User login
Early breast cancer: Updated MINDACT findings back omitting chemotherapy in low genomic risk
Key clinical point: Among women with breast cancer and high clinical risk, a subgroup of patients with low genomic risk identified by the 70-gene signature showed excellent distant metastasis-free survival (DMFS) even without chemotherapy. No benefits of chemotherapy were observed in women older than 50 years.
Major finding: Eight-year DMFS in patients with high clinical but low genomic risk with vs. without chemotherapy was 92.0% vs. 89.4%, with the benefit of adding chemotherapy to endocrine therapy remaining small (absolute difference, 2·6 percentage points). Chemotherapy showed no benefits in older women aged more than 50 years (absolute difference, 0.2 percentage points).
Study details: Findings are from the 8-year analysis of phase 3 MINDACT trial, including women with primary nonmetastatic invasive breast cancer with up to 3 positive lymph nodes. The 70-gene signature (MammaPrint) identified women with high clinical but low genomic risk who were randomly assigned to chemotherapy (n=749) vs. no chemotherapy (n=748).
Disclosures: This study was funded by European Commission Sixth Framework Programme (ECSFP). Some investigators including the lead author reported ties with various pharmaceutical companies and funding agencies including the ECSFP.
Source: Piccart M et al. Lancet Oncol. 2021 Mar 12. doi: 10.1016/S1470-2045(21)00007-3.
Key clinical point: Among women with breast cancer and high clinical risk, a subgroup of patients with low genomic risk identified by the 70-gene signature showed excellent distant metastasis-free survival (DMFS) even without chemotherapy. No benefits of chemotherapy were observed in women older than 50 years.
Major finding: Eight-year DMFS in patients with high clinical but low genomic risk with vs. without chemotherapy was 92.0% vs. 89.4%, with the benefit of adding chemotherapy to endocrine therapy remaining small (absolute difference, 2·6 percentage points). Chemotherapy showed no benefits in older women aged more than 50 years (absolute difference, 0.2 percentage points).
Study details: Findings are from the 8-year analysis of phase 3 MINDACT trial, including women with primary nonmetastatic invasive breast cancer with up to 3 positive lymph nodes. The 70-gene signature (MammaPrint) identified women with high clinical but low genomic risk who were randomly assigned to chemotherapy (n=749) vs. no chemotherapy (n=748).
Disclosures: This study was funded by European Commission Sixth Framework Programme (ECSFP). Some investigators including the lead author reported ties with various pharmaceutical companies and funding agencies including the ECSFP.
Source: Piccart M et al. Lancet Oncol. 2021 Mar 12. doi: 10.1016/S1470-2045(21)00007-3.
Key clinical point: Among women with breast cancer and high clinical risk, a subgroup of patients with low genomic risk identified by the 70-gene signature showed excellent distant metastasis-free survival (DMFS) even without chemotherapy. No benefits of chemotherapy were observed in women older than 50 years.
Major finding: Eight-year DMFS in patients with high clinical but low genomic risk with vs. without chemotherapy was 92.0% vs. 89.4%, with the benefit of adding chemotherapy to endocrine therapy remaining small (absolute difference, 2·6 percentage points). Chemotherapy showed no benefits in older women aged more than 50 years (absolute difference, 0.2 percentage points).
Study details: Findings are from the 8-year analysis of phase 3 MINDACT trial, including women with primary nonmetastatic invasive breast cancer with up to 3 positive lymph nodes. The 70-gene signature (MammaPrint) identified women with high clinical but low genomic risk who were randomly assigned to chemotherapy (n=749) vs. no chemotherapy (n=748).
Disclosures: This study was funded by European Commission Sixth Framework Programme (ECSFP). Some investigators including the lead author reported ties with various pharmaceutical companies and funding agencies including the ECSFP.
Source: Piccart M et al. Lancet Oncol. 2021 Mar 12. doi: 10.1016/S1470-2045(21)00007-3.
Early breast cancer: Updated MINDACT findings back omitting chemotherapy in low genomic risk
Key clinical point: Among women with breast cancer and high clinical risk, a subgroup of patients with low genomic risk identified by the 70-gene signature showed excellent distant metastasis-free survival (DMFS) even without chemotherapy. No benefits of chemotherapy were observed in women older than 50 years.
Major finding: Eight-year DMFS in patients with high clinical but low genomic risk with vs. without chemotherapy was 92.0% vs. 89.4%, with the benefit of adding chemotherapy to endocrine therapy remaining small (absolute difference, 2·6 percentage points). Chemotherapy showed no benefits in older women aged more than 50 years (absolute difference, 0.2 percentage points).
Study details: Findings are from the 8-year analysis of phase 3 MINDACT trial, including women with primary nonmetastatic invasive breast cancer with up to 3 positive lymph nodes. The 70-gene signature (MammaPrint) identified women with high clinical but low genomic risk who were randomly assigned to chemotherapy (n=749) vs. no chemotherapy (n=748).
Disclosures: This study was funded by European Commission Sixth Framework Programme (ECSFP). Some investigators including the lead author reported ties with various pharmaceutical companies and funding agencies including the ECSFP.
Source: Piccart M et al. Lancet Oncol. 2021 Mar 12. doi: 10.1016/S1470-2045(21)00007-3.
Key clinical point: Among women with breast cancer and high clinical risk, a subgroup of patients with low genomic risk identified by the 70-gene signature showed excellent distant metastasis-free survival (DMFS) even without chemotherapy. No benefits of chemotherapy were observed in women older than 50 years.
Major finding: Eight-year DMFS in patients with high clinical but low genomic risk with vs. without chemotherapy was 92.0% vs. 89.4%, with the benefit of adding chemotherapy to endocrine therapy remaining small (absolute difference, 2·6 percentage points). Chemotherapy showed no benefits in older women aged more than 50 years (absolute difference, 0.2 percentage points).
Study details: Findings are from the 8-year analysis of phase 3 MINDACT trial, including women with primary nonmetastatic invasive breast cancer with up to 3 positive lymph nodes. The 70-gene signature (MammaPrint) identified women with high clinical but low genomic risk who were randomly assigned to chemotherapy (n=749) vs. no chemotherapy (n=748).
Disclosures: This study was funded by European Commission Sixth Framework Programme (ECSFP). Some investigators including the lead author reported ties with various pharmaceutical companies and funding agencies including the ECSFP.
Source: Piccart M et al. Lancet Oncol. 2021 Mar 12. doi: 10.1016/S1470-2045(21)00007-3.
Key clinical point: Among women with breast cancer and high clinical risk, a subgroup of patients with low genomic risk identified by the 70-gene signature showed excellent distant metastasis-free survival (DMFS) even without chemotherapy. No benefits of chemotherapy were observed in women older than 50 years.
Major finding: Eight-year DMFS in patients with high clinical but low genomic risk with vs. without chemotherapy was 92.0% vs. 89.4%, with the benefit of adding chemotherapy to endocrine therapy remaining small (absolute difference, 2·6 percentage points). Chemotherapy showed no benefits in older women aged more than 50 years (absolute difference, 0.2 percentage points).
Study details: Findings are from the 8-year analysis of phase 3 MINDACT trial, including women with primary nonmetastatic invasive breast cancer with up to 3 positive lymph nodes. The 70-gene signature (MammaPrint) identified women with high clinical but low genomic risk who were randomly assigned to chemotherapy (n=749) vs. no chemotherapy (n=748).
Disclosures: This study was funded by European Commission Sixth Framework Programme (ECSFP). Some investigators including the lead author reported ties with various pharmaceutical companies and funding agencies including the ECSFP.
Source: Piccart M et al. Lancet Oncol. 2021 Mar 12. doi: 10.1016/S1470-2045(21)00007-3.
Risk-based mammography proposed for times of reduced capacity
Researchers evaluated almost 2 million mammograms that had been performed at more than 90 radiology centers and found that 12% of mammograms with “high” and “very high” cancer risk rates accounted for 55% of detected cancers.
In contrast, 44% of mammograms with very low cancer risk rates accounted for 13% of detected cancers. The study was published online March 25, 2021, in JAMA Network Open.
Cancer screening programs dramatically slowed or even came to a screeching halt during 2020, when restrictions and lockdowns were in place. The American Cancer Society even recommended that “no one should go to a health care facility for routine cancer screening,” as part of COVID-19 precautions.
However, concern was voiced that the pause in screening would allow patients with asymptomatic cancers or precursor lesions to develop into a more serious disease state.
The authors pointed out that several professional associations had posted guidance for scheduling individuals for breast imaging services during the COVID-19 pandemic, but these recommendations were based on expert opinion. The investigators’ goal was to help imaging facilities optimize the number of breast cancers that could be detected during periods of reduced capacity using clinical indication and individual characteristics.
The result was a risk-based strategy for triaging mammograms during periods of decreased capacity, which lead author Diana L. Miglioretti, PhD, explained was feasible to implement. Dr. Miglioretti is division chief of biostatistics in the department of public health sciences at University of California, Davis.
“Our risk model used information that is commonly collected by radiology facilities,” she said in an interview. “Vendors of electronic medical records could create tools that pull the information from the medical record, or could create fields in the scheduling system to efficiently collect this information when the mammogram is scheduled.”
Dr. Miglioretti emphasized that, once the information is collected in a standardized manner, “it would be straightforward to use a computer program to apply our algorithm to rank women based on their likelihood of having a breast cancer detected.”
“I think it is worth the investment to create these electronic tools now, given the potential for future shutdowns or periods of reduced capacity due to a variety of reasons, such as natural disasters and cyberattacks – or another pandemic,” she said.
Some facilities are still working through backlogs of mammograms that need to be rescheduled, which would be another way that this algorithm could be used. “They could use this approach to determine who should be scheduled first by using data available in the electronic medical record,” she added.
Five risk groups
Dr. Miglioretti and colleagues conducted a cohort study using data that was prospectively collected from mammography examinations performed from 2014 to 2019 at 92 radiology facilities in the Breast Cancer Surveillance Consortium. The cohort included 898,415 individuals who contributed to 1.8 million mammograms.
Information that included clinical indication for screening, breast symptoms, personal history of breast cancer, age, time since last mammogram/screening interval, family history of breast cancer, breast density, and history of high-risk breast lesion was collected from self-administered questionnaires at the time of mammography or extracted from electronic health records.
Following analysis, the data was categorized into five risk groups: very high (>50), high (22-50), moderate (10-22), low (5-10), and very low (<5) cancer detection rate per 1,000 mammograms. These thresholds were chosen based on the observed cancer detection rates and clinical expertise.
Of the group, about 1.7 million mammograms were from women without a personal history of breast cancer and 156,104 mammograms were from women with a breast cancer history. Most of the cohort were aged 50-69 years at the time of imaging, and 67.9% were White (11.2% Black, 11.3% Asian or Pacific Islander, 7% Hispanic, and 2.2% were another race/ethnicity or mixed race/ethnicity).
Their results showed that 12% of mammograms with very high (89.6-122.3 cancers detected per 1,000 mammograms) or high (36.1-47.5 cancers detected per 1,000 mammograms) cancer detection rates accounted for 55% of all detected cancers. These included mammograms that were done to evaluate an abnormal test or breast lump in individuals of all ages regardless of breast cancer history.
On the opposite end, 44.2% of mammograms with very low cancer detection rates accounted for 13.1% of detected cancers and that included annual screening tests in women aged 50-69 years (3.8 cancers detected per 1,000 mammograms) and all screening mammograms in individuals younger than 50 years regardless of screening interval (2.8 cancers detected per 1000 mammograms).
Treat with caution
In an accompanying editorial, Sarah M. Friedewald, MD, and Dipti Gupta, MD, both from Northwestern University, Chicago, pointed out that, while the authors examined a large dataset to identify a subgroup of patients who would most likely benefit from breast imaging in a setting where capacity is limited, “these data should be used with caution as the only barometer for whether a patient merits cancer screening during a period of rationing.”
They noted that, in the context of an acute crisis, when patient volume needs to be reduced very quickly, it is often impractical for clinicians to sift through patient records in order to capture the information necessary for triage. In addition, asking nonclinical schedulers to accurately pull data at this level, at the time when the patient calls to make an appointment, is unrealistic.
In the context of the pandemic, the editorialists wrote that, while this model uses risk for breast cancer to prioritize those to be seen in the clinic, the risk for complications from COVID-19 may also be an important factor to consider. For example, an older patient may be at a higher risk for breast cancer but may also face a higher risk for COVID-related complications. Conversely, a younger woman at a lower risk for serious COVID-related disease but who has breast cancer detected early will gain more life-years than an older patient.
There are also no algorithms to account for each patient’s perceived risk for breast cancer or COVID-19, and “the downstream effect of delaying cancer diagnosis may similarly lead to unintended consequences but may take longer to become apparent,” they wrote. “Focusing efforts on the operations of accommodating as many patients as possible, such as extending clinic hours, would be preferable.”
Finally, Dr. Friedewald and Dr. Gupta concluded that “the practicality of this process during the COVID-19 pandemic and extrapolation to other emergent settings are less obvious.”
The study was supported through a Patient-centered Outcomes Research Institute program award. Dr. Miglioretti reported receiving royalties from Elsevier outside the submitted work. Several coauthors report relationships with industry. Dr. Friedewald reported receiving grants from Hologic Research during the conduct of the study. Dr. Gupta disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Researchers evaluated almost 2 million mammograms that had been performed at more than 90 radiology centers and found that 12% of mammograms with “high” and “very high” cancer risk rates accounted for 55% of detected cancers.
In contrast, 44% of mammograms with very low cancer risk rates accounted for 13% of detected cancers. The study was published online March 25, 2021, in JAMA Network Open.
Cancer screening programs dramatically slowed or even came to a screeching halt during 2020, when restrictions and lockdowns were in place. The American Cancer Society even recommended that “no one should go to a health care facility for routine cancer screening,” as part of COVID-19 precautions.
However, concern was voiced that the pause in screening would allow patients with asymptomatic cancers or precursor lesions to develop into a more serious disease state.
The authors pointed out that several professional associations had posted guidance for scheduling individuals for breast imaging services during the COVID-19 pandemic, but these recommendations were based on expert opinion. The investigators’ goal was to help imaging facilities optimize the number of breast cancers that could be detected during periods of reduced capacity using clinical indication and individual characteristics.
The result was a risk-based strategy for triaging mammograms during periods of decreased capacity, which lead author Diana L. Miglioretti, PhD, explained was feasible to implement. Dr. Miglioretti is division chief of biostatistics in the department of public health sciences at University of California, Davis.
“Our risk model used information that is commonly collected by radiology facilities,” she said in an interview. “Vendors of electronic medical records could create tools that pull the information from the medical record, or could create fields in the scheduling system to efficiently collect this information when the mammogram is scheduled.”
Dr. Miglioretti emphasized that, once the information is collected in a standardized manner, “it would be straightforward to use a computer program to apply our algorithm to rank women based on their likelihood of having a breast cancer detected.”
“I think it is worth the investment to create these electronic tools now, given the potential for future shutdowns or periods of reduced capacity due to a variety of reasons, such as natural disasters and cyberattacks – or another pandemic,” she said.
Some facilities are still working through backlogs of mammograms that need to be rescheduled, which would be another way that this algorithm could be used. “They could use this approach to determine who should be scheduled first by using data available in the electronic medical record,” she added.
Five risk groups
Dr. Miglioretti and colleagues conducted a cohort study using data that was prospectively collected from mammography examinations performed from 2014 to 2019 at 92 radiology facilities in the Breast Cancer Surveillance Consortium. The cohort included 898,415 individuals who contributed to 1.8 million mammograms.
Information that included clinical indication for screening, breast symptoms, personal history of breast cancer, age, time since last mammogram/screening interval, family history of breast cancer, breast density, and history of high-risk breast lesion was collected from self-administered questionnaires at the time of mammography or extracted from electronic health records.
Following analysis, the data was categorized into five risk groups: very high (>50), high (22-50), moderate (10-22), low (5-10), and very low (<5) cancer detection rate per 1,000 mammograms. These thresholds were chosen based on the observed cancer detection rates and clinical expertise.
Of the group, about 1.7 million mammograms were from women without a personal history of breast cancer and 156,104 mammograms were from women with a breast cancer history. Most of the cohort were aged 50-69 years at the time of imaging, and 67.9% were White (11.2% Black, 11.3% Asian or Pacific Islander, 7% Hispanic, and 2.2% were another race/ethnicity or mixed race/ethnicity).
Their results showed that 12% of mammograms with very high (89.6-122.3 cancers detected per 1,000 mammograms) or high (36.1-47.5 cancers detected per 1,000 mammograms) cancer detection rates accounted for 55% of all detected cancers. These included mammograms that were done to evaluate an abnormal test or breast lump in individuals of all ages regardless of breast cancer history.
On the opposite end, 44.2% of mammograms with very low cancer detection rates accounted for 13.1% of detected cancers and that included annual screening tests in women aged 50-69 years (3.8 cancers detected per 1,000 mammograms) and all screening mammograms in individuals younger than 50 years regardless of screening interval (2.8 cancers detected per 1000 mammograms).
Treat with caution
In an accompanying editorial, Sarah M. Friedewald, MD, and Dipti Gupta, MD, both from Northwestern University, Chicago, pointed out that, while the authors examined a large dataset to identify a subgroup of patients who would most likely benefit from breast imaging in a setting where capacity is limited, “these data should be used with caution as the only barometer for whether a patient merits cancer screening during a period of rationing.”
They noted that, in the context of an acute crisis, when patient volume needs to be reduced very quickly, it is often impractical for clinicians to sift through patient records in order to capture the information necessary for triage. In addition, asking nonclinical schedulers to accurately pull data at this level, at the time when the patient calls to make an appointment, is unrealistic.
In the context of the pandemic, the editorialists wrote that, while this model uses risk for breast cancer to prioritize those to be seen in the clinic, the risk for complications from COVID-19 may also be an important factor to consider. For example, an older patient may be at a higher risk for breast cancer but may also face a higher risk for COVID-related complications. Conversely, a younger woman at a lower risk for serious COVID-related disease but who has breast cancer detected early will gain more life-years than an older patient.
There are also no algorithms to account for each patient’s perceived risk for breast cancer or COVID-19, and “the downstream effect of delaying cancer diagnosis may similarly lead to unintended consequences but may take longer to become apparent,” they wrote. “Focusing efforts on the operations of accommodating as many patients as possible, such as extending clinic hours, would be preferable.”
Finally, Dr. Friedewald and Dr. Gupta concluded that “the practicality of this process during the COVID-19 pandemic and extrapolation to other emergent settings are less obvious.”
The study was supported through a Patient-centered Outcomes Research Institute program award. Dr. Miglioretti reported receiving royalties from Elsevier outside the submitted work. Several coauthors report relationships with industry. Dr. Friedewald reported receiving grants from Hologic Research during the conduct of the study. Dr. Gupta disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Researchers evaluated almost 2 million mammograms that had been performed at more than 90 radiology centers and found that 12% of mammograms with “high” and “very high” cancer risk rates accounted for 55% of detected cancers.
In contrast, 44% of mammograms with very low cancer risk rates accounted for 13% of detected cancers. The study was published online March 25, 2021, in JAMA Network Open.
Cancer screening programs dramatically slowed or even came to a screeching halt during 2020, when restrictions and lockdowns were in place. The American Cancer Society even recommended that “no one should go to a health care facility for routine cancer screening,” as part of COVID-19 precautions.
However, concern was voiced that the pause in screening would allow patients with asymptomatic cancers or precursor lesions to develop into a more serious disease state.
The authors pointed out that several professional associations had posted guidance for scheduling individuals for breast imaging services during the COVID-19 pandemic, but these recommendations were based on expert opinion. The investigators’ goal was to help imaging facilities optimize the number of breast cancers that could be detected during periods of reduced capacity using clinical indication and individual characteristics.
The result was a risk-based strategy for triaging mammograms during periods of decreased capacity, which lead author Diana L. Miglioretti, PhD, explained was feasible to implement. Dr. Miglioretti is division chief of biostatistics in the department of public health sciences at University of California, Davis.
“Our risk model used information that is commonly collected by radiology facilities,” she said in an interview. “Vendors of electronic medical records could create tools that pull the information from the medical record, or could create fields in the scheduling system to efficiently collect this information when the mammogram is scheduled.”
Dr. Miglioretti emphasized that, once the information is collected in a standardized manner, “it would be straightforward to use a computer program to apply our algorithm to rank women based on their likelihood of having a breast cancer detected.”
“I think it is worth the investment to create these electronic tools now, given the potential for future shutdowns or periods of reduced capacity due to a variety of reasons, such as natural disasters and cyberattacks – or another pandemic,” she said.
Some facilities are still working through backlogs of mammograms that need to be rescheduled, which would be another way that this algorithm could be used. “They could use this approach to determine who should be scheduled first by using data available in the electronic medical record,” she added.
Five risk groups
Dr. Miglioretti and colleagues conducted a cohort study using data that was prospectively collected from mammography examinations performed from 2014 to 2019 at 92 radiology facilities in the Breast Cancer Surveillance Consortium. The cohort included 898,415 individuals who contributed to 1.8 million mammograms.
Information that included clinical indication for screening, breast symptoms, personal history of breast cancer, age, time since last mammogram/screening interval, family history of breast cancer, breast density, and history of high-risk breast lesion was collected from self-administered questionnaires at the time of mammography or extracted from electronic health records.
Following analysis, the data was categorized into five risk groups: very high (>50), high (22-50), moderate (10-22), low (5-10), and very low (<5) cancer detection rate per 1,000 mammograms. These thresholds were chosen based on the observed cancer detection rates and clinical expertise.
Of the group, about 1.7 million mammograms were from women without a personal history of breast cancer and 156,104 mammograms were from women with a breast cancer history. Most of the cohort were aged 50-69 years at the time of imaging, and 67.9% were White (11.2% Black, 11.3% Asian or Pacific Islander, 7% Hispanic, and 2.2% were another race/ethnicity or mixed race/ethnicity).
Their results showed that 12% of mammograms with very high (89.6-122.3 cancers detected per 1,000 mammograms) or high (36.1-47.5 cancers detected per 1,000 mammograms) cancer detection rates accounted for 55% of all detected cancers. These included mammograms that were done to evaluate an abnormal test or breast lump in individuals of all ages regardless of breast cancer history.
On the opposite end, 44.2% of mammograms with very low cancer detection rates accounted for 13.1% of detected cancers and that included annual screening tests in women aged 50-69 years (3.8 cancers detected per 1,000 mammograms) and all screening mammograms in individuals younger than 50 years regardless of screening interval (2.8 cancers detected per 1000 mammograms).
Treat with caution
In an accompanying editorial, Sarah M. Friedewald, MD, and Dipti Gupta, MD, both from Northwestern University, Chicago, pointed out that, while the authors examined a large dataset to identify a subgroup of patients who would most likely benefit from breast imaging in a setting where capacity is limited, “these data should be used with caution as the only barometer for whether a patient merits cancer screening during a period of rationing.”
They noted that, in the context of an acute crisis, when patient volume needs to be reduced very quickly, it is often impractical for clinicians to sift through patient records in order to capture the information necessary for triage. In addition, asking nonclinical schedulers to accurately pull data at this level, at the time when the patient calls to make an appointment, is unrealistic.
In the context of the pandemic, the editorialists wrote that, while this model uses risk for breast cancer to prioritize those to be seen in the clinic, the risk for complications from COVID-19 may also be an important factor to consider. For example, an older patient may be at a higher risk for breast cancer but may also face a higher risk for COVID-related complications. Conversely, a younger woman at a lower risk for serious COVID-related disease but who has breast cancer detected early will gain more life-years than an older patient.
There are also no algorithms to account for each patient’s perceived risk for breast cancer or COVID-19, and “the downstream effect of delaying cancer diagnosis may similarly lead to unintended consequences but may take longer to become apparent,” they wrote. “Focusing efforts on the operations of accommodating as many patients as possible, such as extending clinic hours, would be preferable.”
Finally, Dr. Friedewald and Dr. Gupta concluded that “the practicality of this process during the COVID-19 pandemic and extrapolation to other emergent settings are less obvious.”
The study was supported through a Patient-centered Outcomes Research Institute program award. Dr. Miglioretti reported receiving royalties from Elsevier outside the submitted work. Several coauthors report relationships with industry. Dr. Friedewald reported receiving grants from Hologic Research during the conduct of the study. Dr. Gupta disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Encephalopathy common, often lethal in hospitalized patients with COVID-19
, new research shows. Results of a retrospective study show that of almost 4,500 patients with COVID-19, 12% were diagnosed with TME. Of these, 78% developed encephalopathy immediately prior to hospital admission. Septic encephalopathy, hypoxic-ischemic encephalopathy (HIE), and uremia were the most common causes, although multiple causes were present in close to 80% of patients. TME was also associated with a 24% higher risk of in-hospital death.
“We found that close to one in eight patients who were hospitalized with COVID-19 had TME that was not attributed to the effects of sedatives, and that this is incredibly common among these patients who are critically ill” said lead author Jennifer A. Frontera, MD, New York University.
“The general principle of our findings is to be more aggressive in TME; and from a neurologist perspective, the way to do this is to eliminate the effects of sedation, which is a confounder,” she said.
The study was published online March 16 in Neurocritical Care.
Drilling down
“Many neurological complications of COVID-19 are sequelae of severe illness or secondary effects of multisystem organ failure, but our previous work identified TME as the most common neurological complication,” Dr. Frontera said.
Previous research investigating encephalopathy among patients with COVID-19 included patients who may have been sedated or have had a positive Confusion Assessment Method (CAM) result.
“A lot of the delirium literature is effectively heterogeneous because there are a number of patients who are on sedative medication that, if you could turn it off, these patients would return to normal. Some may have underlying neurological issues that can be addressed, but you can›t get to the bottom of this unless you turn off the sedation,” Dr. Frontera noted.
“We wanted to be specific and try to drill down to see what the underlying cause of the encephalopathy was,” she said.
The researchers retrospectively analyzed data on 4,491 patients (≥ 18 years old) with COVID-19 who were admitted to four New York City hospitals between March 1, 2020, and May 20, 2020. Of these, 559 (12%) with TME were compared with 3,932 patients without TME.
The researchers looked at index admissions and included patients who had:
- New changes in mental status or significant worsening of mental status (in patients with baseline abnormal mental status).
- Hyperglycemia or with transient focal neurologic deficits that resolved with glucose correction.
- An adequate washout of sedating medications (when relevant) prior to mental status assessment.
Potential etiologies included electrolyte abnormalities, organ failure, hypertensive encephalopathy, sepsis or active infection, fever, nutritional deficiency, and environmental injury.
Foreign environment
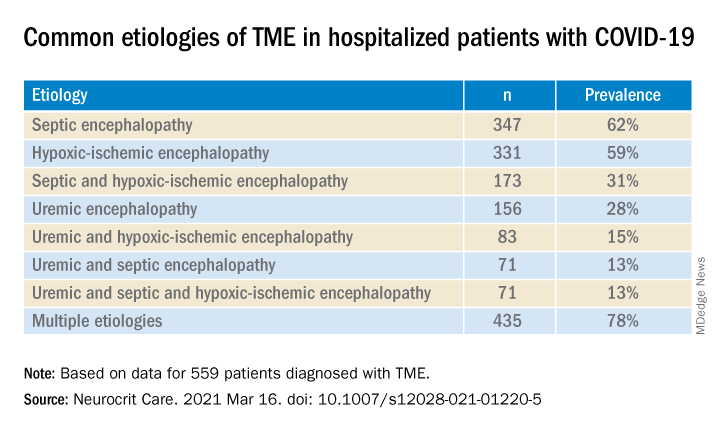
Most (78%) of the 559 patients diagnosed with TME had already developed encephalopathy immediately prior to hospital admission, the authors report. The most common etiologies of TME among hospitalized patients with COVID-19 are listed below.
Compared with patients without TME, those with TME – (all Ps < .001):
- Were older (76 vs. 62 years).
- Had higher rates of dementia (27% vs. 3%).
- Had higher rates of psychiatric history (20% vs. 10%).
- Were more often intubated (37% vs. 20%).
- Had a longer length of hospital stay (7.9 vs. 6.0 days).
- Were less often discharged home (25% vs. 66%).
“It’s no surprise that older patients and people with dementia or psychiatric illness are predisposed to becoming encephalopathic,” said Dr. Frontera. “Being in a foreign environment, such as a hospital, or being sleep-deprived in the ICU is likely to make them more confused during their hospital stay.”
Delirium as a symptom
In-hospital mortality or discharge to hospice was considerably higher in the TME versus non-TME patients (44% vs. 18%, respectively).
When the researchers adjusted for confounders (age, sex, race, worse Sequential Organ Failure Assessment score during hospitalization, ventilator status, study week, hospital location, and ICU care level) and excluded patients receiving only comfort care, they found that TME was associated with a 24% increased risk of in-hospital death (30% in patients with TME vs. 16% in those without TME).
The highest mortality risk was associated with hypoxemia, with 42% of patients with HIE dying during hospitalization, compared with 16% of patients without HIE (adjusted hazard ratio 1.56; 95% confidence interval, 1.21-2.00; P = .001).
“Not all patients who are intubated require sedation, but there’s generally a lot of hesitation in reducing or stopping sedation in some patients,” Dr. Frontera observed.
She acknowledged there are “many extremely sick patients whom you can’t ventilate without sedation.”
Nevertheless, “delirium in and of itself does not cause death. It’s a symptom, not a disease, and we have to figure out what causes it. Delirium might not need to be sedated, and it’s more important to see what the causal problem is.”
Independent predictor of death
Commenting on the study, Panayiotis N. Varelas, MD, PhD, vice president of the Neurocritical Care Society, said the study “approached the TME issue better than previously, namely allowing time for sedatives to wear off to have a better sample of patients with this syndrome.”
Dr. Varelas, who is chairman of the department of neurology and professor of neurology at Albany (N.Y.) Medical College, emphasized that TME “is not benign and, in patients with COVID-19, it is an independent predictor of in-hospital mortality.”
“One should take all possible measures … to avoid desaturation and hypotensive episodes and also aggressively treat SAE and uremic encephalopathy in hopes of improving the outcomes,” added Dr. Varelas, who was not involved with the study.
Also commenting on the study, Mitchell Elkind, MD, professor of neurology and epidemiology at Columbia University in New York, who was not associated with the research, said it “nicely distinguishes among the different causes of encephalopathy, including sepsis, hypoxia, and kidney failure … emphasizing just how sick these patients are.”
The study received no direct funding. Individual investigators were supported by grants from the National Institute on Aging and the National Institute of Neurological Disorders and Stroke. The investigators, Dr. Varelas, and Dr. Elkind have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, new research shows. Results of a retrospective study show that of almost 4,500 patients with COVID-19, 12% were diagnosed with TME. Of these, 78% developed encephalopathy immediately prior to hospital admission. Septic encephalopathy, hypoxic-ischemic encephalopathy (HIE), and uremia were the most common causes, although multiple causes were present in close to 80% of patients. TME was also associated with a 24% higher risk of in-hospital death.
“We found that close to one in eight patients who were hospitalized with COVID-19 had TME that was not attributed to the effects of sedatives, and that this is incredibly common among these patients who are critically ill” said lead author Jennifer A. Frontera, MD, New York University.
“The general principle of our findings is to be more aggressive in TME; and from a neurologist perspective, the way to do this is to eliminate the effects of sedation, which is a confounder,” she said.
The study was published online March 16 in Neurocritical Care.
Drilling down
“Many neurological complications of COVID-19 are sequelae of severe illness or secondary effects of multisystem organ failure, but our previous work identified TME as the most common neurological complication,” Dr. Frontera said.
Previous research investigating encephalopathy among patients with COVID-19 included patients who may have been sedated or have had a positive Confusion Assessment Method (CAM) result.
“A lot of the delirium literature is effectively heterogeneous because there are a number of patients who are on sedative medication that, if you could turn it off, these patients would return to normal. Some may have underlying neurological issues that can be addressed, but you can›t get to the bottom of this unless you turn off the sedation,” Dr. Frontera noted.
“We wanted to be specific and try to drill down to see what the underlying cause of the encephalopathy was,” she said.
The researchers retrospectively analyzed data on 4,491 patients (≥ 18 years old) with COVID-19 who were admitted to four New York City hospitals between March 1, 2020, and May 20, 2020. Of these, 559 (12%) with TME were compared with 3,932 patients without TME.
The researchers looked at index admissions and included patients who had:
- New changes in mental status or significant worsening of mental status (in patients with baseline abnormal mental status).
- Hyperglycemia or with transient focal neurologic deficits that resolved with glucose correction.
- An adequate washout of sedating medications (when relevant) prior to mental status assessment.
Potential etiologies included electrolyte abnormalities, organ failure, hypertensive encephalopathy, sepsis or active infection, fever, nutritional deficiency, and environmental injury.
Foreign environment
Most (78%) of the 559 patients diagnosed with TME had already developed encephalopathy immediately prior to hospital admission, the authors report. The most common etiologies of TME among hospitalized patients with COVID-19 are listed below.
Compared with patients without TME, those with TME – (all Ps < .001):
- Were older (76 vs. 62 years).
- Had higher rates of dementia (27% vs. 3%).
- Had higher rates of psychiatric history (20% vs. 10%).
- Were more often intubated (37% vs. 20%).
- Had a longer length of hospital stay (7.9 vs. 6.0 days).
- Were less often discharged home (25% vs. 66%).
“It’s no surprise that older patients and people with dementia or psychiatric illness are predisposed to becoming encephalopathic,” said Dr. Frontera. “Being in a foreign environment, such as a hospital, or being sleep-deprived in the ICU is likely to make them more confused during their hospital stay.”
Delirium as a symptom
In-hospital mortality or discharge to hospice was considerably higher in the TME versus non-TME patients (44% vs. 18%, respectively).
When the researchers adjusted for confounders (age, sex, race, worse Sequential Organ Failure Assessment score during hospitalization, ventilator status, study week, hospital location, and ICU care level) and excluded patients receiving only comfort care, they found that TME was associated with a 24% increased risk of in-hospital death (30% in patients with TME vs. 16% in those without TME).
The highest mortality risk was associated with hypoxemia, with 42% of patients with HIE dying during hospitalization, compared with 16% of patients without HIE (adjusted hazard ratio 1.56; 95% confidence interval, 1.21-2.00; P = .001).
“Not all patients who are intubated require sedation, but there’s generally a lot of hesitation in reducing or stopping sedation in some patients,” Dr. Frontera observed.
She acknowledged there are “many extremely sick patients whom you can’t ventilate without sedation.”
Nevertheless, “delirium in and of itself does not cause death. It’s a symptom, not a disease, and we have to figure out what causes it. Delirium might not need to be sedated, and it’s more important to see what the causal problem is.”
Independent predictor of death
Commenting on the study, Panayiotis N. Varelas, MD, PhD, vice president of the Neurocritical Care Society, said the study “approached the TME issue better than previously, namely allowing time for sedatives to wear off to have a better sample of patients with this syndrome.”
Dr. Varelas, who is chairman of the department of neurology and professor of neurology at Albany (N.Y.) Medical College, emphasized that TME “is not benign and, in patients with COVID-19, it is an independent predictor of in-hospital mortality.”
“One should take all possible measures … to avoid desaturation and hypotensive episodes and also aggressively treat SAE and uremic encephalopathy in hopes of improving the outcomes,” added Dr. Varelas, who was not involved with the study.
Also commenting on the study, Mitchell Elkind, MD, professor of neurology and epidemiology at Columbia University in New York, who was not associated with the research, said it “nicely distinguishes among the different causes of encephalopathy, including sepsis, hypoxia, and kidney failure … emphasizing just how sick these patients are.”
The study received no direct funding. Individual investigators were supported by grants from the National Institute on Aging and the National Institute of Neurological Disorders and Stroke. The investigators, Dr. Varelas, and Dr. Elkind have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, new research shows. Results of a retrospective study show that of almost 4,500 patients with COVID-19, 12% were diagnosed with TME. Of these, 78% developed encephalopathy immediately prior to hospital admission. Septic encephalopathy, hypoxic-ischemic encephalopathy (HIE), and uremia were the most common causes, although multiple causes were present in close to 80% of patients. TME was also associated with a 24% higher risk of in-hospital death.
“We found that close to one in eight patients who were hospitalized with COVID-19 had TME that was not attributed to the effects of sedatives, and that this is incredibly common among these patients who are critically ill” said lead author Jennifer A. Frontera, MD, New York University.
“The general principle of our findings is to be more aggressive in TME; and from a neurologist perspective, the way to do this is to eliminate the effects of sedation, which is a confounder,” she said.
The study was published online March 16 in Neurocritical Care.
Drilling down
“Many neurological complications of COVID-19 are sequelae of severe illness or secondary effects of multisystem organ failure, but our previous work identified TME as the most common neurological complication,” Dr. Frontera said.
Previous research investigating encephalopathy among patients with COVID-19 included patients who may have been sedated or have had a positive Confusion Assessment Method (CAM) result.
“A lot of the delirium literature is effectively heterogeneous because there are a number of patients who are on sedative medication that, if you could turn it off, these patients would return to normal. Some may have underlying neurological issues that can be addressed, but you can›t get to the bottom of this unless you turn off the sedation,” Dr. Frontera noted.
“We wanted to be specific and try to drill down to see what the underlying cause of the encephalopathy was,” she said.
The researchers retrospectively analyzed data on 4,491 patients (≥ 18 years old) with COVID-19 who were admitted to four New York City hospitals between March 1, 2020, and May 20, 2020. Of these, 559 (12%) with TME were compared with 3,932 patients without TME.
The researchers looked at index admissions and included patients who had:
- New changes in mental status or significant worsening of mental status (in patients with baseline abnormal mental status).
- Hyperglycemia or with transient focal neurologic deficits that resolved with glucose correction.
- An adequate washout of sedating medications (when relevant) prior to mental status assessment.
Potential etiologies included electrolyte abnormalities, organ failure, hypertensive encephalopathy, sepsis or active infection, fever, nutritional deficiency, and environmental injury.
Foreign environment
Most (78%) of the 559 patients diagnosed with TME had already developed encephalopathy immediately prior to hospital admission, the authors report. The most common etiologies of TME among hospitalized patients with COVID-19 are listed below.
Compared with patients without TME, those with TME – (all Ps < .001):
- Were older (76 vs. 62 years).
- Had higher rates of dementia (27% vs. 3%).
- Had higher rates of psychiatric history (20% vs. 10%).
- Were more often intubated (37% vs. 20%).
- Had a longer length of hospital stay (7.9 vs. 6.0 days).
- Were less often discharged home (25% vs. 66%).
“It’s no surprise that older patients and people with dementia or psychiatric illness are predisposed to becoming encephalopathic,” said Dr. Frontera. “Being in a foreign environment, such as a hospital, or being sleep-deprived in the ICU is likely to make them more confused during their hospital stay.”
Delirium as a symptom
In-hospital mortality or discharge to hospice was considerably higher in the TME versus non-TME patients (44% vs. 18%, respectively).
When the researchers adjusted for confounders (age, sex, race, worse Sequential Organ Failure Assessment score during hospitalization, ventilator status, study week, hospital location, and ICU care level) and excluded patients receiving only comfort care, they found that TME was associated with a 24% increased risk of in-hospital death (30% in patients with TME vs. 16% in those without TME).
The highest mortality risk was associated with hypoxemia, with 42% of patients with HIE dying during hospitalization, compared with 16% of patients without HIE (adjusted hazard ratio 1.56; 95% confidence interval, 1.21-2.00; P = .001).
“Not all patients who are intubated require sedation, but there’s generally a lot of hesitation in reducing or stopping sedation in some patients,” Dr. Frontera observed.
She acknowledged there are “many extremely sick patients whom you can’t ventilate without sedation.”
Nevertheless, “delirium in and of itself does not cause death. It’s a symptom, not a disease, and we have to figure out what causes it. Delirium might not need to be sedated, and it’s more important to see what the causal problem is.”
Independent predictor of death
Commenting on the study, Panayiotis N. Varelas, MD, PhD, vice president of the Neurocritical Care Society, said the study “approached the TME issue better than previously, namely allowing time for sedatives to wear off to have a better sample of patients with this syndrome.”
Dr. Varelas, who is chairman of the department of neurology and professor of neurology at Albany (N.Y.) Medical College, emphasized that TME “is not benign and, in patients with COVID-19, it is an independent predictor of in-hospital mortality.”
“One should take all possible measures … to avoid desaturation and hypotensive episodes and also aggressively treat SAE and uremic encephalopathy in hopes of improving the outcomes,” added Dr. Varelas, who was not involved with the study.
Also commenting on the study, Mitchell Elkind, MD, professor of neurology and epidemiology at Columbia University in New York, who was not associated with the research, said it “nicely distinguishes among the different causes of encephalopathy, including sepsis, hypoxia, and kidney failure … emphasizing just how sick these patients are.”
The study received no direct funding. Individual investigators were supported by grants from the National Institute on Aging and the National Institute of Neurological Disorders and Stroke. The investigators, Dr. Varelas, and Dr. Elkind have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM NEUROCRITICAL CARE
Huge, struggling breast cancer screening trial gets lifeline
But mammography trends can’t be ignored.
A controversial, big-budget breast cancer screening trial that has been chronically unable to attract enough female participants since its debut in 2017 got a vote of confidence from a special working group of the National Cancer Institute (NCI) on March 17.
The Tomosynthesis Mammography Imaging Screening Trial (TMIST) should continue, but with modification, the expert group concluded in its report.
The randomized trial, with an estimated cost of $100 million, compares two kinds of mammography screenings for breast cancer in healthy women. One group of patients is screened with digital breast tomosynthesis, also known as 3-D mammography; the other is screened with the older, less expensive 2-D digital technology.
Tomosynthesis is already considered superior in detecting small cancers and reducing callbacks and is increasingly being used in the real world, leading some experts in the field to say that TMIST is critically hampered by women’s and radiologists’ preference for 3-D mammography.
At a meeting of an NCI advisory board in September 2020, there was a suggestion that the federal agency may kill the trial.
But at the latest meeting, the working group proposed that the trial should live on.
One of the main problems with the trial has been recruitment; the recommended changes discussed at the meeting include reducing the number of women needed in the study (from 165,000 to 102,000), which would allow patient “accrual to be completed more quickly,” the working group noted. In addition, the target date for completing patient accrual would be moved to 2023, 3 years after the original completion date of 2020.
The group’s recommendations now go to NCI staff for scientific review. The NCI will then decide about implementing the proposed changes.
The trial, which is the NCI’s largest and most expensive screening study, has never come close to targeted monthly enrollment. It was enrolling fewer than 1,500 patients per month over a 2-year period, instead of the projected 5,500 per month, Philip Castle, PhD, MPH, director of the NCI’s Division of Cancer Prevention, said last year. He called for a review of TMIST’s “feasibility and relevance” in view of the increasing use of tomosynthesis in the United States, as well as other factors.
The new technology has been “rapidly adopted” by facilities in North America, the working group noted. As of December 2020, approximately 74% of breast cancer screening clinics in the United States had at least one tomosynthesis or 3-D system; 42% of the mammography machines were 3-D.
This trend of increasing use of 3-D machines might be too much for TMIST to surmount, said Nancy Davidson, MD, of the Fred Hutchinson Cancer Research Center, in Seattle, who chaired the working group.
“We are worried the challenges [to patient accrual] may persist due to the increasing adoption of three-dimensional breast tomosynthesis in the United States over time,” she said during the working group’s virtual presentation of the report to the NCI’s Clinical Trials and Translational Research Advisory Committee.
Committee member Smita Bhatia, MD, PhD, of the University of Alabama at Birmingham, wondered, “What are the ongoing barriers that [TMIST investigators] are going to face beside recruitment?”
Dr. Davidson answered by speaking, again, about market penetration of tomosynthesis machines and suggested that the recruitment problems and the availability of 3-D mammography are intertwined.
“Is this a technology that has or will arrive, at which point it may not be very easy to put the genie back in the bottle?” she asked.
That question has already been answered – widespread use of tomosynthesis is here to stay, argued Daniel Kopans, MD, of Harvard Medical School, Boston, who invented digital breast tomosynthesis but no longer benefits financially from his invention because the patent has expired.
“The horse is out of the barn,” Dr. Kopans said in an interview. By the time the study results are available, digital mammography will be a tool of the past, he said.
TMIST is a trial “that should never have been started in the first place, and it’s failing,” he said. “I was hoping they [the NCI] would say, ‘Let’s stop this because there’s not enough accrual.’ But it looks like they’re not.”
“TMIST is a waste of money,” said Dr. Kopans, repeating a criticism he has made in the past.
A drop in study power
The new recommendations for TMIST come about 1 year after Medscape Medical News reported that the study was lagging in enrollment of both patients and participating sites/physicians.
Last year, two TMIST study investigators said it had been difficult enlisting sites, in part because many radiologists and facilities – informed by their experience and previous research results – already believe that the 3-D technology is superior.
Currently, most 3-D systems are used in conjunction with 2-D. First, two static images of the breast are taken (2-D), and then the unit moves in an arc, taking multiple images of the breast (3-D). Thus, 3-D is widely described as allowing clinicians to flip through the images like “pages in a book” and as offering a superior read of the breast.
The NCI working group concedes that “there is evidence that screening utilizing tomosynthesis may reduce recall rates and improve cancer detection,” but it says the trial is needed to address “questions that still remain regarding the overall benefit to patients.”
Furthermore, tomosynthesis “may carry higher out-of-pocket costs for women and is more labor intensive and costly for health care systems in that it requires about twice as much reader time for interpretation,” the working group said.
The “main hypothesis of TMIST” is that “tomosynthesis will decrease the cumulative incidence of advanced breast cancers, a surrogate for mortality, compared to standard digital mammography,” posits the group.
Advanced breast cancer is defined in TMIST as invasive breast cancers that meet any of the following criteria:
- Distant metastases
- At least one lymph node macrometastasis
- Tumor size >1 cm and triple-negative or positive for human epidermal growth factor receptor
- Tumor size ≥ 20 mm unless of pure mucinous or other favorable histologies.
In the original study design, the sample size was estimated to be sufficient to provide 90% power to detect a 20% relative reduction in the proportion of advanced cancers in the intervention arm (tomosynthesis, or 3-D) compared to the control arm (digital mammography, or 2-D) 4.5 years from randomization.
Now, with fewer patients and a revised analytic approach, the study’s statistical power will be decreased to 80% from the original 90%.
Also, an advanced cancer is counted “if it occurs at any time while the participant is on study.”
Dr. Kopans says that is a problem.
“That is a huge mistake, since digital breast tomosynthesis cannot impact prevalent cancers. They are already there. This means that their ‘power calculation’ is wrong, and they won’t have the power that they are claiming,” he said.
Dr. Kopans explained that the first screen in TMIST will have “no effect on the number of advanced cancers.” That’s because the cancers will have already grown enough to become advanced, he said.
On the other hand, an initial screening might detect and thus lead to the removal of nonadvanced, smaller cancers, which, had they not been detected and removed, would have grown to become advanced cancers by the next year. Thus, the screenings done after the first year are the ones that potentially prove the effect of the intervention.
However, the working group report says that changes to the study will not affect anything other than a 10% reduction in the study’s power.
The working group is concerned about TMIST going on for years and years. For that reason, they recommended that the NCI establish “strict criteria for termination of the study” if accrual goals are not met. However, those parameters have not been developed, and it was not part of the study group’s mission to establish them.
The working group was sponsored by the NCI. Dr. Kopans reports consulting with DART Imaging in China.
A version of this article first appeared on Medscape.com.
But mammography trends can’t be ignored.
But mammography trends can’t be ignored.
A controversial, big-budget breast cancer screening trial that has been chronically unable to attract enough female participants since its debut in 2017 got a vote of confidence from a special working group of the National Cancer Institute (NCI) on March 17.
The Tomosynthesis Mammography Imaging Screening Trial (TMIST) should continue, but with modification, the expert group concluded in its report.
The randomized trial, with an estimated cost of $100 million, compares two kinds of mammography screenings for breast cancer in healthy women. One group of patients is screened with digital breast tomosynthesis, also known as 3-D mammography; the other is screened with the older, less expensive 2-D digital technology.
Tomosynthesis is already considered superior in detecting small cancers and reducing callbacks and is increasingly being used in the real world, leading some experts in the field to say that TMIST is critically hampered by women’s and radiologists’ preference for 3-D mammography.
At a meeting of an NCI advisory board in September 2020, there was a suggestion that the federal agency may kill the trial.
But at the latest meeting, the working group proposed that the trial should live on.
One of the main problems with the trial has been recruitment; the recommended changes discussed at the meeting include reducing the number of women needed in the study (from 165,000 to 102,000), which would allow patient “accrual to be completed more quickly,” the working group noted. In addition, the target date for completing patient accrual would be moved to 2023, 3 years after the original completion date of 2020.
The group’s recommendations now go to NCI staff for scientific review. The NCI will then decide about implementing the proposed changes.
The trial, which is the NCI’s largest and most expensive screening study, has never come close to targeted monthly enrollment. It was enrolling fewer than 1,500 patients per month over a 2-year period, instead of the projected 5,500 per month, Philip Castle, PhD, MPH, director of the NCI’s Division of Cancer Prevention, said last year. He called for a review of TMIST’s “feasibility and relevance” in view of the increasing use of tomosynthesis in the United States, as well as other factors.
The new technology has been “rapidly adopted” by facilities in North America, the working group noted. As of December 2020, approximately 74% of breast cancer screening clinics in the United States had at least one tomosynthesis or 3-D system; 42% of the mammography machines were 3-D.
This trend of increasing use of 3-D machines might be too much for TMIST to surmount, said Nancy Davidson, MD, of the Fred Hutchinson Cancer Research Center, in Seattle, who chaired the working group.
“We are worried the challenges [to patient accrual] may persist due to the increasing adoption of three-dimensional breast tomosynthesis in the United States over time,” she said during the working group’s virtual presentation of the report to the NCI’s Clinical Trials and Translational Research Advisory Committee.
Committee member Smita Bhatia, MD, PhD, of the University of Alabama at Birmingham, wondered, “What are the ongoing barriers that [TMIST investigators] are going to face beside recruitment?”
Dr. Davidson answered by speaking, again, about market penetration of tomosynthesis machines and suggested that the recruitment problems and the availability of 3-D mammography are intertwined.
“Is this a technology that has or will arrive, at which point it may not be very easy to put the genie back in the bottle?” she asked.
That question has already been answered – widespread use of tomosynthesis is here to stay, argued Daniel Kopans, MD, of Harvard Medical School, Boston, who invented digital breast tomosynthesis but no longer benefits financially from his invention because the patent has expired.
“The horse is out of the barn,” Dr. Kopans said in an interview. By the time the study results are available, digital mammography will be a tool of the past, he said.
TMIST is a trial “that should never have been started in the first place, and it’s failing,” he said. “I was hoping they [the NCI] would say, ‘Let’s stop this because there’s not enough accrual.’ But it looks like they’re not.”
“TMIST is a waste of money,” said Dr. Kopans, repeating a criticism he has made in the past.
A drop in study power
The new recommendations for TMIST come about 1 year after Medscape Medical News reported that the study was lagging in enrollment of both patients and participating sites/physicians.
Last year, two TMIST study investigators said it had been difficult enlisting sites, in part because many radiologists and facilities – informed by their experience and previous research results – already believe that the 3-D technology is superior.
Currently, most 3-D systems are used in conjunction with 2-D. First, two static images of the breast are taken (2-D), and then the unit moves in an arc, taking multiple images of the breast (3-D). Thus, 3-D is widely described as allowing clinicians to flip through the images like “pages in a book” and as offering a superior read of the breast.
The NCI working group concedes that “there is evidence that screening utilizing tomosynthesis may reduce recall rates and improve cancer detection,” but it says the trial is needed to address “questions that still remain regarding the overall benefit to patients.”
Furthermore, tomosynthesis “may carry higher out-of-pocket costs for women and is more labor intensive and costly for health care systems in that it requires about twice as much reader time for interpretation,” the working group said.
The “main hypothesis of TMIST” is that “tomosynthesis will decrease the cumulative incidence of advanced breast cancers, a surrogate for mortality, compared to standard digital mammography,” posits the group.
Advanced breast cancer is defined in TMIST as invasive breast cancers that meet any of the following criteria:
- Distant metastases
- At least one lymph node macrometastasis
- Tumor size >1 cm and triple-negative or positive for human epidermal growth factor receptor
- Tumor size ≥ 20 mm unless of pure mucinous or other favorable histologies.
In the original study design, the sample size was estimated to be sufficient to provide 90% power to detect a 20% relative reduction in the proportion of advanced cancers in the intervention arm (tomosynthesis, or 3-D) compared to the control arm (digital mammography, or 2-D) 4.5 years from randomization.
Now, with fewer patients and a revised analytic approach, the study’s statistical power will be decreased to 80% from the original 90%.
Also, an advanced cancer is counted “if it occurs at any time while the participant is on study.”
Dr. Kopans says that is a problem.
“That is a huge mistake, since digital breast tomosynthesis cannot impact prevalent cancers. They are already there. This means that their ‘power calculation’ is wrong, and they won’t have the power that they are claiming,” he said.
Dr. Kopans explained that the first screen in TMIST will have “no effect on the number of advanced cancers.” That’s because the cancers will have already grown enough to become advanced, he said.
On the other hand, an initial screening might detect and thus lead to the removal of nonadvanced, smaller cancers, which, had they not been detected and removed, would have grown to become advanced cancers by the next year. Thus, the screenings done after the first year are the ones that potentially prove the effect of the intervention.
However, the working group report says that changes to the study will not affect anything other than a 10% reduction in the study’s power.
The working group is concerned about TMIST going on for years and years. For that reason, they recommended that the NCI establish “strict criteria for termination of the study” if accrual goals are not met. However, those parameters have not been developed, and it was not part of the study group’s mission to establish them.
The working group was sponsored by the NCI. Dr. Kopans reports consulting with DART Imaging in China.
A version of this article first appeared on Medscape.com.
A controversial, big-budget breast cancer screening trial that has been chronically unable to attract enough female participants since its debut in 2017 got a vote of confidence from a special working group of the National Cancer Institute (NCI) on March 17.
The Tomosynthesis Mammography Imaging Screening Trial (TMIST) should continue, but with modification, the expert group concluded in its report.
The randomized trial, with an estimated cost of $100 million, compares two kinds of mammography screenings for breast cancer in healthy women. One group of patients is screened with digital breast tomosynthesis, also known as 3-D mammography; the other is screened with the older, less expensive 2-D digital technology.
Tomosynthesis is already considered superior in detecting small cancers and reducing callbacks and is increasingly being used in the real world, leading some experts in the field to say that TMIST is critically hampered by women’s and radiologists’ preference for 3-D mammography.
At a meeting of an NCI advisory board in September 2020, there was a suggestion that the federal agency may kill the trial.
But at the latest meeting, the working group proposed that the trial should live on.
One of the main problems with the trial has been recruitment; the recommended changes discussed at the meeting include reducing the number of women needed in the study (from 165,000 to 102,000), which would allow patient “accrual to be completed more quickly,” the working group noted. In addition, the target date for completing patient accrual would be moved to 2023, 3 years after the original completion date of 2020.
The group’s recommendations now go to NCI staff for scientific review. The NCI will then decide about implementing the proposed changes.
The trial, which is the NCI’s largest and most expensive screening study, has never come close to targeted monthly enrollment. It was enrolling fewer than 1,500 patients per month over a 2-year period, instead of the projected 5,500 per month, Philip Castle, PhD, MPH, director of the NCI’s Division of Cancer Prevention, said last year. He called for a review of TMIST’s “feasibility and relevance” in view of the increasing use of tomosynthesis in the United States, as well as other factors.
The new technology has been “rapidly adopted” by facilities in North America, the working group noted. As of December 2020, approximately 74% of breast cancer screening clinics in the United States had at least one tomosynthesis or 3-D system; 42% of the mammography machines were 3-D.
This trend of increasing use of 3-D machines might be too much for TMIST to surmount, said Nancy Davidson, MD, of the Fred Hutchinson Cancer Research Center, in Seattle, who chaired the working group.
“We are worried the challenges [to patient accrual] may persist due to the increasing adoption of three-dimensional breast tomosynthesis in the United States over time,” she said during the working group’s virtual presentation of the report to the NCI’s Clinical Trials and Translational Research Advisory Committee.
Committee member Smita Bhatia, MD, PhD, of the University of Alabama at Birmingham, wondered, “What are the ongoing barriers that [TMIST investigators] are going to face beside recruitment?”
Dr. Davidson answered by speaking, again, about market penetration of tomosynthesis machines and suggested that the recruitment problems and the availability of 3-D mammography are intertwined.
“Is this a technology that has or will arrive, at which point it may not be very easy to put the genie back in the bottle?” she asked.
That question has already been answered – widespread use of tomosynthesis is here to stay, argued Daniel Kopans, MD, of Harvard Medical School, Boston, who invented digital breast tomosynthesis but no longer benefits financially from his invention because the patent has expired.
“The horse is out of the barn,” Dr. Kopans said in an interview. By the time the study results are available, digital mammography will be a tool of the past, he said.
TMIST is a trial “that should never have been started in the first place, and it’s failing,” he said. “I was hoping they [the NCI] would say, ‘Let’s stop this because there’s not enough accrual.’ But it looks like they’re not.”
“TMIST is a waste of money,” said Dr. Kopans, repeating a criticism he has made in the past.
A drop in study power
The new recommendations for TMIST come about 1 year after Medscape Medical News reported that the study was lagging in enrollment of both patients and participating sites/physicians.
Last year, two TMIST study investigators said it had been difficult enlisting sites, in part because many radiologists and facilities – informed by their experience and previous research results – already believe that the 3-D technology is superior.
Currently, most 3-D systems are used in conjunction with 2-D. First, two static images of the breast are taken (2-D), and then the unit moves in an arc, taking multiple images of the breast (3-D). Thus, 3-D is widely described as allowing clinicians to flip through the images like “pages in a book” and as offering a superior read of the breast.
The NCI working group concedes that “there is evidence that screening utilizing tomosynthesis may reduce recall rates and improve cancer detection,” but it says the trial is needed to address “questions that still remain regarding the overall benefit to patients.”
Furthermore, tomosynthesis “may carry higher out-of-pocket costs for women and is more labor intensive and costly for health care systems in that it requires about twice as much reader time for interpretation,” the working group said.
The “main hypothesis of TMIST” is that “tomosynthesis will decrease the cumulative incidence of advanced breast cancers, a surrogate for mortality, compared to standard digital mammography,” posits the group.
Advanced breast cancer is defined in TMIST as invasive breast cancers that meet any of the following criteria:
- Distant metastases
- At least one lymph node macrometastasis
- Tumor size >1 cm and triple-negative or positive for human epidermal growth factor receptor
- Tumor size ≥ 20 mm unless of pure mucinous or other favorable histologies.
In the original study design, the sample size was estimated to be sufficient to provide 90% power to detect a 20% relative reduction in the proportion of advanced cancers in the intervention arm (tomosynthesis, or 3-D) compared to the control arm (digital mammography, or 2-D) 4.5 years from randomization.
Now, with fewer patients and a revised analytic approach, the study’s statistical power will be decreased to 80% from the original 90%.
Also, an advanced cancer is counted “if it occurs at any time while the participant is on study.”
Dr. Kopans says that is a problem.
“That is a huge mistake, since digital breast tomosynthesis cannot impact prevalent cancers. They are already there. This means that their ‘power calculation’ is wrong, and they won’t have the power that they are claiming,” he said.
Dr. Kopans explained that the first screen in TMIST will have “no effect on the number of advanced cancers.” That’s because the cancers will have already grown enough to become advanced, he said.
On the other hand, an initial screening might detect and thus lead to the removal of nonadvanced, smaller cancers, which, had they not been detected and removed, would have grown to become advanced cancers by the next year. Thus, the screenings done after the first year are the ones that potentially prove the effect of the intervention.
However, the working group report says that changes to the study will not affect anything other than a 10% reduction in the study’s power.
The working group is concerned about TMIST going on for years and years. For that reason, they recommended that the NCI establish “strict criteria for termination of the study” if accrual goals are not met. However, those parameters have not been developed, and it was not part of the study group’s mission to establish them.
The working group was sponsored by the NCI. Dr. Kopans reports consulting with DART Imaging in China.
A version of this article first appeared on Medscape.com.
Could tamoxifen dose be slashed down to 2.5 mg?
Tamoxifen has long been used in breast cancer, both in the adjuvant and preventive setting, but uptake and adherence are notoriously low, mainly because of adverse events.
Using a much lower dose to reduce the incidence of side effects would be a “way forward,” reasoned Swedish researchers. They report that a substantially lower dose of tamoxifen (2.5 mg) may be as effective as the standard dose (20 mg), but reduced by half the incidence of severe vasomotor symptoms, including hot flashes, cold sweats, and night sweats.
The research was published online March 18 in the Journal of Clinical Oncology.
The study involved 1,439 women (aged 40-74 years) who were participating in the Swedish mammography screening program and tested tamoxifen at various doses.
“We performed a dose determination study that we hope will initiate follow-up studies that in turn will influence both adjuvant treatment and prevention of breast cancer,” said lead author Per Hall, MD, PhD, head of the department of medical epidemiology and biostatistics at Karolinska Institutet in Stockholm.
The study measured the effects of the different doses (1, 2.5, 5, 10, and 20 mg) on mammographic breast density.
Dr. Hall emphasized that breast density was used as a proxy for therapy response. “We do not know how that translates to actual clinical effect,” he said in an interview. “This is step one.”
Previous studies have also used breast density changes as a proxy endpoint for tamoxifen therapy response, in both prophylactic and adjuvant settings, the authors note. There is some data to suggest that this does translate to a clinical effect. A recent study showed that tamoxifen at 5 mg/day taken for 3 years reduced the recurrence of breast intraepithelial neoplasia by 50% and contralateral breast cancer by 75%, with a symptom profile similar to placebo (J Clin Oncol. 2019;37:1629-1637).
Lower density, fewer symptoms
In the current study, Dr. Hall and colleagues found that the mammographic breast density (mean overall area) was decreased by 9.6% in the 20 mg tamoxifen group, and similar decreases were seen in the 2.5 and 10 mg dose groups, but not in the placebo and 1 mg dose groups.
These changes were driven primarily by changes observed among premenopausal women where the 20 mg mean decrease was 18.5% (P < .001 for interaction with menopausal status) with decreases of 13.4% in the 2.5 mg group, 19.6% in the 5 mg group, and 17% in the 10 mg group.
The results were quite different in postmenopausal participants, where those who received the 20 mg dose had a density mean decrease of 4%, which was not substantially different to the placebo, 1 mg, 2.5 mg, and 10 mg treatment arms.
The authors point out that the difference in density decrease between premenopausal and postmenopausal women was not dependent on differences in baseline density.
When reviewing adverse events with the various doses, the team found a large decrease in severe vasomotor symptoms with the lower doses of tamoxifen. These adverse events were reported by 34% of women taking 20 mg, 24.4% on 5 mg, 20.5% on 2.5 mg, 18.5% on 1 mg, and 13.7% of women taking placebo. There were no similar trends seen for gynecologic, sexual, or musculoskeletal symptoms.
Future studies should test whether 2.5 mg of tamoxifen reduces the risk of primary breast cancer, Dr. Hall commented.
“We are planning a trial now where women are offered risk assessment when attending mammography screening,” Dr. Hall said. “For those at very high risk, low-dose tamoxifen will be offered.”
The study received support from the Kamprad Foundation, Swedish Research Council, Marit and Hans Rausing’s Initiative Against Breast Cancer, Swedish Cancer Society, and Stockholm County Council.
Dr. Hall reports several relationships with industry, had a pending patent on compositions and methods for prevention of breast cancer with an option to license to Atossa Therapeutics, and has licensed an algorithm for risk prediction based on analyses of mammographic features to iCAD Travel. Several co-authors have also declared relationships with industry.
A version of this article first appeared on Medscape.com.
Tamoxifen has long been used in breast cancer, both in the adjuvant and preventive setting, but uptake and adherence are notoriously low, mainly because of adverse events.
Using a much lower dose to reduce the incidence of side effects would be a “way forward,” reasoned Swedish researchers. They report that a substantially lower dose of tamoxifen (2.5 mg) may be as effective as the standard dose (20 mg), but reduced by half the incidence of severe vasomotor symptoms, including hot flashes, cold sweats, and night sweats.
The research was published online March 18 in the Journal of Clinical Oncology.
The study involved 1,439 women (aged 40-74 years) who were participating in the Swedish mammography screening program and tested tamoxifen at various doses.
“We performed a dose determination study that we hope will initiate follow-up studies that in turn will influence both adjuvant treatment and prevention of breast cancer,” said lead author Per Hall, MD, PhD, head of the department of medical epidemiology and biostatistics at Karolinska Institutet in Stockholm.
The study measured the effects of the different doses (1, 2.5, 5, 10, and 20 mg) on mammographic breast density.
Dr. Hall emphasized that breast density was used as a proxy for therapy response. “We do not know how that translates to actual clinical effect,” he said in an interview. “This is step one.”
Previous studies have also used breast density changes as a proxy endpoint for tamoxifen therapy response, in both prophylactic and adjuvant settings, the authors note. There is some data to suggest that this does translate to a clinical effect. A recent study showed that tamoxifen at 5 mg/day taken for 3 years reduced the recurrence of breast intraepithelial neoplasia by 50% and contralateral breast cancer by 75%, with a symptom profile similar to placebo (J Clin Oncol. 2019;37:1629-1637).
Lower density, fewer symptoms
In the current study, Dr. Hall and colleagues found that the mammographic breast density (mean overall area) was decreased by 9.6% in the 20 mg tamoxifen group, and similar decreases were seen in the 2.5 and 10 mg dose groups, but not in the placebo and 1 mg dose groups.
These changes were driven primarily by changes observed among premenopausal women where the 20 mg mean decrease was 18.5% (P < .001 for interaction with menopausal status) with decreases of 13.4% in the 2.5 mg group, 19.6% in the 5 mg group, and 17% in the 10 mg group.
The results were quite different in postmenopausal participants, where those who received the 20 mg dose had a density mean decrease of 4%, which was not substantially different to the placebo, 1 mg, 2.5 mg, and 10 mg treatment arms.
The authors point out that the difference in density decrease between premenopausal and postmenopausal women was not dependent on differences in baseline density.
When reviewing adverse events with the various doses, the team found a large decrease in severe vasomotor symptoms with the lower doses of tamoxifen. These adverse events were reported by 34% of women taking 20 mg, 24.4% on 5 mg, 20.5% on 2.5 mg, 18.5% on 1 mg, and 13.7% of women taking placebo. There were no similar trends seen for gynecologic, sexual, or musculoskeletal symptoms.
Future studies should test whether 2.5 mg of tamoxifen reduces the risk of primary breast cancer, Dr. Hall commented.
“We are planning a trial now where women are offered risk assessment when attending mammography screening,” Dr. Hall said. “For those at very high risk, low-dose tamoxifen will be offered.”
The study received support from the Kamprad Foundation, Swedish Research Council, Marit and Hans Rausing’s Initiative Against Breast Cancer, Swedish Cancer Society, and Stockholm County Council.
Dr. Hall reports several relationships with industry, had a pending patent on compositions and methods for prevention of breast cancer with an option to license to Atossa Therapeutics, and has licensed an algorithm for risk prediction based on analyses of mammographic features to iCAD Travel. Several co-authors have also declared relationships with industry.
A version of this article first appeared on Medscape.com.
Tamoxifen has long been used in breast cancer, both in the adjuvant and preventive setting, but uptake and adherence are notoriously low, mainly because of adverse events.
Using a much lower dose to reduce the incidence of side effects would be a “way forward,” reasoned Swedish researchers. They report that a substantially lower dose of tamoxifen (2.5 mg) may be as effective as the standard dose (20 mg), but reduced by half the incidence of severe vasomotor symptoms, including hot flashes, cold sweats, and night sweats.
The research was published online March 18 in the Journal of Clinical Oncology.
The study involved 1,439 women (aged 40-74 years) who were participating in the Swedish mammography screening program and tested tamoxifen at various doses.
“We performed a dose determination study that we hope will initiate follow-up studies that in turn will influence both adjuvant treatment and prevention of breast cancer,” said lead author Per Hall, MD, PhD, head of the department of medical epidemiology and biostatistics at Karolinska Institutet in Stockholm.
The study measured the effects of the different doses (1, 2.5, 5, 10, and 20 mg) on mammographic breast density.
Dr. Hall emphasized that breast density was used as a proxy for therapy response. “We do not know how that translates to actual clinical effect,” he said in an interview. “This is step one.”
Previous studies have also used breast density changes as a proxy endpoint for tamoxifen therapy response, in both prophylactic and adjuvant settings, the authors note. There is some data to suggest that this does translate to a clinical effect. A recent study showed that tamoxifen at 5 mg/day taken for 3 years reduced the recurrence of breast intraepithelial neoplasia by 50% and contralateral breast cancer by 75%, with a symptom profile similar to placebo (J Clin Oncol. 2019;37:1629-1637).
Lower density, fewer symptoms
In the current study, Dr. Hall and colleagues found that the mammographic breast density (mean overall area) was decreased by 9.6% in the 20 mg tamoxifen group, and similar decreases were seen in the 2.5 and 10 mg dose groups, but not in the placebo and 1 mg dose groups.
These changes were driven primarily by changes observed among premenopausal women where the 20 mg mean decrease was 18.5% (P < .001 for interaction with menopausal status) with decreases of 13.4% in the 2.5 mg group, 19.6% in the 5 mg group, and 17% in the 10 mg group.
The results were quite different in postmenopausal participants, where those who received the 20 mg dose had a density mean decrease of 4%, which was not substantially different to the placebo, 1 mg, 2.5 mg, and 10 mg treatment arms.
The authors point out that the difference in density decrease between premenopausal and postmenopausal women was not dependent on differences in baseline density.
When reviewing adverse events with the various doses, the team found a large decrease in severe vasomotor symptoms with the lower doses of tamoxifen. These adverse events were reported by 34% of women taking 20 mg, 24.4% on 5 mg, 20.5% on 2.5 mg, 18.5% on 1 mg, and 13.7% of women taking placebo. There were no similar trends seen for gynecologic, sexual, or musculoskeletal symptoms.
Future studies should test whether 2.5 mg of tamoxifen reduces the risk of primary breast cancer, Dr. Hall commented.
“We are planning a trial now where women are offered risk assessment when attending mammography screening,” Dr. Hall said. “For those at very high risk, low-dose tamoxifen will be offered.”
The study received support from the Kamprad Foundation, Swedish Research Council, Marit and Hans Rausing’s Initiative Against Breast Cancer, Swedish Cancer Society, and Stockholm County Council.
Dr. Hall reports several relationships with industry, had a pending patent on compositions and methods for prevention of breast cancer with an option to license to Atossa Therapeutics, and has licensed an algorithm for risk prediction based on analyses of mammographic features to iCAD Travel. Several co-authors have also declared relationships with industry.
A version of this article first appeared on Medscape.com.
Most breast cancer screening centers not following guidelines
, say researchers reporting on a new analysis.
They assessed 606 centers and report that, among the centers that recommended a starting age for screening mammography, nearly 90% advised women to begin screening at age 40 years and to continue annually.
This contrasts with the current recommendations from the U.S. Preventive Services Task Force (USPSTF) on mammography screening, which stipulate starting at age 50 years and continuing every 2 years.
The new analysis was published online in JAMA Internal Medicine.
This may be doing “more harm than good,” warn the authors of an accompanying editorial.
“The recommendation for annual mammography in women younger than 50 years is, at best, confusing for patients and is likely to conflict with advice from their primary care physicians, which can create tension,” write Anand R. Habib, MD, MPhil; Deborah Grady, MD; and Rita F. Redberg, MD, all from the University of California, San Francisco.
“This recommendation can also produce unnecessary testing, invasive procedures, overdiagnosis, and anxiety among women who receive screening,” they write.
“Breast cancer centers with clear financial benefits from increased mammography rates may wish to reconsider offering recommendations that create greater referral volume but conflict with unbiased evidence-based USPSTF guidelines and have the potential to increase harms among women,” the editorialists add.
The age at which to start mammography screening has long been a contentious issue, with some experts and medical societies arguing that it should begin at 40.
The American College of Radiology, the Society of Breast Imaging, and the American Society of Breast Surgeons recommend that women start annual mammography screening at age 40.
The American Cancer Society’s guidelines recommend an initial screening mammogram between ages 45 and 55 and after that, screening every 1-2 years.
One expert who argues for starting at 40 years is Laurie Margolies, MD, chief of breast imaging, Mount Sinai Health System, and professor of radiology, Icahn School of Medicine at Mount Sinai, New York.
In a statement, she noted that 17% of all breast cancers are diagnosed in women in their 40s and that the majority of these women are not considered to be at high risk of developing the disease.
“Our own Mount Sinai research has shown that women with screen-detected breast cancers are less likely to need a mastectomy and are less likely to require chemotherapy or axillary node dissection,” Dr. Margolies said.
“Additionally, women who get regular breast cancer screening have a 47% lower risk of breast cancer death within 20 years of diagnosis than those not regularly screened. Skipping a mammogram can have lethal consequences,” she said.
Details of the analysis
The analysis of recommendations by breast cancer centers regarding screening mammography was carried out by Jennifer L. Marti, MD, from Weill Cornell Medicine, New York, and colleagues.
They examined 606 centers and found that 487 (80.4%) offered screening recommendations on their public websites.
Of 431 centers that recommended a starting age, 376 centers (87.2%) advised women to begin screening at age 40 years; 35 centers (8.1%) recommended beginning at age 45 years; and the remaining 20 centers (4.6%) recommended that screening begin at age 50 years.
A total of 429 centers recommended both a starting age and a screening interval. Of this group, 347 centers (80.9%) advised that annual screening begin at age 40 years. Only 16 centers (3.3%) recommended biennial mammography (as per the USPSTF guidelines). Almost three-quarters (72.7%, n = 354) recommended annual screening; 59 centers (12.1%) recommended annual or biennial screening; and 58 centers (11.9%) recommended a discussion with a physician.
The authors note that there were differences between centers according to National Cancer Institute designation, but these differences did not reach statistical significance.
Dr. Marti and coauthors, Dr. Habib and coauthors, and Dr. Margolies have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, say researchers reporting on a new analysis.
They assessed 606 centers and report that, among the centers that recommended a starting age for screening mammography, nearly 90% advised women to begin screening at age 40 years and to continue annually.
This contrasts with the current recommendations from the U.S. Preventive Services Task Force (USPSTF) on mammography screening, which stipulate starting at age 50 years and continuing every 2 years.
The new analysis was published online in JAMA Internal Medicine.
This may be doing “more harm than good,” warn the authors of an accompanying editorial.
“The recommendation for annual mammography in women younger than 50 years is, at best, confusing for patients and is likely to conflict with advice from their primary care physicians, which can create tension,” write Anand R. Habib, MD, MPhil; Deborah Grady, MD; and Rita F. Redberg, MD, all from the University of California, San Francisco.
“This recommendation can also produce unnecessary testing, invasive procedures, overdiagnosis, and anxiety among women who receive screening,” they write.
“Breast cancer centers with clear financial benefits from increased mammography rates may wish to reconsider offering recommendations that create greater referral volume but conflict with unbiased evidence-based USPSTF guidelines and have the potential to increase harms among women,” the editorialists add.
The age at which to start mammography screening has long been a contentious issue, with some experts and medical societies arguing that it should begin at 40.
The American College of Radiology, the Society of Breast Imaging, and the American Society of Breast Surgeons recommend that women start annual mammography screening at age 40.
The American Cancer Society’s guidelines recommend an initial screening mammogram between ages 45 and 55 and after that, screening every 1-2 years.
One expert who argues for starting at 40 years is Laurie Margolies, MD, chief of breast imaging, Mount Sinai Health System, and professor of radiology, Icahn School of Medicine at Mount Sinai, New York.
In a statement, she noted that 17% of all breast cancers are diagnosed in women in their 40s and that the majority of these women are not considered to be at high risk of developing the disease.
“Our own Mount Sinai research has shown that women with screen-detected breast cancers are less likely to need a mastectomy and are less likely to require chemotherapy or axillary node dissection,” Dr. Margolies said.
“Additionally, women who get regular breast cancer screening have a 47% lower risk of breast cancer death within 20 years of diagnosis than those not regularly screened. Skipping a mammogram can have lethal consequences,” she said.
Details of the analysis
The analysis of recommendations by breast cancer centers regarding screening mammography was carried out by Jennifer L. Marti, MD, from Weill Cornell Medicine, New York, and colleagues.
They examined 606 centers and found that 487 (80.4%) offered screening recommendations on their public websites.
Of 431 centers that recommended a starting age, 376 centers (87.2%) advised women to begin screening at age 40 years; 35 centers (8.1%) recommended beginning at age 45 years; and the remaining 20 centers (4.6%) recommended that screening begin at age 50 years.
A total of 429 centers recommended both a starting age and a screening interval. Of this group, 347 centers (80.9%) advised that annual screening begin at age 40 years. Only 16 centers (3.3%) recommended biennial mammography (as per the USPSTF guidelines). Almost three-quarters (72.7%, n = 354) recommended annual screening; 59 centers (12.1%) recommended annual or biennial screening; and 58 centers (11.9%) recommended a discussion with a physician.
The authors note that there were differences between centers according to National Cancer Institute designation, but these differences did not reach statistical significance.
Dr. Marti and coauthors, Dr. Habib and coauthors, and Dr. Margolies have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, say researchers reporting on a new analysis.
They assessed 606 centers and report that, among the centers that recommended a starting age for screening mammography, nearly 90% advised women to begin screening at age 40 years and to continue annually.
This contrasts with the current recommendations from the U.S. Preventive Services Task Force (USPSTF) on mammography screening, which stipulate starting at age 50 years and continuing every 2 years.
The new analysis was published online in JAMA Internal Medicine.
This may be doing “more harm than good,” warn the authors of an accompanying editorial.
“The recommendation for annual mammography in women younger than 50 years is, at best, confusing for patients and is likely to conflict with advice from their primary care physicians, which can create tension,” write Anand R. Habib, MD, MPhil; Deborah Grady, MD; and Rita F. Redberg, MD, all from the University of California, San Francisco.
“This recommendation can also produce unnecessary testing, invasive procedures, overdiagnosis, and anxiety among women who receive screening,” they write.
“Breast cancer centers with clear financial benefits from increased mammography rates may wish to reconsider offering recommendations that create greater referral volume but conflict with unbiased evidence-based USPSTF guidelines and have the potential to increase harms among women,” the editorialists add.
The age at which to start mammography screening has long been a contentious issue, with some experts and medical societies arguing that it should begin at 40.
The American College of Radiology, the Society of Breast Imaging, and the American Society of Breast Surgeons recommend that women start annual mammography screening at age 40.
The American Cancer Society’s guidelines recommend an initial screening mammogram between ages 45 and 55 and after that, screening every 1-2 years.
One expert who argues for starting at 40 years is Laurie Margolies, MD, chief of breast imaging, Mount Sinai Health System, and professor of radiology, Icahn School of Medicine at Mount Sinai, New York.
In a statement, she noted that 17% of all breast cancers are diagnosed in women in their 40s and that the majority of these women are not considered to be at high risk of developing the disease.
“Our own Mount Sinai research has shown that women with screen-detected breast cancers are less likely to need a mastectomy and are less likely to require chemotherapy or axillary node dissection,” Dr. Margolies said.
“Additionally, women who get regular breast cancer screening have a 47% lower risk of breast cancer death within 20 years of diagnosis than those not regularly screened. Skipping a mammogram can have lethal consequences,” she said.
Details of the analysis
The analysis of recommendations by breast cancer centers regarding screening mammography was carried out by Jennifer L. Marti, MD, from Weill Cornell Medicine, New York, and colleagues.
They examined 606 centers and found that 487 (80.4%) offered screening recommendations on their public websites.
Of 431 centers that recommended a starting age, 376 centers (87.2%) advised women to begin screening at age 40 years; 35 centers (8.1%) recommended beginning at age 45 years; and the remaining 20 centers (4.6%) recommended that screening begin at age 50 years.
A total of 429 centers recommended both a starting age and a screening interval. Of this group, 347 centers (80.9%) advised that annual screening begin at age 40 years. Only 16 centers (3.3%) recommended biennial mammography (as per the USPSTF guidelines). Almost three-quarters (72.7%, n = 354) recommended annual screening; 59 centers (12.1%) recommended annual or biennial screening; and 58 centers (11.9%) recommended a discussion with a physician.
The authors note that there were differences between centers according to National Cancer Institute designation, but these differences did not reach statistical significance.
Dr. Marti and coauthors, Dr. Habib and coauthors, and Dr. Margolies have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FDA scrutinizes cancer therapies granted accelerated approval
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
SNP chips deemed ‘extremely unreliable’ for identifying rare variants
In fact, SNP chips are “extremely unreliable for genotyping very rare pathogenic variants,” and a positive result for such a variant “is more likely to be wrong than right,” researchers reported in the BMJ.
The authors explained that SNP chips are “DNA microarrays that test genetic variation at many hundreds of thousands of specific locations across the genome.” Although SNP chips have proven accurate in identifying common variants, past reports have suggested that SNP chips perform poorly for genotyping rare variants.
To gain more insight, Caroline Wright, PhD, of the University of Exeter (England) and colleagues conducted a large study.
The researchers analyzed data on 49,908 people from the UK Biobank who had SNP chip and next-generation sequencing results, as well as an additional 21 people who purchased consumer genetic tests and shared their data online via the Personal Genome Project.
The researchers compared the SNP chip and sequencing results. They also selected rare pathogenic variants in BRCA1 and BRCA2 for detailed analysis of clinically actionable variants in the UK Biobank, and they assessed BRCA-related cancers in participants using cancer registry data.
Largest evaluation of SNP chips
SNP chips performed well for common variants, the researchers found. Sensitivity, specificity, positive-predictive value, and negative-predictive value all exceeded 99% for 108,574 common variants.
For rare variants, SNP chips performed poorly, with a positive-predictive value of 16% for variants with a frequency below 0.001% in the UK Biobank.
“The study provides the largest evaluation of the performance of SNP chips for genotyping genetic variants at different frequencies in the population, particularly focusing on very rare variants,” Dr. Wright said. “The biggest surprise was how poorly the SNP chips we evaluated performed for rare variants.”
Dr. Wright noted that there is an inherent problem built into using SNP chip technology to genotype very rare variants.
“The SNP chip technology relies on clustering data from multiple individuals in order to determine what genotype each individual has at a specific position in their genome,” Dr. Wright explained. “Although this method works very well for common variants, the rarer the variant, the harder it is to distinguish from experimental noise.”
False positives and cancer: ‘Don’t trust the results’
The researchers found that, for rare BRCA variants (frequency below 0.01%), SNP chips had a sensitivity of 34.6%, specificity of 98.3%, negative-predictive value of 99.9%, and positive-predictive value of 4.2%.
Rates of BRCA-related cancers in patients with positive SNP chip results were similar to rates in age-matched control subjects because “the vast majority of variants were false positives,” the researchers noted.
“If these variants are incorrectly genotyped – that is, false positives detected – a woman could be offered screening or even prophylactic surgery inappropriately when she is more likely to be at population background risk [for BRCA-related cancers],” Dr. Wright said.
“For very-rare-disease–causing genetic variants, don’t trust the results from SNP chips; for example, those from direct-to-consumer genetic tests. Never use them to guide clinical action without diagnostic validation,” she added.
Heather Hampel, a genetic counselor and researcher at the Ohio State University Comprehensive Cancer Center in Columbus, agreed.
“Positive results on SNP-based tests need to be confirmed by medical-grade genetic testing using a sequencing technology,” she said. “Negative results on an SNP- based test cannot be considered to rule out mutations in BRCA1/2 or other cancer-susceptibility genes, so individuals with strong personal and family histories of cancer should be seen by a genetic counselor to consider medical-grade genetic testing using a sequencing technology.”
Practicing oncologists can trust patients’ prior germline genetic test results if the testing was performed in a cancer genetics clinic, which uses sequencing-based technologies, Ms. Hampel noted.
“If the test was performed before 2013, there are likely new genes that have been discovered for which their patient was not tested, and repeat testing may be warranted,” Ms. Hampel said. “A referral to a cancer genetic counselor would be appropriate.”
Ms. Hampel disclosed relationships with Genome Medical, GI OnDemand, Invitae Genetics, and Promega. Dr. Wright and her coauthors disclosed no conflicts of interest. The group’s research was conducted using the UK Biobank and the University of Exeter High-Performance Computing, with funding from the Wellcome Trust and the National Institute for Health Research.
In fact, SNP chips are “extremely unreliable for genotyping very rare pathogenic variants,” and a positive result for such a variant “is more likely to be wrong than right,” researchers reported in the BMJ.
The authors explained that SNP chips are “DNA microarrays that test genetic variation at many hundreds of thousands of specific locations across the genome.” Although SNP chips have proven accurate in identifying common variants, past reports have suggested that SNP chips perform poorly for genotyping rare variants.
To gain more insight, Caroline Wright, PhD, of the University of Exeter (England) and colleagues conducted a large study.
The researchers analyzed data on 49,908 people from the UK Biobank who had SNP chip and next-generation sequencing results, as well as an additional 21 people who purchased consumer genetic tests and shared their data online via the Personal Genome Project.
The researchers compared the SNP chip and sequencing results. They also selected rare pathogenic variants in BRCA1 and BRCA2 for detailed analysis of clinically actionable variants in the UK Biobank, and they assessed BRCA-related cancers in participants using cancer registry data.
Largest evaluation of SNP chips
SNP chips performed well for common variants, the researchers found. Sensitivity, specificity, positive-predictive value, and negative-predictive value all exceeded 99% for 108,574 common variants.
For rare variants, SNP chips performed poorly, with a positive-predictive value of 16% for variants with a frequency below 0.001% in the UK Biobank.
“The study provides the largest evaluation of the performance of SNP chips for genotyping genetic variants at different frequencies in the population, particularly focusing on very rare variants,” Dr. Wright said. “The biggest surprise was how poorly the SNP chips we evaluated performed for rare variants.”
Dr. Wright noted that there is an inherent problem built into using SNP chip technology to genotype very rare variants.
“The SNP chip technology relies on clustering data from multiple individuals in order to determine what genotype each individual has at a specific position in their genome,” Dr. Wright explained. “Although this method works very well for common variants, the rarer the variant, the harder it is to distinguish from experimental noise.”
False positives and cancer: ‘Don’t trust the results’
The researchers found that, for rare BRCA variants (frequency below 0.01%), SNP chips had a sensitivity of 34.6%, specificity of 98.3%, negative-predictive value of 99.9%, and positive-predictive value of 4.2%.
Rates of BRCA-related cancers in patients with positive SNP chip results were similar to rates in age-matched control subjects because “the vast majority of variants were false positives,” the researchers noted.
“If these variants are incorrectly genotyped – that is, false positives detected – a woman could be offered screening or even prophylactic surgery inappropriately when she is more likely to be at population background risk [for BRCA-related cancers],” Dr. Wright said.
“For very-rare-disease–causing genetic variants, don’t trust the results from SNP chips; for example, those from direct-to-consumer genetic tests. Never use them to guide clinical action without diagnostic validation,” she added.
Heather Hampel, a genetic counselor and researcher at the Ohio State University Comprehensive Cancer Center in Columbus, agreed.
“Positive results on SNP-based tests need to be confirmed by medical-grade genetic testing using a sequencing technology,” she said. “Negative results on an SNP- based test cannot be considered to rule out mutations in BRCA1/2 or other cancer-susceptibility genes, so individuals with strong personal and family histories of cancer should be seen by a genetic counselor to consider medical-grade genetic testing using a sequencing technology.”
Practicing oncologists can trust patients’ prior germline genetic test results if the testing was performed in a cancer genetics clinic, which uses sequencing-based technologies, Ms. Hampel noted.
“If the test was performed before 2013, there are likely new genes that have been discovered for which their patient was not tested, and repeat testing may be warranted,” Ms. Hampel said. “A referral to a cancer genetic counselor would be appropriate.”
Ms. Hampel disclosed relationships with Genome Medical, GI OnDemand, Invitae Genetics, and Promega. Dr. Wright and her coauthors disclosed no conflicts of interest. The group’s research was conducted using the UK Biobank and the University of Exeter High-Performance Computing, with funding from the Wellcome Trust and the National Institute for Health Research.
In fact, SNP chips are “extremely unreliable for genotyping very rare pathogenic variants,” and a positive result for such a variant “is more likely to be wrong than right,” researchers reported in the BMJ.
The authors explained that SNP chips are “DNA microarrays that test genetic variation at many hundreds of thousands of specific locations across the genome.” Although SNP chips have proven accurate in identifying common variants, past reports have suggested that SNP chips perform poorly for genotyping rare variants.
To gain more insight, Caroline Wright, PhD, of the University of Exeter (England) and colleagues conducted a large study.
The researchers analyzed data on 49,908 people from the UK Biobank who had SNP chip and next-generation sequencing results, as well as an additional 21 people who purchased consumer genetic tests and shared their data online via the Personal Genome Project.
The researchers compared the SNP chip and sequencing results. They also selected rare pathogenic variants in BRCA1 and BRCA2 for detailed analysis of clinically actionable variants in the UK Biobank, and they assessed BRCA-related cancers in participants using cancer registry data.
Largest evaluation of SNP chips
SNP chips performed well for common variants, the researchers found. Sensitivity, specificity, positive-predictive value, and negative-predictive value all exceeded 99% for 108,574 common variants.
For rare variants, SNP chips performed poorly, with a positive-predictive value of 16% for variants with a frequency below 0.001% in the UK Biobank.
“The study provides the largest evaluation of the performance of SNP chips for genotyping genetic variants at different frequencies in the population, particularly focusing on very rare variants,” Dr. Wright said. “The biggest surprise was how poorly the SNP chips we evaluated performed for rare variants.”
Dr. Wright noted that there is an inherent problem built into using SNP chip technology to genotype very rare variants.
“The SNP chip technology relies on clustering data from multiple individuals in order to determine what genotype each individual has at a specific position in their genome,” Dr. Wright explained. “Although this method works very well for common variants, the rarer the variant, the harder it is to distinguish from experimental noise.”
False positives and cancer: ‘Don’t trust the results’
The researchers found that, for rare BRCA variants (frequency below 0.01%), SNP chips had a sensitivity of 34.6%, specificity of 98.3%, negative-predictive value of 99.9%, and positive-predictive value of 4.2%.
Rates of BRCA-related cancers in patients with positive SNP chip results were similar to rates in age-matched control subjects because “the vast majority of variants were false positives,” the researchers noted.
“If these variants are incorrectly genotyped – that is, false positives detected – a woman could be offered screening or even prophylactic surgery inappropriately when she is more likely to be at population background risk [for BRCA-related cancers],” Dr. Wright said.
“For very-rare-disease–causing genetic variants, don’t trust the results from SNP chips; for example, those from direct-to-consumer genetic tests. Never use them to guide clinical action without diagnostic validation,” she added.
Heather Hampel, a genetic counselor and researcher at the Ohio State University Comprehensive Cancer Center in Columbus, agreed.
“Positive results on SNP-based tests need to be confirmed by medical-grade genetic testing using a sequencing technology,” she said. “Negative results on an SNP- based test cannot be considered to rule out mutations in BRCA1/2 or other cancer-susceptibility genes, so individuals with strong personal and family histories of cancer should be seen by a genetic counselor to consider medical-grade genetic testing using a sequencing technology.”
Practicing oncologists can trust patients’ prior germline genetic test results if the testing was performed in a cancer genetics clinic, which uses sequencing-based technologies, Ms. Hampel noted.
“If the test was performed before 2013, there are likely new genes that have been discovered for which their patient was not tested, and repeat testing may be warranted,” Ms. Hampel said. “A referral to a cancer genetic counselor would be appropriate.”
Ms. Hampel disclosed relationships with Genome Medical, GI OnDemand, Invitae Genetics, and Promega. Dr. Wright and her coauthors disclosed no conflicts of interest. The group’s research was conducted using the UK Biobank and the University of Exeter High-Performance Computing, with funding from the Wellcome Trust and the National Institute for Health Research.
FROM BMJ
Neurologic drug prices jump 50% in five years
, new research shows. Results of the retrospective study also showed that most of the increased costs for these agents were due to rising costs for neuroimmunology drugs, mainly for those used to treat multiple sclerosis (MS).

“The same brand name medication in 2017 cost approximately 50% more than in 2013,” said Adam de Havenon, MD, assistant professor of neurology, University of Utah, Salt Lake City.
“An analogy would be if you bought an iPhone 5 in 2013 for $500, and then in 2017, you were asked to pay $750 for the exact same iPhone 5,” Dr. de Havenon added.
The study findings were published online March 10 in the journal Neurology.
$26 billion in payments
Both neurologists and patients are concerned about the high cost of prescription drugs for neurologic diseases, and Medicare Part D data indicate that these drugs are the most expensive component of neurologic care, the researchers noted. In addition, out-of-pocket costs have increased significantly for patients with neurologic disease such as Parkinson’s disease, epilepsy, and MS.
To understand trends in payments for neurologic drugs, Dr. de Havenon and colleagues analyzed Medicare Part D claims filed from 2013 to 2017. The payments include costs paid by Medicare, the patient, government subsidies, and other third-party payers.
In addition to examining more current Medicare Part D data than previous studies, the current analysis examined all medications prescribed by neurologists that consistently remained branded or generic during the 5-year study period, said Dr. de Havenon. This approach resulted in a large number of claims and a large total cost.
To calculate the percentage change in annual payment claims, the researchers used 2013 prices as a reference point. They identified drugs named in 2013 claims and classified them as generic, brand-name only, or brand-name with generic equivalent. Researchers also divided the drugs by neurologic subspecialty.
The analysis included 520 drugs, all of which were available in each year of the study period. Of these drugs, 322 were generic, 61 were brand-name only, and 137 were brand-name with a generic equivalent. There were 90.7 million total claims.
Results showed total payments amounted to $26.65 billion. Yearly total payments increased from $4.05 billion in 2013 to $6.09 billion in 2017, representing a 50.4% increase, even after adjusting for inflation. Total claims increased by 7.6% – from 17.1 million in 2013 to 18.4 million in 2017.
From 2013 to 2017, claim payments increased by 0.6% for generic drugs, 42.4% for brand-name only drugs, and 45% for brand-name drugs with generic equivalents. The proportion of claims increased from 81.9% to 88% for generic drugs and from 4.9% to 6.2% for brand-name only drugs.
However, the proportion of claims for brand-name drugs with generic equivalents decreased from 13.3% to 5.8%.
Treatment barrier
Neuroimmunologic drugs, most of which were prescribed for MS, had exceptional cost, the researchers noted. These drugs accounted for more than 50% of payments but only 4.3% of claims. Claim payment for these drugs increased by 46.9% during the study period, from $3,337 to $4,902.
When neuroimmunologic drugs were removed from the analysis there was still significant increase in claim payments for brand-name only drugs (50.4%) and brand-name drugs with generic equivalents (45.6%).
Although neuroimmunologic medicines, including monoclonal antibodies, are more expensive to produce, this factor alone does not explain their exceptional cost, said Dr. de Havenon. “The high cost of brand-name drugs in this speciality is likely because the market bears it,” he added. “In other words, MS is a disabling disease and the medications work, so historically the Centers for Medicare & Medicaid Services have been willing to tolerate the high cost of these primarily brand-name medications.”
Several countries have controlled drug costs by negotiating with pharmaceutical companies and through legislation, Dr. de Havenon noted.
“My intent with this article was to raise awareness on the topic, which I struggle with frequently as a clinician. I know I want my patients to have a medication, but the cost prevents it,” he said.
‘Unfettered’ price-setting
Commenting on the findings, Robert J. Fox, MD, vice chair for research at the Neurological Institute of the Cleveland Clinic, said the study “brings into clear light” what neurologists, particularly those who treat MS, have long suspected but did not really know. These neurologists “are typically distanced from the payment aspects of the medications they prescribe,” said Dr. Fox, who was not involved with the research.
Although a particular strength of the study was its comprehensiveness, the researchers excluded infusion claims – which account for a large portion of total patient care costs for many disorders, he noted.
Drugs for MS historically have been expensive, ostensibly because of their high cost of development. In addition, the large and continued price increase that occurs long after these drugs have been approved remains unexplained, said Dr. Fox.
He noted that the study findings might not directly affect clinical practice because neurologists will continue prescribing medications they think are best for their patients. “Instead, I think this is a lesson to lawmakers about the massive error in the Medicare Modernization Act of 2003, where the federal government was prohibited from negotiating drug prices. If the seller is unfettered in setting a price, then no one should be surprised when the price rises,” Dr. Fox said.
Because many new drugs and new generic formulations for treating MS have become available during the past year, “repeating these types of economic studies for the period 2020-2025 will help us understand if generic competition – as well as new laws if they are passed – alter price,” he concluded.
The study was funded by the American Academy of Neurology, which publishes Neurology. Dr. de Havenon has received clinical research funding from AMAG Pharmaceuticals and Regeneron Pharmaceuticals. Dr. Fox receives consulting fees from many pharmaceutical companies involved in the development of therapies for MS.
A version of this article first appeared on Medscape.com.
, new research shows. Results of the retrospective study also showed that most of the increased costs for these agents were due to rising costs for neuroimmunology drugs, mainly for those used to treat multiple sclerosis (MS).
“The same brand name medication in 2017 cost approximately 50% more than in 2013,” said Adam de Havenon, MD, assistant professor of neurology, University of Utah, Salt Lake City.
“An analogy would be if you bought an iPhone 5 in 2013 for $500, and then in 2017, you were asked to pay $750 for the exact same iPhone 5,” Dr. de Havenon added.
The study findings were published online March 10 in the journal Neurology.
$26 billion in payments
Both neurologists and patients are concerned about the high cost of prescription drugs for neurologic diseases, and Medicare Part D data indicate that these drugs are the most expensive component of neurologic care, the researchers noted. In addition, out-of-pocket costs have increased significantly for patients with neurologic disease such as Parkinson’s disease, epilepsy, and MS.
To understand trends in payments for neurologic drugs, Dr. de Havenon and colleagues analyzed Medicare Part D claims filed from 2013 to 2017. The payments include costs paid by Medicare, the patient, government subsidies, and other third-party payers.
In addition to examining more current Medicare Part D data than previous studies, the current analysis examined all medications prescribed by neurologists that consistently remained branded or generic during the 5-year study period, said Dr. de Havenon. This approach resulted in a large number of claims and a large total cost.
To calculate the percentage change in annual payment claims, the researchers used 2013 prices as a reference point. They identified drugs named in 2013 claims and classified them as generic, brand-name only, or brand-name with generic equivalent. Researchers also divided the drugs by neurologic subspecialty.
The analysis included 520 drugs, all of which were available in each year of the study period. Of these drugs, 322 were generic, 61 were brand-name only, and 137 were brand-name with a generic equivalent. There were 90.7 million total claims.
Results showed total payments amounted to $26.65 billion. Yearly total payments increased from $4.05 billion in 2013 to $6.09 billion in 2017, representing a 50.4% increase, even after adjusting for inflation. Total claims increased by 7.6% – from 17.1 million in 2013 to 18.4 million in 2017.
From 2013 to 2017, claim payments increased by 0.6% for generic drugs, 42.4% for brand-name only drugs, and 45% for brand-name drugs with generic equivalents. The proportion of claims increased from 81.9% to 88% for generic drugs and from 4.9% to 6.2% for brand-name only drugs.
However, the proportion of claims for brand-name drugs with generic equivalents decreased from 13.3% to 5.8%.
Treatment barrier
Neuroimmunologic drugs, most of which were prescribed for MS, had exceptional cost, the researchers noted. These drugs accounted for more than 50% of payments but only 4.3% of claims. Claim payment for these drugs increased by 46.9% during the study period, from $3,337 to $4,902.
When neuroimmunologic drugs were removed from the analysis there was still significant increase in claim payments for brand-name only drugs (50.4%) and brand-name drugs with generic equivalents (45.6%).
Although neuroimmunologic medicines, including monoclonal antibodies, are more expensive to produce, this factor alone does not explain their exceptional cost, said Dr. de Havenon. “The high cost of brand-name drugs in this speciality is likely because the market bears it,” he added. “In other words, MS is a disabling disease and the medications work, so historically the Centers for Medicare & Medicaid Services have been willing to tolerate the high cost of these primarily brand-name medications.”
Several countries have controlled drug costs by negotiating with pharmaceutical companies and through legislation, Dr. de Havenon noted.
“My intent with this article was to raise awareness on the topic, which I struggle with frequently as a clinician. I know I want my patients to have a medication, but the cost prevents it,” he said.
‘Unfettered’ price-setting
Commenting on the findings, Robert J. Fox, MD, vice chair for research at the Neurological Institute of the Cleveland Clinic, said the study “brings into clear light” what neurologists, particularly those who treat MS, have long suspected but did not really know. These neurologists “are typically distanced from the payment aspects of the medications they prescribe,” said Dr. Fox, who was not involved with the research.
Although a particular strength of the study was its comprehensiveness, the researchers excluded infusion claims – which account for a large portion of total patient care costs for many disorders, he noted.
Drugs for MS historically have been expensive, ostensibly because of their high cost of development. In addition, the large and continued price increase that occurs long after these drugs have been approved remains unexplained, said Dr. Fox.
He noted that the study findings might not directly affect clinical practice because neurologists will continue prescribing medications they think are best for their patients. “Instead, I think this is a lesson to lawmakers about the massive error in the Medicare Modernization Act of 2003, where the federal government was prohibited from negotiating drug prices. If the seller is unfettered in setting a price, then no one should be surprised when the price rises,” Dr. Fox said.
Because many new drugs and new generic formulations for treating MS have become available during the past year, “repeating these types of economic studies for the period 2020-2025 will help us understand if generic competition – as well as new laws if they are passed – alter price,” he concluded.
The study was funded by the American Academy of Neurology, which publishes Neurology. Dr. de Havenon has received clinical research funding from AMAG Pharmaceuticals and Regeneron Pharmaceuticals. Dr. Fox receives consulting fees from many pharmaceutical companies involved in the development of therapies for MS.
A version of this article first appeared on Medscape.com.
, new research shows. Results of the retrospective study also showed that most of the increased costs for these agents were due to rising costs for neuroimmunology drugs, mainly for those used to treat multiple sclerosis (MS).
“The same brand name medication in 2017 cost approximately 50% more than in 2013,” said Adam de Havenon, MD, assistant professor of neurology, University of Utah, Salt Lake City.
“An analogy would be if you bought an iPhone 5 in 2013 for $500, and then in 2017, you were asked to pay $750 for the exact same iPhone 5,” Dr. de Havenon added.
The study findings were published online March 10 in the journal Neurology.
$26 billion in payments
Both neurologists and patients are concerned about the high cost of prescription drugs for neurologic diseases, and Medicare Part D data indicate that these drugs are the most expensive component of neurologic care, the researchers noted. In addition, out-of-pocket costs have increased significantly for patients with neurologic disease such as Parkinson’s disease, epilepsy, and MS.
To understand trends in payments for neurologic drugs, Dr. de Havenon and colleagues analyzed Medicare Part D claims filed from 2013 to 2017. The payments include costs paid by Medicare, the patient, government subsidies, and other third-party payers.
In addition to examining more current Medicare Part D data than previous studies, the current analysis examined all medications prescribed by neurologists that consistently remained branded or generic during the 5-year study period, said Dr. de Havenon. This approach resulted in a large number of claims and a large total cost.
To calculate the percentage change in annual payment claims, the researchers used 2013 prices as a reference point. They identified drugs named in 2013 claims and classified them as generic, brand-name only, or brand-name with generic equivalent. Researchers also divided the drugs by neurologic subspecialty.
The analysis included 520 drugs, all of which were available in each year of the study period. Of these drugs, 322 were generic, 61 were brand-name only, and 137 were brand-name with a generic equivalent. There were 90.7 million total claims.
Results showed total payments amounted to $26.65 billion. Yearly total payments increased from $4.05 billion in 2013 to $6.09 billion in 2017, representing a 50.4% increase, even after adjusting for inflation. Total claims increased by 7.6% – from 17.1 million in 2013 to 18.4 million in 2017.
From 2013 to 2017, claim payments increased by 0.6% for generic drugs, 42.4% for brand-name only drugs, and 45% for brand-name drugs with generic equivalents. The proportion of claims increased from 81.9% to 88% for generic drugs and from 4.9% to 6.2% for brand-name only drugs.
However, the proportion of claims for brand-name drugs with generic equivalents decreased from 13.3% to 5.8%.
Treatment barrier
Neuroimmunologic drugs, most of which were prescribed for MS, had exceptional cost, the researchers noted. These drugs accounted for more than 50% of payments but only 4.3% of claims. Claim payment for these drugs increased by 46.9% during the study period, from $3,337 to $4,902.
When neuroimmunologic drugs were removed from the analysis there was still significant increase in claim payments for brand-name only drugs (50.4%) and brand-name drugs with generic equivalents (45.6%).
Although neuroimmunologic medicines, including monoclonal antibodies, are more expensive to produce, this factor alone does not explain their exceptional cost, said Dr. de Havenon. “The high cost of brand-name drugs in this speciality is likely because the market bears it,” he added. “In other words, MS is a disabling disease and the medications work, so historically the Centers for Medicare & Medicaid Services have been willing to tolerate the high cost of these primarily brand-name medications.”
Several countries have controlled drug costs by negotiating with pharmaceutical companies and through legislation, Dr. de Havenon noted.
“My intent with this article was to raise awareness on the topic, which I struggle with frequently as a clinician. I know I want my patients to have a medication, but the cost prevents it,” he said.
‘Unfettered’ price-setting
Commenting on the findings, Robert J. Fox, MD, vice chair for research at the Neurological Institute of the Cleveland Clinic, said the study “brings into clear light” what neurologists, particularly those who treat MS, have long suspected but did not really know. These neurologists “are typically distanced from the payment aspects of the medications they prescribe,” said Dr. Fox, who was not involved with the research.
Although a particular strength of the study was its comprehensiveness, the researchers excluded infusion claims – which account for a large portion of total patient care costs for many disorders, he noted.
Drugs for MS historically have been expensive, ostensibly because of their high cost of development. In addition, the large and continued price increase that occurs long after these drugs have been approved remains unexplained, said Dr. Fox.
He noted that the study findings might not directly affect clinical practice because neurologists will continue prescribing medications they think are best for their patients. “Instead, I think this is a lesson to lawmakers about the massive error in the Medicare Modernization Act of 2003, where the federal government was prohibited from negotiating drug prices. If the seller is unfettered in setting a price, then no one should be surprised when the price rises,” Dr. Fox said.
Because many new drugs and new generic formulations for treating MS have become available during the past year, “repeating these types of economic studies for the period 2020-2025 will help us understand if generic competition – as well as new laws if they are passed – alter price,” he concluded.
The study was funded by the American Academy of Neurology, which publishes Neurology. Dr. de Havenon has received clinical research funding from AMAG Pharmaceuticals and Regeneron Pharmaceuticals. Dr. Fox receives consulting fees from many pharmaceutical companies involved in the development of therapies for MS.
A version of this article first appeared on Medscape.com.
FROM NEUROLOGY