User login
A serum urate level greater than approximately 6.8 mg/dL, the saturation point of urate in biological fluids, is the underlying cause of gout. Hyperuricemia, along with other factors (detailed below), over time can result in the deposition of monosodium urate crystals into the joints. Gouty attacks are thought to occur by the abrupt release of these crystals into the joint space, where they may initiate an acute inflammatory reaction recognized as acute gouty arthritis. The acute attack is self-limited, but crystals remain in the joint and low-grade, often subclinical, inflammation persists even between acute attacks. Although acute attacks can be treated with anti-inflammatory medications, the underlying cause of the disease can be treated only by lowering the serum urate level.
CRYSTAL DEPOSITION AND THE DEVELOPMENT OF GOUT
Asymptomatic hyperuricemia is not a disease but rather is the underlying factor that can predispose to gout. A serum urate level of approximately 6.8 mg/dL is the concentration at which monosodium urate crystals begin to precipitate.1,2 Although this level is based on in vitro studies, it suggests a reasonable biological threshold for clinicians assessing patients for hyperuricemia. It should be noted that there are often no manifestations of gout during an extended period of hyperuricemia even though urate crystals are beginning to deposit into joints. The higher the serum urate level, the more likely that crystals will deposit into joints.
Predisposition is not causation
In the Normative Aging Study, 22% of men who had serum urate levels greater than 9 mg/dL developed gout during a 5-year period—a much higher rate than among men with serum urate levels less than 9 mg/dL.3 Nevertheless, a full 78% of the men in this study with serum urate levels greater than 9 mg/dL did not develop gout over the 5-year period, illustrating that while hyperuricemia predisposes to gout, it does not automatically cause gout.
Contributing factors beyond serum urate
Other factors, when combined with hyperuricemia, contribute to crystal deposition and the development of gout.
Trauma or irritation. Patients with hyperuricemia tend to have monosodium urate crystal deposition at sites of trauma or irritation. The first metatarsophalangeal joint is often affected, at least in part because it is a site of mechanical stress. Likewise, mechanical irritation from leaning on the elbow may cause crystals to deposit in the olecranon bursa.
Lower temperatures favor crystal deposition,1,4 which may explain why the helix of the ear and the foot are often sites of crystal deposition and tophus development. Both temperature and mechanical effects probably play a role in crystal deposition, however, as gouty attacks tend to occur at the first metatarsophalangeal joint, not at the interphalangeal joints of the foot, which are at a lower temperature.
Previous disease. Crystals also deposit with an increased incidence in previously diseased joints. The Heberden node is a good example.5 A patient with osteoarthritis in the fingers may experience dramatically increased pain and swelling because of a gout flare superimposed on an osteoarthritic joint.
ACUTE GOUTY ARTHRITIS
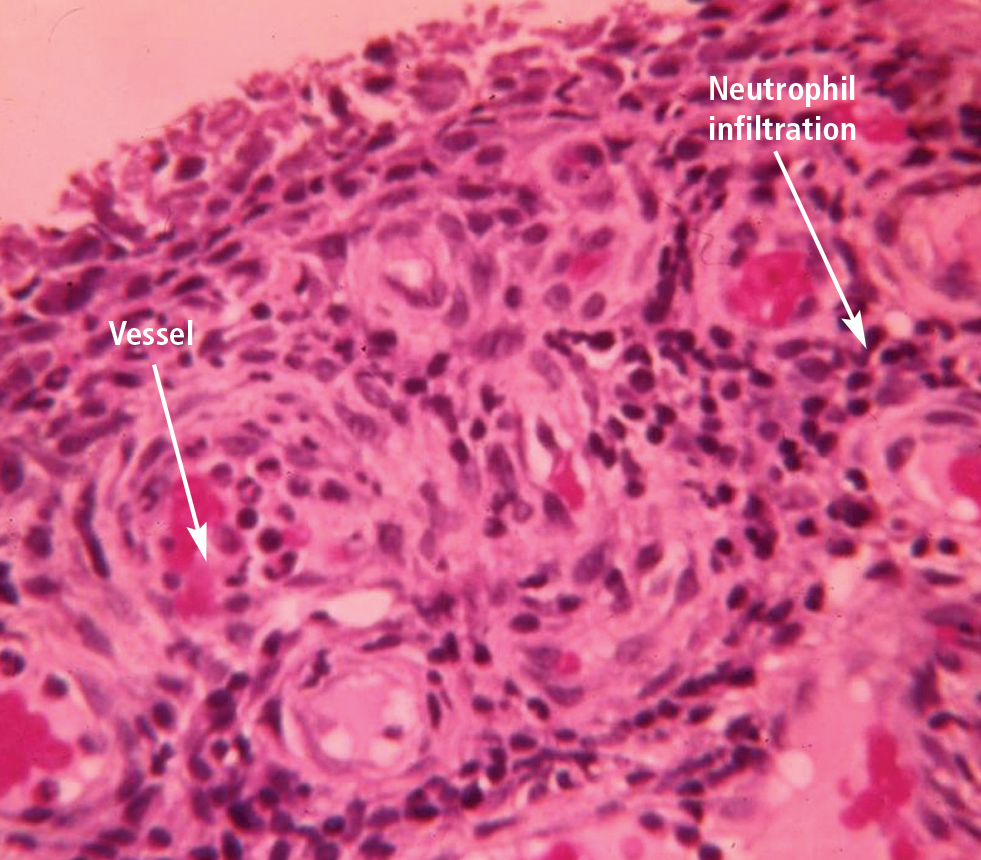
Gout flares may resolve spontaneously
Clinicians should be aware that gout attacks initially subside spontaneously.9 Because acute attacks of gout typically resolve with or without treatment, especially early in the course of the disease,10 it can be difficult to evaluate which treatments actually are effective against acute attacks.
A number of factors have been identified to explain how inflammation in acute attacks can be spontaneously suppressed. Crystals may dissolve or become sequestered in the tissue. Monocytes mature into macrophages, changing their responsiveness to urate crystals, and can begin to produce anti-inflammatory cytokines. In addition, some proteins that exude into the joint space with the attack, such as apolipoprotein B, can coat the crystals and reduce their inflammatory properties.11
Crystals persist during intercritical periods
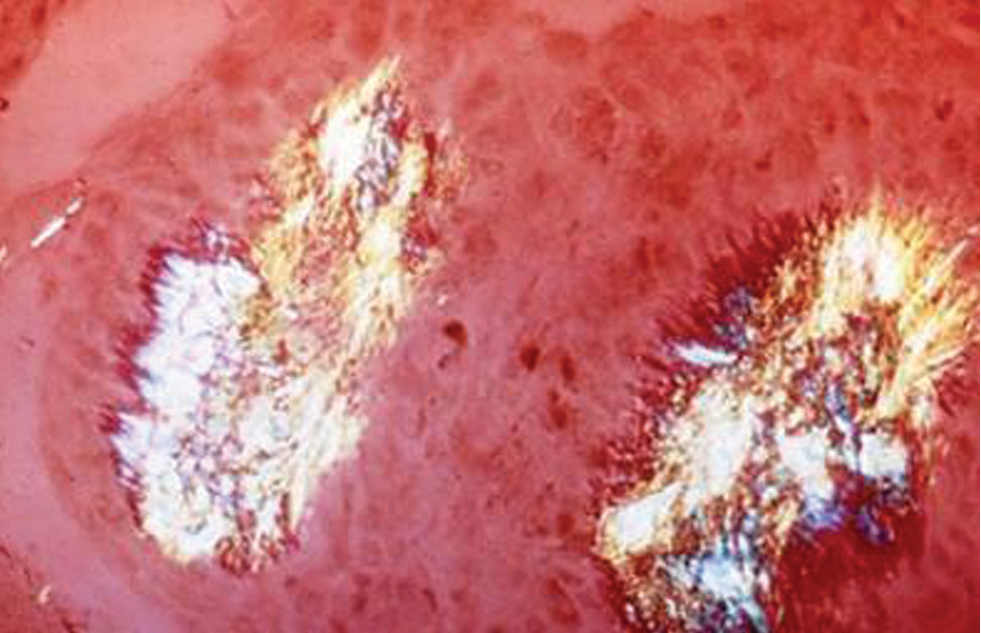
ADVANCED GOUT
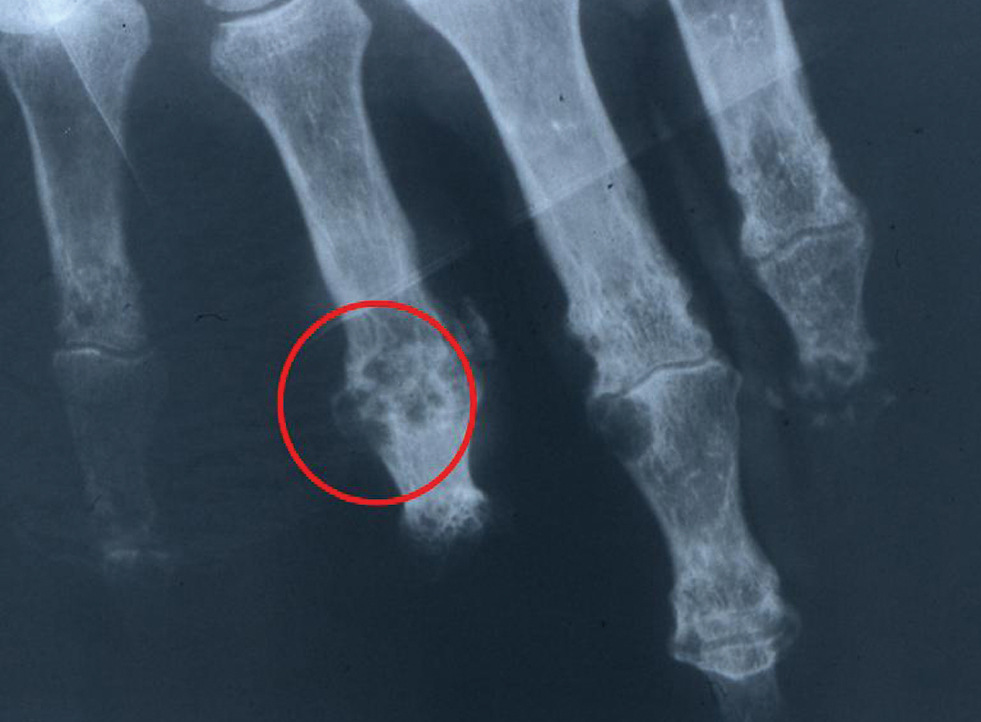
INTERVENTIONS MUST NORMALIZE URATE LEVEL
Acute gout attacks can be treated with anti-inflammatory drugs, but the disease can and often will continue to progress unless the serum urate level is normalized. Two studies of patients whose serum urate levels were successfully reduced to less than 6 mg/dL showed that crystals began to be depleted from the patients’ joint fluid, which should ultimately prevent the risk of progressive gouty arthritis.12,16 Perez-Ruiz and colleagues have shown that tophi can be dissolved by decreasing the serum urate level.17 When tophi are present, aiming for even lower levels of serum urate, such as 4 to 5 mg/dL, may help to promote more rapid dissolution of crystals.17
- Loeb JN. The influence of temperature on the solubility of monosodium urate. Arthritis Rheum 1972; 15:189–192.
- Kippen I, Klinenberg JR, Weinberger A, Wilcox WR. Factors affecting urate solubility in vitro. Ann Rheum Dis 1974; 33:313–317.
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med 1987; 82:421–426.
- Scott JT. New knowledge of the pathogenesis of gout. J Clin Pathol Suppl (R Coll Pathol) 1978; 12:205–213.
- Terkeltaub RA. Pathogenesis of gouty inflammation. In: Klippel JH, Crofford LJ, Stone JH, Weyand CM, eds. Primer on the Rheumatic Diseases. 12th ed. Atlanta, GA: Arthritis Foundation; 2001: 311–312.
- Schumacher HR Jr, Wortmann RL. The pathology of crystal-induced arthropathies. In: Wortman RL, Schumacher HR Jr, Becker MA, Ryan LM, eds. Crystal-Induced Arthropathies. New York, NY: Taylor & Francis Group; 2006:291–319.
- Martinon F, Glimcher LH. Gout: new insights into an old disease. J Clin Invest 2006; 116:2073–2075.
- Chen CJ, Shi Y, Hearn A, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest 2006; 116:2262–2271.
- Landis RC, Yagnik DR, Florey O, et al. Safe disposal of inflammatory monosodium urate monohydrate crystals by differentiated macrophages. Arthritis Rheum 2002; 46:3026–3033.
- Yagnik DR, Evans BJ, Florey O, Mason JC, Landis RC, Haskard DO. Macrophage release of transforming growth factor beta1 during resolution of monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum 2004; 50:2273–2280.
- Cherian PV, Schumacher HR Jr. Immunochemical and ultrastructural characterization of serum proteins associated with monosodium urate crystals (MSU) in synovial fluid cells from patients with gout. Ultrastruct Pathol 1986; 10:209–219.
- Li-Yu J, Clayburne G, Sieck M, et al. Treatment of chronic gout. Can we determine when urate stores are depleted enough to prevent attacks of gout? J Rheumatol 2001; 28:577–580.
- Pascual E, Pedraz T. Gout. Curr Opin Rheumatol 2004; 16: 282–286.
- Pascual E, Batlle-Gualda E, Martinez A, Rosas J, Vela P. Synovial fluid analysis for diagnosis of intercritical gout. Ann Intern Med 1999; 131:756–759.
- Dalbeth N, Clark B, Gregory K, Sheehan T, McQueen F. Clinical images: three-dimensional computed tomography imaging of tophaceous gout. Arthritis Rheum 2007; 56:29.
- Pascual E, Sivera F. Time required for disappearance of urate crystals from synovial fluid after successful hypouricaemic treatment relates to the duration of gout. Ann Rheum Dis 2007; 66: 1056–1058.
- Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum 2002; 47:356–360.
A serum urate level greater than approximately 6.8 mg/dL, the saturation point of urate in biological fluids, is the underlying cause of gout. Hyperuricemia, along with other factors (detailed below), over time can result in the deposition of monosodium urate crystals into the joints. Gouty attacks are thought to occur by the abrupt release of these crystals into the joint space, where they may initiate an acute inflammatory reaction recognized as acute gouty arthritis. The acute attack is self-limited, but crystals remain in the joint and low-grade, often subclinical, inflammation persists even between acute attacks. Although acute attacks can be treated with anti-inflammatory medications, the underlying cause of the disease can be treated only by lowering the serum urate level.
CRYSTAL DEPOSITION AND THE DEVELOPMENT OF GOUT
Asymptomatic hyperuricemia is not a disease but rather is the underlying factor that can predispose to gout. A serum urate level of approximately 6.8 mg/dL is the concentration at which monosodium urate crystals begin to precipitate.1,2 Although this level is based on in vitro studies, it suggests a reasonable biological threshold for clinicians assessing patients for hyperuricemia. It should be noted that there are often no manifestations of gout during an extended period of hyperuricemia even though urate crystals are beginning to deposit into joints. The higher the serum urate level, the more likely that crystals will deposit into joints.
Predisposition is not causation
In the Normative Aging Study, 22% of men who had serum urate levels greater than 9 mg/dL developed gout during a 5-year period—a much higher rate than among men with serum urate levels less than 9 mg/dL.3 Nevertheless, a full 78% of the men in this study with serum urate levels greater than 9 mg/dL did not develop gout over the 5-year period, illustrating that while hyperuricemia predisposes to gout, it does not automatically cause gout.
Contributing factors beyond serum urate
Other factors, when combined with hyperuricemia, contribute to crystal deposition and the development of gout.
Trauma or irritation. Patients with hyperuricemia tend to have monosodium urate crystal deposition at sites of trauma or irritation. The first metatarsophalangeal joint is often affected, at least in part because it is a site of mechanical stress. Likewise, mechanical irritation from leaning on the elbow may cause crystals to deposit in the olecranon bursa.
Lower temperatures favor crystal deposition,1,4 which may explain why the helix of the ear and the foot are often sites of crystal deposition and tophus development. Both temperature and mechanical effects probably play a role in crystal deposition, however, as gouty attacks tend to occur at the first metatarsophalangeal joint, not at the interphalangeal joints of the foot, which are at a lower temperature.
Previous disease. Crystals also deposit with an increased incidence in previously diseased joints. The Heberden node is a good example.5 A patient with osteoarthritis in the fingers may experience dramatically increased pain and swelling because of a gout flare superimposed on an osteoarthritic joint.
ACUTE GOUTY ARTHRITIS
Gout flares may resolve spontaneously
Clinicians should be aware that gout attacks initially subside spontaneously.9 Because acute attacks of gout typically resolve with or without treatment, especially early in the course of the disease,10 it can be difficult to evaluate which treatments actually are effective against acute attacks.
A number of factors have been identified to explain how inflammation in acute attacks can be spontaneously suppressed. Crystals may dissolve or become sequestered in the tissue. Monocytes mature into macrophages, changing their responsiveness to urate crystals, and can begin to produce anti-inflammatory cytokines. In addition, some proteins that exude into the joint space with the attack, such as apolipoprotein B, can coat the crystals and reduce their inflammatory properties.11
Crystals persist during intercritical periods
ADVANCED GOUT
INTERVENTIONS MUST NORMALIZE URATE LEVEL
Acute gout attacks can be treated with anti-inflammatory drugs, but the disease can and often will continue to progress unless the serum urate level is normalized. Two studies of patients whose serum urate levels were successfully reduced to less than 6 mg/dL showed that crystals began to be depleted from the patients’ joint fluid, which should ultimately prevent the risk of progressive gouty arthritis.12,16 Perez-Ruiz and colleagues have shown that tophi can be dissolved by decreasing the serum urate level.17 When tophi are present, aiming for even lower levels of serum urate, such as 4 to 5 mg/dL, may help to promote more rapid dissolution of crystals.17
A serum urate level greater than approximately 6.8 mg/dL, the saturation point of urate in biological fluids, is the underlying cause of gout. Hyperuricemia, along with other factors (detailed below), over time can result in the deposition of monosodium urate crystals into the joints. Gouty attacks are thought to occur by the abrupt release of these crystals into the joint space, where they may initiate an acute inflammatory reaction recognized as acute gouty arthritis. The acute attack is self-limited, but crystals remain in the joint and low-grade, often subclinical, inflammation persists even between acute attacks. Although acute attacks can be treated with anti-inflammatory medications, the underlying cause of the disease can be treated only by lowering the serum urate level.
CRYSTAL DEPOSITION AND THE DEVELOPMENT OF GOUT
Asymptomatic hyperuricemia is not a disease but rather is the underlying factor that can predispose to gout. A serum urate level of approximately 6.8 mg/dL is the concentration at which monosodium urate crystals begin to precipitate.1,2 Although this level is based on in vitro studies, it suggests a reasonable biological threshold for clinicians assessing patients for hyperuricemia. It should be noted that there are often no manifestations of gout during an extended period of hyperuricemia even though urate crystals are beginning to deposit into joints. The higher the serum urate level, the more likely that crystals will deposit into joints.
Predisposition is not causation
In the Normative Aging Study, 22% of men who had serum urate levels greater than 9 mg/dL developed gout during a 5-year period—a much higher rate than among men with serum urate levels less than 9 mg/dL.3 Nevertheless, a full 78% of the men in this study with serum urate levels greater than 9 mg/dL did not develop gout over the 5-year period, illustrating that while hyperuricemia predisposes to gout, it does not automatically cause gout.
Contributing factors beyond serum urate
Other factors, when combined with hyperuricemia, contribute to crystal deposition and the development of gout.
Trauma or irritation. Patients with hyperuricemia tend to have monosodium urate crystal deposition at sites of trauma or irritation. The first metatarsophalangeal joint is often affected, at least in part because it is a site of mechanical stress. Likewise, mechanical irritation from leaning on the elbow may cause crystals to deposit in the olecranon bursa.
Lower temperatures favor crystal deposition,1,4 which may explain why the helix of the ear and the foot are often sites of crystal deposition and tophus development. Both temperature and mechanical effects probably play a role in crystal deposition, however, as gouty attacks tend to occur at the first metatarsophalangeal joint, not at the interphalangeal joints of the foot, which are at a lower temperature.
Previous disease. Crystals also deposit with an increased incidence in previously diseased joints. The Heberden node is a good example.5 A patient with osteoarthritis in the fingers may experience dramatically increased pain and swelling because of a gout flare superimposed on an osteoarthritic joint.
ACUTE GOUTY ARTHRITIS
Gout flares may resolve spontaneously
Clinicians should be aware that gout attacks initially subside spontaneously.9 Because acute attacks of gout typically resolve with or without treatment, especially early in the course of the disease,10 it can be difficult to evaluate which treatments actually are effective against acute attacks.
A number of factors have been identified to explain how inflammation in acute attacks can be spontaneously suppressed. Crystals may dissolve or become sequestered in the tissue. Monocytes mature into macrophages, changing their responsiveness to urate crystals, and can begin to produce anti-inflammatory cytokines. In addition, some proteins that exude into the joint space with the attack, such as apolipoprotein B, can coat the crystals and reduce their inflammatory properties.11
Crystals persist during intercritical periods
ADVANCED GOUT
INTERVENTIONS MUST NORMALIZE URATE LEVEL
Acute gout attacks can be treated with anti-inflammatory drugs, but the disease can and often will continue to progress unless the serum urate level is normalized. Two studies of patients whose serum urate levels were successfully reduced to less than 6 mg/dL showed that crystals began to be depleted from the patients’ joint fluid, which should ultimately prevent the risk of progressive gouty arthritis.12,16 Perez-Ruiz and colleagues have shown that tophi can be dissolved by decreasing the serum urate level.17 When tophi are present, aiming for even lower levels of serum urate, such as 4 to 5 mg/dL, may help to promote more rapid dissolution of crystals.17
- Loeb JN. The influence of temperature on the solubility of monosodium urate. Arthritis Rheum 1972; 15:189–192.
- Kippen I, Klinenberg JR, Weinberger A, Wilcox WR. Factors affecting urate solubility in vitro. Ann Rheum Dis 1974; 33:313–317.
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med 1987; 82:421–426.
- Scott JT. New knowledge of the pathogenesis of gout. J Clin Pathol Suppl (R Coll Pathol) 1978; 12:205–213.
- Terkeltaub RA. Pathogenesis of gouty inflammation. In: Klippel JH, Crofford LJ, Stone JH, Weyand CM, eds. Primer on the Rheumatic Diseases. 12th ed. Atlanta, GA: Arthritis Foundation; 2001: 311–312.
- Schumacher HR Jr, Wortmann RL. The pathology of crystal-induced arthropathies. In: Wortman RL, Schumacher HR Jr, Becker MA, Ryan LM, eds. Crystal-Induced Arthropathies. New York, NY: Taylor & Francis Group; 2006:291–319.
- Martinon F, Glimcher LH. Gout: new insights into an old disease. J Clin Invest 2006; 116:2073–2075.
- Chen CJ, Shi Y, Hearn A, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest 2006; 116:2262–2271.
- Landis RC, Yagnik DR, Florey O, et al. Safe disposal of inflammatory monosodium urate monohydrate crystals by differentiated macrophages. Arthritis Rheum 2002; 46:3026–3033.
- Yagnik DR, Evans BJ, Florey O, Mason JC, Landis RC, Haskard DO. Macrophage release of transforming growth factor beta1 during resolution of monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum 2004; 50:2273–2280.
- Cherian PV, Schumacher HR Jr. Immunochemical and ultrastructural characterization of serum proteins associated with monosodium urate crystals (MSU) in synovial fluid cells from patients with gout. Ultrastruct Pathol 1986; 10:209–219.
- Li-Yu J, Clayburne G, Sieck M, et al. Treatment of chronic gout. Can we determine when urate stores are depleted enough to prevent attacks of gout? J Rheumatol 2001; 28:577–580.
- Pascual E, Pedraz T. Gout. Curr Opin Rheumatol 2004; 16: 282–286.
- Pascual E, Batlle-Gualda E, Martinez A, Rosas J, Vela P. Synovial fluid analysis for diagnosis of intercritical gout. Ann Intern Med 1999; 131:756–759.
- Dalbeth N, Clark B, Gregory K, Sheehan T, McQueen F. Clinical images: three-dimensional computed tomography imaging of tophaceous gout. Arthritis Rheum 2007; 56:29.
- Pascual E, Sivera F. Time required for disappearance of urate crystals from synovial fluid after successful hypouricaemic treatment relates to the duration of gout. Ann Rheum Dis 2007; 66: 1056–1058.
- Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum 2002; 47:356–360.
- Loeb JN. The influence of temperature on the solubility of monosodium urate. Arthritis Rheum 1972; 15:189–192.
- Kippen I, Klinenberg JR, Weinberger A, Wilcox WR. Factors affecting urate solubility in vitro. Ann Rheum Dis 1974; 33:313–317.
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med 1987; 82:421–426.
- Scott JT. New knowledge of the pathogenesis of gout. J Clin Pathol Suppl (R Coll Pathol) 1978; 12:205–213.
- Terkeltaub RA. Pathogenesis of gouty inflammation. In: Klippel JH, Crofford LJ, Stone JH, Weyand CM, eds. Primer on the Rheumatic Diseases. 12th ed. Atlanta, GA: Arthritis Foundation; 2001: 311–312.
- Schumacher HR Jr, Wortmann RL. The pathology of crystal-induced arthropathies. In: Wortman RL, Schumacher HR Jr, Becker MA, Ryan LM, eds. Crystal-Induced Arthropathies. New York, NY: Taylor & Francis Group; 2006:291–319.
- Martinon F, Glimcher LH. Gout: new insights into an old disease. J Clin Invest 2006; 116:2073–2075.
- Chen CJ, Shi Y, Hearn A, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest 2006; 116:2262–2271.
- Landis RC, Yagnik DR, Florey O, et al. Safe disposal of inflammatory monosodium urate monohydrate crystals by differentiated macrophages. Arthritis Rheum 2002; 46:3026–3033.
- Yagnik DR, Evans BJ, Florey O, Mason JC, Landis RC, Haskard DO. Macrophage release of transforming growth factor beta1 during resolution of monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum 2004; 50:2273–2280.
- Cherian PV, Schumacher HR Jr. Immunochemical and ultrastructural characterization of serum proteins associated with monosodium urate crystals (MSU) in synovial fluid cells from patients with gout. Ultrastruct Pathol 1986; 10:209–219.
- Li-Yu J, Clayburne G, Sieck M, et al. Treatment of chronic gout. Can we determine when urate stores are depleted enough to prevent attacks of gout? J Rheumatol 2001; 28:577–580.
- Pascual E, Pedraz T. Gout. Curr Opin Rheumatol 2004; 16: 282–286.
- Pascual E, Batlle-Gualda E, Martinez A, Rosas J, Vela P. Synovial fluid analysis for diagnosis of intercritical gout. Ann Intern Med 1999; 131:756–759.
- Dalbeth N, Clark B, Gregory K, Sheehan T, McQueen F. Clinical images: three-dimensional computed tomography imaging of tophaceous gout. Arthritis Rheum 2007; 56:29.
- Pascual E, Sivera F. Time required for disappearance of urate crystals from synovial fluid after successful hypouricaemic treatment relates to the duration of gout. Ann Rheum Dis 2007; 66: 1056–1058.
- Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum 2002; 47:356–360.
KEY POINTS
- A serum urate level of approximately 6.8 mg/dL is the concentration at which urate crystals begin to precipitate. The higher the urate level, the more likely that crystals will deposit into joints.
- Local factors that combine with hyperuricemia to contribute to the development of gout are trauma, irritation, reduced temperature, and prior joint disease.
- Because acute attacks of gout typically resolve spontaneously, especially early in the disease course, evaluating the efficacy of acute therapies can be difficult.
- Lowering the serum urate to less than 6 mg/dL will dissolve crystals out of the joints, ultimately preventing acute gout attacks and joint damage.