User login
System may better predict thrombosis in lymphoma

DUBROVNIK, CROATIA—An updated scoring system can more accurately identify lymphoma patients who may require thromboprophylaxis, according to researchers.
The revised scoring system, ThroLy, proved more effective than other systems for predicting thromboembolic events in lymphoma patients.
Researchers found the updated ThroLy had a positive predictive value of 22% to 25%, a negative predictive value of 96%, sensitivity of 56% to 57%, and specificity of 85% to 87%.
Darko Antić, MD, PhD, of the University of Belgrade in Serbia, presented these findings at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Dr. Antić said he and his colleagues developed ThroLy because other systems used to predict venous thromboembolism (VTE) are not quite right for lymphoma. He noted that the Padua score is not designed for cancer patients, and the Khorana score is predominantly for solid tumor malignancies.
“It’s good . . . , but it’s not specific for lymphoma patients,” Dr. Antić said.
With this in mind, he and his colleagues developed ThroLy. They based the scoring system on variables used in the Padua and Khorana systems as well as variables that are specific to lymphoma patients.
In a past study*, the researchers found several variables that were independently associated with risk for VTE in lymphoma:
- Previous VTE
- Previous acute myocardial infarction/stroke
- Mediastinal involvement
- Body mass index > 30 kg/m2
- Reduced mobility
- Extranodal localization
- Development of neutropenia
- Hemoglobin level < 100g/L.
Previous VTE, previous acute myocardial infarction/stroke, obesity, and mediastinal involvement were all worth 2 points, and the other factors were worth a single point.
Patients with scores of 0 to 1 were considered low-risk, patients with scores of 2 to 3 were considered intermediate-risk, and patients with scores of 4 or greater were considered high-risk.
Prospective validation
To validate and refine ThroLy, Dr. Antić and his colleagues used it to assess 1723 lymphoma patients treated at 8 institutions in Austria, Croatia, France, Jordan, Macedonia, Spain, Switzerland, and the United States.
Patients had indolent non-Hodgkin lymphoma (n=467), aggressive non-Hodgkin lymphoma (n=647), chronic lymphocytic leukemia/small lymphocytic lymphoma (n=235), and Hodgkin lymphoma (n=366). Most subjects (84%) were outpatients.
Nine percent of patients had thrombosis (n=142), with 7% having VTE (n=121).
ThroLy had a positive predictive value of 17%, compared to 11% with Khorana and 13% with Padua. The negative predictive value was 93%, 92%, and 95%, respectively.
The sensitivity was 51% with ThroLy, 42% with Khorana, and 70% with Padua. The specificity was 72%, 64%, and 52%, respectively.
“The positive predictive value was low [with ThroLy] but definitely higher than the positive predictive value of the other two [scoring systems],” Dr. Antić noted.
Updated models
To further improve ThroLy, the researchers updated the system, creating two new models.
Model 1 included the following variables:
- Type of lymphoma/clinical stage (aggressive/advanced)—1 point
- Previous VTE—5 points
- Reduced mobility—2 points
- Hemoglobin level < 100 g/L—1 point
- Presence of vascular devices—1 point.
Model 2 included all of the aforementioned variables as well as thrombophilic condition, which was worth 1 point.
With these models, patients were divided into two risk groups—low-risk (≤ 2 points) and high-risk (>2 points).
For Model 1, the positive predictive value was 22%, the negative predictive value was 96%, the sensitivity was 56%, and the specificity was 85%.
For Model 2, the positive predictive value was 25%, the negative predictive value was 96%, the sensitivity was 57%, and the specificity was 87%.
Dr. Antić said there were no major differences in model discrimination and calibration according to the country in which a patient was treated or whether patients were treated in inpatient or outpatient settings.
Dr. Antić did not report any conflicts of interest.
*Antić D et al. Am J Hematol. 2016 Oct;91(10):1014-9. doi: 10.1002/ajh.24466.
DUBROVNIK, CROATIA—An updated scoring system can more accurately identify lymphoma patients who may require thromboprophylaxis, according to researchers.
The revised scoring system, ThroLy, proved more effective than other systems for predicting thromboembolic events in lymphoma patients.
Researchers found the updated ThroLy had a positive predictive value of 22% to 25%, a negative predictive value of 96%, sensitivity of 56% to 57%, and specificity of 85% to 87%.
Darko Antić, MD, PhD, of the University of Belgrade in Serbia, presented these findings at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Dr. Antić said he and his colleagues developed ThroLy because other systems used to predict venous thromboembolism (VTE) are not quite right for lymphoma. He noted that the Padua score is not designed for cancer patients, and the Khorana score is predominantly for solid tumor malignancies.
“It’s good . . . , but it’s not specific for lymphoma patients,” Dr. Antić said.
With this in mind, he and his colleagues developed ThroLy. They based the scoring system on variables used in the Padua and Khorana systems as well as variables that are specific to lymphoma patients.
In a past study*, the researchers found several variables that were independently associated with risk for VTE in lymphoma:
- Previous VTE
- Previous acute myocardial infarction/stroke
- Mediastinal involvement
- Body mass index > 30 kg/m2
- Reduced mobility
- Extranodal localization
- Development of neutropenia
- Hemoglobin level < 100g/L.
Previous VTE, previous acute myocardial infarction/stroke, obesity, and mediastinal involvement were all worth 2 points, and the other factors were worth a single point.
Patients with scores of 0 to 1 were considered low-risk, patients with scores of 2 to 3 were considered intermediate-risk, and patients with scores of 4 or greater were considered high-risk.
Prospective validation
To validate and refine ThroLy, Dr. Antić and his colleagues used it to assess 1723 lymphoma patients treated at 8 institutions in Austria, Croatia, France, Jordan, Macedonia, Spain, Switzerland, and the United States.
Patients had indolent non-Hodgkin lymphoma (n=467), aggressive non-Hodgkin lymphoma (n=647), chronic lymphocytic leukemia/small lymphocytic lymphoma (n=235), and Hodgkin lymphoma (n=366). Most subjects (84%) were outpatients.
Nine percent of patients had thrombosis (n=142), with 7% having VTE (n=121).
ThroLy had a positive predictive value of 17%, compared to 11% with Khorana and 13% with Padua. The negative predictive value was 93%, 92%, and 95%, respectively.
The sensitivity was 51% with ThroLy, 42% with Khorana, and 70% with Padua. The specificity was 72%, 64%, and 52%, respectively.
“The positive predictive value was low [with ThroLy] but definitely higher than the positive predictive value of the other two [scoring systems],” Dr. Antić noted.
Updated models
To further improve ThroLy, the researchers updated the system, creating two new models.
Model 1 included the following variables:
- Type of lymphoma/clinical stage (aggressive/advanced)—1 point
- Previous VTE—5 points
- Reduced mobility—2 points
- Hemoglobin level < 100 g/L—1 point
- Presence of vascular devices—1 point.
Model 2 included all of the aforementioned variables as well as thrombophilic condition, which was worth 1 point.
With these models, patients were divided into two risk groups—low-risk (≤ 2 points) and high-risk (>2 points).
For Model 1, the positive predictive value was 22%, the negative predictive value was 96%, the sensitivity was 56%, and the specificity was 85%.
For Model 2, the positive predictive value was 25%, the negative predictive value was 96%, the sensitivity was 57%, and the specificity was 87%.
Dr. Antić said there were no major differences in model discrimination and calibration according to the country in which a patient was treated or whether patients were treated in inpatient or outpatient settings.
Dr. Antić did not report any conflicts of interest.
*Antić D et al. Am J Hematol. 2016 Oct;91(10):1014-9. doi: 10.1002/ajh.24466.
DUBROVNIK, CROATIA—An updated scoring system can more accurately identify lymphoma patients who may require thromboprophylaxis, according to researchers.
The revised scoring system, ThroLy, proved more effective than other systems for predicting thromboembolic events in lymphoma patients.
Researchers found the updated ThroLy had a positive predictive value of 22% to 25%, a negative predictive value of 96%, sensitivity of 56% to 57%, and specificity of 85% to 87%.
Darko Antić, MD, PhD, of the University of Belgrade in Serbia, presented these findings at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Dr. Antić said he and his colleagues developed ThroLy because other systems used to predict venous thromboembolism (VTE) are not quite right for lymphoma. He noted that the Padua score is not designed for cancer patients, and the Khorana score is predominantly for solid tumor malignancies.
“It’s good . . . , but it’s not specific for lymphoma patients,” Dr. Antić said.
With this in mind, he and his colleagues developed ThroLy. They based the scoring system on variables used in the Padua and Khorana systems as well as variables that are specific to lymphoma patients.
In a past study*, the researchers found several variables that were independently associated with risk for VTE in lymphoma:
- Previous VTE
- Previous acute myocardial infarction/stroke
- Mediastinal involvement
- Body mass index > 30 kg/m2
- Reduced mobility
- Extranodal localization
- Development of neutropenia
- Hemoglobin level < 100g/L.
Previous VTE, previous acute myocardial infarction/stroke, obesity, and mediastinal involvement were all worth 2 points, and the other factors were worth a single point.
Patients with scores of 0 to 1 were considered low-risk, patients with scores of 2 to 3 were considered intermediate-risk, and patients with scores of 4 or greater were considered high-risk.
Prospective validation
To validate and refine ThroLy, Dr. Antić and his colleagues used it to assess 1723 lymphoma patients treated at 8 institutions in Austria, Croatia, France, Jordan, Macedonia, Spain, Switzerland, and the United States.
Patients had indolent non-Hodgkin lymphoma (n=467), aggressive non-Hodgkin lymphoma (n=647), chronic lymphocytic leukemia/small lymphocytic lymphoma (n=235), and Hodgkin lymphoma (n=366). Most subjects (84%) were outpatients.
Nine percent of patients had thrombosis (n=142), with 7% having VTE (n=121).
ThroLy had a positive predictive value of 17%, compared to 11% with Khorana and 13% with Padua. The negative predictive value was 93%, 92%, and 95%, respectively.
The sensitivity was 51% with ThroLy, 42% with Khorana, and 70% with Padua. The specificity was 72%, 64%, and 52%, respectively.
“The positive predictive value was low [with ThroLy] but definitely higher than the positive predictive value of the other two [scoring systems],” Dr. Antić noted.
Updated models
To further improve ThroLy, the researchers updated the system, creating two new models.
Model 1 included the following variables:
- Type of lymphoma/clinical stage (aggressive/advanced)—1 point
- Previous VTE—5 points
- Reduced mobility—2 points
- Hemoglobin level < 100 g/L—1 point
- Presence of vascular devices—1 point.
Model 2 included all of the aforementioned variables as well as thrombophilic condition, which was worth 1 point.
With these models, patients were divided into two risk groups—low-risk (≤ 2 points) and high-risk (>2 points).
For Model 1, the positive predictive value was 22%, the negative predictive value was 96%, the sensitivity was 56%, and the specificity was 85%.
For Model 2, the positive predictive value was 25%, the negative predictive value was 96%, the sensitivity was 57%, and the specificity was 87%.
Dr. Antić said there were no major differences in model discrimination and calibration according to the country in which a patient was treated or whether patients were treated in inpatient or outpatient settings.
Dr. Antić did not report any conflicts of interest.
*Antić D et al. Am J Hematol. 2016 Oct;91(10):1014-9. doi: 10.1002/ajh.24466.
Researchers say rethink ‘arbitrary categorization’ of VTE risk

Danish researchers conducted a large, 16-year study of patients with venous thromboembolism (VTE) and found a high recurrence risk in all types of VTE.
At 6 months of follow-up, patients with unprovoked and provoked VTE had similar risk of recurrence, lower than that for patients with cancer-related VTE.
But at a 10-year follow-up, patients with unprovoked VTE had a recurrence risk similar to cancer-related VTE.
Based on these findings, the investigators concluded that risk stratification for these patients needs to be optimized.
“Our findings indicate that we may need to rethink arbitrary categorization, considering the heterogeneity of patients with venous thromboembolism,” they wrote.
They published their findings in The American Journal of Medicine.
The investigators used data from 3 nationwide Danish registries to analyze the risk of recurrent VTE in 73,993 patients with incident VTE. They stratified the patients according to whether the VTE was unprovoked, provoked, or cancer-related.
Provoked VTE occurs in patients without cancer but with other contributing factors such as surgery or trauma, and unprovoked VTE occurs without well-known provoking risk factors. Investigators did not include non-melanoma skin cancer in the cancer-related VTE classification.
Median age of the study population was 62.3 years and 54.1% were women.
During a median follow-up of 3.7 years, 9,205 patients experienced a recurrent event.
At the 6-month follow-up, the recurrence rates per 100 person-years were 6.92 for provoked, 6.80 for unprovoked, and 9.06 for cancer-related VTE.
And at the 10-year follow-up, recurrence rates were 2.22 (provoked), 2.84 (unprovoked), and 3.70 (cancer-related). This corresponded to an 18% higher adjusted relative risk of recurrence for patients with unprovoked VTE than patients with provoked VTE.
The investigators observed that at 10 years, the recurrence risk following an unprovoked VTE resembled the risk of patients with cancer-related VTE.
They suggested the mechanism for this could be a “rebound thrombosis” caused by a discontinuation of oral anticoagulants after an unprovoked VTE.
The investigators noted that the findings persisted through various sensitivity analyses “conducted to challenge the robustness of our finding.”
Two areas of concern, they wrote, arise from these findings: how long to anticoagulate patients and which patients to treat.
"Optimal duration of anticoagulation is a pivotal and an ongoing scientific and clinical concern," explained lead investigator Ida Ehlers Albertsen, MD, of Aalborg University in Denmark.
"The emergence of the non-vitamin K antagonist oral anticoagulants has changed the landscape for prevention of thrombosis, and contemporary risk stratification approaches may need to be adjusted according to these effective and safer agents."
Danish researchers conducted a large, 16-year study of patients with venous thromboembolism (VTE) and found a high recurrence risk in all types of VTE.
At 6 months of follow-up, patients with unprovoked and provoked VTE had similar risk of recurrence, lower than that for patients with cancer-related VTE.
But at a 10-year follow-up, patients with unprovoked VTE had a recurrence risk similar to cancer-related VTE.
Based on these findings, the investigators concluded that risk stratification for these patients needs to be optimized.
“Our findings indicate that we may need to rethink arbitrary categorization, considering the heterogeneity of patients with venous thromboembolism,” they wrote.
They published their findings in The American Journal of Medicine.
The investigators used data from 3 nationwide Danish registries to analyze the risk of recurrent VTE in 73,993 patients with incident VTE. They stratified the patients according to whether the VTE was unprovoked, provoked, or cancer-related.
Provoked VTE occurs in patients without cancer but with other contributing factors such as surgery or trauma, and unprovoked VTE occurs without well-known provoking risk factors. Investigators did not include non-melanoma skin cancer in the cancer-related VTE classification.
Median age of the study population was 62.3 years and 54.1% were women.
During a median follow-up of 3.7 years, 9,205 patients experienced a recurrent event.
At the 6-month follow-up, the recurrence rates per 100 person-years were 6.92 for provoked, 6.80 for unprovoked, and 9.06 for cancer-related VTE.
And at the 10-year follow-up, recurrence rates were 2.22 (provoked), 2.84 (unprovoked), and 3.70 (cancer-related). This corresponded to an 18% higher adjusted relative risk of recurrence for patients with unprovoked VTE than patients with provoked VTE.
The investigators observed that at 10 years, the recurrence risk following an unprovoked VTE resembled the risk of patients with cancer-related VTE.
They suggested the mechanism for this could be a “rebound thrombosis” caused by a discontinuation of oral anticoagulants after an unprovoked VTE.
The investigators noted that the findings persisted through various sensitivity analyses “conducted to challenge the robustness of our finding.”
Two areas of concern, they wrote, arise from these findings: how long to anticoagulate patients and which patients to treat.
"Optimal duration of anticoagulation is a pivotal and an ongoing scientific and clinical concern," explained lead investigator Ida Ehlers Albertsen, MD, of Aalborg University in Denmark.
"The emergence of the non-vitamin K antagonist oral anticoagulants has changed the landscape for prevention of thrombosis, and contemporary risk stratification approaches may need to be adjusted according to these effective and safer agents."
Danish researchers conducted a large, 16-year study of patients with venous thromboembolism (VTE) and found a high recurrence risk in all types of VTE.
At 6 months of follow-up, patients with unprovoked and provoked VTE had similar risk of recurrence, lower than that for patients with cancer-related VTE.
But at a 10-year follow-up, patients with unprovoked VTE had a recurrence risk similar to cancer-related VTE.
Based on these findings, the investigators concluded that risk stratification for these patients needs to be optimized.
“Our findings indicate that we may need to rethink arbitrary categorization, considering the heterogeneity of patients with venous thromboembolism,” they wrote.
They published their findings in The American Journal of Medicine.
The investigators used data from 3 nationwide Danish registries to analyze the risk of recurrent VTE in 73,993 patients with incident VTE. They stratified the patients according to whether the VTE was unprovoked, provoked, or cancer-related.
Provoked VTE occurs in patients without cancer but with other contributing factors such as surgery or trauma, and unprovoked VTE occurs without well-known provoking risk factors. Investigators did not include non-melanoma skin cancer in the cancer-related VTE classification.
Median age of the study population was 62.3 years and 54.1% were women.
During a median follow-up of 3.7 years, 9,205 patients experienced a recurrent event.
At the 6-month follow-up, the recurrence rates per 100 person-years were 6.92 for provoked, 6.80 for unprovoked, and 9.06 for cancer-related VTE.
And at the 10-year follow-up, recurrence rates were 2.22 (provoked), 2.84 (unprovoked), and 3.70 (cancer-related). This corresponded to an 18% higher adjusted relative risk of recurrence for patients with unprovoked VTE than patients with provoked VTE.
The investigators observed that at 10 years, the recurrence risk following an unprovoked VTE resembled the risk of patients with cancer-related VTE.
They suggested the mechanism for this could be a “rebound thrombosis” caused by a discontinuation of oral anticoagulants after an unprovoked VTE.
The investigators noted that the findings persisted through various sensitivity analyses “conducted to challenge the robustness of our finding.”
Two areas of concern, they wrote, arise from these findings: how long to anticoagulate patients and which patients to treat.
"Optimal duration of anticoagulation is a pivotal and an ongoing scientific and clinical concern," explained lead investigator Ida Ehlers Albertsen, MD, of Aalborg University in Denmark.
"The emergence of the non-vitamin K antagonist oral anticoagulants has changed the landscape for prevention of thrombosis, and contemporary risk stratification approaches may need to be adjusted according to these effective and safer agents."
Weighing the costs of CAR T-cell therapy

The cost-effectiveness of tisagenlecleucel (Kymriah) depends on long-term clinical outcomes, which are presently unknown, according to investigators.
If the long-term outcomes are more modest than clinical trials suggest, then payers may be unwilling to cover the costly therapy, reported John K. Lin, MD, of Stanford University, and his colleagues.
Lowering the price or setting up an outcomes-based pricing structure may be necessary to get insurers to cover the therapy.
Tisagenlecleucel is an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy that was approved by the U.S. Food and Drug Administration in August 2017 for relapsed or refractory pediatric B-cell acute lymphoblastic leukemia (ALL).
In 2018, the FDA expanded the indication for tisagenlecleucel to include adults with relapsed or refractory large B-cell lymphoma, though outcomes from lymphoma trials are not analyzed in the current study.
At a wholesale acquisition cost of $475,000 per infusion, it is the most expensive existing oncology therapy to date, and can be accompanied by expensive, potentially fatal adverse effects.
However, clinical trials suggest that tisagenlecleucel can offer years of relapse-free remission, thereby allowing patients to forgo other expensive therapies such as hematopoietic stem cell transplantation (HSCT).
“Although tisagenlecleucel-induced remission rates are promising, compared with those of established therapies (greater than 80% vs. less than 50%), only short-term follow-up data currently exist,” the investigators wrote in the Journal of Clinical Oncology.
“Given the high cost and broad applicability in other malignancies of tisagenlecleucel, a pressing question for policy makers, payers, patients, and clinicians is whether the cost of therapy represents reasonable value.”
The study used a Markov model to assess various long-term clinical outcome rates and cost thresholds of tisagenlecleucel. The lifetime cost of therapy was assessed and compared with costs of existing therapies.
The results showed that a 5-year relapse free survival rate of 40% would make the present cost ($475,000) of tisagenlecleucel economically reasonable. In this scenario, the increased life expectancy would be 12.1 years and would result in an additional 5.07 quality-adjusted life years (QALY) gained at a cost of $61,000 per QALY, compared with blinatumomab.
But if long-term outcomes are less favorable, tisagenlecleucel becomes much less cost effective. A 5-year relapse-free survival rate of 20% would drop increased life expectancy to 3.8 years, resulting in 1.80 QALYs gained and raising the cost to $151,000 per QALY.
“Our results suggest that at tisagenlecleucel’s current price and payment structure, its economic value is uncertain,” the investigators wrote.
They suggested a price drop to $200,000 or $350,000, which would allow the drug to remain cost effective even in a worse-case scenario, in which patients relapse and tisagenlecleucel is a bridge to transplant.
Another option is to move to outcomes-based pricing. Making payment conditional on 7 months of remission would make the treatment cost effective, according to the analysis.
“Price reductions of tisagenlecleucel or payment only for longer-term remissions would favorably influence cost-effectiveness, even if long-term clinical outcomes are modest,” the investigators wrote.
The study was funded by a Veterans Affairs Office of Academic Affiliations advanced fellowship in health service and research development, and a National Center for Advancing Translational Science Clinical and Translational Science Award.
One of the study coauthors reported consulting and research funding from Novartis.
The cost-effectiveness of tisagenlecleucel (Kymriah) depends on long-term clinical outcomes, which are presently unknown, according to investigators.
If the long-term outcomes are more modest than clinical trials suggest, then payers may be unwilling to cover the costly therapy, reported John K. Lin, MD, of Stanford University, and his colleagues.
Lowering the price or setting up an outcomes-based pricing structure may be necessary to get insurers to cover the therapy.
Tisagenlecleucel is an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy that was approved by the U.S. Food and Drug Administration in August 2017 for relapsed or refractory pediatric B-cell acute lymphoblastic leukemia (ALL).
In 2018, the FDA expanded the indication for tisagenlecleucel to include adults with relapsed or refractory large B-cell lymphoma, though outcomes from lymphoma trials are not analyzed in the current study.
At a wholesale acquisition cost of $475,000 per infusion, it is the most expensive existing oncology therapy to date, and can be accompanied by expensive, potentially fatal adverse effects.
However, clinical trials suggest that tisagenlecleucel can offer years of relapse-free remission, thereby allowing patients to forgo other expensive therapies such as hematopoietic stem cell transplantation (HSCT).
“Although tisagenlecleucel-induced remission rates are promising, compared with those of established therapies (greater than 80% vs. less than 50%), only short-term follow-up data currently exist,” the investigators wrote in the Journal of Clinical Oncology.
“Given the high cost and broad applicability in other malignancies of tisagenlecleucel, a pressing question for policy makers, payers, patients, and clinicians is whether the cost of therapy represents reasonable value.”
The study used a Markov model to assess various long-term clinical outcome rates and cost thresholds of tisagenlecleucel. The lifetime cost of therapy was assessed and compared with costs of existing therapies.
The results showed that a 5-year relapse free survival rate of 40% would make the present cost ($475,000) of tisagenlecleucel economically reasonable. In this scenario, the increased life expectancy would be 12.1 years and would result in an additional 5.07 quality-adjusted life years (QALY) gained at a cost of $61,000 per QALY, compared with blinatumomab.
But if long-term outcomes are less favorable, tisagenlecleucel becomes much less cost effective. A 5-year relapse-free survival rate of 20% would drop increased life expectancy to 3.8 years, resulting in 1.80 QALYs gained and raising the cost to $151,000 per QALY.
“Our results suggest that at tisagenlecleucel’s current price and payment structure, its economic value is uncertain,” the investigators wrote.
They suggested a price drop to $200,000 or $350,000, which would allow the drug to remain cost effective even in a worse-case scenario, in which patients relapse and tisagenlecleucel is a bridge to transplant.
Another option is to move to outcomes-based pricing. Making payment conditional on 7 months of remission would make the treatment cost effective, according to the analysis.
“Price reductions of tisagenlecleucel or payment only for longer-term remissions would favorably influence cost-effectiveness, even if long-term clinical outcomes are modest,” the investigators wrote.
The study was funded by a Veterans Affairs Office of Academic Affiliations advanced fellowship in health service and research development, and a National Center for Advancing Translational Science Clinical and Translational Science Award.
One of the study coauthors reported consulting and research funding from Novartis.
The cost-effectiveness of tisagenlecleucel (Kymriah) depends on long-term clinical outcomes, which are presently unknown, according to investigators.
If the long-term outcomes are more modest than clinical trials suggest, then payers may be unwilling to cover the costly therapy, reported John K. Lin, MD, of Stanford University, and his colleagues.
Lowering the price or setting up an outcomes-based pricing structure may be necessary to get insurers to cover the therapy.
Tisagenlecleucel is an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy that was approved by the U.S. Food and Drug Administration in August 2017 for relapsed or refractory pediatric B-cell acute lymphoblastic leukemia (ALL).
In 2018, the FDA expanded the indication for tisagenlecleucel to include adults with relapsed or refractory large B-cell lymphoma, though outcomes from lymphoma trials are not analyzed in the current study.
At a wholesale acquisition cost of $475,000 per infusion, it is the most expensive existing oncology therapy to date, and can be accompanied by expensive, potentially fatal adverse effects.
However, clinical trials suggest that tisagenlecleucel can offer years of relapse-free remission, thereby allowing patients to forgo other expensive therapies such as hematopoietic stem cell transplantation (HSCT).
“Although tisagenlecleucel-induced remission rates are promising, compared with those of established therapies (greater than 80% vs. less than 50%), only short-term follow-up data currently exist,” the investigators wrote in the Journal of Clinical Oncology.
“Given the high cost and broad applicability in other malignancies of tisagenlecleucel, a pressing question for policy makers, payers, patients, and clinicians is whether the cost of therapy represents reasonable value.”
The study used a Markov model to assess various long-term clinical outcome rates and cost thresholds of tisagenlecleucel. The lifetime cost of therapy was assessed and compared with costs of existing therapies.
The results showed that a 5-year relapse free survival rate of 40% would make the present cost ($475,000) of tisagenlecleucel economically reasonable. In this scenario, the increased life expectancy would be 12.1 years and would result in an additional 5.07 quality-adjusted life years (QALY) gained at a cost of $61,000 per QALY, compared with blinatumomab.
But if long-term outcomes are less favorable, tisagenlecleucel becomes much less cost effective. A 5-year relapse-free survival rate of 20% would drop increased life expectancy to 3.8 years, resulting in 1.80 QALYs gained and raising the cost to $151,000 per QALY.
“Our results suggest that at tisagenlecleucel’s current price and payment structure, its economic value is uncertain,” the investigators wrote.
They suggested a price drop to $200,000 or $350,000, which would allow the drug to remain cost effective even in a worse-case scenario, in which patients relapse and tisagenlecleucel is a bridge to transplant.
Another option is to move to outcomes-based pricing. Making payment conditional on 7 months of remission would make the treatment cost effective, according to the analysis.
“Price reductions of tisagenlecleucel or payment only for longer-term remissions would favorably influence cost-effectiveness, even if long-term clinical outcomes are modest,” the investigators wrote.
The study was funded by a Veterans Affairs Office of Academic Affiliations advanced fellowship in health service and research development, and a National Center for Advancing Translational Science Clinical and Translational Science Award.
One of the study coauthors reported consulting and research funding from Novartis.
CAR T therapy being explored in Hodgkin lymphoma

New York—Although the data set is small and not yet mature, chimeric antigen receptor (CAR) T-cell therapy appears to be a promising approach for Hodgkin lymphoma, according to Philippe Armand, MD, PhD, of Dana-Farber/Brigham and Women’s Cancer Center and the Massachusetts General Hospital Cancer Center.
While based on a handful of patients, the data do suggest this approach may play a role either by targeting CD30 or Epstein Barr virus (EBV), Dr. Armand said in a presentation at the NCCN 13th Annual Congress: Hematologic Malignancies.
“Most importantly perhaps, like it's experience outside of Hodgkin lymphoma, it may really have curative potential, based on the long CR rates that have been already exhibited,” he told attendees at the NCCN conference.
Much of the published clinical experience to date is with CD30-directed CAR Ts, Dr. Armand said, noting that in Hodgkin lymphoma, results so far show promise for this particular approach.
In a recent phase 1 dose escalation study, 9 patients with relapsed/refractory Hodgkin lymphoma or anaplastic large-cell lymphoma (ALCL) received infusions of autologous T cells modified to express CD30-specific CAR T cells encoding the CD28 costimulatory domain, with no conditioning regimen.
Out of 7 relapsed Hodgkin patients, one had a complete response (CR) lasting beyond 2.5 years following a second infusion. Another had a CR persisting almost 2 years and 3 had transient stable disease.
One of the 2 ALCL patients had a CR lasting 9 months after a fourth infusion. No toxicities attributable to the therapy were seen, according to investigators.
The CD30 CAR T cells are being evaluated with a conditioning regimen in the phase 1 RELY-30 trial. According to Dr. Armand, preliminary results presented at the EBMT 2018 meeting showed better expansion of CAR T cells and responses in 3 out of 5 patients, including 2 CRs.
A CD30-directed CAR T-cell therapy with a 4-1bb costimulatory domain has also been tested in a small group of Hodgkin patients with a response rate of 35%, including some CRs. Response rates were lower in patients with extranodal involvement, although that needs to be validated with further study, according to Dr. Armand.
A considerable amount of active research is ongoing in China, Dr. Armand said, while a phase 1 study of T cells expressing a fully human anti-CD30 CAR is being evaluated in the United States in CD30-expressing lymphomas, he added.
Among non-CD30-targeted products, a CD19 CAR-T approach has been tried in Hodgkin lymphoma, though preliminary results suggest only transient activity.
An interesting approach has been the targeting of EBV, Dr. Armand noted. Recently reported results showed that two doses of T cells with specificity for EBV-derived tumor antigens induced clinical responses in patients with EBV-positive Hodgkin lymphoma.
The cells were engineered to express dominant-negative TGF-β receptor type 2 (DNRII).
“We know that TGF-β provides a strong immunosuppressant signal in the tumor microenvironment,” Dr. Armand said, noting that some of the responses in the 7 evaluable patients lasted 4 years or more.
New York—Although the data set is small and not yet mature, chimeric antigen receptor (CAR) T-cell therapy appears to be a promising approach for Hodgkin lymphoma, according to Philippe Armand, MD, PhD, of Dana-Farber/Brigham and Women’s Cancer Center and the Massachusetts General Hospital Cancer Center.
While based on a handful of patients, the data do suggest this approach may play a role either by targeting CD30 or Epstein Barr virus (EBV), Dr. Armand said in a presentation at the NCCN 13th Annual Congress: Hematologic Malignancies.
“Most importantly perhaps, like it's experience outside of Hodgkin lymphoma, it may really have curative potential, based on the long CR rates that have been already exhibited,” he told attendees at the NCCN conference.
Much of the published clinical experience to date is with CD30-directed CAR Ts, Dr. Armand said, noting that in Hodgkin lymphoma, results so far show promise for this particular approach.
In a recent phase 1 dose escalation study, 9 patients with relapsed/refractory Hodgkin lymphoma or anaplastic large-cell lymphoma (ALCL) received infusions of autologous T cells modified to express CD30-specific CAR T cells encoding the CD28 costimulatory domain, with no conditioning regimen.
Out of 7 relapsed Hodgkin patients, one had a complete response (CR) lasting beyond 2.5 years following a second infusion. Another had a CR persisting almost 2 years and 3 had transient stable disease.
One of the 2 ALCL patients had a CR lasting 9 months after a fourth infusion. No toxicities attributable to the therapy were seen, according to investigators.
The CD30 CAR T cells are being evaluated with a conditioning regimen in the phase 1 RELY-30 trial. According to Dr. Armand, preliminary results presented at the EBMT 2018 meeting showed better expansion of CAR T cells and responses in 3 out of 5 patients, including 2 CRs.
A CD30-directed CAR T-cell therapy with a 4-1bb costimulatory domain has also been tested in a small group of Hodgkin patients with a response rate of 35%, including some CRs. Response rates were lower in patients with extranodal involvement, although that needs to be validated with further study, according to Dr. Armand.
A considerable amount of active research is ongoing in China, Dr. Armand said, while a phase 1 study of T cells expressing a fully human anti-CD30 CAR is being evaluated in the United States in CD30-expressing lymphomas, he added.
Among non-CD30-targeted products, a CD19 CAR-T approach has been tried in Hodgkin lymphoma, though preliminary results suggest only transient activity.
An interesting approach has been the targeting of EBV, Dr. Armand noted. Recently reported results showed that two doses of T cells with specificity for EBV-derived tumor antigens induced clinical responses in patients with EBV-positive Hodgkin lymphoma.
The cells were engineered to express dominant-negative TGF-β receptor type 2 (DNRII).
“We know that TGF-β provides a strong immunosuppressant signal in the tumor microenvironment,” Dr. Armand said, noting that some of the responses in the 7 evaluable patients lasted 4 years or more.
New York—Although the data set is small and not yet mature, chimeric antigen receptor (CAR) T-cell therapy appears to be a promising approach for Hodgkin lymphoma, according to Philippe Armand, MD, PhD, of Dana-Farber/Brigham and Women’s Cancer Center and the Massachusetts General Hospital Cancer Center.
While based on a handful of patients, the data do suggest this approach may play a role either by targeting CD30 or Epstein Barr virus (EBV), Dr. Armand said in a presentation at the NCCN 13th Annual Congress: Hematologic Malignancies.
“Most importantly perhaps, like it's experience outside of Hodgkin lymphoma, it may really have curative potential, based on the long CR rates that have been already exhibited,” he told attendees at the NCCN conference.
Much of the published clinical experience to date is with CD30-directed CAR Ts, Dr. Armand said, noting that in Hodgkin lymphoma, results so far show promise for this particular approach.
In a recent phase 1 dose escalation study, 9 patients with relapsed/refractory Hodgkin lymphoma or anaplastic large-cell lymphoma (ALCL) received infusions of autologous T cells modified to express CD30-specific CAR T cells encoding the CD28 costimulatory domain, with no conditioning regimen.
Out of 7 relapsed Hodgkin patients, one had a complete response (CR) lasting beyond 2.5 years following a second infusion. Another had a CR persisting almost 2 years and 3 had transient stable disease.
One of the 2 ALCL patients had a CR lasting 9 months after a fourth infusion. No toxicities attributable to the therapy were seen, according to investigators.
The CD30 CAR T cells are being evaluated with a conditioning regimen in the phase 1 RELY-30 trial. According to Dr. Armand, preliminary results presented at the EBMT 2018 meeting showed better expansion of CAR T cells and responses in 3 out of 5 patients, including 2 CRs.
A CD30-directed CAR T-cell therapy with a 4-1bb costimulatory domain has also been tested in a small group of Hodgkin patients with a response rate of 35%, including some CRs. Response rates were lower in patients with extranodal involvement, although that needs to be validated with further study, according to Dr. Armand.
A considerable amount of active research is ongoing in China, Dr. Armand said, while a phase 1 study of T cells expressing a fully human anti-CD30 CAR is being evaluated in the United States in CD30-expressing lymphomas, he added.
Among non-CD30-targeted products, a CD19 CAR-T approach has been tried in Hodgkin lymphoma, though preliminary results suggest only transient activity.
An interesting approach has been the targeting of EBV, Dr. Armand noted. Recently reported results showed that two doses of T cells with specificity for EBV-derived tumor antigens induced clinical responses in patients with EBV-positive Hodgkin lymphoma.
The cells were engineered to express dominant-negative TGF-β receptor type 2 (DNRII).
“We know that TGF-β provides a strong immunosuppressant signal in the tumor microenvironment,” Dr. Armand said, noting that some of the responses in the 7 evaluable patients lasted 4 years or more.
Duvelisib bests ofatumumab as monotherapy for treatment of CLL/SLL

Final analysis of the phase 3 DUO trial has shown monotherapy with oral duvelisib results in a statistically significant improvement in progression-free survival (PFS) and overall response rate (ORR) compared to monotherapy with ofatumumab for patients with relapsed or refractory chronic lymphocytic leukemia/small lympchocytic lymphoma (CLL/SLL).
PFS for all patients as assessed by Independent Review Committee (IRC) was a median 13.3 months with duvelisib compared to 9.9 months with ofatumumab (P<0.0001).
ORR was significantly higher with duvelisib, 74% compared to 45%, P<0.0001, regardless of deletion 17p status.
Duvelisib (Copiktra™) was recently approved by the U.S. Food and Drug Administration for CLL/SLL based in part on this head-to-head trial.
The investigators reported the results in Blood.
"The way we treat patients with CLL is changing rapidly as we move from standard chemotherapy-based approaches to more targeted therapies," said principal investigator Ian W. Flinn, MD, PhD, of Sarah Cannon Research Institute in Nashville.
"Based on these data, duvelisib may offer a new treatment option for patients who otherwise may have limited options."
Duvelisib is an oral, dual inhibitor of phosphoinositide 3-kinase (PI3K)-δ and -γ, which means it blocks the survival and proliferation of malignant B cells and also disrupts the recruitment and differentiation of T cells and macrophages within the tumor microenvironment.
Ofatumumab is a humanized anti-CD20 antibody with single-agent efficacy in refractory CLL. It is approved by the FDA as a treatment option in CLL.
Study design
Investigators randomized 319 relapsed or refractory CLL/SLL patients, 160 to the duvelisib arm and 159 to the ofatumumab arm.
Patients in the duvelisib arm self-administered 25 mg capsules twice daily continuously in 28-day cycles. They could take duvelisib for up to 18 cycles, until disease progression or unacceptable toxicity.
Ofatumumab-treated patients received infusions as approved in the product labeling for monotherapy in relapsed CLL. Dosing of ofatumumab could not exceed 12 doses in 7 cycles.
Prophylaxis for Pneumocystis jirovecci was required for all patients on both treatment arms.
Patients were allowed to crossover to a separate extension study to receive the opposite therapy if they had progressive disease.
They were followed for a median of 22.4 months.
Patient characteristics
According to the investigators, patient characteristics were well balanced between the arms.
The majority (60%) were male and the median age in both arms was 69. Most had an ECOG performance status of 0 or 1; 7% in the duvelisib arm and 10% in the ofatumumab arm had a performance status of 2.
Other patient characteristics in the duvelisib and ofatumumab arms, respectively, were:
- Time from initial diagnosis: 7.5 years, 6.7 years
- CLL/SLL, %: 97/5, 99/2
- Bulky disease: 46%, 45%
- Baseline lymphocyte counts: 38x109/L, 35x109/L
- Deletion 17p and/or TP53 mutation: 31%, 33%
- Median number of prior therapies: 2 in each arm
- Previous alkylating agent: 93%, 95%
- Previous monoclonal antibody: 78%, 83%
- Prior purine analog: 60%, 71%
Of the total patients enrolled, 158 patients in the duvelisib arm and 155 in the ofatumumab arm received treatment, for a median exposure of 50 weeks and 23 weeks, respectively.
Efficacy
In addition to the significantly improved overall PFS and ORR with duvelisib, further analysis revealed that PFS also improved for all predefined subgroups.
High-risk patients with deletion 17p/TP53 mutations also experienced a significant improvement in PFS with duvelisib of 12.7 months compared to 9.0 months with ofatumumab by IRC (P=0.0002).
The estimated probability of being progression-free for these patients at 6 and 12 moths was 73% and 55% with duvelisib and 63% and 30% with ofatumumab.
The investigators pointed out that duvelisib treatment was particularly effective in eliciting a lymph node response—85.0% compared to 15.7% with ofatumumab as assessed by IRC (P<0.0001).
Median overall survival was not reached in either arm. The 12-month probability of survival was 86% for both treatments.
Safety
Median treatment exposure was almost twice as long in the duvelisib arm because ofatumumab treatment was not allowed to exceed 12 doses as specified in the prescribing information.
The investigators explained this resulted in a longer adverse event (AE) reporting period for duvelisib.
One hundred twenty-four duvelisib-treated patients discontinued treatment, most commonly due to AEs (35%), disease progression (22%), subject withdrawal (8%), and death (8%).
All ofatumumab-treated patients discontinued treatment by the time of data cutoff, and 67% had completed treatment as per protocol. Others discontinued due to disease progression (20%), subject withdrawal (5%), and AEs (4%).
Eight (5%) duvelisib patients crossed over to ofatumumab therapy at the time of disease progression, and 89 (57%) ofatumumab-treated patients crossed over to duvelisib.
Nearly all patients in both arms experienced an AE.
The most common hematologic malignancies with duvelisib and ofatumumab, respectively, occurring in 10% or more patients were neutropenia (33%, 21%), anemia (23%, 10%), and thrombocytopenia (15%, 6%).
The most common nonhematologic AES with duvelisib were diarrhea (51%), pyrexia (29%), nausea (23%), and cough (21%).
With ofatumumab, the most common nonhematologic AES were infusion-related reaction (19%), cough (14%), and diarrhea, rash, and fatigue (12% each).
Grade 3 or greater AEs occurred in 87% of duvelisib-treated patients and 48% in the ofatumumab arm.
The most common grade 3 or greater events with duvelisib were neutropenia (30%), diarrhea (15%), pneumonia (14%), and anemia (13%).
With ofatumumab, only neutropenia (17%) of grade 3 or higher occurred in 10% or more patients.
Severe immune-related toxicities with duvelisib included colitis (12%) and pneumonitis, alanine transaminase (ALT) or aspartate transaminase (AST) increase (3% each). The events were managed with dose interruptions and steroid therapy for pneumonitis or colitis. All reported events resolved, and none was fatal.
Infectious AEs occurred more frequently with duvelisib, 69% compared to 43% in the ofatumumab arm. Pneumonia (18%) and upper respiratory tract infection (16%) were the most common events.
Three patients in the duvelisib arm and 1 in the ofatumumab arm contracted Pneumocystis jirovecii.
The most frequently reported serious AE was pneumonia (duvelisib 15%; ofatumumab 3%).
Nineteen fatal AEs occurred in patients on the duvelisib arm, 4 of which were related to the study drug: staphylococcal pneumonia (n = 2), sepsis (n=1), and general health deterioration (n = 1).
Seven fatal AEs occurred in patients on the ofatumumab arm, although none was attributed to ofatumumab.
The DUO trial was sponsored by Verastem Oncology and Infinity Pharmaceuticals , Inc.
Final analysis of the phase 3 DUO trial has shown monotherapy with oral duvelisib results in a statistically significant improvement in progression-free survival (PFS) and overall response rate (ORR) compared to monotherapy with ofatumumab for patients with relapsed or refractory chronic lymphocytic leukemia/small lympchocytic lymphoma (CLL/SLL).
PFS for all patients as assessed by Independent Review Committee (IRC) was a median 13.3 months with duvelisib compared to 9.9 months with ofatumumab (P<0.0001).
ORR was significantly higher with duvelisib, 74% compared to 45%, P<0.0001, regardless of deletion 17p status.
Duvelisib (Copiktra™) was recently approved by the U.S. Food and Drug Administration for CLL/SLL based in part on this head-to-head trial.
The investigators reported the results in Blood.
"The way we treat patients with CLL is changing rapidly as we move from standard chemotherapy-based approaches to more targeted therapies," said principal investigator Ian W. Flinn, MD, PhD, of Sarah Cannon Research Institute in Nashville.
"Based on these data, duvelisib may offer a new treatment option for patients who otherwise may have limited options."
Duvelisib is an oral, dual inhibitor of phosphoinositide 3-kinase (PI3K)-δ and -γ, which means it blocks the survival and proliferation of malignant B cells and also disrupts the recruitment and differentiation of T cells and macrophages within the tumor microenvironment.
Ofatumumab is a humanized anti-CD20 antibody with single-agent efficacy in refractory CLL. It is approved by the FDA as a treatment option in CLL.
Study design
Investigators randomized 319 relapsed or refractory CLL/SLL patients, 160 to the duvelisib arm and 159 to the ofatumumab arm.
Patients in the duvelisib arm self-administered 25 mg capsules twice daily continuously in 28-day cycles. They could take duvelisib for up to 18 cycles, until disease progression or unacceptable toxicity.
Ofatumumab-treated patients received infusions as approved in the product labeling for monotherapy in relapsed CLL. Dosing of ofatumumab could not exceed 12 doses in 7 cycles.
Prophylaxis for Pneumocystis jirovecci was required for all patients on both treatment arms.
Patients were allowed to crossover to a separate extension study to receive the opposite therapy if they had progressive disease.
They were followed for a median of 22.4 months.
Patient characteristics
According to the investigators, patient characteristics were well balanced between the arms.
The majority (60%) were male and the median age in both arms was 69. Most had an ECOG performance status of 0 or 1; 7% in the duvelisib arm and 10% in the ofatumumab arm had a performance status of 2.
Other patient characteristics in the duvelisib and ofatumumab arms, respectively, were:
- Time from initial diagnosis: 7.5 years, 6.7 years
- CLL/SLL, %: 97/5, 99/2
- Bulky disease: 46%, 45%
- Baseline lymphocyte counts: 38x109/L, 35x109/L
- Deletion 17p and/or TP53 mutation: 31%, 33%
- Median number of prior therapies: 2 in each arm
- Previous alkylating agent: 93%, 95%
- Previous monoclonal antibody: 78%, 83%
- Prior purine analog: 60%, 71%
Of the total patients enrolled, 158 patients in the duvelisib arm and 155 in the ofatumumab arm received treatment, for a median exposure of 50 weeks and 23 weeks, respectively.
Efficacy
In addition to the significantly improved overall PFS and ORR with duvelisib, further analysis revealed that PFS also improved for all predefined subgroups.
High-risk patients with deletion 17p/TP53 mutations also experienced a significant improvement in PFS with duvelisib of 12.7 months compared to 9.0 months with ofatumumab by IRC (P=0.0002).
The estimated probability of being progression-free for these patients at 6 and 12 moths was 73% and 55% with duvelisib and 63% and 30% with ofatumumab.
The investigators pointed out that duvelisib treatment was particularly effective in eliciting a lymph node response—85.0% compared to 15.7% with ofatumumab as assessed by IRC (P<0.0001).
Median overall survival was not reached in either arm. The 12-month probability of survival was 86% for both treatments.
Safety
Median treatment exposure was almost twice as long in the duvelisib arm because ofatumumab treatment was not allowed to exceed 12 doses as specified in the prescribing information.
The investigators explained this resulted in a longer adverse event (AE) reporting period for duvelisib.
One hundred twenty-four duvelisib-treated patients discontinued treatment, most commonly due to AEs (35%), disease progression (22%), subject withdrawal (8%), and death (8%).
All ofatumumab-treated patients discontinued treatment by the time of data cutoff, and 67% had completed treatment as per protocol. Others discontinued due to disease progression (20%), subject withdrawal (5%), and AEs (4%).
Eight (5%) duvelisib patients crossed over to ofatumumab therapy at the time of disease progression, and 89 (57%) ofatumumab-treated patients crossed over to duvelisib.
Nearly all patients in both arms experienced an AE.
The most common hematologic malignancies with duvelisib and ofatumumab, respectively, occurring in 10% or more patients were neutropenia (33%, 21%), anemia (23%, 10%), and thrombocytopenia (15%, 6%).
The most common nonhematologic AES with duvelisib were diarrhea (51%), pyrexia (29%), nausea (23%), and cough (21%).
With ofatumumab, the most common nonhematologic AES were infusion-related reaction (19%), cough (14%), and diarrhea, rash, and fatigue (12% each).
Grade 3 or greater AEs occurred in 87% of duvelisib-treated patients and 48% in the ofatumumab arm.
The most common grade 3 or greater events with duvelisib were neutropenia (30%), diarrhea (15%), pneumonia (14%), and anemia (13%).
With ofatumumab, only neutropenia (17%) of grade 3 or higher occurred in 10% or more patients.
Severe immune-related toxicities with duvelisib included colitis (12%) and pneumonitis, alanine transaminase (ALT) or aspartate transaminase (AST) increase (3% each). The events were managed with dose interruptions and steroid therapy for pneumonitis or colitis. All reported events resolved, and none was fatal.
Infectious AEs occurred more frequently with duvelisib, 69% compared to 43% in the ofatumumab arm. Pneumonia (18%) and upper respiratory tract infection (16%) were the most common events.
Three patients in the duvelisib arm and 1 in the ofatumumab arm contracted Pneumocystis jirovecii.
The most frequently reported serious AE was pneumonia (duvelisib 15%; ofatumumab 3%).
Nineteen fatal AEs occurred in patients on the duvelisib arm, 4 of which were related to the study drug: staphylococcal pneumonia (n = 2), sepsis (n=1), and general health deterioration (n = 1).
Seven fatal AEs occurred in patients on the ofatumumab arm, although none was attributed to ofatumumab.
The DUO trial was sponsored by Verastem Oncology and Infinity Pharmaceuticals , Inc.
Final analysis of the phase 3 DUO trial has shown monotherapy with oral duvelisib results in a statistically significant improvement in progression-free survival (PFS) and overall response rate (ORR) compared to monotherapy with ofatumumab for patients with relapsed or refractory chronic lymphocytic leukemia/small lympchocytic lymphoma (CLL/SLL).
PFS for all patients as assessed by Independent Review Committee (IRC) was a median 13.3 months with duvelisib compared to 9.9 months with ofatumumab (P<0.0001).
ORR was significantly higher with duvelisib, 74% compared to 45%, P<0.0001, regardless of deletion 17p status.
Duvelisib (Copiktra™) was recently approved by the U.S. Food and Drug Administration for CLL/SLL based in part on this head-to-head trial.
The investigators reported the results in Blood.
"The way we treat patients with CLL is changing rapidly as we move from standard chemotherapy-based approaches to more targeted therapies," said principal investigator Ian W. Flinn, MD, PhD, of Sarah Cannon Research Institute in Nashville.
"Based on these data, duvelisib may offer a new treatment option for patients who otherwise may have limited options."
Duvelisib is an oral, dual inhibitor of phosphoinositide 3-kinase (PI3K)-δ and -γ, which means it blocks the survival and proliferation of malignant B cells and also disrupts the recruitment and differentiation of T cells and macrophages within the tumor microenvironment.
Ofatumumab is a humanized anti-CD20 antibody with single-agent efficacy in refractory CLL. It is approved by the FDA as a treatment option in CLL.
Study design
Investigators randomized 319 relapsed or refractory CLL/SLL patients, 160 to the duvelisib arm and 159 to the ofatumumab arm.
Patients in the duvelisib arm self-administered 25 mg capsules twice daily continuously in 28-day cycles. They could take duvelisib for up to 18 cycles, until disease progression or unacceptable toxicity.
Ofatumumab-treated patients received infusions as approved in the product labeling for monotherapy in relapsed CLL. Dosing of ofatumumab could not exceed 12 doses in 7 cycles.
Prophylaxis for Pneumocystis jirovecci was required for all patients on both treatment arms.
Patients were allowed to crossover to a separate extension study to receive the opposite therapy if they had progressive disease.
They were followed for a median of 22.4 months.
Patient characteristics
According to the investigators, patient characteristics were well balanced between the arms.
The majority (60%) were male and the median age in both arms was 69. Most had an ECOG performance status of 0 or 1; 7% in the duvelisib arm and 10% in the ofatumumab arm had a performance status of 2.
Other patient characteristics in the duvelisib and ofatumumab arms, respectively, were:
- Time from initial diagnosis: 7.5 years, 6.7 years
- CLL/SLL, %: 97/5, 99/2
- Bulky disease: 46%, 45%
- Baseline lymphocyte counts: 38x109/L, 35x109/L
- Deletion 17p and/or TP53 mutation: 31%, 33%
- Median number of prior therapies: 2 in each arm
- Previous alkylating agent: 93%, 95%
- Previous monoclonal antibody: 78%, 83%
- Prior purine analog: 60%, 71%
Of the total patients enrolled, 158 patients in the duvelisib arm and 155 in the ofatumumab arm received treatment, for a median exposure of 50 weeks and 23 weeks, respectively.
Efficacy
In addition to the significantly improved overall PFS and ORR with duvelisib, further analysis revealed that PFS also improved for all predefined subgroups.
High-risk patients with deletion 17p/TP53 mutations also experienced a significant improvement in PFS with duvelisib of 12.7 months compared to 9.0 months with ofatumumab by IRC (P=0.0002).
The estimated probability of being progression-free for these patients at 6 and 12 moths was 73% and 55% with duvelisib and 63% and 30% with ofatumumab.
The investigators pointed out that duvelisib treatment was particularly effective in eliciting a lymph node response—85.0% compared to 15.7% with ofatumumab as assessed by IRC (P<0.0001).
Median overall survival was not reached in either arm. The 12-month probability of survival was 86% for both treatments.
Safety
Median treatment exposure was almost twice as long in the duvelisib arm because ofatumumab treatment was not allowed to exceed 12 doses as specified in the prescribing information.
The investigators explained this resulted in a longer adverse event (AE) reporting period for duvelisib.
One hundred twenty-four duvelisib-treated patients discontinued treatment, most commonly due to AEs (35%), disease progression (22%), subject withdrawal (8%), and death (8%).
All ofatumumab-treated patients discontinued treatment by the time of data cutoff, and 67% had completed treatment as per protocol. Others discontinued due to disease progression (20%), subject withdrawal (5%), and AEs (4%).
Eight (5%) duvelisib patients crossed over to ofatumumab therapy at the time of disease progression, and 89 (57%) ofatumumab-treated patients crossed over to duvelisib.
Nearly all patients in both arms experienced an AE.
The most common hematologic malignancies with duvelisib and ofatumumab, respectively, occurring in 10% or more patients were neutropenia (33%, 21%), anemia (23%, 10%), and thrombocytopenia (15%, 6%).
The most common nonhematologic AES with duvelisib were diarrhea (51%), pyrexia (29%), nausea (23%), and cough (21%).
With ofatumumab, the most common nonhematologic AES were infusion-related reaction (19%), cough (14%), and diarrhea, rash, and fatigue (12% each).
Grade 3 or greater AEs occurred in 87% of duvelisib-treated patients and 48% in the ofatumumab arm.
The most common grade 3 or greater events with duvelisib were neutropenia (30%), diarrhea (15%), pneumonia (14%), and anemia (13%).
With ofatumumab, only neutropenia (17%) of grade 3 or higher occurred in 10% or more patients.
Severe immune-related toxicities with duvelisib included colitis (12%) and pneumonitis, alanine transaminase (ALT) or aspartate transaminase (AST) increase (3% each). The events were managed with dose interruptions and steroid therapy for pneumonitis or colitis. All reported events resolved, and none was fatal.
Infectious AEs occurred more frequently with duvelisib, 69% compared to 43% in the ofatumumab arm. Pneumonia (18%) and upper respiratory tract infection (16%) were the most common events.
Three patients in the duvelisib arm and 1 in the ofatumumab arm contracted Pneumocystis jirovecii.
The most frequently reported serious AE was pneumonia (duvelisib 15%; ofatumumab 3%).
Nineteen fatal AEs occurred in patients on the duvelisib arm, 4 of which were related to the study drug: staphylococcal pneumonia (n = 2), sepsis (n=1), and general health deterioration (n = 1).
Seven fatal AEs occurred in patients on the ofatumumab arm, although none was attributed to ofatumumab.
The DUO trial was sponsored by Verastem Oncology and Infinity Pharmaceuticals , Inc.
Emicizumab now also approved for hemophilia A without inhibitors

The U.S. Food and Drug Administration (FDA) approved emicizumab-kxwh (Hemlibra) for prophylaxis to prevent or reduce the frequency of bleeding episodes in adult and pediatric patients, including newborns, with hemophilia A with or without factor VIII (FVIII) inhibitors.
Emicizumab is a humanized bispecific factor IXa- and factor X-directed antibody for patients with congenital FVIII deficiency.
It was first approved in 2017 for hemophilia A patients with FVIII inhibitors.
The current approval expands the indication to include patients without FVIII inhibitors and provides new dosing regimens.
The FDA based the current approval on the HAVEN 3 and HAVEN 4 trials.
HAVEN 3 (NCT02847637)
This multicenter trial randomized 89 patients with severe hemophilia A without FVIII inhibitors to receive emicizumab prophylaxis at one of 3 dose levels: 1.5 mg/kg once weekly (ARM A), 3 mg/kg once every two weeks (Arm B), or no prophylaxis (Arm C). Patients had previously received on-demand treatment with FVIII.
Before the start of the trial, investigators stratified patients by 24-week bleed rate—fewer than 9 bleeds and 9 or more bleeds.
Patients were treated with emicizumab for a minimum of 24 weeks.
Patients in Arm A experienced a 96% reduction in annualized bleed rate (ABR) compared to patients with no prophylaxis (ABR ratio=0.04; P<0.0001).
Patients in Arm B had a 97% reduction in ABR compared to patients with no prophylaxis (ABR ratio=0.03; P<0.0001).
The trial met all bleed-related secondary endpoints, such as all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds.
HAVEN 4 (NCT03020160)
This was a single-arm multicenter trial in 48 adult and adolescent males with hemophilia A with or without FVIII inhibitors. The patients had previously received on-demand or prophylactic treatment with FVIII or bypassing agents.
The study was conducted in two parts: a run-in of 7 patients to determine the pharmacokinetics after a single 6 mg/kg dose in four weeks. This was followed by the same dose once every four weeks for at least 24 weeks.
The second part was an expansion cohort of 41 patients who received emicizumab at 3 mg/kg once weekly for the first four weeks followed by 6 mg/kg once every four weeks for at least 24 weeks.
The ABR for treated bleeds was 2.4 (95% CI: 1.38, 4.28) and the median ABR was 0.0 (interquartile range: 0.00, 2.08).
The recommended loading dose is 3 mg/kg once weekly for the first four weeks for all prophylactic regimens.
Safety
The prescribing information for emicizumab includes a warning about thrombotic microangiopathy and thromboembolism.
These events were reported on average when a cumulative amount of >100 U/kg/24 hours of activated prothrombin complex concentrate (aPCC) was administered for 24 hours or more to patients receiving emicizumab prophylaxis.
According to the warning box, patients should be monitored for these events if aPCC is administered. If symptoms occur, aPCC should be discontinued and emicizumab dosing suspended.
The most common adverse reactions reported for emicizumab with an incidence ≥10% were injection site reactions (22%), headache (15%), and arthralgia (15%).
Emicizumab is manufactured by Genentech, Inc., a member of the Roche Group.
Additional data on emicizumab can be found in an earlier Roche media release.
The U.S. Food and Drug Administration (FDA) approved emicizumab-kxwh (Hemlibra) for prophylaxis to prevent or reduce the frequency of bleeding episodes in adult and pediatric patients, including newborns, with hemophilia A with or without factor VIII (FVIII) inhibitors.
Emicizumab is a humanized bispecific factor IXa- and factor X-directed antibody for patients with congenital FVIII deficiency.
It was first approved in 2017 for hemophilia A patients with FVIII inhibitors.
The current approval expands the indication to include patients without FVIII inhibitors and provides new dosing regimens.
The FDA based the current approval on the HAVEN 3 and HAVEN 4 trials.
HAVEN 3 (NCT02847637)
This multicenter trial randomized 89 patients with severe hemophilia A without FVIII inhibitors to receive emicizumab prophylaxis at one of 3 dose levels: 1.5 mg/kg once weekly (ARM A), 3 mg/kg once every two weeks (Arm B), or no prophylaxis (Arm C). Patients had previously received on-demand treatment with FVIII.
Before the start of the trial, investigators stratified patients by 24-week bleed rate—fewer than 9 bleeds and 9 or more bleeds.
Patients were treated with emicizumab for a minimum of 24 weeks.
Patients in Arm A experienced a 96% reduction in annualized bleed rate (ABR) compared to patients with no prophylaxis (ABR ratio=0.04; P<0.0001).
Patients in Arm B had a 97% reduction in ABR compared to patients with no prophylaxis (ABR ratio=0.03; P<0.0001).
The trial met all bleed-related secondary endpoints, such as all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds.
HAVEN 4 (NCT03020160)
This was a single-arm multicenter trial in 48 adult and adolescent males with hemophilia A with or without FVIII inhibitors. The patients had previously received on-demand or prophylactic treatment with FVIII or bypassing agents.
The study was conducted in two parts: a run-in of 7 patients to determine the pharmacokinetics after a single 6 mg/kg dose in four weeks. This was followed by the same dose once every four weeks for at least 24 weeks.
The second part was an expansion cohort of 41 patients who received emicizumab at 3 mg/kg once weekly for the first four weeks followed by 6 mg/kg once every four weeks for at least 24 weeks.
The ABR for treated bleeds was 2.4 (95% CI: 1.38, 4.28) and the median ABR was 0.0 (interquartile range: 0.00, 2.08).
The recommended loading dose is 3 mg/kg once weekly for the first four weeks for all prophylactic regimens.
Safety
The prescribing information for emicizumab includes a warning about thrombotic microangiopathy and thromboembolism.
These events were reported on average when a cumulative amount of >100 U/kg/24 hours of activated prothrombin complex concentrate (aPCC) was administered for 24 hours or more to patients receiving emicizumab prophylaxis.
According to the warning box, patients should be monitored for these events if aPCC is administered. If symptoms occur, aPCC should be discontinued and emicizumab dosing suspended.
The most common adverse reactions reported for emicizumab with an incidence ≥10% were injection site reactions (22%), headache (15%), and arthralgia (15%).
Emicizumab is manufactured by Genentech, Inc., a member of the Roche Group.
Additional data on emicizumab can be found in an earlier Roche media release.
The U.S. Food and Drug Administration (FDA) approved emicizumab-kxwh (Hemlibra) for prophylaxis to prevent or reduce the frequency of bleeding episodes in adult and pediatric patients, including newborns, with hemophilia A with or without factor VIII (FVIII) inhibitors.
Emicizumab is a humanized bispecific factor IXa- and factor X-directed antibody for patients with congenital FVIII deficiency.
It was first approved in 2017 for hemophilia A patients with FVIII inhibitors.
The current approval expands the indication to include patients without FVIII inhibitors and provides new dosing regimens.
The FDA based the current approval on the HAVEN 3 and HAVEN 4 trials.
HAVEN 3 (NCT02847637)
This multicenter trial randomized 89 patients with severe hemophilia A without FVIII inhibitors to receive emicizumab prophylaxis at one of 3 dose levels: 1.5 mg/kg once weekly (ARM A), 3 mg/kg once every two weeks (Arm B), or no prophylaxis (Arm C). Patients had previously received on-demand treatment with FVIII.
Before the start of the trial, investigators stratified patients by 24-week bleed rate—fewer than 9 bleeds and 9 or more bleeds.
Patients were treated with emicizumab for a minimum of 24 weeks.
Patients in Arm A experienced a 96% reduction in annualized bleed rate (ABR) compared to patients with no prophylaxis (ABR ratio=0.04; P<0.0001).
Patients in Arm B had a 97% reduction in ABR compared to patients with no prophylaxis (ABR ratio=0.03; P<0.0001).
The trial met all bleed-related secondary endpoints, such as all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds.
HAVEN 4 (NCT03020160)
This was a single-arm multicenter trial in 48 adult and adolescent males with hemophilia A with or without FVIII inhibitors. The patients had previously received on-demand or prophylactic treatment with FVIII or bypassing agents.
The study was conducted in two parts: a run-in of 7 patients to determine the pharmacokinetics after a single 6 mg/kg dose in four weeks. This was followed by the same dose once every four weeks for at least 24 weeks.
The second part was an expansion cohort of 41 patients who received emicizumab at 3 mg/kg once weekly for the first four weeks followed by 6 mg/kg once every four weeks for at least 24 weeks.
The ABR for treated bleeds was 2.4 (95% CI: 1.38, 4.28) and the median ABR was 0.0 (interquartile range: 0.00, 2.08).
The recommended loading dose is 3 mg/kg once weekly for the first four weeks for all prophylactic regimens.
Safety
The prescribing information for emicizumab includes a warning about thrombotic microangiopathy and thromboembolism.
These events were reported on average when a cumulative amount of >100 U/kg/24 hours of activated prothrombin complex concentrate (aPCC) was administered for 24 hours or more to patients receiving emicizumab prophylaxis.
According to the warning box, patients should be monitored for these events if aPCC is administered. If symptoms occur, aPCC should be discontinued and emicizumab dosing suspended.
The most common adverse reactions reported for emicizumab with an incidence ≥10% were injection site reactions (22%), headache (15%), and arthralgia (15%).
Emicizumab is manufactured by Genentech, Inc., a member of the Roche Group.
Additional data on emicizumab can be found in an earlier Roche media release.
Quadruplet therapy could be the future in MM

New York—Four-drug combinations are holding promise for the treatment of multiple myeloma (MM), although data from additional randomized trials are needed to define their role in clinical practice, according to Natalie S. Callander, MD, of the University of Wisconsin Carbone Cancer Center.
The efficacy of multiple triplet regimens has been well documented, and data are now emerging on the potential of 4-drug combinations.
“Triplet therapy is the standard,” she said during a presentation at the NCCN 13th Annual Congress: Hematologic Malignancies, “and quad therapy may be in the future.”
The study that set the standard for triplets in myeloma, she said, is SWOG 0777, an open label, phase 3 trial that compared bortezomib with lenalidomide and dexamethasone (VRd) to lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma.
Adding bortezomib to lenalidomide and dexamethasone significantly improved both progression-free and overall survival in the 525-patient trial, with a risk-benefit profile that was acceptable. The trial was recently reported in The Lancet.
The median progression free survival (PFS) was 43 months for the triplet, versus 30 months for the two-drug regimen (P=0.0018).
Likewise, median overall survival (OS) was significantly improved, at 75 months versus 64 months for triplet versus doublet therapy (P=0.025).
“Very convincingly, just receiving that short exposure to bortezomib,” she said, “ended up causing a substantial increase of progression-free and overall survival.”
Effective triplet regimens include the combination of carfilzomib, lenalidomide, and dexamethasone (KRd), cyclophosphamide, bortezomib, and dexamethasone (CyBorD), and more recently, ixazomib, lenalidomide, and dexamethasone (IRd).
These regimens have “excellent” response rates and survival data, Dr. Callander noted.
Quadruplet data
The combination of elotuzumab plus VRd produced high response rates that were even higher after transplant, with reasonable toxicity, Dr. Callander said of phase 2 trial data presented at ASCO 2017 (abstract 8002).
Similarly, the combination of daratumumab plus KRd had a 100% rate of partial response or better in phase 2 data also presented at ASCO 2017 (abstract 8000), with rates of very good partial response and complete response that improved with successive cycles of therapy, she said.
Even so, “it remains to be seen whether four drugs will be the new standard,” Dr Callander conjectured.
Four versus three drug strategies are being evaluated in ongoing randomized clinical trials including patients with previously untreated myeloma, she said.
These studies include Cassiopeia (NCT02541383), which is evaluating bortezomib, thalidomide, and dexamethasone with or without daratumumab, and GRIFFIN (NCT02874742), which is looking at VRd with or without daratumumab.
Daratumumab received an additional indication in myeloma, this time as part of a 4-drug regimen, several months ago, Dr. Callander added in a discussion on treatment options for elderly myeloma patients.
The U.S. Food and Drug Administration (FDA) approved the monoclonal antibody in combination with bortezomib, melphalan, and prednisone (VMP) for treatment of newly diagnosed myeloma patients who are transplant ineligible.
That approval was based on results of the multicenter phase 3 ALCYONE (MMY3007) study, published in The New England Journal of Medicine, showing an 18-month PFS of 71.6% for the 4-drug combination versus 50.2% for VMP alone (P<0.001).
New York—Four-drug combinations are holding promise for the treatment of multiple myeloma (MM), although data from additional randomized trials are needed to define their role in clinical practice, according to Natalie S. Callander, MD, of the University of Wisconsin Carbone Cancer Center.
The efficacy of multiple triplet regimens has been well documented, and data are now emerging on the potential of 4-drug combinations.
“Triplet therapy is the standard,” she said during a presentation at the NCCN 13th Annual Congress: Hematologic Malignancies, “and quad therapy may be in the future.”
The study that set the standard for triplets in myeloma, she said, is SWOG 0777, an open label, phase 3 trial that compared bortezomib with lenalidomide and dexamethasone (VRd) to lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma.
Adding bortezomib to lenalidomide and dexamethasone significantly improved both progression-free and overall survival in the 525-patient trial, with a risk-benefit profile that was acceptable. The trial was recently reported in The Lancet.
The median progression free survival (PFS) was 43 months for the triplet, versus 30 months for the two-drug regimen (P=0.0018).
Likewise, median overall survival (OS) was significantly improved, at 75 months versus 64 months for triplet versus doublet therapy (P=0.025).
“Very convincingly, just receiving that short exposure to bortezomib,” she said, “ended up causing a substantial increase of progression-free and overall survival.”
Effective triplet regimens include the combination of carfilzomib, lenalidomide, and dexamethasone (KRd), cyclophosphamide, bortezomib, and dexamethasone (CyBorD), and more recently, ixazomib, lenalidomide, and dexamethasone (IRd).
These regimens have “excellent” response rates and survival data, Dr. Callander noted.
Quadruplet data
The combination of elotuzumab plus VRd produced high response rates that were even higher after transplant, with reasonable toxicity, Dr. Callander said of phase 2 trial data presented at ASCO 2017 (abstract 8002).
Similarly, the combination of daratumumab plus KRd had a 100% rate of partial response or better in phase 2 data also presented at ASCO 2017 (abstract 8000), with rates of very good partial response and complete response that improved with successive cycles of therapy, she said.
Even so, “it remains to be seen whether four drugs will be the new standard,” Dr Callander conjectured.
Four versus three drug strategies are being evaluated in ongoing randomized clinical trials including patients with previously untreated myeloma, she said.
These studies include Cassiopeia (NCT02541383), which is evaluating bortezomib, thalidomide, and dexamethasone with or without daratumumab, and GRIFFIN (NCT02874742), which is looking at VRd with or without daratumumab.
Daratumumab received an additional indication in myeloma, this time as part of a 4-drug regimen, several months ago, Dr. Callander added in a discussion on treatment options for elderly myeloma patients.
The U.S. Food and Drug Administration (FDA) approved the monoclonal antibody in combination with bortezomib, melphalan, and prednisone (VMP) for treatment of newly diagnosed myeloma patients who are transplant ineligible.
That approval was based on results of the multicenter phase 3 ALCYONE (MMY3007) study, published in The New England Journal of Medicine, showing an 18-month PFS of 71.6% for the 4-drug combination versus 50.2% for VMP alone (P<0.001).
New York—Four-drug combinations are holding promise for the treatment of multiple myeloma (MM), although data from additional randomized trials are needed to define their role in clinical practice, according to Natalie S. Callander, MD, of the University of Wisconsin Carbone Cancer Center.
The efficacy of multiple triplet regimens has been well documented, and data are now emerging on the potential of 4-drug combinations.
“Triplet therapy is the standard,” she said during a presentation at the NCCN 13th Annual Congress: Hematologic Malignancies, “and quad therapy may be in the future.”
The study that set the standard for triplets in myeloma, she said, is SWOG 0777, an open label, phase 3 trial that compared bortezomib with lenalidomide and dexamethasone (VRd) to lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma.
Adding bortezomib to lenalidomide and dexamethasone significantly improved both progression-free and overall survival in the 525-patient trial, with a risk-benefit profile that was acceptable. The trial was recently reported in The Lancet.
The median progression free survival (PFS) was 43 months for the triplet, versus 30 months for the two-drug regimen (P=0.0018).
Likewise, median overall survival (OS) was significantly improved, at 75 months versus 64 months for triplet versus doublet therapy (P=0.025).
“Very convincingly, just receiving that short exposure to bortezomib,” she said, “ended up causing a substantial increase of progression-free and overall survival.”
Effective triplet regimens include the combination of carfilzomib, lenalidomide, and dexamethasone (KRd), cyclophosphamide, bortezomib, and dexamethasone (CyBorD), and more recently, ixazomib, lenalidomide, and dexamethasone (IRd).
These regimens have “excellent” response rates and survival data, Dr. Callander noted.
Quadruplet data
The combination of elotuzumab plus VRd produced high response rates that were even higher after transplant, with reasonable toxicity, Dr. Callander said of phase 2 trial data presented at ASCO 2017 (abstract 8002).
Similarly, the combination of daratumumab plus KRd had a 100% rate of partial response or better in phase 2 data also presented at ASCO 2017 (abstract 8000), with rates of very good partial response and complete response that improved with successive cycles of therapy, she said.
Even so, “it remains to be seen whether four drugs will be the new standard,” Dr Callander conjectured.
Four versus three drug strategies are being evaluated in ongoing randomized clinical trials including patients with previously untreated myeloma, she said.
These studies include Cassiopeia (NCT02541383), which is evaluating bortezomib, thalidomide, and dexamethasone with or without daratumumab, and GRIFFIN (NCT02874742), which is looking at VRd with or without daratumumab.
Daratumumab received an additional indication in myeloma, this time as part of a 4-drug regimen, several months ago, Dr. Callander added in a discussion on treatment options for elderly myeloma patients.
The U.S. Food and Drug Administration (FDA) approved the monoclonal antibody in combination with bortezomib, melphalan, and prednisone (VMP) for treatment of newly diagnosed myeloma patients who are transplant ineligible.
That approval was based on results of the multicenter phase 3 ALCYONE (MMY3007) study, published in The New England Journal of Medicine, showing an 18-month PFS of 71.6% for the 4-drug combination versus 50.2% for VMP alone (P<0.001).
5-year remission rates with combo prove durable in MCL
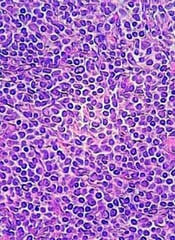
Long-term results of a phase 2 clinical trial of the lenalidomide and rituximab combination as first-line therapy for mantle cell lymphoma (MCL) show continued durable responses with manageable toxicities after 5 years.
With a median follow-up of 64 months, 21 of 33 patients with initial responses remained in durable, minimal residual disease (MRD)-negative remission following induction with lenalidomide and rituximab and maintenance with those same two agents for at least 3 years.
The patients with durable responses included five who opted to discontinue maintenance after 3 years, reported Jia Ruan, MD, PhD, of Cornell University in New York, and her colleagues.
“Our long-term data provide proof of concept that an outpatient-based induction and maintenance strategy free of conventional chemotherapy is effective, safe, and feasible as first-line therapy for MCL,” they wrote.
Their report was published in Blood.
In the multicenter, phase 2 single-arm study (NCT01472562), 38 patients with untreated MCL were enrolled and treated with lenalidomide 20 mg daily on days 1-21 of each 28-day cycle for 12 cycles during induction, followed by dose reduction to 15 mg during the maintenance phase.
Patients also received standard dose rituximab 375 mg/m2 weekly for 4 weeks during cycle 1, then once every other cycle.
Patients remained on treatment until disease progression, unacceptable toxicities, or study withdrawal. Patients who remained in remission after 3 years, based on routine surveillance CT scans, had the option to discontinue maintenance.
Results
Of the original 38 patients enrolled, 36 were evaluable for response, including 23 with a complete response (CR) and 10 with a partial response.
At the 64-month median follow-up, neither the median progression-free survival (PFS) nor duration of response had been reached.
Overall, 21 of the 33 patients with responses (64%) had ongoing responses, including six patients with responses beyond 6 years.
Estimated 3-year and 5-year PFS rates were 80.3% and 63.9%, respectively. Respective estimated 3- and 5-year overall survival rates were 89.5% and 77.4%.
Mantle cell lymphoma international prognostic index (MIPI) scores were not associated with either response or PFS rates, but patients with high-risk MIPI scores were significantly more likely to have worse overall survival (P=0.04).
Safety
Grade 3 or greater hematologic toxicities included neutropenia in 42% of patients in both induction and maintenance, anemia in 8% and 3%, thrombocytopenia in 11% and 5%, and febrile neutropenia in 3% and 5%.
Secondary primary malignancies occurred in six patients. These included five noninvasive skin cancers requiring only local therapy without the need for study interruption.
Two patients, including one with a skin cancer, died from the secondary malignancies, including one from Merkel cell carcinoma and one from pancreatic cancer.
“The efficacy and survival outcome observed in our study compared favorably to those reported with lenalidomide either as single agent, or in combination with rituximab in relapsed and refractory setting,” the investigators wrote, “lending support for prioritizing novel agents such as lenalidomide early in the treatment sequence, to compare to conventional chemotherapy-based approach.”
The study was supported in part by Celgene Corporation, a Clinical Translational Science Center grant, and the Lymphoma Foundation.
Long-term results of a phase 2 clinical trial of the lenalidomide and rituximab combination as first-line therapy for mantle cell lymphoma (MCL) show continued durable responses with manageable toxicities after 5 years.
With a median follow-up of 64 months, 21 of 33 patients with initial responses remained in durable, minimal residual disease (MRD)-negative remission following induction with lenalidomide and rituximab and maintenance with those same two agents for at least 3 years.
The patients with durable responses included five who opted to discontinue maintenance after 3 years, reported Jia Ruan, MD, PhD, of Cornell University in New York, and her colleagues.
“Our long-term data provide proof of concept that an outpatient-based induction and maintenance strategy free of conventional chemotherapy is effective, safe, and feasible as first-line therapy for MCL,” they wrote.
Their report was published in Blood.
In the multicenter, phase 2 single-arm study (NCT01472562), 38 patients with untreated MCL were enrolled and treated with lenalidomide 20 mg daily on days 1-21 of each 28-day cycle for 12 cycles during induction, followed by dose reduction to 15 mg during the maintenance phase.
Patients also received standard dose rituximab 375 mg/m2 weekly for 4 weeks during cycle 1, then once every other cycle.
Patients remained on treatment until disease progression, unacceptable toxicities, or study withdrawal. Patients who remained in remission after 3 years, based on routine surveillance CT scans, had the option to discontinue maintenance.
Results
Of the original 38 patients enrolled, 36 were evaluable for response, including 23 with a complete response (CR) and 10 with a partial response.
At the 64-month median follow-up, neither the median progression-free survival (PFS) nor duration of response had been reached.
Overall, 21 of the 33 patients with responses (64%) had ongoing responses, including six patients with responses beyond 6 years.
Estimated 3-year and 5-year PFS rates were 80.3% and 63.9%, respectively. Respective estimated 3- and 5-year overall survival rates were 89.5% and 77.4%.
Mantle cell lymphoma international prognostic index (MIPI) scores were not associated with either response or PFS rates, but patients with high-risk MIPI scores were significantly more likely to have worse overall survival (P=0.04).
Safety
Grade 3 or greater hematologic toxicities included neutropenia in 42% of patients in both induction and maintenance, anemia in 8% and 3%, thrombocytopenia in 11% and 5%, and febrile neutropenia in 3% and 5%.
Secondary primary malignancies occurred in six patients. These included five noninvasive skin cancers requiring only local therapy without the need for study interruption.
Two patients, including one with a skin cancer, died from the secondary malignancies, including one from Merkel cell carcinoma and one from pancreatic cancer.
“The efficacy and survival outcome observed in our study compared favorably to those reported with lenalidomide either as single agent, or in combination with rituximab in relapsed and refractory setting,” the investigators wrote, “lending support for prioritizing novel agents such as lenalidomide early in the treatment sequence, to compare to conventional chemotherapy-based approach.”
The study was supported in part by Celgene Corporation, a Clinical Translational Science Center grant, and the Lymphoma Foundation.
Long-term results of a phase 2 clinical trial of the lenalidomide and rituximab combination as first-line therapy for mantle cell lymphoma (MCL) show continued durable responses with manageable toxicities after 5 years.
With a median follow-up of 64 months, 21 of 33 patients with initial responses remained in durable, minimal residual disease (MRD)-negative remission following induction with lenalidomide and rituximab and maintenance with those same two agents for at least 3 years.
The patients with durable responses included five who opted to discontinue maintenance after 3 years, reported Jia Ruan, MD, PhD, of Cornell University in New York, and her colleagues.
“Our long-term data provide proof of concept that an outpatient-based induction and maintenance strategy free of conventional chemotherapy is effective, safe, and feasible as first-line therapy for MCL,” they wrote.
Their report was published in Blood.
In the multicenter, phase 2 single-arm study (NCT01472562), 38 patients with untreated MCL were enrolled and treated with lenalidomide 20 mg daily on days 1-21 of each 28-day cycle for 12 cycles during induction, followed by dose reduction to 15 mg during the maintenance phase.
Patients also received standard dose rituximab 375 mg/m2 weekly for 4 weeks during cycle 1, then once every other cycle.
Patients remained on treatment until disease progression, unacceptable toxicities, or study withdrawal. Patients who remained in remission after 3 years, based on routine surveillance CT scans, had the option to discontinue maintenance.
Results
Of the original 38 patients enrolled, 36 were evaluable for response, including 23 with a complete response (CR) and 10 with a partial response.
At the 64-month median follow-up, neither the median progression-free survival (PFS) nor duration of response had been reached.
Overall, 21 of the 33 patients with responses (64%) had ongoing responses, including six patients with responses beyond 6 years.
Estimated 3-year and 5-year PFS rates were 80.3% and 63.9%, respectively. Respective estimated 3- and 5-year overall survival rates were 89.5% and 77.4%.
Mantle cell lymphoma international prognostic index (MIPI) scores were not associated with either response or PFS rates, but patients with high-risk MIPI scores were significantly more likely to have worse overall survival (P=0.04).
Safety
Grade 3 or greater hematologic toxicities included neutropenia in 42% of patients in both induction and maintenance, anemia in 8% and 3%, thrombocytopenia in 11% and 5%, and febrile neutropenia in 3% and 5%.
Secondary primary malignancies occurred in six patients. These included five noninvasive skin cancers requiring only local therapy without the need for study interruption.
Two patients, including one with a skin cancer, died from the secondary malignancies, including one from Merkel cell carcinoma and one from pancreatic cancer.
“The efficacy and survival outcome observed in our study compared favorably to those reported with lenalidomide either as single agent, or in combination with rituximab in relapsed and refractory setting,” the investigators wrote, “lending support for prioritizing novel agents such as lenalidomide early in the treatment sequence, to compare to conventional chemotherapy-based approach.”
The study was supported in part by Celgene Corporation, a Clinical Translational Science Center grant, and the Lymphoma Foundation.
Researchers develop genetics-based prognostic tool for MDS

Researchers have developed a new risk model for primary myelodysplastic syndromes (MDS) that integrates genetic and clinical information.
The research team considered the current standard for prognostication—the revised International Prognostic Scoring System (IPSS-R)—to be too complex, limited to newly diagnosed cases, and missing information on mutations and age.
So they devised a “simpler and more contemporary” prognostic system, the Mayo Alliance Prognostic Model for MDS.
The team, from the Mayo Clinic in Rochester, Minnesota, and the National Taiwan University Hospital (NTUH), described the new model in Mayo Clinic Proceedings.
Lead author Ayalew Tefferi, MD, of the Mayo Clinic, said the new model “is not an enhancement of the international prognostic scoring system tool, it's a complete makeover."
The team analyzed mutation information from 357 patients with primary MDS or leukemic transformation treated at the Mayo Clinic from the end of December 1994 through mid-December 2017.
The patients were a median age of 74 and 70% were males.
They compared the Mayo patients to 328 NTUH patients, who were a median age of 66 and 65% were males.
Multivariate analysis of the Mayo cohort identified the following as predictors of inferior overall survival:
- Monosomal karyotype (hazard ratio [HR], 5.2; 95% CI, 3.1-8.6)
- Non-monosomal karyotype abnormalities other than single/double del(5q) (HR, 1.8; 95% CI, 1.3-2.6)
- RUNX1 (HR, 2.0; 95% CI, 1.2-3.1)
- ASXL1 (HR, 1.7; 95% CI, 1.2-2.3) mutations
- Absence of SF3B1 mutations (HR, 1.6; 95% CI, 1.1-2.4)
- Age greater than 70 years (HR, 2.2; 95% CI, 1.6-3.1)
- Hemoglobin level less than 8 g/dL in women or less than 9 g/dL in men (HR, 2.3; 95% CI, 1.7-3.1)
- Platelet count less than 75 x 109/L (HR, 1.5; 95% CI, 1.1-2.1)
- 10% or more bone marrow blasts (HR, 1.7; 95% CI, 1.1-2.8)
They then provided values to reflect the prognostic contribution of each of the above predictors and devised the new 4-tiered Mayo prognostic model.
Median 5-year overall survival rates in the 4 categories in the Mayo model were 73% (low risk), 34% (intermediate-1), 7% (intermediate-2), and 0% (high risk; 9-month median survival).
The team then validated the Mayo alliance model by using the NTUH cohort and compared it to the IPSS-R.
The investigators were able to confirm superior predictive accuracy of their model and a substantial discordance between the the Mayo model and the IPSS-R in terms of the pattern of risk distribution.
Examples of discordance included:
- More than 25% of patients belonging to the high-risk category according to the Mayo alliance model were classified as IPSS-R low or intermediate risk
- Almost 50% of patients with intermediate-2 risk category according to the Mayo alliance model were classified as IPSS-R very low or low risk
- Almost 50% of patients with IPSS-R very low risk were classified as intermediate-2 or intermediate-1 risk according to the Mayo alliance model
The authors wrote that this “suggests a fundamental and not incremental advantage for the new Mayo alliance model.”
Researchers have developed a new risk model for primary myelodysplastic syndromes (MDS) that integrates genetic and clinical information.
The research team considered the current standard for prognostication—the revised International Prognostic Scoring System (IPSS-R)—to be too complex, limited to newly diagnosed cases, and missing information on mutations and age.
So they devised a “simpler and more contemporary” prognostic system, the Mayo Alliance Prognostic Model for MDS.
The team, from the Mayo Clinic in Rochester, Minnesota, and the National Taiwan University Hospital (NTUH), described the new model in Mayo Clinic Proceedings.
Lead author Ayalew Tefferi, MD, of the Mayo Clinic, said the new model “is not an enhancement of the international prognostic scoring system tool, it's a complete makeover."
The team analyzed mutation information from 357 patients with primary MDS or leukemic transformation treated at the Mayo Clinic from the end of December 1994 through mid-December 2017.
The patients were a median age of 74 and 70% were males.
They compared the Mayo patients to 328 NTUH patients, who were a median age of 66 and 65% were males.
Multivariate analysis of the Mayo cohort identified the following as predictors of inferior overall survival:
- Monosomal karyotype (hazard ratio [HR], 5.2; 95% CI, 3.1-8.6)
- Non-monosomal karyotype abnormalities other than single/double del(5q) (HR, 1.8; 95% CI, 1.3-2.6)
- RUNX1 (HR, 2.0; 95% CI, 1.2-3.1)
- ASXL1 (HR, 1.7; 95% CI, 1.2-2.3) mutations
- Absence of SF3B1 mutations (HR, 1.6; 95% CI, 1.1-2.4)
- Age greater than 70 years (HR, 2.2; 95% CI, 1.6-3.1)
- Hemoglobin level less than 8 g/dL in women or less than 9 g/dL in men (HR, 2.3; 95% CI, 1.7-3.1)
- Platelet count less than 75 x 109/L (HR, 1.5; 95% CI, 1.1-2.1)
- 10% or more bone marrow blasts (HR, 1.7; 95% CI, 1.1-2.8)
They then provided values to reflect the prognostic contribution of each of the above predictors and devised the new 4-tiered Mayo prognostic model.
Median 5-year overall survival rates in the 4 categories in the Mayo model were 73% (low risk), 34% (intermediate-1), 7% (intermediate-2), and 0% (high risk; 9-month median survival).
The team then validated the Mayo alliance model by using the NTUH cohort and compared it to the IPSS-R.
The investigators were able to confirm superior predictive accuracy of their model and a substantial discordance between the the Mayo model and the IPSS-R in terms of the pattern of risk distribution.
Examples of discordance included:
- More than 25% of patients belonging to the high-risk category according to the Mayo alliance model were classified as IPSS-R low or intermediate risk
- Almost 50% of patients with intermediate-2 risk category according to the Mayo alliance model were classified as IPSS-R very low or low risk
- Almost 50% of patients with IPSS-R very low risk were classified as intermediate-2 or intermediate-1 risk according to the Mayo alliance model
The authors wrote that this “suggests a fundamental and not incremental advantage for the new Mayo alliance model.”
Researchers have developed a new risk model for primary myelodysplastic syndromes (MDS) that integrates genetic and clinical information.
The research team considered the current standard for prognostication—the revised International Prognostic Scoring System (IPSS-R)—to be too complex, limited to newly diagnosed cases, and missing information on mutations and age.
So they devised a “simpler and more contemporary” prognostic system, the Mayo Alliance Prognostic Model for MDS.
The team, from the Mayo Clinic in Rochester, Minnesota, and the National Taiwan University Hospital (NTUH), described the new model in Mayo Clinic Proceedings.
Lead author Ayalew Tefferi, MD, of the Mayo Clinic, said the new model “is not an enhancement of the international prognostic scoring system tool, it's a complete makeover."
The team analyzed mutation information from 357 patients with primary MDS or leukemic transformation treated at the Mayo Clinic from the end of December 1994 through mid-December 2017.
The patients were a median age of 74 and 70% were males.
They compared the Mayo patients to 328 NTUH patients, who were a median age of 66 and 65% were males.
Multivariate analysis of the Mayo cohort identified the following as predictors of inferior overall survival:
- Monosomal karyotype (hazard ratio [HR], 5.2; 95% CI, 3.1-8.6)
- Non-monosomal karyotype abnormalities other than single/double del(5q) (HR, 1.8; 95% CI, 1.3-2.6)
- RUNX1 (HR, 2.0; 95% CI, 1.2-3.1)
- ASXL1 (HR, 1.7; 95% CI, 1.2-2.3) mutations
- Absence of SF3B1 mutations (HR, 1.6; 95% CI, 1.1-2.4)
- Age greater than 70 years (HR, 2.2; 95% CI, 1.6-3.1)
- Hemoglobin level less than 8 g/dL in women or less than 9 g/dL in men (HR, 2.3; 95% CI, 1.7-3.1)
- Platelet count less than 75 x 109/L (HR, 1.5; 95% CI, 1.1-2.1)
- 10% or more bone marrow blasts (HR, 1.7; 95% CI, 1.1-2.8)
They then provided values to reflect the prognostic contribution of each of the above predictors and devised the new 4-tiered Mayo prognostic model.
Median 5-year overall survival rates in the 4 categories in the Mayo model were 73% (low risk), 34% (intermediate-1), 7% (intermediate-2), and 0% (high risk; 9-month median survival).
The team then validated the Mayo alliance model by using the NTUH cohort and compared it to the IPSS-R.
The investigators were able to confirm superior predictive accuracy of their model and a substantial discordance between the the Mayo model and the IPSS-R in terms of the pattern of risk distribution.
Examples of discordance included:
- More than 25% of patients belonging to the high-risk category according to the Mayo alliance model were classified as IPSS-R low or intermediate risk
- Almost 50% of patients with intermediate-2 risk category according to the Mayo alliance model were classified as IPSS-R very low or low risk
- Almost 50% of patients with IPSS-R very low risk were classified as intermediate-2 or intermediate-1 risk according to the Mayo alliance model
The authors wrote that this “suggests a fundamental and not incremental advantage for the new Mayo alliance model.”
Carfilzomib receives approval for once-weekly dosing
The U.S. Food and Drug Administration (FDA) has approved carfilzomib (Kyprolis) for a once-weekly dosing option in combination with dexamethasone for patients with relapsed or refractory multiple myeloma (MM).
Carfilzomib administered once-weekly at 70 mg/m2 with dexamethasone achieved a superior progression-free survival (PFS) and overall response rates (ORR) compared to twice-weekly carfilzomib at doses of 27 mg/m2.
Carfilzomib is not, however, approved for the twice-weekly 27 mg/m2 dose with dexamethasone alone, but with dexamethasone and lenalidomide.
The FDA based its approval on data from the phase 3 ARROW trial.
The FDA reviewed and approved the supplemental New Drug Application under its Oncology Center of Excellence Real-Time Oncology Review and Assessment Aid pilot program. The program is exploring a more efficient review process to ensure that safe and effective treatments are available to patients as soon as possible.
The FDA approved the carfilzomib application in just over a month.
ARROW
The ARROW study, reported at the 2018 ASCO annual meeting and published in The Lancet, evaluated 478 patients with relapsed or refractory MM who had received at least two but no more than three prior therapies. Prior therapies could include bortezomib and an immunomodulatory drug.
Patients randomized to the investigational arm receive a 30-minute infusion of once-weekly carfilzomib (20 mg/m2 on day 1 of cycle 1; 70 mg/m2 on days 8 and 15 of cycle 1; and 70 mg/m2 on days 1, 8 and 15 of subsequent cycles) with 40 mg of dexamethasone.
Patients randomized to the comparator arm received a 10-minute infusion of twice-weekly carfilzomib (20 mg/m2 on days 1 and 2 of cycle 1; 27 mg/m2 on days 8, 9, 15 and 16 of cycle 1; and 27 mg/m2 on days 1, 2, 8, 9, 15 and 16 of subsequent cycles) with 40 mg of dexamethasone.
Patients in the once-weekly arm achieved a statistically significant 3.7-month improvement in PFS compared to the twice-weekly regimen. Median PFS was 11.2 months for the once-weekly patients and 7.6 months for the twice-weekly group (P=0.0014).
Patients in the once-weekly group had a 62.9% ORR compared to 40.8% for those treated twice weekly (P<0.0001).
More patients (7.1%) in the once-weekly group had complete responses or better than those in the twice-weekly arm (1.7%).
The safety profile of the two arms were comparable, with no new safety risks identified in the once-weekly arm.
Treatment-emergent adverse events occurring in 20% or more patients in either arm included anemia, diarrhea, fatigue, hypertension, insomnia, and pyrexia.
First approved in 2012, carfilzomib has indications for the following in the U.S.:
- Treatment of patients with relapsed or refractory multiple myeloma who have received one to three lines of therapy in combination with dexamethasone or with lenalidomide plus dexamethasone.
- As a single agent for the treatment of patients with relapsed or refractory multiple myeloma who have received one or more lines of therapy.
Amgen manufactures carfilzomib for Onyx Pharmaceuticals, Inc.
Prescribing information for carfilzomib is available online.
The U.S. Food and Drug Administration (FDA) has approved carfilzomib (Kyprolis) for a once-weekly dosing option in combination with dexamethasone for patients with relapsed or refractory multiple myeloma (MM).
Carfilzomib administered once-weekly at 70 mg/m2 with dexamethasone achieved a superior progression-free survival (PFS) and overall response rates (ORR) compared to twice-weekly carfilzomib at doses of 27 mg/m2.
Carfilzomib is not, however, approved for the twice-weekly 27 mg/m2 dose with dexamethasone alone, but with dexamethasone and lenalidomide.
The FDA based its approval on data from the phase 3 ARROW trial.
The FDA reviewed and approved the supplemental New Drug Application under its Oncology Center of Excellence Real-Time Oncology Review and Assessment Aid pilot program. The program is exploring a more efficient review process to ensure that safe and effective treatments are available to patients as soon as possible.
The FDA approved the carfilzomib application in just over a month.
ARROW
The ARROW study, reported at the 2018 ASCO annual meeting and published in The Lancet, evaluated 478 patients with relapsed or refractory MM who had received at least two but no more than three prior therapies. Prior therapies could include bortezomib and an immunomodulatory drug.
Patients randomized to the investigational arm receive a 30-minute infusion of once-weekly carfilzomib (20 mg/m2 on day 1 of cycle 1; 70 mg/m2 on days 8 and 15 of cycle 1; and 70 mg/m2 on days 1, 8 and 15 of subsequent cycles) with 40 mg of dexamethasone.
Patients randomized to the comparator arm received a 10-minute infusion of twice-weekly carfilzomib (20 mg/m2 on days 1 and 2 of cycle 1; 27 mg/m2 on days 8, 9, 15 and 16 of cycle 1; and 27 mg/m2 on days 1, 2, 8, 9, 15 and 16 of subsequent cycles) with 40 mg of dexamethasone.
Patients in the once-weekly arm achieved a statistically significant 3.7-month improvement in PFS compared to the twice-weekly regimen. Median PFS was 11.2 months for the once-weekly patients and 7.6 months for the twice-weekly group (P=0.0014).
Patients in the once-weekly group had a 62.9% ORR compared to 40.8% for those treated twice weekly (P<0.0001).
More patients (7.1%) in the once-weekly group had complete responses or better than those in the twice-weekly arm (1.7%).
The safety profile of the two arms were comparable, with no new safety risks identified in the once-weekly arm.
Treatment-emergent adverse events occurring in 20% or more patients in either arm included anemia, diarrhea, fatigue, hypertension, insomnia, and pyrexia.
First approved in 2012, carfilzomib has indications for the following in the U.S.:
- Treatment of patients with relapsed or refractory multiple myeloma who have received one to three lines of therapy in combination with dexamethasone or with lenalidomide plus dexamethasone.
- As a single agent for the treatment of patients with relapsed or refractory multiple myeloma who have received one or more lines of therapy.
Amgen manufactures carfilzomib for Onyx Pharmaceuticals, Inc.
Prescribing information for carfilzomib is available online.
The U.S. Food and Drug Administration (FDA) has approved carfilzomib (Kyprolis) for a once-weekly dosing option in combination with dexamethasone for patients with relapsed or refractory multiple myeloma (MM).
Carfilzomib administered once-weekly at 70 mg/m2 with dexamethasone achieved a superior progression-free survival (PFS) and overall response rates (ORR) compared to twice-weekly carfilzomib at doses of 27 mg/m2.
Carfilzomib is not, however, approved for the twice-weekly 27 mg/m2 dose with dexamethasone alone, but with dexamethasone and lenalidomide.
The FDA based its approval on data from the phase 3 ARROW trial.
The FDA reviewed and approved the supplemental New Drug Application under its Oncology Center of Excellence Real-Time Oncology Review and Assessment Aid pilot program. The program is exploring a more efficient review process to ensure that safe and effective treatments are available to patients as soon as possible.
The FDA approved the carfilzomib application in just over a month.
ARROW
The ARROW study, reported at the 2018 ASCO annual meeting and published in The Lancet, evaluated 478 patients with relapsed or refractory MM who had received at least two but no more than three prior therapies. Prior therapies could include bortezomib and an immunomodulatory drug.
Patients randomized to the investigational arm receive a 30-minute infusion of once-weekly carfilzomib (20 mg/m2 on day 1 of cycle 1; 70 mg/m2 on days 8 and 15 of cycle 1; and 70 mg/m2 on days 1, 8 and 15 of subsequent cycles) with 40 mg of dexamethasone.
Patients randomized to the comparator arm received a 10-minute infusion of twice-weekly carfilzomib (20 mg/m2 on days 1 and 2 of cycle 1; 27 mg/m2 on days 8, 9, 15 and 16 of cycle 1; and 27 mg/m2 on days 1, 2, 8, 9, 15 and 16 of subsequent cycles) with 40 mg of dexamethasone.
Patients in the once-weekly arm achieved a statistically significant 3.7-month improvement in PFS compared to the twice-weekly regimen. Median PFS was 11.2 months for the once-weekly patients and 7.6 months for the twice-weekly group (P=0.0014).
Patients in the once-weekly group had a 62.9% ORR compared to 40.8% for those treated twice weekly (P<0.0001).
More patients (7.1%) in the once-weekly group had complete responses or better than those in the twice-weekly arm (1.7%).
The safety profile of the two arms were comparable, with no new safety risks identified in the once-weekly arm.
Treatment-emergent adverse events occurring in 20% or more patients in either arm included anemia, diarrhea, fatigue, hypertension, insomnia, and pyrexia.
First approved in 2012, carfilzomib has indications for the following in the U.S.:
- Treatment of patients with relapsed or refractory multiple myeloma who have received one to three lines of therapy in combination with dexamethasone or with lenalidomide plus dexamethasone.
- As a single agent for the treatment of patients with relapsed or refractory multiple myeloma who have received one or more lines of therapy.
Amgen manufactures carfilzomib for Onyx Pharmaceuticals, Inc.
Prescribing information for carfilzomib is available online.