User login
Breastfeeding with MS: Good for mom, too
BERLIN – In the changing multiple sclerosis landscape, more women are having babies, and more are asking questions. With these women, what’s the best way to address the complicated interplay among pregnancy, relapse risk, breastfeeding, and medication resumption? A starting point is to recognize that “women with MS are very different today than they were 25 years ago,” said Annette Langer-Gould, MD, PhD. Not only have diagnostic criteria changed but also highly effective treatments now exist that were not available when the first pregnancy cohorts were studied, she pointed out, speaking at the annual congress of the European Committee on Treatment and Research in Multiple Sclerosis.

The existing literature, said Dr. Langer-Gould, has addressed one controversy: “Most women with MS can have normal pregnancies – and breastfeed – without incurring harm,” though it’s true that severe rebound relapses are possible if natalizumab (Tysabri) or fingolimod (Gilenya) are stopped before pregnancy. In any case, new small-molecule MS medications need to be stopped during pregnancy and breastfeeding, she pointed out. “We didn’t have to worry about that too much when we only had injectables and monoclonal antibodies because they were larger and didn’t cross the placenta.”
Since the 1980s, the conversation about pregnancy and MS has moved from asking “Is pregnancy bad for women with MS?” to the current MS landscape, in which sicker women are able to become pregnant, Dr. Langer-Gould said, adding that how women with MS fare through pregnancy and in the postpartum period is changing over time as well. She and her colleagues’ experience with pregnancy in a cohort of women with MS in the Kaiser Permanente care system, where she is a clinical neurologist and regional research lead, revealed a relapse rate of 8.4%. “So it was pretty rare for a woman to have a relapse during pregnancy,” Dr. Langer-Gould said.
Most women with MS who become pregnant, whether their care is received in a referral center or is community based, are now doing so while on a disease-modifying therapy (DMT), Dr. Langer-Gould said. On these highly effective treatments, “women who were too sick to get pregnant are now well controlled and having babies.”
As more women with MS become pregnant, more conversations about breastfeeding will inevitably crop up, she said. And the discussion about breastfeeding has now begun to acknowledge the “strong benefits to mom and the baby of not just breastfeeding, but longer breastfeeding,” as well.
“Because of this baby-friendly push in a lot of hospitals in the United States, where they’re trying to encourage all women to breastfeed,” a full 87% of women breastfed their infants at least some of the time, and over a third of women (35%) breastfed exclusively for at least 2 months, Dr. Langer-Gould said.
“There’s no one clear explanation of why the women seem to be healthier and doing better through pregnancy as a group, but it’s probably a combination of having milder disease, breastfeeding more, and they’ve got better controlled disease before pregnancy,” she said.
At least eight studies to date have examined the relationship between postpartum MS relapses and breastfeeding, Dr. Langer-Gould said.
“The thing to take away ... is that, even though we’ve studied this many, many times, no one can show that it’s harmful,” she said. For mothers who want to breastfeed, “you can support them in the breastfeeding choice, because they are not going to have more severe disease because of that.”
Whether breastfeeding is exclusive or not has not always been tracked in studies of childbearing women with MS, but when it was captured in the data, exclusive breastfeeding has exerted a protective effect, with about a 50% reduction in risk for postpartum relapse seen in one study (JAMA Neurol. 2015 Oct;72[10]:1132-8).
There is a hormonal rationale for exclusive breastfeeding exerting a protective effect on MS: With exclusive breastfeeding comes more frequent, intense suckling, with more profound elevations in prolactin, and larger drops in follicle-stimulating hormone, luteinizing hormone, progesterone, and estradiol. All these hormonal changes work together to produce more prolonged amenorrhea and anovulation, Dr. Langer-Gould said, with potentially beneficial immunologic effects.
When other, more general maternal and infant health benefits of breastfeeding also are taken into account, there’s strong evidence for the benefits of breastfeeding for women with MS whose medication profile allows them to breastfeed, she said.
However, the “treatment” effect of exclusive breastfeeding is only effective until the infant starts taking regular supplemental feedings, including the introduction of table food at around 6 months of age. “Once regular supplemental feedings are introduced, relapses return,” Dr. Langer-Gould said.
There is some suggestion that, in women without MS, prolonged breastfeeding may be associated with reduced risk of MS. In the MS Sunshine study, breastfeeding for 15 months or longer decreased the risk of later MS by 23%-53% (Nutrients. 2018 Feb 27;10[3]:268). The investigators, led by Dr. Langer-Gould, summed the total months of breastfeeding across all children, so that the 15-month threshold could be reached by breastfeeding one child for 15 months, or three children for 5 months each. “It’s a single study; I wouldn’t make too much out of it,” Dr. Langer-Gould said.
Open questions still remain, she said: “So far, no one has been able to demonstrate a clear beneficial effect in reducing the risk of postpartum relapse if they resume their DMT early in the postpartum period.” Dr. Langer-Gould noted that the literature in this area is hampered by heterogeneity and by the fact that newer, more highly active DMTs have not been well studied.
Also, the link between postpartum relapses and long-term prognosis is not completely delineated. Indirect evidence, she said, points to a postpartum relapse as being “overall, a low-impact event.”
Dr. Langer-Gould reported that she has been the site principal investigator for clinical trials sponsored by Roche and Biogen.
SOURCE: Langer-Gould A. ECTRIMS 2018, Abstract 5.
BERLIN – In the changing multiple sclerosis landscape, more women are having babies, and more are asking questions. With these women, what’s the best way to address the complicated interplay among pregnancy, relapse risk, breastfeeding, and medication resumption? A starting point is to recognize that “women with MS are very different today than they were 25 years ago,” said Annette Langer-Gould, MD, PhD. Not only have diagnostic criteria changed but also highly effective treatments now exist that were not available when the first pregnancy cohorts were studied, she pointed out, speaking at the annual congress of the European Committee on Treatment and Research in Multiple Sclerosis.
The existing literature, said Dr. Langer-Gould, has addressed one controversy: “Most women with MS can have normal pregnancies – and breastfeed – without incurring harm,” though it’s true that severe rebound relapses are possible if natalizumab (Tysabri) or fingolimod (Gilenya) are stopped before pregnancy. In any case, new small-molecule MS medications need to be stopped during pregnancy and breastfeeding, she pointed out. “We didn’t have to worry about that too much when we only had injectables and monoclonal antibodies because they were larger and didn’t cross the placenta.”
Since the 1980s, the conversation about pregnancy and MS has moved from asking “Is pregnancy bad for women with MS?” to the current MS landscape, in which sicker women are able to become pregnant, Dr. Langer-Gould said, adding that how women with MS fare through pregnancy and in the postpartum period is changing over time as well. She and her colleagues’ experience with pregnancy in a cohort of women with MS in the Kaiser Permanente care system, where she is a clinical neurologist and regional research lead, revealed a relapse rate of 8.4%. “So it was pretty rare for a woman to have a relapse during pregnancy,” Dr. Langer-Gould said.
Most women with MS who become pregnant, whether their care is received in a referral center or is community based, are now doing so while on a disease-modifying therapy (DMT), Dr. Langer-Gould said. On these highly effective treatments, “women who were too sick to get pregnant are now well controlled and having babies.”
As more women with MS become pregnant, more conversations about breastfeeding will inevitably crop up, she said. And the discussion about breastfeeding has now begun to acknowledge the “strong benefits to mom and the baby of not just breastfeeding, but longer breastfeeding,” as well.
“Because of this baby-friendly push in a lot of hospitals in the United States, where they’re trying to encourage all women to breastfeed,” a full 87% of women breastfed their infants at least some of the time, and over a third of women (35%) breastfed exclusively for at least 2 months, Dr. Langer-Gould said.
“There’s no one clear explanation of why the women seem to be healthier and doing better through pregnancy as a group, but it’s probably a combination of having milder disease, breastfeeding more, and they’ve got better controlled disease before pregnancy,” she said.
At least eight studies to date have examined the relationship between postpartum MS relapses and breastfeeding, Dr. Langer-Gould said.
“The thing to take away ... is that, even though we’ve studied this many, many times, no one can show that it’s harmful,” she said. For mothers who want to breastfeed, “you can support them in the breastfeeding choice, because they are not going to have more severe disease because of that.”
Whether breastfeeding is exclusive or not has not always been tracked in studies of childbearing women with MS, but when it was captured in the data, exclusive breastfeeding has exerted a protective effect, with about a 50% reduction in risk for postpartum relapse seen in one study (JAMA Neurol. 2015 Oct;72[10]:1132-8).
There is a hormonal rationale for exclusive breastfeeding exerting a protective effect on MS: With exclusive breastfeeding comes more frequent, intense suckling, with more profound elevations in prolactin, and larger drops in follicle-stimulating hormone, luteinizing hormone, progesterone, and estradiol. All these hormonal changes work together to produce more prolonged amenorrhea and anovulation, Dr. Langer-Gould said, with potentially beneficial immunologic effects.
When other, more general maternal and infant health benefits of breastfeeding also are taken into account, there’s strong evidence for the benefits of breastfeeding for women with MS whose medication profile allows them to breastfeed, she said.
However, the “treatment” effect of exclusive breastfeeding is only effective until the infant starts taking regular supplemental feedings, including the introduction of table food at around 6 months of age. “Once regular supplemental feedings are introduced, relapses return,” Dr. Langer-Gould said.
There is some suggestion that, in women without MS, prolonged breastfeeding may be associated with reduced risk of MS. In the MS Sunshine study, breastfeeding for 15 months or longer decreased the risk of later MS by 23%-53% (Nutrients. 2018 Feb 27;10[3]:268). The investigators, led by Dr. Langer-Gould, summed the total months of breastfeeding across all children, so that the 15-month threshold could be reached by breastfeeding one child for 15 months, or three children for 5 months each. “It’s a single study; I wouldn’t make too much out of it,” Dr. Langer-Gould said.
Open questions still remain, she said: “So far, no one has been able to demonstrate a clear beneficial effect in reducing the risk of postpartum relapse if they resume their DMT early in the postpartum period.” Dr. Langer-Gould noted that the literature in this area is hampered by heterogeneity and by the fact that newer, more highly active DMTs have not been well studied.
Also, the link between postpartum relapses and long-term prognosis is not completely delineated. Indirect evidence, she said, points to a postpartum relapse as being “overall, a low-impact event.”
Dr. Langer-Gould reported that she has been the site principal investigator for clinical trials sponsored by Roche and Biogen.
SOURCE: Langer-Gould A. ECTRIMS 2018, Abstract 5.
BERLIN – In the changing multiple sclerosis landscape, more women are having babies, and more are asking questions. With these women, what’s the best way to address the complicated interplay among pregnancy, relapse risk, breastfeeding, and medication resumption? A starting point is to recognize that “women with MS are very different today than they were 25 years ago,” said Annette Langer-Gould, MD, PhD. Not only have diagnostic criteria changed but also highly effective treatments now exist that were not available when the first pregnancy cohorts were studied, she pointed out, speaking at the annual congress of the European Committee on Treatment and Research in Multiple Sclerosis.
The existing literature, said Dr. Langer-Gould, has addressed one controversy: “Most women with MS can have normal pregnancies – and breastfeed – without incurring harm,” though it’s true that severe rebound relapses are possible if natalizumab (Tysabri) or fingolimod (Gilenya) are stopped before pregnancy. In any case, new small-molecule MS medications need to be stopped during pregnancy and breastfeeding, she pointed out. “We didn’t have to worry about that too much when we only had injectables and monoclonal antibodies because they were larger and didn’t cross the placenta.”
Since the 1980s, the conversation about pregnancy and MS has moved from asking “Is pregnancy bad for women with MS?” to the current MS landscape, in which sicker women are able to become pregnant, Dr. Langer-Gould said, adding that how women with MS fare through pregnancy and in the postpartum period is changing over time as well. She and her colleagues’ experience with pregnancy in a cohort of women with MS in the Kaiser Permanente care system, where she is a clinical neurologist and regional research lead, revealed a relapse rate of 8.4%. “So it was pretty rare for a woman to have a relapse during pregnancy,” Dr. Langer-Gould said.
Most women with MS who become pregnant, whether their care is received in a referral center or is community based, are now doing so while on a disease-modifying therapy (DMT), Dr. Langer-Gould said. On these highly effective treatments, “women who were too sick to get pregnant are now well controlled and having babies.”
As more women with MS become pregnant, more conversations about breastfeeding will inevitably crop up, she said. And the discussion about breastfeeding has now begun to acknowledge the “strong benefits to mom and the baby of not just breastfeeding, but longer breastfeeding,” as well.
“Because of this baby-friendly push in a lot of hospitals in the United States, where they’re trying to encourage all women to breastfeed,” a full 87% of women breastfed their infants at least some of the time, and over a third of women (35%) breastfed exclusively for at least 2 months, Dr. Langer-Gould said.
“There’s no one clear explanation of why the women seem to be healthier and doing better through pregnancy as a group, but it’s probably a combination of having milder disease, breastfeeding more, and they’ve got better controlled disease before pregnancy,” she said.
At least eight studies to date have examined the relationship between postpartum MS relapses and breastfeeding, Dr. Langer-Gould said.
“The thing to take away ... is that, even though we’ve studied this many, many times, no one can show that it’s harmful,” she said. For mothers who want to breastfeed, “you can support them in the breastfeeding choice, because they are not going to have more severe disease because of that.”
Whether breastfeeding is exclusive or not has not always been tracked in studies of childbearing women with MS, but when it was captured in the data, exclusive breastfeeding has exerted a protective effect, with about a 50% reduction in risk for postpartum relapse seen in one study (JAMA Neurol. 2015 Oct;72[10]:1132-8).
There is a hormonal rationale for exclusive breastfeeding exerting a protective effect on MS: With exclusive breastfeeding comes more frequent, intense suckling, with more profound elevations in prolactin, and larger drops in follicle-stimulating hormone, luteinizing hormone, progesterone, and estradiol. All these hormonal changes work together to produce more prolonged amenorrhea and anovulation, Dr. Langer-Gould said, with potentially beneficial immunologic effects.
When other, more general maternal and infant health benefits of breastfeeding also are taken into account, there’s strong evidence for the benefits of breastfeeding for women with MS whose medication profile allows them to breastfeed, she said.
However, the “treatment” effect of exclusive breastfeeding is only effective until the infant starts taking regular supplemental feedings, including the introduction of table food at around 6 months of age. “Once regular supplemental feedings are introduced, relapses return,” Dr. Langer-Gould said.
There is some suggestion that, in women without MS, prolonged breastfeeding may be associated with reduced risk of MS. In the MS Sunshine study, breastfeeding for 15 months or longer decreased the risk of later MS by 23%-53% (Nutrients. 2018 Feb 27;10[3]:268). The investigators, led by Dr. Langer-Gould, summed the total months of breastfeeding across all children, so that the 15-month threshold could be reached by breastfeeding one child for 15 months, or three children for 5 months each. “It’s a single study; I wouldn’t make too much out of it,” Dr. Langer-Gould said.
Open questions still remain, she said: “So far, no one has been able to demonstrate a clear beneficial effect in reducing the risk of postpartum relapse if they resume their DMT early in the postpartum period.” Dr. Langer-Gould noted that the literature in this area is hampered by heterogeneity and by the fact that newer, more highly active DMTs have not been well studied.
Also, the link between postpartum relapses and long-term prognosis is not completely delineated. Indirect evidence, she said, points to a postpartum relapse as being “overall, a low-impact event.”
Dr. Langer-Gould reported that she has been the site principal investigator for clinical trials sponsored by Roche and Biogen.
SOURCE: Langer-Gould A. ECTRIMS 2018, Abstract 5.
REPORTING FROM ECTRIMS 2018

Mutation confers resistance to venetoclax in CLL

SAN DIEGO—A recurrent mutation in BCL2, the therapeutic target of venetoclax, appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, according to Piers Blombery, MBBS, of the Peter MacCallum Cancer Center in Melbourne, Victoria, Australia.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said during the late-breaking abstracts session at the 2018 ASH Annual Meeting.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” Dr. Blombery added.
A paper on the mutation was published in Cancer Discovery to coincide with the presentation at ASH (abstract LBA-7).
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven patients, the investigators identified a novel mutation that showed up at the time of progression but was absent from the pre-venetoclax samples.
The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies.
Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, the investigators determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and, in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage compared with wild-type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
He added that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
Dr. Blombery reported having no relevant disclosures. The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation.
SAN DIEGO—A recurrent mutation in BCL2, the therapeutic target of venetoclax, appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, according to Piers Blombery, MBBS, of the Peter MacCallum Cancer Center in Melbourne, Victoria, Australia.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said during the late-breaking abstracts session at the 2018 ASH Annual Meeting.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” Dr. Blombery added.
A paper on the mutation was published in Cancer Discovery to coincide with the presentation at ASH (abstract LBA-7).
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven patients, the investigators identified a novel mutation that showed up at the time of progression but was absent from the pre-venetoclax samples.
The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies.
Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, the investigators determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and, in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage compared with wild-type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
He added that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
Dr. Blombery reported having no relevant disclosures. The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation.
SAN DIEGO—A recurrent mutation in BCL2, the therapeutic target of venetoclax, appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, according to Piers Blombery, MBBS, of the Peter MacCallum Cancer Center in Melbourne, Victoria, Australia.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said during the late-breaking abstracts session at the 2018 ASH Annual Meeting.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” Dr. Blombery added.
A paper on the mutation was published in Cancer Discovery to coincide with the presentation at ASH (abstract LBA-7).
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven patients, the investigators identified a novel mutation that showed up at the time of progression but was absent from the pre-venetoclax samples.
The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies.
Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, the investigators determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and, in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage compared with wild-type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
He added that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
Dr. Blombery reported having no relevant disclosures. The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation.
Emerging biosimilars market presents opportunities and challenges
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6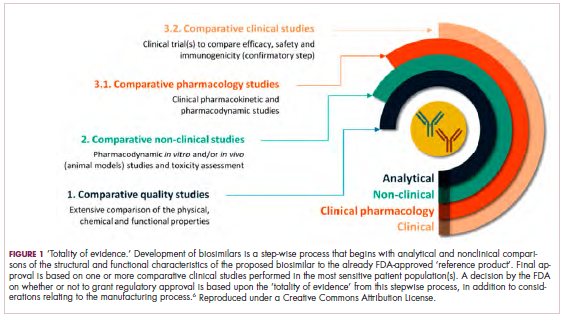
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10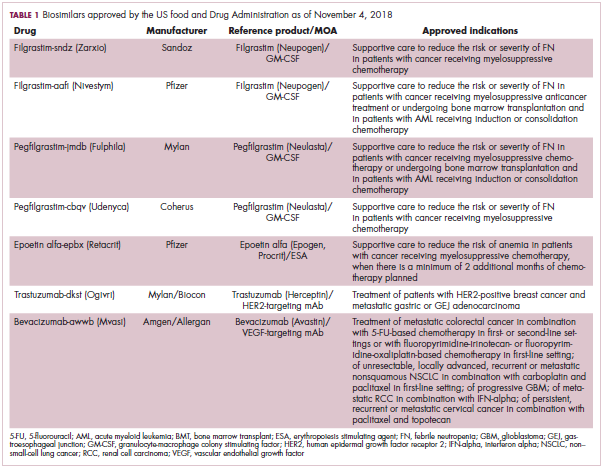
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22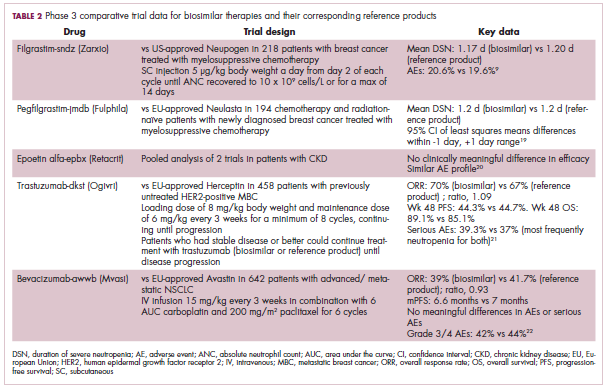
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.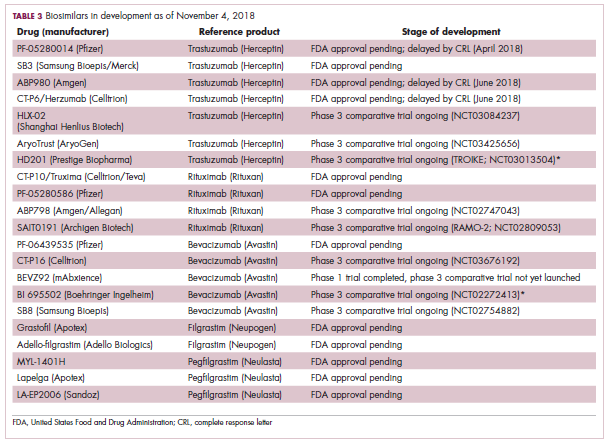
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
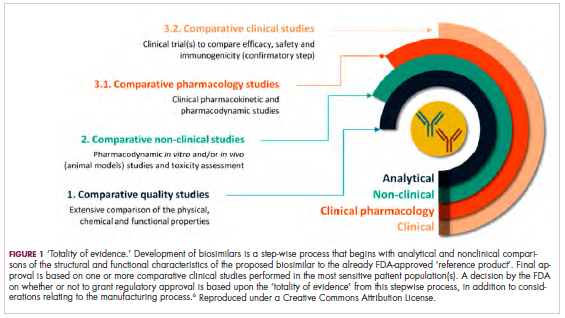
Intravascular large B-cell lymphoma: an elusive diagnosis with challenging management
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.
2018: A banner year for hematology drug approvals
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
REPORTING FROM ASH 2018

Phase 3 study confirms biosimilarity of PF-05280586 with rituximab
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).

The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).
The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).
The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
REPORTING FROM ASH 2018
Key clinical point: PF-05280586 shows biosimilarity to rituximab at up to 26 weeks.
Major finding: ORR at 26 weeks was 75.5% vs. 70.7% with PF-05280586 vs. rituximab, respectively.
Study details: A phase 3 study of 394 patients.
Disclosures: This study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
Source: Sharman J et al. ASH 2018: Abstract 394.
CLL resistance mechanism to venetoclax identified
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The mutation was identified in samples from seven patients after venetoclax therapy, but not in any of the pretherapy samples.
Study details: Genetic analysis of CLL mutations in 15 patients enrolled in clinical trials of venetoclax.
Disclosures: The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
Source: Blombery P et al. ASH 2018, Abstract LBA-7.

Shorter R-CHOP regimen noninferior in certain DLBCL patients

SAN DIEGO—A shortened regimen of four cycles of rituximab (R) plus cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy was noninferior in efficacy to the standard six cycles of R-CHOP in younger patients with favorable-risk diffuse large B-cell lymphoma (DLBCL), according to investigators of the FLYER trial.
In addition, the truncated regimen was associated with about a one-third reduction in non-hematologic adverse events.
Viola Poeschel, MD, of Saarland University Medical School in Homburg/Saar, Germany, reported results of this study on behalf of the German High-Grade Non-Hodgkin’s Lymphoma Study Group/German Lymphoma Alliance at the 2018 ASH Annual Meeting (abstract 781).
Among 588 evaluable patients younger than 60 with favorable-prognosis DLBCL, there were no significant differences in progression-free survival (PFS), event-free survival (EFS), or overall survival (OS) between patients who received four cycles of R-CHOP and those who received six cycles, Dr. Poeschel reported.
“Six cycles of R-CHOP led to a higher toxicity with respect to leukocytopenia and anemia, both of any grade and also of grades 3 to 4, compared to four cycles of R-CHOP,” she said.
The findings suggest that, for younger patients with favorable-prognosis DLBCL—defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm)—four cycles of R-CHOP can be a new standard of care, Dr. Poeschel said.
The investigators were prompted to look at the question of a shorter R-CHOP regimen by results of the MInT trial, in which a subpopulation of favorable-prognosis DLBCL patients had a 3-year PFS rate of 89%.
The FLYER trial (NCT00278421) was designed as a non-inferiority study to see whether, in a similar group of patients, reducing the number of R-CHOP cycles could maintain efficacy while reducing toxicity.
At a median follow-up of 66 months, the PFS rate, the primary endpoint, was 94% in the six-cycle group and 96% for the four-cycle group.
“As the lower limit of the 95% confidence interval of our experimental arm was 94%, it is shown that it is definitely non-inferior to the standard arm, six cycles of R-CHOP,” Dr. Poeschel said.
Similarly, the rate of 3-year OS was 98% in the six-cycle group, compared with 99% in the four-cycle group, and the survival curves were virtually superimposable out to more than 10 years of follow-up.
Treatment with six cycles was associated with more frequent hematologic adverse events than four cycles. Leukopenia of any grade occurred in 237 and 171 patients, respectively. Grade 3-4 leukopenia occurred in 110 and 80 patients, respectively.
Any-grade anemia occurred in 172 patients assigned to six cycles and 107 assigned to four cycles. Rates of grade 3-4 anemia were similar between the groups, as were rates of thrombocytopenia of any grade or grade 3-4.
Non-hematologic adverse events of any grade or grade 3-4 that were more frequent with six cycles included parasthesia, nausea, infection, vomiting, and mucositis.
The total number of non-hematologic adverse events was reduced by about one-third.
“We are certainly always looking for ways to make treatments easier for our patients to reduce adverse effects, and, certainly, for this subgroup of patients, it appears that we can make their treatment shorter and have less burden but equivalent efficacy,” said David Steensma, MD, of the Dana-Farber Cancer Institute/Harvard Cancer Center in Boston, Massachusetts.
Drs. Steensma and Poeschel both cautioned that the results of this study pertain only to those patients with DLBCL who are younger and have favorable-prognosis disease.
“We can’t extend it to other subtypes of large-cell lymphoma, but that’s always a laudable goal, so I think this will immediately influence clinical practice,” Dr. Steensma said.
The study was supported by Deutsche Krebshilfe. Dr. Poeschel disclosed travel grants from Roche and Amgen. Dr. Steensma had no disclosures relevant to the study.
SAN DIEGO—A shortened regimen of four cycles of rituximab (R) plus cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy was noninferior in efficacy to the standard six cycles of R-CHOP in younger patients with favorable-risk diffuse large B-cell lymphoma (DLBCL), according to investigators of the FLYER trial.
In addition, the truncated regimen was associated with about a one-third reduction in non-hematologic adverse events.
Viola Poeschel, MD, of Saarland University Medical School in Homburg/Saar, Germany, reported results of this study on behalf of the German High-Grade Non-Hodgkin’s Lymphoma Study Group/German Lymphoma Alliance at the 2018 ASH Annual Meeting (abstract 781).
Among 588 evaluable patients younger than 60 with favorable-prognosis DLBCL, there were no significant differences in progression-free survival (PFS), event-free survival (EFS), or overall survival (OS) between patients who received four cycles of R-CHOP and those who received six cycles, Dr. Poeschel reported.
“Six cycles of R-CHOP led to a higher toxicity with respect to leukocytopenia and anemia, both of any grade and also of grades 3 to 4, compared to four cycles of R-CHOP,” she said.
The findings suggest that, for younger patients with favorable-prognosis DLBCL—defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm)—four cycles of R-CHOP can be a new standard of care, Dr. Poeschel said.
The investigators were prompted to look at the question of a shorter R-CHOP regimen by results of the MInT trial, in which a subpopulation of favorable-prognosis DLBCL patients had a 3-year PFS rate of 89%.
The FLYER trial (NCT00278421) was designed as a non-inferiority study to see whether, in a similar group of patients, reducing the number of R-CHOP cycles could maintain efficacy while reducing toxicity.
At a median follow-up of 66 months, the PFS rate, the primary endpoint, was 94% in the six-cycle group and 96% for the four-cycle group.
“As the lower limit of the 95% confidence interval of our experimental arm was 94%, it is shown that it is definitely non-inferior to the standard arm, six cycles of R-CHOP,” Dr. Poeschel said.
Similarly, the rate of 3-year OS was 98% in the six-cycle group, compared with 99% in the four-cycle group, and the survival curves were virtually superimposable out to more than 10 years of follow-up.
Treatment with six cycles was associated with more frequent hematologic adverse events than four cycles. Leukopenia of any grade occurred in 237 and 171 patients, respectively. Grade 3-4 leukopenia occurred in 110 and 80 patients, respectively.
Any-grade anemia occurred in 172 patients assigned to six cycles and 107 assigned to four cycles. Rates of grade 3-4 anemia were similar between the groups, as were rates of thrombocytopenia of any grade or grade 3-4.
Non-hematologic adverse events of any grade or grade 3-4 that were more frequent with six cycles included parasthesia, nausea, infection, vomiting, and mucositis.
The total number of non-hematologic adverse events was reduced by about one-third.
“We are certainly always looking for ways to make treatments easier for our patients to reduce adverse effects, and, certainly, for this subgroup of patients, it appears that we can make their treatment shorter and have less burden but equivalent efficacy,” said David Steensma, MD, of the Dana-Farber Cancer Institute/Harvard Cancer Center in Boston, Massachusetts.
Drs. Steensma and Poeschel both cautioned that the results of this study pertain only to those patients with DLBCL who are younger and have favorable-prognosis disease.
“We can’t extend it to other subtypes of large-cell lymphoma, but that’s always a laudable goal, so I think this will immediately influence clinical practice,” Dr. Steensma said.
The study was supported by Deutsche Krebshilfe. Dr. Poeschel disclosed travel grants from Roche and Amgen. Dr. Steensma had no disclosures relevant to the study.
SAN DIEGO—A shortened regimen of four cycles of rituximab (R) plus cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy was noninferior in efficacy to the standard six cycles of R-CHOP in younger patients with favorable-risk diffuse large B-cell lymphoma (DLBCL), according to investigators of the FLYER trial.
In addition, the truncated regimen was associated with about a one-third reduction in non-hematologic adverse events.
Viola Poeschel, MD, of Saarland University Medical School in Homburg/Saar, Germany, reported results of this study on behalf of the German High-Grade Non-Hodgkin’s Lymphoma Study Group/German Lymphoma Alliance at the 2018 ASH Annual Meeting (abstract 781).
Among 588 evaluable patients younger than 60 with favorable-prognosis DLBCL, there were no significant differences in progression-free survival (PFS), event-free survival (EFS), or overall survival (OS) between patients who received four cycles of R-CHOP and those who received six cycles, Dr. Poeschel reported.
“Six cycles of R-CHOP led to a higher toxicity with respect to leukocytopenia and anemia, both of any grade and also of grades 3 to 4, compared to four cycles of R-CHOP,” she said.
The findings suggest that, for younger patients with favorable-prognosis DLBCL—defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm)—four cycles of R-CHOP can be a new standard of care, Dr. Poeschel said.
The investigators were prompted to look at the question of a shorter R-CHOP regimen by results of the MInT trial, in which a subpopulation of favorable-prognosis DLBCL patients had a 3-year PFS rate of 89%.
The FLYER trial (NCT00278421) was designed as a non-inferiority study to see whether, in a similar group of patients, reducing the number of R-CHOP cycles could maintain efficacy while reducing toxicity.
At a median follow-up of 66 months, the PFS rate, the primary endpoint, was 94% in the six-cycle group and 96% for the four-cycle group.
“As the lower limit of the 95% confidence interval of our experimental arm was 94%, it is shown that it is definitely non-inferior to the standard arm, six cycles of R-CHOP,” Dr. Poeschel said.
Similarly, the rate of 3-year OS was 98% in the six-cycle group, compared with 99% in the four-cycle group, and the survival curves were virtually superimposable out to more than 10 years of follow-up.
Treatment with six cycles was associated with more frequent hematologic adverse events than four cycles. Leukopenia of any grade occurred in 237 and 171 patients, respectively. Grade 3-4 leukopenia occurred in 110 and 80 patients, respectively.
Any-grade anemia occurred in 172 patients assigned to six cycles and 107 assigned to four cycles. Rates of grade 3-4 anemia were similar between the groups, as were rates of thrombocytopenia of any grade or grade 3-4.
Non-hematologic adverse events of any grade or grade 3-4 that were more frequent with six cycles included parasthesia, nausea, infection, vomiting, and mucositis.
The total number of non-hematologic adverse events was reduced by about one-third.
“We are certainly always looking for ways to make treatments easier for our patients to reduce adverse effects, and, certainly, for this subgroup of patients, it appears that we can make their treatment shorter and have less burden but equivalent efficacy,” said David Steensma, MD, of the Dana-Farber Cancer Institute/Harvard Cancer Center in Boston, Massachusetts.
Drs. Steensma and Poeschel both cautioned that the results of this study pertain only to those patients with DLBCL who are younger and have favorable-prognosis disease.
“We can’t extend it to other subtypes of large-cell lymphoma, but that’s always a laudable goal, so I think this will immediately influence clinical practice,” Dr. Steensma said.
The study was supported by Deutsche Krebshilfe. Dr. Poeschel disclosed travel grants from Roche and Amgen. Dr. Steensma had no disclosures relevant to the study.

Regimen provides survival benefit in PTCL

SAN DIEGO—A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to a presentation at the 2018 ASH Annual Meeting.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS) compared to patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large-cell lymphoma or other CD30-expressing PTCLs, including angioimmunoblastic T-cell lymphoma and PTCL not otherwise specified.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center in Basking Ridge, New Jersey.
Dr. Horwitz presented results from this trial at ASH as abstract 997. Results were simultaneously published in The Lancet.
Patients and treatment
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive (n=98) or -negative (n=218) systemic anaplastic large-cell lymphoma, PTCL not otherwise specified (n=72), angioimmunoblastic T-cell lymphoma (n=54), enteropathy-associated T-cell lymphoma (n=7), and adult T-cell leukemia/lymphoma (n=3).
Patients were randomized to receive BV-CHP plus placebo (n=226) or CHOP plus placebo (n=226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in both the BV-CHP arm (range, 18-85) and the CHOP arm (range, 18-83). The majority of patients were male—59% in the BV-CHP arm and 67% in the CHOP arm—and most patients had stage III/IV disease—81% and 80%, respectively.
Eighty-nine percent of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
Twenty-seven percent of patients in the BV-CHP arm and 19% in the CHOP arm received consolidation consisting of radiotherapy (6% and 3%, respectively) and/or stem cell transplant (22% and 17%).
Twenty-six percent of patients in the BV-CHP arm and 42% in the CHOP arm received systemic therapy for residual or progressive disease, and 4% of patients in each arm received palliative radiation.
Efficacy
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P=0.0032). The complete response rates were 68% and 56%, respectively (P=0.0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio=0.71, P=0.011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (hazard ratio=0.66, P=0.0244).
Dr. Horwitz noted that this study was not powered to determine differences in OS or PFS according to PTCL subtypes.
Safety
BV-CHP had a comparable safety profile to CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The most common AEs of any grade occurring in at least 20% of patients (in the BV-CHP and CHOP arms, respectively) were:
- Nausea (46% and 38%)
- Peripheral sensory neuropathy (45% and 41%)
- Neutropenia (38% for both)
- Diarrhea (38% and 20%)
- Constipation (29% and 30%)
- Alopecia (26% and 25%)
- Pyrexia (26% and 19%)
- Vomiting (26% and 17%)
- Fatigue (24% and 20%)
- Anemia (21% and 16%).
This research was funded by Seattle Genetics Inc. and Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Dr. Horwitz disclosed relationships with Seattle Genetics, Aileron Therapeutics, Innate Pharma, Millennium/Takeda, Forty Seven, Corvus, Mundipharma, ADC Therapeutics, Trillium, Celgene, Portola, Infinity/Verastem, Spectrum, and Kyowa-Hakka-Kirin.
SAN DIEGO—A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to a presentation at the 2018 ASH Annual Meeting.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS) compared to patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large-cell lymphoma or other CD30-expressing PTCLs, including angioimmunoblastic T-cell lymphoma and PTCL not otherwise specified.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center in Basking Ridge, New Jersey.
Dr. Horwitz presented results from this trial at ASH as abstract 997. Results were simultaneously published in The Lancet.
Patients and treatment
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive (n=98) or -negative (n=218) systemic anaplastic large-cell lymphoma, PTCL not otherwise specified (n=72), angioimmunoblastic T-cell lymphoma (n=54), enteropathy-associated T-cell lymphoma (n=7), and adult T-cell leukemia/lymphoma (n=3).
Patients were randomized to receive BV-CHP plus placebo (n=226) or CHOP plus placebo (n=226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in both the BV-CHP arm (range, 18-85) and the CHOP arm (range, 18-83). The majority of patients were male—59% in the BV-CHP arm and 67% in the CHOP arm—and most patients had stage III/IV disease—81% and 80%, respectively.
Eighty-nine percent of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
Twenty-seven percent of patients in the BV-CHP arm and 19% in the CHOP arm received consolidation consisting of radiotherapy (6% and 3%, respectively) and/or stem cell transplant (22% and 17%).
Twenty-six percent of patients in the BV-CHP arm and 42% in the CHOP arm received systemic therapy for residual or progressive disease, and 4% of patients in each arm received palliative radiation.
Efficacy
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P=0.0032). The complete response rates were 68% and 56%, respectively (P=0.0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio=0.71, P=0.011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (hazard ratio=0.66, P=0.0244).
Dr. Horwitz noted that this study was not powered to determine differences in OS or PFS according to PTCL subtypes.
Safety
BV-CHP had a comparable safety profile to CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The most common AEs of any grade occurring in at least 20% of patients (in the BV-CHP and CHOP arms, respectively) were:
- Nausea (46% and 38%)
- Peripheral sensory neuropathy (45% and 41%)
- Neutropenia (38% for both)
- Diarrhea (38% and 20%)
- Constipation (29% and 30%)
- Alopecia (26% and 25%)
- Pyrexia (26% and 19%)
- Vomiting (26% and 17%)
- Fatigue (24% and 20%)
- Anemia (21% and 16%).
This research was funded by Seattle Genetics Inc. and Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Dr. Horwitz disclosed relationships with Seattle Genetics, Aileron Therapeutics, Innate Pharma, Millennium/Takeda, Forty Seven, Corvus, Mundipharma, ADC Therapeutics, Trillium, Celgene, Portola, Infinity/Verastem, Spectrum, and Kyowa-Hakka-Kirin.
SAN DIEGO—A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to a presentation at the 2018 ASH Annual Meeting.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS) compared to patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large-cell lymphoma or other CD30-expressing PTCLs, including angioimmunoblastic T-cell lymphoma and PTCL not otherwise specified.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center in Basking Ridge, New Jersey.
Dr. Horwitz presented results from this trial at ASH as abstract 997. Results were simultaneously published in The Lancet.
Patients and treatment
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive (n=98) or -negative (n=218) systemic anaplastic large-cell lymphoma, PTCL not otherwise specified (n=72), angioimmunoblastic T-cell lymphoma (n=54), enteropathy-associated T-cell lymphoma (n=7), and adult T-cell leukemia/lymphoma (n=3).
Patients were randomized to receive BV-CHP plus placebo (n=226) or CHOP plus placebo (n=226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in both the BV-CHP arm (range, 18-85) and the CHOP arm (range, 18-83). The majority of patients were male—59% in the BV-CHP arm and 67% in the CHOP arm—and most patients had stage III/IV disease—81% and 80%, respectively.
Eighty-nine percent of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
Twenty-seven percent of patients in the BV-CHP arm and 19% in the CHOP arm received consolidation consisting of radiotherapy (6% and 3%, respectively) and/or stem cell transplant (22% and 17%).
Twenty-six percent of patients in the BV-CHP arm and 42% in the CHOP arm received systemic therapy for residual or progressive disease, and 4% of patients in each arm received palliative radiation.
Efficacy
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P=0.0032). The complete response rates were 68% and 56%, respectively (P=0.0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio=0.71, P=0.011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (hazard ratio=0.66, P=0.0244).
Dr. Horwitz noted that this study was not powered to determine differences in OS or PFS according to PTCL subtypes.
Safety
BV-CHP had a comparable safety profile to CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The most common AEs of any grade occurring in at least 20% of patients (in the BV-CHP and CHOP arms, respectively) were:
- Nausea (46% and 38%)
- Peripheral sensory neuropathy (45% and 41%)
- Neutropenia (38% for both)
- Diarrhea (38% and 20%)
- Constipation (29% and 30%)
- Alopecia (26% and 25%)
- Pyrexia (26% and 19%)
- Vomiting (26% and 17%)
- Fatigue (24% and 20%)
- Anemia (21% and 16%).
This research was funded by Seattle Genetics Inc. and Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Dr. Horwitz disclosed relationships with Seattle Genetics, Aileron Therapeutics, Innate Pharma, Millennium/Takeda, Forty Seven, Corvus, Mundipharma, ADC Therapeutics, Trillium, Celgene, Portola, Infinity/Verastem, Spectrum, and Kyowa-Hakka-Kirin.
Update shows durable responses in rel/ref DLBCL

SAN DIEGO—An updated analysis of the JULIET trial showed that tisagenlecleucel produced a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
After a median follow-up of 19 months, two-thirds of adults with relapsed/refractory DLBCL who had early responses to the chimeric antigen receptor (CAR) T-cell therapy remained in remission with no evidence of minimal residual disease.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression, no treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” said study investigator Richard Thomas Maziarz, MD, of Oregon Health & Science University’s Knight Cancer Institute in Portland.
Dr. Maziarz and his colleagues reported the updated study results at the 2018 ASH Annual Meeting (abstract 1684). Results were published simultaneously in The New England Journal of Medicine. Data reported here are based on the ASH data.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL who had relapsed or were refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant (HSCT) or who experienced disease progression after HSCT.
Interim results of the study were previously reported at the 22nd Congress of the European Hematology Association in 2017.
At that meeting, Gilles Salles, MD, PhD, of the University of Lyon in France, presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up.
In this population, the best overall response rate (ORR) was 59%. Three-month ORR was 45%, consisting of 37% complete responses (CR) and 8% partial responses (PR).
Relapse-free survival at 6 months was 79%, and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
The current analysis was completed after a median time from infusion to data cutoff of 19 months as of May 21, 2018. The analysis included 115 patients who received CAR T-cell infusions, 99 of whom were evaluable for efficacy.
As reported at ASH, the best ORR, the primary endpoint, was 54%, comprised of 40% CR and 13% PR.
Fifty-four percent of patients who had achieved PR converted to CR.
The response rates were consistent across all subgroups, regardless of age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 64%.
The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a CR.
Median overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months and not reached for patients in CR.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to the treatment, CRS, or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
The JULIET trial is supported by Novartis. Dr. Maziarz disclosed honoraria, consultancy fees, and/or research funding from Novartis, Incyte, Juno Therapeutics, and Kite Therapeutics as well as patents/royalties from Athersys, Inc.
SAN DIEGO—An updated analysis of the JULIET trial showed that tisagenlecleucel produced a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
After a median follow-up of 19 months, two-thirds of adults with relapsed/refractory DLBCL who had early responses to the chimeric antigen receptor (CAR) T-cell therapy remained in remission with no evidence of minimal residual disease.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression, no treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” said study investigator Richard Thomas Maziarz, MD, of Oregon Health & Science University’s Knight Cancer Institute in Portland.
Dr. Maziarz and his colleagues reported the updated study results at the 2018 ASH Annual Meeting (abstract 1684). Results were published simultaneously in The New England Journal of Medicine. Data reported here are based on the ASH data.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL who had relapsed or were refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant (HSCT) or who experienced disease progression after HSCT.
Interim results of the study were previously reported at the 22nd Congress of the European Hematology Association in 2017.
At that meeting, Gilles Salles, MD, PhD, of the University of Lyon in France, presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up.
In this population, the best overall response rate (ORR) was 59%. Three-month ORR was 45%, consisting of 37% complete responses (CR) and 8% partial responses (PR).
Relapse-free survival at 6 months was 79%, and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
The current analysis was completed after a median time from infusion to data cutoff of 19 months as of May 21, 2018. The analysis included 115 patients who received CAR T-cell infusions, 99 of whom were evaluable for efficacy.
As reported at ASH, the best ORR, the primary endpoint, was 54%, comprised of 40% CR and 13% PR.
Fifty-four percent of patients who had achieved PR converted to CR.
The response rates were consistent across all subgroups, regardless of age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 64%.
The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a CR.
Median overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months and not reached for patients in CR.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to the treatment, CRS, or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
The JULIET trial is supported by Novartis. Dr. Maziarz disclosed honoraria, consultancy fees, and/or research funding from Novartis, Incyte, Juno Therapeutics, and Kite Therapeutics as well as patents/royalties from Athersys, Inc.
SAN DIEGO—An updated analysis of the JULIET trial showed that tisagenlecleucel produced a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
After a median follow-up of 19 months, two-thirds of adults with relapsed/refractory DLBCL who had early responses to the chimeric antigen receptor (CAR) T-cell therapy remained in remission with no evidence of minimal residual disease.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression, no treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” said study investigator Richard Thomas Maziarz, MD, of Oregon Health & Science University’s Knight Cancer Institute in Portland.
Dr. Maziarz and his colleagues reported the updated study results at the 2018 ASH Annual Meeting (abstract 1684). Results were published simultaneously in The New England Journal of Medicine. Data reported here are based on the ASH data.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL who had relapsed or were refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant (HSCT) or who experienced disease progression after HSCT.
Interim results of the study were previously reported at the 22nd Congress of the European Hematology Association in 2017.
At that meeting, Gilles Salles, MD, PhD, of the University of Lyon in France, presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up.
In this population, the best overall response rate (ORR) was 59%. Three-month ORR was 45%, consisting of 37% complete responses (CR) and 8% partial responses (PR).
Relapse-free survival at 6 months was 79%, and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
The current analysis was completed after a median time from infusion to data cutoff of 19 months as of May 21, 2018. The analysis included 115 patients who received CAR T-cell infusions, 99 of whom were evaluable for efficacy.
As reported at ASH, the best ORR, the primary endpoint, was 54%, comprised of 40% CR and 13% PR.
Fifty-four percent of patients who had achieved PR converted to CR.
The response rates were consistent across all subgroups, regardless of age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 64%.
The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a CR.
Median overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months and not reached for patients in CR.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to the treatment, CRS, or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
The JULIET trial is supported by Novartis. Dr. Maziarz disclosed honoraria, consultancy fees, and/or research funding from Novartis, Incyte, Juno Therapeutics, and Kite Therapeutics as well as patents/royalties from Athersys, Inc.
