User login
CHMP recommends approval of ixazomib for MM

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that ixazomib (NinlaroTM) receive conditional marketing authorization.
The recommendation is for ixazomib in combination with lenalidomide and dexamethasone as a treatment for adults with multiple myeloma (MM) who have received at least 1 prior therapy.
The CHMP’s recommendation will be reviewed by the European Commission (EC).
If the EC grants the authorization, ixazomib will be the first oral proteasome inhibitor approved for use across the European Economic Area.
Ixazomib is being developed by Takeda Pharmaceutical Company Limited.
Phase 3 trial
The CHMP’s positive opinion of ixazomib is based on results from the phase 3 TOURMALINE-MM1 trial, which were presented at the 2015 ASH Annual Meeting.
The trial included 722 patients with relapsed or refractory MM. The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362).
Baseline patient characteristics were similar between the treatment arms. Fifty-nine percent of patients in both arms had received 1 prior line of therapy, and 41% in both arms had 2 or 3 prior lines of therapy.
Seventy-eight percent of patients responded to IRd, and 72% responded to Rd (P=0.035). The rates of complete response were 12% and 7%, respectively (P=0.019).
At a median follow-up of about 15 months, the median progression-free survival was 20.6 months in the IRd arm and 14.7 months in the Rd arm. The hazard ratio was 0.742 (P=0.012).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
The incidence of adverse events (AEs) was 98% in the IRd arm and 99% in the Rd arm. The incidence of grade 3 or higher AEs was 74% and 69%, respectively. The incidence of serious AEs was 47% and 49%, respectively.
Common AEs in the IRd and Rd arms, respectively, were diarrhea (45% vs 39%), constipation (35% vs 26%), nausea (29% vs 22%), vomiting (23% vs 12%), rash (36% vs 23%), back pain (24% vs 17%), upper respiratory tract infection (23% vs 19%), thrombocytopenia (31% vs 16%), peripheral neuropathy (27% vs 22%), peripheral edema (28% vs 20%), thromboembolism (8% vs 11%), and neutropenia (33% vs 31%).
About conditional authorization
Conditional marketing authorization represents an expedited path for approval. The EC grants this type of authorization before pivotal registration studies are completed.
Conditional marketing authorization is granted to products whose benefits are thought to outweigh their risks, products that address unmet needs, and products that are expected to provide a significant public health benefit.
If ixazomib receives conditional marketing authorization, Takeda will be required to provide post-approval updates on safety and efficacy analyses for TOURMALINE-MM1 and some other ongoing studies to demonstrate the treatment’s long-term effects. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that ixazomib (NinlaroTM) receive conditional marketing authorization.
The recommendation is for ixazomib in combination with lenalidomide and dexamethasone as a treatment for adults with multiple myeloma (MM) who have received at least 1 prior therapy.
The CHMP’s recommendation will be reviewed by the European Commission (EC).
If the EC grants the authorization, ixazomib will be the first oral proteasome inhibitor approved for use across the European Economic Area.
Ixazomib is being developed by Takeda Pharmaceutical Company Limited.
Phase 3 trial
The CHMP’s positive opinion of ixazomib is based on results from the phase 3 TOURMALINE-MM1 trial, which were presented at the 2015 ASH Annual Meeting.
The trial included 722 patients with relapsed or refractory MM. The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362).
Baseline patient characteristics were similar between the treatment arms. Fifty-nine percent of patients in both arms had received 1 prior line of therapy, and 41% in both arms had 2 or 3 prior lines of therapy.
Seventy-eight percent of patients responded to IRd, and 72% responded to Rd (P=0.035). The rates of complete response were 12% and 7%, respectively (P=0.019).
At a median follow-up of about 15 months, the median progression-free survival was 20.6 months in the IRd arm and 14.7 months in the Rd arm. The hazard ratio was 0.742 (P=0.012).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
The incidence of adverse events (AEs) was 98% in the IRd arm and 99% in the Rd arm. The incidence of grade 3 or higher AEs was 74% and 69%, respectively. The incidence of serious AEs was 47% and 49%, respectively.
Common AEs in the IRd and Rd arms, respectively, were diarrhea (45% vs 39%), constipation (35% vs 26%), nausea (29% vs 22%), vomiting (23% vs 12%), rash (36% vs 23%), back pain (24% vs 17%), upper respiratory tract infection (23% vs 19%), thrombocytopenia (31% vs 16%), peripheral neuropathy (27% vs 22%), peripheral edema (28% vs 20%), thromboembolism (8% vs 11%), and neutropenia (33% vs 31%).
About conditional authorization
Conditional marketing authorization represents an expedited path for approval. The EC grants this type of authorization before pivotal registration studies are completed.
Conditional marketing authorization is granted to products whose benefits are thought to outweigh their risks, products that address unmet needs, and products that are expected to provide a significant public health benefit.
If ixazomib receives conditional marketing authorization, Takeda will be required to provide post-approval updates on safety and efficacy analyses for TOURMALINE-MM1 and some other ongoing studies to demonstrate the treatment’s long-term effects. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that ixazomib (NinlaroTM) receive conditional marketing authorization.
The recommendation is for ixazomib in combination with lenalidomide and dexamethasone as a treatment for adults with multiple myeloma (MM) who have received at least 1 prior therapy.
The CHMP’s recommendation will be reviewed by the European Commission (EC).
If the EC grants the authorization, ixazomib will be the first oral proteasome inhibitor approved for use across the European Economic Area.
Ixazomib is being developed by Takeda Pharmaceutical Company Limited.
Phase 3 trial
The CHMP’s positive opinion of ixazomib is based on results from the phase 3 TOURMALINE-MM1 trial, which were presented at the 2015 ASH Annual Meeting.
The trial included 722 patients with relapsed or refractory MM. The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362).
Baseline patient characteristics were similar between the treatment arms. Fifty-nine percent of patients in both arms had received 1 prior line of therapy, and 41% in both arms had 2 or 3 prior lines of therapy.
Seventy-eight percent of patients responded to IRd, and 72% responded to Rd (P=0.035). The rates of complete response were 12% and 7%, respectively (P=0.019).
At a median follow-up of about 15 months, the median progression-free survival was 20.6 months in the IRd arm and 14.7 months in the Rd arm. The hazard ratio was 0.742 (P=0.012).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
The incidence of adverse events (AEs) was 98% in the IRd arm and 99% in the Rd arm. The incidence of grade 3 or higher AEs was 74% and 69%, respectively. The incidence of serious AEs was 47% and 49%, respectively.
Common AEs in the IRd and Rd arms, respectively, were diarrhea (45% vs 39%), constipation (35% vs 26%), nausea (29% vs 22%), vomiting (23% vs 12%), rash (36% vs 23%), back pain (24% vs 17%), upper respiratory tract infection (23% vs 19%), thrombocytopenia (31% vs 16%), peripheral neuropathy (27% vs 22%), peripheral edema (28% vs 20%), thromboembolism (8% vs 11%), and neutropenia (33% vs 31%).
About conditional authorization
Conditional marketing authorization represents an expedited path for approval. The EC grants this type of authorization before pivotal registration studies are completed.
Conditional marketing authorization is granted to products whose benefits are thought to outweigh their risks, products that address unmet needs, and products that are expected to provide a significant public health benefit.
If ixazomib receives conditional marketing authorization, Takeda will be required to provide post-approval updates on safety and efficacy analyses for TOURMALINE-MM1 and some other ongoing studies to demonstrate the treatment’s long-term effects. ![]()
HHS, NIH aim to increase clinical trial transparency

tumor in a test tube
Photo by Rhoda Baer
The US Department of Health and Human Services (HHS) and the National Institutes of Health (NIH) have announced new efforts to increase clinical trial transparency.
The HHS has issued a final rule that expands the legal requirements for registering certain clinical trials on ClinicalTrials.gov and providing summary results of these trials on the website.
The NIH has issued a complementary policy for registering and submitting summary results to ClinicalTrials.gov for all NIH-funded trials, including those not subject to the final rule.
Both the HHS rule and the NIH policy will be effective on January 18, 2017.
“Access to more information about clinical trials is good for patients, the public, and science,” said NIH Director Francis S. Collins, MD, PhD.
“The final rule and NIH policy we have issued today will help maximize the value of clinical trials, whether publicly or privately supported, and help us honor our commitments to trial participants who do so much to help society advance knowledge and improve health.”
About the HHS rule
The final rule specifies how and when information collected in a clinical trial must be submitted to ClinicalTrials.gov. It does not dictate how clinical trials should be designed or conducted, or what data must be collected.
Requirements under the rule apply to most interventional studies of drug, biological, and device products regulated by the US Food and Drug Administration (FDA). The requirements do not apply to phase 1 trials of drug and biological products or small feasibility studies of device products.
Elements of the rule include:
- Providing a checklist for evaluating which clinical trials are subject to the regulations and who is responsible for submitting the required information
- Expanding the scope of trials for which summary results information must be submitted to include trials involving FDA-regulated products that have not yet been approved, licensed, or cleared by the FDA
- Requiring additional registration and summary results information data elements to be submitted to ClinicalTrials.gov, including the race and ethnicity of trial participants, if collected, and the full protocol
- Requiring additional types of adverse event information
- Providing a list of potential legal consequences for non-compliance.
About the NIH policy
The NIH policy mandates that investigators conducting clinical trials funded by NIH (in whole or in part) will ensure that the trials are registered at ClinicalTrials.gov and that summary results of these trials are posted to the website within 12 months of the primary completion date (although this can be delayed for up to 2 years).
The policy applies to all NIH-funded trials, including phase 1 trials of FDA-regulated products and small feasibility device trials, as well as trials of products that are not regulated by the FDA, such as behavioral interventions. ![]()
tumor in a test tube
Photo by Rhoda Baer
The US Department of Health and Human Services (HHS) and the National Institutes of Health (NIH) have announced new efforts to increase clinical trial transparency.
The HHS has issued a final rule that expands the legal requirements for registering certain clinical trials on ClinicalTrials.gov and providing summary results of these trials on the website.
The NIH has issued a complementary policy for registering and submitting summary results to ClinicalTrials.gov for all NIH-funded trials, including those not subject to the final rule.
Both the HHS rule and the NIH policy will be effective on January 18, 2017.
“Access to more information about clinical trials is good for patients, the public, and science,” said NIH Director Francis S. Collins, MD, PhD.
“The final rule and NIH policy we have issued today will help maximize the value of clinical trials, whether publicly or privately supported, and help us honor our commitments to trial participants who do so much to help society advance knowledge and improve health.”
About the HHS rule
The final rule specifies how and when information collected in a clinical trial must be submitted to ClinicalTrials.gov. It does not dictate how clinical trials should be designed or conducted, or what data must be collected.
Requirements under the rule apply to most interventional studies of drug, biological, and device products regulated by the US Food and Drug Administration (FDA). The requirements do not apply to phase 1 trials of drug and biological products or small feasibility studies of device products.
Elements of the rule include:
- Providing a checklist for evaluating which clinical trials are subject to the regulations and who is responsible for submitting the required information
- Expanding the scope of trials for which summary results information must be submitted to include trials involving FDA-regulated products that have not yet been approved, licensed, or cleared by the FDA
- Requiring additional registration and summary results information data elements to be submitted to ClinicalTrials.gov, including the race and ethnicity of trial participants, if collected, and the full protocol
- Requiring additional types of adverse event information
- Providing a list of potential legal consequences for non-compliance.
About the NIH policy
The NIH policy mandates that investigators conducting clinical trials funded by NIH (in whole or in part) will ensure that the trials are registered at ClinicalTrials.gov and that summary results of these trials are posted to the website within 12 months of the primary completion date (although this can be delayed for up to 2 years).
The policy applies to all NIH-funded trials, including phase 1 trials of FDA-regulated products and small feasibility device trials, as well as trials of products that are not regulated by the FDA, such as behavioral interventions. ![]()
tumor in a test tube
Photo by Rhoda Baer
The US Department of Health and Human Services (HHS) and the National Institutes of Health (NIH) have announced new efforts to increase clinical trial transparency.
The HHS has issued a final rule that expands the legal requirements for registering certain clinical trials on ClinicalTrials.gov and providing summary results of these trials on the website.
The NIH has issued a complementary policy for registering and submitting summary results to ClinicalTrials.gov for all NIH-funded trials, including those not subject to the final rule.
Both the HHS rule and the NIH policy will be effective on January 18, 2017.
“Access to more information about clinical trials is good for patients, the public, and science,” said NIH Director Francis S. Collins, MD, PhD.
“The final rule and NIH policy we have issued today will help maximize the value of clinical trials, whether publicly or privately supported, and help us honor our commitments to trial participants who do so much to help society advance knowledge and improve health.”
About the HHS rule
The final rule specifies how and when information collected in a clinical trial must be submitted to ClinicalTrials.gov. It does not dictate how clinical trials should be designed or conducted, or what data must be collected.
Requirements under the rule apply to most interventional studies of drug, biological, and device products regulated by the US Food and Drug Administration (FDA). The requirements do not apply to phase 1 trials of drug and biological products or small feasibility studies of device products.
Elements of the rule include:
- Providing a checklist for evaluating which clinical trials are subject to the regulations and who is responsible for submitting the required information
- Expanding the scope of trials for which summary results information must be submitted to include trials involving FDA-regulated products that have not yet been approved, licensed, or cleared by the FDA
- Requiring additional registration and summary results information data elements to be submitted to ClinicalTrials.gov, including the race and ethnicity of trial participants, if collected, and the full protocol
- Requiring additional types of adverse event information
- Providing a list of potential legal consequences for non-compliance.
About the NIH policy
The NIH policy mandates that investigators conducting clinical trials funded by NIH (in whole or in part) will ensure that the trials are registered at ClinicalTrials.gov and that summary results of these trials are posted to the website within 12 months of the primary completion date (although this can be delayed for up to 2 years).
The policy applies to all NIH-funded trials, including phase 1 trials of FDA-regulated products and small feasibility device trials, as well as trials of products that are not regulated by the FDA, such as behavioral interventions. ![]()
MRD status should be endpoint in MM trials, team says

Photo courtesy of the
Dana-Farber Cancer Institute
Patients with newly diagnosed multiple myeloma (MM) have better survival outcomes if they are minimal residual disease (MRD)-negative after treatment, according to research published in JAMA Oncology.
MRD negativity was significantly associated with better progression-free survival (PFS) and overall survival (OS).
Researchers therefore concluded that MRD status after treatment should be considered as an endpoint in clinical trials of MM.
Nikhil C. Munshi, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and his colleagues conducted this study.
The team evaluated the prognostic value of MRD in patients with MM by performing a meta-analysis of 21 studies published between January 1990 and January 2016.
The impact of MRD on PFS was assessed in 14 of the studies (n=1273), and the impact of MRD on OS was assessed in 12 studies (n=1100).
Five of the PFS studies (n=574) had results reported specifically in patients with a complete response (CR), as did 6 of the OS studies (n=616).
Dr Munchi and his colleagues found that MRD negativity was associated with significantly better PFS—both overall and in studies looking specifically at patients with CRs. The hazard ratios were 0.41 (95% CI, 0.36-0.48) and 0.44 (95% CI, 0.34-0.56), respectively (P<0.001 for both).
Likewise, MRD negativity was associated with significantly better OS—both overall and in studies looking at patients with CRs. The hazard ratios were 0.57 (95% CI, 0.46-0.71) and 0.47 (95% CI, 0.33-0.67), respectively (P<0.001 for both).
The researchers said there were no significant differences among the studies for PFS and OS.
The team therefore concluded that this study provides quantitative evidence to support the integration of MRD assessment as an endpoint in trials of MM patients. ![]()
Photo courtesy of the
Dana-Farber Cancer Institute
Patients with newly diagnosed multiple myeloma (MM) have better survival outcomes if they are minimal residual disease (MRD)-negative after treatment, according to research published in JAMA Oncology.
MRD negativity was significantly associated with better progression-free survival (PFS) and overall survival (OS).
Researchers therefore concluded that MRD status after treatment should be considered as an endpoint in clinical trials of MM.
Nikhil C. Munshi, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and his colleagues conducted this study.
The team evaluated the prognostic value of MRD in patients with MM by performing a meta-analysis of 21 studies published between January 1990 and January 2016.
The impact of MRD on PFS was assessed in 14 of the studies (n=1273), and the impact of MRD on OS was assessed in 12 studies (n=1100).
Five of the PFS studies (n=574) had results reported specifically in patients with a complete response (CR), as did 6 of the OS studies (n=616).
Dr Munchi and his colleagues found that MRD negativity was associated with significantly better PFS—both overall and in studies looking specifically at patients with CRs. The hazard ratios were 0.41 (95% CI, 0.36-0.48) and 0.44 (95% CI, 0.34-0.56), respectively (P<0.001 for both).
Likewise, MRD negativity was associated with significantly better OS—both overall and in studies looking at patients with CRs. The hazard ratios were 0.57 (95% CI, 0.46-0.71) and 0.47 (95% CI, 0.33-0.67), respectively (P<0.001 for both).
The researchers said there were no significant differences among the studies for PFS and OS.
The team therefore concluded that this study provides quantitative evidence to support the integration of MRD assessment as an endpoint in trials of MM patients. ![]()
Photo courtesy of the
Dana-Farber Cancer Institute
Patients with newly diagnosed multiple myeloma (MM) have better survival outcomes if they are minimal residual disease (MRD)-negative after treatment, according to research published in JAMA Oncology.
MRD negativity was significantly associated with better progression-free survival (PFS) and overall survival (OS).
Researchers therefore concluded that MRD status after treatment should be considered as an endpoint in clinical trials of MM.
Nikhil C. Munshi, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and his colleagues conducted this study.
The team evaluated the prognostic value of MRD in patients with MM by performing a meta-analysis of 21 studies published between January 1990 and January 2016.
The impact of MRD on PFS was assessed in 14 of the studies (n=1273), and the impact of MRD on OS was assessed in 12 studies (n=1100).
Five of the PFS studies (n=574) had results reported specifically in patients with a complete response (CR), as did 6 of the OS studies (n=616).
Dr Munchi and his colleagues found that MRD negativity was associated with significantly better PFS—both overall and in studies looking specifically at patients with CRs. The hazard ratios were 0.41 (95% CI, 0.36-0.48) and 0.44 (95% CI, 0.34-0.56), respectively (P<0.001 for both).
Likewise, MRD negativity was associated with significantly better OS—both overall and in studies looking at patients with CRs. The hazard ratios were 0.57 (95% CI, 0.46-0.71) and 0.47 (95% CI, 0.33-0.67), respectively (P<0.001 for both).
The researchers said there were no significant differences among the studies for PFS and OS.
The team therefore concluded that this study provides quantitative evidence to support the integration of MRD assessment as an endpoint in trials of MM patients. ![]()
EZH2 inhibitors could improve treatment of CML, studies suggest

Image by Difu Wu
Two preclinical studies published in Cancer Discovery suggest that EZH2 inhibitors might enhance the efficacy of treatment with tyrosine kinase inhibitors (TKIs) in patients with chronic myeloid leukemia (CML).
One study showed that CML leukemic stem cells (LSCs) are dependent upon EZH2.
The other study revealed that epigenetic reprogramming sensitizes CML LSCs to combined treatment with an EZH2 inhibitor and a TKI.
EZH2 dependence
In the first study, in vitro experiments revealed that LSCs have an overabundance of EZH2. In fact, the researchers found that EZH2 helps LSCs survive and give rise to full-fledged CML cells.
Experiments in mice showed that inactivating EZH2 through gene-editing techniques caused LSCs to die, halting CML at its source.
“The stem cells’ dependence on EZH2 suggests they will be especially vulnerable to drugs that target the protein,” said study author Stuart Orkin, MD, of Boston Children’s Hospital in Massachusetts.
“Our findings suggest inhibition of EZH2 should be considered as a way to eradicate CML when used in combination with current targeted therapies. It offers a promising approach to shortening the duration of therapy in order to achieve a cure. If successful, the cost savings of such an approach could also be significant.”
Combination treatment
The second study supports the idea that combining EZH2 inhibitors and TKIs could benefit patients with CML.
Mary T. Scott, of the University of Glasgow in the UK, and her colleagues found that EZH2 and H3K27me3 reprogramming is important for LSC survival and renders the cells sensitive to combined treatment with an EZH2 inhibitor and a TKI.
The researchers treated CML CD34+ cells, normal CD34+ cells, and LSCs with the EZH2 inhibitor GSK343 and the TKI dasatinib, both alone and in combination. The team said that GSK343 selectively targeted the loss of H3K27me3 in the presence of dasatinib.
In addition, combination treatment led to a significant reduction in cell viability (even in “TKI-persistent” cells), an increase in apoptosis, and a reduction in colony-forming cell and granulocyte/erythroid/macrophage/megakaryocyte outputs, when compared to dasatinib alone.
The researchers also evaluated a TKI and an EZH2 inhibitor in a mouse model of CML. The mice received nilotinib and EPZ-6438, both alone and in combination, for either 14 days or 25 days.
After 14 days of treatment, mice that received the combination had significant reductions in levels of leukemic (Ph+) human CD45+ cells, CD45+CD34+ progenitor cells, and primitive CD45+CD34+CD38− stem cells in the bone marrow, when compared to mice that received nilotinib alone.
After 25 days of treatment, mice that received the combination had near-complete elimination of CD45+CD34+ progenitor cells and a greater than 70% reduction of CD45+CD34+CD38− cells, when compared to mice that received nilotinib alone.
The researchers said these results demonstrate a clear rationale for combining TKI treatment with an EZH2 inhibitor, as an EZH2 inhibitor can target primitive CML cells not eradicated by a TKI alone. ![]()
Image by Difu Wu
Two preclinical studies published in Cancer Discovery suggest that EZH2 inhibitors might enhance the efficacy of treatment with tyrosine kinase inhibitors (TKIs) in patients with chronic myeloid leukemia (CML).
One study showed that CML leukemic stem cells (LSCs) are dependent upon EZH2.
The other study revealed that epigenetic reprogramming sensitizes CML LSCs to combined treatment with an EZH2 inhibitor and a TKI.
EZH2 dependence
In the first study, in vitro experiments revealed that LSCs have an overabundance of EZH2. In fact, the researchers found that EZH2 helps LSCs survive and give rise to full-fledged CML cells.
Experiments in mice showed that inactivating EZH2 through gene-editing techniques caused LSCs to die, halting CML at its source.
“The stem cells’ dependence on EZH2 suggests they will be especially vulnerable to drugs that target the protein,” said study author Stuart Orkin, MD, of Boston Children’s Hospital in Massachusetts.
“Our findings suggest inhibition of EZH2 should be considered as a way to eradicate CML when used in combination with current targeted therapies. It offers a promising approach to shortening the duration of therapy in order to achieve a cure. If successful, the cost savings of such an approach could also be significant.”
Combination treatment
The second study supports the idea that combining EZH2 inhibitors and TKIs could benefit patients with CML.
Mary T. Scott, of the University of Glasgow in the UK, and her colleagues found that EZH2 and H3K27me3 reprogramming is important for LSC survival and renders the cells sensitive to combined treatment with an EZH2 inhibitor and a TKI.
The researchers treated CML CD34+ cells, normal CD34+ cells, and LSCs with the EZH2 inhibitor GSK343 and the TKI dasatinib, both alone and in combination. The team said that GSK343 selectively targeted the loss of H3K27me3 in the presence of dasatinib.
In addition, combination treatment led to a significant reduction in cell viability (even in “TKI-persistent” cells), an increase in apoptosis, and a reduction in colony-forming cell and granulocyte/erythroid/macrophage/megakaryocyte outputs, when compared to dasatinib alone.
The researchers also evaluated a TKI and an EZH2 inhibitor in a mouse model of CML. The mice received nilotinib and EPZ-6438, both alone and in combination, for either 14 days or 25 days.
After 14 days of treatment, mice that received the combination had significant reductions in levels of leukemic (Ph+) human CD45+ cells, CD45+CD34+ progenitor cells, and primitive CD45+CD34+CD38− stem cells in the bone marrow, when compared to mice that received nilotinib alone.
After 25 days of treatment, mice that received the combination had near-complete elimination of CD45+CD34+ progenitor cells and a greater than 70% reduction of CD45+CD34+CD38− cells, when compared to mice that received nilotinib alone.
The researchers said these results demonstrate a clear rationale for combining TKI treatment with an EZH2 inhibitor, as an EZH2 inhibitor can target primitive CML cells not eradicated by a TKI alone. ![]()
Image by Difu Wu
Two preclinical studies published in Cancer Discovery suggest that EZH2 inhibitors might enhance the efficacy of treatment with tyrosine kinase inhibitors (TKIs) in patients with chronic myeloid leukemia (CML).
One study showed that CML leukemic stem cells (LSCs) are dependent upon EZH2.
The other study revealed that epigenetic reprogramming sensitizes CML LSCs to combined treatment with an EZH2 inhibitor and a TKI.
EZH2 dependence
In the first study, in vitro experiments revealed that LSCs have an overabundance of EZH2. In fact, the researchers found that EZH2 helps LSCs survive and give rise to full-fledged CML cells.
Experiments in mice showed that inactivating EZH2 through gene-editing techniques caused LSCs to die, halting CML at its source.
“The stem cells’ dependence on EZH2 suggests they will be especially vulnerable to drugs that target the protein,” said study author Stuart Orkin, MD, of Boston Children’s Hospital in Massachusetts.
“Our findings suggest inhibition of EZH2 should be considered as a way to eradicate CML when used in combination with current targeted therapies. It offers a promising approach to shortening the duration of therapy in order to achieve a cure. If successful, the cost savings of such an approach could also be significant.”
Combination treatment
The second study supports the idea that combining EZH2 inhibitors and TKIs could benefit patients with CML.
Mary T. Scott, of the University of Glasgow in the UK, and her colleagues found that EZH2 and H3K27me3 reprogramming is important for LSC survival and renders the cells sensitive to combined treatment with an EZH2 inhibitor and a TKI.
The researchers treated CML CD34+ cells, normal CD34+ cells, and LSCs with the EZH2 inhibitor GSK343 and the TKI dasatinib, both alone and in combination. The team said that GSK343 selectively targeted the loss of H3K27me3 in the presence of dasatinib.
In addition, combination treatment led to a significant reduction in cell viability (even in “TKI-persistent” cells), an increase in apoptosis, and a reduction in colony-forming cell and granulocyte/erythroid/macrophage/megakaryocyte outputs, when compared to dasatinib alone.
The researchers also evaluated a TKI and an EZH2 inhibitor in a mouse model of CML. The mice received nilotinib and EPZ-6438, both alone and in combination, for either 14 days or 25 days.
After 14 days of treatment, mice that received the combination had significant reductions in levels of leukemic (Ph+) human CD45+ cells, CD45+CD34+ progenitor cells, and primitive CD45+CD34+CD38− stem cells in the bone marrow, when compared to mice that received nilotinib alone.
After 25 days of treatment, mice that received the combination had near-complete elimination of CD45+CD34+ progenitor cells and a greater than 70% reduction of CD45+CD34+CD38− cells, when compared to mice that received nilotinib alone.
The researchers said these results demonstrate a clear rationale for combining TKI treatment with an EZH2 inhibitor, as an EZH2 inhibitor can target primitive CML cells not eradicated by a TKI alone. ![]()
Smartphone app measures hemoglobin

Photo by Daniel Sone
HEIDELBERG, GERMANY—Scientists have developed a smartphone application called HemaApp that can be used to measure hemoglobin levels and screen for anemia non-invasively.
HemaApp uses illumination sources from a smartphone, in combination with other light sources, and algorithms that analyze the color of a patient’s blood to estimate hemoglobin levels.
In a trial of 31 patients, HemaApp’s results compared favorably to an approved medical device that measures hemoglobin non-invasively.
Researchers described HemaApp in a paper presented at the Association for Computing Machinery’s 2016 International Joint Conference on Pervasive and Ubiquitous Computing (UbiComp 2016).
“In developing countries, community health workers have so much specialized equipment to monitor different conditions that they literally have whole bags full of devices,” said study author Edward Wang, a doctoral student at the University of Washington in Seattle.
“We are trying to make these screening tools work on one ubiquitous platform—a smartphone.”
How the app works
By shining light from the phone’s camera flash through the patient’s finger, HemaApp analyzes the color of the patient’s blood to estimate hemoglobin concentrations.
HemaApp bombards the finger with different wavelengths of light and infrared energy and creates a series of videos. By analyzing how colors are absorbed and reflected across those wavelengths, it can detect concentrations of hemoglobin and other blood components like plasma.
To ensure that the app works on different skin tones and body masses, the researchers developed processing algorithms that use the patient’s pulse to distinguish between the properties of the patient’s blood and the physical characteristics of his or her finger.
Testing
The researchers tested HemaApp on 31 subjects, including healthy students and staff at the University of Washington, inpatients at a children’s cancer and transfusion clinic, and inpatients at an adult cancer and bone marrow transplant clinic.
The subjects ranged in age from 6 to 77. Their hemoglobin levels ranged from 8 to 16 g/dL, and they had skin tones ranging from pale to dark.
The researchers compared results with HemaApp to results obtained via a complete blood count (CBC) and via the Masimo Pronto 7, a device that non-invasively measures hemoglobin by clipping a sensor onto a person’s finger.
The team tested HemaApp under 3 different scenarios—using the smartphone camera’s flash alone, in combination with a common incandescent lightbulb, and with a low-cost LED lighting attachment.
The additional illumination sources tap into other parts of the electromagnetic spectrum that have useful absorption properties but that aren’t currently found on all smartphone cameras.
“New phones are beginning to have more advanced infrared and multi-color LED capabilities,” said study author Shwetak Patel, PhD, of the University of Washington.
“But what we found is that even if your phone doesn’t have all that, you can put your finger near an external light source like a common lightbulb and boost the accuracy rates.”
Results
HemaApp’s hemoglobin measurements using a smartphone camera alone had a 69% correlation to results with the CBC. The app had a 74% correlation with the CBC when used with an incandescent light bulb and an 82% correlation when used with the LED lights.
In comparison, the Masimo Pronto 7’s measurements had an 81% correlation to the CBC.
When used to screen for anemia, HemaApp had higher sensitivity than the Masimo Pronto but lower specificity.
The app’s sensitivity was 79% using just the phone camera and 86% when used with the incandescent light bulb or LED lights, whereas Masimo Pronto’s sensitivity was 69%.
The app’s specificity was 71% when using just the phone or the incandescent light bulb and 77% with the LED lights, whereas Masimo Pronto’s specificity was 88%.
Next steps
The researchers said HemaApp is not intended to replace blood tests, which remain the most accurate way to measure hemoglobin. But the early test results suggest HemaApp can be an effective and affordable initial screening tool to determine whether further blood testing is warranted.
“Anemia is one of the most common problems affecting adults and children worldwide,” said study author Doug Hawkins, MD, of Seattle Children’s Hospital.
“The ability to screen quickly with a smartphone-based test could be a huge improvement to delivering care in limited-resource environments.”
Next research steps include wider national and international testing of HemaApp, collecting more data to improve accuracy rates, and using smartphones to try to detect abnormal hemoglobin properties that could help screen for sickle cell disease and other blood disorders.
“We’re just starting to scratch the surface here,” Dr Patel said. “There’s a lot that we want to tackle in using phones for non-invasively screening disease.” ![]()
Photo by Daniel Sone
HEIDELBERG, GERMANY—Scientists have developed a smartphone application called HemaApp that can be used to measure hemoglobin levels and screen for anemia non-invasively.
HemaApp uses illumination sources from a smartphone, in combination with other light sources, and algorithms that analyze the color of a patient’s blood to estimate hemoglobin levels.
In a trial of 31 patients, HemaApp’s results compared favorably to an approved medical device that measures hemoglobin non-invasively.
Researchers described HemaApp in a paper presented at the Association for Computing Machinery’s 2016 International Joint Conference on Pervasive and Ubiquitous Computing (UbiComp 2016).
“In developing countries, community health workers have so much specialized equipment to monitor different conditions that they literally have whole bags full of devices,” said study author Edward Wang, a doctoral student at the University of Washington in Seattle.
“We are trying to make these screening tools work on one ubiquitous platform—a smartphone.”
How the app works
By shining light from the phone’s camera flash through the patient’s finger, HemaApp analyzes the color of the patient’s blood to estimate hemoglobin concentrations.
HemaApp bombards the finger with different wavelengths of light and infrared energy and creates a series of videos. By analyzing how colors are absorbed and reflected across those wavelengths, it can detect concentrations of hemoglobin and other blood components like plasma.
To ensure that the app works on different skin tones and body masses, the researchers developed processing algorithms that use the patient’s pulse to distinguish between the properties of the patient’s blood and the physical characteristics of his or her finger.
Testing
The researchers tested HemaApp on 31 subjects, including healthy students and staff at the University of Washington, inpatients at a children’s cancer and transfusion clinic, and inpatients at an adult cancer and bone marrow transplant clinic.
The subjects ranged in age from 6 to 77. Their hemoglobin levels ranged from 8 to 16 g/dL, and they had skin tones ranging from pale to dark.
The researchers compared results with HemaApp to results obtained via a complete blood count (CBC) and via the Masimo Pronto 7, a device that non-invasively measures hemoglobin by clipping a sensor onto a person’s finger.
The team tested HemaApp under 3 different scenarios—using the smartphone camera’s flash alone, in combination with a common incandescent lightbulb, and with a low-cost LED lighting attachment.
The additional illumination sources tap into other parts of the electromagnetic spectrum that have useful absorption properties but that aren’t currently found on all smartphone cameras.
“New phones are beginning to have more advanced infrared and multi-color LED capabilities,” said study author Shwetak Patel, PhD, of the University of Washington.
“But what we found is that even if your phone doesn’t have all that, you can put your finger near an external light source like a common lightbulb and boost the accuracy rates.”
Results
HemaApp’s hemoglobin measurements using a smartphone camera alone had a 69% correlation to results with the CBC. The app had a 74% correlation with the CBC when used with an incandescent light bulb and an 82% correlation when used with the LED lights.
In comparison, the Masimo Pronto 7’s measurements had an 81% correlation to the CBC.
When used to screen for anemia, HemaApp had higher sensitivity than the Masimo Pronto but lower specificity.
The app’s sensitivity was 79% using just the phone camera and 86% when used with the incandescent light bulb or LED lights, whereas Masimo Pronto’s sensitivity was 69%.
The app’s specificity was 71% when using just the phone or the incandescent light bulb and 77% with the LED lights, whereas Masimo Pronto’s specificity was 88%.
Next steps
The researchers said HemaApp is not intended to replace blood tests, which remain the most accurate way to measure hemoglobin. But the early test results suggest HemaApp can be an effective and affordable initial screening tool to determine whether further blood testing is warranted.
“Anemia is one of the most common problems affecting adults and children worldwide,” said study author Doug Hawkins, MD, of Seattle Children’s Hospital.
“The ability to screen quickly with a smartphone-based test could be a huge improvement to delivering care in limited-resource environments.”
Next research steps include wider national and international testing of HemaApp, collecting more data to improve accuracy rates, and using smartphones to try to detect abnormal hemoglobin properties that could help screen for sickle cell disease and other blood disorders.
“We’re just starting to scratch the surface here,” Dr Patel said. “There’s a lot that we want to tackle in using phones for non-invasively screening disease.” ![]()
Photo by Daniel Sone
HEIDELBERG, GERMANY—Scientists have developed a smartphone application called HemaApp that can be used to measure hemoglobin levels and screen for anemia non-invasively.
HemaApp uses illumination sources from a smartphone, in combination with other light sources, and algorithms that analyze the color of a patient’s blood to estimate hemoglobin levels.
In a trial of 31 patients, HemaApp’s results compared favorably to an approved medical device that measures hemoglobin non-invasively.
Researchers described HemaApp in a paper presented at the Association for Computing Machinery’s 2016 International Joint Conference on Pervasive and Ubiquitous Computing (UbiComp 2016).
“In developing countries, community health workers have so much specialized equipment to monitor different conditions that they literally have whole bags full of devices,” said study author Edward Wang, a doctoral student at the University of Washington in Seattle.
“We are trying to make these screening tools work on one ubiquitous platform—a smartphone.”
How the app works
By shining light from the phone’s camera flash through the patient’s finger, HemaApp analyzes the color of the patient’s blood to estimate hemoglobin concentrations.
HemaApp bombards the finger with different wavelengths of light and infrared energy and creates a series of videos. By analyzing how colors are absorbed and reflected across those wavelengths, it can detect concentrations of hemoglobin and other blood components like plasma.
To ensure that the app works on different skin tones and body masses, the researchers developed processing algorithms that use the patient’s pulse to distinguish between the properties of the patient’s blood and the physical characteristics of his or her finger.
Testing
The researchers tested HemaApp on 31 subjects, including healthy students and staff at the University of Washington, inpatients at a children’s cancer and transfusion clinic, and inpatients at an adult cancer and bone marrow transplant clinic.
The subjects ranged in age from 6 to 77. Their hemoglobin levels ranged from 8 to 16 g/dL, and they had skin tones ranging from pale to dark.
The researchers compared results with HemaApp to results obtained via a complete blood count (CBC) and via the Masimo Pronto 7, a device that non-invasively measures hemoglobin by clipping a sensor onto a person’s finger.
The team tested HemaApp under 3 different scenarios—using the smartphone camera’s flash alone, in combination with a common incandescent lightbulb, and with a low-cost LED lighting attachment.
The additional illumination sources tap into other parts of the electromagnetic spectrum that have useful absorption properties but that aren’t currently found on all smartphone cameras.
“New phones are beginning to have more advanced infrared and multi-color LED capabilities,” said study author Shwetak Patel, PhD, of the University of Washington.
“But what we found is that even if your phone doesn’t have all that, you can put your finger near an external light source like a common lightbulb and boost the accuracy rates.”
Results
HemaApp’s hemoglobin measurements using a smartphone camera alone had a 69% correlation to results with the CBC. The app had a 74% correlation with the CBC when used with an incandescent light bulb and an 82% correlation when used with the LED lights.
In comparison, the Masimo Pronto 7’s measurements had an 81% correlation to the CBC.
When used to screen for anemia, HemaApp had higher sensitivity than the Masimo Pronto but lower specificity.
The app’s sensitivity was 79% using just the phone camera and 86% when used with the incandescent light bulb or LED lights, whereas Masimo Pronto’s sensitivity was 69%.
The app’s specificity was 71% when using just the phone or the incandescent light bulb and 77% with the LED lights, whereas Masimo Pronto’s specificity was 88%.
Next steps
The researchers said HemaApp is not intended to replace blood tests, which remain the most accurate way to measure hemoglobin. But the early test results suggest HemaApp can be an effective and affordable initial screening tool to determine whether further blood testing is warranted.
“Anemia is one of the most common problems affecting adults and children worldwide,” said study author Doug Hawkins, MD, of Seattle Children’s Hospital.
“The ability to screen quickly with a smartphone-based test could be a huge improvement to delivering care in limited-resource environments.”
Next research steps include wider national and international testing of HemaApp, collecting more data to improve accuracy rates, and using smartphones to try to detect abnormal hemoglobin properties that could help screen for sickle cell disease and other blood disorders.
“We’re just starting to scratch the surface here,” Dr Patel said. “There’s a lot that we want to tackle in using phones for non-invasively screening disease.” ![]()
Prophylaxis proves safer than screen-and-treat method

Photo by Nina Matthews
A new study suggests that treating pregnant women according to the results of malaria tests does not lower the risk of adverse pregnancy outcomes when compared to treating all women prophylactically.
In fact, the screen-and-treat approach, in which women received dihydroartemisinin-piperaquine (DP) only if they tested positive for malaria, was associated with higher fetal loss and more malaria at delivery than the prophylactic approach, in which women just received treatment with sulfadoxine-pyrimethamine (SP).
This is in spite of the fact that the study was conducted in an area of high SP resistance.
Feiko ter Kuile, MD, PhD, of the Liverpool School of Tropical Medicine in the UK, and his colleagues conducted this study and detailed the results in PLOS Medicine.
During pregnancy, undetected infection with malaria parasites can lead to maternal anemia, low birthweight, and fetal loss.
Therefore, in areas where malaria is endemic, the World Health Organization recommends treating pregnant women with SP. However, in some areas, more than 90% of Plasmodium parasites are resistant to SP.
In the current study, researchers compared this standard of care to a screening approach where pregnant women were tested for malaria using rapid diagnostic tests and treated with DP only if they tested positive for the parasite.
The study included 1873 HIV-negative pregnant women treated at 3 sites in Malawi, Africa. All of the women had 3 or 4 scheduled visits in the second and third trimester, 4 to 6 weeks apart.
The women were randomized to receive SP at each visit (n=921) or to be screened for malaria at every visit and treated with DP if they tested positive (n=923).
The prevalence of malaria at delivery was higher in the screening-DP group than in the SP group—48.7% and 40.8%, respectively (relative risk=1.19; P=0.007).
And fetal loss was higher in the screening-DP group than the SP group—2.6% and 1.3%, respectively (relative risk=2.06; P=0.046).
However, the risk of live adverse birth outcomes was similar between the screening-DP and SP groups—29.9% and 28.8%, respectively (relative risk=1.04, P=0.625).
The researchers said these results suggest that intermittent malaria screening and treatment with DP is not a viable strategy to replace intermittent preventive therapy with SP in malaria-endemic areas in sub-Saharan Africa, despite the high levels of resistance to SP. ![]()
Photo by Nina Matthews
A new study suggests that treating pregnant women according to the results of malaria tests does not lower the risk of adverse pregnancy outcomes when compared to treating all women prophylactically.
In fact, the screen-and-treat approach, in which women received dihydroartemisinin-piperaquine (DP) only if they tested positive for malaria, was associated with higher fetal loss and more malaria at delivery than the prophylactic approach, in which women just received treatment with sulfadoxine-pyrimethamine (SP).
This is in spite of the fact that the study was conducted in an area of high SP resistance.
Feiko ter Kuile, MD, PhD, of the Liverpool School of Tropical Medicine in the UK, and his colleagues conducted this study and detailed the results in PLOS Medicine.
During pregnancy, undetected infection with malaria parasites can lead to maternal anemia, low birthweight, and fetal loss.
Therefore, in areas where malaria is endemic, the World Health Organization recommends treating pregnant women with SP. However, in some areas, more than 90% of Plasmodium parasites are resistant to SP.
In the current study, researchers compared this standard of care to a screening approach where pregnant women were tested for malaria using rapid diagnostic tests and treated with DP only if they tested positive for the parasite.
The study included 1873 HIV-negative pregnant women treated at 3 sites in Malawi, Africa. All of the women had 3 or 4 scheduled visits in the second and third trimester, 4 to 6 weeks apart.
The women were randomized to receive SP at each visit (n=921) or to be screened for malaria at every visit and treated with DP if they tested positive (n=923).
The prevalence of malaria at delivery was higher in the screening-DP group than in the SP group—48.7% and 40.8%, respectively (relative risk=1.19; P=0.007).
And fetal loss was higher in the screening-DP group than the SP group—2.6% and 1.3%, respectively (relative risk=2.06; P=0.046).
However, the risk of live adverse birth outcomes was similar between the screening-DP and SP groups—29.9% and 28.8%, respectively (relative risk=1.04, P=0.625).
The researchers said these results suggest that intermittent malaria screening and treatment with DP is not a viable strategy to replace intermittent preventive therapy with SP in malaria-endemic areas in sub-Saharan Africa, despite the high levels of resistance to SP. ![]()
Photo by Nina Matthews
A new study suggests that treating pregnant women according to the results of malaria tests does not lower the risk of adverse pregnancy outcomes when compared to treating all women prophylactically.
In fact, the screen-and-treat approach, in which women received dihydroartemisinin-piperaquine (DP) only if they tested positive for malaria, was associated with higher fetal loss and more malaria at delivery than the prophylactic approach, in which women just received treatment with sulfadoxine-pyrimethamine (SP).
This is in spite of the fact that the study was conducted in an area of high SP resistance.
Feiko ter Kuile, MD, PhD, of the Liverpool School of Tropical Medicine in the UK, and his colleagues conducted this study and detailed the results in PLOS Medicine.
During pregnancy, undetected infection with malaria parasites can lead to maternal anemia, low birthweight, and fetal loss.
Therefore, in areas where malaria is endemic, the World Health Organization recommends treating pregnant women with SP. However, in some areas, more than 90% of Plasmodium parasites are resistant to SP.
In the current study, researchers compared this standard of care to a screening approach where pregnant women were tested for malaria using rapid diagnostic tests and treated with DP only if they tested positive for the parasite.
The study included 1873 HIV-negative pregnant women treated at 3 sites in Malawi, Africa. All of the women had 3 or 4 scheduled visits in the second and third trimester, 4 to 6 weeks apart.
The women were randomized to receive SP at each visit (n=921) or to be screened for malaria at every visit and treated with DP if they tested positive (n=923).
The prevalence of malaria at delivery was higher in the screening-DP group than in the SP group—48.7% and 40.8%, respectively (relative risk=1.19; P=0.007).
And fetal loss was higher in the screening-DP group than the SP group—2.6% and 1.3%, respectively (relative risk=2.06; P=0.046).
However, the risk of live adverse birth outcomes was similar between the screening-DP and SP groups—29.9% and 28.8%, respectively (relative risk=1.04, P=0.625).
The researchers said these results suggest that intermittent malaria screening and treatment with DP is not a viable strategy to replace intermittent preventive therapy with SP in malaria-endemic areas in sub-Saharan Africa, despite the high levels of resistance to SP. ![]()
Metric measures influence of research

Photo by Rhoda Baer
Researchers have developed a metric that uses citation rates to determine the influence of a scientific article.
The team says the metric, known as the Relative Citation Ratio (RCR), measures a scientific publication’s influence in a way that is article-level and field-independent.
George Santangelo, PhD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues described this metric in PLOS Biology.
The researchers noted that citation is the primary mechanism for scientists to recognize the importance of each other’s work, but citation practices vary widely between fields.
RCR incorporates a novel method for field-normalization: the co-citation network. This network is formed from the reference lists of articles that cite the article in question.
For example, if Article X is cited by Article A, Article B, and Article C, the co-citation network of Article X would contain all the articles from the reference lists of Articles A, B, and C. Comparing the citation rate of Article X to the citation rate in the co-citation network allows each article to create its own individualized field.
In addition to using the co-citation network, RCR is also benchmarked to a peer comparison group so that it’s easy to determine the relative impact of an article.
The researchers said this benchmarking step is particularly important as it allows “apples-to-apples” comparisons for groups of papers; eg, comparing research output between similar types of institutions or between developing nations.
To test RCR, Dr Santangelo and his colleagues analyzed 88,835 articles published between 2003 and 2010.
The team said the National Institutes of Health awardees listed as authors of those articles “occupy relatively stable positions of influence across all disciplines.” Furthermore, the values generated by RCR correlated with the opinions of subject matter experts.
Still, the researchers acknowledged that RCR should not be used as a substitute for expert opinion.
“No number can fully represent the impact of an individual work or investigator,” Dr Santangelo said. “Neither RCR nor any other metric can quantitate the underlying value of a study nor measure the importance of making progress in solving a particular problem.”
Dr Santangelo said that, although expert opinion will remain the gold standard, RCR can assist in “the dissemination of a dynamic way to measure the influence of articles on their respective fields.”
A beta version of “iCite,” a web tool for calculating the RCR of articles listed in PubMed, is available at https://icite.od.nih.gov. ![]()
Photo by Rhoda Baer
Researchers have developed a metric that uses citation rates to determine the influence of a scientific article.
The team says the metric, known as the Relative Citation Ratio (RCR), measures a scientific publication’s influence in a way that is article-level and field-independent.
George Santangelo, PhD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues described this metric in PLOS Biology.
The researchers noted that citation is the primary mechanism for scientists to recognize the importance of each other’s work, but citation practices vary widely between fields.
RCR incorporates a novel method for field-normalization: the co-citation network. This network is formed from the reference lists of articles that cite the article in question.
For example, if Article X is cited by Article A, Article B, and Article C, the co-citation network of Article X would contain all the articles from the reference lists of Articles A, B, and C. Comparing the citation rate of Article X to the citation rate in the co-citation network allows each article to create its own individualized field.
In addition to using the co-citation network, RCR is also benchmarked to a peer comparison group so that it’s easy to determine the relative impact of an article.
The researchers said this benchmarking step is particularly important as it allows “apples-to-apples” comparisons for groups of papers; eg, comparing research output between similar types of institutions or between developing nations.
To test RCR, Dr Santangelo and his colleagues analyzed 88,835 articles published between 2003 and 2010.
The team said the National Institutes of Health awardees listed as authors of those articles “occupy relatively stable positions of influence across all disciplines.” Furthermore, the values generated by RCR correlated with the opinions of subject matter experts.
Still, the researchers acknowledged that RCR should not be used as a substitute for expert opinion.
“No number can fully represent the impact of an individual work or investigator,” Dr Santangelo said. “Neither RCR nor any other metric can quantitate the underlying value of a study nor measure the importance of making progress in solving a particular problem.”
Dr Santangelo said that, although expert opinion will remain the gold standard, RCR can assist in “the dissemination of a dynamic way to measure the influence of articles on their respective fields.”
A beta version of “iCite,” a web tool for calculating the RCR of articles listed in PubMed, is available at https://icite.od.nih.gov. ![]()
Photo by Rhoda Baer
Researchers have developed a metric that uses citation rates to determine the influence of a scientific article.
The team says the metric, known as the Relative Citation Ratio (RCR), measures a scientific publication’s influence in a way that is article-level and field-independent.
George Santangelo, PhD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues described this metric in PLOS Biology.
The researchers noted that citation is the primary mechanism for scientists to recognize the importance of each other’s work, but citation practices vary widely between fields.
RCR incorporates a novel method for field-normalization: the co-citation network. This network is formed from the reference lists of articles that cite the article in question.
For example, if Article X is cited by Article A, Article B, and Article C, the co-citation network of Article X would contain all the articles from the reference lists of Articles A, B, and C. Comparing the citation rate of Article X to the citation rate in the co-citation network allows each article to create its own individualized field.
In addition to using the co-citation network, RCR is also benchmarked to a peer comparison group so that it’s easy to determine the relative impact of an article.
The researchers said this benchmarking step is particularly important as it allows “apples-to-apples” comparisons for groups of papers; eg, comparing research output between similar types of institutions or between developing nations.
To test RCR, Dr Santangelo and his colleagues analyzed 88,835 articles published between 2003 and 2010.
The team said the National Institutes of Health awardees listed as authors of those articles “occupy relatively stable positions of influence across all disciplines.” Furthermore, the values generated by RCR correlated with the opinions of subject matter experts.
Still, the researchers acknowledged that RCR should not be used as a substitute for expert opinion.
“No number can fully represent the impact of an individual work or investigator,” Dr Santangelo said. “Neither RCR nor any other metric can quantitate the underlying value of a study nor measure the importance of making progress in solving a particular problem.”
Dr Santangelo said that, although expert opinion will remain the gold standard, RCR can assist in “the dissemination of a dynamic way to measure the influence of articles on their respective fields.”
A beta version of “iCite,” a web tool for calculating the RCR of articles listed in PubMed, is available at https://icite.od.nih.gov.
Certain nuclear facilities may increase risk of leukemia
![]()
Photo by Rafaël Delaedt
Living near a certain type of nuclear facility may increase a child’s risk of developing acute leukemia, a new study suggests.
The research showed a 2- to 3-fold increased risk of acute leukemia among children living near 1 nuclear facility in Belgium—Mol-Dessel.
However, children who lived near the 3 other Belgian nuclear facilities studied—Doel, Fleurus, and Tihange—had no such increased risk.
Researchers said this study does not confirm a causal relationship between the Mol-Dessel facility and the increased risk of leukemia observed.
However, they did say the facility seems a plausible cause of the increased risk because the data showed significant associations between acute leukemia incidence and 3 surrogate measures of exposure—distance from the facility, prevailing winds, and simulated discharges of the radionuclide Ar-41.
An Van Nieuwenhuyse, MD, PhD, of the Scientific Institute of Public Health in Brussels, Belgium, and colleagues conducted this research and reported the results in the European Journal of Cancer Prevention.
Facilities
The study included 4 nuclear facilities in Belgium—Doel, Mol-Dessel, Fleurus, and Tihange. (The researchers also evaluated a fifth facility, Chooz, but they said it was not possible to draw conclusions regarding this site because the region around it is sparsely populated, which limits the number of cancer cases.)
Doel and Tihange are electricity-generating nuclear power plants that started up in 1975. The nuclear facility at Fleurus has produced radionuclides for medicine and industry since 1971.
The Mol-Dessel facility started up in 1956, and a combination of nuclear activities have taken place there, including scientific and technological research, applied research and metrology, operational waste management, the Belgian Underground Research Laboratory, and the production of fuel assemblies for pressurized-water reactors based on uranium oxide and mixed oxides.
Analysis
The researchers set out to determine whether there was an excess incidence of acute leukemia among children ages 0 to 14 who lived in the vicinity of the aforementioned nuclear facilities. The team analyzed data spanning the period from 2002 to 2008.
For all 4 facilities, the researchers used 2 different measures of surrogate exposure to radionuclide gaseous discharges—residential proximity to the nuclear site and prevailing wind directions.
For the Mol-Dessel site, the researchers also used estimated discharges of the radionuclide Ar-41 as a surrogate measure of exposure.
Results
The data did not show an increased incidence of acute leukemia among children living near the Doel, Tihange, or Fleurus facilities. However, children living within 15 km of the Mol-Dessel site had a significantly increased risk of acute leukemia.
In fact, the researchers said they observed statistically significant associations as a function of distance, prevailing winds, and simulated Ar-41 discharges, which suggests a potential link between acute leukemia incidence and the Mol-Dessel site.
The rate ratios for the Mol-Dessel facility, which were adjusted for age, sex, and socioeconomic status, were:
- 2.70 (95% CI: 1.15–6.33) for children living 0 to 5 km from the site (5 cases of acute leukemia)
- 1.82 (95% CI: 1.02–3.25) for children living 0 to 10 km from the site (11 cases of acute leukemia)
- 1.96 (95% CI: 1.19–3.22) for children living 0 to 15 km from the site (15 cases of acute leukemia)
- 1.09 (95% CI: 0.71–1.61) for children living 0 to 20 km from the site (21 cases of acute leukemia).
The 3 communities lying in the dominant wind direction of the Mol-Dessel nuclear site had rate ratios of 6.81 (95% CI: 2.28–20.32), 4.39 (95% CI: 1.46–13.17), and 3.74 (95% CI: 0.98–14.27).
The researchers calculated P values using Stone’s test, Bithell’s linear risk score test (LRS), and Bithell’s linear risk score test with corresponding ranks as a function of the different measures of surrogate exposure (LRSr).
All 3 tests showed a significant association between acute leukemia incidence and proximity to the Mol-Dessel site (P<0.01 for all). However, only LRS and LRSr showed a significant association for wind direction (P=0.01 and 0.03, respectively) and simulated radioactive discharges by Ar-41 (P<0.01 for both).
The researchers noted that, although these results suggest an association between acute childhood leukemia and the Mol-Dessel site, this study had limitations, and more research is needed.
![]()
Photo by Rafaël Delaedt
Living near a certain type of nuclear facility may increase a child’s risk of developing acute leukemia, a new study suggests.
The research showed a 2- to 3-fold increased risk of acute leukemia among children living near 1 nuclear facility in Belgium—Mol-Dessel.
However, children who lived near the 3 other Belgian nuclear facilities studied—Doel, Fleurus, and Tihange—had no such increased risk.
Researchers said this study does not confirm a causal relationship between the Mol-Dessel facility and the increased risk of leukemia observed.
However, they did say the facility seems a plausible cause of the increased risk because the data showed significant associations between acute leukemia incidence and 3 surrogate measures of exposure—distance from the facility, prevailing winds, and simulated discharges of the radionuclide Ar-41.
An Van Nieuwenhuyse, MD, PhD, of the Scientific Institute of Public Health in Brussels, Belgium, and colleagues conducted this research and reported the results in the European Journal of Cancer Prevention.
Facilities
The study included 4 nuclear facilities in Belgium—Doel, Mol-Dessel, Fleurus, and Tihange. (The researchers also evaluated a fifth facility, Chooz, but they said it was not possible to draw conclusions regarding this site because the region around it is sparsely populated, which limits the number of cancer cases.)
Doel and Tihange are electricity-generating nuclear power plants that started up in 1975. The nuclear facility at Fleurus has produced radionuclides for medicine and industry since 1971.
The Mol-Dessel facility started up in 1956, and a combination of nuclear activities have taken place there, including scientific and technological research, applied research and metrology, operational waste management, the Belgian Underground Research Laboratory, and the production of fuel assemblies for pressurized-water reactors based on uranium oxide and mixed oxides.
Analysis
The researchers set out to determine whether there was an excess incidence of acute leukemia among children ages 0 to 14 who lived in the vicinity of the aforementioned nuclear facilities. The team analyzed data spanning the period from 2002 to 2008.
For all 4 facilities, the researchers used 2 different measures of surrogate exposure to radionuclide gaseous discharges—residential proximity to the nuclear site and prevailing wind directions.
For the Mol-Dessel site, the researchers also used estimated discharges of the radionuclide Ar-41 as a surrogate measure of exposure.
Results
The data did not show an increased incidence of acute leukemia among children living near the Doel, Tihange, or Fleurus facilities. However, children living within 15 km of the Mol-Dessel site had a significantly increased risk of acute leukemia.
In fact, the researchers said they observed statistically significant associations as a function of distance, prevailing winds, and simulated Ar-41 discharges, which suggests a potential link between acute leukemia incidence and the Mol-Dessel site.
The rate ratios for the Mol-Dessel facility, which were adjusted for age, sex, and socioeconomic status, were:
- 2.70 (95% CI: 1.15–6.33) for children living 0 to 5 km from the site (5 cases of acute leukemia)
- 1.82 (95% CI: 1.02–3.25) for children living 0 to 10 km from the site (11 cases of acute leukemia)
- 1.96 (95% CI: 1.19–3.22) for children living 0 to 15 km from the site (15 cases of acute leukemia)
- 1.09 (95% CI: 0.71–1.61) for children living 0 to 20 km from the site (21 cases of acute leukemia).
The 3 communities lying in the dominant wind direction of the Mol-Dessel nuclear site had rate ratios of 6.81 (95% CI: 2.28–20.32), 4.39 (95% CI: 1.46–13.17), and 3.74 (95% CI: 0.98–14.27).
The researchers calculated P values using Stone’s test, Bithell’s linear risk score test (LRS), and Bithell’s linear risk score test with corresponding ranks as a function of the different measures of surrogate exposure (LRSr).
All 3 tests showed a significant association between acute leukemia incidence and proximity to the Mol-Dessel site (P<0.01 for all). However, only LRS and LRSr showed a significant association for wind direction (P=0.01 and 0.03, respectively) and simulated radioactive discharges by Ar-41 (P<0.01 for both).
The researchers noted that, although these results suggest an association between acute childhood leukemia and the Mol-Dessel site, this study had limitations, and more research is needed.
![]()
Photo by Rafaël Delaedt
Living near a certain type of nuclear facility may increase a child’s risk of developing acute leukemia, a new study suggests.
The research showed a 2- to 3-fold increased risk of acute leukemia among children living near 1 nuclear facility in Belgium—Mol-Dessel.
However, children who lived near the 3 other Belgian nuclear facilities studied—Doel, Fleurus, and Tihange—had no such increased risk.
Researchers said this study does not confirm a causal relationship between the Mol-Dessel facility and the increased risk of leukemia observed.
However, they did say the facility seems a plausible cause of the increased risk because the data showed significant associations between acute leukemia incidence and 3 surrogate measures of exposure—distance from the facility, prevailing winds, and simulated discharges of the radionuclide Ar-41.
An Van Nieuwenhuyse, MD, PhD, of the Scientific Institute of Public Health in Brussels, Belgium, and colleagues conducted this research and reported the results in the European Journal of Cancer Prevention.
Facilities
The study included 4 nuclear facilities in Belgium—Doel, Mol-Dessel, Fleurus, and Tihange. (The researchers also evaluated a fifth facility, Chooz, but they said it was not possible to draw conclusions regarding this site because the region around it is sparsely populated, which limits the number of cancer cases.)
Doel and Tihange are electricity-generating nuclear power plants that started up in 1975. The nuclear facility at Fleurus has produced radionuclides for medicine and industry since 1971.
The Mol-Dessel facility started up in 1956, and a combination of nuclear activities have taken place there, including scientific and technological research, applied research and metrology, operational waste management, the Belgian Underground Research Laboratory, and the production of fuel assemblies for pressurized-water reactors based on uranium oxide and mixed oxides.
Analysis
The researchers set out to determine whether there was an excess incidence of acute leukemia among children ages 0 to 14 who lived in the vicinity of the aforementioned nuclear facilities. The team analyzed data spanning the period from 2002 to 2008.
For all 4 facilities, the researchers used 2 different measures of surrogate exposure to radionuclide gaseous discharges—residential proximity to the nuclear site and prevailing wind directions.
For the Mol-Dessel site, the researchers also used estimated discharges of the radionuclide Ar-41 as a surrogate measure of exposure.
Results
The data did not show an increased incidence of acute leukemia among children living near the Doel, Tihange, or Fleurus facilities. However, children living within 15 km of the Mol-Dessel site had a significantly increased risk of acute leukemia.
In fact, the researchers said they observed statistically significant associations as a function of distance, prevailing winds, and simulated Ar-41 discharges, which suggests a potential link between acute leukemia incidence and the Mol-Dessel site.
The rate ratios for the Mol-Dessel facility, which were adjusted for age, sex, and socioeconomic status, were:
- 2.70 (95% CI: 1.15–6.33) for children living 0 to 5 km from the site (5 cases of acute leukemia)
- 1.82 (95% CI: 1.02–3.25) for children living 0 to 10 km from the site (11 cases of acute leukemia)
- 1.96 (95% CI: 1.19–3.22) for children living 0 to 15 km from the site (15 cases of acute leukemia)
- 1.09 (95% CI: 0.71–1.61) for children living 0 to 20 km from the site (21 cases of acute leukemia).
The 3 communities lying in the dominant wind direction of the Mol-Dessel nuclear site had rate ratios of 6.81 (95% CI: 2.28–20.32), 4.39 (95% CI: 1.46–13.17), and 3.74 (95% CI: 0.98–14.27).
The researchers calculated P values using Stone’s test, Bithell’s linear risk score test (LRS), and Bithell’s linear risk score test with corresponding ranks as a function of the different measures of surrogate exposure (LRSr).
All 3 tests showed a significant association between acute leukemia incidence and proximity to the Mol-Dessel site (P<0.01 for all). However, only LRS and LRSr showed a significant association for wind direction (P=0.01 and 0.03, respectively) and simulated radioactive discharges by Ar-41 (P<0.01 for both).
The researchers noted that, although these results suggest an association between acute childhood leukemia and the Mol-Dessel site, this study had limitations, and more research is needed.
WHO certifies Sri Lanka malaria-free
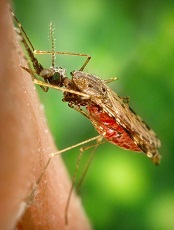
Photo by James Gathany
The World Health Organization (WHO) has certified that Sri Lanka is now free of malaria.
Malaria was on the rise in Sri Lanka in the 1970s and 1980s. So, in the 1990s, the country adjusted its anti-malaria campaign.
The new strategy was to target the malaria parasite as well as the mosquitoes that spread the disease.
Sri Lanka set up mobile malaria clinics in high-transmission areas, which meant that prompt treatment could reduce the parasite reservoir and the possibility of further transmission.
Meanwhile, efforts to enhance surveillance, community engagement, and health education improved the ability of authorities to respond and mobilized popular support for the anti-malaria campaign.
Sri Lanka also received support from partners such as WHO and the Global Fund to Fight AIDS, Tuberculosis and Malaria.
By 2006, Sri Lanka recorded less than 1000 cases of malaria per year. By October 2012, the indigenous cases were down to 0. For the past 3.5 years, no locally transmitted cases have been recorded.
“Sri Lanka’s achievement is truly remarkable,” said Dr Poonam Khetrapal Singh, WHO regional director for Southeast Asia. “In the mid-20th century, it was among the most malaria-affected countries, but now it is malaria-free.”
“This is a testament to the courage and vision of its leaders and signifies the great leaps that can be made when targeted action is taken. It also demonstrates the importance of grass-roots community engagement and a whole-of-society approach when it comes to making dramatic public health gains.”
To maintain elimination and ensure the malaria parasite is not reintroduced to Sri Lanka, the anti-malaria campaign is working closely with local authorities and international partners to maintain surveillance and response capacity and to screen high-risk populations entering the country.
Dr Khetrapal Singh said WHO will continue to support the efforts of Sri Lanka’s health authorities as they relate to malaria, as well as the country’s wider public health mission.
Sri Lanka is the second country in the WHO Southeast Asia region to eliminate malaria—after Maldives.
The announcement that Sri Lanka is malaria-free was made at the Sixty-ninth Session of the WHO Regional Committee for South-East Asia in Colombo, Sri Lanka, in the presence of health ministers and senior health officials from all 11 member states.
Photo by James Gathany
The World Health Organization (WHO) has certified that Sri Lanka is now free of malaria.
Malaria was on the rise in Sri Lanka in the 1970s and 1980s. So, in the 1990s, the country adjusted its anti-malaria campaign.
The new strategy was to target the malaria parasite as well as the mosquitoes that spread the disease.
Sri Lanka set up mobile malaria clinics in high-transmission areas, which meant that prompt treatment could reduce the parasite reservoir and the possibility of further transmission.
Meanwhile, efforts to enhance surveillance, community engagement, and health education improved the ability of authorities to respond and mobilized popular support for the anti-malaria campaign.
Sri Lanka also received support from partners such as WHO and the Global Fund to Fight AIDS, Tuberculosis and Malaria.
By 2006, Sri Lanka recorded less than 1000 cases of malaria per year. By October 2012, the indigenous cases were down to 0. For the past 3.5 years, no locally transmitted cases have been recorded.
“Sri Lanka’s achievement is truly remarkable,” said Dr Poonam Khetrapal Singh, WHO regional director for Southeast Asia. “In the mid-20th century, it was among the most malaria-affected countries, but now it is malaria-free.”
“This is a testament to the courage and vision of its leaders and signifies the great leaps that can be made when targeted action is taken. It also demonstrates the importance of grass-roots community engagement and a whole-of-society approach when it comes to making dramatic public health gains.”
To maintain elimination and ensure the malaria parasite is not reintroduced to Sri Lanka, the anti-malaria campaign is working closely with local authorities and international partners to maintain surveillance and response capacity and to screen high-risk populations entering the country.
Dr Khetrapal Singh said WHO will continue to support the efforts of Sri Lanka’s health authorities as they relate to malaria, as well as the country’s wider public health mission.
Sri Lanka is the second country in the WHO Southeast Asia region to eliminate malaria—after Maldives.
The announcement that Sri Lanka is malaria-free was made at the Sixty-ninth Session of the WHO Regional Committee for South-East Asia in Colombo, Sri Lanka, in the presence of health ministers and senior health officials from all 11 member states.
Photo by James Gathany
The World Health Organization (WHO) has certified that Sri Lanka is now free of malaria.
Malaria was on the rise in Sri Lanka in the 1970s and 1980s. So, in the 1990s, the country adjusted its anti-malaria campaign.
The new strategy was to target the malaria parasite as well as the mosquitoes that spread the disease.
Sri Lanka set up mobile malaria clinics in high-transmission areas, which meant that prompt treatment could reduce the parasite reservoir and the possibility of further transmission.
Meanwhile, efforts to enhance surveillance, community engagement, and health education improved the ability of authorities to respond and mobilized popular support for the anti-malaria campaign.
Sri Lanka also received support from partners such as WHO and the Global Fund to Fight AIDS, Tuberculosis and Malaria.
By 2006, Sri Lanka recorded less than 1000 cases of malaria per year. By October 2012, the indigenous cases were down to 0. For the past 3.5 years, no locally transmitted cases have been recorded.
“Sri Lanka’s achievement is truly remarkable,” said Dr Poonam Khetrapal Singh, WHO regional director for Southeast Asia. “In the mid-20th century, it was among the most malaria-affected countries, but now it is malaria-free.”
“This is a testament to the courage and vision of its leaders and signifies the great leaps that can be made when targeted action is taken. It also demonstrates the importance of grass-roots community engagement and a whole-of-society approach when it comes to making dramatic public health gains.”
To maintain elimination and ensure the malaria parasite is not reintroduced to Sri Lanka, the anti-malaria campaign is working closely with local authorities and international partners to maintain surveillance and response capacity and to screen high-risk populations entering the country.
Dr Khetrapal Singh said WHO will continue to support the efforts of Sri Lanka’s health authorities as they relate to malaria, as well as the country’s wider public health mission.
Sri Lanka is the second country in the WHO Southeast Asia region to eliminate malaria—after Maldives.
The announcement that Sri Lanka is malaria-free was made at the Sixty-ninth Session of the WHO Regional Committee for South-East Asia in Colombo, Sri Lanka, in the presence of health ministers and senior health officials from all 11 member states.
Combo could overcome resistance in Ph+ ALL

Photo by Aaron Logan
Preclinical research suggests that combining a BCL2 inhibitor with a tyrosine kinase inhibitor (TKI) could overcome resistance in Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL).
Investigators found that combining venetoclax and dasatinib increased antitumor activity (when compared to either agent alone) against Ph+ ALL cells in vitro and in a mouse model of the disease.
Jessica Leonard, MD, of Oregon Health & Science University in Portland, and her colleagues reported these findings in Science Translational Medicine.
The researchers said venetoclax and dasatinib demonstrated synergy in vitro, and the combination resulted in a greater degree of apoptosis in Ph+ ALL cells than either agent alone.
The team also observed synergy between venetoclax and cytarabine, dexamethasone, doxorubicin, and vincristine. They said this suggests venetoclax could potentially be used in combination with standard chemotherapy in patients with Ph+ ALL.
The investigators then assessed synergy between venetoclax and other TKIs. Venetoclax demonstrated synergy with imatinib and nilotinib, but the researchers said they saw the most robust synergy when venetoclax was combined with dasatinib or ponatinib.
Further investigation revealed that ponatinib and dasatinib inhibit LYN activity, which impedes STAT5 phosphorylation, which inhibits upregulation of MCL-1.
As upregulation of MCL-1 is a known mechanism of resistance to venetoclax, the researchers believe the addition of either TKI could potentially overcome the development of venetoclax resistance.
Results in a mouse model of Ph+ ALL seemed to support this theory. The researchers injected NSG mice with mononuclear cells from a patient with Ph+ ALL and found that combination treatment with venetoclax and dasatinib prevented engraftment within 90 days in all mice that received the therapy.
In comparison, mice in the control group and those that received single-agent venetoclax engrafted within 60 days. Two of the 5 mice that received dasatinib alone engrafted within 90 days.
In another mouse model, the investigators injected NSG mice with a primary Ph+ ALL sample and found the combination of venetoclax and dasatinib reduced spleen size when compared to no treatment.
In addition, all of the mice that received the combination remained alive during the 4-week treatment period, whereas untreated mice became moribund and were euthanized.
The researchers said these results suggest the combination of dasatinib and venetoclax has the potential to improve the treatment of Ph+ ALL and should be further evaluated for patient care.
Photo by Aaron Logan
Preclinical research suggests that combining a BCL2 inhibitor with a tyrosine kinase inhibitor (TKI) could overcome resistance in Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL).
Investigators found that combining venetoclax and dasatinib increased antitumor activity (when compared to either agent alone) against Ph+ ALL cells in vitro and in a mouse model of the disease.
Jessica Leonard, MD, of Oregon Health & Science University in Portland, and her colleagues reported these findings in Science Translational Medicine.
The researchers said venetoclax and dasatinib demonstrated synergy in vitro, and the combination resulted in a greater degree of apoptosis in Ph+ ALL cells than either agent alone.
The team also observed synergy between venetoclax and cytarabine, dexamethasone, doxorubicin, and vincristine. They said this suggests venetoclax could potentially be used in combination with standard chemotherapy in patients with Ph+ ALL.
The investigators then assessed synergy between venetoclax and other TKIs. Venetoclax demonstrated synergy with imatinib and nilotinib, but the researchers said they saw the most robust synergy when venetoclax was combined with dasatinib or ponatinib.
Further investigation revealed that ponatinib and dasatinib inhibit LYN activity, which impedes STAT5 phosphorylation, which inhibits upregulation of MCL-1.
As upregulation of MCL-1 is a known mechanism of resistance to venetoclax, the researchers believe the addition of either TKI could potentially overcome the development of venetoclax resistance.
Results in a mouse model of Ph+ ALL seemed to support this theory. The researchers injected NSG mice with mononuclear cells from a patient with Ph+ ALL and found that combination treatment with venetoclax and dasatinib prevented engraftment within 90 days in all mice that received the therapy.
In comparison, mice in the control group and those that received single-agent venetoclax engrafted within 60 days. Two of the 5 mice that received dasatinib alone engrafted within 90 days.
In another mouse model, the investigators injected NSG mice with a primary Ph+ ALL sample and found the combination of venetoclax and dasatinib reduced spleen size when compared to no treatment.
In addition, all of the mice that received the combination remained alive during the 4-week treatment period, whereas untreated mice became moribund and were euthanized.
The researchers said these results suggest the combination of dasatinib and venetoclax has the potential to improve the treatment of Ph+ ALL and should be further evaluated for patient care.
Photo by Aaron Logan
Preclinical research suggests that combining a BCL2 inhibitor with a tyrosine kinase inhibitor (TKI) could overcome resistance in Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL).
Investigators found that combining venetoclax and dasatinib increased antitumor activity (when compared to either agent alone) against Ph+ ALL cells in vitro and in a mouse model of the disease.
Jessica Leonard, MD, of Oregon Health & Science University in Portland, and her colleagues reported these findings in Science Translational Medicine.
The researchers said venetoclax and dasatinib demonstrated synergy in vitro, and the combination resulted in a greater degree of apoptosis in Ph+ ALL cells than either agent alone.
The team also observed synergy between venetoclax and cytarabine, dexamethasone, doxorubicin, and vincristine. They said this suggests venetoclax could potentially be used in combination with standard chemotherapy in patients with Ph+ ALL.
The investigators then assessed synergy between venetoclax and other TKIs. Venetoclax demonstrated synergy with imatinib and nilotinib, but the researchers said they saw the most robust synergy when venetoclax was combined with dasatinib or ponatinib.
Further investigation revealed that ponatinib and dasatinib inhibit LYN activity, which impedes STAT5 phosphorylation, which inhibits upregulation of MCL-1.
As upregulation of MCL-1 is a known mechanism of resistance to venetoclax, the researchers believe the addition of either TKI could potentially overcome the development of venetoclax resistance.
Results in a mouse model of Ph+ ALL seemed to support this theory. The researchers injected NSG mice with mononuclear cells from a patient with Ph+ ALL and found that combination treatment with venetoclax and dasatinib prevented engraftment within 90 days in all mice that received the therapy.
In comparison, mice in the control group and those that received single-agent venetoclax engrafted within 60 days. Two of the 5 mice that received dasatinib alone engrafted within 90 days.
In another mouse model, the investigators injected NSG mice with a primary Ph+ ALL sample and found the combination of venetoclax and dasatinib reduced spleen size when compared to no treatment.
In addition, all of the mice that received the combination remained alive during the 4-week treatment period, whereas untreated mice became moribund and were euthanized.
The researchers said these results suggest the combination of dasatinib and venetoclax has the potential to improve the treatment of Ph+ ALL and should be further evaluated for patient care.