User login
Getting high heightens stroke, arrhythmia risks
Stoners, beware: , and people with cannabis use disorder are at a 50% greater risk of being hospitalized for arrhythmias, according to new research presented at the American Heart Association Scientific Sessions 2019.

An analysis of pooled data on nearly 44,000 participants in a cross-sectional survey showed that, among the 13.6% who reported using marijuana within the last 30 days, the adjusted odds ratio for young-onset stroke (aged 18-44 years), compared with non-users, was 2.75, reported Tarang Parekh, MBBS, a health policy researcher of George Mason University in Fairfax, Va., and colleagues.
In a separate study, a retrospective analysis of national inpatient data showed that people diagnosed with cannabis use disorder – a pathological pattern of impaired control, social impairment, risky behavior or physiological adaptation similar in nature to alcoholism – had a 47%-52% increased likelihood of hospitalization for an arrhythmia, reported Rikinkumar S. Patel, MD, a psychiatry resident at Griffin Memorial Hospital in Norman, Okla.
“As these [cannabis] products become increasingly used across the country, getting clearer, scientifically rigorous data is going to be important as we try to understand the overall health effects of cannabis,” said AHA President Robert Harrington, MD, of Stanford (Calif.) University in a statement.
Currently, use of both medical and recreational marijuana is fully legal in 11 U.S. states and the District of Columbia. Medical marijuana is legal with recreational use decriminalized (or penalties reduced) in 28 other states, and totally illegal in 11 other states, according to employee screening firm DISA Global Solutions.
Stroke study
In an oral presentation with simultaneous publication in the AHA journal Stroke, Dr. Parekh and colleagues presented an analysis of pooled data from the Behavioral Risk Factor Surveillance System (BRFSS), a nationally representative cross-sectional survey collected by the Centers for Disease Control and Prevention in 2016 and 2017.
They looked at baseline sociodemographic data and created multivariable logistic regression models with state fixed effects to determine whether marijuana use within the last 30 days was associated with young-onset stroke.
They identified 43,860 participants representing a weighted sample of 35.5 million Americans. Of the sample, 63.3% were male, and 13.6 % of all participants reported using marijuana in the last 30 days.
They found in an unadjusted model that marijuana users had an odds ratio for stroke, compared with nonusers, of 1.59 (P less than.1), and in a model adjusted for demographic factors (gender, race, ethnicity, and education) the OR increased to 1.76 (P less than .05).
When they threw risk behavior into the model (physical activity, body mass index, heavy drinking, and cigarette smoking), they saw that the OR for stroke shot up to 2.75 (P less than .01).
“Physicians should ask patients if they use cannabis and counsel them about its potential stroke risk as part of regular doctor visits,” Dr. Parekh said in a statement.
Arrhythmias study

Based on recent studies suggesting that cannabis use may trigger cardiovascular events, Dr. Patel and colleagues studied whether cannabis use disorder may be related to arrhythmias, approaching the question through hospital records.
“The effects of using cannabis are seen within 15 minutes and last for around 3 hours. At lower doses, it is linked to a rapid heartbeat. At higher doses, it is linked to a too-slow heartbeat,” he said in a statement.
Dr. Patel and colleagues conducted a retrospective analysis of the Nationwide Inpatient Sample from 2010-2014, a period during which medical marijuana became legal in several states and recreational marijuana became legal in Colorado and Washington. The sample is a database maintained by the Healthcare Cost and Utilization Project of the U.S. Office of Disease Prevention and Health Promotion.
They identified 570,557 patients aged 15-54 years with a primary diagnosis of arrhythmia, and compared them with a sample of 67,662,082 patients hospitalized with no arrhythmia diagnosed during the same period.
They found a 2.6% incidence of cannabis use disorder among patients hospitalized for arrhythmias. Patients with cannabis use disorder tended to be younger (15- to 24-years-old; OR, 4.23), male (OR, 1.70) and African American (OR, 2.70).
In regression analysis adjusted for demographics and comorbidities, cannabis use disorder was associated with higher odds of arrhythmia hospitalization in young patients, at 1.28 times among 15- to 24-year-olds (95% confidence interval, 1.229-1.346) and 1.52 times for 25- to 34-year-olds (95% CI, 1.469-1.578).
“As medical and recreational cannabis is legalized in many states, it is important to know the difference between therapeutic cannabis dosing for medical purposes and the consequences of cannabis abuse. We urgently need additional research to understand these issues,” Dr. Patel said.
“It’s not proving that there’s a direct link, but it’s raising a suggestion in an observational analysis that [this] indeed might be the case. What that means for clinicians is that, if you’re seeing a patient who is presenting with a symptomatic arrhythmia, adding cannabis usage to your list of questions as you begin to try to understand possible precipitating factors for this arrhythmia seems to be a reasonable thing to do,” Dr. Harrington commented.
Stoners, beware: , and people with cannabis use disorder are at a 50% greater risk of being hospitalized for arrhythmias, according to new research presented at the American Heart Association Scientific Sessions 2019.
An analysis of pooled data on nearly 44,000 participants in a cross-sectional survey showed that, among the 13.6% who reported using marijuana within the last 30 days, the adjusted odds ratio for young-onset stroke (aged 18-44 years), compared with non-users, was 2.75, reported Tarang Parekh, MBBS, a health policy researcher of George Mason University in Fairfax, Va., and colleagues.
In a separate study, a retrospective analysis of national inpatient data showed that people diagnosed with cannabis use disorder – a pathological pattern of impaired control, social impairment, risky behavior or physiological adaptation similar in nature to alcoholism – had a 47%-52% increased likelihood of hospitalization for an arrhythmia, reported Rikinkumar S. Patel, MD, a psychiatry resident at Griffin Memorial Hospital in Norman, Okla.
“As these [cannabis] products become increasingly used across the country, getting clearer, scientifically rigorous data is going to be important as we try to understand the overall health effects of cannabis,” said AHA President Robert Harrington, MD, of Stanford (Calif.) University in a statement.
Currently, use of both medical and recreational marijuana is fully legal in 11 U.S. states and the District of Columbia. Medical marijuana is legal with recreational use decriminalized (or penalties reduced) in 28 other states, and totally illegal in 11 other states, according to employee screening firm DISA Global Solutions.
Stroke study
In an oral presentation with simultaneous publication in the AHA journal Stroke, Dr. Parekh and colleagues presented an analysis of pooled data from the Behavioral Risk Factor Surveillance System (BRFSS), a nationally representative cross-sectional survey collected by the Centers for Disease Control and Prevention in 2016 and 2017.
They looked at baseline sociodemographic data and created multivariable logistic regression models with state fixed effects to determine whether marijuana use within the last 30 days was associated with young-onset stroke.
They identified 43,860 participants representing a weighted sample of 35.5 million Americans. Of the sample, 63.3% were male, and 13.6 % of all participants reported using marijuana in the last 30 days.
They found in an unadjusted model that marijuana users had an odds ratio for stroke, compared with nonusers, of 1.59 (P less than.1), and in a model adjusted for demographic factors (gender, race, ethnicity, and education) the OR increased to 1.76 (P less than .05).
When they threw risk behavior into the model (physical activity, body mass index, heavy drinking, and cigarette smoking), they saw that the OR for stroke shot up to 2.75 (P less than .01).
“Physicians should ask patients if they use cannabis and counsel them about its potential stroke risk as part of regular doctor visits,” Dr. Parekh said in a statement.
Arrhythmias study
Based on recent studies suggesting that cannabis use may trigger cardiovascular events, Dr. Patel and colleagues studied whether cannabis use disorder may be related to arrhythmias, approaching the question through hospital records.
“The effects of using cannabis are seen within 15 minutes and last for around 3 hours. At lower doses, it is linked to a rapid heartbeat. At higher doses, it is linked to a too-slow heartbeat,” he said in a statement.
Dr. Patel and colleagues conducted a retrospective analysis of the Nationwide Inpatient Sample from 2010-2014, a period during which medical marijuana became legal in several states and recreational marijuana became legal in Colorado and Washington. The sample is a database maintained by the Healthcare Cost and Utilization Project of the U.S. Office of Disease Prevention and Health Promotion.
They identified 570,557 patients aged 15-54 years with a primary diagnosis of arrhythmia, and compared them with a sample of 67,662,082 patients hospitalized with no arrhythmia diagnosed during the same period.
They found a 2.6% incidence of cannabis use disorder among patients hospitalized for arrhythmias. Patients with cannabis use disorder tended to be younger (15- to 24-years-old; OR, 4.23), male (OR, 1.70) and African American (OR, 2.70).
In regression analysis adjusted for demographics and comorbidities, cannabis use disorder was associated with higher odds of arrhythmia hospitalization in young patients, at 1.28 times among 15- to 24-year-olds (95% confidence interval, 1.229-1.346) and 1.52 times for 25- to 34-year-olds (95% CI, 1.469-1.578).
“As medical and recreational cannabis is legalized in many states, it is important to know the difference between therapeutic cannabis dosing for medical purposes and the consequences of cannabis abuse. We urgently need additional research to understand these issues,” Dr. Patel said.
“It’s not proving that there’s a direct link, but it’s raising a suggestion in an observational analysis that [this] indeed might be the case. What that means for clinicians is that, if you’re seeing a patient who is presenting with a symptomatic arrhythmia, adding cannabis usage to your list of questions as you begin to try to understand possible precipitating factors for this arrhythmia seems to be a reasonable thing to do,” Dr. Harrington commented.
Stoners, beware: , and people with cannabis use disorder are at a 50% greater risk of being hospitalized for arrhythmias, according to new research presented at the American Heart Association Scientific Sessions 2019.
An analysis of pooled data on nearly 44,000 participants in a cross-sectional survey showed that, among the 13.6% who reported using marijuana within the last 30 days, the adjusted odds ratio for young-onset stroke (aged 18-44 years), compared with non-users, was 2.75, reported Tarang Parekh, MBBS, a health policy researcher of George Mason University in Fairfax, Va., and colleagues.
In a separate study, a retrospective analysis of national inpatient data showed that people diagnosed with cannabis use disorder – a pathological pattern of impaired control, social impairment, risky behavior or physiological adaptation similar in nature to alcoholism – had a 47%-52% increased likelihood of hospitalization for an arrhythmia, reported Rikinkumar S. Patel, MD, a psychiatry resident at Griffin Memorial Hospital in Norman, Okla.
“As these [cannabis] products become increasingly used across the country, getting clearer, scientifically rigorous data is going to be important as we try to understand the overall health effects of cannabis,” said AHA President Robert Harrington, MD, of Stanford (Calif.) University in a statement.
Currently, use of both medical and recreational marijuana is fully legal in 11 U.S. states and the District of Columbia. Medical marijuana is legal with recreational use decriminalized (or penalties reduced) in 28 other states, and totally illegal in 11 other states, according to employee screening firm DISA Global Solutions.
Stroke study
In an oral presentation with simultaneous publication in the AHA journal Stroke, Dr. Parekh and colleagues presented an analysis of pooled data from the Behavioral Risk Factor Surveillance System (BRFSS), a nationally representative cross-sectional survey collected by the Centers for Disease Control and Prevention in 2016 and 2017.
They looked at baseline sociodemographic data and created multivariable logistic regression models with state fixed effects to determine whether marijuana use within the last 30 days was associated with young-onset stroke.
They identified 43,860 participants representing a weighted sample of 35.5 million Americans. Of the sample, 63.3% were male, and 13.6 % of all participants reported using marijuana in the last 30 days.
They found in an unadjusted model that marijuana users had an odds ratio for stroke, compared with nonusers, of 1.59 (P less than.1), and in a model adjusted for demographic factors (gender, race, ethnicity, and education) the OR increased to 1.76 (P less than .05).
When they threw risk behavior into the model (physical activity, body mass index, heavy drinking, and cigarette smoking), they saw that the OR for stroke shot up to 2.75 (P less than .01).
“Physicians should ask patients if they use cannabis and counsel them about its potential stroke risk as part of regular doctor visits,” Dr. Parekh said in a statement.
Arrhythmias study
Based on recent studies suggesting that cannabis use may trigger cardiovascular events, Dr. Patel and colleagues studied whether cannabis use disorder may be related to arrhythmias, approaching the question through hospital records.
“The effects of using cannabis are seen within 15 minutes and last for around 3 hours. At lower doses, it is linked to a rapid heartbeat. At higher doses, it is linked to a too-slow heartbeat,” he said in a statement.
Dr. Patel and colleagues conducted a retrospective analysis of the Nationwide Inpatient Sample from 2010-2014, a period during which medical marijuana became legal in several states and recreational marijuana became legal in Colorado and Washington. The sample is a database maintained by the Healthcare Cost and Utilization Project of the U.S. Office of Disease Prevention and Health Promotion.
They identified 570,557 patients aged 15-54 years with a primary diagnosis of arrhythmia, and compared them with a sample of 67,662,082 patients hospitalized with no arrhythmia diagnosed during the same period.
They found a 2.6% incidence of cannabis use disorder among patients hospitalized for arrhythmias. Patients with cannabis use disorder tended to be younger (15- to 24-years-old; OR, 4.23), male (OR, 1.70) and African American (OR, 2.70).
In regression analysis adjusted for demographics and comorbidities, cannabis use disorder was associated with higher odds of arrhythmia hospitalization in young patients, at 1.28 times among 15- to 24-year-olds (95% confidence interval, 1.229-1.346) and 1.52 times for 25- to 34-year-olds (95% CI, 1.469-1.578).
“As medical and recreational cannabis is legalized in many states, it is important to know the difference between therapeutic cannabis dosing for medical purposes and the consequences of cannabis abuse. We urgently need additional research to understand these issues,” Dr. Patel said.
“It’s not proving that there’s a direct link, but it’s raising a suggestion in an observational analysis that [this] indeed might be the case. What that means for clinicians is that, if you’re seeing a patient who is presenting with a symptomatic arrhythmia, adding cannabis usage to your list of questions as you begin to try to understand possible precipitating factors for this arrhythmia seems to be a reasonable thing to do,” Dr. Harrington commented.
REPORTING FROM AHA 2019
Best practice alerts really can work
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
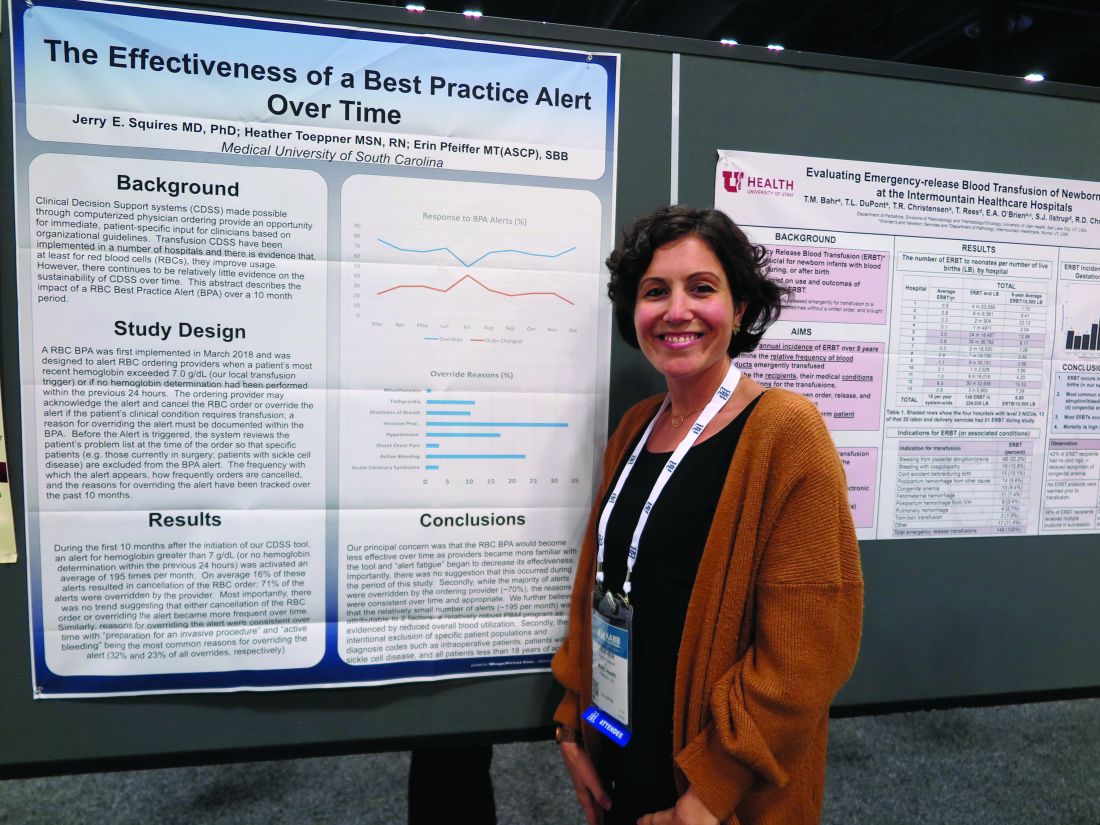
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
REPORTING FROM AABB 2019
First NCCN guideline on hematopoietic cell transplantation focuses on GVHD
Recommendations for the diagnosis and management of acute and chronic graft-versus-host disease (GVHD) are the central focus of the first National Comprehensive Cancer Network (NCCN) guideline on hematopoietic cell transplantation.
The guideline presents detailed recommendations for the evaluation of hematopoietic cell transplant (HCT) recipients, and an extensive section on the diagnosis and workup of GVHD, including information on staging and grading of acute GVHD, grading of chronic GVHD, treatment response criteria, and suggested systemic therapies for steroid-refractory disease.
“We wanted to both build on the commonly used approach to stage and treat graft-versus-host disease, and make sure that this information is readily available for physicians-in-training and young physicians who are learning about transplants,” said guideline committee chair Ayman Saad, MB BCh, of The Ohio State University Comprehensive Cancer Center, James Cancer Hospital and Solove Research Institute in Columbus, Ohio.
In an interview, Dr. Saad emphasized that an important goal of the guidelines is to encourage general oncologists to recognize early signs of GVHD and refer potential candidates to transplant centers for further evaluation.
“We also urge oncologists who may be caring for patients after HCT to familiarize themselves with the varied manifestations of GVHD – a very common and significant posttransplant complication – and to consult with transplant providers to optimize their ongoing care. The guidelines explain how to diagnose and treat this condition in order to achieve the best possible outcomes,” guideline panel member Alison W. Loren, MD, director of blood and marrow transplantation at Abraham Cancer Center, University of Pennsylvania, Philadelphia, said in a statement.
The guideline includes links to other NCCN guidelines for diseases where HCT is a common therapeutic option, including leukemias, myeloid malignancies, lymphomas, central nervous system cancers, and testicular cancer.
The HCT guideline includes:
- Pretransplant recipient evaluation with recommendations for clinical assessment and imaging.
- Diagnosis and workup of GVHD, with separate algorithms for suspected acute or chronic GVHD.
- Specific interventions for management of acute GVHD with corticosteroids or other systemic agents.
- Chronic GVHD diagnosis by organ site and symptoms, with a severity scoring system.
- Chronic GVHD steroid response definitions and criteria.
- Suggested systemic agents for steroid-refractory GVHD.
One feature that is unusual for an NCCN guideline document is a page of photographs to assist clinicians in diagnosing range-of-motion abnormalities in the shoulder, elbow, hand, and ankle of patients with suspected or confirmed GVHD. Dr. Saad said that future iterations of the guideline will include additional photos to help clinicians develop a visual repertoire of potential GVHD signs.
Future versions will also include a discussion section and more comprehensive information on other common complications following HCT transplant, as well as management of posttransplant relapse.
Ideally, the guideline will help clinicians document and justify clinical decisions surrounding HCT and GVHD management in discussions with third-party payers, Dr. Saad said.
“Sometimes we struggle with payers when we want to use a certain modality to treat GVHD, and they respond ‘that’s not approved,’ or ‘that’s not a common indication,’ et cetera,” he said. “What we’re trying to put here are the commonly used therapies that most, but not all, experts agree on, and we can use this to negotiate with payers.”
He also emphasized that the guideline is meant to be instructive rather than prescriptive and is not meant to hinder innovations that may emerge from clinical trials.
“We would appreciate any feedback from non-NCCN centers as well as experts at NCCN centers, and we’ll be more than happy to address any concerns or criticisms they have,” he said.
Dr. Saad reported financial relationships with Actinium and Incysus Biomedical. Dr. Loren has previously reported having no disclosures.
Recommendations for the diagnosis and management of acute and chronic graft-versus-host disease (GVHD) are the central focus of the first National Comprehensive Cancer Network (NCCN) guideline on hematopoietic cell transplantation.
The guideline presents detailed recommendations for the evaluation of hematopoietic cell transplant (HCT) recipients, and an extensive section on the diagnosis and workup of GVHD, including information on staging and grading of acute GVHD, grading of chronic GVHD, treatment response criteria, and suggested systemic therapies for steroid-refractory disease.
“We wanted to both build on the commonly used approach to stage and treat graft-versus-host disease, and make sure that this information is readily available for physicians-in-training and young physicians who are learning about transplants,” said guideline committee chair Ayman Saad, MB BCh, of The Ohio State University Comprehensive Cancer Center, James Cancer Hospital and Solove Research Institute in Columbus, Ohio.
In an interview, Dr. Saad emphasized that an important goal of the guidelines is to encourage general oncologists to recognize early signs of GVHD and refer potential candidates to transplant centers for further evaluation.
“We also urge oncologists who may be caring for patients after HCT to familiarize themselves with the varied manifestations of GVHD – a very common and significant posttransplant complication – and to consult with transplant providers to optimize their ongoing care. The guidelines explain how to diagnose and treat this condition in order to achieve the best possible outcomes,” guideline panel member Alison W. Loren, MD, director of blood and marrow transplantation at Abraham Cancer Center, University of Pennsylvania, Philadelphia, said in a statement.
The guideline includes links to other NCCN guidelines for diseases where HCT is a common therapeutic option, including leukemias, myeloid malignancies, lymphomas, central nervous system cancers, and testicular cancer.
The HCT guideline includes:
- Pretransplant recipient evaluation with recommendations for clinical assessment and imaging.
- Diagnosis and workup of GVHD, with separate algorithms for suspected acute or chronic GVHD.
- Specific interventions for management of acute GVHD with corticosteroids or other systemic agents.
- Chronic GVHD diagnosis by organ site and symptoms, with a severity scoring system.
- Chronic GVHD steroid response definitions and criteria.
- Suggested systemic agents for steroid-refractory GVHD.
One feature that is unusual for an NCCN guideline document is a page of photographs to assist clinicians in diagnosing range-of-motion abnormalities in the shoulder, elbow, hand, and ankle of patients with suspected or confirmed GVHD. Dr. Saad said that future iterations of the guideline will include additional photos to help clinicians develop a visual repertoire of potential GVHD signs.
Future versions will also include a discussion section and more comprehensive information on other common complications following HCT transplant, as well as management of posttransplant relapse.
Ideally, the guideline will help clinicians document and justify clinical decisions surrounding HCT and GVHD management in discussions with third-party payers, Dr. Saad said.
“Sometimes we struggle with payers when we want to use a certain modality to treat GVHD, and they respond ‘that’s not approved,’ or ‘that’s not a common indication,’ et cetera,” he said. “What we’re trying to put here are the commonly used therapies that most, but not all, experts agree on, and we can use this to negotiate with payers.”
He also emphasized that the guideline is meant to be instructive rather than prescriptive and is not meant to hinder innovations that may emerge from clinical trials.
“We would appreciate any feedback from non-NCCN centers as well as experts at NCCN centers, and we’ll be more than happy to address any concerns or criticisms they have,” he said.
Dr. Saad reported financial relationships with Actinium and Incysus Biomedical. Dr. Loren has previously reported having no disclosures.
Recommendations for the diagnosis and management of acute and chronic graft-versus-host disease (GVHD) are the central focus of the first National Comprehensive Cancer Network (NCCN) guideline on hematopoietic cell transplantation.
The guideline presents detailed recommendations for the evaluation of hematopoietic cell transplant (HCT) recipients, and an extensive section on the diagnosis and workup of GVHD, including information on staging and grading of acute GVHD, grading of chronic GVHD, treatment response criteria, and suggested systemic therapies for steroid-refractory disease.
“We wanted to both build on the commonly used approach to stage and treat graft-versus-host disease, and make sure that this information is readily available for physicians-in-training and young physicians who are learning about transplants,” said guideline committee chair Ayman Saad, MB BCh, of The Ohio State University Comprehensive Cancer Center, James Cancer Hospital and Solove Research Institute in Columbus, Ohio.
In an interview, Dr. Saad emphasized that an important goal of the guidelines is to encourage general oncologists to recognize early signs of GVHD and refer potential candidates to transplant centers for further evaluation.
“We also urge oncologists who may be caring for patients after HCT to familiarize themselves with the varied manifestations of GVHD – a very common and significant posttransplant complication – and to consult with transplant providers to optimize their ongoing care. The guidelines explain how to diagnose and treat this condition in order to achieve the best possible outcomes,” guideline panel member Alison W. Loren, MD, director of blood and marrow transplantation at Abraham Cancer Center, University of Pennsylvania, Philadelphia, said in a statement.
The guideline includes links to other NCCN guidelines for diseases where HCT is a common therapeutic option, including leukemias, myeloid malignancies, lymphomas, central nervous system cancers, and testicular cancer.
The HCT guideline includes:
- Pretransplant recipient evaluation with recommendations for clinical assessment and imaging.
- Diagnosis and workup of GVHD, with separate algorithms for suspected acute or chronic GVHD.
- Specific interventions for management of acute GVHD with corticosteroids or other systemic agents.
- Chronic GVHD diagnosis by organ site and symptoms, with a severity scoring system.
- Chronic GVHD steroid response definitions and criteria.
- Suggested systemic agents for steroid-refractory GVHD.
One feature that is unusual for an NCCN guideline document is a page of photographs to assist clinicians in diagnosing range-of-motion abnormalities in the shoulder, elbow, hand, and ankle of patients with suspected or confirmed GVHD. Dr. Saad said that future iterations of the guideline will include additional photos to help clinicians develop a visual repertoire of potential GVHD signs.
Future versions will also include a discussion section and more comprehensive information on other common complications following HCT transplant, as well as management of posttransplant relapse.
Ideally, the guideline will help clinicians document and justify clinical decisions surrounding HCT and GVHD management in discussions with third-party payers, Dr. Saad said.
“Sometimes we struggle with payers when we want to use a certain modality to treat GVHD, and they respond ‘that’s not approved,’ or ‘that’s not a common indication,’ et cetera,” he said. “What we’re trying to put here are the commonly used therapies that most, but not all, experts agree on, and we can use this to negotiate with payers.”
He also emphasized that the guideline is meant to be instructive rather than prescriptive and is not meant to hinder innovations that may emerge from clinical trials.
“We would appreciate any feedback from non-NCCN centers as well as experts at NCCN centers, and we’ll be more than happy to address any concerns or criticisms they have,” he said.
Dr. Saad reported financial relationships with Actinium and Incysus Biomedical. Dr. Loren has previously reported having no disclosures.
Trifluridine/tipiracil effective in metastatic gastric cancer after gastrectomy
Treatment with trifluridine/tipiracil was safe and showed efficacy in patients with metastatic gastric cancer or gastroesophageal junction cancer regardless of their gastrectomy status, results of a preplanned analysis from the phase 3 randomized TAGS trial showed.
Trifluridine/tipiracil (FTD/TPI) was associated with significantly better overall survival (OS) and progression-free survival (PFS) than placebo in patients who had undergone gastrectomy, reported David H. Ilson, MD, PhD, of Memorial Sloan Kettering Cancer Center, New York, and colleagues.
“The benefits of FTD/TPI were especially noteworthy in the subpopulation of patients who had undergone gastrectomy, who tended to be more heavily pretreated and were less tolerant of therapy. The overall safety profile of the drug, including the incidence of severe AEs [adverse events] was similar among patients who had or had not undergone gastrectomy. No new safety concerns were reported in patients who had undergone gastrectomy,” they wrote in JAMA Oncology.
FTD/TPI is an oral drug combining the thymidine analogue trifluridine and tipiracil, an inhibitor of trifluridine degradation. The drug was approved by the FDA in 2015 under the trade name Lonsurf for the treatment of refractory metastatic colorectal cancer, and in 2019 for patients with metastatic gastric cancer or gastroesophageal junction cancer (mGC/GEJC) that had been treated with at least two lines of chemotherapy.
In a phase 2 study conducted in Japan with 29 patients with metastatic gastric cancer that had progressed after chemotherapy with fluoropyrimidine, platinum, taxanes, or irinotecan, FTD/TPI was associated with a median overall survival of 8.7 months and an investigator-assessed disease control rate of 65.5%,
As previously reported, in the randomized, controlled TAGS (TAS-102 Gastric Study), median overall survival, the primary endpoint, was 5.7 months for patients assigned to receive trifluridine/tipiracil, compared with 3.6 months for patients randomized to placebo.
The current study is a preplanned subgroup analysis from the TAGS trial. Of 507 randomized patients, 221 had undergone gastrectomy, and of this group 147 were randomized to FTD/TPI and 74 to placebo. The remaining 286 patients had not undergone gastrectomy, and of this group 190 were randomized to FTD/TPI and 96 to placebo.
Among patients who had undergone gastrectomy, the hazard ratio for OS of patients treated with FTD/TPI vs. placebo was 0.57 (95% confidence interval, 0.41-0.79), and the HR for PFS was 0.48 (95% CI, 0.35-0.65).
In the no-gastrectomy subgroup, the overall survival HR for patients who received FTD/TPI vs. placebo was 0.80 (95% CI , 0.60-1.06), and the HR for PFS was 0.65 (95% CI, 0.49-0.85).
Grade 3 or greater adverse events with FTD/TPI occurred in 84.1% of gastrectomy patients and 76.3% of no-gastrectomy patients. Grade 3 or greater neutropenia occurred in 44.1% and 26.3%, respectively; grade 3 or greater anemia in 21.4% vs. 17.4%; and grade 3 or greater leukopenia was observed in 14.5% vs. 5.3%.
Dose modifications because of adverse events were required for 64.8% of patients who had undergone gastrectomy, and in 53.2% of those who had not.
Treatment discontinuations because of AEs occurred in 10.3% and 14.7%, respectively.
The study was funded by Taiho Oncology and Taiho Pharmaceutical Company. Dr. Islon reported grants and advisory role support from Taiho and others. Multiple coauthors had similar disclosures.
SOURCE: Ilson DH et al. JAMA Oncol. 2019 Oct 10. doi: 10.1001/jamaoncol.2019.3531.
Treatment with trifluridine/tipiracil was safe and showed efficacy in patients with metastatic gastric cancer or gastroesophageal junction cancer regardless of their gastrectomy status, results of a preplanned analysis from the phase 3 randomized TAGS trial showed.
Trifluridine/tipiracil (FTD/TPI) was associated with significantly better overall survival (OS) and progression-free survival (PFS) than placebo in patients who had undergone gastrectomy, reported David H. Ilson, MD, PhD, of Memorial Sloan Kettering Cancer Center, New York, and colleagues.
“The benefits of FTD/TPI were especially noteworthy in the subpopulation of patients who had undergone gastrectomy, who tended to be more heavily pretreated and were less tolerant of therapy. The overall safety profile of the drug, including the incidence of severe AEs [adverse events] was similar among patients who had or had not undergone gastrectomy. No new safety concerns were reported in patients who had undergone gastrectomy,” they wrote in JAMA Oncology.
FTD/TPI is an oral drug combining the thymidine analogue trifluridine and tipiracil, an inhibitor of trifluridine degradation. The drug was approved by the FDA in 2015 under the trade name Lonsurf for the treatment of refractory metastatic colorectal cancer, and in 2019 for patients with metastatic gastric cancer or gastroesophageal junction cancer (mGC/GEJC) that had been treated with at least two lines of chemotherapy.
In a phase 2 study conducted in Japan with 29 patients with metastatic gastric cancer that had progressed after chemotherapy with fluoropyrimidine, platinum, taxanes, or irinotecan, FTD/TPI was associated with a median overall survival of 8.7 months and an investigator-assessed disease control rate of 65.5%,
As previously reported, in the randomized, controlled TAGS (TAS-102 Gastric Study), median overall survival, the primary endpoint, was 5.7 months for patients assigned to receive trifluridine/tipiracil, compared with 3.6 months for patients randomized to placebo.
The current study is a preplanned subgroup analysis from the TAGS trial. Of 507 randomized patients, 221 had undergone gastrectomy, and of this group 147 were randomized to FTD/TPI and 74 to placebo. The remaining 286 patients had not undergone gastrectomy, and of this group 190 were randomized to FTD/TPI and 96 to placebo.
Among patients who had undergone gastrectomy, the hazard ratio for OS of patients treated with FTD/TPI vs. placebo was 0.57 (95% confidence interval, 0.41-0.79), and the HR for PFS was 0.48 (95% CI, 0.35-0.65).
In the no-gastrectomy subgroup, the overall survival HR for patients who received FTD/TPI vs. placebo was 0.80 (95% CI , 0.60-1.06), and the HR for PFS was 0.65 (95% CI, 0.49-0.85).
Grade 3 or greater adverse events with FTD/TPI occurred in 84.1% of gastrectomy patients and 76.3% of no-gastrectomy patients. Grade 3 or greater neutropenia occurred in 44.1% and 26.3%, respectively; grade 3 or greater anemia in 21.4% vs. 17.4%; and grade 3 or greater leukopenia was observed in 14.5% vs. 5.3%.
Dose modifications because of adverse events were required for 64.8% of patients who had undergone gastrectomy, and in 53.2% of those who had not.
Treatment discontinuations because of AEs occurred in 10.3% and 14.7%, respectively.
The study was funded by Taiho Oncology and Taiho Pharmaceutical Company. Dr. Islon reported grants and advisory role support from Taiho and others. Multiple coauthors had similar disclosures.
SOURCE: Ilson DH et al. JAMA Oncol. 2019 Oct 10. doi: 10.1001/jamaoncol.2019.3531.
Treatment with trifluridine/tipiracil was safe and showed efficacy in patients with metastatic gastric cancer or gastroesophageal junction cancer regardless of their gastrectomy status, results of a preplanned analysis from the phase 3 randomized TAGS trial showed.
Trifluridine/tipiracil (FTD/TPI) was associated with significantly better overall survival (OS) and progression-free survival (PFS) than placebo in patients who had undergone gastrectomy, reported David H. Ilson, MD, PhD, of Memorial Sloan Kettering Cancer Center, New York, and colleagues.
“The benefits of FTD/TPI were especially noteworthy in the subpopulation of patients who had undergone gastrectomy, who tended to be more heavily pretreated and were less tolerant of therapy. The overall safety profile of the drug, including the incidence of severe AEs [adverse events] was similar among patients who had or had not undergone gastrectomy. No new safety concerns were reported in patients who had undergone gastrectomy,” they wrote in JAMA Oncology.
FTD/TPI is an oral drug combining the thymidine analogue trifluridine and tipiracil, an inhibitor of trifluridine degradation. The drug was approved by the FDA in 2015 under the trade name Lonsurf for the treatment of refractory metastatic colorectal cancer, and in 2019 for patients with metastatic gastric cancer or gastroesophageal junction cancer (mGC/GEJC) that had been treated with at least two lines of chemotherapy.
In a phase 2 study conducted in Japan with 29 patients with metastatic gastric cancer that had progressed after chemotherapy with fluoropyrimidine, platinum, taxanes, or irinotecan, FTD/TPI was associated with a median overall survival of 8.7 months and an investigator-assessed disease control rate of 65.5%,
As previously reported, in the randomized, controlled TAGS (TAS-102 Gastric Study), median overall survival, the primary endpoint, was 5.7 months for patients assigned to receive trifluridine/tipiracil, compared with 3.6 months for patients randomized to placebo.
The current study is a preplanned subgroup analysis from the TAGS trial. Of 507 randomized patients, 221 had undergone gastrectomy, and of this group 147 were randomized to FTD/TPI and 74 to placebo. The remaining 286 patients had not undergone gastrectomy, and of this group 190 were randomized to FTD/TPI and 96 to placebo.
Among patients who had undergone gastrectomy, the hazard ratio for OS of patients treated with FTD/TPI vs. placebo was 0.57 (95% confidence interval, 0.41-0.79), and the HR for PFS was 0.48 (95% CI, 0.35-0.65).
In the no-gastrectomy subgroup, the overall survival HR for patients who received FTD/TPI vs. placebo was 0.80 (95% CI , 0.60-1.06), and the HR for PFS was 0.65 (95% CI, 0.49-0.85).
Grade 3 or greater adverse events with FTD/TPI occurred in 84.1% of gastrectomy patients and 76.3% of no-gastrectomy patients. Grade 3 or greater neutropenia occurred in 44.1% and 26.3%, respectively; grade 3 or greater anemia in 21.4% vs. 17.4%; and grade 3 or greater leukopenia was observed in 14.5% vs. 5.3%.
Dose modifications because of adverse events were required for 64.8% of patients who had undergone gastrectomy, and in 53.2% of those who had not.
Treatment discontinuations because of AEs occurred in 10.3% and 14.7%, respectively.
The study was funded by Taiho Oncology and Taiho Pharmaceutical Company. Dr. Islon reported grants and advisory role support from Taiho and others. Multiple coauthors had similar disclosures.
SOURCE: Ilson DH et al. JAMA Oncol. 2019 Oct 10. doi: 10.1001/jamaoncol.2019.3531.
FROM JAMA ONCOLOGY
Fresh RBCs offer no benefit over older cells in pediatric ICU
SAN ANTONIO – Fresh red cells were no better than conventional stored red cells when transfused into critically ill children, and there was some evidence in the ABC PICU trial suggesting that fresh red cells could be associated with a higher incidence of posttransfusion organ dysfunction.

Among 1,461 children randomly assigned to receive RBC transfusions with either fresh cells (stored for 7 days or less) or standard-issue cells (stored anywhere from 2-42 days), there were no differences in the primary endpoint of new or progressive multiple organ dysfunction syndrome (NPMODS), reported Philip Spinella, MD from Washington University, St. Louis.
“Our results do not support current blood management policies that recommend providing fresh red cell units to certain populations of children,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The study findings support those of a systematic review (Transfus Med Rev. 2018;32:77-88), whose authors found that “transfusion of fresher RBCs is not associated with decreased risk of death but is associated with higher rates of transfusion reactions and possibly infection.” The authors of the review concluded that “the current evidence does not support a change from current usual transfusion practice.”
Is fresh really better?
The launch of the ABC PICU trial was motivated by laboratory and observational evidence suggesting that older RBCs may be less safe or efficacious than fresh RBCs, especially in vulnerable populations such as critically ill children.
Although physician and institutional practice has been to transfuse fresh RBCs to some pediatric patients, the standard practice among blood banks has been to deliver the oldest stored units first, in an effort to prevent product wastage.
Dr. Spinella and colleagues across 50 centers in the United States, Canada, France, Italy, and Israel enrolled patients who were admitted to a pediatric ICU who received their first RBC transfusion within 7 days of admission and had an expected length of stay after transfusion of more than 24 hours.
The median patient age was 1.8 years for those who received fresh cells, and 1.9 years for those who received usual care.
There were 1,630 transfusions of fresh RBCs stored for a median of 5 days and 1,533 transfusions of standard RBCs stored for a median of 18 days. The median volume of red cell units transfused was 17.5 mL/kg in the fresh group and 16.6 mL/kg in the standard group.
The incidence of NPMODS was 20.2% for fresh-RBC recipients and 18.2% for standard-product recipients. The absolute difference of 2.0% was not statistically significant.
There were also no significant differences in the timing of NPMODS occurrence between the groups, and no significant differences by patient age (28 days or younger, 29-365 days old, or older than 1 year).
Similarly, there were no differences in NPMODS incidence between the groups by country, although in Canada there was a trend toward a higher incidence of organ dysfunction in the group that received fresh RBCs, Dr. Spinella noted.
Additionally, there were no significant differences between the groups by admission to the ICU by medical, surgical, cardiac, or trauma services; no differences by quartile of red cell volume transfused; and no differences in mortality rates either in the ICU or the main hospital, or at 28 or 90 days after discharge.
Why no difference?
Seeking explanations for why fresh RBCs did not perform better than older stored cells, Dr. Spinella suggested that changes such as storage lesions that occur over time may not be as clinically relevant as previously supposed.
“Another possibility is that these study patients didn’t need red cells to begin with to improve oxygen delivery,” he said.
Other potential explanations include the possibility that exposure to fresh red cells may be associated with immune suppression because viable white cells may also be present in the product, and that the chronological age of a stored red cell unit may not equate to its biologic or metabolic age or performance, he added.
ABC PICU was supported by Washington University; the National Heart, Lung, and Blood Institute; the Canadian and French governments; and other groups. Dr. Spinella reported having no relevant conflicts of interest.
SAN ANTONIO – Fresh red cells were no better than conventional stored red cells when transfused into critically ill children, and there was some evidence in the ABC PICU trial suggesting that fresh red cells could be associated with a higher incidence of posttransfusion organ dysfunction.
Among 1,461 children randomly assigned to receive RBC transfusions with either fresh cells (stored for 7 days or less) or standard-issue cells (stored anywhere from 2-42 days), there were no differences in the primary endpoint of new or progressive multiple organ dysfunction syndrome (NPMODS), reported Philip Spinella, MD from Washington University, St. Louis.
“Our results do not support current blood management policies that recommend providing fresh red cell units to certain populations of children,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The study findings support those of a systematic review (Transfus Med Rev. 2018;32:77-88), whose authors found that “transfusion of fresher RBCs is not associated with decreased risk of death but is associated with higher rates of transfusion reactions and possibly infection.” The authors of the review concluded that “the current evidence does not support a change from current usual transfusion practice.”
Is fresh really better?
The launch of the ABC PICU trial was motivated by laboratory and observational evidence suggesting that older RBCs may be less safe or efficacious than fresh RBCs, especially in vulnerable populations such as critically ill children.
Although physician and institutional practice has been to transfuse fresh RBCs to some pediatric patients, the standard practice among blood banks has been to deliver the oldest stored units first, in an effort to prevent product wastage.
Dr. Spinella and colleagues across 50 centers in the United States, Canada, France, Italy, and Israel enrolled patients who were admitted to a pediatric ICU who received their first RBC transfusion within 7 days of admission and had an expected length of stay after transfusion of more than 24 hours.
The median patient age was 1.8 years for those who received fresh cells, and 1.9 years for those who received usual care.
There were 1,630 transfusions of fresh RBCs stored for a median of 5 days and 1,533 transfusions of standard RBCs stored for a median of 18 days. The median volume of red cell units transfused was 17.5 mL/kg in the fresh group and 16.6 mL/kg in the standard group.
The incidence of NPMODS was 20.2% for fresh-RBC recipients and 18.2% for standard-product recipients. The absolute difference of 2.0% was not statistically significant.
There were also no significant differences in the timing of NPMODS occurrence between the groups, and no significant differences by patient age (28 days or younger, 29-365 days old, or older than 1 year).
Similarly, there were no differences in NPMODS incidence between the groups by country, although in Canada there was a trend toward a higher incidence of organ dysfunction in the group that received fresh RBCs, Dr. Spinella noted.
Additionally, there were no significant differences between the groups by admission to the ICU by medical, surgical, cardiac, or trauma services; no differences by quartile of red cell volume transfused; and no differences in mortality rates either in the ICU or the main hospital, or at 28 or 90 days after discharge.
Why no difference?
Seeking explanations for why fresh RBCs did not perform better than older stored cells, Dr. Spinella suggested that changes such as storage lesions that occur over time may not be as clinically relevant as previously supposed.
“Another possibility is that these study patients didn’t need red cells to begin with to improve oxygen delivery,” he said.
Other potential explanations include the possibility that exposure to fresh red cells may be associated with immune suppression because viable white cells may also be present in the product, and that the chronological age of a stored red cell unit may not equate to its biologic or metabolic age or performance, he added.
ABC PICU was supported by Washington University; the National Heart, Lung, and Blood Institute; the Canadian and French governments; and other groups. Dr. Spinella reported having no relevant conflicts of interest.
SAN ANTONIO – Fresh red cells were no better than conventional stored red cells when transfused into critically ill children, and there was some evidence in the ABC PICU trial suggesting that fresh red cells could be associated with a higher incidence of posttransfusion organ dysfunction.
Among 1,461 children randomly assigned to receive RBC transfusions with either fresh cells (stored for 7 days or less) or standard-issue cells (stored anywhere from 2-42 days), there were no differences in the primary endpoint of new or progressive multiple organ dysfunction syndrome (NPMODS), reported Philip Spinella, MD from Washington University, St. Louis.
“Our results do not support current blood management policies that recommend providing fresh red cell units to certain populations of children,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The study findings support those of a systematic review (Transfus Med Rev. 2018;32:77-88), whose authors found that “transfusion of fresher RBCs is not associated with decreased risk of death but is associated with higher rates of transfusion reactions and possibly infection.” The authors of the review concluded that “the current evidence does not support a change from current usual transfusion practice.”
Is fresh really better?
The launch of the ABC PICU trial was motivated by laboratory and observational evidence suggesting that older RBCs may be less safe or efficacious than fresh RBCs, especially in vulnerable populations such as critically ill children.
Although physician and institutional practice has been to transfuse fresh RBCs to some pediatric patients, the standard practice among blood banks has been to deliver the oldest stored units first, in an effort to prevent product wastage.
Dr. Spinella and colleagues across 50 centers in the United States, Canada, France, Italy, and Israel enrolled patients who were admitted to a pediatric ICU who received their first RBC transfusion within 7 days of admission and had an expected length of stay after transfusion of more than 24 hours.
The median patient age was 1.8 years for those who received fresh cells, and 1.9 years for those who received usual care.
There were 1,630 transfusions of fresh RBCs stored for a median of 5 days and 1,533 transfusions of standard RBCs stored for a median of 18 days. The median volume of red cell units transfused was 17.5 mL/kg in the fresh group and 16.6 mL/kg in the standard group.
The incidence of NPMODS was 20.2% for fresh-RBC recipients and 18.2% for standard-product recipients. The absolute difference of 2.0% was not statistically significant.
There were also no significant differences in the timing of NPMODS occurrence between the groups, and no significant differences by patient age (28 days or younger, 29-365 days old, or older than 1 year).
Similarly, there were no differences in NPMODS incidence between the groups by country, although in Canada there was a trend toward a higher incidence of organ dysfunction in the group that received fresh RBCs, Dr. Spinella noted.
Additionally, there were no significant differences between the groups by admission to the ICU by medical, surgical, cardiac, or trauma services; no differences by quartile of red cell volume transfused; and no differences in mortality rates either in the ICU or the main hospital, or at 28 or 90 days after discharge.
Why no difference?
Seeking explanations for why fresh RBCs did not perform better than older stored cells, Dr. Spinella suggested that changes such as storage lesions that occur over time may not be as clinically relevant as previously supposed.
“Another possibility is that these study patients didn’t need red cells to begin with to improve oxygen delivery,” he said.
Other potential explanations include the possibility that exposure to fresh red cells may be associated with immune suppression because viable white cells may also be present in the product, and that the chronological age of a stored red cell unit may not equate to its biologic or metabolic age or performance, he added.
ABC PICU was supported by Washington University; the National Heart, Lung, and Blood Institute; the Canadian and French governments; and other groups. Dr. Spinella reported having no relevant conflicts of interest.
REPORTING FROM AABB 2019
ART traces in donor samples show risk to blood supply
SAN ANTONIO, TEX. – Some HIV-positive individuals on antiretroviral therapy continue to attempt blood donation, for reasons that may include ignorance of the risks of transmission, test-seeking behavior, or other unknown motivations to donate despite knowing their viral status, caution investigators who monitor the nation’s blood supply.

Additionally, evidence of pre-exposure prophylaxis (PrEP) with antiretroviral agents (ARV) in HIV-negative donors raises concerns about possible risk of transmission from HIV-breakthrough infections, reported Brian S. Custer, PhD, of Vitalant Research Institute.
Dr. Custer and colleagues in the U.S. Transfusion Transmissible Infections Monitoring System (TTIMS) evaluated plasma samples from four large blood collection organizations and found metabolites of drugs typically used in ARV regimens, at concentrations indicating that they had been taken within a week of blood donation.
“What are the motivations of these individuals who are ARV-positive and HIV-positive? Are they using the blood center as a place to monitor their infection status? We just do not know the answer to that, but it does bring back this issue of whether there a form of test seeking going on here,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The findings suggest that current donor health questionnaires and pre-donation screening are not adequate for ascertaining disclosure of HIV status or behaviors that could put the safety of the blood supply in doubt.
Does ‘U’ really equal ‘U’?
The public health message that “undetectable” equals “untransmittable” may help to prevent new infections, but may not translate into transfusion safety, Dr. Custer said.
Current HIV treatment guidelines recommend that infected individuals start on ARV immediately upon receiving a diagnosis. ARV drugs both suppress viremia and alter biomarkers of HIV progression, and may delay the time to antibody seroconversion, which could affect the ability to detect HIV through blood donation screening.
To see whether there is renewed cause for concern about safety of the blood supply, the TTIMS investigators tested for ARV metabolites in donated blood samples as a surrogate for HIV infection.
They looked at 299 HIV-positive plasma samples and 300 samples negative for all testable infections using liquid chromatography–mass spectometry for metabolites of 13 ARVs.
No ARV metabolites could be detected in the HIV-negative samples, but in 299 samples from 463 HIV-positive donors they found that 46 (15.4%) tested positive for ARVs. Of these 46 specimens, 43 were from first-time donors, and 34 were from males. Three samples were from repeat donors.
In all, 41 of the 46 ARV-positive donors would have been considered as potential “elite controllers” – HIV-infected individuals with no detectable viral loads in the absence of therapy – based on HIV-positive serology but negative nucleic acid test (NAT) results.
Samples from five first-time donors tested positive for HIV by both serology and NAT, and also contained ARV metabolites, suggesting that the donors had started ARVs recently, had suboptimal therapy, or were poorly adherent to their regimens.
PrEP use among HIV-negative donors
The investigators also looked at PrEP use in 1494 samples from first-time male donors that tested negative for all markers routinely screened: HIV 1/2, hepatitis B virus, hepatitis C virus, HTLV-I/II, West Nile virus, ZIKV, Treponema pallidum and Trypanosoma cruzi.
The donors came from Boston, Los Angeles, Miami, New York, San Francisco, and Washington, all cities with elevated HIV prevalence and active PrEP rollout campaigns.
They tested for analytes to emtricitabine and tenofovir, the two ARV components of the PrEP drug Truvada, and found that nine samples (0.6%) tested positive for PrEP. Of these, three donors had taken PrEP approximately 1 day before donation, two took it 2 days before, and four took it about 4 days before donation.
“There is quite a bit of concern about what this might mean for blood safety.” Dr. Custer said. “There’s a possibility of a breakthrough infection if someone is not PrEP adherent, and would we be able to detect them? Could this be an additional indicator for the risk of transfusion-transmissible infections? Could we identify subgroups of PrEP use in the donor population, and would we see any evidence?”
Mitigation Efforts
The National Heart, Lung, and Blood Institute is supporting a 7-year research project to study the potential impact of antiretroviral therapy and PrEP in the United States and Brazil under the Recipient Epidemiology and Donor Evaluation Study-IV-Pediatric (REDS-IV-P) project.
In addition, an AABB task force has drafted new questions for the donor health questionnaire intended to focus the attention of potential donors on PrEP, postexposure prophylaxis (PEP), and antiretroviral therapy.
In addition to asking about the use of medications on the deferral list, the proposed additions would ask, “In the past 16 weeks, have you taken any medication to prevent an HIV infection?” and “Have you EVER taken any medication to treat an HIV infection?”
TTIMS is supported by the U.S. Food and Drug Administration, NHLBI, and the Office of the Assistant Secretary for Health. Dr. Custer and all coinvestigators reported having no relevant disclosures.
SOURCE: : wit AABB 2019. Abstract PL5-MN4-32
SAN ANTONIO, TEX. – Some HIV-positive individuals on antiretroviral therapy continue to attempt blood donation, for reasons that may include ignorance of the risks of transmission, test-seeking behavior, or other unknown motivations to donate despite knowing their viral status, caution investigators who monitor the nation’s blood supply.
Additionally, evidence of pre-exposure prophylaxis (PrEP) with antiretroviral agents (ARV) in HIV-negative donors raises concerns about possible risk of transmission from HIV-breakthrough infections, reported Brian S. Custer, PhD, of Vitalant Research Institute.
Dr. Custer and colleagues in the U.S. Transfusion Transmissible Infections Monitoring System (TTIMS) evaluated plasma samples from four large blood collection organizations and found metabolites of drugs typically used in ARV regimens, at concentrations indicating that they had been taken within a week of blood donation.
“What are the motivations of these individuals who are ARV-positive and HIV-positive? Are they using the blood center as a place to monitor their infection status? We just do not know the answer to that, but it does bring back this issue of whether there a form of test seeking going on here,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The findings suggest that current donor health questionnaires and pre-donation screening are not adequate for ascertaining disclosure of HIV status or behaviors that could put the safety of the blood supply in doubt.
Does ‘U’ really equal ‘U’?
The public health message that “undetectable” equals “untransmittable” may help to prevent new infections, but may not translate into transfusion safety, Dr. Custer said.
Current HIV treatment guidelines recommend that infected individuals start on ARV immediately upon receiving a diagnosis. ARV drugs both suppress viremia and alter biomarkers of HIV progression, and may delay the time to antibody seroconversion, which could affect the ability to detect HIV through blood donation screening.
To see whether there is renewed cause for concern about safety of the blood supply, the TTIMS investigators tested for ARV metabolites in donated blood samples as a surrogate for HIV infection.
They looked at 299 HIV-positive plasma samples and 300 samples negative for all testable infections using liquid chromatography–mass spectometry for metabolites of 13 ARVs.
No ARV metabolites could be detected in the HIV-negative samples, but in 299 samples from 463 HIV-positive donors they found that 46 (15.4%) tested positive for ARVs. Of these 46 specimens, 43 were from first-time donors, and 34 were from males. Three samples were from repeat donors.
In all, 41 of the 46 ARV-positive donors would have been considered as potential “elite controllers” – HIV-infected individuals with no detectable viral loads in the absence of therapy – based on HIV-positive serology but negative nucleic acid test (NAT) results.
Samples from five first-time donors tested positive for HIV by both serology and NAT, and also contained ARV metabolites, suggesting that the donors had started ARVs recently, had suboptimal therapy, or were poorly adherent to their regimens.
PrEP use among HIV-negative donors
The investigators also looked at PrEP use in 1494 samples from first-time male donors that tested negative for all markers routinely screened: HIV 1/2, hepatitis B virus, hepatitis C virus, HTLV-I/II, West Nile virus, ZIKV, Treponema pallidum and Trypanosoma cruzi.
The donors came from Boston, Los Angeles, Miami, New York, San Francisco, and Washington, all cities with elevated HIV prevalence and active PrEP rollout campaigns.
They tested for analytes to emtricitabine and tenofovir, the two ARV components of the PrEP drug Truvada, and found that nine samples (0.6%) tested positive for PrEP. Of these, three donors had taken PrEP approximately 1 day before donation, two took it 2 days before, and four took it about 4 days before donation.
“There is quite a bit of concern about what this might mean for blood safety.” Dr. Custer said. “There’s a possibility of a breakthrough infection if someone is not PrEP adherent, and would we be able to detect them? Could this be an additional indicator for the risk of transfusion-transmissible infections? Could we identify subgroups of PrEP use in the donor population, and would we see any evidence?”
Mitigation Efforts
The National Heart, Lung, and Blood Institute is supporting a 7-year research project to study the potential impact of antiretroviral therapy and PrEP in the United States and Brazil under the Recipient Epidemiology and Donor Evaluation Study-IV-Pediatric (REDS-IV-P) project.
In addition, an AABB task force has drafted new questions for the donor health questionnaire intended to focus the attention of potential donors on PrEP, postexposure prophylaxis (PEP), and antiretroviral therapy.
In addition to asking about the use of medications on the deferral list, the proposed additions would ask, “In the past 16 weeks, have you taken any medication to prevent an HIV infection?” and “Have you EVER taken any medication to treat an HIV infection?”
TTIMS is supported by the U.S. Food and Drug Administration, NHLBI, and the Office of the Assistant Secretary for Health. Dr. Custer and all coinvestigators reported having no relevant disclosures.
SOURCE: : wit AABB 2019. Abstract PL5-MN4-32
SAN ANTONIO, TEX. – Some HIV-positive individuals on antiretroviral therapy continue to attempt blood donation, for reasons that may include ignorance of the risks of transmission, test-seeking behavior, or other unknown motivations to donate despite knowing their viral status, caution investigators who monitor the nation’s blood supply.
Additionally, evidence of pre-exposure prophylaxis (PrEP) with antiretroviral agents (ARV) in HIV-negative donors raises concerns about possible risk of transmission from HIV-breakthrough infections, reported Brian S. Custer, PhD, of Vitalant Research Institute.
Dr. Custer and colleagues in the U.S. Transfusion Transmissible Infections Monitoring System (TTIMS) evaluated plasma samples from four large blood collection organizations and found metabolites of drugs typically used in ARV regimens, at concentrations indicating that they had been taken within a week of blood donation.
“What are the motivations of these individuals who are ARV-positive and HIV-positive? Are they using the blood center as a place to monitor their infection status? We just do not know the answer to that, but it does bring back this issue of whether there a form of test seeking going on here,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The findings suggest that current donor health questionnaires and pre-donation screening are not adequate for ascertaining disclosure of HIV status or behaviors that could put the safety of the blood supply in doubt.
Does ‘U’ really equal ‘U’?
The public health message that “undetectable” equals “untransmittable” may help to prevent new infections, but may not translate into transfusion safety, Dr. Custer said.
Current HIV treatment guidelines recommend that infected individuals start on ARV immediately upon receiving a diagnosis. ARV drugs both suppress viremia and alter biomarkers of HIV progression, and may delay the time to antibody seroconversion, which could affect the ability to detect HIV through blood donation screening.
To see whether there is renewed cause for concern about safety of the blood supply, the TTIMS investigators tested for ARV metabolites in donated blood samples as a surrogate for HIV infection.
They looked at 299 HIV-positive plasma samples and 300 samples negative for all testable infections using liquid chromatography–mass spectometry for metabolites of 13 ARVs.
No ARV metabolites could be detected in the HIV-negative samples, but in 299 samples from 463 HIV-positive donors they found that 46 (15.4%) tested positive for ARVs. Of these 46 specimens, 43 were from first-time donors, and 34 were from males. Three samples were from repeat donors.
In all, 41 of the 46 ARV-positive donors would have been considered as potential “elite controllers” – HIV-infected individuals with no detectable viral loads in the absence of therapy – based on HIV-positive serology but negative nucleic acid test (NAT) results.
Samples from five first-time donors tested positive for HIV by both serology and NAT, and also contained ARV metabolites, suggesting that the donors had started ARVs recently, had suboptimal therapy, or were poorly adherent to their regimens.
PrEP use among HIV-negative donors
The investigators also looked at PrEP use in 1494 samples from first-time male donors that tested negative for all markers routinely screened: HIV 1/2, hepatitis B virus, hepatitis C virus, HTLV-I/II, West Nile virus, ZIKV, Treponema pallidum and Trypanosoma cruzi.
The donors came from Boston, Los Angeles, Miami, New York, San Francisco, and Washington, all cities with elevated HIV prevalence and active PrEP rollout campaigns.
They tested for analytes to emtricitabine and tenofovir, the two ARV components of the PrEP drug Truvada, and found that nine samples (0.6%) tested positive for PrEP. Of these, three donors had taken PrEP approximately 1 day before donation, two took it 2 days before, and four took it about 4 days before donation.
“There is quite a bit of concern about what this might mean for blood safety.” Dr. Custer said. “There’s a possibility of a breakthrough infection if someone is not PrEP adherent, and would we be able to detect them? Could this be an additional indicator for the risk of transfusion-transmissible infections? Could we identify subgroups of PrEP use in the donor population, and would we see any evidence?”
Mitigation Efforts
The National Heart, Lung, and Blood Institute is supporting a 7-year research project to study the potential impact of antiretroviral therapy and PrEP in the United States and Brazil under the Recipient Epidemiology and Donor Evaluation Study-IV-Pediatric (REDS-IV-P) project.
In addition, an AABB task force has drafted new questions for the donor health questionnaire intended to focus the attention of potential donors on PrEP, postexposure prophylaxis (PEP), and antiretroviral therapy.
In addition to asking about the use of medications on the deferral list, the proposed additions would ask, “In the past 16 weeks, have you taken any medication to prevent an HIV infection?” and “Have you EVER taken any medication to treat an HIV infection?”
TTIMS is supported by the U.S. Food and Drug Administration, NHLBI, and the Office of the Assistant Secretary for Health. Dr. Custer and all coinvestigators reported having no relevant disclosures.
SOURCE: : wit AABB 2019. Abstract PL5-MN4-32
REPORTING FROM AABB 2019
Fibrinogen concentrate effective, safe for postop bleeding
SAN ANTONIO – Fibrinogen concentrate was noninferior to cryoprecipitate for controlling bleeding following cardiac surgery in the randomized FIBRES trial, Canadian investigators reported.

Among 827 patients undergoing cardiopulmonary bypass, there were no significant differences in the use of allogenenic transfusion products within 24 hours of surgery for patients assigned to receive fibrinogen concentrate for control of bleeding, compared with patients who received cryoprecipitate, reported Jeannie Callum, MD, from Sunnybrook Health Sciences Centre in Toronto, on behalf of coinvestigators in the FIBRES trial.
Fibrinogen concentrate, commonly used to control postoperative bleeding in Europe, was associated with numerically, but not statistically, lower incidence of both adverse events and serious adverse events than cryoprecipitate, the current standard of care in North America.
“Given its safety and logistical advantages, fibrinogen concentrate may be considered in bleeding patients with acquired hypofibrinogenemia,” Dr. Callum said at the annual meeting of the AABB, the group formerly known as the American Association of Blood Banks.
Results of the FIBRES trial were published simultaneously in JAMA (2019 Oct 21. doi: 10.1001/jama.2019.17312).
Acquired hypofibrinogenemia, defined as a fibrinogen level below the range of 1.5-2.0 g/L, is a major cause of excess bleeding after cardiac surgery. European guidelines on the management of bleeding following trauma or cardiac surgery recommend the use of either cryoprecipitate or fibrinogen concentrate to control excessive bleeding in patients with acquired hypofibrinogenemia, Dr. Callum noted.
Cryoprecipitate is a pooled plasma–derived product that contains fibrinogen, but also fibronectin, platelet microparticles, coagulation factors VIII and XIII, and von Willebrand factor.
Additionally, fibrinogen levels in cryoprecipitate can range from as low as 3 g/L to as high as 30 g/L, and the product is normally kept and shipped frozen, and is then thawed for use and pooled prior to administration, with a shelf life of just 4-6 hours.
In contrast, fibrinogen concentrates “are pathogen-reduced and purified; have standardized fibrinogen content (20 g/L); are lyophilized, allowing for easy storage, reconstitution, and administration; and have longer shelf life after reconstitution (up to 24 hours),” Dr. Callum and her colleagues reported.
Despite the North American preference for cryoprecipitate and the European preference for fibrinogen concentrate, there have been few studies directly comparing the two products, which prompted the FIBRES investigators to design a head-to-head trial.
The randomized trial was conducted in 11 Canadian hospitals with adults undergoing cardiac surgery with cardiopulmonary bypass for whom fibrinogen supplementation was ordered in accordance with accepted clinical standards.
Patients were randomly assigned to received either 4 g of fibrinogen concentrate or 10 units of cryoprecipitate for 24 hours, with all patients receiving additional cryoprecipitate as needed after the first day.
Of 15,412 cardiac patients treated at the participating sites, 827 patients met the trial criteria and were randomized. Because the trial met the prespecified stopping criterion for noninferiority of fibrinogen at the interim analysis, the trial was halted, leaving the 827 patients as the final analysis population.
The mean number of allogeneic blood component units administered – the primary outcome – was 16.3 units in the fibrinogen concentrate group and 17.0 units in the cryoprecipitate group (mean ratio, 0.96; P for noninferiority less than .001; P for superiority = .50).
Fibrinogen was also noninferior for the secondary outcomes of individual 24-hour and cumulative 7-day blood component transfusions, and in a post-hoc analysis of cumulative transfusions measured from product administration to 24 hours after termination of cardiopulmonary bypass. These endpoints should be interpreted with caution, however, because they were not corrected for type 1 error, the investigators noted.
Fibrinogen concentrate also appeared to be noninferior for all defined subgroups, except for patients who underwent nonelective procedures, which included all patients in critical state before surgery.
Adverse events (AEs) of any kind occurred in 66.7% of patients with fibrinogen concentrate vs. 72.7% of those on cryoprecipitate. Serious AEs occurred in 31.5% vs. 34.7%, respectively.
Thromboembolic events – stroke or transient ischemic attack, amaurosis fugax (temporary vision loss), myocardial infarction, deep-vein thrombosis, pulmonary embolism, other-vessel thrombosis, disseminated intravascular coagulation, or thrombophlebitis – occurred in 7% vs. 9.6%, respectively.
The investigators acknowledged that the study was limited by the inability to blind the clinical team to the product used, by the adult-only population, and by the likelihood of variable dosing in the cryoprecipitate group.
Advantages of fibrinogen concentrate over cryoprecipitate are that the former is pathogen reduced and is easier to deliver, the investigators said.
“One important consideration is the cost differential that currently favors cryoprecipitate, but this varies across regions, and the most recent economic analysis failed to include the costs of future emerging pathogens and did not include comprehensive activity-based costing,” the investigators wrote in JAMA.
The trial was sponsored by Octapharma AG, which also provided fibrinogen concentrate. Cryoprecipitate was provided by the Canadian Blood Services and Héma-Québec. Dr. Callum reported receiving grants from Canadian Blood Services, Octapharma, and CSL Behring during the conduct of the study. Multiple coauthors had similar disclosures.
SOURCE: Callum J et al. JAMA. 2019 Oct 21. doi:10.1001/jama.2019.17312.
SAN ANTONIO – Fibrinogen concentrate was noninferior to cryoprecipitate for controlling bleeding following cardiac surgery in the randomized FIBRES trial, Canadian investigators reported.
Among 827 patients undergoing cardiopulmonary bypass, there were no significant differences in the use of allogenenic transfusion products within 24 hours of surgery for patients assigned to receive fibrinogen concentrate for control of bleeding, compared with patients who received cryoprecipitate, reported Jeannie Callum, MD, from Sunnybrook Health Sciences Centre in Toronto, on behalf of coinvestigators in the FIBRES trial.
Fibrinogen concentrate, commonly used to control postoperative bleeding in Europe, was associated with numerically, but not statistically, lower incidence of both adverse events and serious adverse events than cryoprecipitate, the current standard of care in North America.
“Given its safety and logistical advantages, fibrinogen concentrate may be considered in bleeding patients with acquired hypofibrinogenemia,” Dr. Callum said at the annual meeting of the AABB, the group formerly known as the American Association of Blood Banks.
Results of the FIBRES trial were published simultaneously in JAMA (2019 Oct 21. doi: 10.1001/jama.2019.17312).
Acquired hypofibrinogenemia, defined as a fibrinogen level below the range of 1.5-2.0 g/L, is a major cause of excess bleeding after cardiac surgery. European guidelines on the management of bleeding following trauma or cardiac surgery recommend the use of either cryoprecipitate or fibrinogen concentrate to control excessive bleeding in patients with acquired hypofibrinogenemia, Dr. Callum noted.
Cryoprecipitate is a pooled plasma–derived product that contains fibrinogen, but also fibronectin, platelet microparticles, coagulation factors VIII and XIII, and von Willebrand factor.
Additionally, fibrinogen levels in cryoprecipitate can range from as low as 3 g/L to as high as 30 g/L, and the product is normally kept and shipped frozen, and is then thawed for use and pooled prior to administration, with a shelf life of just 4-6 hours.
In contrast, fibrinogen concentrates “are pathogen-reduced and purified; have standardized fibrinogen content (20 g/L); are lyophilized, allowing for easy storage, reconstitution, and administration; and have longer shelf life after reconstitution (up to 24 hours),” Dr. Callum and her colleagues reported.
Despite the North American preference for cryoprecipitate and the European preference for fibrinogen concentrate, there have been few studies directly comparing the two products, which prompted the FIBRES investigators to design a head-to-head trial.
The randomized trial was conducted in 11 Canadian hospitals with adults undergoing cardiac surgery with cardiopulmonary bypass for whom fibrinogen supplementation was ordered in accordance with accepted clinical standards.
Patients were randomly assigned to received either 4 g of fibrinogen concentrate or 10 units of cryoprecipitate for 24 hours, with all patients receiving additional cryoprecipitate as needed after the first day.
Of 15,412 cardiac patients treated at the participating sites, 827 patients met the trial criteria and were randomized. Because the trial met the prespecified stopping criterion for noninferiority of fibrinogen at the interim analysis, the trial was halted, leaving the 827 patients as the final analysis population.
The mean number of allogeneic blood component units administered – the primary outcome – was 16.3 units in the fibrinogen concentrate group and 17.0 units in the cryoprecipitate group (mean ratio, 0.96; P for noninferiority less than .001; P for superiority = .50).
Fibrinogen was also noninferior for the secondary outcomes of individual 24-hour and cumulative 7-day blood component transfusions, and in a post-hoc analysis of cumulative transfusions measured from product administration to 24 hours after termination of cardiopulmonary bypass. These endpoints should be interpreted with caution, however, because they were not corrected for type 1 error, the investigators noted.
Fibrinogen concentrate also appeared to be noninferior for all defined subgroups, except for patients who underwent nonelective procedures, which included all patients in critical state before surgery.
Adverse events (AEs) of any kind occurred in 66.7% of patients with fibrinogen concentrate vs. 72.7% of those on cryoprecipitate. Serious AEs occurred in 31.5% vs. 34.7%, respectively.
Thromboembolic events – stroke or transient ischemic attack, amaurosis fugax (temporary vision loss), myocardial infarction, deep-vein thrombosis, pulmonary embolism, other-vessel thrombosis, disseminated intravascular coagulation, or thrombophlebitis – occurred in 7% vs. 9.6%, respectively.
The investigators acknowledged that the study was limited by the inability to blind the clinical team to the product used, by the adult-only population, and by the likelihood of variable dosing in the cryoprecipitate group.
Advantages of fibrinogen concentrate over cryoprecipitate are that the former is pathogen reduced and is easier to deliver, the investigators said.
“One important consideration is the cost differential that currently favors cryoprecipitate, but this varies across regions, and the most recent economic analysis failed to include the costs of future emerging pathogens and did not include comprehensive activity-based costing,” the investigators wrote in JAMA.
The trial was sponsored by Octapharma AG, which also provided fibrinogen concentrate. Cryoprecipitate was provided by the Canadian Blood Services and Héma-Québec. Dr. Callum reported receiving grants from Canadian Blood Services, Octapharma, and CSL Behring during the conduct of the study. Multiple coauthors had similar disclosures.
SOURCE: Callum J et al. JAMA. 2019 Oct 21. doi:10.1001/jama.2019.17312.
SAN ANTONIO – Fibrinogen concentrate was noninferior to cryoprecipitate for controlling bleeding following cardiac surgery in the randomized FIBRES trial, Canadian investigators reported.
Among 827 patients undergoing cardiopulmonary bypass, there were no significant differences in the use of allogenenic transfusion products within 24 hours of surgery for patients assigned to receive fibrinogen concentrate for control of bleeding, compared with patients who received cryoprecipitate, reported Jeannie Callum, MD, from Sunnybrook Health Sciences Centre in Toronto, on behalf of coinvestigators in the FIBRES trial.
Fibrinogen concentrate, commonly used to control postoperative bleeding in Europe, was associated with numerically, but not statistically, lower incidence of both adverse events and serious adverse events than cryoprecipitate, the current standard of care in North America.
“Given its safety and logistical advantages, fibrinogen concentrate may be considered in bleeding patients with acquired hypofibrinogenemia,” Dr. Callum said at the annual meeting of the AABB, the group formerly known as the American Association of Blood Banks.
Results of the FIBRES trial were published simultaneously in JAMA (2019 Oct 21. doi: 10.1001/jama.2019.17312).
Acquired hypofibrinogenemia, defined as a fibrinogen level below the range of 1.5-2.0 g/L, is a major cause of excess bleeding after cardiac surgery. European guidelines on the management of bleeding following trauma or cardiac surgery recommend the use of either cryoprecipitate or fibrinogen concentrate to control excessive bleeding in patients with acquired hypofibrinogenemia, Dr. Callum noted.
Cryoprecipitate is a pooled plasma–derived product that contains fibrinogen, but also fibronectin, platelet microparticles, coagulation factors VIII and XIII, and von Willebrand factor.
Additionally, fibrinogen levels in cryoprecipitate can range from as low as 3 g/L to as high as 30 g/L, and the product is normally kept and shipped frozen, and is then thawed for use and pooled prior to administration, with a shelf life of just 4-6 hours.
In contrast, fibrinogen concentrates “are pathogen-reduced and purified; have standardized fibrinogen content (20 g/L); are lyophilized, allowing for easy storage, reconstitution, and administration; and have longer shelf life after reconstitution (up to 24 hours),” Dr. Callum and her colleagues reported.
Despite the North American preference for cryoprecipitate and the European preference for fibrinogen concentrate, there have been few studies directly comparing the two products, which prompted the FIBRES investigators to design a head-to-head trial.
The randomized trial was conducted in 11 Canadian hospitals with adults undergoing cardiac surgery with cardiopulmonary bypass for whom fibrinogen supplementation was ordered in accordance with accepted clinical standards.
Patients were randomly assigned to received either 4 g of fibrinogen concentrate or 10 units of cryoprecipitate for 24 hours, with all patients receiving additional cryoprecipitate as needed after the first day.
Of 15,412 cardiac patients treated at the participating sites, 827 patients met the trial criteria and were randomized. Because the trial met the prespecified stopping criterion for noninferiority of fibrinogen at the interim analysis, the trial was halted, leaving the 827 patients as the final analysis population.
The mean number of allogeneic blood component units administered – the primary outcome – was 16.3 units in the fibrinogen concentrate group and 17.0 units in the cryoprecipitate group (mean ratio, 0.96; P for noninferiority less than .001; P for superiority = .50).
Fibrinogen was also noninferior for the secondary outcomes of individual 24-hour and cumulative 7-day blood component transfusions, and in a post-hoc analysis of cumulative transfusions measured from product administration to 24 hours after termination of cardiopulmonary bypass. These endpoints should be interpreted with caution, however, because they were not corrected for type 1 error, the investigators noted.
Fibrinogen concentrate also appeared to be noninferior for all defined subgroups, except for patients who underwent nonelective procedures, which included all patients in critical state before surgery.
Adverse events (AEs) of any kind occurred in 66.7% of patients with fibrinogen concentrate vs. 72.7% of those on cryoprecipitate. Serious AEs occurred in 31.5% vs. 34.7%, respectively.
Thromboembolic events – stroke or transient ischemic attack, amaurosis fugax (temporary vision loss), myocardial infarction, deep-vein thrombosis, pulmonary embolism, other-vessel thrombosis, disseminated intravascular coagulation, or thrombophlebitis – occurred in 7% vs. 9.6%, respectively.
The investigators acknowledged that the study was limited by the inability to blind the clinical team to the product used, by the adult-only population, and by the likelihood of variable dosing in the cryoprecipitate group.
Advantages of fibrinogen concentrate over cryoprecipitate are that the former is pathogen reduced and is easier to deliver, the investigators said.
“One important consideration is the cost differential that currently favors cryoprecipitate, but this varies across regions, and the most recent economic analysis failed to include the costs of future emerging pathogens and did not include comprehensive activity-based costing,” the investigators wrote in JAMA.
The trial was sponsored by Octapharma AG, which also provided fibrinogen concentrate. Cryoprecipitate was provided by the Canadian Blood Services and Héma-Québec. Dr. Callum reported receiving grants from Canadian Blood Services, Octapharma, and CSL Behring during the conduct of the study. Multiple coauthors had similar disclosures.
SOURCE: Callum J et al. JAMA. 2019 Oct 21. doi:10.1001/jama.2019.17312.
REPORTING FROM AABB 2019
SPARTAN: Apalutamide delays second progression in nonmetastatic CRPC
BARCELONA – The androgen receptor inhibitor apalutamide was associated with a 45% improvement in time to second progression in patients with nonmetastatic, castration-resistant prostate cancer as well as an overall survival edge, although the latter fell just short of statistical significance in an interim analysis of results from the SPARTAN trial.

Among 1,207 men with nonmetastatic castration-resistant prostate cancer (CRPC), those randomly assigned to apalutamide had significantly longer second progression-free survival (PFS2), an exploratory endpoint defined as the time of study entry to progression on subsequent treatment.
In all, 69% of patients on placebo and 40% of those on apalutamide went on to another life-prolonging therapy, usually abiraterone acetate (Zytiga) plus prednisone. The respective median PFS2s were 43.8 and 55.6 months, translating into a hazard ratio for second progression in the apalutamide group of 0.55 (P less than .0001).
Overall survival (OS) also trended toward a benefit for apalutamide, but the difference did not reach the prespecified boundary for significance in this planned interim analysis, reported Matthew R. Smith, MD, PhD, from the Massachusetts General Hospital Cancer Center in Boston.
Among men randomly assigned in the phase 3 trial to receive apalutamide (Erleada), the 4-year OS rate was 72.1%, compared with 64.7% for patients assigned to placebo.
“This OS benefit for apalutamide was observed despite crossover of placebo patients to apalutamide, and higher rates of subsequent life-prolonging therapy in the placebo group,” he said at the European Society for Medical Oncology Congress.
Apalutamide was approved by the Food and Drug Administration in 2018 for treatment of patients with nonmetastatic CRPC who were on androgen deprivation therapy with a rapidly rising prostate-specific antigen level and no detectable distant metastases on conventional imaging.
That approval was based on the primary results from SPARTAN, which showed a median metastasis-free survival for patients taking apalutamide of 40.5 months, compared with 16.2 months for patients taking placebo.
The drug also recently received FDA approval for treatment of men with metastatic CRPC, based on results of the TITAN trial.
Overall survival
At the time of the primary analysis of SPARTAN for metastasis-free survival, the OS data were immature, with only 104 of 427 events required for the prespecified analysis.
At ESMO 2019, Dr. Smith presented results of the second interim survival analysis looking at the effects of apalutamide on OS, as well as time to chemotherapy, PFS2, and safety.
In the trial, men with nonmetastatic CRPC were randomized on a 2:1 basis to receive either apalutamide 240 mg daily (806 patients) or placebo (401) plus androgen deprivation therapy, either with gonadotropin-releasing hormone analogue therapy or with surgical castration.
At a median follow-up of 41 months, median OS was not reached in either arm. As noted before, the 4-year OS rate was higher in the apalutamide group, translating into a HR for death of 0.75. However, the P value of .0197 was higher than the prespecified O’Brien-Fleming boundary of 0.0121, which means that the study follow-up will continue per protocol, with the final analysis for overall survival planned after 427 deaths have occurred, Dr. Smith said.
The survival benefit with apalutamide either trended toward significance across all subgroups, with notable benefits in patients aged younger than 65 years, those with good performance status, prostate-specific antigen doubling time longer than 6 months, and those with locoregional disease status of N1 versus N0.
The secondary endpoint of time to chemotherapy initiation was not formally tested because OS did not reach statistical significance, but proportionally fewer patients on apalutamide had started cytotoxic chemotherapy at the time of the interim analysis (14% vs. 20% of patients on placebo). The median time to initiation of chemotherapy has not been reached in either trial arm, however.
The safety profile of the androgen receptor inhibitor was consistent with that seen in the primary PFS analysis, with any adverse event seen in 97.3% of patients versus 93.7% of patients on placebo, grade 3-4 adverse events in 53.1% versus 36.7%, any serious adverse event in 33.5% versus 24.9%, events leading to treatment discontinuation in 13.8% versus 7.3%, and fatal adverse events in 2.1 versus .5%.
“Of some note, discontinuation due to disease progression was 34% in the apalutamide group, compared to 74% in the placebo group,” Dr. Smith said.
“These results further support apalutamide as a standard-of-care option for men with high-risk, nonmetastatic CRPC,” he concluded.
PFS2 data ‘important’
Invited discussant Karim Fizazi, MD, PhD, from the Institut Gustave Roussy in Villejuif, France, acknowledged it’s too early to draw conclusions about OS, but added that the PFS2 data are important.

The 45% reduction in risk of progression “is particularly important in this trial because abiraterone was provided as a potential salvage treatment in both arms. So in other words, in that trial they were almost truly comparing early AR [androgen receptor] targeting versus deferred AR targeting, at least in some patients, and it seems that [apalutamide] is making a difference in terms of second progression,” he said.
An unanswered question about the trial, however, is how active abiraterone was post apalutamide in the experimental, because there are data suggesting minimal efficacy in sequential therapy of AR-targeted agents, he added.
The SPARTAN trial was funded by Janssen. Dr. Smith disclosed an advisory role, research funding, and travel reimbursement for Janssen and others. Dr. Fizazi disclosed consulting for Janssen and others.
SOURCE: Smith MR et al. ESMO 2019, Abstract 843O
BARCELONA – The androgen receptor inhibitor apalutamide was associated with a 45% improvement in time to second progression in patients with nonmetastatic, castration-resistant prostate cancer as well as an overall survival edge, although the latter fell just short of statistical significance in an interim analysis of results from the SPARTAN trial.
Among 1,207 men with nonmetastatic castration-resistant prostate cancer (CRPC), those randomly assigned to apalutamide had significantly longer second progression-free survival (PFS2), an exploratory endpoint defined as the time of study entry to progression on subsequent treatment.
In all, 69% of patients on placebo and 40% of those on apalutamide went on to another life-prolonging therapy, usually abiraterone acetate (Zytiga) plus prednisone. The respective median PFS2s were 43.8 and 55.6 months, translating into a hazard ratio for second progression in the apalutamide group of 0.55 (P less than .0001).
Overall survival (OS) also trended toward a benefit for apalutamide, but the difference did not reach the prespecified boundary for significance in this planned interim analysis, reported Matthew R. Smith, MD, PhD, from the Massachusetts General Hospital Cancer Center in Boston.
Among men randomly assigned in the phase 3 trial to receive apalutamide (Erleada), the 4-year OS rate was 72.1%, compared with 64.7% for patients assigned to placebo.
“This OS benefit for apalutamide was observed despite crossover of placebo patients to apalutamide, and higher rates of subsequent life-prolonging therapy in the placebo group,” he said at the European Society for Medical Oncology Congress.
Apalutamide was approved by the Food and Drug Administration in 2018 for treatment of patients with nonmetastatic CRPC who were on androgen deprivation therapy with a rapidly rising prostate-specific antigen level and no detectable distant metastases on conventional imaging.
That approval was based on the primary results from SPARTAN, which showed a median metastasis-free survival for patients taking apalutamide of 40.5 months, compared with 16.2 months for patients taking placebo.
The drug also recently received FDA approval for treatment of men with metastatic CRPC, based on results of the TITAN trial.
Overall survival
At the time of the primary analysis of SPARTAN for metastasis-free survival, the OS data were immature, with only 104 of 427 events required for the prespecified analysis.
At ESMO 2019, Dr. Smith presented results of the second interim survival analysis looking at the effects of apalutamide on OS, as well as time to chemotherapy, PFS2, and safety.
In the trial, men with nonmetastatic CRPC were randomized on a 2:1 basis to receive either apalutamide 240 mg daily (806 patients) or placebo (401) plus androgen deprivation therapy, either with gonadotropin-releasing hormone analogue therapy or with surgical castration.
At a median follow-up of 41 months, median OS was not reached in either arm. As noted before, the 4-year OS rate was higher in the apalutamide group, translating into a HR for death of 0.75. However, the P value of .0197 was higher than the prespecified O’Brien-Fleming boundary of 0.0121, which means that the study follow-up will continue per protocol, with the final analysis for overall survival planned after 427 deaths have occurred, Dr. Smith said.
The survival benefit with apalutamide either trended toward significance across all subgroups, with notable benefits in patients aged younger than 65 years, those with good performance status, prostate-specific antigen doubling time longer than 6 months, and those with locoregional disease status of N1 versus N0.
The secondary endpoint of time to chemotherapy initiation was not formally tested because OS did not reach statistical significance, but proportionally fewer patients on apalutamide had started cytotoxic chemotherapy at the time of the interim analysis (14% vs. 20% of patients on placebo). The median time to initiation of chemotherapy has not been reached in either trial arm, however.
The safety profile of the androgen receptor inhibitor was consistent with that seen in the primary PFS analysis, with any adverse event seen in 97.3% of patients versus 93.7% of patients on placebo, grade 3-4 adverse events in 53.1% versus 36.7%, any serious adverse event in 33.5% versus 24.9%, events leading to treatment discontinuation in 13.8% versus 7.3%, and fatal adverse events in 2.1 versus .5%.
“Of some note, discontinuation due to disease progression was 34% in the apalutamide group, compared to 74% in the placebo group,” Dr. Smith said.
“These results further support apalutamide as a standard-of-care option for men with high-risk, nonmetastatic CRPC,” he concluded.
PFS2 data ‘important’
Invited discussant Karim Fizazi, MD, PhD, from the Institut Gustave Roussy in Villejuif, France, acknowledged it’s too early to draw conclusions about OS, but added that the PFS2 data are important.
The 45% reduction in risk of progression “is particularly important in this trial because abiraterone was provided as a potential salvage treatment in both arms. So in other words, in that trial they were almost truly comparing early AR [androgen receptor] targeting versus deferred AR targeting, at least in some patients, and it seems that [apalutamide] is making a difference in terms of second progression,” he said.
An unanswered question about the trial, however, is how active abiraterone was post apalutamide in the experimental, because there are data suggesting minimal efficacy in sequential therapy of AR-targeted agents, he added.
The SPARTAN trial was funded by Janssen. Dr. Smith disclosed an advisory role, research funding, and travel reimbursement for Janssen and others. Dr. Fizazi disclosed consulting for Janssen and others.
SOURCE: Smith MR et al. ESMO 2019, Abstract 843O
BARCELONA – The androgen receptor inhibitor apalutamide was associated with a 45% improvement in time to second progression in patients with nonmetastatic, castration-resistant prostate cancer as well as an overall survival edge, although the latter fell just short of statistical significance in an interim analysis of results from the SPARTAN trial.
Among 1,207 men with nonmetastatic castration-resistant prostate cancer (CRPC), those randomly assigned to apalutamide had significantly longer second progression-free survival (PFS2), an exploratory endpoint defined as the time of study entry to progression on subsequent treatment.
In all, 69% of patients on placebo and 40% of those on apalutamide went on to another life-prolonging therapy, usually abiraterone acetate (Zytiga) plus prednisone. The respective median PFS2s were 43.8 and 55.6 months, translating into a hazard ratio for second progression in the apalutamide group of 0.55 (P less than .0001).
Overall survival (OS) also trended toward a benefit for apalutamide, but the difference did not reach the prespecified boundary for significance in this planned interim analysis, reported Matthew R. Smith, MD, PhD, from the Massachusetts General Hospital Cancer Center in Boston.
Among men randomly assigned in the phase 3 trial to receive apalutamide (Erleada), the 4-year OS rate was 72.1%, compared with 64.7% for patients assigned to placebo.
“This OS benefit for apalutamide was observed despite crossover of placebo patients to apalutamide, and higher rates of subsequent life-prolonging therapy in the placebo group,” he said at the European Society for Medical Oncology Congress.
Apalutamide was approved by the Food and Drug Administration in 2018 for treatment of patients with nonmetastatic CRPC who were on androgen deprivation therapy with a rapidly rising prostate-specific antigen level and no detectable distant metastases on conventional imaging.
That approval was based on the primary results from SPARTAN, which showed a median metastasis-free survival for patients taking apalutamide of 40.5 months, compared with 16.2 months for patients taking placebo.
The drug also recently received FDA approval for treatment of men with metastatic CRPC, based on results of the TITAN trial.
Overall survival
At the time of the primary analysis of SPARTAN for metastasis-free survival, the OS data were immature, with only 104 of 427 events required for the prespecified analysis.
At ESMO 2019, Dr. Smith presented results of the second interim survival analysis looking at the effects of apalutamide on OS, as well as time to chemotherapy, PFS2, and safety.
In the trial, men with nonmetastatic CRPC were randomized on a 2:1 basis to receive either apalutamide 240 mg daily (806 patients) or placebo (401) plus androgen deprivation therapy, either with gonadotropin-releasing hormone analogue therapy or with surgical castration.
At a median follow-up of 41 months, median OS was not reached in either arm. As noted before, the 4-year OS rate was higher in the apalutamide group, translating into a HR for death of 0.75. However, the P value of .0197 was higher than the prespecified O’Brien-Fleming boundary of 0.0121, which means that the study follow-up will continue per protocol, with the final analysis for overall survival planned after 427 deaths have occurred, Dr. Smith said.
The survival benefit with apalutamide either trended toward significance across all subgroups, with notable benefits in patients aged younger than 65 years, those with good performance status, prostate-specific antigen doubling time longer than 6 months, and those with locoregional disease status of N1 versus N0.
The secondary endpoint of time to chemotherapy initiation was not formally tested because OS did not reach statistical significance, but proportionally fewer patients on apalutamide had started cytotoxic chemotherapy at the time of the interim analysis (14% vs. 20% of patients on placebo). The median time to initiation of chemotherapy has not been reached in either trial arm, however.
The safety profile of the androgen receptor inhibitor was consistent with that seen in the primary PFS analysis, with any adverse event seen in 97.3% of patients versus 93.7% of patients on placebo, grade 3-4 adverse events in 53.1% versus 36.7%, any serious adverse event in 33.5% versus 24.9%, events leading to treatment discontinuation in 13.8% versus 7.3%, and fatal adverse events in 2.1 versus .5%.
“Of some note, discontinuation due to disease progression was 34% in the apalutamide group, compared to 74% in the placebo group,” Dr. Smith said.
“These results further support apalutamide as a standard-of-care option for men with high-risk, nonmetastatic CRPC,” he concluded.
PFS2 data ‘important’
Invited discussant Karim Fizazi, MD, PhD, from the Institut Gustave Roussy in Villejuif, France, acknowledged it’s too early to draw conclusions about OS, but added that the PFS2 data are important.
The 45% reduction in risk of progression “is particularly important in this trial because abiraterone was provided as a potential salvage treatment in both arms. So in other words, in that trial they were almost truly comparing early AR [androgen receptor] targeting versus deferred AR targeting, at least in some patients, and it seems that [apalutamide] is making a difference in terms of second progression,” he said.
An unanswered question about the trial, however, is how active abiraterone was post apalutamide in the experimental, because there are data suggesting minimal efficacy in sequential therapy of AR-targeted agents, he added.
The SPARTAN trial was funded by Janssen. Dr. Smith disclosed an advisory role, research funding, and travel reimbursement for Janssen and others. Dr. Fizazi disclosed consulting for Janssen and others.
SOURCE: Smith MR et al. ESMO 2019, Abstract 843O
REPORTING FROM ESMO 2019
Atezolizumab plus chemo gives slight PFS edge in mUC
BARCELONA – Adding the immune checkpoint inhibitor atezolizumab (Tecentriq) to standard platinum-based chemotherapy was associated with a small but statistically significant progression-free survival benefit for patients with previously untreated metastatic urothelial carcinoma, investigators in the IMvigor130 trial found.

Among 1,213 patients with newly diagnosed metastatic urothelial carcinoma assigned to receive either atezolizumab monotherapy or chemotherapy with a platinum compound and gemcitabine plus either atezolizumab or placebo, the median progression-free survival (PFS) at a median follow-up of 11.8 months was 8.2 months with atezolizumab/chemotherapy, compared with 6.3 months with chemotherapy plus placebo, reported Enrique Grande, MD, of MD Anderson Cancer Center Madrid.
“IMvigor130 is the first immune checkpoint inhibitor study to demonstrate an improvement in progression-free survival over the standard of care in first-line metastatic urothelial carcinoma. At this interim analysis, we observed a clinically meaningful improvement in the overall survival for the combination of atezolizumab plus chemotherapy that did not meet the prespecified interim boundary for significance,” he said at the European Society for Medical Oncology Congress.
Median overall survival (OS) at the interim analysis was 16.0 months in the atezolizumab arm, vs. 13.4 months in the placebo arm, translating into a hazard ratio (HR) of 0.83 trending in favor of the combination. But as noted by Dr. Grande, the P value was .027, which did not reach the interim efficacy boundary of .007.
IMvigor130 investigators enrolled patients with locally advanced metastatic urothelial carcinoma with no prior systemic therapy in the metastatic setting who had good performance status (ECOG 2 or less) and were eligible for chemotherapy with either cisplatin or carboplatin plus gemcitabine.
The patients were stratified by programmed death ligand-1 (PD-L1) status, prognostic (Bajorin) risk factor scores, and investigator choice of cisplatin or carboplatin, and then randomized to either atezolizumab plus chemotherapy, atezolizumab monotherapy, or placebo plus chemotherapy.
As noted, the co-primary endpoint of PFS in the chemotherapy arms in the intention-to-treat population was statistically significant at the preplanned interim analysis, but the other primary endpoint of OS had not reached significance.
At the time of the data cutoff in May 2019, the stratified HR for progression with atezolizumab was 0.82 (P = .007).
An analysis by subgroup showed either significant benefit or a trend favoring atezolizumab across all stratification factors, Dr. Grande said.
A hierarchical analysis of atezolizumab monotherapy vs. chemotherapy in the placebo-control arm showed a median OS of 15.7 vs. 13.1 months, respectively, with a hazard ratio of 1.02 (nonsignificant).
The benefit of atezolizumab appeared to be almost entirely among patients whose tumors had higher levels of PD-L1 expression according to immunohistochemistry (IC). The interim OS among patients with PD-L1 IC0/1 was a median of 13.5 months with atezolizumab vs. 12.9 months with chemotherapy alone, with an unstratified HR of 1.07 (nonsignificant). In contrast, among patients with PD-L1 IC2/3 status, the median OS was not reached for patients in the atezolizumab arm, vs. 17.8 months in the chemotherapy alone arm, for a stratified HR of 0.68, although this too did not reach statistical significance.
Responses were similar between the two chemotherapy arms, with an overall response rate (ORR) of 47% with atezolizumab added, vs. 44% with placebo added. The complete response (CR) rates were 13% and 7%, respectively. The ORR in the monotherapy arm was 23%, consisting of 6% complete and 17% partial responses.
Grade 3 or 4 treatment-related adverse events occurred in 81% of patients in each chemotherapy arm, compared with 15% in the atezolizumab monotherapy arm. Nine patients in the atezolizumab/chemotherapy arm died from a treatment-related cause, compared with four in the chemotherapy alone arm, and three in the atezolizumab monotherapy arm.
Adverse events leading to treatment discontinuation occurred in one-fourth of patients in each chemotherapy-containing arm, vs. less than 1% of patients in the monotherapy arm.
“The results from the IMvigor130 trial support the use of atezolizumab in combination with chemotherapy as an important new treatment option for patients with untreated metastatic urothelial carcinoma,” Dr. Grande concluded.
But invited discussant Thomas Powles, MD, of Barts Cancer Institute in London, cautioned that more data are needed to conclude that the addition of atezolizumab should become a standard of care.

“Does this change practice? Well, the for and against: significant delay in PFS, but how meaningful is that? OS trending the right way, but not significant yet. CR rates, yes with 13% CRs, but response rates weren’t very different from one another, and as response rates are similar, it’s hard to argue that the two are synergistic together, for example,” he said.
He added that the adverse event profiles “actually are very acceptable to me, and I’m really looking forward to the quality-of-life data.”
The IMvigor130 study is sponsored by F. Hoffman-La Roche. Dr. Grande disclosed honoraria and research grants from Roche and others. Dr. Powles disclosed research funding, honoraria, and travel costs from Roche and others.
SOURCE: Grande E et al. ESMO 2019. Abstract LBA14_PR.
BARCELONA – Adding the immune checkpoint inhibitor atezolizumab (Tecentriq) to standard platinum-based chemotherapy was associated with a small but statistically significant progression-free survival benefit for patients with previously untreated metastatic urothelial carcinoma, investigators in the IMvigor130 trial found.
Among 1,213 patients with newly diagnosed metastatic urothelial carcinoma assigned to receive either atezolizumab monotherapy or chemotherapy with a platinum compound and gemcitabine plus either atezolizumab or placebo, the median progression-free survival (PFS) at a median follow-up of 11.8 months was 8.2 months with atezolizumab/chemotherapy, compared with 6.3 months with chemotherapy plus placebo, reported Enrique Grande, MD, of MD Anderson Cancer Center Madrid.
“IMvigor130 is the first immune checkpoint inhibitor study to demonstrate an improvement in progression-free survival over the standard of care in first-line metastatic urothelial carcinoma. At this interim analysis, we observed a clinically meaningful improvement in the overall survival for the combination of atezolizumab plus chemotherapy that did not meet the prespecified interim boundary for significance,” he said at the European Society for Medical Oncology Congress.
Median overall survival (OS) at the interim analysis was 16.0 months in the atezolizumab arm, vs. 13.4 months in the placebo arm, translating into a hazard ratio (HR) of 0.83 trending in favor of the combination. But as noted by Dr. Grande, the P value was .027, which did not reach the interim efficacy boundary of .007.
IMvigor130 investigators enrolled patients with locally advanced metastatic urothelial carcinoma with no prior systemic therapy in the metastatic setting who had good performance status (ECOG 2 or less) and were eligible for chemotherapy with either cisplatin or carboplatin plus gemcitabine.
The patients were stratified by programmed death ligand-1 (PD-L1) status, prognostic (Bajorin) risk factor scores, and investigator choice of cisplatin or carboplatin, and then randomized to either atezolizumab plus chemotherapy, atezolizumab monotherapy, or placebo plus chemotherapy.
As noted, the co-primary endpoint of PFS in the chemotherapy arms in the intention-to-treat population was statistically significant at the preplanned interim analysis, but the other primary endpoint of OS had not reached significance.
At the time of the data cutoff in May 2019, the stratified HR for progression with atezolizumab was 0.82 (P = .007).
An analysis by subgroup showed either significant benefit or a trend favoring atezolizumab across all stratification factors, Dr. Grande said.
A hierarchical analysis of atezolizumab monotherapy vs. chemotherapy in the placebo-control arm showed a median OS of 15.7 vs. 13.1 months, respectively, with a hazard ratio of 1.02 (nonsignificant).
The benefit of atezolizumab appeared to be almost entirely among patients whose tumors had higher levels of PD-L1 expression according to immunohistochemistry (IC). The interim OS among patients with PD-L1 IC0/1 was a median of 13.5 months with atezolizumab vs. 12.9 months with chemotherapy alone, with an unstratified HR of 1.07 (nonsignificant). In contrast, among patients with PD-L1 IC2/3 status, the median OS was not reached for patients in the atezolizumab arm, vs. 17.8 months in the chemotherapy alone arm, for a stratified HR of 0.68, although this too did not reach statistical significance.
Responses were similar between the two chemotherapy arms, with an overall response rate (ORR) of 47% with atezolizumab added, vs. 44% with placebo added. The complete response (CR) rates were 13% and 7%, respectively. The ORR in the monotherapy arm was 23%, consisting of 6% complete and 17% partial responses.
Grade 3 or 4 treatment-related adverse events occurred in 81% of patients in each chemotherapy arm, compared with 15% in the atezolizumab monotherapy arm. Nine patients in the atezolizumab/chemotherapy arm died from a treatment-related cause, compared with four in the chemotherapy alone arm, and three in the atezolizumab monotherapy arm.
Adverse events leading to treatment discontinuation occurred in one-fourth of patients in each chemotherapy-containing arm, vs. less than 1% of patients in the monotherapy arm.
“The results from the IMvigor130 trial support the use of atezolizumab in combination with chemotherapy as an important new treatment option for patients with untreated metastatic urothelial carcinoma,” Dr. Grande concluded.
But invited discussant Thomas Powles, MD, of Barts Cancer Institute in London, cautioned that more data are needed to conclude that the addition of atezolizumab should become a standard of care.
“Does this change practice? Well, the for and against: significant delay in PFS, but how meaningful is that? OS trending the right way, but not significant yet. CR rates, yes with 13% CRs, but response rates weren’t very different from one another, and as response rates are similar, it’s hard to argue that the two are synergistic together, for example,” he said.
He added that the adverse event profiles “actually are very acceptable to me, and I’m really looking forward to the quality-of-life data.”
The IMvigor130 study is sponsored by F. Hoffman-La Roche. Dr. Grande disclosed honoraria and research grants from Roche and others. Dr. Powles disclosed research funding, honoraria, and travel costs from Roche and others.
SOURCE: Grande E et al. ESMO 2019. Abstract LBA14_PR.
BARCELONA – Adding the immune checkpoint inhibitor atezolizumab (Tecentriq) to standard platinum-based chemotherapy was associated with a small but statistically significant progression-free survival benefit for patients with previously untreated metastatic urothelial carcinoma, investigators in the IMvigor130 trial found.
Among 1,213 patients with newly diagnosed metastatic urothelial carcinoma assigned to receive either atezolizumab monotherapy or chemotherapy with a platinum compound and gemcitabine plus either atezolizumab or placebo, the median progression-free survival (PFS) at a median follow-up of 11.8 months was 8.2 months with atezolizumab/chemotherapy, compared with 6.3 months with chemotherapy plus placebo, reported Enrique Grande, MD, of MD Anderson Cancer Center Madrid.
“IMvigor130 is the first immune checkpoint inhibitor study to demonstrate an improvement in progression-free survival over the standard of care in first-line metastatic urothelial carcinoma. At this interim analysis, we observed a clinically meaningful improvement in the overall survival for the combination of atezolizumab plus chemotherapy that did not meet the prespecified interim boundary for significance,” he said at the European Society for Medical Oncology Congress.
Median overall survival (OS) at the interim analysis was 16.0 months in the atezolizumab arm, vs. 13.4 months in the placebo arm, translating into a hazard ratio (HR) of 0.83 trending in favor of the combination. But as noted by Dr. Grande, the P value was .027, which did not reach the interim efficacy boundary of .007.
IMvigor130 investigators enrolled patients with locally advanced metastatic urothelial carcinoma with no prior systemic therapy in the metastatic setting who had good performance status (ECOG 2 or less) and were eligible for chemotherapy with either cisplatin or carboplatin plus gemcitabine.
The patients were stratified by programmed death ligand-1 (PD-L1) status, prognostic (Bajorin) risk factor scores, and investigator choice of cisplatin or carboplatin, and then randomized to either atezolizumab plus chemotherapy, atezolizumab monotherapy, or placebo plus chemotherapy.
As noted, the co-primary endpoint of PFS in the chemotherapy arms in the intention-to-treat population was statistically significant at the preplanned interim analysis, but the other primary endpoint of OS had not reached significance.
At the time of the data cutoff in May 2019, the stratified HR for progression with atezolizumab was 0.82 (P = .007).
An analysis by subgroup showed either significant benefit or a trend favoring atezolizumab across all stratification factors, Dr. Grande said.
A hierarchical analysis of atezolizumab monotherapy vs. chemotherapy in the placebo-control arm showed a median OS of 15.7 vs. 13.1 months, respectively, with a hazard ratio of 1.02 (nonsignificant).
The benefit of atezolizumab appeared to be almost entirely among patients whose tumors had higher levels of PD-L1 expression according to immunohistochemistry (IC). The interim OS among patients with PD-L1 IC0/1 was a median of 13.5 months with atezolizumab vs. 12.9 months with chemotherapy alone, with an unstratified HR of 1.07 (nonsignificant). In contrast, among patients with PD-L1 IC2/3 status, the median OS was not reached for patients in the atezolizumab arm, vs. 17.8 months in the chemotherapy alone arm, for a stratified HR of 0.68, although this too did not reach statistical significance.
Responses were similar between the two chemotherapy arms, with an overall response rate (ORR) of 47% with atezolizumab added, vs. 44% with placebo added. The complete response (CR) rates were 13% and 7%, respectively. The ORR in the monotherapy arm was 23%, consisting of 6% complete and 17% partial responses.
Grade 3 or 4 treatment-related adverse events occurred in 81% of patients in each chemotherapy arm, compared with 15% in the atezolizumab monotherapy arm. Nine patients in the atezolizumab/chemotherapy arm died from a treatment-related cause, compared with four in the chemotherapy alone arm, and three in the atezolizumab monotherapy arm.
Adverse events leading to treatment discontinuation occurred in one-fourth of patients in each chemotherapy-containing arm, vs. less than 1% of patients in the monotherapy arm.
“The results from the IMvigor130 trial support the use of atezolizumab in combination with chemotherapy as an important new treatment option for patients with untreated metastatic urothelial carcinoma,” Dr. Grande concluded.
But invited discussant Thomas Powles, MD, of Barts Cancer Institute in London, cautioned that more data are needed to conclude that the addition of atezolizumab should become a standard of care.
“Does this change practice? Well, the for and against: significant delay in PFS, but how meaningful is that? OS trending the right way, but not significant yet. CR rates, yes with 13% CRs, but response rates weren’t very different from one another, and as response rates are similar, it’s hard to argue that the two are synergistic together, for example,” he said.
He added that the adverse event profiles “actually are very acceptable to me, and I’m really looking forward to the quality-of-life data.”
The IMvigor130 study is sponsored by F. Hoffman-La Roche. Dr. Grande disclosed honoraria and research grants from Roche and others. Dr. Powles disclosed research funding, honoraria, and travel costs from Roche and others.
SOURCE: Grande E et al. ESMO 2019. Abstract LBA14_PR.
REPORTING FROM ESMO 2019
Ribociclib/fulvestrant boosts survival in advanced breast cancer
BARCELONA – In postmenopausal women with hormone receptor–positive, HER2-negative advanced breast cancer, the combination of ribociclib (Kisqali) and fulvestrant (Faslodex) was associated with a significant survival benefit, compared with fulvestrant alone, investigators reported.

Among 726 postmenopausal patients in MONALEESA-3 randomly assigned to receive either ribociclib or placebo plus fulvestrant who were followed for a median of 39.4 months, the median overall survival (OS) was not reached for patients assigned to ribociclib, compared with 40 months for patients in the control (fulvestrant-only) arm, reported Dennis J. Slamon, MD, PhD from the University of California, Los Angeles
“The combined data set of MONALEESA-3 and MONALEESA-7 represent some 1,400 patients, and the largest body of evidence for overall survival for any [cyclin-dependent kinases 4/6] inhibitor, and these data demonstrate a consistent, meaningful prolongation of survival irrespective of menopausal status, pre or post, and irrespective of whether the patient is treated in the first line or the second line,” he said at the European Society for Medical Oncology Congress.
Results of the MONALEESA-7 trial, which showed an overall survival advantage for adding ribociclib to endocrine therapy in premenopausal women with hormone receptor–positive, HER2-negative breast cancer, were reported at the 2019 annual meeting of the American Society of Clinical Oncology.
MONALEESA-3 investigators enrolled 726 postmenopausal women with hormone receptor–positive/HER2-negative advanced breast cancer who had received not more than one prior line of endocrine therapy for advanced disease, stratified them by the presence of liver and/or lung metastases and prior endocrine therapy, and then randomly assigned them to receive intramuscular fulvestrant 500 mg every 28 days, with an additional dose on day 15 of cycle one, plus either ribociclib 600 mg per day, 3 weeks on and 1 week off for every cycle, or placebo.
Dr. Slamon presented results of the primary endpoint of progression-free survival at the 2018 ASCO annual meeting, which showed a median PFS with ribociclib plus fulvestrant of 20.5 months versus 12.8 months for fulvestrant alone (hazard ratio, 5.93; P less than .0001). The overall survival data presented by Dr. Slamon at ESMO 2019 were not mature at that time.
As noted, at a median follow-up of 39.4 months, median overall survival was not reached in the combination arm, compared with 40 months in the fulvestrant/placebo arm, translating into a HR for risk of death with ribociclib of 0.724 (P = .00455).
Estimated OS rates at 36 months were 67% in the ribociclib arm versus 58.2% in the placebo arm, and at 42 months were 57.8% and 45.9%, respectively.
The survival benefit appeared consistent both for patients treated in the first line (median OS not reached vs. 45.1 months; HR, 0.70) and for those treated after early relapse of therapy for early breast or in the second line (median OS, 40.2 vs. 32.5 months, respectively; HR, 0.73). In both comparisons, however, the 95% confidence interval crossed 1, indicating that the results were not statistically significant.
An analysis of OS by subgroup indicated significant benefit or trends for most categories except Asian race, and treatment in Asia or Latin America, although the numbers of patients in each of those subgroups was less than 20, making it difficult to draw significant inferences from the findings, Dr. Slamon cautioned.
A descriptive analysis of updated PFS results were very similar to those previously reported, with a median of 20.6 months with ribociclib versus 12.8 months with placebo (HR, 0.587). Median PFS by line of therapy was 33.6 versus 19.2 months, respectively, in the first line, and 14.5 versus 9.1 months in the second line (HR, 0.546 and 0.571; P values not shown).

“CDK4/6 inhibitors not only improve progression-free survival in first- and second-line metastatic breast cancer, it also translates into an overall survival improvement. What more do we really want?” said invited discussant Sybil Loibl, MD, from Goethe University in Frankfurt, Germany.
“Outcome improves irrespective of pretreatment, menopausal status, endocrine sensitivity and site of metastases, which is good knowledge for our patients,” she said.
To date there are no biomarkers identified that can select subgroups that could experience particular benefit from CDK4/6 inhibitors. It may require a meta-analysis of all available clinical trial data on these agents in both the first and second line therapy to reveal potential differences among subgroups, she said.
MONALEESA-3 is supported by Novartis. Dr. Slamon disclosed a consulting or advisory role, research funding, honoraria, and travel expenses from Novartis and others. Dr. Loibl disclosed honoraria and research grants from Novartis and others.
SOURCE: Slamon DJ et al. ESMO 2019, Abstract LBA7_PR.
BARCELONA – In postmenopausal women with hormone receptor–positive, HER2-negative advanced breast cancer, the combination of ribociclib (Kisqali) and fulvestrant (Faslodex) was associated with a significant survival benefit, compared with fulvestrant alone, investigators reported.
Among 726 postmenopausal patients in MONALEESA-3 randomly assigned to receive either ribociclib or placebo plus fulvestrant who were followed for a median of 39.4 months, the median overall survival (OS) was not reached for patients assigned to ribociclib, compared with 40 months for patients in the control (fulvestrant-only) arm, reported Dennis J. Slamon, MD, PhD from the University of California, Los Angeles
“The combined data set of MONALEESA-3 and MONALEESA-7 represent some 1,400 patients, and the largest body of evidence for overall survival for any [cyclin-dependent kinases 4/6] inhibitor, and these data demonstrate a consistent, meaningful prolongation of survival irrespective of menopausal status, pre or post, and irrespective of whether the patient is treated in the first line or the second line,” he said at the European Society for Medical Oncology Congress.
Results of the MONALEESA-7 trial, which showed an overall survival advantage for adding ribociclib to endocrine therapy in premenopausal women with hormone receptor–positive, HER2-negative breast cancer, were reported at the 2019 annual meeting of the American Society of Clinical Oncology.
MONALEESA-3 investigators enrolled 726 postmenopausal women with hormone receptor–positive/HER2-negative advanced breast cancer who had received not more than one prior line of endocrine therapy for advanced disease, stratified them by the presence of liver and/or lung metastases and prior endocrine therapy, and then randomly assigned them to receive intramuscular fulvestrant 500 mg every 28 days, with an additional dose on day 15 of cycle one, plus either ribociclib 600 mg per day, 3 weeks on and 1 week off for every cycle, or placebo.
Dr. Slamon presented results of the primary endpoint of progression-free survival at the 2018 ASCO annual meeting, which showed a median PFS with ribociclib plus fulvestrant of 20.5 months versus 12.8 months for fulvestrant alone (hazard ratio, 5.93; P less than .0001). The overall survival data presented by Dr. Slamon at ESMO 2019 were not mature at that time.
As noted, at a median follow-up of 39.4 months, median overall survival was not reached in the combination arm, compared with 40 months in the fulvestrant/placebo arm, translating into a HR for risk of death with ribociclib of 0.724 (P = .00455).
Estimated OS rates at 36 months were 67% in the ribociclib arm versus 58.2% in the placebo arm, and at 42 months were 57.8% and 45.9%, respectively.
The survival benefit appeared consistent both for patients treated in the first line (median OS not reached vs. 45.1 months; HR, 0.70) and for those treated after early relapse of therapy for early breast or in the second line (median OS, 40.2 vs. 32.5 months, respectively; HR, 0.73). In both comparisons, however, the 95% confidence interval crossed 1, indicating that the results were not statistically significant.
An analysis of OS by subgroup indicated significant benefit or trends for most categories except Asian race, and treatment in Asia or Latin America, although the numbers of patients in each of those subgroups was less than 20, making it difficult to draw significant inferences from the findings, Dr. Slamon cautioned.
A descriptive analysis of updated PFS results were very similar to those previously reported, with a median of 20.6 months with ribociclib versus 12.8 months with placebo (HR, 0.587). Median PFS by line of therapy was 33.6 versus 19.2 months, respectively, in the first line, and 14.5 versus 9.1 months in the second line (HR, 0.546 and 0.571; P values not shown).
“CDK4/6 inhibitors not only improve progression-free survival in first- and second-line metastatic breast cancer, it also translates into an overall survival improvement. What more do we really want?” said invited discussant Sybil Loibl, MD, from Goethe University in Frankfurt, Germany.
“Outcome improves irrespective of pretreatment, menopausal status, endocrine sensitivity and site of metastases, which is good knowledge for our patients,” she said.
To date there are no biomarkers identified that can select subgroups that could experience particular benefit from CDK4/6 inhibitors. It may require a meta-analysis of all available clinical trial data on these agents in both the first and second line therapy to reveal potential differences among subgroups, she said.
MONALEESA-3 is supported by Novartis. Dr. Slamon disclosed a consulting or advisory role, research funding, honoraria, and travel expenses from Novartis and others. Dr. Loibl disclosed honoraria and research grants from Novartis and others.
SOURCE: Slamon DJ et al. ESMO 2019, Abstract LBA7_PR.
BARCELONA – In postmenopausal women with hormone receptor–positive, HER2-negative advanced breast cancer, the combination of ribociclib (Kisqali) and fulvestrant (Faslodex) was associated with a significant survival benefit, compared with fulvestrant alone, investigators reported.
Among 726 postmenopausal patients in MONALEESA-3 randomly assigned to receive either ribociclib or placebo plus fulvestrant who were followed for a median of 39.4 months, the median overall survival (OS) was not reached for patients assigned to ribociclib, compared with 40 months for patients in the control (fulvestrant-only) arm, reported Dennis J. Slamon, MD, PhD from the University of California, Los Angeles
“The combined data set of MONALEESA-3 and MONALEESA-7 represent some 1,400 patients, and the largest body of evidence for overall survival for any [cyclin-dependent kinases 4/6] inhibitor, and these data demonstrate a consistent, meaningful prolongation of survival irrespective of menopausal status, pre or post, and irrespective of whether the patient is treated in the first line or the second line,” he said at the European Society for Medical Oncology Congress.
Results of the MONALEESA-7 trial, which showed an overall survival advantage for adding ribociclib to endocrine therapy in premenopausal women with hormone receptor–positive, HER2-negative breast cancer, were reported at the 2019 annual meeting of the American Society of Clinical Oncology.
MONALEESA-3 investigators enrolled 726 postmenopausal women with hormone receptor–positive/HER2-negative advanced breast cancer who had received not more than one prior line of endocrine therapy for advanced disease, stratified them by the presence of liver and/or lung metastases and prior endocrine therapy, and then randomly assigned them to receive intramuscular fulvestrant 500 mg every 28 days, with an additional dose on day 15 of cycle one, plus either ribociclib 600 mg per day, 3 weeks on and 1 week off for every cycle, or placebo.
Dr. Slamon presented results of the primary endpoint of progression-free survival at the 2018 ASCO annual meeting, which showed a median PFS with ribociclib plus fulvestrant of 20.5 months versus 12.8 months for fulvestrant alone (hazard ratio, 5.93; P less than .0001). The overall survival data presented by Dr. Slamon at ESMO 2019 were not mature at that time.
As noted, at a median follow-up of 39.4 months, median overall survival was not reached in the combination arm, compared with 40 months in the fulvestrant/placebo arm, translating into a HR for risk of death with ribociclib of 0.724 (P = .00455).
Estimated OS rates at 36 months were 67% in the ribociclib arm versus 58.2% in the placebo arm, and at 42 months were 57.8% and 45.9%, respectively.
The survival benefit appeared consistent both for patients treated in the first line (median OS not reached vs. 45.1 months; HR, 0.70) and for those treated after early relapse of therapy for early breast or in the second line (median OS, 40.2 vs. 32.5 months, respectively; HR, 0.73). In both comparisons, however, the 95% confidence interval crossed 1, indicating that the results were not statistically significant.
An analysis of OS by subgroup indicated significant benefit or trends for most categories except Asian race, and treatment in Asia or Latin America, although the numbers of patients in each of those subgroups was less than 20, making it difficult to draw significant inferences from the findings, Dr. Slamon cautioned.
A descriptive analysis of updated PFS results were very similar to those previously reported, with a median of 20.6 months with ribociclib versus 12.8 months with placebo (HR, 0.587). Median PFS by line of therapy was 33.6 versus 19.2 months, respectively, in the first line, and 14.5 versus 9.1 months in the second line (HR, 0.546 and 0.571; P values not shown).
“CDK4/6 inhibitors not only improve progression-free survival in first- and second-line metastatic breast cancer, it also translates into an overall survival improvement. What more do we really want?” said invited discussant Sybil Loibl, MD, from Goethe University in Frankfurt, Germany.
“Outcome improves irrespective of pretreatment, menopausal status, endocrine sensitivity and site of metastases, which is good knowledge for our patients,” she said.
To date there are no biomarkers identified that can select subgroups that could experience particular benefit from CDK4/6 inhibitors. It may require a meta-analysis of all available clinical trial data on these agents in both the first and second line therapy to reveal potential differences among subgroups, she said.
MONALEESA-3 is supported by Novartis. Dr. Slamon disclosed a consulting or advisory role, research funding, honoraria, and travel expenses from Novartis and others. Dr. Loibl disclosed honoraria and research grants from Novartis and others.
SOURCE: Slamon DJ et al. ESMO 2019, Abstract LBA7_PR.
REPORTING FROM ESMO 2019