User login
Continuous Rd should be standard of care, group says

Photo courtesy of Celgene
Updated trial results support continuous treatment with lenalidomide and low-dose dexamethasone (Rd) as a standard of care for patients of all ages who have newly diagnosed multiple myeloma (MM) and are ineligible for stem cell transplant, according to researchers.
In this phase 3 trial, patients who received continuous Rd (until disease progression) had better progression-free survival (PFS) and overall survival (OS) than patients who received 18 cycles of Rd (Rd18) or a combination of melphalan, prednisone, and thalidomide (MPT).
Updated results from this study, known as the FIRST trial, were published in the Journal of Clinical Oncology.
Results were previously published in NEJM in 2014. The study was supported by Intergroupe Francophone du Myélome and Celgene Corporation, the makers of lenalidomide.
Thierry Facon, MD, of Centre Hospitalier Regional Universitaire de Lille in France, and his colleagues enrolled 1623 patients on this trial. They were newly diagnosed with MM and not eligible for stem cell transplant.
Patients were randomized to receive Rd in 28-day cycles until disease progression (n=535), Rd18 for 72 weeks (n=541), or MPT for 72 weeks (n=547).
Response
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median duration of response was 32 months (range, 26-37) in the continuous Rd group, 22 months (range, 19-23) in the Rd18 group, and 22 months (range, 20-25) in the MPT group.
PFS and OS
The median PFS was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rate was 33%, 14%, and 13%, respectively.
The hazard ratio (HR) for continuous Rd vs MPT was 0.69. The HR for continuous Rd vs Rd18 was 0.71. And the HR for Rd18 vs MPT was 0.99.
The median OS was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rate was 60%, 57%, and 51%, respectively.
The HR for continuous Rd vs MPT was 0.75. The HR for continuous Rd vs Rd18 was 0.91. And the HR for Rd18 vs MPT was 0.83.
Safety
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
Grade 3/4 peripheral sensory neuropathy was less frequent in the continuous Rd and Rd18 groups than the MPT group.
The incidence of invasive second primary malignancy was 4% in the continuous Rd group, 6% in the Rd18 group, and 6% in the MPT group. ![]()
Photo courtesy of Celgene
Updated trial results support continuous treatment with lenalidomide and low-dose dexamethasone (Rd) as a standard of care for patients of all ages who have newly diagnosed multiple myeloma (MM) and are ineligible for stem cell transplant, according to researchers.
In this phase 3 trial, patients who received continuous Rd (until disease progression) had better progression-free survival (PFS) and overall survival (OS) than patients who received 18 cycles of Rd (Rd18) or a combination of melphalan, prednisone, and thalidomide (MPT).
Updated results from this study, known as the FIRST trial, were published in the Journal of Clinical Oncology.
Results were previously published in NEJM in 2014. The study was supported by Intergroupe Francophone du Myélome and Celgene Corporation, the makers of lenalidomide.
Thierry Facon, MD, of Centre Hospitalier Regional Universitaire de Lille in France, and his colleagues enrolled 1623 patients on this trial. They were newly diagnosed with MM and not eligible for stem cell transplant.
Patients were randomized to receive Rd in 28-day cycles until disease progression (n=535), Rd18 for 72 weeks (n=541), or MPT for 72 weeks (n=547).
Response
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median duration of response was 32 months (range, 26-37) in the continuous Rd group, 22 months (range, 19-23) in the Rd18 group, and 22 months (range, 20-25) in the MPT group.
PFS and OS
The median PFS was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rate was 33%, 14%, and 13%, respectively.
The hazard ratio (HR) for continuous Rd vs MPT was 0.69. The HR for continuous Rd vs Rd18 was 0.71. And the HR for Rd18 vs MPT was 0.99.
The median OS was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rate was 60%, 57%, and 51%, respectively.
The HR for continuous Rd vs MPT was 0.75. The HR for continuous Rd vs Rd18 was 0.91. And the HR for Rd18 vs MPT was 0.83.
Safety
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
Grade 3/4 peripheral sensory neuropathy was less frequent in the continuous Rd and Rd18 groups than the MPT group.
The incidence of invasive second primary malignancy was 4% in the continuous Rd group, 6% in the Rd18 group, and 6% in the MPT group. ![]()
Photo courtesy of Celgene
Updated trial results support continuous treatment with lenalidomide and low-dose dexamethasone (Rd) as a standard of care for patients of all ages who have newly diagnosed multiple myeloma (MM) and are ineligible for stem cell transplant, according to researchers.
In this phase 3 trial, patients who received continuous Rd (until disease progression) had better progression-free survival (PFS) and overall survival (OS) than patients who received 18 cycles of Rd (Rd18) or a combination of melphalan, prednisone, and thalidomide (MPT).
Updated results from this study, known as the FIRST trial, were published in the Journal of Clinical Oncology.
Results were previously published in NEJM in 2014. The study was supported by Intergroupe Francophone du Myélome and Celgene Corporation, the makers of lenalidomide.
Thierry Facon, MD, of Centre Hospitalier Regional Universitaire de Lille in France, and his colleagues enrolled 1623 patients on this trial. They were newly diagnosed with MM and not eligible for stem cell transplant.
Patients were randomized to receive Rd in 28-day cycles until disease progression (n=535), Rd18 for 72 weeks (n=541), or MPT for 72 weeks (n=547).
Response
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median duration of response was 32 months (range, 26-37) in the continuous Rd group, 22 months (range, 19-23) in the Rd18 group, and 22 months (range, 20-25) in the MPT group.
PFS and OS
The median PFS was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rate was 33%, 14%, and 13%, respectively.
The hazard ratio (HR) for continuous Rd vs MPT was 0.69. The HR for continuous Rd vs Rd18 was 0.71. And the HR for Rd18 vs MPT was 0.99.
The median OS was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rate was 60%, 57%, and 51%, respectively.
The HR for continuous Rd vs MPT was 0.75. The HR for continuous Rd vs Rd18 was 0.91. And the HR for Rd18 vs MPT was 0.83.
Safety
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
Grade 3/4 peripheral sensory neuropathy was less frequent in the continuous Rd and Rd18 groups than the MPT group.
The incidence of invasive second primary malignancy was 4% in the continuous Rd group, 6% in the Rd18 group, and 6% in the MPT group. ![]()
Results support continued study of CMV vaccine

CMV infection
A vaccine designed to control cytomegalovirus (CMV) has produced favorable results in a phase 1 trial of healthy volunteers.
Investigators said the vaccine, known as Triplex, was well-tolerated at multiple dose levels.
The vaccine also generated “robust” and “durable” virus-specific immunity in subjects who were previously infected with CMV and those who were not.
The results of this study were published in Blood.
Triplex is a universal (non-HLA-restricted), recombinant modified vaccinia ankara viral vector vaccine engineered to induce a virus-specific T-cell response to 3 immuno-dominant proteins (UL83 [pp65], UL123 [IE1], and UL122 [IE2]) linked to CMV complications in the post-transplant setting.
Helocyte Inc. is developing the vaccine for control of CMV in recipients of allogeneic hematopoietic stem cell transplant (HSCT) and solid organ transplant.
This study was not funded by Helocyte. However, investigator Don J. Diamond, PhD, of City of Hope Comprehensive Cancer Center in Duarte, California is chair of Helocyte’s scientific advisory board and receives personal service fees from the company.
In this trial, Dr Diamond and his colleagues studied the response to Triplex in 24 healthy volunteers.
Subjects were divided into 3 groups of 8 receiving 3 different doses of the vaccine. The first dose level was 10xE7 plaque-forming units (pfu), the second was 5x10E7 pfu, and the third was 5x10E8 pfu.
The subjects received the vaccine in a volume of 1 mL by intramuscular injection in the upper arm and an identical booster injection 28 days later.
Safety
The investigators said Triplex was well tolerated in most subjects at all dose levels. There were no dose-limiting toxicities and no serious adverse events attributed to the vaccine.
One subject experienced a grade 3 injection site adverse event (erythema), which resolved in 1 day without treatment. In addition, there were 3 mild-to-moderate cutaneous reactions.
The investigators said the most common systemic reaction was mild-to-moderate flu-like symptoms. Most subjects in the highest dose group experienced these symptoms, as did a few subjects from the lower dose groups. All of these events were transient, self-limiting, and resolved.
Immunogenicity
The investigators reported “robust, functional, and durable” expansion of CMV-specific T cells after Triplex vaccination, in subjects with and without prior CMV infection.
At day 42, subjects had experienced a significant increase in pp65-specific T cells from baseline. The P values were 0.0003 for pp65-specific CD137+ CD8+ T cells and 0.001 for CD137+ CD4+ T cells.
Expansion remained above baseline levels until at least day 360 for pp65-specific CD137+ CD8+ T cells and at least until day 482 for pp65-specific CD137+ CD4+ T cells.
IE1-exon4- and IE2-exon5-specific T-cell expansions occurred as well, although the increase from baseline was not significant for IE1-exon4-specific T cells.
The median concentrations of IE2-exon5-specific T cells had increased significantly from baseline at day 42—for both CD137+ CD8+ T cells (P=0.014) and CD137+ CD4+ T cells (P=0.003).
The investigators noted that there was no significant difference in the responses to all 3 CMV libraries according to Triplex dose level, previous smallpox vaccination, or CMV-serostatus.
Next, the team found that Triplex vaccination induced significant expansion of pp65-specific IFN-γ+ CD8+ T cells. They said this suggests the vaccine was able to expand a functional subset of CMV-specific T cells, even in the absence of CMV viremia.
The median concentration of pp65-specific IFN-γ + CD8+ T cells increased significantly from baseline to day 28 (P=0.024), day 56 (P=0.003), day 100 (P=0.011), and day 360 (P=0.085).
There was no significant increase for pp65-specific IFN-γ + CD4+ T cells, IE1-exon4-specific IFN-γ+ T cells, or IE2-exon5-specific IFN-γ + T cells.
The investigators also said the Triplex vaccine induced significant vaccinia-specific T-cell increases by day 42 (P=0.0005), with an estimated decline to pre-vaccination levels at day 274.
These data supported the initiation of an ongoing phase 2 trial in which investigators are evaluating Triplex in patients undergoing allogeneic HSCT (NCT02506933).
“After years of work, it is very gratifying that we are making advancements in helping people worldwide achieve better health outcomes after a transplant procedure,” said Dr Diamond, who led the team that developed Triplex.
“Furthermore, Triplex’s favorable safety and immunogenicity may make the vaccine an ideal therapeutic platform to combat significant complications in many disease areas, like solid organ transplant and glioblastoma.” ![]()
CMV infection
A vaccine designed to control cytomegalovirus (CMV) has produced favorable results in a phase 1 trial of healthy volunteers.
Investigators said the vaccine, known as Triplex, was well-tolerated at multiple dose levels.
The vaccine also generated “robust” and “durable” virus-specific immunity in subjects who were previously infected with CMV and those who were not.
The results of this study were published in Blood.
Triplex is a universal (non-HLA-restricted), recombinant modified vaccinia ankara viral vector vaccine engineered to induce a virus-specific T-cell response to 3 immuno-dominant proteins (UL83 [pp65], UL123 [IE1], and UL122 [IE2]) linked to CMV complications in the post-transplant setting.
Helocyte Inc. is developing the vaccine for control of CMV in recipients of allogeneic hematopoietic stem cell transplant (HSCT) and solid organ transplant.
This study was not funded by Helocyte. However, investigator Don J. Diamond, PhD, of City of Hope Comprehensive Cancer Center in Duarte, California is chair of Helocyte’s scientific advisory board and receives personal service fees from the company.
In this trial, Dr Diamond and his colleagues studied the response to Triplex in 24 healthy volunteers.
Subjects were divided into 3 groups of 8 receiving 3 different doses of the vaccine. The first dose level was 10xE7 plaque-forming units (pfu), the second was 5x10E7 pfu, and the third was 5x10E8 pfu.
The subjects received the vaccine in a volume of 1 mL by intramuscular injection in the upper arm and an identical booster injection 28 days later.
Safety
The investigators said Triplex was well tolerated in most subjects at all dose levels. There were no dose-limiting toxicities and no serious adverse events attributed to the vaccine.
One subject experienced a grade 3 injection site adverse event (erythema), which resolved in 1 day without treatment. In addition, there were 3 mild-to-moderate cutaneous reactions.
The investigators said the most common systemic reaction was mild-to-moderate flu-like symptoms. Most subjects in the highest dose group experienced these symptoms, as did a few subjects from the lower dose groups. All of these events were transient, self-limiting, and resolved.
Immunogenicity
The investigators reported “robust, functional, and durable” expansion of CMV-specific T cells after Triplex vaccination, in subjects with and without prior CMV infection.
At day 42, subjects had experienced a significant increase in pp65-specific T cells from baseline. The P values were 0.0003 for pp65-specific CD137+ CD8+ T cells and 0.001 for CD137+ CD4+ T cells.
Expansion remained above baseline levels until at least day 360 for pp65-specific CD137+ CD8+ T cells and at least until day 482 for pp65-specific CD137+ CD4+ T cells.
IE1-exon4- and IE2-exon5-specific T-cell expansions occurred as well, although the increase from baseline was not significant for IE1-exon4-specific T cells.
The median concentrations of IE2-exon5-specific T cells had increased significantly from baseline at day 42—for both CD137+ CD8+ T cells (P=0.014) and CD137+ CD4+ T cells (P=0.003).
The investigators noted that there was no significant difference in the responses to all 3 CMV libraries according to Triplex dose level, previous smallpox vaccination, or CMV-serostatus.
Next, the team found that Triplex vaccination induced significant expansion of pp65-specific IFN-γ+ CD8+ T cells. They said this suggests the vaccine was able to expand a functional subset of CMV-specific T cells, even in the absence of CMV viremia.
The median concentration of pp65-specific IFN-γ + CD8+ T cells increased significantly from baseline to day 28 (P=0.024), day 56 (P=0.003), day 100 (P=0.011), and day 360 (P=0.085).
There was no significant increase for pp65-specific IFN-γ + CD4+ T cells, IE1-exon4-specific IFN-γ+ T cells, or IE2-exon5-specific IFN-γ + T cells.
The investigators also said the Triplex vaccine induced significant vaccinia-specific T-cell increases by day 42 (P=0.0005), with an estimated decline to pre-vaccination levels at day 274.
These data supported the initiation of an ongoing phase 2 trial in which investigators are evaluating Triplex in patients undergoing allogeneic HSCT (NCT02506933).
“After years of work, it is very gratifying that we are making advancements in helping people worldwide achieve better health outcomes after a transplant procedure,” said Dr Diamond, who led the team that developed Triplex.
“Furthermore, Triplex’s favorable safety and immunogenicity may make the vaccine an ideal therapeutic platform to combat significant complications in many disease areas, like solid organ transplant and glioblastoma.” ![]()
CMV infection
A vaccine designed to control cytomegalovirus (CMV) has produced favorable results in a phase 1 trial of healthy volunteers.
Investigators said the vaccine, known as Triplex, was well-tolerated at multiple dose levels.
The vaccine also generated “robust” and “durable” virus-specific immunity in subjects who were previously infected with CMV and those who were not.
The results of this study were published in Blood.
Triplex is a universal (non-HLA-restricted), recombinant modified vaccinia ankara viral vector vaccine engineered to induce a virus-specific T-cell response to 3 immuno-dominant proteins (UL83 [pp65], UL123 [IE1], and UL122 [IE2]) linked to CMV complications in the post-transplant setting.
Helocyte Inc. is developing the vaccine for control of CMV in recipients of allogeneic hematopoietic stem cell transplant (HSCT) and solid organ transplant.
This study was not funded by Helocyte. However, investigator Don J. Diamond, PhD, of City of Hope Comprehensive Cancer Center in Duarte, California is chair of Helocyte’s scientific advisory board and receives personal service fees from the company.
In this trial, Dr Diamond and his colleagues studied the response to Triplex in 24 healthy volunteers.
Subjects were divided into 3 groups of 8 receiving 3 different doses of the vaccine. The first dose level was 10xE7 plaque-forming units (pfu), the second was 5x10E7 pfu, and the third was 5x10E8 pfu.
The subjects received the vaccine in a volume of 1 mL by intramuscular injection in the upper arm and an identical booster injection 28 days later.
Safety
The investigators said Triplex was well tolerated in most subjects at all dose levels. There were no dose-limiting toxicities and no serious adverse events attributed to the vaccine.
One subject experienced a grade 3 injection site adverse event (erythema), which resolved in 1 day without treatment. In addition, there were 3 mild-to-moderate cutaneous reactions.
The investigators said the most common systemic reaction was mild-to-moderate flu-like symptoms. Most subjects in the highest dose group experienced these symptoms, as did a few subjects from the lower dose groups. All of these events were transient, self-limiting, and resolved.
Immunogenicity
The investigators reported “robust, functional, and durable” expansion of CMV-specific T cells after Triplex vaccination, in subjects with and without prior CMV infection.
At day 42, subjects had experienced a significant increase in pp65-specific T cells from baseline. The P values were 0.0003 for pp65-specific CD137+ CD8+ T cells and 0.001 for CD137+ CD4+ T cells.
Expansion remained above baseline levels until at least day 360 for pp65-specific CD137+ CD8+ T cells and at least until day 482 for pp65-specific CD137+ CD4+ T cells.
IE1-exon4- and IE2-exon5-specific T-cell expansions occurred as well, although the increase from baseline was not significant for IE1-exon4-specific T cells.
The median concentrations of IE2-exon5-specific T cells had increased significantly from baseline at day 42—for both CD137+ CD8+ T cells (P=0.014) and CD137+ CD4+ T cells (P=0.003).
The investigators noted that there was no significant difference in the responses to all 3 CMV libraries according to Triplex dose level, previous smallpox vaccination, or CMV-serostatus.
Next, the team found that Triplex vaccination induced significant expansion of pp65-specific IFN-γ+ CD8+ T cells. They said this suggests the vaccine was able to expand a functional subset of CMV-specific T cells, even in the absence of CMV viremia.
The median concentration of pp65-specific IFN-γ + CD8+ T cells increased significantly from baseline to day 28 (P=0.024), day 56 (P=0.003), day 100 (P=0.011), and day 360 (P=0.085).
There was no significant increase for pp65-specific IFN-γ + CD4+ T cells, IE1-exon4-specific IFN-γ+ T cells, or IE2-exon5-specific IFN-γ + T cells.
The investigators also said the Triplex vaccine induced significant vaccinia-specific T-cell increases by day 42 (P=0.0005), with an estimated decline to pre-vaccination levels at day 274.
These data supported the initiation of an ongoing phase 2 trial in which investigators are evaluating Triplex in patients undergoing allogeneic HSCT (NCT02506933).
“After years of work, it is very gratifying that we are making advancements in helping people worldwide achieve better health outcomes after a transplant procedure,” said Dr Diamond, who led the team that developed Triplex.
“Furthermore, Triplex’s favorable safety and immunogenicity may make the vaccine an ideal therapeutic platform to combat significant complications in many disease areas, like solid organ transplant and glioblastoma.” ![]()
Rethinking testing in multiple myeloma
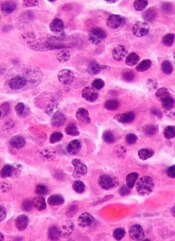
NEW YORK—As the number of therapeutic options for multiple myeloma (MM) increases, so too does the need to reassess prognostic markers for the disease, according to a speaker at Lymphoma & Myeloma 2016.
“A good prognosticator for one patient may have little meaning for another patient,” said Scott Ely, MD, of Weill Cornell Medicine in New York, New York.
“It’s really important before doing any testing to ask, ‘Will the result of this test affect patient care?’”
To answer this question, Dr Ely reviewed the different testing methods used in MM patients and explained the advantages of each.
“[I]t’s really important to understand that a lot of methods are really great for research but don’t work or are not feasible for real-life diagnostic purposes,” he added.
Dr Ely also said it’s important to consider who wants the data, how much the test costs, and who will pay for it, keeping in mind that, these days, the patient’s share of the bill is increasing.
Dr Ely stressed that, until more precise targets or a better understanding of drug mechanisms exist, clinical features—patient age, performance status or frailty, renal function, and disease stage—remain the most important prognosticators.
“But still, 2 patients in the same box based on clinical features will often have very different outcomes,” he said. “So in addition to clinical factors, we need prognosticators for tumor cell behavior. We need to know how fast they are growing and how they will respond to treatment.”
Methods to assess myeloma cell proliferation
Cytogenetics (FISH), gene array technology, and genomics using next-generation sequencing can provide some information, but they are not necessarily good methods to assess proliferation, Dr Ely explained.
To determine the proliferation rate of a patient’s cancer, you can look at tens of thousands of genes by gene array, he said, “or you can just look at one thing, which is Ki67.”
If the cell has Ki67, it’s proliferating, and if it doesn’t have Ki67, it’s not.
“Often, looking at all the other upstream molecules can be confusing and even misleading,” he noted. “So Ki67 is the best way to look for proliferation when it comes to myeloma.”
Other methods include the plasma cell labeling index (PCLI), gene expression profiling, flow cytometry, and multiplex immunohistochemistry (mIHC).
Dr Ely, as a hematopathologist, has found IHC to be the best method to determine proliferation, most likely because the other methods use bone marrow aspirate and IHC uses core biopsy of histologic sections.
It’s the gold standard, he said, for determining the percentage of plasma cells because core biopsy takes a “complete, intact piece of marrow that’s truly representative of what’s going on in the patient.”
In a study of more than 350 bone marrow samples comparing core biopsy with aspirate smears, plasma cells were under-represented in approximately half the aspirate specimens by about 20%.
In addition, Dr Ely noted that myeloma cells die very quickly once they are removed from the stroma.
“So if you take myeloma cells out as an aspirate,” he said, “myeloma cells die and others survive.”
And if the aspirate is sent overnight to the lab, the number of plasma cells in the specimen will already be reduced when the lab gets it.
Aspirates are best for leukemia and myelodysplastic syndromes, Dr Ely said, while core biopsies are best for lymphoma and myeloma.
Plasma cell proliferation indices
Proliferation is a myeloma-defining criterion, Dr Ely said. It predicts an 80% probability of progression in 2 years.
And PCLI has shown conclusively that plasma cell proliferation is a good prognosticator in all types of myeloma patients. However, it is not really feasible to use or easy to perform.
On the other hand, core biopsy combined with IHC is a feasible way of measuring plasma cell proliferation for routine clinical use.
Using standard IHC, it’s difficult to distinguish proliferating from non-proliferating cells, Dr Ely said.
“The solution to this problem is multiplex IHC,” he said, using 3 stains—red for CD138 (the plasma cell marker), brown for Ki67 (the proliferation marker), and blue as a negative nuclear counter stain.
A red membrane around the stained cell indicates a myeloma cell. Non-proliferating MM cells have blue nuclei, and proliferating MM cells have brown nuclei.
This assay is called the plasma cell proliferation index (PCPI).
“[A]ny lab that can do standard IHC can do multiplex IHC,” Dr Ely added.
It uses the same machines, the same reagents, the same expertise, and it’s easy to set up.
Validation studies
Dr Ely and his colleagues performed extensive laboratory validation to make sure the new PCPI correlated with the old PCLI.
They also performed 3 clinical validation studies, the first in bone marrow transplant patients. The investigators followed the patients for 12 years and found the PCPI correlated with survival.
The investigators performed a second clinical validation in 151 newly diagnosed patients. On multivariate analysis, the team found PCPI to be an independent prognostic indicator.
Each 1% increase in PCPI was associated with a 3% increased risk of disease progression (hazard ratio=1.03, 95% CI, 1.01-1.05, P=0.02).
The third clinical validation the investigators conducted was a retrospective cohort study in which they evaluated the effect of rising PCPI at relapse in 37 patients. The team defined rising PCPI as a 5% or greater increase in plasma cells.
Nineteen patients had a rising PCPI, and 17 patients had stable or decreased PCPI.
Patients with a rising PCPI at relapse had a shorter median overall survival than patients with stable or decreased PCPI—72 months and not reached, respectively (P=0.0069).
Patients with a rising PCPI also had a shorter median progression-free survival on first post-relapse treatment compared to patients with stable or decreased PCPI—25 months and 47 months, respectively (P=0.036).
“It’s also important to note that if you’re getting high-risk by PCPI plus β2-microglobulin albumin, I’d advise that, for all high-risk patients, getting the cytogenetics doesn’t really help,” Dr Ely said
Three patients considered high-risk by cytogenetics were standard-risk by PCPI plus β2 microglobulin.
“So we found that cytogenetics isn’t really adding anything except the cost,” Dr Ely asserted.
Other labs are now using PCPI for prognostication, he noted, adding, “We hope PCPI will be incorporated into the International Myeloma Working Group diagnosis of MM.” ![]()
NEW YORK—As the number of therapeutic options for multiple myeloma (MM) increases, so too does the need to reassess prognostic markers for the disease, according to a speaker at Lymphoma & Myeloma 2016.
“A good prognosticator for one patient may have little meaning for another patient,” said Scott Ely, MD, of Weill Cornell Medicine in New York, New York.
“It’s really important before doing any testing to ask, ‘Will the result of this test affect patient care?’”
To answer this question, Dr Ely reviewed the different testing methods used in MM patients and explained the advantages of each.
“[I]t’s really important to understand that a lot of methods are really great for research but don’t work or are not feasible for real-life diagnostic purposes,” he added.
Dr Ely also said it’s important to consider who wants the data, how much the test costs, and who will pay for it, keeping in mind that, these days, the patient’s share of the bill is increasing.
Dr Ely stressed that, until more precise targets or a better understanding of drug mechanisms exist, clinical features—patient age, performance status or frailty, renal function, and disease stage—remain the most important prognosticators.
“But still, 2 patients in the same box based on clinical features will often have very different outcomes,” he said. “So in addition to clinical factors, we need prognosticators for tumor cell behavior. We need to know how fast they are growing and how they will respond to treatment.”
Methods to assess myeloma cell proliferation
Cytogenetics (FISH), gene array technology, and genomics using next-generation sequencing can provide some information, but they are not necessarily good methods to assess proliferation, Dr Ely explained.
To determine the proliferation rate of a patient’s cancer, you can look at tens of thousands of genes by gene array, he said, “or you can just look at one thing, which is Ki67.”
If the cell has Ki67, it’s proliferating, and if it doesn’t have Ki67, it’s not.
“Often, looking at all the other upstream molecules can be confusing and even misleading,” he noted. “So Ki67 is the best way to look for proliferation when it comes to myeloma.”
Other methods include the plasma cell labeling index (PCLI), gene expression profiling, flow cytometry, and multiplex immunohistochemistry (mIHC).
Dr Ely, as a hematopathologist, has found IHC to be the best method to determine proliferation, most likely because the other methods use bone marrow aspirate and IHC uses core biopsy of histologic sections.
It’s the gold standard, he said, for determining the percentage of plasma cells because core biopsy takes a “complete, intact piece of marrow that’s truly representative of what’s going on in the patient.”
In a study of more than 350 bone marrow samples comparing core biopsy with aspirate smears, plasma cells were under-represented in approximately half the aspirate specimens by about 20%.
In addition, Dr Ely noted that myeloma cells die very quickly once they are removed from the stroma.
“So if you take myeloma cells out as an aspirate,” he said, “myeloma cells die and others survive.”
And if the aspirate is sent overnight to the lab, the number of plasma cells in the specimen will already be reduced when the lab gets it.
Aspirates are best for leukemia and myelodysplastic syndromes, Dr Ely said, while core biopsies are best for lymphoma and myeloma.
Plasma cell proliferation indices
Proliferation is a myeloma-defining criterion, Dr Ely said. It predicts an 80% probability of progression in 2 years.
And PCLI has shown conclusively that plasma cell proliferation is a good prognosticator in all types of myeloma patients. However, it is not really feasible to use or easy to perform.
On the other hand, core biopsy combined with IHC is a feasible way of measuring plasma cell proliferation for routine clinical use.
Using standard IHC, it’s difficult to distinguish proliferating from non-proliferating cells, Dr Ely said.
“The solution to this problem is multiplex IHC,” he said, using 3 stains—red for CD138 (the plasma cell marker), brown for Ki67 (the proliferation marker), and blue as a negative nuclear counter stain.
A red membrane around the stained cell indicates a myeloma cell. Non-proliferating MM cells have blue nuclei, and proliferating MM cells have brown nuclei.
This assay is called the plasma cell proliferation index (PCPI).
“[A]ny lab that can do standard IHC can do multiplex IHC,” Dr Ely added.
It uses the same machines, the same reagents, the same expertise, and it’s easy to set up.
Validation studies
Dr Ely and his colleagues performed extensive laboratory validation to make sure the new PCPI correlated with the old PCLI.
They also performed 3 clinical validation studies, the first in bone marrow transplant patients. The investigators followed the patients for 12 years and found the PCPI correlated with survival.
The investigators performed a second clinical validation in 151 newly diagnosed patients. On multivariate analysis, the team found PCPI to be an independent prognostic indicator.
Each 1% increase in PCPI was associated with a 3% increased risk of disease progression (hazard ratio=1.03, 95% CI, 1.01-1.05, P=0.02).
The third clinical validation the investigators conducted was a retrospective cohort study in which they evaluated the effect of rising PCPI at relapse in 37 patients. The team defined rising PCPI as a 5% or greater increase in plasma cells.
Nineteen patients had a rising PCPI, and 17 patients had stable or decreased PCPI.
Patients with a rising PCPI at relapse had a shorter median overall survival than patients with stable or decreased PCPI—72 months and not reached, respectively (P=0.0069).
Patients with a rising PCPI also had a shorter median progression-free survival on first post-relapse treatment compared to patients with stable or decreased PCPI—25 months and 47 months, respectively (P=0.036).
“It’s also important to note that if you’re getting high-risk by PCPI plus β2-microglobulin albumin, I’d advise that, for all high-risk patients, getting the cytogenetics doesn’t really help,” Dr Ely said
Three patients considered high-risk by cytogenetics were standard-risk by PCPI plus β2 microglobulin.
“So we found that cytogenetics isn’t really adding anything except the cost,” Dr Ely asserted.
Other labs are now using PCPI for prognostication, he noted, adding, “We hope PCPI will be incorporated into the International Myeloma Working Group diagnosis of MM.” ![]()
NEW YORK—As the number of therapeutic options for multiple myeloma (MM) increases, so too does the need to reassess prognostic markers for the disease, according to a speaker at Lymphoma & Myeloma 2016.
“A good prognosticator for one patient may have little meaning for another patient,” said Scott Ely, MD, of Weill Cornell Medicine in New York, New York.
“It’s really important before doing any testing to ask, ‘Will the result of this test affect patient care?’”
To answer this question, Dr Ely reviewed the different testing methods used in MM patients and explained the advantages of each.
“[I]t’s really important to understand that a lot of methods are really great for research but don’t work or are not feasible for real-life diagnostic purposes,” he added.
Dr Ely also said it’s important to consider who wants the data, how much the test costs, and who will pay for it, keeping in mind that, these days, the patient’s share of the bill is increasing.
Dr Ely stressed that, until more precise targets or a better understanding of drug mechanisms exist, clinical features—patient age, performance status or frailty, renal function, and disease stage—remain the most important prognosticators.
“But still, 2 patients in the same box based on clinical features will often have very different outcomes,” he said. “So in addition to clinical factors, we need prognosticators for tumor cell behavior. We need to know how fast they are growing and how they will respond to treatment.”
Methods to assess myeloma cell proliferation
Cytogenetics (FISH), gene array technology, and genomics using next-generation sequencing can provide some information, but they are not necessarily good methods to assess proliferation, Dr Ely explained.
To determine the proliferation rate of a patient’s cancer, you can look at tens of thousands of genes by gene array, he said, “or you can just look at one thing, which is Ki67.”
If the cell has Ki67, it’s proliferating, and if it doesn’t have Ki67, it’s not.
“Often, looking at all the other upstream molecules can be confusing and even misleading,” he noted. “So Ki67 is the best way to look for proliferation when it comes to myeloma.”
Other methods include the plasma cell labeling index (PCLI), gene expression profiling, flow cytometry, and multiplex immunohistochemistry (mIHC).
Dr Ely, as a hematopathologist, has found IHC to be the best method to determine proliferation, most likely because the other methods use bone marrow aspirate and IHC uses core biopsy of histologic sections.
It’s the gold standard, he said, for determining the percentage of plasma cells because core biopsy takes a “complete, intact piece of marrow that’s truly representative of what’s going on in the patient.”
In a study of more than 350 bone marrow samples comparing core biopsy with aspirate smears, plasma cells were under-represented in approximately half the aspirate specimens by about 20%.
In addition, Dr Ely noted that myeloma cells die very quickly once they are removed from the stroma.
“So if you take myeloma cells out as an aspirate,” he said, “myeloma cells die and others survive.”
And if the aspirate is sent overnight to the lab, the number of plasma cells in the specimen will already be reduced when the lab gets it.
Aspirates are best for leukemia and myelodysplastic syndromes, Dr Ely said, while core biopsies are best for lymphoma and myeloma.
Plasma cell proliferation indices
Proliferation is a myeloma-defining criterion, Dr Ely said. It predicts an 80% probability of progression in 2 years.
And PCLI has shown conclusively that plasma cell proliferation is a good prognosticator in all types of myeloma patients. However, it is not really feasible to use or easy to perform.
On the other hand, core biopsy combined with IHC is a feasible way of measuring plasma cell proliferation for routine clinical use.
Using standard IHC, it’s difficult to distinguish proliferating from non-proliferating cells, Dr Ely said.
“The solution to this problem is multiplex IHC,” he said, using 3 stains—red for CD138 (the plasma cell marker), brown for Ki67 (the proliferation marker), and blue as a negative nuclear counter stain.
A red membrane around the stained cell indicates a myeloma cell. Non-proliferating MM cells have blue nuclei, and proliferating MM cells have brown nuclei.
This assay is called the plasma cell proliferation index (PCPI).
“[A]ny lab that can do standard IHC can do multiplex IHC,” Dr Ely added.
It uses the same machines, the same reagents, the same expertise, and it’s easy to set up.
Validation studies
Dr Ely and his colleagues performed extensive laboratory validation to make sure the new PCPI correlated with the old PCLI.
They also performed 3 clinical validation studies, the first in bone marrow transplant patients. The investigators followed the patients for 12 years and found the PCPI correlated with survival.
The investigators performed a second clinical validation in 151 newly diagnosed patients. On multivariate analysis, the team found PCPI to be an independent prognostic indicator.
Each 1% increase in PCPI was associated with a 3% increased risk of disease progression (hazard ratio=1.03, 95% CI, 1.01-1.05, P=0.02).
The third clinical validation the investigators conducted was a retrospective cohort study in which they evaluated the effect of rising PCPI at relapse in 37 patients. The team defined rising PCPI as a 5% or greater increase in plasma cells.
Nineteen patients had a rising PCPI, and 17 patients had stable or decreased PCPI.
Patients with a rising PCPI at relapse had a shorter median overall survival than patients with stable or decreased PCPI—72 months and not reached, respectively (P=0.0069).
Patients with a rising PCPI also had a shorter median progression-free survival on first post-relapse treatment compared to patients with stable or decreased PCPI—25 months and 47 months, respectively (P=0.036).
“It’s also important to note that if you’re getting high-risk by PCPI plus β2-microglobulin albumin, I’d advise that, for all high-risk patients, getting the cytogenetics doesn’t really help,” Dr Ely said
Three patients considered high-risk by cytogenetics were standard-risk by PCPI plus β2 microglobulin.
“So we found that cytogenetics isn’t really adding anything except the cost,” Dr Ely asserted.
Other labs are now using PCPI for prognostication, he noted, adding, “We hope PCPI will be incorporated into the International Myeloma Working Group diagnosis of MM.” ![]()
Combo shows promise for treating T-ALL
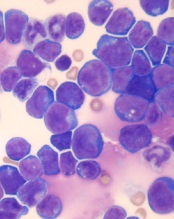
acute lymphoblastic leukemia
Image by Hind Medyou
Preclinical research suggests that 2 investigational drugs synergize to kill T-cell acute lymphoblastic leukemia (T-ALL) cells while having a minimal impact on normal blood cells.
Both drugs—the CK2 inhibitor CX-4945 and the BET inhibitor JQ1—have already been tested as single agents in clinical trials of hematologic malignancies and solid tumors.
However, the effects of the drugs in combination were not known until now.
“Previous studies provided us a rationale to test the combination of CX-4945 and JQ1 on refractory/relapsed T-cell leukemia,” said Hui Feng, MD, PhD, of Boston University School of Medicine in Massachusetts.
“Our findings suggest that the combination treatment of CX-4945 and JQ1 could be an effective strategy to target refractory/relapsed T-cell leukemia.”
Dr Feng and her colleagues reported these findings in Haematologica.
The researchers noted that targeting MYC-mediated transcriptional programs using JQ1 produces anti-leukemic activity in vitro and in vivo. However, global repression of transcription is likely to cause toxicities.
Therefore, the team theorized that finding drugs that synergize with JQ1 might allow them to reduce the dose of JQ1 and therefore decrease the risk of toxicity while enhancing the efficacy of treatment.
For this, the researchers looked to CX-4945, which is currently being investigated in clinical trials of breast cancer and multiple myeloma.
The team said CX-4945 has been shown to significantly reduce the growth and survival of human T-ALL cells on its own.
In a series of experiments, Dr Feng and her colleagues were able to show that CX-4945 destabilizes NOTCH1 and synergizes with JQ1 to induce apoptosis in human T-ALL cells.
The researchers also assessed the effects of JQ1 and CX-4946, alone and in combination, on normal peripheral blood monocytes (PBMs).
PBMs proved less sensitive than ALL-SIL T-ALL cells to each drug alone and to the drugs in combination. In fact, the combination had an antagonistic effect in PBMs.
Dr Feng and her colleagues said this research suggests JQ1 and CX-4946 in combination may be a feasible treatment option for relapsed/refractory T-ALL and other cancers involving CK2 and NOTCH1/MYC. ![]()
acute lymphoblastic leukemia
Image by Hind Medyou
Preclinical research suggests that 2 investigational drugs synergize to kill T-cell acute lymphoblastic leukemia (T-ALL) cells while having a minimal impact on normal blood cells.
Both drugs—the CK2 inhibitor CX-4945 and the BET inhibitor JQ1—have already been tested as single agents in clinical trials of hematologic malignancies and solid tumors.
However, the effects of the drugs in combination were not known until now.
“Previous studies provided us a rationale to test the combination of CX-4945 and JQ1 on refractory/relapsed T-cell leukemia,” said Hui Feng, MD, PhD, of Boston University School of Medicine in Massachusetts.
“Our findings suggest that the combination treatment of CX-4945 and JQ1 could be an effective strategy to target refractory/relapsed T-cell leukemia.”
Dr Feng and her colleagues reported these findings in Haematologica.
The researchers noted that targeting MYC-mediated transcriptional programs using JQ1 produces anti-leukemic activity in vitro and in vivo. However, global repression of transcription is likely to cause toxicities.
Therefore, the team theorized that finding drugs that synergize with JQ1 might allow them to reduce the dose of JQ1 and therefore decrease the risk of toxicity while enhancing the efficacy of treatment.
For this, the researchers looked to CX-4945, which is currently being investigated in clinical trials of breast cancer and multiple myeloma.
The team said CX-4945 has been shown to significantly reduce the growth and survival of human T-ALL cells on its own.
In a series of experiments, Dr Feng and her colleagues were able to show that CX-4945 destabilizes NOTCH1 and synergizes with JQ1 to induce apoptosis in human T-ALL cells.
The researchers also assessed the effects of JQ1 and CX-4946, alone and in combination, on normal peripheral blood monocytes (PBMs).
PBMs proved less sensitive than ALL-SIL T-ALL cells to each drug alone and to the drugs in combination. In fact, the combination had an antagonistic effect in PBMs.
Dr Feng and her colleagues said this research suggests JQ1 and CX-4946 in combination may be a feasible treatment option for relapsed/refractory T-ALL and other cancers involving CK2 and NOTCH1/MYC. ![]()
acute lymphoblastic leukemia
Image by Hind Medyou
Preclinical research suggests that 2 investigational drugs synergize to kill T-cell acute lymphoblastic leukemia (T-ALL) cells while having a minimal impact on normal blood cells.
Both drugs—the CK2 inhibitor CX-4945 and the BET inhibitor JQ1—have already been tested as single agents in clinical trials of hematologic malignancies and solid tumors.
However, the effects of the drugs in combination were not known until now.
“Previous studies provided us a rationale to test the combination of CX-4945 and JQ1 on refractory/relapsed T-cell leukemia,” said Hui Feng, MD, PhD, of Boston University School of Medicine in Massachusetts.
“Our findings suggest that the combination treatment of CX-4945 and JQ1 could be an effective strategy to target refractory/relapsed T-cell leukemia.”
Dr Feng and her colleagues reported these findings in Haematologica.
The researchers noted that targeting MYC-mediated transcriptional programs using JQ1 produces anti-leukemic activity in vitro and in vivo. However, global repression of transcription is likely to cause toxicities.
Therefore, the team theorized that finding drugs that synergize with JQ1 might allow them to reduce the dose of JQ1 and therefore decrease the risk of toxicity while enhancing the efficacy of treatment.
For this, the researchers looked to CX-4945, which is currently being investigated in clinical trials of breast cancer and multiple myeloma.
The team said CX-4945 has been shown to significantly reduce the growth and survival of human T-ALL cells on its own.
In a series of experiments, Dr Feng and her colleagues were able to show that CX-4945 destabilizes NOTCH1 and synergizes with JQ1 to induce apoptosis in human T-ALL cells.
The researchers also assessed the effects of JQ1 and CX-4946, alone and in combination, on normal peripheral blood monocytes (PBMs).
PBMs proved less sensitive than ALL-SIL T-ALL cells to each drug alone and to the drugs in combination. In fact, the combination had an antagonistic effect in PBMs.
Dr Feng and her colleagues said this research suggests JQ1 and CX-4946 in combination may be a feasible treatment option for relapsed/refractory T-ALL and other cancers involving CK2 and NOTCH1/MYC. ![]()
Drug approved to treat hemophilia B in Switzerland

Photo courtesy of Biogen
The Swiss Agency for Therapeutic Products, Swissmedic, has approved eftrenonacog alfa (Alprolix®) for the treatment of hemophilia B.
Eftrenonacog alfa is a recombinant factor IX Fc fusion protein indicated for both on-demand treatment and prophylaxis in previously treated patients with hemophilia B.
For prophylaxis, 1 dose of eftrenonacog alfa can be given every 7 days or every 10 days, with the ability to adjust the dosing interval based on individual response.
Eftrenonacog alfa is the only recombinant factor IX Fc fusion protein therapy approved in Switzerland for the treatment of hemophilia B.
“The Swiss approval of Alprolix is an important milestone for the hemophilia community, offering the opportunity for people with hemophilia B to experience prolonged protection from bleeds,” said Krassimir Mitchev, MD, PhD, vice president and medical therapeutic area head of hemophilia at Sobi, the company developing eftrenonacog alfa in collaboration with Biogen.
“We will now focus on ensuring timely and sustainable access to Alprolix in Switzerland.”
About eftrenonacog alfa
Eftrenonacog alfa is a recombinant clotting factor therapy developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1 (IgG1). This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Eftrenonacog alfa is currently approved for the treatment of hemophilia B in the European Economic Area, the US, Canada, Japan, Australia, New Zealand, and other countries.
Sobi and Biogen collaborate on the development and commercialization of eftrenonacog alfa.
The product has been evaluated in two phase 3 trials of patients with hemophilia B: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced
a bleeding episode while on study. In total, 90.4% of bleeding episodes
were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study. ![]()
Photo courtesy of Biogen
The Swiss Agency for Therapeutic Products, Swissmedic, has approved eftrenonacog alfa (Alprolix®) for the treatment of hemophilia B.
Eftrenonacog alfa is a recombinant factor IX Fc fusion protein indicated for both on-demand treatment and prophylaxis in previously treated patients with hemophilia B.
For prophylaxis, 1 dose of eftrenonacog alfa can be given every 7 days or every 10 days, with the ability to adjust the dosing interval based on individual response.
Eftrenonacog alfa is the only recombinant factor IX Fc fusion protein therapy approved in Switzerland for the treatment of hemophilia B.
“The Swiss approval of Alprolix is an important milestone for the hemophilia community, offering the opportunity for people with hemophilia B to experience prolonged protection from bleeds,” said Krassimir Mitchev, MD, PhD, vice president and medical therapeutic area head of hemophilia at Sobi, the company developing eftrenonacog alfa in collaboration with Biogen.
“We will now focus on ensuring timely and sustainable access to Alprolix in Switzerland.”
About eftrenonacog alfa
Eftrenonacog alfa is a recombinant clotting factor therapy developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1 (IgG1). This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Eftrenonacog alfa is currently approved for the treatment of hemophilia B in the European Economic Area, the US, Canada, Japan, Australia, New Zealand, and other countries.
Sobi and Biogen collaborate on the development and commercialization of eftrenonacog alfa.
The product has been evaluated in two phase 3 trials of patients with hemophilia B: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced
a bleeding episode while on study. In total, 90.4% of bleeding episodes
were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study. ![]()
Photo courtesy of Biogen
The Swiss Agency for Therapeutic Products, Swissmedic, has approved eftrenonacog alfa (Alprolix®) for the treatment of hemophilia B.
Eftrenonacog alfa is a recombinant factor IX Fc fusion protein indicated for both on-demand treatment and prophylaxis in previously treated patients with hemophilia B.
For prophylaxis, 1 dose of eftrenonacog alfa can be given every 7 days or every 10 days, with the ability to adjust the dosing interval based on individual response.
Eftrenonacog alfa is the only recombinant factor IX Fc fusion protein therapy approved in Switzerland for the treatment of hemophilia B.
“The Swiss approval of Alprolix is an important milestone for the hemophilia community, offering the opportunity for people with hemophilia B to experience prolonged protection from bleeds,” said Krassimir Mitchev, MD, PhD, vice president and medical therapeutic area head of hemophilia at Sobi, the company developing eftrenonacog alfa in collaboration with Biogen.
“We will now focus on ensuring timely and sustainable access to Alprolix in Switzerland.”
About eftrenonacog alfa
Eftrenonacog alfa is a recombinant clotting factor therapy developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1 (IgG1). This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Eftrenonacog alfa is currently approved for the treatment of hemophilia B in the European Economic Area, the US, Canada, Japan, Australia, New Zealand, and other countries.
Sobi and Biogen collaborate on the development and commercialization of eftrenonacog alfa.
The product has been evaluated in two phase 3 trials of patients with hemophilia B: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced
a bleeding episode while on study. In total, 90.4% of bleeding episodes
were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study. ![]()
Treatment facility volume linked to survival in MM

Photo courtesy of the CDC
Patients with multiple myeloma (MM) are more likely to live longer if they are treated at a medical center where the staff has more experience with the disease, according to research published in the Journal of Clinical Oncology.
The study showed that patients treated at medical centers seeing 10 new MM patients per year had a 20% higher risk of death than patients treated at centers seeing 40 new MM patients per year.
Most cancer treatment centers in the US see fewer than 10 new MM patients per year.
“It is very difficult to be proficient when doctors are seeing only 1 or 2 new cases of multiple myeloma per year,” said study author Ronald Go, MD, of the Mayo Clinic in Rochester, Minnesota.
“Studies on cancer surgery have shown the more experience the center or practitioner has, the better the outcome. We wanted to see if volume matters when it comes to nonsurgical treatment of rare cancers such as multiple myeloma.”
To investigate, Dr Go and his colleagues used the National Cancer Database, examining outcomes for 94,722 newly diagnosed MM patients treated at 1333 facilities between 2003 and 2011.
The researchers grouped the facilities into quartiles according to the volume of MM patients treated there each year.
The mean number of MM patients treated per year was:
- Less than 3.6 for quartile 1 (Q1)
- 3.6 to 6.1 for Q2
- 6.1 to 10.3 for Q3
- More than 10.3 for Q4.
The majority of patients (60.3%) were treated in Q4 facilities. For all facilities, the median number of new MM patients per year was 6.1 (range, 3.6 to 10.3). The mean was 8.8 ± 9.9.
The researchers calculated the relationship between MM patient volume at these facilities and patient mortality, adjusting for demographic characteristics, socioeconomic factors, geographic factors, comorbidities, and year of diagnosis.
Outcomes
The unadjusted median overall survival was 26.9 months for patients treated at Q1 facilities, 29.1 months for Q2, 31.9 months for Q3, and 49.1 months for Q4 (P<0.001).
The 1-year mortality rate was 33.5% for patients treated at Q1 facilities, 32.3% for Q2, 30.7% for Q3, and 21.9% for Q4.
The researchers’ multivariable analysis showed that facility volume was independently associated with all-cause mortality.
Patients treated at the lower-quartile facilities had a higher risk of death than patients treated at Q4 facilities. The hazard ratios were 1.12 for patients at Q3 facilities, 1.12 for Q2, and 1.22 for Q1.
The researchers performed another analysis in which volume was treated as a continuous variable, and they compared various volume sizes to a reference volume of 10 patients per year.
Compared with facilities treating 10 new MM patients per year, facilities treating 20 MM patients per year had roughly 10% lower overall mortality rates.
Facilities treating 30 MM patients per year had about 15% lower mortality rates. And facilities treating 40 MM patients per year had 20% lower overall mortality rates. ![]()
Photo courtesy of the CDC
Patients with multiple myeloma (MM) are more likely to live longer if they are treated at a medical center where the staff has more experience with the disease, according to research published in the Journal of Clinical Oncology.
The study showed that patients treated at medical centers seeing 10 new MM patients per year had a 20% higher risk of death than patients treated at centers seeing 40 new MM patients per year.
Most cancer treatment centers in the US see fewer than 10 new MM patients per year.
“It is very difficult to be proficient when doctors are seeing only 1 or 2 new cases of multiple myeloma per year,” said study author Ronald Go, MD, of the Mayo Clinic in Rochester, Minnesota.
“Studies on cancer surgery have shown the more experience the center or practitioner has, the better the outcome. We wanted to see if volume matters when it comes to nonsurgical treatment of rare cancers such as multiple myeloma.”
To investigate, Dr Go and his colleagues used the National Cancer Database, examining outcomes for 94,722 newly diagnosed MM patients treated at 1333 facilities between 2003 and 2011.
The researchers grouped the facilities into quartiles according to the volume of MM patients treated there each year.
The mean number of MM patients treated per year was:
- Less than 3.6 for quartile 1 (Q1)
- 3.6 to 6.1 for Q2
- 6.1 to 10.3 for Q3
- More than 10.3 for Q4.
The majority of patients (60.3%) were treated in Q4 facilities. For all facilities, the median number of new MM patients per year was 6.1 (range, 3.6 to 10.3). The mean was 8.8 ± 9.9.
The researchers calculated the relationship between MM patient volume at these facilities and patient mortality, adjusting for demographic characteristics, socioeconomic factors, geographic factors, comorbidities, and year of diagnosis.
Outcomes
The unadjusted median overall survival was 26.9 months for patients treated at Q1 facilities, 29.1 months for Q2, 31.9 months for Q3, and 49.1 months for Q4 (P<0.001).
The 1-year mortality rate was 33.5% for patients treated at Q1 facilities, 32.3% for Q2, 30.7% for Q3, and 21.9% for Q4.
The researchers’ multivariable analysis showed that facility volume was independently associated with all-cause mortality.
Patients treated at the lower-quartile facilities had a higher risk of death than patients treated at Q4 facilities. The hazard ratios were 1.12 for patients at Q3 facilities, 1.12 for Q2, and 1.22 for Q1.
The researchers performed another analysis in which volume was treated as a continuous variable, and they compared various volume sizes to a reference volume of 10 patients per year.
Compared with facilities treating 10 new MM patients per year, facilities treating 20 MM patients per year had roughly 10% lower overall mortality rates.
Facilities treating 30 MM patients per year had about 15% lower mortality rates. And facilities treating 40 MM patients per year had 20% lower overall mortality rates. ![]()
Photo courtesy of the CDC
Patients with multiple myeloma (MM) are more likely to live longer if they are treated at a medical center where the staff has more experience with the disease, according to research published in the Journal of Clinical Oncology.
The study showed that patients treated at medical centers seeing 10 new MM patients per year had a 20% higher risk of death than patients treated at centers seeing 40 new MM patients per year.
Most cancer treatment centers in the US see fewer than 10 new MM patients per year.
“It is very difficult to be proficient when doctors are seeing only 1 or 2 new cases of multiple myeloma per year,” said study author Ronald Go, MD, of the Mayo Clinic in Rochester, Minnesota.
“Studies on cancer surgery have shown the more experience the center or practitioner has, the better the outcome. We wanted to see if volume matters when it comes to nonsurgical treatment of rare cancers such as multiple myeloma.”
To investigate, Dr Go and his colleagues used the National Cancer Database, examining outcomes for 94,722 newly diagnosed MM patients treated at 1333 facilities between 2003 and 2011.
The researchers grouped the facilities into quartiles according to the volume of MM patients treated there each year.
The mean number of MM patients treated per year was:
- Less than 3.6 for quartile 1 (Q1)
- 3.6 to 6.1 for Q2
- 6.1 to 10.3 for Q3
- More than 10.3 for Q4.
The majority of patients (60.3%) were treated in Q4 facilities. For all facilities, the median number of new MM patients per year was 6.1 (range, 3.6 to 10.3). The mean was 8.8 ± 9.9.
The researchers calculated the relationship between MM patient volume at these facilities and patient mortality, adjusting for demographic characteristics, socioeconomic factors, geographic factors, comorbidities, and year of diagnosis.
Outcomes
The unadjusted median overall survival was 26.9 months for patients treated at Q1 facilities, 29.1 months for Q2, 31.9 months for Q3, and 49.1 months for Q4 (P<0.001).
The 1-year mortality rate was 33.5% for patients treated at Q1 facilities, 32.3% for Q2, 30.7% for Q3, and 21.9% for Q4.
The researchers’ multivariable analysis showed that facility volume was independently associated with all-cause mortality.
Patients treated at the lower-quartile facilities had a higher risk of death than patients treated at Q4 facilities. The hazard ratios were 1.12 for patients at Q3 facilities, 1.12 for Q2, and 1.22 for Q1.
The researchers performed another analysis in which volume was treated as a continuous variable, and they compared various volume sizes to a reference volume of 10 patients per year.
Compared with facilities treating 10 new MM patients per year, facilities treating 20 MM patients per year had roughly 10% lower overall mortality rates.
Facilities treating 30 MM patients per year had about 15% lower mortality rates. And facilities treating 40 MM patients per year had 20% lower overall mortality rates. ![]()
Agent could treat hemophilia A and B, team says

A new bypassing agent mimics the pro-clotting activity of factor V Leiden and might prove effective for treating hemophilia A and B, according to preclinical research published in Blood.
“We know that patients who have severe hemophilia and also have mutations that increase clotting, such as factor V Leiden, experience less severe bleeding,” said study author Trevor Baglin, MD, of Addenbrooke’s Hospital in Cambridge, UK.
In patients with factor V Leiden, defects in the anticoagulant activated protein C (APC) mechanism lead to an overactive production of thrombin.
The researchers set out to determine if they could exploit this phenomenon to treat hemophilia by developing a direct inhibitor of APC. They modified serine protease inhibitors, known as serpins, to make them specific and efficient inhibitors of APC.
“We hypothesized that if we targeted the protein C pathway we could prolong thrombin production,” said study author James Huntington, PhD, of the University of Cambridge in the UK.
“We engineered a serpin so that it could selectively prevent APC from shutting down thrombin production before the formation of a stable clot.”
The researchers administered the serpin to mice with hemophilia B and clipped their tails. In this model, the blood loss decreased as the dose increased, with the highest dose reducing bleeding to the level of healthy mice.
Further injury models underscored that the serpin helped the majority of mice form stable clots, with higher doses resulting in quicker clot formation.
The serpin was also able to accelerate clot formation when added to blood samples from patients with hemophilia A.
“It is our understanding that because we are targeting a general anticlotting process, our serpin could effectively treat patients with either hemophilia A or B, including those who develop inhibitors to more traditional therapy,” Dr Huntington said.
“Additionally, we have focused on engineering the serpin to be both subcutaneously delivered and long-acting. This will free patients from the cumbersome thrice-weekly infusions that are necessary under many contemporary therapy regimens.”
“Within 3 years, we hope to be conducting our first-in-man trials of a subcutaneously administered form of our serpin,” Dr Baglin added.
“It is important to remember that the majority of people in the world with hemophilia have no access to therapy. A stable, subcutaneous, long-acting, effective hemostatic agent could bring treatment to a great deal many more hemophilia sufferers.”
This study forms part of a patent application by the authors, and the serpin is being developed into a therapeutic by a start-up company known as ApcinteX, with funding from Medicxi. ![]()
A new bypassing agent mimics the pro-clotting activity of factor V Leiden and might prove effective for treating hemophilia A and B, according to preclinical research published in Blood.
“We know that patients who have severe hemophilia and also have mutations that increase clotting, such as factor V Leiden, experience less severe bleeding,” said study author Trevor Baglin, MD, of Addenbrooke’s Hospital in Cambridge, UK.
In patients with factor V Leiden, defects in the anticoagulant activated protein C (APC) mechanism lead to an overactive production of thrombin.
The researchers set out to determine if they could exploit this phenomenon to treat hemophilia by developing a direct inhibitor of APC. They modified serine protease inhibitors, known as serpins, to make them specific and efficient inhibitors of APC.
“We hypothesized that if we targeted the protein C pathway we could prolong thrombin production,” said study author James Huntington, PhD, of the University of Cambridge in the UK.
“We engineered a serpin so that it could selectively prevent APC from shutting down thrombin production before the formation of a stable clot.”
The researchers administered the serpin to mice with hemophilia B and clipped their tails. In this model, the blood loss decreased as the dose increased, with the highest dose reducing bleeding to the level of healthy mice.
Further injury models underscored that the serpin helped the majority of mice form stable clots, with higher doses resulting in quicker clot formation.
The serpin was also able to accelerate clot formation when added to blood samples from patients with hemophilia A.
“It is our understanding that because we are targeting a general anticlotting process, our serpin could effectively treat patients with either hemophilia A or B, including those who develop inhibitors to more traditional therapy,” Dr Huntington said.
“Additionally, we have focused on engineering the serpin to be both subcutaneously delivered and long-acting. This will free patients from the cumbersome thrice-weekly infusions that are necessary under many contemporary therapy regimens.”
“Within 3 years, we hope to be conducting our first-in-man trials of a subcutaneously administered form of our serpin,” Dr Baglin added.
“It is important to remember that the majority of people in the world with hemophilia have no access to therapy. A stable, subcutaneous, long-acting, effective hemostatic agent could bring treatment to a great deal many more hemophilia sufferers.”
This study forms part of a patent application by the authors, and the serpin is being developed into a therapeutic by a start-up company known as ApcinteX, with funding from Medicxi. ![]()
A new bypassing agent mimics the pro-clotting activity of factor V Leiden and might prove effective for treating hemophilia A and B, according to preclinical research published in Blood.
“We know that patients who have severe hemophilia and also have mutations that increase clotting, such as factor V Leiden, experience less severe bleeding,” said study author Trevor Baglin, MD, of Addenbrooke’s Hospital in Cambridge, UK.
In patients with factor V Leiden, defects in the anticoagulant activated protein C (APC) mechanism lead to an overactive production of thrombin.
The researchers set out to determine if they could exploit this phenomenon to treat hemophilia by developing a direct inhibitor of APC. They modified serine protease inhibitors, known as serpins, to make them specific and efficient inhibitors of APC.
“We hypothesized that if we targeted the protein C pathway we could prolong thrombin production,” said study author James Huntington, PhD, of the University of Cambridge in the UK.
“We engineered a serpin so that it could selectively prevent APC from shutting down thrombin production before the formation of a stable clot.”
The researchers administered the serpin to mice with hemophilia B and clipped their tails. In this model, the blood loss decreased as the dose increased, with the highest dose reducing bleeding to the level of healthy mice.
Further injury models underscored that the serpin helped the majority of mice form stable clots, with higher doses resulting in quicker clot formation.
The serpin was also able to accelerate clot formation when added to blood samples from patients with hemophilia A.
“It is our understanding that because we are targeting a general anticlotting process, our serpin could effectively treat patients with either hemophilia A or B, including those who develop inhibitors to more traditional therapy,” Dr Huntington said.
“Additionally, we have focused on engineering the serpin to be both subcutaneously delivered and long-acting. This will free patients from the cumbersome thrice-weekly infusions that are necessary under many contemporary therapy regimens.”
“Within 3 years, we hope to be conducting our first-in-man trials of a subcutaneously administered form of our serpin,” Dr Baglin added.
“It is important to remember that the majority of people in the world with hemophilia have no access to therapy. A stable, subcutaneous, long-acting, effective hemostatic agent could bring treatment to a great deal many more hemophilia sufferers.”
This study forms part of a patent application by the authors, and the serpin is being developed into a therapeutic by a start-up company known as ApcinteX, with funding from Medicxi.
NORD publishes physician guide to CTCL

mycosis fungoides
The National Organization for Rare Disorders (NORD) has published a guide for physicians treating patients with cutaneous T-cell lymphoma (CTCL).
The guide contains information about disease classification, signs and symptoms of CTCL, methods of diagnosing the disease, standard therapies, and investigational therapies for CTCL.
The guide also includes a list of resources for physicians and patients.
“The NORD Physician Guide to Cutaneous T-Cell Lymphoma (CTCL)” is available for free on the NORD Physician Guides website.
The guide was made possible by an educational grant from Therakos, now a part of Mallinckrodt Pharmaceuticals.
The guide was developed in collaboration with Oleg E. Akilov, MD, PhD, of the University of Pittsburgh School of Medicine in Pennsylvania.
“Eczema and even some cases of psoriasis may look very similar to mycosis fungoides, the most common type of cutaneous T-cell lymphomas,” Dr Akilov noted.
“It is important to be aware of these similarities and to be ready to think about cutaneous lymphoma when a patient with ‘common dermatosis’ does not respond to regular treatments.”
About NORD guides
NORD established its physician guide series as part of a broader strategic initiative to promote earlier diagnosis and state-of-the-art care for people with rare diseases. Each online guide is written or reviewed by a medical professional with expertise on the topic.
Other recent guides in the series include:
- The NORD Physician Guide to Mitochondrial Myopathies
- The NORD Physician Guide to Paroxysmal Nocturnal Hemoglobinuria (PNH)
- The NORD Physician Guide to Atypical Hemolytic Uremic Syndrome (aHUS)
- The NORD Physician Guide to Nontuberculous Mycobacterial Lung Disease.
“People who have rare diseases often go for many years without a diagnosis,” said Marsha Lanes, a genetic counselor in NORD’s Educational Initiatives Department.
“The purpose of NORD’s free online physician guides is to reduce the time to diagnosis and encourage optimal treatment for patients with little-known and little-understood rare diseases.”
mycosis fungoides
The National Organization for Rare Disorders (NORD) has published a guide for physicians treating patients with cutaneous T-cell lymphoma (CTCL).
The guide contains information about disease classification, signs and symptoms of CTCL, methods of diagnosing the disease, standard therapies, and investigational therapies for CTCL.
The guide also includes a list of resources for physicians and patients.
“The NORD Physician Guide to Cutaneous T-Cell Lymphoma (CTCL)” is available for free on the NORD Physician Guides website.
The guide was made possible by an educational grant from Therakos, now a part of Mallinckrodt Pharmaceuticals.
The guide was developed in collaboration with Oleg E. Akilov, MD, PhD, of the University of Pittsburgh School of Medicine in Pennsylvania.
“Eczema and even some cases of psoriasis may look very similar to mycosis fungoides, the most common type of cutaneous T-cell lymphomas,” Dr Akilov noted.
“It is important to be aware of these similarities and to be ready to think about cutaneous lymphoma when a patient with ‘common dermatosis’ does not respond to regular treatments.”
About NORD guides
NORD established its physician guide series as part of a broader strategic initiative to promote earlier diagnosis and state-of-the-art care for people with rare diseases. Each online guide is written or reviewed by a medical professional with expertise on the topic.
Other recent guides in the series include:
- The NORD Physician Guide to Mitochondrial Myopathies
- The NORD Physician Guide to Paroxysmal Nocturnal Hemoglobinuria (PNH)
- The NORD Physician Guide to Atypical Hemolytic Uremic Syndrome (aHUS)
- The NORD Physician Guide to Nontuberculous Mycobacterial Lung Disease.
“People who have rare diseases often go for many years without a diagnosis,” said Marsha Lanes, a genetic counselor in NORD’s Educational Initiatives Department.
“The purpose of NORD’s free online physician guides is to reduce the time to diagnosis and encourage optimal treatment for patients with little-known and little-understood rare diseases.”
mycosis fungoides
The National Organization for Rare Disorders (NORD) has published a guide for physicians treating patients with cutaneous T-cell lymphoma (CTCL).
The guide contains information about disease classification, signs and symptoms of CTCL, methods of diagnosing the disease, standard therapies, and investigational therapies for CTCL.
The guide also includes a list of resources for physicians and patients.
“The NORD Physician Guide to Cutaneous T-Cell Lymphoma (CTCL)” is available for free on the NORD Physician Guides website.
The guide was made possible by an educational grant from Therakos, now a part of Mallinckrodt Pharmaceuticals.
The guide was developed in collaboration with Oleg E. Akilov, MD, PhD, of the University of Pittsburgh School of Medicine in Pennsylvania.
“Eczema and even some cases of psoriasis may look very similar to mycosis fungoides, the most common type of cutaneous T-cell lymphomas,” Dr Akilov noted.
“It is important to be aware of these similarities and to be ready to think about cutaneous lymphoma when a patient with ‘common dermatosis’ does not respond to regular treatments.”
About NORD guides
NORD established its physician guide series as part of a broader strategic initiative to promote earlier diagnosis and state-of-the-art care for people with rare diseases. Each online guide is written or reviewed by a medical professional with expertise on the topic.
Other recent guides in the series include:
- The NORD Physician Guide to Mitochondrial Myopathies
- The NORD Physician Guide to Paroxysmal Nocturnal Hemoglobinuria (PNH)
- The NORD Physician Guide to Atypical Hemolytic Uremic Syndrome (aHUS)
- The NORD Physician Guide to Nontuberculous Mycobacterial Lung Disease.
“People who have rare diseases often go for many years without a diagnosis,” said Marsha Lanes, a genetic counselor in NORD’s Educational Initiatives Department.
“The purpose of NORD’s free online physician guides is to reduce the time to diagnosis and encourage optimal treatment for patients with little-known and little-understood rare diseases.”
Explaining the development of MPNs, leukemia
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation.
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation.
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation.
Genetic screening for CLL premature, speaker says

Photo courtesy of the
National Institute
of General Medical Science
NEW YORK—Research has shown that family history is a strong risk factor for developing chronic lymphocytic leukemia (CLL).
First-degree relatives have an 8.5-fold risk of getting CLL and an increased risk of other lymphoproliferative disorders, according to a study published in 2009.
However, despite the strong evidence of a genetic contribution, one expert believes it’s premature to bring genetic testing into the clinic for screening in CLL.
“At this time, we do not recommend genetic screening,” said Susan Slager, PhD, of the Mayo Clinic in Rochester, Minnesota.
“There’s no known relationship between the inherited variants and treatment response,” she explained, and the relatively low incidence of CLL argues against active screening in affected families at present.
Dr Slager discussed genetic and non-genetic factors associated with CLL and the clinical implications of these factors at Lymphoma & Myeloma 2016.
Demographic risk factors
Dr Slager noted that age, gender, and race are risk factors for CLL.
Individuals aged 65 to 74 have the highest incidence of CLL, at 28%, while the risk is almost non-existent for those under age 20, she said.
There is a higher incidence of CLL in males than in females, and the reason for this gender disparity is unknown.
There is a higher incidence of CLL in Caucasians than Asians, for both males and females.
“Again, it’s unknown why there’s this variability in incidence in CLL,” Dr Slager said. “Obviously, age, sex, and race—these are things you can’t modify. You’re stuck with them.”
However, several studies have been undertaken to look at some of the potentially modifiable factors associated with CLL.
Beyond demographic factors
The International Lymphoma Epidemiology Consortium, known as InterLymph, was initiated in 2001 to evaluate the association of risk factors in CLL. Study centers are located primarily in North America and Europe, with one in Australia.
In one of the larger InterLymph studies, investigators evaluated risk factors—lifestyle exposure, reproductive history, medical history, occupational exposures, farming exposure, and family history—in 2440 CLL patients and 15,186 controls.
The investigators found that sun exposure and atopy—allergies, asthma, eczema, and hay fever—have a protective effect in CLL, while serological hepatitis C virus (HCV) infections, farming exposure, and family history carry an increased risk of CLL.
This confirmed an earlier study conducted in New South Wales, Australia, that had uncovered an inverse association between sun exposure and non-Hodgkin lymphoma (NHL) risk, which fell significantly with increasing recreational sun exposure.
Medical history
Another earlier study from New South Wales revealed a 20% reduction in the risk of NHL for any specific allergy.
However, the investigators of the large, more recent study observed little to no evidence of reduced risk for asthma and eczema.
The underlying biology for atopy or allergies is a hyper-immune system, Dr Slager explained.
“So if you have a hyper-immune system, then we hypothesize that you have protection against CLL,” she said.
Another medical exposure investigators analyzed that impacts CLL risk is HCV. People infected with HCV have an increased risk of CLL, perhaps due to chronic antigen stimulation or possibly disruption of the T-cell function.
Height is also associated with CLL. CLL risk increases with greater height. The concept is that taller individuals have increased exposure to growth hormones that possibly result in cell proliferation.
Another hypothesis supporting the height association is that people of shorter stature experience more infections, which could result in a stronger immune system. And a stronger immune system perhaps protects against NHL.
Occupational exposures
Investigators consistently observed a 20% increased risk of CLL for people living or working on a farm.
Animal farmers, as opposed to crop farmers, experienced some protection. However, the sample size was too small to be conclusive, with only 29 people across all studies being animal farmers.
Among other occupations evaluated, hairdressers also had an increased risk of CLL, although this too was based on a small sample size.
Family history
One of the strongest risk factors for CLL is family history.
Using population-based registry data from Sweden, investigators found that people with a first-degree relative with CLL have an 8.5-fold risk of CLL.
They also have an elevated risk of other lymphoproliferative disorders, including NHL (1.9-fold risk), Waldenström’s macroglobulinemia (4.0-fold risk), hairy cell leukemia (3.3-fold risk), and follicular lymphoma (1.6-fold risk).
GWAS in CLL
Investigators conducted genome-wide association studies (GWAS) to determine what is driving the familial risk.
Dr Slager described these studies as an agnostic approach that looks across the entire genome to determine which regions are associated with a trait of interest.
Typically, many markers are genotyped—somewhere between half a million to 5 million markers—and each is looked at individually with respect to CLL, she said.
Unrelated cases and controls are included in the studies.
The first GWAS study identifying susceptibility loci for CLL was published in 2008. Subsequently, more studies were published with increasing sample sizes—more cases, more controls, and more genetic variants identified.
In the largest meta-analysis for CLL to date (Slager and Houlston et al, not yet published), investigators analyzed 4400 CLL cases and 13,000 controls.
They identified 9 more inherited variances with CLL, for a total of 43 identified to date.
The genes involved follow an apoptosis pathway, the telomere length pathway, and the B-cell lymphocyte development pathway.
“We have to remember, though, that these are non-causal,” Dr Slager cautioned. “We are just identifying the region in the genome that’s associated with CLL . . . . So now we have to dig deeper in these relationships to understand what’s going on.”
Using the identified CLL single-nucleotide polymorphisms, the investigators computed a polygenic risk score. CLL cases in the highest quintile had 2.7-fold increased risk of CLL.
However, the most common GWAS variants explain only 17% of the genetic heritability of CLL, which suggests that more loci are yet to be identified, Dr Slager clarified.
She went on to say that CLL incidence varies by ethnicity. Caucasians have a very high rate of CLL, while Asians have a very low rate. And African Americans have an incidence rate between those of Caucasians and Asians.
Investigators have hypothesized that the differences in incidence are based on the distinct genetic variants that are associated with the ethnicities.
For example, 4 of the variants with more than 20% frequency in Caucasians are quite rare in Chinese individuals and are also quite uncommon in African Americans, with frequencies less than 10%.
Dr Slager suggested that conducting these kinds of studies in Asians and African Americans will take a large sample size and most likely require an international consortium to bring enough CLL cases together.
Impact on clinical practice
Because of the strong genetic risk, patients with CLL naturally want to know about their offspring and their siblings, Dr Slager has found.
Patients who have monoclonal B-cell lymphocytosis (MBL), which is a precursor to CLL, pose the biggest quandary.
MBL is detected in about 5% of people over age 40. However, it’s detected in about 15% to 18% of people with a first-degree relative with CLL.
“These are individuals who have lymphocytosis,” Dr Slager said. “They come to your clinic and have an elevated blood cell count, flow cytometry. [So] you screen them for MBL, and these individuals who have more than 500 cells per microliter, they are the ones who progress to CLL, at 1% per year.”
Individuals who don’t have the elevated blood counts do have the clonal cells, Dr Slager noted.
“They just don’t have the expansion,” she said. “The progression of these individuals to CLL is still yet to be determined.”
For these reasons, Dr Slager believes “it’s still premature to bring genetic testing into clinical practice.”
Future directions include bringing together the non-environmental issues and the inherited issues to create a model that will accurately predict the risk of CLL.
Photo courtesy of the
National Institute
of General Medical Science
NEW YORK—Research has shown that family history is a strong risk factor for developing chronic lymphocytic leukemia (CLL).
First-degree relatives have an 8.5-fold risk of getting CLL and an increased risk of other lymphoproliferative disorders, according to a study published in 2009.
However, despite the strong evidence of a genetic contribution, one expert believes it’s premature to bring genetic testing into the clinic for screening in CLL.
“At this time, we do not recommend genetic screening,” said Susan Slager, PhD, of the Mayo Clinic in Rochester, Minnesota.
“There’s no known relationship between the inherited variants and treatment response,” she explained, and the relatively low incidence of CLL argues against active screening in affected families at present.
Dr Slager discussed genetic and non-genetic factors associated with CLL and the clinical implications of these factors at Lymphoma & Myeloma 2016.
Demographic risk factors
Dr Slager noted that age, gender, and race are risk factors for CLL.
Individuals aged 65 to 74 have the highest incidence of CLL, at 28%, while the risk is almost non-existent for those under age 20, she said.
There is a higher incidence of CLL in males than in females, and the reason for this gender disparity is unknown.
There is a higher incidence of CLL in Caucasians than Asians, for both males and females.
“Again, it’s unknown why there’s this variability in incidence in CLL,” Dr Slager said. “Obviously, age, sex, and race—these are things you can’t modify. You’re stuck with them.”
However, several studies have been undertaken to look at some of the potentially modifiable factors associated with CLL.
Beyond demographic factors
The International Lymphoma Epidemiology Consortium, known as InterLymph, was initiated in 2001 to evaluate the association of risk factors in CLL. Study centers are located primarily in North America and Europe, with one in Australia.
In one of the larger InterLymph studies, investigators evaluated risk factors—lifestyle exposure, reproductive history, medical history, occupational exposures, farming exposure, and family history—in 2440 CLL patients and 15,186 controls.
The investigators found that sun exposure and atopy—allergies, asthma, eczema, and hay fever—have a protective effect in CLL, while serological hepatitis C virus (HCV) infections, farming exposure, and family history carry an increased risk of CLL.
This confirmed an earlier study conducted in New South Wales, Australia, that had uncovered an inverse association between sun exposure and non-Hodgkin lymphoma (NHL) risk, which fell significantly with increasing recreational sun exposure.
Medical history
Another earlier study from New South Wales revealed a 20% reduction in the risk of NHL for any specific allergy.
However, the investigators of the large, more recent study observed little to no evidence of reduced risk for asthma and eczema.
The underlying biology for atopy or allergies is a hyper-immune system, Dr Slager explained.
“So if you have a hyper-immune system, then we hypothesize that you have protection against CLL,” she said.
Another medical exposure investigators analyzed that impacts CLL risk is HCV. People infected with HCV have an increased risk of CLL, perhaps due to chronic antigen stimulation or possibly disruption of the T-cell function.
Height is also associated with CLL. CLL risk increases with greater height. The concept is that taller individuals have increased exposure to growth hormones that possibly result in cell proliferation.
Another hypothesis supporting the height association is that people of shorter stature experience more infections, which could result in a stronger immune system. And a stronger immune system perhaps protects against NHL.
Occupational exposures
Investigators consistently observed a 20% increased risk of CLL for people living or working on a farm.
Animal farmers, as opposed to crop farmers, experienced some protection. However, the sample size was too small to be conclusive, with only 29 people across all studies being animal farmers.
Among other occupations evaluated, hairdressers also had an increased risk of CLL, although this too was based on a small sample size.
Family history
One of the strongest risk factors for CLL is family history.
Using population-based registry data from Sweden, investigators found that people with a first-degree relative with CLL have an 8.5-fold risk of CLL.
They also have an elevated risk of other lymphoproliferative disorders, including NHL (1.9-fold risk), Waldenström’s macroglobulinemia (4.0-fold risk), hairy cell leukemia (3.3-fold risk), and follicular lymphoma (1.6-fold risk).
GWAS in CLL
Investigators conducted genome-wide association studies (GWAS) to determine what is driving the familial risk.
Dr Slager described these studies as an agnostic approach that looks across the entire genome to determine which regions are associated with a trait of interest.
Typically, many markers are genotyped—somewhere between half a million to 5 million markers—and each is looked at individually with respect to CLL, she said.
Unrelated cases and controls are included in the studies.
The first GWAS study identifying susceptibility loci for CLL was published in 2008. Subsequently, more studies were published with increasing sample sizes—more cases, more controls, and more genetic variants identified.
In the largest meta-analysis for CLL to date (Slager and Houlston et al, not yet published), investigators analyzed 4400 CLL cases and 13,000 controls.
They identified 9 more inherited variances with CLL, for a total of 43 identified to date.
The genes involved follow an apoptosis pathway, the telomere length pathway, and the B-cell lymphocyte development pathway.
“We have to remember, though, that these are non-causal,” Dr Slager cautioned. “We are just identifying the region in the genome that’s associated with CLL . . . . So now we have to dig deeper in these relationships to understand what’s going on.”
Using the identified CLL single-nucleotide polymorphisms, the investigators computed a polygenic risk score. CLL cases in the highest quintile had 2.7-fold increased risk of CLL.
However, the most common GWAS variants explain only 17% of the genetic heritability of CLL, which suggests that more loci are yet to be identified, Dr Slager clarified.
She went on to say that CLL incidence varies by ethnicity. Caucasians have a very high rate of CLL, while Asians have a very low rate. And African Americans have an incidence rate between those of Caucasians and Asians.
Investigators have hypothesized that the differences in incidence are based on the distinct genetic variants that are associated with the ethnicities.
For example, 4 of the variants with more than 20% frequency in Caucasians are quite rare in Chinese individuals and are also quite uncommon in African Americans, with frequencies less than 10%.
Dr Slager suggested that conducting these kinds of studies in Asians and African Americans will take a large sample size and most likely require an international consortium to bring enough CLL cases together.
Impact on clinical practice
Because of the strong genetic risk, patients with CLL naturally want to know about their offspring and their siblings, Dr Slager has found.
Patients who have monoclonal B-cell lymphocytosis (MBL), which is a precursor to CLL, pose the biggest quandary.
MBL is detected in about 5% of people over age 40. However, it’s detected in about 15% to 18% of people with a first-degree relative with CLL.
“These are individuals who have lymphocytosis,” Dr Slager said. “They come to your clinic and have an elevated blood cell count, flow cytometry. [So] you screen them for MBL, and these individuals who have more than 500 cells per microliter, they are the ones who progress to CLL, at 1% per year.”
Individuals who don’t have the elevated blood counts do have the clonal cells, Dr Slager noted.
“They just don’t have the expansion,” she said. “The progression of these individuals to CLL is still yet to be determined.”
For these reasons, Dr Slager believes “it’s still premature to bring genetic testing into clinical practice.”
Future directions include bringing together the non-environmental issues and the inherited issues to create a model that will accurately predict the risk of CLL.
Photo courtesy of the
National Institute
of General Medical Science
NEW YORK—Research has shown that family history is a strong risk factor for developing chronic lymphocytic leukemia (CLL).
First-degree relatives have an 8.5-fold risk of getting CLL and an increased risk of other lymphoproliferative disorders, according to a study published in 2009.
However, despite the strong evidence of a genetic contribution, one expert believes it’s premature to bring genetic testing into the clinic for screening in CLL.
“At this time, we do not recommend genetic screening,” said Susan Slager, PhD, of the Mayo Clinic in Rochester, Minnesota.
“There’s no known relationship between the inherited variants and treatment response,” she explained, and the relatively low incidence of CLL argues against active screening in affected families at present.
Dr Slager discussed genetic and non-genetic factors associated with CLL and the clinical implications of these factors at Lymphoma & Myeloma 2016.
Demographic risk factors
Dr Slager noted that age, gender, and race are risk factors for CLL.
Individuals aged 65 to 74 have the highest incidence of CLL, at 28%, while the risk is almost non-existent for those under age 20, she said.
There is a higher incidence of CLL in males than in females, and the reason for this gender disparity is unknown.
There is a higher incidence of CLL in Caucasians than Asians, for both males and females.
“Again, it’s unknown why there’s this variability in incidence in CLL,” Dr Slager said. “Obviously, age, sex, and race—these are things you can’t modify. You’re stuck with them.”
However, several studies have been undertaken to look at some of the potentially modifiable factors associated with CLL.
Beyond demographic factors
The International Lymphoma Epidemiology Consortium, known as InterLymph, was initiated in 2001 to evaluate the association of risk factors in CLL. Study centers are located primarily in North America and Europe, with one in Australia.
In one of the larger InterLymph studies, investigators evaluated risk factors—lifestyle exposure, reproductive history, medical history, occupational exposures, farming exposure, and family history—in 2440 CLL patients and 15,186 controls.
The investigators found that sun exposure and atopy—allergies, asthma, eczema, and hay fever—have a protective effect in CLL, while serological hepatitis C virus (HCV) infections, farming exposure, and family history carry an increased risk of CLL.
This confirmed an earlier study conducted in New South Wales, Australia, that had uncovered an inverse association between sun exposure and non-Hodgkin lymphoma (NHL) risk, which fell significantly with increasing recreational sun exposure.
Medical history
Another earlier study from New South Wales revealed a 20% reduction in the risk of NHL for any specific allergy.
However, the investigators of the large, more recent study observed little to no evidence of reduced risk for asthma and eczema.
The underlying biology for atopy or allergies is a hyper-immune system, Dr Slager explained.
“So if you have a hyper-immune system, then we hypothesize that you have protection against CLL,” she said.
Another medical exposure investigators analyzed that impacts CLL risk is HCV. People infected with HCV have an increased risk of CLL, perhaps due to chronic antigen stimulation or possibly disruption of the T-cell function.
Height is also associated with CLL. CLL risk increases with greater height. The concept is that taller individuals have increased exposure to growth hormones that possibly result in cell proliferation.
Another hypothesis supporting the height association is that people of shorter stature experience more infections, which could result in a stronger immune system. And a stronger immune system perhaps protects against NHL.
Occupational exposures
Investigators consistently observed a 20% increased risk of CLL for people living or working on a farm.
Animal farmers, as opposed to crop farmers, experienced some protection. However, the sample size was too small to be conclusive, with only 29 people across all studies being animal farmers.
Among other occupations evaluated, hairdressers also had an increased risk of CLL, although this too was based on a small sample size.
Family history
One of the strongest risk factors for CLL is family history.
Using population-based registry data from Sweden, investigators found that people with a first-degree relative with CLL have an 8.5-fold risk of CLL.
They also have an elevated risk of other lymphoproliferative disorders, including NHL (1.9-fold risk), Waldenström’s macroglobulinemia (4.0-fold risk), hairy cell leukemia (3.3-fold risk), and follicular lymphoma (1.6-fold risk).
GWAS in CLL
Investigators conducted genome-wide association studies (GWAS) to determine what is driving the familial risk.
Dr Slager described these studies as an agnostic approach that looks across the entire genome to determine which regions are associated with a trait of interest.
Typically, many markers are genotyped—somewhere between half a million to 5 million markers—and each is looked at individually with respect to CLL, she said.
Unrelated cases and controls are included in the studies.
The first GWAS study identifying susceptibility loci for CLL was published in 2008. Subsequently, more studies were published with increasing sample sizes—more cases, more controls, and more genetic variants identified.
In the largest meta-analysis for CLL to date (Slager and Houlston et al, not yet published), investigators analyzed 4400 CLL cases and 13,000 controls.
They identified 9 more inherited variances with CLL, for a total of 43 identified to date.
The genes involved follow an apoptosis pathway, the telomere length pathway, and the B-cell lymphocyte development pathway.
“We have to remember, though, that these are non-causal,” Dr Slager cautioned. “We are just identifying the region in the genome that’s associated with CLL . . . . So now we have to dig deeper in these relationships to understand what’s going on.”
Using the identified CLL single-nucleotide polymorphisms, the investigators computed a polygenic risk score. CLL cases in the highest quintile had 2.7-fold increased risk of CLL.
However, the most common GWAS variants explain only 17% of the genetic heritability of CLL, which suggests that more loci are yet to be identified, Dr Slager clarified.
She went on to say that CLL incidence varies by ethnicity. Caucasians have a very high rate of CLL, while Asians have a very low rate. And African Americans have an incidence rate between those of Caucasians and Asians.
Investigators have hypothesized that the differences in incidence are based on the distinct genetic variants that are associated with the ethnicities.
For example, 4 of the variants with more than 20% frequency in Caucasians are quite rare in Chinese individuals and are also quite uncommon in African Americans, with frequencies less than 10%.
Dr Slager suggested that conducting these kinds of studies in Asians and African Americans will take a large sample size and most likely require an international consortium to bring enough CLL cases together.
Impact on clinical practice
Because of the strong genetic risk, patients with CLL naturally want to know about their offspring and their siblings, Dr Slager has found.
Patients who have monoclonal B-cell lymphocytosis (MBL), which is a precursor to CLL, pose the biggest quandary.
MBL is detected in about 5% of people over age 40. However, it’s detected in about 15% to 18% of people with a first-degree relative with CLL.
“These are individuals who have lymphocytosis,” Dr Slager said. “They come to your clinic and have an elevated blood cell count, flow cytometry. [So] you screen them for MBL, and these individuals who have more than 500 cells per microliter, they are the ones who progress to CLL, at 1% per year.”
Individuals who don’t have the elevated blood counts do have the clonal cells, Dr Slager noted.
“They just don’t have the expansion,” she said. “The progression of these individuals to CLL is still yet to be determined.”
For these reasons, Dr Slager believes “it’s still premature to bring genetic testing into clinical practice.”
Future directions include bringing together the non-environmental issues and the inherited issues to create a model that will accurately predict the risk of CLL.