial roles in skin differentiation and proliferation, cutaneous innate immunity, hair follicle cycling, photoprotection, and wound healing.56 Vitamin D deficiency has been observed in a large range of cutaneous diseases including skin cancer, psoriasis, vitiligo, bullous pemphigoid, atopic dermatitis, and various types of alopecia.56-59 It is unclear whether vitamin D deficiency facilitates these disease processes or is merely the consequence of a disrupted cutaneous surface with the inability to complete the first step in vitamin D processing. A 2014 meta-analysis of 290 prospective cohort studies and 172 randomized trials concluded that 25(OH)D deficiency was associated with ill health and did not find causal evidence for any specific disease, dermatologic or otherwise.60 Calcium deficiency may cause epidermal changes including dry skin, coarse hair, and brittle nails.61
Diagnosis and Monitoring—The ECCO guidelines recommend obtaining serum 25(OH)D levels every 3 months in patients with IBD.62 Levels less than 75 nmol/L are considered deficient, and a value less than 30 nmol/L increases the risk for osteomalacia and nutritional rickets, constituting severe vitamin D deficiency.63-65
An observational study of 325 patients with IBD showed a statistically significant negative correlation between serum vitamin D and fecal calprotectin (r=−0.19; P<.001), a stool-based marker for gut inflammation, supporting vitamin D as a potential biomarker in IBD.66
Evaluation of calcium can be done through serum levels in patients with IBD.67 Patients with IBD are at risk for hypoalbuminemia; therefore, consideration should be taken to ensure calcium levels are corrected, as approximately 50% of calcium is bound to albumin or other ions in the body,68 which can be done by adjusting the calcium concentration by 0.02 mmol/L for every 1 g/L of albumin above or below 40 g/L. In the most critically ill patients, a direct ionized calcium blood level should be used instead because the previously mentioned correction calculations are inaccurate when albumin is critically low.69
Treatment—The ECCO guidelines recommend calcium and vitamin D repletion of 500 to 1000 mg and 800 to 1000 U, respectively, in patients with IBD on systemic corticosteroids to prevent the negative effects of bone loss.62 Calcium repletion in patients with IBD who are not on systemic steroids are the same as for the general population.65
Vitamin D repletion also may help decrease IBD activity. In a prospective study, 10,000 IU/d of vitamin D in 10 patients with IBD—adjusted over 12 weeks to a target of 100 to 125 nmol/L of serum 25(OH)D—showed a significant reduction in clinical Crohn activity (P=.019) over the study period.70 In contrast, 2000 IU/d for 3 months in an RCT of 27 patients with Crohn disease found significantly lower CRP (P=.019) and significantly higher self-reported quality of life (P=.037) but nonsignificant decreases in Crohn activity (P=.082) in patients with 25(OH)D levels of 75 nmol/L or higher compared with those with 25(OH)D levels less than 75 nmol/L.71
These discrepancies illustrate the need for expanded clinical trials to elucidate the optimal vitamin D dosing for patients with IBD. Ultimately, assessing vitamin D and calcium status and considering repletion in patients with IBD, especially those with comorbid dermatologic diseases such as poor wound healing, psoriasis, or atopic dermatitis, is important.
Vitamin B6 (Pyridoxine)
Pathophysiology—Pyridoxine is an important coenzyme for many functions including amino acid transamination, fatty acid metabolism, and conversion of tryptophan to niacin. It is absorbed in the jejunum and ileum and subsequently transported to the liver for rephosphorylation and release into its active form.36 An observational study assessing the nutritional status of patients with IBD found that only 5.7% of 105 patients with food records had inadequate dietary intake of pyridoxine, but 29% of all patients with IBD had subnormal pyridoxine levels.72 Additionally, they found no significant difference in the prevalence of subnormal pyridoxine levels in patients with active IBD vs IBD in remission. The authors suggested that the subnormal pyridoxine levels in patients with IBD likely were multifactorial and resulted from malabsorption due to active disease, inflammation, and inadequate intake.72
Cutaneous Manifestations—Cutaneous findings associated with pyridoxine deficiency include periorificial and perineal dermatitis,73 angular stomatitis, and cheilitis with associated burning, redness, and tongue edema.36 Additionally, pyridoxine is involved in the conversion of tryptophan to niacin, and its deficiency may manifest with pellagralike findings.74
Because pyridoxine is critical to protein metabolism, its deficiency may disrupt key cellular structures that rely on protein concentrations to maintain structural integrity. One such structure in the skin that heavily relies on protein concentrations is the ground substance of the extracellular matrix—the amorphous gelatinous spaces that occupy the areas between the extracellular matrix, which consists of cross-linked glycosaminoglycans and proteins.75 Without protein, ground substance increases in viscosity and can disrupt the epidermal barrier, leading to increased transepidermal water loss and ultimately inflammation.76 Although this theory has yet to be validated fully, this is a potential mechanistic explanation for the inflammation in dermal papillae that leads to dermatitis observed in pyridoxine deficiency.
Diagnosis and Monitoring—Direct biomarkers of pyridoxine status are in serum, plasma, erythrocytes, and urine, with the most common measurement in plasma as pyridoxal 5′-phosphate (PLP).77 Plasma PLP concentrations lower than 20 nmol/L are suggestive of deficiency.78 Plasma PLP has shown inverse relationships with acute phase inflammatory markers CRP79 and AP,78 thereby raising concerns for its validity to assess pyridoxine status in patients with symptomatic IBD.80
Alternative evaluations of pyridoxine include tryptophan and methionine loading tests,36 which are measured via urinary excretion and require normal kidney function to be accurate. They should be considered in IBD if necessary, but routine testing, even in patients with symptomatic IBD, is not recommended in the ECCO guidelines. Additional considerations should be taken in patients with altered nutrient requirements such as those who have undergone bowel resection due to highly active disease or those who receive parenteral nutritional supplementation.81
Treatment—Recommendations for oral pyridoxine supplementation range from 25 to 600 mg daily,82 with symptoms typically improving on 100 mg daily.36 Pyridoxine supplementation may have additional benefits for patients with IBD and potentially modulate disease severity. An IL-10 knockout mouse supplemented with pyridoxine had an approximately 60% reduction (P<.05) in inflammation compared to mice deficient in pyridoxine.83 The authors suggest that PLP-dependent enzymes can inhibit further proinflammatory signaling and T-cell migration that can exacerbate IBD. Ultimately, more data is needed before determining the efficacy of pyridoxine supplementation for active IBD.
Vitamin B12 and Vitamin B9 (Folic Acid)
Pathophysiology—Vitamin B12 is reabsorbed in the terminal ileum, the distal portion of the small intestine. The American Gastroenterological Association recommends that patients with a history of extensive ileal disease or prior ileal surgery, which is the case for many patients with Crohn disease, be monitored for vitamin B12 deficiency.23 Monitoring and rapid supplementation of vitamin B12 can prevent pernicious anemia and irreversible neurologic damage that may result from deficiency.84
Folic acid is primarily absorbed in the duodenum and jejunum of the small intestine. A meta-analysis performed in 2017 assessed studies observing folic acid and vitamin B12 levels in 1086 patients with IBD compared with 1484 healthy controls and found an average difference in serum folate concentration of 0.46 nmol/L (P<.001).84 Interestingly, this study did not find a significant difference in serum vitamin B12 levels between patients with IBD and healthy controls, highlighting the mechanism of vitamin B12 deficiency in IBD because only patients with terminal ileal involvement are at risk for malabsorption and subsequent deficiency.
Cutaneous Manifestations—Both vitamin B12 and folic acid deficiency can manifest as cheilitis, glossitis, and/or generalized hyperpigmentation that is accentuated in the flexural areas, palms, soles, and oral cavity.85,86 Systemic symptoms of patients with vitamin B12 and folic acid deficiency include megaloblastic anemia, pallor, and fatigue. A potential mechanism for the hyperpigmentation observed from vitamin B12 deficiency came from an electron microscope study that showed an increased concentration of melanosomes in a patient with deficiency.87
Diagnosis and Monitoring—In patients with suspected vitamin B12 and/or folic acid deficiency, initial evaluation should include a CBC with peripheral smear and serum vitamin B12 and folate levels. In cases for which the diagnosis still is unclear after initial testing,
Treatment—According to the Centers for Disease Control and Prevention, supplementation of vitamin B12 can be done orally with 1000 µg daily in patients with deficiency. In patients with active IBD, oral reabsorption of vitamin B12 can be less effective, making subcutaneous or intramuscular administration (1000 µg/wk for 8 weeks, then monthly for life) better options.89
Patients with IBD managed with methotrexate should be screened carefully for folate deficiency. Methotrexate is a folate analog that sometimes is used for the treatment of IBD. Reversible competitive inhibition of dihydrofolate reductase can precipitate a systemic folic acid decrease.91 Typically, oral folic acid (1 to 5 mg/d) is sufficient to treat folate deficiency, with the ESPEN recommending 5 mg once weekly 24 to 72 hours after methotrexate treatment or 1 mg daily for 5 days per week in patients with IBD.1 Alternative formulations—IV, subcutaneous, or intramuscular—are available for patients who cannot tolerate oral intake.92
Final Thoughts
Dermatologists can be the first to observe the cutaneous manifestations of micronutrient deficiencies. Although the symptoms of each micronutrient deficiency discussed may overlap, attention to small clinical clues in patients with IBD can improve patient outcomes and quality of life. For example, koilonychia with glossitis and xerosis likely is due to iron deficiency, while zinc deficiency should be suspected in patients with scaly eczematous plaques in skin folds. A high level of suspicion for micronutrient deficiencies in patients with IBD should be followed by a complete patient history, review of systems, and thorough clinical examination. A thorough laboratory evaluation can pinpoint nutritional deficiencies in patients with IBD, keeping in mind that specific biomarkers such as ferritin and serum zinc also act as acute phase reactants and should be interpreted in this context. Co-management with gastroenterologists should be a priority in patients with IBD, as gaining control of inflammatory disease is crucial for the prevention of recurrent vitamin and micronutrient deficiencies in addition to long-term health in this population.
- Bischoff SC, Bager P, Escher J, et al. ESPEN guideline on clinical nutrition in inflammatory bowel disease. Clin Nutr. 2023;42:352-379. doi:10.1016/j.clnu.2022.12.004
- Gerasimidis K, McGrogan P, Edwards CA. The aetiology and impact of malnutrition in paediatric inflammator y bowel disease. J Hum Nutr Diet. 2011;24:313-326. doi:10.1111/j.1365-277X.2011.01171.x
- Mentella MC, Scaldaferri F, Pizzoferrato M, et al. Nutrition, IBD and gut microbiota: a review. Nutrients. 2020;12:944. doi:10.3390/nu12040944
- Bonsack O, Caron B, Baumann C, et al. Food avoidance and fasting in patients with inflammatory bowel disease: experience from the Nancy IBD nutrition clinic. United European Gastroenterol J. 2023;11:361-370. doi:10.1002/ueg2.1238521
- Campmans-Kuijpers MJE, Dijkstra G. Food and food groups in inflammatory bowel disease (IBD): the design of the Groningen Anti-Inflammatory Diet (GrAID). Nutrients. 2021;13:1067. doi:10.3390/nu13041067
- Hwang C, Issokson K, Giguere-Rich C, et al. Development and pilot testing of the inflammatory bowel disease nutrition care pathway. Clin Gastroenterol Hepatol. 2020;18:2645-2649.e4. doi:10.1016/j.cgh.2020.06.039
- Filmann N, Rey J, Schneeweiss S, et al. Prevalence of anemia in inflammatory bowel diseases in European countries: a systematic review and individual patient data meta-analysis. Inflamm Bowel Dis. 2014;20:936-945. doi:10.1097/01.MIB.0000442728.74340.fd
- Stein J, Hartmann F, Dignass AU. Diagnosis and management of iron deficiency anemia in patients with IBD. Nat Rev Gastroenterol Hepatol. 2010;7:599-610. doi:10.1038/nrgastro.2010.151
- Ems T, St Lucia K, Huecker MR. Biochemistry, iron absorption. StatPearls [Internet]. Updated April 17, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448204/
- Evstatiev R, Gasche C. Iron sensing and signalling. Gut. 2012;61:933-952. doi:10.1136/gut.2010.214312
- Przybyszewska J, Zekanowska E. The role of hepcidin, ferroportin, HCP1, and DMT1 protein in iron absorption in the human digestive tract. Prz Gastroenterol. 2014;9:208-213. doi:10.5114/pg.2014.45102
- Weiss G, Gasche C. Pathogenesis and treatment of anemia in inflammatory bowel disease. Haematologica. 2010;95:175-178. doi:10.3324/haematol.2009.017046
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72. doi:10.4291/wjgp.v6.i3.62
- Moiz B. Spoon nails: still seen in today’s world. Clin Case Rep. 2018;6:547-548. doi:10.1002/ccr3.1404
- St Pierre SA, Vercellotti GM, Donovan JC, et al. Iron deficiency and diffuse nonscarring scalp alopecia in women: more pieces to the puzzle. J Am Acad Dermatol. 2010;63:1070-1076. doi:10.1016/j.jaad.2009.05.054
- Chiang CP, Yu-Fong Chang J, Wang YP, et al. Anemia, hematinic deficiencies, hyperhomocysteinemia, and serum gastric parietal cell antibody positivity in atrophic glossitis patients with or without microcytosis. J Formos Med Assoc. 2019;118:1401-1407. doi:10.1016/j.jfma.2019.06.004
- Chiang CP, Chang JY, Wang YP, et al. Atrophic glossitis: Etiology, serum autoantibodies, anemia, hematinic deficiencies, hyperhomocysteinemia, and management. J Formos Med Assoc. 2020;119:774-780. doi:10.1016/j.jfma.2019.04.015
- Walker J, Baran R, Vélez N, et al. Koilonychia: an update on pathophysiology, differential diagnosis and clinical relevance. J Eur Acad Dermatol Venereol. 2016;30:1985-1991. doi:10.1111/jdv.13610
- Guo HF, Tsai CL, Terajima M, et al. Pro-metastatic collagen lysyl hydroxylase dimer assemblies stabilized by Fe2+-binding. Nat Commun. 2018;9:512. doi:10.1038/s41467-018-02859-z
- Saini S, Jain AK, Agarwal S, et al. Iron deficiency and pruritus: a cross-sectional analysis to assess its association and relationship. Indian J Dermatol. 2021;66:705. doi:10.4103/ijd.ijd_326_21
- Du X, She E, Gelbart T, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320:1088-1092. doi:10.1126/science.1157121
- Lee S, Lee H, Lee CH, et al. Comorbidities in alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol. 2019;80:466-477.e16. doi:10.1016/j.jaad.2018.07.013
- Hashash JG, Elkins J, Lewis JD, et al. AGA Clinical Practice Update on diet and nutritional therapies in patients with inflammatory bowel disease: expert review [published online January 23, 2024]. Gastroenterology. doi:10.1053/j.gastro.2023.11.303
- Choudhuri S, Chowdhury IH, Saha A, et al. Acute monocyte pro- inflammatory response predicts higher positive to negative acute phase reactants ratio and severe hemostatic derangement in dengue fever. Cytokine. 2021;146:155644. doi:10.1016/j.cyto.2021.155644
- Dignass AU, Gasche C, Bettenworth D, et al; European Crohn’s and Colitis Organisation. European consensus on the diagnosis and management of iron deficiency and anaemia in inflammatory bowel diseases. J Crohn’s Colitis. 2015;9:211-222. doi:10.1093/ecco-jcc/jju009
- Daude S, Remen T, Chateau T, et al. Comparative accuracy of ferritin, transferrin saturation and soluble transferrin receptor for the diagnosis of iron deficiency in inflammatory bowel disease. Aliment Pharmacol Ther. 2020;51:1087-1095. doi:10.1111/apt.15739
- Pfeiffer CM, Looker AC. Laboratory methodologies for indicators of iron status: strengths, limitations, and analytical challenges. Am J Clin Nutr. 2017;106(suppl 6):1606S-1614S. doi:10.3945/ajcn.117.155887
- Tolkien Z, Stecher L, Mander AP, et al. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: a systematic review and meta-analysis. PLoS One. 2015;10:e0117383. doi:10.1371/journal.pone.0117383
- Evstatiev R, Marteau P, Iqbal T, et al. FERGIcor, a randomized controlled trial on ferric carboxymaltose for iron deficiency anemia in inflammatory bowel disease. Gastroenterology. 2011;141:846-853.e8532. doi:10.1053/j.gastro.2011.06.005
- Zupo R, Sila A, Castellana F, et al. Prevalence of zinc deficiency in inflammatory bowel disease: a systematic review and meta-analysis. Nutrients. 2022;14:4052. doi:10.3390/nu14194052
- Thompson MW. Regulation of zinc-dependent enzymes by metal carrier proteins. Biometals. 2022;35:187-213. doi:10.1007/s10534-022-00373-w
- Maares M, Haase H. A guide to human zinc absorption: general overview and recent advances of in vitro intestinal models. Nutrients. 2020;12:762. doi:10.3390/nu12030762
- Ranaldi G, Ferruzza S, Canali R, et al. Intracellular zinc is required for intestinal cell survival signals triggered by the inflammatory cytokine TNFα. J Nutr Biochem. 2013;24:967-976. doi:10.1016/j.jnutbio.2012.06.020
- Ogawa Y, Kawamura T, Shimada S. Zinc and skin biology. Arch Biochem Biophys. 2016;611:113-119. doi:10.1016/j.abb.2016.06.003
- Wilson D, Varigos G, Ackland ML. Apoptosis may underlie the pathology of zinc-deficient skin. Immunol Cell Biol. 2006;84:28-37. doi:10.1111/j.1440-1711.2005.01391.x
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685. doi:10.1016/j.clindermatol.2010.03.029
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624. doi:10.3390/nu9060624
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U S A. 2005;102:6843-6848. doi:10.1073/pnas.0502257102
- Vermeire S, Van Assche G, Rutgeerts P. Laboratory markers in IBD: useful, magic, or unnecessary toys?. Gut. 2006;55:426-431. doi:10.1136/gut.2005.069476
- Morisaku M, Ito K, Ogiso A, et al. Correlation between serum albumin and serum zinc in malignant lymphoma. Fujita Med J. 2022;8:59-64. doi:10.20407/fmj.2021-006
- Falchuk KH. Effect of acute disease and ACTH on serum zinc proteins. N Engl J Med. 1977:296:1129-1134.
- Naber TH, Baadenhuysen H, Jansen JB, et al. Serum alkaline phosphatase activity during zinc deficiency and long-term inflammatory stress. Clin Chim Acta. 1996;249:109-127. doi:10.1016/0009-8981(96)06281-x
- Lowe D, Sanvictores T, Zubair M, et al. Alkaline phosphatase. StatPearls [Internet]. Updated October 29, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK459201/
- Krebs NF. Update on zinc deficiency and excess in clinical pediatric practice. Ann Nutr Metab. 2013;62 suppl 1:19-29. doi:10.1159/000348261
- Maxfield L, Shukla S, Crane JS. Zinc deficiency. StatPearls [Internet]. Updated June 28, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493231/
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808. doi:10.1016/j.gtc.2017.08.011
- Caviezel D, Maissen S, Niess JH, et al. High prevalence of vitamin D deficiency among patients with inflammatory bowel disease. Inflamm Intest Dis. 2018;2:200-210. doi:10.1159/000489010
- Jasielska M, Grzybowska-Chlebowczyk U. Hypocalcemia and vitamin D deficiency in children with inflammatory bowel diseases and lactose intolerance. Nutrients. 2021;13:2583. doi:10.3390/nu13082583
- Vernia F, Valvano M, Longo S, et al. Vitamin D in inflammatory bowel diseases. Mechanisms of action and therapeutic implications. Nutrients. 2022;14:269. doi:10.3390/nu14020269
- Khazai N, Judd SE, Tangpricha V. Calcium and vitamin D: skeletal and extraskeletal health. Curr Rheumatol Rep. 2008;10:110-117. doi:10.1007/s11926-008-0020-y
- Domazetovic V, Iantomasi T, Bonanomi AG, et al. Vitamin D regulates claudin-2 and claudin-4 expression in active ulcerative colitis by p-Stat-6 and Smad-7 signaling. Int J Colorectal Dis. 2020;35:1231-1242. doi:10.1007/s00384-020-03576-0
- Gubatan J, Chou ND, Nielsen OH, et al. Systematic review with meta-analysis: association of vitamin D status with clinical outcomes in adult patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2019;50:1146-1158. doi:10.1111/apt.15506
- Fakhoury HMA, Kvietys PR, AlKattan W, et al. Vitamin D and intestinal homeostasis: barrier, microbiota, and immune modulation. J Steroid Biochem Mol Biol. 2020;200:105663. doi:10.1016/j.jsbmb.2020.105663
- Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770-1773. doi:10.1126/science.1123933
- Mostafa WZ, Hegazy RA. Vitamin D and the skin: focus on a complex relationship: a review. J Adv Res. 2015;6:793-804. doi:10.1016/j.jare.2014.01.011
- Searing DA, Leung DY. Vitamin D in atopic dermatitis, asthma and allergic diseases. Immunol Allergy Clin North Am. 2010;30:397-409.
- Lee YH, Song GG. Association between circulating 25-hydroxyvitamin D levels and psoriasis, and correlation with disease severity: a meta-analysis. Clin Exp Dermatol. 2018;43:529-535.
- Adorini L, Penna G. Control of autoimmune diseases by the vitamin D endocrine system. Nat Clin Pract Rheumatol. 2008;4:404-412.
- Autier P, Boniol M, Pizot C, et al. Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol. 2014;2:76-89. doi:10.1016/S2213-8587(13)70165-7
- Schafer AL, Shoback DM. Hypocalcemia: diagnosis and treatment. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext [Internet]. Updated January 3, 2016. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK279022/
- Magro F, Gionchetti P, Eliakim R, et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J Crohns Colitis. 2017;11:649-670. doi:10.1093/ecco-jcc/jjx008
- Amrein K, Scherkl M, Hoffmann M, et al. Vitamin D deficiency 2.0: an update on the current status worldwide. Eur J Clin Nutr. 2020;74:1498-1513. doi:10.1038/s41430-020-0558-y
- Munns CF, Shaw N, Kiely M, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. 2016;101:394-415. doi:10.1210/jc.2015-2175
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, Del Valle HB, eds. Dietary Reference Intakes for Calcium and Vitamin D. National Academies Press (US); 2011.
- Yeaman F, Nguyen A, Abasszade J, et al. Assessing vitamin D as a biomarker in inflammatory bowel disease. JGH Open. 2023;7:953-958. doi:10.1002/jgh3.13010
- Vernia P, Loizos P, Di Giuseppantonio I, et al S. Dietary calcium intake in patients with inflammatory bowel disease. J Crohns Colitis. 2014;8:312-317. doi:10.1016/j.crohns.2013.09.008
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ. 2008;336:1298-1302. doi:10.1136/bmj.39582.589433.BE
- Kenny CM, Murphy CE, Boyce DS, et al. Things we do for no reason™: calculating a “corrected calcium” level. J Hosp Med. 2021;16:499-501. doi:10.12788/jhm.3619
- Garg M, Rosella O, Rosella G, et al. Evaluation of a 12-week targeted vitamin D supplementation regimen in patients with active inflammatory bowel disease. Clin Nutr. 2018;37:1375-1382. doi:10.1016/j.clnu.2017.06.011
- Raftery T, Martineau AR, Greiller CL, et al. Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn’s disease: results from a randomised double-blind placebo-controlled study. United European Gastroenterol J. 2015;3:294-302. doi:10.1177/2050640615572176
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319. doi:10.1177/0148607107031004311
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274. doi:10.1016/s0190-9622(86)70301-0
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739. doi:10.3949/ccjm.83a.15061
- Elgharably N, Al Abadie M, Al Abadie M, et al. Vitamin B group levels and supplementations in dermatology. Dermatol Reports. 2022;15:9511. doi:10.4081/dr.2022.9511
- Hołubiec P, Leon´czyk M, Staszewski F, et al. Pathophysiology and clinical management of pellagra—a review. Folia Med Cracov. 2021;61:125-137. doi:10.24425/fmc.2021.138956
- Ink SL, Henderson LM. Vitamin B6 metabolism. Annu Rev Nutr. 1984;4:455-470. doi:10.1146/annurev.nu.04.070184.002323
- Brown MJ, Ameer MA, Daley SF, et al. Vitamin B6 deficiency. StatPearls [Internet]. Updated August 8, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK470579/.
- Vasilaki AT, McMillan DC, Kinsella J, et al. Relation between pyridoxal and pyridoxal phosphate concentrations in plasma, red cells, and white cells in patients with critical illness. Am J Clin Nutr. 2008;88:140-146. doi:10.1093/ajcn/88.1.140
- Chiang EP, Bagley PJ, Selhub J, et al. Abnormal vitamin B(6) status is associated with severity of symptoms in patients with rheumatoid arthritis. Am J Med. 2003;114:283-287. doi:10.1016/s0002-9343(02)01528-0
- Maaser C, Sturm A, Vavricka SR, et al. ECCO-ESGAR guideline for diagnostic assessment in IBD. Part 1: initial diagnosis, monitoring of known IBD, detection of complications. J Crohns Colitis. 2019;13:144-164. doi:10.1093/ecco-jcc/jjy113
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Selhub J, Byun A, Liu Z, et al. Dietary vitamin B6 intake modulates colonic inflammation in the IL10-/- model of inflammatory bowel disease. J Nutr Biochem. 2013;24:2138-2143. doi:10.1016/j.jnutbio.2013.08.005
- Pan Y, Liu Y, Guo H, et al. Associations between folate and vitamin B12 levels and inflammatory bowel disease: a meta-analysis. Nutrients. 2017;9:382. doi:10.3390/nu9040382
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33. doi:10.1007/s40257-014-0107-3
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503. doi:10.1002/ncp.10321
- Mori K, Ando I, Kukita A. Generalized hyperpigmentation of the skin due to vitamin B12 deficiency. J Dermatol. 2001;28:282-285. doi:10.1111/j.1346-8138.2001.tb00134.x
- Green R. Indicators for assessing folate and vitamin B-12 status and for monitoring the efficacy of intervention strategies. Am J Clin Nutr. 2011;94:666S-672S. doi:10.3945/ajcn.110.009613
- NIH Office of Dietary Supplements. Vitamin B12: fact sheet for health professionals. Updated February 27, 2024. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/
- NIH Office of Dietary Supplements. Folate: fact sheet for health professionals. Updated November 20, 2023. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/.
- Saibeni S, Bollani S, Losco A, et al. The use of methotrexate for treatment of inflammatory bowel disease in clinical practice. Dig Liver Dis. 2012;44:123-127. doi:10.1016/j.dld.2011.09.015
- Khan KM, Jialal I. Folic acid deficiency. StatPearls [Internet]. Updated June 26, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK535377/
In 2023, ESPEN (the European Society for Clinical Nutrition and Metabolism) published consensus recommendations highlighting the importance of regular monitoring and treatment of nutrient deficiencies in patients with inflammatory bowel disease (IBD) for improved prognosis, mortality, and quality of life.1 Suboptimal nutrition in patients with IBD predominantly results from inflammation of the gastrointestinal (GI) tract leading to malabsorption; however, medications commonly used to manage IBD also can contribute to malnutrition.2,3 Additionally, patients may develop nausea and food avoidance due to medication or the disease itself, leading to nutritional withdrawal and eventual deficiency.4 Even with the development of diets focused on balancing nutritional needs and decreasing inflammation,5 offsetting this aversion to food can be difficult to overcome.2
Cutaneous manifestations of IBD are multifaceted and can be secondary to the disease, reactive to or associated with IBD, or effects from nutritional deficiencies. The most common vitamin and nutrient deficiencies in patients with IBD include iron; zinc; calcium; vitamin D; and vitamins B6 (pyridoxine), B9 (folic acid), and B12.6 Malnutrition may manifest with cutaneous disease, and dermatologists can be the first to identify and assess for nutritional deficiencies. In this article, we review the mechanisms of these micronutrient depletions in the context of IBD, their subsequent dermatologic manifestations (Table), and treatment and monitoring guidelines for each deficiency.
Iron
A systematic review conducted from 2007 to 2012 in European patients with IBD (N=2192) found the overall prevalence of anemia in this population to be 24% (95% CI, 18%-31%), with 57% of patients with anemia experiencing iron deficiency.7 Anemia is observed more commonly in patients hospitalized with IBD and is common in patients with both Crohn disease and ulcerative colitis.8
Pathophysiology—Iron is critically important in oxygen transportation throughout the body as a major component of hemoglobin. Physiologically, the low pH of the duodenum and proximal jejunum allows divalent metal transporter 1 to transfer dietary Fe3+ into enterocytes, where it is reduced to the transportable Fe2+.9,10 Distribution of Fe2+ ions from enterocytes relies on ferroportin, an iron-transporting protein, which is heavily regulated by the protein hepcidin.11 Hepcidin, a known acute phase reactant, will increase in the setting of active IBD, causing a depletion of ferroportin and an inability of the body to utilize the stored iron in enterocytes.12 This poor utilization of iron stores combined with blood loss caused by inflammation in the GI tract is the proposed primary mechanism of iron-deficiency anemia observed in patients with IBD.13
Cutaneous Manifestations—From a dermatologic perspective, iron-deficiency anemia can manifest with a wide range of symptoms including glossitis, koilonychia, xerosis and/or pruritus, and brittle hair or hair loss.14,15 Although the underlying pathophysiology of these cutaneous manifestations is not fully understood, there are several theories assessing the mechanisms behind the skin findings of iron deficiency.
Atrophic glossitis has been observed in many patients with iron deficiency and is thought to manifest due to low iron concentrations in the blood, thereby decreasing oxygen delivery to the papillae of the dorsal tongue with resultant atrophy.16,17 Similarly, decreased oxygen delivery to the nail bed capillaries may cause deformities in the nail called koilonychia (or “spoon nails”).18 Iron is a key co-factor in collagen lysyl hydroxylase that promotes collagen binding; iron deficiency may lead to disruptions in the epidermal barrier that can cause pruritus and xerosis.19 An observational study of 200 healthy patients with a primary concern of pruritus found a correlation between low serum ferritin and a higher degree of pruritus (r=−0.768; P<.00001).20
Evidence for iron’s role in hair growth comes from a mouse model study with a mutation in the serine protease TMPRSS6—a protein that regulates hepcidin and iron absorption—which caused an increase in hepcidin production and subsequent systemic iron deficiency. Mice at 4 weeks of age were devoid of all body hair but had substantial regrowth after initiation of a 2-week iron-rich diet, which suggests a connection between iron repletion and hair growth in mice with iron deficiency.21 Additionally, a meta-analysis analyzing the comorbidities of patients with alopecia areata found them to have higher odds (odds ratio [OR]=2.78; 95% CI, 1.23-6.29) of iron-deficiency anemia but no association with IBD (OR=1.48; 95% CI, 0.32-6.82).22
Diagnosis and Monitoring—The American Gastroenterological Association recommends a complete blood cell count (CBC), serum ferritin, transferrin saturation (TfS), and C-reactive protein (CRP) as standard evaluations for iron deficiency in patients with IBD. Patients with active IBD should be screened every 3 months,and patients with inactive disease should be screened every 6 to 12 months.23
Although ferritin and TfS often are used as markers for iron status in healthy individuals, they are positive and negative acute phase reactants, respectively. Using them to assess iron status in patients with IBD may inaccurately represent iron status in the setting of inflammation from the disease.24 The European Crohn’s and Colitis Organisation (ECCO) produced guidelines to define iron deficiency as a TfS less than 20% or a ferritin level less than 30 µg/L in patients without evidence of active IBD and a ferritin level less than 100 µg/L for patients with active inflammation.25
A 2020 multicenter observational study of 202 patients with diagnosed IBD found that the ECCO guideline of ferritin less than 30 µg/L had an area under the receiver operating characteristic (AUROC) curve of 0.69, a sensitivity of 0.43, and a specificity of 0.95 in their population.26 In a sensitivity analysis stratifying patients by CRP level (<10 or ≥10 mg/L), the authors found that for patients with ulcerative colitis and a CRP less than 10 mg/L, a cut-off value of ferritin less than 65 µg/L (AUROC=0.78) had a sensitivity of 0.78 and specificity of 0.76, and a TfS value of less than 16% (AUROC=0.88) had a sensitivity of 0.79 and a specificity of 0.9. In patients with a CRP of 10 mg/L or greater, a cut-off value of ferritin 80 µg/L (AUROC=0.76) had a sensitivity of 0.75 and a specificity of 0.82, and a TfS value of less than 11% (AUROC=0.69) had a sensitivity of 0.79 and a specificity of 0.88. There were no ferritin cut-off values associated with good diagnostic performance (defined as both sensitivity and specificity >0.70) for iron deficiency in patients with Crohn disease.26
The authors recommended using an alternative iron measurement such as soluble transferrin receptor (sTfR)/log ferritin ratio (TfR-F) that is not influenced by active inflammation and has a good correlation with ferritin values (TfR-F: r=0.66; P<.001).26 However, both sTfR and TfR-F have high costs and intermethod variability as well as differences in their reference ranges depending on which laboratory performs the analysis, limiting the accessibility and practicality of easily obtaining these tests.27 Although there may be inaccuracies for standard ferritin or TfS under ECCO guidelines, proposed alternatives have their own limitations, which may make ferritin and TfS the most reasonable evaluations of iron status as long as disease activity status at the time of testing is taken into consideration.
Treatment—Treatment of underlying iron deficiency in patients with IBD requires reversing the cause of the deficiency and supplementing iron. In patients with IBD, the options to supplement iron may be limited by active disease, making oral intake less effective. Oral iron supplementation also is associated with notable GI adverse effects that may be exacerbated in patients with IBD. A systematic review of 43 randomized controlled trials (RCTs) evaluating GI adverse effects (eg, nausea, abdominal pain, diarrhea, constipation, and black or tarry stools) of oral ferrous sulfate compared with placebo or intravenous (IV) iron supplementation in healthy nonanemic individuals found a significant increase in GI adverse effects with oral supplementation (placebo: OR=2.32; P<.0001; IV: OR=3.05; P<.0001).28
Therefore, IV iron repletion may be necessary in patients with IBD and may require numerous infusions depending on the formulation of iron. In an RCT conducted in 2011, patients with iron-deficiency anemia with quiescent or mild to moderate IBD were treated with either IV iron sulfate or ferric carboxymaltose.29 With a primary end point of hemoglobin response greater than 2 g/dL, the authors found that 150 of 240 patients responded to ferric carboxymaltose vs 118 of 235 treated with iron sulfate (P=.004). The dosing for ferric carboxymaltose was 1 to 3 infusions of 500 to 1000 mg of iron and for iron sulfate up to 11 infusions of 200 mg of iron.29
Zinc
A systematic review of zinc deficiency in patients with IBD identified 7 studies including 2413 patients and revealed those with Crohn disease had a higher prevalence of zinc deficiency compared with patients with ulcerative colitis (54% vs 41%).30
Pathophysiology—Zinc serves as a catalytic cofactor for enzymatic activity within proteins and immune cells.31 The homeostasis of zinc is tightly regulated within the brush border of the small intestine by zinc transporters ZIP4 and ZIP1 from the lumen of enterocytes into the bloodstream.32 Inflammation in the small intestine due to Crohn disease can result in zinc malabsorption.
Ranaldi et al33 exposed intestinal cells and zinc-depleted intestinal cells to tumor necrosis factor α media to simulate an inflammatory environment. They measured transepithelial electrical resistance as a surrogate for transmembrane permeability and found that zinc-depleted cells had a statistically significantly higher transepithelial electrical resistance percentage (60% reduction after 4 hours; P<1.10–6) when exposed to tumor necrosis factor α signaling compared with normal intestinal cells. They concluded that zinc deficiency can increase intestinal permeability in the presence of inflammation, creating a cycle of further nutrient malabsorption and inflammation exacerbating IBD symptoms.33
Cutaneous Manifestations—After absorption in the small intestine, approximately 5% of zinc resides in the skin, with the highest concentration in the stratum spinosum.34 A cell study found that keratinocytes in zinc-deficient environments had higher rates of apoptosis compared with cells in normal media. The authors proposed that this higher rate of apoptosis and the resulting inflammation could be a mechanism for developing the desquamative or eczematous scaly plaques that are common cutaneous manifestations of zinc deficiency.35
Other cutaneous findings may include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.36 The histopathology of these skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.37
Diagnosis and Monitoring—Assessing serum zinc levels is challenging, as they may decrease during states of inflammation.38 A mouse model study showed a 3.1-fold increase (P<.001) in ZIP14 expression in wild-type mice compared with an IL-6 -/- knock-down model after IL-6 exposure. The authors concluded that the upregulation of ZIP14 in the liver due to inflammatory cytokine upregulation decreases zinc availability in serum.39 Additionally, serum zinc can overestimate the level of deficiency in IBD because approximately 75% of serum zinc is bound to albumin, which decreases in the setting of inflammation.40-42
Alternatively, alkaline phosphatase (AP), a zinc-dependent metalloenzyme, may be a better evaluator of zinc status during periods of inflammation. A study in rats evaluated zinc through serum zinc levels and AP levels after a period of induced stress to mimic a short-term inflammatory state.43 The researchers found that total body stores of zinc were unaffected throughout the experiment; only serum zinc declined throughout the experiment duration while AP did not. Because approximately 75% of serum zinc is bound to serum albumin,42 the researchers concluded the induced inflammatory state depleted serum albumin and redistributed zinc to the liver, causing the observed serum zinc changes, while total body zinc levels and AP were largely unaffected in comparison.43 Comorbid conditions such as liver or bone disease can increase AP levels, which limits the utility of AP as a surrogate for zinc in patients with comorbidities.44 However, even in the context of active IBD, serum zinc still is currently considered the best biomarker to evaluate zinc status.45
Treatment—The recommended dose for zinc supplementation is 20 to 40 mg daily with higher doses (>50 mg/d) for patients with malabsorptive syndromes such as IBD.46 It can be administered orally or parenterally. Although rare, zinc replacement therapy may be associated with diarrhea, nausea, vomiting, mild headaches, and fatigue.46 Additional considerations should be taken when repleting other micronutrients with zinc, as calcium and folate can inhibit zinc reabsorption, while zinc itself can inhibit iron and copper reabsorption.47
Vitamin D and Calcium
Low vitamin D levels (<50 nmol/L) and hypocalcemia (<8.8 mg/dL) are common in patients with IBD.48,49
Pathophysiology—Vitamin D levels are maintained via 2 mechanisms. The first mechanism is through the skin, as keratinocytes produce 7-dehydrocholesterol after exposure to UV light, which is converted into previtamin D3 and then thermally isomerizes into vitamin D3. This vitamin D3 is then transported to the liver on vitamin D–binding protein.50 The second mechanism is through oral vitamin D3 that is absorbed through vitamin D receptors in intestinal epithelium and transported to the liver, where it is hydroxylated into 25-hydroxyvitamin D (25[OH]D), then to the kidneys for hydroxylation to 1,25(OH)2D for redistribution throughout the body.50 This activated form of vitamin D regulates calcium absorption in the intestine, and optimal vitamin D levels are necessary to absorb calcium efficiently.51 Inflammation from IBD within the small intestine can downregulate vitamin D receptors, causing malabsorption and decreased serum vitamin D.52
Vitamin D signaling also is vital to maintaining the tight junctions and adherens junctions of the intestinal epithelium. Weakening the permeability of the epithelium further exacerbates malabsorption and subsequent vitamin D deficiency.52 A meta-analysis of 27 studies including 8316 patients with IBD showed low vitamin D levels were associated with increased odds of disease activity (OR=1.53; 95% CI, 1.32-1.77), mucosal inflammation (OR=1.25; 95% CI, 1.06-1.47), and future clinical relapse (OR=1.23; 95% CI, 1.03-1.47) in patients with Crohn disease. The authors concluded that low levels of vitamin D could be used as a potential biomarker of inflammatory status in Crohn disease.53
Vitamin D and calcium are further implicated in maintaining skeletal health,47 while vitamin D specifically helps maintain intestinal homeostasis54 and immune system modulation in the skin.55
Cutaneous Manifestations—Vitamin D is thought to play crucial roles in skin differentiation and proliferation, cutaneous innate immunity, hair follicle cycling, photoprotection, and wound healing.56 Vitamin D deficiency has been observed in a large range of cutaneous diseases including skin cancer, psoriasis, vitiligo, bullous pemphigoid, atopic dermatitis, and various types of alopecia.56-59 It is unclear whether vitamin D deficiency facilitates these disease processes or is merely the consequence of a disrupted cutaneous surface with the inability to complete the first step in vitamin D processing. A 2014 meta-analysis of 290 prospective cohort studies and 172 randomized trials concluded that 25(OH)D deficiency was associated with ill health and did not find causal evidence for any specific disease, dermatologic or otherwise.60 Calcium deficiency may cause epidermal changes including dry skin, coarse hair, and brittle nails.61
Diagnosis and Monitoring—The ECCO guidelines recommend obtaining serum 25(OH)D levels every 3 months in patients with IBD.62 Levels less than 75 nmol/L are considered deficient, and a value less than 30 nmol/L increases the risk for osteomalacia and nutritional rickets, constituting severe vitamin D deficiency.63-65
An observational study of 325 patients with IBD showed a statistically significant negative correlation between serum vitamin D and fecal calprotectin (r=−0.19; P<.001), a stool-based marker for gut inflammation, supporting vitamin D as a potential biomarker in IBD.66
Evaluation of calcium can be done through serum levels in patients with IBD.67 Patients with IBD are at risk for hypoalbuminemia; therefore, consideration should be taken to ensure calcium levels are corrected, as approximately 50% of calcium is bound to albumin or other ions in the body,68 which can be done by adjusting the calcium concentration by 0.02 mmol/L for every 1 g/L of albumin above or below 40 g/L. In the most critically ill patients, a direct ionized calcium blood level should be used instead because the previously mentioned correction calculations are inaccurate when albumin is critically low.69
Treatment—The ECCO guidelines recommend calcium and vitamin D repletion of 500 to 1000 mg and 800 to 1000 U, respectively, in patients with IBD on systemic corticosteroids to prevent the negative effects of bone loss.62 Calcium repletion in patients with IBD who are not on systemic steroids are the same as for the general population.65
Vitamin D repletion also may help decrease IBD activity. In a prospective study, 10,000 IU/d of vitamin D in 10 patients with IBD—adjusted over 12 weeks to a target of 100 to 125 nmol/L of serum 25(OH)D—showed a significant reduction in clinical Crohn activity (P=.019) over the study period.70 In contrast, 2000 IU/d for 3 months in an RCT of 27 patients with Crohn disease found significantly lower CRP (P=.019) and significantly higher self-reported quality of life (P=.037) but nonsignificant decreases in Crohn activity (P=.082) in patients with 25(OH)D levels of 75 nmol/L or higher compared with those with 25(OH)D levels less than 75 nmol/L.71
These discrepancies illustrate the need for expanded clinical trials to elucidate the optimal vitamin D dosing for patients with IBD. Ultimately, assessing vitamin D and calcium status and considering repletion in patients with IBD, especially those with comorbid dermatologic diseases such as poor wound healing, psoriasis, or atopic dermatitis, is important.
Vitamin B6 (Pyridoxine)
Pathophysiology—Pyridoxine is an important coenzyme for many functions including amino acid transamination, fatty acid metabolism, and conversion of tryptophan to niacin. It is absorbed in the jejunum and ileum and subsequently transported to the liver for rephosphorylation and release into its active form.36 An observational study assessing the nutritional status of patients with IBD found that only 5.7% of 105 patients with food records had inadequate dietary intake of pyridoxine, but 29% of all patients with IBD had subnormal pyridoxine levels.72 Additionally, they found no significant difference in the prevalence of subnormal pyridoxine levels in patients with active IBD vs IBD in remission. The authors suggested that the subnormal pyridoxine levels in patients with IBD likely were multifactorial and resulted from malabsorption due to active disease, inflammation, and inadequate intake.72
Cutaneous Manifestations—Cutaneous findings associated with pyridoxine deficiency include periorificial and perineal dermatitis,73 angular stomatitis, and cheilitis with associated burning, redness, and tongue edema.36 Additionally, pyridoxine is involved in the conversion of tryptophan to niacin, and its deficiency may manifest with pellagralike findings.74
Because pyridoxine is critical to protein metabolism, its deficiency may disrupt key cellular structures that rely on protein concentrations to maintain structural integrity. One such structure in the skin that heavily relies on protein concentrations is the ground substance of the extracellular matrix—the amorphous gelatinous spaces that occupy the areas between the extracellular matrix, which consists of cross-linked glycosaminoglycans and proteins.75 Without protein, ground substance increases in viscosity and can disrupt the epidermal barrier, leading to increased transepidermal water loss and ultimately inflammation.76 Although this theory has yet to be validated fully, this is a potential mechanistic explanation for the inflammation in dermal papillae that leads to dermatitis observed in pyridoxine deficiency.
Diagnosis and Monitoring—Direct biomarkers of pyridoxine status are in serum, plasma, erythrocytes, and urine, with the most common measurement in plasma as pyridoxal 5′-phosphate (PLP).77 Plasma PLP concentrations lower than 20 nmol/L are suggestive of deficiency.78 Plasma PLP has shown inverse relationships with acute phase inflammatory markers CRP79 and AP,78 thereby raising concerns for its validity to assess pyridoxine status in patients with symptomatic IBD.80
Alternative evaluations of pyridoxine include tryptophan and methionine loading tests,36 which are measured via urinary excretion and require normal kidney function to be accurate. They should be considered in IBD if necessary, but routine testing, even in patients with symptomatic IBD, is not recommended in the ECCO guidelines. Additional considerations should be taken in patients with altered nutrient requirements such as those who have undergone bowel resection due to highly active disease or those who receive parenteral nutritional supplementation.81
Treatment—Recommendations for oral pyridoxine supplementation range from 25 to 600 mg daily,82 with symptoms typically improving on 100 mg daily.36 Pyridoxine supplementation may have additional benefits for patients with IBD and potentially modulate disease severity. An IL-10 knockout mouse supplemented with pyridoxine had an approximately 60% reduction (P<.05) in inflammation compared to mice deficient in pyridoxine.83 The authors suggest that PLP-dependent enzymes can inhibit further proinflammatory signaling and T-cell migration that can exacerbate IBD. Ultimately, more data is needed before determining the efficacy of pyridoxine supplementation for active IBD.
Vitamin B12 and Vitamin B9 (Folic Acid)
Pathophysiology—Vitamin B12 is reabsorbed in the terminal ileum, the distal portion of the small intestine. The American Gastroenterological Association recommends that patients with a history of extensive ileal disease or prior ileal surgery, which is the case for many patients with Crohn disease, be monitored for vitamin B12 deficiency.23 Monitoring and rapid supplementation of vitamin B12 can prevent pernicious anemia and irreversible neurologic damage that may result from deficiency.84
Folic acid is primarily absorbed in the duodenum and jejunum of the small intestine. A meta-analysis performed in 2017 assessed studies observing folic acid and vitamin B12 levels in 1086 patients with IBD compared with 1484 healthy controls and found an average difference in serum folate concentration of 0.46 nmol/L (P<.001).84 Interestingly, this study did not find a significant difference in serum vitamin B12 levels between patients with IBD and healthy controls, highlighting the mechanism of vitamin B12 deficiency in IBD because only patients with terminal ileal involvement are at risk for malabsorption and subsequent deficiency.
Cutaneous Manifestations—Both vitamin B12 and folic acid deficiency can manifest as cheilitis, glossitis, and/or generalized hyperpigmentation that is accentuated in the flexural areas, palms, soles, and oral cavity.85,86 Systemic symptoms of patients with vitamin B12 and folic acid deficiency include megaloblastic anemia, pallor, and fatigue. A potential mechanism for the hyperpigmentation observed from vitamin B12 deficiency came from an electron microscope study that showed an increased concentration of melanosomes in a patient with deficiency.87
Diagnosis and Monitoring—In patients with suspected vitamin B12 and/or folic acid deficiency, initial evaluation should include a CBC with peripheral smear and serum vitamin B12 and folate levels. In cases for which the diagnosis still is unclear after initial testing,
Treatment—According to the Centers for Disease Control and Prevention, supplementation of vitamin B12 can be done orally with 1000 µg daily in patients with deficiency. In patients with active IBD, oral reabsorption of vitamin B12 can be less effective, making subcutaneous or intramuscular administration (1000 µg/wk for 8 weeks, then monthly for life) better options.89
Patients with IBD managed with methotrexate should be screened carefully for folate deficiency. Methotrexate is a folate analog that sometimes is used for the treatment of IBD. Reversible competitive inhibition of dihydrofolate reductase can precipitate a systemic folic acid decrease.91 Typically, oral folic acid (1 to 5 mg/d) is sufficient to treat folate deficiency, with the ESPEN recommending 5 mg once weekly 24 to 72 hours after methotrexate treatment or 1 mg daily for 5 days per week in patients with IBD.1 Alternative formulations—IV, subcutaneous, or intramuscular—are available for patients who cannot tolerate oral intake.92
Final Thoughts
Dermatologists can be the first to observe the cutaneous manifestations of micronutrient deficiencies. Although the symptoms of each micronutrient deficiency discussed may overlap, attention to small clinical clues in patients with IBD can improve patient outcomes and quality of life. For example, koilonychia with glossitis and xerosis likely is due to iron deficiency, while zinc deficiency should be suspected in patients with scaly eczematous plaques in skin folds. A high level of suspicion for micronutrient deficiencies in patients with IBD should be followed by a complete patient history, review of systems, and thorough clinical examination. A thorough laboratory evaluation can pinpoint nutritional deficiencies in patients with IBD, keeping in mind that specific biomarkers such as ferritin and serum zinc also act as acute phase reactants and should be interpreted in this context. Co-management with gastroenterologists should be a priority in patients with IBD, as gaining control of inflammatory disease is crucial for the prevention of recurrent vitamin and micronutrient deficiencies in addition to long-term health in this population.
In 2023, ESPEN (the European Society for Clinical Nutrition and Metabolism) published consensus recommendations highlighting the importance of regular monitoring and treatment of nutrient deficiencies in patients with inflammatory bowel disease (IBD) for improved prognosis, mortality, and quality of life.1 Suboptimal nutrition in patients with IBD predominantly results from inflammation of the gastrointestinal (GI) tract leading to malabsorption; however, medications commonly used to manage IBD also can contribute to malnutrition.2,3 Additionally, patients may develop nausea and food avoidance due to medication or the disease itself, leading to nutritional withdrawal and eventual deficiency.4 Even with the development of diets focused on balancing nutritional needs and decreasing inflammation,5 offsetting this aversion to food can be difficult to overcome.2
Cutaneous manifestations of IBD are multifaceted and can be secondary to the disease, reactive to or associated with IBD, or effects from nutritional deficiencies. The most common vitamin and nutrient deficiencies in patients with IBD include iron; zinc; calcium; vitamin D; and vitamins B6 (pyridoxine), B9 (folic acid), and B12.6 Malnutrition may manifest with cutaneous disease, and dermatologists can be the first to identify and assess for nutritional deficiencies. In this article, we review the mechanisms of these micronutrient depletions in the context of IBD, their subsequent dermatologic manifestations (Table), and treatment and monitoring guidelines for each deficiency.
Iron
A systematic review conducted from 2007 to 2012 in European patients with IBD (N=2192) found the overall prevalence of anemia in this population to be 24% (95% CI, 18%-31%), with 57% of patients with anemia experiencing iron deficiency.7 Anemia is observed more commonly in patients hospitalized with IBD and is common in patients with both Crohn disease and ulcerative colitis.8
Pathophysiology—Iron is critically important in oxygen transportation throughout the body as a major component of hemoglobin. Physiologically, the low pH of the duodenum and proximal jejunum allows divalent metal transporter 1 to transfer dietary Fe3+ into enterocytes, where it is reduced to the transportable Fe2+.9,10 Distribution of Fe2+ ions from enterocytes relies on ferroportin, an iron-transporting protein, which is heavily regulated by the protein hepcidin.11 Hepcidin, a known acute phase reactant, will increase in the setting of active IBD, causing a depletion of ferroportin and an inability of the body to utilize the stored iron in enterocytes.12 This poor utilization of iron stores combined with blood loss caused by inflammation in the GI tract is the proposed primary mechanism of iron-deficiency anemia observed in patients with IBD.13
Cutaneous Manifestations—From a dermatologic perspective, iron-deficiency anemia can manifest with a wide range of symptoms including glossitis, koilonychia, xerosis and/or pruritus, and brittle hair or hair loss.14,15 Although the underlying pathophysiology of these cutaneous manifestations is not fully understood, there are several theories assessing the mechanisms behind the skin findings of iron deficiency.
Atrophic glossitis has been observed in many patients with iron deficiency and is thought to manifest due to low iron concentrations in the blood, thereby decreasing oxygen delivery to the papillae of the dorsal tongue with resultant atrophy.16,17 Similarly, decreased oxygen delivery to the nail bed capillaries may cause deformities in the nail called koilonychia (or “spoon nails”).18 Iron is a key co-factor in collagen lysyl hydroxylase that promotes collagen binding; iron deficiency may lead to disruptions in the epidermal barrier that can cause pruritus and xerosis.19 An observational study of 200 healthy patients with a primary concern of pruritus found a correlation between low serum ferritin and a higher degree of pruritus (r=−0.768; P<.00001).20
Evidence for iron’s role in hair growth comes from a mouse model study with a mutation in the serine protease TMPRSS6—a protein that regulates hepcidin and iron absorption—which caused an increase in hepcidin production and subsequent systemic iron deficiency. Mice at 4 weeks of age were devoid of all body hair but had substantial regrowth after initiation of a 2-week iron-rich diet, which suggests a connection between iron repletion and hair growth in mice with iron deficiency.21 Additionally, a meta-analysis analyzing the comorbidities of patients with alopecia areata found them to have higher odds (odds ratio [OR]=2.78; 95% CI, 1.23-6.29) of iron-deficiency anemia but no association with IBD (OR=1.48; 95% CI, 0.32-6.82).22
Diagnosis and Monitoring—The American Gastroenterological Association recommends a complete blood cell count (CBC), serum ferritin, transferrin saturation (TfS), and C-reactive protein (CRP) as standard evaluations for iron deficiency in patients with IBD. Patients with active IBD should be screened every 3 months,and patients with inactive disease should be screened every 6 to 12 months.23
Although ferritin and TfS often are used as markers for iron status in healthy individuals, they are positive and negative acute phase reactants, respectively. Using them to assess iron status in patients with IBD may inaccurately represent iron status in the setting of inflammation from the disease.24 The European Crohn’s and Colitis Organisation (ECCO) produced guidelines to define iron deficiency as a TfS less than 20% or a ferritin level less than 30 µg/L in patients without evidence of active IBD and a ferritin level less than 100 µg/L for patients with active inflammation.25
A 2020 multicenter observational study of 202 patients with diagnosed IBD found that the ECCO guideline of ferritin less than 30 µg/L had an area under the receiver operating characteristic (AUROC) curve of 0.69, a sensitivity of 0.43, and a specificity of 0.95 in their population.26 In a sensitivity analysis stratifying patients by CRP level (<10 or ≥10 mg/L), the authors found that for patients with ulcerative colitis and a CRP less than 10 mg/L, a cut-off value of ferritin less than 65 µg/L (AUROC=0.78) had a sensitivity of 0.78 and specificity of 0.76, and a TfS value of less than 16% (AUROC=0.88) had a sensitivity of 0.79 and a specificity of 0.9. In patients with a CRP of 10 mg/L or greater, a cut-off value of ferritin 80 µg/L (AUROC=0.76) had a sensitivity of 0.75 and a specificity of 0.82, and a TfS value of less than 11% (AUROC=0.69) had a sensitivity of 0.79 and a specificity of 0.88. There were no ferritin cut-off values associated with good diagnostic performance (defined as both sensitivity and specificity >0.70) for iron deficiency in patients with Crohn disease.26
The authors recommended using an alternative iron measurement such as soluble transferrin receptor (sTfR)/log ferritin ratio (TfR-F) that is not influenced by active inflammation and has a good correlation with ferritin values (TfR-F: r=0.66; P<.001).26 However, both sTfR and TfR-F have high costs and intermethod variability as well as differences in their reference ranges depending on which laboratory performs the analysis, limiting the accessibility and practicality of easily obtaining these tests.27 Although there may be inaccuracies for standard ferritin or TfS under ECCO guidelines, proposed alternatives have their own limitations, which may make ferritin and TfS the most reasonable evaluations of iron status as long as disease activity status at the time of testing is taken into consideration.
Treatment—Treatment of underlying iron deficiency in patients with IBD requires reversing the cause of the deficiency and supplementing iron. In patients with IBD, the options to supplement iron may be limited by active disease, making oral intake less effective. Oral iron supplementation also is associated with notable GI adverse effects that may be exacerbated in patients with IBD. A systematic review of 43 randomized controlled trials (RCTs) evaluating GI adverse effects (eg, nausea, abdominal pain, diarrhea, constipation, and black or tarry stools) of oral ferrous sulfate compared with placebo or intravenous (IV) iron supplementation in healthy nonanemic individuals found a significant increase in GI adverse effects with oral supplementation (placebo: OR=2.32; P<.0001; IV: OR=3.05; P<.0001).28
Therefore, IV iron repletion may be necessary in patients with IBD and may require numerous infusions depending on the formulation of iron. In an RCT conducted in 2011, patients with iron-deficiency anemia with quiescent or mild to moderate IBD were treated with either IV iron sulfate or ferric carboxymaltose.29 With a primary end point of hemoglobin response greater than 2 g/dL, the authors found that 150 of 240 patients responded to ferric carboxymaltose vs 118 of 235 treated with iron sulfate (P=.004). The dosing for ferric carboxymaltose was 1 to 3 infusions of 500 to 1000 mg of iron and for iron sulfate up to 11 infusions of 200 mg of iron.29
Zinc
A systematic review of zinc deficiency in patients with IBD identified 7 studies including 2413 patients and revealed those with Crohn disease had a higher prevalence of zinc deficiency compared with patients with ulcerative colitis (54% vs 41%).30
Pathophysiology—Zinc serves as a catalytic cofactor for enzymatic activity within proteins and immune cells.31 The homeostasis of zinc is tightly regulated within the brush border of the small intestine by zinc transporters ZIP4 and ZIP1 from the lumen of enterocytes into the bloodstream.32 Inflammation in the small intestine due to Crohn disease can result in zinc malabsorption.
Ranaldi et al33 exposed intestinal cells and zinc-depleted intestinal cells to tumor necrosis factor α media to simulate an inflammatory environment. They measured transepithelial electrical resistance as a surrogate for transmembrane permeability and found that zinc-depleted cells had a statistically significantly higher transepithelial electrical resistance percentage (60% reduction after 4 hours; P<1.10–6) when exposed to tumor necrosis factor α signaling compared with normal intestinal cells. They concluded that zinc deficiency can increase intestinal permeability in the presence of inflammation, creating a cycle of further nutrient malabsorption and inflammation exacerbating IBD symptoms.33
Cutaneous Manifestations—After absorption in the small intestine, approximately 5% of zinc resides in the skin, with the highest concentration in the stratum spinosum.34 A cell study found that keratinocytes in zinc-deficient environments had higher rates of apoptosis compared with cells in normal media. The authors proposed that this higher rate of apoptosis and the resulting inflammation could be a mechanism for developing the desquamative or eczematous scaly plaques that are common cutaneous manifestations of zinc deficiency.35
Other cutaneous findings may include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.36 The histopathology of these skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.37
Diagnosis and Monitoring—Assessing serum zinc levels is challenging, as they may decrease during states of inflammation.38 A mouse model study showed a 3.1-fold increase (P<.001) in ZIP14 expression in wild-type mice compared with an IL-6 -/- knock-down model after IL-6 exposure. The authors concluded that the upregulation of ZIP14 in the liver due to inflammatory cytokine upregulation decreases zinc availability in serum.39 Additionally, serum zinc can overestimate the level of deficiency in IBD because approximately 75% of serum zinc is bound to albumin, which decreases in the setting of inflammation.40-42
Alternatively, alkaline phosphatase (AP), a zinc-dependent metalloenzyme, may be a better evaluator of zinc status during periods of inflammation. A study in rats evaluated zinc through serum zinc levels and AP levels after a period of induced stress to mimic a short-term inflammatory state.43 The researchers found that total body stores of zinc were unaffected throughout the experiment; only serum zinc declined throughout the experiment duration while AP did not. Because approximately 75% of serum zinc is bound to serum albumin,42 the researchers concluded the induced inflammatory state depleted serum albumin and redistributed zinc to the liver, causing the observed serum zinc changes, while total body zinc levels and AP were largely unaffected in comparison.43 Comorbid conditions such as liver or bone disease can increase AP levels, which limits the utility of AP as a surrogate for zinc in patients with comorbidities.44 However, even in the context of active IBD, serum zinc still is currently considered the best biomarker to evaluate zinc status.45
Treatment—The recommended dose for zinc supplementation is 20 to 40 mg daily with higher doses (>50 mg/d) for patients with malabsorptive syndromes such as IBD.46 It can be administered orally or parenterally. Although rare, zinc replacement therapy may be associated with diarrhea, nausea, vomiting, mild headaches, and fatigue.46 Additional considerations should be taken when repleting other micronutrients with zinc, as calcium and folate can inhibit zinc reabsorption, while zinc itself can inhibit iron and copper reabsorption.47
Vitamin D and Calcium
Low vitamin D levels (<50 nmol/L) and hypocalcemia (<8.8 mg/dL) are common in patients with IBD.48,49
Pathophysiology—Vitamin D levels are maintained via 2 mechanisms. The first mechanism is through the skin, as keratinocytes produce 7-dehydrocholesterol after exposure to UV light, which is converted into previtamin D3 and then thermally isomerizes into vitamin D3. This vitamin D3 is then transported to the liver on vitamin D–binding protein.50 The second mechanism is through oral vitamin D3 that is absorbed through vitamin D receptors in intestinal epithelium and transported to the liver, where it is hydroxylated into 25-hydroxyvitamin D (25[OH]D), then to the kidneys for hydroxylation to 1,25(OH)2D for redistribution throughout the body.50 This activated form of vitamin D regulates calcium absorption in the intestine, and optimal vitamin D levels are necessary to absorb calcium efficiently.51 Inflammation from IBD within the small intestine can downregulate vitamin D receptors, causing malabsorption and decreased serum vitamin D.52
Vitamin D signaling also is vital to maintaining the tight junctions and adherens junctions of the intestinal epithelium. Weakening the permeability of the epithelium further exacerbates malabsorption and subsequent vitamin D deficiency.52 A meta-analysis of 27 studies including 8316 patients with IBD showed low vitamin D levels were associated with increased odds of disease activity (OR=1.53; 95% CI, 1.32-1.77), mucosal inflammation (OR=1.25; 95% CI, 1.06-1.47), and future clinical relapse (OR=1.23; 95% CI, 1.03-1.47) in patients with Crohn disease. The authors concluded that low levels of vitamin D could be used as a potential biomarker of inflammatory status in Crohn disease.53
Vitamin D and calcium are further implicated in maintaining skeletal health,47 while vitamin D specifically helps maintain intestinal homeostasis54 and immune system modulation in the skin.55
Cutaneous Manifestations—Vitamin D is thought to play crucial roles in skin differentiation and proliferation, cutaneous innate immunity, hair follicle cycling, photoprotection, and wound healing.56 Vitamin D deficiency has been observed in a large range of cutaneous diseases including skin cancer, psoriasis, vitiligo, bullous pemphigoid, atopic dermatitis, and various types of alopecia.56-59 It is unclear whether vitamin D deficiency facilitates these disease processes or is merely the consequence of a disrupted cutaneous surface with the inability to complete the first step in vitamin D processing. A 2014 meta-analysis of 290 prospective cohort studies and 172 randomized trials concluded that 25(OH)D deficiency was associated with ill health and did not find causal evidence for any specific disease, dermatologic or otherwise.60 Calcium deficiency may cause epidermal changes including dry skin, coarse hair, and brittle nails.61
Diagnosis and Monitoring—The ECCO guidelines recommend obtaining serum 25(OH)D levels every 3 months in patients with IBD.62 Levels less than 75 nmol/L are considered deficient, and a value less than 30 nmol/L increases the risk for osteomalacia and nutritional rickets, constituting severe vitamin D deficiency.63-65
An observational study of 325 patients with IBD showed a statistically significant negative correlation between serum vitamin D and fecal calprotectin (r=−0.19; P<.001), a stool-based marker for gut inflammation, supporting vitamin D as a potential biomarker in IBD.66
Evaluation of calcium can be done through serum levels in patients with IBD.67 Patients with IBD are at risk for hypoalbuminemia; therefore, consideration should be taken to ensure calcium levels are corrected, as approximately 50% of calcium is bound to albumin or other ions in the body,68 which can be done by adjusting the calcium concentration by 0.02 mmol/L for every 1 g/L of albumin above or below 40 g/L. In the most critically ill patients, a direct ionized calcium blood level should be used instead because the previously mentioned correction calculations are inaccurate when albumin is critically low.69
Treatment—The ECCO guidelines recommend calcium and vitamin D repletion of 500 to 1000 mg and 800 to 1000 U, respectively, in patients with IBD on systemic corticosteroids to prevent the negative effects of bone loss.62 Calcium repletion in patients with IBD who are not on systemic steroids are the same as for the general population.65
Vitamin D repletion also may help decrease IBD activity. In a prospective study, 10,000 IU/d of vitamin D in 10 patients with IBD—adjusted over 12 weeks to a target of 100 to 125 nmol/L of serum 25(OH)D—showed a significant reduction in clinical Crohn activity (P=.019) over the study period.70 In contrast, 2000 IU/d for 3 months in an RCT of 27 patients with Crohn disease found significantly lower CRP (P=.019) and significantly higher self-reported quality of life (P=.037) but nonsignificant decreases in Crohn activity (P=.082) in patients with 25(OH)D levels of 75 nmol/L or higher compared with those with 25(OH)D levels less than 75 nmol/L.71
These discrepancies illustrate the need for expanded clinical trials to elucidate the optimal vitamin D dosing for patients with IBD. Ultimately, assessing vitamin D and calcium status and considering repletion in patients with IBD, especially those with comorbid dermatologic diseases such as poor wound healing, psoriasis, or atopic dermatitis, is important.
Vitamin B6 (Pyridoxine)
Pathophysiology—Pyridoxine is an important coenzyme for many functions including amino acid transamination, fatty acid metabolism, and conversion of tryptophan to niacin. It is absorbed in the jejunum and ileum and subsequently transported to the liver for rephosphorylation and release into its active form.36 An observational study assessing the nutritional status of patients with IBD found that only 5.7% of 105 patients with food records had inadequate dietary intake of pyridoxine, but 29% of all patients with IBD had subnormal pyridoxine levels.72 Additionally, they found no significant difference in the prevalence of subnormal pyridoxine levels in patients with active IBD vs IBD in remission. The authors suggested that the subnormal pyridoxine levels in patients with IBD likely were multifactorial and resulted from malabsorption due to active disease, inflammation, and inadequate intake.72
Cutaneous Manifestations—Cutaneous findings associated with pyridoxine deficiency include periorificial and perineal dermatitis,73 angular stomatitis, and cheilitis with associated burning, redness, and tongue edema.36 Additionally, pyridoxine is involved in the conversion of tryptophan to niacin, and its deficiency may manifest with pellagralike findings.74
Because pyridoxine is critical to protein metabolism, its deficiency may disrupt key cellular structures that rely on protein concentrations to maintain structural integrity. One such structure in the skin that heavily relies on protein concentrations is the ground substance of the extracellular matrix—the amorphous gelatinous spaces that occupy the areas between the extracellular matrix, which consists of cross-linked glycosaminoglycans and proteins.75 Without protein, ground substance increases in viscosity and can disrupt the epidermal barrier, leading to increased transepidermal water loss and ultimately inflammation.76 Although this theory has yet to be validated fully, this is a potential mechanistic explanation for the inflammation in dermal papillae that leads to dermatitis observed in pyridoxine deficiency.
Diagnosis and Monitoring—Direct biomarkers of pyridoxine status are in serum, plasma, erythrocytes, and urine, with the most common measurement in plasma as pyridoxal 5′-phosphate (PLP).77 Plasma PLP concentrations lower than 20 nmol/L are suggestive of deficiency.78 Plasma PLP has shown inverse relationships with acute phase inflammatory markers CRP79 and AP,78 thereby raising concerns for its validity to assess pyridoxine status in patients with symptomatic IBD.80
Alternative evaluations of pyridoxine include tryptophan and methionine loading tests,36 which are measured via urinary excretion and require normal kidney function to be accurate. They should be considered in IBD if necessary, but routine testing, even in patients with symptomatic IBD, is not recommended in the ECCO guidelines. Additional considerations should be taken in patients with altered nutrient requirements such as those who have undergone bowel resection due to highly active disease or those who receive parenteral nutritional supplementation.81
Treatment—Recommendations for oral pyridoxine supplementation range from 25 to 600 mg daily,82 with symptoms typically improving on 100 mg daily.36 Pyridoxine supplementation may have additional benefits for patients with IBD and potentially modulate disease severity. An IL-10 knockout mouse supplemented with pyridoxine had an approximately 60% reduction (P<.05) in inflammation compared to mice deficient in pyridoxine.83 The authors suggest that PLP-dependent enzymes can inhibit further proinflammatory signaling and T-cell migration that can exacerbate IBD. Ultimately, more data is needed before determining the efficacy of pyridoxine supplementation for active IBD.
Vitamin B12 and Vitamin B9 (Folic Acid)
Pathophysiology—Vitamin B12 is reabsorbed in the terminal ileum, the distal portion of the small intestine. The American Gastroenterological Association recommends that patients with a history of extensive ileal disease or prior ileal surgery, which is the case for many patients with Crohn disease, be monitored for vitamin B12 deficiency.23 Monitoring and rapid supplementation of vitamin B12 can prevent pernicious anemia and irreversible neurologic damage that may result from deficiency.84
Folic acid is primarily absorbed in the duodenum and jejunum of the small intestine. A meta-analysis performed in 2017 assessed studies observing folic acid and vitamin B12 levels in 1086 patients with IBD compared with 1484 healthy controls and found an average difference in serum folate concentration of 0.46 nmol/L (P<.001).84 Interestingly, this study did not find a significant difference in serum vitamin B12 levels between patients with IBD and healthy controls, highlighting the mechanism of vitamin B12 deficiency in IBD because only patients with terminal ileal involvement are at risk for malabsorption and subsequent deficiency.
Cutaneous Manifestations—Both vitamin B12 and folic acid deficiency can manifest as cheilitis, glossitis, and/or generalized hyperpigmentation that is accentuated in the flexural areas, palms, soles, and oral cavity.85,86 Systemic symptoms of patients with vitamin B12 and folic acid deficiency include megaloblastic anemia, pallor, and fatigue. A potential mechanism for the hyperpigmentation observed from vitamin B12 deficiency came from an electron microscope study that showed an increased concentration of melanosomes in a patient with deficiency.87
Diagnosis and Monitoring—In patients with suspected vitamin B12 and/or folic acid deficiency, initial evaluation should include a CBC with peripheral smear and serum vitamin B12 and folate levels. In cases for which the diagnosis still is unclear after initial testing,
Treatment—According to the Centers for Disease Control and Prevention, supplementation of vitamin B12 can be done orally with 1000 µg daily in patients with deficiency. In patients with active IBD, oral reabsorption of vitamin B12 can be less effective, making subcutaneous or intramuscular administration (1000 µg/wk for 8 weeks, then monthly for life) better options.89
Patients with IBD managed with methotrexate should be screened carefully for folate deficiency. Methotrexate is a folate analog that sometimes is used for the treatment of IBD. Reversible competitive inhibition of dihydrofolate reductase can precipitate a systemic folic acid decrease.91 Typically, oral folic acid (1 to 5 mg/d) is sufficient to treat folate deficiency, with the ESPEN recommending 5 mg once weekly 24 to 72 hours after methotrexate treatment or 1 mg daily for 5 days per week in patients with IBD.1 Alternative formulations—IV, subcutaneous, or intramuscular—are available for patients who cannot tolerate oral intake.92
Final Thoughts
Dermatologists can be the first to observe the cutaneous manifestations of micronutrient deficiencies. Although the symptoms of each micronutrient deficiency discussed may overlap, attention to small clinical clues in patients with IBD can improve patient outcomes and quality of life. For example, koilonychia with glossitis and xerosis likely is due to iron deficiency, while zinc deficiency should be suspected in patients with scaly eczematous plaques in skin folds. A high level of suspicion for micronutrient deficiencies in patients with IBD should be followed by a complete patient history, review of systems, and thorough clinical examination. A thorough laboratory evaluation can pinpoint nutritional deficiencies in patients with IBD, keeping in mind that specific biomarkers such as ferritin and serum zinc also act as acute phase reactants and should be interpreted in this context. Co-management with gastroenterologists should be a priority in patients with IBD, as gaining control of inflammatory disease is crucial for the prevention of recurrent vitamin and micronutrient deficiencies in addition to long-term health in this population.
- Bischoff SC, Bager P, Escher J, et al. ESPEN guideline on clinical nutrition in inflammatory bowel disease. Clin Nutr. 2023;42:352-379. doi:10.1016/j.clnu.2022.12.004
- Gerasimidis K, McGrogan P, Edwards CA. The aetiology and impact of malnutrition in paediatric inflammator y bowel disease. J Hum Nutr Diet. 2011;24:313-326. doi:10.1111/j.1365-277X.2011.01171.x
- Mentella MC, Scaldaferri F, Pizzoferrato M, et al. Nutrition, IBD and gut microbiota: a review. Nutrients. 2020;12:944. doi:10.3390/nu12040944
- Bonsack O, Caron B, Baumann C, et al. Food avoidance and fasting in patients with inflammatory bowel disease: experience from the Nancy IBD nutrition clinic. United European Gastroenterol J. 2023;11:361-370. doi:10.1002/ueg2.1238521
- Campmans-Kuijpers MJE, Dijkstra G. Food and food groups in inflammatory bowel disease (IBD): the design of the Groningen Anti-Inflammatory Diet (GrAID). Nutrients. 2021;13:1067. doi:10.3390/nu13041067
- Hwang C, Issokson K, Giguere-Rich C, et al. Development and pilot testing of the inflammatory bowel disease nutrition care pathway. Clin Gastroenterol Hepatol. 2020;18:2645-2649.e4. doi:10.1016/j.cgh.2020.06.039
- Filmann N, Rey J, Schneeweiss S, et al. Prevalence of anemia in inflammatory bowel diseases in European countries: a systematic review and individual patient data meta-analysis. Inflamm Bowel Dis. 2014;20:936-945. doi:10.1097/01.MIB.0000442728.74340.fd
- Stein J, Hartmann F, Dignass AU. Diagnosis and management of iron deficiency anemia in patients with IBD. Nat Rev Gastroenterol Hepatol. 2010;7:599-610. doi:10.1038/nrgastro.2010.151
- Ems T, St Lucia K, Huecker MR. Biochemistry, iron absorption. StatPearls [Internet]. Updated April 17, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448204/
- Evstatiev R, Gasche C. Iron sensing and signalling. Gut. 2012;61:933-952. doi:10.1136/gut.2010.214312
- Przybyszewska J, Zekanowska E. The role of hepcidin, ferroportin, HCP1, and DMT1 protein in iron absorption in the human digestive tract. Prz Gastroenterol. 2014;9:208-213. doi:10.5114/pg.2014.45102
- Weiss G, Gasche C. Pathogenesis and treatment of anemia in inflammatory bowel disease. Haematologica. 2010;95:175-178. doi:10.3324/haematol.2009.017046
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72. doi:10.4291/wjgp.v6.i3.62
- Moiz B. Spoon nails: still seen in today’s world. Clin Case Rep. 2018;6:547-548. doi:10.1002/ccr3.1404
- St Pierre SA, Vercellotti GM, Donovan JC, et al. Iron deficiency and diffuse nonscarring scalp alopecia in women: more pieces to the puzzle. J Am Acad Dermatol. 2010;63:1070-1076. doi:10.1016/j.jaad.2009.05.054
- Chiang CP, Yu-Fong Chang J, Wang YP, et al. Anemia, hematinic deficiencies, hyperhomocysteinemia, and serum gastric parietal cell antibody positivity in atrophic glossitis patients with or without microcytosis. J Formos Med Assoc. 2019;118:1401-1407. doi:10.1016/j.jfma.2019.06.004
- Chiang CP, Chang JY, Wang YP, et al. Atrophic glossitis: Etiology, serum autoantibodies, anemia, hematinic deficiencies, hyperhomocysteinemia, and management. J Formos Med Assoc. 2020;119:774-780. doi:10.1016/j.jfma.2019.04.015
- Walker J, Baran R, Vélez N, et al. Koilonychia: an update on pathophysiology, differential diagnosis and clinical relevance. J Eur Acad Dermatol Venereol. 2016;30:1985-1991. doi:10.1111/jdv.13610
- Guo HF, Tsai CL, Terajima M, et al. Pro-metastatic collagen lysyl hydroxylase dimer assemblies stabilized by Fe2+-binding. Nat Commun. 2018;9:512. doi:10.1038/s41467-018-02859-z
- Saini S, Jain AK, Agarwal S, et al. Iron deficiency and pruritus: a cross-sectional analysis to assess its association and relationship. Indian J Dermatol. 2021;66:705. doi:10.4103/ijd.ijd_326_21
- Du X, She E, Gelbart T, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320:1088-1092. doi:10.1126/science.1157121
- Lee S, Lee H, Lee CH, et al. Comorbidities in alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol. 2019;80:466-477.e16. doi:10.1016/j.jaad.2018.07.013
- Hashash JG, Elkins J, Lewis JD, et al. AGA Clinical Practice Update on diet and nutritional therapies in patients with inflammatory bowel disease: expert review [published online January 23, 2024]. Gastroenterology. doi:10.1053/j.gastro.2023.11.303
- Choudhuri S, Chowdhury IH, Saha A, et al. Acute monocyte pro- inflammatory response predicts higher positive to negative acute phase reactants ratio and severe hemostatic derangement in dengue fever. Cytokine. 2021;146:155644. doi:10.1016/j.cyto.2021.155644
- Dignass AU, Gasche C, Bettenworth D, et al; European Crohn’s and Colitis Organisation. European consensus on the diagnosis and management of iron deficiency and anaemia in inflammatory bowel diseases. J Crohn’s Colitis. 2015;9:211-222. doi:10.1093/ecco-jcc/jju009
- Daude S, Remen T, Chateau T, et al. Comparative accuracy of ferritin, transferrin saturation and soluble transferrin receptor for the diagnosis of iron deficiency in inflammatory bowel disease. Aliment Pharmacol Ther. 2020;51:1087-1095. doi:10.1111/apt.15739
- Pfeiffer CM, Looker AC. Laboratory methodologies for indicators of iron status: strengths, limitations, and analytical challenges. Am J Clin Nutr. 2017;106(suppl 6):1606S-1614S. doi:10.3945/ajcn.117.155887
- Tolkien Z, Stecher L, Mander AP, et al. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: a systematic review and meta-analysis. PLoS One. 2015;10:e0117383. doi:10.1371/journal.pone.0117383
- Evstatiev R, Marteau P, Iqbal T, et al. FERGIcor, a randomized controlled trial on ferric carboxymaltose for iron deficiency anemia in inflammatory bowel disease. Gastroenterology. 2011;141:846-853.e8532. doi:10.1053/j.gastro.2011.06.005
- Zupo R, Sila A, Castellana F, et al. Prevalence of zinc deficiency in inflammatory bowel disease: a systematic review and meta-analysis. Nutrients. 2022;14:4052. doi:10.3390/nu14194052
- Thompson MW. Regulation of zinc-dependent enzymes by metal carrier proteins. Biometals. 2022;35:187-213. doi:10.1007/s10534-022-00373-w
- Maares M, Haase H. A guide to human zinc absorption: general overview and recent advances of in vitro intestinal models. Nutrients. 2020;12:762. doi:10.3390/nu12030762
- Ranaldi G, Ferruzza S, Canali R, et al. Intracellular zinc is required for intestinal cell survival signals triggered by the inflammatory cytokine TNFα. J Nutr Biochem. 2013;24:967-976. doi:10.1016/j.jnutbio.2012.06.020
- Ogawa Y, Kawamura T, Shimada S. Zinc and skin biology. Arch Biochem Biophys. 2016;611:113-119. doi:10.1016/j.abb.2016.06.003
- Wilson D, Varigos G, Ackland ML. Apoptosis may underlie the pathology of zinc-deficient skin. Immunol Cell Biol. 2006;84:28-37. doi:10.1111/j.1440-1711.2005.01391.x
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685. doi:10.1016/j.clindermatol.2010.03.029
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624. doi:10.3390/nu9060624
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U S A. 2005;102:6843-6848. doi:10.1073/pnas.0502257102
- Vermeire S, Van Assche G, Rutgeerts P. Laboratory markers in IBD: useful, magic, or unnecessary toys?. Gut. 2006;55:426-431. doi:10.1136/gut.2005.069476
- Morisaku M, Ito K, Ogiso A, et al. Correlation between serum albumin and serum zinc in malignant lymphoma. Fujita Med J. 2022;8:59-64. doi:10.20407/fmj.2021-006
- Falchuk KH. Effect of acute disease and ACTH on serum zinc proteins. N Engl J Med. 1977:296:1129-1134.
- Naber TH, Baadenhuysen H, Jansen JB, et al. Serum alkaline phosphatase activity during zinc deficiency and long-term inflammatory stress. Clin Chim Acta. 1996;249:109-127. doi:10.1016/0009-8981(96)06281-x
- Lowe D, Sanvictores T, Zubair M, et al. Alkaline phosphatase. StatPearls [Internet]. Updated October 29, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK459201/
- Krebs NF. Update on zinc deficiency and excess in clinical pediatric practice. Ann Nutr Metab. 2013;62 suppl 1:19-29. doi:10.1159/000348261
- Maxfield L, Shukla S, Crane JS. Zinc deficiency. StatPearls [Internet]. Updated June 28, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493231/
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808. doi:10.1016/j.gtc.2017.08.011
- Caviezel D, Maissen S, Niess JH, et al. High prevalence of vitamin D deficiency among patients with inflammatory bowel disease. Inflamm Intest Dis. 2018;2:200-210. doi:10.1159/000489010
- Jasielska M, Grzybowska-Chlebowczyk U. Hypocalcemia and vitamin D deficiency in children with inflammatory bowel diseases and lactose intolerance. Nutrients. 2021;13:2583. doi:10.3390/nu13082583
- Vernia F, Valvano M, Longo S, et al. Vitamin D in inflammatory bowel diseases. Mechanisms of action and therapeutic implications. Nutrients. 2022;14:269. doi:10.3390/nu14020269
- Khazai N, Judd SE, Tangpricha V. Calcium and vitamin D: skeletal and extraskeletal health. Curr Rheumatol Rep. 2008;10:110-117. doi:10.1007/s11926-008-0020-y
- Domazetovic V, Iantomasi T, Bonanomi AG, et al. Vitamin D regulates claudin-2 and claudin-4 expression in active ulcerative colitis by p-Stat-6 and Smad-7 signaling. Int J Colorectal Dis. 2020;35:1231-1242. doi:10.1007/s00384-020-03576-0
- Gubatan J, Chou ND, Nielsen OH, et al. Systematic review with meta-analysis: association of vitamin D status with clinical outcomes in adult patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2019;50:1146-1158. doi:10.1111/apt.15506
- Fakhoury HMA, Kvietys PR, AlKattan W, et al. Vitamin D and intestinal homeostasis: barrier, microbiota, and immune modulation. J Steroid Biochem Mol Biol. 2020;200:105663. doi:10.1016/j.jsbmb.2020.105663
- Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770-1773. doi:10.1126/science.1123933
- Mostafa WZ, Hegazy RA. Vitamin D and the skin: focus on a complex relationship: a review. J Adv Res. 2015;6:793-804. doi:10.1016/j.jare.2014.01.011
- Searing DA, Leung DY. Vitamin D in atopic dermatitis, asthma and allergic diseases. Immunol Allergy Clin North Am. 2010;30:397-409.
- Lee YH, Song GG. Association between circulating 25-hydroxyvitamin D levels and psoriasis, and correlation with disease severity: a meta-analysis. Clin Exp Dermatol. 2018;43:529-535.
- Adorini L, Penna G. Control of autoimmune diseases by the vitamin D endocrine system. Nat Clin Pract Rheumatol. 2008;4:404-412.
- Autier P, Boniol M, Pizot C, et al. Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol. 2014;2:76-89. doi:10.1016/S2213-8587(13)70165-7
- Schafer AL, Shoback DM. Hypocalcemia: diagnosis and treatment. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext [Internet]. Updated January 3, 2016. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK279022/
- Magro F, Gionchetti P, Eliakim R, et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J Crohns Colitis. 2017;11:649-670. doi:10.1093/ecco-jcc/jjx008
- Amrein K, Scherkl M, Hoffmann M, et al. Vitamin D deficiency 2.0: an update on the current status worldwide. Eur J Clin Nutr. 2020;74:1498-1513. doi:10.1038/s41430-020-0558-y
- Munns CF, Shaw N, Kiely M, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. 2016;101:394-415. doi:10.1210/jc.2015-2175
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, Del Valle HB, eds. Dietary Reference Intakes for Calcium and Vitamin D. National Academies Press (US); 2011.
- Yeaman F, Nguyen A, Abasszade J, et al. Assessing vitamin D as a biomarker in inflammatory bowel disease. JGH Open. 2023;7:953-958. doi:10.1002/jgh3.13010
- Vernia P, Loizos P, Di Giuseppantonio I, et al S. Dietary calcium intake in patients with inflammatory bowel disease. J Crohns Colitis. 2014;8:312-317. doi:10.1016/j.crohns.2013.09.008
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ. 2008;336:1298-1302. doi:10.1136/bmj.39582.589433.BE
- Kenny CM, Murphy CE, Boyce DS, et al. Things we do for no reason™: calculating a “corrected calcium” level. J Hosp Med. 2021;16:499-501. doi:10.12788/jhm.3619
- Garg M, Rosella O, Rosella G, et al. Evaluation of a 12-week targeted vitamin D supplementation regimen in patients with active inflammatory bowel disease. Clin Nutr. 2018;37:1375-1382. doi:10.1016/j.clnu.2017.06.011
- Raftery T, Martineau AR, Greiller CL, et al. Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn’s disease: results from a randomised double-blind placebo-controlled study. United European Gastroenterol J. 2015;3:294-302. doi:10.1177/2050640615572176
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319. doi:10.1177/0148607107031004311
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274. doi:10.1016/s0190-9622(86)70301-0
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739. doi:10.3949/ccjm.83a.15061
- Elgharably N, Al Abadie M, Al Abadie M, et al. Vitamin B group levels and supplementations in dermatology. Dermatol Reports. 2022;15:9511. doi:10.4081/dr.2022.9511
- Hołubiec P, Leon´czyk M, Staszewski F, et al. Pathophysiology and clinical management of pellagra—a review. Folia Med Cracov. 2021;61:125-137. doi:10.24425/fmc.2021.138956
- Ink SL, Henderson LM. Vitamin B6 metabolism. Annu Rev Nutr. 1984;4:455-470. doi:10.1146/annurev.nu.04.070184.002323
- Brown MJ, Ameer MA, Daley SF, et al. Vitamin B6 deficiency. StatPearls [Internet]. Updated August 8, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK470579/.
- Vasilaki AT, McMillan DC, Kinsella J, et al. Relation between pyridoxal and pyridoxal phosphate concentrations in plasma, red cells, and white cells in patients with critical illness. Am J Clin Nutr. 2008;88:140-146. doi:10.1093/ajcn/88.1.140
- Chiang EP, Bagley PJ, Selhub J, et al. Abnormal vitamin B(6) status is associated with severity of symptoms in patients with rheumatoid arthritis. Am J Med. 2003;114:283-287. doi:10.1016/s0002-9343(02)01528-0
- Maaser C, Sturm A, Vavricka SR, et al. ECCO-ESGAR guideline for diagnostic assessment in IBD. Part 1: initial diagnosis, monitoring of known IBD, detection of complications. J Crohns Colitis. 2019;13:144-164. doi:10.1093/ecco-jcc/jjy113
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Selhub J, Byun A, Liu Z, et al. Dietary vitamin B6 intake modulates colonic inflammation in the IL10-/- model of inflammatory bowel disease. J Nutr Biochem. 2013;24:2138-2143. doi:10.1016/j.jnutbio.2013.08.005
- Pan Y, Liu Y, Guo H, et al. Associations between folate and vitamin B12 levels and inflammatory bowel disease: a meta-analysis. Nutrients. 2017;9:382. doi:10.3390/nu9040382
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33. doi:10.1007/s40257-014-0107-3
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503. doi:10.1002/ncp.10321
- Mori K, Ando I, Kukita A. Generalized hyperpigmentation of the skin due to vitamin B12 deficiency. J Dermatol. 2001;28:282-285. doi:10.1111/j.1346-8138.2001.tb00134.x
- Green R. Indicators for assessing folate and vitamin B-12 status and for monitoring the efficacy of intervention strategies. Am J Clin Nutr. 2011;94:666S-672S. doi:10.3945/ajcn.110.009613
- NIH Office of Dietary Supplements. Vitamin B12: fact sheet for health professionals. Updated February 27, 2024. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/
- NIH Office of Dietary Supplements. Folate: fact sheet for health professionals. Updated November 20, 2023. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/.
- Saibeni S, Bollani S, Losco A, et al. The use of methotrexate for treatment of inflammatory bowel disease in clinical practice. Dig Liver Dis. 2012;44:123-127. doi:10.1016/j.dld.2011.09.015
- Khan KM, Jialal I. Folic acid deficiency. StatPearls [Internet]. Updated June 26, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK535377/
- Bischoff SC, Bager P, Escher J, et al. ESPEN guideline on clinical nutrition in inflammatory bowel disease. Clin Nutr. 2023;42:352-379. doi:10.1016/j.clnu.2022.12.004
- Gerasimidis K, McGrogan P, Edwards CA. The aetiology and impact of malnutrition in paediatric inflammator y bowel disease. J Hum Nutr Diet. 2011;24:313-326. doi:10.1111/j.1365-277X.2011.01171.x
- Mentella MC, Scaldaferri F, Pizzoferrato M, et al. Nutrition, IBD and gut microbiota: a review. Nutrients. 2020;12:944. doi:10.3390/nu12040944
- Bonsack O, Caron B, Baumann C, et al. Food avoidance and fasting in patients with inflammatory bowel disease: experience from the Nancy IBD nutrition clinic. United European Gastroenterol J. 2023;11:361-370. doi:10.1002/ueg2.1238521
- Campmans-Kuijpers MJE, Dijkstra G. Food and food groups in inflammatory bowel disease (IBD): the design of the Groningen Anti-Inflammatory Diet (GrAID). Nutrients. 2021;13:1067. doi:10.3390/nu13041067
- Hwang C, Issokson K, Giguere-Rich C, et al. Development and pilot testing of the inflammatory bowel disease nutrition care pathway. Clin Gastroenterol Hepatol. 2020;18:2645-2649.e4. doi:10.1016/j.cgh.2020.06.039
- Filmann N, Rey J, Schneeweiss S, et al. Prevalence of anemia in inflammatory bowel diseases in European countries: a systematic review and individual patient data meta-analysis. Inflamm Bowel Dis. 2014;20:936-945. doi:10.1097/01.MIB.0000442728.74340.fd
- Stein J, Hartmann F, Dignass AU. Diagnosis and management of iron deficiency anemia in patients with IBD. Nat Rev Gastroenterol Hepatol. 2010;7:599-610. doi:10.1038/nrgastro.2010.151
- Ems T, St Lucia K, Huecker MR. Biochemistry, iron absorption. StatPearls [Internet]. Updated April 17, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448204/
- Evstatiev R, Gasche C. Iron sensing and signalling. Gut. 2012;61:933-952. doi:10.1136/gut.2010.214312
- Przybyszewska J, Zekanowska E. The role of hepcidin, ferroportin, HCP1, and DMT1 protein in iron absorption in the human digestive tract. Prz Gastroenterol. 2014;9:208-213. doi:10.5114/pg.2014.45102
- Weiss G, Gasche C. Pathogenesis and treatment of anemia in inflammatory bowel disease. Haematologica. 2010;95:175-178. doi:10.3324/haematol.2009.017046
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72. doi:10.4291/wjgp.v6.i3.62
- Moiz B. Spoon nails: still seen in today’s world. Clin Case Rep. 2018;6:547-548. doi:10.1002/ccr3.1404
- St Pierre SA, Vercellotti GM, Donovan JC, et al. Iron deficiency and diffuse nonscarring scalp alopecia in women: more pieces to the puzzle. J Am Acad Dermatol. 2010;63:1070-1076. doi:10.1016/j.jaad.2009.05.054
- Chiang CP, Yu-Fong Chang J, Wang YP, et al. Anemia, hematinic deficiencies, hyperhomocysteinemia, and serum gastric parietal cell antibody positivity in atrophic glossitis patients with or without microcytosis. J Formos Med Assoc. 2019;118:1401-1407. doi:10.1016/j.jfma.2019.06.004
- Chiang CP, Chang JY, Wang YP, et al. Atrophic glossitis: Etiology, serum autoantibodies, anemia, hematinic deficiencies, hyperhomocysteinemia, and management. J Formos Med Assoc. 2020;119:774-780. doi:10.1016/j.jfma.2019.04.015
- Walker J, Baran R, Vélez N, et al. Koilonychia: an update on pathophysiology, differential diagnosis and clinical relevance. J Eur Acad Dermatol Venereol. 2016;30:1985-1991. doi:10.1111/jdv.13610
- Guo HF, Tsai CL, Terajima M, et al. Pro-metastatic collagen lysyl hydroxylase dimer assemblies stabilized by Fe2+-binding. Nat Commun. 2018;9:512. doi:10.1038/s41467-018-02859-z
- Saini S, Jain AK, Agarwal S, et al. Iron deficiency and pruritus: a cross-sectional analysis to assess its association and relationship. Indian J Dermatol. 2021;66:705. doi:10.4103/ijd.ijd_326_21
- Du X, She E, Gelbart T, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320:1088-1092. doi:10.1126/science.1157121
- Lee S, Lee H, Lee CH, et al. Comorbidities in alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol. 2019;80:466-477.e16. doi:10.1016/j.jaad.2018.07.013
- Hashash JG, Elkins J, Lewis JD, et al. AGA Clinical Practice Update on diet and nutritional therapies in patients with inflammatory bowel disease: expert review [published online January 23, 2024]. Gastroenterology. doi:10.1053/j.gastro.2023.11.303
- Choudhuri S, Chowdhury IH, Saha A, et al. Acute monocyte pro- inflammatory response predicts higher positive to negative acute phase reactants ratio and severe hemostatic derangement in dengue fever. Cytokine. 2021;146:155644. doi:10.1016/j.cyto.2021.155644
- Dignass AU, Gasche C, Bettenworth D, et al; European Crohn’s and Colitis Organisation. European consensus on the diagnosis and management of iron deficiency and anaemia in inflammatory bowel diseases. J Crohn’s Colitis. 2015;9:211-222. doi:10.1093/ecco-jcc/jju009
- Daude S, Remen T, Chateau T, et al. Comparative accuracy of ferritin, transferrin saturation and soluble transferrin receptor for the diagnosis of iron deficiency in inflammatory bowel disease. Aliment Pharmacol Ther. 2020;51:1087-1095. doi:10.1111/apt.15739
- Pfeiffer CM, Looker AC. Laboratory methodologies for indicators of iron status: strengths, limitations, and analytical challenges. Am J Clin Nutr. 2017;106(suppl 6):1606S-1614S. doi:10.3945/ajcn.117.155887
- Tolkien Z, Stecher L, Mander AP, et al. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: a systematic review and meta-analysis. PLoS One. 2015;10:e0117383. doi:10.1371/journal.pone.0117383
- Evstatiev R, Marteau P, Iqbal T, et al. FERGIcor, a randomized controlled trial on ferric carboxymaltose for iron deficiency anemia in inflammatory bowel disease. Gastroenterology. 2011;141:846-853.e8532. doi:10.1053/j.gastro.2011.06.005
- Zupo R, Sila A, Castellana F, et al. Prevalence of zinc deficiency in inflammatory bowel disease: a systematic review and meta-analysis. Nutrients. 2022;14:4052. doi:10.3390/nu14194052
- Thompson MW. Regulation of zinc-dependent enzymes by metal carrier proteins. Biometals. 2022;35:187-213. doi:10.1007/s10534-022-00373-w
- Maares M, Haase H. A guide to human zinc absorption: general overview and recent advances of in vitro intestinal models. Nutrients. 2020;12:762. doi:10.3390/nu12030762
- Ranaldi G, Ferruzza S, Canali R, et al. Intracellular zinc is required for intestinal cell survival signals triggered by the inflammatory cytokine TNFα. J Nutr Biochem. 2013;24:967-976. doi:10.1016/j.jnutbio.2012.06.020
- Ogawa Y, Kawamura T, Shimada S. Zinc and skin biology. Arch Biochem Biophys. 2016;611:113-119. doi:10.1016/j.abb.2016.06.003
- Wilson D, Varigos G, Ackland ML. Apoptosis may underlie the pathology of zinc-deficient skin. Immunol Cell Biol. 2006;84:28-37. doi:10.1111/j.1440-1711.2005.01391.x
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685. doi:10.1016/j.clindermatol.2010.03.029
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624. doi:10.3390/nu9060624
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U S A. 2005;102:6843-6848. doi:10.1073/pnas.0502257102
- Vermeire S, Van Assche G, Rutgeerts P. Laboratory markers in IBD: useful, magic, or unnecessary toys?. Gut. 2006;55:426-431. doi:10.1136/gut.2005.069476
- Morisaku M, Ito K, Ogiso A, et al. Correlation between serum albumin and serum zinc in malignant lymphoma. Fujita Med J. 2022;8:59-64. doi:10.20407/fmj.2021-006
- Falchuk KH. Effect of acute disease and ACTH on serum zinc proteins. N Engl J Med. 1977:296:1129-1134.
- Naber TH, Baadenhuysen H, Jansen JB, et al. Serum alkaline phosphatase activity during zinc deficiency and long-term inflammatory stress. Clin Chim Acta. 1996;249:109-127. doi:10.1016/0009-8981(96)06281-x
- Lowe D, Sanvictores T, Zubair M, et al. Alkaline phosphatase. StatPearls [Internet]. Updated October 29, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK459201/
- Krebs NF. Update on zinc deficiency and excess in clinical pediatric practice. Ann Nutr Metab. 2013;62 suppl 1:19-29. doi:10.1159/000348261
- Maxfield L, Shukla S, Crane JS. Zinc deficiency. StatPearls [Internet]. Updated June 28, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493231/
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808. doi:10.1016/j.gtc.2017.08.011
- Caviezel D, Maissen S, Niess JH, et al. High prevalence of vitamin D deficiency among patients with inflammatory bowel disease. Inflamm Intest Dis. 2018;2:200-210. doi:10.1159/000489010
- Jasielska M, Grzybowska-Chlebowczyk U. Hypocalcemia and vitamin D deficiency in children with inflammatory bowel diseases and lactose intolerance. Nutrients. 2021;13:2583. doi:10.3390/nu13082583
- Vernia F, Valvano M, Longo S, et al. Vitamin D in inflammatory bowel diseases. Mechanisms of action and therapeutic implications. Nutrients. 2022;14:269. doi:10.3390/nu14020269
- Khazai N, Judd SE, Tangpricha V. Calcium and vitamin D: skeletal and extraskeletal health. Curr Rheumatol Rep. 2008;10:110-117. doi:10.1007/s11926-008-0020-y
- Domazetovic V, Iantomasi T, Bonanomi AG, et al. Vitamin D regulates claudin-2 and claudin-4 expression in active ulcerative colitis by p-Stat-6 and Smad-7 signaling. Int J Colorectal Dis. 2020;35:1231-1242. doi:10.1007/s00384-020-03576-0
- Gubatan J, Chou ND, Nielsen OH, et al. Systematic review with meta-analysis: association of vitamin D status with clinical outcomes in adult patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2019;50:1146-1158. doi:10.1111/apt.15506
- Fakhoury HMA, Kvietys PR, AlKattan W, et al. Vitamin D and intestinal homeostasis: barrier, microbiota, and immune modulation. J Steroid Biochem Mol Biol. 2020;200:105663. doi:10.1016/j.jsbmb.2020.105663
- Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770-1773. doi:10.1126/science.1123933
- Mostafa WZ, Hegazy RA. Vitamin D and the skin: focus on a complex relationship: a review. J Adv Res. 2015;6:793-804. doi:10.1016/j.jare.2014.01.011
- Searing DA, Leung DY. Vitamin D in atopic dermatitis, asthma and allergic diseases. Immunol Allergy Clin North Am. 2010;30:397-409.
- Lee YH, Song GG. Association between circulating 25-hydroxyvitamin D levels and psoriasis, and correlation with disease severity: a meta-analysis. Clin Exp Dermatol. 2018;43:529-535.
- Adorini L, Penna G. Control of autoimmune diseases by the vitamin D endocrine system. Nat Clin Pract Rheumatol. 2008;4:404-412.
- Autier P, Boniol M, Pizot C, et al. Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol. 2014;2:76-89. doi:10.1016/S2213-8587(13)70165-7
- Schafer AL, Shoback DM. Hypocalcemia: diagnosis and treatment. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext [Internet]. Updated January 3, 2016. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK279022/
- Magro F, Gionchetti P, Eliakim R, et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J Crohns Colitis. 2017;11:649-670. doi:10.1093/ecco-jcc/jjx008
- Amrein K, Scherkl M, Hoffmann M, et al. Vitamin D deficiency 2.0: an update on the current status worldwide. Eur J Clin Nutr. 2020;74:1498-1513. doi:10.1038/s41430-020-0558-y
- Munns CF, Shaw N, Kiely M, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. 2016;101:394-415. doi:10.1210/jc.2015-2175
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, Del Valle HB, eds. Dietary Reference Intakes for Calcium and Vitamin D. National Academies Press (US); 2011.
- Yeaman F, Nguyen A, Abasszade J, et al. Assessing vitamin D as a biomarker in inflammatory bowel disease. JGH Open. 2023;7:953-958. doi:10.1002/jgh3.13010
- Vernia P, Loizos P, Di Giuseppantonio I, et al S. Dietary calcium intake in patients with inflammatory bowel disease. J Crohns Colitis. 2014;8:312-317. doi:10.1016/j.crohns.2013.09.008
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ. 2008;336:1298-1302. doi:10.1136/bmj.39582.589433.BE
- Kenny CM, Murphy CE, Boyce DS, et al. Things we do for no reason™: calculating a “corrected calcium” level. J Hosp Med. 2021;16:499-501. doi:10.12788/jhm.3619
- Garg M, Rosella O, Rosella G, et al. Evaluation of a 12-week targeted vitamin D supplementation regimen in patients with active inflammatory bowel disease. Clin Nutr. 2018;37:1375-1382. doi:10.1016/j.clnu.2017.06.011
- Raftery T, Martineau AR, Greiller CL, et al. Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn’s disease: results from a randomised double-blind placebo-controlled study. United European Gastroenterol J. 2015;3:294-302. doi:10.1177/2050640615572176
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319. doi:10.1177/0148607107031004311
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274. doi:10.1016/s0190-9622(86)70301-0
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739. doi:10.3949/ccjm.83a.15061
- Elgharably N, Al Abadie M, Al Abadie M, et al. Vitamin B group levels and supplementations in dermatology. Dermatol Reports. 2022;15:9511. doi:10.4081/dr.2022.9511
- Hołubiec P, Leon´czyk M, Staszewski F, et al. Pathophysiology and clinical management of pellagra—a review. Folia Med Cracov. 2021;61:125-137. doi:10.24425/fmc.2021.138956
- Ink SL, Henderson LM. Vitamin B6 metabolism. Annu Rev Nutr. 1984;4:455-470. doi:10.1146/annurev.nu.04.070184.002323
- Brown MJ, Ameer MA, Daley SF, et al. Vitamin B6 deficiency. StatPearls [Internet]. Updated August 8, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK470579/.
- Vasilaki AT, McMillan DC, Kinsella J, et al. Relation between pyridoxal and pyridoxal phosphate concentrations in plasma, red cells, and white cells in patients with critical illness. Am J Clin Nutr. 2008;88:140-146. doi:10.1093/ajcn/88.1.140
- Chiang EP, Bagley PJ, Selhub J, et al. Abnormal vitamin B(6) status is associated with severity of symptoms in patients with rheumatoid arthritis. Am J Med. 2003;114:283-287. doi:10.1016/s0002-9343(02)01528-0
- Maaser C, Sturm A, Vavricka SR, et al. ECCO-ESGAR guideline for diagnostic assessment in IBD. Part 1: initial diagnosis, monitoring of known IBD, detection of complications. J Crohns Colitis. 2019;13:144-164. doi:10.1093/ecco-jcc/jjy113
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Selhub J, Byun A, Liu Z, et al. Dietary vitamin B6 intake modulates colonic inflammation in the IL10-/- model of inflammatory bowel disease. J Nutr Biochem. 2013;24:2138-2143. doi:10.1016/j.jnutbio.2013.08.005
- Pan Y, Liu Y, Guo H, et al. Associations between folate and vitamin B12 levels and inflammatory bowel disease: a meta-analysis. Nutrients. 2017;9:382. doi:10.3390/nu9040382
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33. doi:10.1007/s40257-014-0107-3
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503. doi:10.1002/ncp.10321
- Mori K, Ando I, Kukita A. Generalized hyperpigmentation of the skin due to vitamin B12 deficiency. J Dermatol. 2001;28:282-285. doi:10.1111/j.1346-8138.2001.tb00134.x
- Green R. Indicators for assessing folate and vitamin B-12 status and for monitoring the efficacy of intervention strategies. Am J Clin Nutr. 2011;94:666S-672S. doi:10.3945/ajcn.110.009613
- NIH Office of Dietary Supplements. Vitamin B12: fact sheet for health professionals. Updated February 27, 2024. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/
- NIH Office of Dietary Supplements. Folate: fact sheet for health professionals. Updated November 20, 2023. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/.
- Saibeni S, Bollani S, Losco A, et al. The use of methotrexate for treatment of inflammatory bowel disease in clinical practice. Dig Liver Dis. 2012;44:123-127. doi:10.1016/j.dld.2011.09.015
- Khan KM, Jialal I. Folic acid deficiency. StatPearls [Internet]. Updated June 26, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK535377/
Practice Points
- Patients with inflammatory bowel disease (IBD) are at increased risk for vitamin and nutrient deficiencies that may be identified first through cutaneous manifestations.
- Because active inflammation in IBD may skew routine laboratory values used for screening of micronutrient deficiencies, be cautious when interpreting these values.
- Patients taking systemic therapies for IBD such as corticosteroids and methotrexate are at higher risk for nutritional deficiencies.
Recurrent Aphthous Stomatitis: Clinical Experience From a University Hospital in Brazil
To the Editor:
Recurrent aphthous stomatitis (RAS) is a mucocutaneous condition characterized by single or multiple, painful,1,2 round ulcerations of variable sizes with a tendency for recurrence, most commonly located in nonkeratinized areas of the oral mucosa. Pathergy commonly is observed.3 Although many authors consider the terms RAS andaphtha to be synonymous,4,5 differentiating the clinical lesion (aphthous ulceration) from the disease (aphtha or RAS) can be useful, as several other diseases can at times manifest with similar ulcers (called aphthoid lesions), such as pemphigus vulgaris, mucous membrane pemphigoid, and erythema multiforme.6
It is estimated that approximately 20% of individuals worldwide have at least one episode of aphtha during their lifetime,7 and it is considered the most common disease of the oral mucosa.8,9 However, only patients presenting with severe acute outbreaks or frequent relapses typically seek medical treatment. Clinically, aphthous ulcers are classified as aphtha minor (small number of small lesions), aphtha major (large deep lesions that also can affect the minor salivary glands with intense necrosis, difficulty in healing, and mucosal scarring), and aphtha herpetiformis (innumerous tiny lesions that reappear in recurring outbreaks).1-3 The term complex aphthosis was introduced in 198510 and is defined as recurrent oral and genital aphthous ulcerations or recurring multiple oral aphthous ulcers in the absence of systemic manifestations or Behçet disease11,12; however, complex aphthosis also has been reported as frequent episodes of ulcerations that may be associated with systemic diseases including Behçet disease.13,14
Currently, RAS is considered an immunologically mediated alteration in cutaneous mucosal reactivity with a multifactorial systemic cause. Underlying conditions such as Behçet disease, inflammatory bowel disease (IBD), iatrogenic immunosuppression (eg, following solid organ transplantation), AIDS, and cyclic neutropenia may or may not be detected.11-13
Our retrospective study explored the systemic nature of RAS. We reviewed patient records to evaluate underlying systemic conditions associated with the diagnosis of RAS and the use of oral medications in managing the disease. Medical records from the Department of Dermatology of the University of São Paulo, Brazil, from 2003 to 2017 were reviewed to identify patients with a diagnosis of RAS. Clinical classification of RAS—minor, major, or herpetiform—as well as the presence of aphthous lesions in other locations and the presence of other associated inflammatory cutaneous manifestations also were noted. Associated systemic diseases and treatments for RAS were recorded. Patients for whom the diagnosis of RAS was changed during follow-up were excluded. Because this was a retrospective analysis of medical records and without any patient risk, informed consent was not needed.
Medical records for 125 patients were reviewed; 63 were male (50.4%), and 62 were female (49.6%). The age at onset of symptoms, which ranged from a few months after birth to 74 years, was reported in only 92 (73.6%) patient medical records. Of these, 30 (32.6%) reported onset before 20 years of age, 39 (42.4%) between 20 and 39 years, 17 (18.5%) between 40 and 59 years, and 6 (6.5%) at 60 years or older. Morphologically, 72 (57.6%) had minor, 42 (33.6%) had major, and 11 (8.8%) had herpetiform aphthous ulcers. None of the patients presented with sporadic lesions; the disease was long-standing and persistent in all cases (complex aphthosis).
Regarding the location of the ulcers, 92 (73.6%) patients had lesions on the oral mucosa only. Some patients had lesions in more than one site in addition to the oral mucosa: 32 (25.6%) had aphthae in the genital/groin region and 4 (3.2%) presented with perianal/anal aphthae. Nineteen patients (19.2%) presented other cutaneous manifestations in addition to aphthae: 11 (45.8%) had folliculitis/pseudofolliculitis, and 8 (33.3%) had erythema nodosum (EN). Eight patients (33.3%) presented with uveitis, and 6 (25%) presented with concomitant arthralgia/arthritis. Fifty-four patients (43.2%) had confirmed or suspected associated disease: Behçet disease (21 [38.9%]), IBD (10 [18.5%]), solid organ transplantation (7 [13.0%])(kidney, 4 [57.1%]; heart, 2 [28.6%]; liver, 1 [14.3%]), HIV infection (6 [11.1%]), lymphoma (1 [1.9%]), aplastic anemia (1 [1.9%]), or myelodysplastic syndrome (1 [1.9%]). Ten patients (18.5%) presented with other diseases under investigation (eg, unidentified rheumatologic disease, unexplained neutropenia, undiagnosed immunodeficiencies, autoinflammatory syndromes, possible cyclic neutropenia).
Biopsies of the oral mucosa were performed in 31 patients. Histopathologic findings will be discussed in a future publication (unpublished data).
Five patients (4.0%) were lost to follow-up and did not receive treatment; 10 (8.0%) received only topical treatment (analgesics and/or corticosteroids). All 9 (7.2%) patients undergoing intralesional corticosteroid injections also were on a systemic treatment. One hundred ten (88.0%) patients were treated systemically—with colchicine (84/110 [76.4%]), thalidomide (43/110 [39.1%]), small pulses of oral corticosteroids (26 [23.6%]), dapsone (12/110 [10.9%]), or pentoxifylline (3 [2.7%]). Furthermore, in patients with associated diseases, treatment of the underlying condition was conducted when available, and follow-up was carried out in conjunction with the appropriate specialists. For treatment of the associated disease, patients received other medications such as methotrexate, azathioprine, cyclophosphamide, intravenous corticosteroid pulse, and immunobiologics.
The prevalence of RAS between sexes in our study population was similar (50.4% male; 49.6% female). Results from prior studies have been mixed; some reported a higher prevalence in females,15-18 while others found no predilection for sex among patients diagnosed with RAS.19,20 In our analysis, 75% of patients experienced symptoms of RAS before 40 years of age; in prior studies, up to 56% of patients experienced symptoms between the ages of 20 and 40 years.21,22
In our study, 26.4% of patients had extraoral aphthae. Genital lesions have been described as infrequent,23 and lesions manifesting in other mucous membranes or on the skin are rare.24 A study reported genital involvement in 8% to 13% of patients with oral aphtha.25 We observed genital involvement in 25.6% of patients. Likewise, this higher value may be due to our study population of patients referred to our university hospital. In our study, 19.2% of patients presented with other inflammatory manifestations in addition to aphthous ulcerations (eg, folliculitis, EN, uveitis, arthritis). As dermatologists in a tertiary reference hospital, we actively look for such associations in every aphtha patient, which may not be the case in many nondermatologic oral care services.
In our study population, 43.2% of patients were diagnosed with or were under investigation for systemic diseases known to be associated with RAS. We found associations with Behçet disease most frequently, followed by IBD,26 solid organ transplantation, and HIV. In this group of patients, the respective systemic disease was active or poorly controlled. In transplant recipients, aphtha major was the most common type, similar to other studies.27 We observed no notable difference in the clinical picture of the oral ulcers in patients with a well-established systemic disease vs those without.
Most of our cases did not present findings other than aphtha, indicating that the intrinsic defect that predisposes to RAS is always systemic. Even mild and sporadic cases may be attributable to a systemic disorder of cutaneous-mucosal reactivity. The predisposition to RAS never originates in the oral cavity, hence the confusion caused and the uselessness of studies that relate aphthae to factors such as local food allergies, pH changes, or local infection with microorganisms.5,28 The disease course (reducing the frequency of lesion appearance and accelerating the healing of extensive lesions) is only modified with systemic treatment, with local measures proving to be only moderately useful to relieve pain. We believe that RAS can in many ways be compared to EN and pyoderma gangrenosum (PG): some systemic conditions that predispose patients to EN and PG also may predispose them to RAS (eg, IBD, hematologic disorders). Similar to RAS, many cases of EN and PG are idiopathic. In addition, pathergy also occurs in PG.11,13
We were unable to observe or establish any predictive clinical element that could indicate a better or worse response to the prescribed treatments, which also has been noted by other authors.3,4 Treatment of RAS is empiric, generally starting with drugs that are easier to prescribe and with fewer adverse effects, then progressing to more complex drugs when a good response is not obtained. Colchicine was the most commonly prescribed medication (76.4% [84/110]). It has been proposed by several authors3,4 as a first-line systemic medication for the treatment of recurrent aphthae, as it has been shown to be effective and safe. The dosage ranged from 0.5 mg twice daily to 0.5 mg 4 times daily. Dapsone is an established drug for aphtha29,30 and was used in 12 of our patients. The dosage used in our patients ranged from 50 to 100 mg/d. Adverse effects such as hemolytic anemia frequently are seen, and one of the patients in our study developed DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome in response to dapsone. In 7 cases, colchicine and dapsone were used together, which is believed to potentiate the therapeutic effects. This combination may be useful in patients for whom thalidomide cannot be used or those who have not improved with monotherapy.29 Thalidomide is considered one of the most effective drugs for RAS.30,31 Forty-three patients in our analysis were treated with thalidomide, usually as a first choice. The dosage ranged from 100 to 200 mg/d. It was mainly chosen in disabling pediatric cases, adult men with aphthous major, and women with no risk for pregnancy. Due to its potential adverse effects, thalidomide has been recommended when there is no response with other medications that are dose dependent; severe adverse effects such as thromboembolism and peripheral neuropathy are rare.31 Oral corticosteroids were used in 26 patients, aiming at rapid improvement in very symptomatic cases; however, due to the potential for long-term adverse effects, in all cases they were prescribed in combination with another medication that was maintained after the corticosteroid was discontinued.
We highlight the systemic nature of RAS as well as its frequent association with systemic diseases and other correlated manifestations (pustules, EN, arthralgia). We also emphasize the importance of using oral medications to adequately control the disease and do not recommend topical medications aimed at treating local causes. Dermatologists should be consulted in managing severe cases of RAS.
- Buño IJ, Huff JC, Weston WL, et al. Elevated levels of interferon gamma, tumor necrosis factor alpha, interleukins 2, 4, and 5, but not interleukin 10, are present in recurrent aphthous stomatitis. Arch Dermatol. 1998;134:827-831.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728- 732.
- Natah SS, Konttinen YT, Enattah NS, et al. Recurrent aphthous ulcers today: a review of the growing knowledge. Int J Oral Maxillofac Surg. 2004;33:221-234.
- Zunt SL. Recurrent aphthous stomatitis. Dermatol Clin. 2003;21:33-39.
- Jurge S, Kuffer R, Scully C, et al. Mucosal disease series. number VI. recurrent aphthous stomatitis. Oral Dis. 2006;12:1-21.
- Chams-Davatchi C, Shizarpour M, Davatchi F, et al. Comparison of oral aphthae in Behçet’s disease and idiopathic recurrent aphthous stomatitis. Adv Exp Med Biol. 2003;528:317-320.
- Schemel-Suárez M, López-López J, Chimenos-Küstner E. Oral ulcers: differential diagnosis and treatment [in Spanish]. Med Clin (Barc). 2015;145:499-503.
- S´lebioda Z, Szponar E, Kowalska A. Etiopathogenesis of recurrent aphthous stomatitis and the role of immunologic aspects: literature review. Arch Immunol Ther Exp (Warsz). 2014;62:205-215.
- Edgar NR, Saleh D, Miller RA. Recurrent aphthous stomatitis: a review. J Clin Aesthet Dermatol. 2017;10:26-36.
- Jorizzo JL, Taylor RS, Schmalstieg FC, et al. Complex aphthosis: a forme fruste of Behçet’s syndrome? J Am Acad Dermatol. 1985;13:80-84.
- McCarty MA, Garton RA, Jorizzo JL. Complex aphthosis and Behçet’s disease. Dermatol Clin. 2003;21:41-48.
- Bulur I, Melrem O. Behçet disease: new aspects. Clin Dermatol. 2017;35:421-434.
- Cui RZ, Rogers RS 3rd. Recurrent aphthous stomatitis. Clin Dermatol. 2016;34:475-481.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728-732.
- Ship II. Epidemiologic aspects of recurrent aphthous ulcerations. Oral Surg Oral Med Oral Pathol. 1972;33:400-406.
- Ship JA. Recurrent aphthous stomatitis. an update. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:141-147.
- Wilhelmsen NS, Weber R, Monteiro F, et al. Correlation between histocompatibility antigens and recurrent aphthous stomatitis in the Brazilian population. Braz J Otorhinolaryngol. 2009;75:426-431.
- S´lebioda Z, Dorocka-Bobkowska B. Systemic and environmental risk factors for recurrent aphthous stomatitis in a Polish cohort of patients. Postepy Dermatol Alergol. 2019;36:196-201.
- Ship JA, Chavez EM, Doerr PA, et al. Recurrent aphthous stomatitis. Quintessence Int. 2000;31:95-112.
- Brocklehurst P, Tickle M, Glenny AM, et al. Systemic interventions for recurrent aphthous stomatitis (mouth ulcers). Cochrane Database Syst Rev. 2012;12:CD005411.
- Belenguer-Guallar I, Jiménez-Soriano Y, Ariadna Claramunt-Lozano A. Treatment of recurrent aphthous stomatitis. a literature review. J Clin Exp Dent. 2014;6:E168-E174.
- Bagán JV, Sanchis JM, Milián MA, et al. Recurrent aphthous stomatitis. a study of the clinical characteristics of lesions in 93 cases. J Oral Pathol Med. 1991;20:395-397.
- Huppert JS, Gerber MA, Deitch HR, et al. Vulvar ulcers in young females: a manifestation of aphthosis. J Pediatr Adolesc Gynecol. 2006;19:195-204.
- Scully C, Porter S. Recurrent aphthous stomatitis: current concepts of etiology, pathogenesis and management. J Oral Pathol Med. 1989;18:21-27
- Chapel TA. Origins of penile ulcerations. Arch Androl. 1979; 3: 351-357.
- Lourenço SV, Hussein TP, Bologna SB, et al. Oral manifestations of inflammatory bowel disease: a review based on the observation of six cases. J Eur Acad Dermatol Venereol. 2010;24:204-207.
- Nico MM, Brito AE, Martins LE, et al. Oral ulcers in an immunosuppressed 5-year-old boy. Clin Exp Dermatol. 2008;33:367-368.
- Trakji B, Baroudi K, Kharma Y. The effect of dietary habits on the development of the recurrent aphthous stomatitis. Niger Med J. 2012;53:9-11.
- Lynde CB, Bruce AJ, Rogers RS 3rd. Successful treatment of complex aphthosis with colchicine and dapsone. Arch Dermatol. 2009;145:273-276.
- Letsinger JA, McCarty MA, Jorizzo JL. Complex aphthosis: a large case series with evaluation algorithm and therapeutic ladder from topicals to thalidomide. J Am Acad Dermatol. 2005(3 pt 1);52:500-508.
- Hello M, Barbarot S, Bastuji-Garin S, et al. Use of thalidomide for severe recurrent aphthous stomatitis: a multicenter cohort analysis. Medicine (Baltimore). 2010;89:176-182.
To the Editor:
Recurrent aphthous stomatitis (RAS) is a mucocutaneous condition characterized by single or multiple, painful,1,2 round ulcerations of variable sizes with a tendency for recurrence, most commonly located in nonkeratinized areas of the oral mucosa. Pathergy commonly is observed.3 Although many authors consider the terms RAS andaphtha to be synonymous,4,5 differentiating the clinical lesion (aphthous ulceration) from the disease (aphtha or RAS) can be useful, as several other diseases can at times manifest with similar ulcers (called aphthoid lesions), such as pemphigus vulgaris, mucous membrane pemphigoid, and erythema multiforme.6
It is estimated that approximately 20% of individuals worldwide have at least one episode of aphtha during their lifetime,7 and it is considered the most common disease of the oral mucosa.8,9 However, only patients presenting with severe acute outbreaks or frequent relapses typically seek medical treatment. Clinically, aphthous ulcers are classified as aphtha minor (small number of small lesions), aphtha major (large deep lesions that also can affect the minor salivary glands with intense necrosis, difficulty in healing, and mucosal scarring), and aphtha herpetiformis (innumerous tiny lesions that reappear in recurring outbreaks).1-3 The term complex aphthosis was introduced in 198510 and is defined as recurrent oral and genital aphthous ulcerations or recurring multiple oral aphthous ulcers in the absence of systemic manifestations or Behçet disease11,12; however, complex aphthosis also has been reported as frequent episodes of ulcerations that may be associated with systemic diseases including Behçet disease.13,14
Currently, RAS is considered an immunologically mediated alteration in cutaneous mucosal reactivity with a multifactorial systemic cause. Underlying conditions such as Behçet disease, inflammatory bowel disease (IBD), iatrogenic immunosuppression (eg, following solid organ transplantation), AIDS, and cyclic neutropenia may or may not be detected.11-13
Our retrospective study explored the systemic nature of RAS. We reviewed patient records to evaluate underlying systemic conditions associated with the diagnosis of RAS and the use of oral medications in managing the disease. Medical records from the Department of Dermatology of the University of São Paulo, Brazil, from 2003 to 2017 were reviewed to identify patients with a diagnosis of RAS. Clinical classification of RAS—minor, major, or herpetiform—as well as the presence of aphthous lesions in other locations and the presence of other associated inflammatory cutaneous manifestations also were noted. Associated systemic diseases and treatments for RAS were recorded. Patients for whom the diagnosis of RAS was changed during follow-up were excluded. Because this was a retrospective analysis of medical records and without any patient risk, informed consent was not needed.
Medical records for 125 patients were reviewed; 63 were male (50.4%), and 62 were female (49.6%). The age at onset of symptoms, which ranged from a few months after birth to 74 years, was reported in only 92 (73.6%) patient medical records. Of these, 30 (32.6%) reported onset before 20 years of age, 39 (42.4%) between 20 and 39 years, 17 (18.5%) between 40 and 59 years, and 6 (6.5%) at 60 years or older. Morphologically, 72 (57.6%) had minor, 42 (33.6%) had major, and 11 (8.8%) had herpetiform aphthous ulcers. None of the patients presented with sporadic lesions; the disease was long-standing and persistent in all cases (complex aphthosis).
Regarding the location of the ulcers, 92 (73.6%) patients had lesions on the oral mucosa only. Some patients had lesions in more than one site in addition to the oral mucosa: 32 (25.6%) had aphthae in the genital/groin region and 4 (3.2%) presented with perianal/anal aphthae. Nineteen patients (19.2%) presented other cutaneous manifestations in addition to aphthae: 11 (45.8%) had folliculitis/pseudofolliculitis, and 8 (33.3%) had erythema nodosum (EN). Eight patients (33.3%) presented with uveitis, and 6 (25%) presented with concomitant arthralgia/arthritis. Fifty-four patients (43.2%) had confirmed or suspected associated disease: Behçet disease (21 [38.9%]), IBD (10 [18.5%]), solid organ transplantation (7 [13.0%])(kidney, 4 [57.1%]; heart, 2 [28.6%]; liver, 1 [14.3%]), HIV infection (6 [11.1%]), lymphoma (1 [1.9%]), aplastic anemia (1 [1.9%]), or myelodysplastic syndrome (1 [1.9%]). Ten patients (18.5%) presented with other diseases under investigation (eg, unidentified rheumatologic disease, unexplained neutropenia, undiagnosed immunodeficiencies, autoinflammatory syndromes, possible cyclic neutropenia).
Biopsies of the oral mucosa were performed in 31 patients. Histopathologic findings will be discussed in a future publication (unpublished data).
Five patients (4.0%) were lost to follow-up and did not receive treatment; 10 (8.0%) received only topical treatment (analgesics and/or corticosteroids). All 9 (7.2%) patients undergoing intralesional corticosteroid injections also were on a systemic treatment. One hundred ten (88.0%) patients were treated systemically—with colchicine (84/110 [76.4%]), thalidomide (43/110 [39.1%]), small pulses of oral corticosteroids (26 [23.6%]), dapsone (12/110 [10.9%]), or pentoxifylline (3 [2.7%]). Furthermore, in patients with associated diseases, treatment of the underlying condition was conducted when available, and follow-up was carried out in conjunction with the appropriate specialists. For treatment of the associated disease, patients received other medications such as methotrexate, azathioprine, cyclophosphamide, intravenous corticosteroid pulse, and immunobiologics.
The prevalence of RAS between sexes in our study population was similar (50.4% male; 49.6% female). Results from prior studies have been mixed; some reported a higher prevalence in females,15-18 while others found no predilection for sex among patients diagnosed with RAS.19,20 In our analysis, 75% of patients experienced symptoms of RAS before 40 years of age; in prior studies, up to 56% of patients experienced symptoms between the ages of 20 and 40 years.21,22
In our study, 26.4% of patients had extraoral aphthae. Genital lesions have been described as infrequent,23 and lesions manifesting in other mucous membranes or on the skin are rare.24 A study reported genital involvement in 8% to 13% of patients with oral aphtha.25 We observed genital involvement in 25.6% of patients. Likewise, this higher value may be due to our study population of patients referred to our university hospital. In our study, 19.2% of patients presented with other inflammatory manifestations in addition to aphthous ulcerations (eg, folliculitis, EN, uveitis, arthritis). As dermatologists in a tertiary reference hospital, we actively look for such associations in every aphtha patient, which may not be the case in many nondermatologic oral care services.
In our study population, 43.2% of patients were diagnosed with or were under investigation for systemic diseases known to be associated with RAS. We found associations with Behçet disease most frequently, followed by IBD,26 solid organ transplantation, and HIV. In this group of patients, the respective systemic disease was active or poorly controlled. In transplant recipients, aphtha major was the most common type, similar to other studies.27 We observed no notable difference in the clinical picture of the oral ulcers in patients with a well-established systemic disease vs those without.
Most of our cases did not present findings other than aphtha, indicating that the intrinsic defect that predisposes to RAS is always systemic. Even mild and sporadic cases may be attributable to a systemic disorder of cutaneous-mucosal reactivity. The predisposition to RAS never originates in the oral cavity, hence the confusion caused and the uselessness of studies that relate aphthae to factors such as local food allergies, pH changes, or local infection with microorganisms.5,28 The disease course (reducing the frequency of lesion appearance and accelerating the healing of extensive lesions) is only modified with systemic treatment, with local measures proving to be only moderately useful to relieve pain. We believe that RAS can in many ways be compared to EN and pyoderma gangrenosum (PG): some systemic conditions that predispose patients to EN and PG also may predispose them to RAS (eg, IBD, hematologic disorders). Similar to RAS, many cases of EN and PG are idiopathic. In addition, pathergy also occurs in PG.11,13
We were unable to observe or establish any predictive clinical element that could indicate a better or worse response to the prescribed treatments, which also has been noted by other authors.3,4 Treatment of RAS is empiric, generally starting with drugs that are easier to prescribe and with fewer adverse effects, then progressing to more complex drugs when a good response is not obtained. Colchicine was the most commonly prescribed medication (76.4% [84/110]). It has been proposed by several authors3,4 as a first-line systemic medication for the treatment of recurrent aphthae, as it has been shown to be effective and safe. The dosage ranged from 0.5 mg twice daily to 0.5 mg 4 times daily. Dapsone is an established drug for aphtha29,30 and was used in 12 of our patients. The dosage used in our patients ranged from 50 to 100 mg/d. Adverse effects such as hemolytic anemia frequently are seen, and one of the patients in our study developed DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome in response to dapsone. In 7 cases, colchicine and dapsone were used together, which is believed to potentiate the therapeutic effects. This combination may be useful in patients for whom thalidomide cannot be used or those who have not improved with monotherapy.29 Thalidomide is considered one of the most effective drugs for RAS.30,31 Forty-three patients in our analysis were treated with thalidomide, usually as a first choice. The dosage ranged from 100 to 200 mg/d. It was mainly chosen in disabling pediatric cases, adult men with aphthous major, and women with no risk for pregnancy. Due to its potential adverse effects, thalidomide has been recommended when there is no response with other medications that are dose dependent; severe adverse effects such as thromboembolism and peripheral neuropathy are rare.31 Oral corticosteroids were used in 26 patients, aiming at rapid improvement in very symptomatic cases; however, due to the potential for long-term adverse effects, in all cases they were prescribed in combination with another medication that was maintained after the corticosteroid was discontinued.
We highlight the systemic nature of RAS as well as its frequent association with systemic diseases and other correlated manifestations (pustules, EN, arthralgia). We also emphasize the importance of using oral medications to adequately control the disease and do not recommend topical medications aimed at treating local causes. Dermatologists should be consulted in managing severe cases of RAS.
To the Editor:
Recurrent aphthous stomatitis (RAS) is a mucocutaneous condition characterized by single or multiple, painful,1,2 round ulcerations of variable sizes with a tendency for recurrence, most commonly located in nonkeratinized areas of the oral mucosa. Pathergy commonly is observed.3 Although many authors consider the terms RAS andaphtha to be synonymous,4,5 differentiating the clinical lesion (aphthous ulceration) from the disease (aphtha or RAS) can be useful, as several other diseases can at times manifest with similar ulcers (called aphthoid lesions), such as pemphigus vulgaris, mucous membrane pemphigoid, and erythema multiforme.6
It is estimated that approximately 20% of individuals worldwide have at least one episode of aphtha during their lifetime,7 and it is considered the most common disease of the oral mucosa.8,9 However, only patients presenting with severe acute outbreaks or frequent relapses typically seek medical treatment. Clinically, aphthous ulcers are classified as aphtha minor (small number of small lesions), aphtha major (large deep lesions that also can affect the minor salivary glands with intense necrosis, difficulty in healing, and mucosal scarring), and aphtha herpetiformis (innumerous tiny lesions that reappear in recurring outbreaks).1-3 The term complex aphthosis was introduced in 198510 and is defined as recurrent oral and genital aphthous ulcerations or recurring multiple oral aphthous ulcers in the absence of systemic manifestations or Behçet disease11,12; however, complex aphthosis also has been reported as frequent episodes of ulcerations that may be associated with systemic diseases including Behçet disease.13,14
Currently, RAS is considered an immunologically mediated alteration in cutaneous mucosal reactivity with a multifactorial systemic cause. Underlying conditions such as Behçet disease, inflammatory bowel disease (IBD), iatrogenic immunosuppression (eg, following solid organ transplantation), AIDS, and cyclic neutropenia may or may not be detected.11-13
Our retrospective study explored the systemic nature of RAS. We reviewed patient records to evaluate underlying systemic conditions associated with the diagnosis of RAS and the use of oral medications in managing the disease. Medical records from the Department of Dermatology of the University of São Paulo, Brazil, from 2003 to 2017 were reviewed to identify patients with a diagnosis of RAS. Clinical classification of RAS—minor, major, or herpetiform—as well as the presence of aphthous lesions in other locations and the presence of other associated inflammatory cutaneous manifestations also were noted. Associated systemic diseases and treatments for RAS were recorded. Patients for whom the diagnosis of RAS was changed during follow-up were excluded. Because this was a retrospective analysis of medical records and without any patient risk, informed consent was not needed.
Medical records for 125 patients were reviewed; 63 were male (50.4%), and 62 were female (49.6%). The age at onset of symptoms, which ranged from a few months after birth to 74 years, was reported in only 92 (73.6%) patient medical records. Of these, 30 (32.6%) reported onset before 20 years of age, 39 (42.4%) between 20 and 39 years, 17 (18.5%) between 40 and 59 years, and 6 (6.5%) at 60 years or older. Morphologically, 72 (57.6%) had minor, 42 (33.6%) had major, and 11 (8.8%) had herpetiform aphthous ulcers. None of the patients presented with sporadic lesions; the disease was long-standing and persistent in all cases (complex aphthosis).
Regarding the location of the ulcers, 92 (73.6%) patients had lesions on the oral mucosa only. Some patients had lesions in more than one site in addition to the oral mucosa: 32 (25.6%) had aphthae in the genital/groin region and 4 (3.2%) presented with perianal/anal aphthae. Nineteen patients (19.2%) presented other cutaneous manifestations in addition to aphthae: 11 (45.8%) had folliculitis/pseudofolliculitis, and 8 (33.3%) had erythema nodosum (EN). Eight patients (33.3%) presented with uveitis, and 6 (25%) presented with concomitant arthralgia/arthritis. Fifty-four patients (43.2%) had confirmed or suspected associated disease: Behçet disease (21 [38.9%]), IBD (10 [18.5%]), solid organ transplantation (7 [13.0%])(kidney, 4 [57.1%]; heart, 2 [28.6%]; liver, 1 [14.3%]), HIV infection (6 [11.1%]), lymphoma (1 [1.9%]), aplastic anemia (1 [1.9%]), or myelodysplastic syndrome (1 [1.9%]). Ten patients (18.5%) presented with other diseases under investigation (eg, unidentified rheumatologic disease, unexplained neutropenia, undiagnosed immunodeficiencies, autoinflammatory syndromes, possible cyclic neutropenia).
Biopsies of the oral mucosa were performed in 31 patients. Histopathologic findings will be discussed in a future publication (unpublished data).
Five patients (4.0%) were lost to follow-up and did not receive treatment; 10 (8.0%) received only topical treatment (analgesics and/or corticosteroids). All 9 (7.2%) patients undergoing intralesional corticosteroid injections also were on a systemic treatment. One hundred ten (88.0%) patients were treated systemically—with colchicine (84/110 [76.4%]), thalidomide (43/110 [39.1%]), small pulses of oral corticosteroids (26 [23.6%]), dapsone (12/110 [10.9%]), or pentoxifylline (3 [2.7%]). Furthermore, in patients with associated diseases, treatment of the underlying condition was conducted when available, and follow-up was carried out in conjunction with the appropriate specialists. For treatment of the associated disease, patients received other medications such as methotrexate, azathioprine, cyclophosphamide, intravenous corticosteroid pulse, and immunobiologics.
The prevalence of RAS between sexes in our study population was similar (50.4% male; 49.6% female). Results from prior studies have been mixed; some reported a higher prevalence in females,15-18 while others found no predilection for sex among patients diagnosed with RAS.19,20 In our analysis, 75% of patients experienced symptoms of RAS before 40 years of age; in prior studies, up to 56% of patients experienced symptoms between the ages of 20 and 40 years.21,22
In our study, 26.4% of patients had extraoral aphthae. Genital lesions have been described as infrequent,23 and lesions manifesting in other mucous membranes or on the skin are rare.24 A study reported genital involvement in 8% to 13% of patients with oral aphtha.25 We observed genital involvement in 25.6% of patients. Likewise, this higher value may be due to our study population of patients referred to our university hospital. In our study, 19.2% of patients presented with other inflammatory manifestations in addition to aphthous ulcerations (eg, folliculitis, EN, uveitis, arthritis). As dermatologists in a tertiary reference hospital, we actively look for such associations in every aphtha patient, which may not be the case in many nondermatologic oral care services.
In our study population, 43.2% of patients were diagnosed with or were under investigation for systemic diseases known to be associated with RAS. We found associations with Behçet disease most frequently, followed by IBD,26 solid organ transplantation, and HIV. In this group of patients, the respective systemic disease was active or poorly controlled. In transplant recipients, aphtha major was the most common type, similar to other studies.27 We observed no notable difference in the clinical picture of the oral ulcers in patients with a well-established systemic disease vs those without.
Most of our cases did not present findings other than aphtha, indicating that the intrinsic defect that predisposes to RAS is always systemic. Even mild and sporadic cases may be attributable to a systemic disorder of cutaneous-mucosal reactivity. The predisposition to RAS never originates in the oral cavity, hence the confusion caused and the uselessness of studies that relate aphthae to factors such as local food allergies, pH changes, or local infection with microorganisms.5,28 The disease course (reducing the frequency of lesion appearance and accelerating the healing of extensive lesions) is only modified with systemic treatment, with local measures proving to be only moderately useful to relieve pain. We believe that RAS can in many ways be compared to EN and pyoderma gangrenosum (PG): some systemic conditions that predispose patients to EN and PG also may predispose them to RAS (eg, IBD, hematologic disorders). Similar to RAS, many cases of EN and PG are idiopathic. In addition, pathergy also occurs in PG.11,13
We were unable to observe or establish any predictive clinical element that could indicate a better or worse response to the prescribed treatments, which also has been noted by other authors.3,4 Treatment of RAS is empiric, generally starting with drugs that are easier to prescribe and with fewer adverse effects, then progressing to more complex drugs when a good response is not obtained. Colchicine was the most commonly prescribed medication (76.4% [84/110]). It has been proposed by several authors3,4 as a first-line systemic medication for the treatment of recurrent aphthae, as it has been shown to be effective and safe. The dosage ranged from 0.5 mg twice daily to 0.5 mg 4 times daily. Dapsone is an established drug for aphtha29,30 and was used in 12 of our patients. The dosage used in our patients ranged from 50 to 100 mg/d. Adverse effects such as hemolytic anemia frequently are seen, and one of the patients in our study developed DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome in response to dapsone. In 7 cases, colchicine and dapsone were used together, which is believed to potentiate the therapeutic effects. This combination may be useful in patients for whom thalidomide cannot be used or those who have not improved with monotherapy.29 Thalidomide is considered one of the most effective drugs for RAS.30,31 Forty-three patients in our analysis were treated with thalidomide, usually as a first choice. The dosage ranged from 100 to 200 mg/d. It was mainly chosen in disabling pediatric cases, adult men with aphthous major, and women with no risk for pregnancy. Due to its potential adverse effects, thalidomide has been recommended when there is no response with other medications that are dose dependent; severe adverse effects such as thromboembolism and peripheral neuropathy are rare.31 Oral corticosteroids were used in 26 patients, aiming at rapid improvement in very symptomatic cases; however, due to the potential for long-term adverse effects, in all cases they were prescribed in combination with another medication that was maintained after the corticosteroid was discontinued.
We highlight the systemic nature of RAS as well as its frequent association with systemic diseases and other correlated manifestations (pustules, EN, arthralgia). We also emphasize the importance of using oral medications to adequately control the disease and do not recommend topical medications aimed at treating local causes. Dermatologists should be consulted in managing severe cases of RAS.
- Buño IJ, Huff JC, Weston WL, et al. Elevated levels of interferon gamma, tumor necrosis factor alpha, interleukins 2, 4, and 5, but not interleukin 10, are present in recurrent aphthous stomatitis. Arch Dermatol. 1998;134:827-831.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728- 732.
- Natah SS, Konttinen YT, Enattah NS, et al. Recurrent aphthous ulcers today: a review of the growing knowledge. Int J Oral Maxillofac Surg. 2004;33:221-234.
- Zunt SL. Recurrent aphthous stomatitis. Dermatol Clin. 2003;21:33-39.
- Jurge S, Kuffer R, Scully C, et al. Mucosal disease series. number VI. recurrent aphthous stomatitis. Oral Dis. 2006;12:1-21.
- Chams-Davatchi C, Shizarpour M, Davatchi F, et al. Comparison of oral aphthae in Behçet’s disease and idiopathic recurrent aphthous stomatitis. Adv Exp Med Biol. 2003;528:317-320.
- Schemel-Suárez M, López-López J, Chimenos-Küstner E. Oral ulcers: differential diagnosis and treatment [in Spanish]. Med Clin (Barc). 2015;145:499-503.
- S´lebioda Z, Szponar E, Kowalska A. Etiopathogenesis of recurrent aphthous stomatitis and the role of immunologic aspects: literature review. Arch Immunol Ther Exp (Warsz). 2014;62:205-215.
- Edgar NR, Saleh D, Miller RA. Recurrent aphthous stomatitis: a review. J Clin Aesthet Dermatol. 2017;10:26-36.
- Jorizzo JL, Taylor RS, Schmalstieg FC, et al. Complex aphthosis: a forme fruste of Behçet’s syndrome? J Am Acad Dermatol. 1985;13:80-84.
- McCarty MA, Garton RA, Jorizzo JL. Complex aphthosis and Behçet’s disease. Dermatol Clin. 2003;21:41-48.
- Bulur I, Melrem O. Behçet disease: new aspects. Clin Dermatol. 2017;35:421-434.
- Cui RZ, Rogers RS 3rd. Recurrent aphthous stomatitis. Clin Dermatol. 2016;34:475-481.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728-732.
- Ship II. Epidemiologic aspects of recurrent aphthous ulcerations. Oral Surg Oral Med Oral Pathol. 1972;33:400-406.
- Ship JA. Recurrent aphthous stomatitis. an update. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:141-147.
- Wilhelmsen NS, Weber R, Monteiro F, et al. Correlation between histocompatibility antigens and recurrent aphthous stomatitis in the Brazilian population. Braz J Otorhinolaryngol. 2009;75:426-431.
- S´lebioda Z, Dorocka-Bobkowska B. Systemic and environmental risk factors for recurrent aphthous stomatitis in a Polish cohort of patients. Postepy Dermatol Alergol. 2019;36:196-201.
- Ship JA, Chavez EM, Doerr PA, et al. Recurrent aphthous stomatitis. Quintessence Int. 2000;31:95-112.
- Brocklehurst P, Tickle M, Glenny AM, et al. Systemic interventions for recurrent aphthous stomatitis (mouth ulcers). Cochrane Database Syst Rev. 2012;12:CD005411.
- Belenguer-Guallar I, Jiménez-Soriano Y, Ariadna Claramunt-Lozano A. Treatment of recurrent aphthous stomatitis. a literature review. J Clin Exp Dent. 2014;6:E168-E174.
- Bagán JV, Sanchis JM, Milián MA, et al. Recurrent aphthous stomatitis. a study of the clinical characteristics of lesions in 93 cases. J Oral Pathol Med. 1991;20:395-397.
- Huppert JS, Gerber MA, Deitch HR, et al. Vulvar ulcers in young females: a manifestation of aphthosis. J Pediatr Adolesc Gynecol. 2006;19:195-204.
- Scully C, Porter S. Recurrent aphthous stomatitis: current concepts of etiology, pathogenesis and management. J Oral Pathol Med. 1989;18:21-27
- Chapel TA. Origins of penile ulcerations. Arch Androl. 1979; 3: 351-357.
- Lourenço SV, Hussein TP, Bologna SB, et al. Oral manifestations of inflammatory bowel disease: a review based on the observation of six cases. J Eur Acad Dermatol Venereol. 2010;24:204-207.
- Nico MM, Brito AE, Martins LE, et al. Oral ulcers in an immunosuppressed 5-year-old boy. Clin Exp Dermatol. 2008;33:367-368.
- Trakji B, Baroudi K, Kharma Y. The effect of dietary habits on the development of the recurrent aphthous stomatitis. Niger Med J. 2012;53:9-11.
- Lynde CB, Bruce AJ, Rogers RS 3rd. Successful treatment of complex aphthosis with colchicine and dapsone. Arch Dermatol. 2009;145:273-276.
- Letsinger JA, McCarty MA, Jorizzo JL. Complex aphthosis: a large case series with evaluation algorithm and therapeutic ladder from topicals to thalidomide. J Am Acad Dermatol. 2005(3 pt 1);52:500-508.
- Hello M, Barbarot S, Bastuji-Garin S, et al. Use of thalidomide for severe recurrent aphthous stomatitis: a multicenter cohort analysis. Medicine (Baltimore). 2010;89:176-182.
- Buño IJ, Huff JC, Weston WL, et al. Elevated levels of interferon gamma, tumor necrosis factor alpha, interleukins 2, 4, and 5, but not interleukin 10, are present in recurrent aphthous stomatitis. Arch Dermatol. 1998;134:827-831.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728- 732.
- Natah SS, Konttinen YT, Enattah NS, et al. Recurrent aphthous ulcers today: a review of the growing knowledge. Int J Oral Maxillofac Surg. 2004;33:221-234.
- Zunt SL. Recurrent aphthous stomatitis. Dermatol Clin. 2003;21:33-39.
- Jurge S, Kuffer R, Scully C, et al. Mucosal disease series. number VI. recurrent aphthous stomatitis. Oral Dis. 2006;12:1-21.
- Chams-Davatchi C, Shizarpour M, Davatchi F, et al. Comparison of oral aphthae in Behçet’s disease and idiopathic recurrent aphthous stomatitis. Adv Exp Med Biol. 2003;528:317-320.
- Schemel-Suárez M, López-López J, Chimenos-Küstner E. Oral ulcers: differential diagnosis and treatment [in Spanish]. Med Clin (Barc). 2015;145:499-503.
- S´lebioda Z, Szponar E, Kowalska A. Etiopathogenesis of recurrent aphthous stomatitis and the role of immunologic aspects: literature review. Arch Immunol Ther Exp (Warsz). 2014;62:205-215.
- Edgar NR, Saleh D, Miller RA. Recurrent aphthous stomatitis: a review. J Clin Aesthet Dermatol. 2017;10:26-36.
- Jorizzo JL, Taylor RS, Schmalstieg FC, et al. Complex aphthosis: a forme fruste of Behçet’s syndrome? J Am Acad Dermatol. 1985;13:80-84.
- McCarty MA, Garton RA, Jorizzo JL. Complex aphthosis and Behçet’s disease. Dermatol Clin. 2003;21:41-48.
- Bulur I, Melrem O. Behçet disease: new aspects. Clin Dermatol. 2017;35:421-434.
- Cui RZ, Rogers RS 3rd. Recurrent aphthous stomatitis. Clin Dermatol. 2016;34:475-481.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728-732.
- Ship II. Epidemiologic aspects of recurrent aphthous ulcerations. Oral Surg Oral Med Oral Pathol. 1972;33:400-406.
- Ship JA. Recurrent aphthous stomatitis. an update. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:141-147.
- Wilhelmsen NS, Weber R, Monteiro F, et al. Correlation between histocompatibility antigens and recurrent aphthous stomatitis in the Brazilian population. Braz J Otorhinolaryngol. 2009;75:426-431.
- S´lebioda Z, Dorocka-Bobkowska B. Systemic and environmental risk factors for recurrent aphthous stomatitis in a Polish cohort of patients. Postepy Dermatol Alergol. 2019;36:196-201.
- Ship JA, Chavez EM, Doerr PA, et al. Recurrent aphthous stomatitis. Quintessence Int. 2000;31:95-112.
- Brocklehurst P, Tickle M, Glenny AM, et al. Systemic interventions for recurrent aphthous stomatitis (mouth ulcers). Cochrane Database Syst Rev. 2012;12:CD005411.
- Belenguer-Guallar I, Jiménez-Soriano Y, Ariadna Claramunt-Lozano A. Treatment of recurrent aphthous stomatitis. a literature review. J Clin Exp Dent. 2014;6:E168-E174.
- Bagán JV, Sanchis JM, Milián MA, et al. Recurrent aphthous stomatitis. a study of the clinical characteristics of lesions in 93 cases. J Oral Pathol Med. 1991;20:395-397.
- Huppert JS, Gerber MA, Deitch HR, et al. Vulvar ulcers in young females: a manifestation of aphthosis. J Pediatr Adolesc Gynecol. 2006;19:195-204.
- Scully C, Porter S. Recurrent aphthous stomatitis: current concepts of etiology, pathogenesis and management. J Oral Pathol Med. 1989;18:21-27
- Chapel TA. Origins of penile ulcerations. Arch Androl. 1979; 3: 351-357.
- Lourenço SV, Hussein TP, Bologna SB, et al. Oral manifestations of inflammatory bowel disease: a review based on the observation of six cases. J Eur Acad Dermatol Venereol. 2010;24:204-207.
- Nico MM, Brito AE, Martins LE, et al. Oral ulcers in an immunosuppressed 5-year-old boy. Clin Exp Dermatol. 2008;33:367-368.
- Trakji B, Baroudi K, Kharma Y. The effect of dietary habits on the development of the recurrent aphthous stomatitis. Niger Med J. 2012;53:9-11.
- Lynde CB, Bruce AJ, Rogers RS 3rd. Successful treatment of complex aphthosis with colchicine and dapsone. Arch Dermatol. 2009;145:273-276.
- Letsinger JA, McCarty MA, Jorizzo JL. Complex aphthosis: a large case series with evaluation algorithm and therapeutic ladder from topicals to thalidomide. J Am Acad Dermatol. 2005(3 pt 1);52:500-508.
- Hello M, Barbarot S, Bastuji-Garin S, et al. Use of thalidomide for severe recurrent aphthous stomatitis: a multicenter cohort analysis. Medicine (Baltimore). 2010;89:176-182.
Practice Points
- The process that leads to the formation of aphthous ulcerations is always systemic, not local, even in the absence of a diagnosable systemic disease.
- Relapsing cases of aphthae should be treated with systemic medication.
Advancements in Targeted Therapies for Vitiligo: Prioritizing Equity in Drug Development
Vitiligo is a common acquired autoimmune disease that causes depigmented patches to develop throughout the skin , with descriptions dating back more than 3000 years to the earliest known Indian and Egyptian texts. Approximately 1.4% of the worldwide population has vitiligo,1 and onset follows a bimodal age distribution with an early-onset population (mean age at onset, 10.3 years) as well as an adult-onset population (mean age at onset, 34 years).2 Vitiligo manifests as well-defined, irregular, depigmented macules and patches surrounded by normal skin. The patches can vary in size from a few millimeters to several centimeters. There may be signs of inflammation, and the lesions can be itchy, but in most cases vitiligo is asymptomatic. In nonsegmental vitiligo, the depigmented patches are ymmetrical, can appear in any area of the body, and commonly progress slowly. In segmental vitiligo, the patches are unilateral, rarely cross the midline of the body, and are localized to one area. Segmental vitiligo commonly appears in childhood and progresses rapidly but stops abruptly within 6 to 12 months and remains stable, usually for life.3 Although the condition may be more apparent in patients with skin of color, vitiligo manifests at a similar rate in individuals of all races and ethnicities.4
Similar to most autoimmune diseases, vitiligo has a strong genetic predisposition. Although the overall prevalence of vitiligo is less than 2%, having a family history of vitiligo (ie, a first-degree relative with vitiligo) increases an individual’s risk to 6%, while concordance in identical twins is 23%.5 Beyond genetic predisposition, there is strong evidence that environmental exposures, such as hair dyes, contribute to risk for disease.6 Interestingly, vitiligo is associated with polyautoimmunity—the presence of multiple autoimmune diseases in a single patient,7 such as type 1 diabetes mellitus, rheumatoid arthritis, autoimmune thyroid disease, pernicious anemia, and Addison disease. Similar to vitiligo itself, polyautoimmunity likely is driven by a combination of genetic and environmental factors.5
We provide a brief overview of clinical trial results of Janus kinase (JAK) inhibitors for treating vitiligo and discuss the trial cohorts, with an emphasis on the impact of cohort demographic composition for individuals with skin of color. We recommend factors that investigators should consider to ensure equitable representation of individuals with skin of color in future clinical trials.
Autoimmune Pathogenesis and Treatment With JAK Inhibitors
Vitiligo is driven by autoreactive CD8+ T cells that target melanocytes and secrete IFN-g. Signaling of IFN-g occurs through the JAK–signal transducer and activator of transcription (JAK-STAT) pathway, leading to transcriptional changes that activate proinflammatory genes such as the chemokine CXCL10, which is required for the directed accumulation of melanocyte-specific CD8+ T cells at the epidermis where melanocytes reside.8 Once vitiligo has been initiated, the disease persists due to the presence of resident memory T cells that remain in the skin and destroy new melanocytes.9,10
Given the central role of IFN-g signaling in the pathogenesis of vitiligo, drugs that inhibit JAK signaling are appealing to treat the disease. These JAK inhibitors bind to the kinase domain of JAK to prevent its activation, thus preventing downstream signaling events including STAT phosphorylation and its translocation to the nucleus, which ultimately stops the upregulation of inflammatory gene transcription. This process attenuates the autoimmune response in the skin and results in repigmentation of vitiligo lesions. In 2022, the US Food and Drug Administration approved the topical JAK inhibitor ruxolitinib for the treatment of vitiligo. Additional clinical trials have been initiated to test oral JAK inhibitors—ritlecitinib (ClinicalTrials.gov identifiers NCT06163326, NCT06072183, NCT05583526), povorcitinib (NCT04818346, NCT06113445, NCT06113471), and upadacitinib (NCT04927975, NCT06118411)—with strong results reported so far.11
The effects of JAK inhibitors can be striking, as shown in the Figure. A patient of one of the authors (J.E.H.) used topical ruxolitinib on only the left arm for approximately 36 weeks and results were as expected—strong repigmentation of only the treated area, which is possible with JAK inhibitors. Indeed, 2 phase 3 studies—Topical Ruxolitinib Evaluation in Vitiligo (TRuE-V1 and TRuE-V2)—showed that approximately 30% of participants in TRuE-V1 (N=330) and 30.9% of participants in TRuE-V2 (N=344) achieved at least 75% improvement over baseline in the facial vitiligo area scoring index (VASI).12 In the oral ritlecitinib phase 2b study, 12.1% of the 187 participants on the highest tested dose of ritlecitinib (loading dose of 200 mg/d for 28 days, followed by 50 mg/d maintenance dose) achieved at least 75% improvement over baseline in the VASI at 24 weeks.11 Although this rate is lower than for topical ruxolitinib, this trial required all participants to have active disease (unlike the TRuE-V trials of ruxolitinib), which likely created a higher bar for repigmentation and thus resulted in fewer participants achieving the primary outcome at the early 6-month end point. Extension of treatment through 48 weeks demonstrated continued improvement over baseline without any evidence of plateau.11 Although treatment with JAK inhibitors can result in dramatic repigmentation of vitiligo patches, it falls short of providing a permanent cure, as stopping treatment results in relapse (ie, the return of depigmented lesions).

Racial Disparities in Clinical Trials
Even though vitiligo affects all skin types and races/ethnicities with similar prevalence and severity, the proportion of individuals with darker skin types enrolled in these clinical trials fails to match their representation in the population as a whole. A study examining the prevalence of vitiligo in the United States reported that Black or African American individuals represented 15.8% of vitiligo diagnoses in the United States4 even though they are only 12.7% of the total US population. However, Black or African American individuals comprised only 5% of the combined participants in the TRuE-V clinical trials for topical ruxolitinib12 and 2.7% of the participants in the phase 2b study of oral ritlecitinib.11 This lack of appropriate representation is not unique to JAK inhibitors or other vitiligo trials. Indeed, the US Food and Drug Administration reported that Black or African American individuals comprised only 8% of participants for all clinical trials in 2020.13
Efficacy Metrics Beyond Repigmentation
Disparities in quality-of-life (QOL) metrics in diseases affecting individuals with skin of color also exist. In vitiligo, the contrast between affected and unaffected skin is greater in patients with skin of color, which means that for a given VASI score, the visibility of depigmentation as well as repigmentation may be variable among patients. Additionally, there is evidence that QOL concerns vary between patients with skin of color and those with lighter skin types. Ezzedine et al14 found that QOL concerns in vitiligo patients with darker skin focused more on appearance, while concerns in vitiligo patients with lighter skin focused more on skin cancer risk. In addition to QOL differences among individuals with different skin types, there also are well-documented differences in attitudes to vitiligo among certain ethnic or cultural groups.15 For example, the Rigveda (an ancient Hindu text) indicates that individuals with vitiligo and their progeny are disqualified from marriage. Although the JAK inhibitor clinical trials for vitiligo did not appear to show differences in the degree of repigmentation among different skin types or races/ethnicities, QOL measures were not collected as a secondary end point in these studies—despite the fact that at least 1 study had documented that QOL measures were not uniform across patients when stratified by age and extent of disease.1,11,12 This same study also presented limited data suggestive of lower QOL in patients with the darkest skin phototype.1
Considerations for Future Clinical Trials
It is logical to assume that every clinical trialist in dermatology seeks equitable representation among a diverse set of races, ethnicities, and skin types, but achieving this goal remains elusive. Two recent publications16,17 outlined the challenges and examined solutions to address enrollment disparities, including several barriers to diversity among clinical trial participants: awareness of the clinical trials among minority populations; easy access to clinical trial sites; reluctance to participate because of prior experiences of discrimination, even if unrelated to clinical trials; and a lack of workforce diversity among the clinical trialist teams. To overcome these barriers, a multifaceted approach is needed that requires action at the level of the patient, provider, community, and institution. Once diverse representation is achieved, investigators should consider the need for QOL metrics as a secondary outcome in their trials, which will ensure that the intended clinical effect is matched by patient expectations across different races and ethnicities based on the potential differential impact that diseases such as vitiligo can have on patients with skin of color.
- Bibeau K, Pandya AG, Ezzedine K, et al. Vitiligo prevalence and quality of life among adults in Europe, Japan and the USA. J Eur Acad Dermatol Venereol. 2022;36:1831-1844.
- Jin Y, Roberts GHL, Ferrara TM, et al. Early-onset autoimmune vitiligo associated with an enhancer variant haplotype that upregulates class II HLA expression. Nat Commun. 2019;10:391.
- Rodrigues M, Ezzedine K, Hamzavi I, et al; Vitiligo Working Group. New discoveries in the pathogenesis and classification of vitiligo. J Am Acad Dermatol. 2017;77:1-13.
- Gandhi K, Ezzedine K, Anastassopoulos KP, et al. Prevalence of vitiligo among adults in the United States. JAMA Dermatol. 2022;158:43-50.
- Spritz RA, Santorico SA. The genetic basis of vitiligo. J Invest Dermatol. 2021;141:265-73.
- Harris JE. Chemical-induced vitiligo. Dermatol Clin. 2017;35:151-161.
- Ahmed F, Moseley I, Ragi SD, et al. Vitiligo in underrepresented communities: an all of us database analysis. J Am Acad Dermatol. 2023;88:945-948.
- Frisoli ML, Essien K, Harris JE. Vitiligo: mechanisms of pathogenesis and treatment. Annu Rev Immunol. 2020;38:621-648.
- Richmond JM, Strassner JP, Zapata L Jr, et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. 2018;10:eaam7710.
- Richmond JM, Strassner JP, Rashighi M, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol. 2019;139:769-778.
- Ezzedine K, Peeva E, Yamaguchi Y, et al. Efficacy and safety of oral ritlecitinib for the treatment of active nonsegmental vitiligo: a randomized phase 2b clinical trial. J Am Acad Dermatol. 2023;88:395-403.
- Rosmarin D, Passeron T, Pandya AG, et al. Two phase 3, randomized, controlled trials of ruxolitinib cream for vitiligo. N Engl J Med. 2022;387:1445-1455.
- Cavazzoni P, Anagnostiadis E, Lolic M. Drug trials snapshots summary report. US Food and Drug Administration website. Accessed March 19, 2024. https://www.fda.gov/media/145718/download
- Ezzedine K, Grimes PE, Meurant JM, et al. Living with vitiligo: results from a national survey indicate differences between skin phototypes. Br J Dermatol. 2015;173:607-609.
- Elbuluk N, Ezzedine K. Quality of life, burden of disease, co-morbidities, and systemic effects in vitiligo patients. Dermatol Clin. 2017;35:117-128.
- Kahn JM, Gray DM 2nd, Oliveri JM, et al. Strategies to improve diversity, equity, and inclusion in clinical trials. Cancer. 2022;128:216-221.
- Nolan TS, McKoy A, Gray DM 2nd, et al. Virtual community engagement for retention of black men in clinical research. Am J Mens Health. 2023;17:15579883221147767.
Vitiligo is a common acquired autoimmune disease that causes depigmented patches to develop throughout the skin , with descriptions dating back more than 3000 years to the earliest known Indian and Egyptian texts. Approximately 1.4% of the worldwide population has vitiligo,1 and onset follows a bimodal age distribution with an early-onset population (mean age at onset, 10.3 years) as well as an adult-onset population (mean age at onset, 34 years).2 Vitiligo manifests as well-defined, irregular, depigmented macules and patches surrounded by normal skin. The patches can vary in size from a few millimeters to several centimeters. There may be signs of inflammation, and the lesions can be itchy, but in most cases vitiligo is asymptomatic. In nonsegmental vitiligo, the depigmented patches are ymmetrical, can appear in any area of the body, and commonly progress slowly. In segmental vitiligo, the patches are unilateral, rarely cross the midline of the body, and are localized to one area. Segmental vitiligo commonly appears in childhood and progresses rapidly but stops abruptly within 6 to 12 months and remains stable, usually for life.3 Although the condition may be more apparent in patients with skin of color, vitiligo manifests at a similar rate in individuals of all races and ethnicities.4
Similar to most autoimmune diseases, vitiligo has a strong genetic predisposition. Although the overall prevalence of vitiligo is less than 2%, having a family history of vitiligo (ie, a first-degree relative with vitiligo) increases an individual’s risk to 6%, while concordance in identical twins is 23%.5 Beyond genetic predisposition, there is strong evidence that environmental exposures, such as hair dyes, contribute to risk for disease.6 Interestingly, vitiligo is associated with polyautoimmunity—the presence of multiple autoimmune diseases in a single patient,7 such as type 1 diabetes mellitus, rheumatoid arthritis, autoimmune thyroid disease, pernicious anemia, and Addison disease. Similar to vitiligo itself, polyautoimmunity likely is driven by a combination of genetic and environmental factors.5
We provide a brief overview of clinical trial results of Janus kinase (JAK) inhibitors for treating vitiligo and discuss the trial cohorts, with an emphasis on the impact of cohort demographic composition for individuals with skin of color. We recommend factors that investigators should consider to ensure equitable representation of individuals with skin of color in future clinical trials.
Autoimmune Pathogenesis and Treatment With JAK Inhibitors
Vitiligo is driven by autoreactive CD8+ T cells that target melanocytes and secrete IFN-g. Signaling of IFN-g occurs through the JAK–signal transducer and activator of transcription (JAK-STAT) pathway, leading to transcriptional changes that activate proinflammatory genes such as the chemokine CXCL10, which is required for the directed accumulation of melanocyte-specific CD8+ T cells at the epidermis where melanocytes reside.8 Once vitiligo has been initiated, the disease persists due to the presence of resident memory T cells that remain in the skin and destroy new melanocytes.9,10
Given the central role of IFN-g signaling in the pathogenesis of vitiligo, drugs that inhibit JAK signaling are appealing to treat the disease. These JAK inhibitors bind to the kinase domain of JAK to prevent its activation, thus preventing downstream signaling events including STAT phosphorylation and its translocation to the nucleus, which ultimately stops the upregulation of inflammatory gene transcription. This process attenuates the autoimmune response in the skin and results in repigmentation of vitiligo lesions. In 2022, the US Food and Drug Administration approved the topical JAK inhibitor ruxolitinib for the treatment of vitiligo. Additional clinical trials have been initiated to test oral JAK inhibitors—ritlecitinib (ClinicalTrials.gov identifiers NCT06163326, NCT06072183, NCT05583526), povorcitinib (NCT04818346, NCT06113445, NCT06113471), and upadacitinib (NCT04927975, NCT06118411)—with strong results reported so far.11
The effects of JAK inhibitors can be striking, as shown in the Figure. A patient of one of the authors (J.E.H.) used topical ruxolitinib on only the left arm for approximately 36 weeks and results were as expected—strong repigmentation of only the treated area, which is possible with JAK inhibitors. Indeed, 2 phase 3 studies—Topical Ruxolitinib Evaluation in Vitiligo (TRuE-V1 and TRuE-V2)—showed that approximately 30% of participants in TRuE-V1 (N=330) and 30.9% of participants in TRuE-V2 (N=344) achieved at least 75% improvement over baseline in the facial vitiligo area scoring index (VASI).12 In the oral ritlecitinib phase 2b study, 12.1% of the 187 participants on the highest tested dose of ritlecitinib (loading dose of 200 mg/d for 28 days, followed by 50 mg/d maintenance dose) achieved at least 75% improvement over baseline in the VASI at 24 weeks.11 Although this rate is lower than for topical ruxolitinib, this trial required all participants to have active disease (unlike the TRuE-V trials of ruxolitinib), which likely created a higher bar for repigmentation and thus resulted in fewer participants achieving the primary outcome at the early 6-month end point. Extension of treatment through 48 weeks demonstrated continued improvement over baseline without any evidence of plateau.11 Although treatment with JAK inhibitors can result in dramatic repigmentation of vitiligo patches, it falls short of providing a permanent cure, as stopping treatment results in relapse (ie, the return of depigmented lesions).
Racial Disparities in Clinical Trials
Even though vitiligo affects all skin types and races/ethnicities with similar prevalence and severity, the proportion of individuals with darker skin types enrolled in these clinical trials fails to match their representation in the population as a whole. A study examining the prevalence of vitiligo in the United States reported that Black or African American individuals represented 15.8% of vitiligo diagnoses in the United States4 even though they are only 12.7% of the total US population. However, Black or African American individuals comprised only 5% of the combined participants in the TRuE-V clinical trials for topical ruxolitinib12 and 2.7% of the participants in the phase 2b study of oral ritlecitinib.11 This lack of appropriate representation is not unique to JAK inhibitors or other vitiligo trials. Indeed, the US Food and Drug Administration reported that Black or African American individuals comprised only 8% of participants for all clinical trials in 2020.13
Efficacy Metrics Beyond Repigmentation
Disparities in quality-of-life (QOL) metrics in diseases affecting individuals with skin of color also exist. In vitiligo, the contrast between affected and unaffected skin is greater in patients with skin of color, which means that for a given VASI score, the visibility of depigmentation as well as repigmentation may be variable among patients. Additionally, there is evidence that QOL concerns vary between patients with skin of color and those with lighter skin types. Ezzedine et al14 found that QOL concerns in vitiligo patients with darker skin focused more on appearance, while concerns in vitiligo patients with lighter skin focused more on skin cancer risk. In addition to QOL differences among individuals with different skin types, there also are well-documented differences in attitudes to vitiligo among certain ethnic or cultural groups.15 For example, the Rigveda (an ancient Hindu text) indicates that individuals with vitiligo and their progeny are disqualified from marriage. Although the JAK inhibitor clinical trials for vitiligo did not appear to show differences in the degree of repigmentation among different skin types or races/ethnicities, QOL measures were not collected as a secondary end point in these studies—despite the fact that at least 1 study had documented that QOL measures were not uniform across patients when stratified by age and extent of disease.1,11,12 This same study also presented limited data suggestive of lower QOL in patients with the darkest skin phototype.1
Considerations for Future Clinical Trials
It is logical to assume that every clinical trialist in dermatology seeks equitable representation among a diverse set of races, ethnicities, and skin types, but achieving this goal remains elusive. Two recent publications16,17 outlined the challenges and examined solutions to address enrollment disparities, including several barriers to diversity among clinical trial participants: awareness of the clinical trials among minority populations; easy access to clinical trial sites; reluctance to participate because of prior experiences of discrimination, even if unrelated to clinical trials; and a lack of workforce diversity among the clinical trialist teams. To overcome these barriers, a multifaceted approach is needed that requires action at the level of the patient, provider, community, and institution. Once diverse representation is achieved, investigators should consider the need for QOL metrics as a secondary outcome in their trials, which will ensure that the intended clinical effect is matched by patient expectations across different races and ethnicities based on the potential differential impact that diseases such as vitiligo can have on patients with skin of color.
Vitiligo is a common acquired autoimmune disease that causes depigmented patches to develop throughout the skin , with descriptions dating back more than 3000 years to the earliest known Indian and Egyptian texts. Approximately 1.4% of the worldwide population has vitiligo,1 and onset follows a bimodal age distribution with an early-onset population (mean age at onset, 10.3 years) as well as an adult-onset population (mean age at onset, 34 years).2 Vitiligo manifests as well-defined, irregular, depigmented macules and patches surrounded by normal skin. The patches can vary in size from a few millimeters to several centimeters. There may be signs of inflammation, and the lesions can be itchy, but in most cases vitiligo is asymptomatic. In nonsegmental vitiligo, the depigmented patches are ymmetrical, can appear in any area of the body, and commonly progress slowly. In segmental vitiligo, the patches are unilateral, rarely cross the midline of the body, and are localized to one area. Segmental vitiligo commonly appears in childhood and progresses rapidly but stops abruptly within 6 to 12 months and remains stable, usually for life.3 Although the condition may be more apparent in patients with skin of color, vitiligo manifests at a similar rate in individuals of all races and ethnicities.4
Similar to most autoimmune diseases, vitiligo has a strong genetic predisposition. Although the overall prevalence of vitiligo is less than 2%, having a family history of vitiligo (ie, a first-degree relative with vitiligo) increases an individual’s risk to 6%, while concordance in identical twins is 23%.5 Beyond genetic predisposition, there is strong evidence that environmental exposures, such as hair dyes, contribute to risk for disease.6 Interestingly, vitiligo is associated with polyautoimmunity—the presence of multiple autoimmune diseases in a single patient,7 such as type 1 diabetes mellitus, rheumatoid arthritis, autoimmune thyroid disease, pernicious anemia, and Addison disease. Similar to vitiligo itself, polyautoimmunity likely is driven by a combination of genetic and environmental factors.5
We provide a brief overview of clinical trial results of Janus kinase (JAK) inhibitors for treating vitiligo and discuss the trial cohorts, with an emphasis on the impact of cohort demographic composition for individuals with skin of color. We recommend factors that investigators should consider to ensure equitable representation of individuals with skin of color in future clinical trials.
Autoimmune Pathogenesis and Treatment With JAK Inhibitors
Vitiligo is driven by autoreactive CD8+ T cells that target melanocytes and secrete IFN-g. Signaling of IFN-g occurs through the JAK–signal transducer and activator of transcription (JAK-STAT) pathway, leading to transcriptional changes that activate proinflammatory genes such as the chemokine CXCL10, which is required for the directed accumulation of melanocyte-specific CD8+ T cells at the epidermis where melanocytes reside.8 Once vitiligo has been initiated, the disease persists due to the presence of resident memory T cells that remain in the skin and destroy new melanocytes.9,10
Given the central role of IFN-g signaling in the pathogenesis of vitiligo, drugs that inhibit JAK signaling are appealing to treat the disease. These JAK inhibitors bind to the kinase domain of JAK to prevent its activation, thus preventing downstream signaling events including STAT phosphorylation and its translocation to the nucleus, which ultimately stops the upregulation of inflammatory gene transcription. This process attenuates the autoimmune response in the skin and results in repigmentation of vitiligo lesions. In 2022, the US Food and Drug Administration approved the topical JAK inhibitor ruxolitinib for the treatment of vitiligo. Additional clinical trials have been initiated to test oral JAK inhibitors—ritlecitinib (ClinicalTrials.gov identifiers NCT06163326, NCT06072183, NCT05583526), povorcitinib (NCT04818346, NCT06113445, NCT06113471), and upadacitinib (NCT04927975, NCT06118411)—with strong results reported so far.11
The effects of JAK inhibitors can be striking, as shown in the Figure. A patient of one of the authors (J.E.H.) used topical ruxolitinib on only the left arm for approximately 36 weeks and results were as expected—strong repigmentation of only the treated area, which is possible with JAK inhibitors. Indeed, 2 phase 3 studies—Topical Ruxolitinib Evaluation in Vitiligo (TRuE-V1 and TRuE-V2)—showed that approximately 30% of participants in TRuE-V1 (N=330) and 30.9% of participants in TRuE-V2 (N=344) achieved at least 75% improvement over baseline in the facial vitiligo area scoring index (VASI).12 In the oral ritlecitinib phase 2b study, 12.1% of the 187 participants on the highest tested dose of ritlecitinib (loading dose of 200 mg/d for 28 days, followed by 50 mg/d maintenance dose) achieved at least 75% improvement over baseline in the VASI at 24 weeks.11 Although this rate is lower than for topical ruxolitinib, this trial required all participants to have active disease (unlike the TRuE-V trials of ruxolitinib), which likely created a higher bar for repigmentation and thus resulted in fewer participants achieving the primary outcome at the early 6-month end point. Extension of treatment through 48 weeks demonstrated continued improvement over baseline without any evidence of plateau.11 Although treatment with JAK inhibitors can result in dramatic repigmentation of vitiligo patches, it falls short of providing a permanent cure, as stopping treatment results in relapse (ie, the return of depigmented lesions).
Racial Disparities in Clinical Trials
Even though vitiligo affects all skin types and races/ethnicities with similar prevalence and severity, the proportion of individuals with darker skin types enrolled in these clinical trials fails to match their representation in the population as a whole. A study examining the prevalence of vitiligo in the United States reported that Black or African American individuals represented 15.8% of vitiligo diagnoses in the United States4 even though they are only 12.7% of the total US population. However, Black or African American individuals comprised only 5% of the combined participants in the TRuE-V clinical trials for topical ruxolitinib12 and 2.7% of the participants in the phase 2b study of oral ritlecitinib.11 This lack of appropriate representation is not unique to JAK inhibitors or other vitiligo trials. Indeed, the US Food and Drug Administration reported that Black or African American individuals comprised only 8% of participants for all clinical trials in 2020.13
Efficacy Metrics Beyond Repigmentation
Disparities in quality-of-life (QOL) metrics in diseases affecting individuals with skin of color also exist. In vitiligo, the contrast between affected and unaffected skin is greater in patients with skin of color, which means that for a given VASI score, the visibility of depigmentation as well as repigmentation may be variable among patients. Additionally, there is evidence that QOL concerns vary between patients with skin of color and those with lighter skin types. Ezzedine et al14 found that QOL concerns in vitiligo patients with darker skin focused more on appearance, while concerns in vitiligo patients with lighter skin focused more on skin cancer risk. In addition to QOL differences among individuals with different skin types, there also are well-documented differences in attitudes to vitiligo among certain ethnic or cultural groups.15 For example, the Rigveda (an ancient Hindu text) indicates that individuals with vitiligo and their progeny are disqualified from marriage. Although the JAK inhibitor clinical trials for vitiligo did not appear to show differences in the degree of repigmentation among different skin types or races/ethnicities, QOL measures were not collected as a secondary end point in these studies—despite the fact that at least 1 study had documented that QOL measures were not uniform across patients when stratified by age and extent of disease.1,11,12 This same study also presented limited data suggestive of lower QOL in patients with the darkest skin phototype.1
Considerations for Future Clinical Trials
It is logical to assume that every clinical trialist in dermatology seeks equitable representation among a diverse set of races, ethnicities, and skin types, but achieving this goal remains elusive. Two recent publications16,17 outlined the challenges and examined solutions to address enrollment disparities, including several barriers to diversity among clinical trial participants: awareness of the clinical trials among minority populations; easy access to clinical trial sites; reluctance to participate because of prior experiences of discrimination, even if unrelated to clinical trials; and a lack of workforce diversity among the clinical trialist teams. To overcome these barriers, a multifaceted approach is needed that requires action at the level of the patient, provider, community, and institution. Once diverse representation is achieved, investigators should consider the need for QOL metrics as a secondary outcome in their trials, which will ensure that the intended clinical effect is matched by patient expectations across different races and ethnicities based on the potential differential impact that diseases such as vitiligo can have on patients with skin of color.
- Bibeau K, Pandya AG, Ezzedine K, et al. Vitiligo prevalence and quality of life among adults in Europe, Japan and the USA. J Eur Acad Dermatol Venereol. 2022;36:1831-1844.
- Jin Y, Roberts GHL, Ferrara TM, et al. Early-onset autoimmune vitiligo associated with an enhancer variant haplotype that upregulates class II HLA expression. Nat Commun. 2019;10:391.
- Rodrigues M, Ezzedine K, Hamzavi I, et al; Vitiligo Working Group. New discoveries in the pathogenesis and classification of vitiligo. J Am Acad Dermatol. 2017;77:1-13.
- Gandhi K, Ezzedine K, Anastassopoulos KP, et al. Prevalence of vitiligo among adults in the United States. JAMA Dermatol. 2022;158:43-50.
- Spritz RA, Santorico SA. The genetic basis of vitiligo. J Invest Dermatol. 2021;141:265-73.
- Harris JE. Chemical-induced vitiligo. Dermatol Clin. 2017;35:151-161.
- Ahmed F, Moseley I, Ragi SD, et al. Vitiligo in underrepresented communities: an all of us database analysis. J Am Acad Dermatol. 2023;88:945-948.
- Frisoli ML, Essien K, Harris JE. Vitiligo: mechanisms of pathogenesis and treatment. Annu Rev Immunol. 2020;38:621-648.
- Richmond JM, Strassner JP, Zapata L Jr, et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. 2018;10:eaam7710.
- Richmond JM, Strassner JP, Rashighi M, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol. 2019;139:769-778.
- Ezzedine K, Peeva E, Yamaguchi Y, et al. Efficacy and safety of oral ritlecitinib for the treatment of active nonsegmental vitiligo: a randomized phase 2b clinical trial. J Am Acad Dermatol. 2023;88:395-403.
- Rosmarin D, Passeron T, Pandya AG, et al. Two phase 3, randomized, controlled trials of ruxolitinib cream for vitiligo. N Engl J Med. 2022;387:1445-1455.
- Cavazzoni P, Anagnostiadis E, Lolic M. Drug trials snapshots summary report. US Food and Drug Administration website. Accessed March 19, 2024. https://www.fda.gov/media/145718/download
- Ezzedine K, Grimes PE, Meurant JM, et al. Living with vitiligo: results from a national survey indicate differences between skin phototypes. Br J Dermatol. 2015;173:607-609.
- Elbuluk N, Ezzedine K. Quality of life, burden of disease, co-morbidities, and systemic effects in vitiligo patients. Dermatol Clin. 2017;35:117-128.
- Kahn JM, Gray DM 2nd, Oliveri JM, et al. Strategies to improve diversity, equity, and inclusion in clinical trials. Cancer. 2022;128:216-221.
- Nolan TS, McKoy A, Gray DM 2nd, et al. Virtual community engagement for retention of black men in clinical research. Am J Mens Health. 2023;17:15579883221147767.
- Bibeau K, Pandya AG, Ezzedine K, et al. Vitiligo prevalence and quality of life among adults in Europe, Japan and the USA. J Eur Acad Dermatol Venereol. 2022;36:1831-1844.
- Jin Y, Roberts GHL, Ferrara TM, et al. Early-onset autoimmune vitiligo associated with an enhancer variant haplotype that upregulates class II HLA expression. Nat Commun. 2019;10:391.
- Rodrigues M, Ezzedine K, Hamzavi I, et al; Vitiligo Working Group. New discoveries in the pathogenesis and classification of vitiligo. J Am Acad Dermatol. 2017;77:1-13.
- Gandhi K, Ezzedine K, Anastassopoulos KP, et al. Prevalence of vitiligo among adults in the United States. JAMA Dermatol. 2022;158:43-50.
- Spritz RA, Santorico SA. The genetic basis of vitiligo. J Invest Dermatol. 2021;141:265-73.
- Harris JE. Chemical-induced vitiligo. Dermatol Clin. 2017;35:151-161.
- Ahmed F, Moseley I, Ragi SD, et al. Vitiligo in underrepresented communities: an all of us database analysis. J Am Acad Dermatol. 2023;88:945-948.
- Frisoli ML, Essien K, Harris JE. Vitiligo: mechanisms of pathogenesis and treatment. Annu Rev Immunol. 2020;38:621-648.
- Richmond JM, Strassner JP, Zapata L Jr, et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. 2018;10:eaam7710.
- Richmond JM, Strassner JP, Rashighi M, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol. 2019;139:769-778.
- Ezzedine K, Peeva E, Yamaguchi Y, et al. Efficacy and safety of oral ritlecitinib for the treatment of active nonsegmental vitiligo: a randomized phase 2b clinical trial. J Am Acad Dermatol. 2023;88:395-403.
- Rosmarin D, Passeron T, Pandya AG, et al. Two phase 3, randomized, controlled trials of ruxolitinib cream for vitiligo. N Engl J Med. 2022;387:1445-1455.
- Cavazzoni P, Anagnostiadis E, Lolic M. Drug trials snapshots summary report. US Food and Drug Administration website. Accessed March 19, 2024. https://www.fda.gov/media/145718/download
- Ezzedine K, Grimes PE, Meurant JM, et al. Living with vitiligo: results from a national survey indicate differences between skin phototypes. Br J Dermatol. 2015;173:607-609.
- Elbuluk N, Ezzedine K. Quality of life, burden of disease, co-morbidities, and systemic effects in vitiligo patients. Dermatol Clin. 2017;35:117-128.
- Kahn JM, Gray DM 2nd, Oliveri JM, et al. Strategies to improve diversity, equity, and inclusion in clinical trials. Cancer. 2022;128:216-221.
- Nolan TS, McKoy A, Gray DM 2nd, et al. Virtual community engagement for retention of black men in clinical research. Am J Mens Health. 2023;17:15579883221147767.
Practice Points
- Vitiligo is an autoimmune disease of the skin that affects all skin types but can be particularly disfiguring in those with skin of color.
- Ruxolitinib, a topical Janus kinase (JAK) inhibitor, is the only US Food and Drug Administration–approved treatment to repigment the skin in vitiligo and has shown efficacy for individuals with all skin phototypes.
- Individuals with skin of color are underrepresented in patient cohorts for JAK inhibitor clinical trials for vitiligo, mirroring a phenomenon seen in the majority of clinical trials. Ensuring diverse participant enrollment and measuring quality-of-life metrics will strengthen future clinical trials for treatment of vitiligo and other skin diseases impacting patients with skin of color.
