User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Urine drug screens: Not just for job applicants
Although urine drug screens (UDS) are most commonly used to screen job applicants, some clinicians have started to use them as a tool for improving their patients’ clinical outcomes.1 Recently, some clinicians have begun using UDS to help patients who experience chronic pain and dependency (mainly on opioids) and for those who use diverted drugs to relieve these conditions. Many psychiatrists are concerned about the high cost of drug diversion, as well as the possibility of diversion-related patient mortality. Clinicians should therefore consider using UDS as a tool to help address these challenges.
Consider individualized UDS monitoring
The standard 5-substance UDS test panel consists of tetrahydrocannabinol, opiates, amphetamines, cocaine, and phencyclidine. Although this panel was sufficient for an employment screening-related UDS, the American Society of Addiction Medicine (ASAM) has rejected its use for patients with substance abuse. As part of its emphasis on the importance of incorporating preventative procedures, diagnostics, and surveillance protocols, the ASAM advocates using a rotating test panel in conjunction with a patient-specific UDS.2 This type of patient-specific regimen would take into account the dynamic nature of a patient’s health profile factors, including comorbid and psychosocial status, subjective pain features, and diverted drug use. Furthermore, the ASAM recommends evaluating patients for the concurrent use of other substances and agents, such as benzodiazepines, sleep-inducing medications, stimulants, and alcohol, because these can interact with opioids.
Consider extending individualized monitoring by implementing standard “cutoff” values for each drug; patients whose levels of a specific substance are above the established cutoff value are categorized as testing positive for the use of that substance. The Substance Abuse Mental Health Services Administration favors adjusting UDS cutoffs, specifically the use of decreased cutoffs, to improve patient compliance.3 However, standardized drug concentration cutoff values may not be applicable for each patient; therefore, such values may need to be carefully tailored to each patient.
Additional drug monitoring techniques
Existing UDS practices, such as medication adherence and compliance, can be supplemented or alternately used with UDS panels that are modified to account for a patient’s fluctuating clinical conditions and concurrent medications. Point-of-care immunoassays, which provide accurate screening for medication compliance and adherence and possible drug diversion, should be used for routine monitoring. Using DNA-authenticated UDS also adds further control in monitoring a patient’s use of different drugs.4,5
In addition to being helpful for monitoring opioid use, a DNA-verified UDS can be used to evaluate for the presence of synthetic urine substitutes.6-8 Diversion remains a growing epidemiologic concern, and the number of cases is vastly underreported in the literature. The DNA-authenticated UDS can give clinicians greater precision in identifying synthetic and substituted urine among patient-provided samples.4
Using a combination of the methods described here can help expand a clinician’s ability to perform individualized drug monitoring, and verify whether a patient is adhering to his or her treatment regimen.
1. Choudhry Z, Islam F, Siddiqui W, et al. UDS in mental health: is it time to move forward? J Psychiatry. 2015;18(5): doi: 10.4172/2378-5756.1000319.
2. Drug testing: a white paper of the American Society of Addiction Medicine. Chevy Chase, MD: American Society of Addiction Medicine; https://www.asam.org/docs/default-source/public-policy-statements/drug-testing-a-white-paper-by-asam.pdf. Published October 26, 2013. Accessed November 13, 2018.
3. Substance Abuse Mental Health Services Administration (SAMHSA). Technical Assistance Publication Series, TAP 32. Clinical drug testing in primary care. Rockville, MD: U.S. Department of Health and Human Services; 2012.
4. Genotox Laboratories. DNA Authenticated Drug Screen (ToxProtect). https://genotoxlabs.com/. Accessed October 11, 2018
5. 3RX Holdings Inc. 3RX Toxicology Urinary Drug Testing. http://3rxholdings.com/. Accessed October 11, 2018.
6. Genetic testing to confirm the identity of laboratory specimens. Document No GENE.00041. Medical Policy. Virginia Beach, VA: Amerigroup; 2018.
7. UnitedHealthcare Services. Drug Testing Policy. Reimbursement policy No 2018R6005A. https://www.uhcprovider.com/content/dam/provider/docs/public/policies/comm-reimbursement/COMM-Drug-Testing-Policy.pdf. Accessed October 12, 2018.
8. OzMed Laboratory Services. DNA-Verified Urine Drug Testing. http://www.ozmed.org/. Accessed October 11, 2018.
Although urine drug screens (UDS) are most commonly used to screen job applicants, some clinicians have started to use them as a tool for improving their patients’ clinical outcomes.1 Recently, some clinicians have begun using UDS to help patients who experience chronic pain and dependency (mainly on opioids) and for those who use diverted drugs to relieve these conditions. Many psychiatrists are concerned about the high cost of drug diversion, as well as the possibility of diversion-related patient mortality. Clinicians should therefore consider using UDS as a tool to help address these challenges.
Consider individualized UDS monitoring
The standard 5-substance UDS test panel consists of tetrahydrocannabinol, opiates, amphetamines, cocaine, and phencyclidine. Although this panel was sufficient for an employment screening-related UDS, the American Society of Addiction Medicine (ASAM) has rejected its use for patients with substance abuse. As part of its emphasis on the importance of incorporating preventative procedures, diagnostics, and surveillance protocols, the ASAM advocates using a rotating test panel in conjunction with a patient-specific UDS.2 This type of patient-specific regimen would take into account the dynamic nature of a patient’s health profile factors, including comorbid and psychosocial status, subjective pain features, and diverted drug use. Furthermore, the ASAM recommends evaluating patients for the concurrent use of other substances and agents, such as benzodiazepines, sleep-inducing medications, stimulants, and alcohol, because these can interact with opioids.
Consider extending individualized monitoring by implementing standard “cutoff” values for each drug; patients whose levels of a specific substance are above the established cutoff value are categorized as testing positive for the use of that substance. The Substance Abuse Mental Health Services Administration favors adjusting UDS cutoffs, specifically the use of decreased cutoffs, to improve patient compliance.3 However, standardized drug concentration cutoff values may not be applicable for each patient; therefore, such values may need to be carefully tailored to each patient.
Additional drug monitoring techniques
Existing UDS practices, such as medication adherence and compliance, can be supplemented or alternately used with UDS panels that are modified to account for a patient’s fluctuating clinical conditions and concurrent medications. Point-of-care immunoassays, which provide accurate screening for medication compliance and adherence and possible drug diversion, should be used for routine monitoring. Using DNA-authenticated UDS also adds further control in monitoring a patient’s use of different drugs.4,5
In addition to being helpful for monitoring opioid use, a DNA-verified UDS can be used to evaluate for the presence of synthetic urine substitutes.6-8 Diversion remains a growing epidemiologic concern, and the number of cases is vastly underreported in the literature. The DNA-authenticated UDS can give clinicians greater precision in identifying synthetic and substituted urine among patient-provided samples.4
Using a combination of the methods described here can help expand a clinician’s ability to perform individualized drug monitoring, and verify whether a patient is adhering to his or her treatment regimen.
Although urine drug screens (UDS) are most commonly used to screen job applicants, some clinicians have started to use them as a tool for improving their patients’ clinical outcomes.1 Recently, some clinicians have begun using UDS to help patients who experience chronic pain and dependency (mainly on opioids) and for those who use diverted drugs to relieve these conditions. Many psychiatrists are concerned about the high cost of drug diversion, as well as the possibility of diversion-related patient mortality. Clinicians should therefore consider using UDS as a tool to help address these challenges.
Consider individualized UDS monitoring
The standard 5-substance UDS test panel consists of tetrahydrocannabinol, opiates, amphetamines, cocaine, and phencyclidine. Although this panel was sufficient for an employment screening-related UDS, the American Society of Addiction Medicine (ASAM) has rejected its use for patients with substance abuse. As part of its emphasis on the importance of incorporating preventative procedures, diagnostics, and surveillance protocols, the ASAM advocates using a rotating test panel in conjunction with a patient-specific UDS.2 This type of patient-specific regimen would take into account the dynamic nature of a patient’s health profile factors, including comorbid and psychosocial status, subjective pain features, and diverted drug use. Furthermore, the ASAM recommends evaluating patients for the concurrent use of other substances and agents, such as benzodiazepines, sleep-inducing medications, stimulants, and alcohol, because these can interact with opioids.
Consider extending individualized monitoring by implementing standard “cutoff” values for each drug; patients whose levels of a specific substance are above the established cutoff value are categorized as testing positive for the use of that substance. The Substance Abuse Mental Health Services Administration favors adjusting UDS cutoffs, specifically the use of decreased cutoffs, to improve patient compliance.3 However, standardized drug concentration cutoff values may not be applicable for each patient; therefore, such values may need to be carefully tailored to each patient.
Additional drug monitoring techniques
Existing UDS practices, such as medication adherence and compliance, can be supplemented or alternately used with UDS panels that are modified to account for a patient’s fluctuating clinical conditions and concurrent medications. Point-of-care immunoassays, which provide accurate screening for medication compliance and adherence and possible drug diversion, should be used for routine monitoring. Using DNA-authenticated UDS also adds further control in monitoring a patient’s use of different drugs.4,5
In addition to being helpful for monitoring opioid use, a DNA-verified UDS can be used to evaluate for the presence of synthetic urine substitutes.6-8 Diversion remains a growing epidemiologic concern, and the number of cases is vastly underreported in the literature. The DNA-authenticated UDS can give clinicians greater precision in identifying synthetic and substituted urine among patient-provided samples.4
Using a combination of the methods described here can help expand a clinician’s ability to perform individualized drug monitoring, and verify whether a patient is adhering to his or her treatment regimen.
1. Choudhry Z, Islam F, Siddiqui W, et al. UDS in mental health: is it time to move forward? J Psychiatry. 2015;18(5): doi: 10.4172/2378-5756.1000319.
2. Drug testing: a white paper of the American Society of Addiction Medicine. Chevy Chase, MD: American Society of Addiction Medicine; https://www.asam.org/docs/default-source/public-policy-statements/drug-testing-a-white-paper-by-asam.pdf. Published October 26, 2013. Accessed November 13, 2018.
3. Substance Abuse Mental Health Services Administration (SAMHSA). Technical Assistance Publication Series, TAP 32. Clinical drug testing in primary care. Rockville, MD: U.S. Department of Health and Human Services; 2012.
4. Genotox Laboratories. DNA Authenticated Drug Screen (ToxProtect). https://genotoxlabs.com/. Accessed October 11, 2018
5. 3RX Holdings Inc. 3RX Toxicology Urinary Drug Testing. http://3rxholdings.com/. Accessed October 11, 2018.
6. Genetic testing to confirm the identity of laboratory specimens. Document No GENE.00041. Medical Policy. Virginia Beach, VA: Amerigroup; 2018.
7. UnitedHealthcare Services. Drug Testing Policy. Reimbursement policy No 2018R6005A. https://www.uhcprovider.com/content/dam/provider/docs/public/policies/comm-reimbursement/COMM-Drug-Testing-Policy.pdf. Accessed October 12, 2018.
8. OzMed Laboratory Services. DNA-Verified Urine Drug Testing. http://www.ozmed.org/. Accessed October 11, 2018.
1. Choudhry Z, Islam F, Siddiqui W, et al. UDS in mental health: is it time to move forward? J Psychiatry. 2015;18(5): doi: 10.4172/2378-5756.1000319.
2. Drug testing: a white paper of the American Society of Addiction Medicine. Chevy Chase, MD: American Society of Addiction Medicine; https://www.asam.org/docs/default-source/public-policy-statements/drug-testing-a-white-paper-by-asam.pdf. Published October 26, 2013. Accessed November 13, 2018.
3. Substance Abuse Mental Health Services Administration (SAMHSA). Technical Assistance Publication Series, TAP 32. Clinical drug testing in primary care. Rockville, MD: U.S. Department of Health and Human Services; 2012.
4. Genotox Laboratories. DNA Authenticated Drug Screen (ToxProtect). https://genotoxlabs.com/. Accessed October 11, 2018
5. 3RX Holdings Inc. 3RX Toxicology Urinary Drug Testing. http://3rxholdings.com/. Accessed October 11, 2018.
6. Genetic testing to confirm the identity of laboratory specimens. Document No GENE.00041. Medical Policy. Virginia Beach, VA: Amerigroup; 2018.
7. UnitedHealthcare Services. Drug Testing Policy. Reimbursement policy No 2018R6005A. https://www.uhcprovider.com/content/dam/provider/docs/public/policies/comm-reimbursement/COMM-Drug-Testing-Policy.pdf. Accessed October 12, 2018.
8. OzMed Laboratory Services. DNA-Verified Urine Drug Testing. http://www.ozmed.org/. Accessed October 11, 2018.
Psychopharmacology 3.0
There is little doubt that the psychopharmacology revolution has been transformational for psychiatry and is also credited for sparking the momentous neuroscience advances of the past half century.
The field of psychiatry, dominated by Freudian psychology for decades, radically evolved from psychoanalysis to pharmacotherapy with the discovery that serious mental disorders are treatable with medications, thus dispensing with the couch.
Prior to 1952, the prevailing dogma was that “madness is irreversible.” That’s why millions of patients with various psychiatric disorders were locked up in institutions, which added to the stigma of mental illness. Then came the first antipsychotic drug, chlorpromazine, which “magically” eliminated the delusions and hallucinations of patients who had been hospitalized for years. That serendipitous and historic discovery was as transformational for psychiatry as penicillin was for infections (yet inexplicably, only the discovery of penicillin received a Nobel Prize). Most people today do not know that before chlorpromazine, 50% of all hospital beds in the U.S. were occupied by psychiatric patients. The massive shuttering of state hospitals in the 1970s and ’80s was a direct consequence of the widespread use of chlorpromazine and its cohort of first-generation antipsychotics (FGAs).
That was Psychopharmacology 1.0, spanning the period 1952 to 1987. It included dozens of FGAs belonging to 6 classes: phenothiazines, thioxanthenes, butyrophenones, dibenzazepines, dihydroindolones, and dibenzodiazepines. Psychopharmacology 1.0 also included monoamine oxidase inhibitors and tricyclic antidepressants for depression, and lithium for bipolar mania. Ironically, clozapine, the incognito seed template of the second-generation antipsychotic (SGA) class, was synthesized in 1959 with the early wave of FGAs, and launched in Europe in 1972, only to be withdrawn in 1974 due to agranulocytosis-induced deaths not recognized during the clinical trials.
The late 1980s ushered in Psychopharmacology 2.0, which was also transformative. It began in 1987 with the introduction of fluoxetine, the first selective serotonin receptor inhibitor. Then clozapine was resurrected in 1988 as the first FDA-approved drug for refractory schizophrenia. Being the first SGA (no acute extrapyramidal side effects at all, in contrast to all FGAs), it became the “mechanistic model” for all other SGA agents, which were introduced starting in 1993. All SGAs were designed by pharmaceutical companies’ medicinal chemists to mimic clozapine’s receptor profile: far stronger affinity to serotonin 5HT-2A receptors than to dopamine D2 receptors. Three partial agonists and several heterocyclic antidepressants were also introduced during this 2.0 era, which continued until approximately 2017. Of the 11 SGAs that were initially approved for schizophrenia, 7 also were approved for bipolar mania, and 2 received an FDA indication for bipolar depression, thus addressing a glaring unmet need.
Psychopharmacology 3.0 has already begun. Its seeds started sprouting over the past few years with the landmark studies of intravenous ketamine, which was demonstrated to reverse severe and refractory depression and suicidal urges within hours of injection. The first ketamine product, esketamine, an intranasal formulation, is expected to be approved by the FDA soon. In the same vein, other rapid-acting antidepressants, a welcome paradigm shift, are being developed, including IV scopolamine, IV rapastinel, and inhalable nitrous oxide.
Three novel and important pharmacologic agents have arrived in this 3.0 era:
- Pimavanserin, a serotonin 5HT-2A inverse agonist, the first and only non-dopamine–blocking antipsychotic approved by the FDA for the delusions and hallucinations of Parkinson’s disease psychosis. It is currently in clinical trials for schizophrenia and Alzheimer’s disease psychosis (for which nothing is yet approved).
- Valbenazine, the first drug approved for tardive dyskinesia (TD), the treatment of which had been elusive and remained a huge unmet need for 60 years. Its novel mechanism of action is inhibition of vesicular monoamine transporter 2 (VMAT2), which reduces the putative dopamine supersensitivity of TD.
- Deutetrabenazine, which was also approved for TD a few months after valbenazine, and has the same mechanism of action. It also was approved for Huntington’s chorea.
Continue to: Another important feature...
Another important feature of Psychopharmacology 3.0 is the repurposing of hallucinogens into novel therapies for posttraumatic stress disorder, anxiety, and depression.1 The opioid system is being recognized as another key player in depression, with many studies showing buprenorphine has antidepressant and anti-suicidal properties2 and the recent finding that pre-treatment with naloxone blocks the rapid antidepressive effects of ketamine.3 This finding casts doubt on the notion that the antidepressant mechanism of action of ketamine is solely mediated via its antagonism of the glutamate N-methyl-
These early developments in Psychopharmacology 3.0 augur well for the future. Companies in the pharmaceutical industry (which are hated by many, and even demonized and kept at arm’s length by major medical schools) are, in fact, the only entities in the world that develop new medications for psychiatric disorders, 82% of which still have no FDA-approved drug.4 Psychiatric researchers and clinicians should collaborate and advise the pharmaceutical companies about the urgent or unmet needs of psychiatric patients so they can target those unmet needs with their massive R&D resources.
In that spirit, here is my wish list of therapeutic targets that I hope will emerge during the Psychopharmacology 3.0 era and beyond:
1. New mechanisms of action for antipsychotics, based on emerging neurobiological research in schizophrenia and related psychoses, such as:
- Inhibit microglia activation
- Repair mitochondrial dysfunction
- Modulate the hypofunctional NMDA receptors
- Inhibit apoptosis
- Enhance neurogenesis
- Repair myelin pathology
- Inhibit neuroinflammation and oxidative stress
- Increase neurotropic growth factors
- Neurosteroid therapies (including estrogen)
- Exploit the microbiome influence on both the enteric and cephalic brains
2. Long-acting injectable antidepressants and mood stabilizers, because there is a malignant transformation into treatment-resistance in mood disorders after recurrent episodes due to nonadherence.5
3. Treatments for personality disorders, especially borderline and antisocial personality disorders.
4. An effective treatment for alcoholism.
5. Pharmacotherapy for aggression.
6. Vaccines for substance use.
7. Stage-specific pharmacotherapies (because the neurobiology of prodromal, first-episode, and multiple-episode patients have been shown to be quite different).
8. Drugs for epigenetic modulation to inhibit risk genes and to over-express protective genes.
It may take decades and hundreds of billions (even trillions) of R&D investment to accomplish the above, but I remain excited about the prospects of astounding psychopharmacologic advances to treat the disorders of the mind. Precision psychiatry advances will also expedite the selection of the right medication for each patient by employing predictive biomarkers. Breakthrough methodologies, such as pluripotent stem cells, opto-genetics, and clustered regularly interspaced short palindromic repeats (CRISPR), promise to revolutionize the biology, diagnosis, treatment, and prevention of various neuropsychiatric disorders.
The future of psychopharmacology is bright, if adequate resources are invested. The current direct and indirect costs of mental disorders and addictions are in the hundreds of billions of dollars annually. Only intensive research and disruptive discoveries will have the salutary dual effect of healing disease and reducing the economic burden of neuropsychiatric disorders. Psychopharmacology 3.0 advances, along with nonpharmacologic therapies such as neuromodulation (electroconvulsive therapy, transcranial magnetic stimulation, vagus nerve stimulation, and a dozen other techniques in development). Together with the indispensable evidence-based psychotherapies such as cognitive-behavioral therapy, dialectical behavior therapy, and interpersonal psychotherapy, psychopharmacology represents the leading edge of progress in psychiatric treatment. The psychiatrists of 1952 could only fantasize about what has since become a reality in healing ailing minds.
To comment on this editorial or other topics of interest: [email protected]
1. Nasrallah, HA. Maddening therapies: How hallucinogens morphed into novel treatments. Current Psychiatry. 2017;16(1):19-21.
2. Serafini G, Adavastro G, Canepa G, et al. The efficacy of buprenorphine in major depression, treatment-resistant depression and suicidal behavior: a systematic review. Int J Mol Sci. 2018;19(8). doi: 10.3390/ijms19082410.
3. Williams NR, Heifets BD, Blasey C, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am J Psychiatry. 2018. doi: 10.1176/appi.ajp.2018.18020138. [Epub ahead of print].
4. Devulapalli KK, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders. The majority of psychiatric diagnoses have no approved drug. Asian J Psychiatr. 2009;2(1):29-36.
5. Post RM. Preventing the malignant transformation of bipolar disorder. JAMA. 2018;319(12):1197-1198.
There is little doubt that the psychopharmacology revolution has been transformational for psychiatry and is also credited for sparking the momentous neuroscience advances of the past half century.
The field of psychiatry, dominated by Freudian psychology for decades, radically evolved from psychoanalysis to pharmacotherapy with the discovery that serious mental disorders are treatable with medications, thus dispensing with the couch.
Prior to 1952, the prevailing dogma was that “madness is irreversible.” That’s why millions of patients with various psychiatric disorders were locked up in institutions, which added to the stigma of mental illness. Then came the first antipsychotic drug, chlorpromazine, which “magically” eliminated the delusions and hallucinations of patients who had been hospitalized for years. That serendipitous and historic discovery was as transformational for psychiatry as penicillin was for infections (yet inexplicably, only the discovery of penicillin received a Nobel Prize). Most people today do not know that before chlorpromazine, 50% of all hospital beds in the U.S. were occupied by psychiatric patients. The massive shuttering of state hospitals in the 1970s and ’80s was a direct consequence of the widespread use of chlorpromazine and its cohort of first-generation antipsychotics (FGAs).
That was Psychopharmacology 1.0, spanning the period 1952 to 1987. It included dozens of FGAs belonging to 6 classes: phenothiazines, thioxanthenes, butyrophenones, dibenzazepines, dihydroindolones, and dibenzodiazepines. Psychopharmacology 1.0 also included monoamine oxidase inhibitors and tricyclic antidepressants for depression, and lithium for bipolar mania. Ironically, clozapine, the incognito seed template of the second-generation antipsychotic (SGA) class, was synthesized in 1959 with the early wave of FGAs, and launched in Europe in 1972, only to be withdrawn in 1974 due to agranulocytosis-induced deaths not recognized during the clinical trials.
The late 1980s ushered in Psychopharmacology 2.0, which was also transformative. It began in 1987 with the introduction of fluoxetine, the first selective serotonin receptor inhibitor. Then clozapine was resurrected in 1988 as the first FDA-approved drug for refractory schizophrenia. Being the first SGA (no acute extrapyramidal side effects at all, in contrast to all FGAs), it became the “mechanistic model” for all other SGA agents, which were introduced starting in 1993. All SGAs were designed by pharmaceutical companies’ medicinal chemists to mimic clozapine’s receptor profile: far stronger affinity to serotonin 5HT-2A receptors than to dopamine D2 receptors. Three partial agonists and several heterocyclic antidepressants were also introduced during this 2.0 era, which continued until approximately 2017. Of the 11 SGAs that were initially approved for schizophrenia, 7 also were approved for bipolar mania, and 2 received an FDA indication for bipolar depression, thus addressing a glaring unmet need.
Psychopharmacology 3.0 has already begun. Its seeds started sprouting over the past few years with the landmark studies of intravenous ketamine, which was demonstrated to reverse severe and refractory depression and suicidal urges within hours of injection. The first ketamine product, esketamine, an intranasal formulation, is expected to be approved by the FDA soon. In the same vein, other rapid-acting antidepressants, a welcome paradigm shift, are being developed, including IV scopolamine, IV rapastinel, and inhalable nitrous oxide.
Three novel and important pharmacologic agents have arrived in this 3.0 era:
- Pimavanserin, a serotonin 5HT-2A inverse agonist, the first and only non-dopamine–blocking antipsychotic approved by the FDA for the delusions and hallucinations of Parkinson’s disease psychosis. It is currently in clinical trials for schizophrenia and Alzheimer’s disease psychosis (for which nothing is yet approved).
- Valbenazine, the first drug approved for tardive dyskinesia (TD), the treatment of which had been elusive and remained a huge unmet need for 60 years. Its novel mechanism of action is inhibition of vesicular monoamine transporter 2 (VMAT2), which reduces the putative dopamine supersensitivity of TD.
- Deutetrabenazine, which was also approved for TD a few months after valbenazine, and has the same mechanism of action. It also was approved for Huntington’s chorea.
Continue to: Another important feature...
Another important feature of Psychopharmacology 3.0 is the repurposing of hallucinogens into novel therapies for posttraumatic stress disorder, anxiety, and depression.1 The opioid system is being recognized as another key player in depression, with many studies showing buprenorphine has antidepressant and anti-suicidal properties2 and the recent finding that pre-treatment with naloxone blocks the rapid antidepressive effects of ketamine.3 This finding casts doubt on the notion that the antidepressant mechanism of action of ketamine is solely mediated via its antagonism of the glutamate N-methyl-
These early developments in Psychopharmacology 3.0 augur well for the future. Companies in the pharmaceutical industry (which are hated by many, and even demonized and kept at arm’s length by major medical schools) are, in fact, the only entities in the world that develop new medications for psychiatric disorders, 82% of which still have no FDA-approved drug.4 Psychiatric researchers and clinicians should collaborate and advise the pharmaceutical companies about the urgent or unmet needs of psychiatric patients so they can target those unmet needs with their massive R&D resources.
In that spirit, here is my wish list of therapeutic targets that I hope will emerge during the Psychopharmacology 3.0 era and beyond:
1. New mechanisms of action for antipsychotics, based on emerging neurobiological research in schizophrenia and related psychoses, such as:
- Inhibit microglia activation
- Repair mitochondrial dysfunction
- Modulate the hypofunctional NMDA receptors
- Inhibit apoptosis
- Enhance neurogenesis
- Repair myelin pathology
- Inhibit neuroinflammation and oxidative stress
- Increase neurotropic growth factors
- Neurosteroid therapies (including estrogen)
- Exploit the microbiome influence on both the enteric and cephalic brains
2. Long-acting injectable antidepressants and mood stabilizers, because there is a malignant transformation into treatment-resistance in mood disorders after recurrent episodes due to nonadherence.5
3. Treatments for personality disorders, especially borderline and antisocial personality disorders.
4. An effective treatment for alcoholism.
5. Pharmacotherapy for aggression.
6. Vaccines for substance use.
7. Stage-specific pharmacotherapies (because the neurobiology of prodromal, first-episode, and multiple-episode patients have been shown to be quite different).
8. Drugs for epigenetic modulation to inhibit risk genes and to over-express protective genes.
It may take decades and hundreds of billions (even trillions) of R&D investment to accomplish the above, but I remain excited about the prospects of astounding psychopharmacologic advances to treat the disorders of the mind. Precision psychiatry advances will also expedite the selection of the right medication for each patient by employing predictive biomarkers. Breakthrough methodologies, such as pluripotent stem cells, opto-genetics, and clustered regularly interspaced short palindromic repeats (CRISPR), promise to revolutionize the biology, diagnosis, treatment, and prevention of various neuropsychiatric disorders.
The future of psychopharmacology is bright, if adequate resources are invested. The current direct and indirect costs of mental disorders and addictions are in the hundreds of billions of dollars annually. Only intensive research and disruptive discoveries will have the salutary dual effect of healing disease and reducing the economic burden of neuropsychiatric disorders. Psychopharmacology 3.0 advances, along with nonpharmacologic therapies such as neuromodulation (electroconvulsive therapy, transcranial magnetic stimulation, vagus nerve stimulation, and a dozen other techniques in development). Together with the indispensable evidence-based psychotherapies such as cognitive-behavioral therapy, dialectical behavior therapy, and interpersonal psychotherapy, psychopharmacology represents the leading edge of progress in psychiatric treatment. The psychiatrists of 1952 could only fantasize about what has since become a reality in healing ailing minds.
To comment on this editorial or other topics of interest: [email protected]
There is little doubt that the psychopharmacology revolution has been transformational for psychiatry and is also credited for sparking the momentous neuroscience advances of the past half century.
The field of psychiatry, dominated by Freudian psychology for decades, radically evolved from psychoanalysis to pharmacotherapy with the discovery that serious mental disorders are treatable with medications, thus dispensing with the couch.
Prior to 1952, the prevailing dogma was that “madness is irreversible.” That’s why millions of patients with various psychiatric disorders were locked up in institutions, which added to the stigma of mental illness. Then came the first antipsychotic drug, chlorpromazine, which “magically” eliminated the delusions and hallucinations of patients who had been hospitalized for years. That serendipitous and historic discovery was as transformational for psychiatry as penicillin was for infections (yet inexplicably, only the discovery of penicillin received a Nobel Prize). Most people today do not know that before chlorpromazine, 50% of all hospital beds in the U.S. were occupied by psychiatric patients. The massive shuttering of state hospitals in the 1970s and ’80s was a direct consequence of the widespread use of chlorpromazine and its cohort of first-generation antipsychotics (FGAs).
That was Psychopharmacology 1.0, spanning the period 1952 to 1987. It included dozens of FGAs belonging to 6 classes: phenothiazines, thioxanthenes, butyrophenones, dibenzazepines, dihydroindolones, and dibenzodiazepines. Psychopharmacology 1.0 also included monoamine oxidase inhibitors and tricyclic antidepressants for depression, and lithium for bipolar mania. Ironically, clozapine, the incognito seed template of the second-generation antipsychotic (SGA) class, was synthesized in 1959 with the early wave of FGAs, and launched in Europe in 1972, only to be withdrawn in 1974 due to agranulocytosis-induced deaths not recognized during the clinical trials.
The late 1980s ushered in Psychopharmacology 2.0, which was also transformative. It began in 1987 with the introduction of fluoxetine, the first selective serotonin receptor inhibitor. Then clozapine was resurrected in 1988 as the first FDA-approved drug for refractory schizophrenia. Being the first SGA (no acute extrapyramidal side effects at all, in contrast to all FGAs), it became the “mechanistic model” for all other SGA agents, which were introduced starting in 1993. All SGAs were designed by pharmaceutical companies’ medicinal chemists to mimic clozapine’s receptor profile: far stronger affinity to serotonin 5HT-2A receptors than to dopamine D2 receptors. Three partial agonists and several heterocyclic antidepressants were also introduced during this 2.0 era, which continued until approximately 2017. Of the 11 SGAs that were initially approved for schizophrenia, 7 also were approved for bipolar mania, and 2 received an FDA indication for bipolar depression, thus addressing a glaring unmet need.
Psychopharmacology 3.0 has already begun. Its seeds started sprouting over the past few years with the landmark studies of intravenous ketamine, which was demonstrated to reverse severe and refractory depression and suicidal urges within hours of injection. The first ketamine product, esketamine, an intranasal formulation, is expected to be approved by the FDA soon. In the same vein, other rapid-acting antidepressants, a welcome paradigm shift, are being developed, including IV scopolamine, IV rapastinel, and inhalable nitrous oxide.
Three novel and important pharmacologic agents have arrived in this 3.0 era:
- Pimavanserin, a serotonin 5HT-2A inverse agonist, the first and only non-dopamine–blocking antipsychotic approved by the FDA for the delusions and hallucinations of Parkinson’s disease psychosis. It is currently in clinical trials for schizophrenia and Alzheimer’s disease psychosis (for which nothing is yet approved).
- Valbenazine, the first drug approved for tardive dyskinesia (TD), the treatment of which had been elusive and remained a huge unmet need for 60 years. Its novel mechanism of action is inhibition of vesicular monoamine transporter 2 (VMAT2), which reduces the putative dopamine supersensitivity of TD.
- Deutetrabenazine, which was also approved for TD a few months after valbenazine, and has the same mechanism of action. It also was approved for Huntington’s chorea.
Continue to: Another important feature...
Another important feature of Psychopharmacology 3.0 is the repurposing of hallucinogens into novel therapies for posttraumatic stress disorder, anxiety, and depression.1 The opioid system is being recognized as another key player in depression, with many studies showing buprenorphine has antidepressant and anti-suicidal properties2 and the recent finding that pre-treatment with naloxone blocks the rapid antidepressive effects of ketamine.3 This finding casts doubt on the notion that the antidepressant mechanism of action of ketamine is solely mediated via its antagonism of the glutamate N-methyl-
These early developments in Psychopharmacology 3.0 augur well for the future. Companies in the pharmaceutical industry (which are hated by many, and even demonized and kept at arm’s length by major medical schools) are, in fact, the only entities in the world that develop new medications for psychiatric disorders, 82% of which still have no FDA-approved drug.4 Psychiatric researchers and clinicians should collaborate and advise the pharmaceutical companies about the urgent or unmet needs of psychiatric patients so they can target those unmet needs with their massive R&D resources.
In that spirit, here is my wish list of therapeutic targets that I hope will emerge during the Psychopharmacology 3.0 era and beyond:
1. New mechanisms of action for antipsychotics, based on emerging neurobiological research in schizophrenia and related psychoses, such as:
- Inhibit microglia activation
- Repair mitochondrial dysfunction
- Modulate the hypofunctional NMDA receptors
- Inhibit apoptosis
- Enhance neurogenesis
- Repair myelin pathology
- Inhibit neuroinflammation and oxidative stress
- Increase neurotropic growth factors
- Neurosteroid therapies (including estrogen)
- Exploit the microbiome influence on both the enteric and cephalic brains
2. Long-acting injectable antidepressants and mood stabilizers, because there is a malignant transformation into treatment-resistance in mood disorders after recurrent episodes due to nonadherence.5
3. Treatments for personality disorders, especially borderline and antisocial personality disorders.
4. An effective treatment for alcoholism.
5. Pharmacotherapy for aggression.
6. Vaccines for substance use.
7. Stage-specific pharmacotherapies (because the neurobiology of prodromal, first-episode, and multiple-episode patients have been shown to be quite different).
8. Drugs for epigenetic modulation to inhibit risk genes and to over-express protective genes.
It may take decades and hundreds of billions (even trillions) of R&D investment to accomplish the above, but I remain excited about the prospects of astounding psychopharmacologic advances to treat the disorders of the mind. Precision psychiatry advances will also expedite the selection of the right medication for each patient by employing predictive biomarkers. Breakthrough methodologies, such as pluripotent stem cells, opto-genetics, and clustered regularly interspaced short palindromic repeats (CRISPR), promise to revolutionize the biology, diagnosis, treatment, and prevention of various neuropsychiatric disorders.
The future of psychopharmacology is bright, if adequate resources are invested. The current direct and indirect costs of mental disorders and addictions are in the hundreds of billions of dollars annually. Only intensive research and disruptive discoveries will have the salutary dual effect of healing disease and reducing the economic burden of neuropsychiatric disorders. Psychopharmacology 3.0 advances, along with nonpharmacologic therapies such as neuromodulation (electroconvulsive therapy, transcranial magnetic stimulation, vagus nerve stimulation, and a dozen other techniques in development). Together with the indispensable evidence-based psychotherapies such as cognitive-behavioral therapy, dialectical behavior therapy, and interpersonal psychotherapy, psychopharmacology represents the leading edge of progress in psychiatric treatment. The psychiatrists of 1952 could only fantasize about what has since become a reality in healing ailing minds.
To comment on this editorial or other topics of interest: [email protected]
1. Nasrallah, HA. Maddening therapies: How hallucinogens morphed into novel treatments. Current Psychiatry. 2017;16(1):19-21.
2. Serafini G, Adavastro G, Canepa G, et al. The efficacy of buprenorphine in major depression, treatment-resistant depression and suicidal behavior: a systematic review. Int J Mol Sci. 2018;19(8). doi: 10.3390/ijms19082410.
3. Williams NR, Heifets BD, Blasey C, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am J Psychiatry. 2018. doi: 10.1176/appi.ajp.2018.18020138. [Epub ahead of print].
4. Devulapalli KK, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders. The majority of psychiatric diagnoses have no approved drug. Asian J Psychiatr. 2009;2(1):29-36.
5. Post RM. Preventing the malignant transformation of bipolar disorder. JAMA. 2018;319(12):1197-1198.
1. Nasrallah, HA. Maddening therapies: How hallucinogens morphed into novel treatments. Current Psychiatry. 2017;16(1):19-21.
2. Serafini G, Adavastro G, Canepa G, et al. The efficacy of buprenorphine in major depression, treatment-resistant depression and suicidal behavior: a systematic review. Int J Mol Sci. 2018;19(8). doi: 10.3390/ijms19082410.
3. Williams NR, Heifets BD, Blasey C, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am J Psychiatry. 2018. doi: 10.1176/appi.ajp.2018.18020138. [Epub ahead of print].
4. Devulapalli KK, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders. The majority of psychiatric diagnoses have no approved drug. Asian J Psychiatr. 2009;2(1):29-36.
5. Post RM. Preventing the malignant transformation of bipolar disorder. JAMA. 2018;319(12):1197-1198.

Treatment-resistant OCD: There’s more we can do
Treatment-resistant OCD can be a debilitating condition. Diagnostic clarity is crucial to fully elicit symptoms and identify comorbid conditions in order to develop practical, evidence-based treatment strategies and improve the patient’s and family’s quality of life. In this article, we delineate first-line strategies for treatment-resistant OCD and then review augmentation strategies, with an emphasis on glutamate-modulating agents.
Making the diagnosis
The diagnosis of OCD is made when a patient meets DSM-5 criteria for the presence of obsessions and/or compulsions, which are defined as unwanted, distressing, intrusive, recurrent thoughts or images (obsessions) and repetitive behaviors or mental acts (compulsions).1 OCD is considered a chronic waxing and waning disorder; stress and lack of sleep lead to worsening symptoms. The hidden nature of symptoms and the reinforcement provided by the reduction in anxiety after performing a compulsion contribute to sustained illness. Eliciting symptoms from patients may be challenging due to the shame they may feel. When reviewing symptoms on the Y-BOCS, it is helpful to preface questions with statements such as “Many people report excessive concern or disgust with…” to help the patient feel understood and less anxious, rather than using direct queries, such as “Are you bothered by…?”

Consider comorbid conditions
After making the initial diagnosis of OCD, it is important to assess whether the symptoms are better accounted for by another condition, and whether comorbid conditions are present (Table 1).
CASE CONTINUED
Ruling out other diagnoses
_
Initial treatment: CBT
Cognitive-behavioral therapy with exposures and response prevention (from here on referred to as CBT) has been established as a first-line, evidence-based treatment for OCD in both children and adults.2,3 For patients with treatment-resistant OCD, intensive daily CBT in a partial hospitalization or inpatient setting that is a tailor-made, patient-specific program is one of the most effective treatments, with response rates of up to 70%4-8 CBT’s advantages over medication include lower relapse rates and no known adverse effects. Unfortunately, CBT is underused9-11 due in part to a shortage of trained clinicians, and because patients may favor the ease of taking medication over the time, effort, and cost involved in CBT.
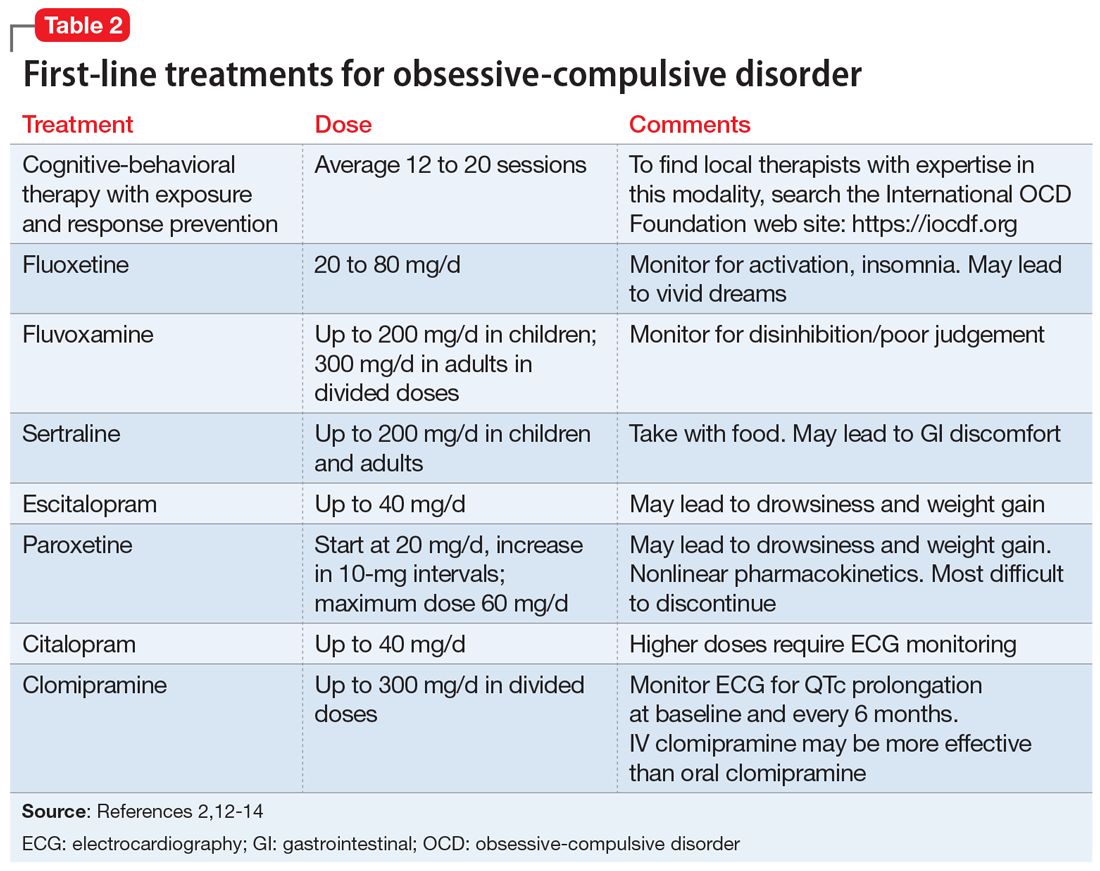
First-line pharmacologic options for treating OCD are SSRIs and clomipramine, as supported by multiple randomized controlled trials (RCTs), meta-analyses, expert guidelines, and consensus statements (Table 22,12-14). No significant difference has been found among SSRIs for the treatment of OCD in a review of 17 studies that included more than 3,000 patients.15 Treatment with SSRIs or clomipramine is effective for 50% to 60% of patients.16 Many clinicians view the combination of an SSRI and CBT as the treatment of choice for OCD.2
Continue to: Reluctance to engage in CBT
CASE CONTINUED
Reluctance to engage in CBT
To determine the next course of action, you review Mr. S’s treatment history. He has received adequate doses of 2 SSRIs and currently is taking clomipramine, 100 mg twice daily. He recently began CBT, which includes homework to help face his fears; however, Mr. S is reluctant to complete the exposure assignments, and after pausing for a few seconds as he tries to resist sending an apology email to his coworkers, he then returns to his compulsive behavior.
Facing treatment resistance
Although currently there isn’t a cure to resolve all traces of OCD, the goal of treatment is to decrease distress, interference, and the frequency of symptoms to a minimal level such that only the patients themselves are aware of symptoms. In broad terms, “response” has been defined as a decrease in symptoms, and “remission” has been defined as minimal symptoms after treatment.
Close to half of adults treated for OCD respond well to standard-of-care treatment (CBT and/or an SSRI), while the other 50% are considered partial responders or nonresponders.2 For patients with OCD, researchers often define “treatment response” as a ≥25% reduction in symptom severity score on the Y-BOCS. Approximately 30% of adults with OCD do not respond substantially to the first-line treatments, and even those who are defined as “responders” in research studies typically continue to have significant symptoms that impact their quality of life.2 In children, a clinical definition for treatment-refractory OCD has been presented as failing to achieve adequate symptom relief despite receiving an adequate course of CBT and at least 2 adequate trials of an SSRI or clomipramine.17 In the Pediatric OCD Treatment Study (POTS) trial, >46% of youth did not achieve remission from their OCD symptoms, even after receiving evidence-based care provided by experienced clinicians (combined treatment with CBT and an SSRI).18
_
Challenges in psychotherapy
Compassion is a key element in developing rapport with patients to help them face increasingly more challenging exposures. Making OCD the problem, not the person, is an essential element in helping patients move forward. Some clinicians may become frustrated with patients when treatment is not moving along well, referring to resistance, denial, or sabotage. According to March and Mulle,19 these terms lack the recognition and compassion that exposures are inherently difficult.19
Another challenge for therapists is if the patient’s presenting symptoms are personally offensive or a sensitive topic. For example, a therapist who is disgusted by public restrooms will find it difficult to tolerate the risks associated with exposure to germs and support a patient in touching objects in the restroom. Therapists also may be challenged when the patient’s fears align with the therapist’s religious beliefs. In these situations, consider transferring care to another therapist.
Family members need to learn about the nature of the illness and their roles in helping patients improve. Family members may unknowingly enable symptoms or criticize patients for their lack of motivation, which can lead to conflict in the home. Family dysfunction can in turn worsen OCD symptoms.
The most likely cause of lack of response to therapy is inexpert CBT.19 Deep breathing and relaxation training have been used as an active placebo in studies20; in a meta-analysis examining the effective components of CBT, studies that added relaxation training were not more effective than those that employed exposures alone.21 Patients receiving CBT should be able to articulate the hierarchical approach used to gradually face their fears.
Continue to: Pharmacologic augmentation strategies
Pharmacologic augmentation strategies
Selective serotonin reuptake inhibitors. While most OCD research trials have assessed SSRIs in 12-week studies, clinicians may consider extending SSRI treatment for an additional 12 weeks for nonresponders because some patients will continue to make gains. In the past, it was generally believed that higher doses of SSRIs are needed for treating OCD than for treating major depressive disorder. For instance, greater improvement was seen with 250 to 400 mg/d of sertraline compared with 200 mg/d22 and with escitalopram after an increase of dose up to 50 mg/d.23 However, more recently, this notion of higher doses being necessary for treatment response has been called into question. For example, a study of escitalopram found similar responses to 10 mg/d vs 20 mg/d after 24 weeks.24 A meta-analysis of adult studies of SSRIs for OCD supported higher doses as being more effective, but noted that the drop-out rate from treatment was greater in patients treated with higher doses.25 As a note of caution, long-term, high-dose maintenance therapy increases the risk of adverse reactions.26
Following a failed treatment with a first SSRI, it remains debatable as to what ought to be the second pharmacologic treatment. Although clomipramine is often reserved for treatment after 2 failed trials of an SSRI due to its greater risk of adverse effects, in an open-label study, switching from an SSRI to clomipramine led to greater response than switching from one SSRI to another.27 On the other hand, while meta-analyses have reported greater treatment effect for oral clomipramine than for SSRIs, direct head-to-head comparisons have not supported this notion.28 To get the best of both worlds, some clinicians employ a strategy of combining clomipramine with an SSRI, while monitoring for adverse effects and interactions such as serotonin syndrome.29-31
Benzodiazepines. Although benzodiazepines are useful for brief treatment of an anxiety disorder (eg, for a person with a fear of heights who needs to take an airplane),32 they have not been shown to be effective for OCD33 or as augmentation to an SSRI.34
N-acetylcysteine (NAC). Two RCTs of adults with OCD who received adjunctive NAC, 3 g/d in divided doses, found no significant difference in the treatment arms by the conclusion of 16 weeks—either both groups improved, or both groups failed to improve.35,36 In a 10-week study of patients with moderate to severe OCD symptoms, NAC, 2 g/d, as augmentation to fluvoxamine, 200 mg/d, showed a significant time x interaction in the treatment group.37 No follow-up information is available, however.
In a multicenter RCT of NAC given to children and adolescents with OCD as augmentation to citalopram, symptoms decreased and the quality-of-life score improved, with a large treatment effect size in the NAC group.38 However, in a study aimed at examining NAC in youth with Tourette syndrome, OCD symptoms were measured as a secondary outcome and there was no benefit of NAC over placebo.39
Memantine. Four 8- to 12-week RCTs in adults with OCD favored adjunctive memantine, 20 mg/d, taken with an SSRI, over placebo.40-43 A small study suggests that patients with OCD may be more likely to respond to memantine than patients with generalized anxiety disorder.44 Case reports have noted that memantine has been beneficial for pediatric patients with refractory OCD.45
Continue to: Topiramate
Topiramate. Three 12-week RCTs examined topiramate augmentation at 100 to 400 mg/d in patients with OCD who had failed at least 1 previous trial of an SSRI. The earliest study was most encouraging: Y-BOCS scores decreased by 32% in the topiramate group but by only 2.4% in the placebo group.46 However, the other 2 studies found no difference in the final OCD symptom severity score between active treatment and placebo groups,47,48 and the use of topiramate, particularly at higher doses, was limited by its adverse effects.
Lamotrigine. Initially, lamotrigine augmentation of SSRIs in OCD did not appear to be helpful.49 More recently, several case studies reported that lamotrigine, 100 to 200 mg/d, added to paroxetine or clomipramine, resulted in dramatic improvement in Y-BOCS scores for patients with long-standing refractory symptoms.50,51 In a retrospective review of 22 patients who received augmentation with lamotrigine, 150 mg/d, 20 had a significant response; the mean decrease in Y-BOCS score was 67%.52 Finally, in a 16-week RCT, lamotrigine, 100 mg/d, added to an SSRI led to a significant decrease in both Y-BOCS score and depressive symptoms while also improving semantic fluency.53
Ketamine. Ketamine is drawing increased attention for its nearly instantaneous antidepressant effect that lasts for up to 2 weeks after a single infusion.54 In a study of 15 medication-free adults with continuous intrusive obsessions, 4 of 8 patients who received a single IV infusion of ketamine, 0.5 mg/kg, met the criteria for treatment response (>35% reduction in Y-BOCS score measured 1 week later); none of the patients who received a placebo infusion of saline met this criteria.55 A small open-label trial of 10 treatment-refractory patients found that an infusion of ketamine, 0.5 mg/kg, was beneficial for comorbid depression but had only a minimal effect on OCD symptoms measured 3 days post-infusion.56 A short-term follow-up on these patients revealed dysphoria in some responders.57
D-cycloserine. The idea of using a pharmacologic agent to increase the speed or efficacy of behavioral therapy is intriguing. Proof of concept was demonstrated in a study that found that giving D-cycloserine prior to computerized exposure therapy significantly improved clinical response in patients with acrophobia.58 However, using this approach to treating OCD netted mixed results; D-cycloserine was found to be most helpful during early stages of treatment.59,60
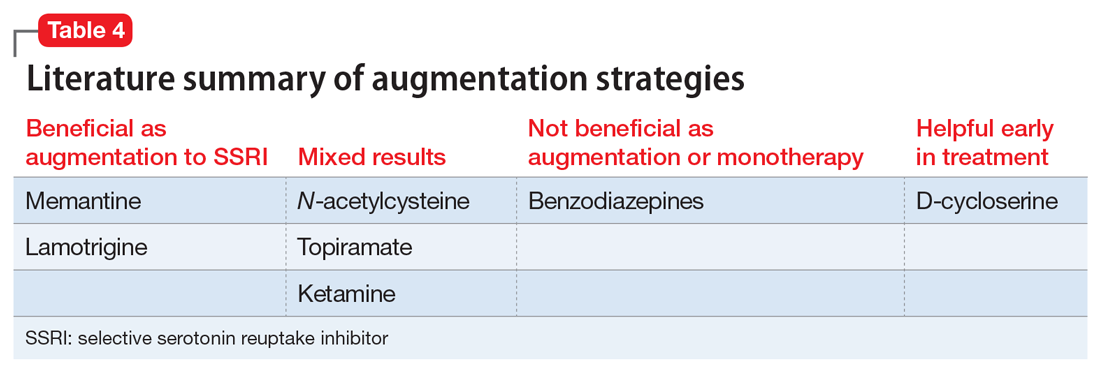
Table 3 outlines the mechanisms of action and common uses for NAC, memantine, ketamine, topiramate, lamotrigine, and D-cycloserine. Table 4 summarizes the literature on the efficacy of some of the augmentation strategies for treating OCD described in this article.

Continue to: Alternative strategies
Alternative strategies
Augmentation strategies with neuroleptics,61 transcranial magnetic stimulation,62 and deep brain stimulation63 have recently been reviewed. Space limitations preclude a comprehensive review of these strategies, but in a cross-sectional study of augmentation strategies in OCD, no difference was found in terms of symptom severity between those prescribed SSRI monotherapy or augmentation with neuroleptics, benzodiazepines, or antidepressants.64
CASE CONTINUED
Progress in CBT
Mr. S agrees to a trial of NAC as an augmentation strategy, but after 8 weeks of treatment with NAC, 600 mg twice daily, his Y-BOCS had declined by only 2 points. He also complains of nausea and does not want to increase the dose. You discontinue NAC and opt to further explore his reaction to CBT. Mr. S shares that he has been seeing his psychologist only once every 3 weeks because he does not want to miss work. You encourage him to increase to weekly CBT sessions, and you obtain his permission to contact his therapist and his family members. Fortunately, his therapist is highly qualified, but unfortunately, Mr. S’s father has been sending him multiple critical emails about not advancing at his job and for being “lazy” at work. You schedule a session with Mr. S and his father. Great progress is made after Mr. S and his father both share their frustrations and come to understand and appreciate each other’s struggles. Four weeks later, after weekly CBT appointments, Mr. S has a Y-BOCS of 18 and spends <2 hours/d checking emails for errors and apologizing.
Bottom Line
It is unrealistic to expect OCD symptoms to be cured. Many ‘treatment-resistant’ patients have not received properly delivered cognitive-behavioral therapy, and this first-line treatment modality should be considered in every eligible patient, and augmented with a selective serotonin reuptake inhibitor (SSRI) when needed. Glutamatergic agents, in turn, can augment SSRIs.
Related Resources
- Yale-Brown Obsessive-Compulsive Scale. https://iocdf.org/ wp-content/uploads/2014/08/Assessment-Tools.pdf.
- The International OCD Foundation. https://iocdf.org.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Ketamine • Ketalar
Lamotrigine • Lamictal
Memantine • Namenda
Paroxetine • Paxil
Sertraline • Zoloft
Topiramate • Topomax
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Koran LM, Hanna GL, Hollander E, et al. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry; 2007;164(suppl 7):5-53.
3. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
4. Bystritsky A, Munford PR, Rosen RM, et al. A preliminary study of partial hospital management of severe obsessive-compulsive disorder. Psychiatr Serv. 1996;47(2):170-174.
5. Calvocoressi L, McDougle CI, Wasylink S, et al. Inpatient treatment of patients with severe obsessive-compulsive disorder. Hosp Community Psychiatry. 1993;44(12):1150-1154.
6. Eddy KT, Dutra L, Bradley R, et al. A multidimensional meta-analysis of psychotherapy and pharmacotherapy for obsessive-compulsive disorder. Clin Psychol Rev. 2004;24(8):1011-1030.
7. Abramowitz JS. The psychological treatment of obsessive-compulsive disorder. Can J Psychiatry. 2006;51(7):407-416.
8. Simpson HB, Huppert JD, Petkova E, et al. Response versus remission in obsessive-compulsive disorder. J Clin Psychiatry. 2006;67(2):269-276.
9. Marques L, LeBlanc NJ, Weingarden HM, et al. Barriers to treatment and service utilization in an internet sample of individuals with obsessive-compulsive symptoms. Depress Anxiety. 2010;27(5):470-475.
10. Goodwin R, Koenen KC, Hellman F, et al. Helpseeking and access to mental health treatment for obsessive-compulsive disorder. Acta Psychiatr Scand. 2002;106(2):143-149.
11. Kohn R, Saxena S, Levav I, et al. The treatment gap in mental health care. Bull World Health Organ. 2004;82(11):858-866.
12. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439.
13. Lovell K, Bee P. Implementing the NICE OCD/BDD guidelines. Psychol Psychother. 2008;81(Pt 4):365-376.
14. Bandelow B, Sher L, Bunevicius R, et al. Guidelines for the pharmacological treatment of anxiety disorders, obsessive-compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract. 2012;16(2):77-84.
15. Soomro GM, Altman D, Rajagopal S, et al. Selective serotonin re-uptake inhibitors (SSRIs) versus placebo for obsessive compulsive disorder (OCD). Cochrane Database Syst Rev. 2008;(1):CD001765.
16. Pittenger C, Bloch MH. Pharmacological treatment of obsessive-compulsive disorder. Psychiatr Clin North Am. 2014;37(3):375-391.
17. Bloch MH, Storch EA. Assessment and management of treatment-refractory obsessive-compulsive disorder in children. J Am Acad Child Adolesc Psychiatry. 2015;54(4):251-262.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
19. March JS, Mulle K. OCD in children and adolescents: a cognitive-behavioral treatment manual. New York, NY: Guilford Press; 1998.
20. Marks IM. Fears, phobias, and rituals: Panic, anxiety, and their disorders. 1987, New York, NY: Oxford University Press; 1987.
21. Ale CM, McCarthy DM, Rothschild LM, et al. Components of cognitive behavioral therapy related to outcome in childhood anxiety disorders. Clin Child Fam Psychol Rev. 2015;18(3):240-251.
22. Ninan PT, Koran LM, Kiev A, et al. High-dose sertraline strategy for nonresponders to acute treatment for obsessive-compulsive disorder: a multicenter double-blind trial. J Clin Psychiatry. 2006;67(1):15-22.
23. Rabinowitz I, Baruch Y, Barak Y. High-dose escitalopram for the treatment of obsessive-compulsive disorder. Int Clin Psychopharmacol. 2008;23(1):49-53.
24. Stein DJ, Andersen EW, Tonnoir B, et al. Escitalopram in obsessive-compulsive disorder: a randomized, placebo-controlled, paroxetine-referenced, fixed-dose, 24-week study. Curr Med Res Opin. 2007;23(4):701-711.
25. Bloch MH, McGuire J, Landeros-Weisenberger A, et al. Meta-analysis of the dose-response relationship of SSRI in obsessive-compulsive disorder. Mol Psychiatry. 2010;15(8):850-855.
26. Sayyah M, Majzoob S, Sayyah M. Metabolic and toxicological considerations for obsessive-compulsive disorder drug therapy. Expert Opin Drug Metab Toxicol. 2013;9(6):657-673.
27. Hollander E, Bienstock CA, Koran LM, et al. Refractory obsessive-compulsive disorder: state-of-the-art treatment. J Clin Psychiatry. 2002;63(suppl 6):20-29.
28. Fineberg NA, Gale TM. Evidence-based pharmacotherapy of obsessive-compulsive disorder. Int J Neuropsychopharmacol. 2005;8(1):107-129.
29. Marazziti D, Golia F, Consoli G, et al. Effectiveness of long-term augmentation with citalopram to clomipramine in treatment-resistant OCD patients. CNS Spectr. 2008;13(11):971-976.
30. Browne M, Horn E, Jones TT. The benefits of clomipramine-fluoxetine combination in obsessive compulsive disorder. Can J Psychiatry. 1993;38(4):242-243.
31. Ravizza L, Barzega G, Bellino S, et al. Drug treatment of obsessive-compulsive disorder (OCD): long-term trial with clomipramine and selective serotonin reuptake inhibitors (SSRIs). Psychopharmacol Bull. 1996;32(1):167-173.
32. Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci. 2011;13(4):423-437.
33. Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
34. Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam with sertraline in obsessive-compulsive disorder. Ann Clin Psychiatry. 2004;16(3):127-132.
35. Costa DLC, Diniz JB, Requena G, et al. Randomized, double-blind, placebo-controlled trial of n-acetylcysteine augmentation for treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2017;78(7):e766-e773.
36. Sarris J, Oliver G, Camfield DA, et al. N-Acetyl Cysteine (NAC) in the treatment of obsessive-compulsive disorder: a 16-week, double-blind, randomised, placebo-controlled study. CNS Drugs. 2015;29(9):801-809.
37. Paydary K, Akamaloo A, Ahmadipour A, et al. N-acetylcysteine augmentation therapy for moderate-to-severe obsessive-compulsive disorder: randomized, double-blind, placebo-controlled trial. J Clin Pharm Ther. 2016;41(2):214-219.
38. Ghanizadeh A, Mohammadi MR, Bahraini S, et al. Efficacy of N-acetylcysteine augmentation on obsessive compulsive disorder: a multicenter randomized double blind placebo controlled clinical trial. Iran J Psychiatry. 2017;12(2):134-141.
39. Bloch MH, Panza KE, Yaffa A, et al. N-acetylcysteine in the treatment of pediatric tourette syndrome: randomized, double-blind, placebo-controlled add-on trial. J Child Adolesc Psychopharmacol. 2016;26(4):327-334.
40. Ghaleiha A, Entezari N, Modabbernia A, et al. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47(2):175-180.
41. Stewart SE, Jenike EA, Hezel DM, et al. A single-blinded case-control study of memantine in severe obsessive-compulsive disorder. J Clin Psychopharmacol. 2010;30(1):34-39.
42. Modarresi A, Sayyah M, Razooghi S, et al. Memantine augmentation improves symptoms in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder: a randomized controlled trial. Pharmacopsychiatry. 2017. doi: 10.1055/s-0043-120268. [Epub ahead of print].
43. Haghighi M, Jahangard L, Mohammad-Beigi H, et al. In a double-blind, randomized and placebo-controlled trial, adjuvant memantine improved symptoms in inpatients suffering from refractory obsessive-compulsive disorders (OCD). Psychopharmacology (Berl). 2013;228(4):633-640.
44. Feusner JD, Kerwin L, Saxena S, et al. Differential efficacy of memantine for obsessive-compulsive disorder vs. generalized anxiety disorder: an open-label trial. Psychopharmacol Bull. 2009;42(1):81-93.
45. Hezel DM, Beattie K, Stewart SE. Memantine as an augmenting agent for severe pediatric OCD. Am J Psychiatry. 2009;166(2):237.
46. Mowla A, Khajeian AM, Sahraian A, et al. topiramate augmentation in resistant ocd: a double-blind placebo-controlled clinical trial. CNS Spectr. 2010;15(11):613-617.
47. Berlin H, Koran LM, Jenike MA, et al. Double-blind, placebo-controlled trial of topiramate augmentation in treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2011;72(5):716-721.
48. Afshar H, Akuchekian S, Mahaky B, et al. Topiramate augmentation in refractory obsessive-compulsive disorder: A randomized, double-blind, placebo-controlled trial. J Res Med Sci. 2014;19(10):976-981.
49. Kumar TC, Khanna S. Lamotrigine augmentation of serotonin re-uptake inhibitors in obsessive-compulsive disorder. Aust N Z J Psychiatry. 2000;34(3):527-528.
50. Arrojo-Romero M, Tajes Alonso M, de Leon J. Lamotrigine augmentation of serotonin reuptake inhibitors in severe and long-term treatment-resistant obsessive-compulsive disorder. Case Rep Psychiatry. 2013;2013:612459.
51. Uzun O. Lamotrigine as an augmentation agent in treatment-resistant obsessive-compulsive disorder: a case report. J Psychopharmacol. 2010;24(3):425-427.
52. Hussain A, Dar MA, Wani RA, et al. Role of lamotrigine augmentation in treatment-resistant obsessive compulsive disorder: a retrospective case review from South Asia. Indian J Psychol Med. 2015;37(2):154-158.
53. Bruno A, Micò U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456-1462.
54. Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73(12):113311-41.
55. Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38(12):2475-2483.
56. Bloch MH, Wasylink S, Landeros-Weisenberger A,, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biol Psychiatry. 2012;72(11):964-970.
57. Niciu MJ, Grunschel BD, Corlett PR, et al. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
58. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61(11):1136-1144.
59. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
60. Xia J, Du Y, Han J, et al. D-cycloserine augmentation in behavioral therapy for obsessive-compulsive disorder: a meta-analysis. Drug Des Devel Ther. 2015;9:2101-2117.
61. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and meta-analysis. BMC Psychiatry. 2014;14:317.
62. Guo Q, Li C, Wang J. Updated review on the clinical use of repetitive transcranial magnetic stimulation in psychiatric disorders. Neurosci Bull. 2017;33(6):747-756.
63. Naesström, M, Blomstedt P, Bodlund O. A systematic review of psychiatric indications for deep brain stimulation, with focus on major depressive and obsessive-compulsive disorder. Nord J Psychiatry. 2016;70(7):483-491.
64. Van Ameringen M, Simpson W, Patterson B, et al. Pharmacological treatment strategies in obsessive compulsive disorder: A cross-sectional view in nine international OCD centers. J Psychopharmacol, 2014;28(6):596-602.
Treatment-resistant OCD can be a debilitating condition. Diagnostic clarity is crucial to fully elicit symptoms and identify comorbid conditions in order to develop practical, evidence-based treatment strategies and improve the patient’s and family’s quality of life. In this article, we delineate first-line strategies for treatment-resistant OCD and then review augmentation strategies, with an emphasis on glutamate-modulating agents.
Making the diagnosis
The diagnosis of OCD is made when a patient meets DSM-5 criteria for the presence of obsessions and/or compulsions, which are defined as unwanted, distressing, intrusive, recurrent thoughts or images (obsessions) and repetitive behaviors or mental acts (compulsions).1 OCD is considered a chronic waxing and waning disorder; stress and lack of sleep lead to worsening symptoms. The hidden nature of symptoms and the reinforcement provided by the reduction in anxiety after performing a compulsion contribute to sustained illness. Eliciting symptoms from patients may be challenging due to the shame they may feel. When reviewing symptoms on the Y-BOCS, it is helpful to preface questions with statements such as “Many people report excessive concern or disgust with…” to help the patient feel understood and less anxious, rather than using direct queries, such as “Are you bothered by…?”
Consider comorbid conditions
After making the initial diagnosis of OCD, it is important to assess whether the symptoms are better accounted for by another condition, and whether comorbid conditions are present (Table 1).
CASE CONTINUED
Ruling out other diagnoses
_
Initial treatment: CBT
Cognitive-behavioral therapy with exposures and response prevention (from here on referred to as CBT) has been established as a first-line, evidence-based treatment for OCD in both children and adults.2,3 For patients with treatment-resistant OCD, intensive daily CBT in a partial hospitalization or inpatient setting that is a tailor-made, patient-specific program is one of the most effective treatments, with response rates of up to 70%4-8 CBT’s advantages over medication include lower relapse rates and no known adverse effects. Unfortunately, CBT is underused9-11 due in part to a shortage of trained clinicians, and because patients may favor the ease of taking medication over the time, effort, and cost involved in CBT.
First-line pharmacologic options for treating OCD are SSRIs and clomipramine, as supported by multiple randomized controlled trials (RCTs), meta-analyses, expert guidelines, and consensus statements (Table 22,12-14). No significant difference has been found among SSRIs for the treatment of OCD in a review of 17 studies that included more than 3,000 patients.15 Treatment with SSRIs or clomipramine is effective for 50% to 60% of patients.16 Many clinicians view the combination of an SSRI and CBT as the treatment of choice for OCD.2
Continue to: Reluctance to engage in CBT
CASE CONTINUED
Reluctance to engage in CBT
To determine the next course of action, you review Mr. S’s treatment history. He has received adequate doses of 2 SSRIs and currently is taking clomipramine, 100 mg twice daily. He recently began CBT, which includes homework to help face his fears; however, Mr. S is reluctant to complete the exposure assignments, and after pausing for a few seconds as he tries to resist sending an apology email to his coworkers, he then returns to his compulsive behavior.
Facing treatment resistance
Although currently there isn’t a cure to resolve all traces of OCD, the goal of treatment is to decrease distress, interference, and the frequency of symptoms to a minimal level such that only the patients themselves are aware of symptoms. In broad terms, “response” has been defined as a decrease in symptoms, and “remission” has been defined as minimal symptoms after treatment.
Close to half of adults treated for OCD respond well to standard-of-care treatment (CBT and/or an SSRI), while the other 50% are considered partial responders or nonresponders.2 For patients with OCD, researchers often define “treatment response” as a ≥25% reduction in symptom severity score on the Y-BOCS. Approximately 30% of adults with OCD do not respond substantially to the first-line treatments, and even those who are defined as “responders” in research studies typically continue to have significant symptoms that impact their quality of life.2 In children, a clinical definition for treatment-refractory OCD has been presented as failing to achieve adequate symptom relief despite receiving an adequate course of CBT and at least 2 adequate trials of an SSRI or clomipramine.17 In the Pediatric OCD Treatment Study (POTS) trial, >46% of youth did not achieve remission from their OCD symptoms, even after receiving evidence-based care provided by experienced clinicians (combined treatment with CBT and an SSRI).18
_
Challenges in psychotherapy
Compassion is a key element in developing rapport with patients to help them face increasingly more challenging exposures. Making OCD the problem, not the person, is an essential element in helping patients move forward. Some clinicians may become frustrated with patients when treatment is not moving along well, referring to resistance, denial, or sabotage. According to March and Mulle,19 these terms lack the recognition and compassion that exposures are inherently difficult.19
Another challenge for therapists is if the patient’s presenting symptoms are personally offensive or a sensitive topic. For example, a therapist who is disgusted by public restrooms will find it difficult to tolerate the risks associated with exposure to germs and support a patient in touching objects in the restroom. Therapists also may be challenged when the patient’s fears align with the therapist’s religious beliefs. In these situations, consider transferring care to another therapist.
Family members need to learn about the nature of the illness and their roles in helping patients improve. Family members may unknowingly enable symptoms or criticize patients for their lack of motivation, which can lead to conflict in the home. Family dysfunction can in turn worsen OCD symptoms.
The most likely cause of lack of response to therapy is inexpert CBT.19 Deep breathing and relaxation training have been used as an active placebo in studies20; in a meta-analysis examining the effective components of CBT, studies that added relaxation training were not more effective than those that employed exposures alone.21 Patients receiving CBT should be able to articulate the hierarchical approach used to gradually face their fears.
Continue to: Pharmacologic augmentation strategies
Pharmacologic augmentation strategies
Selective serotonin reuptake inhibitors. While most OCD research trials have assessed SSRIs in 12-week studies, clinicians may consider extending SSRI treatment for an additional 12 weeks for nonresponders because some patients will continue to make gains. In the past, it was generally believed that higher doses of SSRIs are needed for treating OCD than for treating major depressive disorder. For instance, greater improvement was seen with 250 to 400 mg/d of sertraline compared with 200 mg/d22 and with escitalopram after an increase of dose up to 50 mg/d.23 However, more recently, this notion of higher doses being necessary for treatment response has been called into question. For example, a study of escitalopram found similar responses to 10 mg/d vs 20 mg/d after 24 weeks.24 A meta-analysis of adult studies of SSRIs for OCD supported higher doses as being more effective, but noted that the drop-out rate from treatment was greater in patients treated with higher doses.25 As a note of caution, long-term, high-dose maintenance therapy increases the risk of adverse reactions.26
Following a failed treatment with a first SSRI, it remains debatable as to what ought to be the second pharmacologic treatment. Although clomipramine is often reserved for treatment after 2 failed trials of an SSRI due to its greater risk of adverse effects, in an open-label study, switching from an SSRI to clomipramine led to greater response than switching from one SSRI to another.27 On the other hand, while meta-analyses have reported greater treatment effect for oral clomipramine than for SSRIs, direct head-to-head comparisons have not supported this notion.28 To get the best of both worlds, some clinicians employ a strategy of combining clomipramine with an SSRI, while monitoring for adverse effects and interactions such as serotonin syndrome.29-31
Benzodiazepines. Although benzodiazepines are useful for brief treatment of an anxiety disorder (eg, for a person with a fear of heights who needs to take an airplane),32 they have not been shown to be effective for OCD33 or as augmentation to an SSRI.34
N-acetylcysteine (NAC). Two RCTs of adults with OCD who received adjunctive NAC, 3 g/d in divided doses, found no significant difference in the treatment arms by the conclusion of 16 weeks—either both groups improved, or both groups failed to improve.35,36 In a 10-week study of patients with moderate to severe OCD symptoms, NAC, 2 g/d, as augmentation to fluvoxamine, 200 mg/d, showed a significant time x interaction in the treatment group.37 No follow-up information is available, however.
In a multicenter RCT of NAC given to children and adolescents with OCD as augmentation to citalopram, symptoms decreased and the quality-of-life score improved, with a large treatment effect size in the NAC group.38 However, in a study aimed at examining NAC in youth with Tourette syndrome, OCD symptoms were measured as a secondary outcome and there was no benefit of NAC over placebo.39
Memantine. Four 8- to 12-week RCTs in adults with OCD favored adjunctive memantine, 20 mg/d, taken with an SSRI, over placebo.40-43 A small study suggests that patients with OCD may be more likely to respond to memantine than patients with generalized anxiety disorder.44 Case reports have noted that memantine has been beneficial for pediatric patients with refractory OCD.45
Continue to: Topiramate
Topiramate. Three 12-week RCTs examined topiramate augmentation at 100 to 400 mg/d in patients with OCD who had failed at least 1 previous trial of an SSRI. The earliest study was most encouraging: Y-BOCS scores decreased by 32% in the topiramate group but by only 2.4% in the placebo group.46 However, the other 2 studies found no difference in the final OCD symptom severity score between active treatment and placebo groups,47,48 and the use of topiramate, particularly at higher doses, was limited by its adverse effects.
Lamotrigine. Initially, lamotrigine augmentation of SSRIs in OCD did not appear to be helpful.49 More recently, several case studies reported that lamotrigine, 100 to 200 mg/d, added to paroxetine or clomipramine, resulted in dramatic improvement in Y-BOCS scores for patients with long-standing refractory symptoms.50,51 In a retrospective review of 22 patients who received augmentation with lamotrigine, 150 mg/d, 20 had a significant response; the mean decrease in Y-BOCS score was 67%.52 Finally, in a 16-week RCT, lamotrigine, 100 mg/d, added to an SSRI led to a significant decrease in both Y-BOCS score and depressive symptoms while also improving semantic fluency.53
Ketamine. Ketamine is drawing increased attention for its nearly instantaneous antidepressant effect that lasts for up to 2 weeks after a single infusion.54 In a study of 15 medication-free adults with continuous intrusive obsessions, 4 of 8 patients who received a single IV infusion of ketamine, 0.5 mg/kg, met the criteria for treatment response (>35% reduction in Y-BOCS score measured 1 week later); none of the patients who received a placebo infusion of saline met this criteria.55 A small open-label trial of 10 treatment-refractory patients found that an infusion of ketamine, 0.5 mg/kg, was beneficial for comorbid depression but had only a minimal effect on OCD symptoms measured 3 days post-infusion.56 A short-term follow-up on these patients revealed dysphoria in some responders.57
D-cycloserine. The idea of using a pharmacologic agent to increase the speed or efficacy of behavioral therapy is intriguing. Proof of concept was demonstrated in a study that found that giving D-cycloserine prior to computerized exposure therapy significantly improved clinical response in patients with acrophobia.58 However, using this approach to treating OCD netted mixed results; D-cycloserine was found to be most helpful during early stages of treatment.59,60
Table 3 outlines the mechanisms of action and common uses for NAC, memantine, ketamine, topiramate, lamotrigine, and D-cycloserine. Table 4 summarizes the literature on the efficacy of some of the augmentation strategies for treating OCD described in this article.
Continue to: Alternative strategies
Alternative strategies
Augmentation strategies with neuroleptics,61 transcranial magnetic stimulation,62 and deep brain stimulation63 have recently been reviewed. Space limitations preclude a comprehensive review of these strategies, but in a cross-sectional study of augmentation strategies in OCD, no difference was found in terms of symptom severity between those prescribed SSRI monotherapy or augmentation with neuroleptics, benzodiazepines, or antidepressants.64
CASE CONTINUED
Progress in CBT
Mr. S agrees to a trial of NAC as an augmentation strategy, but after 8 weeks of treatment with NAC, 600 mg twice daily, his Y-BOCS had declined by only 2 points. He also complains of nausea and does not want to increase the dose. You discontinue NAC and opt to further explore his reaction to CBT. Mr. S shares that he has been seeing his psychologist only once every 3 weeks because he does not want to miss work. You encourage him to increase to weekly CBT sessions, and you obtain his permission to contact his therapist and his family members. Fortunately, his therapist is highly qualified, but unfortunately, Mr. S’s father has been sending him multiple critical emails about not advancing at his job and for being “lazy” at work. You schedule a session with Mr. S and his father. Great progress is made after Mr. S and his father both share their frustrations and come to understand and appreciate each other’s struggles. Four weeks later, after weekly CBT appointments, Mr. S has a Y-BOCS of 18 and spends <2 hours/d checking emails for errors and apologizing.
Bottom Line
It is unrealistic to expect OCD symptoms to be cured. Many ‘treatment-resistant’ patients have not received properly delivered cognitive-behavioral therapy, and this first-line treatment modality should be considered in every eligible patient, and augmented with a selective serotonin reuptake inhibitor (SSRI) when needed. Glutamatergic agents, in turn, can augment SSRIs.
Related Resources
- Yale-Brown Obsessive-Compulsive Scale. https://iocdf.org/ wp-content/uploads/2014/08/Assessment-Tools.pdf.
- The International OCD Foundation. https://iocdf.org.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Ketamine • Ketalar
Lamotrigine • Lamictal
Memantine • Namenda
Paroxetine • Paxil
Sertraline • Zoloft
Topiramate • Topomax
Treatment-resistant OCD can be a debilitating condition. Diagnostic clarity is crucial to fully elicit symptoms and identify comorbid conditions in order to develop practical, evidence-based treatment strategies and improve the patient’s and family’s quality of life. In this article, we delineate first-line strategies for treatment-resistant OCD and then review augmentation strategies, with an emphasis on glutamate-modulating agents.
Making the diagnosis
The diagnosis of OCD is made when a patient meets DSM-5 criteria for the presence of obsessions and/or compulsions, which are defined as unwanted, distressing, intrusive, recurrent thoughts or images (obsessions) and repetitive behaviors or mental acts (compulsions).1 OCD is considered a chronic waxing and waning disorder; stress and lack of sleep lead to worsening symptoms. The hidden nature of symptoms and the reinforcement provided by the reduction in anxiety after performing a compulsion contribute to sustained illness. Eliciting symptoms from patients may be challenging due to the shame they may feel. When reviewing symptoms on the Y-BOCS, it is helpful to preface questions with statements such as “Many people report excessive concern or disgust with…” to help the patient feel understood and less anxious, rather than using direct queries, such as “Are you bothered by…?”
Consider comorbid conditions
After making the initial diagnosis of OCD, it is important to assess whether the symptoms are better accounted for by another condition, and whether comorbid conditions are present (Table 1).
CASE CONTINUED
Ruling out other diagnoses
_
Initial treatment: CBT
Cognitive-behavioral therapy with exposures and response prevention (from here on referred to as CBT) has been established as a first-line, evidence-based treatment for OCD in both children and adults.2,3 For patients with treatment-resistant OCD, intensive daily CBT in a partial hospitalization or inpatient setting that is a tailor-made, patient-specific program is one of the most effective treatments, with response rates of up to 70%4-8 CBT’s advantages over medication include lower relapse rates and no known adverse effects. Unfortunately, CBT is underused9-11 due in part to a shortage of trained clinicians, and because patients may favor the ease of taking medication over the time, effort, and cost involved in CBT.
First-line pharmacologic options for treating OCD are SSRIs and clomipramine, as supported by multiple randomized controlled trials (RCTs), meta-analyses, expert guidelines, and consensus statements (Table 22,12-14). No significant difference has been found among SSRIs for the treatment of OCD in a review of 17 studies that included more than 3,000 patients.15 Treatment with SSRIs or clomipramine is effective for 50% to 60% of patients.16 Many clinicians view the combination of an SSRI and CBT as the treatment of choice for OCD.2
Continue to: Reluctance to engage in CBT
CASE CONTINUED
Reluctance to engage in CBT
To determine the next course of action, you review Mr. S’s treatment history. He has received adequate doses of 2 SSRIs and currently is taking clomipramine, 100 mg twice daily. He recently began CBT, which includes homework to help face his fears; however, Mr. S is reluctant to complete the exposure assignments, and after pausing for a few seconds as he tries to resist sending an apology email to his coworkers, he then returns to his compulsive behavior.
Facing treatment resistance
Although currently there isn’t a cure to resolve all traces of OCD, the goal of treatment is to decrease distress, interference, and the frequency of symptoms to a minimal level such that only the patients themselves are aware of symptoms. In broad terms, “response” has been defined as a decrease in symptoms, and “remission” has been defined as minimal symptoms after treatment.
Close to half of adults treated for OCD respond well to standard-of-care treatment (CBT and/or an SSRI), while the other 50% are considered partial responders or nonresponders.2 For patients with OCD, researchers often define “treatment response” as a ≥25% reduction in symptom severity score on the Y-BOCS. Approximately 30% of adults with OCD do not respond substantially to the first-line treatments, and even those who are defined as “responders” in research studies typically continue to have significant symptoms that impact their quality of life.2 In children, a clinical definition for treatment-refractory OCD has been presented as failing to achieve adequate symptom relief despite receiving an adequate course of CBT and at least 2 adequate trials of an SSRI or clomipramine.17 In the Pediatric OCD Treatment Study (POTS) trial, >46% of youth did not achieve remission from their OCD symptoms, even after receiving evidence-based care provided by experienced clinicians (combined treatment with CBT and an SSRI).18
_
Challenges in psychotherapy
Compassion is a key element in developing rapport with patients to help them face increasingly more challenging exposures. Making OCD the problem, not the person, is an essential element in helping patients move forward. Some clinicians may become frustrated with patients when treatment is not moving along well, referring to resistance, denial, or sabotage. According to March and Mulle,19 these terms lack the recognition and compassion that exposures are inherently difficult.19
Another challenge for therapists is if the patient’s presenting symptoms are personally offensive or a sensitive topic. For example, a therapist who is disgusted by public restrooms will find it difficult to tolerate the risks associated with exposure to germs and support a patient in touching objects in the restroom. Therapists also may be challenged when the patient’s fears align with the therapist’s religious beliefs. In these situations, consider transferring care to another therapist.
Family members need to learn about the nature of the illness and their roles in helping patients improve. Family members may unknowingly enable symptoms or criticize patients for their lack of motivation, which can lead to conflict in the home. Family dysfunction can in turn worsen OCD symptoms.
The most likely cause of lack of response to therapy is inexpert CBT.19 Deep breathing and relaxation training have been used as an active placebo in studies20; in a meta-analysis examining the effective components of CBT, studies that added relaxation training were not more effective than those that employed exposures alone.21 Patients receiving CBT should be able to articulate the hierarchical approach used to gradually face their fears.
Continue to: Pharmacologic augmentation strategies
Pharmacologic augmentation strategies
Selective serotonin reuptake inhibitors. While most OCD research trials have assessed SSRIs in 12-week studies, clinicians may consider extending SSRI treatment for an additional 12 weeks for nonresponders because some patients will continue to make gains. In the past, it was generally believed that higher doses of SSRIs are needed for treating OCD than for treating major depressive disorder. For instance, greater improvement was seen with 250 to 400 mg/d of sertraline compared with 200 mg/d22 and with escitalopram after an increase of dose up to 50 mg/d.23 However, more recently, this notion of higher doses being necessary for treatment response has been called into question. For example, a study of escitalopram found similar responses to 10 mg/d vs 20 mg/d after 24 weeks.24 A meta-analysis of adult studies of SSRIs for OCD supported higher doses as being more effective, but noted that the drop-out rate from treatment was greater in patients treated with higher doses.25 As a note of caution, long-term, high-dose maintenance therapy increases the risk of adverse reactions.26
Following a failed treatment with a first SSRI, it remains debatable as to what ought to be the second pharmacologic treatment. Although clomipramine is often reserved for treatment after 2 failed trials of an SSRI due to its greater risk of adverse effects, in an open-label study, switching from an SSRI to clomipramine led to greater response than switching from one SSRI to another.27 On the other hand, while meta-analyses have reported greater treatment effect for oral clomipramine than for SSRIs, direct head-to-head comparisons have not supported this notion.28 To get the best of both worlds, some clinicians employ a strategy of combining clomipramine with an SSRI, while monitoring for adverse effects and interactions such as serotonin syndrome.29-31
Benzodiazepines. Although benzodiazepines are useful for brief treatment of an anxiety disorder (eg, for a person with a fear of heights who needs to take an airplane),32 they have not been shown to be effective for OCD33 or as augmentation to an SSRI.34
N-acetylcysteine (NAC). Two RCTs of adults with OCD who received adjunctive NAC, 3 g/d in divided doses, found no significant difference in the treatment arms by the conclusion of 16 weeks—either both groups improved, or both groups failed to improve.35,36 In a 10-week study of patients with moderate to severe OCD symptoms, NAC, 2 g/d, as augmentation to fluvoxamine, 200 mg/d, showed a significant time x interaction in the treatment group.37 No follow-up information is available, however.
In a multicenter RCT of NAC given to children and adolescents with OCD as augmentation to citalopram, symptoms decreased and the quality-of-life score improved, with a large treatment effect size in the NAC group.38 However, in a study aimed at examining NAC in youth with Tourette syndrome, OCD symptoms were measured as a secondary outcome and there was no benefit of NAC over placebo.39
Memantine. Four 8- to 12-week RCTs in adults with OCD favored adjunctive memantine, 20 mg/d, taken with an SSRI, over placebo.40-43 A small study suggests that patients with OCD may be more likely to respond to memantine than patients with generalized anxiety disorder.44 Case reports have noted that memantine has been beneficial for pediatric patients with refractory OCD.45
Continue to: Topiramate
Topiramate. Three 12-week RCTs examined topiramate augmentation at 100 to 400 mg/d in patients with OCD who had failed at least 1 previous trial of an SSRI. The earliest study was most encouraging: Y-BOCS scores decreased by 32% in the topiramate group but by only 2.4% in the placebo group.46 However, the other 2 studies found no difference in the final OCD symptom severity score between active treatment and placebo groups,47,48 and the use of topiramate, particularly at higher doses, was limited by its adverse effects.
Lamotrigine. Initially, lamotrigine augmentation of SSRIs in OCD did not appear to be helpful.49 More recently, several case studies reported that lamotrigine, 100 to 200 mg/d, added to paroxetine or clomipramine, resulted in dramatic improvement in Y-BOCS scores for patients with long-standing refractory symptoms.50,51 In a retrospective review of 22 patients who received augmentation with lamotrigine, 150 mg/d, 20 had a significant response; the mean decrease in Y-BOCS score was 67%.52 Finally, in a 16-week RCT, lamotrigine, 100 mg/d, added to an SSRI led to a significant decrease in both Y-BOCS score and depressive symptoms while also improving semantic fluency.53
Ketamine. Ketamine is drawing increased attention for its nearly instantaneous antidepressant effect that lasts for up to 2 weeks after a single infusion.54 In a study of 15 medication-free adults with continuous intrusive obsessions, 4 of 8 patients who received a single IV infusion of ketamine, 0.5 mg/kg, met the criteria for treatment response (>35% reduction in Y-BOCS score measured 1 week later); none of the patients who received a placebo infusion of saline met this criteria.55 A small open-label trial of 10 treatment-refractory patients found that an infusion of ketamine, 0.5 mg/kg, was beneficial for comorbid depression but had only a minimal effect on OCD symptoms measured 3 days post-infusion.56 A short-term follow-up on these patients revealed dysphoria in some responders.57
D-cycloserine. The idea of using a pharmacologic agent to increase the speed or efficacy of behavioral therapy is intriguing. Proof of concept was demonstrated in a study that found that giving D-cycloserine prior to computerized exposure therapy significantly improved clinical response in patients with acrophobia.58 However, using this approach to treating OCD netted mixed results; D-cycloserine was found to be most helpful during early stages of treatment.59,60
Table 3 outlines the mechanisms of action and common uses for NAC, memantine, ketamine, topiramate, lamotrigine, and D-cycloserine. Table 4 summarizes the literature on the efficacy of some of the augmentation strategies for treating OCD described in this article.
Continue to: Alternative strategies
Alternative strategies
Augmentation strategies with neuroleptics,61 transcranial magnetic stimulation,62 and deep brain stimulation63 have recently been reviewed. Space limitations preclude a comprehensive review of these strategies, but in a cross-sectional study of augmentation strategies in OCD, no difference was found in terms of symptom severity between those prescribed SSRI monotherapy or augmentation with neuroleptics, benzodiazepines, or antidepressants.64
CASE CONTINUED
Progress in CBT
Mr. S agrees to a trial of NAC as an augmentation strategy, but after 8 weeks of treatment with NAC, 600 mg twice daily, his Y-BOCS had declined by only 2 points. He also complains of nausea and does not want to increase the dose. You discontinue NAC and opt to further explore his reaction to CBT. Mr. S shares that he has been seeing his psychologist only once every 3 weeks because he does not want to miss work. You encourage him to increase to weekly CBT sessions, and you obtain his permission to contact his therapist and his family members. Fortunately, his therapist is highly qualified, but unfortunately, Mr. S’s father has been sending him multiple critical emails about not advancing at his job and for being “lazy” at work. You schedule a session with Mr. S and his father. Great progress is made after Mr. S and his father both share their frustrations and come to understand and appreciate each other’s struggles. Four weeks later, after weekly CBT appointments, Mr. S has a Y-BOCS of 18 and spends <2 hours/d checking emails for errors and apologizing.
Bottom Line
It is unrealistic to expect OCD symptoms to be cured. Many ‘treatment-resistant’ patients have not received properly delivered cognitive-behavioral therapy, and this first-line treatment modality should be considered in every eligible patient, and augmented with a selective serotonin reuptake inhibitor (SSRI) when needed. Glutamatergic agents, in turn, can augment SSRIs.
Related Resources
- Yale-Brown Obsessive-Compulsive Scale. https://iocdf.org/ wp-content/uploads/2014/08/Assessment-Tools.pdf.
- The International OCD Foundation. https://iocdf.org.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Ketamine • Ketalar
Lamotrigine • Lamictal
Memantine • Namenda
Paroxetine • Paxil
Sertraline • Zoloft
Topiramate • Topomax
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Koran LM, Hanna GL, Hollander E, et al. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry; 2007;164(suppl 7):5-53.
3. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
4. Bystritsky A, Munford PR, Rosen RM, et al. A preliminary study of partial hospital management of severe obsessive-compulsive disorder. Psychiatr Serv. 1996;47(2):170-174.
5. Calvocoressi L, McDougle CI, Wasylink S, et al. Inpatient treatment of patients with severe obsessive-compulsive disorder. Hosp Community Psychiatry. 1993;44(12):1150-1154.
6. Eddy KT, Dutra L, Bradley R, et al. A multidimensional meta-analysis of psychotherapy and pharmacotherapy for obsessive-compulsive disorder. Clin Psychol Rev. 2004;24(8):1011-1030.
7. Abramowitz JS. The psychological treatment of obsessive-compulsive disorder. Can J Psychiatry. 2006;51(7):407-416.
8. Simpson HB, Huppert JD, Petkova E, et al. Response versus remission in obsessive-compulsive disorder. J Clin Psychiatry. 2006;67(2):269-276.
9. Marques L, LeBlanc NJ, Weingarden HM, et al. Barriers to treatment and service utilization in an internet sample of individuals with obsessive-compulsive symptoms. Depress Anxiety. 2010;27(5):470-475.
10. Goodwin R, Koenen KC, Hellman F, et al. Helpseeking and access to mental health treatment for obsessive-compulsive disorder. Acta Psychiatr Scand. 2002;106(2):143-149.
11. Kohn R, Saxena S, Levav I, et al. The treatment gap in mental health care. Bull World Health Organ. 2004;82(11):858-866.
12. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439.
13. Lovell K, Bee P. Implementing the NICE OCD/BDD guidelines. Psychol Psychother. 2008;81(Pt 4):365-376.
14. Bandelow B, Sher L, Bunevicius R, et al. Guidelines for the pharmacological treatment of anxiety disorders, obsessive-compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract. 2012;16(2):77-84.
15. Soomro GM, Altman D, Rajagopal S, et al. Selective serotonin re-uptake inhibitors (SSRIs) versus placebo for obsessive compulsive disorder (OCD). Cochrane Database Syst Rev. 2008;(1):CD001765.
16. Pittenger C, Bloch MH. Pharmacological treatment of obsessive-compulsive disorder. Psychiatr Clin North Am. 2014;37(3):375-391.
17. Bloch MH, Storch EA. Assessment and management of treatment-refractory obsessive-compulsive disorder in children. J Am Acad Child Adolesc Psychiatry. 2015;54(4):251-262.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
19. March JS, Mulle K. OCD in children and adolescents: a cognitive-behavioral treatment manual. New York, NY: Guilford Press; 1998.
20. Marks IM. Fears, phobias, and rituals: Panic, anxiety, and their disorders. 1987, New York, NY: Oxford University Press; 1987.
21. Ale CM, McCarthy DM, Rothschild LM, et al. Components of cognitive behavioral therapy related to outcome in childhood anxiety disorders. Clin Child Fam Psychol Rev. 2015;18(3):240-251.
22. Ninan PT, Koran LM, Kiev A, et al. High-dose sertraline strategy for nonresponders to acute treatment for obsessive-compulsive disorder: a multicenter double-blind trial. J Clin Psychiatry. 2006;67(1):15-22.
23. Rabinowitz I, Baruch Y, Barak Y. High-dose escitalopram for the treatment of obsessive-compulsive disorder. Int Clin Psychopharmacol. 2008;23(1):49-53.
24. Stein DJ, Andersen EW, Tonnoir B, et al. Escitalopram in obsessive-compulsive disorder: a randomized, placebo-controlled, paroxetine-referenced, fixed-dose, 24-week study. Curr Med Res Opin. 2007;23(4):701-711.
25. Bloch MH, McGuire J, Landeros-Weisenberger A, et al. Meta-analysis of the dose-response relationship of SSRI in obsessive-compulsive disorder. Mol Psychiatry. 2010;15(8):850-855.
26. Sayyah M, Majzoob S, Sayyah M. Metabolic and toxicological considerations for obsessive-compulsive disorder drug therapy. Expert Opin Drug Metab Toxicol. 2013;9(6):657-673.
27. Hollander E, Bienstock CA, Koran LM, et al. Refractory obsessive-compulsive disorder: state-of-the-art treatment. J Clin Psychiatry. 2002;63(suppl 6):20-29.
28. Fineberg NA, Gale TM. Evidence-based pharmacotherapy of obsessive-compulsive disorder. Int J Neuropsychopharmacol. 2005;8(1):107-129.
29. Marazziti D, Golia F, Consoli G, et al. Effectiveness of long-term augmentation with citalopram to clomipramine in treatment-resistant OCD patients. CNS Spectr. 2008;13(11):971-976.
30. Browne M, Horn E, Jones TT. The benefits of clomipramine-fluoxetine combination in obsessive compulsive disorder. Can J Psychiatry. 1993;38(4):242-243.
31. Ravizza L, Barzega G, Bellino S, et al. Drug treatment of obsessive-compulsive disorder (OCD): long-term trial with clomipramine and selective serotonin reuptake inhibitors (SSRIs). Psychopharmacol Bull. 1996;32(1):167-173.
32. Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci. 2011;13(4):423-437.
33. Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
34. Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam with sertraline in obsessive-compulsive disorder. Ann Clin Psychiatry. 2004;16(3):127-132.
35. Costa DLC, Diniz JB, Requena G, et al. Randomized, double-blind, placebo-controlled trial of n-acetylcysteine augmentation for treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2017;78(7):e766-e773.
36. Sarris J, Oliver G, Camfield DA, et al. N-Acetyl Cysteine (NAC) in the treatment of obsessive-compulsive disorder: a 16-week, double-blind, randomised, placebo-controlled study. CNS Drugs. 2015;29(9):801-809.
37. Paydary K, Akamaloo A, Ahmadipour A, et al. N-acetylcysteine augmentation therapy for moderate-to-severe obsessive-compulsive disorder: randomized, double-blind, placebo-controlled trial. J Clin Pharm Ther. 2016;41(2):214-219.
38. Ghanizadeh A, Mohammadi MR, Bahraini S, et al. Efficacy of N-acetylcysteine augmentation on obsessive compulsive disorder: a multicenter randomized double blind placebo controlled clinical trial. Iran J Psychiatry. 2017;12(2):134-141.
39. Bloch MH, Panza KE, Yaffa A, et al. N-acetylcysteine in the treatment of pediatric tourette syndrome: randomized, double-blind, placebo-controlled add-on trial. J Child Adolesc Psychopharmacol. 2016;26(4):327-334.
40. Ghaleiha A, Entezari N, Modabbernia A, et al. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47(2):175-180.
41. Stewart SE, Jenike EA, Hezel DM, et al. A single-blinded case-control study of memantine in severe obsessive-compulsive disorder. J Clin Psychopharmacol. 2010;30(1):34-39.
42. Modarresi A, Sayyah M, Razooghi S, et al. Memantine augmentation improves symptoms in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder: a randomized controlled trial. Pharmacopsychiatry. 2017. doi: 10.1055/s-0043-120268. [Epub ahead of print].
43. Haghighi M, Jahangard L, Mohammad-Beigi H, et al. In a double-blind, randomized and placebo-controlled trial, adjuvant memantine improved symptoms in inpatients suffering from refractory obsessive-compulsive disorders (OCD). Psychopharmacology (Berl). 2013;228(4):633-640.
44. Feusner JD, Kerwin L, Saxena S, et al. Differential efficacy of memantine for obsessive-compulsive disorder vs. generalized anxiety disorder: an open-label trial. Psychopharmacol Bull. 2009;42(1):81-93.
45. Hezel DM, Beattie K, Stewart SE. Memantine as an augmenting agent for severe pediatric OCD. Am J Psychiatry. 2009;166(2):237.
46. Mowla A, Khajeian AM, Sahraian A, et al. topiramate augmentation in resistant ocd: a double-blind placebo-controlled clinical trial. CNS Spectr. 2010;15(11):613-617.
47. Berlin H, Koran LM, Jenike MA, et al. Double-blind, placebo-controlled trial of topiramate augmentation in treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2011;72(5):716-721.
48. Afshar H, Akuchekian S, Mahaky B, et al. Topiramate augmentation in refractory obsessive-compulsive disorder: A randomized, double-blind, placebo-controlled trial. J Res Med Sci. 2014;19(10):976-981.
49. Kumar TC, Khanna S. Lamotrigine augmentation of serotonin re-uptake inhibitors in obsessive-compulsive disorder. Aust N Z J Psychiatry. 2000;34(3):527-528.
50. Arrojo-Romero M, Tajes Alonso M, de Leon J. Lamotrigine augmentation of serotonin reuptake inhibitors in severe and long-term treatment-resistant obsessive-compulsive disorder. Case Rep Psychiatry. 2013;2013:612459.
51. Uzun O. Lamotrigine as an augmentation agent in treatment-resistant obsessive-compulsive disorder: a case report. J Psychopharmacol. 2010;24(3):425-427.
52. Hussain A, Dar MA, Wani RA, et al. Role of lamotrigine augmentation in treatment-resistant obsessive compulsive disorder: a retrospective case review from South Asia. Indian J Psychol Med. 2015;37(2):154-158.
53. Bruno A, Micò U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456-1462.
54. Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73(12):113311-41.
55. Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38(12):2475-2483.
56. Bloch MH, Wasylink S, Landeros-Weisenberger A,, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biol Psychiatry. 2012;72(11):964-970.
57. Niciu MJ, Grunschel BD, Corlett PR, et al. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
58. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61(11):1136-1144.
59. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
60. Xia J, Du Y, Han J, et al. D-cycloserine augmentation in behavioral therapy for obsessive-compulsive disorder: a meta-analysis. Drug Des Devel Ther. 2015;9:2101-2117.
61. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and meta-analysis. BMC Psychiatry. 2014;14:317.
62. Guo Q, Li C, Wang J. Updated review on the clinical use of repetitive transcranial magnetic stimulation in psychiatric disorders. Neurosci Bull. 2017;33(6):747-756.
63. Naesström, M, Blomstedt P, Bodlund O. A systematic review of psychiatric indications for deep brain stimulation, with focus on major depressive and obsessive-compulsive disorder. Nord J Psychiatry. 2016;70(7):483-491.
64. Van Ameringen M, Simpson W, Patterson B, et al. Pharmacological treatment strategies in obsessive compulsive disorder: A cross-sectional view in nine international OCD centers. J Psychopharmacol, 2014;28(6):596-602.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Koran LM, Hanna GL, Hollander E, et al. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry; 2007;164(suppl 7):5-53.
3. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
4. Bystritsky A, Munford PR, Rosen RM, et al. A preliminary study of partial hospital management of severe obsessive-compulsive disorder. Psychiatr Serv. 1996;47(2):170-174.
5. Calvocoressi L, McDougle CI, Wasylink S, et al. Inpatient treatment of patients with severe obsessive-compulsive disorder. Hosp Community Psychiatry. 1993;44(12):1150-1154.
6. Eddy KT, Dutra L, Bradley R, et al. A multidimensional meta-analysis of psychotherapy and pharmacotherapy for obsessive-compulsive disorder. Clin Psychol Rev. 2004;24(8):1011-1030.
7. Abramowitz JS. The psychological treatment of obsessive-compulsive disorder. Can J Psychiatry. 2006;51(7):407-416.
8. Simpson HB, Huppert JD, Petkova E, et al. Response versus remission in obsessive-compulsive disorder. J Clin Psychiatry. 2006;67(2):269-276.
9. Marques L, LeBlanc NJ, Weingarden HM, et al. Barriers to treatment and service utilization in an internet sample of individuals with obsessive-compulsive symptoms. Depress Anxiety. 2010;27(5):470-475.
10. Goodwin R, Koenen KC, Hellman F, et al. Helpseeking and access to mental health treatment for obsessive-compulsive disorder. Acta Psychiatr Scand. 2002;106(2):143-149.
11. Kohn R, Saxena S, Levav I, et al. The treatment gap in mental health care. Bull World Health Organ. 2004;82(11):858-866.
12. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439.
13. Lovell K, Bee P. Implementing the NICE OCD/BDD guidelines. Psychol Psychother. 2008;81(Pt 4):365-376.
14. Bandelow B, Sher L, Bunevicius R, et al. Guidelines for the pharmacological treatment of anxiety disorders, obsessive-compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract. 2012;16(2):77-84.
15. Soomro GM, Altman D, Rajagopal S, et al. Selective serotonin re-uptake inhibitors (SSRIs) versus placebo for obsessive compulsive disorder (OCD). Cochrane Database Syst Rev. 2008;(1):CD001765.
16. Pittenger C, Bloch MH. Pharmacological treatment of obsessive-compulsive disorder. Psychiatr Clin North Am. 2014;37(3):375-391.
17. Bloch MH, Storch EA. Assessment and management of treatment-refractory obsessive-compulsive disorder in children. J Am Acad Child Adolesc Psychiatry. 2015;54(4):251-262.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
19. March JS, Mulle K. OCD in children and adolescents: a cognitive-behavioral treatment manual. New York, NY: Guilford Press; 1998.
20. Marks IM. Fears, phobias, and rituals: Panic, anxiety, and their disorders. 1987, New York, NY: Oxford University Press; 1987.
21. Ale CM, McCarthy DM, Rothschild LM, et al. Components of cognitive behavioral therapy related to outcome in childhood anxiety disorders. Clin Child Fam Psychol Rev. 2015;18(3):240-251.
22. Ninan PT, Koran LM, Kiev A, et al. High-dose sertraline strategy for nonresponders to acute treatment for obsessive-compulsive disorder: a multicenter double-blind trial. J Clin Psychiatry. 2006;67(1):15-22.
23. Rabinowitz I, Baruch Y, Barak Y. High-dose escitalopram for the treatment of obsessive-compulsive disorder. Int Clin Psychopharmacol. 2008;23(1):49-53.
24. Stein DJ, Andersen EW, Tonnoir B, et al. Escitalopram in obsessive-compulsive disorder: a randomized, placebo-controlled, paroxetine-referenced, fixed-dose, 24-week study. Curr Med Res Opin. 2007;23(4):701-711.
25. Bloch MH, McGuire J, Landeros-Weisenberger A, et al. Meta-analysis of the dose-response relationship of SSRI in obsessive-compulsive disorder. Mol Psychiatry. 2010;15(8):850-855.
26. Sayyah M, Majzoob S, Sayyah M. Metabolic and toxicological considerations for obsessive-compulsive disorder drug therapy. Expert Opin Drug Metab Toxicol. 2013;9(6):657-673.
27. Hollander E, Bienstock CA, Koran LM, et al. Refractory obsessive-compulsive disorder: state-of-the-art treatment. J Clin Psychiatry. 2002;63(suppl 6):20-29.
28. Fineberg NA, Gale TM. Evidence-based pharmacotherapy of obsessive-compulsive disorder. Int J Neuropsychopharmacol. 2005;8(1):107-129.
29. Marazziti D, Golia F, Consoli G, et al. Effectiveness of long-term augmentation with citalopram to clomipramine in treatment-resistant OCD patients. CNS Spectr. 2008;13(11):971-976.
30. Browne M, Horn E, Jones TT. The benefits of clomipramine-fluoxetine combination in obsessive compulsive disorder. Can J Psychiatry. 1993;38(4):242-243.
31. Ravizza L, Barzega G, Bellino S, et al. Drug treatment of obsessive-compulsive disorder (OCD): long-term trial with clomipramine and selective serotonin reuptake inhibitors (SSRIs). Psychopharmacol Bull. 1996;32(1):167-173.
32. Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci. 2011;13(4):423-437.
33. Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
34. Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam with sertraline in obsessive-compulsive disorder. Ann Clin Psychiatry. 2004;16(3):127-132.
35. Costa DLC, Diniz JB, Requena G, et al. Randomized, double-blind, placebo-controlled trial of n-acetylcysteine augmentation for treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2017;78(7):e766-e773.
36. Sarris J, Oliver G, Camfield DA, et al. N-Acetyl Cysteine (NAC) in the treatment of obsessive-compulsive disorder: a 16-week, double-blind, randomised, placebo-controlled study. CNS Drugs. 2015;29(9):801-809.
37. Paydary K, Akamaloo A, Ahmadipour A, et al. N-acetylcysteine augmentation therapy for moderate-to-severe obsessive-compulsive disorder: randomized, double-blind, placebo-controlled trial. J Clin Pharm Ther. 2016;41(2):214-219.
38. Ghanizadeh A, Mohammadi MR, Bahraini S, et al. Efficacy of N-acetylcysteine augmentation on obsessive compulsive disorder: a multicenter randomized double blind placebo controlled clinical trial. Iran J Psychiatry. 2017;12(2):134-141.
39. Bloch MH, Panza KE, Yaffa A, et al. N-acetylcysteine in the treatment of pediatric tourette syndrome: randomized, double-blind, placebo-controlled add-on trial. J Child Adolesc Psychopharmacol. 2016;26(4):327-334.
40. Ghaleiha A, Entezari N, Modabbernia A, et al. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47(2):175-180.
41. Stewart SE, Jenike EA, Hezel DM, et al. A single-blinded case-control study of memantine in severe obsessive-compulsive disorder. J Clin Psychopharmacol. 2010;30(1):34-39.
42. Modarresi A, Sayyah M, Razooghi S, et al. Memantine augmentation improves symptoms in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder: a randomized controlled trial. Pharmacopsychiatry. 2017. doi: 10.1055/s-0043-120268. [Epub ahead of print].
43. Haghighi M, Jahangard L, Mohammad-Beigi H, et al. In a double-blind, randomized and placebo-controlled trial, adjuvant memantine improved symptoms in inpatients suffering from refractory obsessive-compulsive disorders (OCD). Psychopharmacology (Berl). 2013;228(4):633-640.
44. Feusner JD, Kerwin L, Saxena S, et al. Differential efficacy of memantine for obsessive-compulsive disorder vs. generalized anxiety disorder: an open-label trial. Psychopharmacol Bull. 2009;42(1):81-93.
45. Hezel DM, Beattie K, Stewart SE. Memantine as an augmenting agent for severe pediatric OCD. Am J Psychiatry. 2009;166(2):237.
46. Mowla A, Khajeian AM, Sahraian A, et al. topiramate augmentation in resistant ocd: a double-blind placebo-controlled clinical trial. CNS Spectr. 2010;15(11):613-617.
47. Berlin H, Koran LM, Jenike MA, et al. Double-blind, placebo-controlled trial of topiramate augmentation in treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2011;72(5):716-721.
48. Afshar H, Akuchekian S, Mahaky B, et al. Topiramate augmentation in refractory obsessive-compulsive disorder: A randomized, double-blind, placebo-controlled trial. J Res Med Sci. 2014;19(10):976-981.
49. Kumar TC, Khanna S. Lamotrigine augmentation of serotonin re-uptake inhibitors in obsessive-compulsive disorder. Aust N Z J Psychiatry. 2000;34(3):527-528.
50. Arrojo-Romero M, Tajes Alonso M, de Leon J. Lamotrigine augmentation of serotonin reuptake inhibitors in severe and long-term treatment-resistant obsessive-compulsive disorder. Case Rep Psychiatry. 2013;2013:612459.
51. Uzun O. Lamotrigine as an augmentation agent in treatment-resistant obsessive-compulsive disorder: a case report. J Psychopharmacol. 2010;24(3):425-427.
52. Hussain A, Dar MA, Wani RA, et al. Role of lamotrigine augmentation in treatment-resistant obsessive compulsive disorder: a retrospective case review from South Asia. Indian J Psychol Med. 2015;37(2):154-158.
53. Bruno A, Micò U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456-1462.
54. Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73(12):113311-41.
55. Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38(12):2475-2483.
56. Bloch MH, Wasylink S, Landeros-Weisenberger A,, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biol Psychiatry. 2012;72(11):964-970.
57. Niciu MJ, Grunschel BD, Corlett PR, et al. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
58. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61(11):1136-1144.
59. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
60. Xia J, Du Y, Han J, et al. D-cycloserine augmentation in behavioral therapy for obsessive-compulsive disorder: a meta-analysis. Drug Des Devel Ther. 2015;9:2101-2117.
61. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and meta-analysis. BMC Psychiatry. 2014;14:317.
62. Guo Q, Li C, Wang J. Updated review on the clinical use of repetitive transcranial magnetic stimulation in psychiatric disorders. Neurosci Bull. 2017;33(6):747-756.
63. Naesström, M, Blomstedt P, Bodlund O. A systematic review of psychiatric indications for deep brain stimulation, with focus on major depressive and obsessive-compulsive disorder. Nord J Psychiatry. 2016;70(7):483-491.
64. Van Ameringen M, Simpson W, Patterson B, et al. Pharmacological treatment strategies in obsessive compulsive disorder: A cross-sectional view in nine international OCD centers. J Psychopharmacol, 2014;28(6):596-602.

Disruptive mood dysregulation disorder: A better understanding
Disruptive mood dysregulation disorder (DMDD)—a childhood condition of extreme irritability, anger, and frequent, intense temper outbursts—has been a source of controversy among clinicians in the field of pediatric mental health. Before DSM-5 was published, the validity of DMDD had been questioned because DMDD had failed a field trial; agreement between clinicians on the diagnosis of DMDD was poor.1 Axelson2 and Birmaher et al3 examined its validity in their COBY (Course and Outcome of Bipolar Youth) sample. They concluded that only 19% met the criteria for DMDD in 3 times of follow-up. Furthermore, most DMDD criteria overlap with those of other common pediatric psychiatric disorders, including oppositional defiant disorder (ODD), attention-deficit/hyperactivity disorder (ADHD), and pediatric bipolar disorder (BD). Because diagnosis of pediatric BD increased drastically from 2.9% to 15.1% between 1990 and 2000,4 it was believed that introducing DMDD as a diagnosis might lessen the overdiagnosis of pediatric BD by identifying children with chronic irritability and temper tantrums who previously would have been diagnosed with BD.
It is important to recognize that in pediatric patients, mood disorders present differently than they do in adults.5 In children/adolescents, mood disorders are less likely to present as distinct episodes (narrow band), but more likely to present as chronic, broad symptoms. Also, irritability is a common presentation in many pediatric psychiatric disorders, such as ODD, BD (irritability without elation),6 and depression. Thus, for many clinicians, determining the correct mood disorder diagnosis in pediatric patients can be challenging.
This article describes the diagnosis of DMDD, and how its presentation is similar to—and different from—those of other common pediatric psychiatric disorders.
_
The origin of DMDD
Many researchers have investigated the broadband phenotypical presentation of pediatric mood disorders, which have been mostly diagnosed in the psychiatric community as pediatric BD. Leibenluft7 identified a subtype of mood disorder that they termed “severe mood dysregulation” (SMD). Compared with the narrow-band, clearly episodic BD, SMD has a different trajectory, outcome, and findings on brain imaging. SMD is characterized by chronic irritability with abnormal mood (anger or sadness) at least half of the day on most days, with 3 hyperarousal symptoms, including pressured speech, racing thoughts or flight of ideas, intrusiveness, distractibility, insomnia, and agitation.8 Eventually, SMD became the foundation of the development of DMDD.
DSM-5 diagnostic criteria for DMDD include severe recurrent temper outbursts that are out of proportion to the situation, inconsistent with developmental level, and occurring on average ≥3 times per week, plus persistently irritable or angry mood for most of the day nearly every day.9 Additional criteria include the presence of symptoms for at least 12 months (without a symptom-free period of at least 3 consecutive months) in ≥2 settings (at home, at school, or with peers) with onset before age 10. The course of DMDD typically is chronic with accompanying severe temperament. The estimated 6-month to 1-year prevalence is 2% to 5%; the diagnosis is more common among males and school-age children than it is in females and adolescents.9,10
_
DMDD or bipolar disorder?
A patient cannot be dually diagnosed with both disorders. If a patient exhibits a manic episode for more than 1 day, that would null and void the DMDD diagnosis. However, in a study to evaluate BD in pediatric patients, researchers divided BD symptoms into BD-specific categories (elevated mood, grandiosity, and increased goal-directed activity) and nonspecific symptoms such as irritability and talkativeness, distractibility, and flight of ideas or racing thoughts. They found that in the absence of specific symptoms, a diagnosis of BD is unlikely to be the correct diagnosis.11 Hence, as a nonspecific symptom, chronic irritability should be attributed to the symptom count for DMDD, rather than BD. Most epidemiologic studies have concluded that depression and anxiety, and not irritability, are typically the preceeding presentations prior to the development of BD in young adults.12 Chronic irritability, however, predicts major depressive disorder and anxiety disorders in later adolescence and one’s early twenties.13 Furthermore, BD commonly presents with infrequent and discrete episodes and a later age of onset, while DMDD presents with chronic and frequent, severe temper outbursts. Some differences between DMDD and BD are illustrated in Table 1.11-13
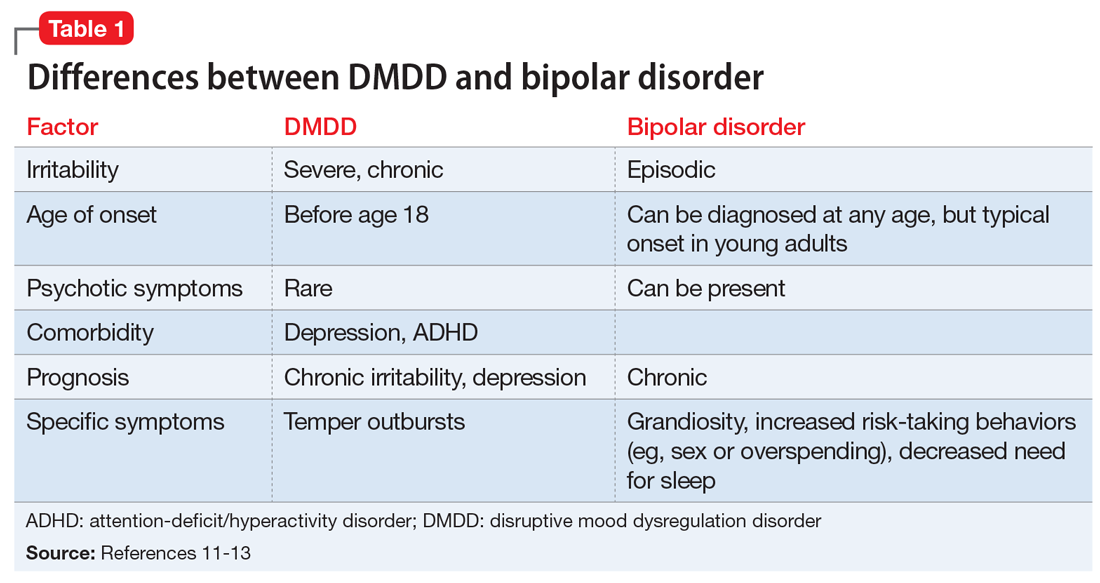
Continue to: CASE 1
CASE 1
Irritable and taking risks
Ms. N, age 16, is brought to the outpatient psychiatry clinic by her parents for evaluation of mood symptoms, including irritability. Her mother claims her daughter was an introverted, anxious, shy child, but by the beginning of middle school, she began to feel irritable and frequently stayed up at night with little sleep. In high school, Ms. N had displayed several episodes of risk-taking behaviors, including taking her father’s vehicle for a drive despite not having a driver’s permit, running away for 2 days, and having unprotected sex.
During her assessment, Ms. N is pleasant and claims she usually has a great mood. She fought with her mother several times this year, which led her to run away. Her parents had divorced when Ms. N was 5 years old and have shared custody. Ms. N is doing well in school despite her parents’ concerns.
Diagnosis. The most likely diagnosis is emerging BD. Notice that Ms. N may have had anxiety symptoms before she developed irritability. She had a relatively late onset of symptoms that were episodic in nature, which further supports a diagnosis of BD.
_
>
DMDD or oppositional defiant disorder?
DMDD and ODD cannot be dually diagnosed. However, if a patient meets the criteria for both DMDD and ODD, only the DMDD diagnosis should be considered. One of many issues of DMDD is its similarity to ODD. In fact, more than 70% of patients with DMDD also meet the diagnostic criteria for ODD.10,14 Some researchers have conceptualized DMDD as a severe form of ODD. However, there are a few differences that clinicians can use to distinguish the 2 disorders.
Compared with patients with ODD, those with DMDD more frequently experience severe irritability.15 Patients with ODD may present with delinquent behaviors and trouble with authority figures. Moreover, comorbidity with ADHD is twice as common in ODD; more than 65% of patients with ADHD have ODD vs 30% who have DMDD.10,16 Finally, in general, children with DMDD have more social impairments compared with those with ODD. Differences between DMDD and BD are illustrated in Table 2.10,14-16
Continue to: CASE 2
CASE 2
Angry and defiant
Mr. R, age 14, is brought to the emergency department (ED) by his parents after becoming very aggressive with them. He punched a wall and vandalized his room after his parents grounded him because of his previous defiant behavior. He had been suspended from school that day for disrespecting his teacher after he was caught fighting another student.
His parents describe Mr. R as a strong-willed, stubborn child. He has difficulty with rules and refuses to follow them. He is grouchy and irritable around adults, including the ED staff. Mr. R enjoys being with his friends and playing video games. He had been diagnosed with ADHD when he was in kindergarten, when his teacher noticed he had a poor attention span and could not sit still. According to his parents, Mr. R has “blown up” a few times before, smashing items in his room and shouting obscenities. Mr. R’s parents noticed that he is more defiant in concurrence with discontinuing his ADHD stimulant medication.
Diagnosis. The most likely diagnosis for Mr. R is ODD. Notice the comorbidity of ADHD, which is more commonly associated with ODD. The frequency and severity of his outbursts and irritability symptoms were lower than that typically associated with DMDD.
_
Treatment strategies for DMDD
Management of DMDD should focus on helping children and adolescents improve their emotional dysregulation.
Clinicians should always consider behavioral therapy as a first-line intervention. The behavioral planning team may include (but is not limited to) a behavior specialist, child psychiatrist, psychologist, therapist, skills trainer, teachers, and the caregiver(s). The plan should be implemented across all settings, including home and school. Furthermore, social skills training is necessary for many children with DMDD, who may require intensive behavioral modification planning. Comorbidity with ADHD should be addressed with a combination of behavioral planning and stimulant medications.17 If available, parent training and parent-child interactive therapy can help to improve defiant behavior.
Pharmacotherapy
Currently, no medications are FDA-approved for treating DMDD. Most pharmacologic trials that included patients with DMDD focused on managing chronic irritability and/or stabilizing comorbid disorders (ie, ADHD, depression, and anxiety).
Continue to: Stimulants
Stimulants. Previous trials examined the benefit of CNS stimulant medications, alone or in conjunction with behavioral therapy, in treating DMDD and comorbid ADHD. Methylphenidate results in a significant reduction in aggression18 with a dosing recommendation range from 1 to 1.2 mg/kg/d. CNS stimulants should be considered as first-line pharmacotherapy for DMDD, especially for patients with comorbid ADHD.
Anticonvulsants. Divalproex sodium is superior to placebo in treating aggression in children and adolescents.19 Trials found that divalproex sodium reduces irritability and aggression whether it is prescribed as monotherapy or combined with stimulant medications.19
Lithium is one of the main treatment options for mania in BD. The benefits of lithium for controlling aggression in DMDD are still under investigation. Earlier studies found that lithium significantly improves aggressive behavior in hospitalized pediatric with conduct disorder.20,21 However, a later study that evaluated lithium vs placebo for children with SMD (which arguably is phenotypically related to the DMDD) found there were no significant differences in improvement of irritability symptoms between groups.22 More research is needed to determine if lithium may play a role in treating patients with DMDD.
Antipsychotics. Aripiprazole and risperidone are FDA-approved for treating irritability in autism. A 2017 meta-analysis found both medications were effective in controlling irritability and aggression in other diagnoses as well.23 Other antipsychotic medications did not show sufficient benefits in treating irritability.23 When considering antipsychotics, clinicians should weigh the risks of metabolic adverse effects and follow practice guidelines.
Antidepressants. A systematic review did not find that selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors effectively reduce irritability.24 However, in most of the studies evaluated, irritability was not the primary outcome measure.24
Other medications. Alpha-2 agonists (guanfacine, clonidine), and atomoxetine may help irritability indirectly by improving ADHD symptoms.25
Bottom Line
Disruptive mood dysregulation disorder (DMDD), bipolar disorder, and oppositional defiant disorder have similar presentations and diagnostic criteria. The frequency and severity of irritability can be a distinguishing factor. Behavioral therapy is a first-line treatment. No medications are FDA-approved for treating DMDD, but pharmacotherapy may help reduce irritability and aggression.
Related Resources
- Rao U. DSM-5: disruptive mood dysregulation disorder. Asian J Psychiatr. 2014;11:119-123.
- Roy AK, Lopes V, Klein RG. Disruptive mood dysregulation disorder: a new diagnostic approach to chronic irritability in youth. Am J Psychiatry. 2014;171(9):918-924.
Drug Brand Names
Aripiprazole • Abilify
Atomoxetine • Strattera
Clonidine • Catapres
Divalproex sodium • Depakote, Depakote ER
Guanfacine • Intuniv, Tenex
Lithium • Eskalith, Lithobid
Methylphenidate • Concerta, Ritalin
Risperidone • Risperdal
1. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
2. Axelson D. Taking disruptive mood dysregulation disorder out for a test drive. Am J Psychiatry. 2013;170(2):136-139.
3. Birmaher B, Axelson D, Goldstein B, et al. Four-year longitudinal course of children and adolescents with bipolar spectrum disorders: the Course and Outcome of Bipolar Youth (COBY) study. Am J Psychiatry. 2009;166(7):795-804.
4. Case BG, Olfson M, Marcus SC, et al. Trends in the inpatient mental health treatment of children and adolescents in US community hospitals between 1990 and 2000. Arch Gen Psychiatry. 2007;64(1):89-96.
5. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
6. Hunt J, Birmaher B, Leonard H, et al. Irritability without elation in a large bipolar youth sample: frequency and clinical description. J Am Acad Child Adolesc Psychiatry. 2009;48(7):730-739.
7. Leibenluft E. Severe mood dysregulation, irritability, and the diagnostic boundaries of bipolar disorder in youths. Am J Psychiatry. 2011;168(2):129-142.
8. Rich BA, Carver FW, Holroyd T, et al. Different neural pathways to negative affect in youth with pediatric bipolar disorder and severe mood dysregulation. J Psychiatr Res. 2011;45(10):1283-1294.
9. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
10. Copeland WE, Angold A, Costello EJ, et al. Prevalence, comorbidity, and correlates of DSM-5 proposed disruptive mood dysregulation disorder. Am J Psychiatry. 2013;170(2):173-179.
11. Elmaadawi AZ, Jensen PS, Arnold LE, et al. Risk for emerging bipolar disorder, variants, and symptoms in children with attention deficit hyperactivity disorder, now grown up. World J Psychiatry. 2015;5(4):412-424.
12. Duffy A. The early natural history of bipolar disorder: what we have learned from longitudinal high-risk research. Can J Psychiatry. 2010;55(8):477-485.
13. Stringaris A, Cohen P, Pine DS, et al. Adult outcomes of youth irritability: a 20-year prospective community-based study. Am J Psychiatry. 2009;166(9):1048-1054.
14. Mayes SD, Waxmonsky JD, Calhoun SL, et al. Disruptive mood dysregulation disorder symptoms and association with oppositional defiant and other disorders in a general population child sample. J Child Adolesc Psychopharmacol. 2016;26(2):101-106.
15. Stringaris A, Vidal-Ribas P, Brotman MA, et al. Practitioner review: definition, recognition, and treatment challenges of irritability in young people. J Child Psychol Psychiatry. 2018;59(7):721-739.
16. Angold A, Costello EJ, Erkanli A. Comorbidity. J Child Psychol Psychiatry. 1999;40(1):57-87.
17. Fernandez de la Cruz L, Simonoff E, McGough JJ, et al. Treatment of children with attention-deficit/hyperactivity disorder (ADHD) and irritability: results from the multimodal treatment study of children with ADHD (MTA). J Am Acad Child Adolesc Psychiatry. 2015;54(1):62-70.
18. Pappadopulos E, Woolston S, Chait A, et al. Pharmacotherapy of aggression in children and adolescents: efficacy and effect size. J Can Acad Child Adolesc Psychiatry. 2006;15(1):27-39.
19. Donovan SJ, Stewart JW, Nunes EV, et al. Divalproex treatment for youth with explosive temper and mood lability: a double-blind, placebo-controlled crossover design. Am J Psychiatry. 2000;157(5):818-820.
20. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
21. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
22. Dickstein DP, Towbin KE, Van Der Veen JW, et al. Randomized double-blind placebo-controlled trial of lithium in youths with severe mood dysregulation. J Child Adolesc Psychopharmacol. 2009;19(1):61-73.
23. van Schalkwyk GI, Lewis AS, Beyer C, et al. Efficacy of antipsychotics for irritability and aggression in children: a meta-analysis. Expert Rev Neurother. 2017;17(10):1045-1053.
24. Kim S, Boylan K. Effectiveness of antidepressant medications for symptoms of irritability and disruptive behaviors in children and adolescents. J Child Adolesc Psychopharmacol. 2016;26(8):694-704.
25. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158(7):1067-1074.
Disruptive mood dysregulation disorder (DMDD)—a childhood condition of extreme irritability, anger, and frequent, intense temper outbursts—has been a source of controversy among clinicians in the field of pediatric mental health. Before DSM-5 was published, the validity of DMDD had been questioned because DMDD had failed a field trial; agreement between clinicians on the diagnosis of DMDD was poor.1 Axelson2 and Birmaher et al3 examined its validity in their COBY (Course and Outcome of Bipolar Youth) sample. They concluded that only 19% met the criteria for DMDD in 3 times of follow-up. Furthermore, most DMDD criteria overlap with those of other common pediatric psychiatric disorders, including oppositional defiant disorder (ODD), attention-deficit/hyperactivity disorder (ADHD), and pediatric bipolar disorder (BD). Because diagnosis of pediatric BD increased drastically from 2.9% to 15.1% between 1990 and 2000,4 it was believed that introducing DMDD as a diagnosis might lessen the overdiagnosis of pediatric BD by identifying children with chronic irritability and temper tantrums who previously would have been diagnosed with BD.
It is important to recognize that in pediatric patients, mood disorders present differently than they do in adults.5 In children/adolescents, mood disorders are less likely to present as distinct episodes (narrow band), but more likely to present as chronic, broad symptoms. Also, irritability is a common presentation in many pediatric psychiatric disorders, such as ODD, BD (irritability without elation),6 and depression. Thus, for many clinicians, determining the correct mood disorder diagnosis in pediatric patients can be challenging.
This article describes the diagnosis of DMDD, and how its presentation is similar to—and different from—those of other common pediatric psychiatric disorders.
_
The origin of DMDD
Many researchers have investigated the broadband phenotypical presentation of pediatric mood disorders, which have been mostly diagnosed in the psychiatric community as pediatric BD. Leibenluft7 identified a subtype of mood disorder that they termed “severe mood dysregulation” (SMD). Compared with the narrow-band, clearly episodic BD, SMD has a different trajectory, outcome, and findings on brain imaging. SMD is characterized by chronic irritability with abnormal mood (anger or sadness) at least half of the day on most days, with 3 hyperarousal symptoms, including pressured speech, racing thoughts or flight of ideas, intrusiveness, distractibility, insomnia, and agitation.8 Eventually, SMD became the foundation of the development of DMDD.
DSM-5 diagnostic criteria for DMDD include severe recurrent temper outbursts that are out of proportion to the situation, inconsistent with developmental level, and occurring on average ≥3 times per week, plus persistently irritable or angry mood for most of the day nearly every day.9 Additional criteria include the presence of symptoms for at least 12 months (without a symptom-free period of at least 3 consecutive months) in ≥2 settings (at home, at school, or with peers) with onset before age 10. The course of DMDD typically is chronic with accompanying severe temperament. The estimated 6-month to 1-year prevalence is 2% to 5%; the diagnosis is more common among males and school-age children than it is in females and adolescents.9,10
_
DMDD or bipolar disorder?
A patient cannot be dually diagnosed with both disorders. If a patient exhibits a manic episode for more than 1 day, that would null and void the DMDD diagnosis. However, in a study to evaluate BD in pediatric patients, researchers divided BD symptoms into BD-specific categories (elevated mood, grandiosity, and increased goal-directed activity) and nonspecific symptoms such as irritability and talkativeness, distractibility, and flight of ideas or racing thoughts. They found that in the absence of specific symptoms, a diagnosis of BD is unlikely to be the correct diagnosis.11 Hence, as a nonspecific symptom, chronic irritability should be attributed to the symptom count for DMDD, rather than BD. Most epidemiologic studies have concluded that depression and anxiety, and not irritability, are typically the preceeding presentations prior to the development of BD in young adults.12 Chronic irritability, however, predicts major depressive disorder and anxiety disorders in later adolescence and one’s early twenties.13 Furthermore, BD commonly presents with infrequent and discrete episodes and a later age of onset, while DMDD presents with chronic and frequent, severe temper outbursts. Some differences between DMDD and BD are illustrated in Table 1.11-13
Continue to: CASE 1
CASE 1
Irritable and taking risks
Ms. N, age 16, is brought to the outpatient psychiatry clinic by her parents for evaluation of mood symptoms, including irritability. Her mother claims her daughter was an introverted, anxious, shy child, but by the beginning of middle school, she began to feel irritable and frequently stayed up at night with little sleep. In high school, Ms. N had displayed several episodes of risk-taking behaviors, including taking her father’s vehicle for a drive despite not having a driver’s permit, running away for 2 days, and having unprotected sex.
During her assessment, Ms. N is pleasant and claims she usually has a great mood. She fought with her mother several times this year, which led her to run away. Her parents had divorced when Ms. N was 5 years old and have shared custody. Ms. N is doing well in school despite her parents’ concerns.
Diagnosis. The most likely diagnosis is emerging BD. Notice that Ms. N may have had anxiety symptoms before she developed irritability. She had a relatively late onset of symptoms that were episodic in nature, which further supports a diagnosis of BD.
_
>
DMDD or oppositional defiant disorder?
DMDD and ODD cannot be dually diagnosed. However, if a patient meets the criteria for both DMDD and ODD, only the DMDD diagnosis should be considered. One of many issues of DMDD is its similarity to ODD. In fact, more than 70% of patients with DMDD also meet the diagnostic criteria for ODD.10,14 Some researchers have conceptualized DMDD as a severe form of ODD. However, there are a few differences that clinicians can use to distinguish the 2 disorders.
Compared with patients with ODD, those with DMDD more frequently experience severe irritability.15 Patients with ODD may present with delinquent behaviors and trouble with authority figures. Moreover, comorbidity with ADHD is twice as common in ODD; more than 65% of patients with ADHD have ODD vs 30% who have DMDD.10,16 Finally, in general, children with DMDD have more social impairments compared with those with ODD. Differences between DMDD and BD are illustrated in Table 2.10,14-16
Continue to: CASE 2
CASE 2
Angry and defiant
Mr. R, age 14, is brought to the emergency department (ED) by his parents after becoming very aggressive with them. He punched a wall and vandalized his room after his parents grounded him because of his previous defiant behavior. He had been suspended from school that day for disrespecting his teacher after he was caught fighting another student.
His parents describe Mr. R as a strong-willed, stubborn child. He has difficulty with rules and refuses to follow them. He is grouchy and irritable around adults, including the ED staff. Mr. R enjoys being with his friends and playing video games. He had been diagnosed with ADHD when he was in kindergarten, when his teacher noticed he had a poor attention span and could not sit still. According to his parents, Mr. R has “blown up” a few times before, smashing items in his room and shouting obscenities. Mr. R’s parents noticed that he is more defiant in concurrence with discontinuing his ADHD stimulant medication.
Diagnosis. The most likely diagnosis for Mr. R is ODD. Notice the comorbidity of ADHD, which is more commonly associated with ODD. The frequency and severity of his outbursts and irritability symptoms were lower than that typically associated with DMDD.
_
Treatment strategies for DMDD
Management of DMDD should focus on helping children and adolescents improve their emotional dysregulation.
Clinicians should always consider behavioral therapy as a first-line intervention. The behavioral planning team may include (but is not limited to) a behavior specialist, child psychiatrist, psychologist, therapist, skills trainer, teachers, and the caregiver(s). The plan should be implemented across all settings, including home and school. Furthermore, social skills training is necessary for many children with DMDD, who may require intensive behavioral modification planning. Comorbidity with ADHD should be addressed with a combination of behavioral planning and stimulant medications.17 If available, parent training and parent-child interactive therapy can help to improve defiant behavior.
Pharmacotherapy
Currently, no medications are FDA-approved for treating DMDD. Most pharmacologic trials that included patients with DMDD focused on managing chronic irritability and/or stabilizing comorbid disorders (ie, ADHD, depression, and anxiety).
Continue to: Stimulants
Stimulants. Previous trials examined the benefit of CNS stimulant medications, alone or in conjunction with behavioral therapy, in treating DMDD and comorbid ADHD. Methylphenidate results in a significant reduction in aggression18 with a dosing recommendation range from 1 to 1.2 mg/kg/d. CNS stimulants should be considered as first-line pharmacotherapy for DMDD, especially for patients with comorbid ADHD.
Anticonvulsants. Divalproex sodium is superior to placebo in treating aggression in children and adolescents.19 Trials found that divalproex sodium reduces irritability and aggression whether it is prescribed as monotherapy or combined with stimulant medications.19
Lithium is one of the main treatment options for mania in BD. The benefits of lithium for controlling aggression in DMDD are still under investigation. Earlier studies found that lithium significantly improves aggressive behavior in hospitalized pediatric with conduct disorder.20,21 However, a later study that evaluated lithium vs placebo for children with SMD (which arguably is phenotypically related to the DMDD) found there were no significant differences in improvement of irritability symptoms between groups.22 More research is needed to determine if lithium may play a role in treating patients with DMDD.
Antipsychotics. Aripiprazole and risperidone are FDA-approved for treating irritability in autism. A 2017 meta-analysis found both medications were effective in controlling irritability and aggression in other diagnoses as well.23 Other antipsychotic medications did not show sufficient benefits in treating irritability.23 When considering antipsychotics, clinicians should weigh the risks of metabolic adverse effects and follow practice guidelines.
Antidepressants. A systematic review did not find that selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors effectively reduce irritability.24 However, in most of the studies evaluated, irritability was not the primary outcome measure.24
Other medications. Alpha-2 agonists (guanfacine, clonidine), and atomoxetine may help irritability indirectly by improving ADHD symptoms.25
Bottom Line
Disruptive mood dysregulation disorder (DMDD), bipolar disorder, and oppositional defiant disorder have similar presentations and diagnostic criteria. The frequency and severity of irritability can be a distinguishing factor. Behavioral therapy is a first-line treatment. No medications are FDA-approved for treating DMDD, but pharmacotherapy may help reduce irritability and aggression.
Related Resources
- Rao U. DSM-5: disruptive mood dysregulation disorder. Asian J Psychiatr. 2014;11:119-123.
- Roy AK, Lopes V, Klein RG. Disruptive mood dysregulation disorder: a new diagnostic approach to chronic irritability in youth. Am J Psychiatry. 2014;171(9):918-924.
Drug Brand Names
Aripiprazole • Abilify
Atomoxetine • Strattera
Clonidine • Catapres
Divalproex sodium • Depakote, Depakote ER
Guanfacine • Intuniv, Tenex
Lithium • Eskalith, Lithobid
Methylphenidate • Concerta, Ritalin
Risperidone • Risperdal
Disruptive mood dysregulation disorder (DMDD)—a childhood condition of extreme irritability, anger, and frequent, intense temper outbursts—has been a source of controversy among clinicians in the field of pediatric mental health. Before DSM-5 was published, the validity of DMDD had been questioned because DMDD had failed a field trial; agreement between clinicians on the diagnosis of DMDD was poor.1 Axelson2 and Birmaher et al3 examined its validity in their COBY (Course and Outcome of Bipolar Youth) sample. They concluded that only 19% met the criteria for DMDD in 3 times of follow-up. Furthermore, most DMDD criteria overlap with those of other common pediatric psychiatric disorders, including oppositional defiant disorder (ODD), attention-deficit/hyperactivity disorder (ADHD), and pediatric bipolar disorder (BD). Because diagnosis of pediatric BD increased drastically from 2.9% to 15.1% between 1990 and 2000,4 it was believed that introducing DMDD as a diagnosis might lessen the overdiagnosis of pediatric BD by identifying children with chronic irritability and temper tantrums who previously would have been diagnosed with BD.
It is important to recognize that in pediatric patients, mood disorders present differently than they do in adults.5 In children/adolescents, mood disorders are less likely to present as distinct episodes (narrow band), but more likely to present as chronic, broad symptoms. Also, irritability is a common presentation in many pediatric psychiatric disorders, such as ODD, BD (irritability without elation),6 and depression. Thus, for many clinicians, determining the correct mood disorder diagnosis in pediatric patients can be challenging.
This article describes the diagnosis of DMDD, and how its presentation is similar to—and different from—those of other common pediatric psychiatric disorders.
_
The origin of DMDD
Many researchers have investigated the broadband phenotypical presentation of pediatric mood disorders, which have been mostly diagnosed in the psychiatric community as pediatric BD. Leibenluft7 identified a subtype of mood disorder that they termed “severe mood dysregulation” (SMD). Compared with the narrow-band, clearly episodic BD, SMD has a different trajectory, outcome, and findings on brain imaging. SMD is characterized by chronic irritability with abnormal mood (anger or sadness) at least half of the day on most days, with 3 hyperarousal symptoms, including pressured speech, racing thoughts or flight of ideas, intrusiveness, distractibility, insomnia, and agitation.8 Eventually, SMD became the foundation of the development of DMDD.
DSM-5 diagnostic criteria for DMDD include severe recurrent temper outbursts that are out of proportion to the situation, inconsistent with developmental level, and occurring on average ≥3 times per week, plus persistently irritable or angry mood for most of the day nearly every day.9 Additional criteria include the presence of symptoms for at least 12 months (without a symptom-free period of at least 3 consecutive months) in ≥2 settings (at home, at school, or with peers) with onset before age 10. The course of DMDD typically is chronic with accompanying severe temperament. The estimated 6-month to 1-year prevalence is 2% to 5%; the diagnosis is more common among males and school-age children than it is in females and adolescents.9,10
_
DMDD or bipolar disorder?
A patient cannot be dually diagnosed with both disorders. If a patient exhibits a manic episode for more than 1 day, that would null and void the DMDD diagnosis. However, in a study to evaluate BD in pediatric patients, researchers divided BD symptoms into BD-specific categories (elevated mood, grandiosity, and increased goal-directed activity) and nonspecific symptoms such as irritability and talkativeness, distractibility, and flight of ideas or racing thoughts. They found that in the absence of specific symptoms, a diagnosis of BD is unlikely to be the correct diagnosis.11 Hence, as a nonspecific symptom, chronic irritability should be attributed to the symptom count for DMDD, rather than BD. Most epidemiologic studies have concluded that depression and anxiety, and not irritability, are typically the preceeding presentations prior to the development of BD in young adults.12 Chronic irritability, however, predicts major depressive disorder and anxiety disorders in later adolescence and one’s early twenties.13 Furthermore, BD commonly presents with infrequent and discrete episodes and a later age of onset, while DMDD presents with chronic and frequent, severe temper outbursts. Some differences between DMDD and BD are illustrated in Table 1.11-13
Continue to: CASE 1
CASE 1
Irritable and taking risks
Ms. N, age 16, is brought to the outpatient psychiatry clinic by her parents for evaluation of mood symptoms, including irritability. Her mother claims her daughter was an introverted, anxious, shy child, but by the beginning of middle school, she began to feel irritable and frequently stayed up at night with little sleep. In high school, Ms. N had displayed several episodes of risk-taking behaviors, including taking her father’s vehicle for a drive despite not having a driver’s permit, running away for 2 days, and having unprotected sex.
During her assessment, Ms. N is pleasant and claims she usually has a great mood. She fought with her mother several times this year, which led her to run away. Her parents had divorced when Ms. N was 5 years old and have shared custody. Ms. N is doing well in school despite her parents’ concerns.
Diagnosis. The most likely diagnosis is emerging BD. Notice that Ms. N may have had anxiety symptoms before she developed irritability. She had a relatively late onset of symptoms that were episodic in nature, which further supports a diagnosis of BD.
_
>
DMDD or oppositional defiant disorder?
DMDD and ODD cannot be dually diagnosed. However, if a patient meets the criteria for both DMDD and ODD, only the DMDD diagnosis should be considered. One of many issues of DMDD is its similarity to ODD. In fact, more than 70% of patients with DMDD also meet the diagnostic criteria for ODD.10,14 Some researchers have conceptualized DMDD as a severe form of ODD. However, there are a few differences that clinicians can use to distinguish the 2 disorders.
Compared with patients with ODD, those with DMDD more frequently experience severe irritability.15 Patients with ODD may present with delinquent behaviors and trouble with authority figures. Moreover, comorbidity with ADHD is twice as common in ODD; more than 65% of patients with ADHD have ODD vs 30% who have DMDD.10,16 Finally, in general, children with DMDD have more social impairments compared with those with ODD. Differences between DMDD and BD are illustrated in Table 2.10,14-16
Continue to: CASE 2
CASE 2
Angry and defiant
Mr. R, age 14, is brought to the emergency department (ED) by his parents after becoming very aggressive with them. He punched a wall and vandalized his room after his parents grounded him because of his previous defiant behavior. He had been suspended from school that day for disrespecting his teacher after he was caught fighting another student.
His parents describe Mr. R as a strong-willed, stubborn child. He has difficulty with rules and refuses to follow them. He is grouchy and irritable around adults, including the ED staff. Mr. R enjoys being with his friends and playing video games. He had been diagnosed with ADHD when he was in kindergarten, when his teacher noticed he had a poor attention span and could not sit still. According to his parents, Mr. R has “blown up” a few times before, smashing items in his room and shouting obscenities. Mr. R’s parents noticed that he is more defiant in concurrence with discontinuing his ADHD stimulant medication.
Diagnosis. The most likely diagnosis for Mr. R is ODD. Notice the comorbidity of ADHD, which is more commonly associated with ODD. The frequency and severity of his outbursts and irritability symptoms were lower than that typically associated with DMDD.
_
Treatment strategies for DMDD
Management of DMDD should focus on helping children and adolescents improve their emotional dysregulation.
Clinicians should always consider behavioral therapy as a first-line intervention. The behavioral planning team may include (but is not limited to) a behavior specialist, child psychiatrist, psychologist, therapist, skills trainer, teachers, and the caregiver(s). The plan should be implemented across all settings, including home and school. Furthermore, social skills training is necessary for many children with DMDD, who may require intensive behavioral modification planning. Comorbidity with ADHD should be addressed with a combination of behavioral planning and stimulant medications.17 If available, parent training and parent-child interactive therapy can help to improve defiant behavior.
Pharmacotherapy
Currently, no medications are FDA-approved for treating DMDD. Most pharmacologic trials that included patients with DMDD focused on managing chronic irritability and/or stabilizing comorbid disorders (ie, ADHD, depression, and anxiety).
Continue to: Stimulants
Stimulants. Previous trials examined the benefit of CNS stimulant medications, alone or in conjunction with behavioral therapy, in treating DMDD and comorbid ADHD. Methylphenidate results in a significant reduction in aggression18 with a dosing recommendation range from 1 to 1.2 mg/kg/d. CNS stimulants should be considered as first-line pharmacotherapy for DMDD, especially for patients with comorbid ADHD.
Anticonvulsants. Divalproex sodium is superior to placebo in treating aggression in children and adolescents.19 Trials found that divalproex sodium reduces irritability and aggression whether it is prescribed as monotherapy or combined with stimulant medications.19
Lithium is one of the main treatment options for mania in BD. The benefits of lithium for controlling aggression in DMDD are still under investigation. Earlier studies found that lithium significantly improves aggressive behavior in hospitalized pediatric with conduct disorder.20,21 However, a later study that evaluated lithium vs placebo for children with SMD (which arguably is phenotypically related to the DMDD) found there were no significant differences in improvement of irritability symptoms between groups.22 More research is needed to determine if lithium may play a role in treating patients with DMDD.
Antipsychotics. Aripiprazole and risperidone are FDA-approved for treating irritability in autism. A 2017 meta-analysis found both medications were effective in controlling irritability and aggression in other diagnoses as well.23 Other antipsychotic medications did not show sufficient benefits in treating irritability.23 When considering antipsychotics, clinicians should weigh the risks of metabolic adverse effects and follow practice guidelines.
Antidepressants. A systematic review did not find that selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors effectively reduce irritability.24 However, in most of the studies evaluated, irritability was not the primary outcome measure.24
Other medications. Alpha-2 agonists (guanfacine, clonidine), and atomoxetine may help irritability indirectly by improving ADHD symptoms.25
Bottom Line
Disruptive mood dysregulation disorder (DMDD), bipolar disorder, and oppositional defiant disorder have similar presentations and diagnostic criteria. The frequency and severity of irritability can be a distinguishing factor. Behavioral therapy is a first-line treatment. No medications are FDA-approved for treating DMDD, but pharmacotherapy may help reduce irritability and aggression.
Related Resources
- Rao U. DSM-5: disruptive mood dysregulation disorder. Asian J Psychiatr. 2014;11:119-123.
- Roy AK, Lopes V, Klein RG. Disruptive mood dysregulation disorder: a new diagnostic approach to chronic irritability in youth. Am J Psychiatry. 2014;171(9):918-924.
Drug Brand Names
Aripiprazole • Abilify
Atomoxetine • Strattera
Clonidine • Catapres
Divalproex sodium • Depakote, Depakote ER
Guanfacine • Intuniv, Tenex
Lithium • Eskalith, Lithobid
Methylphenidate • Concerta, Ritalin
Risperidone • Risperdal
1. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
2. Axelson D. Taking disruptive mood dysregulation disorder out for a test drive. Am J Psychiatry. 2013;170(2):136-139.
3. Birmaher B, Axelson D, Goldstein B, et al. Four-year longitudinal course of children and adolescents with bipolar spectrum disorders: the Course and Outcome of Bipolar Youth (COBY) study. Am J Psychiatry. 2009;166(7):795-804.
4. Case BG, Olfson M, Marcus SC, et al. Trends in the inpatient mental health treatment of children and adolescents in US community hospitals between 1990 and 2000. Arch Gen Psychiatry. 2007;64(1):89-96.
5. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
6. Hunt J, Birmaher B, Leonard H, et al. Irritability without elation in a large bipolar youth sample: frequency and clinical description. J Am Acad Child Adolesc Psychiatry. 2009;48(7):730-739.
7. Leibenluft E. Severe mood dysregulation, irritability, and the diagnostic boundaries of bipolar disorder in youths. Am J Psychiatry. 2011;168(2):129-142.
8. Rich BA, Carver FW, Holroyd T, et al. Different neural pathways to negative affect in youth with pediatric bipolar disorder and severe mood dysregulation. J Psychiatr Res. 2011;45(10):1283-1294.
9. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
10. Copeland WE, Angold A, Costello EJ, et al. Prevalence, comorbidity, and correlates of DSM-5 proposed disruptive mood dysregulation disorder. Am J Psychiatry. 2013;170(2):173-179.
11. Elmaadawi AZ, Jensen PS, Arnold LE, et al. Risk for emerging bipolar disorder, variants, and symptoms in children with attention deficit hyperactivity disorder, now grown up. World J Psychiatry. 2015;5(4):412-424.
12. Duffy A. The early natural history of bipolar disorder: what we have learned from longitudinal high-risk research. Can J Psychiatry. 2010;55(8):477-485.
13. Stringaris A, Cohen P, Pine DS, et al. Adult outcomes of youth irritability: a 20-year prospective community-based study. Am J Psychiatry. 2009;166(9):1048-1054.
14. Mayes SD, Waxmonsky JD, Calhoun SL, et al. Disruptive mood dysregulation disorder symptoms and association with oppositional defiant and other disorders in a general population child sample. J Child Adolesc Psychopharmacol. 2016;26(2):101-106.
15. Stringaris A, Vidal-Ribas P, Brotman MA, et al. Practitioner review: definition, recognition, and treatment challenges of irritability in young people. J Child Psychol Psychiatry. 2018;59(7):721-739.
16. Angold A, Costello EJ, Erkanli A. Comorbidity. J Child Psychol Psychiatry. 1999;40(1):57-87.
17. Fernandez de la Cruz L, Simonoff E, McGough JJ, et al. Treatment of children with attention-deficit/hyperactivity disorder (ADHD) and irritability: results from the multimodal treatment study of children with ADHD (MTA). J Am Acad Child Adolesc Psychiatry. 2015;54(1):62-70.
18. Pappadopulos E, Woolston S, Chait A, et al. Pharmacotherapy of aggression in children and adolescents: efficacy and effect size. J Can Acad Child Adolesc Psychiatry. 2006;15(1):27-39.
19. Donovan SJ, Stewart JW, Nunes EV, et al. Divalproex treatment for youth with explosive temper and mood lability: a double-blind, placebo-controlled crossover design. Am J Psychiatry. 2000;157(5):818-820.
20. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
21. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
22. Dickstein DP, Towbin KE, Van Der Veen JW, et al. Randomized double-blind placebo-controlled trial of lithium in youths with severe mood dysregulation. J Child Adolesc Psychopharmacol. 2009;19(1):61-73.
23. van Schalkwyk GI, Lewis AS, Beyer C, et al. Efficacy of antipsychotics for irritability and aggression in children: a meta-analysis. Expert Rev Neurother. 2017;17(10):1045-1053.
24. Kim S, Boylan K. Effectiveness of antidepressant medications for symptoms of irritability and disruptive behaviors in children and adolescents. J Child Adolesc Psychopharmacol. 2016;26(8):694-704.
25. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158(7):1067-1074.
1. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
2. Axelson D. Taking disruptive mood dysregulation disorder out for a test drive. Am J Psychiatry. 2013;170(2):136-139.
3. Birmaher B, Axelson D, Goldstein B, et al. Four-year longitudinal course of children and adolescents with bipolar spectrum disorders: the Course and Outcome of Bipolar Youth (COBY) study. Am J Psychiatry. 2009;166(7):795-804.
4. Case BG, Olfson M, Marcus SC, et al. Trends in the inpatient mental health treatment of children and adolescents in US community hospitals between 1990 and 2000. Arch Gen Psychiatry. 2007;64(1):89-96.
5. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
6. Hunt J, Birmaher B, Leonard H, et al. Irritability without elation in a large bipolar youth sample: frequency and clinical description. J Am Acad Child Adolesc Psychiatry. 2009;48(7):730-739.
7. Leibenluft E. Severe mood dysregulation, irritability, and the diagnostic boundaries of bipolar disorder in youths. Am J Psychiatry. 2011;168(2):129-142.
8. Rich BA, Carver FW, Holroyd T, et al. Different neural pathways to negative affect in youth with pediatric bipolar disorder and severe mood dysregulation. J Psychiatr Res. 2011;45(10):1283-1294.
9. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
10. Copeland WE, Angold A, Costello EJ, et al. Prevalence, comorbidity, and correlates of DSM-5 proposed disruptive mood dysregulation disorder. Am J Psychiatry. 2013;170(2):173-179.
11. Elmaadawi AZ, Jensen PS, Arnold LE, et al. Risk for emerging bipolar disorder, variants, and symptoms in children with attention deficit hyperactivity disorder, now grown up. World J Psychiatry. 2015;5(4):412-424.
12. Duffy A. The early natural history of bipolar disorder: what we have learned from longitudinal high-risk research. Can J Psychiatry. 2010;55(8):477-485.
13. Stringaris A, Cohen P, Pine DS, et al. Adult outcomes of youth irritability: a 20-year prospective community-based study. Am J Psychiatry. 2009;166(9):1048-1054.
14. Mayes SD, Waxmonsky JD, Calhoun SL, et al. Disruptive mood dysregulation disorder symptoms and association with oppositional defiant and other disorders in a general population child sample. J Child Adolesc Psychopharmacol. 2016;26(2):101-106.
15. Stringaris A, Vidal-Ribas P, Brotman MA, et al. Practitioner review: definition, recognition, and treatment challenges of irritability in young people. J Child Psychol Psychiatry. 2018;59(7):721-739.
16. Angold A, Costello EJ, Erkanli A. Comorbidity. J Child Psychol Psychiatry. 1999;40(1):57-87.
17. Fernandez de la Cruz L, Simonoff E, McGough JJ, et al. Treatment of children with attention-deficit/hyperactivity disorder (ADHD) and irritability: results from the multimodal treatment study of children with ADHD (MTA). J Am Acad Child Adolesc Psychiatry. 2015;54(1):62-70.
18. Pappadopulos E, Woolston S, Chait A, et al. Pharmacotherapy of aggression in children and adolescents: efficacy and effect size. J Can Acad Child Adolesc Psychiatry. 2006;15(1):27-39.
19. Donovan SJ, Stewart JW, Nunes EV, et al. Divalproex treatment for youth with explosive temper and mood lability: a double-blind, placebo-controlled crossover design. Am J Psychiatry. 2000;157(5):818-820.
20. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
21. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
22. Dickstein DP, Towbin KE, Van Der Veen JW, et al. Randomized double-blind placebo-controlled trial of lithium in youths with severe mood dysregulation. J Child Adolesc Psychopharmacol. 2009;19(1):61-73.
23. van Schalkwyk GI, Lewis AS, Beyer C, et al. Efficacy of antipsychotics for irritability and aggression in children: a meta-analysis. Expert Rev Neurother. 2017;17(10):1045-1053.
24. Kim S, Boylan K. Effectiveness of antidepressant medications for symptoms of irritability and disruptive behaviors in children and adolescents. J Child Adolesc Psychopharmacol. 2016;26(8):694-704.
25. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158(7):1067-1074.

Bright light therapy for bipolar depression
Bright light therapy (BLT) refers to the use of bright light to treat symptoms of depression. BLT was initially prescribed as a treatment for patients with seasonal affective disorder.1 It was later found helpful for nonseasonal depression,2 premenstrual dysphoric disorder, postpartum depression, and phase shift circadian disorders, including for patients with dementia whose cognitive function improved after treatment with BLT.3 More recent studies suggest year-round benefit for nonseasonal depression.2 The American Psychiatric Association practice guidelines for the treatment of depression list BLT as an alternative and/or addition to pharmacologic and psychological treatment.4 BLT also may be beneficial for patients who are in the depressive phase of bipolar illness.
This article describes the evidence, rationale for use, mechanism of action, benefits, and safety profile of BLT for treating patients with bipolar depression.
Circadian rhythm disruption in bipolar disorder
Clinical manifestation. Patients with bipolar disorder (BD) spend more time in depression than in mania.5 Sleep disturbance is a core symptom of BD; patients typically have little need for sleep during a manic episode, and excess sleepiness during a depressive episode. Sleep complaints can be both precipitating factors and consequences of mood disorders. Patients with seasonal depression have excess sleepiness and weight gain in the winter followed by hypomanic-like symptoms in the spring, including decreased need for sleep and weight loss with psychomotor activation. In a recent review of sleep problems in patients with BD, Steinan et al6 reported that 20% of patients with euthymic mood in bipolar disorder experience a sleep disorder. Circadian disruption and “eveningness” (being more active during the evening) have been associated with mood episodes, functional impairment, poor quality of life, and treatment resistance.7-10
Pathophysiology. Existing hypotheses for the biological mechanism underlying dysregulation of circadian rhythm in BD include changes in melatonin levels, expression of melatonin receptors in the CNS, and daily cortisol profiles.11 Genetic evidence also links circadian rhythm dysregulation with BD. Two polymorphisms on the circadian locomotor output cycles kaput (CLOCK) gene that control circadian rhythm—aryl hydrocarbon receptor nuclear translocator-like (ARNTL) and timeless circadian clock (TIMELESS)—have been linked to lithium responsiveness in BD.12 In addition, Per2, Cry1, and Rev-Erbα expression—all components of the circadian clock—have been found to increase individual susceptibility to the therapeutic effects of lithium in mice.13 In addition, circadian rhythm dysregulation is associated with metabolic problems encountered by patients with BD, including weight gain, diabetes mellitus, and cardiovascular disease.14
Rationale for use
Regulation of a patient’s circadian rhythm disruption is a potential treatment for BD. Hashimoto et al15 demonstrated that midday bright light exposure can phase advance and increase the amplitude of nocturnal melatonin production in healthy individuals. Morning light therapy has been shown to increase blood serotonin throughout the day in both healthy individuals and in patients with nonseasonal depression; the effect was apparent with light intensities as low as 50 lux.16 Lithium may exert its therapeutic effect through its influence on the retino-hypothalamic-pineal tract and thus its effect on melatonin secretion.17
BLT is a logical choice to treat the depression phase of BD when exposure to sunlight is not feasible due to geographical location, season, or other factor. For patients who live in areas that receive frequent sunshine, an outside stroll for half an hour will likely achieve similar benefit to BLT.
The precise mechanism of action of BLT for bipolar depression has not yet been determined. It may be attributed to a phase-resetting effect via melanopsin and the suprachiasmatic nucleus (Box18-24).
Box
Bright light therapy: How it works
The mechanism of action of bright light therapy is yet to be elucidated. The suprachiasmatic nucleus (SCN) in the hypothalamus is the center of circadian rhythm regulation and receives direct input from the retina through the retinohypothalamic tract.18 Melanopsin, a short-wavelength, light-sensitive G-protein–coupled receptor located in human retinal ganglion cells, is known to transduce short-wavelength light signals into neural signals.19 Since melanopsin is primarily responsible for resetting the timing of the SCN, suppressing pineal gland melatonin secretion and improving alertness and electroencephalogram-derived correlates of arousal,20 short-wavelength light with a low light intensity might be a better stimulator for melanopsin-containing retinal ganglion cells and the behaviors mediated via this photoreceptor system.21,22 Whether the antidepressant effect of light is also related to its alerting property is unclear.23 However, the acute alerting and performance-enhancing effects of light are increasingly taken into account for the design of indoor light standards in office environments.24 Response to light therapy is thus attributed to its phase-resetting effect.
Continue to: BLT for BD...
BLT for BD: What’s the evidence?
Several studies and case reports have evaluated the use of BLT for bipolar depression. The number of participants in these studies is small, and there is no uniformity of methodology or patient selection.
Dauphinais et al (2012)25 randomly assigned 44 patients with bipolar depression to BLT or a high-density or low-density negative ion generator for 8 weeks. They reported no difference in outcome between the various groups (50% vs 55.6%, remission and response rate). Only one patient in each group showed a switch to hypomania.
Carmadese et al (2015)26 reported an open-label study of adjunctive BLT in 31 difficult-to-treat patients with depression (16 unipolar and 15 bipolar). Significant improvement was noted within 3 weeks and was sustained in 1 patient with bipolar depression 5 weeks after cessation of BLT.
Papatheodorou and Kutcher (1995)27 treated 7 adolescents with bipolar depression with adjunctive BLT (10,000 lux twice per day). Three patients showed a marked response (>70% decrease from baseline Beck Depression Inventory and Symptom Check List scores). Two patients had a moderate response (40% to 47% decrease) and 2 patients obtained mild to no response. There were no reported adverse effects.
Benedetti et al (2014)28 studied 141 patients with treatment-resistant bipolar depression. Approximately one-quarter (23%) had a history of attempted suicide, and 83% had a history of drug resistance. The authors found a combination of total sleep deprivation, BLT, and lithium rapidly decreased suicidality and improved patients’ depressive symptoms.
Liebenluft et al (1995)29 administered 13 trials of BLT to 9 patients with rapid-cycling BD during a 3-month period. Five patients received the treatment in the morning, 5 around midday, and 3 in the evening. Patients who received BLT at midday had the best outcome, while 3 of the 5 patients who received morning BLT developed unstable mood. The authors recommended titrating the duration of light exposure so that patients could skip a treatment if their mood was trending toward hypomania.
Sit et al (2007)30 evaluated BLT in a case series of 9 women with bipolar I or II disorder in the depression phase. Patients were exposed to 50 lux of red light for 2 weeks, and then they received 7,000 lux BLT for 15, 30, and 45 minutes daily for 2 weeks (4 patients received morning light and 5 received midday light). Mood was assessed using the Structured Interview Guide for the Hamilton Depression Rating Scale with Atypical Depression Supplement and the Mania Rating Scale. Of the 4 patients receiving morning BLT, one patient had a full response and the other 3 developed hypomania. Of the 5 patients who received midday BLT, 2 achieved full response, 2 showed early improvement but required a dose increase, and one remained depressed but had a full response when she was switched to morning BLT.
Tseng et al (2016)31 reported a meta-analysis of BLT for bipolar depression that included a total of 567 patients from 11 studies. They reported significant improvement with BLT alone or in combination with antidepressants or total sleep deprivation. They also reported significant improvement with BLT in 130 patients who were not receiving other treatments. There was no difference in the frequency of mood shifts between patients on BLT alone or in combination with other modalities. The authors reported no mood shift in any of the patients receiving concurrent mood stabilizers. They also reported no difference with the color of light, gender, or duration of illness.
Yorguner et al (2017)32 conducted a 2-week randomized, single-blind study of BLT as an add-on treatment for 32 patients with bipolar depression. Patients were randomly assigned to BLT or dim light, which they were administered each morning for 30 mins for 2 weeks. Sixteen patients who received BLT showed a significantly greater reduction in Hamilton Depression Rating Scale scores (mean score of 24 at baseline down to 12) compared with 16 patients who received dim light (mean score of 24 at baseline down to 18). The authors also reported remission in 4 out of 4 patients who had seasonal depression, compared with 3 out of 12 who did not have seasonal depression (the other 9 showed response but not remission).
Zhou et al (2018)33 conducted a multi-center, randomized, single-blind clinical trial of 63 patients with bipolar depression. Thirty-three patients received morning BLT, and 30 received dim red light therapy (control group). The authors reported a significantly higher response rate in the BLT group (78%) compared with the control group (43%).
Sit et al (2018)34 conducted a 6-week randomized, double-blind, placebo-controlled trial of BLT vs dim red light in patients with bipolar I or II depression. Twenty-three patients were administered 7,000 lux bright white light, and 23 patients received 50 lux dim red light, at midday 5 days a week. The light dose was increased by 15 minutes every week up to 60 minutes by Week 4, unless the patient achieved remission. Patients were maintained on their usual medications, which included mood stabilizers and/or antidepressants. At Week 6, the group randomized to BLT had a significantly higher remission rate (68%) compared with patients who received dim red light (22%). Improvement was noted by Week 4. Patients receiving BLT also had significantly fewer depressive symptoms, and no mood polarity switch was noted.
Prescribing bright light therapy
Light box selection criteria. When selecting a light box or related BLT treatment apparatus, the Center for Environmental Therapeutics recommends consideration of the following factors35:
- clinical efficacy
- ocular and dermatologic safety
- visual comfort.
Selecting a dose. The dose received is determined by the intensity emitted from the light source, distance from the light box, and duration of exposure.36 Begin with midday light therapy between 12 noon and 2
Monitor for adverse effects. Generally, BLT is well tolerated.37 Adverse effects are rare; the most common ones include headache, eyestrain, nausea, and agitation.38 One study found no adverse ocular effects from light therapy after 5 years of treatment.39 Adverse effects tend to remit spontaneously or after dose reduction.35 Evening administration of BLT may increase the incidence of sleep disturbances.40 Like other biologic treatments for bipolar depression, BLT can precipitate manic/hypomanic and mixed states in susceptible patients, although the light dose can be titrated against emergent symptoms of hypomania.41
Bottom Line
Evidence suggests that bright light therapy is an effective, well tolerated, and affordable adjunct treatment for bipolar depression. Exposure to 5,000 to 7,000 lux around noon for 15 to 60 minutes will enhance the remission rate.
Related Resource
Mostert M, Dubovsky S. When bipolar treatment fails: what’s your next step? Current Psychiatry. 2008;7(1):39-46.
Drug Brand Name
Lithium • Eskalith, Lithobid
1. Pjrek E, Winkler D, Stastny J, et al. Bright light therapy in seasonal affective disorder--does it suffice? Eur Neuropsychopharmacol. 2004.14(4):347-351.
2. Al-Karawi D, Jubair L. Bright light therapy for nonseasonal depression: meta-analysis of clinical trials. J Affect Disord. 2016;198:64-71.
3. Sekiguchi H, Iritani S, Fujita K. Bright light therapy for sleep disturbance in dementia is most effective for mild to moderate Alzheimer’s type dementia: a case series. Psychogeriatrics, 2017;17(5):275-281.
4. Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder, third edition. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf American Psychiatric Association. 2010. Accessed August, 10, 2017.
5. Kupka RW, Altshuler LL, Nolen WA, et al. Three times more days depressed than manic or hypomanic in both bipolar I and bipolar II disorder. Bipolar Disord. 2007;9(5):531-535.
6. Steinan MK, Krane-Gartiser K, Morken G, et al. Sleep problems in euthymic bipolar disorders: a review of clinical studies. Current Psychiatry Reviews. 2015;11:235-243.
7. Cudney LE, Frey BN, Streiner D, et al. Biological rhythms are independently associated with quality of life in bipolar disorder. Int J Bipolar Disord. 2016;4(1):9.
8. Duarte FA, Cardoso TA, Campos MT, et al. Biological rhythms in bipolar and depressive disorders: a community study with drug-naive young adults. J Affect Disord, 2015;186:145-148.
9. Pinho M, Sehmbi M, Cudney LE, et al. The association between biological rhythms, depression, and functioning in bipolar disorder: a large multi-center study. Acta Psychiatr Scand. 2015:133(2);102-108.
10. Ng TH, Chung KF, Lee CT, et al. Eveningness and its associated impairments in remitted bipolar disorder. Behav Sleep Med. 2016:14(6):650-664.
11. Wu YH, Ursinus J, Zahn JN, et al. Alterations of melatonin receptors MT1 and MT2 in the hypothalamic suprachiasmatic nucleus during depression. J Affect Disord, 2013:148(2-3):357-367.
12. Rybakowski JK, Dmitrzak-Weglar M, Kliwicki S, et al. Polymorphism of circadian clock genes and prophylactic lithium response. Bipolar Disord. 2014;16(2):151-158.
13. Schnell A, Sandrelli F, Ranc V, et al. Mice lacking circadian clock components display different mood-related behaviors and do not respond uniformly to chronic lithium treatment. Chronobiol Int. 2015;32(8):1075-1089.
14. Kim Y, Santos R, Gage FH, et al. Molecular mechanisms of bipolar disorder: progress made and future challenges. Front Cell Neurosci. 2017;11:30.
15. Hashimoto S, Kohsaka M, Nakamura K. Midday exposure to bright light changes the circadian organization of plasma melatonin rhythm in humans. Neurosci Lett. 1997;221(2-3):
89-92.
16. Rao ML, Müller-Oerlinghausen B, Mackert A, et al. The influence of phototherapy on serotonin and melatonin in non-seasonal depression. Pharmacopsychiatry.1990;23(3):155-158.
17. Moreira J, Geoffroy PA. Lithium and bipolar disorder: impacts from molecular to behavioural circadian rhythms. Chronobiol Int. 2016;33(4):351-373.
18. Oldham MA, Ciraulo DA. Bright light therapy for depression: a review of its effects on chronobiology and the autonomic nervous system. Chronobiol Int. 2014;31(3):305-319.
19. Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295(5557):1070-1073.
20. Peirson S, Foster RG. Melanopsin: another way of signaling light. Neuron. 2006;49(3):331-339.
21. Anderson JL, Glod CA, Dai J, et al. Lux vs. wavelength in light treatment of seasonal affective disorder. Acta Psychiatr Scand. 2009;120(3):203-212.
22. Wirz-Justice A, Graw P, Kräuchi K, et al. Effect of light on unmasked circadian rhythms in winter depression. In: Wetterberg L, ed. Light and biological rhythms in man. Oxford, United Kingdom:Pergamon Press;1993:385-393.
23. Cajochen C. Alerting effects of light. Sleep Med Rev. 2007;11(6):453-464.
24. Aries MBC. Human lighting demands: healthy lighting in an office environment. Eindhoven, Eindhoven University Press. 2005;158. doi:10.6100/IR594257.
25. Dauphinais DR, Rosenthal JZ, Terman M, et al. Controlled trial of safety and efficacy of bright light therapy vs. negative air ions in patients with bipolar depression. Psychiatry Res. 2012;196(1):57-61.
26. Camardese G, Leone B, Serrani R, et al. Augmentation of light therapy in difficult-to-treat depressed patients: an open-label trial in both unipolar and bipolar patients. Neuropsychiatr Dis Treat. 2015;11:2331-2338.
27. Papatheodorou G, Kutcher S. The effect of adjunctive light therapy on ameliorating breakthrough depressive symptoms in adolescent-onset bipolar disorder.
J Psychiatry Neurosci. 1995;20(3):226-232.
28. Benedetti F, Riccaboni R, Locatelli C, et al. Rapid treatment response of suicidal symptoms to lithium, sleep deprivation, and light therapy (chronotherapeutics) in drug-resistant bipolar depression. J Clin Psychiatry. 2014;75(2):133-140.
29. Liebenluft E, Turner EH, Felman-Naim S, et al. Light therapy in patients with rapid cycling bipolar disorder: preliminary results. Psychopharmacol Bull. 1995;31(4):
705-710.
30. Sit DK, Wisner KL, Hanusa BH, et al. Light therapy for bipolar disorder: a case series in women. Bipolar Disord. 2007;9(8):918-927.
31. Tseng PT, Chen YW, Tu KY, et al. Light therapy in the treatment of patients with bipolar depression: a meta-analytic study. Eur Neuropsychopharmacol. 2016;26(6):
1037-1047.
32. Yorguner KN, Bulut NS, Carkaxhiu BG, et al. Efficacy of bright light therapy in bipolar depression. Psychiatry Res. 2017;260:432-438.
33. Zhou TH, Dang WM, Ma YT, et al. Clinical efficacy, onset time and safety of bright light therapy in acute bipolar depression as an adjunctive therapy: a randomized controlled trial. J Affect Disord. 2018;227:90-96.
34. Sit DK, McGowan J, Wiltrout C, et al. Adjunctive bright light therapy for bipolar depression: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2018;175(2):
131-139.
35. Center for Environmental Therapeutics. https://www.cet.org/. Center for Environmental Therapeutics. Accessed November 15, 2017.
36. Lam RW, Levitt AJ. Canadian consensus guidelines for the treatment of seasonal affective disorder. https://mdsc.ca/documents/Consumer%20and%20Family%20Support/CCG_on_Seasonal_Affective_Disorder.pdf. 1999. Accessed August 2, 2017.
37. Terman M, Terman JS. Bright light therapy: side effects and benefits across the symptom spectrum. J Clin Psychiatry. 1999; 60(11):799-808;quiz 809.
38. Labbate LA, et al. Side effects induced by bright light treatment for seasonal affective disorder. J Clin Psychiatry. 1994; 55(5):189-191.
39. Gallin PF, et al. Ophthalmologic examination of patients with seasonal affective disorder, before and after bright light therapy. Am J Ophthalmol. 1995;119(2):202-210.
40. Chan PK, Lam RW, Perry KF. Mania precipitated by light therapy for patients with SAD. J Clin Psychiatry. 1994;55(10):454.
41. Kripke DF. Timing of phototherapy and occurrence of mania. Biol Psychiatry. 1991; 29(11):1156-1157.
Bright light therapy (BLT) refers to the use of bright light to treat symptoms of depression. BLT was initially prescribed as a treatment for patients with seasonal affective disorder.1 It was later found helpful for nonseasonal depression,2 premenstrual dysphoric disorder, postpartum depression, and phase shift circadian disorders, including for patients with dementia whose cognitive function improved after treatment with BLT.3 More recent studies suggest year-round benefit for nonseasonal depression.2 The American Psychiatric Association practice guidelines for the treatment of depression list BLT as an alternative and/or addition to pharmacologic and psychological treatment.4 BLT also may be beneficial for patients who are in the depressive phase of bipolar illness.
This article describes the evidence, rationale for use, mechanism of action, benefits, and safety profile of BLT for treating patients with bipolar depression.
Circadian rhythm disruption in bipolar disorder
Clinical manifestation. Patients with bipolar disorder (BD) spend more time in depression than in mania.5 Sleep disturbance is a core symptom of BD; patients typically have little need for sleep during a manic episode, and excess sleepiness during a depressive episode. Sleep complaints can be both precipitating factors and consequences of mood disorders. Patients with seasonal depression have excess sleepiness and weight gain in the winter followed by hypomanic-like symptoms in the spring, including decreased need for sleep and weight loss with psychomotor activation. In a recent review of sleep problems in patients with BD, Steinan et al6 reported that 20% of patients with euthymic mood in bipolar disorder experience a sleep disorder. Circadian disruption and “eveningness” (being more active during the evening) have been associated with mood episodes, functional impairment, poor quality of life, and treatment resistance.7-10
Pathophysiology. Existing hypotheses for the biological mechanism underlying dysregulation of circadian rhythm in BD include changes in melatonin levels, expression of melatonin receptors in the CNS, and daily cortisol profiles.11 Genetic evidence also links circadian rhythm dysregulation with BD. Two polymorphisms on the circadian locomotor output cycles kaput (CLOCK) gene that control circadian rhythm—aryl hydrocarbon receptor nuclear translocator-like (ARNTL) and timeless circadian clock (TIMELESS)—have been linked to lithium responsiveness in BD.12 In addition, Per2, Cry1, and Rev-Erbα expression—all components of the circadian clock—have been found to increase individual susceptibility to the therapeutic effects of lithium in mice.13 In addition, circadian rhythm dysregulation is associated with metabolic problems encountered by patients with BD, including weight gain, diabetes mellitus, and cardiovascular disease.14
Rationale for use
Regulation of a patient’s circadian rhythm disruption is a potential treatment for BD. Hashimoto et al15 demonstrated that midday bright light exposure can phase advance and increase the amplitude of nocturnal melatonin production in healthy individuals. Morning light therapy has been shown to increase blood serotonin throughout the day in both healthy individuals and in patients with nonseasonal depression; the effect was apparent with light intensities as low as 50 lux.16 Lithium may exert its therapeutic effect through its influence on the retino-hypothalamic-pineal tract and thus its effect on melatonin secretion.17
BLT is a logical choice to treat the depression phase of BD when exposure to sunlight is not feasible due to geographical location, season, or other factor. For patients who live in areas that receive frequent sunshine, an outside stroll for half an hour will likely achieve similar benefit to BLT.
The precise mechanism of action of BLT for bipolar depression has not yet been determined. It may be attributed to a phase-resetting effect via melanopsin and the suprachiasmatic nucleus (Box18-24).
Box
Bright light therapy: How it works
The mechanism of action of bright light therapy is yet to be elucidated. The suprachiasmatic nucleus (SCN) in the hypothalamus is the center of circadian rhythm regulation and receives direct input from the retina through the retinohypothalamic tract.18 Melanopsin, a short-wavelength, light-sensitive G-protein–coupled receptor located in human retinal ganglion cells, is known to transduce short-wavelength light signals into neural signals.19 Since melanopsin is primarily responsible for resetting the timing of the SCN, suppressing pineal gland melatonin secretion and improving alertness and electroencephalogram-derived correlates of arousal,20 short-wavelength light with a low light intensity might be a better stimulator for melanopsin-containing retinal ganglion cells and the behaviors mediated via this photoreceptor system.21,22 Whether the antidepressant effect of light is also related to its alerting property is unclear.23 However, the acute alerting and performance-enhancing effects of light are increasingly taken into account for the design of indoor light standards in office environments.24 Response to light therapy is thus attributed to its phase-resetting effect.
Continue to: BLT for BD...
BLT for BD: What’s the evidence?
Several studies and case reports have evaluated the use of BLT for bipolar depression. The number of participants in these studies is small, and there is no uniformity of methodology or patient selection.
Dauphinais et al (2012)25 randomly assigned 44 patients with bipolar depression to BLT or a high-density or low-density negative ion generator for 8 weeks. They reported no difference in outcome between the various groups (50% vs 55.6%, remission and response rate). Only one patient in each group showed a switch to hypomania.
Carmadese et al (2015)26 reported an open-label study of adjunctive BLT in 31 difficult-to-treat patients with depression (16 unipolar and 15 bipolar). Significant improvement was noted within 3 weeks and was sustained in 1 patient with bipolar depression 5 weeks after cessation of BLT.
Papatheodorou and Kutcher (1995)27 treated 7 adolescents with bipolar depression with adjunctive BLT (10,000 lux twice per day). Three patients showed a marked response (>70% decrease from baseline Beck Depression Inventory and Symptom Check List scores). Two patients had a moderate response (40% to 47% decrease) and 2 patients obtained mild to no response. There were no reported adverse effects.
Benedetti et al (2014)28 studied 141 patients with treatment-resistant bipolar depression. Approximately one-quarter (23%) had a history of attempted suicide, and 83% had a history of drug resistance. The authors found a combination of total sleep deprivation, BLT, and lithium rapidly decreased suicidality and improved patients’ depressive symptoms.
Liebenluft et al (1995)29 administered 13 trials of BLT to 9 patients with rapid-cycling BD during a 3-month period. Five patients received the treatment in the morning, 5 around midday, and 3 in the evening. Patients who received BLT at midday had the best outcome, while 3 of the 5 patients who received morning BLT developed unstable mood. The authors recommended titrating the duration of light exposure so that patients could skip a treatment if their mood was trending toward hypomania.
Sit et al (2007)30 evaluated BLT in a case series of 9 women with bipolar I or II disorder in the depression phase. Patients were exposed to 50 lux of red light for 2 weeks, and then they received 7,000 lux BLT for 15, 30, and 45 minutes daily for 2 weeks (4 patients received morning light and 5 received midday light). Mood was assessed using the Structured Interview Guide for the Hamilton Depression Rating Scale with Atypical Depression Supplement and the Mania Rating Scale. Of the 4 patients receiving morning BLT, one patient had a full response and the other 3 developed hypomania. Of the 5 patients who received midday BLT, 2 achieved full response, 2 showed early improvement but required a dose increase, and one remained depressed but had a full response when she was switched to morning BLT.
Tseng et al (2016)31 reported a meta-analysis of BLT for bipolar depression that included a total of 567 patients from 11 studies. They reported significant improvement with BLT alone or in combination with antidepressants or total sleep deprivation. They also reported significant improvement with BLT in 130 patients who were not receiving other treatments. There was no difference in the frequency of mood shifts between patients on BLT alone or in combination with other modalities. The authors reported no mood shift in any of the patients receiving concurrent mood stabilizers. They also reported no difference with the color of light, gender, or duration of illness.
Yorguner et al (2017)32 conducted a 2-week randomized, single-blind study of BLT as an add-on treatment for 32 patients with bipolar depression. Patients were randomly assigned to BLT or dim light, which they were administered each morning for 30 mins for 2 weeks. Sixteen patients who received BLT showed a significantly greater reduction in Hamilton Depression Rating Scale scores (mean score of 24 at baseline down to 12) compared with 16 patients who received dim light (mean score of 24 at baseline down to 18). The authors also reported remission in 4 out of 4 patients who had seasonal depression, compared with 3 out of 12 who did not have seasonal depression (the other 9 showed response but not remission).
Zhou et al (2018)33 conducted a multi-center, randomized, single-blind clinical trial of 63 patients with bipolar depression. Thirty-three patients received morning BLT, and 30 received dim red light therapy (control group). The authors reported a significantly higher response rate in the BLT group (78%) compared with the control group (43%).
Sit et al (2018)34 conducted a 6-week randomized, double-blind, placebo-controlled trial of BLT vs dim red light in patients with bipolar I or II depression. Twenty-three patients were administered 7,000 lux bright white light, and 23 patients received 50 lux dim red light, at midday 5 days a week. The light dose was increased by 15 minutes every week up to 60 minutes by Week 4, unless the patient achieved remission. Patients were maintained on their usual medications, which included mood stabilizers and/or antidepressants. At Week 6, the group randomized to BLT had a significantly higher remission rate (68%) compared with patients who received dim red light (22%). Improvement was noted by Week 4. Patients receiving BLT also had significantly fewer depressive symptoms, and no mood polarity switch was noted.
Prescribing bright light therapy
Light box selection criteria. When selecting a light box or related BLT treatment apparatus, the Center for Environmental Therapeutics recommends consideration of the following factors35:
- clinical efficacy
- ocular and dermatologic safety
- visual comfort.
Selecting a dose. The dose received is determined by the intensity emitted from the light source, distance from the light box, and duration of exposure.36 Begin with midday light therapy between 12 noon and 2
Monitor for adverse effects. Generally, BLT is well tolerated.37 Adverse effects are rare; the most common ones include headache, eyestrain, nausea, and agitation.38 One study found no adverse ocular effects from light therapy after 5 years of treatment.39 Adverse effects tend to remit spontaneously or after dose reduction.35 Evening administration of BLT may increase the incidence of sleep disturbances.40 Like other biologic treatments for bipolar depression, BLT can precipitate manic/hypomanic and mixed states in susceptible patients, although the light dose can be titrated against emergent symptoms of hypomania.41
Bottom Line
Evidence suggests that bright light therapy is an effective, well tolerated, and affordable adjunct treatment for bipolar depression. Exposure to 5,000 to 7,000 lux around noon for 15 to 60 minutes will enhance the remission rate.
Related Resource
Mostert M, Dubovsky S. When bipolar treatment fails: what’s your next step? Current Psychiatry. 2008;7(1):39-46.
Drug Brand Name
Lithium • Eskalith, Lithobid
Bright light therapy (BLT) refers to the use of bright light to treat symptoms of depression. BLT was initially prescribed as a treatment for patients with seasonal affective disorder.1 It was later found helpful for nonseasonal depression,2 premenstrual dysphoric disorder, postpartum depression, and phase shift circadian disorders, including for patients with dementia whose cognitive function improved after treatment with BLT.3 More recent studies suggest year-round benefit for nonseasonal depression.2 The American Psychiatric Association practice guidelines for the treatment of depression list BLT as an alternative and/or addition to pharmacologic and psychological treatment.4 BLT also may be beneficial for patients who are in the depressive phase of bipolar illness.
This article describes the evidence, rationale for use, mechanism of action, benefits, and safety profile of BLT for treating patients with bipolar depression.
Circadian rhythm disruption in bipolar disorder
Clinical manifestation. Patients with bipolar disorder (BD) spend more time in depression than in mania.5 Sleep disturbance is a core symptom of BD; patients typically have little need for sleep during a manic episode, and excess sleepiness during a depressive episode. Sleep complaints can be both precipitating factors and consequences of mood disorders. Patients with seasonal depression have excess sleepiness and weight gain in the winter followed by hypomanic-like symptoms in the spring, including decreased need for sleep and weight loss with psychomotor activation. In a recent review of sleep problems in patients with BD, Steinan et al6 reported that 20% of patients with euthymic mood in bipolar disorder experience a sleep disorder. Circadian disruption and “eveningness” (being more active during the evening) have been associated with mood episodes, functional impairment, poor quality of life, and treatment resistance.7-10
Pathophysiology. Existing hypotheses for the biological mechanism underlying dysregulation of circadian rhythm in BD include changes in melatonin levels, expression of melatonin receptors in the CNS, and daily cortisol profiles.11 Genetic evidence also links circadian rhythm dysregulation with BD. Two polymorphisms on the circadian locomotor output cycles kaput (CLOCK) gene that control circadian rhythm—aryl hydrocarbon receptor nuclear translocator-like (ARNTL) and timeless circadian clock (TIMELESS)—have been linked to lithium responsiveness in BD.12 In addition, Per2, Cry1, and Rev-Erbα expression—all components of the circadian clock—have been found to increase individual susceptibility to the therapeutic effects of lithium in mice.13 In addition, circadian rhythm dysregulation is associated with metabolic problems encountered by patients with BD, including weight gain, diabetes mellitus, and cardiovascular disease.14
Rationale for use
Regulation of a patient’s circadian rhythm disruption is a potential treatment for BD. Hashimoto et al15 demonstrated that midday bright light exposure can phase advance and increase the amplitude of nocturnal melatonin production in healthy individuals. Morning light therapy has been shown to increase blood serotonin throughout the day in both healthy individuals and in patients with nonseasonal depression; the effect was apparent with light intensities as low as 50 lux.16 Lithium may exert its therapeutic effect through its influence on the retino-hypothalamic-pineal tract and thus its effect on melatonin secretion.17
BLT is a logical choice to treat the depression phase of BD when exposure to sunlight is not feasible due to geographical location, season, or other factor. For patients who live in areas that receive frequent sunshine, an outside stroll for half an hour will likely achieve similar benefit to BLT.
The precise mechanism of action of BLT for bipolar depression has not yet been determined. It may be attributed to a phase-resetting effect via melanopsin and the suprachiasmatic nucleus (Box18-24).
Box
Bright light therapy: How it works
The mechanism of action of bright light therapy is yet to be elucidated. The suprachiasmatic nucleus (SCN) in the hypothalamus is the center of circadian rhythm regulation and receives direct input from the retina through the retinohypothalamic tract.18 Melanopsin, a short-wavelength, light-sensitive G-protein–coupled receptor located in human retinal ganglion cells, is known to transduce short-wavelength light signals into neural signals.19 Since melanopsin is primarily responsible for resetting the timing of the SCN, suppressing pineal gland melatonin secretion and improving alertness and electroencephalogram-derived correlates of arousal,20 short-wavelength light with a low light intensity might be a better stimulator for melanopsin-containing retinal ganglion cells and the behaviors mediated via this photoreceptor system.21,22 Whether the antidepressant effect of light is also related to its alerting property is unclear.23 However, the acute alerting and performance-enhancing effects of light are increasingly taken into account for the design of indoor light standards in office environments.24 Response to light therapy is thus attributed to its phase-resetting effect.
Continue to: BLT for BD...
BLT for BD: What’s the evidence?
Several studies and case reports have evaluated the use of BLT for bipolar depression. The number of participants in these studies is small, and there is no uniformity of methodology or patient selection.
Dauphinais et al (2012)25 randomly assigned 44 patients with bipolar depression to BLT or a high-density or low-density negative ion generator for 8 weeks. They reported no difference in outcome between the various groups (50% vs 55.6%, remission and response rate). Only one patient in each group showed a switch to hypomania.
Carmadese et al (2015)26 reported an open-label study of adjunctive BLT in 31 difficult-to-treat patients with depression (16 unipolar and 15 bipolar). Significant improvement was noted within 3 weeks and was sustained in 1 patient with bipolar depression 5 weeks after cessation of BLT.
Papatheodorou and Kutcher (1995)27 treated 7 adolescents with bipolar depression with adjunctive BLT (10,000 lux twice per day). Three patients showed a marked response (>70% decrease from baseline Beck Depression Inventory and Symptom Check List scores). Two patients had a moderate response (40% to 47% decrease) and 2 patients obtained mild to no response. There were no reported adverse effects.
Benedetti et al (2014)28 studied 141 patients with treatment-resistant bipolar depression. Approximately one-quarter (23%) had a history of attempted suicide, and 83% had a history of drug resistance. The authors found a combination of total sleep deprivation, BLT, and lithium rapidly decreased suicidality and improved patients’ depressive symptoms.
Liebenluft et al (1995)29 administered 13 trials of BLT to 9 patients with rapid-cycling BD during a 3-month period. Five patients received the treatment in the morning, 5 around midday, and 3 in the evening. Patients who received BLT at midday had the best outcome, while 3 of the 5 patients who received morning BLT developed unstable mood. The authors recommended titrating the duration of light exposure so that patients could skip a treatment if their mood was trending toward hypomania.
Sit et al (2007)30 evaluated BLT in a case series of 9 women with bipolar I or II disorder in the depression phase. Patients were exposed to 50 lux of red light for 2 weeks, and then they received 7,000 lux BLT for 15, 30, and 45 minutes daily for 2 weeks (4 patients received morning light and 5 received midday light). Mood was assessed using the Structured Interview Guide for the Hamilton Depression Rating Scale with Atypical Depression Supplement and the Mania Rating Scale. Of the 4 patients receiving morning BLT, one patient had a full response and the other 3 developed hypomania. Of the 5 patients who received midday BLT, 2 achieved full response, 2 showed early improvement but required a dose increase, and one remained depressed but had a full response when she was switched to morning BLT.
Tseng et al (2016)31 reported a meta-analysis of BLT for bipolar depression that included a total of 567 patients from 11 studies. They reported significant improvement with BLT alone or in combination with antidepressants or total sleep deprivation. They also reported significant improvement with BLT in 130 patients who were not receiving other treatments. There was no difference in the frequency of mood shifts between patients on BLT alone or in combination with other modalities. The authors reported no mood shift in any of the patients receiving concurrent mood stabilizers. They also reported no difference with the color of light, gender, or duration of illness.
Yorguner et al (2017)32 conducted a 2-week randomized, single-blind study of BLT as an add-on treatment for 32 patients with bipolar depression. Patients were randomly assigned to BLT or dim light, which they were administered each morning for 30 mins for 2 weeks. Sixteen patients who received BLT showed a significantly greater reduction in Hamilton Depression Rating Scale scores (mean score of 24 at baseline down to 12) compared with 16 patients who received dim light (mean score of 24 at baseline down to 18). The authors also reported remission in 4 out of 4 patients who had seasonal depression, compared with 3 out of 12 who did not have seasonal depression (the other 9 showed response but not remission).
Zhou et al (2018)33 conducted a multi-center, randomized, single-blind clinical trial of 63 patients with bipolar depression. Thirty-three patients received morning BLT, and 30 received dim red light therapy (control group). The authors reported a significantly higher response rate in the BLT group (78%) compared with the control group (43%).
Sit et al (2018)34 conducted a 6-week randomized, double-blind, placebo-controlled trial of BLT vs dim red light in patients with bipolar I or II depression. Twenty-three patients were administered 7,000 lux bright white light, and 23 patients received 50 lux dim red light, at midday 5 days a week. The light dose was increased by 15 minutes every week up to 60 minutes by Week 4, unless the patient achieved remission. Patients were maintained on their usual medications, which included mood stabilizers and/or antidepressants. At Week 6, the group randomized to BLT had a significantly higher remission rate (68%) compared with patients who received dim red light (22%). Improvement was noted by Week 4. Patients receiving BLT also had significantly fewer depressive symptoms, and no mood polarity switch was noted.
Prescribing bright light therapy
Light box selection criteria. When selecting a light box or related BLT treatment apparatus, the Center for Environmental Therapeutics recommends consideration of the following factors35:
- clinical efficacy
- ocular and dermatologic safety
- visual comfort.
Selecting a dose. The dose received is determined by the intensity emitted from the light source, distance from the light box, and duration of exposure.36 Begin with midday light therapy between 12 noon and 2
Monitor for adverse effects. Generally, BLT is well tolerated.37 Adverse effects are rare; the most common ones include headache, eyestrain, nausea, and agitation.38 One study found no adverse ocular effects from light therapy after 5 years of treatment.39 Adverse effects tend to remit spontaneously or after dose reduction.35 Evening administration of BLT may increase the incidence of sleep disturbances.40 Like other biologic treatments for bipolar depression, BLT can precipitate manic/hypomanic and mixed states in susceptible patients, although the light dose can be titrated against emergent symptoms of hypomania.41
Bottom Line
Evidence suggests that bright light therapy is an effective, well tolerated, and affordable adjunct treatment for bipolar depression. Exposure to 5,000 to 7,000 lux around noon for 15 to 60 minutes will enhance the remission rate.
Related Resource
Mostert M, Dubovsky S. When bipolar treatment fails: what’s your next step? Current Psychiatry. 2008;7(1):39-46.
Drug Brand Name
Lithium • Eskalith, Lithobid
1. Pjrek E, Winkler D, Stastny J, et al. Bright light therapy in seasonal affective disorder--does it suffice? Eur Neuropsychopharmacol. 2004.14(4):347-351.
2. Al-Karawi D, Jubair L. Bright light therapy for nonseasonal depression: meta-analysis of clinical trials. J Affect Disord. 2016;198:64-71.
3. Sekiguchi H, Iritani S, Fujita K. Bright light therapy for sleep disturbance in dementia is most effective for mild to moderate Alzheimer’s type dementia: a case series. Psychogeriatrics, 2017;17(5):275-281.
4. Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder, third edition. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf American Psychiatric Association. 2010. Accessed August, 10, 2017.
5. Kupka RW, Altshuler LL, Nolen WA, et al. Three times more days depressed than manic or hypomanic in both bipolar I and bipolar II disorder. Bipolar Disord. 2007;9(5):531-535.
6. Steinan MK, Krane-Gartiser K, Morken G, et al. Sleep problems in euthymic bipolar disorders: a review of clinical studies. Current Psychiatry Reviews. 2015;11:235-243.
7. Cudney LE, Frey BN, Streiner D, et al. Biological rhythms are independently associated with quality of life in bipolar disorder. Int J Bipolar Disord. 2016;4(1):9.
8. Duarte FA, Cardoso TA, Campos MT, et al. Biological rhythms in bipolar and depressive disorders: a community study with drug-naive young adults. J Affect Disord, 2015;186:145-148.
9. Pinho M, Sehmbi M, Cudney LE, et al. The association between biological rhythms, depression, and functioning in bipolar disorder: a large multi-center study. Acta Psychiatr Scand. 2015:133(2);102-108.
10. Ng TH, Chung KF, Lee CT, et al. Eveningness and its associated impairments in remitted bipolar disorder. Behav Sleep Med. 2016:14(6):650-664.
11. Wu YH, Ursinus J, Zahn JN, et al. Alterations of melatonin receptors MT1 and MT2 in the hypothalamic suprachiasmatic nucleus during depression. J Affect Disord, 2013:148(2-3):357-367.
12. Rybakowski JK, Dmitrzak-Weglar M, Kliwicki S, et al. Polymorphism of circadian clock genes and prophylactic lithium response. Bipolar Disord. 2014;16(2):151-158.
13. Schnell A, Sandrelli F, Ranc V, et al. Mice lacking circadian clock components display different mood-related behaviors and do not respond uniformly to chronic lithium treatment. Chronobiol Int. 2015;32(8):1075-1089.
14. Kim Y, Santos R, Gage FH, et al. Molecular mechanisms of bipolar disorder: progress made and future challenges. Front Cell Neurosci. 2017;11:30.
15. Hashimoto S, Kohsaka M, Nakamura K. Midday exposure to bright light changes the circadian organization of plasma melatonin rhythm in humans. Neurosci Lett. 1997;221(2-3):
89-92.
16. Rao ML, Müller-Oerlinghausen B, Mackert A, et al. The influence of phototherapy on serotonin and melatonin in non-seasonal depression. Pharmacopsychiatry.1990;23(3):155-158.
17. Moreira J, Geoffroy PA. Lithium and bipolar disorder: impacts from molecular to behavioural circadian rhythms. Chronobiol Int. 2016;33(4):351-373.
18. Oldham MA, Ciraulo DA. Bright light therapy for depression: a review of its effects on chronobiology and the autonomic nervous system. Chronobiol Int. 2014;31(3):305-319.
19. Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295(5557):1070-1073.
20. Peirson S, Foster RG. Melanopsin: another way of signaling light. Neuron. 2006;49(3):331-339.
21. Anderson JL, Glod CA, Dai J, et al. Lux vs. wavelength in light treatment of seasonal affective disorder. Acta Psychiatr Scand. 2009;120(3):203-212.
22. Wirz-Justice A, Graw P, Kräuchi K, et al. Effect of light on unmasked circadian rhythms in winter depression. In: Wetterberg L, ed. Light and biological rhythms in man. Oxford, United Kingdom:Pergamon Press;1993:385-393.
23. Cajochen C. Alerting effects of light. Sleep Med Rev. 2007;11(6):453-464.
24. Aries MBC. Human lighting demands: healthy lighting in an office environment. Eindhoven, Eindhoven University Press. 2005;158. doi:10.6100/IR594257.
25. Dauphinais DR, Rosenthal JZ, Terman M, et al. Controlled trial of safety and efficacy of bright light therapy vs. negative air ions in patients with bipolar depression. Psychiatry Res. 2012;196(1):57-61.
26. Camardese G, Leone B, Serrani R, et al. Augmentation of light therapy in difficult-to-treat depressed patients: an open-label trial in both unipolar and bipolar patients. Neuropsychiatr Dis Treat. 2015;11:2331-2338.
27. Papatheodorou G, Kutcher S. The effect of adjunctive light therapy on ameliorating breakthrough depressive symptoms in adolescent-onset bipolar disorder.
J Psychiatry Neurosci. 1995;20(3):226-232.
28. Benedetti F, Riccaboni R, Locatelli C, et al. Rapid treatment response of suicidal symptoms to lithium, sleep deprivation, and light therapy (chronotherapeutics) in drug-resistant bipolar depression. J Clin Psychiatry. 2014;75(2):133-140.
29. Liebenluft E, Turner EH, Felman-Naim S, et al. Light therapy in patients with rapid cycling bipolar disorder: preliminary results. Psychopharmacol Bull. 1995;31(4):
705-710.
30. Sit DK, Wisner KL, Hanusa BH, et al. Light therapy for bipolar disorder: a case series in women. Bipolar Disord. 2007;9(8):918-927.
31. Tseng PT, Chen YW, Tu KY, et al. Light therapy in the treatment of patients with bipolar depression: a meta-analytic study. Eur Neuropsychopharmacol. 2016;26(6):
1037-1047.
32. Yorguner KN, Bulut NS, Carkaxhiu BG, et al. Efficacy of bright light therapy in bipolar depression. Psychiatry Res. 2017;260:432-438.
33. Zhou TH, Dang WM, Ma YT, et al. Clinical efficacy, onset time and safety of bright light therapy in acute bipolar depression as an adjunctive therapy: a randomized controlled trial. J Affect Disord. 2018;227:90-96.
34. Sit DK, McGowan J, Wiltrout C, et al. Adjunctive bright light therapy for bipolar depression: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2018;175(2):
131-139.
35. Center for Environmental Therapeutics. https://www.cet.org/. Center for Environmental Therapeutics. Accessed November 15, 2017.
36. Lam RW, Levitt AJ. Canadian consensus guidelines for the treatment of seasonal affective disorder. https://mdsc.ca/documents/Consumer%20and%20Family%20Support/CCG_on_Seasonal_Affective_Disorder.pdf. 1999. Accessed August 2, 2017.
37. Terman M, Terman JS. Bright light therapy: side effects and benefits across the symptom spectrum. J Clin Psychiatry. 1999; 60(11):799-808;quiz 809.
38. Labbate LA, et al. Side effects induced by bright light treatment for seasonal affective disorder. J Clin Psychiatry. 1994; 55(5):189-191.
39. Gallin PF, et al. Ophthalmologic examination of patients with seasonal affective disorder, before and after bright light therapy. Am J Ophthalmol. 1995;119(2):202-210.
40. Chan PK, Lam RW, Perry KF. Mania precipitated by light therapy for patients with SAD. J Clin Psychiatry. 1994;55(10):454.
41. Kripke DF. Timing of phototherapy and occurrence of mania. Biol Psychiatry. 1991; 29(11):1156-1157.
1. Pjrek E, Winkler D, Stastny J, et al. Bright light therapy in seasonal affective disorder--does it suffice? Eur Neuropsychopharmacol. 2004.14(4):347-351.
2. Al-Karawi D, Jubair L. Bright light therapy for nonseasonal depression: meta-analysis of clinical trials. J Affect Disord. 2016;198:64-71.
3. Sekiguchi H, Iritani S, Fujita K. Bright light therapy for sleep disturbance in dementia is most effective for mild to moderate Alzheimer’s type dementia: a case series. Psychogeriatrics, 2017;17(5):275-281.
4. Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder, third edition. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf American Psychiatric Association. 2010. Accessed August, 10, 2017.
5. Kupka RW, Altshuler LL, Nolen WA, et al. Three times more days depressed than manic or hypomanic in both bipolar I and bipolar II disorder. Bipolar Disord. 2007;9(5):531-535.
6. Steinan MK, Krane-Gartiser K, Morken G, et al. Sleep problems in euthymic bipolar disorders: a review of clinical studies. Current Psychiatry Reviews. 2015;11:235-243.
7. Cudney LE, Frey BN, Streiner D, et al. Biological rhythms are independently associated with quality of life in bipolar disorder. Int J Bipolar Disord. 2016;4(1):9.
8. Duarte FA, Cardoso TA, Campos MT, et al. Biological rhythms in bipolar and depressive disorders: a community study with drug-naive young adults. J Affect Disord, 2015;186:145-148.
9. Pinho M, Sehmbi M, Cudney LE, et al. The association between biological rhythms, depression, and functioning in bipolar disorder: a large multi-center study. Acta Psychiatr Scand. 2015:133(2);102-108.
10. Ng TH, Chung KF, Lee CT, et al. Eveningness and its associated impairments in remitted bipolar disorder. Behav Sleep Med. 2016:14(6):650-664.
11. Wu YH, Ursinus J, Zahn JN, et al. Alterations of melatonin receptors MT1 and MT2 in the hypothalamic suprachiasmatic nucleus during depression. J Affect Disord, 2013:148(2-3):357-367.
12. Rybakowski JK, Dmitrzak-Weglar M, Kliwicki S, et al. Polymorphism of circadian clock genes and prophylactic lithium response. Bipolar Disord. 2014;16(2):151-158.
13. Schnell A, Sandrelli F, Ranc V, et al. Mice lacking circadian clock components display different mood-related behaviors and do not respond uniformly to chronic lithium treatment. Chronobiol Int. 2015;32(8):1075-1089.
14. Kim Y, Santos R, Gage FH, et al. Molecular mechanisms of bipolar disorder: progress made and future challenges. Front Cell Neurosci. 2017;11:30.
15. Hashimoto S, Kohsaka M, Nakamura K. Midday exposure to bright light changes the circadian organization of plasma melatonin rhythm in humans. Neurosci Lett. 1997;221(2-3):
89-92.
16. Rao ML, Müller-Oerlinghausen B, Mackert A, et al. The influence of phototherapy on serotonin and melatonin in non-seasonal depression. Pharmacopsychiatry.1990;23(3):155-158.
17. Moreira J, Geoffroy PA. Lithium and bipolar disorder: impacts from molecular to behavioural circadian rhythms. Chronobiol Int. 2016;33(4):351-373.
18. Oldham MA, Ciraulo DA. Bright light therapy for depression: a review of its effects on chronobiology and the autonomic nervous system. Chronobiol Int. 2014;31(3):305-319.
19. Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295(5557):1070-1073.
20. Peirson S, Foster RG. Melanopsin: another way of signaling light. Neuron. 2006;49(3):331-339.
21. Anderson JL, Glod CA, Dai J, et al. Lux vs. wavelength in light treatment of seasonal affective disorder. Acta Psychiatr Scand. 2009;120(3):203-212.
22. Wirz-Justice A, Graw P, Kräuchi K, et al. Effect of light on unmasked circadian rhythms in winter depression. In: Wetterberg L, ed. Light and biological rhythms in man. Oxford, United Kingdom:Pergamon Press;1993:385-393.
23. Cajochen C. Alerting effects of light. Sleep Med Rev. 2007;11(6):453-464.
24. Aries MBC. Human lighting demands: healthy lighting in an office environment. Eindhoven, Eindhoven University Press. 2005;158. doi:10.6100/IR594257.
25. Dauphinais DR, Rosenthal JZ, Terman M, et al. Controlled trial of safety and efficacy of bright light therapy vs. negative air ions in patients with bipolar depression. Psychiatry Res. 2012;196(1):57-61.
26. Camardese G, Leone B, Serrani R, et al. Augmentation of light therapy in difficult-to-treat depressed patients: an open-label trial in both unipolar and bipolar patients. Neuropsychiatr Dis Treat. 2015;11:2331-2338.
27. Papatheodorou G, Kutcher S. The effect of adjunctive light therapy on ameliorating breakthrough depressive symptoms in adolescent-onset bipolar disorder.
J Psychiatry Neurosci. 1995;20(3):226-232.
28. Benedetti F, Riccaboni R, Locatelli C, et al. Rapid treatment response of suicidal symptoms to lithium, sleep deprivation, and light therapy (chronotherapeutics) in drug-resistant bipolar depression. J Clin Psychiatry. 2014;75(2):133-140.
29. Liebenluft E, Turner EH, Felman-Naim S, et al. Light therapy in patients with rapid cycling bipolar disorder: preliminary results. Psychopharmacol Bull. 1995;31(4):
705-710.
30. Sit DK, Wisner KL, Hanusa BH, et al. Light therapy for bipolar disorder: a case series in women. Bipolar Disord. 2007;9(8):918-927.
31. Tseng PT, Chen YW, Tu KY, et al. Light therapy in the treatment of patients with bipolar depression: a meta-analytic study. Eur Neuropsychopharmacol. 2016;26(6):
1037-1047.
32. Yorguner KN, Bulut NS, Carkaxhiu BG, et al. Efficacy of bright light therapy in bipolar depression. Psychiatry Res. 2017;260:432-438.
33. Zhou TH, Dang WM, Ma YT, et al. Clinical efficacy, onset time and safety of bright light therapy in acute bipolar depression as an adjunctive therapy: a randomized controlled trial. J Affect Disord. 2018;227:90-96.
34. Sit DK, McGowan J, Wiltrout C, et al. Adjunctive bright light therapy for bipolar depression: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2018;175(2):
131-139.
35. Center for Environmental Therapeutics. https://www.cet.org/. Center for Environmental Therapeutics. Accessed November 15, 2017.
36. Lam RW, Levitt AJ. Canadian consensus guidelines for the treatment of seasonal affective disorder. https://mdsc.ca/documents/Consumer%20and%20Family%20Support/CCG_on_Seasonal_Affective_Disorder.pdf. 1999. Accessed August 2, 2017.
37. Terman M, Terman JS. Bright light therapy: side effects and benefits across the symptom spectrum. J Clin Psychiatry. 1999; 60(11):799-808;quiz 809.
38. Labbate LA, et al. Side effects induced by bright light treatment for seasonal affective disorder. J Clin Psychiatry. 1994; 55(5):189-191.
39. Gallin PF, et al. Ophthalmologic examination of patients with seasonal affective disorder, before and after bright light therapy. Am J Ophthalmol. 1995;119(2):202-210.
40. Chan PK, Lam RW, Perry KF. Mania precipitated by light therapy for patients with SAD. J Clin Psychiatry. 1994;55(10):454.
41. Kripke DF. Timing of phototherapy and occurrence of mania. Biol Psychiatry. 1991; 29(11):1156-1157.

Xenomelia and sexuality
Your patient traced the history of his desire to amputate his leg (as do other individuals with xenomelia) to age 4, when he saw a man with a missing limb, which made a vivid impression on him. As you discuss, this was probably a moment when he had an intensely psychosexual imprinting of this perception. However, the actual memory of the man with the amputation may well have been a screen memory for other more arousing and traumatic experiences that the patient experienced at this early age, such as castration anxiety with or without actual overstimulation of the physical body.
Nathan Szajnberg, MD, and I reported a case of a man who desired that his partner pretend to be an amputee in order to strengthen sexual arousal, an arousal that he recalled having as early as age 5 or 6.1 The report traced this fetish back to research films of his upbringing, which indicated heightened physical stimulation in very early life.2 As we wrote, “The case provides unusual information about the manner in which early childhood events interdigitate with intrapsychic processes and mental structuralisation.” This has led me to wonder if similar mental processes are at work in the current wave of young people who are convinced that they are a different gender than the one indicated by their anatomy.
1. Massie H, Szajnberg N. The ontogeny of a sexual fetish from birth to age 30 and memory processes—a research and case report from a prospective longitudinal study. Int J Psychoanal. 1997;78(pt 4):755-771.
2. Massie H, Szajnberg N. Lives across time/growing up: paths to emotional health and emotional illness from birth to 30 in 76 people. London, UK: Kamac Books; 2008.
Your patient traced the history of his desire to amputate his leg (as do other individuals with xenomelia) to age 4, when he saw a man with a missing limb, which made a vivid impression on him. As you discuss, this was probably a moment when he had an intensely psychosexual imprinting of this perception. However, the actual memory of the man with the amputation may well have been a screen memory for other more arousing and traumatic experiences that the patient experienced at this early age, such as castration anxiety with or without actual overstimulation of the physical body.
Nathan Szajnberg, MD, and I reported a case of a man who desired that his partner pretend to be an amputee in order to strengthen sexual arousal, an arousal that he recalled having as early as age 5 or 6.1 The report traced this fetish back to research films of his upbringing, which indicated heightened physical stimulation in very early life.2 As we wrote, “The case provides unusual information about the manner in which early childhood events interdigitate with intrapsychic processes and mental structuralisation.” This has led me to wonder if similar mental processes are at work in the current wave of young people who are convinced that they are a different gender than the one indicated by their anatomy.
Your patient traced the history of his desire to amputate his leg (as do other individuals with xenomelia) to age 4, when he saw a man with a missing limb, which made a vivid impression on him. As you discuss, this was probably a moment when he had an intensely psychosexual imprinting of this perception. However, the actual memory of the man with the amputation may well have been a screen memory for other more arousing and traumatic experiences that the patient experienced at this early age, such as castration anxiety with or without actual overstimulation of the physical body.
Nathan Szajnberg, MD, and I reported a case of a man who desired that his partner pretend to be an amputee in order to strengthen sexual arousal, an arousal that he recalled having as early as age 5 or 6.1 The report traced this fetish back to research films of his upbringing, which indicated heightened physical stimulation in very early life.2 As we wrote, “The case provides unusual information about the manner in which early childhood events interdigitate with intrapsychic processes and mental structuralisation.” This has led me to wonder if similar mental processes are at work in the current wave of young people who are convinced that they are a different gender than the one indicated by their anatomy.
1. Massie H, Szajnberg N. The ontogeny of a sexual fetish from birth to age 30 and memory processes—a research and case report from a prospective longitudinal study. Int J Psychoanal. 1997;78(pt 4):755-771.
2. Massie H, Szajnberg N. Lives across time/growing up: paths to emotional health and emotional illness from birth to 30 in 76 people. London, UK: Kamac Books; 2008.
1. Massie H, Szajnberg N. The ontogeny of a sexual fetish from birth to age 30 and memory processes—a research and case report from a prospective longitudinal study. Int J Psychoanal. 1997;78(pt 4):755-771.
2. Massie H, Szajnberg N. Lives across time/growing up: paths to emotional health and emotional illness from birth to 30 in 76 people. London, UK: Kamac Books; 2008.
The teenager who couldn’t stay awake
CASE Somnolent, confused, and hungry
Mr. G, age 14, presents to the emergency department (ED) for acute-onset hypersomnia that has gradually worsened over the course of a few days. Mr. G now sleeps most of the day, has altered mental status, and is experiencing emotional dysregulation with no clear etiology. His mother, who accompanies him to the ED, says that prior to the onset of these symptoms her son had been healthy. She notes that he has been eating more than usual, which she assumes is due to a growth spurt.
Mr. G’s symptoms began 4 days ago when he became increasingly fatigued, sleeping for 11 to 12 hours per day, with intermittent episodes of staring and unresponsiveness from which he rapidly returned to baseline. During the next 3 days, he became more confused and somnolent, and began to display bizarre behavior, including eating food out of the trash and attempting to microwave a full metal pot. He exhibited unexplained crying spells, calling out for his “mommy,” and saying he was “afraid I’m dying.”
During the 2 days before he came to our ED, Mr. G was seen at 2 other hospitals. Following extensive imaging and laboratory work-up, clinicians at these facilities attributed his symptoms to intoxication from an unknown substance. Mr. G has a history of marijuana use, and his mother reports that his friends had recently been using synthetic marijuana. However, no intoxicant was identified on urine or gas chromatography drug screening.
Mr. G’s history includes oppositional behavior, and a brief psychiatric hospitalization at age 5 for aggression. He has otherwise been healthy. His family history is significant for maternal substance use and anxiety disorders. In addition to sporadic cannabis use, Mr. G’s social history includes multiple recent family losses, previous foster care placement, and recent declining academic performance.
[polldaddy:10148168]
EVALUATION No red flags
On admission, Mr. G appears somnolent and displays disorganized speech, impulsivity, frequent disorientation, and intermittent agitation/anxiety; he sleeps 16 to 18 hours per day. Mr. G is admitted with a presumptive diagnosis of substance intoxication and transferred to the general pediatric inpatient unit. Upon arrival, he is found to be bradycardic (42 beats per minute), although afebrile with otherwise age-appropriate vitals. On exam, he is somnolent but arousable and follows simple commands.
Continue to: Mr. G undergoes a Monospot test...
Mr. G undergoes a Monospot test, which is positive, with subsequent evidence of a prior, but not active, Epstein-Barr virus (EBV) infection. He also has a mildly elevated CSF protein level, but subsequent CSF labs are negative for both infectious and non-infectious processes. An EEG reveals focal neuronal slowing.
During brief periods of wakefulness, Mr. G calls out to his mother and says, “I’m not going to make it to my birthday,” and “You’re going to have to let me go.” He occasionally becomes combative, pulling at IV lines and swearing at staff. His bradycardia resolves without intervention during his admission. On Day 8 of his hospitalization, Mr. G displays hypersexuality and makes sexually suggestive comments toward female staff members. He also experiences recurrence of hyperphagia.
On Day 10 of his stay on the pediatric unit, because of the emergence of hypersexuality and hyperphagia, along with a largely negative medical evaluation, Mr. G is transferred to the pediatric psychiatric unit for ongoing evaluation and management.
[polldaddy:10148172]
The authors’ observations
Given Mr. G’s rapid onset of confusion, hypersomnia, and emotional dysregulation, our differential diagnosis included delirium of unclear etiology, substance intoxication, autoimmune encephalitis, viral meningitis, heavy metal intoxication, primary psychotic disorder, and KLS. Mr. G underwent an extensive diagnostic evaluation, which was largely unremarkable (Table). He had a mildly elevated CSF protein level, but subsequent CSF labs were negative for both infectious and non-infectious processes. When Mr. G was transferred to the pediatric inpatient psychiatric unit on Day 10, the presumptive diagnosis was KLS.
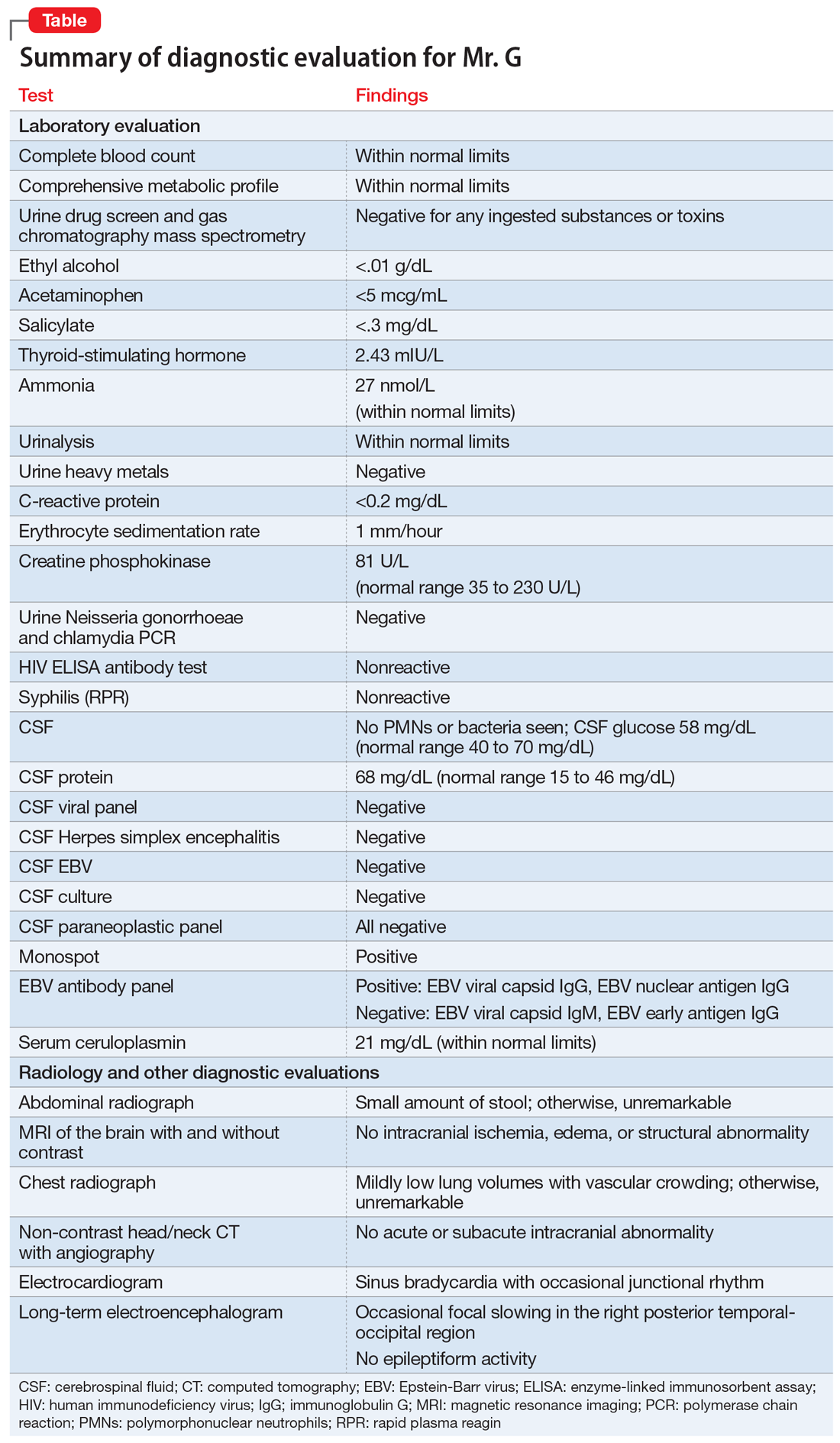
KLS is a rare neurologic disorder, with an incidence of 1 to 5 in 1 million and a 4:1 male-to-female predominance.1 It poses a diagnostic challenge due to its low prevalence and broad differential. The disorder typically presents in early adolescence and is characterized by episodes of severe hypersomnia with associated cognitive and/or behavioral disturbance2 (Box2-4). Bradycardia, as seen in Mr. G, and other forms of autonomic dysregulation have been reported in the literature, as has the focal neuronal slowing noted on Mr. G’s EEG.3

[polldaddy:10148174]
Continue to: TREATMENT Methylphenidate and a safety plan
TREATMENT Methylphenidate and a safety plan
On Day 11 of hospitalization, Mr. G is started on methylphenidate, 10 mg in the morning and 5 mg in the afternoon. After starting methylphenidate, he sustains more regular wakefulness, with improved thought organization, engagement, and fewer disruptive behaviors. He receives infrequent, as-needed doses of olanzapine, and by Day 14, he returns to his baseline behavior and cognition.
A safety plan is created for the family to address worsening symptoms or future episodes. The safety plan is developed with Mr. G and input from his family. It is to be administered in all settings and we particularly emphasized using it in the school setting, where staff may not be familiar with KLS. The safety plan involves a description of KLS, its symptoms, the risks for hypersomnolence, hypersexuality, and psychotic symptoms or behavioral dysregulation. It stresses close supervision of Mr. G, not allowing him to be unsupervised or unchaperoned on school trips or other outings, and lethal means restriction. It outlines a detailed plan if Mr. G’s behavior decompensates or escalates, including a step-wise approach to engaging psychological interventions and mental health resources, and securing crisis services as needed.
On Day 15, he is discharged to home in stable condition with outpatient mental health follow-up and continues to take the prescribed methylphenidate.
The authors’ observations
Management of KLS is primarily supportive. Stimulants may help reduce hypersomnia, impulsivity, and inattention early in the disease course.1 However, in a systematic review, 89% of patients with KLS who received methylphenidate experienced worsening or no improvement, and 11% showed only partial improvement.2 Amantadine was more promising, with 29% of patients with KLS showing partial benefit and 12% showing significant benefit.2 Multiple other pharmacologic agents have been described with varying efficacy, including lithium, valproate, risperidone, bupropion, and immunoglobulins.2 Furthermore, lithium and valproate have been suggested to be helpful in preventing recurrences in some cases.6
The circumstances surrounding Mr. G’s symptom onset are unclear and may have been multifactorial. It is possible that his prior EBV infection was a trigger for this KLS-associated episode, as EBV is a known precipitant for KLS episodes.3 Mr. G’s history of cannabis use may also have served as an early trigger for KLS.3
This case highlights the importance of multidisciplinary collaboration in a diagnostically challenging case. It emphasizes the need for a broad differential and the importance of challenging a previous diagnosis in the face of mounting evidence to the contrary. In this case, the patient’s history of irritability, aggression, and cannabis use resulted in multiple clinicians misattributing his symptoms to substance use or a primary psychiatric disorder. However, given his symptom acuity, progression, and the lack of findings on diagnostic evaluation to explain his presentation, these initial diagnoses did not explain the severity, nature, or duration of his symptoms. Keeping KLS in the differential is particularly important for patients with a prior history of psychiatric illness or substance use, because these patients are at higher risk for misattribution of symptoms to pre-existing psychiatric illness. Evolution of symptoms, a negative diagnostic evaluation, and maintaining a broad differential resulted in eventually reaching the final diagnosis of KLS and development of a longitudinal management plan.
While further work must be done to clearly define the pharmacologic approach to acute management of KLS episodes, nonpharmacologic aspects of care must not be neglected. Behavioral planning, adjustment of the environment, engagement with schools/community supports, and family education are valuable tools for facilitating the patient’s de-escalation, avoiding unneeded polypharmacy, reducing anxieties, and safeguarding the patient from unnecessary harm.7 Clinicians can support their patients’ transitions back into the community by ensuring careful outpatient follow-up for symptom monitoring and by communicating with patients’ schools and employers.
OUTCOME Asymptomatic; no recurrence of symptoms
Forty-six days after his symptoms began and 31 days after hospital discharge, Mr. G is asymptomatic with no recurrence of symptoms.
Bottom Line
Kleine-Levin syndrome (KLS) is a rare, often-overlooked condition that should be considered in the differential diagnosis for patients who present with hypersomnolence and altered mental status without a clear etiology. Rapid recognition of KLS can prevent misattribution of symptoms, unnecessary treatment, and missed opportunities for care.
1. Billiard M, Jaussent I, Dauvilliers Y, et al. Recurrent hypersomnia: a review of 339 cases. Sleep Med. 2011;15(4):247-257.
2. Arnulf I, Lin L, Gadoth N, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol. 2008;63(4):482-493.
3. Arnulf I. Kleine-Levin syndrome: a systematic review of 186 cases in the literature. Brain. 2005;128(12):2763-2776.
4. Lisk R. Kleine-Levin syndrome. Pract Neurol. 2009;9(1);42-45.
5. de Araújo Lima TF, da Silva Behrens NS, Lopes E, et al. Kleine–Levin Syndrome: a case report. Sleep Sci. 2014;7(2):122-125.
6. Goldberg MA. The treatment of Kleine-Levin syndrome with lithium. Can J Psychiatry. 1983;28:491-493.
7. Gadoth N, Kesler A, Vainstein G, et al. Clinical and polysomnographic characteristics of 34 patients with Kleine-Levin syndrome. J Sleep Res. 2001;10(4):337-341.
CASE Somnolent, confused, and hungry
Mr. G, age 14, presents to the emergency department (ED) for acute-onset hypersomnia that has gradually worsened over the course of a few days. Mr. G now sleeps most of the day, has altered mental status, and is experiencing emotional dysregulation with no clear etiology. His mother, who accompanies him to the ED, says that prior to the onset of these symptoms her son had been healthy. She notes that he has been eating more than usual, which she assumes is due to a growth spurt.
Mr. G’s symptoms began 4 days ago when he became increasingly fatigued, sleeping for 11 to 12 hours per day, with intermittent episodes of staring and unresponsiveness from which he rapidly returned to baseline. During the next 3 days, he became more confused and somnolent, and began to display bizarre behavior, including eating food out of the trash and attempting to microwave a full metal pot. He exhibited unexplained crying spells, calling out for his “mommy,” and saying he was “afraid I’m dying.”
During the 2 days before he came to our ED, Mr. G was seen at 2 other hospitals. Following extensive imaging and laboratory work-up, clinicians at these facilities attributed his symptoms to intoxication from an unknown substance. Mr. G has a history of marijuana use, and his mother reports that his friends had recently been using synthetic marijuana. However, no intoxicant was identified on urine or gas chromatography drug screening.
Mr. G’s history includes oppositional behavior, and a brief psychiatric hospitalization at age 5 for aggression. He has otherwise been healthy. His family history is significant for maternal substance use and anxiety disorders. In addition to sporadic cannabis use, Mr. G’s social history includes multiple recent family losses, previous foster care placement, and recent declining academic performance.
[polldaddy:10148168]
EVALUATION No red flags
On admission, Mr. G appears somnolent and displays disorganized speech, impulsivity, frequent disorientation, and intermittent agitation/anxiety; he sleeps 16 to 18 hours per day. Mr. G is admitted with a presumptive diagnosis of substance intoxication and transferred to the general pediatric inpatient unit. Upon arrival, he is found to be bradycardic (42 beats per minute), although afebrile with otherwise age-appropriate vitals. On exam, he is somnolent but arousable and follows simple commands.
Continue to: Mr. G undergoes a Monospot test...
Mr. G undergoes a Monospot test, which is positive, with subsequent evidence of a prior, but not active, Epstein-Barr virus (EBV) infection. He also has a mildly elevated CSF protein level, but subsequent CSF labs are negative for both infectious and non-infectious processes. An EEG reveals focal neuronal slowing.
During brief periods of wakefulness, Mr. G calls out to his mother and says, “I’m not going to make it to my birthday,” and “You’re going to have to let me go.” He occasionally becomes combative, pulling at IV lines and swearing at staff. His bradycardia resolves without intervention during his admission. On Day 8 of his hospitalization, Mr. G displays hypersexuality and makes sexually suggestive comments toward female staff members. He also experiences recurrence of hyperphagia.
On Day 10 of his stay on the pediatric unit, because of the emergence of hypersexuality and hyperphagia, along with a largely negative medical evaluation, Mr. G is transferred to the pediatric psychiatric unit for ongoing evaluation and management.
[polldaddy:10148172]
The authors’ observations
Given Mr. G’s rapid onset of confusion, hypersomnia, and emotional dysregulation, our differential diagnosis included delirium of unclear etiology, substance intoxication, autoimmune encephalitis, viral meningitis, heavy metal intoxication, primary psychotic disorder, and KLS. Mr. G underwent an extensive diagnostic evaluation, which was largely unremarkable (Table). He had a mildly elevated CSF protein level, but subsequent CSF labs were negative for both infectious and non-infectious processes. When Mr. G was transferred to the pediatric inpatient psychiatric unit on Day 10, the presumptive diagnosis was KLS.
KLS is a rare neurologic disorder, with an incidence of 1 to 5 in 1 million and a 4:1 male-to-female predominance.1 It poses a diagnostic challenge due to its low prevalence and broad differential. The disorder typically presents in early adolescence and is characterized by episodes of severe hypersomnia with associated cognitive and/or behavioral disturbance2 (Box2-4). Bradycardia, as seen in Mr. G, and other forms of autonomic dysregulation have been reported in the literature, as has the focal neuronal slowing noted on Mr. G’s EEG.3
[polldaddy:10148174]
Continue to: TREATMENT Methylphenidate and a safety plan
TREATMENT Methylphenidate and a safety plan
On Day 11 of hospitalization, Mr. G is started on methylphenidate, 10 mg in the morning and 5 mg in the afternoon. After starting methylphenidate, he sustains more regular wakefulness, with improved thought organization, engagement, and fewer disruptive behaviors. He receives infrequent, as-needed doses of olanzapine, and by Day 14, he returns to his baseline behavior and cognition.
A safety plan is created for the family to address worsening symptoms or future episodes. The safety plan is developed with Mr. G and input from his family. It is to be administered in all settings and we particularly emphasized using it in the school setting, where staff may not be familiar with KLS. The safety plan involves a description of KLS, its symptoms, the risks for hypersomnolence, hypersexuality, and psychotic symptoms or behavioral dysregulation. It stresses close supervision of Mr. G, not allowing him to be unsupervised or unchaperoned on school trips or other outings, and lethal means restriction. It outlines a detailed plan if Mr. G’s behavior decompensates or escalates, including a step-wise approach to engaging psychological interventions and mental health resources, and securing crisis services as needed.
On Day 15, he is discharged to home in stable condition with outpatient mental health follow-up and continues to take the prescribed methylphenidate.
The authors’ observations
Management of KLS is primarily supportive. Stimulants may help reduce hypersomnia, impulsivity, and inattention early in the disease course.1 However, in a systematic review, 89% of patients with KLS who received methylphenidate experienced worsening or no improvement, and 11% showed only partial improvement.2 Amantadine was more promising, with 29% of patients with KLS showing partial benefit and 12% showing significant benefit.2 Multiple other pharmacologic agents have been described with varying efficacy, including lithium, valproate, risperidone, bupropion, and immunoglobulins.2 Furthermore, lithium and valproate have been suggested to be helpful in preventing recurrences in some cases.6
The circumstances surrounding Mr. G’s symptom onset are unclear and may have been multifactorial. It is possible that his prior EBV infection was a trigger for this KLS-associated episode, as EBV is a known precipitant for KLS episodes.3 Mr. G’s history of cannabis use may also have served as an early trigger for KLS.3
This case highlights the importance of multidisciplinary collaboration in a diagnostically challenging case. It emphasizes the need for a broad differential and the importance of challenging a previous diagnosis in the face of mounting evidence to the contrary. In this case, the patient’s history of irritability, aggression, and cannabis use resulted in multiple clinicians misattributing his symptoms to substance use or a primary psychiatric disorder. However, given his symptom acuity, progression, and the lack of findings on diagnostic evaluation to explain his presentation, these initial diagnoses did not explain the severity, nature, or duration of his symptoms. Keeping KLS in the differential is particularly important for patients with a prior history of psychiatric illness or substance use, because these patients are at higher risk for misattribution of symptoms to pre-existing psychiatric illness. Evolution of symptoms, a negative diagnostic evaluation, and maintaining a broad differential resulted in eventually reaching the final diagnosis of KLS and development of a longitudinal management plan.
While further work must be done to clearly define the pharmacologic approach to acute management of KLS episodes, nonpharmacologic aspects of care must not be neglected. Behavioral planning, adjustment of the environment, engagement with schools/community supports, and family education are valuable tools for facilitating the patient’s de-escalation, avoiding unneeded polypharmacy, reducing anxieties, and safeguarding the patient from unnecessary harm.7 Clinicians can support their patients’ transitions back into the community by ensuring careful outpatient follow-up for symptom monitoring and by communicating with patients’ schools and employers.
OUTCOME Asymptomatic; no recurrence of symptoms
Forty-six days after his symptoms began and 31 days after hospital discharge, Mr. G is asymptomatic with no recurrence of symptoms.
Bottom Line
Kleine-Levin syndrome (KLS) is a rare, often-overlooked condition that should be considered in the differential diagnosis for patients who present with hypersomnolence and altered mental status without a clear etiology. Rapid recognition of KLS can prevent misattribution of symptoms, unnecessary treatment, and missed opportunities for care.
CASE Somnolent, confused, and hungry
Mr. G, age 14, presents to the emergency department (ED) for acute-onset hypersomnia that has gradually worsened over the course of a few days. Mr. G now sleeps most of the day, has altered mental status, and is experiencing emotional dysregulation with no clear etiology. His mother, who accompanies him to the ED, says that prior to the onset of these symptoms her son had been healthy. She notes that he has been eating more than usual, which she assumes is due to a growth spurt.
Mr. G’s symptoms began 4 days ago when he became increasingly fatigued, sleeping for 11 to 12 hours per day, with intermittent episodes of staring and unresponsiveness from which he rapidly returned to baseline. During the next 3 days, he became more confused and somnolent, and began to display bizarre behavior, including eating food out of the trash and attempting to microwave a full metal pot. He exhibited unexplained crying spells, calling out for his “mommy,” and saying he was “afraid I’m dying.”
During the 2 days before he came to our ED, Mr. G was seen at 2 other hospitals. Following extensive imaging and laboratory work-up, clinicians at these facilities attributed his symptoms to intoxication from an unknown substance. Mr. G has a history of marijuana use, and his mother reports that his friends had recently been using synthetic marijuana. However, no intoxicant was identified on urine or gas chromatography drug screening.
Mr. G’s history includes oppositional behavior, and a brief psychiatric hospitalization at age 5 for aggression. He has otherwise been healthy. His family history is significant for maternal substance use and anxiety disorders. In addition to sporadic cannabis use, Mr. G’s social history includes multiple recent family losses, previous foster care placement, and recent declining academic performance.
[polldaddy:10148168]
EVALUATION No red flags
On admission, Mr. G appears somnolent and displays disorganized speech, impulsivity, frequent disorientation, and intermittent agitation/anxiety; he sleeps 16 to 18 hours per day. Mr. G is admitted with a presumptive diagnosis of substance intoxication and transferred to the general pediatric inpatient unit. Upon arrival, he is found to be bradycardic (42 beats per minute), although afebrile with otherwise age-appropriate vitals. On exam, he is somnolent but arousable and follows simple commands.
Continue to: Mr. G undergoes a Monospot test...
Mr. G undergoes a Monospot test, which is positive, with subsequent evidence of a prior, but not active, Epstein-Barr virus (EBV) infection. He also has a mildly elevated CSF protein level, but subsequent CSF labs are negative for both infectious and non-infectious processes. An EEG reveals focal neuronal slowing.
During brief periods of wakefulness, Mr. G calls out to his mother and says, “I’m not going to make it to my birthday,” and “You’re going to have to let me go.” He occasionally becomes combative, pulling at IV lines and swearing at staff. His bradycardia resolves without intervention during his admission. On Day 8 of his hospitalization, Mr. G displays hypersexuality and makes sexually suggestive comments toward female staff members. He also experiences recurrence of hyperphagia.
On Day 10 of his stay on the pediatric unit, because of the emergence of hypersexuality and hyperphagia, along with a largely negative medical evaluation, Mr. G is transferred to the pediatric psychiatric unit for ongoing evaluation and management.
[polldaddy:10148172]
The authors’ observations
Given Mr. G’s rapid onset of confusion, hypersomnia, and emotional dysregulation, our differential diagnosis included delirium of unclear etiology, substance intoxication, autoimmune encephalitis, viral meningitis, heavy metal intoxication, primary psychotic disorder, and KLS. Mr. G underwent an extensive diagnostic evaluation, which was largely unremarkable (Table). He had a mildly elevated CSF protein level, but subsequent CSF labs were negative for both infectious and non-infectious processes. When Mr. G was transferred to the pediatric inpatient psychiatric unit on Day 10, the presumptive diagnosis was KLS.
KLS is a rare neurologic disorder, with an incidence of 1 to 5 in 1 million and a 4:1 male-to-female predominance.1 It poses a diagnostic challenge due to its low prevalence and broad differential. The disorder typically presents in early adolescence and is characterized by episodes of severe hypersomnia with associated cognitive and/or behavioral disturbance2 (Box2-4). Bradycardia, as seen in Mr. G, and other forms of autonomic dysregulation have been reported in the literature, as has the focal neuronal slowing noted on Mr. G’s EEG.3
[polldaddy:10148174]
Continue to: TREATMENT Methylphenidate and a safety plan
TREATMENT Methylphenidate and a safety plan
On Day 11 of hospitalization, Mr. G is started on methylphenidate, 10 mg in the morning and 5 mg in the afternoon. After starting methylphenidate, he sustains more regular wakefulness, with improved thought organization, engagement, and fewer disruptive behaviors. He receives infrequent, as-needed doses of olanzapine, and by Day 14, he returns to his baseline behavior and cognition.
A safety plan is created for the family to address worsening symptoms or future episodes. The safety plan is developed with Mr. G and input from his family. It is to be administered in all settings and we particularly emphasized using it in the school setting, where staff may not be familiar with KLS. The safety plan involves a description of KLS, its symptoms, the risks for hypersomnolence, hypersexuality, and psychotic symptoms or behavioral dysregulation. It stresses close supervision of Mr. G, not allowing him to be unsupervised or unchaperoned on school trips or other outings, and lethal means restriction. It outlines a detailed plan if Mr. G’s behavior decompensates or escalates, including a step-wise approach to engaging psychological interventions and mental health resources, and securing crisis services as needed.
On Day 15, he is discharged to home in stable condition with outpatient mental health follow-up and continues to take the prescribed methylphenidate.
The authors’ observations
Management of KLS is primarily supportive. Stimulants may help reduce hypersomnia, impulsivity, and inattention early in the disease course.1 However, in a systematic review, 89% of patients with KLS who received methylphenidate experienced worsening or no improvement, and 11% showed only partial improvement.2 Amantadine was more promising, with 29% of patients with KLS showing partial benefit and 12% showing significant benefit.2 Multiple other pharmacologic agents have been described with varying efficacy, including lithium, valproate, risperidone, bupropion, and immunoglobulins.2 Furthermore, lithium and valproate have been suggested to be helpful in preventing recurrences in some cases.6
The circumstances surrounding Mr. G’s symptom onset are unclear and may have been multifactorial. It is possible that his prior EBV infection was a trigger for this KLS-associated episode, as EBV is a known precipitant for KLS episodes.3 Mr. G’s history of cannabis use may also have served as an early trigger for KLS.3
This case highlights the importance of multidisciplinary collaboration in a diagnostically challenging case. It emphasizes the need for a broad differential and the importance of challenging a previous diagnosis in the face of mounting evidence to the contrary. In this case, the patient’s history of irritability, aggression, and cannabis use resulted in multiple clinicians misattributing his symptoms to substance use or a primary psychiatric disorder. However, given his symptom acuity, progression, and the lack of findings on diagnostic evaluation to explain his presentation, these initial diagnoses did not explain the severity, nature, or duration of his symptoms. Keeping KLS in the differential is particularly important for patients with a prior history of psychiatric illness or substance use, because these patients are at higher risk for misattribution of symptoms to pre-existing psychiatric illness. Evolution of symptoms, a negative diagnostic evaluation, and maintaining a broad differential resulted in eventually reaching the final diagnosis of KLS and development of a longitudinal management plan.
While further work must be done to clearly define the pharmacologic approach to acute management of KLS episodes, nonpharmacologic aspects of care must not be neglected. Behavioral planning, adjustment of the environment, engagement with schools/community supports, and family education are valuable tools for facilitating the patient’s de-escalation, avoiding unneeded polypharmacy, reducing anxieties, and safeguarding the patient from unnecessary harm.7 Clinicians can support their patients’ transitions back into the community by ensuring careful outpatient follow-up for symptom monitoring and by communicating with patients’ schools and employers.
OUTCOME Asymptomatic; no recurrence of symptoms
Forty-six days after his symptoms began and 31 days after hospital discharge, Mr. G is asymptomatic with no recurrence of symptoms.
Bottom Line
Kleine-Levin syndrome (KLS) is a rare, often-overlooked condition that should be considered in the differential diagnosis for patients who present with hypersomnolence and altered mental status without a clear etiology. Rapid recognition of KLS can prevent misattribution of symptoms, unnecessary treatment, and missed opportunities for care.
1. Billiard M, Jaussent I, Dauvilliers Y, et al. Recurrent hypersomnia: a review of 339 cases. Sleep Med. 2011;15(4):247-257.
2. Arnulf I, Lin L, Gadoth N, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol. 2008;63(4):482-493.
3. Arnulf I. Kleine-Levin syndrome: a systematic review of 186 cases in the literature. Brain. 2005;128(12):2763-2776.
4. Lisk R. Kleine-Levin syndrome. Pract Neurol. 2009;9(1);42-45.
5. de Araújo Lima TF, da Silva Behrens NS, Lopes E, et al. Kleine–Levin Syndrome: a case report. Sleep Sci. 2014;7(2):122-125.
6. Goldberg MA. The treatment of Kleine-Levin syndrome with lithium. Can J Psychiatry. 1983;28:491-493.
7. Gadoth N, Kesler A, Vainstein G, et al. Clinical and polysomnographic characteristics of 34 patients with Kleine-Levin syndrome. J Sleep Res. 2001;10(4):337-341.
1. Billiard M, Jaussent I, Dauvilliers Y, et al. Recurrent hypersomnia: a review of 339 cases. Sleep Med. 2011;15(4):247-257.
2. Arnulf I, Lin L, Gadoth N, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol. 2008;63(4):482-493.
3. Arnulf I. Kleine-Levin syndrome: a systematic review of 186 cases in the literature. Brain. 2005;128(12):2763-2776.
4. Lisk R. Kleine-Levin syndrome. Pract Neurol. 2009;9(1);42-45.
5. de Araújo Lima TF, da Silva Behrens NS, Lopes E, et al. Kleine–Levin Syndrome: a case report. Sleep Sci. 2014;7(2):122-125.
6. Goldberg MA. The treatment of Kleine-Levin syndrome with lithium. Can J Psychiatry. 1983;28:491-493.
7. Gadoth N, Kesler A, Vainstein G, et al. Clinical and polysomnographic characteristics of 34 patients with Kleine-Levin syndrome. J Sleep Res. 2001;10(4):337-341.
COMPRESS: Key questions to ask during shift changes in a psychiatric ER
Clinical errors are common during shift changes in a hospital setting.1-3 Clinicians on the outgoing shift may forget to communicate important details, such as medication dosages, critical laboratory orders, or other interventions, to the clinicians in the next shift. To help myself formally structure the sign-out process for each patient during a shift change in a psychiatric emergency room, I came up with the acronym COMPRESS for key questions to ask the outgoing provider:
Communicate. Did you communicate with this patient in any way at any time during your shift?
Orders. Did you write any orders for this patient? If not, had another clinician already written orders for this patient?
Medications. Did you review and reconcile the medication list for this patient? If not, had another clinician already reviewed and reconciled the medication list for this patient?
PRogrESs. Did you write a progress note for this patient? If not, had the attending clinician written a progress note for this patient within the last 24 hours?
Sign. Did you sign all of your orders and progress notes for this patient?
In my experience in the psychiatric emergency room, COMPRESS has helped me efficiently structure the outgoing clinicians’ reports about my patients by having them provide vital clinical sign-out information before they leave. I hope that other clinicians working in this setting also find these questions useful.
1. Dubosh NM, Carney D, Fisher J, et al. Implementation of an emergency department sign-out checklist improves transfer of information at shift change. J Emerg Med. 2014;47(5):580-585.
2. Estryn-Behar MR, Milanini-Magny G, Chaumon E, et al. Shift change handovers and subsequent interruptions: potential impacts on quality of care. J Patient Saf. 2014;10(1):29-44.
3. Mardis T, Mardis M, Davis J, et al. Bedside shift-to-shift handoffs: a systematic review of the literature. J Nurs Care Qual. 2016;31(1):54-60.
Clinical errors are common during shift changes in a hospital setting.1-3 Clinicians on the outgoing shift may forget to communicate important details, such as medication dosages, critical laboratory orders, or other interventions, to the clinicians in the next shift. To help myself formally structure the sign-out process for each patient during a shift change in a psychiatric emergency room, I came up with the acronym COMPRESS for key questions to ask the outgoing provider:
Communicate. Did you communicate with this patient in any way at any time during your shift?
Orders. Did you write any orders for this patient? If not, had another clinician already written orders for this patient?
Medications. Did you review and reconcile the medication list for this patient? If not, had another clinician already reviewed and reconciled the medication list for this patient?
PRogrESs. Did you write a progress note for this patient? If not, had the attending clinician written a progress note for this patient within the last 24 hours?
Sign. Did you sign all of your orders and progress notes for this patient?
In my experience in the psychiatric emergency room, COMPRESS has helped me efficiently structure the outgoing clinicians’ reports about my patients by having them provide vital clinical sign-out information before they leave. I hope that other clinicians working in this setting also find these questions useful.
Clinical errors are common during shift changes in a hospital setting.1-3 Clinicians on the outgoing shift may forget to communicate important details, such as medication dosages, critical laboratory orders, or other interventions, to the clinicians in the next shift. To help myself formally structure the sign-out process for each patient during a shift change in a psychiatric emergency room, I came up with the acronym COMPRESS for key questions to ask the outgoing provider:
Communicate. Did you communicate with this patient in any way at any time during your shift?
Orders. Did you write any orders for this patient? If not, had another clinician already written orders for this patient?
Medications. Did you review and reconcile the medication list for this patient? If not, had another clinician already reviewed and reconciled the medication list for this patient?
PRogrESs. Did you write a progress note for this patient? If not, had the attending clinician written a progress note for this patient within the last 24 hours?
Sign. Did you sign all of your orders and progress notes for this patient?
In my experience in the psychiatric emergency room, COMPRESS has helped me efficiently structure the outgoing clinicians’ reports about my patients by having them provide vital clinical sign-out information before they leave. I hope that other clinicians working in this setting also find these questions useful.
1. Dubosh NM, Carney D, Fisher J, et al. Implementation of an emergency department sign-out checklist improves transfer of information at shift change. J Emerg Med. 2014;47(5):580-585.
2. Estryn-Behar MR, Milanini-Magny G, Chaumon E, et al. Shift change handovers and subsequent interruptions: potential impacts on quality of care. J Patient Saf. 2014;10(1):29-44.
3. Mardis T, Mardis M, Davis J, et al. Bedside shift-to-shift handoffs: a systematic review of the literature. J Nurs Care Qual. 2016;31(1):54-60.
1. Dubosh NM, Carney D, Fisher J, et al. Implementation of an emergency department sign-out checklist improves transfer of information at shift change. J Emerg Med. 2014;47(5):580-585.
2. Estryn-Behar MR, Milanini-Magny G, Chaumon E, et al. Shift change handovers and subsequent interruptions: potential impacts on quality of care. J Patient Saf. 2014;10(1):29-44.
3. Mardis T, Mardis M, Davis J, et al. Bedside shift-to-shift handoffs: a systematic review of the literature. J Nurs Care Qual. 2016;31(1):54-60.