User login
Study highlights need for induction strategy in elderly, frail MM patients

ATLANTA—Initial results of the phase 2 HOVON-126 trial in newly diagnosed multiple myeloma (MM) patients have highlighted the need for an induction strategy in elderly and frail patients.
The trial showed high overall response rates (ORRs) after induction with ixazomib, thalidomide, and low-dose dexamethasone.
However, 62% of patients older than 75 and 60% of frail patients discontinued therapy prior to starting maintenance.
HOVON-126 was designed to determine the ORR of induction therapy with ixazomib, thalidomide, and dexamethasone but also compare progression-free survival in patients who received ixazomib maintenance and those who received placebo.
Sonja Zweegman, MD, of VUmc in Amsterdam, The Netherlands, presented induction results from HOVON-126 at the 2017 ASH Annual Meeting (abstract 433).
The study was supported by Takeda and the Dutch Cancer Society. Dr Zweegman disclosed research funding from, and advisory board participation for, Takeda.
Study design
Investigators enrolled patients with previously untreated, symptomatic MM who were not eligible for stem cell transplant. Patients had to have measurable disease and a WHO performance status of 0 to 3 for patients younger than 75 and 0 to 2 for patients 75 or older.
Patients were not eligible if they had grade 3 neuropathy or grade 2 with pain. They were also ineligible if their creatinine clearance was less than 30 mL/minute.
All patients received ixazomib at 4 mg on days 1, 8, and 15; thalidomide at 100 mg on days 1 to 28; and dexamethasone at 40 mg on days 1, 8, 15, and 22 for nine 28-day cycles.
They could then be randomized to ixazomib maintenance (on the aforementioned schedule) or placebo for 28-day cycles until progression.
Investigators performed subgroup analyses based on cytogenetic risk and frailty.
They defined frailty according to the modified IMWG frailty index, which takes into account age, the Charlson Comorbidity Index, and the WHO performance scale as a proxy for Activities of Daily Living.
They defined high-risk cytogenetics as del17p, t(4;14), or t(14;16).
Investigators planned to enroll 142 patients and expected 94 patients to be randomized.
Patient demographics
The first 120 patients enrolled had a median age of 74 (range, 64–90). Thirty percent (n=38) were older than 75, and 8% (n=10) were older than 80.
More than two-thirds had an ISS score of I or II, and three-quarters had a WHO performance status of 0 or 1. Twenty-four percent had a performance status of 2, and 1% had a performance status of 3.
Eighty percent had lytic bone disease.
One hundred thirteen patients (94%) had FISH analysis performed. Of those, 10% had del17p, 7% had t(4;14), and 1% had t(14;16).
Eighty-one percent of patients fell into the standard-risk category and 19% into the high-risk category.
Almost half of patients (47%) were considered frail, 28% unfit, 21% fit, and 4% unknown.
Response
The ORR for induction was 81%. Ten percent of patients achieved a complete response (CR), 34% had a very good partial response (VGPR), and 37% had a partial response (PR).
The median time to response was 1.1 months, and the median time to maximum response was 4.7 months.
The response rate was independent of cytogenetic risk. Standard-risk patients achieved an ORR of 84%, a VGPR rate of 48%, and a CR rate of 10%. High-risk patients had an ORR of 79%, VGPR of 42%, and CR of 11%.
The response rate was also independent of frailty. Fit patients had an ORR of 88%, unfit patients 85%, and frail patients 75%. The VGPR rate was 36% for fit, 53% for unfit, and 43% for frail patients. The CR rate was 16% for fit, 9% for unfit, and 9% for frail patients.
Safety
“Grade 3 and 4 toxicities were found to be limited, with mainly infections, [gastrointestinal], and skin toxicity,” Dr Zweegman noted. “There was also a very low incidence of neuropathy, with only 3% grade 3 neuropathy and no grade 4 neuropathy.”
Grade 3 adverse events (AEs) occurred in 50% of patients and grade 4 in 11%.
Hematologic AEs of grade 3 and 4, respectively, included anemia (5%, 1%), thrombocytopenia (3%, 1%), and neutropenia (1%, 0).
Nonhematologic AEs of grade 3 and 4, respectively, included infections (12%, 3%), neuropathy (3%, 0), cardiac events (7%, 3%), gastrointestinal events (8%, 0), skin AEs (10%, 0), and venous thromboembolism (0, 2%).
The incidence of severe neuropathy was low. Fifty-eight percent of patients had grade 0 neuropathy, 24% grade 1, 14% grade 2, 3% grade 3, and no grade 4.
Discontinuation
Fifty-four patients (45%) discontinued therapy. The reasons for discontinuation were:
- Progressive disease, 13%
- Toxicity, 15%
- Death, 4%
- Noncompliance, 8%
- Not eligible for randomization, 0.8%
- Other, 4%.
“And when looking in detail into the toxicity, it was shown that it was mainly asthenia and neuropathy being judged by the treating physicians as caused by thalidomide,” Dr Zweegman explained.
Investigators also evaluated discontinuation according to age and found that 35% of patients 75 or younger discontinued therapy, compared with 62% of those older than 75.
However, there was no significant difference in discontinuation rate during the first 6 cycles. Seventy-seven percent of the younger patients and 69% of the older group completed 6 cycles.
Older patients who discontinued early had rates of progressive disease and toxicity comparable to the younger patients, but “there was a difference in early mortality,” Dr Zweegman added.
Nine percent of older patients discontinued before maintenance due to early mortality, compared with 1% of younger patients. And mortality in the older group was mainly due to infections and 1 cardiac arrest.
“So I think that highlights the need for antibiotic prophylaxis, which was not mandatory in this study,” Dr Zweegman said.
And finally, the investigators evaluated discontinuation according to frailty. Twenty-four percent of fit patients discontinued prior to maintenance, 32% of unfit, and 60% of frail.
Again, investigators found no significant difference in discontinuation rate during the first 6 cycles of induction. Eighty percent of fit patients completed 6 cycles, as did 79% of unfit patients and 70% of frail patients.
Despite the feasibility of the treatment and an ORR of 81%, the investigators say novel approaches are needed for frail patients and those older than 75.
“One possibility is to limit the duration of induction therapy . . . ,” Dr Zweegman said. “That would allow the start of long-term administration of maintenance treatment.”
The investigators also suggest evaluating less toxic combinations, such as ixazomib and daratumumab with lower doses of dexamethasone, the combination used in the HOVON-143 study.
Ixazomib is approved by the US Food and Drug Administration, Health Canada, and conditionally approved by the European Commission for use in combination with lenalidomide and dexamethasone to treat MM patients who have received at least 1 prior therapy. ![]()
ATLANTA—Initial results of the phase 2 HOVON-126 trial in newly diagnosed multiple myeloma (MM) patients have highlighted the need for an induction strategy in elderly and frail patients.
The trial showed high overall response rates (ORRs) after induction with ixazomib, thalidomide, and low-dose dexamethasone.
However, 62% of patients older than 75 and 60% of frail patients discontinued therapy prior to starting maintenance.
HOVON-126 was designed to determine the ORR of induction therapy with ixazomib, thalidomide, and dexamethasone but also compare progression-free survival in patients who received ixazomib maintenance and those who received placebo.
Sonja Zweegman, MD, of VUmc in Amsterdam, The Netherlands, presented induction results from HOVON-126 at the 2017 ASH Annual Meeting (abstract 433).
The study was supported by Takeda and the Dutch Cancer Society. Dr Zweegman disclosed research funding from, and advisory board participation for, Takeda.
Study design
Investigators enrolled patients with previously untreated, symptomatic MM who were not eligible for stem cell transplant. Patients had to have measurable disease and a WHO performance status of 0 to 3 for patients younger than 75 and 0 to 2 for patients 75 or older.
Patients were not eligible if they had grade 3 neuropathy or grade 2 with pain. They were also ineligible if their creatinine clearance was less than 30 mL/minute.
All patients received ixazomib at 4 mg on days 1, 8, and 15; thalidomide at 100 mg on days 1 to 28; and dexamethasone at 40 mg on days 1, 8, 15, and 22 for nine 28-day cycles.
They could then be randomized to ixazomib maintenance (on the aforementioned schedule) or placebo for 28-day cycles until progression.
Investigators performed subgroup analyses based on cytogenetic risk and frailty.
They defined frailty according to the modified IMWG frailty index, which takes into account age, the Charlson Comorbidity Index, and the WHO performance scale as a proxy for Activities of Daily Living.
They defined high-risk cytogenetics as del17p, t(4;14), or t(14;16).
Investigators planned to enroll 142 patients and expected 94 patients to be randomized.
Patient demographics
The first 120 patients enrolled had a median age of 74 (range, 64–90). Thirty percent (n=38) were older than 75, and 8% (n=10) were older than 80.
More than two-thirds had an ISS score of I or II, and three-quarters had a WHO performance status of 0 or 1. Twenty-four percent had a performance status of 2, and 1% had a performance status of 3.
Eighty percent had lytic bone disease.
One hundred thirteen patients (94%) had FISH analysis performed. Of those, 10% had del17p, 7% had t(4;14), and 1% had t(14;16).
Eighty-one percent of patients fell into the standard-risk category and 19% into the high-risk category.
Almost half of patients (47%) were considered frail, 28% unfit, 21% fit, and 4% unknown.
Response
The ORR for induction was 81%. Ten percent of patients achieved a complete response (CR), 34% had a very good partial response (VGPR), and 37% had a partial response (PR).
The median time to response was 1.1 months, and the median time to maximum response was 4.7 months.
The response rate was independent of cytogenetic risk. Standard-risk patients achieved an ORR of 84%, a VGPR rate of 48%, and a CR rate of 10%. High-risk patients had an ORR of 79%, VGPR of 42%, and CR of 11%.
The response rate was also independent of frailty. Fit patients had an ORR of 88%, unfit patients 85%, and frail patients 75%. The VGPR rate was 36% for fit, 53% for unfit, and 43% for frail patients. The CR rate was 16% for fit, 9% for unfit, and 9% for frail patients.
Safety
“Grade 3 and 4 toxicities were found to be limited, with mainly infections, [gastrointestinal], and skin toxicity,” Dr Zweegman noted. “There was also a very low incidence of neuropathy, with only 3% grade 3 neuropathy and no grade 4 neuropathy.”
Grade 3 adverse events (AEs) occurred in 50% of patients and grade 4 in 11%.
Hematologic AEs of grade 3 and 4, respectively, included anemia (5%, 1%), thrombocytopenia (3%, 1%), and neutropenia (1%, 0).
Nonhematologic AEs of grade 3 and 4, respectively, included infections (12%, 3%), neuropathy (3%, 0), cardiac events (7%, 3%), gastrointestinal events (8%, 0), skin AEs (10%, 0), and venous thromboembolism (0, 2%).
The incidence of severe neuropathy was low. Fifty-eight percent of patients had grade 0 neuropathy, 24% grade 1, 14% grade 2, 3% grade 3, and no grade 4.
Discontinuation
Fifty-four patients (45%) discontinued therapy. The reasons for discontinuation were:
- Progressive disease, 13%
- Toxicity, 15%
- Death, 4%
- Noncompliance, 8%
- Not eligible for randomization, 0.8%
- Other, 4%.
“And when looking in detail into the toxicity, it was shown that it was mainly asthenia and neuropathy being judged by the treating physicians as caused by thalidomide,” Dr Zweegman explained.
Investigators also evaluated discontinuation according to age and found that 35% of patients 75 or younger discontinued therapy, compared with 62% of those older than 75.
However, there was no significant difference in discontinuation rate during the first 6 cycles. Seventy-seven percent of the younger patients and 69% of the older group completed 6 cycles.
Older patients who discontinued early had rates of progressive disease and toxicity comparable to the younger patients, but “there was a difference in early mortality,” Dr Zweegman added.
Nine percent of older patients discontinued before maintenance due to early mortality, compared with 1% of younger patients. And mortality in the older group was mainly due to infections and 1 cardiac arrest.
“So I think that highlights the need for antibiotic prophylaxis, which was not mandatory in this study,” Dr Zweegman said.
And finally, the investigators evaluated discontinuation according to frailty. Twenty-four percent of fit patients discontinued prior to maintenance, 32% of unfit, and 60% of frail.
Again, investigators found no significant difference in discontinuation rate during the first 6 cycles of induction. Eighty percent of fit patients completed 6 cycles, as did 79% of unfit patients and 70% of frail patients.
Despite the feasibility of the treatment and an ORR of 81%, the investigators say novel approaches are needed for frail patients and those older than 75.
“One possibility is to limit the duration of induction therapy . . . ,” Dr Zweegman said. “That would allow the start of long-term administration of maintenance treatment.”
The investigators also suggest evaluating less toxic combinations, such as ixazomib and daratumumab with lower doses of dexamethasone, the combination used in the HOVON-143 study.
Ixazomib is approved by the US Food and Drug Administration, Health Canada, and conditionally approved by the European Commission for use in combination with lenalidomide and dexamethasone to treat MM patients who have received at least 1 prior therapy. ![]()
ATLANTA—Initial results of the phase 2 HOVON-126 trial in newly diagnosed multiple myeloma (MM) patients have highlighted the need for an induction strategy in elderly and frail patients.
The trial showed high overall response rates (ORRs) after induction with ixazomib, thalidomide, and low-dose dexamethasone.
However, 62% of patients older than 75 and 60% of frail patients discontinued therapy prior to starting maintenance.
HOVON-126 was designed to determine the ORR of induction therapy with ixazomib, thalidomide, and dexamethasone but also compare progression-free survival in patients who received ixazomib maintenance and those who received placebo.
Sonja Zweegman, MD, of VUmc in Amsterdam, The Netherlands, presented induction results from HOVON-126 at the 2017 ASH Annual Meeting (abstract 433).
The study was supported by Takeda and the Dutch Cancer Society. Dr Zweegman disclosed research funding from, and advisory board participation for, Takeda.
Study design
Investigators enrolled patients with previously untreated, symptomatic MM who were not eligible for stem cell transplant. Patients had to have measurable disease and a WHO performance status of 0 to 3 for patients younger than 75 and 0 to 2 for patients 75 or older.
Patients were not eligible if they had grade 3 neuropathy or grade 2 with pain. They were also ineligible if their creatinine clearance was less than 30 mL/minute.
All patients received ixazomib at 4 mg on days 1, 8, and 15; thalidomide at 100 mg on days 1 to 28; and dexamethasone at 40 mg on days 1, 8, 15, and 22 for nine 28-day cycles.
They could then be randomized to ixazomib maintenance (on the aforementioned schedule) or placebo for 28-day cycles until progression.
Investigators performed subgroup analyses based on cytogenetic risk and frailty.
They defined frailty according to the modified IMWG frailty index, which takes into account age, the Charlson Comorbidity Index, and the WHO performance scale as a proxy for Activities of Daily Living.
They defined high-risk cytogenetics as del17p, t(4;14), or t(14;16).
Investigators planned to enroll 142 patients and expected 94 patients to be randomized.
Patient demographics
The first 120 patients enrolled had a median age of 74 (range, 64–90). Thirty percent (n=38) were older than 75, and 8% (n=10) were older than 80.
More than two-thirds had an ISS score of I or II, and three-quarters had a WHO performance status of 0 or 1. Twenty-four percent had a performance status of 2, and 1% had a performance status of 3.
Eighty percent had lytic bone disease.
One hundred thirteen patients (94%) had FISH analysis performed. Of those, 10% had del17p, 7% had t(4;14), and 1% had t(14;16).
Eighty-one percent of patients fell into the standard-risk category and 19% into the high-risk category.
Almost half of patients (47%) were considered frail, 28% unfit, 21% fit, and 4% unknown.
Response
The ORR for induction was 81%. Ten percent of patients achieved a complete response (CR), 34% had a very good partial response (VGPR), and 37% had a partial response (PR).
The median time to response was 1.1 months, and the median time to maximum response was 4.7 months.
The response rate was independent of cytogenetic risk. Standard-risk patients achieved an ORR of 84%, a VGPR rate of 48%, and a CR rate of 10%. High-risk patients had an ORR of 79%, VGPR of 42%, and CR of 11%.
The response rate was also independent of frailty. Fit patients had an ORR of 88%, unfit patients 85%, and frail patients 75%. The VGPR rate was 36% for fit, 53% for unfit, and 43% for frail patients. The CR rate was 16% for fit, 9% for unfit, and 9% for frail patients.
Safety
“Grade 3 and 4 toxicities were found to be limited, with mainly infections, [gastrointestinal], and skin toxicity,” Dr Zweegman noted. “There was also a very low incidence of neuropathy, with only 3% grade 3 neuropathy and no grade 4 neuropathy.”
Grade 3 adverse events (AEs) occurred in 50% of patients and grade 4 in 11%.
Hematologic AEs of grade 3 and 4, respectively, included anemia (5%, 1%), thrombocytopenia (3%, 1%), and neutropenia (1%, 0).
Nonhematologic AEs of grade 3 and 4, respectively, included infections (12%, 3%), neuropathy (3%, 0), cardiac events (7%, 3%), gastrointestinal events (8%, 0), skin AEs (10%, 0), and venous thromboembolism (0, 2%).
The incidence of severe neuropathy was low. Fifty-eight percent of patients had grade 0 neuropathy, 24% grade 1, 14% grade 2, 3% grade 3, and no grade 4.
Discontinuation
Fifty-four patients (45%) discontinued therapy. The reasons for discontinuation were:
- Progressive disease, 13%
- Toxicity, 15%
- Death, 4%
- Noncompliance, 8%
- Not eligible for randomization, 0.8%
- Other, 4%.
“And when looking in detail into the toxicity, it was shown that it was mainly asthenia and neuropathy being judged by the treating physicians as caused by thalidomide,” Dr Zweegman explained.
Investigators also evaluated discontinuation according to age and found that 35% of patients 75 or younger discontinued therapy, compared with 62% of those older than 75.
However, there was no significant difference in discontinuation rate during the first 6 cycles. Seventy-seven percent of the younger patients and 69% of the older group completed 6 cycles.
Older patients who discontinued early had rates of progressive disease and toxicity comparable to the younger patients, but “there was a difference in early mortality,” Dr Zweegman added.
Nine percent of older patients discontinued before maintenance due to early mortality, compared with 1% of younger patients. And mortality in the older group was mainly due to infections and 1 cardiac arrest.
“So I think that highlights the need for antibiotic prophylaxis, which was not mandatory in this study,” Dr Zweegman said.
And finally, the investigators evaluated discontinuation according to frailty. Twenty-four percent of fit patients discontinued prior to maintenance, 32% of unfit, and 60% of frail.
Again, investigators found no significant difference in discontinuation rate during the first 6 cycles of induction. Eighty percent of fit patients completed 6 cycles, as did 79% of unfit patients and 70% of frail patients.
Despite the feasibility of the treatment and an ORR of 81%, the investigators say novel approaches are needed for frail patients and those older than 75.
“One possibility is to limit the duration of induction therapy . . . ,” Dr Zweegman said. “That would allow the start of long-term administration of maintenance treatment.”
The investigators also suggest evaluating less toxic combinations, such as ixazomib and daratumumab with lower doses of dexamethasone, the combination used in the HOVON-143 study.
Ixazomib is approved by the US Food and Drug Administration, Health Canada, and conditionally approved by the European Commission for use in combination with lenalidomide and dexamethasone to treat MM patients who have received at least 1 prior therapy. ![]()
Iron chelating agent could enhance chemo in AML

Chemotherapy for acute myeloid leukemia (AML) might be improved by the addition of deferoxamine, according to preclinical research published in Cell Stem Cell.
Researchers found that, when certain areas of the bone marrow are overtaken by AML cells, hematopoietic stem cells (HSCs) are lost, and the delivery of chemotherapy may be compromised.
However, the team also discovered that deferoxamine, a drug already approved to treat iron overload, can protect these areas of the bone marrow, allowing HSCs to survive and improving the efficacy of chemotherapy.
“Since the drug is already approved for human use for a different condition, we already know that it is safe,” said study author Cristina Lo Celso, PhD, of Imperial College London in the UK.
“We still need to test it in the context of leukemia and chemotherapy, but, because it is already in use, we can progress to clinical trials much quicker than we could with a brand-new drug.”
For the current study, Dr Lo Celso and her colleagues used intravital microscopy to study AML cells, healthy hematopoietic cells, and the bone marrow microenvironment in mice.
The researchers found the endosteal microenvironment was hit particularly hard by AML. Specifically, AML progression led to endosteal remodeling, with AML cells degrading endosteal endothelium, stromal cells, and osteoblastic cells.
This remodeling resulted in the loss of nonleukemic HSCs, which hindered hematopoiesis. However, preserving endosteal vessels prevented the loss of HSCs.
Previous research had shown that deferoxamine could induce endosteal vessel expansion through enhancement of hypoxia-inducible factor 1a stability and activity. So the researchers administered deferoxamine to mice with AML.
The drug had a protective effect on endosteal vessels, which were able to support healthy HSCs and improve HSC homing.
The researchers also found that enhanced endosteal vessels improved the efficacy of chemotherapy (cytarabine and doxorubicin) in mice with AML.
The team compared Fbxw7iΔEC-mutant mice, in which the administration of tamoxifen increases the number of endosteal vessels and arterioles, to control mice. Both sets of mice had AML.
After confirming the mutant mice had increased numbers of endosteal vessels, the researchers treated the mutant mice and controls with cytarabine and doxorubicin.
Both sets of mice had significant chemotherapy-induced damage to the bone marrow vasculature, including endosteal vessels.
However, after treatment, the Fbxw7iΔEC-mutant mice had lower numbers of surviving AML cells in the bone marrow, delayed relapse, and longer survival than control mice.
The researchers therefore concluded that rescuing endosteal vessels before starting chemotherapy can improve the efficacy of treatment in AML.
“Our work suggests that therapies targeting these blood vessels may improve existing therapeutic regimens for AML and perhaps other leukemias too,” said study author Delfim Duarte, MD, of Imperial College London.
Based on this work, the researchers are hoping to start trials of deferoxamine in patients with AML. ![]()
Chemotherapy for acute myeloid leukemia (AML) might be improved by the addition of deferoxamine, according to preclinical research published in Cell Stem Cell.
Researchers found that, when certain areas of the bone marrow are overtaken by AML cells, hematopoietic stem cells (HSCs) are lost, and the delivery of chemotherapy may be compromised.
However, the team also discovered that deferoxamine, a drug already approved to treat iron overload, can protect these areas of the bone marrow, allowing HSCs to survive and improving the efficacy of chemotherapy.
“Since the drug is already approved for human use for a different condition, we already know that it is safe,” said study author Cristina Lo Celso, PhD, of Imperial College London in the UK.
“We still need to test it in the context of leukemia and chemotherapy, but, because it is already in use, we can progress to clinical trials much quicker than we could with a brand-new drug.”
For the current study, Dr Lo Celso and her colleagues used intravital microscopy to study AML cells, healthy hematopoietic cells, and the bone marrow microenvironment in mice.
The researchers found the endosteal microenvironment was hit particularly hard by AML. Specifically, AML progression led to endosteal remodeling, with AML cells degrading endosteal endothelium, stromal cells, and osteoblastic cells.
This remodeling resulted in the loss of nonleukemic HSCs, which hindered hematopoiesis. However, preserving endosteal vessels prevented the loss of HSCs.
Previous research had shown that deferoxamine could induce endosteal vessel expansion through enhancement of hypoxia-inducible factor 1a stability and activity. So the researchers administered deferoxamine to mice with AML.
The drug had a protective effect on endosteal vessels, which were able to support healthy HSCs and improve HSC homing.
The researchers also found that enhanced endosteal vessels improved the efficacy of chemotherapy (cytarabine and doxorubicin) in mice with AML.
The team compared Fbxw7iΔEC-mutant mice, in which the administration of tamoxifen increases the number of endosteal vessels and arterioles, to control mice. Both sets of mice had AML.
After confirming the mutant mice had increased numbers of endosteal vessels, the researchers treated the mutant mice and controls with cytarabine and doxorubicin.
Both sets of mice had significant chemotherapy-induced damage to the bone marrow vasculature, including endosteal vessels.
However, after treatment, the Fbxw7iΔEC-mutant mice had lower numbers of surviving AML cells in the bone marrow, delayed relapse, and longer survival than control mice.
The researchers therefore concluded that rescuing endosteal vessels before starting chemotherapy can improve the efficacy of treatment in AML.
“Our work suggests that therapies targeting these blood vessels may improve existing therapeutic regimens for AML and perhaps other leukemias too,” said study author Delfim Duarte, MD, of Imperial College London.
Based on this work, the researchers are hoping to start trials of deferoxamine in patients with AML. ![]()
Chemotherapy for acute myeloid leukemia (AML) might be improved by the addition of deferoxamine, according to preclinical research published in Cell Stem Cell.
Researchers found that, when certain areas of the bone marrow are overtaken by AML cells, hematopoietic stem cells (HSCs) are lost, and the delivery of chemotherapy may be compromised.
However, the team also discovered that deferoxamine, a drug already approved to treat iron overload, can protect these areas of the bone marrow, allowing HSCs to survive and improving the efficacy of chemotherapy.
“Since the drug is already approved for human use for a different condition, we already know that it is safe,” said study author Cristina Lo Celso, PhD, of Imperial College London in the UK.
“We still need to test it in the context of leukemia and chemotherapy, but, because it is already in use, we can progress to clinical trials much quicker than we could with a brand-new drug.”
For the current study, Dr Lo Celso and her colleagues used intravital microscopy to study AML cells, healthy hematopoietic cells, and the bone marrow microenvironment in mice.
The researchers found the endosteal microenvironment was hit particularly hard by AML. Specifically, AML progression led to endosteal remodeling, with AML cells degrading endosteal endothelium, stromal cells, and osteoblastic cells.
This remodeling resulted in the loss of nonleukemic HSCs, which hindered hematopoiesis. However, preserving endosteal vessels prevented the loss of HSCs.
Previous research had shown that deferoxamine could induce endosteal vessel expansion through enhancement of hypoxia-inducible factor 1a stability and activity. So the researchers administered deferoxamine to mice with AML.
The drug had a protective effect on endosteal vessels, which were able to support healthy HSCs and improve HSC homing.
The researchers also found that enhanced endosteal vessels improved the efficacy of chemotherapy (cytarabine and doxorubicin) in mice with AML.
The team compared Fbxw7iΔEC-mutant mice, in which the administration of tamoxifen increases the number of endosteal vessels and arterioles, to control mice. Both sets of mice had AML.
After confirming the mutant mice had increased numbers of endosteal vessels, the researchers treated the mutant mice and controls with cytarabine and doxorubicin.
Both sets of mice had significant chemotherapy-induced damage to the bone marrow vasculature, including endosteal vessels.
However, after treatment, the Fbxw7iΔEC-mutant mice had lower numbers of surviving AML cells in the bone marrow, delayed relapse, and longer survival than control mice.
The researchers therefore concluded that rescuing endosteal vessels before starting chemotherapy can improve the efficacy of treatment in AML.
“Our work suggests that therapies targeting these blood vessels may improve existing therapeutic regimens for AML and perhaps other leukemias too,” said study author Delfim Duarte, MD, of Imperial College London.
Based on this work, the researchers are hoping to start trials of deferoxamine in patients with AML. ![]()
Research explains why cisplatin causes hearing loss
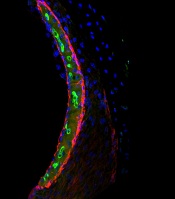
Researchers have gained new insight into hearing loss caused by cisplatin.
By measuring and mapping cisplatin retention in mouse and human inner ear tissues, the researchers found that cisplatin builds up in the inner ear and can remain there for years.
The team also found that a region in the inner ear called the stria vascularis could be targeted to prevent hearing loss resulting from cisplatin.
Lisa L. Cunningham, PhD, of the National Institute on Deafness and other Communications Disorders (NIDCD) in Bethesda, Maryland, and her colleagues reported these findings in Nature Communications.
The researchers noted that cisplatin can cause permanent hearing loss in 40% to 80% of treated patients. The team’s new findings help explain why.
The researchers found that, in most areas of the body, cisplatin is eliminated within days or weeks of treatment, but, in the inner ear, the drug remains much longer.
The team developed a mouse model that represents cisplatin-induced hearing loss seen in human patients.
By looking at inner ear tissue of mice after the first, second, and third cisplatin treatment, the researchers saw that cisplatin remained in the mouse inner ear much longer than in most other body tissues, and the drug builds up with each successive treatment.
The team also studied inner ear tissue donated by deceased adults who had been treated with cisplatin and found the drug is retained in the inner ear months or years after treatment.
When the researchers examined inner ear tissue from a child, they found cisplatin buildup that was even higher than that seen in adults.
Taken together, these results suggest the inner ear readily takes up cisplatin but has limited ability to remove the drug.
In mice and human tissues, the researchers saw the highest buildup of cisplatin in a part of the inner ear called the stria vascularis, which helps maintain the positive electrical charge in inner ear fluid that certain cells need to detect sound.
The team found the accumulation of cisplatin in the stria vascularis contributed to cisplatin-related hearing loss.
“Our findings suggest that if we can prevent cisplatin from entering the stria vascularis in the inner ear during treatment, we may be able to protect cancer patients from developing cisplatin-induced hearing loss,” Dr Cunningham said. ![]()
Researchers have gained new insight into hearing loss caused by cisplatin.
By measuring and mapping cisplatin retention in mouse and human inner ear tissues, the researchers found that cisplatin builds up in the inner ear and can remain there for years.
The team also found that a region in the inner ear called the stria vascularis could be targeted to prevent hearing loss resulting from cisplatin.
Lisa L. Cunningham, PhD, of the National Institute on Deafness and other Communications Disorders (NIDCD) in Bethesda, Maryland, and her colleagues reported these findings in Nature Communications.
The researchers noted that cisplatin can cause permanent hearing loss in 40% to 80% of treated patients. The team’s new findings help explain why.
The researchers found that, in most areas of the body, cisplatin is eliminated within days or weeks of treatment, but, in the inner ear, the drug remains much longer.
The team developed a mouse model that represents cisplatin-induced hearing loss seen in human patients.
By looking at inner ear tissue of mice after the first, second, and third cisplatin treatment, the researchers saw that cisplatin remained in the mouse inner ear much longer than in most other body tissues, and the drug builds up with each successive treatment.
The team also studied inner ear tissue donated by deceased adults who had been treated with cisplatin and found the drug is retained in the inner ear months or years after treatment.
When the researchers examined inner ear tissue from a child, they found cisplatin buildup that was even higher than that seen in adults.
Taken together, these results suggest the inner ear readily takes up cisplatin but has limited ability to remove the drug.
In mice and human tissues, the researchers saw the highest buildup of cisplatin in a part of the inner ear called the stria vascularis, which helps maintain the positive electrical charge in inner ear fluid that certain cells need to detect sound.
The team found the accumulation of cisplatin in the stria vascularis contributed to cisplatin-related hearing loss.
“Our findings suggest that if we can prevent cisplatin from entering the stria vascularis in the inner ear during treatment, we may be able to protect cancer patients from developing cisplatin-induced hearing loss,” Dr Cunningham said. ![]()
Researchers have gained new insight into hearing loss caused by cisplatin.
By measuring and mapping cisplatin retention in mouse and human inner ear tissues, the researchers found that cisplatin builds up in the inner ear and can remain there for years.
The team also found that a region in the inner ear called the stria vascularis could be targeted to prevent hearing loss resulting from cisplatin.
Lisa L. Cunningham, PhD, of the National Institute on Deafness and other Communications Disorders (NIDCD) in Bethesda, Maryland, and her colleagues reported these findings in Nature Communications.
The researchers noted that cisplatin can cause permanent hearing loss in 40% to 80% of treated patients. The team’s new findings help explain why.
The researchers found that, in most areas of the body, cisplatin is eliminated within days or weeks of treatment, but, in the inner ear, the drug remains much longer.
The team developed a mouse model that represents cisplatin-induced hearing loss seen in human patients.
By looking at inner ear tissue of mice after the first, second, and third cisplatin treatment, the researchers saw that cisplatin remained in the mouse inner ear much longer than in most other body tissues, and the drug builds up with each successive treatment.
The team also studied inner ear tissue donated by deceased adults who had been treated with cisplatin and found the drug is retained in the inner ear months or years after treatment.
When the researchers examined inner ear tissue from a child, they found cisplatin buildup that was even higher than that seen in adults.
Taken together, these results suggest the inner ear readily takes up cisplatin but has limited ability to remove the drug.
In mice and human tissues, the researchers saw the highest buildup of cisplatin in a part of the inner ear called the stria vascularis, which helps maintain the positive electrical charge in inner ear fluid that certain cells need to detect sound.
The team found the accumulation of cisplatin in the stria vascularis contributed to cisplatin-related hearing loss.
“Our findings suggest that if we can prevent cisplatin from entering the stria vascularis in the inner ear during treatment, we may be able to protect cancer patients from developing cisplatin-induced hearing loss,” Dr Cunningham said. ![]()
Team develops new scoring systems for PMF

ATLANTA—Two novel prognostic scoring systems can help clinicians decide how to treat certain patients with primary myelofibrosis (PMF), according to a new study.
The scoring systems, which build upon the International Prognostic Scoring System (IPSS), were developed for use in PMF patients age 70 and younger who are potential candidates for hematopoietic stem cell transplant (HSCT).
One of the scoring systems—MIPSS70—integrates clinical, histologic, and molecular information. The other—MIPSS70-plus—also includes cytogenetic information.
Alessandro M. Vannucchi, MD, of the University of Florence in Italy, presented details on these prognostic scoring systems at the 2017 ASH Annual Meeting (abstract 200*).
The information was published simultaneously in the Journal of Clinical Oncology.
Dr Vannucchi noted that, in PMF, survival is currently predicted by the IPSS, the dynamic IPSS, and the dynamic IPSS-plus.
“The IPSS score is used at the time of diagnosis, while the dynamic IPSS or dynamic IPSS-plus are used to provide survival estimates at the time of patient referral,” he explained. “In clinical practice, these prognostic risk scores are mainly used for [HSCT] decision-making in younger patients.”
Driver mutations and other myeloid neoplasm-associated mutations provide prognostic information that is independent of the IPSS/dynamic IPSS/dynamic IPSS-plus scoring systems.
The degree of bone marrow fibrosis and cytogenetic abnormalities configuring an unfavorable category also contribute prognostic information that is independent of these scoring systems.
With this in mind, Dr Vannucchi and his colleagues set out to develop an updated prognostic score that included molecular information (MIPSS70) and, if possible, cytogenetic information (MIPSS70-plus) for PMF patients age 70 and younger who are potential candidates for HSCT.
The researchers developed 2 prognostic models using a training/validation cohort approach.
For MIPSS70, the training cohort included 490 patients from 6 Italian institutions associated with the Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative project (AGIMM group), and the validation cohort included 211 patients from the Mayo Clinic in Rochester, Minnesota.
For MIPSS70-plus, the training cohort included 315 patients from the Mayo Clinic, and the validation cohort included 261 patients from the AGIMM group.
Using the MIPSS70 risk score in the validation cohort, the 5-year overall survival rate was 96% in low-risk, 67% in intermediate-risk, and 34% in high-risk patients.
“MIPSS70 performed better than IPSS in predicting survival,” Dr Vannucchi said. “About 30% of patients who were high-risk with MIPPS70 were missed by IPSS.”
Using the MIPSS70-plus risk score in the validation cohort, the 5-year overall survival rate was 100% in low-risk, 90% in intermediate-risk, 76% in high-risk, and 46.5% in very high-risk patients.
The MIPSS70-plus risk score also identified patients at very high risk for leukemic transformation, Dr Vannucchi said.
Furthermore, both MIPSS70 and MIPSS70-plus remained predictive of survival when the researchers evaluated patients older than 70 years of age.
“The new MIPSS70 and MIPSS70-plus scores include modern disease-associated risk variables pertinent to both pre-PMF and overt-PMF according to the 2016 WHO classification,” Dr Vannucchi said. “They integrate prognostically relevant clinical, cytogenetic, and mutation data and provide complementary systems of improved risk stratification for transplantation-age patients with PMF.”
Dr Vannucchi disclosed membership in speaker’s bureaus with Gilead, Shire, and Novartis, and research funding and membership on Board of Directors or advisory committees with Novartis. ![]()
*Data in the presentation differ from the abstract.
ATLANTA—Two novel prognostic scoring systems can help clinicians decide how to treat certain patients with primary myelofibrosis (PMF), according to a new study.
The scoring systems, which build upon the International Prognostic Scoring System (IPSS), were developed for use in PMF patients age 70 and younger who are potential candidates for hematopoietic stem cell transplant (HSCT).
One of the scoring systems—MIPSS70—integrates clinical, histologic, and molecular information. The other—MIPSS70-plus—also includes cytogenetic information.
Alessandro M. Vannucchi, MD, of the University of Florence in Italy, presented details on these prognostic scoring systems at the 2017 ASH Annual Meeting (abstract 200*).
The information was published simultaneously in the Journal of Clinical Oncology.
Dr Vannucchi noted that, in PMF, survival is currently predicted by the IPSS, the dynamic IPSS, and the dynamic IPSS-plus.
“The IPSS score is used at the time of diagnosis, while the dynamic IPSS or dynamic IPSS-plus are used to provide survival estimates at the time of patient referral,” he explained. “In clinical practice, these prognostic risk scores are mainly used for [HSCT] decision-making in younger patients.”
Driver mutations and other myeloid neoplasm-associated mutations provide prognostic information that is independent of the IPSS/dynamic IPSS/dynamic IPSS-plus scoring systems.
The degree of bone marrow fibrosis and cytogenetic abnormalities configuring an unfavorable category also contribute prognostic information that is independent of these scoring systems.
With this in mind, Dr Vannucchi and his colleagues set out to develop an updated prognostic score that included molecular information (MIPSS70) and, if possible, cytogenetic information (MIPSS70-plus) for PMF patients age 70 and younger who are potential candidates for HSCT.
The researchers developed 2 prognostic models using a training/validation cohort approach.
For MIPSS70, the training cohort included 490 patients from 6 Italian institutions associated with the Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative project (AGIMM group), and the validation cohort included 211 patients from the Mayo Clinic in Rochester, Minnesota.
For MIPSS70-plus, the training cohort included 315 patients from the Mayo Clinic, and the validation cohort included 261 patients from the AGIMM group.
Using the MIPSS70 risk score in the validation cohort, the 5-year overall survival rate was 96% in low-risk, 67% in intermediate-risk, and 34% in high-risk patients.
“MIPSS70 performed better than IPSS in predicting survival,” Dr Vannucchi said. “About 30% of patients who were high-risk with MIPPS70 were missed by IPSS.”
Using the MIPSS70-plus risk score in the validation cohort, the 5-year overall survival rate was 100% in low-risk, 90% in intermediate-risk, 76% in high-risk, and 46.5% in very high-risk patients.
The MIPSS70-plus risk score also identified patients at very high risk for leukemic transformation, Dr Vannucchi said.
Furthermore, both MIPSS70 and MIPSS70-plus remained predictive of survival when the researchers evaluated patients older than 70 years of age.
“The new MIPSS70 and MIPSS70-plus scores include modern disease-associated risk variables pertinent to both pre-PMF and overt-PMF according to the 2016 WHO classification,” Dr Vannucchi said. “They integrate prognostically relevant clinical, cytogenetic, and mutation data and provide complementary systems of improved risk stratification for transplantation-age patients with PMF.”
Dr Vannucchi disclosed membership in speaker’s bureaus with Gilead, Shire, and Novartis, and research funding and membership on Board of Directors or advisory committees with Novartis. ![]()
*Data in the presentation differ from the abstract.
ATLANTA—Two novel prognostic scoring systems can help clinicians decide how to treat certain patients with primary myelofibrosis (PMF), according to a new study.
The scoring systems, which build upon the International Prognostic Scoring System (IPSS), were developed for use in PMF patients age 70 and younger who are potential candidates for hematopoietic stem cell transplant (HSCT).
One of the scoring systems—MIPSS70—integrates clinical, histologic, and molecular information. The other—MIPSS70-plus—also includes cytogenetic information.
Alessandro M. Vannucchi, MD, of the University of Florence in Italy, presented details on these prognostic scoring systems at the 2017 ASH Annual Meeting (abstract 200*).
The information was published simultaneously in the Journal of Clinical Oncology.
Dr Vannucchi noted that, in PMF, survival is currently predicted by the IPSS, the dynamic IPSS, and the dynamic IPSS-plus.
“The IPSS score is used at the time of diagnosis, while the dynamic IPSS or dynamic IPSS-plus are used to provide survival estimates at the time of patient referral,” he explained. “In clinical practice, these prognostic risk scores are mainly used for [HSCT] decision-making in younger patients.”
Driver mutations and other myeloid neoplasm-associated mutations provide prognostic information that is independent of the IPSS/dynamic IPSS/dynamic IPSS-plus scoring systems.
The degree of bone marrow fibrosis and cytogenetic abnormalities configuring an unfavorable category also contribute prognostic information that is independent of these scoring systems.
With this in mind, Dr Vannucchi and his colleagues set out to develop an updated prognostic score that included molecular information (MIPSS70) and, if possible, cytogenetic information (MIPSS70-plus) for PMF patients age 70 and younger who are potential candidates for HSCT.
The researchers developed 2 prognostic models using a training/validation cohort approach.
For MIPSS70, the training cohort included 490 patients from 6 Italian institutions associated with the Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative project (AGIMM group), and the validation cohort included 211 patients from the Mayo Clinic in Rochester, Minnesota.
For MIPSS70-plus, the training cohort included 315 patients from the Mayo Clinic, and the validation cohort included 261 patients from the AGIMM group.
Using the MIPSS70 risk score in the validation cohort, the 5-year overall survival rate was 96% in low-risk, 67% in intermediate-risk, and 34% in high-risk patients.
“MIPSS70 performed better than IPSS in predicting survival,” Dr Vannucchi said. “About 30% of patients who were high-risk with MIPPS70 were missed by IPSS.”
Using the MIPSS70-plus risk score in the validation cohort, the 5-year overall survival rate was 100% in low-risk, 90% in intermediate-risk, 76% in high-risk, and 46.5% in very high-risk patients.
The MIPSS70-plus risk score also identified patients at very high risk for leukemic transformation, Dr Vannucchi said.
Furthermore, both MIPSS70 and MIPSS70-plus remained predictive of survival when the researchers evaluated patients older than 70 years of age.
“The new MIPSS70 and MIPSS70-plus scores include modern disease-associated risk variables pertinent to both pre-PMF and overt-PMF according to the 2016 WHO classification,” Dr Vannucchi said. “They integrate prognostically relevant clinical, cytogenetic, and mutation data and provide complementary systems of improved risk stratification for transplantation-age patients with PMF.”
Dr Vannucchi disclosed membership in speaker’s bureaus with Gilead, Shire, and Novartis, and research funding and membership on Board of Directors or advisory committees with Novartis. ![]()
*Data in the presentation differ from the abstract.
Drug receives fast track, orphan designations for PTCL

The US Food and Drug Administration (FDA) has granted orphan drug and fast track designations to tenalisib (RP6530) for the treatment of peripheral T-cell lymphoma (PTCL).
Tenalisib is a dual PI3K delta/gamma inhibitor being developed by Rhizen Pharmaceuticals.
Research has shown that tenalisib inhibits the growth of immortalized cancerous cell lines and primary leukemia/lymphoma cells.
In preclinical studies, tenalisib reprogrammed macrophages from an immunosuppressive M2-like phenotype (pro-tumor) to an inflammatory M1-like state (anti-tumor).
Researchers are currently conducting a phase 1 study of tenalisib in patients with relapsed/refractory PTCL. Results from this study were presented at the 2017 ASH Annual Meeting (abstract 2791*).
The presentation included data on 50 patients—24 with PTCL and 26 with cutaneous T-cell lymphoma (CTCL).
For the PTCL patients, the median age was 63 (range, 40-89), and 67% were male. The median number of prior therapies was 3 (range, 1-7). All patients had an ECOG status of 0 (n=14) or 1 (n=10). More patients had relapsed disease (n=17, 58%) than refractory disease (n=10, 42%).
For the CTCL patients, the median age was 67 (range, 37-84), and 46% were male. The median number of prior therapies was 5.5 (range, 2-15). All patients had an ECOG status of 0 (n=23) or 1 (n=3). More patients had refractory disease (n=15, 58%) than relapsed disease (n=11, 42%).
In the dose-escalation portion of the study, patients received tenalisib at 200 mg twice daily (BID), 400 mg BID, 800 mg BID fasting, or 800 mg BID fed. The maximum tolerated dose was 800 mg BID fasting, so this dose is being used in the expansion cohort.
Twelve PTCL patients were evaluable for efficacy. The overall response rate in these patients was 58% (7/12), with a 25% complete response rate (3/12).
Sixteen CTCL patients were evaluable for efficacy. The overall response rate was 56% (9/16). All responders had partial responses.
In both PTCL and CTCL patients, treatment-related grade 3 or higher adverse events (AEs) included transaminitis (22%), rash (6%), neutropenia (6%), hypophosphatemia (2%), increased international normalized ratio (2%), diplopia secondary to neuropathy (2%), and sepsis (2%).
Treatment-related serious AEs included sepsis, increased international normalized ratio, diplopia secondary to neuropathy, and pyrexia. Five patients discontinued treatment due to AEs.
About orphan and fast track designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of new drug applications for medicines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss all aspects of development to support a drug’s approval, and also provides the opportunity to submit sections of a new drug application on a rolling basis as data become available. ![]()
*Data in the abstract differ from the presentation.
The US Food and Drug Administration (FDA) has granted orphan drug and fast track designations to tenalisib (RP6530) for the treatment of peripheral T-cell lymphoma (PTCL).
Tenalisib is a dual PI3K delta/gamma inhibitor being developed by Rhizen Pharmaceuticals.
Research has shown that tenalisib inhibits the growth of immortalized cancerous cell lines and primary leukemia/lymphoma cells.
In preclinical studies, tenalisib reprogrammed macrophages from an immunosuppressive M2-like phenotype (pro-tumor) to an inflammatory M1-like state (anti-tumor).
Researchers are currently conducting a phase 1 study of tenalisib in patients with relapsed/refractory PTCL. Results from this study were presented at the 2017 ASH Annual Meeting (abstract 2791*).
The presentation included data on 50 patients—24 with PTCL and 26 with cutaneous T-cell lymphoma (CTCL).
For the PTCL patients, the median age was 63 (range, 40-89), and 67% were male. The median number of prior therapies was 3 (range, 1-7). All patients had an ECOG status of 0 (n=14) or 1 (n=10). More patients had relapsed disease (n=17, 58%) than refractory disease (n=10, 42%).
For the CTCL patients, the median age was 67 (range, 37-84), and 46% were male. The median number of prior therapies was 5.5 (range, 2-15). All patients had an ECOG status of 0 (n=23) or 1 (n=3). More patients had refractory disease (n=15, 58%) than relapsed disease (n=11, 42%).
In the dose-escalation portion of the study, patients received tenalisib at 200 mg twice daily (BID), 400 mg BID, 800 mg BID fasting, or 800 mg BID fed. The maximum tolerated dose was 800 mg BID fasting, so this dose is being used in the expansion cohort.
Twelve PTCL patients were evaluable for efficacy. The overall response rate in these patients was 58% (7/12), with a 25% complete response rate (3/12).
Sixteen CTCL patients were evaluable for efficacy. The overall response rate was 56% (9/16). All responders had partial responses.
In both PTCL and CTCL patients, treatment-related grade 3 or higher adverse events (AEs) included transaminitis (22%), rash (6%), neutropenia (6%), hypophosphatemia (2%), increased international normalized ratio (2%), diplopia secondary to neuropathy (2%), and sepsis (2%).
Treatment-related serious AEs included sepsis, increased international normalized ratio, diplopia secondary to neuropathy, and pyrexia. Five patients discontinued treatment due to AEs.
About orphan and fast track designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of new drug applications for medicines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss all aspects of development to support a drug’s approval, and also provides the opportunity to submit sections of a new drug application on a rolling basis as data become available. ![]()
*Data in the abstract differ from the presentation.
The US Food and Drug Administration (FDA) has granted orphan drug and fast track designations to tenalisib (RP6530) for the treatment of peripheral T-cell lymphoma (PTCL).
Tenalisib is a dual PI3K delta/gamma inhibitor being developed by Rhizen Pharmaceuticals.
Research has shown that tenalisib inhibits the growth of immortalized cancerous cell lines and primary leukemia/lymphoma cells.
In preclinical studies, tenalisib reprogrammed macrophages from an immunosuppressive M2-like phenotype (pro-tumor) to an inflammatory M1-like state (anti-tumor).
Researchers are currently conducting a phase 1 study of tenalisib in patients with relapsed/refractory PTCL. Results from this study were presented at the 2017 ASH Annual Meeting (abstract 2791*).
The presentation included data on 50 patients—24 with PTCL and 26 with cutaneous T-cell lymphoma (CTCL).
For the PTCL patients, the median age was 63 (range, 40-89), and 67% were male. The median number of prior therapies was 3 (range, 1-7). All patients had an ECOG status of 0 (n=14) or 1 (n=10). More patients had relapsed disease (n=17, 58%) than refractory disease (n=10, 42%).
For the CTCL patients, the median age was 67 (range, 37-84), and 46% were male. The median number of prior therapies was 5.5 (range, 2-15). All patients had an ECOG status of 0 (n=23) or 1 (n=3). More patients had refractory disease (n=15, 58%) than relapsed disease (n=11, 42%).
In the dose-escalation portion of the study, patients received tenalisib at 200 mg twice daily (BID), 400 mg BID, 800 mg BID fasting, or 800 mg BID fed. The maximum tolerated dose was 800 mg BID fasting, so this dose is being used in the expansion cohort.
Twelve PTCL patients were evaluable for efficacy. The overall response rate in these patients was 58% (7/12), with a 25% complete response rate (3/12).
Sixteen CTCL patients were evaluable for efficacy. The overall response rate was 56% (9/16). All responders had partial responses.
In both PTCL and CTCL patients, treatment-related grade 3 or higher adverse events (AEs) included transaminitis (22%), rash (6%), neutropenia (6%), hypophosphatemia (2%), increased international normalized ratio (2%), diplopia secondary to neuropathy (2%), and sepsis (2%).
Treatment-related serious AEs included sepsis, increased international normalized ratio, diplopia secondary to neuropathy, and pyrexia. Five patients discontinued treatment due to AEs.
About orphan and fast track designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of new drug applications for medicines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss all aspects of development to support a drug’s approval, and also provides the opportunity to submit sections of a new drug application on a rolling basis as data become available. ![]()
*Data in the abstract differ from the presentation.
FDA grants orphan designation to drug for AML

The US Food and Drug Administration (FDA) has granted orphan drug designation to CG’806 for the treatment of patients with acute myeloid leukemia (AML).
CG’806 is an oral, first-in-class pan-FLT3/pan-BTK inhibitor being developed by Aptose Biosciences Inc.
In preclinical studies, CG’806 inhibited all wild-type and mutant forms of FLT3 tested, suppressed multiple oncogenic pathways operative in AML, and eliminated AML tumors (without toxicity) in murine xenograft models.
In addition, CG’806 demonstrated non-covalent inhibition of the wild-type and Cys481Ser mutant forms of the BTK enzyme, as well as other oncogenic kinases operative in B-cell malignancies.
Preclinical results with CG’806 were presented as posters at the AACR conference “Hematologic Malignancies: Translating Discoveries to Novel Therapies,” which took place last May.
“Results from non-clinical studies that we and our research collaborators have generated are promising and give reason for our eagerness to begin clinical trials in both AML and B-cell malignancies in 2018,” said William G. Rice, PhD, chairman, president, and chief executive officer at Aptose.
“We are pleased that the FDA has recognized the unique potential of CG’806 to address AML and has assigned CG’806 the status of orphan drug designation.”
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to CG’806 for the treatment of patients with acute myeloid leukemia (AML).
CG’806 is an oral, first-in-class pan-FLT3/pan-BTK inhibitor being developed by Aptose Biosciences Inc.
In preclinical studies, CG’806 inhibited all wild-type and mutant forms of FLT3 tested, suppressed multiple oncogenic pathways operative in AML, and eliminated AML tumors (without toxicity) in murine xenograft models.
In addition, CG’806 demonstrated non-covalent inhibition of the wild-type and Cys481Ser mutant forms of the BTK enzyme, as well as other oncogenic kinases operative in B-cell malignancies.
Preclinical results with CG’806 were presented as posters at the AACR conference “Hematologic Malignancies: Translating Discoveries to Novel Therapies,” which took place last May.
“Results from non-clinical studies that we and our research collaborators have generated are promising and give reason for our eagerness to begin clinical trials in both AML and B-cell malignancies in 2018,” said William G. Rice, PhD, chairman, president, and chief executive officer at Aptose.
“We are pleased that the FDA has recognized the unique potential of CG’806 to address AML and has assigned CG’806 the status of orphan drug designation.”
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to CG’806 for the treatment of patients with acute myeloid leukemia (AML).
CG’806 is an oral, first-in-class pan-FLT3/pan-BTK inhibitor being developed by Aptose Biosciences Inc.
In preclinical studies, CG’806 inhibited all wild-type and mutant forms of FLT3 tested, suppressed multiple oncogenic pathways operative in AML, and eliminated AML tumors (without toxicity) in murine xenograft models.
In addition, CG’806 demonstrated non-covalent inhibition of the wild-type and Cys481Ser mutant forms of the BTK enzyme, as well as other oncogenic kinases operative in B-cell malignancies.
Preclinical results with CG’806 were presented as posters at the AACR conference “Hematologic Malignancies: Translating Discoveries to Novel Therapies,” which took place last May.
“Results from non-clinical studies that we and our research collaborators have generated are promising and give reason for our eagerness to begin clinical trials in both AML and B-cell malignancies in 2018,” said William G. Rice, PhD, chairman, president, and chief executive officer at Aptose.
“We are pleased that the FDA has recognized the unique potential of CG’806 to address AML and has assigned CG’806 the status of orphan drug designation.”
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
bb2121 induces durable, deepening responses in MM patients

ATLANTA—Updated results from a phase 1 trial have shown that bb2121, a chimeric antigen receptor (CAR) T-cell product, can induce durable, deepening responses in patients with relapsed/refractory multiple myeloma (MM).
Responses continue to improve from very good partial responses to complete responses (CRs), even 15 months after infusion.
In 5 months, the CR rate increased from 27% to 56%, and ongoing responses have now surpassed 1 year.
The overall response rate (ORR) stands at 94%, and the median progression-free survival (PFS) has not been reached with a follow-up of 40 weeks.
bb2121 is a second-generation CAR T-cell product that targets the B-cell maturation antigen (BCMA).
BCMA is expressed nearly universally on MM cells, and its expression is largely restricted to plasma cells and some mature B cells, making it “an attractive target for immunotherapies,” said James N. Kochenderfer, MD, of the National Cancer Institute/National Institutes of Health in Bethesda, Maryland.
Dr Kochenderfer reported results from the phase 1 study of bb2121 (NCT02658929) at the 2017 ASH Annual Meeting (abstract 740*).
Study sponsors and collaborators were bluebird bio and Celgene Corporation. Dr Kochenderfer disclosed that he has multiple patents in the CAR field and has received research funding from bluebird bio and Kite Pharma.
Study design
Patients with relapsed or refractory MM who had 3 or more prior lines of therapy, including a proteasome inhibitor and immunomodulatory drug, or who were double refractory were eligible for the dose-escalation cohort of the study. They had to have measurable disease, 50% or more BCMA expression, and adequate bone marrow, renal, and hepatic function.
BCMA expression was not required for the dose-expansion cohort. For this cohort, patients must have received daratumumab and have been refractory to their last line of therapy.
The dose-escalation cohort was a standard 3 + 3 design and included CAR T-cell doses of 50 x 106, 150 x 106, 450 x 106, and 800 x 106.
Patients were screened, underwent leukapheresis, and waited for the manufacture of their CAR T cells. One centralized manufacturing site produced the T-cell products for the 9 US clinical study sites.
“We had a manufacturing success rate of 100%,” Dr Kochenderfer noted, and the manufacturing took 10 days.
Five days prior to bb2121 infusion, patients received lymphodepletion with fludarabine (30 mg/m2) and cyclophosphamide (300 mg/m2).
Patient characteristics
Investigators dosed 21 patients as of the data cut-off of October 2.
Their median age was 58 (range, 37–74), 62% were male, and they had a median time since diagnosis of 4 years.
All patients had an ECOG performance status of 0 or 1, and 43% had high-risk cytogenetics, defined as del17p, t(4;14), and t(14;16).
“One of the most impressive things about our study was how heavily pretreated the patients were,” Dr Kochenderfer noted. “These patients had a median of 7 prior lines of therapy, and 100% of the patients had a prior autologous stem cell transplant.”
All patients were exposed to bortezomib and lenalidomide, and 67% and 86%, respectively, were refractory to those agents. Patients were also exposed to carfilzomib (91%), pomalidomide (91%), and daratumumab (71%) and had varying degrees of refractoriness to those agents.
Safety
“In general, the treatment was very well tolerated,” Dr Kochenderfer said. “[It was] well tolerated compared with other T-cell products I’ve had experience with.”
The investigators observed no dose-limiting toxicities.
Cytokine release syndrome (CRS) of all grades occurred in 15 (71%) patients, and grade 3 or higher CRS occurred in 2 (10%) patients. The latter resolved within 24 hours.
Five (24%) patients experienced neurotoxicity, none grade 3 or higher.
Dr Kochenderfer described 1 case of delayed-onset, grade 4, reversible neurotoxicity that was associated with tumor lysis syndrome (TLS) and CRS.
The patient had no toxicity until day 10. By day 12, magnetic resonance imaging showed cerebral edema.
The patient was transferred to the intensive care unit and required intubation. She was treated with high-dose methylprednisolone and tocilizumab. She also received hemodialysis for TLS.
“By day 17, she dramatically improved,” Dr Kochenderfer said.
Her mental status cleared, TLS resolved, she was extubated, and she was doing much better, he reported.
On day 30, the patient was out of the intensive care unit.
“So the whole course was fairly brief,” Dr Kochenderfer said. “And, today, she’s doing well. She’s actually asymptomatic.”
Cytopenias—neutropenia, thrombocytopenia, and anemia—were primarily related to the lymphodepleting drugs, and patients recovered to grade 3 or lower by month 2 after the infusion.
Fourteen patients experienced 1 or more serious adverse events. Four had grade 1-2 CRS that required hospitalization per protocol, and 2 had pyrexia.
Five patients died, 3 due to disease progression, all who received treatment at the lowest dose.
Two patients treated at active doses were in CR when they died. One had a cardiac arrest, and the other had myelodysplastic syndrome following discontinuation.
Efficacy
In addition to the high ORR (94%) and CR rate (56%) in this study, 9 of 10 patients evaluated for minimal residual disease were negative.
The median time to first response was 1.02 months, and median time to best response was 3.74 months. The median time to CR was 3.84 months.
The median duration of response and PFS have not been reached. The PFS rate was 81% at 6 months and 71% at 9 months.
“We found that all the doses between 150 million and 450 million were effective,” Dr Kochenderfer noted. “We didn’t see a clear difference in efficacy between those doses, so we’ve chosen to use the 150 – 300 million dose range for the follow-up study.”
The investigators observed robust expansion of bb2121, which peaked in the first week after the infusion. Six of 13 patients had evident CAR T cells at 6 months. One patient has persistence over 12 months.
The investigators also observed a robust decrease in M protein and rapid clearance of serum-free light chains and serum BCMA. They noted that the activity of the CAR-positive T cells was not inhibited by high baseline serum BCMA.
Four patients progressed. The investigators analyzed the patients’ tumor burden, bb2121 dose, best response, time to progression, BCMA expression, grades of CRS, and bb2121 persistence. And progression was independent of these factors.
“So we can’t pick out a very good factor of why they progressed,” Dr Kochenderfer said.
However, he noted that the patients are eligible for re-treatment.
Investigators have opened a global trial of bb2121 (NCT03361748) given at doses ranging from 150 – 300 million CAR T cells.
The US Food and Drug Administration and the European Medicines Agency recently fast-tracked bb2121. ![]()
*Data presented differ from the abstract.
ATLANTA—Updated results from a phase 1 trial have shown that bb2121, a chimeric antigen receptor (CAR) T-cell product, can induce durable, deepening responses in patients with relapsed/refractory multiple myeloma (MM).
Responses continue to improve from very good partial responses to complete responses (CRs), even 15 months after infusion.
In 5 months, the CR rate increased from 27% to 56%, and ongoing responses have now surpassed 1 year.
The overall response rate (ORR) stands at 94%, and the median progression-free survival (PFS) has not been reached with a follow-up of 40 weeks.
bb2121 is a second-generation CAR T-cell product that targets the B-cell maturation antigen (BCMA).
BCMA is expressed nearly universally on MM cells, and its expression is largely restricted to plasma cells and some mature B cells, making it “an attractive target for immunotherapies,” said James N. Kochenderfer, MD, of the National Cancer Institute/National Institutes of Health in Bethesda, Maryland.
Dr Kochenderfer reported results from the phase 1 study of bb2121 (NCT02658929) at the 2017 ASH Annual Meeting (abstract 740*).
Study sponsors and collaborators were bluebird bio and Celgene Corporation. Dr Kochenderfer disclosed that he has multiple patents in the CAR field and has received research funding from bluebird bio and Kite Pharma.
Study design
Patients with relapsed or refractory MM who had 3 or more prior lines of therapy, including a proteasome inhibitor and immunomodulatory drug, or who were double refractory were eligible for the dose-escalation cohort of the study. They had to have measurable disease, 50% or more BCMA expression, and adequate bone marrow, renal, and hepatic function.
BCMA expression was not required for the dose-expansion cohort. For this cohort, patients must have received daratumumab and have been refractory to their last line of therapy.
The dose-escalation cohort was a standard 3 + 3 design and included CAR T-cell doses of 50 x 106, 150 x 106, 450 x 106, and 800 x 106.
Patients were screened, underwent leukapheresis, and waited for the manufacture of their CAR T cells. One centralized manufacturing site produced the T-cell products for the 9 US clinical study sites.
“We had a manufacturing success rate of 100%,” Dr Kochenderfer noted, and the manufacturing took 10 days.
Five days prior to bb2121 infusion, patients received lymphodepletion with fludarabine (30 mg/m2) and cyclophosphamide (300 mg/m2).
Patient characteristics
Investigators dosed 21 patients as of the data cut-off of October 2.
Their median age was 58 (range, 37–74), 62% were male, and they had a median time since diagnosis of 4 years.
All patients had an ECOG performance status of 0 or 1, and 43% had high-risk cytogenetics, defined as del17p, t(4;14), and t(14;16).
“One of the most impressive things about our study was how heavily pretreated the patients were,” Dr Kochenderfer noted. “These patients had a median of 7 prior lines of therapy, and 100% of the patients had a prior autologous stem cell transplant.”
All patients were exposed to bortezomib and lenalidomide, and 67% and 86%, respectively, were refractory to those agents. Patients were also exposed to carfilzomib (91%), pomalidomide (91%), and daratumumab (71%) and had varying degrees of refractoriness to those agents.
Safety
“In general, the treatment was very well tolerated,” Dr Kochenderfer said. “[It was] well tolerated compared with other T-cell products I’ve had experience with.”
The investigators observed no dose-limiting toxicities.
Cytokine release syndrome (CRS) of all grades occurred in 15 (71%) patients, and grade 3 or higher CRS occurred in 2 (10%) patients. The latter resolved within 24 hours.
Five (24%) patients experienced neurotoxicity, none grade 3 or higher.
Dr Kochenderfer described 1 case of delayed-onset, grade 4, reversible neurotoxicity that was associated with tumor lysis syndrome (TLS) and CRS.
The patient had no toxicity until day 10. By day 12, magnetic resonance imaging showed cerebral edema.
The patient was transferred to the intensive care unit and required intubation. She was treated with high-dose methylprednisolone and tocilizumab. She also received hemodialysis for TLS.
“By day 17, she dramatically improved,” Dr Kochenderfer said.
Her mental status cleared, TLS resolved, she was extubated, and she was doing much better, he reported.
On day 30, the patient was out of the intensive care unit.
“So the whole course was fairly brief,” Dr Kochenderfer said. “And, today, she’s doing well. She’s actually asymptomatic.”
Cytopenias—neutropenia, thrombocytopenia, and anemia—were primarily related to the lymphodepleting drugs, and patients recovered to grade 3 or lower by month 2 after the infusion.
Fourteen patients experienced 1 or more serious adverse events. Four had grade 1-2 CRS that required hospitalization per protocol, and 2 had pyrexia.
Five patients died, 3 due to disease progression, all who received treatment at the lowest dose.
Two patients treated at active doses were in CR when they died. One had a cardiac arrest, and the other had myelodysplastic syndrome following discontinuation.
Efficacy
In addition to the high ORR (94%) and CR rate (56%) in this study, 9 of 10 patients evaluated for minimal residual disease were negative.
The median time to first response was 1.02 months, and median time to best response was 3.74 months. The median time to CR was 3.84 months.
The median duration of response and PFS have not been reached. The PFS rate was 81% at 6 months and 71% at 9 months.
“We found that all the doses between 150 million and 450 million were effective,” Dr Kochenderfer noted. “We didn’t see a clear difference in efficacy between those doses, so we’ve chosen to use the 150 – 300 million dose range for the follow-up study.”
The investigators observed robust expansion of bb2121, which peaked in the first week after the infusion. Six of 13 patients had evident CAR T cells at 6 months. One patient has persistence over 12 months.
The investigators also observed a robust decrease in M protein and rapid clearance of serum-free light chains and serum BCMA. They noted that the activity of the CAR-positive T cells was not inhibited by high baseline serum BCMA.
Four patients progressed. The investigators analyzed the patients’ tumor burden, bb2121 dose, best response, time to progression, BCMA expression, grades of CRS, and bb2121 persistence. And progression was independent of these factors.
“So we can’t pick out a very good factor of why they progressed,” Dr Kochenderfer said.
However, he noted that the patients are eligible for re-treatment.
Investigators have opened a global trial of bb2121 (NCT03361748) given at doses ranging from 150 – 300 million CAR T cells.
The US Food and Drug Administration and the European Medicines Agency recently fast-tracked bb2121. ![]()
*Data presented differ from the abstract.
ATLANTA—Updated results from a phase 1 trial have shown that bb2121, a chimeric antigen receptor (CAR) T-cell product, can induce durable, deepening responses in patients with relapsed/refractory multiple myeloma (MM).
Responses continue to improve from very good partial responses to complete responses (CRs), even 15 months after infusion.
In 5 months, the CR rate increased from 27% to 56%, and ongoing responses have now surpassed 1 year.
The overall response rate (ORR) stands at 94%, and the median progression-free survival (PFS) has not been reached with a follow-up of 40 weeks.
bb2121 is a second-generation CAR T-cell product that targets the B-cell maturation antigen (BCMA).
BCMA is expressed nearly universally on MM cells, and its expression is largely restricted to plasma cells and some mature B cells, making it “an attractive target for immunotherapies,” said James N. Kochenderfer, MD, of the National Cancer Institute/National Institutes of Health in Bethesda, Maryland.
Dr Kochenderfer reported results from the phase 1 study of bb2121 (NCT02658929) at the 2017 ASH Annual Meeting (abstract 740*).
Study sponsors and collaborators were bluebird bio and Celgene Corporation. Dr Kochenderfer disclosed that he has multiple patents in the CAR field and has received research funding from bluebird bio and Kite Pharma.
Study design
Patients with relapsed or refractory MM who had 3 or more prior lines of therapy, including a proteasome inhibitor and immunomodulatory drug, or who were double refractory were eligible for the dose-escalation cohort of the study. They had to have measurable disease, 50% or more BCMA expression, and adequate bone marrow, renal, and hepatic function.
BCMA expression was not required for the dose-expansion cohort. For this cohort, patients must have received daratumumab and have been refractory to their last line of therapy.
The dose-escalation cohort was a standard 3 + 3 design and included CAR T-cell doses of 50 x 106, 150 x 106, 450 x 106, and 800 x 106.
Patients were screened, underwent leukapheresis, and waited for the manufacture of their CAR T cells. One centralized manufacturing site produced the T-cell products for the 9 US clinical study sites.
“We had a manufacturing success rate of 100%,” Dr Kochenderfer noted, and the manufacturing took 10 days.
Five days prior to bb2121 infusion, patients received lymphodepletion with fludarabine (30 mg/m2) and cyclophosphamide (300 mg/m2).
Patient characteristics
Investigators dosed 21 patients as of the data cut-off of October 2.
Their median age was 58 (range, 37–74), 62% were male, and they had a median time since diagnosis of 4 years.
All patients had an ECOG performance status of 0 or 1, and 43% had high-risk cytogenetics, defined as del17p, t(4;14), and t(14;16).
“One of the most impressive things about our study was how heavily pretreated the patients were,” Dr Kochenderfer noted. “These patients had a median of 7 prior lines of therapy, and 100% of the patients had a prior autologous stem cell transplant.”
All patients were exposed to bortezomib and lenalidomide, and 67% and 86%, respectively, were refractory to those agents. Patients were also exposed to carfilzomib (91%), pomalidomide (91%), and daratumumab (71%) and had varying degrees of refractoriness to those agents.
Safety
“In general, the treatment was very well tolerated,” Dr Kochenderfer said. “[It was] well tolerated compared with other T-cell products I’ve had experience with.”
The investigators observed no dose-limiting toxicities.
Cytokine release syndrome (CRS) of all grades occurred in 15 (71%) patients, and grade 3 or higher CRS occurred in 2 (10%) patients. The latter resolved within 24 hours.
Five (24%) patients experienced neurotoxicity, none grade 3 or higher.
Dr Kochenderfer described 1 case of delayed-onset, grade 4, reversible neurotoxicity that was associated with tumor lysis syndrome (TLS) and CRS.
The patient had no toxicity until day 10. By day 12, magnetic resonance imaging showed cerebral edema.
The patient was transferred to the intensive care unit and required intubation. She was treated with high-dose methylprednisolone and tocilizumab. She also received hemodialysis for TLS.
“By day 17, she dramatically improved,” Dr Kochenderfer said.
Her mental status cleared, TLS resolved, she was extubated, and she was doing much better, he reported.
On day 30, the patient was out of the intensive care unit.
“So the whole course was fairly brief,” Dr Kochenderfer said. “And, today, she’s doing well. She’s actually asymptomatic.”
Cytopenias—neutropenia, thrombocytopenia, and anemia—were primarily related to the lymphodepleting drugs, and patients recovered to grade 3 or lower by month 2 after the infusion.
Fourteen patients experienced 1 or more serious adverse events. Four had grade 1-2 CRS that required hospitalization per protocol, and 2 had pyrexia.
Five patients died, 3 due to disease progression, all who received treatment at the lowest dose.
Two patients treated at active doses were in CR when they died. One had a cardiac arrest, and the other had myelodysplastic syndrome following discontinuation.
Efficacy
In addition to the high ORR (94%) and CR rate (56%) in this study, 9 of 10 patients evaluated for minimal residual disease were negative.
The median time to first response was 1.02 months, and median time to best response was 3.74 months. The median time to CR was 3.84 months.
The median duration of response and PFS have not been reached. The PFS rate was 81% at 6 months and 71% at 9 months.
“We found that all the doses between 150 million and 450 million were effective,” Dr Kochenderfer noted. “We didn’t see a clear difference in efficacy between those doses, so we’ve chosen to use the 150 – 300 million dose range for the follow-up study.”
The investigators observed robust expansion of bb2121, which peaked in the first week after the infusion. Six of 13 patients had evident CAR T cells at 6 months. One patient has persistence over 12 months.
The investigators also observed a robust decrease in M protein and rapid clearance of serum-free light chains and serum BCMA. They noted that the activity of the CAR-positive T cells was not inhibited by high baseline serum BCMA.
Four patients progressed. The investigators analyzed the patients’ tumor burden, bb2121 dose, best response, time to progression, BCMA expression, grades of CRS, and bb2121 persistence. And progression was independent of these factors.
“So we can’t pick out a very good factor of why they progressed,” Dr Kochenderfer said.
However, he noted that the patients are eligible for re-treatment.
Investigators have opened a global trial of bb2121 (NCT03361748) given at doses ranging from 150 – 300 million CAR T cells.
The US Food and Drug Administration and the European Medicines Agency recently fast-tracked bb2121.
*Data presented differ from the abstract.
Drug approved for kids with sickle cell anemia

The US Food and Drug Administration (FDA) has approved a hydroxyurea product (Addmedica’s Siklos) for use in pediatric patients with sickle cell anemia.
Siklos is intended to reduce the frequency of painful crises and the need for blood transfusions in pediatric patients age 2 and older who have sickle cell anemia and recurrent moderate to severe painful crises.
The recommended dose of Siklos is 20 mg/kg once daily.
The FDA granted priority review and orphan drug designation to the application for Siklos.
The agency’s approval of Siklos was based on data from the ESCORT HU study (NCT02516579). The trial was an evaluation of Siklos in 405 patients, ages 2 to 18, with sickle cell disease (SCD).
Thirty-five percent of these patients (n=141) had not received hydroxyurea prior to study enrollment and were therefore evaluable for efficacy. The median follow-up was 23 months (range, 12 to 80 months).
The researchers found that Siklos prompted an increase in fetal hemoglobin. Median fetal hemoglobin percentages were 5.6% (range, 1.3 to 15.0) at baseline and 12.8% (range, 2.1 to 37.2) at around 6 months after Siklos initiation (the value closest to 6 months collected between 5 and 14 months).
In addition, the percentage of patients with at least 1 vaso-occlusive episode, 1 episode of acute chest syndrome, 1 hospitalization due to SCD, or 1 blood transfusion decreased after 12 months of Siklos treatment.
The proportion of patients with at least 1 vaso-occlusive episode was 69.2% at baseline and 42.5% at 12 months. The proportion with at least 1 episode of acute chest syndrome was 23.6% and 5.7%, respectively.
The proportion with at least 1 hospitalization due to SCD was 75.5% and 41.8%, respectively. And the proportion with at least 1 blood transfusion was 45.9% and 23.0%, respectively.
The most common adverse events (occurring in at least 10% of patients) were infections (39.8%), gastrointestinal disorders (13.1%), neutropenia (12.6%), nervous system disorders (11.1%), and metabolic and nutrition disorders (10.9%).
Full prescribing information for Siklos is available on the FDA website.
The US Food and Drug Administration (FDA) has approved a hydroxyurea product (Addmedica’s Siklos) for use in pediatric patients with sickle cell anemia.
Siklos is intended to reduce the frequency of painful crises and the need for blood transfusions in pediatric patients age 2 and older who have sickle cell anemia and recurrent moderate to severe painful crises.
The recommended dose of Siklos is 20 mg/kg once daily.
The FDA granted priority review and orphan drug designation to the application for Siklos.
The agency’s approval of Siklos was based on data from the ESCORT HU study (NCT02516579). The trial was an evaluation of Siklos in 405 patients, ages 2 to 18, with sickle cell disease (SCD).
Thirty-five percent of these patients (n=141) had not received hydroxyurea prior to study enrollment and were therefore evaluable for efficacy. The median follow-up was 23 months (range, 12 to 80 months).
The researchers found that Siklos prompted an increase in fetal hemoglobin. Median fetal hemoglobin percentages were 5.6% (range, 1.3 to 15.0) at baseline and 12.8% (range, 2.1 to 37.2) at around 6 months after Siklos initiation (the value closest to 6 months collected between 5 and 14 months).
In addition, the percentage of patients with at least 1 vaso-occlusive episode, 1 episode of acute chest syndrome, 1 hospitalization due to SCD, or 1 blood transfusion decreased after 12 months of Siklos treatment.
The proportion of patients with at least 1 vaso-occlusive episode was 69.2% at baseline and 42.5% at 12 months. The proportion with at least 1 episode of acute chest syndrome was 23.6% and 5.7%, respectively.
The proportion with at least 1 hospitalization due to SCD was 75.5% and 41.8%, respectively. And the proportion with at least 1 blood transfusion was 45.9% and 23.0%, respectively.
The most common adverse events (occurring in at least 10% of patients) were infections (39.8%), gastrointestinal disorders (13.1%), neutropenia (12.6%), nervous system disorders (11.1%), and metabolic and nutrition disorders (10.9%).
Full prescribing information for Siklos is available on the FDA website.
The US Food and Drug Administration (FDA) has approved a hydroxyurea product (Addmedica’s Siklos) for use in pediatric patients with sickle cell anemia.
Siklos is intended to reduce the frequency of painful crises and the need for blood transfusions in pediatric patients age 2 and older who have sickle cell anemia and recurrent moderate to severe painful crises.
The recommended dose of Siklos is 20 mg/kg once daily.
The FDA granted priority review and orphan drug designation to the application for Siklos.
The agency’s approval of Siklos was based on data from the ESCORT HU study (NCT02516579). The trial was an evaluation of Siklos in 405 patients, ages 2 to 18, with sickle cell disease (SCD).
Thirty-five percent of these patients (n=141) had not received hydroxyurea prior to study enrollment and were therefore evaluable for efficacy. The median follow-up was 23 months (range, 12 to 80 months).
The researchers found that Siklos prompted an increase in fetal hemoglobin. Median fetal hemoglobin percentages were 5.6% (range, 1.3 to 15.0) at baseline and 12.8% (range, 2.1 to 37.2) at around 6 months after Siklos initiation (the value closest to 6 months collected between 5 and 14 months).
In addition, the percentage of patients with at least 1 vaso-occlusive episode, 1 episode of acute chest syndrome, 1 hospitalization due to SCD, or 1 blood transfusion decreased after 12 months of Siklos treatment.
The proportion of patients with at least 1 vaso-occlusive episode was 69.2% at baseline and 42.5% at 12 months. The proportion with at least 1 episode of acute chest syndrome was 23.6% and 5.7%, respectively.
The proportion with at least 1 hospitalization due to SCD was 75.5% and 41.8%, respectively. And the proportion with at least 1 blood transfusion was 45.9% and 23.0%, respectively.
The most common adverse events (occurring in at least 10% of patients) were infections (39.8%), gastrointestinal disorders (13.1%), neutropenia (12.6%), nervous system disorders (11.1%), and metabolic and nutrition disorders (10.9%).
Full prescribing information for Siklos is available on the FDA website.
Nilotinib label updated with info on discontinuation

The US Food and Drug Administration (FDA) has approved an update to the product label for nilotinib (Tasigna) that includes information about how to discontinue the drug in certain patients.
Nilotinib, which was first approved by the FDA in 2007, is indicated for the treatment of patients with Philadelphia chromosome positive (Ph+) chronic myeloid leukemia (CML).
The updated prescribing information for the drug now outlines which of these patients may be eligible to stop receiving nilotinib.
Patients with chronic phase, Ph+ CML who have been taking nilotinib for 3 years or more (as first-line treatment or after failure with imatinib) and have achieved a sustained deep molecular response may be eligible to stop treatment.
Patients must have maintained a molecular response of at least MR4.0 for 1 year prior to discontinuation and achieved an MR4.5 at their last assessment.
Patients must have typical BCR-ABL transcripts (e13a2/b2a2 or e14a2/b3a2), no history of accelerated phase or blast crisis, and no past attempts at treatment discontinuation that resulted in relapse.
After discontinuation, patients must be monitored for possible loss of major molecular response (MMR). BCR-ABL transcript levels must be assessed and a complete blood count with differential performed monthly for 1 year, then every 6 weeks for the second year, and every 12 weeks thereafter.
BCR-ABL transcript levels should be measured using the MolecularMD MRDxTM BCR-ABL test, an FDA-authorized companion diagnostic validated to measure down to MR4.5.
If patients lose MMR, they must restart nilotinib within 4 weeks and receive the dose level they were receiving prior to discontinuation.
BCR-ABL transcript levels should be monitored every 2 weeks until they are lower than MMR for 4 consecutive measurements. Patients can then return to the original monitoring schedule.
The update of nilotinib’s label to include this information was based on results from 2 single-arm trials—ENESTfreedom and ENESTop.
ENESTfreedom
This phase 2 trial included 215 patients with Ph+, chronic phase CML. Researchers evaluated stopping treatment in 190 patients who had achieved a response of MR4.5 with nilotinib as first-line treatment. The patients had sustained deep molecular response for 1 year prior to treatment discontinuation.
Ninety-six weeks after stopping treatment, 48.9% of patients were still in MMR, also known as treatment-free remission (TFR).
Of the 88 patients who restarted nilotinib due to loss of MMR by the cut-off date, 98.9% were able to regain MMR (n=87). One patient discontinued the study at 7.1 weeks without regaining MMR after reinitiating treatment with nilotinib. Eighty-one patients (92.0%) regained MR4.5 by the cut-off date.
Common adverse events (AEs) in patients who discontinued nilotinib included musculoskeletal symptoms such as body aches, bone pain, and pain in extremities. However, these AEs decreased over time.
The incidence of musculoskeletal pain-related AEs decreased from 34.0% to 9.0% during the first and second 48 weeks of the TFR phase, respectively. In comparison, the incidence of such AEs was 17.0% during the treatment consolidation phase.
ENESTop
This phase 2 trial included 163 patients with Ph+, chronic phase CML. Researchers evaluated stopping treatment in 126 patients who had previously received imatinib, then switched to nilotinib and sustained a molecular response for 1 year prior to stopping nilotinib.
At 96 weeks, 53.2% of patients were still in TFR.
Fifty-six patients with confirmed loss of MR4.0 or loss of MMR restarted nilotinib by the cut-off date. Of these patients, 92.9% (n=52) regained both MR4.0 and MR4.5.
As in ENESTfreedom, patients who discontinued nilotinib had musculoskeletal symptoms that decreased over time.
The incidence of musculoskeletal pain-related AEs decreased from 47.9% to 15.1% during the first and second 48 weeks of the TFR phase, respectively. In comparison, the incidence of such AEs was 13.7% during the treatment consolidation phase.
Additional data from ENESTop and ENESTfreedom, as well as the recommendations for stopping nilotinib, are included in the updated prescribing information, which is available at https://www.us.tasigna.com/.
The US Food and Drug Administration (FDA) has approved an update to the product label for nilotinib (Tasigna) that includes information about how to discontinue the drug in certain patients.
Nilotinib, which was first approved by the FDA in 2007, is indicated for the treatment of patients with Philadelphia chromosome positive (Ph+) chronic myeloid leukemia (CML).
The updated prescribing information for the drug now outlines which of these patients may be eligible to stop receiving nilotinib.
Patients with chronic phase, Ph+ CML who have been taking nilotinib for 3 years or more (as first-line treatment or after failure with imatinib) and have achieved a sustained deep molecular response may be eligible to stop treatment.
Patients must have maintained a molecular response of at least MR4.0 for 1 year prior to discontinuation and achieved an MR4.5 at their last assessment.
Patients must have typical BCR-ABL transcripts (e13a2/b2a2 or e14a2/b3a2), no history of accelerated phase or blast crisis, and no past attempts at treatment discontinuation that resulted in relapse.
After discontinuation, patients must be monitored for possible loss of major molecular response (MMR). BCR-ABL transcript levels must be assessed and a complete blood count with differential performed monthly for 1 year, then every 6 weeks for the second year, and every 12 weeks thereafter.
BCR-ABL transcript levels should be measured using the MolecularMD MRDxTM BCR-ABL test, an FDA-authorized companion diagnostic validated to measure down to MR4.5.
If patients lose MMR, they must restart nilotinib within 4 weeks and receive the dose level they were receiving prior to discontinuation.
BCR-ABL transcript levels should be monitored every 2 weeks until they are lower than MMR for 4 consecutive measurements. Patients can then return to the original monitoring schedule.
The update of nilotinib’s label to include this information was based on results from 2 single-arm trials—ENESTfreedom and ENESTop.
ENESTfreedom
This phase 2 trial included 215 patients with Ph+, chronic phase CML. Researchers evaluated stopping treatment in 190 patients who had achieved a response of MR4.5 with nilotinib as first-line treatment. The patients had sustained deep molecular response for 1 year prior to treatment discontinuation.
Ninety-six weeks after stopping treatment, 48.9% of patients were still in MMR, also known as treatment-free remission (TFR).
Of the 88 patients who restarted nilotinib due to loss of MMR by the cut-off date, 98.9% were able to regain MMR (n=87). One patient discontinued the study at 7.1 weeks without regaining MMR after reinitiating treatment with nilotinib. Eighty-one patients (92.0%) regained MR4.5 by the cut-off date.
Common adverse events (AEs) in patients who discontinued nilotinib included musculoskeletal symptoms such as body aches, bone pain, and pain in extremities. However, these AEs decreased over time.
The incidence of musculoskeletal pain-related AEs decreased from 34.0% to 9.0% during the first and second 48 weeks of the TFR phase, respectively. In comparison, the incidence of such AEs was 17.0% during the treatment consolidation phase.
ENESTop
This phase 2 trial included 163 patients with Ph+, chronic phase CML. Researchers evaluated stopping treatment in 126 patients who had previously received imatinib, then switched to nilotinib and sustained a molecular response for 1 year prior to stopping nilotinib.
At 96 weeks, 53.2% of patients were still in TFR.
Fifty-six patients with confirmed loss of MR4.0 or loss of MMR restarted nilotinib by the cut-off date. Of these patients, 92.9% (n=52) regained both MR4.0 and MR4.5.
As in ENESTfreedom, patients who discontinued nilotinib had musculoskeletal symptoms that decreased over time.
The incidence of musculoskeletal pain-related AEs decreased from 47.9% to 15.1% during the first and second 48 weeks of the TFR phase, respectively. In comparison, the incidence of such AEs was 13.7% during the treatment consolidation phase.
Additional data from ENESTop and ENESTfreedom, as well as the recommendations for stopping nilotinib, are included in the updated prescribing information, which is available at https://www.us.tasigna.com/.
The US Food and Drug Administration (FDA) has approved an update to the product label for nilotinib (Tasigna) that includes information about how to discontinue the drug in certain patients.
Nilotinib, which was first approved by the FDA in 2007, is indicated for the treatment of patients with Philadelphia chromosome positive (Ph+) chronic myeloid leukemia (CML).
The updated prescribing information for the drug now outlines which of these patients may be eligible to stop receiving nilotinib.
Patients with chronic phase, Ph+ CML who have been taking nilotinib for 3 years or more (as first-line treatment or after failure with imatinib) and have achieved a sustained deep molecular response may be eligible to stop treatment.
Patients must have maintained a molecular response of at least MR4.0 for 1 year prior to discontinuation and achieved an MR4.5 at their last assessment.
Patients must have typical BCR-ABL transcripts (e13a2/b2a2 or e14a2/b3a2), no history of accelerated phase or blast crisis, and no past attempts at treatment discontinuation that resulted in relapse.
After discontinuation, patients must be monitored for possible loss of major molecular response (MMR). BCR-ABL transcript levels must be assessed and a complete blood count with differential performed monthly for 1 year, then every 6 weeks for the second year, and every 12 weeks thereafter.
BCR-ABL transcript levels should be measured using the MolecularMD MRDxTM BCR-ABL test, an FDA-authorized companion diagnostic validated to measure down to MR4.5.
If patients lose MMR, they must restart nilotinib within 4 weeks and receive the dose level they were receiving prior to discontinuation.
BCR-ABL transcript levels should be monitored every 2 weeks until they are lower than MMR for 4 consecutive measurements. Patients can then return to the original monitoring schedule.
The update of nilotinib’s label to include this information was based on results from 2 single-arm trials—ENESTfreedom and ENESTop.
ENESTfreedom
This phase 2 trial included 215 patients with Ph+, chronic phase CML. Researchers evaluated stopping treatment in 190 patients who had achieved a response of MR4.5 with nilotinib as first-line treatment. The patients had sustained deep molecular response for 1 year prior to treatment discontinuation.
Ninety-six weeks after stopping treatment, 48.9% of patients were still in MMR, also known as treatment-free remission (TFR).
Of the 88 patients who restarted nilotinib due to loss of MMR by the cut-off date, 98.9% were able to regain MMR (n=87). One patient discontinued the study at 7.1 weeks without regaining MMR after reinitiating treatment with nilotinib. Eighty-one patients (92.0%) regained MR4.5 by the cut-off date.
Common adverse events (AEs) in patients who discontinued nilotinib included musculoskeletal symptoms such as body aches, bone pain, and pain in extremities. However, these AEs decreased over time.
The incidence of musculoskeletal pain-related AEs decreased from 34.0% to 9.0% during the first and second 48 weeks of the TFR phase, respectively. In comparison, the incidence of such AEs was 17.0% during the treatment consolidation phase.
ENESTop
This phase 2 trial included 163 patients with Ph+, chronic phase CML. Researchers evaluated stopping treatment in 126 patients who had previously received imatinib, then switched to nilotinib and sustained a molecular response for 1 year prior to stopping nilotinib.
At 96 weeks, 53.2% of patients were still in TFR.
Fifty-six patients with confirmed loss of MR4.0 or loss of MMR restarted nilotinib by the cut-off date. Of these patients, 92.9% (n=52) regained both MR4.0 and MR4.5.
As in ENESTfreedom, patients who discontinued nilotinib had musculoskeletal symptoms that decreased over time.
The incidence of musculoskeletal pain-related AEs decreased from 47.9% to 15.1% during the first and second 48 weeks of the TFR phase, respectively. In comparison, the incidence of such AEs was 13.7% during the treatment consolidation phase.
Additional data from ENESTop and ENESTfreedom, as well as the recommendations for stopping nilotinib, are included in the updated prescribing information, which is available at https://www.us.tasigna.com/.
Chemo-free combo should be option for rel/ref CLL, doc says

ATLANTA—The combination of venetoclax and rituximab (VR) should be a standard treatment option for adults with relapsed/refractory chronic lymphocytic leukemia (CLL), according to a speaker at the 2017 ASH Annual Meeting.
Data from the phase 3 MURANO study showed that patients with relapsed/refractory CLL who received VR had significantly longer progression-free survival (PFS) than those who received bendamustine and rituximab (BR).
In addition, “secondary endpoints were consistently in favor of venetoclax-rituximab,” said study investigator John F. Seymour, MBBS, PhD, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
Adverse events (AEs) were largely consistent with the known safety profiles of the drugs studied, but tumor lysis syndrome (TLS) was infrequent and occurred at a similar frequency in both treatment arms.
“Thus, overall, I believe venetoclax and rituximab should be considered as a suitable standard therapeutic option in patients with relapsed/refractory CLL,” Dr Seymour said.
It is important to note, however, that patients in the VR arm of this study could receive venetoclax for up to 2 years, whereas patients in the BR arm received study treatment for a maximum of six 28-day cycles.
Dr Seymour presented results from MURANO as a late-breaking abstract at ASH (LBA-2). The study was sponsored by Hoffman-La Roche and AbbVie.
MURANO enrolled 389 CLL patients who had received 1 to 3 prior therapies. Patients were randomized to receive VR (n=194) or BR (n=195). Baseline characteristics were similar between the treatment arms.
In both arms, patients received a single monthly dose of rituximab for 6 cycles. The first dose was 375 mg/m2, and all subsequent doses were 500 mg/m2.
In the VR arm, patients received a 4-week or 5-week dose ramp-up of venetoclax from 20 mg to 400 mg daily. This was intended to mitigate the risk of TLS, which has been observed in previous studies of venetoclax.
Patients in the VR arm continued with daily venetoclax at 400 mg for a maximum of 2 years or until disease progression or cessation due to toxicity. They started receiving rituximab after the ramp-up period (at week 6).
In the BR arm, patients received bendamustine at 70 mg/m2 on days 1 and 2 of each 28-day cycle for 6 cycles. Patients could proceed to subsequent therapy if they progressed.
The median follow-up was 23.8 months (range, 0-37.4 months).
Twenty-five percent of patients in the VR arm and 17% in the BR arm discontinued treatment ahead of schedule. Reasons for discontinuation (in the VR and BR arms, respectively) were disease progression (5% and 3%), AEs (12% and 6%), death (1% and 2%), and “other” (6% and 7%).
Survival
The study’s primary endpoint was investigator-assessed PFS. PFS according to an independent review committee (IRC) was a secondary endpoint.
According to investigators, the median PFS was not reached in the VR arm and was 17.0 months in the BR arm (hazard ratio [HR]=0.17, P<0.0001). According to the IRC, the median PFS was not reached in the VR arm and was 18.1 months in the BR arm (HR=0.17, P<0.0001).
According to investigators, the estimated PFS at 24 months was 84.9% in the VR arm and 36.3% in the BR arm. According to the IRC, the 24-month PFS was 82.8% and 37.4%, respectively.
The benefit with VR was consistent across subgroups. Patients had a PFS benefit regardless of their number of prior therapies, deletion 17p status, TP53 mutational status, baseline IGHV mutational status, and whether they had relapsed or refractory disease.
Dr Seymour acknowledged that the differences in treatment duration between the BR and VR arms may have affected the interpretation of these results.
“[T]he treatment duration differed, although, of course, the capacity to deliver more than 6 cycles of bendamustine-rituximab would have been problematic,” he said. “There is some data that antibody treatment may prolong progression-free survival. However, when this study was designed, in 2013, that data was certainly not available. And I believe, currently, maintenance antibody is not an accepted standard of treatment.”
The median overall survival (OS) was not reached in either treatment arm. The 1-year OS rate was 95.9% in the VR arm and 91.1% in the BR arm. The 2-year OS rate was 91.9% and 86.6%, respectively (HR=0.48, P=0.0186).
“[W]ith median follow-up of just on 2 years, there is already a clinically meaningful difference [in OS between the treatment arms],” Dr Seymour said.
“This is not attributable to any difference in availability of novel therapies. Of the 54 patients who received subsequent therapy after progression on the bendamustine-rituximab arm, 40 of those received novel targeted agents.”
Response and MRD
According to investigators, the overall response rate was 93.3% (181/194) in the VR arm and 67.7% (312/195) in the BR arm (P<0.0001). According to the IRC, the overall response rate was 92.3% (179/194) and 72.3% (141/195), respectively (P<0.0001).
According to investigators, the rate of complete response (CR) or CR with incomplete marrow recovery (CRi) was 26.8% (n=52) in the VR arm and 8.2% (n=16) in the BR arm. According to the IRC, the CR/CRi rate was 8.2% (n=16) and 3.6% (n=7), respectively.
Dr Seymour acknowledged the differences in CR/CRi between investigator and IRC assessments. He said 28 of the 42 discrepancies in the VR arm “were attributable to residual CT scan nodal abnormalities in the 16- to 30-mm size.” However, he also noted that 88% of these patients were negative for minimal residual disease (MRD) in the peripheral blood at that time point.
MRD was assessed every 3 months. Patients were counted as MRD-positive if they were positive by either allele-specific oligonucleotide polymerase chain reaction or multicolor flow cytometry. Patients were also counted as MRD-positive if there was a failure to collect a sample.
The proportion of patients who were MRD-negative in the VR and BR arms, respectively, was:
- 45% and 6% at 4 months
- 62% and 13% at 9 months
- 60% and 10% at 12 months
- 57% and 9% at 15 months
- 60% and 5% at 18 months.
Dr Seymour pointed out that 65 patients in the VR arm surpassed the maximum treatment duration for venetoclax (2 years) and therefore stopped receiving the drug, but only 12 of these patients have follow-up beyond 3 months.
“So information about the durability of response after cessation remains immature at the moment,” he said.
Safety
All patients in the VR arm and 98% in the BR arm had at least 1 AE. The rate of serious AEs was 46% and 43%, respectively. The rate of grade 3/4 AEs was 82% and 70%, respectively.
Grade 3/4 AEs with at least a 2% difference in incidence between the treatment arms (in the VR and BR arms, respectively) were neutropenia (58% and 39%), anemia (11% and 14%), thrombocytopenia (6% and 10%), febrile neutropenia (4% and 10%), pneumonia (5% and 8%), infusion-related reactions (2% and 5%), TLS (3% and 1%), hypotension (0% and 3%), hyperglycemia (2% and 0%), and hypogammaglobulinemia (2% and 0%).
The rate of grade 5 AEs was 5% in the VR arm and 6% in the BR arm.
Grade 5 AEs in the VR arm were pneumonia (n=3), sepsis (n=1), cardiac failure (n=1), myocardial infarction (n=1), sudden cardiac death (n=1), colorectal cancer (n=1), status epilepticus (n=1), and acute respiratory failure (n=1).
Grade 5 AEs in the BR arm included sepsis (n=2), lung cancer (n=2), Listeria sepsis (n=1), Scedosporium infection (n=1), lymphoma (n=1), hemorrhagic stroke (n=1), pulmonary embolism (n=1), acute myeloid leukemia (n=1), and sudden death (n=1).
ATLANTA—The combination of venetoclax and rituximab (VR) should be a standard treatment option for adults with relapsed/refractory chronic lymphocytic leukemia (CLL), according to a speaker at the 2017 ASH Annual Meeting.
Data from the phase 3 MURANO study showed that patients with relapsed/refractory CLL who received VR had significantly longer progression-free survival (PFS) than those who received bendamustine and rituximab (BR).
In addition, “secondary endpoints were consistently in favor of venetoclax-rituximab,” said study investigator John F. Seymour, MBBS, PhD, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
Adverse events (AEs) were largely consistent with the known safety profiles of the drugs studied, but tumor lysis syndrome (TLS) was infrequent and occurred at a similar frequency in both treatment arms.
“Thus, overall, I believe venetoclax and rituximab should be considered as a suitable standard therapeutic option in patients with relapsed/refractory CLL,” Dr Seymour said.
It is important to note, however, that patients in the VR arm of this study could receive venetoclax for up to 2 years, whereas patients in the BR arm received study treatment for a maximum of six 28-day cycles.
Dr Seymour presented results from MURANO as a late-breaking abstract at ASH (LBA-2). The study was sponsored by Hoffman-La Roche and AbbVie.
MURANO enrolled 389 CLL patients who had received 1 to 3 prior therapies. Patients were randomized to receive VR (n=194) or BR (n=195). Baseline characteristics were similar between the treatment arms.
In both arms, patients received a single monthly dose of rituximab for 6 cycles. The first dose was 375 mg/m2, and all subsequent doses were 500 mg/m2.
In the VR arm, patients received a 4-week or 5-week dose ramp-up of venetoclax from 20 mg to 400 mg daily. This was intended to mitigate the risk of TLS, which has been observed in previous studies of venetoclax.
Patients in the VR arm continued with daily venetoclax at 400 mg for a maximum of 2 years or until disease progression or cessation due to toxicity. They started receiving rituximab after the ramp-up period (at week 6).
In the BR arm, patients received bendamustine at 70 mg/m2 on days 1 and 2 of each 28-day cycle for 6 cycles. Patients could proceed to subsequent therapy if they progressed.
The median follow-up was 23.8 months (range, 0-37.4 months).
Twenty-five percent of patients in the VR arm and 17% in the BR arm discontinued treatment ahead of schedule. Reasons for discontinuation (in the VR and BR arms, respectively) were disease progression (5% and 3%), AEs (12% and 6%), death (1% and 2%), and “other” (6% and 7%).
Survival
The study’s primary endpoint was investigator-assessed PFS. PFS according to an independent review committee (IRC) was a secondary endpoint.
According to investigators, the median PFS was not reached in the VR arm and was 17.0 months in the BR arm (hazard ratio [HR]=0.17, P<0.0001). According to the IRC, the median PFS was not reached in the VR arm and was 18.1 months in the BR arm (HR=0.17, P<0.0001).
According to investigators, the estimated PFS at 24 months was 84.9% in the VR arm and 36.3% in the BR arm. According to the IRC, the 24-month PFS was 82.8% and 37.4%, respectively.
The benefit with VR was consistent across subgroups. Patients had a PFS benefit regardless of their number of prior therapies, deletion 17p status, TP53 mutational status, baseline IGHV mutational status, and whether they had relapsed or refractory disease.
Dr Seymour acknowledged that the differences in treatment duration between the BR and VR arms may have affected the interpretation of these results.
“[T]he treatment duration differed, although, of course, the capacity to deliver more than 6 cycles of bendamustine-rituximab would have been problematic,” he said. “There is some data that antibody treatment may prolong progression-free survival. However, when this study was designed, in 2013, that data was certainly not available. And I believe, currently, maintenance antibody is not an accepted standard of treatment.”
The median overall survival (OS) was not reached in either treatment arm. The 1-year OS rate was 95.9% in the VR arm and 91.1% in the BR arm. The 2-year OS rate was 91.9% and 86.6%, respectively (HR=0.48, P=0.0186).
“[W]ith median follow-up of just on 2 years, there is already a clinically meaningful difference [in OS between the treatment arms],” Dr Seymour said.
“This is not attributable to any difference in availability of novel therapies. Of the 54 patients who received subsequent therapy after progression on the bendamustine-rituximab arm, 40 of those received novel targeted agents.”
Response and MRD
According to investigators, the overall response rate was 93.3% (181/194) in the VR arm and 67.7% (312/195) in the BR arm (P<0.0001). According to the IRC, the overall response rate was 92.3% (179/194) and 72.3% (141/195), respectively (P<0.0001).
According to investigators, the rate of complete response (CR) or CR with incomplete marrow recovery (CRi) was 26.8% (n=52) in the VR arm and 8.2% (n=16) in the BR arm. According to the IRC, the CR/CRi rate was 8.2% (n=16) and 3.6% (n=7), respectively.
Dr Seymour acknowledged the differences in CR/CRi between investigator and IRC assessments. He said 28 of the 42 discrepancies in the VR arm “were attributable to residual CT scan nodal abnormalities in the 16- to 30-mm size.” However, he also noted that 88% of these patients were negative for minimal residual disease (MRD) in the peripheral blood at that time point.
MRD was assessed every 3 months. Patients were counted as MRD-positive if they were positive by either allele-specific oligonucleotide polymerase chain reaction or multicolor flow cytometry. Patients were also counted as MRD-positive if there was a failure to collect a sample.
The proportion of patients who were MRD-negative in the VR and BR arms, respectively, was:
- 45% and 6% at 4 months
- 62% and 13% at 9 months
- 60% and 10% at 12 months
- 57% and 9% at 15 months
- 60% and 5% at 18 months.
Dr Seymour pointed out that 65 patients in the VR arm surpassed the maximum treatment duration for venetoclax (2 years) and therefore stopped receiving the drug, but only 12 of these patients have follow-up beyond 3 months.
“So information about the durability of response after cessation remains immature at the moment,” he said.
Safety
All patients in the VR arm and 98% in the BR arm had at least 1 AE. The rate of serious AEs was 46% and 43%, respectively. The rate of grade 3/4 AEs was 82% and 70%, respectively.
Grade 3/4 AEs with at least a 2% difference in incidence between the treatment arms (in the VR and BR arms, respectively) were neutropenia (58% and 39%), anemia (11% and 14%), thrombocytopenia (6% and 10%), febrile neutropenia (4% and 10%), pneumonia (5% and 8%), infusion-related reactions (2% and 5%), TLS (3% and 1%), hypotension (0% and 3%), hyperglycemia (2% and 0%), and hypogammaglobulinemia (2% and 0%).
The rate of grade 5 AEs was 5% in the VR arm and 6% in the BR arm.
Grade 5 AEs in the VR arm were pneumonia (n=3), sepsis (n=1), cardiac failure (n=1), myocardial infarction (n=1), sudden cardiac death (n=1), colorectal cancer (n=1), status epilepticus (n=1), and acute respiratory failure (n=1).
Grade 5 AEs in the BR arm included sepsis (n=2), lung cancer (n=2), Listeria sepsis (n=1), Scedosporium infection (n=1), lymphoma (n=1), hemorrhagic stroke (n=1), pulmonary embolism (n=1), acute myeloid leukemia (n=1), and sudden death (n=1).
ATLANTA—The combination of venetoclax and rituximab (VR) should be a standard treatment option for adults with relapsed/refractory chronic lymphocytic leukemia (CLL), according to a speaker at the 2017 ASH Annual Meeting.
Data from the phase 3 MURANO study showed that patients with relapsed/refractory CLL who received VR had significantly longer progression-free survival (PFS) than those who received bendamustine and rituximab (BR).
In addition, “secondary endpoints were consistently in favor of venetoclax-rituximab,” said study investigator John F. Seymour, MBBS, PhD, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
Adverse events (AEs) were largely consistent with the known safety profiles of the drugs studied, but tumor lysis syndrome (TLS) was infrequent and occurred at a similar frequency in both treatment arms.
“Thus, overall, I believe venetoclax and rituximab should be considered as a suitable standard therapeutic option in patients with relapsed/refractory CLL,” Dr Seymour said.
It is important to note, however, that patients in the VR arm of this study could receive venetoclax for up to 2 years, whereas patients in the BR arm received study treatment for a maximum of six 28-day cycles.
Dr Seymour presented results from MURANO as a late-breaking abstract at ASH (LBA-2). The study was sponsored by Hoffman-La Roche and AbbVie.
MURANO enrolled 389 CLL patients who had received 1 to 3 prior therapies. Patients were randomized to receive VR (n=194) or BR (n=195). Baseline characteristics were similar between the treatment arms.
In both arms, patients received a single monthly dose of rituximab for 6 cycles. The first dose was 375 mg/m2, and all subsequent doses were 500 mg/m2.
In the VR arm, patients received a 4-week or 5-week dose ramp-up of venetoclax from 20 mg to 400 mg daily. This was intended to mitigate the risk of TLS, which has been observed in previous studies of venetoclax.
Patients in the VR arm continued with daily venetoclax at 400 mg for a maximum of 2 years or until disease progression or cessation due to toxicity. They started receiving rituximab after the ramp-up period (at week 6).
In the BR arm, patients received bendamustine at 70 mg/m2 on days 1 and 2 of each 28-day cycle for 6 cycles. Patients could proceed to subsequent therapy if they progressed.
The median follow-up was 23.8 months (range, 0-37.4 months).
Twenty-five percent of patients in the VR arm and 17% in the BR arm discontinued treatment ahead of schedule. Reasons for discontinuation (in the VR and BR arms, respectively) were disease progression (5% and 3%), AEs (12% and 6%), death (1% and 2%), and “other” (6% and 7%).
Survival
The study’s primary endpoint was investigator-assessed PFS. PFS according to an independent review committee (IRC) was a secondary endpoint.
According to investigators, the median PFS was not reached in the VR arm and was 17.0 months in the BR arm (hazard ratio [HR]=0.17, P<0.0001). According to the IRC, the median PFS was not reached in the VR arm and was 18.1 months in the BR arm (HR=0.17, P<0.0001).
According to investigators, the estimated PFS at 24 months was 84.9% in the VR arm and 36.3% in the BR arm. According to the IRC, the 24-month PFS was 82.8% and 37.4%, respectively.
The benefit with VR was consistent across subgroups. Patients had a PFS benefit regardless of their number of prior therapies, deletion 17p status, TP53 mutational status, baseline IGHV mutational status, and whether they had relapsed or refractory disease.
Dr Seymour acknowledged that the differences in treatment duration between the BR and VR arms may have affected the interpretation of these results.
“[T]he treatment duration differed, although, of course, the capacity to deliver more than 6 cycles of bendamustine-rituximab would have been problematic,” he said. “There is some data that antibody treatment may prolong progression-free survival. However, when this study was designed, in 2013, that data was certainly not available. And I believe, currently, maintenance antibody is not an accepted standard of treatment.”
The median overall survival (OS) was not reached in either treatment arm. The 1-year OS rate was 95.9% in the VR arm and 91.1% in the BR arm. The 2-year OS rate was 91.9% and 86.6%, respectively (HR=0.48, P=0.0186).
“[W]ith median follow-up of just on 2 years, there is already a clinically meaningful difference [in OS between the treatment arms],” Dr Seymour said.
“This is not attributable to any difference in availability of novel therapies. Of the 54 patients who received subsequent therapy after progression on the bendamustine-rituximab arm, 40 of those received novel targeted agents.”
Response and MRD
According to investigators, the overall response rate was 93.3% (181/194) in the VR arm and 67.7% (312/195) in the BR arm (P<0.0001). According to the IRC, the overall response rate was 92.3% (179/194) and 72.3% (141/195), respectively (P<0.0001).
According to investigators, the rate of complete response (CR) or CR with incomplete marrow recovery (CRi) was 26.8% (n=52) in the VR arm and 8.2% (n=16) in the BR arm. According to the IRC, the CR/CRi rate was 8.2% (n=16) and 3.6% (n=7), respectively.
Dr Seymour acknowledged the differences in CR/CRi between investigator and IRC assessments. He said 28 of the 42 discrepancies in the VR arm “were attributable to residual CT scan nodal abnormalities in the 16- to 30-mm size.” However, he also noted that 88% of these patients were negative for minimal residual disease (MRD) in the peripheral blood at that time point.
MRD was assessed every 3 months. Patients were counted as MRD-positive if they were positive by either allele-specific oligonucleotide polymerase chain reaction or multicolor flow cytometry. Patients were also counted as MRD-positive if there was a failure to collect a sample.
The proportion of patients who were MRD-negative in the VR and BR arms, respectively, was:
- 45% and 6% at 4 months
- 62% and 13% at 9 months
- 60% and 10% at 12 months
- 57% and 9% at 15 months
- 60% and 5% at 18 months.
Dr Seymour pointed out that 65 patients in the VR arm surpassed the maximum treatment duration for venetoclax (2 years) and therefore stopped receiving the drug, but only 12 of these patients have follow-up beyond 3 months.
“So information about the durability of response after cessation remains immature at the moment,” he said.
Safety
All patients in the VR arm and 98% in the BR arm had at least 1 AE. The rate of serious AEs was 46% and 43%, respectively. The rate of grade 3/4 AEs was 82% and 70%, respectively.
Grade 3/4 AEs with at least a 2% difference in incidence between the treatment arms (in the VR and BR arms, respectively) were neutropenia (58% and 39%), anemia (11% and 14%), thrombocytopenia (6% and 10%), febrile neutropenia (4% and 10%), pneumonia (5% and 8%), infusion-related reactions (2% and 5%), TLS (3% and 1%), hypotension (0% and 3%), hyperglycemia (2% and 0%), and hypogammaglobulinemia (2% and 0%).
The rate of grade 5 AEs was 5% in the VR arm and 6% in the BR arm.
Grade 5 AEs in the VR arm were pneumonia (n=3), sepsis (n=1), cardiac failure (n=1), myocardial infarction (n=1), sudden cardiac death (n=1), colorectal cancer (n=1), status epilepticus (n=1), and acute respiratory failure (n=1).
Grade 5 AEs in the BR arm included sepsis (n=2), lung cancer (n=2), Listeria sepsis (n=1), Scedosporium infection (n=1), lymphoma (n=1), hemorrhagic stroke (n=1), pulmonary embolism (n=1), acute myeloid leukemia (n=1), and sudden death (n=1).