User login
Cardiac inflammation can be present after mild COVID infection
Myocardial inflammation is present in a small proportion of patients who have recovered from relatively mild cases of COVID-19 infection, a new study shows.
“Our findings suggest that even in patients who have had relatively mild cases of COVID-19, some will have inflammatory changes to the heart, and these changes can be present without any cardiac symptoms,” senior author, Paaladinesh Thavendiranathan, MD, University of Toronto, told this news organization.
“While our data suggest that this inflammation improves over time, and the outcomes seem positive, we don’t know if there will be any long-term consequences,” he added.
Noting that even a short period of inflammation in the heart may be associated with symptoms or arrhythmias in the longer term, Dr. Thavendiranathan said: “I would recommend that it is best to avoid getting the infection if there is any chance of heart inflammation.”
The study was published online in JAMA Cardiology on Jan. 12.
The authors explain that among patients hospitalized with COVID, early studies suggested that approximately one in four experience cardiovascular injury, defined as an elevation in troponin levels, which was associated with a 5- to 10-fold increase in the risk for death. But there is limited information on cardiac injury in patients who do not require hospitalization.
Although a broad range of abnormal myocardial tissue has been reported in several cardiac MRI studies of patients recovered from COVID infection, there is little understanding of persistent changes in myocardial metabolism in recovered patients, which is a potential concern, given that COVID-19 is associated with systemic inflammation during the acute illness, they say.
For the current study, the researchers examined myocardial inflammation measured using two different methods – cardiac MRI and fluorodeoxyglucose–positron emission tomography (FDG-PET) – in individuals who had recovered from COVID-19 infection and looked at how this related to changes in inflammatory blood markers.
Lead author Kate Hanneman, MD, also from the University of Toronto, explained that FDG-PET imaging is more sensitive than MRI in detecting active inflammation. “Inflammatory cells have a higher uptake of glucose, and FDG-PET imaging is used to look for metabolically active inflammatory tissue that takes up glucose. It gives complementary information to MRI. Cardiac MRI shows structural or functional changes, such as scarring or edema, whereas FDG-PET imaging directly measures metabolic activity related to inflammatory cells.”
The study involved 47 individuals, 51% female, with a mean age of 43 years, who had recently recovered from COVID-19 infection. Of these, the majority had had relatively mild COVID disease, with 85% not requiring hospitalization.
Cardiac imaging was performed a mean of 67 days after the diagnosis of COVID-19. At the time of imaging, 19 participants (40%) reported at least one cardiac symptom, including palpitations, chest pain, and shortness of breath.
Results showed that eight patients (17%) had focal FDG uptake on PET consistent with myocardial inflammation. Compared with those without FDG uptake, patients with focal FDG uptake had higher regional T2, T1, and extracellular volume (colocalizing with focal FDG uptake), higher prevalence of late gadolinium enhancement indicating fibrosis, lower left ventricular ejection fraction, worse global longitudinal and circumferential strain, and higher systemic inflammatory blood markers, including interleukin (IL)-6, IL- 8, an high-sensitivity C-reactive protein.
Of the 47 patients in the study, 13 had received at least one dose of a COVID-19 vaccine. There was no significant difference in the proportion of patients who were PET-positive among those who had received a COVID-19 vaccine and those who had not.
There was also no difference in inflammation in patients who had been hospitalized with COVID-19 and those who had managed their infection at home.
Among patients with focal FDG uptake, PET, MRI, and inflammatory blood markers improved at follow-up imaging performed a mean of 52 days after the first imaging. The authors say this suggests that these abnormalities were not related to pre-existing cardiovascular disease.
Of the eight patients with positive FDG-PET results, two did not show any MRI abnormalities. These two patients also had elevated inflammatory biomarkers. “PET is a more sensitive method of measuring cardiac inflammation, and our results show that these changes may not always translate into functional changes seen on MRI,” Dr. Thavendiranathan noted.
The only cardiac risk factor that was more common in participants with FDG uptake was hypertension. Although cardiac symptoms were nearly twice as common in participants with focal FDG uptake, this difference was not statistically significant.
“Given the growing number of survivors with similar symptoms, these interesting findings warrant further investigation,” the authors say.
Noting that FDG uptake correlated with elevations in systemic inflammatory biomarkers, the researchers suggest that “a more intense systemic inflammatory process may be contributing to cardiac inflammation and the consequential alteration to regional and global myocardial function in PET-positive participants.”
On repeat imaging 2 months later, all eight patients who showed FDG uptake showed improvement or resolution of inflammation without any treatment, although two patients still had some signs of inflammation. Blood biomarkers also improved on follow-up.
“This is encouraging information, but we need longer-term data to see if there are any long-term repercussions of this inflammation,” Dr. Hanneman said.
“Overall, the study findings suggest an imaging phenotype that is expected to have good prognosis. However, longer-term follow-up studies are required to understand the need for ongoing cardiac surveillance, relationship to cardiac symptoms, guidance for safe return to exercise and sports participation, and long-term cardiovascular disease risk,” the researchers state.
This study was funded by grants from the Joint Department of Medical Imaging Academic Incentive Fund, Peter Munk Cardiac Center Innovation Committee, and Ted Rogers Center for Heart Research. Dr. Hanneman reports personal fees from Sanofi Genzyme, Amicus, and Medscape outside the submitted work.
A version of this article first appeared on Medscape.com.
Myocardial inflammation is present in a small proportion of patients who have recovered from relatively mild cases of COVID-19 infection, a new study shows.
“Our findings suggest that even in patients who have had relatively mild cases of COVID-19, some will have inflammatory changes to the heart, and these changes can be present without any cardiac symptoms,” senior author, Paaladinesh Thavendiranathan, MD, University of Toronto, told this news organization.
“While our data suggest that this inflammation improves over time, and the outcomes seem positive, we don’t know if there will be any long-term consequences,” he added.
Noting that even a short period of inflammation in the heart may be associated with symptoms or arrhythmias in the longer term, Dr. Thavendiranathan said: “I would recommend that it is best to avoid getting the infection if there is any chance of heart inflammation.”
The study was published online in JAMA Cardiology on Jan. 12.
The authors explain that among patients hospitalized with COVID, early studies suggested that approximately one in four experience cardiovascular injury, defined as an elevation in troponin levels, which was associated with a 5- to 10-fold increase in the risk for death. But there is limited information on cardiac injury in patients who do not require hospitalization.
Although a broad range of abnormal myocardial tissue has been reported in several cardiac MRI studies of patients recovered from COVID infection, there is little understanding of persistent changes in myocardial metabolism in recovered patients, which is a potential concern, given that COVID-19 is associated with systemic inflammation during the acute illness, they say.
For the current study, the researchers examined myocardial inflammation measured using two different methods – cardiac MRI and fluorodeoxyglucose–positron emission tomography (FDG-PET) – in individuals who had recovered from COVID-19 infection and looked at how this related to changes in inflammatory blood markers.
Lead author Kate Hanneman, MD, also from the University of Toronto, explained that FDG-PET imaging is more sensitive than MRI in detecting active inflammation. “Inflammatory cells have a higher uptake of glucose, and FDG-PET imaging is used to look for metabolically active inflammatory tissue that takes up glucose. It gives complementary information to MRI. Cardiac MRI shows structural or functional changes, such as scarring or edema, whereas FDG-PET imaging directly measures metabolic activity related to inflammatory cells.”
The study involved 47 individuals, 51% female, with a mean age of 43 years, who had recently recovered from COVID-19 infection. Of these, the majority had had relatively mild COVID disease, with 85% not requiring hospitalization.
Cardiac imaging was performed a mean of 67 days after the diagnosis of COVID-19. At the time of imaging, 19 participants (40%) reported at least one cardiac symptom, including palpitations, chest pain, and shortness of breath.
Results showed that eight patients (17%) had focal FDG uptake on PET consistent with myocardial inflammation. Compared with those without FDG uptake, patients with focal FDG uptake had higher regional T2, T1, and extracellular volume (colocalizing with focal FDG uptake), higher prevalence of late gadolinium enhancement indicating fibrosis, lower left ventricular ejection fraction, worse global longitudinal and circumferential strain, and higher systemic inflammatory blood markers, including interleukin (IL)-6, IL- 8, an high-sensitivity C-reactive protein.
Of the 47 patients in the study, 13 had received at least one dose of a COVID-19 vaccine. There was no significant difference in the proportion of patients who were PET-positive among those who had received a COVID-19 vaccine and those who had not.
There was also no difference in inflammation in patients who had been hospitalized with COVID-19 and those who had managed their infection at home.
Among patients with focal FDG uptake, PET, MRI, and inflammatory blood markers improved at follow-up imaging performed a mean of 52 days after the first imaging. The authors say this suggests that these abnormalities were not related to pre-existing cardiovascular disease.
Of the eight patients with positive FDG-PET results, two did not show any MRI abnormalities. These two patients also had elevated inflammatory biomarkers. “PET is a more sensitive method of measuring cardiac inflammation, and our results show that these changes may not always translate into functional changes seen on MRI,” Dr. Thavendiranathan noted.
The only cardiac risk factor that was more common in participants with FDG uptake was hypertension. Although cardiac symptoms were nearly twice as common in participants with focal FDG uptake, this difference was not statistically significant.
“Given the growing number of survivors with similar symptoms, these interesting findings warrant further investigation,” the authors say.
Noting that FDG uptake correlated with elevations in systemic inflammatory biomarkers, the researchers suggest that “a more intense systemic inflammatory process may be contributing to cardiac inflammation and the consequential alteration to regional and global myocardial function in PET-positive participants.”
On repeat imaging 2 months later, all eight patients who showed FDG uptake showed improvement or resolution of inflammation without any treatment, although two patients still had some signs of inflammation. Blood biomarkers also improved on follow-up.
“This is encouraging information, but we need longer-term data to see if there are any long-term repercussions of this inflammation,” Dr. Hanneman said.
“Overall, the study findings suggest an imaging phenotype that is expected to have good prognosis. However, longer-term follow-up studies are required to understand the need for ongoing cardiac surveillance, relationship to cardiac symptoms, guidance for safe return to exercise and sports participation, and long-term cardiovascular disease risk,” the researchers state.
This study was funded by grants from the Joint Department of Medical Imaging Academic Incentive Fund, Peter Munk Cardiac Center Innovation Committee, and Ted Rogers Center for Heart Research. Dr. Hanneman reports personal fees from Sanofi Genzyme, Amicus, and Medscape outside the submitted work.
A version of this article first appeared on Medscape.com.
Myocardial inflammation is present in a small proportion of patients who have recovered from relatively mild cases of COVID-19 infection, a new study shows.
“Our findings suggest that even in patients who have had relatively mild cases of COVID-19, some will have inflammatory changes to the heart, and these changes can be present without any cardiac symptoms,” senior author, Paaladinesh Thavendiranathan, MD, University of Toronto, told this news organization.
“While our data suggest that this inflammation improves over time, and the outcomes seem positive, we don’t know if there will be any long-term consequences,” he added.
Noting that even a short period of inflammation in the heart may be associated with symptoms or arrhythmias in the longer term, Dr. Thavendiranathan said: “I would recommend that it is best to avoid getting the infection if there is any chance of heart inflammation.”
The study was published online in JAMA Cardiology on Jan. 12.
The authors explain that among patients hospitalized with COVID, early studies suggested that approximately one in four experience cardiovascular injury, defined as an elevation in troponin levels, which was associated with a 5- to 10-fold increase in the risk for death. But there is limited information on cardiac injury in patients who do not require hospitalization.
Although a broad range of abnormal myocardial tissue has been reported in several cardiac MRI studies of patients recovered from COVID infection, there is little understanding of persistent changes in myocardial metabolism in recovered patients, which is a potential concern, given that COVID-19 is associated with systemic inflammation during the acute illness, they say.
For the current study, the researchers examined myocardial inflammation measured using two different methods – cardiac MRI and fluorodeoxyglucose–positron emission tomography (FDG-PET) – in individuals who had recovered from COVID-19 infection and looked at how this related to changes in inflammatory blood markers.
Lead author Kate Hanneman, MD, also from the University of Toronto, explained that FDG-PET imaging is more sensitive than MRI in detecting active inflammation. “Inflammatory cells have a higher uptake of glucose, and FDG-PET imaging is used to look for metabolically active inflammatory tissue that takes up glucose. It gives complementary information to MRI. Cardiac MRI shows structural or functional changes, such as scarring or edema, whereas FDG-PET imaging directly measures metabolic activity related to inflammatory cells.”
The study involved 47 individuals, 51% female, with a mean age of 43 years, who had recently recovered from COVID-19 infection. Of these, the majority had had relatively mild COVID disease, with 85% not requiring hospitalization.
Cardiac imaging was performed a mean of 67 days after the diagnosis of COVID-19. At the time of imaging, 19 participants (40%) reported at least one cardiac symptom, including palpitations, chest pain, and shortness of breath.
Results showed that eight patients (17%) had focal FDG uptake on PET consistent with myocardial inflammation. Compared with those without FDG uptake, patients with focal FDG uptake had higher regional T2, T1, and extracellular volume (colocalizing with focal FDG uptake), higher prevalence of late gadolinium enhancement indicating fibrosis, lower left ventricular ejection fraction, worse global longitudinal and circumferential strain, and higher systemic inflammatory blood markers, including interleukin (IL)-6, IL- 8, an high-sensitivity C-reactive protein.
Of the 47 patients in the study, 13 had received at least one dose of a COVID-19 vaccine. There was no significant difference in the proportion of patients who were PET-positive among those who had received a COVID-19 vaccine and those who had not.
There was also no difference in inflammation in patients who had been hospitalized with COVID-19 and those who had managed their infection at home.
Among patients with focal FDG uptake, PET, MRI, and inflammatory blood markers improved at follow-up imaging performed a mean of 52 days after the first imaging. The authors say this suggests that these abnormalities were not related to pre-existing cardiovascular disease.
Of the eight patients with positive FDG-PET results, two did not show any MRI abnormalities. These two patients also had elevated inflammatory biomarkers. “PET is a more sensitive method of measuring cardiac inflammation, and our results show that these changes may not always translate into functional changes seen on MRI,” Dr. Thavendiranathan noted.
The only cardiac risk factor that was more common in participants with FDG uptake was hypertension. Although cardiac symptoms were nearly twice as common in participants with focal FDG uptake, this difference was not statistically significant.
“Given the growing number of survivors with similar symptoms, these interesting findings warrant further investigation,” the authors say.
Noting that FDG uptake correlated with elevations in systemic inflammatory biomarkers, the researchers suggest that “a more intense systemic inflammatory process may be contributing to cardiac inflammation and the consequential alteration to regional and global myocardial function in PET-positive participants.”
On repeat imaging 2 months later, all eight patients who showed FDG uptake showed improvement or resolution of inflammation without any treatment, although two patients still had some signs of inflammation. Blood biomarkers also improved on follow-up.
“This is encouraging information, but we need longer-term data to see if there are any long-term repercussions of this inflammation,” Dr. Hanneman said.
“Overall, the study findings suggest an imaging phenotype that is expected to have good prognosis. However, longer-term follow-up studies are required to understand the need for ongoing cardiac surveillance, relationship to cardiac symptoms, guidance for safe return to exercise and sports participation, and long-term cardiovascular disease risk,” the researchers state.
This study was funded by grants from the Joint Department of Medical Imaging Academic Incentive Fund, Peter Munk Cardiac Center Innovation Committee, and Ted Rogers Center for Heart Research. Dr. Hanneman reports personal fees from Sanofi Genzyme, Amicus, and Medscape outside the submitted work.
A version of this article first appeared on Medscape.com.
Bleeding after reperfusion contributes to cardiac injury in MI
The damage to the heart caused by a myocardial infarction is not just a result of ischemia caused by the blocked artery but is also brought about by bleeding in the myocardium after the artery has been opened, a new study suggests.
This observation is leading to new approaches to limiting infarct size and treating MI.
“In MI treatment, we have always focused on opening up the artery as quickly as possible to limit the myocardial damage caused by ischemia,” the study’s senior author, Rohan Dharmakumar, PhD, Indiana University, Indianapolis, told this news organization.
“We are pursuing a completely new approach focusing on limiting the damage after revascularization,” he said. “We are totally rethinking what a myocardial infarction is – what causes the injury and the time course of the injury – our results suggest that it’s not just ischemic damage and a lot of the harm is caused by hemorrhage after reperfusion.”
It has been known for many years that hemorrhage is often seen in the myocardium in large MIs, but it has not been established before now whether it contributes to the injury or not, Dr. Dharmakumar explained.
“This study was done to look at that – and we found that the hemorrhage drives a second layer of injury on top of the ischemia.”
Dr. Dharmakumar said this hemorrhage is part of the phenomenon known as reperfusion injury. “This has been known to exist for many years, but we haven’t fully understood all the factors contributing to it. Our results suggest that hemorrhage is a major component of reperfusion injury – probably the dominant factor,” he said.
The researchers are now working on therapeutic approaches to try to prevent this hemorrhage and/or to minimize its effect.
“We are studying how hemorrhage drives damage and how to block these biological processes,” Dr. Dharmakumar said. “Our studies suggest that hemorrhage could account for up to half of the damage caused by a myocardial infarction. If we can limit that, we should be able to reduce the size of the infarct and this should translate into better long-term outcomes.
“I’m very excited about these results,” he added. “We are already seeing a remarkable improvement in animal models with some of the potential therapeutic approaches we are working on.”
The current study is published in the January 2022 issue of the Journal of the American College of Cardiology (JACC).
The authors explain that it is now recognized that reperfusion injury can contribute to increasing infarct size, which they refer to as “infarct surge.” Previous studies have also shown that reperfusion injury can contribute to as much as 50% of the final infarct size, but the factors contributing to the observed variability are not known, and previous attempts to limit infarct surge from reperfusion injury have failed.
They noted that after reperfusion, microvessels can remain obstructed, resulting in intramyocardial hemorrhage. They conducted the current study to investigate whether such hemorrhage causes expansion of the infarct.
They studied 70 patients with ST-segment elevation MI who were categorized with cardiovascular MRI to have intramyocardial hemorrhage or not following primary PCI, and for whom serial cardiac troponin measures were used to assess infarct size.
Results showed that while troponin levels were not different before reperfusion, patients with intramyocardial hemorrhage had significantly higher cardiac troponin levels after reperfusion and these levels peaked earlier than in patients without hemorrhage.
In animal models, those with intramyocardial hemorrhage had a more rapid expansion of myocardial necrosis than did those without hemorrhage, and within 72 hours of reperfusion, a fourfold greater loss in salvageable myocardium was evident in hemorrhagic MIs.
“We have shown that damage to the heart continues after revascularization as measured by rapidly increasing troponin levels in the hearts that have had a hemorrhage,” Dr. Dharmakumar said.
“Hemorrhage in the myocardium was associated with larger infarctions, and in infarcts causing the same area of myocardium to be at risk, those with hemorrhage after revascularization lost a lot more of the salvageable myocardium than those without hemorrhage,” he added.
Dr. Dharmakumar estimates that such hemorrhage occurs in about half of MIs after revascularization, with risk factors including male gender, anterior wall MIs, and smoking.
He pointed out that previous attempts to treat or prevent reperfusion injury have not been successful, probably because they have not been addressing the key mechanism. “We have not been looking at hemorrhage in this regard until now. This is because it is only recently that we have had the tools to be able to identify hemorrhage in the heart with the use of cardiac MRI.”
Final frontier
In an accompanying editorial, Colin Berry, MBChB, University of Glasgow, and Borja Ibáñez, MD, Jiménez Díaz Foundation University Hospital, Madrid, said they applaud the investigators for providing new, mechanistic insights into a difficult clinical problem that has an unmet therapeutic need.
But they pointed out that it is difficult to completely dissect the impact of hemorrhage versus MI size on adverse remodeling, noting that it might be the case that more severe ischemia/reperfusion events are associated with large MI sizes and higher degree of hemorrhage.
However, they concluded that: “Intramyocardial hemorrhage represents the final frontier for preventing heart failure post-MI. It is readily detected using CMR, and clinical research of novel therapeutic approaches merits prioritization.”
This work was supported by grants from National Institutes of Health/National Heart, Lung, and Blood Institute. Dr. Dharmakumar and coauthor Robert Finney, PhD, have ownership interest in Cardiotheranostics. Dr. Berry is employed by the University of Glasgow, which holds consultancy and research agreements for his work with Abbott Vascular, AstraZeneca, Boehringer Ingelheim, Causeway Therapeutics, Coroventis, Genentech, GlaxoSmithKline, HeartFlow, Menarini, Neovasc, Siemens Healthcare, and Valo Health.
A version of this article first appeared on Medscape.com.
The damage to the heart caused by a myocardial infarction is not just a result of ischemia caused by the blocked artery but is also brought about by bleeding in the myocardium after the artery has been opened, a new study suggests.
This observation is leading to new approaches to limiting infarct size and treating MI.
“In MI treatment, we have always focused on opening up the artery as quickly as possible to limit the myocardial damage caused by ischemia,” the study’s senior author, Rohan Dharmakumar, PhD, Indiana University, Indianapolis, told this news organization.
“We are pursuing a completely new approach focusing on limiting the damage after revascularization,” he said. “We are totally rethinking what a myocardial infarction is – what causes the injury and the time course of the injury – our results suggest that it’s not just ischemic damage and a lot of the harm is caused by hemorrhage after reperfusion.”
It has been known for many years that hemorrhage is often seen in the myocardium in large MIs, but it has not been established before now whether it contributes to the injury or not, Dr. Dharmakumar explained.
“This study was done to look at that – and we found that the hemorrhage drives a second layer of injury on top of the ischemia.”
Dr. Dharmakumar said this hemorrhage is part of the phenomenon known as reperfusion injury. “This has been known to exist for many years, but we haven’t fully understood all the factors contributing to it. Our results suggest that hemorrhage is a major component of reperfusion injury – probably the dominant factor,” he said.
The researchers are now working on therapeutic approaches to try to prevent this hemorrhage and/or to minimize its effect.
“We are studying how hemorrhage drives damage and how to block these biological processes,” Dr. Dharmakumar said. “Our studies suggest that hemorrhage could account for up to half of the damage caused by a myocardial infarction. If we can limit that, we should be able to reduce the size of the infarct and this should translate into better long-term outcomes.
“I’m very excited about these results,” he added. “We are already seeing a remarkable improvement in animal models with some of the potential therapeutic approaches we are working on.”
The current study is published in the January 2022 issue of the Journal of the American College of Cardiology (JACC).
The authors explain that it is now recognized that reperfusion injury can contribute to increasing infarct size, which they refer to as “infarct surge.” Previous studies have also shown that reperfusion injury can contribute to as much as 50% of the final infarct size, but the factors contributing to the observed variability are not known, and previous attempts to limit infarct surge from reperfusion injury have failed.
They noted that after reperfusion, microvessels can remain obstructed, resulting in intramyocardial hemorrhage. They conducted the current study to investigate whether such hemorrhage causes expansion of the infarct.
They studied 70 patients with ST-segment elevation MI who were categorized with cardiovascular MRI to have intramyocardial hemorrhage or not following primary PCI, and for whom serial cardiac troponin measures were used to assess infarct size.
Results showed that while troponin levels were not different before reperfusion, patients with intramyocardial hemorrhage had significantly higher cardiac troponin levels after reperfusion and these levels peaked earlier than in patients without hemorrhage.
In animal models, those with intramyocardial hemorrhage had a more rapid expansion of myocardial necrosis than did those without hemorrhage, and within 72 hours of reperfusion, a fourfold greater loss in salvageable myocardium was evident in hemorrhagic MIs.
“We have shown that damage to the heart continues after revascularization as measured by rapidly increasing troponin levels in the hearts that have had a hemorrhage,” Dr. Dharmakumar said.
“Hemorrhage in the myocardium was associated with larger infarctions, and in infarcts causing the same area of myocardium to be at risk, those with hemorrhage after revascularization lost a lot more of the salvageable myocardium than those without hemorrhage,” he added.
Dr. Dharmakumar estimates that such hemorrhage occurs in about half of MIs after revascularization, with risk factors including male gender, anterior wall MIs, and smoking.
He pointed out that previous attempts to treat or prevent reperfusion injury have not been successful, probably because they have not been addressing the key mechanism. “We have not been looking at hemorrhage in this regard until now. This is because it is only recently that we have had the tools to be able to identify hemorrhage in the heart with the use of cardiac MRI.”
Final frontier
In an accompanying editorial, Colin Berry, MBChB, University of Glasgow, and Borja Ibáñez, MD, Jiménez Díaz Foundation University Hospital, Madrid, said they applaud the investigators for providing new, mechanistic insights into a difficult clinical problem that has an unmet therapeutic need.
But they pointed out that it is difficult to completely dissect the impact of hemorrhage versus MI size on adverse remodeling, noting that it might be the case that more severe ischemia/reperfusion events are associated with large MI sizes and higher degree of hemorrhage.
However, they concluded that: “Intramyocardial hemorrhage represents the final frontier for preventing heart failure post-MI. It is readily detected using CMR, and clinical research of novel therapeutic approaches merits prioritization.”
This work was supported by grants from National Institutes of Health/National Heart, Lung, and Blood Institute. Dr. Dharmakumar and coauthor Robert Finney, PhD, have ownership interest in Cardiotheranostics. Dr. Berry is employed by the University of Glasgow, which holds consultancy and research agreements for his work with Abbott Vascular, AstraZeneca, Boehringer Ingelheim, Causeway Therapeutics, Coroventis, Genentech, GlaxoSmithKline, HeartFlow, Menarini, Neovasc, Siemens Healthcare, and Valo Health.
A version of this article first appeared on Medscape.com.
The damage to the heart caused by a myocardial infarction is not just a result of ischemia caused by the blocked artery but is also brought about by bleeding in the myocardium after the artery has been opened, a new study suggests.
This observation is leading to new approaches to limiting infarct size and treating MI.
“In MI treatment, we have always focused on opening up the artery as quickly as possible to limit the myocardial damage caused by ischemia,” the study’s senior author, Rohan Dharmakumar, PhD, Indiana University, Indianapolis, told this news organization.
“We are pursuing a completely new approach focusing on limiting the damage after revascularization,” he said. “We are totally rethinking what a myocardial infarction is – what causes the injury and the time course of the injury – our results suggest that it’s not just ischemic damage and a lot of the harm is caused by hemorrhage after reperfusion.”
It has been known for many years that hemorrhage is often seen in the myocardium in large MIs, but it has not been established before now whether it contributes to the injury or not, Dr. Dharmakumar explained.
“This study was done to look at that – and we found that the hemorrhage drives a second layer of injury on top of the ischemia.”
Dr. Dharmakumar said this hemorrhage is part of the phenomenon known as reperfusion injury. “This has been known to exist for many years, but we haven’t fully understood all the factors contributing to it. Our results suggest that hemorrhage is a major component of reperfusion injury – probably the dominant factor,” he said.
The researchers are now working on therapeutic approaches to try to prevent this hemorrhage and/or to minimize its effect.
“We are studying how hemorrhage drives damage and how to block these biological processes,” Dr. Dharmakumar said. “Our studies suggest that hemorrhage could account for up to half of the damage caused by a myocardial infarction. If we can limit that, we should be able to reduce the size of the infarct and this should translate into better long-term outcomes.
“I’m very excited about these results,” he added. “We are already seeing a remarkable improvement in animal models with some of the potential therapeutic approaches we are working on.”
The current study is published in the January 2022 issue of the Journal of the American College of Cardiology (JACC).
The authors explain that it is now recognized that reperfusion injury can contribute to increasing infarct size, which they refer to as “infarct surge.” Previous studies have also shown that reperfusion injury can contribute to as much as 50% of the final infarct size, but the factors contributing to the observed variability are not known, and previous attempts to limit infarct surge from reperfusion injury have failed.
They noted that after reperfusion, microvessels can remain obstructed, resulting in intramyocardial hemorrhage. They conducted the current study to investigate whether such hemorrhage causes expansion of the infarct.
They studied 70 patients with ST-segment elevation MI who were categorized with cardiovascular MRI to have intramyocardial hemorrhage or not following primary PCI, and for whom serial cardiac troponin measures were used to assess infarct size.
Results showed that while troponin levels were not different before reperfusion, patients with intramyocardial hemorrhage had significantly higher cardiac troponin levels after reperfusion and these levels peaked earlier than in patients without hemorrhage.
In animal models, those with intramyocardial hemorrhage had a more rapid expansion of myocardial necrosis than did those without hemorrhage, and within 72 hours of reperfusion, a fourfold greater loss in salvageable myocardium was evident in hemorrhagic MIs.
“We have shown that damage to the heart continues after revascularization as measured by rapidly increasing troponin levels in the hearts that have had a hemorrhage,” Dr. Dharmakumar said.
“Hemorrhage in the myocardium was associated with larger infarctions, and in infarcts causing the same area of myocardium to be at risk, those with hemorrhage after revascularization lost a lot more of the salvageable myocardium than those without hemorrhage,” he added.
Dr. Dharmakumar estimates that such hemorrhage occurs in about half of MIs after revascularization, with risk factors including male gender, anterior wall MIs, and smoking.
He pointed out that previous attempts to treat or prevent reperfusion injury have not been successful, probably because they have not been addressing the key mechanism. “We have not been looking at hemorrhage in this regard until now. This is because it is only recently that we have had the tools to be able to identify hemorrhage in the heart with the use of cardiac MRI.”
Final frontier
In an accompanying editorial, Colin Berry, MBChB, University of Glasgow, and Borja Ibáñez, MD, Jiménez Díaz Foundation University Hospital, Madrid, said they applaud the investigators for providing new, mechanistic insights into a difficult clinical problem that has an unmet therapeutic need.
But they pointed out that it is difficult to completely dissect the impact of hemorrhage versus MI size on adverse remodeling, noting that it might be the case that more severe ischemia/reperfusion events are associated with large MI sizes and higher degree of hemorrhage.
However, they concluded that: “Intramyocardial hemorrhage represents the final frontier for preventing heart failure post-MI. It is readily detected using CMR, and clinical research of novel therapeutic approaches merits prioritization.”
This work was supported by grants from National Institutes of Health/National Heart, Lung, and Blood Institute. Dr. Dharmakumar and coauthor Robert Finney, PhD, have ownership interest in Cardiotheranostics. Dr. Berry is employed by the University of Glasgow, which holds consultancy and research agreements for his work with Abbott Vascular, AstraZeneca, Boehringer Ingelheim, Causeway Therapeutics, Coroventis, Genentech, GlaxoSmithKline, HeartFlow, Menarini, Neovasc, Siemens Healthcare, and Valo Health.
A version of this article first appeared on Medscape.com.
Bullying a ‘persistent, important’ problem for cardiology trainees
A high rate of bullying towards U.K. cardiology trainees by their superiors has been revealed in a new survey. More than 10% of doctors training to be cardiologists in the United Kingdom say they have been bullied in the last 4 weeks, and one-third report having witnessed bullying on a cardiology rotation, the survey reports.
In addition, 33% of cardiology trainees said they had been on the receiving end of inappropriate behavior, including having their opinions and views ignored, being made to feel worthless/useless, and being shouted at or targeted with spontaneous anger.
Senior doctors (consultants) in cardiology were cited as the main perpetrators of such bullying and inappropriate behavior.
Women trainee cardiologists and those who attended medical school outside the United Kingdom were more likely to report having been bullied, suggesting a sexist and racist element.
“In this large and repeated survey of British cardiology trainees, we have shown a persistent and important problem with bullying,” the authors conclude.
Results of the survey from the British Junior Cardiologists’ Association, were published online in a paper in Heart on Dec. 6, 2021.
Examples of such bullying behavior by consultant cardiologists toward their trainees reported in the survey included being belittled in front of others, having their filing cabinet drawer thrown across the room, use of foul language toward trainees, and using previous mistakes as an excuse to humiliate and ridicule them, lead author of the survey, Christian Fielder Camm, MD, Keble College, Oxford (England) University, told this news organization.
Trainees also reported being made to feel inadequate when struggling to achieve unrealistic tasks and being pressured into not taking holidays and study leave.
Many respondents said they did not report such behavior for fear of repercussions and in some cases because the perpetrators were known for their bullying behavior, and previous attempts to intervene had not resulted in any change.
Dr. Camm, who is a cardiology trainee himself, says he has not personally been the victim of bullying, but as secretary of the BJCA he regularly receives reports about it happening.
“We wanted to look at this issue in our survey as we had been hearing anecdotal reports of bullying from cardiology trainees for a number of years,” Dr. Camm said. “We wanted to put it out there that this is not just an isolated issue and shine a light on the problem.”
Noting that the U.K. General Medical Council’s annual survey has found that bullying is seen across all disciplines in medicine, Dr. Camm said that cardiology has the highest reported rate of bullying among the medical specialties.
“This is not a new story – it has been played out throughout history, but things are not magically improving. Over the 4 years of our survey, rates of reported bullying have stayed the same,” he noted. “Our survey is asking more questions about bullying to find more detail on what is happening.”
The current data come from the BJCA annual survey from 2017 to 2020. As part of the survey, trainee cardiologists were asked about direct and indirect experiences of bullying and inappropriate language/behavior in cardiology departments in the preceding 4 weeks.
In all, 2,057 responses were received, 73% were from men, and the average age of respondents was 33 years. Over half (59%) were working in a specialist center for cardiology (tertiary referral center), and 94% had a national training number, which guarantees a continued place on a training program, subject to performance.
“This project has upset me to realize what my colleagues are experiencing. This is the working environment we are creating, and it is not good enough,” Dr. Camm said.
At present, the bullying behavior is not often reported. “Usually, the only person to report it to is the senior person in the department, who is frequently the cause of the problem, so most people just put up with it until they move on to their next training rotation. The working environment should not be so difficult,” he said.
The authors noted that bullying has been shown to significantly affect trainees, with those subject to bullying being 70% more likely to report serious or potentially serious medical errors, and more likely to take time off work and cease direct patient care.
They stressed that addressing bullying of trainees needs to be a priority both to ensure patient safety and to reduce trainee attrition in a time of unprecedented workforce pressures.
Dr. Camm believes a national plan needs to be put in place to deal with this issue and said the BJCA is keen to work with the British Cardiovascular Society to develop a zero-tolerance policy, with a clear strategy to address allegations of bullying.
“The world is changing. Hopefully this publication will be the start of some change,” he added.
Bullying culture is ‘endemic’
In an accompanying editorial, consultant cardiologist Resham Baruah, MBBS, PhD, of Chelsea and Westminster Healthcare NHS Trust, London, and independent professional coach Emma Sedgwick said the findings offer a “sobering insight into current practice” and indicate that “a bullying culture is endemic in many U.K. cardiology departments.”
“These trainee cardiologists are actually quite senior doctors with many years of experience. They work extremely hard. These surveys show that we are not valuing them enough,” Dr. Baruah said in an interview.
“Cardiology is a really competitive specialty. There is a lot of pressure. All the way through training the message is competitive,” she said. “Being collegiate and working as a team is not rewarded. We have to rethink this as we train the next generation.”
“We believe that publishing these data acknowledges bullying is not going unnoticed, although this alone is not enough,” the editorialists said.
Noting that labels matter, they propose a rejection of the term “juniors” and a return to the old system of calling colleagues senior house officers, registrars, and senior registrars.
They also proposed sanctions for institutions that ignore bullying, but stress that better working conditions for all staff are needed.
“Bullies are usually feeling defensive, overwhelmed, and stressed and take these feelings out on others,” Dr. Baruah commented. “I think what we are seeing in this paper is not just restricted to cardiology but happens all though the NHS and is related to workload, lack of autonomy, and burnout. Work culture is crucial to well-being and job satisfaction. Nobody wants to work in a toxic environment.”
She emphasized that bullying behaviors must not be accepted. “They can have catastrophic consequences for the trainees and for patient safety. While working in high-pressure specialties and emergency situations may foster such behavior, it is vitally important to arm trainees with the recognition of bullying and how to deal with it. They must feel empowered to speak up in an appropriate way.”
The editorialists noted that the Royal College of Obstetricians and Gynaecologists responded to high levels of bullying by creating behavior toolkits, workshops, and behavior champions. “This survey should act as a call to arms for cardiology to introduce similar initiatives,” they stated.
“While times are changing, the corporate environment has moved forward in encouraging positive workplace behavior faster than is happening in medicine,” Dr. Baruah said. “But there is an appetite for change. We have to have an environment where people can speak up.”
The study received no specific funding. The authors reported no competing interests.
A version of this article first appeared on Medscape.com.
A high rate of bullying towards U.K. cardiology trainees by their superiors has been revealed in a new survey. More than 10% of doctors training to be cardiologists in the United Kingdom say they have been bullied in the last 4 weeks, and one-third report having witnessed bullying on a cardiology rotation, the survey reports.
In addition, 33% of cardiology trainees said they had been on the receiving end of inappropriate behavior, including having their opinions and views ignored, being made to feel worthless/useless, and being shouted at or targeted with spontaneous anger.
Senior doctors (consultants) in cardiology were cited as the main perpetrators of such bullying and inappropriate behavior.
Women trainee cardiologists and those who attended medical school outside the United Kingdom were more likely to report having been bullied, suggesting a sexist and racist element.
“In this large and repeated survey of British cardiology trainees, we have shown a persistent and important problem with bullying,” the authors conclude.
Results of the survey from the British Junior Cardiologists’ Association, were published online in a paper in Heart on Dec. 6, 2021.
Examples of such bullying behavior by consultant cardiologists toward their trainees reported in the survey included being belittled in front of others, having their filing cabinet drawer thrown across the room, use of foul language toward trainees, and using previous mistakes as an excuse to humiliate and ridicule them, lead author of the survey, Christian Fielder Camm, MD, Keble College, Oxford (England) University, told this news organization.
Trainees also reported being made to feel inadequate when struggling to achieve unrealistic tasks and being pressured into not taking holidays and study leave.
Many respondents said they did not report such behavior for fear of repercussions and in some cases because the perpetrators were known for their bullying behavior, and previous attempts to intervene had not resulted in any change.
Dr. Camm, who is a cardiology trainee himself, says he has not personally been the victim of bullying, but as secretary of the BJCA he regularly receives reports about it happening.
“We wanted to look at this issue in our survey as we had been hearing anecdotal reports of bullying from cardiology trainees for a number of years,” Dr. Camm said. “We wanted to put it out there that this is not just an isolated issue and shine a light on the problem.”
Noting that the U.K. General Medical Council’s annual survey has found that bullying is seen across all disciplines in medicine, Dr. Camm said that cardiology has the highest reported rate of bullying among the medical specialties.
“This is not a new story – it has been played out throughout history, but things are not magically improving. Over the 4 years of our survey, rates of reported bullying have stayed the same,” he noted. “Our survey is asking more questions about bullying to find more detail on what is happening.”
The current data come from the BJCA annual survey from 2017 to 2020. As part of the survey, trainee cardiologists were asked about direct and indirect experiences of bullying and inappropriate language/behavior in cardiology departments in the preceding 4 weeks.
In all, 2,057 responses were received, 73% were from men, and the average age of respondents was 33 years. Over half (59%) were working in a specialist center for cardiology (tertiary referral center), and 94% had a national training number, which guarantees a continued place on a training program, subject to performance.
“This project has upset me to realize what my colleagues are experiencing. This is the working environment we are creating, and it is not good enough,” Dr. Camm said.
At present, the bullying behavior is not often reported. “Usually, the only person to report it to is the senior person in the department, who is frequently the cause of the problem, so most people just put up with it until they move on to their next training rotation. The working environment should not be so difficult,” he said.
The authors noted that bullying has been shown to significantly affect trainees, with those subject to bullying being 70% more likely to report serious or potentially serious medical errors, and more likely to take time off work and cease direct patient care.
They stressed that addressing bullying of trainees needs to be a priority both to ensure patient safety and to reduce trainee attrition in a time of unprecedented workforce pressures.
Dr. Camm believes a national plan needs to be put in place to deal with this issue and said the BJCA is keen to work with the British Cardiovascular Society to develop a zero-tolerance policy, with a clear strategy to address allegations of bullying.
“The world is changing. Hopefully this publication will be the start of some change,” he added.
Bullying culture is ‘endemic’
In an accompanying editorial, consultant cardiologist Resham Baruah, MBBS, PhD, of Chelsea and Westminster Healthcare NHS Trust, London, and independent professional coach Emma Sedgwick said the findings offer a “sobering insight into current practice” and indicate that “a bullying culture is endemic in many U.K. cardiology departments.”
“These trainee cardiologists are actually quite senior doctors with many years of experience. They work extremely hard. These surveys show that we are not valuing them enough,” Dr. Baruah said in an interview.
“Cardiology is a really competitive specialty. There is a lot of pressure. All the way through training the message is competitive,” she said. “Being collegiate and working as a team is not rewarded. We have to rethink this as we train the next generation.”
“We believe that publishing these data acknowledges bullying is not going unnoticed, although this alone is not enough,” the editorialists said.
Noting that labels matter, they propose a rejection of the term “juniors” and a return to the old system of calling colleagues senior house officers, registrars, and senior registrars.
They also proposed sanctions for institutions that ignore bullying, but stress that better working conditions for all staff are needed.
“Bullies are usually feeling defensive, overwhelmed, and stressed and take these feelings out on others,” Dr. Baruah commented. “I think what we are seeing in this paper is not just restricted to cardiology but happens all though the NHS and is related to workload, lack of autonomy, and burnout. Work culture is crucial to well-being and job satisfaction. Nobody wants to work in a toxic environment.”
She emphasized that bullying behaviors must not be accepted. “They can have catastrophic consequences for the trainees and for patient safety. While working in high-pressure specialties and emergency situations may foster such behavior, it is vitally important to arm trainees with the recognition of bullying and how to deal with it. They must feel empowered to speak up in an appropriate way.”
The editorialists noted that the Royal College of Obstetricians and Gynaecologists responded to high levels of bullying by creating behavior toolkits, workshops, and behavior champions. “This survey should act as a call to arms for cardiology to introduce similar initiatives,” they stated.
“While times are changing, the corporate environment has moved forward in encouraging positive workplace behavior faster than is happening in medicine,” Dr. Baruah said. “But there is an appetite for change. We have to have an environment where people can speak up.”
The study received no specific funding. The authors reported no competing interests.
A version of this article first appeared on Medscape.com.
A high rate of bullying towards U.K. cardiology trainees by their superiors has been revealed in a new survey. More than 10% of doctors training to be cardiologists in the United Kingdom say they have been bullied in the last 4 weeks, and one-third report having witnessed bullying on a cardiology rotation, the survey reports.
In addition, 33% of cardiology trainees said they had been on the receiving end of inappropriate behavior, including having their opinions and views ignored, being made to feel worthless/useless, and being shouted at or targeted with spontaneous anger.
Senior doctors (consultants) in cardiology were cited as the main perpetrators of such bullying and inappropriate behavior.
Women trainee cardiologists and those who attended medical school outside the United Kingdom were more likely to report having been bullied, suggesting a sexist and racist element.
“In this large and repeated survey of British cardiology trainees, we have shown a persistent and important problem with bullying,” the authors conclude.
Results of the survey from the British Junior Cardiologists’ Association, were published online in a paper in Heart on Dec. 6, 2021.
Examples of such bullying behavior by consultant cardiologists toward their trainees reported in the survey included being belittled in front of others, having their filing cabinet drawer thrown across the room, use of foul language toward trainees, and using previous mistakes as an excuse to humiliate and ridicule them, lead author of the survey, Christian Fielder Camm, MD, Keble College, Oxford (England) University, told this news organization.
Trainees also reported being made to feel inadequate when struggling to achieve unrealistic tasks and being pressured into not taking holidays and study leave.
Many respondents said they did not report such behavior for fear of repercussions and in some cases because the perpetrators were known for their bullying behavior, and previous attempts to intervene had not resulted in any change.
Dr. Camm, who is a cardiology trainee himself, says he has not personally been the victim of bullying, but as secretary of the BJCA he regularly receives reports about it happening.
“We wanted to look at this issue in our survey as we had been hearing anecdotal reports of bullying from cardiology trainees for a number of years,” Dr. Camm said. “We wanted to put it out there that this is not just an isolated issue and shine a light on the problem.”
Noting that the U.K. General Medical Council’s annual survey has found that bullying is seen across all disciplines in medicine, Dr. Camm said that cardiology has the highest reported rate of bullying among the medical specialties.
“This is not a new story – it has been played out throughout history, but things are not magically improving. Over the 4 years of our survey, rates of reported bullying have stayed the same,” he noted. “Our survey is asking more questions about bullying to find more detail on what is happening.”
The current data come from the BJCA annual survey from 2017 to 2020. As part of the survey, trainee cardiologists were asked about direct and indirect experiences of bullying and inappropriate language/behavior in cardiology departments in the preceding 4 weeks.
In all, 2,057 responses were received, 73% were from men, and the average age of respondents was 33 years. Over half (59%) were working in a specialist center for cardiology (tertiary referral center), and 94% had a national training number, which guarantees a continued place on a training program, subject to performance.
“This project has upset me to realize what my colleagues are experiencing. This is the working environment we are creating, and it is not good enough,” Dr. Camm said.
At present, the bullying behavior is not often reported. “Usually, the only person to report it to is the senior person in the department, who is frequently the cause of the problem, so most people just put up with it until they move on to their next training rotation. The working environment should not be so difficult,” he said.
The authors noted that bullying has been shown to significantly affect trainees, with those subject to bullying being 70% more likely to report serious or potentially serious medical errors, and more likely to take time off work and cease direct patient care.
They stressed that addressing bullying of trainees needs to be a priority both to ensure patient safety and to reduce trainee attrition in a time of unprecedented workforce pressures.
Dr. Camm believes a national plan needs to be put in place to deal with this issue and said the BJCA is keen to work with the British Cardiovascular Society to develop a zero-tolerance policy, with a clear strategy to address allegations of bullying.
“The world is changing. Hopefully this publication will be the start of some change,” he added.
Bullying culture is ‘endemic’
In an accompanying editorial, consultant cardiologist Resham Baruah, MBBS, PhD, of Chelsea and Westminster Healthcare NHS Trust, London, and independent professional coach Emma Sedgwick said the findings offer a “sobering insight into current practice” and indicate that “a bullying culture is endemic in many U.K. cardiology departments.”
“These trainee cardiologists are actually quite senior doctors with many years of experience. They work extremely hard. These surveys show that we are not valuing them enough,” Dr. Baruah said in an interview.
“Cardiology is a really competitive specialty. There is a lot of pressure. All the way through training the message is competitive,” she said. “Being collegiate and working as a team is not rewarded. We have to rethink this as we train the next generation.”
“We believe that publishing these data acknowledges bullying is not going unnoticed, although this alone is not enough,” the editorialists said.
Noting that labels matter, they propose a rejection of the term “juniors” and a return to the old system of calling colleagues senior house officers, registrars, and senior registrars.
They also proposed sanctions for institutions that ignore bullying, but stress that better working conditions for all staff are needed.
“Bullies are usually feeling defensive, overwhelmed, and stressed and take these feelings out on others,” Dr. Baruah commented. “I think what we are seeing in this paper is not just restricted to cardiology but happens all though the NHS and is related to workload, lack of autonomy, and burnout. Work culture is crucial to well-being and job satisfaction. Nobody wants to work in a toxic environment.”
She emphasized that bullying behaviors must not be accepted. “They can have catastrophic consequences for the trainees and for patient safety. While working in high-pressure specialties and emergency situations may foster such behavior, it is vitally important to arm trainees with the recognition of bullying and how to deal with it. They must feel empowered to speak up in an appropriate way.”
The editorialists noted that the Royal College of Obstetricians and Gynaecologists responded to high levels of bullying by creating behavior toolkits, workshops, and behavior champions. “This survey should act as a call to arms for cardiology to introduce similar initiatives,” they stated.
“While times are changing, the corporate environment has moved forward in encouraging positive workplace behavior faster than is happening in medicine,” Dr. Baruah said. “But there is an appetite for change. We have to have an environment where people can speak up.”
The study received no specific funding. The authors reported no competing interests.
A version of this article first appeared on Medscape.com.
FROM HEART
Cardiologist positive for Omicron after London conference
Elad Maor, MD, an interventional cardiologist at Sheba Medical Centre near Tel Aviv, posted on Twitter on Nov. 30: “What a mess! Came back from a conference in London. With a mask and 3 Pfizer vaccines I managed to get Omicron.”
Dr. Maor traveled to London on November 19 to attend the PCR London Valves 2021 conference held at the ExCeL Centre Nov. 21-23. He stayed four nights at a hotel in north London and took public transport to and from the ExCeL Centre in East London each day of the meeting. He returned to Israel on the evening of Nov. 23.
Dr. Maor, 45, who has received three doses of the Pfizer COVID-19 vaccine, had two PCR tests in the United Kingdom – on November 20 and 21 in line with travel requirements – and another PCR test upon arriving back in Israel in the early hours of Nov. 24. All three tests were negative.
He began experiencing symptoms within days and tested positive on Nov. 27. His symptoms have been mild so far, and he said he was feeling “better” at the time of his tweet on Nov. 30.
Dr. Maor believes he was infected during his trip to London. “The only reasonable explanation is that I got infected on the last day of the meeting – maybe at the airport, maybe at the meeting,” he told The Guardian newspaper.
Although his wife accompanied him to London, neither she nor any of his 3 children have experienced symptoms or tested positive for COVID-19. But Dr. Maor believes he has passed the infection to a 69-year-old colleague in Israel who has since tested positive for the Omicron variant. The colleague, who has also received three vaccine doses, is understood to have mild symptoms at present.
The case suggests that the Omicron variant of COVID-19 may have been circulating in the United Kingdom earlier than previously thought.
Implications for in-person conferences
It will also inevitably lead to questions about the safety of face-to-face conferences, which are only just starting to get underway again.
The PCR Valves 2021 meeting had more than 1,250 on-site attendees as well as 2,400 or more joining online, according to figures on its website. Dr. Maor said he did not have any issues with the conference organizers, who required proof of vaccination before entry. But he posted a photograph on his Twitter account of a crowded auditorium with many delegates not wearing masks.
The conference subsequently posted an announcement on its website alerting delegates that one of the attendees had tested positive for COVID-19 after returning to their home country. It reads: “Since the reported case comes less than a week after the end of PCR London Valves, we want to inform you so that you may decide the best course of action, for yourself, if any.” It does not mention that the case was the Omicron variant.
Patrick Jolly, strategic and market development director of the conference, commented: “As you may imagine, the health, safety and well-being of everyone who visited PCR London Valves was our number-one priority. All protocols mandated by the U.K. government were put in place. Anyone entering the congress center had to present a valid health pass and were requested to wear a mask. Hydro-alcoholic gel and masks were made readily available for all participants and disposal bins for used protective equipment were provided.”
Mr. Jolly also noted: “To date – more than 9 days after the end of PCR London Valves – we have had no report of any other case of participants testing positive who attended PCR London Valves.”
He said the EuroPCR organization believes that medical conferences are safe to be held in person.
“With the above sanitary requirements and protocols, and no complacency in their enforcement, we believe strongly that medical conferences can take place, as the benefits of in-person medical conferences are obvious for the concerned medical communities,” Mr. Jolly added.
But what about other meetings happening imminently and planning in-person attendance?
Eileen Murray, executive director of the American Epilepsy Society (AES), whose annual 5-day meeting starts today at Chicago’s McCormick Place Convention Center, said in an interview that the health, safety, and well-being of everyone attending is a priority.
“Vaccinations are required, with no exceptions, to anyone attending the in-person event,” Ms. Murray said. “AES is using the CLEAR HealthPass to verify identity and vaccination status for our attendees. No one who cannot verify identity and vaccination requirement will be permitted to attend the in-person event.”
She noted that masks will also be required except in limited circumstances when actively eating or drinking, or for a faculty member when actively presenting at a lecture or panel. “Anyone not adhering to the mask policy will be asked to leave the meeting and will be denied readmission to the meeting with no refund,” she said.
“These guidelines were developed in accordance with the latest public health guidance and AES will continue to follow that guidance as any updates are made with the emergence of the Omicron variant,” Ms. Murray added.
Also commenting on this issue, a spokesperson for the American Heart Association, which has its large annual international stroke meeting planned for in-person attendance in New Orleans in February, said: “As we have throughout the pandemic, the American Heart Association is closely monitoring conditions and following the guidance of the CDC as well as state and local health departments related to all in-person meetings.”
“Our upcoming International Stroke Conference, February 9-11, is planned as an in-person and digital experience which allows us the ultimate flexibility to address changing pandemic conditions. The health, safety, and well-being of our volunteers, members, and attendees from around the world remains our number-one priority,” the AHA spokesperson added.
But some COVID-19 experts are taking a more cautious view.
Rowland Kao, PhD, an expert in infectious disease dynamics at the University of Edinburgh, United Kingdom, expressed concern about such large in-person conferences.
“We know that the Omicron variant appears to be spreading rapidly, with a recent preprint also telling us that the reinfection rate appears to be higher in South Africa. Should this be borne out, then the evidence would support that our reliance on a combination of vaccine-induced and natural immunity may be compromised by the Omicron variant,” he commented.
“We already know that extended contact indoors provides an additional risk, and so large meetings of this type have the potential to create extended risks. Until we know the extent to which Omicron causes severe illness, we should be extra cautious about these high-risk settings,” Dr. Kao commented.
A version of this article first appeared on Medscape.com.
Elad Maor, MD, an interventional cardiologist at Sheba Medical Centre near Tel Aviv, posted on Twitter on Nov. 30: “What a mess! Came back from a conference in London. With a mask and 3 Pfizer vaccines I managed to get Omicron.”
Dr. Maor traveled to London on November 19 to attend the PCR London Valves 2021 conference held at the ExCeL Centre Nov. 21-23. He stayed four nights at a hotel in north London and took public transport to and from the ExCeL Centre in East London each day of the meeting. He returned to Israel on the evening of Nov. 23.
Dr. Maor, 45, who has received three doses of the Pfizer COVID-19 vaccine, had two PCR tests in the United Kingdom – on November 20 and 21 in line with travel requirements – and another PCR test upon arriving back in Israel in the early hours of Nov. 24. All three tests were negative.
He began experiencing symptoms within days and tested positive on Nov. 27. His symptoms have been mild so far, and he said he was feeling “better” at the time of his tweet on Nov. 30.
Dr. Maor believes he was infected during his trip to London. “The only reasonable explanation is that I got infected on the last day of the meeting – maybe at the airport, maybe at the meeting,” he told The Guardian newspaper.
Although his wife accompanied him to London, neither she nor any of his 3 children have experienced symptoms or tested positive for COVID-19. But Dr. Maor believes he has passed the infection to a 69-year-old colleague in Israel who has since tested positive for the Omicron variant. The colleague, who has also received three vaccine doses, is understood to have mild symptoms at present.
The case suggests that the Omicron variant of COVID-19 may have been circulating in the United Kingdom earlier than previously thought.
Implications for in-person conferences
It will also inevitably lead to questions about the safety of face-to-face conferences, which are only just starting to get underway again.
The PCR Valves 2021 meeting had more than 1,250 on-site attendees as well as 2,400 or more joining online, according to figures on its website. Dr. Maor said he did not have any issues with the conference organizers, who required proof of vaccination before entry. But he posted a photograph on his Twitter account of a crowded auditorium with many delegates not wearing masks.
The conference subsequently posted an announcement on its website alerting delegates that one of the attendees had tested positive for COVID-19 after returning to their home country. It reads: “Since the reported case comes less than a week after the end of PCR London Valves, we want to inform you so that you may decide the best course of action, for yourself, if any.” It does not mention that the case was the Omicron variant.
Patrick Jolly, strategic and market development director of the conference, commented: “As you may imagine, the health, safety and well-being of everyone who visited PCR London Valves was our number-one priority. All protocols mandated by the U.K. government were put in place. Anyone entering the congress center had to present a valid health pass and were requested to wear a mask. Hydro-alcoholic gel and masks were made readily available for all participants and disposal bins for used protective equipment were provided.”
Mr. Jolly also noted: “To date – more than 9 days after the end of PCR London Valves – we have had no report of any other case of participants testing positive who attended PCR London Valves.”
He said the EuroPCR organization believes that medical conferences are safe to be held in person.
“With the above sanitary requirements and protocols, and no complacency in their enforcement, we believe strongly that medical conferences can take place, as the benefits of in-person medical conferences are obvious for the concerned medical communities,” Mr. Jolly added.
But what about other meetings happening imminently and planning in-person attendance?
Eileen Murray, executive director of the American Epilepsy Society (AES), whose annual 5-day meeting starts today at Chicago’s McCormick Place Convention Center, said in an interview that the health, safety, and well-being of everyone attending is a priority.
“Vaccinations are required, with no exceptions, to anyone attending the in-person event,” Ms. Murray said. “AES is using the CLEAR HealthPass to verify identity and vaccination status for our attendees. No one who cannot verify identity and vaccination requirement will be permitted to attend the in-person event.”
She noted that masks will also be required except in limited circumstances when actively eating or drinking, or for a faculty member when actively presenting at a lecture or panel. “Anyone not adhering to the mask policy will be asked to leave the meeting and will be denied readmission to the meeting with no refund,” she said.
“These guidelines were developed in accordance with the latest public health guidance and AES will continue to follow that guidance as any updates are made with the emergence of the Omicron variant,” Ms. Murray added.
Also commenting on this issue, a spokesperson for the American Heart Association, which has its large annual international stroke meeting planned for in-person attendance in New Orleans in February, said: “As we have throughout the pandemic, the American Heart Association is closely monitoring conditions and following the guidance of the CDC as well as state and local health departments related to all in-person meetings.”
“Our upcoming International Stroke Conference, February 9-11, is planned as an in-person and digital experience which allows us the ultimate flexibility to address changing pandemic conditions. The health, safety, and well-being of our volunteers, members, and attendees from around the world remains our number-one priority,” the AHA spokesperson added.
But some COVID-19 experts are taking a more cautious view.
Rowland Kao, PhD, an expert in infectious disease dynamics at the University of Edinburgh, United Kingdom, expressed concern about such large in-person conferences.
“We know that the Omicron variant appears to be spreading rapidly, with a recent preprint also telling us that the reinfection rate appears to be higher in South Africa. Should this be borne out, then the evidence would support that our reliance on a combination of vaccine-induced and natural immunity may be compromised by the Omicron variant,” he commented.
“We already know that extended contact indoors provides an additional risk, and so large meetings of this type have the potential to create extended risks. Until we know the extent to which Omicron causes severe illness, we should be extra cautious about these high-risk settings,” Dr. Kao commented.
A version of this article first appeared on Medscape.com.
Elad Maor, MD, an interventional cardiologist at Sheba Medical Centre near Tel Aviv, posted on Twitter on Nov. 30: “What a mess! Came back from a conference in London. With a mask and 3 Pfizer vaccines I managed to get Omicron.”
Dr. Maor traveled to London on November 19 to attend the PCR London Valves 2021 conference held at the ExCeL Centre Nov. 21-23. He stayed four nights at a hotel in north London and took public transport to and from the ExCeL Centre in East London each day of the meeting. He returned to Israel on the evening of Nov. 23.
Dr. Maor, 45, who has received three doses of the Pfizer COVID-19 vaccine, had two PCR tests in the United Kingdom – on November 20 and 21 in line with travel requirements – and another PCR test upon arriving back in Israel in the early hours of Nov. 24. All three tests were negative.
He began experiencing symptoms within days and tested positive on Nov. 27. His symptoms have been mild so far, and he said he was feeling “better” at the time of his tweet on Nov. 30.
Dr. Maor believes he was infected during his trip to London. “The only reasonable explanation is that I got infected on the last day of the meeting – maybe at the airport, maybe at the meeting,” he told The Guardian newspaper.
Although his wife accompanied him to London, neither she nor any of his 3 children have experienced symptoms or tested positive for COVID-19. But Dr. Maor believes he has passed the infection to a 69-year-old colleague in Israel who has since tested positive for the Omicron variant. The colleague, who has also received three vaccine doses, is understood to have mild symptoms at present.
The case suggests that the Omicron variant of COVID-19 may have been circulating in the United Kingdom earlier than previously thought.
Implications for in-person conferences
It will also inevitably lead to questions about the safety of face-to-face conferences, which are only just starting to get underway again.
The PCR Valves 2021 meeting had more than 1,250 on-site attendees as well as 2,400 or more joining online, according to figures on its website. Dr. Maor said he did not have any issues with the conference organizers, who required proof of vaccination before entry. But he posted a photograph on his Twitter account of a crowded auditorium with many delegates not wearing masks.
The conference subsequently posted an announcement on its website alerting delegates that one of the attendees had tested positive for COVID-19 after returning to their home country. It reads: “Since the reported case comes less than a week after the end of PCR London Valves, we want to inform you so that you may decide the best course of action, for yourself, if any.” It does not mention that the case was the Omicron variant.
Patrick Jolly, strategic and market development director of the conference, commented: “As you may imagine, the health, safety and well-being of everyone who visited PCR London Valves was our number-one priority. All protocols mandated by the U.K. government were put in place. Anyone entering the congress center had to present a valid health pass and were requested to wear a mask. Hydro-alcoholic gel and masks were made readily available for all participants and disposal bins for used protective equipment were provided.”
Mr. Jolly also noted: “To date – more than 9 days after the end of PCR London Valves – we have had no report of any other case of participants testing positive who attended PCR London Valves.”
He said the EuroPCR organization believes that medical conferences are safe to be held in person.
“With the above sanitary requirements and protocols, and no complacency in their enforcement, we believe strongly that medical conferences can take place, as the benefits of in-person medical conferences are obvious for the concerned medical communities,” Mr. Jolly added.
But what about other meetings happening imminently and planning in-person attendance?
Eileen Murray, executive director of the American Epilepsy Society (AES), whose annual 5-day meeting starts today at Chicago’s McCormick Place Convention Center, said in an interview that the health, safety, and well-being of everyone attending is a priority.
“Vaccinations are required, with no exceptions, to anyone attending the in-person event,” Ms. Murray said. “AES is using the CLEAR HealthPass to verify identity and vaccination status for our attendees. No one who cannot verify identity and vaccination requirement will be permitted to attend the in-person event.”
She noted that masks will also be required except in limited circumstances when actively eating or drinking, or for a faculty member when actively presenting at a lecture or panel. “Anyone not adhering to the mask policy will be asked to leave the meeting and will be denied readmission to the meeting with no refund,” she said.
“These guidelines were developed in accordance with the latest public health guidance and AES will continue to follow that guidance as any updates are made with the emergence of the Omicron variant,” Ms. Murray added.
Also commenting on this issue, a spokesperson for the American Heart Association, which has its large annual international stroke meeting planned for in-person attendance in New Orleans in February, said: “As we have throughout the pandemic, the American Heart Association is closely monitoring conditions and following the guidance of the CDC as well as state and local health departments related to all in-person meetings.”
“Our upcoming International Stroke Conference, February 9-11, is planned as an in-person and digital experience which allows us the ultimate flexibility to address changing pandemic conditions. The health, safety, and well-being of our volunteers, members, and attendees from around the world remains our number-one priority,” the AHA spokesperson added.
But some COVID-19 experts are taking a more cautious view.
Rowland Kao, PhD, an expert in infectious disease dynamics at the University of Edinburgh, United Kingdom, expressed concern about such large in-person conferences.
“We know that the Omicron variant appears to be spreading rapidly, with a recent preprint also telling us that the reinfection rate appears to be higher in South Africa. Should this be borne out, then the evidence would support that our reliance on a combination of vaccine-induced and natural immunity may be compromised by the Omicron variant,” he commented.
“We already know that extended contact indoors provides an additional risk, and so large meetings of this type have the potential to create extended risks. Until we know the extent to which Omicron causes severe illness, we should be extra cautious about these high-risk settings,” Dr. Kao commented.
A version of this article first appeared on Medscape.com.
New CETP inhibitor impresses in LDL lowering
A new lipid-lowering agent in a class that had been written off by many is being developed by a group of academic experts, with new data showing large LDL reductions on top of high-intensity statins.
Obicetrapib is a member of the cholesteryl ester transfer protein (CETP) inhibitor class, which had fallen out of favor after several disappointments with previous drugs in this class.
These agents were initially developed for their ability to raise HDL cholesterol, which was thought to be beneficial. But that approach has now been virtually abandoned after several studies failed to show a link between raising HDL and a reduction in subsequent cardiovascular events.
However, obicetrapib, which is said to be the most potent CETP inhibitor to date, has been shown to produce impressive LDL reductions, and it’s this important data that has caused several lipid experts to want to continue its development.
New data, presented at the recent American Heart Association scientific sessions, show that obicetrapib reduces LDL by 50% when given in addition to high-intensity statins, which could place it as competition for PCSK9 inhibitors or the new agent, inclisiran, but with the advantage of oral dosing.
The drug was in development by Amgen, but the company decided to discontinue its development in 2017 after disappointing results had been seen with several other CETP inhibitors and interest in this class of agent was waning.
But academic experts in the lipid field, led by John Kastelein, MD, PhD, professor of medicine at the Academic Medical Center, University of Amsterdam, and Michael Davidson, MD, clinical professor of medicine at University of Chicago, believed the drug had potential and have acquired obicetrapib from Amgen.
Dr. Kastelein and Dr. Davidson have set up a new company – New Amsterdam Pharma – to further develop obicetrapib, and have raised $200 million from venture capital funding to complete phase 2 and phase 3 studies.
The company has a heavyweight academic advisory board including Stephen Nicholls, MD, Monash University, Clayton, Australia; Kausik Ray, MD, Imperial College London; and Christie Ballantyne, MD, Baylor College of Medicine, Houston.
“We wanted to develop obicetrapib further because of its amazing LDL-lowering properties,” Dr. Kastelein said in an interview.
“No one has paid much attention to CETP inhibitors after the HDL hypothesis was disregarded, as everyone thought these drugs were just about raising HDL. But actually, they can also lower LDL, and this particular agent reduces LDL very effectively,” Dr. Kastelein said.
ROSE study
Dr. Nicholls presented the latest data on obicetrapib at the AHA meeting.
“Despite the use of high-intensity statins, two-thirds of patients do not reach their target LDL level, so we have a need for new therapies that lower LDL and can be used in combination with high-intensity statins,” he explained.

He noted that earlier studies with obicetrapib showed a 45% lowering of LDL with monotherapy.
Dr. Nicholls reported that recent evidence has emerged that increases interest in inhibiting CETP to be potentially cardioprotective.
To begin, genetic studies have shown that genetic polymorphisms associated with lower levels of CETP appear to be cardioprotective, and this is associated with lower levels of LDL rather than higher levels of HDL.
Furthermore, the REVEAL cardiovascular outcomes trial with anacetrapib (also a CETP inhibitor) in 2017 showed a significant 9% reduction in major adverse cardiac events (MACE) after 4 years of follow-up. “This was exactly predicted by the 11 mg/dL drop in absolute LDL cholesterol level. It was not predicted or associated with the increase in HDL level observed with that agent,” Dr. Nicholls said.
The objective of the current ROSE study was to evaluate the lipid-lowering ability, safety, and tolerability of obicetrapib in patients on high-intensity statins.
The study included 120 patients who had been treated on a stable dose of high-intensity statins (atorvastatin at a dose of at least 40 mg daily or rosuvastatin at a dose of 20 mg daily) for at least 8 weeks. All patients were required to have a fasting LDL of at least 70 mg/dL and the median baseline LDL was 90 mg/dL. They were randomly assigned to obicetrapib (5 mg or 10 mg daily) or placebo.
The primary endpoint was the difference between groups in percentage change in LDL from baseline to week 8, with LDL levels measured by two different techniques.
Results showed a “robust” 51% reduction in LDL with the 10-mg dose of obicetrapib, and a 42% reduction with the 5-mg dose, Dr. Nicholls reported.
These effects were comparable regardless of baseline LDL and were similar with both methods of LDL measurement.
Almost all patients demonstrated some degree of LDL cholesterol lowering, with only three patients on the 5-mg dose and one patient on the 10-mg dose not showing any reduction in LDL.
Other results showed a dose-dependent lowering of Apo B of up to 30%, and a reduction of non-HDL cholesterol of up to 44%.
“Predictably, there were also increases of HDL cholesterol,” Dr. Nicholls said. “At the 10-mg dose, we see a 165% increase in HDL levels. That is associated with a 48% increase in Apo A1 levels. This is very consistent with findings from the previous monotherapy study.”
There was a 56% reduction in Lp(a) levels, and a modest 11% reduction in triglycerides.
Both doses of obicetrapib were well tolerated, with no increase in the rate of adverse events. Only one patient discontinued the study drug because of an adverse event and that patient was in the placebo group, Dr. Nicholls noted.
“Blood pressure is an important adverse event to look at in the CETP class given the challenges seen with the first CETP evaluated – torcetrapib,” Dr. Nicholls said. “But in the three clinical trials with obicetrapib conducted to date, reassuringly, we see no increase in either systolic or diastolic blood pressure with either the 5-mg or 10-mg dose.”
He concluded that obicetrapib “could be a valuable addition to high-risk patients with atherosclerotic cardiovascular disease who do not achieve their target LDL level despite use of high-intensity statin therapy.”
Differences from other CETP inhibitors
Asked how obicetrapib differs from other agents in the CETP inhibitor class, Dr. Nicholls replied that obicetrapib is much more potent, as shown by the large lipid changes seen with very small quantities of this drug, 5 mg or 10 mg, whereas prior CETP inhibitors showed smaller changes with much higher doses.
“We are giving very small amounts of obicetrapib and seeing very robust effects on both atherogenic and lipid parameters,” he said.
“The other major point with this class of agent is that the first drug, torcetrapib, had toxicity, which resulted in increased cardiovascular events. But it has now been established that torcetrapib had a number of off-target effects that have not been seen with subsequent agents in this class,” he said.
Studies so far show that obicetrapib does not have torcetrapib-like effects. “That is encouraging. This, and the impressive LDL lowering effects, certainly lay the foundation for larger studies moving forward,” he added.
“This has been an intriguing field to many of us involved from the start. We started with a very disappointing result with torcetrapib. Then a couple of studies looked to be clinically futile, but we were encouraged by the REVEAL study which suggested that there might be benefit,” Dr. Nicholls said.
“If we combined the REVEAL results with the genetic data, it has actually flipped the whole CETP story upside down. We started thinking that inhibiting CETP was all about raising HDL, but it turns out that it is about LDL lowering,” he said. “And that is not only important in terms of the lipid effects but also the trials and the way they are designed.
“I think you’ll find that the future trials in this class and with this agent will have LDL very much in mind and that will very much influence the study design,” he said, adding that a larger cardiovascular outcome trial is now being planned.
“The regulatory perspective is that LDL is a pretty trusted surrogate ... but I think an outcomes trial will be important to reinforce and reassure on safety and outline cost-effectiveness, which will help us understand where the sweet spot for using this agent in the clinic will be,” Dr. Nicholls noted.
Dr. Kastelein explained that it has taken some time to realize that CETP inhibitors may be valuable for reducing LDL.
“The first agent, torcetrapib, had an off-target toxicity that led to increased blood pressure but a specific part of the torcetrapib molecule was subsequently identified that was responsible for that, and subsequent agents in the CETP inhibitor class did not have such adverse effects,” he said.
“The next agent, dalcetrapib (Roche), raised HDL but didn’t move LDL, and an outcomes trial with evacetrapib (Lilly) was stopped after 2 years because of futility, but we now believe that lipid lowering trials need longer term follow-up – up to 5 years – to see a benefit,” he noted.
Dr. Kastelein reports that anacetrapib (Merck) has been the most powerful CETP inhibitor until now, giving an LDL reduction of about 20%, which was associated with a 10% reduction in cardiovascular events in first 4 years of follow-up.
“Oxford academic researchers decided to continue follow-up in this trial without Merck and showed a 20% reduction in cardiovascular events by 6 years. This has been the strongest rationale for our investors,” Dr. Kastelein said.
He pointed out that obicetrapib is much more potent than anacetrapib. “Obicetrapib reduces LDL by 50% at just a 10-mg dose, whereas anacetrapib was used at a dose of 100 mg to give a 17%-20% LDL reduction.”
Could HDL increase be beneficial after all?
Although increasing HDL is currently not thought to bring about a direct reduction in cardiovascular events, there is new evidence emerging that increasing HDL may confer some benefit in protecting against the development of type 2 diabetes, Dr. Kastelein noted.
“We know that statins can increase risk of developing type 2 diabetes, and post hoc analyses of previous trials with CETP inhibitors suggest that these drugs have the opposite effect,” he said. “We will investigate this protectively in our phase 3 outcomes trial. If this is a true effect, it should eventually translate into a reduction in cardiovascular outcomes, but this could take a longer time to see than the benefits of lowering LDL.”
Commenting on the current data, Steven Nissen, MD, of Cleveland Clinic, said: “The results are truly impressive – a nearly 50% LDL reduction on a background of statins with a once-daily oral agent. While PCSK9 inhibitors can achieve similar results, they are injectable and costly.
“Since anacetrapib, a much weaker CETP inhibitor, was successful at reducing major adverse cardiac events, the likelihood that obicetrapib would reduce MACE even more substantially is very high,” he added.
Dr. Nissen said he has been aware of this drug for some time and has advised the company about development options and regulatory strategy. “I have encouraged this company to develop this very promising drug,” he said.
The current study was funded by New Amsterdam Pharma. Dr. Nicholls reports grants from AstraZeneca, Amgen, Anthera, Eli Lilly, Esperion, Novartis, Cerenis, The Medicines Company, Resverlogix, Infraredx, Roche, Sanofi-Regeneron and LipoScience, and honoraria from New Amsterdam Pharma, AstraZeneca, Akcea, Eli Lilly, Anthera, Omthera, Merck, Takeda, Resverlogix, Sanofi-Regeneron, CSL Behring, Esperion, and Boehringer Ingelheim. Dr. Kastelein is chief scientific officer of New Amsterdam Pharma.
A version of this article first appeared on Medscape.com.
A new lipid-lowering agent in a class that had been written off by many is being developed by a group of academic experts, with new data showing large LDL reductions on top of high-intensity statins.
Obicetrapib is a member of the cholesteryl ester transfer protein (CETP) inhibitor class, which had fallen out of favor after several disappointments with previous drugs in this class.
These agents were initially developed for their ability to raise HDL cholesterol, which was thought to be beneficial. But that approach has now been virtually abandoned after several studies failed to show a link between raising HDL and a reduction in subsequent cardiovascular events.
However, obicetrapib, which is said to be the most potent CETP inhibitor to date, has been shown to produce impressive LDL reductions, and it’s this important data that has caused several lipid experts to want to continue its development.
New data, presented at the recent American Heart Association scientific sessions, show that obicetrapib reduces LDL by 50% when given in addition to high-intensity statins, which could place it as competition for PCSK9 inhibitors or the new agent, inclisiran, but with the advantage of oral dosing.
The drug was in development by Amgen, but the company decided to discontinue its development in 2017 after disappointing results had been seen with several other CETP inhibitors and interest in this class of agent was waning.
But academic experts in the lipid field, led by John Kastelein, MD, PhD, professor of medicine at the Academic Medical Center, University of Amsterdam, and Michael Davidson, MD, clinical professor of medicine at University of Chicago, believed the drug had potential and have acquired obicetrapib from Amgen.
Dr. Kastelein and Dr. Davidson have set up a new company – New Amsterdam Pharma – to further develop obicetrapib, and have raised $200 million from venture capital funding to complete phase 2 and phase 3 studies.
The company has a heavyweight academic advisory board including Stephen Nicholls, MD, Monash University, Clayton, Australia; Kausik Ray, MD, Imperial College London; and Christie Ballantyne, MD, Baylor College of Medicine, Houston.
“We wanted to develop obicetrapib further because of its amazing LDL-lowering properties,” Dr. Kastelein said in an interview.
“No one has paid much attention to CETP inhibitors after the HDL hypothesis was disregarded, as everyone thought these drugs were just about raising HDL. But actually, they can also lower LDL, and this particular agent reduces LDL very effectively,” Dr. Kastelein said.
ROSE study
Dr. Nicholls presented the latest data on obicetrapib at the AHA meeting.
“Despite the use of high-intensity statins, two-thirds of patients do not reach their target LDL level, so we have a need for new therapies that lower LDL and can be used in combination with high-intensity statins,” he explained.
He noted that earlier studies with obicetrapib showed a 45% lowering of LDL with monotherapy.
Dr. Nicholls reported that recent evidence has emerged that increases interest in inhibiting CETP to be potentially cardioprotective.
To begin, genetic studies have shown that genetic polymorphisms associated with lower levels of CETP appear to be cardioprotective, and this is associated with lower levels of LDL rather than higher levels of HDL.
Furthermore, the REVEAL cardiovascular outcomes trial with anacetrapib (also a CETP inhibitor) in 2017 showed a significant 9% reduction in major adverse cardiac events (MACE) after 4 years of follow-up. “This was exactly predicted by the 11 mg/dL drop in absolute LDL cholesterol level. It was not predicted or associated with the increase in HDL level observed with that agent,” Dr. Nicholls said.
The objective of the current ROSE study was to evaluate the lipid-lowering ability, safety, and tolerability of obicetrapib in patients on high-intensity statins.
The study included 120 patients who had been treated on a stable dose of high-intensity statins (atorvastatin at a dose of at least 40 mg daily or rosuvastatin at a dose of 20 mg daily) for at least 8 weeks. All patients were required to have a fasting LDL of at least 70 mg/dL and the median baseline LDL was 90 mg/dL. They were randomly assigned to obicetrapib (5 mg or 10 mg daily) or placebo.
The primary endpoint was the difference between groups in percentage change in LDL from baseline to week 8, with LDL levels measured by two different techniques.
Results showed a “robust” 51% reduction in LDL with the 10-mg dose of obicetrapib, and a 42% reduction with the 5-mg dose, Dr. Nicholls reported.
These effects were comparable regardless of baseline LDL and were similar with both methods of LDL measurement.
Almost all patients demonstrated some degree of LDL cholesterol lowering, with only three patients on the 5-mg dose and one patient on the 10-mg dose not showing any reduction in LDL.
Other results showed a dose-dependent lowering of Apo B of up to 30%, and a reduction of non-HDL cholesterol of up to 44%.
“Predictably, there were also increases of HDL cholesterol,” Dr. Nicholls said. “At the 10-mg dose, we see a 165% increase in HDL levels. That is associated with a 48% increase in Apo A1 levels. This is very consistent with findings from the previous monotherapy study.”
There was a 56% reduction in Lp(a) levels, and a modest 11% reduction in triglycerides.
Both doses of obicetrapib were well tolerated, with no increase in the rate of adverse events. Only one patient discontinued the study drug because of an adverse event and that patient was in the placebo group, Dr. Nicholls noted.
“Blood pressure is an important adverse event to look at in the CETP class given the challenges seen with the first CETP evaluated – torcetrapib,” Dr. Nicholls said. “But in the three clinical trials with obicetrapib conducted to date, reassuringly, we see no increase in either systolic or diastolic blood pressure with either the 5-mg or 10-mg dose.”
He concluded that obicetrapib “could be a valuable addition to high-risk patients with atherosclerotic cardiovascular disease who do not achieve their target LDL level despite use of high-intensity statin therapy.”
Differences from other CETP inhibitors
Asked how obicetrapib differs from other agents in the CETP inhibitor class, Dr. Nicholls replied that obicetrapib is much more potent, as shown by the large lipid changes seen with very small quantities of this drug, 5 mg or 10 mg, whereas prior CETP inhibitors showed smaller changes with much higher doses.
“We are giving very small amounts of obicetrapib and seeing very robust effects on both atherogenic and lipid parameters,” he said.
“The other major point with this class of agent is that the first drug, torcetrapib, had toxicity, which resulted in increased cardiovascular events. But it has now been established that torcetrapib had a number of off-target effects that have not been seen with subsequent agents in this class,” he said.
Studies so far show that obicetrapib does not have torcetrapib-like effects. “That is encouraging. This, and the impressive LDL lowering effects, certainly lay the foundation for larger studies moving forward,” he added.
“This has been an intriguing field to many of us involved from the start. We started with a very disappointing result with torcetrapib. Then a couple of studies looked to be clinically futile, but we were encouraged by the REVEAL study which suggested that there might be benefit,” Dr. Nicholls said.
“If we combined the REVEAL results with the genetic data, it has actually flipped the whole CETP story upside down. We started thinking that inhibiting CETP was all about raising HDL, but it turns out that it is about LDL lowering,” he said. “And that is not only important in terms of the lipid effects but also the trials and the way they are designed.
“I think you’ll find that the future trials in this class and with this agent will have LDL very much in mind and that will very much influence the study design,” he said, adding that a larger cardiovascular outcome trial is now being planned.
“The regulatory perspective is that LDL is a pretty trusted surrogate ... but I think an outcomes trial will be important to reinforce and reassure on safety and outline cost-effectiveness, which will help us understand where the sweet spot for using this agent in the clinic will be,” Dr. Nicholls noted.
Dr. Kastelein explained that it has taken some time to realize that CETP inhibitors may be valuable for reducing LDL.
“The first agent, torcetrapib, had an off-target toxicity that led to increased blood pressure but a specific part of the torcetrapib molecule was subsequently identified that was responsible for that, and subsequent agents in the CETP inhibitor class did not have such adverse effects,” he said.
“The next agent, dalcetrapib (Roche), raised HDL but didn’t move LDL, and an outcomes trial with evacetrapib (Lilly) was stopped after 2 years because of futility, but we now believe that lipid lowering trials need longer term follow-up – up to 5 years – to see a benefit,” he noted.
Dr. Kastelein reports that anacetrapib (Merck) has been the most powerful CETP inhibitor until now, giving an LDL reduction of about 20%, which was associated with a 10% reduction in cardiovascular events in first 4 years of follow-up.
“Oxford academic researchers decided to continue follow-up in this trial without Merck and showed a 20% reduction in cardiovascular events by 6 years. This has been the strongest rationale for our investors,” Dr. Kastelein said.
He pointed out that obicetrapib is much more potent than anacetrapib. “Obicetrapib reduces LDL by 50% at just a 10-mg dose, whereas anacetrapib was used at a dose of 100 mg to give a 17%-20% LDL reduction.”
Could HDL increase be beneficial after all?
Although increasing HDL is currently not thought to bring about a direct reduction in cardiovascular events, there is new evidence emerging that increasing HDL may confer some benefit in protecting against the development of type 2 diabetes, Dr. Kastelein noted.
“We know that statins can increase risk of developing type 2 diabetes, and post hoc analyses of previous trials with CETP inhibitors suggest that these drugs have the opposite effect,” he said. “We will investigate this protectively in our phase 3 outcomes trial. If this is a true effect, it should eventually translate into a reduction in cardiovascular outcomes, but this could take a longer time to see than the benefits of lowering LDL.”
Commenting on the current data, Steven Nissen, MD, of Cleveland Clinic, said: “The results are truly impressive – a nearly 50% LDL reduction on a background of statins with a once-daily oral agent. While PCSK9 inhibitors can achieve similar results, they are injectable and costly.
“Since anacetrapib, a much weaker CETP inhibitor, was successful at reducing major adverse cardiac events, the likelihood that obicetrapib would reduce MACE even more substantially is very high,” he added.
Dr. Nissen said he has been aware of this drug for some time and has advised the company about development options and regulatory strategy. “I have encouraged this company to develop this very promising drug,” he said.
The current study was funded by New Amsterdam Pharma. Dr. Nicholls reports grants from AstraZeneca, Amgen, Anthera, Eli Lilly, Esperion, Novartis, Cerenis, The Medicines Company, Resverlogix, Infraredx, Roche, Sanofi-Regeneron and LipoScience, and honoraria from New Amsterdam Pharma, AstraZeneca, Akcea, Eli Lilly, Anthera, Omthera, Merck, Takeda, Resverlogix, Sanofi-Regeneron, CSL Behring, Esperion, and Boehringer Ingelheim. Dr. Kastelein is chief scientific officer of New Amsterdam Pharma.
A version of this article first appeared on Medscape.com.
A new lipid-lowering agent in a class that had been written off by many is being developed by a group of academic experts, with new data showing large LDL reductions on top of high-intensity statins.
Obicetrapib is a member of the cholesteryl ester transfer protein (CETP) inhibitor class, which had fallen out of favor after several disappointments with previous drugs in this class.
These agents were initially developed for their ability to raise HDL cholesterol, which was thought to be beneficial. But that approach has now been virtually abandoned after several studies failed to show a link between raising HDL and a reduction in subsequent cardiovascular events.
However, obicetrapib, which is said to be the most potent CETP inhibitor to date, has been shown to produce impressive LDL reductions, and it’s this important data that has caused several lipid experts to want to continue its development.
New data, presented at the recent American Heart Association scientific sessions, show that obicetrapib reduces LDL by 50% when given in addition to high-intensity statins, which could place it as competition for PCSK9 inhibitors or the new agent, inclisiran, but with the advantage of oral dosing.
The drug was in development by Amgen, but the company decided to discontinue its development in 2017 after disappointing results had been seen with several other CETP inhibitors and interest in this class of agent was waning.
But academic experts in the lipid field, led by John Kastelein, MD, PhD, professor of medicine at the Academic Medical Center, University of Amsterdam, and Michael Davidson, MD, clinical professor of medicine at University of Chicago, believed the drug had potential and have acquired obicetrapib from Amgen.
Dr. Kastelein and Dr. Davidson have set up a new company – New Amsterdam Pharma – to further develop obicetrapib, and have raised $200 million from venture capital funding to complete phase 2 and phase 3 studies.
The company has a heavyweight academic advisory board including Stephen Nicholls, MD, Monash University, Clayton, Australia; Kausik Ray, MD, Imperial College London; and Christie Ballantyne, MD, Baylor College of Medicine, Houston.
“We wanted to develop obicetrapib further because of its amazing LDL-lowering properties,” Dr. Kastelein said in an interview.
“No one has paid much attention to CETP inhibitors after the HDL hypothesis was disregarded, as everyone thought these drugs were just about raising HDL. But actually, they can also lower LDL, and this particular agent reduces LDL very effectively,” Dr. Kastelein said.
ROSE study
Dr. Nicholls presented the latest data on obicetrapib at the AHA meeting.
“Despite the use of high-intensity statins, two-thirds of patients do not reach their target LDL level, so we have a need for new therapies that lower LDL and can be used in combination with high-intensity statins,” he explained.
He noted that earlier studies with obicetrapib showed a 45% lowering of LDL with monotherapy.
Dr. Nicholls reported that recent evidence has emerged that increases interest in inhibiting CETP to be potentially cardioprotective.
To begin, genetic studies have shown that genetic polymorphisms associated with lower levels of CETP appear to be cardioprotective, and this is associated with lower levels of LDL rather than higher levels of HDL.
Furthermore, the REVEAL cardiovascular outcomes trial with anacetrapib (also a CETP inhibitor) in 2017 showed a significant 9% reduction in major adverse cardiac events (MACE) after 4 years of follow-up. “This was exactly predicted by the 11 mg/dL drop in absolute LDL cholesterol level. It was not predicted or associated with the increase in HDL level observed with that agent,” Dr. Nicholls said.
The objective of the current ROSE study was to evaluate the lipid-lowering ability, safety, and tolerability of obicetrapib in patients on high-intensity statins.
The study included 120 patients who had been treated on a stable dose of high-intensity statins (atorvastatin at a dose of at least 40 mg daily or rosuvastatin at a dose of 20 mg daily) for at least 8 weeks. All patients were required to have a fasting LDL of at least 70 mg/dL and the median baseline LDL was 90 mg/dL. They were randomly assigned to obicetrapib (5 mg or 10 mg daily) or placebo.
The primary endpoint was the difference between groups in percentage change in LDL from baseline to week 8, with LDL levels measured by two different techniques.
Results showed a “robust” 51% reduction in LDL with the 10-mg dose of obicetrapib, and a 42% reduction with the 5-mg dose, Dr. Nicholls reported.
These effects were comparable regardless of baseline LDL and were similar with both methods of LDL measurement.
Almost all patients demonstrated some degree of LDL cholesterol lowering, with only three patients on the 5-mg dose and one patient on the 10-mg dose not showing any reduction in LDL.
Other results showed a dose-dependent lowering of Apo B of up to 30%, and a reduction of non-HDL cholesterol of up to 44%.
“Predictably, there were also increases of HDL cholesterol,” Dr. Nicholls said. “At the 10-mg dose, we see a 165% increase in HDL levels. That is associated with a 48% increase in Apo A1 levels. This is very consistent with findings from the previous monotherapy study.”
There was a 56% reduction in Lp(a) levels, and a modest 11% reduction in triglycerides.
Both doses of obicetrapib were well tolerated, with no increase in the rate of adverse events. Only one patient discontinued the study drug because of an adverse event and that patient was in the placebo group, Dr. Nicholls noted.
“Blood pressure is an important adverse event to look at in the CETP class given the challenges seen with the first CETP evaluated – torcetrapib,” Dr. Nicholls said. “But in the three clinical trials with obicetrapib conducted to date, reassuringly, we see no increase in either systolic or diastolic blood pressure with either the 5-mg or 10-mg dose.”
He concluded that obicetrapib “could be a valuable addition to high-risk patients with atherosclerotic cardiovascular disease who do not achieve their target LDL level despite use of high-intensity statin therapy.”
Differences from other CETP inhibitors
Asked how obicetrapib differs from other agents in the CETP inhibitor class, Dr. Nicholls replied that obicetrapib is much more potent, as shown by the large lipid changes seen with very small quantities of this drug, 5 mg or 10 mg, whereas prior CETP inhibitors showed smaller changes with much higher doses.
“We are giving very small amounts of obicetrapib and seeing very robust effects on both atherogenic and lipid parameters,” he said.
“The other major point with this class of agent is that the first drug, torcetrapib, had toxicity, which resulted in increased cardiovascular events. But it has now been established that torcetrapib had a number of off-target effects that have not been seen with subsequent agents in this class,” he said.
Studies so far show that obicetrapib does not have torcetrapib-like effects. “That is encouraging. This, and the impressive LDL lowering effects, certainly lay the foundation for larger studies moving forward,” he added.
“This has been an intriguing field to many of us involved from the start. We started with a very disappointing result with torcetrapib. Then a couple of studies looked to be clinically futile, but we were encouraged by the REVEAL study which suggested that there might be benefit,” Dr. Nicholls said.
“If we combined the REVEAL results with the genetic data, it has actually flipped the whole CETP story upside down. We started thinking that inhibiting CETP was all about raising HDL, but it turns out that it is about LDL lowering,” he said. “And that is not only important in terms of the lipid effects but also the trials and the way they are designed.
“I think you’ll find that the future trials in this class and with this agent will have LDL very much in mind and that will very much influence the study design,” he said, adding that a larger cardiovascular outcome trial is now being planned.
“The regulatory perspective is that LDL is a pretty trusted surrogate ... but I think an outcomes trial will be important to reinforce and reassure on safety and outline cost-effectiveness, which will help us understand where the sweet spot for using this agent in the clinic will be,” Dr. Nicholls noted.
Dr. Kastelein explained that it has taken some time to realize that CETP inhibitors may be valuable for reducing LDL.
“The first agent, torcetrapib, had an off-target toxicity that led to increased blood pressure but a specific part of the torcetrapib molecule was subsequently identified that was responsible for that, and subsequent agents in the CETP inhibitor class did not have such adverse effects,” he said.
“The next agent, dalcetrapib (Roche), raised HDL but didn’t move LDL, and an outcomes trial with evacetrapib (Lilly) was stopped after 2 years because of futility, but we now believe that lipid lowering trials need longer term follow-up – up to 5 years – to see a benefit,” he noted.
Dr. Kastelein reports that anacetrapib (Merck) has been the most powerful CETP inhibitor until now, giving an LDL reduction of about 20%, which was associated with a 10% reduction in cardiovascular events in first 4 years of follow-up.
“Oxford academic researchers decided to continue follow-up in this trial without Merck and showed a 20% reduction in cardiovascular events by 6 years. This has been the strongest rationale for our investors,” Dr. Kastelein said.
He pointed out that obicetrapib is much more potent than anacetrapib. “Obicetrapib reduces LDL by 50% at just a 10-mg dose, whereas anacetrapib was used at a dose of 100 mg to give a 17%-20% LDL reduction.”
Could HDL increase be beneficial after all?
Although increasing HDL is currently not thought to bring about a direct reduction in cardiovascular events, there is new evidence emerging that increasing HDL may confer some benefit in protecting against the development of type 2 diabetes, Dr. Kastelein noted.
“We know that statins can increase risk of developing type 2 diabetes, and post hoc analyses of previous trials with CETP inhibitors suggest that these drugs have the opposite effect,” he said. “We will investigate this protectively in our phase 3 outcomes trial. If this is a true effect, it should eventually translate into a reduction in cardiovascular outcomes, but this could take a longer time to see than the benefits of lowering LDL.”
Commenting on the current data, Steven Nissen, MD, of Cleveland Clinic, said: “The results are truly impressive – a nearly 50% LDL reduction on a background of statins with a once-daily oral agent. While PCSK9 inhibitors can achieve similar results, they are injectable and costly.
“Since anacetrapib, a much weaker CETP inhibitor, was successful at reducing major adverse cardiac events, the likelihood that obicetrapib would reduce MACE even more substantially is very high,” he added.
Dr. Nissen said he has been aware of this drug for some time and has advised the company about development options and regulatory strategy. “I have encouraged this company to develop this very promising drug,” he said.
The current study was funded by New Amsterdam Pharma. Dr. Nicholls reports grants from AstraZeneca, Amgen, Anthera, Eli Lilly, Esperion, Novartis, Cerenis, The Medicines Company, Resverlogix, Infraredx, Roche, Sanofi-Regeneron and LipoScience, and honoraria from New Amsterdam Pharma, AstraZeneca, Akcea, Eli Lilly, Anthera, Omthera, Merck, Takeda, Resverlogix, Sanofi-Regeneron, CSL Behring, Esperion, and Boehringer Ingelheim. Dr. Kastelein is chief scientific officer of New Amsterdam Pharma.
A version of this article first appeared on Medscape.com.
FROM AHA 2021
High-dose fish oil: ‘Intriguing’ results in COVID-19
A high dose of the purified form of eicosapentaenoic acid, icosapent ethyl (Vascepa, Amarin), failed to significantly reduce hospitalizations or death in patients infected with COVID-19 in the PREPARE-IT 2 study.
The study did, however, show a favorable trend, with a 16% reduction in the primary endpoint of death or an indication for hospitalization. All secondary endpoints were also numerically reduced, but none reached statistical significance.
The product was also well tolerated over the 28 days of the study period, even though a new high-loading dose was used, with no increase in atrial fibrillation or bleeding or other adverse events versus placebo, although there was a slightly higher rate of discontinuation.
The trial was presented at the American Heart Association scientific sessions on Nov. 15 by Rafael Díaz, MD, director of Estudios Clínicos Latinoamérica in Rosario, Argentina.
“Larger, randomized trials powered for a relative risk reduction of around 15% with icosapent ethyl are needed to establish whether or not this product may have a role in the management of COVID-positive outpatients,” Dr. Diaz concluded.
‘Intriguing signals’
Commenting on the study, Manesh Patel, MD, chief of the division of cardiology and codirector of the Heart Center at Duke University, Durham, N.C., and chair of the Scientific Sessions scientific program, said that: “Certainly there are some intriguing signals.”
“I think the trend is valuable, but do we need a larger trial to confirm a benefit? I will leave that to the clinical community to decide,” Dr. Patel added. “But it is hard to power a trial to get that answer, and the world of COVID has changed since this trial started with vaccines now available and new therapeutics coming. So, there’s going to be a competing landscape.”
Discussing the trial at an AHA news briefing, Erin Michos, MD, associate professor of medicine within the division of cardiology at Johns Hopkins University, Baltimore, said: “Results showed that everything trended in the right direction, but did not reach statistical significance largely because there were fewer events than anticipated. COVID hospitalizations are going down because of the broad adoption of vaccines, which meant that this study didn’t quite meet its endpoint.”
But, she added: “Reassuringly, even with the higher loading dose, there was no increased risk of [atrial fibrillation] when used for just 28 days, and no increased risk in bleeding, so there was very good safety.”
“We need a larger trial to really definitely show whether icosapent ethyl can or cannot help COVID-positive outpatients, but I think a better prevention strategy would be the broad adoption of vaccinations globally,” Dr. Michos concluded.
‘A pretty big ask’
Donald Lloyd-Jones, MD, AHA president and designated discussant at the late-breaking science session, congratulated the investigators on conducting “a very nice pragmatic trial in the midst of the COVID pandemic.”
Dr. Lloyd-Jones concluded that the broad range of potentially beneficial actions of icosapent ethyl – including antitriglyceride, anti-inflammatory, antioxidant, and antithrombotic effects – leads to the possibility of it helping in COVID, but he added that “this is a pretty big ask for a fish oil supplement given short term.”
Presenting the study, Dr. Diaz noted that there are limited options for the outpatient treatment of patients with COVID-19 infection, and it is believed that inflammation plays a major role in worsening the severity of the infection.
He pointed out that previous data support a potential role of omega-3 fatty acids in reducing inflammation and infection, and that icosapent ethyl has shown a reduction in major cardiovascular events in the REDUCE-IT trial, with the mechanism thought to involve anti-inflammatory effects.
In the first trial to investigate the role of icosapent ethyl in COVID-19, PREPARE-IT, the product did not prevent uninfected individuals at risk from COVID from becoming infected with the virus, but there was no increase in side effects versus placebo with use over a 60-day period.
A small study last year in 100 COVID-positive patients showed icosapent ethyl reduced C-reactive protein, an inflammatory marker, and also improved symptoms.
PREPARE-IT 2, a pragmatic web-based trial, was conducted to investigate whether icosapent ethyl in nonhospitalized patients with a positive diagnosis of COVID-19 could reduce hospitalization rates and complications.
The trial enrolled 2,052 patients (mean age, 50 years), of whom 1,010 were allocated to the active group and 1,042 to the placebo group. Inclusion criteria included individuals aged 40 years or older with a confirmed COVID-19 diagnosis and no more than 7 days from the onset of symptoms and without a clear indication for hospitalization.
Patients who were allocated to the active arm received icosapent ethyl at a dose of 8 g (four capsules every 12 hours, morning and evening) for the first 3 days, followed by 4 g (two capsules every 12 hours) thereafter (days 4-28).
The primary outcome, COVID-19–related hospitalization (indication for hospitalization or hospitalization) or death at 28 days, occurred in 11.16% of the active group and 13.69% of the placebo group, giving a hazard ratio of 0.84 (95% confidence interval, 0.65-1.08; P = .166)
Secondary outcomes showed similar positive trends, but none were significant. These included: death or still hospitalized at 28 days (HR, 0.74), major events (MI, stroke, death; HR, 0.38), and total mortality (HR, 0.52).
In terms of safety, there was no significant difference in total adverse events between the two groups (16.5% in the active group vs. 14.8% in the placebo group). The most common adverse effects were constipation (2.7%), diarrhea (7.2%), and nausea (4%), but these were not significantly different from placebo. There were, however, more discontinuations in the active group (7% vs. 4%).
Dr. Diaz pointed out that the PREPARE-IT 2 trial was started in May 2020, when there wasn’t much known about the COVID-19 condition, and there were no vaccines or treatments, so hospitalization rates were high.
“We were hoping to see a 25%-30% reduction in hospitalizations with icosapent ethyl, and the trial was powered for that sort of reduction, but today we know we can expect a more modest reduction of about 15%,” Dr. Diaz concluded. “But to show that, we need a much larger trial with 8,000 or 9,000 patients, and that will be much more difficult to conduct.”
The PREPARE-IT 2 study was funded by Amarin. Dr. Diaz has received grants from Dalcor, Amarin, PHRI, and Lepetit.
A version of this article first appeared on Medscape.com.
A high dose of the purified form of eicosapentaenoic acid, icosapent ethyl (Vascepa, Amarin), failed to significantly reduce hospitalizations or death in patients infected with COVID-19 in the PREPARE-IT 2 study.
The study did, however, show a favorable trend, with a 16% reduction in the primary endpoint of death or an indication for hospitalization. All secondary endpoints were also numerically reduced, but none reached statistical significance.
The product was also well tolerated over the 28 days of the study period, even though a new high-loading dose was used, with no increase in atrial fibrillation or bleeding or other adverse events versus placebo, although there was a slightly higher rate of discontinuation.
The trial was presented at the American Heart Association scientific sessions on Nov. 15 by Rafael Díaz, MD, director of Estudios Clínicos Latinoamérica in Rosario, Argentina.
“Larger, randomized trials powered for a relative risk reduction of around 15% with icosapent ethyl are needed to establish whether or not this product may have a role in the management of COVID-positive outpatients,” Dr. Diaz concluded.
‘Intriguing signals’
Commenting on the study, Manesh Patel, MD, chief of the division of cardiology and codirector of the Heart Center at Duke University, Durham, N.C., and chair of the Scientific Sessions scientific program, said that: “Certainly there are some intriguing signals.”
“I think the trend is valuable, but do we need a larger trial to confirm a benefit? I will leave that to the clinical community to decide,” Dr. Patel added. “But it is hard to power a trial to get that answer, and the world of COVID has changed since this trial started with vaccines now available and new therapeutics coming. So, there’s going to be a competing landscape.”
Discussing the trial at an AHA news briefing, Erin Michos, MD, associate professor of medicine within the division of cardiology at Johns Hopkins University, Baltimore, said: “Results showed that everything trended in the right direction, but did not reach statistical significance largely because there were fewer events than anticipated. COVID hospitalizations are going down because of the broad adoption of vaccines, which meant that this study didn’t quite meet its endpoint.”
But, she added: “Reassuringly, even with the higher loading dose, there was no increased risk of [atrial fibrillation] when used for just 28 days, and no increased risk in bleeding, so there was very good safety.”
“We need a larger trial to really definitely show whether icosapent ethyl can or cannot help COVID-positive outpatients, but I think a better prevention strategy would be the broad adoption of vaccinations globally,” Dr. Michos concluded.
‘A pretty big ask’
Donald Lloyd-Jones, MD, AHA president and designated discussant at the late-breaking science session, congratulated the investigators on conducting “a very nice pragmatic trial in the midst of the COVID pandemic.”
Dr. Lloyd-Jones concluded that the broad range of potentially beneficial actions of icosapent ethyl – including antitriglyceride, anti-inflammatory, antioxidant, and antithrombotic effects – leads to the possibility of it helping in COVID, but he added that “this is a pretty big ask for a fish oil supplement given short term.”
Presenting the study, Dr. Diaz noted that there are limited options for the outpatient treatment of patients with COVID-19 infection, and it is believed that inflammation plays a major role in worsening the severity of the infection.
He pointed out that previous data support a potential role of omega-3 fatty acids in reducing inflammation and infection, and that icosapent ethyl has shown a reduction in major cardiovascular events in the REDUCE-IT trial, with the mechanism thought to involve anti-inflammatory effects.
In the first trial to investigate the role of icosapent ethyl in COVID-19, PREPARE-IT, the product did not prevent uninfected individuals at risk from COVID from becoming infected with the virus, but there was no increase in side effects versus placebo with use over a 60-day period.
A small study last year in 100 COVID-positive patients showed icosapent ethyl reduced C-reactive protein, an inflammatory marker, and also improved symptoms.
PREPARE-IT 2, a pragmatic web-based trial, was conducted to investigate whether icosapent ethyl in nonhospitalized patients with a positive diagnosis of COVID-19 could reduce hospitalization rates and complications.
The trial enrolled 2,052 patients (mean age, 50 years), of whom 1,010 were allocated to the active group and 1,042 to the placebo group. Inclusion criteria included individuals aged 40 years or older with a confirmed COVID-19 diagnosis and no more than 7 days from the onset of symptoms and without a clear indication for hospitalization.
Patients who were allocated to the active arm received icosapent ethyl at a dose of 8 g (four capsules every 12 hours, morning and evening) for the first 3 days, followed by 4 g (two capsules every 12 hours) thereafter (days 4-28).
The primary outcome, COVID-19–related hospitalization (indication for hospitalization or hospitalization) or death at 28 days, occurred in 11.16% of the active group and 13.69% of the placebo group, giving a hazard ratio of 0.84 (95% confidence interval, 0.65-1.08; P = .166)
Secondary outcomes showed similar positive trends, but none were significant. These included: death or still hospitalized at 28 days (HR, 0.74), major events (MI, stroke, death; HR, 0.38), and total mortality (HR, 0.52).
In terms of safety, there was no significant difference in total adverse events between the two groups (16.5% in the active group vs. 14.8% in the placebo group). The most common adverse effects were constipation (2.7%), diarrhea (7.2%), and nausea (4%), but these were not significantly different from placebo. There were, however, more discontinuations in the active group (7% vs. 4%).
Dr. Diaz pointed out that the PREPARE-IT 2 trial was started in May 2020, when there wasn’t much known about the COVID-19 condition, and there were no vaccines or treatments, so hospitalization rates were high.
“We were hoping to see a 25%-30% reduction in hospitalizations with icosapent ethyl, and the trial was powered for that sort of reduction, but today we know we can expect a more modest reduction of about 15%,” Dr. Diaz concluded. “But to show that, we need a much larger trial with 8,000 or 9,000 patients, and that will be much more difficult to conduct.”
The PREPARE-IT 2 study was funded by Amarin. Dr. Diaz has received grants from Dalcor, Amarin, PHRI, and Lepetit.
A version of this article first appeared on Medscape.com.
A high dose of the purified form of eicosapentaenoic acid, icosapent ethyl (Vascepa, Amarin), failed to significantly reduce hospitalizations or death in patients infected with COVID-19 in the PREPARE-IT 2 study.
The study did, however, show a favorable trend, with a 16% reduction in the primary endpoint of death or an indication for hospitalization. All secondary endpoints were also numerically reduced, but none reached statistical significance.
The product was also well tolerated over the 28 days of the study period, even though a new high-loading dose was used, with no increase in atrial fibrillation or bleeding or other adverse events versus placebo, although there was a slightly higher rate of discontinuation.
The trial was presented at the American Heart Association scientific sessions on Nov. 15 by Rafael Díaz, MD, director of Estudios Clínicos Latinoamérica in Rosario, Argentina.
“Larger, randomized trials powered for a relative risk reduction of around 15% with icosapent ethyl are needed to establish whether or not this product may have a role in the management of COVID-positive outpatients,” Dr. Diaz concluded.
‘Intriguing signals’
Commenting on the study, Manesh Patel, MD, chief of the division of cardiology and codirector of the Heart Center at Duke University, Durham, N.C., and chair of the Scientific Sessions scientific program, said that: “Certainly there are some intriguing signals.”
“I think the trend is valuable, but do we need a larger trial to confirm a benefit? I will leave that to the clinical community to decide,” Dr. Patel added. “But it is hard to power a trial to get that answer, and the world of COVID has changed since this trial started with vaccines now available and new therapeutics coming. So, there’s going to be a competing landscape.”
Discussing the trial at an AHA news briefing, Erin Michos, MD, associate professor of medicine within the division of cardiology at Johns Hopkins University, Baltimore, said: “Results showed that everything trended in the right direction, but did not reach statistical significance largely because there were fewer events than anticipated. COVID hospitalizations are going down because of the broad adoption of vaccines, which meant that this study didn’t quite meet its endpoint.”
But, she added: “Reassuringly, even with the higher loading dose, there was no increased risk of [atrial fibrillation] when used for just 28 days, and no increased risk in bleeding, so there was very good safety.”
“We need a larger trial to really definitely show whether icosapent ethyl can or cannot help COVID-positive outpatients, but I think a better prevention strategy would be the broad adoption of vaccinations globally,” Dr. Michos concluded.
‘A pretty big ask’
Donald Lloyd-Jones, MD, AHA president and designated discussant at the late-breaking science session, congratulated the investigators on conducting “a very nice pragmatic trial in the midst of the COVID pandemic.”
Dr. Lloyd-Jones concluded that the broad range of potentially beneficial actions of icosapent ethyl – including antitriglyceride, anti-inflammatory, antioxidant, and antithrombotic effects – leads to the possibility of it helping in COVID, but he added that “this is a pretty big ask for a fish oil supplement given short term.”
Presenting the study, Dr. Diaz noted that there are limited options for the outpatient treatment of patients with COVID-19 infection, and it is believed that inflammation plays a major role in worsening the severity of the infection.
He pointed out that previous data support a potential role of omega-3 fatty acids in reducing inflammation and infection, and that icosapent ethyl has shown a reduction in major cardiovascular events in the REDUCE-IT trial, with the mechanism thought to involve anti-inflammatory effects.
In the first trial to investigate the role of icosapent ethyl in COVID-19, PREPARE-IT, the product did not prevent uninfected individuals at risk from COVID from becoming infected with the virus, but there was no increase in side effects versus placebo with use over a 60-day period.
A small study last year in 100 COVID-positive patients showed icosapent ethyl reduced C-reactive protein, an inflammatory marker, and also improved symptoms.
PREPARE-IT 2, a pragmatic web-based trial, was conducted to investigate whether icosapent ethyl in nonhospitalized patients with a positive diagnosis of COVID-19 could reduce hospitalization rates and complications.
The trial enrolled 2,052 patients (mean age, 50 years), of whom 1,010 were allocated to the active group and 1,042 to the placebo group. Inclusion criteria included individuals aged 40 years or older with a confirmed COVID-19 diagnosis and no more than 7 days from the onset of symptoms and without a clear indication for hospitalization.
Patients who were allocated to the active arm received icosapent ethyl at a dose of 8 g (four capsules every 12 hours, morning and evening) for the first 3 days, followed by 4 g (two capsules every 12 hours) thereafter (days 4-28).
The primary outcome, COVID-19–related hospitalization (indication for hospitalization or hospitalization) or death at 28 days, occurred in 11.16% of the active group and 13.69% of the placebo group, giving a hazard ratio of 0.84 (95% confidence interval, 0.65-1.08; P = .166)
Secondary outcomes showed similar positive trends, but none were significant. These included: death or still hospitalized at 28 days (HR, 0.74), major events (MI, stroke, death; HR, 0.38), and total mortality (HR, 0.52).
In terms of safety, there was no significant difference in total adverse events between the two groups (16.5% in the active group vs. 14.8% in the placebo group). The most common adverse effects were constipation (2.7%), diarrhea (7.2%), and nausea (4%), but these were not significantly different from placebo. There were, however, more discontinuations in the active group (7% vs. 4%).
Dr. Diaz pointed out that the PREPARE-IT 2 trial was started in May 2020, when there wasn’t much known about the COVID-19 condition, and there were no vaccines or treatments, so hospitalization rates were high.
“We were hoping to see a 25%-30% reduction in hospitalizations with icosapent ethyl, and the trial was powered for that sort of reduction, but today we know we can expect a more modest reduction of about 15%,” Dr. Diaz concluded. “But to show that, we need a much larger trial with 8,000 or 9,000 patients, and that will be much more difficult to conduct.”
The PREPARE-IT 2 study was funded by Amarin. Dr. Diaz has received grants from Dalcor, Amarin, PHRI, and Lepetit.
A version of this article first appeared on Medscape.com.
FROM AHA 2021
CABG safe 3 days after stopping ticagrelor: RAPID CABG
Patients with acute coronary syndromes who have been taking the antiplatelet medication, ticagrelor, and who need coronary artery bypass surgery (CABG) may be able to safely have the procedure earlier than typically recommended, a new randomized trial suggests.

The RAPID CABG trial found that early surgery 2-3 days after ticagrelor cessation was noninferior in incurring severe or massive perioperative bleeding, compared with waiting 5-7 days. There was also no significant difference in TIMI CABG or Bleeding Academic Research Consortium (BARC) type 4 or 5 bleeding.
Patients in the delayed group had a numerically higher number of ischemic events requiring earlier surgery and had a longer hospital stay.
The study was presented at the American Heart Association scientific sessions.
“RAPID CABG is the first and only randomized controlled trial evaluating the safety of early surgery in patients taking ticagrelor,” said lead investigator Derek So, MD.
Dr. So, a cardiologist at the University of Ottawa Heart Institute and a professor at the University of Ottawa, explained that ticagrelor is a first-line antiplatelet agent for patients with acute coronary syndromes (ACS), but around 10% of patients presenting with ACS require CABG surgery.
A major concern among patients requiring bypass surgery is perioperative bleeding, and it has been shown that patients undergoing urgent bypass within 24 hours of the last dose of ticagrelor have increased mortality. Accordingly, guidelines suggest a waiting period for patients not requiring urgent bypass surgery, Dr. So noted.
Current North American guidelines suggest a waiting period of at least 5 days after stopping ticagrelor before bypass surgery. In contrast, the updated European and Japanese guidelines suggest a waiting period of 3 days.
Dr. So noted that all of the guidelines are based on cohort studies and pharmacodynamic studies, with no randomized evidence. Pharmacodynamic studies have shown that at 48 hours after the last dose of ticagrelor, the level of platelet inhibition drops to the same levels seen with long-term treatment with clopidogrel, a weaker antiplatelet drug, and after 120 hours (5 days) the effect has completely worn off.
Dr. So concluded that these new results from the RAPID CABG trial “may influence future iterations of North American guidelines with reduced waiting prior to bypass surgery” for patients receiving ticagrelor, and “they could also strengthen the level of evidence in European and Asian guidelines.”

Designated discussant of the RAPID CABG trial, Roxana Mehran, MD, professor of medicine at the Icahn School of Medicine at Mount Sinai, New York, said this was a “very important study,” being the only randomized trial to look at this issue to date.
Dr. Mehran noted that the results showed a similar number of major life-threatening bleeding events in the early and delayed groups and met the noninferiority endpoint, but she pointed out that the trial had a small sample size and a small number of events. “Therefore, larger trials are needed to verify these important and encouraging results.”
However, she concluded that these results should be considered in decisions about the timing of bypass surgery in patients receiving ticagrelor. “I will be changing my practice and sending patients earlier based on this data,” she said.
RAPID CABG
RAPID CABG was a physician-initiated multicenter randomized study evaluating the safety of early surgery at 2-3 days after ticagrelor cessation, compared with a delay of 5-7 days among patients presenting with ACS who required nonemergency CABG surgery.
The study enrolled 143 patients with ACS who were receiving ticagrelor and needed CABG surgery. Patients with stenting for culprit lesions, those requiring urgent surgery (less than 24 hours after presentation), and those requiring valve surgery were excluded.
Three patients declined surgery, and several others underwent surgery outside the assigned time window, so the results were based on the per protocol analysis of patients who actually had CABG in the assigned time window: 65 patients in the early CABG group and 58 in the delayed group.
The mean time from last ticagrelor dose to surgery was 3 days in the early group and 6 days in the delayed group.
Platelet reactivity on the VerifyNow test showed more residual antiplatelet activity in the early group, with P2Y12 reaction unit (PRU) levels of 200 (vs. 251 in the delayed group). This test measures the extent of platelet aggregation in the presence of P2Y12-inhibitor drugs, with lower PRU levels showing stronger antiplatelet effects.
The primary outcome of the study was severe or massive bleeding by Universal Definition of Perioperative Bleeding (UDPB) class 3 or 4. This is defined as a blood transfusions of more than 5 units of red blood cells or plasma within 24 hours of surgical closure, chest tube drainage of over 1,000 mL in the first 12 hours, and reoperation for bleeding.
Results showed that 4.6% of the early-surgery group had a primary outcome bleeding event, compared with 5.2% of the delayed surgery group, meeting the criteria for noninferiority (P = .0253 for noninferiority).
Individual components of the primary endpoint showed three class 3 (severe) bleeding events in both groups and no class 4 (massive) bleeding events in either group.
In terms of other bleeding outcomes, TIMI CABG bleeding occurred in two patients (3.1%) in the early-surgery group vs. no patients in the delayed group; BARC 4 bleeding occurred in two patients (3.1%) in the early group versus none in the delayed group, and there were no BARC 5 bleeding events in either group.
In the intention-to-treat analysis, ischemic events before surgery occurred in six patients (8.7%) in the delayed group (one myocardial infarction, four cases of recurrent ischemia, and one ventricular tachycardia) versus none in the early group.
Cumulative 6-month ischemic events occurred in nine patients (13.0%) in the delayed group vs. four patients (5.6%) in the early group, the difference being driven by nonfatal MI and recurrent ischemia.
There were no cardiovascular deaths in either group and one all-cause death in both groups.
Patients undergoing early surgery also had a shorter hospitalization, with a median length of stay of 9 days versus 12 days in the delayed group.
Larger trial needed
Commenting on the RAPID CABG study at an AHA press conference, Joanna Chikwe, MD, chair of the cardiac surgery department at Cedars-Sinai Medical Center, Los Angeles, said the results were in line with her practice.
“These results confirm what I already think is safe,” she said. “I’m comfortable going within 48 hours. But we individualize our approach, so it was helpful that the study investigators included platelet reactivity data. The interesting thing for me in this study was the number of adverse events in patients who waited longer.”
Dr. Chikwe said her top-line message was that “Surgery looked incredibly safe; there was amazingly low mortality. And if a patient has an indication for surgery, waiting does not serve you well.”
However, she also cautioned that the trial was somewhat underpowered, with a small number of events that drove the primary outcome, leading to some uncertainty on the results.
“The RAPID trial was helpful, and although it confirms my practice, I think physicians may want to see a larger-powered trial to be convincingly compelled that they should change their practice,” Dr. Chikwe noted.
She added that clinical trials in cardiac surgery are driven by inherent challenges. “Cardiac surgery is not very common, and it is hard to recruit patients into these trials, so you are generally tied to a small number of patients, and you therefore have to be extremely thoughtful about the study design. It is almost a given that you will need to use surrogate endpoints, and the choice of the surrogate endpoint can determine which way the trial goes.”
The RAPID CABG study was funded by the Canadian Institutes of Health Research. Dr. So reports research support, consultancy, or speaker’s fees from AggreDyne, Roche Diagnostics, Fujimori Kogyo, and AstraZeneca Canada. Dr. Mehran reports that her institution has received significant trial funding from AstraZeneca (the manufacturer of ticagrelor).
A version of this article first appeared on Medscape.com.
Patients with acute coronary syndromes who have been taking the antiplatelet medication, ticagrelor, and who need coronary artery bypass surgery (CABG) may be able to safely have the procedure earlier than typically recommended, a new randomized trial suggests.
The RAPID CABG trial found that early surgery 2-3 days after ticagrelor cessation was noninferior in incurring severe or massive perioperative bleeding, compared with waiting 5-7 days. There was also no significant difference in TIMI CABG or Bleeding Academic Research Consortium (BARC) type 4 or 5 bleeding.
Patients in the delayed group had a numerically higher number of ischemic events requiring earlier surgery and had a longer hospital stay.
The study was presented at the American Heart Association scientific sessions.
“RAPID CABG is the first and only randomized controlled trial evaluating the safety of early surgery in patients taking ticagrelor,” said lead investigator Derek So, MD.
Dr. So, a cardiologist at the University of Ottawa Heart Institute and a professor at the University of Ottawa, explained that ticagrelor is a first-line antiplatelet agent for patients with acute coronary syndromes (ACS), but around 10% of patients presenting with ACS require CABG surgery.
A major concern among patients requiring bypass surgery is perioperative bleeding, and it has been shown that patients undergoing urgent bypass within 24 hours of the last dose of ticagrelor have increased mortality. Accordingly, guidelines suggest a waiting period for patients not requiring urgent bypass surgery, Dr. So noted.
Current North American guidelines suggest a waiting period of at least 5 days after stopping ticagrelor before bypass surgery. In contrast, the updated European and Japanese guidelines suggest a waiting period of 3 days.
Dr. So noted that all of the guidelines are based on cohort studies and pharmacodynamic studies, with no randomized evidence. Pharmacodynamic studies have shown that at 48 hours after the last dose of ticagrelor, the level of platelet inhibition drops to the same levels seen with long-term treatment with clopidogrel, a weaker antiplatelet drug, and after 120 hours (5 days) the effect has completely worn off.
Dr. So concluded that these new results from the RAPID CABG trial “may influence future iterations of North American guidelines with reduced waiting prior to bypass surgery” for patients receiving ticagrelor, and “they could also strengthen the level of evidence in European and Asian guidelines.”
Designated discussant of the RAPID CABG trial, Roxana Mehran, MD, professor of medicine at the Icahn School of Medicine at Mount Sinai, New York, said this was a “very important study,” being the only randomized trial to look at this issue to date.
Dr. Mehran noted that the results showed a similar number of major life-threatening bleeding events in the early and delayed groups and met the noninferiority endpoint, but she pointed out that the trial had a small sample size and a small number of events. “Therefore, larger trials are needed to verify these important and encouraging results.”
However, she concluded that these results should be considered in decisions about the timing of bypass surgery in patients receiving ticagrelor. “I will be changing my practice and sending patients earlier based on this data,” she said.
RAPID CABG
RAPID CABG was a physician-initiated multicenter randomized study evaluating the safety of early surgery at 2-3 days after ticagrelor cessation, compared with a delay of 5-7 days among patients presenting with ACS who required nonemergency CABG surgery.
The study enrolled 143 patients with ACS who were receiving ticagrelor and needed CABG surgery. Patients with stenting for culprit lesions, those requiring urgent surgery (less than 24 hours after presentation), and those requiring valve surgery were excluded.
Three patients declined surgery, and several others underwent surgery outside the assigned time window, so the results were based on the per protocol analysis of patients who actually had CABG in the assigned time window: 65 patients in the early CABG group and 58 in the delayed group.
The mean time from last ticagrelor dose to surgery was 3 days in the early group and 6 days in the delayed group.
Platelet reactivity on the VerifyNow test showed more residual antiplatelet activity in the early group, with P2Y12 reaction unit (PRU) levels of 200 (vs. 251 in the delayed group). This test measures the extent of platelet aggregation in the presence of P2Y12-inhibitor drugs, with lower PRU levels showing stronger antiplatelet effects.
The primary outcome of the study was severe or massive bleeding by Universal Definition of Perioperative Bleeding (UDPB) class 3 or 4. This is defined as a blood transfusions of more than 5 units of red blood cells or plasma within 24 hours of surgical closure, chest tube drainage of over 1,000 mL in the first 12 hours, and reoperation for bleeding.
Results showed that 4.6% of the early-surgery group had a primary outcome bleeding event, compared with 5.2% of the delayed surgery group, meeting the criteria for noninferiority (P = .0253 for noninferiority).
Individual components of the primary endpoint showed three class 3 (severe) bleeding events in both groups and no class 4 (massive) bleeding events in either group.
In terms of other bleeding outcomes, TIMI CABG bleeding occurred in two patients (3.1%) in the early-surgery group vs. no patients in the delayed group; BARC 4 bleeding occurred in two patients (3.1%) in the early group versus none in the delayed group, and there were no BARC 5 bleeding events in either group.
In the intention-to-treat analysis, ischemic events before surgery occurred in six patients (8.7%) in the delayed group (one myocardial infarction, four cases of recurrent ischemia, and one ventricular tachycardia) versus none in the early group.
Cumulative 6-month ischemic events occurred in nine patients (13.0%) in the delayed group vs. four patients (5.6%) in the early group, the difference being driven by nonfatal MI and recurrent ischemia.
There were no cardiovascular deaths in either group and one all-cause death in both groups.
Patients undergoing early surgery also had a shorter hospitalization, with a median length of stay of 9 days versus 12 days in the delayed group.
Larger trial needed
Commenting on the RAPID CABG study at an AHA press conference, Joanna Chikwe, MD, chair of the cardiac surgery department at Cedars-Sinai Medical Center, Los Angeles, said the results were in line with her practice.
“These results confirm what I already think is safe,” she said. “I’m comfortable going within 48 hours. But we individualize our approach, so it was helpful that the study investigators included platelet reactivity data. The interesting thing for me in this study was the number of adverse events in patients who waited longer.”
Dr. Chikwe said her top-line message was that “Surgery looked incredibly safe; there was amazingly low mortality. And if a patient has an indication for surgery, waiting does not serve you well.”
However, she also cautioned that the trial was somewhat underpowered, with a small number of events that drove the primary outcome, leading to some uncertainty on the results.
“The RAPID trial was helpful, and although it confirms my practice, I think physicians may want to see a larger-powered trial to be convincingly compelled that they should change their practice,” Dr. Chikwe noted.
She added that clinical trials in cardiac surgery are driven by inherent challenges. “Cardiac surgery is not very common, and it is hard to recruit patients into these trials, so you are generally tied to a small number of patients, and you therefore have to be extremely thoughtful about the study design. It is almost a given that you will need to use surrogate endpoints, and the choice of the surrogate endpoint can determine which way the trial goes.”
The RAPID CABG study was funded by the Canadian Institutes of Health Research. Dr. So reports research support, consultancy, or speaker’s fees from AggreDyne, Roche Diagnostics, Fujimori Kogyo, and AstraZeneca Canada. Dr. Mehran reports that her institution has received significant trial funding from AstraZeneca (the manufacturer of ticagrelor).
A version of this article first appeared on Medscape.com.
Patients with acute coronary syndromes who have been taking the antiplatelet medication, ticagrelor, and who need coronary artery bypass surgery (CABG) may be able to safely have the procedure earlier than typically recommended, a new randomized trial suggests.
The RAPID CABG trial found that early surgery 2-3 days after ticagrelor cessation was noninferior in incurring severe or massive perioperative bleeding, compared with waiting 5-7 days. There was also no significant difference in TIMI CABG or Bleeding Academic Research Consortium (BARC) type 4 or 5 bleeding.
Patients in the delayed group had a numerically higher number of ischemic events requiring earlier surgery and had a longer hospital stay.
The study was presented at the American Heart Association scientific sessions.
“RAPID CABG is the first and only randomized controlled trial evaluating the safety of early surgery in patients taking ticagrelor,” said lead investigator Derek So, MD.
Dr. So, a cardiologist at the University of Ottawa Heart Institute and a professor at the University of Ottawa, explained that ticagrelor is a first-line antiplatelet agent for patients with acute coronary syndromes (ACS), but around 10% of patients presenting with ACS require CABG surgery.
A major concern among patients requiring bypass surgery is perioperative bleeding, and it has been shown that patients undergoing urgent bypass within 24 hours of the last dose of ticagrelor have increased mortality. Accordingly, guidelines suggest a waiting period for patients not requiring urgent bypass surgery, Dr. So noted.
Current North American guidelines suggest a waiting period of at least 5 days after stopping ticagrelor before bypass surgery. In contrast, the updated European and Japanese guidelines suggest a waiting period of 3 days.
Dr. So noted that all of the guidelines are based on cohort studies and pharmacodynamic studies, with no randomized evidence. Pharmacodynamic studies have shown that at 48 hours after the last dose of ticagrelor, the level of platelet inhibition drops to the same levels seen with long-term treatment with clopidogrel, a weaker antiplatelet drug, and after 120 hours (5 days) the effect has completely worn off.
Dr. So concluded that these new results from the RAPID CABG trial “may influence future iterations of North American guidelines with reduced waiting prior to bypass surgery” for patients receiving ticagrelor, and “they could also strengthen the level of evidence in European and Asian guidelines.”
Designated discussant of the RAPID CABG trial, Roxana Mehran, MD, professor of medicine at the Icahn School of Medicine at Mount Sinai, New York, said this was a “very important study,” being the only randomized trial to look at this issue to date.
Dr. Mehran noted that the results showed a similar number of major life-threatening bleeding events in the early and delayed groups and met the noninferiority endpoint, but she pointed out that the trial had a small sample size and a small number of events. “Therefore, larger trials are needed to verify these important and encouraging results.”
However, she concluded that these results should be considered in decisions about the timing of bypass surgery in patients receiving ticagrelor. “I will be changing my practice and sending patients earlier based on this data,” she said.
RAPID CABG
RAPID CABG was a physician-initiated multicenter randomized study evaluating the safety of early surgery at 2-3 days after ticagrelor cessation, compared with a delay of 5-7 days among patients presenting with ACS who required nonemergency CABG surgery.
The study enrolled 143 patients with ACS who were receiving ticagrelor and needed CABG surgery. Patients with stenting for culprit lesions, those requiring urgent surgery (less than 24 hours after presentation), and those requiring valve surgery were excluded.
Three patients declined surgery, and several others underwent surgery outside the assigned time window, so the results were based on the per protocol analysis of patients who actually had CABG in the assigned time window: 65 patients in the early CABG group and 58 in the delayed group.
The mean time from last ticagrelor dose to surgery was 3 days in the early group and 6 days in the delayed group.
Platelet reactivity on the VerifyNow test showed more residual antiplatelet activity in the early group, with P2Y12 reaction unit (PRU) levels of 200 (vs. 251 in the delayed group). This test measures the extent of platelet aggregation in the presence of P2Y12-inhibitor drugs, with lower PRU levels showing stronger antiplatelet effects.
The primary outcome of the study was severe or massive bleeding by Universal Definition of Perioperative Bleeding (UDPB) class 3 or 4. This is defined as a blood transfusions of more than 5 units of red blood cells or plasma within 24 hours of surgical closure, chest tube drainage of over 1,000 mL in the first 12 hours, and reoperation for bleeding.
Results showed that 4.6% of the early-surgery group had a primary outcome bleeding event, compared with 5.2% of the delayed surgery group, meeting the criteria for noninferiority (P = .0253 for noninferiority).
Individual components of the primary endpoint showed three class 3 (severe) bleeding events in both groups and no class 4 (massive) bleeding events in either group.
In terms of other bleeding outcomes, TIMI CABG bleeding occurred in two patients (3.1%) in the early-surgery group vs. no patients in the delayed group; BARC 4 bleeding occurred in two patients (3.1%) in the early group versus none in the delayed group, and there were no BARC 5 bleeding events in either group.
In the intention-to-treat analysis, ischemic events before surgery occurred in six patients (8.7%) in the delayed group (one myocardial infarction, four cases of recurrent ischemia, and one ventricular tachycardia) versus none in the early group.
Cumulative 6-month ischemic events occurred in nine patients (13.0%) in the delayed group vs. four patients (5.6%) in the early group, the difference being driven by nonfatal MI and recurrent ischemia.
There were no cardiovascular deaths in either group and one all-cause death in both groups.
Patients undergoing early surgery also had a shorter hospitalization, with a median length of stay of 9 days versus 12 days in the delayed group.
Larger trial needed
Commenting on the RAPID CABG study at an AHA press conference, Joanna Chikwe, MD, chair of the cardiac surgery department at Cedars-Sinai Medical Center, Los Angeles, said the results were in line with her practice.
“These results confirm what I already think is safe,” she said. “I’m comfortable going within 48 hours. But we individualize our approach, so it was helpful that the study investigators included platelet reactivity data. The interesting thing for me in this study was the number of adverse events in patients who waited longer.”
Dr. Chikwe said her top-line message was that “Surgery looked incredibly safe; there was amazingly low mortality. And if a patient has an indication for surgery, waiting does not serve you well.”
However, she also cautioned that the trial was somewhat underpowered, with a small number of events that drove the primary outcome, leading to some uncertainty on the results.
“The RAPID trial was helpful, and although it confirms my practice, I think physicians may want to see a larger-powered trial to be convincingly compelled that they should change their practice,” Dr. Chikwe noted.
She added that clinical trials in cardiac surgery are driven by inherent challenges. “Cardiac surgery is not very common, and it is hard to recruit patients into these trials, so you are generally tied to a small number of patients, and you therefore have to be extremely thoughtful about the study design. It is almost a given that you will need to use surrogate endpoints, and the choice of the surrogate endpoint can determine which way the trial goes.”
The RAPID CABG study was funded by the Canadian Institutes of Health Research. Dr. So reports research support, consultancy, or speaker’s fees from AggreDyne, Roche Diagnostics, Fujimori Kogyo, and AstraZeneca Canada. Dr. Mehran reports that her institution has received significant trial funding from AstraZeneca (the manufacturer of ticagrelor).
A version of this article first appeared on Medscape.com.
FROM AHA 2021
How applicable is ISCHEMIA trial to U.S. clinical practice?
The applicability of the results of the ISCHEMIA trial to real-world clinical practice in the United States has been called into question by a new study showing that less than a third of U.S. patients with stable ischemic heart disease (IHD) who currently undergo intervention would meet the trial’s inclusion criteria.
The ISCHEMIA trial, first reported in 2019, showed that, for stable patients with moderate to severe ischemia, an invasive approach using percutaneous coronary intervention (PCI) did not significantly reduce major cardiovascular events after a median 3.2 years of follow-up, compared with a conservative medical strategy.
For the current study, a group of interventionalists analyzed contemporary U.S. data on patients undergoing PCI and found that a large proportion of patients receiving PCI for stable ischemic heart disease in the United States would not have met criteria of the ISCHEMIA trial population.
The study was published online Nov. 1 in JACC: Cardiovascular Interventions).
“While ISCHEMIA was a very well-conducted trial, our results show that it only applies to about one-third of stable IHD patients undergoing intervention in U.S. clinical practice in the real world. In this group, while ISCHEMIA did not show a reduction in event rate in the intervention group, there was a reduction in symptoms,” lead author Saurav Chatterjee, MD, Long Island Jewish Medical Center, New York, told this news organization.
“But ISCHEMIA did not really answer the question for 67% of stable IHD in current U.S. practice. We may be abIe to defer PCI in these patients, but we don’t know that from the ISCHEMIA trial, as these patients were not included in the trial,” Chatterjee said.
“There is some concern that people will accept the ISCHEMIA results as being universal, but we cannot apply these results to all stable IHD patients who currently undergo intervention,” he added. “We believe that patients who do not fall into the ISCHEMIA population need a nuanced individual approach, taking into account symptom burden and patient preferences.”
In the new report, Dr. Chatterjee and associates note that the applicability of the ISCHEMIA findings to contemporary practice has been questioned by some, because of the exclusion of a significant proportion of patients that are routinely considered for revascularization, both within and outside of the United States.
They point out that the ISCHEMIA trial recruited 16.5% of its participants from the United States, and the proportion of patients in contemporary U.S. practice that would have qualified for the trial is not clear.
They therefore examined the proportion of stable IHD patients meeting inclusion criteria for the ISCHEMIA trial in a U.S. nationwide PCI registry.
The researchers used data from the National Cardiovascular Data Registry (NCDR) CathPCI Registry, which includes patients undergoing PCI at 1,662 institutions and accounts for more than 90% of PCI-capable hospitals in the United States.
All PCI procedures performed at institutions participating in the NCDR CathPCI Registry from October 2017 to June 2019 were identified. Patients presenting with acute coronary syndrome (ACS), cardiogenic shock, or cardiac arrest were excluded, as there is significant evidence in favor of revascularization in these groups, and they were not included in the ISCHEMIA trial.
Subsequently, all remaining stable IHD patients were classified into one of four groups.
- ISCHEMIA-like: These patients had intermediate- or high-risk findings on a stress test but no high-risk features that would have excluded enrollment into the ISCHEMIA trial
- High risk: This group comprised patients with stable IHD and left ventricular ejection fraction less than 35%, significant unprotected left main stenosis (>50%), preexisting dialysis, recent heart-failure exacerbation, or heart transplant. These patients would have met exclusion criteria for the ISCHEMIA trial
- Low risk: This group included patients with stable and negative or low-risk findings on stress test and would have met exclusion criteria for the ISCHEMIA trial
- Not classifiable: This group comprised patients with stable IHD not fitting any of the other cohorts, including no stress test or extent of ischemia not reported on stress testing. These patients would have not had enough information to clearly meet inclusion or exclusion criteria for the ISCHEMIA trial
Results showed that during the study period 927,011 patients underwent PCI as recorded in the NCDR CathPCI Registry. Of these, 58% had ACS, cardiogenic shock, or cardiac arrest and were excluded; the remaining 388,212 patients who underwent PCI for stable IHD comprised the study population.
Of these, 125,302 (32.28%) had a moderate- or high-risk stress test without high-risk anatomic or clinical features and met ISCHEMIA trial inclusion criteria.
Among stable IHD patients not meeting ISCHEMIA trial inclusion criteria, 71,852 (18.51%) had high-risk criteria that would have excluded them from the ISCHEMIA trial, a total of 67,159 (17.29%) patients had low-risk criteria that would have excluded them from the ISCHEMIA trial, and 123,899 (31.92%) were unclassifiable, either owing to lack of stress testing or the extent of ischemia not being reported on stress testing.
The authors suggest that the unclassifiable patients appear to represent a “higher-risk” population than those closely resembling the ISCHEMIA trial population, with more prior myocardial infarction and heart failure.
ISCHEMIA investigators respond
In an accompanying editorial, ISCHEMIA investigators David J. Maron, MD, Stanford (Calif.) University, and Sripal Bangalore, MD, and Judith S. Hochman, MD, New York University, argue that many of the patients highlighted by Dr. Chatterjee and associates were excluded from the ISCHEMIA trial for good reason.
They explain that ISCHEMIA was designed under the premise that prior stable IHD strategy trials such as COURAGE and BARI 2D included lower-risk patients, and the remaining gap was to evaluate the utility of invasive management in those at higher risk with moderate or severe stress-induced ischemia.
They point out that, among the NCDR patients with stable IHD in the current study by Dr. Chatterjee and associates who did not meet ISCHEMIA entry criteria, 18.5% had high-risk features, including 35.2% with left main coronary artery disease, 43.7% with left ventricular systolic dysfunction, and 16.8% with end-stage renal disease.
Although ISCHEMIA results do not apply to patients who were excluded from the trial, there is little controversy regarding the benefit of revascularization in patients with stable IHD with left main coronary artery disease or left ventricular ejection fraction <35%, which is why they were excluded from ISCHEMIA, the editorialists note.
They also report that patients with end-stage renal disease, who were also designated as not meeting ISCHEMIA inclusion criteria, were included in the companion ISCHEMIA CKD trial.
They further point out that, at the other end of the risk spectrum, 17.3% of stable IHD patients in the current analysis had negative or low-risk functional testing, and these patients were excluded from ISCHEMIA because they were shown in COURAGE and BARI 2D to not benefit from revascularization, and they do not meet guideline recommendations for elective PCI in the absence of symptoms.
Of the 31.9% of stable IHD patients who had missing data on ischemic burden, the ISCHEMIA investigators say that some of these would have qualified for the trial, although it is not possible to say how many. They suggest a conservative estimate of 50%.
Taking these arguments into account, the editorialists recalculated the proportion of NCDR PCI patients with stable IHD who would have been included in ISCHEMIA as between 62.1% and 68.6% of patients.
They say the current NCDR analysis by Dr. Chatterjee and associates should be interpreted as indicating, at worst, that the ISCHEMIA trial results apply to only 32% of patients undergoing elective PCI in the United States, and at best “that the results apply to a far higher proportion, excluding only those at high risk (18.5%) or with unacceptable symptoms despite maximal medical therapy (percentage unknown), for whom PCI is clearly indicated.”
The editorialists conclude: “The purpose of the analysis by Chatterjee et al. is to inform the cardiovascular community of the proportion of patients with stable IHD in clinical practice who would have been excluded from ISCHEMIA without regard for the logic of each exclusion criterion. The purpose of this editorial is to provide context for the analysis, admittedly from the perspective of ISCHEMIA investigators, with the hope that this helps readers clearly see the relevance of the trial to patients under their care.”
They add: “For practical and ethical reasons, ISCHEMIA excluded stable patients with high-risk features, angina inadequately controlled by medication, and low-risk features who do not meet evidence-based guidelines for revascularization. That leaves a large percentage of patients for whom the ISCHEMIA trial is highly relevant; exactly what percentage on the basis of NCDR data is hard to say.”
The ISCHEMIA trial was supported by the National Heart, Lung, and Blood Institute.
A version of this article first appeared on Medscape.com.
The applicability of the results of the ISCHEMIA trial to real-world clinical practice in the United States has been called into question by a new study showing that less than a third of U.S. patients with stable ischemic heart disease (IHD) who currently undergo intervention would meet the trial’s inclusion criteria.
The ISCHEMIA trial, first reported in 2019, showed that, for stable patients with moderate to severe ischemia, an invasive approach using percutaneous coronary intervention (PCI) did not significantly reduce major cardiovascular events after a median 3.2 years of follow-up, compared with a conservative medical strategy.
For the current study, a group of interventionalists analyzed contemporary U.S. data on patients undergoing PCI and found that a large proportion of patients receiving PCI for stable ischemic heart disease in the United States would not have met criteria of the ISCHEMIA trial population.
The study was published online Nov. 1 in JACC: Cardiovascular Interventions).
“While ISCHEMIA was a very well-conducted trial, our results show that it only applies to about one-third of stable IHD patients undergoing intervention in U.S. clinical practice in the real world. In this group, while ISCHEMIA did not show a reduction in event rate in the intervention group, there was a reduction in symptoms,” lead author Saurav Chatterjee, MD, Long Island Jewish Medical Center, New York, told this news organization.
“But ISCHEMIA did not really answer the question for 67% of stable IHD in current U.S. practice. We may be abIe to defer PCI in these patients, but we don’t know that from the ISCHEMIA trial, as these patients were not included in the trial,” Chatterjee said.
“There is some concern that people will accept the ISCHEMIA results as being universal, but we cannot apply these results to all stable IHD patients who currently undergo intervention,” he added. “We believe that patients who do not fall into the ISCHEMIA population need a nuanced individual approach, taking into account symptom burden and patient preferences.”
In the new report, Dr. Chatterjee and associates note that the applicability of the ISCHEMIA findings to contemporary practice has been questioned by some, because of the exclusion of a significant proportion of patients that are routinely considered for revascularization, both within and outside of the United States.
They point out that the ISCHEMIA trial recruited 16.5% of its participants from the United States, and the proportion of patients in contemporary U.S. practice that would have qualified for the trial is not clear.
They therefore examined the proportion of stable IHD patients meeting inclusion criteria for the ISCHEMIA trial in a U.S. nationwide PCI registry.
The researchers used data from the National Cardiovascular Data Registry (NCDR) CathPCI Registry, which includes patients undergoing PCI at 1,662 institutions and accounts for more than 90% of PCI-capable hospitals in the United States.
All PCI procedures performed at institutions participating in the NCDR CathPCI Registry from October 2017 to June 2019 were identified. Patients presenting with acute coronary syndrome (ACS), cardiogenic shock, or cardiac arrest were excluded, as there is significant evidence in favor of revascularization in these groups, and they were not included in the ISCHEMIA trial.
Subsequently, all remaining stable IHD patients were classified into one of four groups.
- ISCHEMIA-like: These patients had intermediate- or high-risk findings on a stress test but no high-risk features that would have excluded enrollment into the ISCHEMIA trial
- High risk: This group comprised patients with stable IHD and left ventricular ejection fraction less than 35%, significant unprotected left main stenosis (>50%), preexisting dialysis, recent heart-failure exacerbation, or heart transplant. These patients would have met exclusion criteria for the ISCHEMIA trial
- Low risk: This group included patients with stable and negative or low-risk findings on stress test and would have met exclusion criteria for the ISCHEMIA trial
- Not classifiable: This group comprised patients with stable IHD not fitting any of the other cohorts, including no stress test or extent of ischemia not reported on stress testing. These patients would have not had enough information to clearly meet inclusion or exclusion criteria for the ISCHEMIA trial
Results showed that during the study period 927,011 patients underwent PCI as recorded in the NCDR CathPCI Registry. Of these, 58% had ACS, cardiogenic shock, or cardiac arrest and were excluded; the remaining 388,212 patients who underwent PCI for stable IHD comprised the study population.
Of these, 125,302 (32.28%) had a moderate- or high-risk stress test without high-risk anatomic or clinical features and met ISCHEMIA trial inclusion criteria.
Among stable IHD patients not meeting ISCHEMIA trial inclusion criteria, 71,852 (18.51%) had high-risk criteria that would have excluded them from the ISCHEMIA trial, a total of 67,159 (17.29%) patients had low-risk criteria that would have excluded them from the ISCHEMIA trial, and 123,899 (31.92%) were unclassifiable, either owing to lack of stress testing or the extent of ischemia not being reported on stress testing.
The authors suggest that the unclassifiable patients appear to represent a “higher-risk” population than those closely resembling the ISCHEMIA trial population, with more prior myocardial infarction and heart failure.
ISCHEMIA investigators respond
In an accompanying editorial, ISCHEMIA investigators David J. Maron, MD, Stanford (Calif.) University, and Sripal Bangalore, MD, and Judith S. Hochman, MD, New York University, argue that many of the patients highlighted by Dr. Chatterjee and associates were excluded from the ISCHEMIA trial for good reason.
They explain that ISCHEMIA was designed under the premise that prior stable IHD strategy trials such as COURAGE and BARI 2D included lower-risk patients, and the remaining gap was to evaluate the utility of invasive management in those at higher risk with moderate or severe stress-induced ischemia.
They point out that, among the NCDR patients with stable IHD in the current study by Dr. Chatterjee and associates who did not meet ISCHEMIA entry criteria, 18.5% had high-risk features, including 35.2% with left main coronary artery disease, 43.7% with left ventricular systolic dysfunction, and 16.8% with end-stage renal disease.
Although ISCHEMIA results do not apply to patients who were excluded from the trial, there is little controversy regarding the benefit of revascularization in patients with stable IHD with left main coronary artery disease or left ventricular ejection fraction <35%, which is why they were excluded from ISCHEMIA, the editorialists note.
They also report that patients with end-stage renal disease, who were also designated as not meeting ISCHEMIA inclusion criteria, were included in the companion ISCHEMIA CKD trial.
They further point out that, at the other end of the risk spectrum, 17.3% of stable IHD patients in the current analysis had negative or low-risk functional testing, and these patients were excluded from ISCHEMIA because they were shown in COURAGE and BARI 2D to not benefit from revascularization, and they do not meet guideline recommendations for elective PCI in the absence of symptoms.
Of the 31.9% of stable IHD patients who had missing data on ischemic burden, the ISCHEMIA investigators say that some of these would have qualified for the trial, although it is not possible to say how many. They suggest a conservative estimate of 50%.
Taking these arguments into account, the editorialists recalculated the proportion of NCDR PCI patients with stable IHD who would have been included in ISCHEMIA as between 62.1% and 68.6% of patients.
They say the current NCDR analysis by Dr. Chatterjee and associates should be interpreted as indicating, at worst, that the ISCHEMIA trial results apply to only 32% of patients undergoing elective PCI in the United States, and at best “that the results apply to a far higher proportion, excluding only those at high risk (18.5%) or with unacceptable symptoms despite maximal medical therapy (percentage unknown), for whom PCI is clearly indicated.”
The editorialists conclude: “The purpose of the analysis by Chatterjee et al. is to inform the cardiovascular community of the proportion of patients with stable IHD in clinical practice who would have been excluded from ISCHEMIA without regard for the logic of each exclusion criterion. The purpose of this editorial is to provide context for the analysis, admittedly from the perspective of ISCHEMIA investigators, with the hope that this helps readers clearly see the relevance of the trial to patients under their care.”
They add: “For practical and ethical reasons, ISCHEMIA excluded stable patients with high-risk features, angina inadequately controlled by medication, and low-risk features who do not meet evidence-based guidelines for revascularization. That leaves a large percentage of patients for whom the ISCHEMIA trial is highly relevant; exactly what percentage on the basis of NCDR data is hard to say.”
The ISCHEMIA trial was supported by the National Heart, Lung, and Blood Institute.
A version of this article first appeared on Medscape.com.
The applicability of the results of the ISCHEMIA trial to real-world clinical practice in the United States has been called into question by a new study showing that less than a third of U.S. patients with stable ischemic heart disease (IHD) who currently undergo intervention would meet the trial’s inclusion criteria.
The ISCHEMIA trial, first reported in 2019, showed that, for stable patients with moderate to severe ischemia, an invasive approach using percutaneous coronary intervention (PCI) did not significantly reduce major cardiovascular events after a median 3.2 years of follow-up, compared with a conservative medical strategy.
For the current study, a group of interventionalists analyzed contemporary U.S. data on patients undergoing PCI and found that a large proportion of patients receiving PCI for stable ischemic heart disease in the United States would not have met criteria of the ISCHEMIA trial population.
The study was published online Nov. 1 in JACC: Cardiovascular Interventions).
“While ISCHEMIA was a very well-conducted trial, our results show that it only applies to about one-third of stable IHD patients undergoing intervention in U.S. clinical practice in the real world. In this group, while ISCHEMIA did not show a reduction in event rate in the intervention group, there was a reduction in symptoms,” lead author Saurav Chatterjee, MD, Long Island Jewish Medical Center, New York, told this news organization.
“But ISCHEMIA did not really answer the question for 67% of stable IHD in current U.S. practice. We may be abIe to defer PCI in these patients, but we don’t know that from the ISCHEMIA trial, as these patients were not included in the trial,” Chatterjee said.
“There is some concern that people will accept the ISCHEMIA results as being universal, but we cannot apply these results to all stable IHD patients who currently undergo intervention,” he added. “We believe that patients who do not fall into the ISCHEMIA population need a nuanced individual approach, taking into account symptom burden and patient preferences.”
In the new report, Dr. Chatterjee and associates note that the applicability of the ISCHEMIA findings to contemporary practice has been questioned by some, because of the exclusion of a significant proportion of patients that are routinely considered for revascularization, both within and outside of the United States.
They point out that the ISCHEMIA trial recruited 16.5% of its participants from the United States, and the proportion of patients in contemporary U.S. practice that would have qualified for the trial is not clear.
They therefore examined the proportion of stable IHD patients meeting inclusion criteria for the ISCHEMIA trial in a U.S. nationwide PCI registry.
The researchers used data from the National Cardiovascular Data Registry (NCDR) CathPCI Registry, which includes patients undergoing PCI at 1,662 institutions and accounts for more than 90% of PCI-capable hospitals in the United States.
All PCI procedures performed at institutions participating in the NCDR CathPCI Registry from October 2017 to June 2019 were identified. Patients presenting with acute coronary syndrome (ACS), cardiogenic shock, or cardiac arrest were excluded, as there is significant evidence in favor of revascularization in these groups, and they were not included in the ISCHEMIA trial.
Subsequently, all remaining stable IHD patients were classified into one of four groups.
- ISCHEMIA-like: These patients had intermediate- or high-risk findings on a stress test but no high-risk features that would have excluded enrollment into the ISCHEMIA trial
- High risk: This group comprised patients with stable IHD and left ventricular ejection fraction less than 35%, significant unprotected left main stenosis (>50%), preexisting dialysis, recent heart-failure exacerbation, or heart transplant. These patients would have met exclusion criteria for the ISCHEMIA trial
- Low risk: This group included patients with stable and negative or low-risk findings on stress test and would have met exclusion criteria for the ISCHEMIA trial
- Not classifiable: This group comprised patients with stable IHD not fitting any of the other cohorts, including no stress test or extent of ischemia not reported on stress testing. These patients would have not had enough information to clearly meet inclusion or exclusion criteria for the ISCHEMIA trial
Results showed that during the study period 927,011 patients underwent PCI as recorded in the NCDR CathPCI Registry. Of these, 58% had ACS, cardiogenic shock, or cardiac arrest and were excluded; the remaining 388,212 patients who underwent PCI for stable IHD comprised the study population.
Of these, 125,302 (32.28%) had a moderate- or high-risk stress test without high-risk anatomic or clinical features and met ISCHEMIA trial inclusion criteria.
Among stable IHD patients not meeting ISCHEMIA trial inclusion criteria, 71,852 (18.51%) had high-risk criteria that would have excluded them from the ISCHEMIA trial, a total of 67,159 (17.29%) patients had low-risk criteria that would have excluded them from the ISCHEMIA trial, and 123,899 (31.92%) were unclassifiable, either owing to lack of stress testing or the extent of ischemia not being reported on stress testing.
The authors suggest that the unclassifiable patients appear to represent a “higher-risk” population than those closely resembling the ISCHEMIA trial population, with more prior myocardial infarction and heart failure.
ISCHEMIA investigators respond
In an accompanying editorial, ISCHEMIA investigators David J. Maron, MD, Stanford (Calif.) University, and Sripal Bangalore, MD, and Judith S. Hochman, MD, New York University, argue that many of the patients highlighted by Dr. Chatterjee and associates were excluded from the ISCHEMIA trial for good reason.
They explain that ISCHEMIA was designed under the premise that prior stable IHD strategy trials such as COURAGE and BARI 2D included lower-risk patients, and the remaining gap was to evaluate the utility of invasive management in those at higher risk with moderate or severe stress-induced ischemia.
They point out that, among the NCDR patients with stable IHD in the current study by Dr. Chatterjee and associates who did not meet ISCHEMIA entry criteria, 18.5% had high-risk features, including 35.2% with left main coronary artery disease, 43.7% with left ventricular systolic dysfunction, and 16.8% with end-stage renal disease.
Although ISCHEMIA results do not apply to patients who were excluded from the trial, there is little controversy regarding the benefit of revascularization in patients with stable IHD with left main coronary artery disease or left ventricular ejection fraction <35%, which is why they were excluded from ISCHEMIA, the editorialists note.
They also report that patients with end-stage renal disease, who were also designated as not meeting ISCHEMIA inclusion criteria, were included in the companion ISCHEMIA CKD trial.
They further point out that, at the other end of the risk spectrum, 17.3% of stable IHD patients in the current analysis had negative or low-risk functional testing, and these patients were excluded from ISCHEMIA because they were shown in COURAGE and BARI 2D to not benefit from revascularization, and they do not meet guideline recommendations for elective PCI in the absence of symptoms.
Of the 31.9% of stable IHD patients who had missing data on ischemic burden, the ISCHEMIA investigators say that some of these would have qualified for the trial, although it is not possible to say how many. They suggest a conservative estimate of 50%.
Taking these arguments into account, the editorialists recalculated the proportion of NCDR PCI patients with stable IHD who would have been included in ISCHEMIA as between 62.1% and 68.6% of patients.
They say the current NCDR analysis by Dr. Chatterjee and associates should be interpreted as indicating, at worst, that the ISCHEMIA trial results apply to only 32% of patients undergoing elective PCI in the United States, and at best “that the results apply to a far higher proportion, excluding only those at high risk (18.5%) or with unacceptable symptoms despite maximal medical therapy (percentage unknown), for whom PCI is clearly indicated.”
The editorialists conclude: “The purpose of the analysis by Chatterjee et al. is to inform the cardiovascular community of the proportion of patients with stable IHD in clinical practice who would have been excluded from ISCHEMIA without regard for the logic of each exclusion criterion. The purpose of this editorial is to provide context for the analysis, admittedly from the perspective of ISCHEMIA investigators, with the hope that this helps readers clearly see the relevance of the trial to patients under their care.”
They add: “For practical and ethical reasons, ISCHEMIA excluded stable patients with high-risk features, angina inadequately controlled by medication, and low-risk features who do not meet evidence-based guidelines for revascularization. That leaves a large percentage of patients for whom the ISCHEMIA trial is highly relevant; exactly what percentage on the basis of NCDR data is hard to say.”
The ISCHEMIA trial was supported by the National Heart, Lung, and Blood Institute.
A version of this article first appeared on Medscape.com.
Which agent is best for neuromyelitis optica?
The Alexion-sponsored study was presented at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) by Dean Wingerchuk, MD, of the Mayo Clinic in Scottsdale, Ariz.
Other experts in the field have highlighted limitations to the analysis and pointed out that all three agents are very effective in treating AQP4+ NMOSD, and many other considerations need to be taken into account as well as time to first relapse when selecting a therapy, leaving the door open for all three agents.
Dr. Wingerchuk explained that NMOSD is a rare severely disabling complement-mediated autoimmune neuroinflammatory disease of the central nervous system, characterized by devastating and unpredictable attacks (relapses) that can cause immediate and irreversible damage.
There are three recently approved monoclonal antibody treatment options in the United States for adults with AQP4+ NMOSD: eculizumab (Soliris, Alexion), inebilizumab (Uplizna, Horizon), and satralizumab (Enspryng, Genentech). A comparison of the relative treatment effects of these drugs would facilitate the treatment selection process, Dr. Wingerchuk said.
The objective of this study was to perform an indirect treatment comparison on the efficacy of these three FDA-approved treatment options for adults with AQP4+ NMOSD, in the absence of any head-to-head studies.
Using published data from randomized controlled trials, which were identified by a systematic literature review in September 2020, the researchers performed a Bayesian network meta-analysis to estimate the relative effects between eculizumab, inebilizumab, and satralizumab.
Network meta-analyses were performed for clinically relevant subpopulations based on three treatment networks: (1) patients who received monotherapy with one of the monoclonal antibodies or in combination with an immunosuppressant therapy; (2) patients who received monotherapy with the monoclonal antibody alone; and (3) patients who received a combination of both the monoclonal antibody and immunosuppressant therapy.
Time to first relapse was the primary efficacy outcome assessed. Relative treatment effects were expressed as hazard ratios and the probability that a treatment was the best at delaying time to first relapse was also evaluated.
In the systematic literature review, 29 publications from four unique clinical trials were identified and include in the network meta-analysis. These included publications from congress proceedings and peer-reviewed journals.
The four clinical trials were the N-MOmentum trial of inebilizumab versus placebo; the PREVENT trial of eculizumab with or without immunosuppressant therapy versus placebo with or without immunosuppressant therapy; the SAkuraSky trial of satralizumab plus immunosuppressant therapy versus placebo plus immunosuppressant therapy; and the SAkuraStar trial of satralizumab versus placebo.
Results showed that for the first analysis of mono or combination therapy, patients treated with eculizumab with or without immunosuppressant therapy were 76% less likely to experience a first relapse when compared with patients treated with satralizumab with or without immunosuppressant therapy.
In the monotherapy network, patients on eculizumab were 90% less likely to experience a first relapse when compared with patients treated with satralizumab, and patients on eculizumab were 89% less likely to experience a first relapse when compared with patients treated with inebilizumab.
In the third network analysis – a comparison of eculizumab plus immunosuppressant therapy with inebilizumab plus immunosuppressant therapy (Table 1) – the point estimate appeared to favor eculizumab but the confidence intervals were wide and not significant.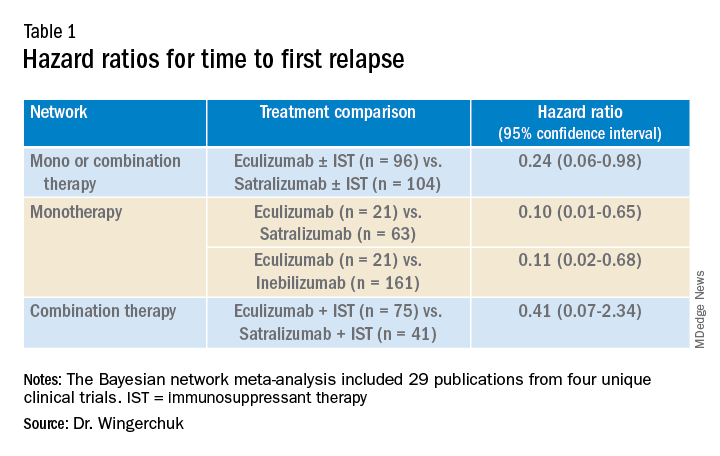
A subsequent analysis looked at the rank order of the best treatment option, with eculizumab coming out first in all three networks (Table 2).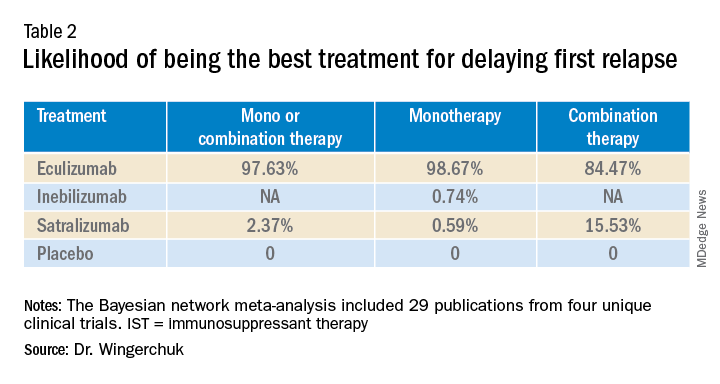
Dr. Wingerchuk acknowledged that there were many limitations to this study, including that analyses for annualized relapse rate, disability, and quality of life were not included because of a lack of consistent outcome reporting by AQP4+ status in the randomized trials.
Safety outcomes were excluded because of a lack of standardized baseline risks and inconsistent reporting by AQP4+ status across trials.
Because this study focused on drugs approved in the United States in a rare disease area, there were a limited number of studies with intervention effects.
There were differences in follow-up durations across the different trials, with N-MOmentum having a follow-up of 197 days compared with 144 weeks for other trials.
“In the absence of head-to-head trials, this network meta-analysis provides important evidence on the relative efficacy of eculizumab versus satralizumab or inebilizumab for the treatment of patients with AQP4+ NMOSD, with significant differences in two out of the three treatment comparison scenarios observed,” Dr. Wingerchuk concluded.
“Based on current evidence, monotherapy and mono-combination therapy with eculizumab appear to more efficacious at preventing relapses than satralizumab or inebilizumab for the treatment of adults with AQP4+ NMOSD. These findings appear to suggest that C5 complement inhibition with treatments such as eculizumab appear to prevent relapses more effectively than other mechanisms involving IL-6 receptor or CD19 inhibition among adults with AQP4+ NMOSD,” he added.
Experts respond
Commenting on the study, several experts in the field provided some balancing views.
Bruce Cree, MD, University of California San Francisco, who was the chief investigator of the N-MOmentum study with inebilizumab, said he was skeptical about this new indirect comparison. “The results of this study seem too good to be true; a 90% difference between agents has to be an overestimate,” he said.
“We know from independent studies that all three drugs are very effective. If we take each trial separately, eculizumab reduced attack risk by 90% versus placebo; and the other two drugs by 77% to 78% versus placebo. For eculizumab to be 90% better than inebilizumab or satralizumab its basically like saying these drugs perform like placebo, but we know that is not the case,” Dr. Cree argued.
He pointed out that when comparing results across studies there are many factors that have to be considered, including the different patient populations included in the different studies, with the characteristic of each population in each trial being unique to that dataset.
In addition, Dr. Cree suggested that all the studies included in the comparison were relatively small for this type of analysis. “Normally this type of analysis is done with much larger studies, so the resulting database is closer to a representation of the disease state itself,” he said.
Dr. Cree also questioned the role of the sponsor in this meta-analysis. “The analysis was sponsored by Alexion and several coauthors were employees of Alexion. There was not much description available of how the statistics were done. I am concerned that the company was involved in the analysis, which could introduce bias. I look forward to seeing details of the statistical methodology,” he said.
“This is definitely a provocative study. They have thrown down the gauntlet. If they are so confident in the results they should now do a head-to-head study to back this result up. If they don’t do that, then I think physicians should ignore it as there are just too many problems with this analysis,” Dr. Cree stated.
Dr. Cree acknowledged that when looking at the four trials separately, eculizumab does look a little better than the other two agents in delaying time to first relapse. “But there are some caveats. Despite a larger reduction in relapse rate there was no reduction in disability in the eculizumab trial. Whereas the inebilizumab trial did show a reduction in disability. And while the PREVENT trial with eculizumab was a good study, during the course of the trial the definition of clinical relapse was changed, and as a consequence that increased the product’s performance – that’s a little bit curious,” he added.
How to choose?
On how to choose between the three agents, Dr. Cree said they are all “extraordinarily effective” at reducing relapse activity. “They are all ‘home run’ products, but they have differences in safety,” he said.
“Inebilizumab is linked to hypogammaglobulinemia over time – we haven’t seen an increase in infection risk linked to this, but with enough time, I would expect that there probably will be. But inebilizumab is a B-cell-depleting agent like the agents used in MS, and we now have a lot of experiences with this type of product, which gives us more confidence on the safety profile,” Dr. Cree noted.
“Eculizumab was linked to a risk of meningococcal meningitis and other bacterial infections, and satralizumab seems to [be] overall well tolerated with no obvious safety concerns to date, but the studies have been quite small,” he added.
On routes of administration and frequency of dosing, Dr. Cree pointed out that while all three drugs have an intensive loading schedule, for maintenance, eculizumab needs to be given as an IV infusion every 2 weeks. Inebilizumab needs just two infusions per year for maintenance, while satralizumab is given by subcutaneous injection once per month.
“It may be that eculizumab could be used at the time of an acute attack but then treatment could be switched to one of the other two for long-term maintenance,” he suggested.
But Dr. Cree pointed out that the biggest challenge for all three agents is access. “The costs are astronomically high ($200,000-$770,000). They are prohibitively expensive and very few insurance companies are covering them.”
Also commenting, Brian Weinshenker, MD, from the Mayo Clinic in Rochester, Minn., who was a member of the attack adjudication committee for both PREVENT and N-MOmentum studies, pointed out that as well as differences in the populations enrolled, and study designs, the studies with the three different drugs also had differences in attack adjudication criteria.
“These factors make it very difficult to compare across studies, which is what was done in this analysis, so I would be reluctant to reach many conclusions about differences.”
Dr. Weinshenker added: “All three treatments provided strong benefit. We are still learning about long-term benefits, but emerging data have suggested that all three seem to provide persistent benefits for the length of the open-label extension study. We don’t have much evidence about the severity of the attacks that did occur, although some limited data suggest that both eculizumab and inebilizumab reduce attack severity.”
Dennis Bourdette, MD, professor emeritus, department of neurology, Oregon Health & Science University, Portland, who was not involved in any of the studies, said he thought the new analysis was “a worthwhile effort to determine the relative effectiveness of the three different drugs in treating AQP4+ NMOSD.
“Given the rarity of APQ4+ NMOSD, it will be difficult to perform randomized head-to-head clinical trials of the agents, so this type of comparison is the best we can do at this time,” he said.
While Dr. Bourdette feels this study supports the notion that eculizumab is more effective at delaying time to first relapse than inebilizumab and satralizumab, he does not believe the results should have a major impact on decisions about which agent to use in clinical practice.
“A difference in delaying time to first relapse tells us little about the relative effectiveness of the long-term benefit of these [agents], particularly with regards to permanent disability or frequency of relapses. However, it is possible that the difference reflects the efficacy kinetics of the agents with eculizumab working faster than the other two agents, which would be useful in making a decision about a patient with very active NMOSD where one wants to get the disease under control as quickly as possible,” Dr. Bourdette noted.
But he added that neurologists should also consider safety profile, convenience, and contraindications. “Eculizumab is clearly less convenient in terms of dosing schedule than the other two agents, and patient convenience is important for long-term compliance.”
Dr. Bourdette pointed out that another consideration is prior treatment. “Many patients with NMOSD will receive the anti-CD20 monoclonal antibody, rituximab – which depletes B cells – off label. Inebilizumab also depletes B cells, so a patient who has had continued NMOSD disease activity on rituximab probably should not be treated with inebilizumab, making eculizumab or satralizumab preferable,” he suggested.
Finally, Dr. Bourdette highlighted the sponsorship of the current study by the manufacturer of eculizumab, Alexion, and that all of the authors have some financial relationship with Alexion as described in their disclosures. “Whether this resulted in any biases about the design, conduct, or interpretation of the study is uncertain but is always a concern,” he said.
Company statements
The companies selling inebilizumab and satralizumab sent statements on the new analysis and repeated many of the above points.
Genentech noted that new longer-term data presented at ECTRIMS show that satralizumab is effective in significantly reducing relapses over 4 years of treatment in people with AQP4+ NMOSD, with a favorable safety profile both as a monotherapy and in conjunction with immunosuppressive therapy. More than 70% of people treated with satralizumab remained relapse free after 4 years in the SAkuraStar (73%) and SAkuraSky (71%) open-label extension studies, and 90% and 91%, respectively, were free from severe relapse, the company reported.
Horizon said: “We are confident in the efficacy and safety of Uplizna (inebilizumab) – a convenient, twice-annual monotherapy – that was studied in the largest randomized, placebo-controlled, global trial of a monotherapy in NMOSD. The endpoints in this trial were prospectively defined and assessed by an adjudication committee as published in The Lancet, with long-term follow-up data now published in the Multiple Sclerosis Journal that further support the efficacy and safety.”
The current study was funded by Alexion–AstraZeneca Rare Disease. Dr. Wingerchuk has participated on data safety monitoring or advisory boards for Roche, Viela Bio, Genentech, Biogen, Reistone, TG Therapeutics, Celgene, Novartis, and Alexion–AstraZeneca Rare Disease. He has received grants for clinical trials through Alexion–AstraZeneca Rare Disease and Terumo BCT, and has been paid consulting fees by Mitsubishi Tanabe. Several coauthors of this study are employees of Alexion Pharmaceutics. Dr. Cree was principal investigator on the N-MOmentum study with inebilizumab. He has a grant from Genentech for MS research, and has consulted for Alexion in the past. Dr. Weinshenker has served as a member of the attack adjudication committee for both PREVENT and N-MOmentum studies and has financial relationships with the manufacturers of all three drugs. Dr. Bourdette has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The Alexion-sponsored study was presented at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) by Dean Wingerchuk, MD, of the Mayo Clinic in Scottsdale, Ariz.
Other experts in the field have highlighted limitations to the analysis and pointed out that all three agents are very effective in treating AQP4+ NMOSD, and many other considerations need to be taken into account as well as time to first relapse when selecting a therapy, leaving the door open for all three agents.
Dr. Wingerchuk explained that NMOSD is a rare severely disabling complement-mediated autoimmune neuroinflammatory disease of the central nervous system, characterized by devastating and unpredictable attacks (relapses) that can cause immediate and irreversible damage.
There are three recently approved monoclonal antibody treatment options in the United States for adults with AQP4+ NMOSD: eculizumab (Soliris, Alexion), inebilizumab (Uplizna, Horizon), and satralizumab (Enspryng, Genentech). A comparison of the relative treatment effects of these drugs would facilitate the treatment selection process, Dr. Wingerchuk said.
The objective of this study was to perform an indirect treatment comparison on the efficacy of these three FDA-approved treatment options for adults with AQP4+ NMOSD, in the absence of any head-to-head studies.
Using published data from randomized controlled trials, which were identified by a systematic literature review in September 2020, the researchers performed a Bayesian network meta-analysis to estimate the relative effects between eculizumab, inebilizumab, and satralizumab.
Network meta-analyses were performed for clinically relevant subpopulations based on three treatment networks: (1) patients who received monotherapy with one of the monoclonal antibodies or in combination with an immunosuppressant therapy; (2) patients who received monotherapy with the monoclonal antibody alone; and (3) patients who received a combination of both the monoclonal antibody and immunosuppressant therapy.
Time to first relapse was the primary efficacy outcome assessed. Relative treatment effects were expressed as hazard ratios and the probability that a treatment was the best at delaying time to first relapse was also evaluated.
In the systematic literature review, 29 publications from four unique clinical trials were identified and include in the network meta-analysis. These included publications from congress proceedings and peer-reviewed journals.
The four clinical trials were the N-MOmentum trial of inebilizumab versus placebo; the PREVENT trial of eculizumab with or without immunosuppressant therapy versus placebo with or without immunosuppressant therapy; the SAkuraSky trial of satralizumab plus immunosuppressant therapy versus placebo plus immunosuppressant therapy; and the SAkuraStar trial of satralizumab versus placebo.
Results showed that for the first analysis of mono or combination therapy, patients treated with eculizumab with or without immunosuppressant therapy were 76% less likely to experience a first relapse when compared with patients treated with satralizumab with or without immunosuppressant therapy.
In the monotherapy network, patients on eculizumab were 90% less likely to experience a first relapse when compared with patients treated with satralizumab, and patients on eculizumab were 89% less likely to experience a first relapse when compared with patients treated with inebilizumab.
In the third network analysis – a comparison of eculizumab plus immunosuppressant therapy with inebilizumab plus immunosuppressant therapy (Table 1) – the point estimate appeared to favor eculizumab but the confidence intervals were wide and not significant.
A subsequent analysis looked at the rank order of the best treatment option, with eculizumab coming out first in all three networks (Table 2).
Dr. Wingerchuk acknowledged that there were many limitations to this study, including that analyses for annualized relapse rate, disability, and quality of life were not included because of a lack of consistent outcome reporting by AQP4+ status in the randomized trials.
Safety outcomes were excluded because of a lack of standardized baseline risks and inconsistent reporting by AQP4+ status across trials.
Because this study focused on drugs approved in the United States in a rare disease area, there were a limited number of studies with intervention effects.
There were differences in follow-up durations across the different trials, with N-MOmentum having a follow-up of 197 days compared with 144 weeks for other trials.
“In the absence of head-to-head trials, this network meta-analysis provides important evidence on the relative efficacy of eculizumab versus satralizumab or inebilizumab for the treatment of patients with AQP4+ NMOSD, with significant differences in two out of the three treatment comparison scenarios observed,” Dr. Wingerchuk concluded.
“Based on current evidence, monotherapy and mono-combination therapy with eculizumab appear to more efficacious at preventing relapses than satralizumab or inebilizumab for the treatment of adults with AQP4+ NMOSD. These findings appear to suggest that C5 complement inhibition with treatments such as eculizumab appear to prevent relapses more effectively than other mechanisms involving IL-6 receptor or CD19 inhibition among adults with AQP4+ NMOSD,” he added.
Experts respond
Commenting on the study, several experts in the field provided some balancing views.
Bruce Cree, MD, University of California San Francisco, who was the chief investigator of the N-MOmentum study with inebilizumab, said he was skeptical about this new indirect comparison. “The results of this study seem too good to be true; a 90% difference between agents has to be an overestimate,” he said.
“We know from independent studies that all three drugs are very effective. If we take each trial separately, eculizumab reduced attack risk by 90% versus placebo; and the other two drugs by 77% to 78% versus placebo. For eculizumab to be 90% better than inebilizumab or satralizumab its basically like saying these drugs perform like placebo, but we know that is not the case,” Dr. Cree argued.
He pointed out that when comparing results across studies there are many factors that have to be considered, including the different patient populations included in the different studies, with the characteristic of each population in each trial being unique to that dataset.
In addition, Dr. Cree suggested that all the studies included in the comparison were relatively small for this type of analysis. “Normally this type of analysis is done with much larger studies, so the resulting database is closer to a representation of the disease state itself,” he said.
Dr. Cree also questioned the role of the sponsor in this meta-analysis. “The analysis was sponsored by Alexion and several coauthors were employees of Alexion. There was not much description available of how the statistics were done. I am concerned that the company was involved in the analysis, which could introduce bias. I look forward to seeing details of the statistical methodology,” he said.
“This is definitely a provocative study. They have thrown down the gauntlet. If they are so confident in the results they should now do a head-to-head study to back this result up. If they don’t do that, then I think physicians should ignore it as there are just too many problems with this analysis,” Dr. Cree stated.
Dr. Cree acknowledged that when looking at the four trials separately, eculizumab does look a little better than the other two agents in delaying time to first relapse. “But there are some caveats. Despite a larger reduction in relapse rate there was no reduction in disability in the eculizumab trial. Whereas the inebilizumab trial did show a reduction in disability. And while the PREVENT trial with eculizumab was a good study, during the course of the trial the definition of clinical relapse was changed, and as a consequence that increased the product’s performance – that’s a little bit curious,” he added.
How to choose?
On how to choose between the three agents, Dr. Cree said they are all “extraordinarily effective” at reducing relapse activity. “They are all ‘home run’ products, but they have differences in safety,” he said.
“Inebilizumab is linked to hypogammaglobulinemia over time – we haven’t seen an increase in infection risk linked to this, but with enough time, I would expect that there probably will be. But inebilizumab is a B-cell-depleting agent like the agents used in MS, and we now have a lot of experiences with this type of product, which gives us more confidence on the safety profile,” Dr. Cree noted.
“Eculizumab was linked to a risk of meningococcal meningitis and other bacterial infections, and satralizumab seems to [be] overall well tolerated with no obvious safety concerns to date, but the studies have been quite small,” he added.
On routes of administration and frequency of dosing, Dr. Cree pointed out that while all three drugs have an intensive loading schedule, for maintenance, eculizumab needs to be given as an IV infusion every 2 weeks. Inebilizumab needs just two infusions per year for maintenance, while satralizumab is given by subcutaneous injection once per month.
“It may be that eculizumab could be used at the time of an acute attack but then treatment could be switched to one of the other two for long-term maintenance,” he suggested.
But Dr. Cree pointed out that the biggest challenge for all three agents is access. “The costs are astronomically high ($200,000-$770,000). They are prohibitively expensive and very few insurance companies are covering them.”
Also commenting, Brian Weinshenker, MD, from the Mayo Clinic in Rochester, Minn., who was a member of the attack adjudication committee for both PREVENT and N-MOmentum studies, pointed out that as well as differences in the populations enrolled, and study designs, the studies with the three different drugs also had differences in attack adjudication criteria.
“These factors make it very difficult to compare across studies, which is what was done in this analysis, so I would be reluctant to reach many conclusions about differences.”
Dr. Weinshenker added: “All three treatments provided strong benefit. We are still learning about long-term benefits, but emerging data have suggested that all three seem to provide persistent benefits for the length of the open-label extension study. We don’t have much evidence about the severity of the attacks that did occur, although some limited data suggest that both eculizumab and inebilizumab reduce attack severity.”
Dennis Bourdette, MD, professor emeritus, department of neurology, Oregon Health & Science University, Portland, who was not involved in any of the studies, said he thought the new analysis was “a worthwhile effort to determine the relative effectiveness of the three different drugs in treating AQP4+ NMOSD.
“Given the rarity of APQ4+ NMOSD, it will be difficult to perform randomized head-to-head clinical trials of the agents, so this type of comparison is the best we can do at this time,” he said.
While Dr. Bourdette feels this study supports the notion that eculizumab is more effective at delaying time to first relapse than inebilizumab and satralizumab, he does not believe the results should have a major impact on decisions about which agent to use in clinical practice.
“A difference in delaying time to first relapse tells us little about the relative effectiveness of the long-term benefit of these [agents], particularly with regards to permanent disability or frequency of relapses. However, it is possible that the difference reflects the efficacy kinetics of the agents with eculizumab working faster than the other two agents, which would be useful in making a decision about a patient with very active NMOSD where one wants to get the disease under control as quickly as possible,” Dr. Bourdette noted.
But he added that neurologists should also consider safety profile, convenience, and contraindications. “Eculizumab is clearly less convenient in terms of dosing schedule than the other two agents, and patient convenience is important for long-term compliance.”
Dr. Bourdette pointed out that another consideration is prior treatment. “Many patients with NMOSD will receive the anti-CD20 monoclonal antibody, rituximab – which depletes B cells – off label. Inebilizumab also depletes B cells, so a patient who has had continued NMOSD disease activity on rituximab probably should not be treated with inebilizumab, making eculizumab or satralizumab preferable,” he suggested.
Finally, Dr. Bourdette highlighted the sponsorship of the current study by the manufacturer of eculizumab, Alexion, and that all of the authors have some financial relationship with Alexion as described in their disclosures. “Whether this resulted in any biases about the design, conduct, or interpretation of the study is uncertain but is always a concern,” he said.
Company statements
The companies selling inebilizumab and satralizumab sent statements on the new analysis and repeated many of the above points.
Genentech noted that new longer-term data presented at ECTRIMS show that satralizumab is effective in significantly reducing relapses over 4 years of treatment in people with AQP4+ NMOSD, with a favorable safety profile both as a monotherapy and in conjunction with immunosuppressive therapy. More than 70% of people treated with satralizumab remained relapse free after 4 years in the SAkuraStar (73%) and SAkuraSky (71%) open-label extension studies, and 90% and 91%, respectively, were free from severe relapse, the company reported.
Horizon said: “We are confident in the efficacy and safety of Uplizna (inebilizumab) – a convenient, twice-annual monotherapy – that was studied in the largest randomized, placebo-controlled, global trial of a monotherapy in NMOSD. The endpoints in this trial were prospectively defined and assessed by an adjudication committee as published in The Lancet, with long-term follow-up data now published in the Multiple Sclerosis Journal that further support the efficacy and safety.”
The current study was funded by Alexion–AstraZeneca Rare Disease. Dr. Wingerchuk has participated on data safety monitoring or advisory boards for Roche, Viela Bio, Genentech, Biogen, Reistone, TG Therapeutics, Celgene, Novartis, and Alexion–AstraZeneca Rare Disease. He has received grants for clinical trials through Alexion–AstraZeneca Rare Disease and Terumo BCT, and has been paid consulting fees by Mitsubishi Tanabe. Several coauthors of this study are employees of Alexion Pharmaceutics. Dr. Cree was principal investigator on the N-MOmentum study with inebilizumab. He has a grant from Genentech for MS research, and has consulted for Alexion in the past. Dr. Weinshenker has served as a member of the attack adjudication committee for both PREVENT and N-MOmentum studies and has financial relationships with the manufacturers of all three drugs. Dr. Bourdette has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The Alexion-sponsored study was presented at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) by Dean Wingerchuk, MD, of the Mayo Clinic in Scottsdale, Ariz.
Other experts in the field have highlighted limitations to the analysis and pointed out that all three agents are very effective in treating AQP4+ NMOSD, and many other considerations need to be taken into account as well as time to first relapse when selecting a therapy, leaving the door open for all three agents.
Dr. Wingerchuk explained that NMOSD is a rare severely disabling complement-mediated autoimmune neuroinflammatory disease of the central nervous system, characterized by devastating and unpredictable attacks (relapses) that can cause immediate and irreversible damage.
There are three recently approved monoclonal antibody treatment options in the United States for adults with AQP4+ NMOSD: eculizumab (Soliris, Alexion), inebilizumab (Uplizna, Horizon), and satralizumab (Enspryng, Genentech). A comparison of the relative treatment effects of these drugs would facilitate the treatment selection process, Dr. Wingerchuk said.
The objective of this study was to perform an indirect treatment comparison on the efficacy of these three FDA-approved treatment options for adults with AQP4+ NMOSD, in the absence of any head-to-head studies.
Using published data from randomized controlled trials, which were identified by a systematic literature review in September 2020, the researchers performed a Bayesian network meta-analysis to estimate the relative effects between eculizumab, inebilizumab, and satralizumab.
Network meta-analyses were performed for clinically relevant subpopulations based on three treatment networks: (1) patients who received monotherapy with one of the monoclonal antibodies or in combination with an immunosuppressant therapy; (2) patients who received monotherapy with the monoclonal antibody alone; and (3) patients who received a combination of both the monoclonal antibody and immunosuppressant therapy.
Time to first relapse was the primary efficacy outcome assessed. Relative treatment effects were expressed as hazard ratios and the probability that a treatment was the best at delaying time to first relapse was also evaluated.
In the systematic literature review, 29 publications from four unique clinical trials were identified and include in the network meta-analysis. These included publications from congress proceedings and peer-reviewed journals.
The four clinical trials were the N-MOmentum trial of inebilizumab versus placebo; the PREVENT trial of eculizumab with or without immunosuppressant therapy versus placebo with or without immunosuppressant therapy; the SAkuraSky trial of satralizumab plus immunosuppressant therapy versus placebo plus immunosuppressant therapy; and the SAkuraStar trial of satralizumab versus placebo.
Results showed that for the first analysis of mono or combination therapy, patients treated with eculizumab with or without immunosuppressant therapy were 76% less likely to experience a first relapse when compared with patients treated with satralizumab with or without immunosuppressant therapy.
In the monotherapy network, patients on eculizumab were 90% less likely to experience a first relapse when compared with patients treated with satralizumab, and patients on eculizumab were 89% less likely to experience a first relapse when compared with patients treated with inebilizumab.
In the third network analysis – a comparison of eculizumab plus immunosuppressant therapy with inebilizumab plus immunosuppressant therapy (Table 1) – the point estimate appeared to favor eculizumab but the confidence intervals were wide and not significant.
A subsequent analysis looked at the rank order of the best treatment option, with eculizumab coming out first in all three networks (Table 2).
Dr. Wingerchuk acknowledged that there were many limitations to this study, including that analyses for annualized relapse rate, disability, and quality of life were not included because of a lack of consistent outcome reporting by AQP4+ status in the randomized trials.
Safety outcomes were excluded because of a lack of standardized baseline risks and inconsistent reporting by AQP4+ status across trials.
Because this study focused on drugs approved in the United States in a rare disease area, there were a limited number of studies with intervention effects.
There were differences in follow-up durations across the different trials, with N-MOmentum having a follow-up of 197 days compared with 144 weeks for other trials.
“In the absence of head-to-head trials, this network meta-analysis provides important evidence on the relative efficacy of eculizumab versus satralizumab or inebilizumab for the treatment of patients with AQP4+ NMOSD, with significant differences in two out of the three treatment comparison scenarios observed,” Dr. Wingerchuk concluded.
“Based on current evidence, monotherapy and mono-combination therapy with eculizumab appear to more efficacious at preventing relapses than satralizumab or inebilizumab for the treatment of adults with AQP4+ NMOSD. These findings appear to suggest that C5 complement inhibition with treatments such as eculizumab appear to prevent relapses more effectively than other mechanisms involving IL-6 receptor or CD19 inhibition among adults with AQP4+ NMOSD,” he added.
Experts respond
Commenting on the study, several experts in the field provided some balancing views.
Bruce Cree, MD, University of California San Francisco, who was the chief investigator of the N-MOmentum study with inebilizumab, said he was skeptical about this new indirect comparison. “The results of this study seem too good to be true; a 90% difference between agents has to be an overestimate,” he said.
“We know from independent studies that all three drugs are very effective. If we take each trial separately, eculizumab reduced attack risk by 90% versus placebo; and the other two drugs by 77% to 78% versus placebo. For eculizumab to be 90% better than inebilizumab or satralizumab its basically like saying these drugs perform like placebo, but we know that is not the case,” Dr. Cree argued.
He pointed out that when comparing results across studies there are many factors that have to be considered, including the different patient populations included in the different studies, with the characteristic of each population in each trial being unique to that dataset.
In addition, Dr. Cree suggested that all the studies included in the comparison were relatively small for this type of analysis. “Normally this type of analysis is done with much larger studies, so the resulting database is closer to a representation of the disease state itself,” he said.
Dr. Cree also questioned the role of the sponsor in this meta-analysis. “The analysis was sponsored by Alexion and several coauthors were employees of Alexion. There was not much description available of how the statistics were done. I am concerned that the company was involved in the analysis, which could introduce bias. I look forward to seeing details of the statistical methodology,” he said.
“This is definitely a provocative study. They have thrown down the gauntlet. If they are so confident in the results they should now do a head-to-head study to back this result up. If they don’t do that, then I think physicians should ignore it as there are just too many problems with this analysis,” Dr. Cree stated.
Dr. Cree acknowledged that when looking at the four trials separately, eculizumab does look a little better than the other two agents in delaying time to first relapse. “But there are some caveats. Despite a larger reduction in relapse rate there was no reduction in disability in the eculizumab trial. Whereas the inebilizumab trial did show a reduction in disability. And while the PREVENT trial with eculizumab was a good study, during the course of the trial the definition of clinical relapse was changed, and as a consequence that increased the product’s performance – that’s a little bit curious,” he added.
How to choose?
On how to choose between the three agents, Dr. Cree said they are all “extraordinarily effective” at reducing relapse activity. “They are all ‘home run’ products, but they have differences in safety,” he said.
“Inebilizumab is linked to hypogammaglobulinemia over time – we haven’t seen an increase in infection risk linked to this, but with enough time, I would expect that there probably will be. But inebilizumab is a B-cell-depleting agent like the agents used in MS, and we now have a lot of experiences with this type of product, which gives us more confidence on the safety profile,” Dr. Cree noted.
“Eculizumab was linked to a risk of meningococcal meningitis and other bacterial infections, and satralizumab seems to [be] overall well tolerated with no obvious safety concerns to date, but the studies have been quite small,” he added.
On routes of administration and frequency of dosing, Dr. Cree pointed out that while all three drugs have an intensive loading schedule, for maintenance, eculizumab needs to be given as an IV infusion every 2 weeks. Inebilizumab needs just two infusions per year for maintenance, while satralizumab is given by subcutaneous injection once per month.
“It may be that eculizumab could be used at the time of an acute attack but then treatment could be switched to one of the other two for long-term maintenance,” he suggested.
But Dr. Cree pointed out that the biggest challenge for all three agents is access. “The costs are astronomically high ($200,000-$770,000). They are prohibitively expensive and very few insurance companies are covering them.”
Also commenting, Brian Weinshenker, MD, from the Mayo Clinic in Rochester, Minn., who was a member of the attack adjudication committee for both PREVENT and N-MOmentum studies, pointed out that as well as differences in the populations enrolled, and study designs, the studies with the three different drugs also had differences in attack adjudication criteria.
“These factors make it very difficult to compare across studies, which is what was done in this analysis, so I would be reluctant to reach many conclusions about differences.”
Dr. Weinshenker added: “All three treatments provided strong benefit. We are still learning about long-term benefits, but emerging data have suggested that all three seem to provide persistent benefits for the length of the open-label extension study. We don’t have much evidence about the severity of the attacks that did occur, although some limited data suggest that both eculizumab and inebilizumab reduce attack severity.”
Dennis Bourdette, MD, professor emeritus, department of neurology, Oregon Health & Science University, Portland, who was not involved in any of the studies, said he thought the new analysis was “a worthwhile effort to determine the relative effectiveness of the three different drugs in treating AQP4+ NMOSD.
“Given the rarity of APQ4+ NMOSD, it will be difficult to perform randomized head-to-head clinical trials of the agents, so this type of comparison is the best we can do at this time,” he said.
While Dr. Bourdette feels this study supports the notion that eculizumab is more effective at delaying time to first relapse than inebilizumab and satralizumab, he does not believe the results should have a major impact on decisions about which agent to use in clinical practice.
“A difference in delaying time to first relapse tells us little about the relative effectiveness of the long-term benefit of these [agents], particularly with regards to permanent disability or frequency of relapses. However, it is possible that the difference reflects the efficacy kinetics of the agents with eculizumab working faster than the other two agents, which would be useful in making a decision about a patient with very active NMOSD where one wants to get the disease under control as quickly as possible,” Dr. Bourdette noted.
But he added that neurologists should also consider safety profile, convenience, and contraindications. “Eculizumab is clearly less convenient in terms of dosing schedule than the other two agents, and patient convenience is important for long-term compliance.”
Dr. Bourdette pointed out that another consideration is prior treatment. “Many patients with NMOSD will receive the anti-CD20 monoclonal antibody, rituximab – which depletes B cells – off label. Inebilizumab also depletes B cells, so a patient who has had continued NMOSD disease activity on rituximab probably should not be treated with inebilizumab, making eculizumab or satralizumab preferable,” he suggested.
Finally, Dr. Bourdette highlighted the sponsorship of the current study by the manufacturer of eculizumab, Alexion, and that all of the authors have some financial relationship with Alexion as described in their disclosures. “Whether this resulted in any biases about the design, conduct, or interpretation of the study is uncertain but is always a concern,” he said.
Company statements
The companies selling inebilizumab and satralizumab sent statements on the new analysis and repeated many of the above points.
Genentech noted that new longer-term data presented at ECTRIMS show that satralizumab is effective in significantly reducing relapses over 4 years of treatment in people with AQP4+ NMOSD, with a favorable safety profile both as a monotherapy and in conjunction with immunosuppressive therapy. More than 70% of people treated with satralizumab remained relapse free after 4 years in the SAkuraStar (73%) and SAkuraSky (71%) open-label extension studies, and 90% and 91%, respectively, were free from severe relapse, the company reported.
Horizon said: “We are confident in the efficacy and safety of Uplizna (inebilizumab) – a convenient, twice-annual monotherapy – that was studied in the largest randomized, placebo-controlled, global trial of a monotherapy in NMOSD. The endpoints in this trial were prospectively defined and assessed by an adjudication committee as published in The Lancet, with long-term follow-up data now published in the Multiple Sclerosis Journal that further support the efficacy and safety.”
The current study was funded by Alexion–AstraZeneca Rare Disease. Dr. Wingerchuk has participated on data safety monitoring or advisory boards for Roche, Viela Bio, Genentech, Biogen, Reistone, TG Therapeutics, Celgene, Novartis, and Alexion–AstraZeneca Rare Disease. He has received grants for clinical trials through Alexion–AstraZeneca Rare Disease and Terumo BCT, and has been paid consulting fees by Mitsubishi Tanabe. Several coauthors of this study are employees of Alexion Pharmaceutics. Dr. Cree was principal investigator on the N-MOmentum study with inebilizumab. He has a grant from Genentech for MS research, and has consulted for Alexion in the past. Dr. Weinshenker has served as a member of the attack adjudication committee for both PREVENT and N-MOmentum studies and has financial relationships with the manufacturers of all three drugs. Dr. Bourdette has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ECTRIMS 2021
Stem cell transplant benefits in secondary progressive MS
, a new Italian study suggests.
In the study, stem cell transplant was associated with a slowing of disability progression and a higher likelihood of disability improvement in patients with secondary progressive MS compared with other disease-modifying therapies.
“Our study shows that, although limited, sustained disability improvement is still possible during early active secondary progressive MS and seems to be more likely with stem cell transplant than other disease-modifying treatments,” said lead author Giacomo Boffa, MD, University of Genoa, Italy.
“Brain penetrating–intent immune suppression in long-term immunological reconstitution within the central nervous system could be responsible for this clinical efficacy,” he added.
Dr. Boffa presented the research at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS).
He explained that compartmentalized inflation in the brain parenchyma, left meninges, and the cerebral spinal fluid (CSF) is a key driver of disability worsening in secondary progressive MS.
Although initial studies did not reveal an effect of disease-modifying therapies in secondary progressive MS, recent randomized trials have established some benefit of siponimod in reducing the risk of disability worsening, and consistent with this finding, observational studies have suggested that other available immunotherapies may also be beneficial for active secondary progressive MS, Dr. Boffa noted.
“Autologous hematopoietic stem cell transplantation has been widely investigated for the treatment of refractory MS, and although the ideal candidate for this procedure is a young patient with relapsing-remitting MS, there is some evidence to suggest that the procedure could also slow down neurological disability in patients with secondary progressive MS,” Dr. Boffa said.
“Indeed, all the drugs used in the transplant technology share the ability to cross the blood-brain barrier and exert a strong immunosuppressant effect within the brain parenchyma and CSF,” he added.
Comparing treatment regimens
The aim of the current study was to compare the effect of autologous hematopoietic stem cell transplantation with other immunotherapies on disability worsening in patients with active secondary progressive MS.
Study endpoints included the Expanded Disability Status Scale (EDSS) score, 6-month worsening and improvement in disability, and sustained disability improvement over time.
The researchers studied patients with secondary progressive MS who had received autologous hematopoietic stem cell transplantation and were included in the Italian Bone Marrow Transplant Group. They were compared with a control group of patients in the Italian MS registry who had started a nontransplant disease-modifying therapy after the diagnosis of secondary progressive MS.
To control for many different variables, two separate analyses were preformed. One analyses was a propensity-score approach (patients were matched based on their propensity to receive bone marrow transplant or one of the other disease-modifying therapies). The other analysis used an overlap weighting approach (each patient was given a weight proportional to the probability of them belonging to the other treatment group).
The final cohort consisted of 79 bone marrow transplant recipients and 1,975 patients who had received other disease-modifying therapies.
Before matching, patients in the control group were older, had a longer disease duration, and had a lower annualized relapse rate than transplanted patients.
After propensity-score matching, there were 69 transplanted patients and 217 control patients who were well balanced in terms of clinical and demographic characteristics. After overlap weighting, the entire cohort was also well balanced for these variables.
In terms of the primary endpoint, stem cell transplant stabilized the EDSS score over time, while patients treated with other disease-modifying therapies had continuous progression of the EDSS score over time.
In the propensity-matched analysis, the EDSS score improved by 0.013 points per year in the stem cell transplant group compared with a worsening of 0.157 points per year in the control group. Similar results were seen in the overlap-weighting analysis.
The effect of stem cell transplant on EDSS score translated into a significantly delayed time to confirmed disability progression in the stem cell transplant group compared with the control group (HR, 0.5; P = .005), Dr. Boffa reported.
Five years after the procedure, 62% of the transplant group were free of disability progression, compared with around 20% of the control group.
Patients in the transplanted group were also more likely to show disability improvement over time. Five years after the procedure, almost 20% of the stem cell transplant group still maintained a disability improvement compared with only 4% of patients treated with other disease-modifying therapies.
“Our study population was composed of relatively young patients (average age 38 years) with clinical activity during secondary progressive MS, and the results of this study would not be applicable to patients with secondary progressive MS patients without signs of inflammatory activity,” Dr. Boffa commented.
“But on the other hand, our results reinforce the notion that ongoing inflammation during progressive MS requires adequate immunotherapy,” he added.
A version of this article first appeared on Medscape.com.
, a new Italian study suggests.
In the study, stem cell transplant was associated with a slowing of disability progression and a higher likelihood of disability improvement in patients with secondary progressive MS compared with other disease-modifying therapies.
“Our study shows that, although limited, sustained disability improvement is still possible during early active secondary progressive MS and seems to be more likely with stem cell transplant than other disease-modifying treatments,” said lead author Giacomo Boffa, MD, University of Genoa, Italy.
“Brain penetrating–intent immune suppression in long-term immunological reconstitution within the central nervous system could be responsible for this clinical efficacy,” he added.
Dr. Boffa presented the research at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS).
He explained that compartmentalized inflation in the brain parenchyma, left meninges, and the cerebral spinal fluid (CSF) is a key driver of disability worsening in secondary progressive MS.
Although initial studies did not reveal an effect of disease-modifying therapies in secondary progressive MS, recent randomized trials have established some benefit of siponimod in reducing the risk of disability worsening, and consistent with this finding, observational studies have suggested that other available immunotherapies may also be beneficial for active secondary progressive MS, Dr. Boffa noted.
“Autologous hematopoietic stem cell transplantation has been widely investigated for the treatment of refractory MS, and although the ideal candidate for this procedure is a young patient with relapsing-remitting MS, there is some evidence to suggest that the procedure could also slow down neurological disability in patients with secondary progressive MS,” Dr. Boffa said.
“Indeed, all the drugs used in the transplant technology share the ability to cross the blood-brain barrier and exert a strong immunosuppressant effect within the brain parenchyma and CSF,” he added.
Comparing treatment regimens
The aim of the current study was to compare the effect of autologous hematopoietic stem cell transplantation with other immunotherapies on disability worsening in patients with active secondary progressive MS.
Study endpoints included the Expanded Disability Status Scale (EDSS) score, 6-month worsening and improvement in disability, and sustained disability improvement over time.
The researchers studied patients with secondary progressive MS who had received autologous hematopoietic stem cell transplantation and were included in the Italian Bone Marrow Transplant Group. They were compared with a control group of patients in the Italian MS registry who had started a nontransplant disease-modifying therapy after the diagnosis of secondary progressive MS.
To control for many different variables, two separate analyses were preformed. One analyses was a propensity-score approach (patients were matched based on their propensity to receive bone marrow transplant or one of the other disease-modifying therapies). The other analysis used an overlap weighting approach (each patient was given a weight proportional to the probability of them belonging to the other treatment group).
The final cohort consisted of 79 bone marrow transplant recipients and 1,975 patients who had received other disease-modifying therapies.
Before matching, patients in the control group were older, had a longer disease duration, and had a lower annualized relapse rate than transplanted patients.
After propensity-score matching, there were 69 transplanted patients and 217 control patients who were well balanced in terms of clinical and demographic characteristics. After overlap weighting, the entire cohort was also well balanced for these variables.
In terms of the primary endpoint, stem cell transplant stabilized the EDSS score over time, while patients treated with other disease-modifying therapies had continuous progression of the EDSS score over time.
In the propensity-matched analysis, the EDSS score improved by 0.013 points per year in the stem cell transplant group compared with a worsening of 0.157 points per year in the control group. Similar results were seen in the overlap-weighting analysis.
The effect of stem cell transplant on EDSS score translated into a significantly delayed time to confirmed disability progression in the stem cell transplant group compared with the control group (HR, 0.5; P = .005), Dr. Boffa reported.
Five years after the procedure, 62% of the transplant group were free of disability progression, compared with around 20% of the control group.
Patients in the transplanted group were also more likely to show disability improvement over time. Five years after the procedure, almost 20% of the stem cell transplant group still maintained a disability improvement compared with only 4% of patients treated with other disease-modifying therapies.
“Our study population was composed of relatively young patients (average age 38 years) with clinical activity during secondary progressive MS, and the results of this study would not be applicable to patients with secondary progressive MS patients without signs of inflammatory activity,” Dr. Boffa commented.
“But on the other hand, our results reinforce the notion that ongoing inflammation during progressive MS requires adequate immunotherapy,” he added.
A version of this article first appeared on Medscape.com.
, a new Italian study suggests.
In the study, stem cell transplant was associated with a slowing of disability progression and a higher likelihood of disability improvement in patients with secondary progressive MS compared with other disease-modifying therapies.
“Our study shows that, although limited, sustained disability improvement is still possible during early active secondary progressive MS and seems to be more likely with stem cell transplant than other disease-modifying treatments,” said lead author Giacomo Boffa, MD, University of Genoa, Italy.
“Brain penetrating–intent immune suppression in long-term immunological reconstitution within the central nervous system could be responsible for this clinical efficacy,” he added.
Dr. Boffa presented the research at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS).
He explained that compartmentalized inflation in the brain parenchyma, left meninges, and the cerebral spinal fluid (CSF) is a key driver of disability worsening in secondary progressive MS.
Although initial studies did not reveal an effect of disease-modifying therapies in secondary progressive MS, recent randomized trials have established some benefit of siponimod in reducing the risk of disability worsening, and consistent with this finding, observational studies have suggested that other available immunotherapies may also be beneficial for active secondary progressive MS, Dr. Boffa noted.
“Autologous hematopoietic stem cell transplantation has been widely investigated for the treatment of refractory MS, and although the ideal candidate for this procedure is a young patient with relapsing-remitting MS, there is some evidence to suggest that the procedure could also slow down neurological disability in patients with secondary progressive MS,” Dr. Boffa said.
“Indeed, all the drugs used in the transplant technology share the ability to cross the blood-brain barrier and exert a strong immunosuppressant effect within the brain parenchyma and CSF,” he added.
Comparing treatment regimens
The aim of the current study was to compare the effect of autologous hematopoietic stem cell transplantation with other immunotherapies on disability worsening in patients with active secondary progressive MS.
Study endpoints included the Expanded Disability Status Scale (EDSS) score, 6-month worsening and improvement in disability, and sustained disability improvement over time.
The researchers studied patients with secondary progressive MS who had received autologous hematopoietic stem cell transplantation and were included in the Italian Bone Marrow Transplant Group. They were compared with a control group of patients in the Italian MS registry who had started a nontransplant disease-modifying therapy after the diagnosis of secondary progressive MS.
To control for many different variables, two separate analyses were preformed. One analyses was a propensity-score approach (patients were matched based on their propensity to receive bone marrow transplant or one of the other disease-modifying therapies). The other analysis used an overlap weighting approach (each patient was given a weight proportional to the probability of them belonging to the other treatment group).
The final cohort consisted of 79 bone marrow transplant recipients and 1,975 patients who had received other disease-modifying therapies.
Before matching, patients in the control group were older, had a longer disease duration, and had a lower annualized relapse rate than transplanted patients.
After propensity-score matching, there were 69 transplanted patients and 217 control patients who were well balanced in terms of clinical and demographic characteristics. After overlap weighting, the entire cohort was also well balanced for these variables.
In terms of the primary endpoint, stem cell transplant stabilized the EDSS score over time, while patients treated with other disease-modifying therapies had continuous progression of the EDSS score over time.
In the propensity-matched analysis, the EDSS score improved by 0.013 points per year in the stem cell transplant group compared with a worsening of 0.157 points per year in the control group. Similar results were seen in the overlap-weighting analysis.
The effect of stem cell transplant on EDSS score translated into a significantly delayed time to confirmed disability progression in the stem cell transplant group compared with the control group (HR, 0.5; P = .005), Dr. Boffa reported.
Five years after the procedure, 62% of the transplant group were free of disability progression, compared with around 20% of the control group.
Patients in the transplanted group were also more likely to show disability improvement over time. Five years after the procedure, almost 20% of the stem cell transplant group still maintained a disability improvement compared with only 4% of patients treated with other disease-modifying therapies.
“Our study population was composed of relatively young patients (average age 38 years) with clinical activity during secondary progressive MS, and the results of this study would not be applicable to patients with secondary progressive MS patients without signs of inflammatory activity,” Dr. Boffa commented.
“But on the other hand, our results reinforce the notion that ongoing inflammation during progressive MS requires adequate immunotherapy,” he added.
A version of this article first appeared on Medscape.com.
FROM ECTRIMS 2021