User login
Orelabrutinib could be ‘preferred’ BTK inhibitor for MCL
ORLANDO – A novel Bruton tyrosine kinase inhibitor has produced favorable results in patients with relapsed or refractory mantle cell lymphoma, according to findings presented at the annual meeting of the American Society of Hematology.

In a phase 2 trial, orelabrutinib produced an overall response rate of 86% and a 12-month progression-free survival rate of 64%. Safety results with orelabrutinib were superior to historical results with ibrutinib.
The efficacy and safety profile of orelabrutinib, as well as its “convenient” dosing, may make it the “preferred therapeutic choice for B-cell malignancy,” said Lijuan Deng, MD, PhD, of Peking University Cancer Hospital & Institute, Beijing, who presented the phase 2 trial of orelabrutinib at ASH 2019.
The trial enrolled 106 patients with relapsed/refractory mantle cell lymphoma who were treated at 22 centers in China. At baseline, the patients had a median age of 62 years (range, 37-73 years), and 79.2% were men. Most patients (94.4%) had stage III-IV disease.
Prior therapies included CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone)-based (69.8%), EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin)-based (22.6%), DHAP (dexamethasone, cytarabine, and cisplatin)-based (22.6%), CVAD (cyclophosphamide, vincristine, doxorubicin, dexamethasone)-based (12.3%), and ESHAP (etoposide, methylprednisolone, cytarabine, and cisplatin)-based (4.7%) regimens, and 88.7% of patients had received prior anti-CD20 therapy.
Patients received orelabrutinib at 100 mg twice daily (n = 20) or 150 mg once a day (n = 86). All 106 patients were evaluable for safety, and 99 were evaluable for efficacy.
Efficacy
“Orelabrutinib achieved high response and durable remissions,” Dr. Deng said.
The overall response rate was 85.9% in the evaluable efficacy population and 83.5% in the 150-mg dosing arm. The complete response rates were 27.3% and 29.1%, respectively. The median time to response, overall, was 1.9 months.
The median duration of response and median progression-free survival were not reached at a median follow-up of 10.5 months. At 12 months, 74.3% of patients were still in response, and the progression-free survival rate was 64%.
Safety
Most adverse events were grade 1-2 in nature. The most common grade 3 or higher events were platelet count decrease (11.3%), neutrophil count decrease (8.5%), anemia (7.5%), hypertension (3.8%), pneumonia (2.8%), white blood count decrease (1.9%), and hypokalemia (1.9%).
Adverse events of interest included grade 3 or higher hypertension (3.8%), diarrhea (6.6%), and infection (10.4%), as well as secondary malignancy (0.9%, n = 1). There were no cases of grade 3 or higher hemorrhage, grade 3 or higher atrial fibrillation/flutter, or grade 5 treatment-related adverse events.
Dr. Deng noted that rates of grade 3 or higher hemorrhage, atrial fibrillation, diarrhea, and infection, as well as rates of secondary malignancies, have historically been higher with ibrutinib (Blood. 2015 Aug 6;126[6]:739-45; Lancet. 2016 Feb 20;387[10020]:770-8).
“Orelabrutinib has an improved safety profile in patients with relapsed or refractory mantle cell lymphoma,” Dr. Deng said. “The most common adverse events were cytopenia and infections, which are considered mechanism based.”
The study was sponsored by InnoCare Pharma. Dr. Deng reported having no conflicts of interest.
SOURCE: Deng L et al. ASH 2019, Abstract 755.
ORLANDO – A novel Bruton tyrosine kinase inhibitor has produced favorable results in patients with relapsed or refractory mantle cell lymphoma, according to findings presented at the annual meeting of the American Society of Hematology.
In a phase 2 trial, orelabrutinib produced an overall response rate of 86% and a 12-month progression-free survival rate of 64%. Safety results with orelabrutinib were superior to historical results with ibrutinib.
The efficacy and safety profile of orelabrutinib, as well as its “convenient” dosing, may make it the “preferred therapeutic choice for B-cell malignancy,” said Lijuan Deng, MD, PhD, of Peking University Cancer Hospital & Institute, Beijing, who presented the phase 2 trial of orelabrutinib at ASH 2019.
The trial enrolled 106 patients with relapsed/refractory mantle cell lymphoma who were treated at 22 centers in China. At baseline, the patients had a median age of 62 years (range, 37-73 years), and 79.2% were men. Most patients (94.4%) had stage III-IV disease.
Prior therapies included CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone)-based (69.8%), EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin)-based (22.6%), DHAP (dexamethasone, cytarabine, and cisplatin)-based (22.6%), CVAD (cyclophosphamide, vincristine, doxorubicin, dexamethasone)-based (12.3%), and ESHAP (etoposide, methylprednisolone, cytarabine, and cisplatin)-based (4.7%) regimens, and 88.7% of patients had received prior anti-CD20 therapy.
Patients received orelabrutinib at 100 mg twice daily (n = 20) or 150 mg once a day (n = 86). All 106 patients were evaluable for safety, and 99 were evaluable for efficacy.
Efficacy
“Orelabrutinib achieved high response and durable remissions,” Dr. Deng said.
The overall response rate was 85.9% in the evaluable efficacy population and 83.5% in the 150-mg dosing arm. The complete response rates were 27.3% and 29.1%, respectively. The median time to response, overall, was 1.9 months.
The median duration of response and median progression-free survival were not reached at a median follow-up of 10.5 months. At 12 months, 74.3% of patients were still in response, and the progression-free survival rate was 64%.
Safety
Most adverse events were grade 1-2 in nature. The most common grade 3 or higher events were platelet count decrease (11.3%), neutrophil count decrease (8.5%), anemia (7.5%), hypertension (3.8%), pneumonia (2.8%), white blood count decrease (1.9%), and hypokalemia (1.9%).
Adverse events of interest included grade 3 or higher hypertension (3.8%), diarrhea (6.6%), and infection (10.4%), as well as secondary malignancy (0.9%, n = 1). There were no cases of grade 3 or higher hemorrhage, grade 3 or higher atrial fibrillation/flutter, or grade 5 treatment-related adverse events.
Dr. Deng noted that rates of grade 3 or higher hemorrhage, atrial fibrillation, diarrhea, and infection, as well as rates of secondary malignancies, have historically been higher with ibrutinib (Blood. 2015 Aug 6;126[6]:739-45; Lancet. 2016 Feb 20;387[10020]:770-8).
“Orelabrutinib has an improved safety profile in patients with relapsed or refractory mantle cell lymphoma,” Dr. Deng said. “The most common adverse events were cytopenia and infections, which are considered mechanism based.”
The study was sponsored by InnoCare Pharma. Dr. Deng reported having no conflicts of interest.
SOURCE: Deng L et al. ASH 2019, Abstract 755.
ORLANDO – A novel Bruton tyrosine kinase inhibitor has produced favorable results in patients with relapsed or refractory mantle cell lymphoma, according to findings presented at the annual meeting of the American Society of Hematology.
In a phase 2 trial, orelabrutinib produced an overall response rate of 86% and a 12-month progression-free survival rate of 64%. Safety results with orelabrutinib were superior to historical results with ibrutinib.
The efficacy and safety profile of orelabrutinib, as well as its “convenient” dosing, may make it the “preferred therapeutic choice for B-cell malignancy,” said Lijuan Deng, MD, PhD, of Peking University Cancer Hospital & Institute, Beijing, who presented the phase 2 trial of orelabrutinib at ASH 2019.
The trial enrolled 106 patients with relapsed/refractory mantle cell lymphoma who were treated at 22 centers in China. At baseline, the patients had a median age of 62 years (range, 37-73 years), and 79.2% were men. Most patients (94.4%) had stage III-IV disease.
Prior therapies included CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone)-based (69.8%), EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin)-based (22.6%), DHAP (dexamethasone, cytarabine, and cisplatin)-based (22.6%), CVAD (cyclophosphamide, vincristine, doxorubicin, dexamethasone)-based (12.3%), and ESHAP (etoposide, methylprednisolone, cytarabine, and cisplatin)-based (4.7%) regimens, and 88.7% of patients had received prior anti-CD20 therapy.
Patients received orelabrutinib at 100 mg twice daily (n = 20) or 150 mg once a day (n = 86). All 106 patients were evaluable for safety, and 99 were evaluable for efficacy.
Efficacy
“Orelabrutinib achieved high response and durable remissions,” Dr. Deng said.
The overall response rate was 85.9% in the evaluable efficacy population and 83.5% in the 150-mg dosing arm. The complete response rates were 27.3% and 29.1%, respectively. The median time to response, overall, was 1.9 months.
The median duration of response and median progression-free survival were not reached at a median follow-up of 10.5 months. At 12 months, 74.3% of patients were still in response, and the progression-free survival rate was 64%.
Safety
Most adverse events were grade 1-2 in nature. The most common grade 3 or higher events were platelet count decrease (11.3%), neutrophil count decrease (8.5%), anemia (7.5%), hypertension (3.8%), pneumonia (2.8%), white blood count decrease (1.9%), and hypokalemia (1.9%).
Adverse events of interest included grade 3 or higher hypertension (3.8%), diarrhea (6.6%), and infection (10.4%), as well as secondary malignancy (0.9%, n = 1). There were no cases of grade 3 or higher hemorrhage, grade 3 or higher atrial fibrillation/flutter, or grade 5 treatment-related adverse events.
Dr. Deng noted that rates of grade 3 or higher hemorrhage, atrial fibrillation, diarrhea, and infection, as well as rates of secondary malignancies, have historically been higher with ibrutinib (Blood. 2015 Aug 6;126[6]:739-45; Lancet. 2016 Feb 20;387[10020]:770-8).
“Orelabrutinib has an improved safety profile in patients with relapsed or refractory mantle cell lymphoma,” Dr. Deng said. “The most common adverse events were cytopenia and infections, which are considered mechanism based.”
The study was sponsored by InnoCare Pharma. Dr. Deng reported having no conflicts of interest.
SOURCE: Deng L et al. ASH 2019, Abstract 755.
REPORTING FROM ASH 2019
Efficacy of postvenetoclax therapy may depend on prior agent exposure in CLL
ORLANDO – For a patient with chronic lymphocytic leukemia (CLL) who has discontinued venetoclax, choosing the best next therapy may depend on what novel agents the patient was exposed to and why they discontinued them, according to Anthony R. Mato, MD, with the Center for CLL at Memorial Sloan Kettering Cancer Center in New York.

If the patient is Bruton tyrosine kinase (BTK) inhibitor naive, then use of a BTK inhibitor after venetoclax would be supported, Dr. Mato said, by the high overall response rates and durable remissions that he and his coinvestigators documented in a retrospective, multicenter study designed specifically to address the gap in knowledge regarding what to use after venetoclax.
If the patient is BTK inhibitor exposed, then the reason for discontinuation needs to be considered before going with that venetoclax-to-BTK inhibitor sequence, Dr. Mato said during an oral presentation at the annual meeting of the American Society of Hematology.
“In patients with resistance to a BTK inhibitor, the sequence was not supported – it did not appear to be effective,” he said. “However, in the setting of intolerance, an alternate BTK inhibitor could be considered.”
The study did not support a venetoclax-to-PI3K inhibitor sequence in PI3K-naive patients, he added, noting that remissions did not appear to be durable, suggesting a potential overlap in resistance mechanisms between agents.
All told, the most effective therapies for in the postvenetoclax setting included the use of a BTK inhibitor in BTK inhibitor–naive or previously responsive patients, and allogeneic transplant following double novel-agent exposure.
“These data may provide support for venetoclax’s earlier use in the course of CLL, and may guide clinical practice and aid in the design of future clinical trials to address sequencing of novel agents,” Dr. Mato told attendees.
While prospective and real-world data clearly show that venetoclax is active in ibrutinib- or idelalisib-exposed patients, data are conversely “variable and limited” with regard to outcomes for next therapies following venetoclax.
“Current data addressing this key sequencing question, I feel, is a major limitation in supporting the sequence of venetoclax to a BTK inhibitor,” Dr. Mato said.
Accordingly, Dr. Mato and colleagues at 31 centers internationally planned and conducted this study, which included data on 326 patients treated with venetoclax who then discontinued for any reason.
“I wanted to highlight that 50% of the sites for this trial were recruited by a single tweet,” said Dr. Mato, adding that he and his coauthors received no funding to conduct this study and volunteered their time to complete it.
They found that, in BTK inhibitor–naive patients who discontinued venetoclax, subsequent BTK inhibitor treatment was associated with a high overall response rate and durable remissions, with a median progression-free survival (PFS) of 32 months.
In BTK inhibitor–exposed patients, response to postvenetoclax BTK inhibitor treatment depended on the reason for discontinuation, with a favorable result (PFS not reached with a mean follow-up of 7.7 months) in patients who were intolerant of the prior BTK inhibitor. By contrast, median PFS was only about 4 months for patients who were resistant to the prior BTK inhibitor.
PI3K inhibitors did not produce durable remissions after venetoclax, with a median PFS also of just 4 months, Dr. Mato reported.
However, cellular therapies appeared to be effective after venetoclax. Allogeneic hematopoietic stem cell transplantation was particularly effective, with the median PFS not reached, while chimeric antigen receptor T-cell therapy produced a PFS of 9 months.
Dr. Mato emphasized that the results of the retrospective trial were “hypothesis generating” and noted that patients in the study had received a median of 3, and up to 11, prior therapies. “This population are probably not our patients receiving venetoclax in clinical practice. They’re more heavily pretreated.”
Dr. Mato reported disclosures related to Gilead, AstraZeneca, AbbVie, Sunesis, Johnson & Johnson, TG Therapeutics, Loxo Oncology, DTRM Biopharma, Genentech, Janssen, Acerta Pharma, Pharmacyclics, and Celgene.
SOURCE: Mato AR et al. ASH 2019, Abstract 502.
ORLANDO – For a patient with chronic lymphocytic leukemia (CLL) who has discontinued venetoclax, choosing the best next therapy may depend on what novel agents the patient was exposed to and why they discontinued them, according to Anthony R. Mato, MD, with the Center for CLL at Memorial Sloan Kettering Cancer Center in New York.
If the patient is Bruton tyrosine kinase (BTK) inhibitor naive, then use of a BTK inhibitor after venetoclax would be supported, Dr. Mato said, by the high overall response rates and durable remissions that he and his coinvestigators documented in a retrospective, multicenter study designed specifically to address the gap in knowledge regarding what to use after venetoclax.
If the patient is BTK inhibitor exposed, then the reason for discontinuation needs to be considered before going with that venetoclax-to-BTK inhibitor sequence, Dr. Mato said during an oral presentation at the annual meeting of the American Society of Hematology.
“In patients with resistance to a BTK inhibitor, the sequence was not supported – it did not appear to be effective,” he said. “However, in the setting of intolerance, an alternate BTK inhibitor could be considered.”
The study did not support a venetoclax-to-PI3K inhibitor sequence in PI3K-naive patients, he added, noting that remissions did not appear to be durable, suggesting a potential overlap in resistance mechanisms between agents.
All told, the most effective therapies for in the postvenetoclax setting included the use of a BTK inhibitor in BTK inhibitor–naive or previously responsive patients, and allogeneic transplant following double novel-agent exposure.
“These data may provide support for venetoclax’s earlier use in the course of CLL, and may guide clinical practice and aid in the design of future clinical trials to address sequencing of novel agents,” Dr. Mato told attendees.
While prospective and real-world data clearly show that venetoclax is active in ibrutinib- or idelalisib-exposed patients, data are conversely “variable and limited” with regard to outcomes for next therapies following venetoclax.
“Current data addressing this key sequencing question, I feel, is a major limitation in supporting the sequence of venetoclax to a BTK inhibitor,” Dr. Mato said.
Accordingly, Dr. Mato and colleagues at 31 centers internationally planned and conducted this study, which included data on 326 patients treated with venetoclax who then discontinued for any reason.
“I wanted to highlight that 50% of the sites for this trial were recruited by a single tweet,” said Dr. Mato, adding that he and his coauthors received no funding to conduct this study and volunteered their time to complete it.
They found that, in BTK inhibitor–naive patients who discontinued venetoclax, subsequent BTK inhibitor treatment was associated with a high overall response rate and durable remissions, with a median progression-free survival (PFS) of 32 months.
In BTK inhibitor–exposed patients, response to postvenetoclax BTK inhibitor treatment depended on the reason for discontinuation, with a favorable result (PFS not reached with a mean follow-up of 7.7 months) in patients who were intolerant of the prior BTK inhibitor. By contrast, median PFS was only about 4 months for patients who were resistant to the prior BTK inhibitor.
PI3K inhibitors did not produce durable remissions after venetoclax, with a median PFS also of just 4 months, Dr. Mato reported.
However, cellular therapies appeared to be effective after venetoclax. Allogeneic hematopoietic stem cell transplantation was particularly effective, with the median PFS not reached, while chimeric antigen receptor T-cell therapy produced a PFS of 9 months.
Dr. Mato emphasized that the results of the retrospective trial were “hypothesis generating” and noted that patients in the study had received a median of 3, and up to 11, prior therapies. “This population are probably not our patients receiving venetoclax in clinical practice. They’re more heavily pretreated.”
Dr. Mato reported disclosures related to Gilead, AstraZeneca, AbbVie, Sunesis, Johnson & Johnson, TG Therapeutics, Loxo Oncology, DTRM Biopharma, Genentech, Janssen, Acerta Pharma, Pharmacyclics, and Celgene.
SOURCE: Mato AR et al. ASH 2019, Abstract 502.
ORLANDO – For a patient with chronic lymphocytic leukemia (CLL) who has discontinued venetoclax, choosing the best next therapy may depend on what novel agents the patient was exposed to and why they discontinued them, according to Anthony R. Mato, MD, with the Center for CLL at Memorial Sloan Kettering Cancer Center in New York.
If the patient is Bruton tyrosine kinase (BTK) inhibitor naive, then use of a BTK inhibitor after venetoclax would be supported, Dr. Mato said, by the high overall response rates and durable remissions that he and his coinvestigators documented in a retrospective, multicenter study designed specifically to address the gap in knowledge regarding what to use after venetoclax.
If the patient is BTK inhibitor exposed, then the reason for discontinuation needs to be considered before going with that venetoclax-to-BTK inhibitor sequence, Dr. Mato said during an oral presentation at the annual meeting of the American Society of Hematology.
“In patients with resistance to a BTK inhibitor, the sequence was not supported – it did not appear to be effective,” he said. “However, in the setting of intolerance, an alternate BTK inhibitor could be considered.”
The study did not support a venetoclax-to-PI3K inhibitor sequence in PI3K-naive patients, he added, noting that remissions did not appear to be durable, suggesting a potential overlap in resistance mechanisms between agents.
All told, the most effective therapies for in the postvenetoclax setting included the use of a BTK inhibitor in BTK inhibitor–naive or previously responsive patients, and allogeneic transplant following double novel-agent exposure.
“These data may provide support for venetoclax’s earlier use in the course of CLL, and may guide clinical practice and aid in the design of future clinical trials to address sequencing of novel agents,” Dr. Mato told attendees.
While prospective and real-world data clearly show that venetoclax is active in ibrutinib- or idelalisib-exposed patients, data are conversely “variable and limited” with regard to outcomes for next therapies following venetoclax.
“Current data addressing this key sequencing question, I feel, is a major limitation in supporting the sequence of venetoclax to a BTK inhibitor,” Dr. Mato said.
Accordingly, Dr. Mato and colleagues at 31 centers internationally planned and conducted this study, which included data on 326 patients treated with venetoclax who then discontinued for any reason.
“I wanted to highlight that 50% of the sites for this trial were recruited by a single tweet,” said Dr. Mato, adding that he and his coauthors received no funding to conduct this study and volunteered their time to complete it.
They found that, in BTK inhibitor–naive patients who discontinued venetoclax, subsequent BTK inhibitor treatment was associated with a high overall response rate and durable remissions, with a median progression-free survival (PFS) of 32 months.
In BTK inhibitor–exposed patients, response to postvenetoclax BTK inhibitor treatment depended on the reason for discontinuation, with a favorable result (PFS not reached with a mean follow-up of 7.7 months) in patients who were intolerant of the prior BTK inhibitor. By contrast, median PFS was only about 4 months for patients who were resistant to the prior BTK inhibitor.
PI3K inhibitors did not produce durable remissions after venetoclax, with a median PFS also of just 4 months, Dr. Mato reported.
However, cellular therapies appeared to be effective after venetoclax. Allogeneic hematopoietic stem cell transplantation was particularly effective, with the median PFS not reached, while chimeric antigen receptor T-cell therapy produced a PFS of 9 months.
Dr. Mato emphasized that the results of the retrospective trial were “hypothesis generating” and noted that patients in the study had received a median of 3, and up to 11, prior therapies. “This population are probably not our patients receiving venetoclax in clinical practice. They’re more heavily pretreated.”
Dr. Mato reported disclosures related to Gilead, AstraZeneca, AbbVie, Sunesis, Johnson & Johnson, TG Therapeutics, Loxo Oncology, DTRM Biopharma, Genentech, Janssen, Acerta Pharma, Pharmacyclics, and Celgene.
SOURCE: Mato AR et al. ASH 2019, Abstract 502.
REPORTING FROM ASH 2019
Care coordination, equity can eliminate disparities for nonwhite patients with DLBCL
ORLANDO – Patients with diffuse large B-cell lymphoma (DLBCL) who are members of an ethnic or racial minority do not have worse outcomes than whites when they receive appropriate treatment and institutional support, a study on disparities in cancer care shows.

Although previous studies have shown that minorities with DLBCL have worse outcomes than do whites, results of a study comparing outcomes from 155 patients of white heritage with those of 41 patients from black, Hispanic, or other minority backgrounds found no significant differences in either progression-free survival (PFS) or overall survival in 2 years over follow-up, reported Nilanjan Ghosh, MD, PhD, from the Levine Cancer Institute, Atrium Health, in Charlotte, N.C.
He attributes the results to his center’s robust nurse navigation program, equal access among all patients – regardless of ability to pay – to standard treatments, and to the availability of clinical trial participation and stem cell transplantation.
“I think a key message is that if you are able to offer the same treatment and clinical trials to people irrespective of their race or socioeconomic status and can provide support, you can get equal outcomes as long as the biology is the same in both groups,” he said at a briefing prior to presentation of data in an oral abstract session at the annual meeting of the American Society of Hematology.
Dr. Ghosh pointed to four separate studies that showed that minority populations with DLBCL have worse outcomes than did whites, and noted that both uninsured and Medicaid-insured patients have also been shown to have poorer results, suggesting a role of socioeconomic factors in determining who gets optimum care and who does not.
The investigators compared PFS and OS among white and nonwhite patients with DLBCL treated in their institution, which has a safety-net cancer center. They also looked at the frequencies of clinical trial participation and stem cell transplantation between the groups.
The study included all patients with de novo DLBCL who presented to their center during January 2016–January 2019. They used patient-reported descriptors of race/ethnicity to create one of two cohorts: either self-identified whites (155 patients) or nonwhites (41), a group that included black patients, Hispanic patients, Asian Americans, and Native Americans.
The authors collected data on demographics, disease characteristics (including revised International Prognostic Index and double-hit status), insurance data, treatment, trial enrollment, progression, and death.
They found that nonwhites were significantly younger at diagnosis (median 56 vs. 64 years; P = .007), with an even distribution between the sexes in each group.
Two-thirds of both white and nonwhite patients had government insurance (Medicare or Medicaid). Of the remaining patients, 33% of white had private insurance, compared with 27% of nonwhites. No whites were uninsured, but 3 of the 41 nonwhites (7%) had no insurance.
Of the 155 white patients, 121 (86%) received nurse navigation services, as did 33 of 41 (81%) of nonwhites. The services include lodging assistance for homeless patients, transportation services for patients without cars, and care coordination among primary care physicians, oncologists, and other specialists. The services are part of the center’s standard practice, with excess costs, if any, folded into the budget, Dr. Ghosh said.
Looking at disease characteristics and treatment, the investigators found that risk profiles were similar between the groups. A higher percentage of whites had double-hit lymphoma (11% vs. 7%), but this difference was not statistically significant.
The investigators also found that in their program race was not a barrier to optimum therapy, with 96% of whites and 98% of nonwhites receiving frontline therapy with an anthracycline and rituximab-based regimen, and 4% and 2%, respectively received a non–anthracycline based regimen.
In each group, 39% of patients had disease that either relapsed or was refractory to frontline therapy.
In all, 11% of whites and 12% of nonwhites enrolled in clinical trials, 11% and 19%, respectively, underwent stem cell transplantation.
For patients with relapsed/refractory disease, the 2-year PFS rates were 60% for whites, and 63% for nonwhites, and the 2-year OS rates were 74% and 81%, respectively.
Dr. Ghosh and colleagues concluded that “our safety net cancer center, with extensive nurse navigator support and access to standard treatments, stem cell transplants, and cutting-edge clinical trials may abrogate the inferior outcomes in minority populations that have been previously reported.”
The study was internally funded. Dr. Ghosh reported consulting fees, research funding, speakers bureau activity, and/or honoraria from multiple companies.
SOURCE: Hu B et al. ASH 2019. Abstract 425.
ORLANDO – Patients with diffuse large B-cell lymphoma (DLBCL) who are members of an ethnic or racial minority do not have worse outcomes than whites when they receive appropriate treatment and institutional support, a study on disparities in cancer care shows.
Although previous studies have shown that minorities with DLBCL have worse outcomes than do whites, results of a study comparing outcomes from 155 patients of white heritage with those of 41 patients from black, Hispanic, or other minority backgrounds found no significant differences in either progression-free survival (PFS) or overall survival in 2 years over follow-up, reported Nilanjan Ghosh, MD, PhD, from the Levine Cancer Institute, Atrium Health, in Charlotte, N.C.
He attributes the results to his center’s robust nurse navigation program, equal access among all patients – regardless of ability to pay – to standard treatments, and to the availability of clinical trial participation and stem cell transplantation.
“I think a key message is that if you are able to offer the same treatment and clinical trials to people irrespective of their race or socioeconomic status and can provide support, you can get equal outcomes as long as the biology is the same in both groups,” he said at a briefing prior to presentation of data in an oral abstract session at the annual meeting of the American Society of Hematology.
Dr. Ghosh pointed to four separate studies that showed that minority populations with DLBCL have worse outcomes than did whites, and noted that both uninsured and Medicaid-insured patients have also been shown to have poorer results, suggesting a role of socioeconomic factors in determining who gets optimum care and who does not.
The investigators compared PFS and OS among white and nonwhite patients with DLBCL treated in their institution, which has a safety-net cancer center. They also looked at the frequencies of clinical trial participation and stem cell transplantation between the groups.
The study included all patients with de novo DLBCL who presented to their center during January 2016–January 2019. They used patient-reported descriptors of race/ethnicity to create one of two cohorts: either self-identified whites (155 patients) or nonwhites (41), a group that included black patients, Hispanic patients, Asian Americans, and Native Americans.
The authors collected data on demographics, disease characteristics (including revised International Prognostic Index and double-hit status), insurance data, treatment, trial enrollment, progression, and death.
They found that nonwhites were significantly younger at diagnosis (median 56 vs. 64 years; P = .007), with an even distribution between the sexes in each group.
Two-thirds of both white and nonwhite patients had government insurance (Medicare or Medicaid). Of the remaining patients, 33% of white had private insurance, compared with 27% of nonwhites. No whites were uninsured, but 3 of the 41 nonwhites (7%) had no insurance.
Of the 155 white patients, 121 (86%) received nurse navigation services, as did 33 of 41 (81%) of nonwhites. The services include lodging assistance for homeless patients, transportation services for patients without cars, and care coordination among primary care physicians, oncologists, and other specialists. The services are part of the center’s standard practice, with excess costs, if any, folded into the budget, Dr. Ghosh said.
Looking at disease characteristics and treatment, the investigators found that risk profiles were similar between the groups. A higher percentage of whites had double-hit lymphoma (11% vs. 7%), but this difference was not statistically significant.
The investigators also found that in their program race was not a barrier to optimum therapy, with 96% of whites and 98% of nonwhites receiving frontline therapy with an anthracycline and rituximab-based regimen, and 4% and 2%, respectively received a non–anthracycline based regimen.
In each group, 39% of patients had disease that either relapsed or was refractory to frontline therapy.
In all, 11% of whites and 12% of nonwhites enrolled in clinical trials, 11% and 19%, respectively, underwent stem cell transplantation.
For patients with relapsed/refractory disease, the 2-year PFS rates were 60% for whites, and 63% for nonwhites, and the 2-year OS rates were 74% and 81%, respectively.
Dr. Ghosh and colleagues concluded that “our safety net cancer center, with extensive nurse navigator support and access to standard treatments, stem cell transplants, and cutting-edge clinical trials may abrogate the inferior outcomes in minority populations that have been previously reported.”
The study was internally funded. Dr. Ghosh reported consulting fees, research funding, speakers bureau activity, and/or honoraria from multiple companies.
SOURCE: Hu B et al. ASH 2019. Abstract 425.
ORLANDO – Patients with diffuse large B-cell lymphoma (DLBCL) who are members of an ethnic or racial minority do not have worse outcomes than whites when they receive appropriate treatment and institutional support, a study on disparities in cancer care shows.
Although previous studies have shown that minorities with DLBCL have worse outcomes than do whites, results of a study comparing outcomes from 155 patients of white heritage with those of 41 patients from black, Hispanic, or other minority backgrounds found no significant differences in either progression-free survival (PFS) or overall survival in 2 years over follow-up, reported Nilanjan Ghosh, MD, PhD, from the Levine Cancer Institute, Atrium Health, in Charlotte, N.C.
He attributes the results to his center’s robust nurse navigation program, equal access among all patients – regardless of ability to pay – to standard treatments, and to the availability of clinical trial participation and stem cell transplantation.
“I think a key message is that if you are able to offer the same treatment and clinical trials to people irrespective of their race or socioeconomic status and can provide support, you can get equal outcomes as long as the biology is the same in both groups,” he said at a briefing prior to presentation of data in an oral abstract session at the annual meeting of the American Society of Hematology.
Dr. Ghosh pointed to four separate studies that showed that minority populations with DLBCL have worse outcomes than did whites, and noted that both uninsured and Medicaid-insured patients have also been shown to have poorer results, suggesting a role of socioeconomic factors in determining who gets optimum care and who does not.
The investigators compared PFS and OS among white and nonwhite patients with DLBCL treated in their institution, which has a safety-net cancer center. They also looked at the frequencies of clinical trial participation and stem cell transplantation between the groups.
The study included all patients with de novo DLBCL who presented to their center during January 2016–January 2019. They used patient-reported descriptors of race/ethnicity to create one of two cohorts: either self-identified whites (155 patients) or nonwhites (41), a group that included black patients, Hispanic patients, Asian Americans, and Native Americans.
The authors collected data on demographics, disease characteristics (including revised International Prognostic Index and double-hit status), insurance data, treatment, trial enrollment, progression, and death.
They found that nonwhites were significantly younger at diagnosis (median 56 vs. 64 years; P = .007), with an even distribution between the sexes in each group.
Two-thirds of both white and nonwhite patients had government insurance (Medicare or Medicaid). Of the remaining patients, 33% of white had private insurance, compared with 27% of nonwhites. No whites were uninsured, but 3 of the 41 nonwhites (7%) had no insurance.
Of the 155 white patients, 121 (86%) received nurse navigation services, as did 33 of 41 (81%) of nonwhites. The services include lodging assistance for homeless patients, transportation services for patients without cars, and care coordination among primary care physicians, oncologists, and other specialists. The services are part of the center’s standard practice, with excess costs, if any, folded into the budget, Dr. Ghosh said.
Looking at disease characteristics and treatment, the investigators found that risk profiles were similar between the groups. A higher percentage of whites had double-hit lymphoma (11% vs. 7%), but this difference was not statistically significant.
The investigators also found that in their program race was not a barrier to optimum therapy, with 96% of whites and 98% of nonwhites receiving frontline therapy with an anthracycline and rituximab-based regimen, and 4% and 2%, respectively received a non–anthracycline based regimen.
In each group, 39% of patients had disease that either relapsed or was refractory to frontline therapy.
In all, 11% of whites and 12% of nonwhites enrolled in clinical trials, 11% and 19%, respectively, underwent stem cell transplantation.
For patients with relapsed/refractory disease, the 2-year PFS rates were 60% for whites, and 63% for nonwhites, and the 2-year OS rates were 74% and 81%, respectively.
Dr. Ghosh and colleagues concluded that “our safety net cancer center, with extensive nurse navigator support and access to standard treatments, stem cell transplants, and cutting-edge clinical trials may abrogate the inferior outcomes in minority populations that have been previously reported.”
The study was internally funded. Dr. Ghosh reported consulting fees, research funding, speakers bureau activity, and/or honoraria from multiple companies.
SOURCE: Hu B et al. ASH 2019. Abstract 425.
REPORTING FROM ASH 2019
Off-the-shelf cellular therapy shows promise in the lab
ORLANDO – A cellular therapy called FT596 is active against B-cell malignancies and, when combined with rituximab, can be more effective than traditional chimeric antigen receptor (CAR) T cells, preclinical research findings suggest.

FT596 is a universal, anti-CD19 CAR natural killer (NK) cell therapy derived from a master induced pluripotent stem cell (iPSC) line.
FT596 reduced tumor growth in mouse models of leukemia and lymphoma. When combined with rituximab, FT596 was able to overcome CD19 antigen escape.
Jode P. Goodridge, PhD, of Fate Therapeutics in San Diego, presented these results at the annual meeting of the American Society of Hematology.
Dr. Goodridge explained that FT596 begins with a source material, such as a fibroblast, that is reprogrammed into an iPSC progenitor cell. That cell is sorted and expanded into a renewable, homogeneous, pluripotent master iPSC line. The iPSCs are differentiated into CD34 cells, which are differentiated into NK cells. The iPSC-derived NK cells are then modified with the following:
- An anti-CD19 CAR that is optimized for NK-cell biology and contains an NKG2D transmembrane domain, a 2B4 costimulatory domain, and a CD3-zeta signaling domain.
- An interleukin-15 receptor fusion that promotes cell survival and reduces the need for cytokine support.
- A high-affinity 158V, noncleavable CD16 Fc receptor that enhances antibody-dependent cellular cytotoxicity when FT596 is combined with a monoclonal antibody such as rituximab.
Dr. Goodridge presented results with FT596, both alone and in combination with rituximab, in vitro and in vivo.
When compared with no treatment, three doses of FT596 monotherapy reduced tumor growth in a mouse model of leukemia (Nalm6). FT596 plus rituximab reduced tumor growth in a mouse model of lymphoma (Raji), when compared with no treatment or rituximab alone.
Three doses of FT596 proved more effective than a single dose of CD19 CAR T-cell therapy in a mouse model of lymphoma (Raji). FT596 both reduced tumor growth and prolonged survival in the mice.
Lastly, in vitro experiments in Raji cells showed that FT596 plus rituximab can produce deeper responses than primary CAR-T cells, and the combination can prevent antigen escape.
Dr. Goodridge said these results support the phase 1 study of FT596, given as monotherapy or in combination with rituximab or obinutuzumab, in patients with relapsed/refractory B-cell lymphomas or chronic lymphocytic leukemia.
Dr. Goodridge is employed by Fate Therapeutics, the company developing FT596.
SOURCE: Goodridge JP et al. ASH 2019. Abstract 301.
ORLANDO – A cellular therapy called FT596 is active against B-cell malignancies and, when combined with rituximab, can be more effective than traditional chimeric antigen receptor (CAR) T cells, preclinical research findings suggest.
FT596 is a universal, anti-CD19 CAR natural killer (NK) cell therapy derived from a master induced pluripotent stem cell (iPSC) line.
FT596 reduced tumor growth in mouse models of leukemia and lymphoma. When combined with rituximab, FT596 was able to overcome CD19 antigen escape.
Jode P. Goodridge, PhD, of Fate Therapeutics in San Diego, presented these results at the annual meeting of the American Society of Hematology.
Dr. Goodridge explained that FT596 begins with a source material, such as a fibroblast, that is reprogrammed into an iPSC progenitor cell. That cell is sorted and expanded into a renewable, homogeneous, pluripotent master iPSC line. The iPSCs are differentiated into CD34 cells, which are differentiated into NK cells. The iPSC-derived NK cells are then modified with the following:
- An anti-CD19 CAR that is optimized for NK-cell biology and contains an NKG2D transmembrane domain, a 2B4 costimulatory domain, and a CD3-zeta signaling domain.
- An interleukin-15 receptor fusion that promotes cell survival and reduces the need for cytokine support.
- A high-affinity 158V, noncleavable CD16 Fc receptor that enhances antibody-dependent cellular cytotoxicity when FT596 is combined with a monoclonal antibody such as rituximab.
Dr. Goodridge presented results with FT596, both alone and in combination with rituximab, in vitro and in vivo.
When compared with no treatment, three doses of FT596 monotherapy reduced tumor growth in a mouse model of leukemia (Nalm6). FT596 plus rituximab reduced tumor growth in a mouse model of lymphoma (Raji), when compared with no treatment or rituximab alone.
Three doses of FT596 proved more effective than a single dose of CD19 CAR T-cell therapy in a mouse model of lymphoma (Raji). FT596 both reduced tumor growth and prolonged survival in the mice.
Lastly, in vitro experiments in Raji cells showed that FT596 plus rituximab can produce deeper responses than primary CAR-T cells, and the combination can prevent antigen escape.
Dr. Goodridge said these results support the phase 1 study of FT596, given as monotherapy or in combination with rituximab or obinutuzumab, in patients with relapsed/refractory B-cell lymphomas or chronic lymphocytic leukemia.
Dr. Goodridge is employed by Fate Therapeutics, the company developing FT596.
SOURCE: Goodridge JP et al. ASH 2019. Abstract 301.
ORLANDO – A cellular therapy called FT596 is active against B-cell malignancies and, when combined with rituximab, can be more effective than traditional chimeric antigen receptor (CAR) T cells, preclinical research findings suggest.
FT596 is a universal, anti-CD19 CAR natural killer (NK) cell therapy derived from a master induced pluripotent stem cell (iPSC) line.
FT596 reduced tumor growth in mouse models of leukemia and lymphoma. When combined with rituximab, FT596 was able to overcome CD19 antigen escape.
Jode P. Goodridge, PhD, of Fate Therapeutics in San Diego, presented these results at the annual meeting of the American Society of Hematology.
Dr. Goodridge explained that FT596 begins with a source material, such as a fibroblast, that is reprogrammed into an iPSC progenitor cell. That cell is sorted and expanded into a renewable, homogeneous, pluripotent master iPSC line. The iPSCs are differentiated into CD34 cells, which are differentiated into NK cells. The iPSC-derived NK cells are then modified with the following:
- An anti-CD19 CAR that is optimized for NK-cell biology and contains an NKG2D transmembrane domain, a 2B4 costimulatory domain, and a CD3-zeta signaling domain.
- An interleukin-15 receptor fusion that promotes cell survival and reduces the need for cytokine support.
- A high-affinity 158V, noncleavable CD16 Fc receptor that enhances antibody-dependent cellular cytotoxicity when FT596 is combined with a monoclonal antibody such as rituximab.
Dr. Goodridge presented results with FT596, both alone and in combination with rituximab, in vitro and in vivo.
When compared with no treatment, three doses of FT596 monotherapy reduced tumor growth in a mouse model of leukemia (Nalm6). FT596 plus rituximab reduced tumor growth in a mouse model of lymphoma (Raji), when compared with no treatment or rituximab alone.
Three doses of FT596 proved more effective than a single dose of CD19 CAR T-cell therapy in a mouse model of lymphoma (Raji). FT596 both reduced tumor growth and prolonged survival in the mice.
Lastly, in vitro experiments in Raji cells showed that FT596 plus rituximab can produce deeper responses than primary CAR-T cells, and the combination can prevent antigen escape.
Dr. Goodridge said these results support the phase 1 study of FT596, given as monotherapy or in combination with rituximab or obinutuzumab, in patients with relapsed/refractory B-cell lymphomas or chronic lymphocytic leukemia.
Dr. Goodridge is employed by Fate Therapeutics, the company developing FT596.
SOURCE: Goodridge JP et al. ASH 2019. Abstract 301.
REPORTING FROM ASH 2019
Newly identified genetic changes contribute to transformation of follicular lymphoma
A molecular analysis of a patient with follicular lymphoma (FL) that became B-lymphoblastic leukemia/lymphoma (B-ALL/LBL) uncovered genetic changes that may improve understanding of this transformation.
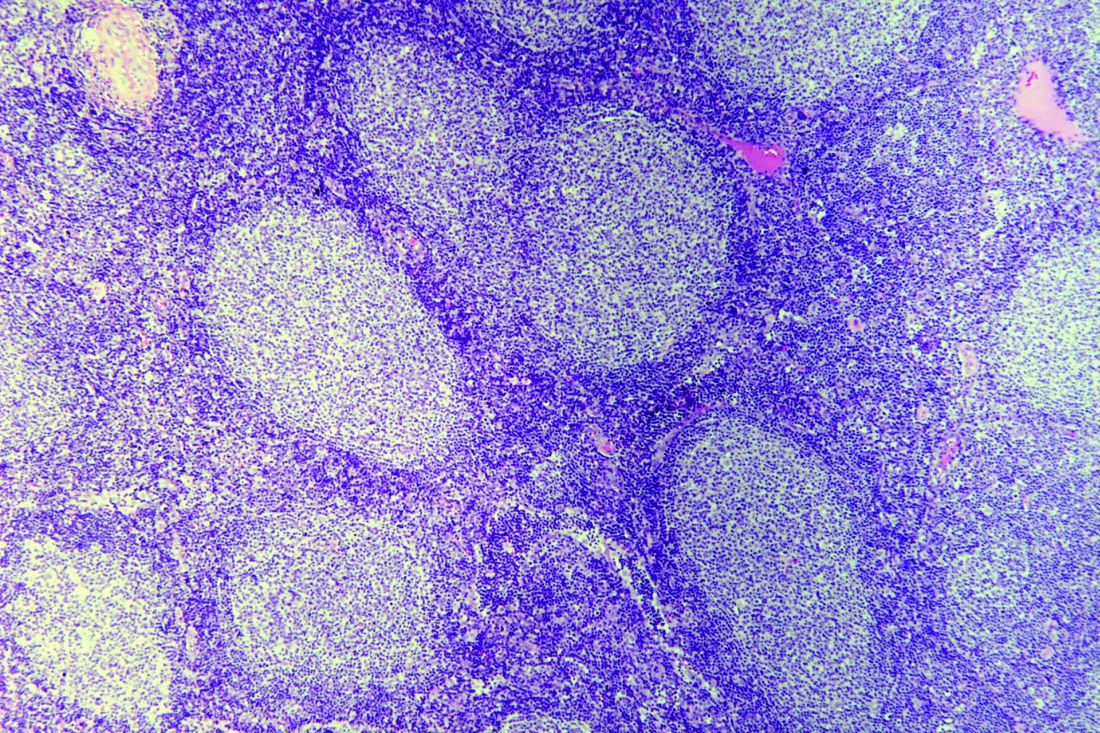
“The study provides new insights into the pathogenesis of FL-B-ALL/LBL transformation and suggests novel, disease biology–based therapeutic approaches to this aggressive and currently incurable disease,” wrote Jonathan Belman, MD, PhD, of the Hospital of the University of Pennsylvania, Philadelphia, and coauthors. Their findings were published in Cold Spring Harbor Molecular Case Studies.
To further understand the rare occasions when FL transforms into a more aggressive form of lymphoma, the researchers investigated a 36-year-old man with low-grade FL that became B-ALL/LBL roughly 1 year after diagnosis. Their analysis included immunoglobulin (Ig) gene rearrangement studies, cytogenetic analysis, and whole exome sequencing of the patient’s FL, B-ALL/LBL, and normal cells.
Next generation sequencing of Ig rearrangements from normal, FL, and B-ALL/LBL specimens revealed considerable somatic hypermutation (SHM) in a single neoplastic clone – IgHV4-34_JH6 – in the FL cells. By comparison, though no dominant clone was found in the B-ALL/LBL specimen, there was even more extensive SHM, along with clones that could be traced to the FL lineage.
In addition, fluorescence in situ hybridization (FISH) studies on the FL specimen were positive for rearrangements of BCL2 and BCL6 genes; a rearrangement of BCL6 is associated with clinical aggressiveness. Those same two rearrangements were found in the B-ALL/LBL specimen, along with a MYC gene rearrangement unseen in the FL.
“MYC translocations are well known to contribute to high aggressiveness of lymphomas, and are hallmark of FL-B-ALL/LBL transformation,” the researchers wrote.
Finally, comparative whole exome sequencing of normal tissue plus the cancerous specimens identified 751 single nucleotide variants. The normal tissue contained 111 of those mutations, while the FL specimens contained 116 mutations – 11 of which were shared solely with B-ALL/LBL – and the B-ALL/LBL specimens contained a striking 575 unique mutations. Notably, a nonsense mutation in the KMT2D gene that was shared by FL and B-ALL/LBL may have contributed to lymphomagenesis.
The study was funded by a grant from the National Institutes of Health, the Abramson Cancer Center Translational Center in Lymphoma, the Daniel B. Allanoff Foundation, and a Hematopathology Divisional Training grant.
SOURCE: Belman JP et al. Cold Spring Harb Mol Case Stud. 2019 Nov 27. doi: 10.1101/mcs.a004614.
A molecular analysis of a patient with follicular lymphoma (FL) that became B-lymphoblastic leukemia/lymphoma (B-ALL/LBL) uncovered genetic changes that may improve understanding of this transformation.
“The study provides new insights into the pathogenesis of FL-B-ALL/LBL transformation and suggests novel, disease biology–based therapeutic approaches to this aggressive and currently incurable disease,” wrote Jonathan Belman, MD, PhD, of the Hospital of the University of Pennsylvania, Philadelphia, and coauthors. Their findings were published in Cold Spring Harbor Molecular Case Studies.
To further understand the rare occasions when FL transforms into a more aggressive form of lymphoma, the researchers investigated a 36-year-old man with low-grade FL that became B-ALL/LBL roughly 1 year after diagnosis. Their analysis included immunoglobulin (Ig) gene rearrangement studies, cytogenetic analysis, and whole exome sequencing of the patient’s FL, B-ALL/LBL, and normal cells.
Next generation sequencing of Ig rearrangements from normal, FL, and B-ALL/LBL specimens revealed considerable somatic hypermutation (SHM) in a single neoplastic clone – IgHV4-34_JH6 – in the FL cells. By comparison, though no dominant clone was found in the B-ALL/LBL specimen, there was even more extensive SHM, along with clones that could be traced to the FL lineage.
In addition, fluorescence in situ hybridization (FISH) studies on the FL specimen were positive for rearrangements of BCL2 and BCL6 genes; a rearrangement of BCL6 is associated with clinical aggressiveness. Those same two rearrangements were found in the B-ALL/LBL specimen, along with a MYC gene rearrangement unseen in the FL.
“MYC translocations are well known to contribute to high aggressiveness of lymphomas, and are hallmark of FL-B-ALL/LBL transformation,” the researchers wrote.
Finally, comparative whole exome sequencing of normal tissue plus the cancerous specimens identified 751 single nucleotide variants. The normal tissue contained 111 of those mutations, while the FL specimens contained 116 mutations – 11 of which were shared solely with B-ALL/LBL – and the B-ALL/LBL specimens contained a striking 575 unique mutations. Notably, a nonsense mutation in the KMT2D gene that was shared by FL and B-ALL/LBL may have contributed to lymphomagenesis.
The study was funded by a grant from the National Institutes of Health, the Abramson Cancer Center Translational Center in Lymphoma, the Daniel B. Allanoff Foundation, and a Hematopathology Divisional Training grant.
SOURCE: Belman JP et al. Cold Spring Harb Mol Case Stud. 2019 Nov 27. doi: 10.1101/mcs.a004614.
A molecular analysis of a patient with follicular lymphoma (FL) that became B-lymphoblastic leukemia/lymphoma (B-ALL/LBL) uncovered genetic changes that may improve understanding of this transformation.
“The study provides new insights into the pathogenesis of FL-B-ALL/LBL transformation and suggests novel, disease biology–based therapeutic approaches to this aggressive and currently incurable disease,” wrote Jonathan Belman, MD, PhD, of the Hospital of the University of Pennsylvania, Philadelphia, and coauthors. Their findings were published in Cold Spring Harbor Molecular Case Studies.
To further understand the rare occasions when FL transforms into a more aggressive form of lymphoma, the researchers investigated a 36-year-old man with low-grade FL that became B-ALL/LBL roughly 1 year after diagnosis. Their analysis included immunoglobulin (Ig) gene rearrangement studies, cytogenetic analysis, and whole exome sequencing of the patient’s FL, B-ALL/LBL, and normal cells.
Next generation sequencing of Ig rearrangements from normal, FL, and B-ALL/LBL specimens revealed considerable somatic hypermutation (SHM) in a single neoplastic clone – IgHV4-34_JH6 – in the FL cells. By comparison, though no dominant clone was found in the B-ALL/LBL specimen, there was even more extensive SHM, along with clones that could be traced to the FL lineage.
In addition, fluorescence in situ hybridization (FISH) studies on the FL specimen were positive for rearrangements of BCL2 and BCL6 genes; a rearrangement of BCL6 is associated with clinical aggressiveness. Those same two rearrangements were found in the B-ALL/LBL specimen, along with a MYC gene rearrangement unseen in the FL.
“MYC translocations are well known to contribute to high aggressiveness of lymphomas, and are hallmark of FL-B-ALL/LBL transformation,” the researchers wrote.
Finally, comparative whole exome sequencing of normal tissue plus the cancerous specimens identified 751 single nucleotide variants. The normal tissue contained 111 of those mutations, while the FL specimens contained 116 mutations – 11 of which were shared solely with B-ALL/LBL – and the B-ALL/LBL specimens contained a striking 575 unique mutations. Notably, a nonsense mutation in the KMT2D gene that was shared by FL and B-ALL/LBL may have contributed to lymphomagenesis.
The study was funded by a grant from the National Institutes of Health, the Abramson Cancer Center Translational Center in Lymphoma, the Daniel B. Allanoff Foundation, and a Hematopathology Divisional Training grant.
SOURCE: Belman JP et al. Cold Spring Harb Mol Case Stud. 2019 Nov 27. doi: 10.1101/mcs.a004614.
FROM COLD SPRING HARBOR MOLECULAR CASE STUDIES
First generics for Gilenya approved by FDA
The Food and Drug Administration has approved the first generics of fingolimod (Gilenya) for the treatment of relapsing forms of multiple sclerosis.
The three generic fingolimod applications came from HEC Pharm, Biocon, and Sun Pharmaceutical Industries.
Fingolimod is a widely used, orally administered treatment option for relapsing forms of multiple sclerosis in adults. The most common adverse events associated with fingolimod in clinical trials include headache, elevation of liver enzymes, diarrhea, cough, influenza, sinusitis, back pain, abdominal pain, and pain in the extremities.
The drug must be dispensed with a medication guide that contains important information on its usage and risk, the FDA noted. Serious risks associated with fingolimod include slowing of the heart rate, vision problems, posterior reversible encephalopathy syndrome, respiratory problems, liver injury, increased blood pressure, skin cancer, and risk of serious infection including a rare and often deadly brain infection called progressive multifocal leukoencephalopathy. Fingolimod can also cause harm to a developing fetus.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generics of fingolimod (Gilenya) for the treatment of relapsing forms of multiple sclerosis.
The three generic fingolimod applications came from HEC Pharm, Biocon, and Sun Pharmaceutical Industries.
Fingolimod is a widely used, orally administered treatment option for relapsing forms of multiple sclerosis in adults. The most common adverse events associated with fingolimod in clinical trials include headache, elevation of liver enzymes, diarrhea, cough, influenza, sinusitis, back pain, abdominal pain, and pain in the extremities.
The drug must be dispensed with a medication guide that contains important information on its usage and risk, the FDA noted. Serious risks associated with fingolimod include slowing of the heart rate, vision problems, posterior reversible encephalopathy syndrome, respiratory problems, liver injury, increased blood pressure, skin cancer, and risk of serious infection including a rare and often deadly brain infection called progressive multifocal leukoencephalopathy. Fingolimod can also cause harm to a developing fetus.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generics of fingolimod (Gilenya) for the treatment of relapsing forms of multiple sclerosis.
The three generic fingolimod applications came from HEC Pharm, Biocon, and Sun Pharmaceutical Industries.
Fingolimod is a widely used, orally administered treatment option for relapsing forms of multiple sclerosis in adults. The most common adverse events associated with fingolimod in clinical trials include headache, elevation of liver enzymes, diarrhea, cough, influenza, sinusitis, back pain, abdominal pain, and pain in the extremities.
The drug must be dispensed with a medication guide that contains important information on its usage and risk, the FDA noted. Serious risks associated with fingolimod include slowing of the heart rate, vision problems, posterior reversible encephalopathy syndrome, respiratory problems, liver injury, increased blood pressure, skin cancer, and risk of serious infection including a rare and often deadly brain infection called progressive multifocal leukoencephalopathy. Fingolimod can also cause harm to a developing fetus.
Find the full press release on the FDA website.
The clinical impact of new approvals in sickle cell, MCL
In this edition of “How I Will Treat My Next Patient,” I highlight two recent drug approvals by the Food and Drug Administration – crizanlizumab for sickle cell patients with painful crises and zanubrutinib for mantle cell lymphoma (MCL) patients in relapse.

Crizanlizumab
P-selectin is an adhesion molecule expressed on activated vascular endothelial cells and platelets. It is a key molecule in the initiation of leukocyte rolling on vessel walls and promotes firm attachment and extravasation to underlying tissues during inflammation. Up-regulation of P-selectin on endothelial cells and platelets contributes to the cell-cell interactions involved in the pathogenesis of sickle cell pain crises.
The SUSTAIN study was a multisite, placebo-controlled, randomized phase 2 trial of two different dosage levels of intravenous crizanlizumab (2.5 mg/kg or 5 mg/kg for 52 weeks), a humanized anti–P-selectin antibody, examining its effect on pain crises in patients with sickle cell disease. The primary endpoint was the annual rate of sickle cell pain crises, with a variety of clinically relevant secondary endpoints. The target population had 2-10 pain crises in the 12 months before enrollment. Patients on a stable dose of hydroxyurea for at least the most recent 3 months were allowed to enter, but if patients were not receiving hydroxyurea, it could not be initiated during the trial. Patients who were undergoing chronic red-cell transfusion therapy were excluded.
Among 198 enrolled patients, 35% did not complete the 52 weeks of treatment. Discontinuations were equally balanced among patients assigned to the high-dose, low-dose, and placebo cohorts. Adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. Serious adverse events occurred in 55 patients, with 5 deaths, all of which were unrelated to treatment. Crizanlizumab did not augment hemolysis or bacterial infections.
In the efficacy analysis, patients receiving high-dose crizanlizumab had a median annual rate of 1.63 health care visits for sickle cell pain crises, compared with 2.98 visits for placebo patients (P = .01). In comparison with placebo, high-dose crizanlizumab also delayed the first pain crisis after starting treatment (4.1 months vs. 1.4 months), delayed the median time to a second pain crisis, and decreased the median number of pain crises annually.
More than twice as many high-dose crizanlizumab patients had no pain crisis episodes, compared with placebo patients. In general, differences were more striking in patients who were not taking hydroxyurea and who had non–hemoglobin SS disease. Differences in the primary endpoint between low-dose crizanlizumab and placebo were numerically, but not statistically, different.
How these results influence practice
It has been over 20 years since a new agent (hydroxyurea) was approved for sickle cell patients and, despite its use, sickle cell pain crises remain a frequent problem. Pain crises are associated with worse quality of life and increased risk of death. A promising advance is badly needed, especially in an era in which sensitivity to providers’ role in the opioid addiction crisis is highly scrutinized and may contribute to future undertreatment of pain episodes. This is especially true for patients from areas with high levels of opioid misuse.
The SUSTAIN trial was international, multi-institutional, placebo-controlled, and inclusive. These attributes enhance the likelihood that crizanlizumab will enhance patient care in routine practice. As an intravenous agent, monitoring adherence and toxicity are less challenging than with hydroxyurea. Despite these factors, however, there are some concerns. Crizanlizumab was not free of toxicity, quality of life via the Brief Pain Inventory used in the trial was not improved, and changes in the pain-severity and pain-interference domains were small. Treatment in SUSTAIN ensued for 52 weeks, so the emergence of late neutralizing antibodies and late toxicities with longer-term therapy will require careful postmarketing assessment.
These concerns notwithstanding, anyone who has cared for sickle cell patients would be excited about the potential benefits crizanlizumab could bring to patient care.
Zanubrutinib
The FDA has approved zanubrutinib for the treatment of MCL in adult patients who have received at least one prior therapy. The approval is based on the results of two studies in which overall response rate was the primary endpoint.
BGB-3111-206 (NCT03206970) was a phase 2, open-label, multicenter, single-arm trial of 86 patients with MCL who received at least one prior therapy. Zanubrutinib was given orally at 160 mg twice daily until disease progression or unacceptable toxicity. BGB-3111-AU-003 (NCT 02343120) was a phase 1/2, open-label, dose-escalation trial of B-cell malignancies, including 32 previously treated MCL patients treated with zanubrutinib at 160 mg twice daily or 320 mg once daily.
In the phase 2 trial, 18fluorodeoxyglucose (FDG)–PET scans were required and the ORR was 84% (95% confidence interval, 74%-91%), with a complete response rate of 59% (95% CI, 48%-70%) and a median response duration of 19.5 months (95% CI, 16.6% to not estimable). In the phase 1/2 dose-escalation trial, FDG-PET scans were not required and the ORR was 84% (95% CI, 67%-95%), with a complete response rate of 22% (95% CI, 9%-40%) and a median response duration of 18.5 months (95% CI, 12.6% to not estimable). In both trials, median follow-up on study was about 18 months.
The most common adverse reactions were cytopenias, upper respiratory tract infection, rash, bruising, diarrhea, and cough. The most common serious adverse reactions were pneumonia in 11% and hemorrhage in 5% of patients. Of 118 MCL patients, 8 stopped therapy because of an adverse event, most frequently pneumonia (3.4%).
How these results influence practice
Unfortunately, the therapy of recurrent MCL is noncurative, because of the rapid development of treatment resistance. There are multiple single-and multiagent chemotherapy regimens that may be tried, many incorporating immunotherapy options such as anti-CD20- or Bruton tyrosine kinase (BTK)–targeted agents. Given the limited efficacy of these agents, temporary nature of remissions, and paucity of data comparing these various treatment options, participation in clinical trials is encouraged whenever possible.
Outside of a clinical trial, zanubrutinib joins ibrutinib and acalabrutinib as approved single-agent BTK inhibitors for adult MCL patients in relapse. The impressive ORR and response duration reported for zanubrutinib are similar to the results achieved with the other agents, but the toxicity pattern may be slightly different.
As in the treatment of hormonally sensitive breast cancer, clinicians and patients benefit when they have multiple similar, equally efficacious oral agents with slightly different toxicity patterns so that quality of life can be improved and treatment duration maximized before treatment resistance develops and a more toxic and/or inconvenient therapy needs to be employed.
Whether zanubrutinib has benefits beyond those for MCL patients in relapse will depend on the results of confirmatory trials and patient-reported outcome data.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
In this edition of “How I Will Treat My Next Patient,” I highlight two recent drug approvals by the Food and Drug Administration – crizanlizumab for sickle cell patients with painful crises and zanubrutinib for mantle cell lymphoma (MCL) patients in relapse.
Crizanlizumab
P-selectin is an adhesion molecule expressed on activated vascular endothelial cells and platelets. It is a key molecule in the initiation of leukocyte rolling on vessel walls and promotes firm attachment and extravasation to underlying tissues during inflammation. Up-regulation of P-selectin on endothelial cells and platelets contributes to the cell-cell interactions involved in the pathogenesis of sickle cell pain crises.
The SUSTAIN study was a multisite, placebo-controlled, randomized phase 2 trial of two different dosage levels of intravenous crizanlizumab (2.5 mg/kg or 5 mg/kg for 52 weeks), a humanized anti–P-selectin antibody, examining its effect on pain crises in patients with sickle cell disease. The primary endpoint was the annual rate of sickle cell pain crises, with a variety of clinically relevant secondary endpoints. The target population had 2-10 pain crises in the 12 months before enrollment. Patients on a stable dose of hydroxyurea for at least the most recent 3 months were allowed to enter, but if patients were not receiving hydroxyurea, it could not be initiated during the trial. Patients who were undergoing chronic red-cell transfusion therapy were excluded.
Among 198 enrolled patients, 35% did not complete the 52 weeks of treatment. Discontinuations were equally balanced among patients assigned to the high-dose, low-dose, and placebo cohorts. Adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. Serious adverse events occurred in 55 patients, with 5 deaths, all of which were unrelated to treatment. Crizanlizumab did not augment hemolysis or bacterial infections.
In the efficacy analysis, patients receiving high-dose crizanlizumab had a median annual rate of 1.63 health care visits for sickle cell pain crises, compared with 2.98 visits for placebo patients (P = .01). In comparison with placebo, high-dose crizanlizumab also delayed the first pain crisis after starting treatment (4.1 months vs. 1.4 months), delayed the median time to a second pain crisis, and decreased the median number of pain crises annually.
More than twice as many high-dose crizanlizumab patients had no pain crisis episodes, compared with placebo patients. In general, differences were more striking in patients who were not taking hydroxyurea and who had non–hemoglobin SS disease. Differences in the primary endpoint between low-dose crizanlizumab and placebo were numerically, but not statistically, different.
How these results influence practice
It has been over 20 years since a new agent (hydroxyurea) was approved for sickle cell patients and, despite its use, sickle cell pain crises remain a frequent problem. Pain crises are associated with worse quality of life and increased risk of death. A promising advance is badly needed, especially in an era in which sensitivity to providers’ role in the opioid addiction crisis is highly scrutinized and may contribute to future undertreatment of pain episodes. This is especially true for patients from areas with high levels of opioid misuse.
The SUSTAIN trial was international, multi-institutional, placebo-controlled, and inclusive. These attributes enhance the likelihood that crizanlizumab will enhance patient care in routine practice. As an intravenous agent, monitoring adherence and toxicity are less challenging than with hydroxyurea. Despite these factors, however, there are some concerns. Crizanlizumab was not free of toxicity, quality of life via the Brief Pain Inventory used in the trial was not improved, and changes in the pain-severity and pain-interference domains were small. Treatment in SUSTAIN ensued for 52 weeks, so the emergence of late neutralizing antibodies and late toxicities with longer-term therapy will require careful postmarketing assessment.
These concerns notwithstanding, anyone who has cared for sickle cell patients would be excited about the potential benefits crizanlizumab could bring to patient care.
Zanubrutinib
The FDA has approved zanubrutinib for the treatment of MCL in adult patients who have received at least one prior therapy. The approval is based on the results of two studies in which overall response rate was the primary endpoint.
BGB-3111-206 (NCT03206970) was a phase 2, open-label, multicenter, single-arm trial of 86 patients with MCL who received at least one prior therapy. Zanubrutinib was given orally at 160 mg twice daily until disease progression or unacceptable toxicity. BGB-3111-AU-003 (NCT 02343120) was a phase 1/2, open-label, dose-escalation trial of B-cell malignancies, including 32 previously treated MCL patients treated with zanubrutinib at 160 mg twice daily or 320 mg once daily.
In the phase 2 trial, 18fluorodeoxyglucose (FDG)–PET scans were required and the ORR was 84% (95% confidence interval, 74%-91%), with a complete response rate of 59% (95% CI, 48%-70%) and a median response duration of 19.5 months (95% CI, 16.6% to not estimable). In the phase 1/2 dose-escalation trial, FDG-PET scans were not required and the ORR was 84% (95% CI, 67%-95%), with a complete response rate of 22% (95% CI, 9%-40%) and a median response duration of 18.5 months (95% CI, 12.6% to not estimable). In both trials, median follow-up on study was about 18 months.
The most common adverse reactions were cytopenias, upper respiratory tract infection, rash, bruising, diarrhea, and cough. The most common serious adverse reactions were pneumonia in 11% and hemorrhage in 5% of patients. Of 118 MCL patients, 8 stopped therapy because of an adverse event, most frequently pneumonia (3.4%).
How these results influence practice
Unfortunately, the therapy of recurrent MCL is noncurative, because of the rapid development of treatment resistance. There are multiple single-and multiagent chemotherapy regimens that may be tried, many incorporating immunotherapy options such as anti-CD20- or Bruton tyrosine kinase (BTK)–targeted agents. Given the limited efficacy of these agents, temporary nature of remissions, and paucity of data comparing these various treatment options, participation in clinical trials is encouraged whenever possible.
Outside of a clinical trial, zanubrutinib joins ibrutinib and acalabrutinib as approved single-agent BTK inhibitors for adult MCL patients in relapse. The impressive ORR and response duration reported for zanubrutinib are similar to the results achieved with the other agents, but the toxicity pattern may be slightly different.
As in the treatment of hormonally sensitive breast cancer, clinicians and patients benefit when they have multiple similar, equally efficacious oral agents with slightly different toxicity patterns so that quality of life can be improved and treatment duration maximized before treatment resistance develops and a more toxic and/or inconvenient therapy needs to be employed.
Whether zanubrutinib has benefits beyond those for MCL patients in relapse will depend on the results of confirmatory trials and patient-reported outcome data.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
In this edition of “How I Will Treat My Next Patient,” I highlight two recent drug approvals by the Food and Drug Administration – crizanlizumab for sickle cell patients with painful crises and zanubrutinib for mantle cell lymphoma (MCL) patients in relapse.
Crizanlizumab
P-selectin is an adhesion molecule expressed on activated vascular endothelial cells and platelets. It is a key molecule in the initiation of leukocyte rolling on vessel walls and promotes firm attachment and extravasation to underlying tissues during inflammation. Up-regulation of P-selectin on endothelial cells and platelets contributes to the cell-cell interactions involved in the pathogenesis of sickle cell pain crises.
The SUSTAIN study was a multisite, placebo-controlled, randomized phase 2 trial of two different dosage levels of intravenous crizanlizumab (2.5 mg/kg or 5 mg/kg for 52 weeks), a humanized anti–P-selectin antibody, examining its effect on pain crises in patients with sickle cell disease. The primary endpoint was the annual rate of sickle cell pain crises, with a variety of clinically relevant secondary endpoints. The target population had 2-10 pain crises in the 12 months before enrollment. Patients on a stable dose of hydroxyurea for at least the most recent 3 months were allowed to enter, but if patients were not receiving hydroxyurea, it could not be initiated during the trial. Patients who were undergoing chronic red-cell transfusion therapy were excluded.
Among 198 enrolled patients, 35% did not complete the 52 weeks of treatment. Discontinuations were equally balanced among patients assigned to the high-dose, low-dose, and placebo cohorts. Adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. Serious adverse events occurred in 55 patients, with 5 deaths, all of which were unrelated to treatment. Crizanlizumab did not augment hemolysis or bacterial infections.
In the efficacy analysis, patients receiving high-dose crizanlizumab had a median annual rate of 1.63 health care visits for sickle cell pain crises, compared with 2.98 visits for placebo patients (P = .01). In comparison with placebo, high-dose crizanlizumab also delayed the first pain crisis after starting treatment (4.1 months vs. 1.4 months), delayed the median time to a second pain crisis, and decreased the median number of pain crises annually.
More than twice as many high-dose crizanlizumab patients had no pain crisis episodes, compared with placebo patients. In general, differences were more striking in patients who were not taking hydroxyurea and who had non–hemoglobin SS disease. Differences in the primary endpoint between low-dose crizanlizumab and placebo were numerically, but not statistically, different.
How these results influence practice
It has been over 20 years since a new agent (hydroxyurea) was approved for sickle cell patients and, despite its use, sickle cell pain crises remain a frequent problem. Pain crises are associated with worse quality of life and increased risk of death. A promising advance is badly needed, especially in an era in which sensitivity to providers’ role in the opioid addiction crisis is highly scrutinized and may contribute to future undertreatment of pain episodes. This is especially true for patients from areas with high levels of opioid misuse.
The SUSTAIN trial was international, multi-institutional, placebo-controlled, and inclusive. These attributes enhance the likelihood that crizanlizumab will enhance patient care in routine practice. As an intravenous agent, monitoring adherence and toxicity are less challenging than with hydroxyurea. Despite these factors, however, there are some concerns. Crizanlizumab was not free of toxicity, quality of life via the Brief Pain Inventory used in the trial was not improved, and changes in the pain-severity and pain-interference domains were small. Treatment in SUSTAIN ensued for 52 weeks, so the emergence of late neutralizing antibodies and late toxicities with longer-term therapy will require careful postmarketing assessment.
These concerns notwithstanding, anyone who has cared for sickle cell patients would be excited about the potential benefits crizanlizumab could bring to patient care.
Zanubrutinib
The FDA has approved zanubrutinib for the treatment of MCL in adult patients who have received at least one prior therapy. The approval is based on the results of two studies in which overall response rate was the primary endpoint.
BGB-3111-206 (NCT03206970) was a phase 2, open-label, multicenter, single-arm trial of 86 patients with MCL who received at least one prior therapy. Zanubrutinib was given orally at 160 mg twice daily until disease progression or unacceptable toxicity. BGB-3111-AU-003 (NCT 02343120) was a phase 1/2, open-label, dose-escalation trial of B-cell malignancies, including 32 previously treated MCL patients treated with zanubrutinib at 160 mg twice daily or 320 mg once daily.
In the phase 2 trial, 18fluorodeoxyglucose (FDG)–PET scans were required and the ORR was 84% (95% confidence interval, 74%-91%), with a complete response rate of 59% (95% CI, 48%-70%) and a median response duration of 19.5 months (95% CI, 16.6% to not estimable). In the phase 1/2 dose-escalation trial, FDG-PET scans were not required and the ORR was 84% (95% CI, 67%-95%), with a complete response rate of 22% (95% CI, 9%-40%) and a median response duration of 18.5 months (95% CI, 12.6% to not estimable). In both trials, median follow-up on study was about 18 months.
The most common adverse reactions were cytopenias, upper respiratory tract infection, rash, bruising, diarrhea, and cough. The most common serious adverse reactions were pneumonia in 11% and hemorrhage in 5% of patients. Of 118 MCL patients, 8 stopped therapy because of an adverse event, most frequently pneumonia (3.4%).
How these results influence practice
Unfortunately, the therapy of recurrent MCL is noncurative, because of the rapid development of treatment resistance. There are multiple single-and multiagent chemotherapy regimens that may be tried, many incorporating immunotherapy options such as anti-CD20- or Bruton tyrosine kinase (BTK)–targeted agents. Given the limited efficacy of these agents, temporary nature of remissions, and paucity of data comparing these various treatment options, participation in clinical trials is encouraged whenever possible.
Outside of a clinical trial, zanubrutinib joins ibrutinib and acalabrutinib as approved single-agent BTK inhibitors for adult MCL patients in relapse. The impressive ORR and response duration reported for zanubrutinib are similar to the results achieved with the other agents, but the toxicity pattern may be slightly different.
As in the treatment of hormonally sensitive breast cancer, clinicians and patients benefit when they have multiple similar, equally efficacious oral agents with slightly different toxicity patterns so that quality of life can be improved and treatment duration maximized before treatment resistance develops and a more toxic and/or inconvenient therapy needs to be employed.
Whether zanubrutinib has benefits beyond those for MCL patients in relapse will depend on the results of confirmatory trials and patient-reported outcome data.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
More phase 3 ubrogepant data published as FDA decision nears
published Dec. 4 in the New England Journal of Medicine. In addition, about 38% of patients who receive ubrogepant no longer have their most bothersome migraine-associated symptom, such as photophobia, phonophobia, or nausea, at 2 hours, compared with 28% of patients who receive placebo, said David W. Dodick, MD, and colleagues.

Dr. Dodick, professor of neurology at the Mayo Clinic in Phoenix, and his coauthors described efficacy and safety results from the ACHIEVE I trial. Another phase 3 study of ubrogepant, ACHIEVE II, was published in JAMA in November. That trial evaluated 25- and 50-mg doses of ubrogepant versus placebo and found rates of pain freedom and absence of the most bothersome symptom in the placebo and active treatment arms that were similar to those in ACHIEVE I.
Assessing a gepant for acute migraine treatment
Ubrogepant is an oral calcitonin gene–related peptide (CGRP) receptor antagonist. Allergan, the company developing the drug, has said it expects the Food and Drug Administration to decide in December whether to approve the drug.
To compare ubrogepant 50 mg, ubrogepant 100 mg, and placebo for the acute treatment of migraine, investigators conducted the randomized ACHIEVE I trial. Researchers enrolled 1,672 adults with migraine with or without aura. They excluded patients with clinically significant cardiovascular or cerebrovascular disease. During the trial, patients treated a single migraine attack, and they had the option to take a second dose. In all, 1,436 participants took an initial dose. Patients had an average age of 40.5 years, about 88% were women, and 82% were white.
In ACHIEVE I, the most common adverse events within 48 hours of treatment were nausea, somnolence, and dry mouth, and these events occurred more frequently in the 100-mg–dose group, Dr. Dodick and colleagues reported. Among patients who received ubrogepant, serious adverse events more than 48 hours after treatment but within 30 days of treatment included appendicitis, spontaneous abortion, pericardial effusion, and seizure. No serious adverse events occurred in the placebo group.
The authors noted that, “there was no active comparator and no evaluation of consistency of effect across multiple migraine attacks; therefore it is not possible to determine whether the drug is more or less effective than standard therapies or consistently effective with repeated use.” In addition, “safety and side-effect data from this trial were based on evaluation of a single attack, and therefore safety after repeated use cannot be inferred; an extension trial has assessed the long-term safety of ubrogepant,” they said.
The present trial was performed well, commented Alan M. Rapoport, MD. “The coprimary endpoints of pain freedom and most bothersome symptom freedom, both at 2 hours after dosing, were statistically superior for both doses of ubrogepant versus placebo,” he said. “Some of the secondary endpoints, such as pain relief at 2 hours post dose and sustained pain relief from 2 to 24 hours, were statistically better than placebo.”

“Based on this data, I suspect that the FDA would approve this gepant after appropriate safety data,” said Dr. Rapoport, clinical professor of neurology at the University of California, Los Angeles and editor-in-chief of Neurology Reviews. “Many more patients need to take this drug before we can be sure it is safe and effective.”
The CGRP therapeutic landscape
“Other gepants have been shown to be effective, although some have caused a degree of liver toxicity,” said Dr. Rapoport. “Blocking the effect of CGRP on the migraine peripheral nervous system, in this case by preventing the ligand from docking at its receptor by administering an oral CGRP receptor blocker, appears to be effective.” Researchers are studying another oral gepant for similar approval, he added.
Ubrogepant stands to join other treatments targeting CGRP.
“There are currently three, and soon to be four, injectable monoclonal antibodies against CGRP functionality, which are preventive, not acute-care drugs,” Dr. Rapoport said. “The first released was a subcutaneous injection of a CGRP receptor blocker, and the other two are subcutaneous injections of CGRP ligand blockers. The last drug will be an intravenous infusion of a ligand blocker. These recently approved migraine treatments have greatly improved the lives of many of our patients, even when other preventives have failed. I expect ubrogepant and other gepants will do the same for the acute care of migraine.”
Allergan funded the trials of ubrogepant, and some of the authors are Allergan employees and stockholders. Dr. Dodick reported consulting fees and advisory board fees from Allergan and various pharmaceutical companies.
SOURCE: Dodick DW et al. N Engl J Med. 2019;381(23):2230-41. doi: 10.1056/NEJMoa1813049.
published Dec. 4 in the New England Journal of Medicine. In addition, about 38% of patients who receive ubrogepant no longer have their most bothersome migraine-associated symptom, such as photophobia, phonophobia, or nausea, at 2 hours, compared with 28% of patients who receive placebo, said David W. Dodick, MD, and colleagues.
Dr. Dodick, professor of neurology at the Mayo Clinic in Phoenix, and his coauthors described efficacy and safety results from the ACHIEVE I trial. Another phase 3 study of ubrogepant, ACHIEVE II, was published in JAMA in November. That trial evaluated 25- and 50-mg doses of ubrogepant versus placebo and found rates of pain freedom and absence of the most bothersome symptom in the placebo and active treatment arms that were similar to those in ACHIEVE I.
Assessing a gepant for acute migraine treatment
Ubrogepant is an oral calcitonin gene–related peptide (CGRP) receptor antagonist. Allergan, the company developing the drug, has said it expects the Food and Drug Administration to decide in December whether to approve the drug.
To compare ubrogepant 50 mg, ubrogepant 100 mg, and placebo for the acute treatment of migraine, investigators conducted the randomized ACHIEVE I trial. Researchers enrolled 1,672 adults with migraine with or without aura. They excluded patients with clinically significant cardiovascular or cerebrovascular disease. During the trial, patients treated a single migraine attack, and they had the option to take a second dose. In all, 1,436 participants took an initial dose. Patients had an average age of 40.5 years, about 88% were women, and 82% were white.
In ACHIEVE I, the most common adverse events within 48 hours of treatment were nausea, somnolence, and dry mouth, and these events occurred more frequently in the 100-mg–dose group, Dr. Dodick and colleagues reported. Among patients who received ubrogepant, serious adverse events more than 48 hours after treatment but within 30 days of treatment included appendicitis, spontaneous abortion, pericardial effusion, and seizure. No serious adverse events occurred in the placebo group.
The authors noted that, “there was no active comparator and no evaluation of consistency of effect across multiple migraine attacks; therefore it is not possible to determine whether the drug is more or less effective than standard therapies or consistently effective with repeated use.” In addition, “safety and side-effect data from this trial were based on evaluation of a single attack, and therefore safety after repeated use cannot be inferred; an extension trial has assessed the long-term safety of ubrogepant,” they said.
The present trial was performed well, commented Alan M. Rapoport, MD. “The coprimary endpoints of pain freedom and most bothersome symptom freedom, both at 2 hours after dosing, were statistically superior for both doses of ubrogepant versus placebo,” he said. “Some of the secondary endpoints, such as pain relief at 2 hours post dose and sustained pain relief from 2 to 24 hours, were statistically better than placebo.”
“Based on this data, I suspect that the FDA would approve this gepant after appropriate safety data,” said Dr. Rapoport, clinical professor of neurology at the University of California, Los Angeles and editor-in-chief of Neurology Reviews. “Many more patients need to take this drug before we can be sure it is safe and effective.”
The CGRP therapeutic landscape
“Other gepants have been shown to be effective, although some have caused a degree of liver toxicity,” said Dr. Rapoport. “Blocking the effect of CGRP on the migraine peripheral nervous system, in this case by preventing the ligand from docking at its receptor by administering an oral CGRP receptor blocker, appears to be effective.” Researchers are studying another oral gepant for similar approval, he added.
Ubrogepant stands to join other treatments targeting CGRP.
“There are currently three, and soon to be four, injectable monoclonal antibodies against CGRP functionality, which are preventive, not acute-care drugs,” Dr. Rapoport said. “The first released was a subcutaneous injection of a CGRP receptor blocker, and the other two are subcutaneous injections of CGRP ligand blockers. The last drug will be an intravenous infusion of a ligand blocker. These recently approved migraine treatments have greatly improved the lives of many of our patients, even when other preventives have failed. I expect ubrogepant and other gepants will do the same for the acute care of migraine.”
Allergan funded the trials of ubrogepant, and some of the authors are Allergan employees and stockholders. Dr. Dodick reported consulting fees and advisory board fees from Allergan and various pharmaceutical companies.
SOURCE: Dodick DW et al. N Engl J Med. 2019;381(23):2230-41. doi: 10.1056/NEJMoa1813049.
published Dec. 4 in the New England Journal of Medicine. In addition, about 38% of patients who receive ubrogepant no longer have their most bothersome migraine-associated symptom, such as photophobia, phonophobia, or nausea, at 2 hours, compared with 28% of patients who receive placebo, said David W. Dodick, MD, and colleagues.
Dr. Dodick, professor of neurology at the Mayo Clinic in Phoenix, and his coauthors described efficacy and safety results from the ACHIEVE I trial. Another phase 3 study of ubrogepant, ACHIEVE II, was published in JAMA in November. That trial evaluated 25- and 50-mg doses of ubrogepant versus placebo and found rates of pain freedom and absence of the most bothersome symptom in the placebo and active treatment arms that were similar to those in ACHIEVE I.
Assessing a gepant for acute migraine treatment
Ubrogepant is an oral calcitonin gene–related peptide (CGRP) receptor antagonist. Allergan, the company developing the drug, has said it expects the Food and Drug Administration to decide in December whether to approve the drug.
To compare ubrogepant 50 mg, ubrogepant 100 mg, and placebo for the acute treatment of migraine, investigators conducted the randomized ACHIEVE I trial. Researchers enrolled 1,672 adults with migraine with or without aura. They excluded patients with clinically significant cardiovascular or cerebrovascular disease. During the trial, patients treated a single migraine attack, and they had the option to take a second dose. In all, 1,436 participants took an initial dose. Patients had an average age of 40.5 years, about 88% were women, and 82% were white.
In ACHIEVE I, the most common adverse events within 48 hours of treatment were nausea, somnolence, and dry mouth, and these events occurred more frequently in the 100-mg–dose group, Dr. Dodick and colleagues reported. Among patients who received ubrogepant, serious adverse events more than 48 hours after treatment but within 30 days of treatment included appendicitis, spontaneous abortion, pericardial effusion, and seizure. No serious adverse events occurred in the placebo group.
The authors noted that, “there was no active comparator and no evaluation of consistency of effect across multiple migraine attacks; therefore it is not possible to determine whether the drug is more or less effective than standard therapies or consistently effective with repeated use.” In addition, “safety and side-effect data from this trial were based on evaluation of a single attack, and therefore safety after repeated use cannot be inferred; an extension trial has assessed the long-term safety of ubrogepant,” they said.
The present trial was performed well, commented Alan M. Rapoport, MD. “The coprimary endpoints of pain freedom and most bothersome symptom freedom, both at 2 hours after dosing, were statistically superior for both doses of ubrogepant versus placebo,” he said. “Some of the secondary endpoints, such as pain relief at 2 hours post dose and sustained pain relief from 2 to 24 hours, were statistically better than placebo.”
“Based on this data, I suspect that the FDA would approve this gepant after appropriate safety data,” said Dr. Rapoport, clinical professor of neurology at the University of California, Los Angeles and editor-in-chief of Neurology Reviews. “Many more patients need to take this drug before we can be sure it is safe and effective.”
The CGRP therapeutic landscape
“Other gepants have been shown to be effective, although some have caused a degree of liver toxicity,” said Dr. Rapoport. “Blocking the effect of CGRP on the migraine peripheral nervous system, in this case by preventing the ligand from docking at its receptor by administering an oral CGRP receptor blocker, appears to be effective.” Researchers are studying another oral gepant for similar approval, he added.
Ubrogepant stands to join other treatments targeting CGRP.
“There are currently three, and soon to be four, injectable monoclonal antibodies against CGRP functionality, which are preventive, not acute-care drugs,” Dr. Rapoport said. “The first released was a subcutaneous injection of a CGRP receptor blocker, and the other two are subcutaneous injections of CGRP ligand blockers. The last drug will be an intravenous infusion of a ligand blocker. These recently approved migraine treatments have greatly improved the lives of many of our patients, even when other preventives have failed. I expect ubrogepant and other gepants will do the same for the acute care of migraine.”
Allergan funded the trials of ubrogepant, and some of the authors are Allergan employees and stockholders. Dr. Dodick reported consulting fees and advisory board fees from Allergan and various pharmaceutical companies.
SOURCE: Dodick DW et al. N Engl J Med. 2019;381(23):2230-41. doi: 10.1056/NEJMoa1813049.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Compared with placebo, ubrogepant tablets result in higher rates of pain freedom and freedom from the most bothersome migraine-associated symptom at 2 hours following treatment.
Major finding: About 20% of patients who receive tablets containing 50 mg or 100 mg of ubrogepant for the acute treatment of migraine are pain free 2 hours later, compared with 12% of patients who receive placebo. In addition, about 38% of patients who receive ubrogepant no longer have their most bothersome migraine-associated symptom, such as photophobia, phonophobia, or nausea, at 2 hours, compared with 28% of patients who receive placebo.
Study details: A randomized trial that enrolled 1,672 adults with migraine with or without aura. Participants treated a single migraine attack.
Disclosures: Allergan funded the trial, and some of the authors are Allergan employees and stockholders. Dr. Dodick reported consulting fees and advisory board fees from Allergan and various pharmaceutical companies.
Source: Dodick DW et al. N Engl J Med. 2019;381(23):2230-41. doi: 10.1056/NEJMoa1813049.
SOX11 shows value as diagnostic marker in MCL
SOX11 may be an accurate diagnostic marker for mantle cell lymphoma (MCL), allowing clinicians to distinguish it from other lymphoproliferative disorders, according to findings from a meta-analysis of 14 case-control studies.

Woojoo Lee, PhD, of Inha University, Incheon, Republic of Korea, and coinvestigators, evaluated the diagnostic accuracy of SOX11 immunohistochemistry for the diagnosis of MCL. The results were published in PLoS One.
The researchers searched major databases for studies that evaluated the use of SOX11 immunohistochemistry in patients with MCL and other lymphoproliferative disorders. After applying the search parameters, the team identified 383 studies, 14 of which were included in the meta-analysis. Various data were extracted from eligible studies, including the type and clonality of anti-SOX11 antibody, number of SOX11-positive lymphomas (MCLs and other lymphoproliferative disorders), specificity, sensitivity, and others. After combining the data, the investigators calculated pooled sensitivity, specificity, and area under the curve. Among the included studies, hairy cell leukemia, Burkitt’s lymphoma, and lymphoblastic lymphoma were common among patient populations. In total, clone MRQ-58 mouse antibodies were used in five study populations.
The researchers found that the pooled specificity was 0.95 (95% CI, 0.9-0.97), and sensitivity was 0.9 (95% CI, 0.86-0.92). There was statistically significant substantial heterogeneity observed for specificity, but not for sensitivity, the investigators reported.
“The results demonstrated that SOX11 was a potential diagnostic marker for MCL,” they wrote. “Meta-regression revealed a significant inverse relationship between specificity and proportions of [Burkitt’s lymphoma, lymphoblastic lymphoma, and hairy cell leukemia].”
With respect to antibody type, the clone MRQ-58 mouse antibody showed consistently high specificity in the clinical setting, despite observed heterogeneity.
“Future studies using MRQ-58 are needed to improve our understanding of the diagnostic accuracy of SOX11 immunohistochemistry for MCL,” the investigators wrote.
The study was funded by the Next-Generation BioGreen 21 program (Republic of Korea). The authors reported having no conflicts of interest.
SOURCE: Lee W et al. PLoS One. 2019 Nov 12. doi: 10.1371/journal.pone.0225096.
SOX11 may be an accurate diagnostic marker for mantle cell lymphoma (MCL), allowing clinicians to distinguish it from other lymphoproliferative disorders, according to findings from a meta-analysis of 14 case-control studies.
Woojoo Lee, PhD, of Inha University, Incheon, Republic of Korea, and coinvestigators, evaluated the diagnostic accuracy of SOX11 immunohistochemistry for the diagnosis of MCL. The results were published in PLoS One.
The researchers searched major databases for studies that evaluated the use of SOX11 immunohistochemistry in patients with MCL and other lymphoproliferative disorders. After applying the search parameters, the team identified 383 studies, 14 of which were included in the meta-analysis. Various data were extracted from eligible studies, including the type and clonality of anti-SOX11 antibody, number of SOX11-positive lymphomas (MCLs and other lymphoproliferative disorders), specificity, sensitivity, and others. After combining the data, the investigators calculated pooled sensitivity, specificity, and area under the curve. Among the included studies, hairy cell leukemia, Burkitt’s lymphoma, and lymphoblastic lymphoma were common among patient populations. In total, clone MRQ-58 mouse antibodies were used in five study populations.
The researchers found that the pooled specificity was 0.95 (95% CI, 0.9-0.97), and sensitivity was 0.9 (95% CI, 0.86-0.92). There was statistically significant substantial heterogeneity observed for specificity, but not for sensitivity, the investigators reported.
“The results demonstrated that SOX11 was a potential diagnostic marker for MCL,” they wrote. “Meta-regression revealed a significant inverse relationship between specificity and proportions of [Burkitt’s lymphoma, lymphoblastic lymphoma, and hairy cell leukemia].”
With respect to antibody type, the clone MRQ-58 mouse antibody showed consistently high specificity in the clinical setting, despite observed heterogeneity.
“Future studies using MRQ-58 are needed to improve our understanding of the diagnostic accuracy of SOX11 immunohistochemistry for MCL,” the investigators wrote.
The study was funded by the Next-Generation BioGreen 21 program (Republic of Korea). The authors reported having no conflicts of interest.
SOURCE: Lee W et al. PLoS One. 2019 Nov 12. doi: 10.1371/journal.pone.0225096.
SOX11 may be an accurate diagnostic marker for mantle cell lymphoma (MCL), allowing clinicians to distinguish it from other lymphoproliferative disorders, according to findings from a meta-analysis of 14 case-control studies.
Woojoo Lee, PhD, of Inha University, Incheon, Republic of Korea, and coinvestigators, evaluated the diagnostic accuracy of SOX11 immunohistochemistry for the diagnosis of MCL. The results were published in PLoS One.
The researchers searched major databases for studies that evaluated the use of SOX11 immunohistochemistry in patients with MCL and other lymphoproliferative disorders. After applying the search parameters, the team identified 383 studies, 14 of which were included in the meta-analysis. Various data were extracted from eligible studies, including the type and clonality of anti-SOX11 antibody, number of SOX11-positive lymphomas (MCLs and other lymphoproliferative disorders), specificity, sensitivity, and others. After combining the data, the investigators calculated pooled sensitivity, specificity, and area under the curve. Among the included studies, hairy cell leukemia, Burkitt’s lymphoma, and lymphoblastic lymphoma were common among patient populations. In total, clone MRQ-58 mouse antibodies were used in five study populations.
The researchers found that the pooled specificity was 0.95 (95% CI, 0.9-0.97), and sensitivity was 0.9 (95% CI, 0.86-0.92). There was statistically significant substantial heterogeneity observed for specificity, but not for sensitivity, the investigators reported.
“The results demonstrated that SOX11 was a potential diagnostic marker for MCL,” they wrote. “Meta-regression revealed a significant inverse relationship between specificity and proportions of [Burkitt’s lymphoma, lymphoblastic lymphoma, and hairy cell leukemia].”
With respect to antibody type, the clone MRQ-58 mouse antibody showed consistently high specificity in the clinical setting, despite observed heterogeneity.
“Future studies using MRQ-58 are needed to improve our understanding of the diagnostic accuracy of SOX11 immunohistochemistry for MCL,” the investigators wrote.
The study was funded by the Next-Generation BioGreen 21 program (Republic of Korea). The authors reported having no conflicts of interest.
SOURCE: Lee W et al. PLoS One. 2019 Nov 12. doi: 10.1371/journal.pone.0225096.
FROM PLOS ONE

Acalabrutinib may outperform other targeted therapies in MCL
For patients with relapsed or refractory mantle cell lymphoma (MCL), second generation Bruton’s tyrosine kinase (BTK) inhibitor acalabrutinib may offer increased response rates and better tolerability compared with other single-agent targeted therapies, based on a recent analysis.

Improved safety could lead to long-term benefits resulting from extended treatment duration, according to lead author Claire Telford, PhD, of AstraZeneca in Gaithersburg, Md., and colleagues. AstraZeneca manufactures acalabrutinib (Calquence).
Currently, treatment for MCL is guided by a number of clinical considerations, the investigators explained in Clinical Therapeutics.
“The type of treatment recommended for relapsed/refractory MCL depends on multiple factors, namely time to the relapse, extent of disease, previous regimens, candidacy for allogeneic stem cell transplantation, and the patient’s overall health,” they wrote.
To determine how acalabrutinib stacks up with other options, the investigators drew data from the phase 2 ACE-LY-004 trial, which tested acalabrutinib in 124 patients with relapsed or refractory MCL. After matching, the investigators compared the ACE-LY-004 outcomes from 12 other trials, in which patients received different targeted therapies.
Results pointed to higher overall response and complete response rates for acalabrutinib, compared with other single-agent targeted therapy. Specifically, acalabrutinib had a higher overall response rate, compared with ibrutinib (9.3% higher), lenalidomide (38.1% higher), temsirolimus (40.7% higher), and bortezomib (50.6% higher). For each of these, complete responses also were higher.
There was no significant difference in overall response or complete response rates between acalabrutinib and rituximab combinations – bendamustine plus rituximab, ibrutinib plus rituximab, and lenalidomide plus rituximab.
The investigators also highlighted a number of safety advantages. Compared with ibrutinib, acalabrutinib was associated with significantly fewer instances of grade 3 or 4 atrial fibrillation. Risk of grade 3 or 4 thrombocytopenia was significantly lower with acalabrutinib than with ibrutinib, bortezomib, lenalidomide, and temsirolimus.
Still, in some instances, acalabrutinib was comparatively less tolerable. It was associated with a higher risk of grade 3 or 4 infections than bendamustine plus rituximab; and anemia was more common among patients receiving acalabrutinib than among those who had lenalidomide plus rituximab or ibrutinib plus rituximab.
“This comparison of targeted therapies used in the treatment of relapsed/refractory MCL has shown that acalabrutinib has the potential to provide higher response rates, with trends for longer [progression-free survival] and [overall survival], and an improved safety profile,” the investigators wrote.
The study was funded by AstraZeneca. Dr. Telford is an employee of AstraZeneca and other authors reported financial relationships with the company.
SOURCE: Telford C et al. Clin Ther. 2019 Nov 4. doi: 10.1016/j.clinthera.2019.09.012 .
For patients with relapsed or refractory mantle cell lymphoma (MCL), second generation Bruton’s tyrosine kinase (BTK) inhibitor acalabrutinib may offer increased response rates and better tolerability compared with other single-agent targeted therapies, based on a recent analysis.
Improved safety could lead to long-term benefits resulting from extended treatment duration, according to lead author Claire Telford, PhD, of AstraZeneca in Gaithersburg, Md., and colleagues. AstraZeneca manufactures acalabrutinib (Calquence).
Currently, treatment for MCL is guided by a number of clinical considerations, the investigators explained in Clinical Therapeutics.
“The type of treatment recommended for relapsed/refractory MCL depends on multiple factors, namely time to the relapse, extent of disease, previous regimens, candidacy for allogeneic stem cell transplantation, and the patient’s overall health,” they wrote.
To determine how acalabrutinib stacks up with other options, the investigators drew data from the phase 2 ACE-LY-004 trial, which tested acalabrutinib in 124 patients with relapsed or refractory MCL. After matching, the investigators compared the ACE-LY-004 outcomes from 12 other trials, in which patients received different targeted therapies.
Results pointed to higher overall response and complete response rates for acalabrutinib, compared with other single-agent targeted therapy. Specifically, acalabrutinib had a higher overall response rate, compared with ibrutinib (9.3% higher), lenalidomide (38.1% higher), temsirolimus (40.7% higher), and bortezomib (50.6% higher). For each of these, complete responses also were higher.
There was no significant difference in overall response or complete response rates between acalabrutinib and rituximab combinations – bendamustine plus rituximab, ibrutinib plus rituximab, and lenalidomide plus rituximab.
The investigators also highlighted a number of safety advantages. Compared with ibrutinib, acalabrutinib was associated with significantly fewer instances of grade 3 or 4 atrial fibrillation. Risk of grade 3 or 4 thrombocytopenia was significantly lower with acalabrutinib than with ibrutinib, bortezomib, lenalidomide, and temsirolimus.
Still, in some instances, acalabrutinib was comparatively less tolerable. It was associated with a higher risk of grade 3 or 4 infections than bendamustine plus rituximab; and anemia was more common among patients receiving acalabrutinib than among those who had lenalidomide plus rituximab or ibrutinib plus rituximab.
“This comparison of targeted therapies used in the treatment of relapsed/refractory MCL has shown that acalabrutinib has the potential to provide higher response rates, with trends for longer [progression-free survival] and [overall survival], and an improved safety profile,” the investigators wrote.
The study was funded by AstraZeneca. Dr. Telford is an employee of AstraZeneca and other authors reported financial relationships with the company.
SOURCE: Telford C et al. Clin Ther. 2019 Nov 4. doi: 10.1016/j.clinthera.2019.09.012 .
For patients with relapsed or refractory mantle cell lymphoma (MCL), second generation Bruton’s tyrosine kinase (BTK) inhibitor acalabrutinib may offer increased response rates and better tolerability compared with other single-agent targeted therapies, based on a recent analysis.
Improved safety could lead to long-term benefits resulting from extended treatment duration, according to lead author Claire Telford, PhD, of AstraZeneca in Gaithersburg, Md., and colleagues. AstraZeneca manufactures acalabrutinib (Calquence).
Currently, treatment for MCL is guided by a number of clinical considerations, the investigators explained in Clinical Therapeutics.
“The type of treatment recommended for relapsed/refractory MCL depends on multiple factors, namely time to the relapse, extent of disease, previous regimens, candidacy for allogeneic stem cell transplantation, and the patient’s overall health,” they wrote.
To determine how acalabrutinib stacks up with other options, the investigators drew data from the phase 2 ACE-LY-004 trial, which tested acalabrutinib in 124 patients with relapsed or refractory MCL. After matching, the investigators compared the ACE-LY-004 outcomes from 12 other trials, in which patients received different targeted therapies.
Results pointed to higher overall response and complete response rates for acalabrutinib, compared with other single-agent targeted therapy. Specifically, acalabrutinib had a higher overall response rate, compared with ibrutinib (9.3% higher), lenalidomide (38.1% higher), temsirolimus (40.7% higher), and bortezomib (50.6% higher). For each of these, complete responses also were higher.
There was no significant difference in overall response or complete response rates between acalabrutinib and rituximab combinations – bendamustine plus rituximab, ibrutinib plus rituximab, and lenalidomide plus rituximab.
The investigators also highlighted a number of safety advantages. Compared with ibrutinib, acalabrutinib was associated with significantly fewer instances of grade 3 or 4 atrial fibrillation. Risk of grade 3 or 4 thrombocytopenia was significantly lower with acalabrutinib than with ibrutinib, bortezomib, lenalidomide, and temsirolimus.
Still, in some instances, acalabrutinib was comparatively less tolerable. It was associated with a higher risk of grade 3 or 4 infections than bendamustine plus rituximab; and anemia was more common among patients receiving acalabrutinib than among those who had lenalidomide plus rituximab or ibrutinib plus rituximab.
“This comparison of targeted therapies used in the treatment of relapsed/refractory MCL has shown that acalabrutinib has the potential to provide higher response rates, with trends for longer [progression-free survival] and [overall survival], and an improved safety profile,” the investigators wrote.
The study was funded by AstraZeneca. Dr. Telford is an employee of AstraZeneca and other authors reported financial relationships with the company.
SOURCE: Telford C et al. Clin Ther. 2019 Nov 4. doi: 10.1016/j.clinthera.2019.09.012 .
FROM CLINICAL THERAPEUTICS