User login
The Epidemiology and Clinical Associations of Portal Vein Thrombosis in Hospitalized Patients With Cirrhosis: A Nationwide Analysis From the National Inpatient Sample
Portal vein thrombosis (PVT) is thought to be rare in the general population and is most commonly found among patients with cirrhosis.1-3 The risk of developing PVT in patients with cirrhosis has been correlated with the severity of hepatic impairment.4,5 There is a lack of national-level data on the epidemiology of PVT and its related outcomes in the inpatient setting. The aim of our study was to describe the prevalence of PVT in hospitalized patients with cirrhosis in the United States. Using the National Inpatient Sample (NIS) database, we described the differences in hepatic decompensation, length of stay, in-hospital mortality, and total charges between patients with cirrhosis with PVT and those without.
METHODS
This study was performed using the 2012 NIS to assess the relationship between PVT and cirrhosis-related outcomes. The NIS has been used reliably to make national estimates of healthcare utilization and estimate disease burden, charges, and outcomes.6 All admissions with either a primary or secondary discharge diagnosis of an International Classification of Diseases, 9th Revision–Clinical Modification (ICD-9-CM) code for PVT (452) and cirrhosis (571.2, 571.5, and 571.6) were identified from the NIS and correlated with age, gender, inpatient length of stay, in-hospital mortality, total charges, and commonly associated diagnoses. Complications of cirrhosis, such as hepatic encephalopathy (572.2), abdominal ascites (789.5), and gastrointestinal bleeding (456 and 456.2), were also identified. Data were assessed using IBM Statistical Package for the Social Sciences Statistics version 19.0 (Chicago, IL). Statistical significance was defined as a P value < .05.
RESULTS
There were 7,296,968 total unweighted admissions in the 2012 NIS, which included 113,766 (1.6%) inpatient admissions for cirrhosis, with 61,867 for nonalcoholic cirrhosis, 49,698 for alcoholic cirrhosis, and 2202 for biliary cirrhosis. The prevalence of PVT among all inpatient admissions was 0.07% (n = 5046) and 1.8% (n = 2046) in patients with cirrhosis (P < .001). On univariate analysis, patients who had a diagnosis of both cirrhosis and PVT had higher proportions of hepatic encephalopathy (22.5% vs 17.7%; P < .00001) as well as gastrointestinal bleeding (11.6% vs 5.7%; P < .00001) as compared with patients with cirrhosis without PVT (Figure). 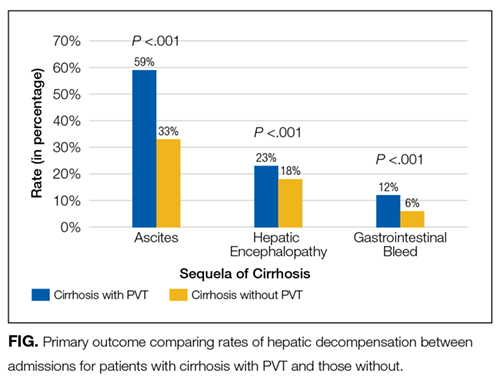
DISCUSSION
We found that hospitalized patients with concurrent diagnoses of cirrhosis and PVT had longer hospital length of stay, higher mean hospital charges, and a higher proportion of cirrhosis-related complications. Our study represents the largest examination of hospitalized patients with cirrhosis and PVT to date and contributes to the evolving understanding of PVT in end-stage liver disease. The relationship between cirrhotic complications and PVT may be independent, but the 2 have similar underlying etiologic processes. Thus, given our findings, intervening to address the underlying factors leading to microvascular and/or PVT or mitigating the propagation of PVT in patients with cirrhosis may be beneficial to reducing morbidity and mortality in these patients. In addition, the prevalence of PVT in the overall hospitalized patient population in our study (0.07%) was similar to the 0.05% to 0.5% previously described in a US autopsy series, which should decrease the likelihood that PVT was missed in the cirrhotic population, which is more likely to have inpatient ultrasound imaging.2 Our study is limited by its retrospective nature, dependency on ICD-9-CM codes for extracting data, and lack of clinical, physical exam, and laboratory results to allow for the calculation of a model for the end-stage liver disease and Child-Pugh score. Also, the study was not designed to evaluate causation, and it is possible that patients with more severe cirrhosis were more likely to be diagnosed with PVT. Further prospective studies directed not only toward the mechanism and treatment of both micro- and macrovascular thrombosis but also at examining the prevention of PVT and attendant benefits are greatly needed.
Disclosure
The authors have nothing to disclose. The contents of this work do not represent the views of the Department of Veterans Affairs or the United States Government.
1. Kumar A, Sharma P, Arora A. Review article: portal vein obstruction—epidemiology, pathogenesis, natural history, prognosis and treatment. Aliment Pharmacol Ther. 2015;41(3):276-292. PubMed
2. Ogren M, Bergqvist D, Björck M, et al. Portal vein thrombosis: prevalence, patient characteristics and lifetime risk: a population study based on 23,796 consecutive autopsies. World J Gastroenterol. 2006;12(13):2115-2119. PubMed
3. Ponziani FR, Zocco MA, Garcovich M, et al. What we should know about portal vein thrombosis in cirrhotic patients: a changing perspective. World J Gastroenterol. 2012;18(36):5014-5020. PubMed
4. Francoz C, Belghiti J, Vilgrain V, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut. 2005;54(5):691-697. PubMed
5. Okuda K, Ohnishi K, Kimura K, et al. Incidence of portal vein thrombosis in liver cirrhosis. An angiographic study in 708 patients. Gastroenterology. 1985;89(2):279-286. PubMed
6. Agency for Healthcare Research and Quality Introduction to the HCUP Nationwide Inpatient Sample 2011. Healthcare Cost and Utilization Project (HCUP) website. https://www.hcup-us.ahrq.gov/reports/methods/2014-04.pdf. Accessed January 30, 2017.
Portal vein thrombosis (PVT) is thought to be rare in the general population and is most commonly found among patients with cirrhosis.1-3 The risk of developing PVT in patients with cirrhosis has been correlated with the severity of hepatic impairment.4,5 There is a lack of national-level data on the epidemiology of PVT and its related outcomes in the inpatient setting. The aim of our study was to describe the prevalence of PVT in hospitalized patients with cirrhosis in the United States. Using the National Inpatient Sample (NIS) database, we described the differences in hepatic decompensation, length of stay, in-hospital mortality, and total charges between patients with cirrhosis with PVT and those without.
METHODS
This study was performed using the 2012 NIS to assess the relationship between PVT and cirrhosis-related outcomes. The NIS has been used reliably to make national estimates of healthcare utilization and estimate disease burden, charges, and outcomes.6 All admissions with either a primary or secondary discharge diagnosis of an International Classification of Diseases, 9th Revision–Clinical Modification (ICD-9-CM) code for PVT (452) and cirrhosis (571.2, 571.5, and 571.6) were identified from the NIS and correlated with age, gender, inpatient length of stay, in-hospital mortality, total charges, and commonly associated diagnoses. Complications of cirrhosis, such as hepatic encephalopathy (572.2), abdominal ascites (789.5), and gastrointestinal bleeding (456 and 456.2), were also identified. Data were assessed using IBM Statistical Package for the Social Sciences Statistics version 19.0 (Chicago, IL). Statistical significance was defined as a P value < .05.
RESULTS
There were 7,296,968 total unweighted admissions in the 2012 NIS, which included 113,766 (1.6%) inpatient admissions for cirrhosis, with 61,867 for nonalcoholic cirrhosis, 49,698 for alcoholic cirrhosis, and 2202 for biliary cirrhosis. The prevalence of PVT among all inpatient admissions was 0.07% (n = 5046) and 1.8% (n = 2046) in patients with cirrhosis (P < .001). On univariate analysis, patients who had a diagnosis of both cirrhosis and PVT had higher proportions of hepatic encephalopathy (22.5% vs 17.7%; P < .00001) as well as gastrointestinal bleeding (11.6% vs 5.7%; P < .00001) as compared with patients with cirrhosis without PVT (Figure).
DISCUSSION
We found that hospitalized patients with concurrent diagnoses of cirrhosis and PVT had longer hospital length of stay, higher mean hospital charges, and a higher proportion of cirrhosis-related complications. Our study represents the largest examination of hospitalized patients with cirrhosis and PVT to date and contributes to the evolving understanding of PVT in end-stage liver disease. The relationship between cirrhotic complications and PVT may be independent, but the 2 have similar underlying etiologic processes. Thus, given our findings, intervening to address the underlying factors leading to microvascular and/or PVT or mitigating the propagation of PVT in patients with cirrhosis may be beneficial to reducing morbidity and mortality in these patients. In addition, the prevalence of PVT in the overall hospitalized patient population in our study (0.07%) was similar to the 0.05% to 0.5% previously described in a US autopsy series, which should decrease the likelihood that PVT was missed in the cirrhotic population, which is more likely to have inpatient ultrasound imaging.2 Our study is limited by its retrospective nature, dependency on ICD-9-CM codes for extracting data, and lack of clinical, physical exam, and laboratory results to allow for the calculation of a model for the end-stage liver disease and Child-Pugh score. Also, the study was not designed to evaluate causation, and it is possible that patients with more severe cirrhosis were more likely to be diagnosed with PVT. Further prospective studies directed not only toward the mechanism and treatment of both micro- and macrovascular thrombosis but also at examining the prevention of PVT and attendant benefits are greatly needed.
Disclosure
The authors have nothing to disclose. The contents of this work do not represent the views of the Department of Veterans Affairs or the United States Government.
Portal vein thrombosis (PVT) is thought to be rare in the general population and is most commonly found among patients with cirrhosis.1-3 The risk of developing PVT in patients with cirrhosis has been correlated with the severity of hepatic impairment.4,5 There is a lack of national-level data on the epidemiology of PVT and its related outcomes in the inpatient setting. The aim of our study was to describe the prevalence of PVT in hospitalized patients with cirrhosis in the United States. Using the National Inpatient Sample (NIS) database, we described the differences in hepatic decompensation, length of stay, in-hospital mortality, and total charges between patients with cirrhosis with PVT and those without.
METHODS
This study was performed using the 2012 NIS to assess the relationship between PVT and cirrhosis-related outcomes. The NIS has been used reliably to make national estimates of healthcare utilization and estimate disease burden, charges, and outcomes.6 All admissions with either a primary or secondary discharge diagnosis of an International Classification of Diseases, 9th Revision–Clinical Modification (ICD-9-CM) code for PVT (452) and cirrhosis (571.2, 571.5, and 571.6) were identified from the NIS and correlated with age, gender, inpatient length of stay, in-hospital mortality, total charges, and commonly associated diagnoses. Complications of cirrhosis, such as hepatic encephalopathy (572.2), abdominal ascites (789.5), and gastrointestinal bleeding (456 and 456.2), were also identified. Data were assessed using IBM Statistical Package for the Social Sciences Statistics version 19.0 (Chicago, IL). Statistical significance was defined as a P value < .05.
RESULTS
There were 7,296,968 total unweighted admissions in the 2012 NIS, which included 113,766 (1.6%) inpatient admissions for cirrhosis, with 61,867 for nonalcoholic cirrhosis, 49,698 for alcoholic cirrhosis, and 2202 for biliary cirrhosis. The prevalence of PVT among all inpatient admissions was 0.07% (n = 5046) and 1.8% (n = 2046) in patients with cirrhosis (P < .001). On univariate analysis, patients who had a diagnosis of both cirrhosis and PVT had higher proportions of hepatic encephalopathy (22.5% vs 17.7%; P < .00001) as well as gastrointestinal bleeding (11.6% vs 5.7%; P < .00001) as compared with patients with cirrhosis without PVT (Figure).
DISCUSSION
We found that hospitalized patients with concurrent diagnoses of cirrhosis and PVT had longer hospital length of stay, higher mean hospital charges, and a higher proportion of cirrhosis-related complications. Our study represents the largest examination of hospitalized patients with cirrhosis and PVT to date and contributes to the evolving understanding of PVT in end-stage liver disease. The relationship between cirrhotic complications and PVT may be independent, but the 2 have similar underlying etiologic processes. Thus, given our findings, intervening to address the underlying factors leading to microvascular and/or PVT or mitigating the propagation of PVT in patients with cirrhosis may be beneficial to reducing morbidity and mortality in these patients. In addition, the prevalence of PVT in the overall hospitalized patient population in our study (0.07%) was similar to the 0.05% to 0.5% previously described in a US autopsy series, which should decrease the likelihood that PVT was missed in the cirrhotic population, which is more likely to have inpatient ultrasound imaging.2 Our study is limited by its retrospective nature, dependency on ICD-9-CM codes for extracting data, and lack of clinical, physical exam, and laboratory results to allow for the calculation of a model for the end-stage liver disease and Child-Pugh score. Also, the study was not designed to evaluate causation, and it is possible that patients with more severe cirrhosis were more likely to be diagnosed with PVT. Further prospective studies directed not only toward the mechanism and treatment of both micro- and macrovascular thrombosis but also at examining the prevention of PVT and attendant benefits are greatly needed.
Disclosure
The authors have nothing to disclose. The contents of this work do not represent the views of the Department of Veterans Affairs or the United States Government.
1. Kumar A, Sharma P, Arora A. Review article: portal vein obstruction—epidemiology, pathogenesis, natural history, prognosis and treatment. Aliment Pharmacol Ther. 2015;41(3):276-292. PubMed
2. Ogren M, Bergqvist D, Björck M, et al. Portal vein thrombosis: prevalence, patient characteristics and lifetime risk: a population study based on 23,796 consecutive autopsies. World J Gastroenterol. 2006;12(13):2115-2119. PubMed
3. Ponziani FR, Zocco MA, Garcovich M, et al. What we should know about portal vein thrombosis in cirrhotic patients: a changing perspective. World J Gastroenterol. 2012;18(36):5014-5020. PubMed
4. Francoz C, Belghiti J, Vilgrain V, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut. 2005;54(5):691-697. PubMed
5. Okuda K, Ohnishi K, Kimura K, et al. Incidence of portal vein thrombosis in liver cirrhosis. An angiographic study in 708 patients. Gastroenterology. 1985;89(2):279-286. PubMed
6. Agency for Healthcare Research and Quality Introduction to the HCUP Nationwide Inpatient Sample 2011. Healthcare Cost and Utilization Project (HCUP) website. https://www.hcup-us.ahrq.gov/reports/methods/2014-04.pdf. Accessed January 30, 2017.
1. Kumar A, Sharma P, Arora A. Review article: portal vein obstruction—epidemiology, pathogenesis, natural history, prognosis and treatment. Aliment Pharmacol Ther. 2015;41(3):276-292. PubMed
2. Ogren M, Bergqvist D, Björck M, et al. Portal vein thrombosis: prevalence, patient characteristics and lifetime risk: a population study based on 23,796 consecutive autopsies. World J Gastroenterol. 2006;12(13):2115-2119. PubMed
3. Ponziani FR, Zocco MA, Garcovich M, et al. What we should know about portal vein thrombosis in cirrhotic patients: a changing perspective. World J Gastroenterol. 2012;18(36):5014-5020. PubMed
4. Francoz C, Belghiti J, Vilgrain V, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut. 2005;54(5):691-697. PubMed
5. Okuda K, Ohnishi K, Kimura K, et al. Incidence of portal vein thrombosis in liver cirrhosis. An angiographic study in 708 patients. Gastroenterology. 1985;89(2):279-286. PubMed
6. Agency for Healthcare Research and Quality Introduction to the HCUP Nationwide Inpatient Sample 2011. Healthcare Cost and Utilization Project (HCUP) website. https://www.hcup-us.ahrq.gov/reports/methods/2014-04.pdf. Accessed January 30, 2017.
© 2017 Society of Hospital Medicine
The pediatrician detective and high lead levels
I am not going to tell you about the dangers of lead, as it is well known and publicized, but I will tell you my family’s story with lead.
In 2012, 1 year after my younger daughter was born, I took her for her 1-year checkup. As I would do with any of my pediatric patients at this age, I took her for a lead level check. Never during my residency training or my first few years of practice as a pediatrician have I encountered a positive lead level. So when I opened the lab result sheet, I thought I would be shredding it the next moment. Well, that didn’t happen. It turned out that her lead level was 7 mcg/dL! Not too high, but detectable. The only question that kept on coming back over the next month or so was a big WHY? Why my child? Now my older daughter’s lead level was normal at her 1-year visit. We had just moved into a new house before my youngest daughter was born. I thought, it has to do with the house, and since my 1-year-old was putting everything in her mouth at this stage, then she must be getting the lead that way.

So it was not the house or the wall pipes that were contaminated with lead. It was not our food that we cooked, otherwise my nanny’s daughter would have had a high lead level, as she ate the same food we ate almost daily. Our family did not travel recently. So what was it that my family had or ate that my neighbor or nanny’s child did not?
The answer was thyme. It is an herb that we mix with olive oil and spread on dough – I call it Lebanese pizza. That is one thing that my nanny and her child never ate, but we did. It was a long painful month of investigation, elimination, and anxiety. I called the public health department in Phoenix and they stated that lots of imported spices were contaminated with lead. There were two theories as to why this might happen. The first one is that the spice dealers would add lead to increase the weight of the spices to get more money. The second is that the spice fields were close to factories that used lead in their manufacturing, and somehow the lead would contaminate the nearby fields where the spices grew.
The type of thyme we used was bought in Syria and packaged in Lebanon. It was not the pure organic type that we usually got from our grandparents in our southern Lebanese village. This packaged thyme had lot of nuts added to it to give it more flavor.
The public health department official asked that I send her some samples of all the spices that I had. I packed up to ten different spice bags including the thyme. Two weeks later she called me, stating that the lead level allowable in spices must be less than 10, and that our thyme’s lead level was 900!
We got rid of all the spices, and have never eaten that packaged spice again. My kids’ lead levels dropped nicely afterward and back to normal. That is our story with lead. Now it seems like a mini-detective story and even fun, but the anxiety that I experienced until we figured out the cause was not!
Dr. Faddoul is a private practice pediatrician in La Canada Flintridge, Calif.
I am not going to tell you about the dangers of lead, as it is well known and publicized, but I will tell you my family’s story with lead.
In 2012, 1 year after my younger daughter was born, I took her for her 1-year checkup. As I would do with any of my pediatric patients at this age, I took her for a lead level check. Never during my residency training or my first few years of practice as a pediatrician have I encountered a positive lead level. So when I opened the lab result sheet, I thought I would be shredding it the next moment. Well, that didn’t happen. It turned out that her lead level was 7 mcg/dL! Not too high, but detectable. The only question that kept on coming back over the next month or so was a big WHY? Why my child? Now my older daughter’s lead level was normal at her 1-year visit. We had just moved into a new house before my youngest daughter was born. I thought, it has to do with the house, and since my 1-year-old was putting everything in her mouth at this stage, then she must be getting the lead that way.
So it was not the house or the wall pipes that were contaminated with lead. It was not our food that we cooked, otherwise my nanny’s daughter would have had a high lead level, as she ate the same food we ate almost daily. Our family did not travel recently. So what was it that my family had or ate that my neighbor or nanny’s child did not?
The answer was thyme. It is an herb that we mix with olive oil and spread on dough – I call it Lebanese pizza. That is one thing that my nanny and her child never ate, but we did. It was a long painful month of investigation, elimination, and anxiety. I called the public health department in Phoenix and they stated that lots of imported spices were contaminated with lead. There were two theories as to why this might happen. The first one is that the spice dealers would add lead to increase the weight of the spices to get more money. The second is that the spice fields were close to factories that used lead in their manufacturing, and somehow the lead would contaminate the nearby fields where the spices grew.
The type of thyme we used was bought in Syria and packaged in Lebanon. It was not the pure organic type that we usually got from our grandparents in our southern Lebanese village. This packaged thyme had lot of nuts added to it to give it more flavor.
The public health department official asked that I send her some samples of all the spices that I had. I packed up to ten different spice bags including the thyme. Two weeks later she called me, stating that the lead level allowable in spices must be less than 10, and that our thyme’s lead level was 900!
We got rid of all the spices, and have never eaten that packaged spice again. My kids’ lead levels dropped nicely afterward and back to normal. That is our story with lead. Now it seems like a mini-detective story and even fun, but the anxiety that I experienced until we figured out the cause was not!
Dr. Faddoul is a private practice pediatrician in La Canada Flintridge, Calif.
I am not going to tell you about the dangers of lead, as it is well known and publicized, but I will tell you my family’s story with lead.
In 2012, 1 year after my younger daughter was born, I took her for her 1-year checkup. As I would do with any of my pediatric patients at this age, I took her for a lead level check. Never during my residency training or my first few years of practice as a pediatrician have I encountered a positive lead level. So when I opened the lab result sheet, I thought I would be shredding it the next moment. Well, that didn’t happen. It turned out that her lead level was 7 mcg/dL! Not too high, but detectable. The only question that kept on coming back over the next month or so was a big WHY? Why my child? Now my older daughter’s lead level was normal at her 1-year visit. We had just moved into a new house before my youngest daughter was born. I thought, it has to do with the house, and since my 1-year-old was putting everything in her mouth at this stage, then she must be getting the lead that way.
So it was not the house or the wall pipes that were contaminated with lead. It was not our food that we cooked, otherwise my nanny’s daughter would have had a high lead level, as she ate the same food we ate almost daily. Our family did not travel recently. So what was it that my family had or ate that my neighbor or nanny’s child did not?
The answer was thyme. It is an herb that we mix with olive oil and spread on dough – I call it Lebanese pizza. That is one thing that my nanny and her child never ate, but we did. It was a long painful month of investigation, elimination, and anxiety. I called the public health department in Phoenix and they stated that lots of imported spices were contaminated with lead. There were two theories as to why this might happen. The first one is that the spice dealers would add lead to increase the weight of the spices to get more money. The second is that the spice fields were close to factories that used lead in their manufacturing, and somehow the lead would contaminate the nearby fields where the spices grew.
The type of thyme we used was bought in Syria and packaged in Lebanon. It was not the pure organic type that we usually got from our grandparents in our southern Lebanese village. This packaged thyme had lot of nuts added to it to give it more flavor.
The public health department official asked that I send her some samples of all the spices that I had. I packed up to ten different spice bags including the thyme. Two weeks later she called me, stating that the lead level allowable in spices must be less than 10, and that our thyme’s lead level was 900!
We got rid of all the spices, and have never eaten that packaged spice again. My kids’ lead levels dropped nicely afterward and back to normal. That is our story with lead. Now it seems like a mini-detective story and even fun, but the anxiety that I experienced until we figured out the cause was not!
Dr. Faddoul is a private practice pediatrician in La Canada Flintridge, Calif.
Introducing the VA Boston Medical Forum
The case history has been the cornerstone of clinical learning since the first record of medical encounters in ancient Egypt.1 The methodical process of taking a patient history by Hippocratic physicians enabled an empirical approach to medicine centuries before the scientific revolution. From Freud in psychiatry to Giovanni Morgagni in pathology—case reports have been the time-honored and time-tested vehicle for teaching medicine.2
Most American physicians grew up reading the most famous modern series of histories, the “Case Records of the Massachusetts General Hospital,” published in that pinnacle of medical scholarship, The New England Journal of Medicine. Now, also from Boston, I’m proud to announce that Federal Practitioner has its own case series, The VA Boston Medical Forum (HIV-Positive Veteran With Progressive Visual Changes, page 18).
The VA Boston Medical Forum is a printed (and electronic, these days) version of the case conferences held at the flagship VA Boston Healthcare System (VABHS), which has academic affiliations with the Boston Medical Center, Beth Israel Deaconess Medical Center, and Brigham and Women’s Hospital. Brian Hoffman, professor emeritus at Harvard Medical School, who previously served as the chief of internal medicine at the VABHS, founded the series, which has continued for more than 10 years.
The didactic driving force of this medical forum are the VABHS chief medical residents and their director of residency education. It is—as you will see in this issue—a case report taken from a weekly multidisciplinary conference. We feel the authors have captured much of the interactive ambience of those case conferences, including laboratory values, medical images, extensive references, and takeaway points, as though you were there at morning rounds.
Each case involves a VA patient and presents in traditional case history format a discussion of the diagnosis and treatment of a challenging patient. Just as they do at the actual case conferences, the chief medical residents moderate these discussions, which also feature expert opinions from nationally recognized leaders in their respective medical specialties.
From the many cases they present, the chief medical residents and their director of residency education will select cases that focus on clinical problems relevant to those caring for veterans, such as homelessness, comorbid substance use disorders, along with thought provoking and complex medical presentations that will test the clinical reasoning of the most experienced diagnostician.
Over many years as a medical educator, I have come to believe that whether it is ethics or surgery, we all learn best from an interesting case history and a good medical mystery. We hope to provide both in this conversational, question-and-answer format. Think back to your days on the wards: You can have all that intellectual stimulation without the night call and “pimping.” So from the comfort of your favorite reading spot, we invite you to sit back and enjoy. This is continuing medical education at its best, and I am proud to welcome our readers to the inaugural case of what we at Federal Practitioner hope will be an enduring feature. We thank the authors of the Boston Medical Forum for their dedication to enhancing VA academic medicine and, most important, helping us all to be smarter caregivers for our veterans.
1. Nissen T, Wynn R. The history of the case report: a selective review. JRSM Open. 2014;5(4): 2054270414523410.
2. Nuland SB. Doctors: The Biography of Medicine. New York: Alfred Knopf, 1988.
The case history has been the cornerstone of clinical learning since the first record of medical encounters in ancient Egypt.1 The methodical process of taking a patient history by Hippocratic physicians enabled an empirical approach to medicine centuries before the scientific revolution. From Freud in psychiatry to Giovanni Morgagni in pathology—case reports have been the time-honored and time-tested vehicle for teaching medicine.2
Most American physicians grew up reading the most famous modern series of histories, the “Case Records of the Massachusetts General Hospital,” published in that pinnacle of medical scholarship, The New England Journal of Medicine. Now, also from Boston, I’m proud to announce that Federal Practitioner has its own case series, The VA Boston Medical Forum (HIV-Positive Veteran With Progressive Visual Changes, page 18).
The VA Boston Medical Forum is a printed (and electronic, these days) version of the case conferences held at the flagship VA Boston Healthcare System (VABHS), which has academic affiliations with the Boston Medical Center, Beth Israel Deaconess Medical Center, and Brigham and Women’s Hospital. Brian Hoffman, professor emeritus at Harvard Medical School, who previously served as the chief of internal medicine at the VABHS, founded the series, which has continued for more than 10 years.
The didactic driving force of this medical forum are the VABHS chief medical residents and their director of residency education. It is—as you will see in this issue—a case report taken from a weekly multidisciplinary conference. We feel the authors have captured much of the interactive ambience of those case conferences, including laboratory values, medical images, extensive references, and takeaway points, as though you were there at morning rounds.
Each case involves a VA patient and presents in traditional case history format a discussion of the diagnosis and treatment of a challenging patient. Just as they do at the actual case conferences, the chief medical residents moderate these discussions, which also feature expert opinions from nationally recognized leaders in their respective medical specialties.
From the many cases they present, the chief medical residents and their director of residency education will select cases that focus on clinical problems relevant to those caring for veterans, such as homelessness, comorbid substance use disorders, along with thought provoking and complex medical presentations that will test the clinical reasoning of the most experienced diagnostician.
Over many years as a medical educator, I have come to believe that whether it is ethics or surgery, we all learn best from an interesting case history and a good medical mystery. We hope to provide both in this conversational, question-and-answer format. Think back to your days on the wards: You can have all that intellectual stimulation without the night call and “pimping.” So from the comfort of your favorite reading spot, we invite you to sit back and enjoy. This is continuing medical education at its best, and I am proud to welcome our readers to the inaugural case of what we at Federal Practitioner hope will be an enduring feature. We thank the authors of the Boston Medical Forum for their dedication to enhancing VA academic medicine and, most important, helping us all to be smarter caregivers for our veterans.
The case history has been the cornerstone of clinical learning since the first record of medical encounters in ancient Egypt.1 The methodical process of taking a patient history by Hippocratic physicians enabled an empirical approach to medicine centuries before the scientific revolution. From Freud in psychiatry to Giovanni Morgagni in pathology—case reports have been the time-honored and time-tested vehicle for teaching medicine.2
Most American physicians grew up reading the most famous modern series of histories, the “Case Records of the Massachusetts General Hospital,” published in that pinnacle of medical scholarship, The New England Journal of Medicine. Now, also from Boston, I’m proud to announce that Federal Practitioner has its own case series, The VA Boston Medical Forum (HIV-Positive Veteran With Progressive Visual Changes, page 18).
The VA Boston Medical Forum is a printed (and electronic, these days) version of the case conferences held at the flagship VA Boston Healthcare System (VABHS), which has academic affiliations with the Boston Medical Center, Beth Israel Deaconess Medical Center, and Brigham and Women’s Hospital. Brian Hoffman, professor emeritus at Harvard Medical School, who previously served as the chief of internal medicine at the VABHS, founded the series, which has continued for more than 10 years.
The didactic driving force of this medical forum are the VABHS chief medical residents and their director of residency education. It is—as you will see in this issue—a case report taken from a weekly multidisciplinary conference. We feel the authors have captured much of the interactive ambience of those case conferences, including laboratory values, medical images, extensive references, and takeaway points, as though you were there at morning rounds.
Each case involves a VA patient and presents in traditional case history format a discussion of the diagnosis and treatment of a challenging patient. Just as they do at the actual case conferences, the chief medical residents moderate these discussions, which also feature expert opinions from nationally recognized leaders in their respective medical specialties.
From the many cases they present, the chief medical residents and their director of residency education will select cases that focus on clinical problems relevant to those caring for veterans, such as homelessness, comorbid substance use disorders, along with thought provoking and complex medical presentations that will test the clinical reasoning of the most experienced diagnostician.
Over many years as a medical educator, I have come to believe that whether it is ethics or surgery, we all learn best from an interesting case history and a good medical mystery. We hope to provide both in this conversational, question-and-answer format. Think back to your days on the wards: You can have all that intellectual stimulation without the night call and “pimping.” So from the comfort of your favorite reading spot, we invite you to sit back and enjoy. This is continuing medical education at its best, and I am proud to welcome our readers to the inaugural case of what we at Federal Practitioner hope will be an enduring feature. We thank the authors of the Boston Medical Forum for their dedication to enhancing VA academic medicine and, most important, helping us all to be smarter caregivers for our veterans.
1. Nissen T, Wynn R. The history of the case report: a selective review. JRSM Open. 2014;5(4): 2054270414523410.
2. Nuland SB. Doctors: The Biography of Medicine. New York: Alfred Knopf, 1988.
1. Nissen T, Wynn R. The history of the case report: a selective review. JRSM Open. 2014;5(4): 2054270414523410.
2. Nuland SB. Doctors: The Biography of Medicine. New York: Alfred Knopf, 1988.
Advanced Stage and Relapsed/Refractory Hodgkin Lymphoma
INTRODUCTION
Hodgkin lymphoma, previously known as Hodgkin’s disease, is a B-cell lymphoproliferative disease characterized by a unique set of pathologic and epidemiologic features. The disease is characterized by the presence of multinucleate giant cells called Hodgkin Reed-Sternberg (HRS) cells.1 Hodgkin lymphoma is unique compared to other B-cell lymphomas because of the relative rarity of the malignant cells within affected tissues. The HRS cells, which usually account for only 0.1% to 10% of the cells, induce accumulation of nonmalignant lymphocytes, macrophages, granulocytes, eosinophils, plasma cells, and histiocytes, which then constitute the majority of tumor cellularity.2 Although the disease was first described by Sir Thomas Hodgkin in 1832, in part because of this unique histopathology, it was not until the 1990s that it was conclusively demonstrated that HRS cells are in fact monoclonal germinal center–derived B cells.
Due to the development of highly effective therapies for Hodgkin lymphoma, cure is a reasonable goal for most patients. Because of the high cure rate, late complications of therapy must be considered when selecting treatment. This article reviews the clinical features and treatment options for advanced stage and relapsed/refractory Hodgkin lymphoma. A previously published article reviewed the epidemiology, etiology/pathogenesis, pathologic classification, initial workup, and staging evaluation of Hodgkin lymphoma, as well as the prognostic stratification and treatment of patients with early-stage Hodgkin lymphoma.3
PRESENTATION, INITIAL EVALUATION, AND PROGNOSIS
Overall, classical Hodgkin lymphoma (cHL) usually presents with asymptomatic mediastinal or cervical lymphadenopathy. At least 50% of patients will have stage I or II disease.4 A mediastinal mass is seen in most patients with nodular sclerosis cHL, at times showing the characteristics of bulky (> 10 cm) disease. Constitutional, or B, symptoms (fever, night sweats, and weight loss) are present in approximately 25% of all patients with cHL, but 50% of advanced stage patients. Between 10% and 15% of patients will have extranodal disease, most commonly involving lung, bone, and liver. Lymphocyte-predominant Hodgkin lymphoma (LPHL) is a rare histological subtype of Hodgkin lymphoma that is differentiated from cHL by distinct clinicopathological features. The clinical course and treatment approach for LPHL are dependent upon the stage of disease. The clinicopathological features of LPHL are discussed in the early-stage Hodgkin lymphoma article.3
For the purposes of prognosis and selection of treatment, Hodgkin lymphoma is commonly classified as early stage favorable, early stage unfavorable, and advanced stage. For advanced stage Hodgkin lymphoma patients, prognosis can be defined using a tool commonly referred to as the International Prognostic Score (IPS). This index consists of 7 factors: male gender, age 45 years or older, stage IV disease, hemoglobin < 10.5 g/dL, white blood cell (WBC) count > 15,000/μL, lymphopenia (absolute lymphocyte count < 600 cells/μL or lymphocytes < 8% of WBC count), and serum albumin < 4 g/dL.5 In the original study by Hasenclever et al,5 the 5-year freedom from progression (FFP) ranged from 42% to 84% and the 5-year overall survival (OS) ranged from 56% to 90%, depending on the number of factors present. This scoring system, however, was developed using a patient population treated prior to 1992. Using a more recently treated patient population, the British Columbia Cancer Agency (BCCA) found that the IPS is still valid for prognostication, but outcomes have improved across all IPS groups, with 5-year FFP now ranging from 62% to 88% and 5-year OS ranging from 67% to 98%.6 This improvement is likely a reflection of improved therapy and supportive care. Table 1 shows the PFS and OS within each IPS group, comparing the data from the German Hodgkin Study Group (GHSG) and BCCA group.5,6 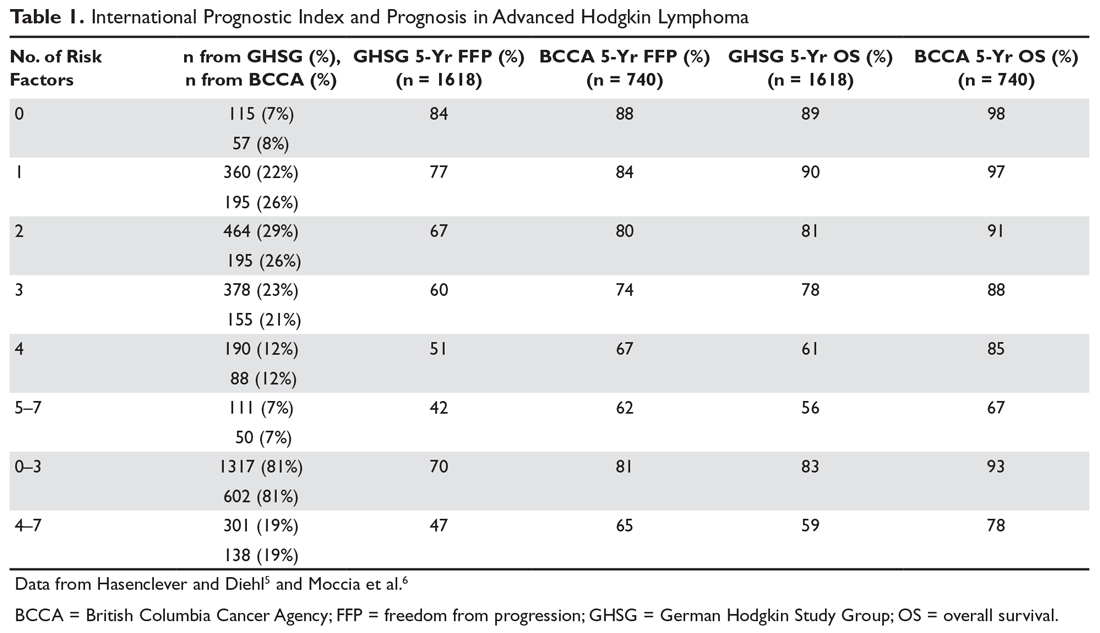
High expression of CD68 is associated with adverse outcomes, whereas high FOXP3 and CD20 expression on tumor cells are predictors of superior outcomes.8 A recent study found that CD68 expression was associated with OS. Five-year OS was 88% in those with less than 25% CD68 expression, versus 63% in those with greater than 25% CD68 expression.9
Roemer and colleagues evaluated 108 newly diagnosed cHL biopsy specimens and found that almost all cHL patients had concordant alteration of PD-L1 (programmed death ligand-1) and PD-L2 loci, with a spectrum of 9p24.1 alterations ranging from low level polysomy to near uniform 9p24.1 amplification. PD-L1/PD-L2 copy number alterations are therefore a defining pathobiological feature of cHL.10 PFS was significantly shorter for patients with 9p24.1 amplification, and those patients were likely to have advanced disease suggesting that 9p24.1 amplification is associated with less favorable prognosis.10 This may change with the increasing use of PD-1 inhibitors in the treatment of cHL.
High baseline metabolic tumor volume and total lesion glycolysis have also been associated with adverse outcomes in cHL. While not routinely assessed in practice currently, these tools may ultimately be used to assess prognosis and guide therapy in clinical practice.11
ADVANCED STAGE HODGKIN LYMPHOMA
FRONTLINE THERAPY
First-line Chemotherapy
Chemotherapy plays an essential role in the treatment of advanced stage Hodgkin lymphoma. In the 1960s, the MOPP regimen (nitrogen mustard, vincristine, procarbazine, prednisone) was developed, with a 10-year OS of 50% and a progression-free survival (PFS) of 52% reported in advanced stage patients. The complete remission (CR) rate was 81%, and 36% of patients who achieved CR relapsed later.12 This chemotherapy regimen is associated with a significant rate of myelosuppression and infertility as well as long-term risk of secondary myelodysplasia and acute leukemias.13,14 This led to the development of newer regimens such as ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine).15 In a randomized trial, ABVD showed improved failure-free survival (FFS) over MOPP (61% versus 50% at 5 years) but similar OS (66%–73%).16 In light of these findings, and considering the lower rate of infertility and myelotoxicity, ABVD became the standard of care for advanced stage cHL in the United States.
The Stanford V regimen was developed in an attempt to further minimize toxicity.17 Stanford V is a condensed, 12-week chemotherapy regimen that includes mechlorethamine, doxorubicin, vinblastine, etoposide, prednisone, vincristine, and bleomycin, followed by involved-field radiation therapy (IFRT). Subsequent trials compared the Stanford V and ABVD regimens and showed similar OS, freedom from treatment failure (FFTF), and response rates.18,19 The ABVD regimen was noted to have higher pulmonary toxicity, while other toxicities such as lymphopenia and neuropathy were higher with the Stanford V regimen. In addition, Stanford V requires patients to receive radiation therapy (RT) to original sites of disease larger than 5 cm in size and contiguous sites.
Another regimen which has been studied extensively for advanced stage Hodgkin lymphoma, and is considered a standard of care in some parts of the world, is escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone). In the HD9 study (n = 1196), the GHSG evaluated BEACOPP, escalated BEACOPP, and COPP/ABVD in advanced stage Hodgkin lymphoma.20 All arms of the study included 30 Gy RT to sites of bulky disease or residual disease. This study showed improved OS and FFTF with escalated BEACOPP, but at the cost of higher rates of toxicity. At 10 years, FFTF was 64%, 70%, and 82% with OS rates of 75%, 80%, and 86% for COPP/ABVD, baseline BEACOPP, and escalated BEACOPP, respectively (P < 0.001). The rate of secondary acute leukemia 10 years after treatment was 0.4% for COPP/ABVD, 1.5% for BEACOPP, and 3.0% for escalated BEACOPP. However, 3 subsequent randomized trials did not confirm a survival benefit with escalated BEACOPP relative to ABVD. In the HD 2000 trial (n = 295)21 and in a trial by Viviani and colleagues (n = 331),22 an improvement in OS was not demonstrated in favor of escalated BEACOPP. These studies also confirmed a higher rate of toxicities as well as secondary malignancies associated with the escalated BEACOPP regimen. In the EORTC20012 Intergroup trial (n = 549), 8 cycles of ABVD was compared with 4 cycles of escalated BEACOPP followed by 4 cycles of baseline BEACOPP, without radiation, in patients with clinical stage III or IV Hodgkin lymphoma with IPS score ≥ 3. Both regimens resulted in statistically similar FFS (63.7% in ABVD × 8 versus 69.3% in BEACOPP 4+4) and OS (86.7% in ABVD × 8 vs 90.3% in BEACOPP 4+4).23
In the United States, ABVD (6–8 cycles) is commonly used, although escalated BEACOPP (particularly for patients with an IPS of 4 or higher) and Stanford V are considered appropriate as well.24 In the North American Intergroup study comparing ABVD to Stanford V, and in the trial by Viviani et al, ABVD was associated with a 5- to 7-year FFS of 73% to 79% and OS of 84% to 92%.19,22 Given these excellent results, as well as the potential to cure patients with second-line therapy consisting of autologous hematopoietic cell transplantation (auto-HCT), the general consensus among most U.S. hematologists and oncologists is that ABVD remains the treatment of choice, and that the improved FFS/PFS with escalated BEACOPP is not outweighed by the additional toxicity associated with the regimen. There may, however, be a role for escalated BEACOPP in select patients who have a suboptimal response to ABVD as defined by interim positron emission tomography (iPET) scan (see below).
Brentuximab vedotin is an anti-CD30 antibody-drug conjugate (ADC) consisting of an anti-CD30 antibody linked to monomethyl auristatin E (MMAE), a potent antitubulin agent. CD30 is highly expressed on HRS cells and also in anaplastic large cell lymphoma. Upon binding to CD30, the ADC/CD30 complex is then internalized and directed to the lysosome, where the ADC is proteolytically cleaved, releasing MMAE from the antibody. MMAE then disrupts microtubule networks within the cell, leading to G2/M cycle arrest and apoptosis. CD30 is consistently expressed on HRS cells. In addition to being studied in the relapsed/refractory setting (described below), brentuximab has been studied in the first-line setting. In a phase 1 trial, brentuximab combined with ABVD was associated with increased pulmonary toxicity, while brentuximab + AVD had no significant pulmonary toxicity, with an excellent CR rate (96%), suggesting that substituting brentuximab for bleomycin may be an effective strategy. In addition to possibly being more efficacious, this strategy would also have the benefit of eliminating the risk of bleomycin pulmonary toxicity.25 Based on this data, a large international phase 3 study (the ECHELON-1 trial) comparing ABVD versus brentuximab + AVD in advanced stage cHL patients was recently completed. This study enrolled 1334 patients, and preliminary results were recently announced. With a median follow-up of 24 months, the brentuximab + AVD arm had a 4.9% absolute improvement in PFS relative to the ABVD arm (82.1% versus 77.2%). The brentuximab + AVD arm had an increased incidence of febrile neutropenia, managed with growth factors and peripheral neuropathy requiring dose adjustments, whereas the ABVD arm had an increased rate and severity of pulmonary toxicity.26 Further follow-up will be required to determine whether this will translate into a survival benefit. See Table 2 for a summary of recent large randomized prospective phase 3 trials in advanced stage Hodgkin lymphoma. 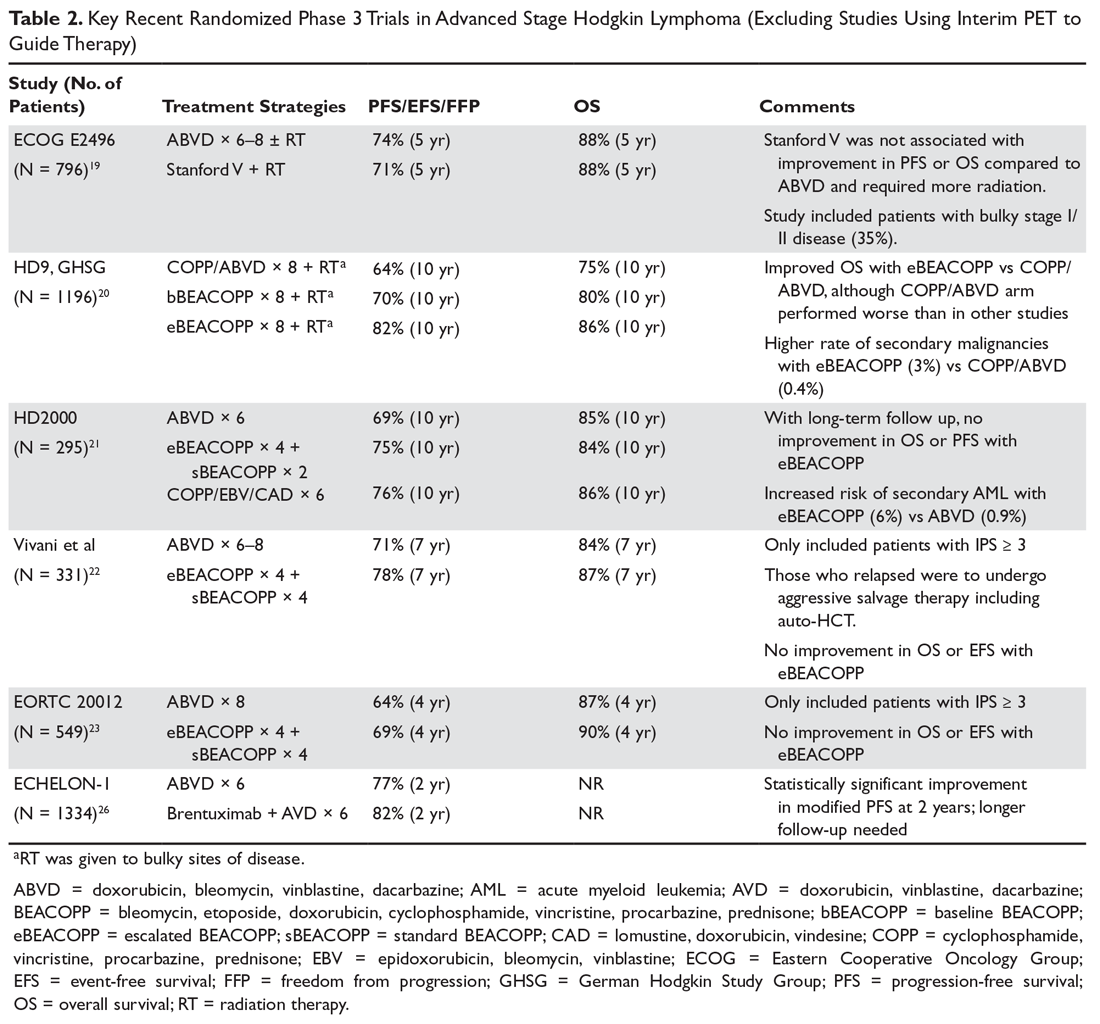
Alternative Regimens in Older Patients
Patients older than 60 years of age often have poor tolerance for ABVD and especially escalated BEACOPP. This results in increased treatment-related mortality and reduced overall dose intensity, with higher relapse rates and poor OS. In an attempt to improve on the results of treatment of elderly patients with Hodgkin lymphoma, alternative regimens have been explored. One example is PVAG (prednisone, vinblastine, doxorubicin, gemcitabine). With this regimen, the 3-year OS was 66% and PFS was 58%. One patient out of 59 died from treatment-related toxicity, which is much improved over the historical figures for elderly patients with Hodgkin lymphoma.27 Another commonly used approach in practice is to simply omit bleomycin from ABVD. In the early-stage setting (GHSG HD-13 trial), this regimen (referred to as AVD) led to 89.6% PFS at 5 years, compared to 93.5% with ABVD.28 It therefore stands to reason that this should be a reasonable option in older or more frail advanced stage cHL patients as well.
Brentuximab has been evaluated as a single-agent therapy for first-line therapy of elderly patients with Hodgkin lymphoma. In a phase 2 study, 27 patients (63% with advanced stage disease) were treated, with a 92% overall response rate and 73% CR rate. However the median duration of remission was disappointing at only 9.1 months.29 Based on this data, single-agent brentuximab appears to be a reasonable and well tolerated option for frail or elderly patients, although with the caveat that long-term disease control is relatively uncommon.
RESPONSE-ADAPTED FRONTLINE THERAPY USING INTERIM PET SCAN
In recent years, response-adapted treatment approaches have been extensively researched in cHL using iPET. The goal is to reduce toxicity by minimizing therapy in those who achieve negative iPET and/or to intensify treatment for patients with suboptimal response on iPET. Gallamini et al evaluated the prognostic role of an early iPET scan in advanced Hodgkin lymphoma patients (n = 190) treated with ABVD. This study found that patients with positive iPET had a 2-year PFS of 12.8% versus 95.0% in patients with negative iPET. This result was highly statistically significant (P < 0.0001). This study also showed that PET-2 (iPET after 2 cycles of ABVD) superseded the prognostic value of the IPS at diagnosis.30 As a result, numerous subsequent studies have been pursued using iPET for risk-adapted treatment in cHL.
A critical element to the conduct of iPET risk-adapted treatment for cHL is the interpretation of the iPET. In hopes of standardizing iPET interpretation in clinical trials, a scoring system called the Deauville score was developed. The Deauville score ranges from 1 to 5 (Table 3). 
The SWOG (Southwest Oncology Group) S0816 trial (n = 358) evaluated iPET-adapted treatment after 2 cycles of ABVD in stage III or IV Hodgkin lymphoma patients. Patients with positive iPET (Deauville score 4 to 5; n = 60) received escalated BEACOPP for 6 cycles, whereas iPET-negative (Deauville score 1 to 3; n = 271) patients continued to receive 4 more cycles of ABVD. The 2-year PFS was 64% for iPET-positive patients.33 This PFS was much higher than the expected 15% to 30% from prior studies such as Gallamini et al,30 suggesting that the treatment intensification may have been of benefit.
In the HD0801 study (n = 519), newly diagnosed advanced Hodgkin lymphoma patients with positive iPET after 2 cycles of ABVD (n = 103) received early ifosfamide-containing salvage therapy followed by high-dose therapy with autologous stem cell rescue. The 2-year PFS was 76% for PET-2–positive patients, comparable with PET-2–negative patients who had PFS of 81%.34 Again, this result for iPET-positive patients was much better than expected based on the historical control from Gallamini et al, suggesting that the treatment intensification may have been beneficial. It should be emphasized, however, that neither HD0801 nor S0816 were randomized prospective trials; rather, all iPET-positive patients were assigned to an intensified treatment approach.
In the HD18 trial (n = 1100), patients with advanced stage cHL started therapy with escalated BEACOPP and underwent an iPET after 2 cycles. For those with a positive iPET, rituximab was added to escalated BEACOPP in the experimental arm (n = 220) for cycles 3 through 8. The control group (n = 220) continued to receive 6 more cycles of escalated BEACOPP. In the 2 groups, the 3-year PFS was similar (91.4% in escalated BEACOPP, 93% in rituximab + escalated BEACOPP), suggesting no significant benefit with addition of rituximab.35 This study also calls into question whether iPET provides useful information for patients receiving intensive therapy such as escalated BEACOPP, and indicates that the historical control data for iPET-positive patients from Gallamini et al may not be consistently reproduced in other prospective trials. As a result, nonrandomized trials that implement an iPET risk-adapted approach should be interpreted with caution. See Table 4 for a summary of recent trials in advanced stage Hodgkin lymphoma using iPET scan to guide therapy. 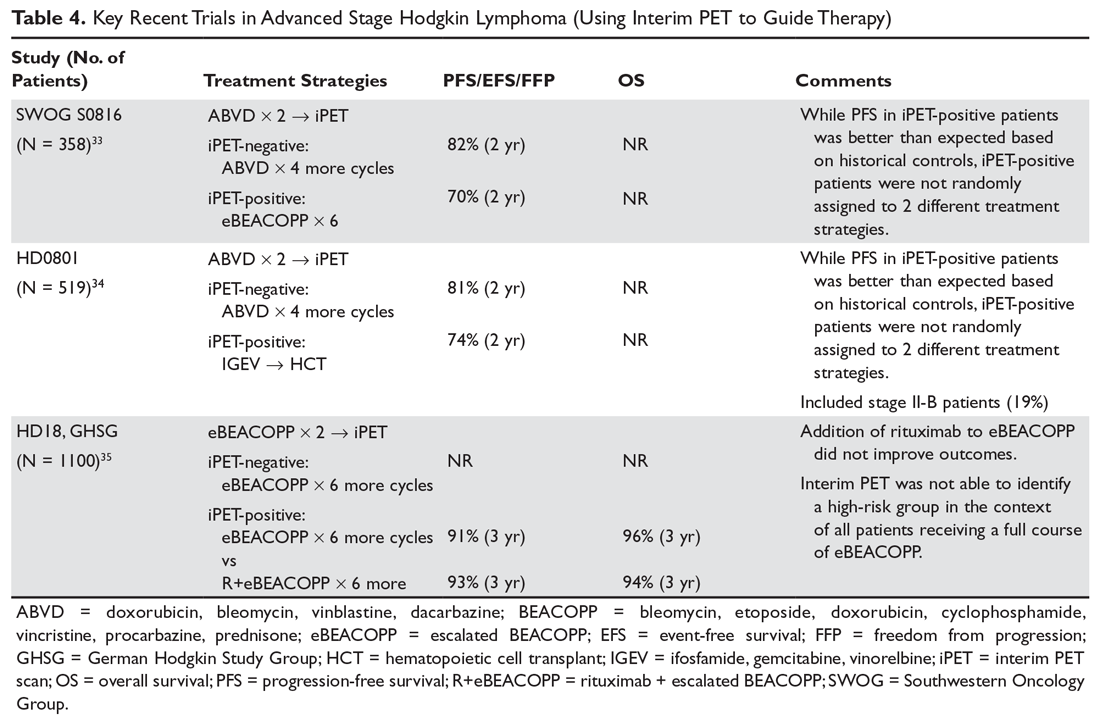
RADIATION THERAPY IN FRONTLINE TREATMENT
In patients with advanced stage Hodgkin lymphoma, IFRT to initial bulky sites of disease may be incorporated into frontline therapy to improve local control. However, whether this provides a survival benefit and which patients benefit most from consolidative RT remain unclear.
The European Organization for Research and Treatment of Cancer (EORTC) completed a randomized study in advanced stage Hodgkin lymphoma patients who achieved complete or partial remission after MOPP-ABV.36 Patients in CR were randomly assigned to receive no further treatment versus IFRT (24 Gy to all initially involved nodal areas and 16 to 24 Gy to all initially involved extranodal sites). Patients in partial remission (PR) were treated with 30 Gy to nodal areas and 18 to 24 Gy to extranodal sites. Among the CR patients, the 5-year event-free survival (EFS) was 79% to 84% and did not differ for those who received radiation versus those who did not. Five-year OS was 85% to 91% and also did not differ between the 2 groups. However, among the patients in PR after chemotherapy, the 5-year EFS was 79% and the 5-year OS was 87%, which is better than expected for PR patients, indicating a possible benefit to RT in patients with a partial response after chemotherapy. In the GHSG HD12 trial, patients with advanced stage Hodgkin lymphoma who had a residual lesion by computed tomography (CT) (but not analyzed by PET) had a very subtle improvement in FFTF (90% versus 87%) in favor of consolidation with IFRT, but again no survival benefit was seen.37
The EORTC and HD12 studies described above utilized CT scan for assigning remission status following chemotherapy, and it is now well known that many patients with residual masses (by CT) after chemotherapy may in fact be cured, as such residual radiographic abnormalities may simply be composed of fibrosis. PET scan is more accurate than CT in identifying patients who truly have residual active disease following chemotherapy. As a result, the EORTC study discussed above and the GHSG HD12 trial are of limited relevance in the modern era, in which patients routinely undergo PET scan at the end of therapy. Restricting IFRT to sites that remain PET-positive after completing chemotherapy may be a reasonable strategy that would allow for the avoidance of RT in many patients, and may obviate the need for aggressive second-line therapy (eg, high-dose therapy and autologous hematopoietic cell transplant [auto-HCT]). This approach was taken in the GHSG HD15 trial (n = 2182) in which advanced stage patients were treated with 3 variations on the BEACOPP regimen (8 cycles of escalated BEACOPP, 6 cycles of escalated BEACOPP, or 8 cycles of baseline BEACOPP, randomized in a 1:1:1 ratio). Patients with a residual mass of 2.5 cm or greater on CT scan then underwent a PET scan; if the lesion was PET positive, it was treated with 30 Gy of IFRT. This overall strategy was very effective, with 5-year FFTF rates of 84.4%, 89.3%, and 85.4%, respectively. The OS rates were 91.9%, 95.3%, and 94.5%, respectively. For patients with lesions that remained PET positive after chemotherapy, the PFS rate was 86.2% at 48 months, whereas patients in PR with persistent mass ≥ 2.5 cm but with negative PET had a PFS of 92.6%, similar to that of patients in CR.38 With this approach of BEACOPP followed by PET-guided radiation, the proportion of patients receiving RT was reduced from 71% (in the HD9 study) to only 11% in the HD15 study,38 with no apparent loss in overall efficacy when comparing the results of the 2 studies.
UPFRONT STEM CELL TRANSPLANTATION
To further improve outcomes of patients with advanced Hodgkin lymphoma with high-risk disease, high-dose therapy with auto-HCT has been explored as part of frontline therapy. While this has been shown to be feasible in such patients,39 randomized trials have not shown a clear benefit in terms of FFS or OS with upfront auto-HCT. 40,41 Therefore, auto-HCT is not considered a standard component of frontline therapy for cHL patients who achieve CR by PET/CT scan.
RELAPSED AND REFRACTORY HODGKIN LYMPHOMA
Depending on the stage, risk factors, and frontline regimen utilized, between 5% and 40% of patients with Hodgkin lymphoma can be expected to experience either primary induction failure or a relapse after attaining remission with frontline therapy.3 Primary refractory Hodgkin lymphoma, which occurs in up to 5% to 10% of patients, is defined as progression or no response during induction treatment or within 90 days of completing treatment. In cases where remission status is in question, an updated tissue biopsy is recommended. Biopsy is also recommended in cases in which new sites of disease have appeared or if relapse has occurred after a durable period of remission. Restaging is recommended at the time of relapse.
For younger patients with relapsed/refractory Hodgkin lymphoma, the standard of care in most cases is second-line (or salvage) chemotherapy followed by high-dose therapy and auto-HCT. For patients not felt to be candidates for auto-HCT, options include conventional second-line chemotherapy alone, salvage radiotherapy, novel agents such as brentuximab or immune checkpoint inhibitors, and/or participation in clinical trials.
CONVENTIONAL MULTI-AGENT CHEMOTHERAPY REGIMENS
Numerous conventional regimens have been shown in phase 2 studies to be active in relapsed and refractory Hodgkin lymphoma. These include platinum-based regimens, gemcitabine-based regimens, and alkylator-based regimens. No randomized trials in Hodgkin lymphoma have been conducted comparing these regimens. In general, regimens are chosen based on the patient’s age, performance status, comorbidities, and whether auto-HCT is being considered.
In the United States, platinum-based regimens such as ICE (ifosfamide, carboplatin, etoposide),42 DHAP (dexamethasone, cisplatin, high-dose cytarabine),43 ESHAP (etoposide, methylprednisolone, high-dose cytarabine, cisplatin),44 GDP (gemcitabine, cisplatin, dexamethasone),45 and GCD (gemcitabine, carboplatin, dexamethasone)46 are all considered appropriate second-line therapy options for patients being considered for auto-HCT, due to their high response rates and because autologous hematopoietic stem cell collection remains feasible after these regimens. Response rates range from 60% to 88%, with CR rates between 17% and 41%, and toxic death rates generally well below 5%.
Other gemcitabine-based regimens such as IGEV (ifosfamide, gemcitabine, vinorelbine) and GVD (gemcitabine, vinorelbine, liposomal doxorubicin) are also effective.47,48 GVD is an excellent choice since it is a generally well-tolerated outpatient regimen with a 60% response rate even in heavily pretreated patients.48 Stem cell collection remains feasible after both IGEV and GVD as well. ABVD can produce CR in approximately 20% to 50% of patients initially treated with MOPP.49–51 In practice, however, most patients today with relapsed or refractory Hodgkin lymphoma have already received ABVD as part of their first-line therapy, and retreatment with ABVD is not a good option because it would be associated with prohibitively high cumulative doses of doxorubicin.
These multi-agent chemotherapy regimens may not be tolerated well in patients over age 65 to 70 years or those with significant underlying comorbidities. In recent years, bendamustine has emerged as one of the most active conventional agents for cHL, with overall response rates of 53% to 58% in heavily pre-treated patients.52,53 Bendamustine can generally be tolerated even in elderly patients as well.
Some centers, particularly in Europe, investigated aggressive salvage regimens such as mini-BEAM (carmustine, etoposide, cytarabine, melphalan)54 or dexa-BEAM (BEAM plus dexamethasone).55 These regimens, however, are associated with significant hematologic toxicity and high (2%–5%) treatment-related mortality. As a result, these are rarely used in the United States.
For patients who have progressed after (or are not candidates for) platinum- and/or gemcitabine-based therapy, older alkylator-based regimens such as MOPP, C-MOPP, or ChlVPP (chlorambucil, vinblastine, procarbazine, prednisone) can be considered.56–58 However, these regimens are associated with significant bone marrow suppression, and autologous hematopoietic stem cell collection may no longer be feasible after such regimens. Therefore, these regimens should only be given to patients not felt to be auto-HCT candidates, or patients for whom autologous hematopoietic stem cell collection has already been completed. Weekly vinblastine or single-agent gemcitabine are palliative chemotherapy options, with response rates in the 60% to 80% range. Patients can sometimes be maintained on such low-intensity palliative regimens for 6 to 12 months or longer.59,60
BRENTUXIMAB VEDOTIN
Several trials are evaluating incorporation of brentuximab into second-line therapy in transplant-eligible patients. These approaches have used brentuximab prior to, concurrent with, or following platinum-based chemotherapy.61 While there is currently no consensus on the optimal way to incorporate brentuximab into salvage therapy, it is possible that the use of brentuximab or other novel agents in salvage therapy may allow for avoidance of conventional chemotherapy in some patients. In addition, this may translate into more patients proceeding to auto-HCT in a PET negative state. PET negativity prior to auto-HCT is a powerful predictor of long-term remission after auto-HCT, so any intervention that increases the rate of PET negativity prior to auto-HCT would be expected to improve outcomes with auto-HCT.62–65
For patients not being considered for autoHCT, or those for whom platinum-based salvage therapy was ineffective, single-agent brentuximab is an excellent option. In 2 phase 2 studies, an overall response rate (ORR) of 60% to 75% (including a CR rate of 22%–34%) was seen in relapsed and refractory Hodgkin lymphoma patients.66 The US Food and Drug Administration (FDA) approved brentuximab vedotin in August 2011 for treatment of relapsed and refractory Hodgkin lymphoma, after a failed auto-HCT, or in patients who are not auto-HCT candidates and who have received at least 2 prior chemotherapy regimens. With more extended follow-up, it has become clear that a proportion of patients who achieve CR to brentuximab may maintain remission long-term—58% at 3 years and 38% at 5 years.67 These patients may in fact be cured, in many cases without having undergone allogeneic HCT (allo-HCT) after brentuximab.
PD-1 (IMMUNE CHECKPOINT) INHIBITORS
As discussed earlier, PD-L1/PD-L2 copy number alterations represent a disease-defining feature of cHL. Alterations in chromosome 9p24.1 increase the expression of PD-1 ligands PD-L1 and PD-L2. Nivolumab and pembrolizumab are PD-1-blocking antibodies, which have recently been FDA approved for relapsed and refractory cHL. In a study with 23 patients, with 78% of them relapsing after auto-HCT and 78% relapsing after brentuximab, nivolumab produced an objective response in 87% of the patients, with 17% achieving CR and 70% achieving PR. The rate of PFS was 86% at 24 weeks.68 Pembrolizumab, another PD-1 antagonist, was also tested in relapsed and refractory Hodgkin lymphoma. In the KEYNOTE-087 study (n = 210), pembrolizumab produced an ORR of 64% to 70% in 3 different cohorts of relapsed and refractory cHL patients. Overall CR rate was 22%.69 In general, these agents are well tolerated, although patients must be monitored closely for
inflammatory/autoimmune-type toxicities including skin rash, diarrhea/colitis, transaminitis, endocrine abnormalities, and pneumonitis. Prompt recognition and initiation of corticosteroids is essential in managing these toxicities. Of note, PD-1 inhibitors should be given very cautiously to patients with a prior history of allo-HCT, since 30% to 55% of such patients will experience acute graft-versus-host disease (GVHD) in this setting. In 2 retrospective studies, the response rate was very high at 77% to 95%; however, 10% to 26% of all patients treated with PD-1 inhibitors post-allo-HCT died from GVHD induced by PD-1 inhibition.70,71 These risks and benefits therefore need to be carefully weighed in the post-allo-HCT setting. In another recent study, the outcomes were reported for 39 patients who underwent allo-HCT after prior therapy with a PD-1 inhibitor. Three patients (7.7%) developed lethal acute GVHD, suggesting there may be an increased risk of GVHD in patients undergoing allo-HCT after prior PD-1 inhibitor therapy.72
AUTOLOGOUS STEM CELL TRANSPLANTATION
Several studies have shown an improved disease-free survival (DFS) or FFS in patients with relapsed cHL treated by auto-HCT as compared to those receiving conventional chemotherapy alone.55,73,74 Overall, for relapsed disease, one can expect an approximately 50% to 60% chance for DFS at 5 years post-transplant. In a retrospective, matched-pair analysis, FFP was 62% for auto-HCT patients, compared to 32% for conventional chemotherapy patients. OS, however, was similar for the 2 groups (47%–54%). Patients failing induction therapy or relapsing within 1 year were seen to benefit the most from auto-HCT, including an OS benefit.74
A European prospective randomized trial was conducted comparing conventional salvage therapy to auto-HCT. In this study, 161 patients with relapsed Hodgkin lymphoma were treated with 2 cycles of dexa-BEAM. Those with chemo-sensitive disease were then randomized to either 2 more cycles of dexa-BEAM or high-dose BEAM with auto-HCT. Auto-HCT was associated with an approximately 55% FFTF at 3 years, versus 34% with conventional chemotherapy alone.55 This benefit again was most apparent for patients relapsing within 1 year of completion of primary therapy, although an OS benefit was not seen with auto-HCT. For patients with late relapse (>1 year after completion of primary therapy), auto-HCT was associated with an approximately 75% FFTF at 3 years, versus 50% with chemotherapy alone. One other small randomized trial of auto-HCT in relapsed and refractory Hodgkin lymphoma also showed an improved 3-year EFS in favor of auto-HCT (53% versus 10%), again with no difference in OS.73
The lack of OS benefit seen in these studies suggests that auto-HCT at first or second relapse provides comparable outcomes. Auto-HCT offers the benefit of avoiding the long-term toxicities associated with multiple salvage regimens and the anxiety associated with multiple relapses. In addition, the treatment-related mortality with auto-HCT is now in the 1% to 2% range in younger patients, at centers that perform the procedure routinely. For all of these reasons, auto-HCT is commonly recommended by physicians for Hodgkin lymphoma patients in first or second relapse. In most cases, transplant is favored in first relapse, since waiting until second relapse may be associated with a lower chance of achieving CR and difficulty collecting sufficient hematopoietic stem cells. For patients with early relapse or primary refractory disease, an even stronger case can be made for auto-HCT as the best option to achieve sustained control of the disease. For patients with late relapse, conventional salvage therapy alone may be a reasonable option, particularly in older or frail patients, or those with significant comorbid conditions.
The optimal conditioning regimen for autoHCT for relapsed and refractory Hodgkin lymphoma remains undefined. No randomized trials have been performed comparing conditioning regimens for relapsed and refractory Hodgkin lymphoma. One retrospective study compared 92 patients with Hodgkin lymphoma who underwent auto-HCT using a total-body irradiation (TBI) regimen versus a chemotherapy-alone regimen. No difference in 5-year OS or EFS was seen.75 Given the lack of benefit seen with TBI, along with reports of increased rates of secondary malignancies and myelodysplasia with TBI,76 chemotherapy-alone conditioning regimens are most widely employed. For example, in the United States, either the BEAM or CBV (cyclophosphamide, carmustine, etoposide) regimens are used in over 80% of cases.77 This practice was justified in a Center for International Blood and Marrow Transplant Research (CIBMTR) retrospective study comparing outcomes by conditioning regimens, in which no regimen performed better than BEAM or CBV.78
IFRT is often given as an adjunctive therapy to sites of initial and/or relapsed disease following auto-HCT. Although a relatively common practice, whether this truly enhances outcomes beyond that obtained with auto-HCT alone is unclear. Two retrospective studies have shown some benefit in terms of improvement in OS at 3 to 5 years in the group that received IFRT (70%–73% versus 40%–56%).79,80 Given the retrospective nature and small size of these studies, a prospective study would be needed to properly define the potential role for IFRT following auto-HCT in relapsed/refractory Hodgkin lymphoma. Another retrospective study (n = 73) that evaluated peri-transplant IFRT in Hodgkin lymphoma patients receiving auto transplant found no improvement in survival for patients who received peri-transplant IFRT. This study, however, did show a survival benefit in the subgroup of patients with limited stage disease.81
Prognostic Factors Associated with Outcome with Auto-HCT
The factor most consistently associated with improved outcome for patients with relapsed and refractory Hodgkin lymphoma who undergo auto-HCT is the disease status at transplant.63,77 Those in a second CR, versus a chemo-sensitive relapse (but not CR), versus a chemo-refractory relapse have DFS rates of 60% to 70%, 30% to 40%, and 10% to 20%, respectively.63 The duration between remission and relapse also has important prognostic significance. Late relapse (> 1 year after completion of frontline therapy) is associated with better outcomes as compared to early relapse.55 Other factors with prognostic significance at relapse include anemia, time to relapse and clinical stage, B symptoms, extranodal disease, number of prior chemotherapy regimens, and performance status.42,82 The prognostic impact of pretransplant disease status has been confirmed by studies using functional imaging (eg, FDG-PET or gallium scans). In a report by Moskowitz et al, patients with negative functional imaging following second-line therapy had a 77% EFS post-auto-HCT versus 33% in those whose functional imaging remained positive.62 Very similar findings have been reported by other groups.63–65
Post-Auto-HCT Brentuximab Maintenance
In the multicenter, randomized, double-blinded phase 3 AETHERA trial (n = 329), brentuximab (n = 165) was compared with placebo (n = 164) in patients with unfavorable risk relapsed or primary refractory cHL who had undergone autologous transplant. Eligible patients had at least 1 of the following risk factors for progression after auto-HCT: primary refractory Hodgkin lymphoma (failure to achieve complete remission), relapsed Hodgkin lymphoma with an initial remission duration of less than 12 months, or extranodal involvement at the start of pre-transplantation salvage chemotherapy. Patients were required to have CR, PR, or stable disease after pretransplant salvage chemotherapy with adequate kidney, liver, and bone marrow function. Patients who previously received brentuximab were excluded. Patients received 16 cycles of brentuximab or placebo once every 3 weeks starting 30 to 45 days after transplant. The PFS was significantly improved in the brentuximab group when compared to the placebo group (hazard ratio 0.57; P = 0.0013) after a median observation time of 30 months. Median PFS was 42.9 months in the brentuximab group versus 24.1 months in the placebo group; estimated 2-year PFS rates were 63% in the brentuximab group and 51% in the placebo group. OS was not significantly different between the study groups (~85%), presumably due to the fact that patients in the control group who relapsed likely went on to receive brentuximab as a subsequent therapy.83
PRIMARY REFRACTORY HODGKIN LYMPHOMA
Patients with primary refractory Hodgkin lymphoma have a poor outcome. Salvage therapy using conventional chemotherapy and/or RT results in long-term DFS in 10% or fewer of such patients.13,84 Given these poor outcomes with conventional salvage therapy, auto-HCT is considered to be the standard of care for this subset of patients. The GHSG retrospectively analyzed the prognostic factors and outcomes of patients with primary refractory Hodgkin lymphoma. The 5-year freedom-from-second-failure and the 5-year OS were reported to be 31% and 43%, respectively, for those patients treated with auto-HCT. Patients with poor functional status at time of transplant, age greater than 50 years, and failure to attain a temporary remission had a 0% 5-year OS, as compared to 55% in patients without any of these risk factors.85 A large retrospective European study showed that patients with chemo-resistant disease who underwent transplant had a 19% survival at 5 years.63 Hence, even patients with primary refractory Hodgkin lymphoma have some chance of achieving long-term survival following auto-HCT.
SALVAGE RADIOTHERAPY
The GHSG performed a retrospective analysis of the efficacy of salvage RT in patients with refractory or first-relapsed Hodgkin lymphoma. Five-year FFTF and OS rates were 28% and 51%, respectively. Patients with a limited-stage relapse and without B symptoms were more likely to benefit from salvage RT.86 Campbell et al reported on 81 patients undergoing salvage RT for persistent or recurrent Hodgkin lymphoma after chemotherapy. The 10-year FFTF and OS rates were 33% and 46%, respectively.87 Similarly, Wirth et al reported a 5-year FFS of 26% and 5-year OS of 57%. These figures were 36% and 75%, respectively, in patients whose relapse was limited to supradiaphragmatic nodal sites without B symptoms.88 RT therefore may be a useful strategy for a subset of patients who relapse following chemotherapy, particularly those with a limited-stage relapse, without B symptoms, and those with relapsed disease after a CR, as opposed to those with a partial response or lack of response to the prior chemotherapy regimen.
INVESTIGATIONAL AGENTS AND NOVEL COMBINATIONS
Several biological therapies are emerging as options for the treatment of refractory or relapsed disease. These therapies consist of monoclonal antibodies and ADCs that target cell surface antigens, or small molecules that inhibit key intracellular pathways within neoplastic cells.
Rituximab
Rituximab is a chimeric anti-CD20 monoclonal antibody used widely in B-cell non-Hodgkin lymphomas. The CD20 molecule is typically highly expressed in nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL). Two studies (one in relapsed patients, the other in a mixture of relapsed and previously untreated patients) showed significant activity of rituximab in relapsed NLPHL, with ORRs ranging from 94% to 100%, CR rates ranging from 41% to 53%, and median duration of remission in the 10- to 33-month range.89,90 In cHL, CD20 is expressed in HRS cells in 20% to 30% of cases. In such cases, single-agent rituximab has also shown activity. There is also evidence that rituximab may be effective even in cases in which the HRS cells are CD20-negative, presumably by virtue of depleting reactive B lymphocytes from the microenvironment, which may enhance anti-tumor immunity, or by eliminating a putative CD20-expressing Hodgkin lymphoma stem cell.91,92
Lenalidomide
Lenalidomide is an immunomodulatory drug that has multiple modes of action, including direct induction of apoptosis in tumor cells, antiangiogenic effects, and the activation of immune cells, such as natural killer cells and T cells. Lenalidomide has been shown to modify many features of the microenvironment of HRS cells and has demonstrated activity in other B-cell neoplasms. As a result, lenalidomide has been evaluated in relapsed and refractory Hodgkin lymphoma patients. A multicenter phase 2 study by Fehniger et al included 35 patients, 87% of whom had previously undergone HCT and 55% of whom were refractory to the last therapy.93 All patients were given lenalidomide 25 mg/day from days 1 to 21 of a 28-day cycle until disease progression. One patient was noted to achieve CR, 6 achieved PR, and 5 had stable disease lasting more than 6 months, for an ORR of 19% and a “cytostatic overall response rate” of 33%. The median duration of CR/partial remission was 6 months, with the median time-to-treatment failure in responders (including those with stable disease > 6 months) being 15 months. Similarly, in another study, Böll et al evaluated 12 patients across 4 German centers with relapsed or refractory disease who were treated with oral lenalidomide for 21 days in a 28-day cycle. No radiological evidence of disease progression after 2 cycles of lenalidomide was seen in any of the enrolled patients. ORR was noted to be 50%, with 6 patients with stable disease and 5 patients achieving PR after 2 cycles.94
Novel Brentuximab Combination Therapies
Brentuximab plus bendamustine. The combination of brentuximab and bendamustine was tested as an outpatient regimen in a phase 1/2 study (n = 55) in primary refractory Hodgkin lymphoma or after first relapse. The CR rate of the combination was 74%, with an overall objective response (CR + PR) of 93%. The CR rates were 64% and 84%, respectively, for refractory and relapsed patients. The PFS at 12 months was 80%, establishing this combination therapy as an effective salvage regimen with durable response.95
Brentuximab plus nivolumab. Preliminary results have recently been presented from 2 studies96,97 evaluating the combination of brentuximab and nivolumab. While this combination would still be considered investigational, these studies showed very encouraging ORRs of 90% to 100% and a CR rate of 62% to 66%. Longer follow-up is needed to determine whether these responses are durable and to document the toxicity profile of this combination.
Mammalian Target of Rapamycin Inhibitors
Two mammalian target of rapamycin (mTOR) inhibitors, everolimus and temsirolimus, are currently available in the United States. While neither drug currently has FDA approval for Hodgkin lymphoma, everolimus was evaluated in a phase 2 trial in a heavily pretreated group of relapsed/refractory patients. An ORR of 47% was seen, with a median time to progression of 7.2 months.98
ALLOGENEIC STEM CELL TRANSPLANTATION
Historically, patients who relapse after having an auto-HCT generally had a poor outcome, with a median survival of 2 to 3 years after failure of auto-HCT.99 These patients may be offered palliative chemotherapy (see above), treatment with novel agents (see above), or enrollment in a clinical trial. Select patients may benefit from a second hematopoietic stem cell transplant, most commonly an allo-HCT. However, rare patients with late relapse after auto-HCT may be considered for a second auto-HCT, with a minority of such patients achieving a durable remission after the second auto-HCT.100,101 Because relapse or progressive disease occurs most commonly in the first several months following auto-HCT, patients are more often considered for allo-HCT than a second auto-HCT. In addition, a second auto-HCT may not be feasible due to impaired bone marrow reserve and/or concerns for development of secondary myelodysplasia or acute myeloid leukemia.
Several studies have evaluated allo-HCT in relapsed/ refractory Hodgkin lymphoma. Early studies evaluating myeloablative allo-HCT for Hodgkin lymphoma showed excessive treatment-related mortality (up to 50%) and disappointingly low rates of long-term survival (< 25%).102,103 This was likely related to the fact that, in that era, most of the patients with Hodgkin lymphoma evaluated for allo-HCT were heavily pretreated and therefore at a higher risk for toxicity as well as lymphoma progression.
More recently, several studies have focused on the use of reduced-intensity conditioning (RIC) allo-HCT for relapsed and refractory Hodgkin lymphoma. This approach relies more on a “graft-versus-lymphoma” effect, the existence of which has been debated in Hodgkin lymphoma. Three single-center studies of RIC allo-HCT in patients with multiply recurrent Hodgkin lymphoma showed improved rates of treatment-related mortality (8%–16%) but still relatively low rates of long-term PFS (23%–39% at 2 to 4 years).104–106 Interestingly, in one of these studies the outcomes were more favorable for patients who underwent haploidentical (versus matched sibling or matched unrelated donor) transplants.105
Two large registry studies have also reported on the outcomes of RIC allo-HCT in patients with relapsed and refractory Hodgkin lymphoma.107,108 These studies also confirmed a modest improvement in outcomes compared with those seen historically with myeloablative transplants. Treatment-related mortality at 1 to 2 years was 23% to 33%, depending on whether a matched sibling donor versus an unrelated donor was used. However, long-term PFS (18%–20% at 2 to 5 years) and OS (28%–37% at 2 to 5 years) remained poor, primarily due to high rates of progressive lymphoma post-transplant. In both of these studies, patients were heavily pretreated (84%–96% had received 3 or more prior lines of chemotherapy, and 62%–89% received a prior auto-HCT), with 47% to 55% of patients chemo-resistant prior to transplant. Of note, both of these registry studies reflect patients who underwent transplant prior to the widespread use of brentuximab and PD-1 inhibitors.
Based on the single-center and registry data above, a prospective multicenter European phase 2 trial was conducted to evaluate the benefit of RIC allo-HCT in Hodgkin lymphoma.109 Ninety-two patients (86% with prior auto-HCT, 90% with 3 or more prior lines of therapy) were enrolled and given salvage therapy. Those who had stable disease or better following salvage therapy remained on protocol (n = 78) and underwent RIC with fludarabine and melphalan, followed by allo-HCT (70% with matched sibling donors). Treatment-related mortality was 15% at 1 year. Relapse or progression occurred in 49% at 2 years (35% if chemo-sensitive prior to transplant). Chronic GVHD was associated with a decreased rate of relapse, supporting the existence of a graft-versus-lymphoma effect in Hodgkin lymphoma. Unfortunately, PFS among all allografted patients was still relatively poor (24% at 4 years). However, among patients in CR prior to allo-HCT, a 50% PFS was seen at 4 years. Therefore, even in a prospective multicenter study, RIC allo-HCT offered significant benefit with manageable toxicity in relapsed and refractory Hodgkin lymphoma patients with chemo-sensitive disease.
These studies suggest that outcomes with allo-HCT would improve further if implemented earlier in the course of disease and/or with a lower burden of disease at transplant. It has therefore been suggested that allo-HCT should be considered soon after failure of auto-HCT is documented. In a retrospective study by Sarina et al, 185 Hodgkin lymphoma patients who relapsed following auto-HCT were then immediately considered for reduced-intensity allo-HCT.110 Of these, 122 had a donor identified, and 104 (85%) actually underwent allo-HCT. These 104 patients were then compared to the other 81 patients who either had no donor identified or had a donor but did not receive the planned allo-HCT. Two-year PFS and OS were superior in the patients undergoing allo-HCT (39% versus 14% and 66% versus 42%, respectively, P < 0.001), with a median follow-up of 4 years. The presence of chronic GVHD again was associated with improved PFS and OS. Disease status prior to transplant remained highly predictive of PFS and OS by multivariate analysis. Two other smaller retrospective studies similarly found a survival benefit associated with allo-HCT compared with patients who underwent conventional salvage therapies alone.111,112 These studies, although subject to the usual limitations of retrospective analyses, suggest that the results with reduced-intensity allo-HCT are in fact enhanced if applied earlier in the disease course, and are superior to those with conventional therapy alone.
Currently, the exact role of allo-HSCT, including the optimal timing and optimal donor source (matched sibling versus haploidentical sibling versus matched unrelated donor), remain undefined for relapsed and refractory Hodgkin lymphoma. As discussed earlier, brentuximab is highly active in relapsed Hodgkin lymphoma patients, with a subset of patients still in CR at 5 years.67 For such patients, avoiding the risks of allo-HCT is a desirable goal.
For those who relapse or progress after auto-HCT, a reasonable strategy therefore is to treat initially with brentuximab, unless the patient is already known to have responded poorly to brentuximab, or already has significant neuropathy. Those who achieve a CR to brentuximab are then observed. A subset of those patients will remain in remission at 5 years without further therapy. For those who relapse, or who achieve less than a CR to brentuximab, additional treatment (with brentuximab re-treatment being one option) followed by a reduced-intensity allo-HCT is a reasonable consideration. This approach has the theoretical advantages of (1) avoiding the risk of allo-HCT in the subset potentially cured by brentuximab, (2) getting patients to allo-HCT with fewer comorbidities (due to a lower total exposure to conventional chemotherapy pre-transplant), and (3) applying allo-HCT in the setting of sensitive disease/lower disease burden (due to the high efficacy of brentuximab). The results of a small study suggest that brentuximab may in fact be a very effective “bridge” to allotransplant. Chen et al113 reported on 18 patients with relapsed/refractory Hodgkin lymphoma (17 of whom had previously undergone auto-HCT) who were treated on brentuximab vedotin clinical trials. The data were retrospectively evaluated to determine the efficacy and safety of subsequent reduced-intensity allo-HCT. Remarkably, at 1 year the OS was 100%, PFS was 92%, and nonrelapse mortality was 0% with a median follow-up of 14 months. Hence, brentuximab is safe for use prior to reduced-intensity allo-HCT in heavily pre-treated patients and appears to be associated with very favorable post-transplant outcomes, particularly in comparison to older studies of allo-HCT in the era prior to brentuximab.
SUMMARY
Currently, cure is possible for the majority of patients diagnosed with advanced stage Hodgkin lymphoma. The challenge to the clinician is to provide curative treatment with the lowest risk of serious toxicities. Which regimen will best provide this balance of risk and benefit needs to be assessed based on the relapse risk, age, frailty, and comorbidity profile for an individual patient. For many patients with relapsed or refractory Hodgkin lymphoma, cure remains possible using approaches based on hematopoietic stem cell transplantation, RT, and/or brentuximab. In addition, there are now numerous conventional chemotherapy agents, RT strategies, and exciting newer agents such as PD-1 inhibitors, that can provide significant clinical benefit even when cure is not feasible.
1. Kuppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 1994;91:10962–6.
2. Kuppers R. The biology of Hodgkin’s lymphoma. Nature Rev Cancer 2009;9:15–27.
3. Narra RK, Pingali SR, Fenske TS. Early-stage Hodgkin’s lymphoma. Hospital Physician Hematology-Oncology Board Review Manual. 2017;12(Part 3).
4. Mauch PM, Kalish LA, Kadin M, et al. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993;71:2062–71.
5. Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med 1998;339:1506–14.
6. Moccia AA, Donaldson J, Chhanabhai M, et al. International Prognostic Score in advanced-stage Hodgkin’s lymphoma: altered utility in the modern era. J Clin Oncol 2012;30:3383–8.
7. Diefenbach CS, Li H, Hong F, et al. Evaluation of the International Prognostic Score (IPS-7) and a Simpler Prognostic Score (IPS-3) for advanced Hodgkin lymphoma in the modern era. Br J Haematol 2015;171:530–8.
8. Greaves P, Clear A, Coutinho R, et al. Expression of FOXP3, CD68, and CD20 at diagnosis in the microenvironment of cassical hodgkin lymphoma is predictive of outcome. J Clin Oncol 2013;31:256–62.
9. Touati M, Delage-Corre M, Monteil J, et al. CD68-positive tumor-associated macrophages predict unfavorable treatment outcomes in classical Hodgkin lymphoma in correlation with interim fluorodeoxyglucose-positron emission tomography assessment. Leuk Lymphoma 2015;56:332–41.
10. Roemer MG, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–7.
11. Pike LC, Kirkwood AA, Patrick P, et al. Can baseline PET-CT features predict outcomes in advanced hodgkin lymphoma? A prospective evaluation of UK patients in The RATHL Trial (CRUK/07/033). Hematological Oncology 2017;35(Supplement S2):37–8.
12. DeVita VT Jr, Simon RM, Hubbard SM, et al. Curability of advanced Hodgkin’s disease with chemotherapy. Long-term follow-up of MOPP-treated patients at the National Cancer Institute. Ann Intern Med 1980;92:587–95.
13. Longo DL, Duffey PL, Young RC, et al. Conventional-dose salvage combination chemotherapy in patients relapsing with Hodgkin’s disease after combination chemotherapy: the low probability for cure. J Clin Oncol 1992;10:210–8.
14. Kaldor JM, Day NE, Clarke EA, et al. Leukemia following Hodgkin’s disease. N Engl J Med 1990;322:7–13.
15. Bonadonna G, Zucali R, Monfardini S, et al. Combination chemotherapy of Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide versus MOPP. Cancer 1975;36:252–9.
16. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–84.
17. Horning SJ, Williams J, Bartlett NL, et al. Assessment of the stanford V regimen and consolidative radiotherapy for bulky and advanced Hodgkin’s disease: Eastern Cooperative Oncology Group pilot study E1492. J Clin Oncol 2000;18:972–80.
18. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the stanford V regimen and ABVD in the treatment of advanced Hodgkin’s Lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRCTN 64141244. J Clin Oncol 2009;27:5390–6.
19. Gordon LI, Hong F, Fisher RI, et al. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: an intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496). J Clin Oncol 2013;31:684–91.
20. Engert A, Diehl V, Franklin J, et al. Escalated-dose BEACOPP in the treatment of patients with advanced-stage Hodgkin’s lymphoma: 10 years of follow-up of the GHSG HD9 study. J Clin Oncol 2009;27:4548–54.
21. Merli F, Luminari S, Gobbi PG, et al. Long-term results of the HD2000 trial comparing ABVD versus BEACOPP versus COPP-EBV-CAD in untreated patients with advanced Hodgkin lymphoma: a study by Fondazione Italiana Linfomi. J Clin Oncol 2016;34:1175–81.
22. Viviani S, Zinzani PL, Rambaldi A, et al. ABVD versus BEACOPP for Hodgkin’s lymphoma when high-dose salvage is planned. N Engl J Med 2011;365:203–12.
23. Carde P, Karrasch M, Fortpied C, et al. Eight cycles of ABVD versus four cycles of BEACOPPescalated plus four cycles of BEACOPPbaseline in stage III to IV, International Prognostic Score >/= 3, high-risk Hodgkin lymphoma: first results of the phase III EORTC 20012 intergroup trial. J Clin Oncol 2016;34:2028–36.
24. National Comprehensive Cancer Network I. NCCN Guidelines Version 1.2017 Hodgkin Lymphoma. Accessed July 20, 2017.
25. Younes A, Connors JM, Park SI, et al. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncology 2013;14:1348–56.
26. Takeda and Seattle Genetics announce positive results from phase 3 ECHELON-1 clinical trial evaluating ADCETRIS® (brentuximab vedotin) in frontline advanced Hodgkin lymphoma [press release]. Cambridge, MA: Takeda Oncology; June 26, 2017.
27. Boll B, Bredenfeld H, Gorgen H, et al. Phase 2 study of PVAG (prednisone, vinblastine, doxorubicin, gemcitabine) in elderly patients with early unfavorable or advanced stage Hodgkin lymphoma. Blood 2011;118:6292–8.
28. Behringer K, Goergen H, Hitz F, et al. Omission of dacarbazine or bleomycin, or both, from the ABVD regimen in treatment of early-stage favourable Hodgkin’s lymphoma (GHSG HD13): an open-label, randomised, non-inferiority trial. Lancet 2015;385:1418–27.
29. Forero-Torres A, Holkova B, Goldschmidt J, et al. Phase 2 study of frontline brentuximab vedotin monotherapy in Hodgkin lymphoma patients aged 60 years and older. Blood 2015;126:2798–804.
30. Gallamini A, Hutchings M, Rigacci L, et al. Early interim 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol 2007;25:3746–52.
31. Meignan M, Gallamini A, Meignan M, et al. Report on the First International Workshop on Interim-PET-Scan in Lymphoma. Leuk Lymphoma 2009;50:1257–60.
32. Barrington SF, Mikhaeel NG, Kostakoglu L, et al. Role of Imaging in the Staging and Response Assessment of Lymphoma: Consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 2014;32:3048–58.
33. Press OW, Li H, Schoder H, et al. US Intergroup trial of response-adapted therapy for stage III to IV Hodgkin lymphoma using early interim fluorodeoxyglucose-positron emission tomography imaging: Southwest Oncology Group S0816. J Clin Oncol 2016;34:2020–7.
34. Zinzani PL, Broccoli A, Gioia DM, et al. Interim positron emission tomography response-adapted therapy in advanced-stage Hodgkin lymphoma: final results of the pPhase II part of the HD0801 study. J Clin Oncol 2016;34:1376–85.
35. Borchmann P, Haverkamp H, Lohri A, et al. Progression-free survival of early interim PET-positive patients with advanced stage Hodgkin’s lymphoma treated with BEACOPPescalated alone or in combination with rituximab (HD18): an open-label, international, randomised phase 3 study by the German Hodgkin Study Group. Lancet Oncology 2017;18:454–63.
36. Aleman BM, Raemaekers JM, Tirelli U, et al. Involved-field radiotherapy for advanced Hodgkin’s lymphoma. N Engl J Med 2003;348:2396–406.
37. Borchmann P, Haverkamp H, Diehl V, et al. Eight cycles of escalated-dose BEACOPP compared with four cycles of escalated-dose BEACOPP followed by four cycles of baseline-dose BEACOPP with or without radiotherapy in patients with advanced-stage hodgkin’s lymphoma: final analysis of the HD12 trial of the German Hodgkin Study Group. J Clin Oncol 2011;29:4234–42.
38. Engert A, Haverkamp H, Kobe C, et al. Reduced-intensity chemotherapy and PET-guided radiotherapy in patients with advanced stage Hodgkin’s lymphoma (HD15 trial): a randomised, open-label, phase 3 non-inferiority trial. Lancet 2012;379:1791–9.
39. Nademanee A, Molina A, Fung H, et al. High-dose chemo/radiotherapy and autologous bone marrow or stem cell transplantation for poor-risk advanced-stage Hodgkin’s disease during first partial or complete remission. Biol Blood Marrow Transplant 1999;5:292–8.
40. Federico M, Bellei M, Brice P, et al. High-dose therapy and autologous stem-cell transplantation versus conventional therapy for patients with advanced Hodgkin’s lymphoma responding to front-line therapy. J Clin Oncol 2003;21:2320–5.
41. Arakelyan N, Berthou C, Desablens B, et al. Early versus late intensification for patients with high-risk Hodgkin lymphoma-3 cycles of intensive chemotherapy plus low-dose lymph node radiation therapy versus 4 cycles of combined doxorubicin, bleomycin, vinblastine, and dacarbazine plus myeloablative chemotherapy with autologous stem cell transplantation: five-year results of a randomized trial on behalf of the GOELAMS Group. Cancer 2008;113:3323–30.
42. Moskowitz CH, Nimer SD, Zelenetz AD, et al. A 2-step comprehensive high-dose chemoradiotherapy second-line program for relapsed and refractory Hodgkin disease: analysis by intent to treat and development of a prognostic model. Blood 2001;97:616–23.
43. Josting A, Rudolph C, Reiser M, et al. Time-intensified dexamethasone/cisplatin/cytarabine: an effective salvage therapy with low toxicity in patients with relapsed and refractory Hodgkin’s disease. Ann Oncol 2002;13:1628–35.
44. Aparicio J, Segura A, Garcera S, et al. ESHAP is an active regimen for relapsing Hodgkin’s disease. Ann Oncol 1999;10:593–5.
45. Baetz T, Belch A, Couban S, et al. Gemcitabine, dexamethasone and cisplatin is an active and non-toxic chemotherapy regimen in relapsed or refractory Hodgkin’s disease: a phase II study by the National Cancer Institute of Canada Clinical Trials Group. Ann Oncol 2003;14:1762–7.
46. Gopal AK, Press OW, Shustov AR, et al. Efficacy and safety of gemcitabine, carboplatin, dexamethasone, and rituximab in patients with relapsed/refractory lymphoma: a prospective multi-center phase II study by the Puget Sound Oncology Consortium. Leuk Lymphoma 2010;51:1523–9.
47. Santoro A, Magagnoli M, Spina M, et al. Ifosfamide, gemcitabine, and vinorelbine: a new induction regimen for refractory and relapsed Hodgkin’s lymphoma. Haematologica 2007;92:35–41.
48. Bartlett NL, Niedzwiecki D, Johnson JL, et al. Gemcitabine, vinorelbine, and pegylated liposomal doxorubicin (GVD), a salvage regimen in relapsed Hodgkin’s lymphoma: CALGB 59804. Ann Oncol 2007;18:1071–9.
49. Santoro A, Bonadonna G. Prolonged disease-free survival in MOPP-resistant Hodgkin’s disease after treatment with adriamycin, bleomycin, vinblastine and dacarbazine (ABVD). Cancer Chemother Pharmacol 1979;2:101–5.
50. Krikorian JG, Portlock CS, Rosenberg SA. Treatment of advanced Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide (ABVD) after failure of MOPP therapy. Cancer 1978;41:2107–11.
51. Piga A, Ambrosetti A, Todeschini G, et al. Doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) salvage of mechlorethamine, vincristine, prednisone, and procarbazine (MOPP)-resistant advanced Hodgkin’s disease. Cancer Treat Rep 1984;68:947–51.
52. Moskowitz AJ, Hamlin PA Jr, Perales MA, et al. Phase II study of bendamustine in relapsed and refractory Hodgkin lymphoma. J Clin Oncol 2013;31:456–60.
53. Anastasia A, Carlo-Stella C, Corradini P, et al. Bendamustine for relapsed/refractory classical Hodgkin lymphoma after high dose chemotherapy and or allogeneic transplant: a study of Fondazione Italiana Linfomi (FIL). Blood 2012;120:3652.
54. Martin A, Fernandez-Jimenez MC, Caballero MD, et al. Long-term follow-up in patients treated with Mini-BEAM as salvage therapy for relapsed or refractory Hodgkin’s disease. Br J Haematol 2001;113:161–71.
55. Schmitz N, Pfistner B, Sextro M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet 2002;359:2065–71.
56. Fisher RI, DeVita VT, Hubbard SP, et al. Prolonged disease-free survival in Hodgkin’s disease with MOPP reinduction after first relapse. Ann Intern Med 1979;90:761–3.
57. ChlVPP therapy for Hodgkin’s disease: experience of 960 patients. The International ChlVPP Treatment Group. Ann Oncol 1995;6:167–72.
58. Selby P, Patel P, Milan S, et al. ChlVPP combination chemotherapy for Hodgkin’s disease: long-term results. Br J Cancer 1990;62:279–85.
59. Santoro A, Bredenfeld H, Devizzi L, et al. Gemcitabine in the treatment of refractory Hodgkin’s disease: results of a multicenter phase II study. J Clin Oncol 2000;18:2615–9.
60. Little R, Wittes RE, Longo DL, Wilson WH. Vinblastine for recurrent Hodgkin’s disease following autologous bone marrow transplant. J Clin Oncol 1998;16:584–8.
61. Chen R, Palmer JM, Martin P, et al. Results of a multicenter phase II trial of brentuximab vedotin as second-line therapy before autologous transplantation in relapsed/refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2015;21:2136–40.
62. Moskowitz CH, Yahalom J, Zelenetz AD, et al. High-dose chemo-radiotherapy for relapsed or refractory Hodgkin lymphoma and the significance of pre-transplant functional imaging. Br J Haematol 2010;148:890–7.
63. Sureda A, Constans M, Iriondo A, et al. Prognostic factors affecting long-term outcome after stem cell transplantation in Hodgkin’s lymphoma autografted after a first relapse. Ann Oncol 2005;16:625–33.
64. Crocchiolo R, Canevari C, Assanelli A, et al. Pre-transplant 18FDG-PET predicts outcome in lymphoma patients treated with high-dose sequential chemotherapy followed by autologous stem cell transplantation. Leuk Lymphoma 2008;49:727–33.
65. Mocikova H, Pytlik R, Markova J, et al. Pre-transplant positron emission tomography in patients with relapsed Hodgkin lymphoma. Leuk Lymphoma 2011;52:1668–74.
66. Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol 2012;30:2183–9.
67. Gopal AK, Chen R, Smith SE, et al. Durable remissions in a pivotal phase 2 study of brentuximab vedotin in relapsed or refractory Hodgkin lymphoma. Blood 2015;125:1236–43.
68. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma.N Engl J Med 2015;372:311–9.
69. Chen R, Zinzani PL, Fanale MA, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol 2017;35:2125–32.
70. Haverkos BM, Abbott D, Hamadani M, et al. PD-1 blockade for relapsed lymphoma post-allogeneic hematopoietic cell transplant: high response rate but frequent GVHD. Blood 2017;130:221–8.
71. Herbaux C, Gauthier J, Brice P, et al. Efficacy and tolerability of nivolumab after allogeneic transplantation for relapsed Hodgkin lymphoma. Blood 2017;129:2471–8.
72. Merryman RW, Kim HT, Zinzani PL et al. Safety and efficacy of allogeneic hematopoietic stem cell transplant after PD-1 blockage in relapsed/refractory lymphoma. Blood 2017;129:1380–8.
73. Linch DC, Winfield D, Goldstone AH, et al. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin’s disease: results of a BNLI randomised trial. Lancet 1993;341:1051–4.
74. Yuen AR, Rosenberg SA, Hoppe RT, et al. Comparison between conventional salvage therapy and high-dose therapy with autografting for recurrent or refractory Hodgkin’s disease. Blood 1997;89:814–22.
75. Gutierrez-Delgado F, Holmberg L, Hooper H, et al. Autologous stem cell transplantation for Hodgkin’s disease: busulfan, melphalan and thiotepa compared to a radiation-based regimen. Bone Marrow Transplant 2003;32:279–85.
76. Hake CR, Graubert TA, Fenske TS. Does autologous transplantation directly increase the risk of secondary leukemia in lymphoma patients? Bone Marrow Transplant 2007;39:59–70.
77. Hahn T, McCarthy PL, Carreras J, et al. Comparison of prognostic models for autologous hematopoietic stem cell transplantation (AHCT) for relapsed Hodgkin lymphoma. Blood 2009;114:1215.
78. Chen Y-B, Lane AA, Logan BR, et al. Impact of conditioning regimen on outcomes for patients with lymphoma undergoing high-dose therapy with autologous hematopoietic cell transplantation. Biology Blood Marrow Transplant 2015;21:1046–53.
79. Wendland MM, Asch JD, Pulsipher MA, et al. The impact of involved field radiation therapy for patients receiving high-dose chemotherapy followed by hematopoietic progenitor cell transplant for the treatment of relapsed or refractory Hodgkin disease. Am J Clin Oncol 2006;29:189–95.
80. Biswas T, Culakova E, Friedberg JW, et al. Involved field radiation therapy following high dose chemotherapy and autologous stem cell transplant benefits local control and survival in refractory or recurrent Hodgkin lymphoma. Radiother Oncol 2012;103:367–72.
81. Levis M, Piva C, Filippi AR, et al. Potential benefit of involved-field radiotherapy for patients with Relapsed-refractory Hodgkin’s lymphoma with incomplete response before autologous stem cell transplantation. Clin Lymphoma Myeloma Leuk 2017;17:14–22.
82. Josting A, Franklin J, May M, et al. New prognostic score based on treatment outcome of patients with relapsed Hodgkin’s lymphoma registered in the database of the German Hodgkin’s lymphoma study group. J Clin Oncol 2002;20:221–30.
83. Moskowitz CH, Nademanee A, Masszi T, et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015;385:1853–62.
84. Bonfante V, Santoro A, Viviani S, et al. Outcome of patients with Hodgkin’s disease failing after primary MOPP-ABVD. J Clin Oncol 1997;15:528–34.
85. Josting A, Rueffer U, Franklin J, et al. Prognostic factors and treatment outcome in primary progressive Hodgkin lymphoma: a report from the German Hodgkin Lymphoma Study Group. Blood 2000;96:1280–6.
86. Josting A, Nogova L, Franklin J, et al. Salvage radiotherapy in patients with relapsed and refractory Hodgkin’s lymphoma: a retrospective analysis from the German Hodgkin Lymphoma Study Group. J Clin Oncol 2005;23:1522–9.
87. Campbell B WA, Milner A, Di Iulio J, et al. Long-term follow-up of salvage radiotherapy in Hodgkin’s lymphoma after chemotherapy failure. Int J Radiat Oncol Biol Phys 2005;63:1538–45.
88. Wirth A, Corry J, Laidlaw C, et al. Salvage radiotherapy for Hodgkin’s disease following chemotherapy failure. Int J Radiat Oncol Biol Phys 1997;39:599–607.
89. Schulz H, Rehwald U, Morschhauser F, et al. Rituximab in relapsed lymphocyte-predominant Hodgkin lymphoma: long-term results of a phase 2 trial by the German Hodgkin Lymphoma Study Group (GHSG). Blood 2008;111:109–11.
90. Ekstrand BC, Lucas JB, Horwitz SM, et al. Rituximab in lymphocyte-predominant Hodgkin disease: results of a phase 2 trial. Blood 2003;101:4285–9.
91. Younes A, Romaguera J, Hagemeister F, et al. A pilot study of rituximab in patients with recurrent, classic Hodgkin disease. Cancer 2003;98:310–4.
92. Rehwald U, Schulz H, Reiser M, et al. Treatment of relapsed CD20+ Hodgkin lymphoma with the monoclonal antibody rituximab is effective and well tolerated: results of a phase 2 trial of the German Hodgkin Lymphoma Study Group. Blood 2003;101:420–4.
93. Fehniger TA, Larson S, Trinkaus K, et al. A phase 2 multicenter study of lenalidomide in relapsed or refractory classical Hodgkin lymphoma. Blood 2011;118:5119–25.
94. Boll B, Borchmann P, Topp MS, et al. Lenalidomide in patients with refractory or multiple relapsed Hodgkin lymphoma. Br J Haematol 2010;148:480–2.
95. LaCasce AS, Bociek G, Sawas A, et al. Brentuximab vedotin plus bendamustine: a highly active salvage treatment regimen for patients with relapsed or refractory Hodgkin lymphoma. Blood 2015;126:3982.
96. Diefenbach CS, Hong F, David KA, et al. A phase I study with an expansion cohort of the combination of ipilimumab and nivolumab and brentuximab vedotin in patients with relapsed/refractory Hodgkin lymphoma: A trial of the ECOG-ACRIN Cancer Research Group (E4412 Arms D and E). Blood 2016;128:1106.
97. Herrera AF, Bartlett NL, Ramchandren R, et al. Preliminary results from a phase 1/2 study of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2016;128:1105.
98. Johnston PB, Inwards DJ, Colgan JP, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol 2010;85:320–4.
99. Kewalramani T, Nimer SD, Zelenetz AD, et al. Progressive disease following autologous transplantation in patients with chemosensitive relapsed or primary refractory Hodgkin’s disease or aggressive non-Hodgkin’s lymphoma. Bone Marrow Transplant 2003;32:673–9.
100. Lin TS, Avalos BR, Penza SL, et al. Second autologous stem cell transplant for multiply relapsed Hodgkin’s disease. Bone Marrow Transplant 2002;29:763–7.
101. Smith SM, van Besien K, Carreras J, et al. Second autologous stem cell transplantation for relapsed lymphoma after a prior autologous transplant. Biol Blood Marrow Transplant 2008;14:904–12.
102. Gajewski JL, Phillips GL, Sobocinski KA, et al. Bone marrow transplants from HLA-identical siblings in advanced Hodgkin’s disease. J Clin Oncol 1996;14:572–8.
103. Peniket AJ, Ruiz de Elvira MC, Taghipour G, et al. An EBMT registry matched study of allogeneic stem cell transplants for lymphoma: allogeneic transplantation is associated with a lower relapse rate but a higher procedure-related mortality rate than autologous transplantation. Bone Marrow Transplant 2003;31:667–78.
104. Anderlini P, Saliba R, Acholonu S, et al. Fludarabine-melphalan as a preparative regimen for reduced-intensity conditioning allogeneic stem cell transplantation in relapsed and refractory Hodgkin’s lymphoma: the updated M.D. Anderson Cancer Center experience. Haematologica 2008;93:257–64.
105. Burroughs LM, O’Donnell PV, Sandmaier BM, et al. Comparison of outcomes of HLA-matched related, unrelated, or HLA-haploidentical related hematopoietic cell transplantation following nonmyeloablative conditioning for relapsed or refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2008;14:1279–87.
106. Peggs KS, Hunter A, Chopra R, et al. Clinical evidence of a graft-versus-Hodgkin’s-lymphoma effect after reduced-intensity allogeneic transplantation. Lancet 2005;365:1934–41.
107. Sureda A, Robinson S, Canals C, et al. Reduced-intensity conditioning compared with conventional allogeneic stem-cell transplantation in relapsed or refractory Hodgkin’s lymphoma: an analysis from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2008;26:455–62.
108. Devetten MP, Hari PN, Carreras J, et al. Unrelated donor reduced-intensity allogeneic hematopoietic stem cell transplantation for relapsed and refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:109–17.
109. Sureda A, Canals C, Arranz R, et al. Allogeneic stem cell transplantation after reduced intensity conditioning in patients with relapsed or refractory Hodgkin’s lymphoma. Results of the HDR-ALLO study - a prospective clinical trial by the Grupo Espanol de Linfomas/Trasplante de Medula Osea (GEL/TAMO) and the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. Haematologica 2012;97:310–7.
110. Sarina B, Castagna L, Farina L, et al. Allogeneic transplantation improves the overall and progression-free survival of Hodgkin lymphoma patients relapsing after autologous transplantation: a retrospective study based on the time of HLA typing and donor availability. Blood 2010;115:3671–7.
111. Castagna L, Sarina B, Todisco E, et al. Allogeneic stem cell transplantation compared with chemotherapy for poor-risk Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:432–8.
112. Thomson KJ, Peggs KS, Smith P, et al. Superiority of reduced-intensity allogeneic transplantation over conventional treatment for relapse of Hodgkin’s lymphoma following autologous stem cell transplantation. Bone Marrow Transplant 2008;41:765–70.
113. Chen R, Palmer JM, Thomas SH, et al. Brentuximab vedotin enables successful reduced-intensity allogeneic hematopoietic cell transplantation in patients with relapsed or refractory Hodgkin lymphoma. Blood 2012;119:6379–81.
INTRODUCTION
Hodgkin lymphoma, previously known as Hodgkin’s disease, is a B-cell lymphoproliferative disease characterized by a unique set of pathologic and epidemiologic features. The disease is characterized by the presence of multinucleate giant cells called Hodgkin Reed-Sternberg (HRS) cells.1 Hodgkin lymphoma is unique compared to other B-cell lymphomas because of the relative rarity of the malignant cells within affected tissues. The HRS cells, which usually account for only 0.1% to 10% of the cells, induce accumulation of nonmalignant lymphocytes, macrophages, granulocytes, eosinophils, plasma cells, and histiocytes, which then constitute the majority of tumor cellularity.2 Although the disease was first described by Sir Thomas Hodgkin in 1832, in part because of this unique histopathology, it was not until the 1990s that it was conclusively demonstrated that HRS cells are in fact monoclonal germinal center–derived B cells.
Due to the development of highly effective therapies for Hodgkin lymphoma, cure is a reasonable goal for most patients. Because of the high cure rate, late complications of therapy must be considered when selecting treatment. This article reviews the clinical features and treatment options for advanced stage and relapsed/refractory Hodgkin lymphoma. A previously published article reviewed the epidemiology, etiology/pathogenesis, pathologic classification, initial workup, and staging evaluation of Hodgkin lymphoma, as well as the prognostic stratification and treatment of patients with early-stage Hodgkin lymphoma.3
PRESENTATION, INITIAL EVALUATION, AND PROGNOSIS
Overall, classical Hodgkin lymphoma (cHL) usually presents with asymptomatic mediastinal or cervical lymphadenopathy. At least 50% of patients will have stage I or II disease.4 A mediastinal mass is seen in most patients with nodular sclerosis cHL, at times showing the characteristics of bulky (> 10 cm) disease. Constitutional, or B, symptoms (fever, night sweats, and weight loss) are present in approximately 25% of all patients with cHL, but 50% of advanced stage patients. Between 10% and 15% of patients will have extranodal disease, most commonly involving lung, bone, and liver. Lymphocyte-predominant Hodgkin lymphoma (LPHL) is a rare histological subtype of Hodgkin lymphoma that is differentiated from cHL by distinct clinicopathological features. The clinical course and treatment approach for LPHL are dependent upon the stage of disease. The clinicopathological features of LPHL are discussed in the early-stage Hodgkin lymphoma article.3
For the purposes of prognosis and selection of treatment, Hodgkin lymphoma is commonly classified as early stage favorable, early stage unfavorable, and advanced stage. For advanced stage Hodgkin lymphoma patients, prognosis can be defined using a tool commonly referred to as the International Prognostic Score (IPS). This index consists of 7 factors: male gender, age 45 years or older, stage IV disease, hemoglobin < 10.5 g/dL, white blood cell (WBC) count > 15,000/μL, lymphopenia (absolute lymphocyte count < 600 cells/μL or lymphocytes < 8% of WBC count), and serum albumin < 4 g/dL.5 In the original study by Hasenclever et al,5 the 5-year freedom from progression (FFP) ranged from 42% to 84% and the 5-year overall survival (OS) ranged from 56% to 90%, depending on the number of factors present. This scoring system, however, was developed using a patient population treated prior to 1992. Using a more recently treated patient population, the British Columbia Cancer Agency (BCCA) found that the IPS is still valid for prognostication, but outcomes have improved across all IPS groups, with 5-year FFP now ranging from 62% to 88% and 5-year OS ranging from 67% to 98%.6 This improvement is likely a reflection of improved therapy and supportive care. Table 1 shows the PFS and OS within each IPS group, comparing the data from the German Hodgkin Study Group (GHSG) and BCCA group.5,6
High expression of CD68 is associated with adverse outcomes, whereas high FOXP3 and CD20 expression on tumor cells are predictors of superior outcomes.8 A recent study found that CD68 expression was associated with OS. Five-year OS was 88% in those with less than 25% CD68 expression, versus 63% in those with greater than 25% CD68 expression.9
Roemer and colleagues evaluated 108 newly diagnosed cHL biopsy specimens and found that almost all cHL patients had concordant alteration of PD-L1 (programmed death ligand-1) and PD-L2 loci, with a spectrum of 9p24.1 alterations ranging from low level polysomy to near uniform 9p24.1 amplification. PD-L1/PD-L2 copy number alterations are therefore a defining pathobiological feature of cHL.10 PFS was significantly shorter for patients with 9p24.1 amplification, and those patients were likely to have advanced disease suggesting that 9p24.1 amplification is associated with less favorable prognosis.10 This may change with the increasing use of PD-1 inhibitors in the treatment of cHL.
High baseline metabolic tumor volume and total lesion glycolysis have also been associated with adverse outcomes in cHL. While not routinely assessed in practice currently, these tools may ultimately be used to assess prognosis and guide therapy in clinical practice.11
ADVANCED STAGE HODGKIN LYMPHOMA
FRONTLINE THERAPY
First-line Chemotherapy
Chemotherapy plays an essential role in the treatment of advanced stage Hodgkin lymphoma. In the 1960s, the MOPP regimen (nitrogen mustard, vincristine, procarbazine, prednisone) was developed, with a 10-year OS of 50% and a progression-free survival (PFS) of 52% reported in advanced stage patients. The complete remission (CR) rate was 81%, and 36% of patients who achieved CR relapsed later.12 This chemotherapy regimen is associated with a significant rate of myelosuppression and infertility as well as long-term risk of secondary myelodysplasia and acute leukemias.13,14 This led to the development of newer regimens such as ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine).15 In a randomized trial, ABVD showed improved failure-free survival (FFS) over MOPP (61% versus 50% at 5 years) but similar OS (66%–73%).16 In light of these findings, and considering the lower rate of infertility and myelotoxicity, ABVD became the standard of care for advanced stage cHL in the United States.
The Stanford V regimen was developed in an attempt to further minimize toxicity.17 Stanford V is a condensed, 12-week chemotherapy regimen that includes mechlorethamine, doxorubicin, vinblastine, etoposide, prednisone, vincristine, and bleomycin, followed by involved-field radiation therapy (IFRT). Subsequent trials compared the Stanford V and ABVD regimens and showed similar OS, freedom from treatment failure (FFTF), and response rates.18,19 The ABVD regimen was noted to have higher pulmonary toxicity, while other toxicities such as lymphopenia and neuropathy were higher with the Stanford V regimen. In addition, Stanford V requires patients to receive radiation therapy (RT) to original sites of disease larger than 5 cm in size and contiguous sites.
Another regimen which has been studied extensively for advanced stage Hodgkin lymphoma, and is considered a standard of care in some parts of the world, is escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone). In the HD9 study (n = 1196), the GHSG evaluated BEACOPP, escalated BEACOPP, and COPP/ABVD in advanced stage Hodgkin lymphoma.20 All arms of the study included 30 Gy RT to sites of bulky disease or residual disease. This study showed improved OS and FFTF with escalated BEACOPP, but at the cost of higher rates of toxicity. At 10 years, FFTF was 64%, 70%, and 82% with OS rates of 75%, 80%, and 86% for COPP/ABVD, baseline BEACOPP, and escalated BEACOPP, respectively (P < 0.001). The rate of secondary acute leukemia 10 years after treatment was 0.4% for COPP/ABVD, 1.5% for BEACOPP, and 3.0% for escalated BEACOPP. However, 3 subsequent randomized trials did not confirm a survival benefit with escalated BEACOPP relative to ABVD. In the HD 2000 trial (n = 295)21 and in a trial by Viviani and colleagues (n = 331),22 an improvement in OS was not demonstrated in favor of escalated BEACOPP. These studies also confirmed a higher rate of toxicities as well as secondary malignancies associated with the escalated BEACOPP regimen. In the EORTC20012 Intergroup trial (n = 549), 8 cycles of ABVD was compared with 4 cycles of escalated BEACOPP followed by 4 cycles of baseline BEACOPP, without radiation, in patients with clinical stage III or IV Hodgkin lymphoma with IPS score ≥ 3. Both regimens resulted in statistically similar FFS (63.7% in ABVD × 8 versus 69.3% in BEACOPP 4+4) and OS (86.7% in ABVD × 8 vs 90.3% in BEACOPP 4+4).23
In the United States, ABVD (6–8 cycles) is commonly used, although escalated BEACOPP (particularly for patients with an IPS of 4 or higher) and Stanford V are considered appropriate as well.24 In the North American Intergroup study comparing ABVD to Stanford V, and in the trial by Viviani et al, ABVD was associated with a 5- to 7-year FFS of 73% to 79% and OS of 84% to 92%.19,22 Given these excellent results, as well as the potential to cure patients with second-line therapy consisting of autologous hematopoietic cell transplantation (auto-HCT), the general consensus among most U.S. hematologists and oncologists is that ABVD remains the treatment of choice, and that the improved FFS/PFS with escalated BEACOPP is not outweighed by the additional toxicity associated with the regimen. There may, however, be a role for escalated BEACOPP in select patients who have a suboptimal response to ABVD as defined by interim positron emission tomography (iPET) scan (see below).
Brentuximab vedotin is an anti-CD30 antibody-drug conjugate (ADC) consisting of an anti-CD30 antibody linked to monomethyl auristatin E (MMAE), a potent antitubulin agent. CD30 is highly expressed on HRS cells and also in anaplastic large cell lymphoma. Upon binding to CD30, the ADC/CD30 complex is then internalized and directed to the lysosome, where the ADC is proteolytically cleaved, releasing MMAE from the antibody. MMAE then disrupts microtubule networks within the cell, leading to G2/M cycle arrest and apoptosis. CD30 is consistently expressed on HRS cells. In addition to being studied in the relapsed/refractory setting (described below), brentuximab has been studied in the first-line setting. In a phase 1 trial, brentuximab combined with ABVD was associated with increased pulmonary toxicity, while brentuximab + AVD had no significant pulmonary toxicity, with an excellent CR rate (96%), suggesting that substituting brentuximab for bleomycin may be an effective strategy. In addition to possibly being more efficacious, this strategy would also have the benefit of eliminating the risk of bleomycin pulmonary toxicity.25 Based on this data, a large international phase 3 study (the ECHELON-1 trial) comparing ABVD versus brentuximab + AVD in advanced stage cHL patients was recently completed. This study enrolled 1334 patients, and preliminary results were recently announced. With a median follow-up of 24 months, the brentuximab + AVD arm had a 4.9% absolute improvement in PFS relative to the ABVD arm (82.1% versus 77.2%). The brentuximab + AVD arm had an increased incidence of febrile neutropenia, managed with growth factors and peripheral neuropathy requiring dose adjustments, whereas the ABVD arm had an increased rate and severity of pulmonary toxicity.26 Further follow-up will be required to determine whether this will translate into a survival benefit. See Table 2 for a summary of recent large randomized prospective phase 3 trials in advanced stage Hodgkin lymphoma.
Alternative Regimens in Older Patients
Patients older than 60 years of age often have poor tolerance for ABVD and especially escalated BEACOPP. This results in increased treatment-related mortality and reduced overall dose intensity, with higher relapse rates and poor OS. In an attempt to improve on the results of treatment of elderly patients with Hodgkin lymphoma, alternative regimens have been explored. One example is PVAG (prednisone, vinblastine, doxorubicin, gemcitabine). With this regimen, the 3-year OS was 66% and PFS was 58%. One patient out of 59 died from treatment-related toxicity, which is much improved over the historical figures for elderly patients with Hodgkin lymphoma.27 Another commonly used approach in practice is to simply omit bleomycin from ABVD. In the early-stage setting (GHSG HD-13 trial), this regimen (referred to as AVD) led to 89.6% PFS at 5 years, compared to 93.5% with ABVD.28 It therefore stands to reason that this should be a reasonable option in older or more frail advanced stage cHL patients as well.
Brentuximab has been evaluated as a single-agent therapy for first-line therapy of elderly patients with Hodgkin lymphoma. In a phase 2 study, 27 patients (63% with advanced stage disease) were treated, with a 92% overall response rate and 73% CR rate. However the median duration of remission was disappointing at only 9.1 months.29 Based on this data, single-agent brentuximab appears to be a reasonable and well tolerated option for frail or elderly patients, although with the caveat that long-term disease control is relatively uncommon.
RESPONSE-ADAPTED FRONTLINE THERAPY USING INTERIM PET SCAN
In recent years, response-adapted treatment approaches have been extensively researched in cHL using iPET. The goal is to reduce toxicity by minimizing therapy in those who achieve negative iPET and/or to intensify treatment for patients with suboptimal response on iPET. Gallamini et al evaluated the prognostic role of an early iPET scan in advanced Hodgkin lymphoma patients (n = 190) treated with ABVD. This study found that patients with positive iPET had a 2-year PFS of 12.8% versus 95.0% in patients with negative iPET. This result was highly statistically significant (P < 0.0001). This study also showed that PET-2 (iPET after 2 cycles of ABVD) superseded the prognostic value of the IPS at diagnosis.30 As a result, numerous subsequent studies have been pursued using iPET for risk-adapted treatment in cHL.
A critical element to the conduct of iPET risk-adapted treatment for cHL is the interpretation of the iPET. In hopes of standardizing iPET interpretation in clinical trials, a scoring system called the Deauville score was developed. The Deauville score ranges from 1 to 5 (Table 3).
The SWOG (Southwest Oncology Group) S0816 trial (n = 358) evaluated iPET-adapted treatment after 2 cycles of ABVD in stage III or IV Hodgkin lymphoma patients. Patients with positive iPET (Deauville score 4 to 5; n = 60) received escalated BEACOPP for 6 cycles, whereas iPET-negative (Deauville score 1 to 3; n = 271) patients continued to receive 4 more cycles of ABVD. The 2-year PFS was 64% for iPET-positive patients.33 This PFS was much higher than the expected 15% to 30% from prior studies such as Gallamini et al,30 suggesting that the treatment intensification may have been of benefit.
In the HD0801 study (n = 519), newly diagnosed advanced Hodgkin lymphoma patients with positive iPET after 2 cycles of ABVD (n = 103) received early ifosfamide-containing salvage therapy followed by high-dose therapy with autologous stem cell rescue. The 2-year PFS was 76% for PET-2–positive patients, comparable with PET-2–negative patients who had PFS of 81%.34 Again, this result for iPET-positive patients was much better than expected based on the historical control from Gallamini et al, suggesting that the treatment intensification may have been beneficial. It should be emphasized, however, that neither HD0801 nor S0816 were randomized prospective trials; rather, all iPET-positive patients were assigned to an intensified treatment approach.
In the HD18 trial (n = 1100), patients with advanced stage cHL started therapy with escalated BEACOPP and underwent an iPET after 2 cycles. For those with a positive iPET, rituximab was added to escalated BEACOPP in the experimental arm (n = 220) for cycles 3 through 8. The control group (n = 220) continued to receive 6 more cycles of escalated BEACOPP. In the 2 groups, the 3-year PFS was similar (91.4% in escalated BEACOPP, 93% in rituximab + escalated BEACOPP), suggesting no significant benefit with addition of rituximab.35 This study also calls into question whether iPET provides useful information for patients receiving intensive therapy such as escalated BEACOPP, and indicates that the historical control data for iPET-positive patients from Gallamini et al may not be consistently reproduced in other prospective trials. As a result, nonrandomized trials that implement an iPET risk-adapted approach should be interpreted with caution. See Table 4 for a summary of recent trials in advanced stage Hodgkin lymphoma using iPET scan to guide therapy.
RADIATION THERAPY IN FRONTLINE TREATMENT
In patients with advanced stage Hodgkin lymphoma, IFRT to initial bulky sites of disease may be incorporated into frontline therapy to improve local control. However, whether this provides a survival benefit and which patients benefit most from consolidative RT remain unclear.
The European Organization for Research and Treatment of Cancer (EORTC) completed a randomized study in advanced stage Hodgkin lymphoma patients who achieved complete or partial remission after MOPP-ABV.36 Patients in CR were randomly assigned to receive no further treatment versus IFRT (24 Gy to all initially involved nodal areas and 16 to 24 Gy to all initially involved extranodal sites). Patients in partial remission (PR) were treated with 30 Gy to nodal areas and 18 to 24 Gy to extranodal sites. Among the CR patients, the 5-year event-free survival (EFS) was 79% to 84% and did not differ for those who received radiation versus those who did not. Five-year OS was 85% to 91% and also did not differ between the 2 groups. However, among the patients in PR after chemotherapy, the 5-year EFS was 79% and the 5-year OS was 87%, which is better than expected for PR patients, indicating a possible benefit to RT in patients with a partial response after chemotherapy. In the GHSG HD12 trial, patients with advanced stage Hodgkin lymphoma who had a residual lesion by computed tomography (CT) (but not analyzed by PET) had a very subtle improvement in FFTF (90% versus 87%) in favor of consolidation with IFRT, but again no survival benefit was seen.37
The EORTC and HD12 studies described above utilized CT scan for assigning remission status following chemotherapy, and it is now well known that many patients with residual masses (by CT) after chemotherapy may in fact be cured, as such residual radiographic abnormalities may simply be composed of fibrosis. PET scan is more accurate than CT in identifying patients who truly have residual active disease following chemotherapy. As a result, the EORTC study discussed above and the GHSG HD12 trial are of limited relevance in the modern era, in which patients routinely undergo PET scan at the end of therapy. Restricting IFRT to sites that remain PET-positive after completing chemotherapy may be a reasonable strategy that would allow for the avoidance of RT in many patients, and may obviate the need for aggressive second-line therapy (eg, high-dose therapy and autologous hematopoietic cell transplant [auto-HCT]). This approach was taken in the GHSG HD15 trial (n = 2182) in which advanced stage patients were treated with 3 variations on the BEACOPP regimen (8 cycles of escalated BEACOPP, 6 cycles of escalated BEACOPP, or 8 cycles of baseline BEACOPP, randomized in a 1:1:1 ratio). Patients with a residual mass of 2.5 cm or greater on CT scan then underwent a PET scan; if the lesion was PET positive, it was treated with 30 Gy of IFRT. This overall strategy was very effective, with 5-year FFTF rates of 84.4%, 89.3%, and 85.4%, respectively. The OS rates were 91.9%, 95.3%, and 94.5%, respectively. For patients with lesions that remained PET positive after chemotherapy, the PFS rate was 86.2% at 48 months, whereas patients in PR with persistent mass ≥ 2.5 cm but with negative PET had a PFS of 92.6%, similar to that of patients in CR.38 With this approach of BEACOPP followed by PET-guided radiation, the proportion of patients receiving RT was reduced from 71% (in the HD9 study) to only 11% in the HD15 study,38 with no apparent loss in overall efficacy when comparing the results of the 2 studies.
UPFRONT STEM CELL TRANSPLANTATION
To further improve outcomes of patients with advanced Hodgkin lymphoma with high-risk disease, high-dose therapy with auto-HCT has been explored as part of frontline therapy. While this has been shown to be feasible in such patients,39 randomized trials have not shown a clear benefit in terms of FFS or OS with upfront auto-HCT. 40,41 Therefore, auto-HCT is not considered a standard component of frontline therapy for cHL patients who achieve CR by PET/CT scan.
RELAPSED AND REFRACTORY HODGKIN LYMPHOMA
Depending on the stage, risk factors, and frontline regimen utilized, between 5% and 40% of patients with Hodgkin lymphoma can be expected to experience either primary induction failure or a relapse after attaining remission with frontline therapy.3 Primary refractory Hodgkin lymphoma, which occurs in up to 5% to 10% of patients, is defined as progression or no response during induction treatment or within 90 days of completing treatment. In cases where remission status is in question, an updated tissue biopsy is recommended. Biopsy is also recommended in cases in which new sites of disease have appeared or if relapse has occurred after a durable period of remission. Restaging is recommended at the time of relapse.
For younger patients with relapsed/refractory Hodgkin lymphoma, the standard of care in most cases is second-line (or salvage) chemotherapy followed by high-dose therapy and auto-HCT. For patients not felt to be candidates for auto-HCT, options include conventional second-line chemotherapy alone, salvage radiotherapy, novel agents such as brentuximab or immune checkpoint inhibitors, and/or participation in clinical trials.
CONVENTIONAL MULTI-AGENT CHEMOTHERAPY REGIMENS
Numerous conventional regimens have been shown in phase 2 studies to be active in relapsed and refractory Hodgkin lymphoma. These include platinum-based regimens, gemcitabine-based regimens, and alkylator-based regimens. No randomized trials in Hodgkin lymphoma have been conducted comparing these regimens. In general, regimens are chosen based on the patient’s age, performance status, comorbidities, and whether auto-HCT is being considered.
In the United States, platinum-based regimens such as ICE (ifosfamide, carboplatin, etoposide),42 DHAP (dexamethasone, cisplatin, high-dose cytarabine),43 ESHAP (etoposide, methylprednisolone, high-dose cytarabine, cisplatin),44 GDP (gemcitabine, cisplatin, dexamethasone),45 and GCD (gemcitabine, carboplatin, dexamethasone)46 are all considered appropriate second-line therapy options for patients being considered for auto-HCT, due to their high response rates and because autologous hematopoietic stem cell collection remains feasible after these regimens. Response rates range from 60% to 88%, with CR rates between 17% and 41%, and toxic death rates generally well below 5%.
Other gemcitabine-based regimens such as IGEV (ifosfamide, gemcitabine, vinorelbine) and GVD (gemcitabine, vinorelbine, liposomal doxorubicin) are also effective.47,48 GVD is an excellent choice since it is a generally well-tolerated outpatient regimen with a 60% response rate even in heavily pretreated patients.48 Stem cell collection remains feasible after both IGEV and GVD as well. ABVD can produce CR in approximately 20% to 50% of patients initially treated with MOPP.49–51 In practice, however, most patients today with relapsed or refractory Hodgkin lymphoma have already received ABVD as part of their first-line therapy, and retreatment with ABVD is not a good option because it would be associated with prohibitively high cumulative doses of doxorubicin.
These multi-agent chemotherapy regimens may not be tolerated well in patients over age 65 to 70 years or those with significant underlying comorbidities. In recent years, bendamustine has emerged as one of the most active conventional agents for cHL, with overall response rates of 53% to 58% in heavily pre-treated patients.52,53 Bendamustine can generally be tolerated even in elderly patients as well.
Some centers, particularly in Europe, investigated aggressive salvage regimens such as mini-BEAM (carmustine, etoposide, cytarabine, melphalan)54 or dexa-BEAM (BEAM plus dexamethasone).55 These regimens, however, are associated with significant hematologic toxicity and high (2%–5%) treatment-related mortality. As a result, these are rarely used in the United States.
For patients who have progressed after (or are not candidates for) platinum- and/or gemcitabine-based therapy, older alkylator-based regimens such as MOPP, C-MOPP, or ChlVPP (chlorambucil, vinblastine, procarbazine, prednisone) can be considered.56–58 However, these regimens are associated with significant bone marrow suppression, and autologous hematopoietic stem cell collection may no longer be feasible after such regimens. Therefore, these regimens should only be given to patients not felt to be auto-HCT candidates, or patients for whom autologous hematopoietic stem cell collection has already been completed. Weekly vinblastine or single-agent gemcitabine are palliative chemotherapy options, with response rates in the 60% to 80% range. Patients can sometimes be maintained on such low-intensity palliative regimens for 6 to 12 months or longer.59,60
BRENTUXIMAB VEDOTIN
Several trials are evaluating incorporation of brentuximab into second-line therapy in transplant-eligible patients. These approaches have used brentuximab prior to, concurrent with, or following platinum-based chemotherapy.61 While there is currently no consensus on the optimal way to incorporate brentuximab into salvage therapy, it is possible that the use of brentuximab or other novel agents in salvage therapy may allow for avoidance of conventional chemotherapy in some patients. In addition, this may translate into more patients proceeding to auto-HCT in a PET negative state. PET negativity prior to auto-HCT is a powerful predictor of long-term remission after auto-HCT, so any intervention that increases the rate of PET negativity prior to auto-HCT would be expected to improve outcomes with auto-HCT.62–65
For patients not being considered for autoHCT, or those for whom platinum-based salvage therapy was ineffective, single-agent brentuximab is an excellent option. In 2 phase 2 studies, an overall response rate (ORR) of 60% to 75% (including a CR rate of 22%–34%) was seen in relapsed and refractory Hodgkin lymphoma patients.66 The US Food and Drug Administration (FDA) approved brentuximab vedotin in August 2011 for treatment of relapsed and refractory Hodgkin lymphoma, after a failed auto-HCT, or in patients who are not auto-HCT candidates and who have received at least 2 prior chemotherapy regimens. With more extended follow-up, it has become clear that a proportion of patients who achieve CR to brentuximab may maintain remission long-term—58% at 3 years and 38% at 5 years.67 These patients may in fact be cured, in many cases without having undergone allogeneic HCT (allo-HCT) after brentuximab.
PD-1 (IMMUNE CHECKPOINT) INHIBITORS
As discussed earlier, PD-L1/PD-L2 copy number alterations represent a disease-defining feature of cHL. Alterations in chromosome 9p24.1 increase the expression of PD-1 ligands PD-L1 and PD-L2. Nivolumab and pembrolizumab are PD-1-blocking antibodies, which have recently been FDA approved for relapsed and refractory cHL. In a study with 23 patients, with 78% of them relapsing after auto-HCT and 78% relapsing after brentuximab, nivolumab produced an objective response in 87% of the patients, with 17% achieving CR and 70% achieving PR. The rate of PFS was 86% at 24 weeks.68 Pembrolizumab, another PD-1 antagonist, was also tested in relapsed and refractory Hodgkin lymphoma. In the KEYNOTE-087 study (n = 210), pembrolizumab produced an ORR of 64% to 70% in 3 different cohorts of relapsed and refractory cHL patients. Overall CR rate was 22%.69 In general, these agents are well tolerated, although patients must be monitored closely for
inflammatory/autoimmune-type toxicities including skin rash, diarrhea/colitis, transaminitis, endocrine abnormalities, and pneumonitis. Prompt recognition and initiation of corticosteroids is essential in managing these toxicities. Of note, PD-1 inhibitors should be given very cautiously to patients with a prior history of allo-HCT, since 30% to 55% of such patients will experience acute graft-versus-host disease (GVHD) in this setting. In 2 retrospective studies, the response rate was very high at 77% to 95%; however, 10% to 26% of all patients treated with PD-1 inhibitors post-allo-HCT died from GVHD induced by PD-1 inhibition.70,71 These risks and benefits therefore need to be carefully weighed in the post-allo-HCT setting. In another recent study, the outcomes were reported for 39 patients who underwent allo-HCT after prior therapy with a PD-1 inhibitor. Three patients (7.7%) developed lethal acute GVHD, suggesting there may be an increased risk of GVHD in patients undergoing allo-HCT after prior PD-1 inhibitor therapy.72
AUTOLOGOUS STEM CELL TRANSPLANTATION
Several studies have shown an improved disease-free survival (DFS) or FFS in patients with relapsed cHL treated by auto-HCT as compared to those receiving conventional chemotherapy alone.55,73,74 Overall, for relapsed disease, one can expect an approximately 50% to 60% chance for DFS at 5 years post-transplant. In a retrospective, matched-pair analysis, FFP was 62% for auto-HCT patients, compared to 32% for conventional chemotherapy patients. OS, however, was similar for the 2 groups (47%–54%). Patients failing induction therapy or relapsing within 1 year were seen to benefit the most from auto-HCT, including an OS benefit.74
A European prospective randomized trial was conducted comparing conventional salvage therapy to auto-HCT. In this study, 161 patients with relapsed Hodgkin lymphoma were treated with 2 cycles of dexa-BEAM. Those with chemo-sensitive disease were then randomized to either 2 more cycles of dexa-BEAM or high-dose BEAM with auto-HCT. Auto-HCT was associated with an approximately 55% FFTF at 3 years, versus 34% with conventional chemotherapy alone.55 This benefit again was most apparent for patients relapsing within 1 year of completion of primary therapy, although an OS benefit was not seen with auto-HCT. For patients with late relapse (>1 year after completion of primary therapy), auto-HCT was associated with an approximately 75% FFTF at 3 years, versus 50% with chemotherapy alone. One other small randomized trial of auto-HCT in relapsed and refractory Hodgkin lymphoma also showed an improved 3-year EFS in favor of auto-HCT (53% versus 10%), again with no difference in OS.73
The lack of OS benefit seen in these studies suggests that auto-HCT at first or second relapse provides comparable outcomes. Auto-HCT offers the benefit of avoiding the long-term toxicities associated with multiple salvage regimens and the anxiety associated with multiple relapses. In addition, the treatment-related mortality with auto-HCT is now in the 1% to 2% range in younger patients, at centers that perform the procedure routinely. For all of these reasons, auto-HCT is commonly recommended by physicians for Hodgkin lymphoma patients in first or second relapse. In most cases, transplant is favored in first relapse, since waiting until second relapse may be associated with a lower chance of achieving CR and difficulty collecting sufficient hematopoietic stem cells. For patients with early relapse or primary refractory disease, an even stronger case can be made for auto-HCT as the best option to achieve sustained control of the disease. For patients with late relapse, conventional salvage therapy alone may be a reasonable option, particularly in older or frail patients, or those with significant comorbid conditions.
The optimal conditioning regimen for autoHCT for relapsed and refractory Hodgkin lymphoma remains undefined. No randomized trials have been performed comparing conditioning regimens for relapsed and refractory Hodgkin lymphoma. One retrospective study compared 92 patients with Hodgkin lymphoma who underwent auto-HCT using a total-body irradiation (TBI) regimen versus a chemotherapy-alone regimen. No difference in 5-year OS or EFS was seen.75 Given the lack of benefit seen with TBI, along with reports of increased rates of secondary malignancies and myelodysplasia with TBI,76 chemotherapy-alone conditioning regimens are most widely employed. For example, in the United States, either the BEAM or CBV (cyclophosphamide, carmustine, etoposide) regimens are used in over 80% of cases.77 This practice was justified in a Center for International Blood and Marrow Transplant Research (CIBMTR) retrospective study comparing outcomes by conditioning regimens, in which no regimen performed better than BEAM or CBV.78
IFRT is often given as an adjunctive therapy to sites of initial and/or relapsed disease following auto-HCT. Although a relatively common practice, whether this truly enhances outcomes beyond that obtained with auto-HCT alone is unclear. Two retrospective studies have shown some benefit in terms of improvement in OS at 3 to 5 years in the group that received IFRT (70%–73% versus 40%–56%).79,80 Given the retrospective nature and small size of these studies, a prospective study would be needed to properly define the potential role for IFRT following auto-HCT in relapsed/refractory Hodgkin lymphoma. Another retrospective study (n = 73) that evaluated peri-transplant IFRT in Hodgkin lymphoma patients receiving auto transplant found no improvement in survival for patients who received peri-transplant IFRT. This study, however, did show a survival benefit in the subgroup of patients with limited stage disease.81
Prognostic Factors Associated with Outcome with Auto-HCT
The factor most consistently associated with improved outcome for patients with relapsed and refractory Hodgkin lymphoma who undergo auto-HCT is the disease status at transplant.63,77 Those in a second CR, versus a chemo-sensitive relapse (but not CR), versus a chemo-refractory relapse have DFS rates of 60% to 70%, 30% to 40%, and 10% to 20%, respectively.63 The duration between remission and relapse also has important prognostic significance. Late relapse (> 1 year after completion of frontline therapy) is associated with better outcomes as compared to early relapse.55 Other factors with prognostic significance at relapse include anemia, time to relapse and clinical stage, B symptoms, extranodal disease, number of prior chemotherapy regimens, and performance status.42,82 The prognostic impact of pretransplant disease status has been confirmed by studies using functional imaging (eg, FDG-PET or gallium scans). In a report by Moskowitz et al, patients with negative functional imaging following second-line therapy had a 77% EFS post-auto-HCT versus 33% in those whose functional imaging remained positive.62 Very similar findings have been reported by other groups.63–65
Post-Auto-HCT Brentuximab Maintenance
In the multicenter, randomized, double-blinded phase 3 AETHERA trial (n = 329), brentuximab (n = 165) was compared with placebo (n = 164) in patients with unfavorable risk relapsed or primary refractory cHL who had undergone autologous transplant. Eligible patients had at least 1 of the following risk factors for progression after auto-HCT: primary refractory Hodgkin lymphoma (failure to achieve complete remission), relapsed Hodgkin lymphoma with an initial remission duration of less than 12 months, or extranodal involvement at the start of pre-transplantation salvage chemotherapy. Patients were required to have CR, PR, or stable disease after pretransplant salvage chemotherapy with adequate kidney, liver, and bone marrow function. Patients who previously received brentuximab were excluded. Patients received 16 cycles of brentuximab or placebo once every 3 weeks starting 30 to 45 days after transplant. The PFS was significantly improved in the brentuximab group when compared to the placebo group (hazard ratio 0.57; P = 0.0013) after a median observation time of 30 months. Median PFS was 42.9 months in the brentuximab group versus 24.1 months in the placebo group; estimated 2-year PFS rates were 63% in the brentuximab group and 51% in the placebo group. OS was not significantly different between the study groups (~85%), presumably due to the fact that patients in the control group who relapsed likely went on to receive brentuximab as a subsequent therapy.83
PRIMARY REFRACTORY HODGKIN LYMPHOMA
Patients with primary refractory Hodgkin lymphoma have a poor outcome. Salvage therapy using conventional chemotherapy and/or RT results in long-term DFS in 10% or fewer of such patients.13,84 Given these poor outcomes with conventional salvage therapy, auto-HCT is considered to be the standard of care for this subset of patients. The GHSG retrospectively analyzed the prognostic factors and outcomes of patients with primary refractory Hodgkin lymphoma. The 5-year freedom-from-second-failure and the 5-year OS were reported to be 31% and 43%, respectively, for those patients treated with auto-HCT. Patients with poor functional status at time of transplant, age greater than 50 years, and failure to attain a temporary remission had a 0% 5-year OS, as compared to 55% in patients without any of these risk factors.85 A large retrospective European study showed that patients with chemo-resistant disease who underwent transplant had a 19% survival at 5 years.63 Hence, even patients with primary refractory Hodgkin lymphoma have some chance of achieving long-term survival following auto-HCT.
SALVAGE RADIOTHERAPY
The GHSG performed a retrospective analysis of the efficacy of salvage RT in patients with refractory or first-relapsed Hodgkin lymphoma. Five-year FFTF and OS rates were 28% and 51%, respectively. Patients with a limited-stage relapse and without B symptoms were more likely to benefit from salvage RT.86 Campbell et al reported on 81 patients undergoing salvage RT for persistent or recurrent Hodgkin lymphoma after chemotherapy. The 10-year FFTF and OS rates were 33% and 46%, respectively.87 Similarly, Wirth et al reported a 5-year FFS of 26% and 5-year OS of 57%. These figures were 36% and 75%, respectively, in patients whose relapse was limited to supradiaphragmatic nodal sites without B symptoms.88 RT therefore may be a useful strategy for a subset of patients who relapse following chemotherapy, particularly those with a limited-stage relapse, without B symptoms, and those with relapsed disease after a CR, as opposed to those with a partial response or lack of response to the prior chemotherapy regimen.
INVESTIGATIONAL AGENTS AND NOVEL COMBINATIONS
Several biological therapies are emerging as options for the treatment of refractory or relapsed disease. These therapies consist of monoclonal antibodies and ADCs that target cell surface antigens, or small molecules that inhibit key intracellular pathways within neoplastic cells.
Rituximab
Rituximab is a chimeric anti-CD20 monoclonal antibody used widely in B-cell non-Hodgkin lymphomas. The CD20 molecule is typically highly expressed in nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL). Two studies (one in relapsed patients, the other in a mixture of relapsed and previously untreated patients) showed significant activity of rituximab in relapsed NLPHL, with ORRs ranging from 94% to 100%, CR rates ranging from 41% to 53%, and median duration of remission in the 10- to 33-month range.89,90 In cHL, CD20 is expressed in HRS cells in 20% to 30% of cases. In such cases, single-agent rituximab has also shown activity. There is also evidence that rituximab may be effective even in cases in which the HRS cells are CD20-negative, presumably by virtue of depleting reactive B lymphocytes from the microenvironment, which may enhance anti-tumor immunity, or by eliminating a putative CD20-expressing Hodgkin lymphoma stem cell.91,92
Lenalidomide
Lenalidomide is an immunomodulatory drug that has multiple modes of action, including direct induction of apoptosis in tumor cells, antiangiogenic effects, and the activation of immune cells, such as natural killer cells and T cells. Lenalidomide has been shown to modify many features of the microenvironment of HRS cells and has demonstrated activity in other B-cell neoplasms. As a result, lenalidomide has been evaluated in relapsed and refractory Hodgkin lymphoma patients. A multicenter phase 2 study by Fehniger et al included 35 patients, 87% of whom had previously undergone HCT and 55% of whom were refractory to the last therapy.93 All patients were given lenalidomide 25 mg/day from days 1 to 21 of a 28-day cycle until disease progression. One patient was noted to achieve CR, 6 achieved PR, and 5 had stable disease lasting more than 6 months, for an ORR of 19% and a “cytostatic overall response rate” of 33%. The median duration of CR/partial remission was 6 months, with the median time-to-treatment failure in responders (including those with stable disease > 6 months) being 15 months. Similarly, in another study, Böll et al evaluated 12 patients across 4 German centers with relapsed or refractory disease who were treated with oral lenalidomide for 21 days in a 28-day cycle. No radiological evidence of disease progression after 2 cycles of lenalidomide was seen in any of the enrolled patients. ORR was noted to be 50%, with 6 patients with stable disease and 5 patients achieving PR after 2 cycles.94
Novel Brentuximab Combination Therapies
Brentuximab plus bendamustine. The combination of brentuximab and bendamustine was tested as an outpatient regimen in a phase 1/2 study (n = 55) in primary refractory Hodgkin lymphoma or after first relapse. The CR rate of the combination was 74%, with an overall objective response (CR + PR) of 93%. The CR rates were 64% and 84%, respectively, for refractory and relapsed patients. The PFS at 12 months was 80%, establishing this combination therapy as an effective salvage regimen with durable response.95
Brentuximab plus nivolumab. Preliminary results have recently been presented from 2 studies96,97 evaluating the combination of brentuximab and nivolumab. While this combination would still be considered investigational, these studies showed very encouraging ORRs of 90% to 100% and a CR rate of 62% to 66%. Longer follow-up is needed to determine whether these responses are durable and to document the toxicity profile of this combination.
Mammalian Target of Rapamycin Inhibitors
Two mammalian target of rapamycin (mTOR) inhibitors, everolimus and temsirolimus, are currently available in the United States. While neither drug currently has FDA approval for Hodgkin lymphoma, everolimus was evaluated in a phase 2 trial in a heavily pretreated group of relapsed/refractory patients. An ORR of 47% was seen, with a median time to progression of 7.2 months.98
ALLOGENEIC STEM CELL TRANSPLANTATION
Historically, patients who relapse after having an auto-HCT generally had a poor outcome, with a median survival of 2 to 3 years after failure of auto-HCT.99 These patients may be offered palliative chemotherapy (see above), treatment with novel agents (see above), or enrollment in a clinical trial. Select patients may benefit from a second hematopoietic stem cell transplant, most commonly an allo-HCT. However, rare patients with late relapse after auto-HCT may be considered for a second auto-HCT, with a minority of such patients achieving a durable remission after the second auto-HCT.100,101 Because relapse or progressive disease occurs most commonly in the first several months following auto-HCT, patients are more often considered for allo-HCT than a second auto-HCT. In addition, a second auto-HCT may not be feasible due to impaired bone marrow reserve and/or concerns for development of secondary myelodysplasia or acute myeloid leukemia.
Several studies have evaluated allo-HCT in relapsed/ refractory Hodgkin lymphoma. Early studies evaluating myeloablative allo-HCT for Hodgkin lymphoma showed excessive treatment-related mortality (up to 50%) and disappointingly low rates of long-term survival (< 25%).102,103 This was likely related to the fact that, in that era, most of the patients with Hodgkin lymphoma evaluated for allo-HCT were heavily pretreated and therefore at a higher risk for toxicity as well as lymphoma progression.
More recently, several studies have focused on the use of reduced-intensity conditioning (RIC) allo-HCT for relapsed and refractory Hodgkin lymphoma. This approach relies more on a “graft-versus-lymphoma” effect, the existence of which has been debated in Hodgkin lymphoma. Three single-center studies of RIC allo-HCT in patients with multiply recurrent Hodgkin lymphoma showed improved rates of treatment-related mortality (8%–16%) but still relatively low rates of long-term PFS (23%–39% at 2 to 4 years).104–106 Interestingly, in one of these studies the outcomes were more favorable for patients who underwent haploidentical (versus matched sibling or matched unrelated donor) transplants.105
Two large registry studies have also reported on the outcomes of RIC allo-HCT in patients with relapsed and refractory Hodgkin lymphoma.107,108 These studies also confirmed a modest improvement in outcomes compared with those seen historically with myeloablative transplants. Treatment-related mortality at 1 to 2 years was 23% to 33%, depending on whether a matched sibling donor versus an unrelated donor was used. However, long-term PFS (18%–20% at 2 to 5 years) and OS (28%–37% at 2 to 5 years) remained poor, primarily due to high rates of progressive lymphoma post-transplant. In both of these studies, patients were heavily pretreated (84%–96% had received 3 or more prior lines of chemotherapy, and 62%–89% received a prior auto-HCT), with 47% to 55% of patients chemo-resistant prior to transplant. Of note, both of these registry studies reflect patients who underwent transplant prior to the widespread use of brentuximab and PD-1 inhibitors.
Based on the single-center and registry data above, a prospective multicenter European phase 2 trial was conducted to evaluate the benefit of RIC allo-HCT in Hodgkin lymphoma.109 Ninety-two patients (86% with prior auto-HCT, 90% with 3 or more prior lines of therapy) were enrolled and given salvage therapy. Those who had stable disease or better following salvage therapy remained on protocol (n = 78) and underwent RIC with fludarabine and melphalan, followed by allo-HCT (70% with matched sibling donors). Treatment-related mortality was 15% at 1 year. Relapse or progression occurred in 49% at 2 years (35% if chemo-sensitive prior to transplant). Chronic GVHD was associated with a decreased rate of relapse, supporting the existence of a graft-versus-lymphoma effect in Hodgkin lymphoma. Unfortunately, PFS among all allografted patients was still relatively poor (24% at 4 years). However, among patients in CR prior to allo-HCT, a 50% PFS was seen at 4 years. Therefore, even in a prospective multicenter study, RIC allo-HCT offered significant benefit with manageable toxicity in relapsed and refractory Hodgkin lymphoma patients with chemo-sensitive disease.
These studies suggest that outcomes with allo-HCT would improve further if implemented earlier in the course of disease and/or with a lower burden of disease at transplant. It has therefore been suggested that allo-HCT should be considered soon after failure of auto-HCT is documented. In a retrospective study by Sarina et al, 185 Hodgkin lymphoma patients who relapsed following auto-HCT were then immediately considered for reduced-intensity allo-HCT.110 Of these, 122 had a donor identified, and 104 (85%) actually underwent allo-HCT. These 104 patients were then compared to the other 81 patients who either had no donor identified or had a donor but did not receive the planned allo-HCT. Two-year PFS and OS were superior in the patients undergoing allo-HCT (39% versus 14% and 66% versus 42%, respectively, P < 0.001), with a median follow-up of 4 years. The presence of chronic GVHD again was associated with improved PFS and OS. Disease status prior to transplant remained highly predictive of PFS and OS by multivariate analysis. Two other smaller retrospective studies similarly found a survival benefit associated with allo-HCT compared with patients who underwent conventional salvage therapies alone.111,112 These studies, although subject to the usual limitations of retrospective analyses, suggest that the results with reduced-intensity allo-HCT are in fact enhanced if applied earlier in the disease course, and are superior to those with conventional therapy alone.
Currently, the exact role of allo-HSCT, including the optimal timing and optimal donor source (matched sibling versus haploidentical sibling versus matched unrelated donor), remain undefined for relapsed and refractory Hodgkin lymphoma. As discussed earlier, brentuximab is highly active in relapsed Hodgkin lymphoma patients, with a subset of patients still in CR at 5 years.67 For such patients, avoiding the risks of allo-HCT is a desirable goal.
For those who relapse or progress after auto-HCT, a reasonable strategy therefore is to treat initially with brentuximab, unless the patient is already known to have responded poorly to brentuximab, or already has significant neuropathy. Those who achieve a CR to brentuximab are then observed. A subset of those patients will remain in remission at 5 years without further therapy. For those who relapse, or who achieve less than a CR to brentuximab, additional treatment (with brentuximab re-treatment being one option) followed by a reduced-intensity allo-HCT is a reasonable consideration. This approach has the theoretical advantages of (1) avoiding the risk of allo-HCT in the subset potentially cured by brentuximab, (2) getting patients to allo-HCT with fewer comorbidities (due to a lower total exposure to conventional chemotherapy pre-transplant), and (3) applying allo-HCT in the setting of sensitive disease/lower disease burden (due to the high efficacy of brentuximab). The results of a small study suggest that brentuximab may in fact be a very effective “bridge” to allotransplant. Chen et al113 reported on 18 patients with relapsed/refractory Hodgkin lymphoma (17 of whom had previously undergone auto-HCT) who were treated on brentuximab vedotin clinical trials. The data were retrospectively evaluated to determine the efficacy and safety of subsequent reduced-intensity allo-HCT. Remarkably, at 1 year the OS was 100%, PFS was 92%, and nonrelapse mortality was 0% with a median follow-up of 14 months. Hence, brentuximab is safe for use prior to reduced-intensity allo-HCT in heavily pre-treated patients and appears to be associated with very favorable post-transplant outcomes, particularly in comparison to older studies of allo-HCT in the era prior to brentuximab.
SUMMARY
Currently, cure is possible for the majority of patients diagnosed with advanced stage Hodgkin lymphoma. The challenge to the clinician is to provide curative treatment with the lowest risk of serious toxicities. Which regimen will best provide this balance of risk and benefit needs to be assessed based on the relapse risk, age, frailty, and comorbidity profile for an individual patient. For many patients with relapsed or refractory Hodgkin lymphoma, cure remains possible using approaches based on hematopoietic stem cell transplantation, RT, and/or brentuximab. In addition, there are now numerous conventional chemotherapy agents, RT strategies, and exciting newer agents such as PD-1 inhibitors, that can provide significant clinical benefit even when cure is not feasible.
INTRODUCTION
Hodgkin lymphoma, previously known as Hodgkin’s disease, is a B-cell lymphoproliferative disease characterized by a unique set of pathologic and epidemiologic features. The disease is characterized by the presence of multinucleate giant cells called Hodgkin Reed-Sternberg (HRS) cells.1 Hodgkin lymphoma is unique compared to other B-cell lymphomas because of the relative rarity of the malignant cells within affected tissues. The HRS cells, which usually account for only 0.1% to 10% of the cells, induce accumulation of nonmalignant lymphocytes, macrophages, granulocytes, eosinophils, plasma cells, and histiocytes, which then constitute the majority of tumor cellularity.2 Although the disease was first described by Sir Thomas Hodgkin in 1832, in part because of this unique histopathology, it was not until the 1990s that it was conclusively demonstrated that HRS cells are in fact monoclonal germinal center–derived B cells.
Due to the development of highly effective therapies for Hodgkin lymphoma, cure is a reasonable goal for most patients. Because of the high cure rate, late complications of therapy must be considered when selecting treatment. This article reviews the clinical features and treatment options for advanced stage and relapsed/refractory Hodgkin lymphoma. A previously published article reviewed the epidemiology, etiology/pathogenesis, pathologic classification, initial workup, and staging evaluation of Hodgkin lymphoma, as well as the prognostic stratification and treatment of patients with early-stage Hodgkin lymphoma.3
PRESENTATION, INITIAL EVALUATION, AND PROGNOSIS
Overall, classical Hodgkin lymphoma (cHL) usually presents with asymptomatic mediastinal or cervical lymphadenopathy. At least 50% of patients will have stage I or II disease.4 A mediastinal mass is seen in most patients with nodular sclerosis cHL, at times showing the characteristics of bulky (> 10 cm) disease. Constitutional, or B, symptoms (fever, night sweats, and weight loss) are present in approximately 25% of all patients with cHL, but 50% of advanced stage patients. Between 10% and 15% of patients will have extranodal disease, most commonly involving lung, bone, and liver. Lymphocyte-predominant Hodgkin lymphoma (LPHL) is a rare histological subtype of Hodgkin lymphoma that is differentiated from cHL by distinct clinicopathological features. The clinical course and treatment approach for LPHL are dependent upon the stage of disease. The clinicopathological features of LPHL are discussed in the early-stage Hodgkin lymphoma article.3
For the purposes of prognosis and selection of treatment, Hodgkin lymphoma is commonly classified as early stage favorable, early stage unfavorable, and advanced stage. For advanced stage Hodgkin lymphoma patients, prognosis can be defined using a tool commonly referred to as the International Prognostic Score (IPS). This index consists of 7 factors: male gender, age 45 years or older, stage IV disease, hemoglobin < 10.5 g/dL, white blood cell (WBC) count > 15,000/μL, lymphopenia (absolute lymphocyte count < 600 cells/μL or lymphocytes < 8% of WBC count), and serum albumin < 4 g/dL.5 In the original study by Hasenclever et al,5 the 5-year freedom from progression (FFP) ranged from 42% to 84% and the 5-year overall survival (OS) ranged from 56% to 90%, depending on the number of factors present. This scoring system, however, was developed using a patient population treated prior to 1992. Using a more recently treated patient population, the British Columbia Cancer Agency (BCCA) found that the IPS is still valid for prognostication, but outcomes have improved across all IPS groups, with 5-year FFP now ranging from 62% to 88% and 5-year OS ranging from 67% to 98%.6 This improvement is likely a reflection of improved therapy and supportive care. Table 1 shows the PFS and OS within each IPS group, comparing the data from the German Hodgkin Study Group (GHSG) and BCCA group.5,6
High expression of CD68 is associated with adverse outcomes, whereas high FOXP3 and CD20 expression on tumor cells are predictors of superior outcomes.8 A recent study found that CD68 expression was associated with OS. Five-year OS was 88% in those with less than 25% CD68 expression, versus 63% in those with greater than 25% CD68 expression.9
Roemer and colleagues evaluated 108 newly diagnosed cHL biopsy specimens and found that almost all cHL patients had concordant alteration of PD-L1 (programmed death ligand-1) and PD-L2 loci, with a spectrum of 9p24.1 alterations ranging from low level polysomy to near uniform 9p24.1 amplification. PD-L1/PD-L2 copy number alterations are therefore a defining pathobiological feature of cHL.10 PFS was significantly shorter for patients with 9p24.1 amplification, and those patients were likely to have advanced disease suggesting that 9p24.1 amplification is associated with less favorable prognosis.10 This may change with the increasing use of PD-1 inhibitors in the treatment of cHL.
High baseline metabolic tumor volume and total lesion glycolysis have also been associated with adverse outcomes in cHL. While not routinely assessed in practice currently, these tools may ultimately be used to assess prognosis and guide therapy in clinical practice.11
ADVANCED STAGE HODGKIN LYMPHOMA
FRONTLINE THERAPY
First-line Chemotherapy
Chemotherapy plays an essential role in the treatment of advanced stage Hodgkin lymphoma. In the 1960s, the MOPP regimen (nitrogen mustard, vincristine, procarbazine, prednisone) was developed, with a 10-year OS of 50% and a progression-free survival (PFS) of 52% reported in advanced stage patients. The complete remission (CR) rate was 81%, and 36% of patients who achieved CR relapsed later.12 This chemotherapy regimen is associated with a significant rate of myelosuppression and infertility as well as long-term risk of secondary myelodysplasia and acute leukemias.13,14 This led to the development of newer regimens such as ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine).15 In a randomized trial, ABVD showed improved failure-free survival (FFS) over MOPP (61% versus 50% at 5 years) but similar OS (66%–73%).16 In light of these findings, and considering the lower rate of infertility and myelotoxicity, ABVD became the standard of care for advanced stage cHL in the United States.
The Stanford V regimen was developed in an attempt to further minimize toxicity.17 Stanford V is a condensed, 12-week chemotherapy regimen that includes mechlorethamine, doxorubicin, vinblastine, etoposide, prednisone, vincristine, and bleomycin, followed by involved-field radiation therapy (IFRT). Subsequent trials compared the Stanford V and ABVD regimens and showed similar OS, freedom from treatment failure (FFTF), and response rates.18,19 The ABVD regimen was noted to have higher pulmonary toxicity, while other toxicities such as lymphopenia and neuropathy were higher with the Stanford V regimen. In addition, Stanford V requires patients to receive radiation therapy (RT) to original sites of disease larger than 5 cm in size and contiguous sites.
Another regimen which has been studied extensively for advanced stage Hodgkin lymphoma, and is considered a standard of care in some parts of the world, is escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone). In the HD9 study (n = 1196), the GHSG evaluated BEACOPP, escalated BEACOPP, and COPP/ABVD in advanced stage Hodgkin lymphoma.20 All arms of the study included 30 Gy RT to sites of bulky disease or residual disease. This study showed improved OS and FFTF with escalated BEACOPP, but at the cost of higher rates of toxicity. At 10 years, FFTF was 64%, 70%, and 82% with OS rates of 75%, 80%, and 86% for COPP/ABVD, baseline BEACOPP, and escalated BEACOPP, respectively (P < 0.001). The rate of secondary acute leukemia 10 years after treatment was 0.4% for COPP/ABVD, 1.5% for BEACOPP, and 3.0% for escalated BEACOPP. However, 3 subsequent randomized trials did not confirm a survival benefit with escalated BEACOPP relative to ABVD. In the HD 2000 trial (n = 295)21 and in a trial by Viviani and colleagues (n = 331),22 an improvement in OS was not demonstrated in favor of escalated BEACOPP. These studies also confirmed a higher rate of toxicities as well as secondary malignancies associated with the escalated BEACOPP regimen. In the EORTC20012 Intergroup trial (n = 549), 8 cycles of ABVD was compared with 4 cycles of escalated BEACOPP followed by 4 cycles of baseline BEACOPP, without radiation, in patients with clinical stage III or IV Hodgkin lymphoma with IPS score ≥ 3. Both regimens resulted in statistically similar FFS (63.7% in ABVD × 8 versus 69.3% in BEACOPP 4+4) and OS (86.7% in ABVD × 8 vs 90.3% in BEACOPP 4+4).23
In the United States, ABVD (6–8 cycles) is commonly used, although escalated BEACOPP (particularly for patients with an IPS of 4 or higher) and Stanford V are considered appropriate as well.24 In the North American Intergroup study comparing ABVD to Stanford V, and in the trial by Viviani et al, ABVD was associated with a 5- to 7-year FFS of 73% to 79% and OS of 84% to 92%.19,22 Given these excellent results, as well as the potential to cure patients with second-line therapy consisting of autologous hematopoietic cell transplantation (auto-HCT), the general consensus among most U.S. hematologists and oncologists is that ABVD remains the treatment of choice, and that the improved FFS/PFS with escalated BEACOPP is not outweighed by the additional toxicity associated with the regimen. There may, however, be a role for escalated BEACOPP in select patients who have a suboptimal response to ABVD as defined by interim positron emission tomography (iPET) scan (see below).
Brentuximab vedotin is an anti-CD30 antibody-drug conjugate (ADC) consisting of an anti-CD30 antibody linked to monomethyl auristatin E (MMAE), a potent antitubulin agent. CD30 is highly expressed on HRS cells and also in anaplastic large cell lymphoma. Upon binding to CD30, the ADC/CD30 complex is then internalized and directed to the lysosome, where the ADC is proteolytically cleaved, releasing MMAE from the antibody. MMAE then disrupts microtubule networks within the cell, leading to G2/M cycle arrest and apoptosis. CD30 is consistently expressed on HRS cells. In addition to being studied in the relapsed/refractory setting (described below), brentuximab has been studied in the first-line setting. In a phase 1 trial, brentuximab combined with ABVD was associated with increased pulmonary toxicity, while brentuximab + AVD had no significant pulmonary toxicity, with an excellent CR rate (96%), suggesting that substituting brentuximab for bleomycin may be an effective strategy. In addition to possibly being more efficacious, this strategy would also have the benefit of eliminating the risk of bleomycin pulmonary toxicity.25 Based on this data, a large international phase 3 study (the ECHELON-1 trial) comparing ABVD versus brentuximab + AVD in advanced stage cHL patients was recently completed. This study enrolled 1334 patients, and preliminary results were recently announced. With a median follow-up of 24 months, the brentuximab + AVD arm had a 4.9% absolute improvement in PFS relative to the ABVD arm (82.1% versus 77.2%). The brentuximab + AVD arm had an increased incidence of febrile neutropenia, managed with growth factors and peripheral neuropathy requiring dose adjustments, whereas the ABVD arm had an increased rate and severity of pulmonary toxicity.26 Further follow-up will be required to determine whether this will translate into a survival benefit. See Table 2 for a summary of recent large randomized prospective phase 3 trials in advanced stage Hodgkin lymphoma.
Alternative Regimens in Older Patients
Patients older than 60 years of age often have poor tolerance for ABVD and especially escalated BEACOPP. This results in increased treatment-related mortality and reduced overall dose intensity, with higher relapse rates and poor OS. In an attempt to improve on the results of treatment of elderly patients with Hodgkin lymphoma, alternative regimens have been explored. One example is PVAG (prednisone, vinblastine, doxorubicin, gemcitabine). With this regimen, the 3-year OS was 66% and PFS was 58%. One patient out of 59 died from treatment-related toxicity, which is much improved over the historical figures for elderly patients with Hodgkin lymphoma.27 Another commonly used approach in practice is to simply omit bleomycin from ABVD. In the early-stage setting (GHSG HD-13 trial), this regimen (referred to as AVD) led to 89.6% PFS at 5 years, compared to 93.5% with ABVD.28 It therefore stands to reason that this should be a reasonable option in older or more frail advanced stage cHL patients as well.
Brentuximab has been evaluated as a single-agent therapy for first-line therapy of elderly patients with Hodgkin lymphoma. In a phase 2 study, 27 patients (63% with advanced stage disease) were treated, with a 92% overall response rate and 73% CR rate. However the median duration of remission was disappointing at only 9.1 months.29 Based on this data, single-agent brentuximab appears to be a reasonable and well tolerated option for frail or elderly patients, although with the caveat that long-term disease control is relatively uncommon.
RESPONSE-ADAPTED FRONTLINE THERAPY USING INTERIM PET SCAN
In recent years, response-adapted treatment approaches have been extensively researched in cHL using iPET. The goal is to reduce toxicity by minimizing therapy in those who achieve negative iPET and/or to intensify treatment for patients with suboptimal response on iPET. Gallamini et al evaluated the prognostic role of an early iPET scan in advanced Hodgkin lymphoma patients (n = 190) treated with ABVD. This study found that patients with positive iPET had a 2-year PFS of 12.8% versus 95.0% in patients with negative iPET. This result was highly statistically significant (P < 0.0001). This study also showed that PET-2 (iPET after 2 cycles of ABVD) superseded the prognostic value of the IPS at diagnosis.30 As a result, numerous subsequent studies have been pursued using iPET for risk-adapted treatment in cHL.
A critical element to the conduct of iPET risk-adapted treatment for cHL is the interpretation of the iPET. In hopes of standardizing iPET interpretation in clinical trials, a scoring system called the Deauville score was developed. The Deauville score ranges from 1 to 5 (Table 3).
The SWOG (Southwest Oncology Group) S0816 trial (n = 358) evaluated iPET-adapted treatment after 2 cycles of ABVD in stage III or IV Hodgkin lymphoma patients. Patients with positive iPET (Deauville score 4 to 5; n = 60) received escalated BEACOPP for 6 cycles, whereas iPET-negative (Deauville score 1 to 3; n = 271) patients continued to receive 4 more cycles of ABVD. The 2-year PFS was 64% for iPET-positive patients.33 This PFS was much higher than the expected 15% to 30% from prior studies such as Gallamini et al,30 suggesting that the treatment intensification may have been of benefit.
In the HD0801 study (n = 519), newly diagnosed advanced Hodgkin lymphoma patients with positive iPET after 2 cycles of ABVD (n = 103) received early ifosfamide-containing salvage therapy followed by high-dose therapy with autologous stem cell rescue. The 2-year PFS was 76% for PET-2–positive patients, comparable with PET-2–negative patients who had PFS of 81%.34 Again, this result for iPET-positive patients was much better than expected based on the historical control from Gallamini et al, suggesting that the treatment intensification may have been beneficial. It should be emphasized, however, that neither HD0801 nor S0816 were randomized prospective trials; rather, all iPET-positive patients were assigned to an intensified treatment approach.
In the HD18 trial (n = 1100), patients with advanced stage cHL started therapy with escalated BEACOPP and underwent an iPET after 2 cycles. For those with a positive iPET, rituximab was added to escalated BEACOPP in the experimental arm (n = 220) for cycles 3 through 8. The control group (n = 220) continued to receive 6 more cycles of escalated BEACOPP. In the 2 groups, the 3-year PFS was similar (91.4% in escalated BEACOPP, 93% in rituximab + escalated BEACOPP), suggesting no significant benefit with addition of rituximab.35 This study also calls into question whether iPET provides useful information for patients receiving intensive therapy such as escalated BEACOPP, and indicates that the historical control data for iPET-positive patients from Gallamini et al may not be consistently reproduced in other prospective trials. As a result, nonrandomized trials that implement an iPET risk-adapted approach should be interpreted with caution. See Table 4 for a summary of recent trials in advanced stage Hodgkin lymphoma using iPET scan to guide therapy.
RADIATION THERAPY IN FRONTLINE TREATMENT
In patients with advanced stage Hodgkin lymphoma, IFRT to initial bulky sites of disease may be incorporated into frontline therapy to improve local control. However, whether this provides a survival benefit and which patients benefit most from consolidative RT remain unclear.
The European Organization for Research and Treatment of Cancer (EORTC) completed a randomized study in advanced stage Hodgkin lymphoma patients who achieved complete or partial remission after MOPP-ABV.36 Patients in CR were randomly assigned to receive no further treatment versus IFRT (24 Gy to all initially involved nodal areas and 16 to 24 Gy to all initially involved extranodal sites). Patients in partial remission (PR) were treated with 30 Gy to nodal areas and 18 to 24 Gy to extranodal sites. Among the CR patients, the 5-year event-free survival (EFS) was 79% to 84% and did not differ for those who received radiation versus those who did not. Five-year OS was 85% to 91% and also did not differ between the 2 groups. However, among the patients in PR after chemotherapy, the 5-year EFS was 79% and the 5-year OS was 87%, which is better than expected for PR patients, indicating a possible benefit to RT in patients with a partial response after chemotherapy. In the GHSG HD12 trial, patients with advanced stage Hodgkin lymphoma who had a residual lesion by computed tomography (CT) (but not analyzed by PET) had a very subtle improvement in FFTF (90% versus 87%) in favor of consolidation with IFRT, but again no survival benefit was seen.37
The EORTC and HD12 studies described above utilized CT scan for assigning remission status following chemotherapy, and it is now well known that many patients with residual masses (by CT) after chemotherapy may in fact be cured, as such residual radiographic abnormalities may simply be composed of fibrosis. PET scan is more accurate than CT in identifying patients who truly have residual active disease following chemotherapy. As a result, the EORTC study discussed above and the GHSG HD12 trial are of limited relevance in the modern era, in which patients routinely undergo PET scan at the end of therapy. Restricting IFRT to sites that remain PET-positive after completing chemotherapy may be a reasonable strategy that would allow for the avoidance of RT in many patients, and may obviate the need for aggressive second-line therapy (eg, high-dose therapy and autologous hematopoietic cell transplant [auto-HCT]). This approach was taken in the GHSG HD15 trial (n = 2182) in which advanced stage patients were treated with 3 variations on the BEACOPP regimen (8 cycles of escalated BEACOPP, 6 cycles of escalated BEACOPP, or 8 cycles of baseline BEACOPP, randomized in a 1:1:1 ratio). Patients with a residual mass of 2.5 cm or greater on CT scan then underwent a PET scan; if the lesion was PET positive, it was treated with 30 Gy of IFRT. This overall strategy was very effective, with 5-year FFTF rates of 84.4%, 89.3%, and 85.4%, respectively. The OS rates were 91.9%, 95.3%, and 94.5%, respectively. For patients with lesions that remained PET positive after chemotherapy, the PFS rate was 86.2% at 48 months, whereas patients in PR with persistent mass ≥ 2.5 cm but with negative PET had a PFS of 92.6%, similar to that of patients in CR.38 With this approach of BEACOPP followed by PET-guided radiation, the proportion of patients receiving RT was reduced from 71% (in the HD9 study) to only 11% in the HD15 study,38 with no apparent loss in overall efficacy when comparing the results of the 2 studies.
UPFRONT STEM CELL TRANSPLANTATION
To further improve outcomes of patients with advanced Hodgkin lymphoma with high-risk disease, high-dose therapy with auto-HCT has been explored as part of frontline therapy. While this has been shown to be feasible in such patients,39 randomized trials have not shown a clear benefit in terms of FFS or OS with upfront auto-HCT. 40,41 Therefore, auto-HCT is not considered a standard component of frontline therapy for cHL patients who achieve CR by PET/CT scan.
RELAPSED AND REFRACTORY HODGKIN LYMPHOMA
Depending on the stage, risk factors, and frontline regimen utilized, between 5% and 40% of patients with Hodgkin lymphoma can be expected to experience either primary induction failure or a relapse after attaining remission with frontline therapy.3 Primary refractory Hodgkin lymphoma, which occurs in up to 5% to 10% of patients, is defined as progression or no response during induction treatment or within 90 days of completing treatment. In cases where remission status is in question, an updated tissue biopsy is recommended. Biopsy is also recommended in cases in which new sites of disease have appeared or if relapse has occurred after a durable period of remission. Restaging is recommended at the time of relapse.
For younger patients with relapsed/refractory Hodgkin lymphoma, the standard of care in most cases is second-line (or salvage) chemotherapy followed by high-dose therapy and auto-HCT. For patients not felt to be candidates for auto-HCT, options include conventional second-line chemotherapy alone, salvage radiotherapy, novel agents such as brentuximab or immune checkpoint inhibitors, and/or participation in clinical trials.
CONVENTIONAL MULTI-AGENT CHEMOTHERAPY REGIMENS
Numerous conventional regimens have been shown in phase 2 studies to be active in relapsed and refractory Hodgkin lymphoma. These include platinum-based regimens, gemcitabine-based regimens, and alkylator-based regimens. No randomized trials in Hodgkin lymphoma have been conducted comparing these regimens. In general, regimens are chosen based on the patient’s age, performance status, comorbidities, and whether auto-HCT is being considered.
In the United States, platinum-based regimens such as ICE (ifosfamide, carboplatin, etoposide),42 DHAP (dexamethasone, cisplatin, high-dose cytarabine),43 ESHAP (etoposide, methylprednisolone, high-dose cytarabine, cisplatin),44 GDP (gemcitabine, cisplatin, dexamethasone),45 and GCD (gemcitabine, carboplatin, dexamethasone)46 are all considered appropriate second-line therapy options for patients being considered for auto-HCT, due to their high response rates and because autologous hematopoietic stem cell collection remains feasible after these regimens. Response rates range from 60% to 88%, with CR rates between 17% and 41%, and toxic death rates generally well below 5%.
Other gemcitabine-based regimens such as IGEV (ifosfamide, gemcitabine, vinorelbine) and GVD (gemcitabine, vinorelbine, liposomal doxorubicin) are also effective.47,48 GVD is an excellent choice since it is a generally well-tolerated outpatient regimen with a 60% response rate even in heavily pretreated patients.48 Stem cell collection remains feasible after both IGEV and GVD as well. ABVD can produce CR in approximately 20% to 50% of patients initially treated with MOPP.49–51 In practice, however, most patients today with relapsed or refractory Hodgkin lymphoma have already received ABVD as part of their first-line therapy, and retreatment with ABVD is not a good option because it would be associated with prohibitively high cumulative doses of doxorubicin.
These multi-agent chemotherapy regimens may not be tolerated well in patients over age 65 to 70 years or those with significant underlying comorbidities. In recent years, bendamustine has emerged as one of the most active conventional agents for cHL, with overall response rates of 53% to 58% in heavily pre-treated patients.52,53 Bendamustine can generally be tolerated even in elderly patients as well.
Some centers, particularly in Europe, investigated aggressive salvage regimens such as mini-BEAM (carmustine, etoposide, cytarabine, melphalan)54 or dexa-BEAM (BEAM plus dexamethasone).55 These regimens, however, are associated with significant hematologic toxicity and high (2%–5%) treatment-related mortality. As a result, these are rarely used in the United States.
For patients who have progressed after (or are not candidates for) platinum- and/or gemcitabine-based therapy, older alkylator-based regimens such as MOPP, C-MOPP, or ChlVPP (chlorambucil, vinblastine, procarbazine, prednisone) can be considered.56–58 However, these regimens are associated with significant bone marrow suppression, and autologous hematopoietic stem cell collection may no longer be feasible after such regimens. Therefore, these regimens should only be given to patients not felt to be auto-HCT candidates, or patients for whom autologous hematopoietic stem cell collection has already been completed. Weekly vinblastine or single-agent gemcitabine are palliative chemotherapy options, with response rates in the 60% to 80% range. Patients can sometimes be maintained on such low-intensity palliative regimens for 6 to 12 months or longer.59,60
BRENTUXIMAB VEDOTIN
Several trials are evaluating incorporation of brentuximab into second-line therapy in transplant-eligible patients. These approaches have used brentuximab prior to, concurrent with, or following platinum-based chemotherapy.61 While there is currently no consensus on the optimal way to incorporate brentuximab into salvage therapy, it is possible that the use of brentuximab or other novel agents in salvage therapy may allow for avoidance of conventional chemotherapy in some patients. In addition, this may translate into more patients proceeding to auto-HCT in a PET negative state. PET negativity prior to auto-HCT is a powerful predictor of long-term remission after auto-HCT, so any intervention that increases the rate of PET negativity prior to auto-HCT would be expected to improve outcomes with auto-HCT.62–65
For patients not being considered for autoHCT, or those for whom platinum-based salvage therapy was ineffective, single-agent brentuximab is an excellent option. In 2 phase 2 studies, an overall response rate (ORR) of 60% to 75% (including a CR rate of 22%–34%) was seen in relapsed and refractory Hodgkin lymphoma patients.66 The US Food and Drug Administration (FDA) approved brentuximab vedotin in August 2011 for treatment of relapsed and refractory Hodgkin lymphoma, after a failed auto-HCT, or in patients who are not auto-HCT candidates and who have received at least 2 prior chemotherapy regimens. With more extended follow-up, it has become clear that a proportion of patients who achieve CR to brentuximab may maintain remission long-term—58% at 3 years and 38% at 5 years.67 These patients may in fact be cured, in many cases without having undergone allogeneic HCT (allo-HCT) after brentuximab.
PD-1 (IMMUNE CHECKPOINT) INHIBITORS
As discussed earlier, PD-L1/PD-L2 copy number alterations represent a disease-defining feature of cHL. Alterations in chromosome 9p24.1 increase the expression of PD-1 ligands PD-L1 and PD-L2. Nivolumab and pembrolizumab are PD-1-blocking antibodies, which have recently been FDA approved for relapsed and refractory cHL. In a study with 23 patients, with 78% of them relapsing after auto-HCT and 78% relapsing after brentuximab, nivolumab produced an objective response in 87% of the patients, with 17% achieving CR and 70% achieving PR. The rate of PFS was 86% at 24 weeks.68 Pembrolizumab, another PD-1 antagonist, was also tested in relapsed and refractory Hodgkin lymphoma. In the KEYNOTE-087 study (n = 210), pembrolizumab produced an ORR of 64% to 70% in 3 different cohorts of relapsed and refractory cHL patients. Overall CR rate was 22%.69 In general, these agents are well tolerated, although patients must be monitored closely for
inflammatory/autoimmune-type toxicities including skin rash, diarrhea/colitis, transaminitis, endocrine abnormalities, and pneumonitis. Prompt recognition and initiation of corticosteroids is essential in managing these toxicities. Of note, PD-1 inhibitors should be given very cautiously to patients with a prior history of allo-HCT, since 30% to 55% of such patients will experience acute graft-versus-host disease (GVHD) in this setting. In 2 retrospective studies, the response rate was very high at 77% to 95%; however, 10% to 26% of all patients treated with PD-1 inhibitors post-allo-HCT died from GVHD induced by PD-1 inhibition.70,71 These risks and benefits therefore need to be carefully weighed in the post-allo-HCT setting. In another recent study, the outcomes were reported for 39 patients who underwent allo-HCT after prior therapy with a PD-1 inhibitor. Three patients (7.7%) developed lethal acute GVHD, suggesting there may be an increased risk of GVHD in patients undergoing allo-HCT after prior PD-1 inhibitor therapy.72
AUTOLOGOUS STEM CELL TRANSPLANTATION
Several studies have shown an improved disease-free survival (DFS) or FFS in patients with relapsed cHL treated by auto-HCT as compared to those receiving conventional chemotherapy alone.55,73,74 Overall, for relapsed disease, one can expect an approximately 50% to 60% chance for DFS at 5 years post-transplant. In a retrospective, matched-pair analysis, FFP was 62% for auto-HCT patients, compared to 32% for conventional chemotherapy patients. OS, however, was similar for the 2 groups (47%–54%). Patients failing induction therapy or relapsing within 1 year were seen to benefit the most from auto-HCT, including an OS benefit.74
A European prospective randomized trial was conducted comparing conventional salvage therapy to auto-HCT. In this study, 161 patients with relapsed Hodgkin lymphoma were treated with 2 cycles of dexa-BEAM. Those with chemo-sensitive disease were then randomized to either 2 more cycles of dexa-BEAM or high-dose BEAM with auto-HCT. Auto-HCT was associated with an approximately 55% FFTF at 3 years, versus 34% with conventional chemotherapy alone.55 This benefit again was most apparent for patients relapsing within 1 year of completion of primary therapy, although an OS benefit was not seen with auto-HCT. For patients with late relapse (>1 year after completion of primary therapy), auto-HCT was associated with an approximately 75% FFTF at 3 years, versus 50% with chemotherapy alone. One other small randomized trial of auto-HCT in relapsed and refractory Hodgkin lymphoma also showed an improved 3-year EFS in favor of auto-HCT (53% versus 10%), again with no difference in OS.73
The lack of OS benefit seen in these studies suggests that auto-HCT at first or second relapse provides comparable outcomes. Auto-HCT offers the benefit of avoiding the long-term toxicities associated with multiple salvage regimens and the anxiety associated with multiple relapses. In addition, the treatment-related mortality with auto-HCT is now in the 1% to 2% range in younger patients, at centers that perform the procedure routinely. For all of these reasons, auto-HCT is commonly recommended by physicians for Hodgkin lymphoma patients in first or second relapse. In most cases, transplant is favored in first relapse, since waiting until second relapse may be associated with a lower chance of achieving CR and difficulty collecting sufficient hematopoietic stem cells. For patients with early relapse or primary refractory disease, an even stronger case can be made for auto-HCT as the best option to achieve sustained control of the disease. For patients with late relapse, conventional salvage therapy alone may be a reasonable option, particularly in older or frail patients, or those with significant comorbid conditions.
The optimal conditioning regimen for autoHCT for relapsed and refractory Hodgkin lymphoma remains undefined. No randomized trials have been performed comparing conditioning regimens for relapsed and refractory Hodgkin lymphoma. One retrospective study compared 92 patients with Hodgkin lymphoma who underwent auto-HCT using a total-body irradiation (TBI) regimen versus a chemotherapy-alone regimen. No difference in 5-year OS or EFS was seen.75 Given the lack of benefit seen with TBI, along with reports of increased rates of secondary malignancies and myelodysplasia with TBI,76 chemotherapy-alone conditioning regimens are most widely employed. For example, in the United States, either the BEAM or CBV (cyclophosphamide, carmustine, etoposide) regimens are used in over 80% of cases.77 This practice was justified in a Center for International Blood and Marrow Transplant Research (CIBMTR) retrospective study comparing outcomes by conditioning regimens, in which no regimen performed better than BEAM or CBV.78
IFRT is often given as an adjunctive therapy to sites of initial and/or relapsed disease following auto-HCT. Although a relatively common practice, whether this truly enhances outcomes beyond that obtained with auto-HCT alone is unclear. Two retrospective studies have shown some benefit in terms of improvement in OS at 3 to 5 years in the group that received IFRT (70%–73% versus 40%–56%).79,80 Given the retrospective nature and small size of these studies, a prospective study would be needed to properly define the potential role for IFRT following auto-HCT in relapsed/refractory Hodgkin lymphoma. Another retrospective study (n = 73) that evaluated peri-transplant IFRT in Hodgkin lymphoma patients receiving auto transplant found no improvement in survival for patients who received peri-transplant IFRT. This study, however, did show a survival benefit in the subgroup of patients with limited stage disease.81
Prognostic Factors Associated with Outcome with Auto-HCT
The factor most consistently associated with improved outcome for patients with relapsed and refractory Hodgkin lymphoma who undergo auto-HCT is the disease status at transplant.63,77 Those in a second CR, versus a chemo-sensitive relapse (but not CR), versus a chemo-refractory relapse have DFS rates of 60% to 70%, 30% to 40%, and 10% to 20%, respectively.63 The duration between remission and relapse also has important prognostic significance. Late relapse (> 1 year after completion of frontline therapy) is associated with better outcomes as compared to early relapse.55 Other factors with prognostic significance at relapse include anemia, time to relapse and clinical stage, B symptoms, extranodal disease, number of prior chemotherapy regimens, and performance status.42,82 The prognostic impact of pretransplant disease status has been confirmed by studies using functional imaging (eg, FDG-PET or gallium scans). In a report by Moskowitz et al, patients with negative functional imaging following second-line therapy had a 77% EFS post-auto-HCT versus 33% in those whose functional imaging remained positive.62 Very similar findings have been reported by other groups.63–65
Post-Auto-HCT Brentuximab Maintenance
In the multicenter, randomized, double-blinded phase 3 AETHERA trial (n = 329), brentuximab (n = 165) was compared with placebo (n = 164) in patients with unfavorable risk relapsed or primary refractory cHL who had undergone autologous transplant. Eligible patients had at least 1 of the following risk factors for progression after auto-HCT: primary refractory Hodgkin lymphoma (failure to achieve complete remission), relapsed Hodgkin lymphoma with an initial remission duration of less than 12 months, or extranodal involvement at the start of pre-transplantation salvage chemotherapy. Patients were required to have CR, PR, or stable disease after pretransplant salvage chemotherapy with adequate kidney, liver, and bone marrow function. Patients who previously received brentuximab were excluded. Patients received 16 cycles of brentuximab or placebo once every 3 weeks starting 30 to 45 days after transplant. The PFS was significantly improved in the brentuximab group when compared to the placebo group (hazard ratio 0.57; P = 0.0013) after a median observation time of 30 months. Median PFS was 42.9 months in the brentuximab group versus 24.1 months in the placebo group; estimated 2-year PFS rates were 63% in the brentuximab group and 51% in the placebo group. OS was not significantly different between the study groups (~85%), presumably due to the fact that patients in the control group who relapsed likely went on to receive brentuximab as a subsequent therapy.83
PRIMARY REFRACTORY HODGKIN LYMPHOMA
Patients with primary refractory Hodgkin lymphoma have a poor outcome. Salvage therapy using conventional chemotherapy and/or RT results in long-term DFS in 10% or fewer of such patients.13,84 Given these poor outcomes with conventional salvage therapy, auto-HCT is considered to be the standard of care for this subset of patients. The GHSG retrospectively analyzed the prognostic factors and outcomes of patients with primary refractory Hodgkin lymphoma. The 5-year freedom-from-second-failure and the 5-year OS were reported to be 31% and 43%, respectively, for those patients treated with auto-HCT. Patients with poor functional status at time of transplant, age greater than 50 years, and failure to attain a temporary remission had a 0% 5-year OS, as compared to 55% in patients without any of these risk factors.85 A large retrospective European study showed that patients with chemo-resistant disease who underwent transplant had a 19% survival at 5 years.63 Hence, even patients with primary refractory Hodgkin lymphoma have some chance of achieving long-term survival following auto-HCT.
SALVAGE RADIOTHERAPY
The GHSG performed a retrospective analysis of the efficacy of salvage RT in patients with refractory or first-relapsed Hodgkin lymphoma. Five-year FFTF and OS rates were 28% and 51%, respectively. Patients with a limited-stage relapse and without B symptoms were more likely to benefit from salvage RT.86 Campbell et al reported on 81 patients undergoing salvage RT for persistent or recurrent Hodgkin lymphoma after chemotherapy. The 10-year FFTF and OS rates were 33% and 46%, respectively.87 Similarly, Wirth et al reported a 5-year FFS of 26% and 5-year OS of 57%. These figures were 36% and 75%, respectively, in patients whose relapse was limited to supradiaphragmatic nodal sites without B symptoms.88 RT therefore may be a useful strategy for a subset of patients who relapse following chemotherapy, particularly those with a limited-stage relapse, without B symptoms, and those with relapsed disease after a CR, as opposed to those with a partial response or lack of response to the prior chemotherapy regimen.
INVESTIGATIONAL AGENTS AND NOVEL COMBINATIONS
Several biological therapies are emerging as options for the treatment of refractory or relapsed disease. These therapies consist of monoclonal antibodies and ADCs that target cell surface antigens, or small molecules that inhibit key intracellular pathways within neoplastic cells.
Rituximab
Rituximab is a chimeric anti-CD20 monoclonal antibody used widely in B-cell non-Hodgkin lymphomas. The CD20 molecule is typically highly expressed in nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL). Two studies (one in relapsed patients, the other in a mixture of relapsed and previously untreated patients) showed significant activity of rituximab in relapsed NLPHL, with ORRs ranging from 94% to 100%, CR rates ranging from 41% to 53%, and median duration of remission in the 10- to 33-month range.89,90 In cHL, CD20 is expressed in HRS cells in 20% to 30% of cases. In such cases, single-agent rituximab has also shown activity. There is also evidence that rituximab may be effective even in cases in which the HRS cells are CD20-negative, presumably by virtue of depleting reactive B lymphocytes from the microenvironment, which may enhance anti-tumor immunity, or by eliminating a putative CD20-expressing Hodgkin lymphoma stem cell.91,92
Lenalidomide
Lenalidomide is an immunomodulatory drug that has multiple modes of action, including direct induction of apoptosis in tumor cells, antiangiogenic effects, and the activation of immune cells, such as natural killer cells and T cells. Lenalidomide has been shown to modify many features of the microenvironment of HRS cells and has demonstrated activity in other B-cell neoplasms. As a result, lenalidomide has been evaluated in relapsed and refractory Hodgkin lymphoma patients. A multicenter phase 2 study by Fehniger et al included 35 patients, 87% of whom had previously undergone HCT and 55% of whom were refractory to the last therapy.93 All patients were given lenalidomide 25 mg/day from days 1 to 21 of a 28-day cycle until disease progression. One patient was noted to achieve CR, 6 achieved PR, and 5 had stable disease lasting more than 6 months, for an ORR of 19% and a “cytostatic overall response rate” of 33%. The median duration of CR/partial remission was 6 months, with the median time-to-treatment failure in responders (including those with stable disease > 6 months) being 15 months. Similarly, in another study, Böll et al evaluated 12 patients across 4 German centers with relapsed or refractory disease who were treated with oral lenalidomide for 21 days in a 28-day cycle. No radiological evidence of disease progression after 2 cycles of lenalidomide was seen in any of the enrolled patients. ORR was noted to be 50%, with 6 patients with stable disease and 5 patients achieving PR after 2 cycles.94
Novel Brentuximab Combination Therapies
Brentuximab plus bendamustine. The combination of brentuximab and bendamustine was tested as an outpatient regimen in a phase 1/2 study (n = 55) in primary refractory Hodgkin lymphoma or after first relapse. The CR rate of the combination was 74%, with an overall objective response (CR + PR) of 93%. The CR rates were 64% and 84%, respectively, for refractory and relapsed patients. The PFS at 12 months was 80%, establishing this combination therapy as an effective salvage regimen with durable response.95
Brentuximab plus nivolumab. Preliminary results have recently been presented from 2 studies96,97 evaluating the combination of brentuximab and nivolumab. While this combination would still be considered investigational, these studies showed very encouraging ORRs of 90% to 100% and a CR rate of 62% to 66%. Longer follow-up is needed to determine whether these responses are durable and to document the toxicity profile of this combination.
Mammalian Target of Rapamycin Inhibitors
Two mammalian target of rapamycin (mTOR) inhibitors, everolimus and temsirolimus, are currently available in the United States. While neither drug currently has FDA approval for Hodgkin lymphoma, everolimus was evaluated in a phase 2 trial in a heavily pretreated group of relapsed/refractory patients. An ORR of 47% was seen, with a median time to progression of 7.2 months.98
ALLOGENEIC STEM CELL TRANSPLANTATION
Historically, patients who relapse after having an auto-HCT generally had a poor outcome, with a median survival of 2 to 3 years after failure of auto-HCT.99 These patients may be offered palliative chemotherapy (see above), treatment with novel agents (see above), or enrollment in a clinical trial. Select patients may benefit from a second hematopoietic stem cell transplant, most commonly an allo-HCT. However, rare patients with late relapse after auto-HCT may be considered for a second auto-HCT, with a minority of such patients achieving a durable remission after the second auto-HCT.100,101 Because relapse or progressive disease occurs most commonly in the first several months following auto-HCT, patients are more often considered for allo-HCT than a second auto-HCT. In addition, a second auto-HCT may not be feasible due to impaired bone marrow reserve and/or concerns for development of secondary myelodysplasia or acute myeloid leukemia.
Several studies have evaluated allo-HCT in relapsed/ refractory Hodgkin lymphoma. Early studies evaluating myeloablative allo-HCT for Hodgkin lymphoma showed excessive treatment-related mortality (up to 50%) and disappointingly low rates of long-term survival (< 25%).102,103 This was likely related to the fact that, in that era, most of the patients with Hodgkin lymphoma evaluated for allo-HCT were heavily pretreated and therefore at a higher risk for toxicity as well as lymphoma progression.
More recently, several studies have focused on the use of reduced-intensity conditioning (RIC) allo-HCT for relapsed and refractory Hodgkin lymphoma. This approach relies more on a “graft-versus-lymphoma” effect, the existence of which has been debated in Hodgkin lymphoma. Three single-center studies of RIC allo-HCT in patients with multiply recurrent Hodgkin lymphoma showed improved rates of treatment-related mortality (8%–16%) but still relatively low rates of long-term PFS (23%–39% at 2 to 4 years).104–106 Interestingly, in one of these studies the outcomes were more favorable for patients who underwent haploidentical (versus matched sibling or matched unrelated donor) transplants.105
Two large registry studies have also reported on the outcomes of RIC allo-HCT in patients with relapsed and refractory Hodgkin lymphoma.107,108 These studies also confirmed a modest improvement in outcomes compared with those seen historically with myeloablative transplants. Treatment-related mortality at 1 to 2 years was 23% to 33%, depending on whether a matched sibling donor versus an unrelated donor was used. However, long-term PFS (18%–20% at 2 to 5 years) and OS (28%–37% at 2 to 5 years) remained poor, primarily due to high rates of progressive lymphoma post-transplant. In both of these studies, patients were heavily pretreated (84%–96% had received 3 or more prior lines of chemotherapy, and 62%–89% received a prior auto-HCT), with 47% to 55% of patients chemo-resistant prior to transplant. Of note, both of these registry studies reflect patients who underwent transplant prior to the widespread use of brentuximab and PD-1 inhibitors.
Based on the single-center and registry data above, a prospective multicenter European phase 2 trial was conducted to evaluate the benefit of RIC allo-HCT in Hodgkin lymphoma.109 Ninety-two patients (86% with prior auto-HCT, 90% with 3 or more prior lines of therapy) were enrolled and given salvage therapy. Those who had stable disease or better following salvage therapy remained on protocol (n = 78) and underwent RIC with fludarabine and melphalan, followed by allo-HCT (70% with matched sibling donors). Treatment-related mortality was 15% at 1 year. Relapse or progression occurred in 49% at 2 years (35% if chemo-sensitive prior to transplant). Chronic GVHD was associated with a decreased rate of relapse, supporting the existence of a graft-versus-lymphoma effect in Hodgkin lymphoma. Unfortunately, PFS among all allografted patients was still relatively poor (24% at 4 years). However, among patients in CR prior to allo-HCT, a 50% PFS was seen at 4 years. Therefore, even in a prospective multicenter study, RIC allo-HCT offered significant benefit with manageable toxicity in relapsed and refractory Hodgkin lymphoma patients with chemo-sensitive disease.
These studies suggest that outcomes with allo-HCT would improve further if implemented earlier in the course of disease and/or with a lower burden of disease at transplant. It has therefore been suggested that allo-HCT should be considered soon after failure of auto-HCT is documented. In a retrospective study by Sarina et al, 185 Hodgkin lymphoma patients who relapsed following auto-HCT were then immediately considered for reduced-intensity allo-HCT.110 Of these, 122 had a donor identified, and 104 (85%) actually underwent allo-HCT. These 104 patients were then compared to the other 81 patients who either had no donor identified or had a donor but did not receive the planned allo-HCT. Two-year PFS and OS were superior in the patients undergoing allo-HCT (39% versus 14% and 66% versus 42%, respectively, P < 0.001), with a median follow-up of 4 years. The presence of chronic GVHD again was associated with improved PFS and OS. Disease status prior to transplant remained highly predictive of PFS and OS by multivariate analysis. Two other smaller retrospective studies similarly found a survival benefit associated with allo-HCT compared with patients who underwent conventional salvage therapies alone.111,112 These studies, although subject to the usual limitations of retrospective analyses, suggest that the results with reduced-intensity allo-HCT are in fact enhanced if applied earlier in the disease course, and are superior to those with conventional therapy alone.
Currently, the exact role of allo-HSCT, including the optimal timing and optimal donor source (matched sibling versus haploidentical sibling versus matched unrelated donor), remain undefined for relapsed and refractory Hodgkin lymphoma. As discussed earlier, brentuximab is highly active in relapsed Hodgkin lymphoma patients, with a subset of patients still in CR at 5 years.67 For such patients, avoiding the risks of allo-HCT is a desirable goal.
For those who relapse or progress after auto-HCT, a reasonable strategy therefore is to treat initially with brentuximab, unless the patient is already known to have responded poorly to brentuximab, or already has significant neuropathy. Those who achieve a CR to brentuximab are then observed. A subset of those patients will remain in remission at 5 years without further therapy. For those who relapse, or who achieve less than a CR to brentuximab, additional treatment (with brentuximab re-treatment being one option) followed by a reduced-intensity allo-HCT is a reasonable consideration. This approach has the theoretical advantages of (1) avoiding the risk of allo-HCT in the subset potentially cured by brentuximab, (2) getting patients to allo-HCT with fewer comorbidities (due to a lower total exposure to conventional chemotherapy pre-transplant), and (3) applying allo-HCT in the setting of sensitive disease/lower disease burden (due to the high efficacy of brentuximab). The results of a small study suggest that brentuximab may in fact be a very effective “bridge” to allotransplant. Chen et al113 reported on 18 patients with relapsed/refractory Hodgkin lymphoma (17 of whom had previously undergone auto-HCT) who were treated on brentuximab vedotin clinical trials. The data were retrospectively evaluated to determine the efficacy and safety of subsequent reduced-intensity allo-HCT. Remarkably, at 1 year the OS was 100%, PFS was 92%, and nonrelapse mortality was 0% with a median follow-up of 14 months. Hence, brentuximab is safe for use prior to reduced-intensity allo-HCT in heavily pre-treated patients and appears to be associated with very favorable post-transplant outcomes, particularly in comparison to older studies of allo-HCT in the era prior to brentuximab.
SUMMARY
Currently, cure is possible for the majority of patients diagnosed with advanced stage Hodgkin lymphoma. The challenge to the clinician is to provide curative treatment with the lowest risk of serious toxicities. Which regimen will best provide this balance of risk and benefit needs to be assessed based on the relapse risk, age, frailty, and comorbidity profile for an individual patient. For many patients with relapsed or refractory Hodgkin lymphoma, cure remains possible using approaches based on hematopoietic stem cell transplantation, RT, and/or brentuximab. In addition, there are now numerous conventional chemotherapy agents, RT strategies, and exciting newer agents such as PD-1 inhibitors, that can provide significant clinical benefit even when cure is not feasible.
1. Kuppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 1994;91:10962–6.
2. Kuppers R. The biology of Hodgkin’s lymphoma. Nature Rev Cancer 2009;9:15–27.
3. Narra RK, Pingali SR, Fenske TS. Early-stage Hodgkin’s lymphoma. Hospital Physician Hematology-Oncology Board Review Manual. 2017;12(Part 3).
4. Mauch PM, Kalish LA, Kadin M, et al. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993;71:2062–71.
5. Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med 1998;339:1506–14.
6. Moccia AA, Donaldson J, Chhanabhai M, et al. International Prognostic Score in advanced-stage Hodgkin’s lymphoma: altered utility in the modern era. J Clin Oncol 2012;30:3383–8.
7. Diefenbach CS, Li H, Hong F, et al. Evaluation of the International Prognostic Score (IPS-7) and a Simpler Prognostic Score (IPS-3) for advanced Hodgkin lymphoma in the modern era. Br J Haematol 2015;171:530–8.
8. Greaves P, Clear A, Coutinho R, et al. Expression of FOXP3, CD68, and CD20 at diagnosis in the microenvironment of cassical hodgkin lymphoma is predictive of outcome. J Clin Oncol 2013;31:256–62.
9. Touati M, Delage-Corre M, Monteil J, et al. CD68-positive tumor-associated macrophages predict unfavorable treatment outcomes in classical Hodgkin lymphoma in correlation with interim fluorodeoxyglucose-positron emission tomography assessment. Leuk Lymphoma 2015;56:332–41.
10. Roemer MG, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–7.
11. Pike LC, Kirkwood AA, Patrick P, et al. Can baseline PET-CT features predict outcomes in advanced hodgkin lymphoma? A prospective evaluation of UK patients in The RATHL Trial (CRUK/07/033). Hematological Oncology 2017;35(Supplement S2):37–8.
12. DeVita VT Jr, Simon RM, Hubbard SM, et al. Curability of advanced Hodgkin’s disease with chemotherapy. Long-term follow-up of MOPP-treated patients at the National Cancer Institute. Ann Intern Med 1980;92:587–95.
13. Longo DL, Duffey PL, Young RC, et al. Conventional-dose salvage combination chemotherapy in patients relapsing with Hodgkin’s disease after combination chemotherapy: the low probability for cure. J Clin Oncol 1992;10:210–8.
14. Kaldor JM, Day NE, Clarke EA, et al. Leukemia following Hodgkin’s disease. N Engl J Med 1990;322:7–13.
15. Bonadonna G, Zucali R, Monfardini S, et al. Combination chemotherapy of Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide versus MOPP. Cancer 1975;36:252–9.
16. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–84.
17. Horning SJ, Williams J, Bartlett NL, et al. Assessment of the stanford V regimen and consolidative radiotherapy for bulky and advanced Hodgkin’s disease: Eastern Cooperative Oncology Group pilot study E1492. J Clin Oncol 2000;18:972–80.
18. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the stanford V regimen and ABVD in the treatment of advanced Hodgkin’s Lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRCTN 64141244. J Clin Oncol 2009;27:5390–6.
19. Gordon LI, Hong F, Fisher RI, et al. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: an intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496). J Clin Oncol 2013;31:684–91.
20. Engert A, Diehl V, Franklin J, et al. Escalated-dose BEACOPP in the treatment of patients with advanced-stage Hodgkin’s lymphoma: 10 years of follow-up of the GHSG HD9 study. J Clin Oncol 2009;27:4548–54.
21. Merli F, Luminari S, Gobbi PG, et al. Long-term results of the HD2000 trial comparing ABVD versus BEACOPP versus COPP-EBV-CAD in untreated patients with advanced Hodgkin lymphoma: a study by Fondazione Italiana Linfomi. J Clin Oncol 2016;34:1175–81.
22. Viviani S, Zinzani PL, Rambaldi A, et al. ABVD versus BEACOPP for Hodgkin’s lymphoma when high-dose salvage is planned. N Engl J Med 2011;365:203–12.
23. Carde P, Karrasch M, Fortpied C, et al. Eight cycles of ABVD versus four cycles of BEACOPPescalated plus four cycles of BEACOPPbaseline in stage III to IV, International Prognostic Score >/= 3, high-risk Hodgkin lymphoma: first results of the phase III EORTC 20012 intergroup trial. J Clin Oncol 2016;34:2028–36.
24. National Comprehensive Cancer Network I. NCCN Guidelines Version 1.2017 Hodgkin Lymphoma. Accessed July 20, 2017.
25. Younes A, Connors JM, Park SI, et al. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncology 2013;14:1348–56.
26. Takeda and Seattle Genetics announce positive results from phase 3 ECHELON-1 clinical trial evaluating ADCETRIS® (brentuximab vedotin) in frontline advanced Hodgkin lymphoma [press release]. Cambridge, MA: Takeda Oncology; June 26, 2017.
27. Boll B, Bredenfeld H, Gorgen H, et al. Phase 2 study of PVAG (prednisone, vinblastine, doxorubicin, gemcitabine) in elderly patients with early unfavorable or advanced stage Hodgkin lymphoma. Blood 2011;118:6292–8.
28. Behringer K, Goergen H, Hitz F, et al. Omission of dacarbazine or bleomycin, or both, from the ABVD regimen in treatment of early-stage favourable Hodgkin’s lymphoma (GHSG HD13): an open-label, randomised, non-inferiority trial. Lancet 2015;385:1418–27.
29. Forero-Torres A, Holkova B, Goldschmidt J, et al. Phase 2 study of frontline brentuximab vedotin monotherapy in Hodgkin lymphoma patients aged 60 years and older. Blood 2015;126:2798–804.
30. Gallamini A, Hutchings M, Rigacci L, et al. Early interim 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol 2007;25:3746–52.
31. Meignan M, Gallamini A, Meignan M, et al. Report on the First International Workshop on Interim-PET-Scan in Lymphoma. Leuk Lymphoma 2009;50:1257–60.
32. Barrington SF, Mikhaeel NG, Kostakoglu L, et al. Role of Imaging in the Staging and Response Assessment of Lymphoma: Consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 2014;32:3048–58.
33. Press OW, Li H, Schoder H, et al. US Intergroup trial of response-adapted therapy for stage III to IV Hodgkin lymphoma using early interim fluorodeoxyglucose-positron emission tomography imaging: Southwest Oncology Group S0816. J Clin Oncol 2016;34:2020–7.
34. Zinzani PL, Broccoli A, Gioia DM, et al. Interim positron emission tomography response-adapted therapy in advanced-stage Hodgkin lymphoma: final results of the pPhase II part of the HD0801 study. J Clin Oncol 2016;34:1376–85.
35. Borchmann P, Haverkamp H, Lohri A, et al. Progression-free survival of early interim PET-positive patients with advanced stage Hodgkin’s lymphoma treated with BEACOPPescalated alone or in combination with rituximab (HD18): an open-label, international, randomised phase 3 study by the German Hodgkin Study Group. Lancet Oncology 2017;18:454–63.
36. Aleman BM, Raemaekers JM, Tirelli U, et al. Involved-field radiotherapy for advanced Hodgkin’s lymphoma. N Engl J Med 2003;348:2396–406.
37. Borchmann P, Haverkamp H, Diehl V, et al. Eight cycles of escalated-dose BEACOPP compared with four cycles of escalated-dose BEACOPP followed by four cycles of baseline-dose BEACOPP with or without radiotherapy in patients with advanced-stage hodgkin’s lymphoma: final analysis of the HD12 trial of the German Hodgkin Study Group. J Clin Oncol 2011;29:4234–42.
38. Engert A, Haverkamp H, Kobe C, et al. Reduced-intensity chemotherapy and PET-guided radiotherapy in patients with advanced stage Hodgkin’s lymphoma (HD15 trial): a randomised, open-label, phase 3 non-inferiority trial. Lancet 2012;379:1791–9.
39. Nademanee A, Molina A, Fung H, et al. High-dose chemo/radiotherapy and autologous bone marrow or stem cell transplantation for poor-risk advanced-stage Hodgkin’s disease during first partial or complete remission. Biol Blood Marrow Transplant 1999;5:292–8.
40. Federico M, Bellei M, Brice P, et al. High-dose therapy and autologous stem-cell transplantation versus conventional therapy for patients with advanced Hodgkin’s lymphoma responding to front-line therapy. J Clin Oncol 2003;21:2320–5.
41. Arakelyan N, Berthou C, Desablens B, et al. Early versus late intensification for patients with high-risk Hodgkin lymphoma-3 cycles of intensive chemotherapy plus low-dose lymph node radiation therapy versus 4 cycles of combined doxorubicin, bleomycin, vinblastine, and dacarbazine plus myeloablative chemotherapy with autologous stem cell transplantation: five-year results of a randomized trial on behalf of the GOELAMS Group. Cancer 2008;113:3323–30.
42. Moskowitz CH, Nimer SD, Zelenetz AD, et al. A 2-step comprehensive high-dose chemoradiotherapy second-line program for relapsed and refractory Hodgkin disease: analysis by intent to treat and development of a prognostic model. Blood 2001;97:616–23.
43. Josting A, Rudolph C, Reiser M, et al. Time-intensified dexamethasone/cisplatin/cytarabine: an effective salvage therapy with low toxicity in patients with relapsed and refractory Hodgkin’s disease. Ann Oncol 2002;13:1628–35.
44. Aparicio J, Segura A, Garcera S, et al. ESHAP is an active regimen for relapsing Hodgkin’s disease. Ann Oncol 1999;10:593–5.
45. Baetz T, Belch A, Couban S, et al. Gemcitabine, dexamethasone and cisplatin is an active and non-toxic chemotherapy regimen in relapsed or refractory Hodgkin’s disease: a phase II study by the National Cancer Institute of Canada Clinical Trials Group. Ann Oncol 2003;14:1762–7.
46. Gopal AK, Press OW, Shustov AR, et al. Efficacy and safety of gemcitabine, carboplatin, dexamethasone, and rituximab in patients with relapsed/refractory lymphoma: a prospective multi-center phase II study by the Puget Sound Oncology Consortium. Leuk Lymphoma 2010;51:1523–9.
47. Santoro A, Magagnoli M, Spina M, et al. Ifosfamide, gemcitabine, and vinorelbine: a new induction regimen for refractory and relapsed Hodgkin’s lymphoma. Haematologica 2007;92:35–41.
48. Bartlett NL, Niedzwiecki D, Johnson JL, et al. Gemcitabine, vinorelbine, and pegylated liposomal doxorubicin (GVD), a salvage regimen in relapsed Hodgkin’s lymphoma: CALGB 59804. Ann Oncol 2007;18:1071–9.
49. Santoro A, Bonadonna G. Prolonged disease-free survival in MOPP-resistant Hodgkin’s disease after treatment with adriamycin, bleomycin, vinblastine and dacarbazine (ABVD). Cancer Chemother Pharmacol 1979;2:101–5.
50. Krikorian JG, Portlock CS, Rosenberg SA. Treatment of advanced Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide (ABVD) after failure of MOPP therapy. Cancer 1978;41:2107–11.
51. Piga A, Ambrosetti A, Todeschini G, et al. Doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) salvage of mechlorethamine, vincristine, prednisone, and procarbazine (MOPP)-resistant advanced Hodgkin’s disease. Cancer Treat Rep 1984;68:947–51.
52. Moskowitz AJ, Hamlin PA Jr, Perales MA, et al. Phase II study of bendamustine in relapsed and refractory Hodgkin lymphoma. J Clin Oncol 2013;31:456–60.
53. Anastasia A, Carlo-Stella C, Corradini P, et al. Bendamustine for relapsed/refractory classical Hodgkin lymphoma after high dose chemotherapy and or allogeneic transplant: a study of Fondazione Italiana Linfomi (FIL). Blood 2012;120:3652.
54. Martin A, Fernandez-Jimenez MC, Caballero MD, et al. Long-term follow-up in patients treated with Mini-BEAM as salvage therapy for relapsed or refractory Hodgkin’s disease. Br J Haematol 2001;113:161–71.
55. Schmitz N, Pfistner B, Sextro M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet 2002;359:2065–71.
56. Fisher RI, DeVita VT, Hubbard SP, et al. Prolonged disease-free survival in Hodgkin’s disease with MOPP reinduction after first relapse. Ann Intern Med 1979;90:761–3.
57. ChlVPP therapy for Hodgkin’s disease: experience of 960 patients. The International ChlVPP Treatment Group. Ann Oncol 1995;6:167–72.
58. Selby P, Patel P, Milan S, et al. ChlVPP combination chemotherapy for Hodgkin’s disease: long-term results. Br J Cancer 1990;62:279–85.
59. Santoro A, Bredenfeld H, Devizzi L, et al. Gemcitabine in the treatment of refractory Hodgkin’s disease: results of a multicenter phase II study. J Clin Oncol 2000;18:2615–9.
60. Little R, Wittes RE, Longo DL, Wilson WH. Vinblastine for recurrent Hodgkin’s disease following autologous bone marrow transplant. J Clin Oncol 1998;16:584–8.
61. Chen R, Palmer JM, Martin P, et al. Results of a multicenter phase II trial of brentuximab vedotin as second-line therapy before autologous transplantation in relapsed/refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2015;21:2136–40.
62. Moskowitz CH, Yahalom J, Zelenetz AD, et al. High-dose chemo-radiotherapy for relapsed or refractory Hodgkin lymphoma and the significance of pre-transplant functional imaging. Br J Haematol 2010;148:890–7.
63. Sureda A, Constans M, Iriondo A, et al. Prognostic factors affecting long-term outcome after stem cell transplantation in Hodgkin’s lymphoma autografted after a first relapse. Ann Oncol 2005;16:625–33.
64. Crocchiolo R, Canevari C, Assanelli A, et al. Pre-transplant 18FDG-PET predicts outcome in lymphoma patients treated with high-dose sequential chemotherapy followed by autologous stem cell transplantation. Leuk Lymphoma 2008;49:727–33.
65. Mocikova H, Pytlik R, Markova J, et al. Pre-transplant positron emission tomography in patients with relapsed Hodgkin lymphoma. Leuk Lymphoma 2011;52:1668–74.
66. Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol 2012;30:2183–9.
67. Gopal AK, Chen R, Smith SE, et al. Durable remissions in a pivotal phase 2 study of brentuximab vedotin in relapsed or refractory Hodgkin lymphoma. Blood 2015;125:1236–43.
68. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma.N Engl J Med 2015;372:311–9.
69. Chen R, Zinzani PL, Fanale MA, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol 2017;35:2125–32.
70. Haverkos BM, Abbott D, Hamadani M, et al. PD-1 blockade for relapsed lymphoma post-allogeneic hematopoietic cell transplant: high response rate but frequent GVHD. Blood 2017;130:221–8.
71. Herbaux C, Gauthier J, Brice P, et al. Efficacy and tolerability of nivolumab after allogeneic transplantation for relapsed Hodgkin lymphoma. Blood 2017;129:2471–8.
72. Merryman RW, Kim HT, Zinzani PL et al. Safety and efficacy of allogeneic hematopoietic stem cell transplant after PD-1 blockage in relapsed/refractory lymphoma. Blood 2017;129:1380–8.
73. Linch DC, Winfield D, Goldstone AH, et al. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin’s disease: results of a BNLI randomised trial. Lancet 1993;341:1051–4.
74. Yuen AR, Rosenberg SA, Hoppe RT, et al. Comparison between conventional salvage therapy and high-dose therapy with autografting for recurrent or refractory Hodgkin’s disease. Blood 1997;89:814–22.
75. Gutierrez-Delgado F, Holmberg L, Hooper H, et al. Autologous stem cell transplantation for Hodgkin’s disease: busulfan, melphalan and thiotepa compared to a radiation-based regimen. Bone Marrow Transplant 2003;32:279–85.
76. Hake CR, Graubert TA, Fenske TS. Does autologous transplantation directly increase the risk of secondary leukemia in lymphoma patients? Bone Marrow Transplant 2007;39:59–70.
77. Hahn T, McCarthy PL, Carreras J, et al. Comparison of prognostic models for autologous hematopoietic stem cell transplantation (AHCT) for relapsed Hodgkin lymphoma. Blood 2009;114:1215.
78. Chen Y-B, Lane AA, Logan BR, et al. Impact of conditioning regimen on outcomes for patients with lymphoma undergoing high-dose therapy with autologous hematopoietic cell transplantation. Biology Blood Marrow Transplant 2015;21:1046–53.
79. Wendland MM, Asch JD, Pulsipher MA, et al. The impact of involved field radiation therapy for patients receiving high-dose chemotherapy followed by hematopoietic progenitor cell transplant for the treatment of relapsed or refractory Hodgkin disease. Am J Clin Oncol 2006;29:189–95.
80. Biswas T, Culakova E, Friedberg JW, et al. Involved field radiation therapy following high dose chemotherapy and autologous stem cell transplant benefits local control and survival in refractory or recurrent Hodgkin lymphoma. Radiother Oncol 2012;103:367–72.
81. Levis M, Piva C, Filippi AR, et al. Potential benefit of involved-field radiotherapy for patients with Relapsed-refractory Hodgkin’s lymphoma with incomplete response before autologous stem cell transplantation. Clin Lymphoma Myeloma Leuk 2017;17:14–22.
82. Josting A, Franklin J, May M, et al. New prognostic score based on treatment outcome of patients with relapsed Hodgkin’s lymphoma registered in the database of the German Hodgkin’s lymphoma study group. J Clin Oncol 2002;20:221–30.
83. Moskowitz CH, Nademanee A, Masszi T, et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015;385:1853–62.
84. Bonfante V, Santoro A, Viviani S, et al. Outcome of patients with Hodgkin’s disease failing after primary MOPP-ABVD. J Clin Oncol 1997;15:528–34.
85. Josting A, Rueffer U, Franklin J, et al. Prognostic factors and treatment outcome in primary progressive Hodgkin lymphoma: a report from the German Hodgkin Lymphoma Study Group. Blood 2000;96:1280–6.
86. Josting A, Nogova L, Franklin J, et al. Salvage radiotherapy in patients with relapsed and refractory Hodgkin’s lymphoma: a retrospective analysis from the German Hodgkin Lymphoma Study Group. J Clin Oncol 2005;23:1522–9.
87. Campbell B WA, Milner A, Di Iulio J, et al. Long-term follow-up of salvage radiotherapy in Hodgkin’s lymphoma after chemotherapy failure. Int J Radiat Oncol Biol Phys 2005;63:1538–45.
88. Wirth A, Corry J, Laidlaw C, et al. Salvage radiotherapy for Hodgkin’s disease following chemotherapy failure. Int J Radiat Oncol Biol Phys 1997;39:599–607.
89. Schulz H, Rehwald U, Morschhauser F, et al. Rituximab in relapsed lymphocyte-predominant Hodgkin lymphoma: long-term results of a phase 2 trial by the German Hodgkin Lymphoma Study Group (GHSG). Blood 2008;111:109–11.
90. Ekstrand BC, Lucas JB, Horwitz SM, et al. Rituximab in lymphocyte-predominant Hodgkin disease: results of a phase 2 trial. Blood 2003;101:4285–9.
91. Younes A, Romaguera J, Hagemeister F, et al. A pilot study of rituximab in patients with recurrent, classic Hodgkin disease. Cancer 2003;98:310–4.
92. Rehwald U, Schulz H, Reiser M, et al. Treatment of relapsed CD20+ Hodgkin lymphoma with the monoclonal antibody rituximab is effective and well tolerated: results of a phase 2 trial of the German Hodgkin Lymphoma Study Group. Blood 2003;101:420–4.
93. Fehniger TA, Larson S, Trinkaus K, et al. A phase 2 multicenter study of lenalidomide in relapsed or refractory classical Hodgkin lymphoma. Blood 2011;118:5119–25.
94. Boll B, Borchmann P, Topp MS, et al. Lenalidomide in patients with refractory or multiple relapsed Hodgkin lymphoma. Br J Haematol 2010;148:480–2.
95. LaCasce AS, Bociek G, Sawas A, et al. Brentuximab vedotin plus bendamustine: a highly active salvage treatment regimen for patients with relapsed or refractory Hodgkin lymphoma. Blood 2015;126:3982.
96. Diefenbach CS, Hong F, David KA, et al. A phase I study with an expansion cohort of the combination of ipilimumab and nivolumab and brentuximab vedotin in patients with relapsed/refractory Hodgkin lymphoma: A trial of the ECOG-ACRIN Cancer Research Group (E4412 Arms D and E). Blood 2016;128:1106.
97. Herrera AF, Bartlett NL, Ramchandren R, et al. Preliminary results from a phase 1/2 study of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2016;128:1105.
98. Johnston PB, Inwards DJ, Colgan JP, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol 2010;85:320–4.
99. Kewalramani T, Nimer SD, Zelenetz AD, et al. Progressive disease following autologous transplantation in patients with chemosensitive relapsed or primary refractory Hodgkin’s disease or aggressive non-Hodgkin’s lymphoma. Bone Marrow Transplant 2003;32:673–9.
100. Lin TS, Avalos BR, Penza SL, et al. Second autologous stem cell transplant for multiply relapsed Hodgkin’s disease. Bone Marrow Transplant 2002;29:763–7.
101. Smith SM, van Besien K, Carreras J, et al. Second autologous stem cell transplantation for relapsed lymphoma after a prior autologous transplant. Biol Blood Marrow Transplant 2008;14:904–12.
102. Gajewski JL, Phillips GL, Sobocinski KA, et al. Bone marrow transplants from HLA-identical siblings in advanced Hodgkin’s disease. J Clin Oncol 1996;14:572–8.
103. Peniket AJ, Ruiz de Elvira MC, Taghipour G, et al. An EBMT registry matched study of allogeneic stem cell transplants for lymphoma: allogeneic transplantation is associated with a lower relapse rate but a higher procedure-related mortality rate than autologous transplantation. Bone Marrow Transplant 2003;31:667–78.
104. Anderlini P, Saliba R, Acholonu S, et al. Fludarabine-melphalan as a preparative regimen for reduced-intensity conditioning allogeneic stem cell transplantation in relapsed and refractory Hodgkin’s lymphoma: the updated M.D. Anderson Cancer Center experience. Haematologica 2008;93:257–64.
105. Burroughs LM, O’Donnell PV, Sandmaier BM, et al. Comparison of outcomes of HLA-matched related, unrelated, or HLA-haploidentical related hematopoietic cell transplantation following nonmyeloablative conditioning for relapsed or refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2008;14:1279–87.
106. Peggs KS, Hunter A, Chopra R, et al. Clinical evidence of a graft-versus-Hodgkin’s-lymphoma effect after reduced-intensity allogeneic transplantation. Lancet 2005;365:1934–41.
107. Sureda A, Robinson S, Canals C, et al. Reduced-intensity conditioning compared with conventional allogeneic stem-cell transplantation in relapsed or refractory Hodgkin’s lymphoma: an analysis from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2008;26:455–62.
108. Devetten MP, Hari PN, Carreras J, et al. Unrelated donor reduced-intensity allogeneic hematopoietic stem cell transplantation for relapsed and refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:109–17.
109. Sureda A, Canals C, Arranz R, et al. Allogeneic stem cell transplantation after reduced intensity conditioning in patients with relapsed or refractory Hodgkin’s lymphoma. Results of the HDR-ALLO study - a prospective clinical trial by the Grupo Espanol de Linfomas/Trasplante de Medula Osea (GEL/TAMO) and the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. Haematologica 2012;97:310–7.
110. Sarina B, Castagna L, Farina L, et al. Allogeneic transplantation improves the overall and progression-free survival of Hodgkin lymphoma patients relapsing after autologous transplantation: a retrospective study based on the time of HLA typing and donor availability. Blood 2010;115:3671–7.
111. Castagna L, Sarina B, Todisco E, et al. Allogeneic stem cell transplantation compared with chemotherapy for poor-risk Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:432–8.
112. Thomson KJ, Peggs KS, Smith P, et al. Superiority of reduced-intensity allogeneic transplantation over conventional treatment for relapse of Hodgkin’s lymphoma following autologous stem cell transplantation. Bone Marrow Transplant 2008;41:765–70.
113. Chen R, Palmer JM, Thomas SH, et al. Brentuximab vedotin enables successful reduced-intensity allogeneic hematopoietic cell transplantation in patients with relapsed or refractory Hodgkin lymphoma. Blood 2012;119:6379–81.
1. Kuppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 1994;91:10962–6.
2. Kuppers R. The biology of Hodgkin’s lymphoma. Nature Rev Cancer 2009;9:15–27.
3. Narra RK, Pingali SR, Fenske TS. Early-stage Hodgkin’s lymphoma. Hospital Physician Hematology-Oncology Board Review Manual. 2017;12(Part 3).
4. Mauch PM, Kalish LA, Kadin M, et al. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993;71:2062–71.
5. Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med 1998;339:1506–14.
6. Moccia AA, Donaldson J, Chhanabhai M, et al. International Prognostic Score in advanced-stage Hodgkin’s lymphoma: altered utility in the modern era. J Clin Oncol 2012;30:3383–8.
7. Diefenbach CS, Li H, Hong F, et al. Evaluation of the International Prognostic Score (IPS-7) and a Simpler Prognostic Score (IPS-3) for advanced Hodgkin lymphoma in the modern era. Br J Haematol 2015;171:530–8.
8. Greaves P, Clear A, Coutinho R, et al. Expression of FOXP3, CD68, and CD20 at diagnosis in the microenvironment of cassical hodgkin lymphoma is predictive of outcome. J Clin Oncol 2013;31:256–62.
9. Touati M, Delage-Corre M, Monteil J, et al. CD68-positive tumor-associated macrophages predict unfavorable treatment outcomes in classical Hodgkin lymphoma in correlation with interim fluorodeoxyglucose-positron emission tomography assessment. Leuk Lymphoma 2015;56:332–41.
10. Roemer MG, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–7.
11. Pike LC, Kirkwood AA, Patrick P, et al. Can baseline PET-CT features predict outcomes in advanced hodgkin lymphoma? A prospective evaluation of UK patients in The RATHL Trial (CRUK/07/033). Hematological Oncology 2017;35(Supplement S2):37–8.
12. DeVita VT Jr, Simon RM, Hubbard SM, et al. Curability of advanced Hodgkin’s disease with chemotherapy. Long-term follow-up of MOPP-treated patients at the National Cancer Institute. Ann Intern Med 1980;92:587–95.
13. Longo DL, Duffey PL, Young RC, et al. Conventional-dose salvage combination chemotherapy in patients relapsing with Hodgkin’s disease after combination chemotherapy: the low probability for cure. J Clin Oncol 1992;10:210–8.
14. Kaldor JM, Day NE, Clarke EA, et al. Leukemia following Hodgkin’s disease. N Engl J Med 1990;322:7–13.
15. Bonadonna G, Zucali R, Monfardini S, et al. Combination chemotherapy of Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide versus MOPP. Cancer 1975;36:252–9.
16. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–84.
17. Horning SJ, Williams J, Bartlett NL, et al. Assessment of the stanford V regimen and consolidative radiotherapy for bulky and advanced Hodgkin’s disease: Eastern Cooperative Oncology Group pilot study E1492. J Clin Oncol 2000;18:972–80.
18. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the stanford V regimen and ABVD in the treatment of advanced Hodgkin’s Lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRCTN 64141244. J Clin Oncol 2009;27:5390–6.
19. Gordon LI, Hong F, Fisher RI, et al. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: an intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496). J Clin Oncol 2013;31:684–91.
20. Engert A, Diehl V, Franklin J, et al. Escalated-dose BEACOPP in the treatment of patients with advanced-stage Hodgkin’s lymphoma: 10 years of follow-up of the GHSG HD9 study. J Clin Oncol 2009;27:4548–54.
21. Merli F, Luminari S, Gobbi PG, et al. Long-term results of the HD2000 trial comparing ABVD versus BEACOPP versus COPP-EBV-CAD in untreated patients with advanced Hodgkin lymphoma: a study by Fondazione Italiana Linfomi. J Clin Oncol 2016;34:1175–81.
22. Viviani S, Zinzani PL, Rambaldi A, et al. ABVD versus BEACOPP for Hodgkin’s lymphoma when high-dose salvage is planned. N Engl J Med 2011;365:203–12.
23. Carde P, Karrasch M, Fortpied C, et al. Eight cycles of ABVD versus four cycles of BEACOPPescalated plus four cycles of BEACOPPbaseline in stage III to IV, International Prognostic Score >/= 3, high-risk Hodgkin lymphoma: first results of the phase III EORTC 20012 intergroup trial. J Clin Oncol 2016;34:2028–36.
24. National Comprehensive Cancer Network I. NCCN Guidelines Version 1.2017 Hodgkin Lymphoma. Accessed July 20, 2017.
25. Younes A, Connors JM, Park SI, et al. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncology 2013;14:1348–56.
26. Takeda and Seattle Genetics announce positive results from phase 3 ECHELON-1 clinical trial evaluating ADCETRIS® (brentuximab vedotin) in frontline advanced Hodgkin lymphoma [press release]. Cambridge, MA: Takeda Oncology; June 26, 2017.
27. Boll B, Bredenfeld H, Gorgen H, et al. Phase 2 study of PVAG (prednisone, vinblastine, doxorubicin, gemcitabine) in elderly patients with early unfavorable or advanced stage Hodgkin lymphoma. Blood 2011;118:6292–8.
28. Behringer K, Goergen H, Hitz F, et al. Omission of dacarbazine or bleomycin, or both, from the ABVD regimen in treatment of early-stage favourable Hodgkin’s lymphoma (GHSG HD13): an open-label, randomised, non-inferiority trial. Lancet 2015;385:1418–27.
29. Forero-Torres A, Holkova B, Goldschmidt J, et al. Phase 2 study of frontline brentuximab vedotin monotherapy in Hodgkin lymphoma patients aged 60 years and older. Blood 2015;126:2798–804.
30. Gallamini A, Hutchings M, Rigacci L, et al. Early interim 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol 2007;25:3746–52.
31. Meignan M, Gallamini A, Meignan M, et al. Report on the First International Workshop on Interim-PET-Scan in Lymphoma. Leuk Lymphoma 2009;50:1257–60.
32. Barrington SF, Mikhaeel NG, Kostakoglu L, et al. Role of Imaging in the Staging and Response Assessment of Lymphoma: Consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 2014;32:3048–58.
33. Press OW, Li H, Schoder H, et al. US Intergroup trial of response-adapted therapy for stage III to IV Hodgkin lymphoma using early interim fluorodeoxyglucose-positron emission tomography imaging: Southwest Oncology Group S0816. J Clin Oncol 2016;34:2020–7.
34. Zinzani PL, Broccoli A, Gioia DM, et al. Interim positron emission tomography response-adapted therapy in advanced-stage Hodgkin lymphoma: final results of the pPhase II part of the HD0801 study. J Clin Oncol 2016;34:1376–85.
35. Borchmann P, Haverkamp H, Lohri A, et al. Progression-free survival of early interim PET-positive patients with advanced stage Hodgkin’s lymphoma treated with BEACOPPescalated alone or in combination with rituximab (HD18): an open-label, international, randomised phase 3 study by the German Hodgkin Study Group. Lancet Oncology 2017;18:454–63.
36. Aleman BM, Raemaekers JM, Tirelli U, et al. Involved-field radiotherapy for advanced Hodgkin’s lymphoma. N Engl J Med 2003;348:2396–406.
37. Borchmann P, Haverkamp H, Diehl V, et al. Eight cycles of escalated-dose BEACOPP compared with four cycles of escalated-dose BEACOPP followed by four cycles of baseline-dose BEACOPP with or without radiotherapy in patients with advanced-stage hodgkin’s lymphoma: final analysis of the HD12 trial of the German Hodgkin Study Group. J Clin Oncol 2011;29:4234–42.
38. Engert A, Haverkamp H, Kobe C, et al. Reduced-intensity chemotherapy and PET-guided radiotherapy in patients with advanced stage Hodgkin’s lymphoma (HD15 trial): a randomised, open-label, phase 3 non-inferiority trial. Lancet 2012;379:1791–9.
39. Nademanee A, Molina A, Fung H, et al. High-dose chemo/radiotherapy and autologous bone marrow or stem cell transplantation for poor-risk advanced-stage Hodgkin’s disease during first partial or complete remission. Biol Blood Marrow Transplant 1999;5:292–8.
40. Federico M, Bellei M, Brice P, et al. High-dose therapy and autologous stem-cell transplantation versus conventional therapy for patients with advanced Hodgkin’s lymphoma responding to front-line therapy. J Clin Oncol 2003;21:2320–5.
41. Arakelyan N, Berthou C, Desablens B, et al. Early versus late intensification for patients with high-risk Hodgkin lymphoma-3 cycles of intensive chemotherapy plus low-dose lymph node radiation therapy versus 4 cycles of combined doxorubicin, bleomycin, vinblastine, and dacarbazine plus myeloablative chemotherapy with autologous stem cell transplantation: five-year results of a randomized trial on behalf of the GOELAMS Group. Cancer 2008;113:3323–30.
42. Moskowitz CH, Nimer SD, Zelenetz AD, et al. A 2-step comprehensive high-dose chemoradiotherapy second-line program for relapsed and refractory Hodgkin disease: analysis by intent to treat and development of a prognostic model. Blood 2001;97:616–23.
43. Josting A, Rudolph C, Reiser M, et al. Time-intensified dexamethasone/cisplatin/cytarabine: an effective salvage therapy with low toxicity in patients with relapsed and refractory Hodgkin’s disease. Ann Oncol 2002;13:1628–35.
44. Aparicio J, Segura A, Garcera S, et al. ESHAP is an active regimen for relapsing Hodgkin’s disease. Ann Oncol 1999;10:593–5.
45. Baetz T, Belch A, Couban S, et al. Gemcitabine, dexamethasone and cisplatin is an active and non-toxic chemotherapy regimen in relapsed or refractory Hodgkin’s disease: a phase II study by the National Cancer Institute of Canada Clinical Trials Group. Ann Oncol 2003;14:1762–7.
46. Gopal AK, Press OW, Shustov AR, et al. Efficacy and safety of gemcitabine, carboplatin, dexamethasone, and rituximab in patients with relapsed/refractory lymphoma: a prospective multi-center phase II study by the Puget Sound Oncology Consortium. Leuk Lymphoma 2010;51:1523–9.
47. Santoro A, Magagnoli M, Spina M, et al. Ifosfamide, gemcitabine, and vinorelbine: a new induction regimen for refractory and relapsed Hodgkin’s lymphoma. Haematologica 2007;92:35–41.
48. Bartlett NL, Niedzwiecki D, Johnson JL, et al. Gemcitabine, vinorelbine, and pegylated liposomal doxorubicin (GVD), a salvage regimen in relapsed Hodgkin’s lymphoma: CALGB 59804. Ann Oncol 2007;18:1071–9.
49. Santoro A, Bonadonna G. Prolonged disease-free survival in MOPP-resistant Hodgkin’s disease after treatment with adriamycin, bleomycin, vinblastine and dacarbazine (ABVD). Cancer Chemother Pharmacol 1979;2:101–5.
50. Krikorian JG, Portlock CS, Rosenberg SA. Treatment of advanced Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide (ABVD) after failure of MOPP therapy. Cancer 1978;41:2107–11.
51. Piga A, Ambrosetti A, Todeschini G, et al. Doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) salvage of mechlorethamine, vincristine, prednisone, and procarbazine (MOPP)-resistant advanced Hodgkin’s disease. Cancer Treat Rep 1984;68:947–51.
52. Moskowitz AJ, Hamlin PA Jr, Perales MA, et al. Phase II study of bendamustine in relapsed and refractory Hodgkin lymphoma. J Clin Oncol 2013;31:456–60.
53. Anastasia A, Carlo-Stella C, Corradini P, et al. Bendamustine for relapsed/refractory classical Hodgkin lymphoma after high dose chemotherapy and or allogeneic transplant: a study of Fondazione Italiana Linfomi (FIL). Blood 2012;120:3652.
54. Martin A, Fernandez-Jimenez MC, Caballero MD, et al. Long-term follow-up in patients treated with Mini-BEAM as salvage therapy for relapsed or refractory Hodgkin’s disease. Br J Haematol 2001;113:161–71.
55. Schmitz N, Pfistner B, Sextro M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet 2002;359:2065–71.
56. Fisher RI, DeVita VT, Hubbard SP, et al. Prolonged disease-free survival in Hodgkin’s disease with MOPP reinduction after first relapse. Ann Intern Med 1979;90:761–3.
57. ChlVPP therapy for Hodgkin’s disease: experience of 960 patients. The International ChlVPP Treatment Group. Ann Oncol 1995;6:167–72.
58. Selby P, Patel P, Milan S, et al. ChlVPP combination chemotherapy for Hodgkin’s disease: long-term results. Br J Cancer 1990;62:279–85.
59. Santoro A, Bredenfeld H, Devizzi L, et al. Gemcitabine in the treatment of refractory Hodgkin’s disease: results of a multicenter phase II study. J Clin Oncol 2000;18:2615–9.
60. Little R, Wittes RE, Longo DL, Wilson WH. Vinblastine for recurrent Hodgkin’s disease following autologous bone marrow transplant. J Clin Oncol 1998;16:584–8.
61. Chen R, Palmer JM, Martin P, et al. Results of a multicenter phase II trial of brentuximab vedotin as second-line therapy before autologous transplantation in relapsed/refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2015;21:2136–40.
62. Moskowitz CH, Yahalom J, Zelenetz AD, et al. High-dose chemo-radiotherapy for relapsed or refractory Hodgkin lymphoma and the significance of pre-transplant functional imaging. Br J Haematol 2010;148:890–7.
63. Sureda A, Constans M, Iriondo A, et al. Prognostic factors affecting long-term outcome after stem cell transplantation in Hodgkin’s lymphoma autografted after a first relapse. Ann Oncol 2005;16:625–33.
64. Crocchiolo R, Canevari C, Assanelli A, et al. Pre-transplant 18FDG-PET predicts outcome in lymphoma patients treated with high-dose sequential chemotherapy followed by autologous stem cell transplantation. Leuk Lymphoma 2008;49:727–33.
65. Mocikova H, Pytlik R, Markova J, et al. Pre-transplant positron emission tomography in patients with relapsed Hodgkin lymphoma. Leuk Lymphoma 2011;52:1668–74.
66. Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol 2012;30:2183–9.
67. Gopal AK, Chen R, Smith SE, et al. Durable remissions in a pivotal phase 2 study of brentuximab vedotin in relapsed or refractory Hodgkin lymphoma. Blood 2015;125:1236–43.
68. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma.N Engl J Med 2015;372:311–9.
69. Chen R, Zinzani PL, Fanale MA, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol 2017;35:2125–32.
70. Haverkos BM, Abbott D, Hamadani M, et al. PD-1 blockade for relapsed lymphoma post-allogeneic hematopoietic cell transplant: high response rate but frequent GVHD. Blood 2017;130:221–8.
71. Herbaux C, Gauthier J, Brice P, et al. Efficacy and tolerability of nivolumab after allogeneic transplantation for relapsed Hodgkin lymphoma. Blood 2017;129:2471–8.
72. Merryman RW, Kim HT, Zinzani PL et al. Safety and efficacy of allogeneic hematopoietic stem cell transplant after PD-1 blockage in relapsed/refractory lymphoma. Blood 2017;129:1380–8.
73. Linch DC, Winfield D, Goldstone AH, et al. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin’s disease: results of a BNLI randomised trial. Lancet 1993;341:1051–4.
74. Yuen AR, Rosenberg SA, Hoppe RT, et al. Comparison between conventional salvage therapy and high-dose therapy with autografting for recurrent or refractory Hodgkin’s disease. Blood 1997;89:814–22.
75. Gutierrez-Delgado F, Holmberg L, Hooper H, et al. Autologous stem cell transplantation for Hodgkin’s disease: busulfan, melphalan and thiotepa compared to a radiation-based regimen. Bone Marrow Transplant 2003;32:279–85.
76. Hake CR, Graubert TA, Fenske TS. Does autologous transplantation directly increase the risk of secondary leukemia in lymphoma patients? Bone Marrow Transplant 2007;39:59–70.
77. Hahn T, McCarthy PL, Carreras J, et al. Comparison of prognostic models for autologous hematopoietic stem cell transplantation (AHCT) for relapsed Hodgkin lymphoma. Blood 2009;114:1215.
78. Chen Y-B, Lane AA, Logan BR, et al. Impact of conditioning regimen on outcomes for patients with lymphoma undergoing high-dose therapy with autologous hematopoietic cell transplantation. Biology Blood Marrow Transplant 2015;21:1046–53.
79. Wendland MM, Asch JD, Pulsipher MA, et al. The impact of involved field radiation therapy for patients receiving high-dose chemotherapy followed by hematopoietic progenitor cell transplant for the treatment of relapsed or refractory Hodgkin disease. Am J Clin Oncol 2006;29:189–95.
80. Biswas T, Culakova E, Friedberg JW, et al. Involved field radiation therapy following high dose chemotherapy and autologous stem cell transplant benefits local control and survival in refractory or recurrent Hodgkin lymphoma. Radiother Oncol 2012;103:367–72.
81. Levis M, Piva C, Filippi AR, et al. Potential benefit of involved-field radiotherapy for patients with Relapsed-refractory Hodgkin’s lymphoma with incomplete response before autologous stem cell transplantation. Clin Lymphoma Myeloma Leuk 2017;17:14–22.
82. Josting A, Franklin J, May M, et al. New prognostic score based on treatment outcome of patients with relapsed Hodgkin’s lymphoma registered in the database of the German Hodgkin’s lymphoma study group. J Clin Oncol 2002;20:221–30.
83. Moskowitz CH, Nademanee A, Masszi T, et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015;385:1853–62.
84. Bonfante V, Santoro A, Viviani S, et al. Outcome of patients with Hodgkin’s disease failing after primary MOPP-ABVD. J Clin Oncol 1997;15:528–34.
85. Josting A, Rueffer U, Franklin J, et al. Prognostic factors and treatment outcome in primary progressive Hodgkin lymphoma: a report from the German Hodgkin Lymphoma Study Group. Blood 2000;96:1280–6.
86. Josting A, Nogova L, Franklin J, et al. Salvage radiotherapy in patients with relapsed and refractory Hodgkin’s lymphoma: a retrospective analysis from the German Hodgkin Lymphoma Study Group. J Clin Oncol 2005;23:1522–9.
87. Campbell B WA, Milner A, Di Iulio J, et al. Long-term follow-up of salvage radiotherapy in Hodgkin’s lymphoma after chemotherapy failure. Int J Radiat Oncol Biol Phys 2005;63:1538–45.
88. Wirth A, Corry J, Laidlaw C, et al. Salvage radiotherapy for Hodgkin’s disease following chemotherapy failure. Int J Radiat Oncol Biol Phys 1997;39:599–607.
89. Schulz H, Rehwald U, Morschhauser F, et al. Rituximab in relapsed lymphocyte-predominant Hodgkin lymphoma: long-term results of a phase 2 trial by the German Hodgkin Lymphoma Study Group (GHSG). Blood 2008;111:109–11.
90. Ekstrand BC, Lucas JB, Horwitz SM, et al. Rituximab in lymphocyte-predominant Hodgkin disease: results of a phase 2 trial. Blood 2003;101:4285–9.
91. Younes A, Romaguera J, Hagemeister F, et al. A pilot study of rituximab in patients with recurrent, classic Hodgkin disease. Cancer 2003;98:310–4.
92. Rehwald U, Schulz H, Reiser M, et al. Treatment of relapsed CD20+ Hodgkin lymphoma with the monoclonal antibody rituximab is effective and well tolerated: results of a phase 2 trial of the German Hodgkin Lymphoma Study Group. Blood 2003;101:420–4.
93. Fehniger TA, Larson S, Trinkaus K, et al. A phase 2 multicenter study of lenalidomide in relapsed or refractory classical Hodgkin lymphoma. Blood 2011;118:5119–25.
94. Boll B, Borchmann P, Topp MS, et al. Lenalidomide in patients with refractory or multiple relapsed Hodgkin lymphoma. Br J Haematol 2010;148:480–2.
95. LaCasce AS, Bociek G, Sawas A, et al. Brentuximab vedotin plus bendamustine: a highly active salvage treatment regimen for patients with relapsed or refractory Hodgkin lymphoma. Blood 2015;126:3982.
96. Diefenbach CS, Hong F, David KA, et al. A phase I study with an expansion cohort of the combination of ipilimumab and nivolumab and brentuximab vedotin in patients with relapsed/refractory Hodgkin lymphoma: A trial of the ECOG-ACRIN Cancer Research Group (E4412 Arms D and E). Blood 2016;128:1106.
97. Herrera AF, Bartlett NL, Ramchandren R, et al. Preliminary results from a phase 1/2 study of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2016;128:1105.
98. Johnston PB, Inwards DJ, Colgan JP, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol 2010;85:320–4.
99. Kewalramani T, Nimer SD, Zelenetz AD, et al. Progressive disease following autologous transplantation in patients with chemosensitive relapsed or primary refractory Hodgkin’s disease or aggressive non-Hodgkin’s lymphoma. Bone Marrow Transplant 2003;32:673–9.
100. Lin TS, Avalos BR, Penza SL, et al. Second autologous stem cell transplant for multiply relapsed Hodgkin’s disease. Bone Marrow Transplant 2002;29:763–7.
101. Smith SM, van Besien K, Carreras J, et al. Second autologous stem cell transplantation for relapsed lymphoma after a prior autologous transplant. Biol Blood Marrow Transplant 2008;14:904–12.
102. Gajewski JL, Phillips GL, Sobocinski KA, et al. Bone marrow transplants from HLA-identical siblings in advanced Hodgkin’s disease. J Clin Oncol 1996;14:572–8.
103. Peniket AJ, Ruiz de Elvira MC, Taghipour G, et al. An EBMT registry matched study of allogeneic stem cell transplants for lymphoma: allogeneic transplantation is associated with a lower relapse rate but a higher procedure-related mortality rate than autologous transplantation. Bone Marrow Transplant 2003;31:667–78.
104. Anderlini P, Saliba R, Acholonu S, et al. Fludarabine-melphalan as a preparative regimen for reduced-intensity conditioning allogeneic stem cell transplantation in relapsed and refractory Hodgkin’s lymphoma: the updated M.D. Anderson Cancer Center experience. Haematologica 2008;93:257–64.
105. Burroughs LM, O’Donnell PV, Sandmaier BM, et al. Comparison of outcomes of HLA-matched related, unrelated, or HLA-haploidentical related hematopoietic cell transplantation following nonmyeloablative conditioning for relapsed or refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2008;14:1279–87.
106. Peggs KS, Hunter A, Chopra R, et al. Clinical evidence of a graft-versus-Hodgkin’s-lymphoma effect after reduced-intensity allogeneic transplantation. Lancet 2005;365:1934–41.
107. Sureda A, Robinson S, Canals C, et al. Reduced-intensity conditioning compared with conventional allogeneic stem-cell transplantation in relapsed or refractory Hodgkin’s lymphoma: an analysis from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2008;26:455–62.
108. Devetten MP, Hari PN, Carreras J, et al. Unrelated donor reduced-intensity allogeneic hematopoietic stem cell transplantation for relapsed and refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:109–17.
109. Sureda A, Canals C, Arranz R, et al. Allogeneic stem cell transplantation after reduced intensity conditioning in patients with relapsed or refractory Hodgkin’s lymphoma. Results of the HDR-ALLO study - a prospective clinical trial by the Grupo Espanol de Linfomas/Trasplante de Medula Osea (GEL/TAMO) and the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. Haematologica 2012;97:310–7.
110. Sarina B, Castagna L, Farina L, et al. Allogeneic transplantation improves the overall and progression-free survival of Hodgkin lymphoma patients relapsing after autologous transplantation: a retrospective study based on the time of HLA typing and donor availability. Blood 2010;115:3671–7.
111. Castagna L, Sarina B, Todisco E, et al. Allogeneic stem cell transplantation compared with chemotherapy for poor-risk Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:432–8.
112. Thomson KJ, Peggs KS, Smith P, et al. Superiority of reduced-intensity allogeneic transplantation over conventional treatment for relapse of Hodgkin’s lymphoma following autologous stem cell transplantation. Bone Marrow Transplant 2008;41:765–70.
113. Chen R, Palmer JM, Thomas SH, et al. Brentuximab vedotin enables successful reduced-intensity allogeneic hematopoietic cell transplantation in patients with relapsed or refractory Hodgkin lymphoma. Blood 2012;119:6379–81.
Pediatric Dermatology Consult - August 2017
BY AYAN KUSARI AND CATALINA MATIZ, MD
The patient was diagnosed with eruptive vellus hair cysts (EVHC). Treatment with a keratolytic, such as 12% lactic acid cream, was recommended. Hydrocortisone 2.5% once daily as needed also was recommended to treat the patient’s itch.
EVHC are benign middermal cysts characterized by epidermoid keratinization of the cyst wall, as well as lamellar keratin and vellus hairs within the cyst.1 The term “eruptive vellus hair cysts” was first used to describe a longstanding hyperpigmented, monomorphous papular eruption in two children by Esterly, Fretzin, and Pinkus in 1977.2 Clinically, EVHC present as 1- to 3-mm follicular, dome-shaped papules that are often skin-colored but also have been described as being brown, gray, green or black colored.3,4 They appear suddenly and sometimes are associated with mild tenderness and pruritus.1,5 EVHC most commonly present on the anterior chest but also can present on the upper and lower extremities, face, neck, axillae, and buttocks.4
Furthermore, although spontaneous resolution is possible through transepidermal elimination of cyst products, cases may persist for years in the absence of treatment.1
Accurate diagnosis of eruptive vellus hair cysts is important to guide therapy.
Keratosis pilaris consists of follicular-based papules with variable erythema.4 It may be widespread – including over the anterior chest – but is most commonly seen on the cheeks, extensor surfaces of proximal upper extremities, and the anterior thighs.4 It is related to excessive keratinization, which leads to formation of horny plugs within hair-follicle orifices.1
Steatocystoma multiplex is typically characterized by firm, yellow-to-flesh–colored dermal cysts ranging from a few millimeters to 1 cm in size.1 They are sometimes clinically hard to distinguish from EVHC, and both are associated with keratin 17 gene mutations and type 2 pachyonychia congenita.1 Nonetheless, this patient’s lesions did not have any features – such as size or drainage – that would point toward steatocystoma multiplex or other skin findings suggestive of pachyonychia congenita.
Superficial folliculitis, also known as Bockhart’s impetigo, is an infection of the follicular ostium and typically presents with perifollicular pustules on an erythematous base that may be painful or pruritic and can occur throughout the corpus, including the anterior trunk.1
Acne vulgaris is a very common disease that involves the pilosebaceous unit and occurs most frequently on the face, back, upper arms, and chest. However, it is characterized by open and closed comedones, papules, and pustules and, in severe cases, nodules and cysts that may leave postinflammatory hyperpigmentation and scarring. Tiny, hyperpigmented, dome-shaped macules occurring exclusively on the chest would not be characteristic.
Patients with hypohidrotic ectodermal dysplasia, also known as Christ-Siemens-Touraine syndrome, can present with EVHC. This condition is characterized by a triad of fair, sparse short hair; hyperthermia related to decreased sweating; and missing teeth.4 Although EVHC have been reported in association with hypohidrotic ectodermal dysplasia, this patient does not have any of the dysmorphic features associated with this syndrome.11
Patients with pachyonychia congenita (type 2) also may have EVHC as part of their presentation, but this patient does not have nail dystrophy, focal palmoplantar keratoderma, follicular keratoses, or multiple steatocysts which also are features of this condition.4
Treatment may be offered to patients who are distressed by the lesions or seek cosmesis. A 2012 review of 220 cases of EVHC found that topical retinoic acid, incision/excision, CO2 laser, erbium:yttrium-aluminum-garnet laser, needle evacuation, dermabrasion, and 10% urea cream were each associated with successful treatment in multiple cases.3
Forty years have passed since EVHC was identified as a distinct disease entity. Despite this, eruptive vellus hair cysts remains somewhat understudied, and further research is needed to determine an ideal treatment algorithm for patients with this condition. Our approach was to attempt noninvasive keratolytic therapy before considering retinoids or surgical options; we also recommended steroid treatment for symptom relief. Providers should keep EVHC in the differential for eruptions consisting of tiny papules so that appropriate treatment may be offered.
Dr. Matiz is a pediatric dermatologist at Rady Children’s Hospital, San Diego, and an assistant clinical professor in the department of pediatric and adolescent dermatology at the University of California, San Diego. Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures.
Email them at [email protected].
References
1. “Dermatology.” 3rd ed. (Philadelphia: Saunders, 2012).
2. Arch Dermatol. 1977 Apr;113(4):500-3.
3. Am J Clin Dermatol. 2012 Feb 1;13(1):19-28.
5. Indian Dermatol Online J. 2013 Jul;4(3):213-5.
6. J Am Acad Dermatol. 1980 Oct;3(4):425-9.
7. Am J Dermatopathol. 1997 Jun;19(3):250-3.
8. Hum Mol Genet. 1998 Jul;7(7):1143-8.
9. Dermatology. 1998;196(4):392-6.
BY AYAN KUSARI AND CATALINA MATIZ, MD
The patient was diagnosed with eruptive vellus hair cysts (EVHC). Treatment with a keratolytic, such as 12% lactic acid cream, was recommended. Hydrocortisone 2.5% once daily as needed also was recommended to treat the patient’s itch.
EVHC are benign middermal cysts characterized by epidermoid keratinization of the cyst wall, as well as lamellar keratin and vellus hairs within the cyst.1 The term “eruptive vellus hair cysts” was first used to describe a longstanding hyperpigmented, monomorphous papular eruption in two children by Esterly, Fretzin, and Pinkus in 1977.2 Clinically, EVHC present as 1- to 3-mm follicular, dome-shaped papules that are often skin-colored but also have been described as being brown, gray, green or black colored.3,4 They appear suddenly and sometimes are associated with mild tenderness and pruritus.1,5 EVHC most commonly present on the anterior chest but also can present on the upper and lower extremities, face, neck, axillae, and buttocks.4
Furthermore, although spontaneous resolution is possible through transepidermal elimination of cyst products, cases may persist for years in the absence of treatment.1
Accurate diagnosis of eruptive vellus hair cysts is important to guide therapy.
Keratosis pilaris consists of follicular-based papules with variable erythema.4 It may be widespread – including over the anterior chest – but is most commonly seen on the cheeks, extensor surfaces of proximal upper extremities, and the anterior thighs.4 It is related to excessive keratinization, which leads to formation of horny plugs within hair-follicle orifices.1
Steatocystoma multiplex is typically characterized by firm, yellow-to-flesh–colored dermal cysts ranging from a few millimeters to 1 cm in size.1 They are sometimes clinically hard to distinguish from EVHC, and both are associated with keratin 17 gene mutations and type 2 pachyonychia congenita.1 Nonetheless, this patient’s lesions did not have any features – such as size or drainage – that would point toward steatocystoma multiplex or other skin findings suggestive of pachyonychia congenita.
Superficial folliculitis, also known as Bockhart’s impetigo, is an infection of the follicular ostium and typically presents with perifollicular pustules on an erythematous base that may be painful or pruritic and can occur throughout the corpus, including the anterior trunk.1
Acne vulgaris is a very common disease that involves the pilosebaceous unit and occurs most frequently on the face, back, upper arms, and chest. However, it is characterized by open and closed comedones, papules, and pustules and, in severe cases, nodules and cysts that may leave postinflammatory hyperpigmentation and scarring. Tiny, hyperpigmented, dome-shaped macules occurring exclusively on the chest would not be characteristic.
Patients with hypohidrotic ectodermal dysplasia, also known as Christ-Siemens-Touraine syndrome, can present with EVHC. This condition is characterized by a triad of fair, sparse short hair; hyperthermia related to decreased sweating; and missing teeth.4 Although EVHC have been reported in association with hypohidrotic ectodermal dysplasia, this patient does not have any of the dysmorphic features associated with this syndrome.11
Patients with pachyonychia congenita (type 2) also may have EVHC as part of their presentation, but this patient does not have nail dystrophy, focal palmoplantar keratoderma, follicular keratoses, or multiple steatocysts which also are features of this condition.4
Treatment may be offered to patients who are distressed by the lesions or seek cosmesis. A 2012 review of 220 cases of EVHC found that topical retinoic acid, incision/excision, CO2 laser, erbium:yttrium-aluminum-garnet laser, needle evacuation, dermabrasion, and 10% urea cream were each associated with successful treatment in multiple cases.3
Forty years have passed since EVHC was identified as a distinct disease entity. Despite this, eruptive vellus hair cysts remains somewhat understudied, and further research is needed to determine an ideal treatment algorithm for patients with this condition. Our approach was to attempt noninvasive keratolytic therapy before considering retinoids or surgical options; we also recommended steroid treatment for symptom relief. Providers should keep EVHC in the differential for eruptions consisting of tiny papules so that appropriate treatment may be offered.
Dr. Matiz is a pediatric dermatologist at Rady Children’s Hospital, San Diego, and an assistant clinical professor in the department of pediatric and adolescent dermatology at the University of California, San Diego. Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures.
Email them at [email protected].
References
1. “Dermatology.” 3rd ed. (Philadelphia: Saunders, 2012).
2. Arch Dermatol. 1977 Apr;113(4):500-3.
3. Am J Clin Dermatol. 2012 Feb 1;13(1):19-28.
5. Indian Dermatol Online J. 2013 Jul;4(3):213-5.
6. J Am Acad Dermatol. 1980 Oct;3(4):425-9.
7. Am J Dermatopathol. 1997 Jun;19(3):250-3.
8. Hum Mol Genet. 1998 Jul;7(7):1143-8.
9. Dermatology. 1998;196(4):392-6.
BY AYAN KUSARI AND CATALINA MATIZ, MD
The patient was diagnosed with eruptive vellus hair cysts (EVHC). Treatment with a keratolytic, such as 12% lactic acid cream, was recommended. Hydrocortisone 2.5% once daily as needed also was recommended to treat the patient’s itch.
EVHC are benign middermal cysts characterized by epidermoid keratinization of the cyst wall, as well as lamellar keratin and vellus hairs within the cyst.1 The term “eruptive vellus hair cysts” was first used to describe a longstanding hyperpigmented, monomorphous papular eruption in two children by Esterly, Fretzin, and Pinkus in 1977.2 Clinically, EVHC present as 1- to 3-mm follicular, dome-shaped papules that are often skin-colored but also have been described as being brown, gray, green or black colored.3,4 They appear suddenly and sometimes are associated with mild tenderness and pruritus.1,5 EVHC most commonly present on the anterior chest but also can present on the upper and lower extremities, face, neck, axillae, and buttocks.4
Furthermore, although spontaneous resolution is possible through transepidermal elimination of cyst products, cases may persist for years in the absence of treatment.1
Accurate diagnosis of eruptive vellus hair cysts is important to guide therapy.
Keratosis pilaris consists of follicular-based papules with variable erythema.4 It may be widespread – including over the anterior chest – but is most commonly seen on the cheeks, extensor surfaces of proximal upper extremities, and the anterior thighs.4 It is related to excessive keratinization, which leads to formation of horny plugs within hair-follicle orifices.1
Steatocystoma multiplex is typically characterized by firm, yellow-to-flesh–colored dermal cysts ranging from a few millimeters to 1 cm in size.1 They are sometimes clinically hard to distinguish from EVHC, and both are associated with keratin 17 gene mutations and type 2 pachyonychia congenita.1 Nonetheless, this patient’s lesions did not have any features – such as size or drainage – that would point toward steatocystoma multiplex or other skin findings suggestive of pachyonychia congenita.
Superficial folliculitis, also known as Bockhart’s impetigo, is an infection of the follicular ostium and typically presents with perifollicular pustules on an erythematous base that may be painful or pruritic and can occur throughout the corpus, including the anterior trunk.1
Acne vulgaris is a very common disease that involves the pilosebaceous unit and occurs most frequently on the face, back, upper arms, and chest. However, it is characterized by open and closed comedones, papules, and pustules and, in severe cases, nodules and cysts that may leave postinflammatory hyperpigmentation and scarring. Tiny, hyperpigmented, dome-shaped macules occurring exclusively on the chest would not be characteristic.
Patients with hypohidrotic ectodermal dysplasia, also known as Christ-Siemens-Touraine syndrome, can present with EVHC. This condition is characterized by a triad of fair, sparse short hair; hyperthermia related to decreased sweating; and missing teeth.4 Although EVHC have been reported in association with hypohidrotic ectodermal dysplasia, this patient does not have any of the dysmorphic features associated with this syndrome.11
Patients with pachyonychia congenita (type 2) also may have EVHC as part of their presentation, but this patient does not have nail dystrophy, focal palmoplantar keratoderma, follicular keratoses, or multiple steatocysts which also are features of this condition.4
Treatment may be offered to patients who are distressed by the lesions or seek cosmesis. A 2012 review of 220 cases of EVHC found that topical retinoic acid, incision/excision, CO2 laser, erbium:yttrium-aluminum-garnet laser, needle evacuation, dermabrasion, and 10% urea cream were each associated with successful treatment in multiple cases.3
Forty years have passed since EVHC was identified as a distinct disease entity. Despite this, eruptive vellus hair cysts remains somewhat understudied, and further research is needed to determine an ideal treatment algorithm for patients with this condition. Our approach was to attempt noninvasive keratolytic therapy before considering retinoids or surgical options; we also recommended steroid treatment for symptom relief. Providers should keep EVHC in the differential for eruptions consisting of tiny papules so that appropriate treatment may be offered.
Dr. Matiz is a pediatric dermatologist at Rady Children’s Hospital, San Diego, and an assistant clinical professor in the department of pediatric and adolescent dermatology at the University of California, San Diego. Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures.
Email them at [email protected].
References
1. “Dermatology.” 3rd ed. (Philadelphia: Saunders, 2012).
2. Arch Dermatol. 1977 Apr;113(4):500-3.
3. Am J Clin Dermatol. 2012 Feb 1;13(1):19-28.
5. Indian Dermatol Online J. 2013 Jul;4(3):213-5.
6. J Am Acad Dermatol. 1980 Oct;3(4):425-9.
7. Am J Dermatopathol. 1997 Jun;19(3):250-3.
8. Hum Mol Genet. 1998 Jul;7(7):1143-8.
9. Dermatology. 1998;196(4):392-6.
A 6-year-old boy presents with bumps on his chest and lower abdomen that have been present for 6 months. The patient’s mother states that the bumps are occasionally pruritic but not painful. She reports that the bumps first appeared on the chest and subsequently spread downward to involve the upper abdomen.
The patient is otherwise healthy. No similar lesions are present beyond the trunk. The patient’s past medical history and developmental history are unremarkable aside from bilateral amblyopia and high myopia. The patient’s mother denies any other family members with similar lesions. There is no history of teeth or nail abnormalities.
On exam, you find symmetrically distributed, firm, nontender, tiny 1- to 2-mm hyperpigmented dome-shaped papules on the anterior chest with no similar lesions elsewhere on the body. The remainder of the physical exam discloses no abnormalities.
Florence A. Blanchfield: A Lifetime of Nursing Leadership
The U.S. Army hospital at Fort Campbell, Kentucky, was named for army nurse, Colonel Florence A. Blanchfield—making it the only current army hospital named for a nurse.
Florence Aby Blanchfield was born into a large family in Shepherdstown, West Virginia, in 1882. Her mother was a nurse, and her father was a mason and stonecutter. She grew up in Oranda, Virginia, and attended both public and private schools. Following in her mother’s footsteps to become a nurse, she attended Southside Hospital Training School in Pittsburgh, Pennsylvania, and graduated in 1906. She moved to Baltimore after graduation and worked with Howard Atwood Kelly, one of the “Big Four” along with William Osler, William Henry Welch, and William Stewart Halsted who were known as the founding physicians of the Johns Hopkins Hospital. 
After what must have been a remarkable experience with the innovative Kelly (inventor of many groundbreaking medical instruments and procedures, including the Kelly clamp), Blanchfield returned to Pittsburgh. She held positions of increasing responsibility over several years, including operating room supervisor at Southside Hospital and Montefiore Hospital and superintendent of the training school at Suburban General Hospital. Looking for adventure as well as service, she gave up her positions of leadership and headed to Panama in 1913 to become an operating room nurse and an anesthetist at Ancon Hospital in the U.S. Canal Zone.
As America prepared for its probable entry into World War I, Blanchfield joined the Army Nurse Corps (ANC) at age 35 to serve with the Medical School Unit of the University of Pittsburgh’s Base Hospital 27. She arrived in France in October 1917 and became acting chief nurse of Base Hospital 27 in Angers, Maine et Loire department. She also served as acting chief nurse of Camp Hospital 15 at Coëtquidan, Ille et Vil department.
Blanchfield returned to civilian life following World War I for a short period but returned to active duty in 1920. Over the next 15 years, she had several assignments within the continental U.S. and overseas in the Philippines and in Tianjin, China (formally known in English as Tientsin). In 1935, Blanchfield joined the Office of the Army Surgeon General in Washington, DC, where she was assigned to work on personnel matters in the office of the superintendent of the ANC. She became assistant superintendent in 1939, acting superintendent in 1942, and served as superintendent from June 1943 until September 1947. During World War II, she presided over the growth of the ANC from about 7,000 nurses on the day Pearl Harbor was attacked to more than 50,000 by the end of the war. She was awarded the Distinguished Service Medal for her contributions and accomplishments during World War II.
Blanchfield, a long-time senior leader in the ANC, was instrumental in many of the significant changes that took place during and after World War II, including nurses gaining full rank and benefits. This was an incremental process that culminated with passage of the Army and Navy Nurse Corps Act of April 1947, with nurses being granted full commissioned status. As a result of this act, she became a lieutenant colonel and the first woman to receive a commission in the regular army. 
Blanchfield remained active in national nursing affairs after her retirement from the U.S. Army. At a time when many believed that nurses did not need specialty training, she promoted the establishment of specialized courses of study. In 1951, she received the Florence Nightingale Medal of the International Red Cross.
Blanchfield died on May 12, 1971, and was buried in the nurse’s section of Arlington National Cemetery with full military honors. In 1978, ANC leadership began a drive to memorialize Blanchfield by naming the new hospital at Fort Campbell, Kentucky, in her honor. A successful letter writing campaign by army nurses inundated the senior commander at Fort Campbell. The Colonel Florence A. Blanchfield Army Community Hospital, which was dedicated in her memory on September 17, 1982.
About this column
This column provides biographical sketches of the namesakes of military and VA health care facilities. To learn more about the individual your facility was named for or to offer a topic suggestion, contact us at [email protected] or on Facebook.
The U.S. Army hospital at Fort Campbell, Kentucky, was named for army nurse, Colonel Florence A. Blanchfield—making it the only current army hospital named for a nurse.
Florence Aby Blanchfield was born into a large family in Shepherdstown, West Virginia, in 1882. Her mother was a nurse, and her father was a mason and stonecutter. She grew up in Oranda, Virginia, and attended both public and private schools. Following in her mother’s footsteps to become a nurse, she attended Southside Hospital Training School in Pittsburgh, Pennsylvania, and graduated in 1906. She moved to Baltimore after graduation and worked with Howard Atwood Kelly, one of the “Big Four” along with William Osler, William Henry Welch, and William Stewart Halsted who were known as the founding physicians of the Johns Hopkins Hospital.
After what must have been a remarkable experience with the innovative Kelly (inventor of many groundbreaking medical instruments and procedures, including the Kelly clamp), Blanchfield returned to Pittsburgh. She held positions of increasing responsibility over several years, including operating room supervisor at Southside Hospital and Montefiore Hospital and superintendent of the training school at Suburban General Hospital. Looking for adventure as well as service, she gave up her positions of leadership and headed to Panama in 1913 to become an operating room nurse and an anesthetist at Ancon Hospital in the U.S. Canal Zone.
As America prepared for its probable entry into World War I, Blanchfield joined the Army Nurse Corps (ANC) at age 35 to serve with the Medical School Unit of the University of Pittsburgh’s Base Hospital 27. She arrived in France in October 1917 and became acting chief nurse of Base Hospital 27 in Angers, Maine et Loire department. She also served as acting chief nurse of Camp Hospital 15 at Coëtquidan, Ille et Vil department.
Blanchfield returned to civilian life following World War I for a short period but returned to active duty in 1920. Over the next 15 years, she had several assignments within the continental U.S. and overseas in the Philippines and in Tianjin, China (formally known in English as Tientsin). In 1935, Blanchfield joined the Office of the Army Surgeon General in Washington, DC, where she was assigned to work on personnel matters in the office of the superintendent of the ANC. She became assistant superintendent in 1939, acting superintendent in 1942, and served as superintendent from June 1943 until September 1947. During World War II, she presided over the growth of the ANC from about 7,000 nurses on the day Pearl Harbor was attacked to more than 50,000 by the end of the war. She was awarded the Distinguished Service Medal for her contributions and accomplishments during World War II.
Blanchfield, a long-time senior leader in the ANC, was instrumental in many of the significant changes that took place during and after World War II, including nurses gaining full rank and benefits. This was an incremental process that culminated with passage of the Army and Navy Nurse Corps Act of April 1947, with nurses being granted full commissioned status. As a result of this act, she became a lieutenant colonel and the first woman to receive a commission in the regular army.
Blanchfield remained active in national nursing affairs after her retirement from the U.S. Army. At a time when many believed that nurses did not need specialty training, she promoted the establishment of specialized courses of study. In 1951, she received the Florence Nightingale Medal of the International Red Cross.
Blanchfield died on May 12, 1971, and was buried in the nurse’s section of Arlington National Cemetery with full military honors. In 1978, ANC leadership began a drive to memorialize Blanchfield by naming the new hospital at Fort Campbell, Kentucky, in her honor. A successful letter writing campaign by army nurses inundated the senior commander at Fort Campbell. The Colonel Florence A. Blanchfield Army Community Hospital, which was dedicated in her memory on September 17, 1982.
About this column
This column provides biographical sketches of the namesakes of military and VA health care facilities. To learn more about the individual your facility was named for or to offer a topic suggestion, contact us at [email protected] or on Facebook.
The U.S. Army hospital at Fort Campbell, Kentucky, was named for army nurse, Colonel Florence A. Blanchfield—making it the only current army hospital named for a nurse.
Florence Aby Blanchfield was born into a large family in Shepherdstown, West Virginia, in 1882. Her mother was a nurse, and her father was a mason and stonecutter. She grew up in Oranda, Virginia, and attended both public and private schools. Following in her mother’s footsteps to become a nurse, she attended Southside Hospital Training School in Pittsburgh, Pennsylvania, and graduated in 1906. She moved to Baltimore after graduation and worked with Howard Atwood Kelly, one of the “Big Four” along with William Osler, William Henry Welch, and William Stewart Halsted who were known as the founding physicians of the Johns Hopkins Hospital.
After what must have been a remarkable experience with the innovative Kelly (inventor of many groundbreaking medical instruments and procedures, including the Kelly clamp), Blanchfield returned to Pittsburgh. She held positions of increasing responsibility over several years, including operating room supervisor at Southside Hospital and Montefiore Hospital and superintendent of the training school at Suburban General Hospital. Looking for adventure as well as service, she gave up her positions of leadership and headed to Panama in 1913 to become an operating room nurse and an anesthetist at Ancon Hospital in the U.S. Canal Zone.
As America prepared for its probable entry into World War I, Blanchfield joined the Army Nurse Corps (ANC) at age 35 to serve with the Medical School Unit of the University of Pittsburgh’s Base Hospital 27. She arrived in France in October 1917 and became acting chief nurse of Base Hospital 27 in Angers, Maine et Loire department. She also served as acting chief nurse of Camp Hospital 15 at Coëtquidan, Ille et Vil department.
Blanchfield returned to civilian life following World War I for a short period but returned to active duty in 1920. Over the next 15 years, she had several assignments within the continental U.S. and overseas in the Philippines and in Tianjin, China (formally known in English as Tientsin). In 1935, Blanchfield joined the Office of the Army Surgeon General in Washington, DC, where she was assigned to work on personnel matters in the office of the superintendent of the ANC. She became assistant superintendent in 1939, acting superintendent in 1942, and served as superintendent from June 1943 until September 1947. During World War II, she presided over the growth of the ANC from about 7,000 nurses on the day Pearl Harbor was attacked to more than 50,000 by the end of the war. She was awarded the Distinguished Service Medal for her contributions and accomplishments during World War II.
Blanchfield, a long-time senior leader in the ANC, was instrumental in many of the significant changes that took place during and after World War II, including nurses gaining full rank and benefits. This was an incremental process that culminated with passage of the Army and Navy Nurse Corps Act of April 1947, with nurses being granted full commissioned status. As a result of this act, she became a lieutenant colonel and the first woman to receive a commission in the regular army.
Blanchfield remained active in national nursing affairs after her retirement from the U.S. Army. At a time when many believed that nurses did not need specialty training, she promoted the establishment of specialized courses of study. In 1951, she received the Florence Nightingale Medal of the International Red Cross.
Blanchfield died on May 12, 1971, and was buried in the nurse’s section of Arlington National Cemetery with full military honors. In 1978, ANC leadership began a drive to memorialize Blanchfield by naming the new hospital at Fort Campbell, Kentucky, in her honor. A successful letter writing campaign by army nurses inundated the senior commander at Fort Campbell. The Colonel Florence A. Blanchfield Army Community Hospital, which was dedicated in her memory on September 17, 1982.
About this column
This column provides biographical sketches of the namesakes of military and VA health care facilities. To learn more about the individual your facility was named for or to offer a topic suggestion, contact us at [email protected] or on Facebook.
An ASCO 2017 recap: significant advances continue
As we head into vacation season and the dog days of summer, let’s reflect for a few minutes on some of the very important advances we heard about at this year’s annual meeting of the American Society of Clinical Oncology in Chicago. Nearly 40,000 individuals registered for the conference, an indication of both the interest and the excitement around the new agents and the emerging clinical trial data. Scientific sessions dedicated to the use of combination immunotherapy, the role of antibody drug conjugates, and targeting molecular aberrations with small molecules were among the most popular (p. e236).

In the setting of metastatic breast cancer, several trials produced highly significant results that will positively affect the duration and quality of life for our patients. The use of PARP inhibitors in BRCA-mutated cancers has been shown to be effective in a few areas, particularly advanced ovarian cancer. The OlympiAD study evaluated olaparib monotherapy and a physician’s choice arm (capecitabine, eribulin, or vinorelbine) in BRCA-mutated, HER2-negative metastatic breast cancer. The 2:1 design enrolled 302 patients and demonstrated a 3-month improvement in progression-free survival (PFS) for olaparib compared with the control arm (7.0 vs 4.2 months, respectively; P = .0009). The patient population for this BRCA-mutated trial was relatively young, with a median age of 45 years, and 50% of the women were hormone positive and 30%, platinum resistant.
The CDK4/6 inhibitors continue to be impressive, with the recently reported results from the MONARCH 2 trial showing encouraging PFS and overall response rate results with the addition of the CDK4/6 inhibitor abemaciclib to fulvestrant, a selective estrogen-receptor degrader. In this study, hormone-positive, HER2-negative women who had progressed on previous endocrine therapy were randomized 2:1 to abemaciclib plus fulvestrant or placebo plus fulvestrant. A total of 669 patients were accrued, and after a median follow-up of 19 months, a highly significant PFS difference of 7 months between the abemaciclib–fulvestrant and fulvestrant–only groups was observed (16.4 vs 9.3 months, respectively; P < .0000001) along with an overall response rate of 48.1 months, compared with 21.3 months. Previous findings have demonstrated monotherapy activity for abemaciclib, and the comparisons with palbociclib and ribociclib will be forthcoming, although no comparative trials are underway. These agents will be extensively assessed in a variety of settings, including adjuvantly.
The results of the much anticipated APHINITY study, which evaluated the addition of pertuzumab to trastuzumab in the adjuvant HER2-positive setting, were met with mixed reviews. Patients were included if they had node-positive invasive breast cancer or node-negative tumors of >1.0 cm. A total of 4,804 patients (37% node negative) were enrolled in the study. The intent-to-treat primary endpoint of invasive disease-free survival (DFS) was statistically positive (P = .045), although the 3-year absolute percentages for the pertuzumab–trastuzumab and trastuzumab-only groups were 94.1% and 93.2%, respectively. It should be noted that the planned statistical assumption was for a delta of 2.6% – 91.8% and 89.2%, respectively. Thus, both arms actually did better than had been planned, which was based on historical comparisons, and the node-positive and hormone-negative subgroups trended toward a greater benefit with the addition of pertuzumab. There was, and will continue to be, much debate around the cost–benefit ratio and which patients should be offered the combination. The outstanding results with the addition of pertuzumab in the neoadjuvant setting will continue to be the setting in which the greatest absolute clinical benefit will be seen. It is unusual in this era to see trials this large planned to identify a small difference, and it is likely that resource constraints will make such studies a thing of the past.
The very active hormonal therapies, abiraterone and enzalutimide, for castrate-resistant prostate cancer remain of high interest in the area of clinical trials. The LATITUDE study evaluated a straightforward design that compared abiraterone with placebo in patients who were newly diagnosed with high-risk, metastatic hormone-naïve prostate cancer. Patients in both arms received androgen-deprivation therapy and high risk was defined by having 2 of 3 criteria: a Gleason score of ≥8; 3 or more bone lesions; or visceral disease. Of note is that 1,199 patients were enrolled before publication of the CHAARTED or STAMPEDE results, which established docetaxel as a standard for these patients. The median age in the LATITUDE trial was 68 years, with 17% of patients having visceral disease and 48% having nodal disease, making it a similar patient population to those in the docetaxel studies. The results favoring abiraterone were strikingly positive, with a 38% reduction in the risk of death (P < .0001) and a 53% reduction in the risk of radiographic progression or death (P < .0001). The regimen was well tolerated overall, and it is clear that this option will be widely considered by physicians and their patients.
Two studies addressing the importance of managing symptoms and improving outcomes were also part of the plenary session. The IDEA Collaboration conducted a prospective pooled analysis of 6 phase 3 studies that assessed 3 and 6 months of oxaliplatin-based regimens for stage 3 colon cancer. FOLFOX and CAPOX given to 12,834 patients in 6 studies from the United States, European Union, Canada, Australia, New Zealand, and Japan were evaluated for DFS, treatment compliance, and adverse events. As would be anticipated, fewer side effects, particularly neurotoxicity, and greater compliance were observed in the 3-month group. Although DFS noninferiority for 3 months of therapy was not established statistically, the overall data led the investigators to issue a consensus statement advocating for a risk-based approach in deciding the duration of therapy and recommending 3 months of therapy for patients with stage 3, T1-3N1 disease, and consideration of 6 months therapy for T4 and/ or N2 disease. The investigators also acknowledged the leader and creator of IDEA, the late Daniel Sargent, PhD, of the Mayo Clinic, who passed away far too young after a brief illness last fall (1970-2016).
The second symptom-based study was performed at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and designed by a group of investigators from the Dana-Farber Cancer Institute in Boston; the Mayo Clinic in Rochester, Minnesota; the University of North Carolina in Chapel Hill; and MSKCC (p. e236). The hypothesis was simply that proactive symptom monitoring during chemotherapy would improve symptom management and lead to better outcomes. For the study, 766 patients with advanced solid tumors who were receiving outpatient chemotherapy were randomized to a control arm with standard follow-up or to the intervention arm, on which patients self-reported on 12 common symptoms before and between visits using a web-based tool and received weekly e-mail reminders and nursing alerts. At 6 months, and compared with baseline, the self-reporting patients in the intervention arm experienced an improved quality of life (P < .001). In addition, 7% fewer of the self-reporting patients visited the emergency department (P = .02), and they experienced longer survival by 5 months compared with the standard follow-up group (31.2 vs 26.0 months, respectively; P = .03). Although there are limitations to such a study, the growth in technological advances should create the opportunity to expand on this strategy in further trials and in practice. With such an emphasis in the Medicare Oncology Home Model on decreasing hospital admissions and visits to the emergency department, there should great motivation for all involved to consider incorporating self-reporting into their patterns of care.
A continued emphasis on molecular profiling, personalized and/or precision medicine, and identifying or matching the patient to the best possible therapy or the most appropriate clinical trial remains vital to improving outcomes. Just before the ASCO meeting, the US Food and Drug Administration approved pembrolizumab for the treatment of patients with high-level microsatellite instability (MSI-H) and mismatch-repair deficient (dMMR) cancers, regardless of the site of origin. The approval was based on data from 149 patients with MSI-H or dMMR cancers, which showed a 40% response rate in this group of patients, two-thirds of whom had previously treated colon cancer. This landmark approval of a cancer therapy for a specific molecular profile and not the site of the disease, will certainly shape the future of oncology drug development. One of the highlighted stories at ASCO was the success of the larotrectinib (LOXO 101) tropomyosin receptor kinase inhibitor in patients with the TRK fusion mutations (p. e237). The data, including waterfall charts, swimmer plots, and computed-tomography scans, were impressive in this targeted population with a 76% response rate and a 91% duration of response at 6 months with a mild side effect profile.
In summary, across a variety of cancers, with treatment strategies of an equally diverse nature, we saw practice-changing data from the ASCO meeting that will benefit our patients. Continuing to seek out clinical trial options for patients will be critical in answering the many questions that have emerged and the substantial number of studies that are ongoing with combination immunotherapies, targeted small molecules, and a growing armamentarium of monoclonal antibodies.
As we head into vacation season and the dog days of summer, let’s reflect for a few minutes on some of the very important advances we heard about at this year’s annual meeting of the American Society of Clinical Oncology in Chicago. Nearly 40,000 individuals registered for the conference, an indication of both the interest and the excitement around the new agents and the emerging clinical trial data. Scientific sessions dedicated to the use of combination immunotherapy, the role of antibody drug conjugates, and targeting molecular aberrations with small molecules were among the most popular (p. e236).
In the setting of metastatic breast cancer, several trials produced highly significant results that will positively affect the duration and quality of life for our patients. The use of PARP inhibitors in BRCA-mutated cancers has been shown to be effective in a few areas, particularly advanced ovarian cancer. The OlympiAD study evaluated olaparib monotherapy and a physician’s choice arm (capecitabine, eribulin, or vinorelbine) in BRCA-mutated, HER2-negative metastatic breast cancer. The 2:1 design enrolled 302 patients and demonstrated a 3-month improvement in progression-free survival (PFS) for olaparib compared with the control arm (7.0 vs 4.2 months, respectively; P = .0009). The patient population for this BRCA-mutated trial was relatively young, with a median age of 45 years, and 50% of the women were hormone positive and 30%, platinum resistant.
The CDK4/6 inhibitors continue to be impressive, with the recently reported results from the MONARCH 2 trial showing encouraging PFS and overall response rate results with the addition of the CDK4/6 inhibitor abemaciclib to fulvestrant, a selective estrogen-receptor degrader. In this study, hormone-positive, HER2-negative women who had progressed on previous endocrine therapy were randomized 2:1 to abemaciclib plus fulvestrant or placebo plus fulvestrant. A total of 669 patients were accrued, and after a median follow-up of 19 months, a highly significant PFS difference of 7 months between the abemaciclib–fulvestrant and fulvestrant–only groups was observed (16.4 vs 9.3 months, respectively; P < .0000001) along with an overall response rate of 48.1 months, compared with 21.3 months. Previous findings have demonstrated monotherapy activity for abemaciclib, and the comparisons with palbociclib and ribociclib will be forthcoming, although no comparative trials are underway. These agents will be extensively assessed in a variety of settings, including adjuvantly.
The results of the much anticipated APHINITY study, which evaluated the addition of pertuzumab to trastuzumab in the adjuvant HER2-positive setting, were met with mixed reviews. Patients were included if they had node-positive invasive breast cancer or node-negative tumors of >1.0 cm. A total of 4,804 patients (37% node negative) were enrolled in the study. The intent-to-treat primary endpoint of invasive disease-free survival (DFS) was statistically positive (P = .045), although the 3-year absolute percentages for the pertuzumab–trastuzumab and trastuzumab-only groups were 94.1% and 93.2%, respectively. It should be noted that the planned statistical assumption was for a delta of 2.6% – 91.8% and 89.2%, respectively. Thus, both arms actually did better than had been planned, which was based on historical comparisons, and the node-positive and hormone-negative subgroups trended toward a greater benefit with the addition of pertuzumab. There was, and will continue to be, much debate around the cost–benefit ratio and which patients should be offered the combination. The outstanding results with the addition of pertuzumab in the neoadjuvant setting will continue to be the setting in which the greatest absolute clinical benefit will be seen. It is unusual in this era to see trials this large planned to identify a small difference, and it is likely that resource constraints will make such studies a thing of the past.
The very active hormonal therapies, abiraterone and enzalutimide, for castrate-resistant prostate cancer remain of high interest in the area of clinical trials. The LATITUDE study evaluated a straightforward design that compared abiraterone with placebo in patients who were newly diagnosed with high-risk, metastatic hormone-naïve prostate cancer. Patients in both arms received androgen-deprivation therapy and high risk was defined by having 2 of 3 criteria: a Gleason score of ≥8; 3 or more bone lesions; or visceral disease. Of note is that 1,199 patients were enrolled before publication of the CHAARTED or STAMPEDE results, which established docetaxel as a standard for these patients. The median age in the LATITUDE trial was 68 years, with 17% of patients having visceral disease and 48% having nodal disease, making it a similar patient population to those in the docetaxel studies. The results favoring abiraterone were strikingly positive, with a 38% reduction in the risk of death (P < .0001) and a 53% reduction in the risk of radiographic progression or death (P < .0001). The regimen was well tolerated overall, and it is clear that this option will be widely considered by physicians and their patients.
Two studies addressing the importance of managing symptoms and improving outcomes were also part of the plenary session. The IDEA Collaboration conducted a prospective pooled analysis of 6 phase 3 studies that assessed 3 and 6 months of oxaliplatin-based regimens for stage 3 colon cancer. FOLFOX and CAPOX given to 12,834 patients in 6 studies from the United States, European Union, Canada, Australia, New Zealand, and Japan were evaluated for DFS, treatment compliance, and adverse events. As would be anticipated, fewer side effects, particularly neurotoxicity, and greater compliance were observed in the 3-month group. Although DFS noninferiority for 3 months of therapy was not established statistically, the overall data led the investigators to issue a consensus statement advocating for a risk-based approach in deciding the duration of therapy and recommending 3 months of therapy for patients with stage 3, T1-3N1 disease, and consideration of 6 months therapy for T4 and/ or N2 disease. The investigators also acknowledged the leader and creator of IDEA, the late Daniel Sargent, PhD, of the Mayo Clinic, who passed away far too young after a brief illness last fall (1970-2016).
The second symptom-based study was performed at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and designed by a group of investigators from the Dana-Farber Cancer Institute in Boston; the Mayo Clinic in Rochester, Minnesota; the University of North Carolina in Chapel Hill; and MSKCC (p. e236). The hypothesis was simply that proactive symptom monitoring during chemotherapy would improve symptom management and lead to better outcomes. For the study, 766 patients with advanced solid tumors who were receiving outpatient chemotherapy were randomized to a control arm with standard follow-up or to the intervention arm, on which patients self-reported on 12 common symptoms before and between visits using a web-based tool and received weekly e-mail reminders and nursing alerts. At 6 months, and compared with baseline, the self-reporting patients in the intervention arm experienced an improved quality of life (P < .001). In addition, 7% fewer of the self-reporting patients visited the emergency department (P = .02), and they experienced longer survival by 5 months compared with the standard follow-up group (31.2 vs 26.0 months, respectively; P = .03). Although there are limitations to such a study, the growth in technological advances should create the opportunity to expand on this strategy in further trials and in practice. With such an emphasis in the Medicare Oncology Home Model on decreasing hospital admissions and visits to the emergency department, there should great motivation for all involved to consider incorporating self-reporting into their patterns of care.
A continued emphasis on molecular profiling, personalized and/or precision medicine, and identifying or matching the patient to the best possible therapy or the most appropriate clinical trial remains vital to improving outcomes. Just before the ASCO meeting, the US Food and Drug Administration approved pembrolizumab for the treatment of patients with high-level microsatellite instability (MSI-H) and mismatch-repair deficient (dMMR) cancers, regardless of the site of origin. The approval was based on data from 149 patients with MSI-H or dMMR cancers, which showed a 40% response rate in this group of patients, two-thirds of whom had previously treated colon cancer. This landmark approval of a cancer therapy for a specific molecular profile and not the site of the disease, will certainly shape the future of oncology drug development. One of the highlighted stories at ASCO was the success of the larotrectinib (LOXO 101) tropomyosin receptor kinase inhibitor in patients with the TRK fusion mutations (p. e237). The data, including waterfall charts, swimmer plots, and computed-tomography scans, were impressive in this targeted population with a 76% response rate and a 91% duration of response at 6 months with a mild side effect profile.
In summary, across a variety of cancers, with treatment strategies of an equally diverse nature, we saw practice-changing data from the ASCO meeting that will benefit our patients. Continuing to seek out clinical trial options for patients will be critical in answering the many questions that have emerged and the substantial number of studies that are ongoing with combination immunotherapies, targeted small molecules, and a growing armamentarium of monoclonal antibodies.
As we head into vacation season and the dog days of summer, let’s reflect for a few minutes on some of the very important advances we heard about at this year’s annual meeting of the American Society of Clinical Oncology in Chicago. Nearly 40,000 individuals registered for the conference, an indication of both the interest and the excitement around the new agents and the emerging clinical trial data. Scientific sessions dedicated to the use of combination immunotherapy, the role of antibody drug conjugates, and targeting molecular aberrations with small molecules were among the most popular (p. e236).
In the setting of metastatic breast cancer, several trials produced highly significant results that will positively affect the duration and quality of life for our patients. The use of PARP inhibitors in BRCA-mutated cancers has been shown to be effective in a few areas, particularly advanced ovarian cancer. The OlympiAD study evaluated olaparib monotherapy and a physician’s choice arm (capecitabine, eribulin, or vinorelbine) in BRCA-mutated, HER2-negative metastatic breast cancer. The 2:1 design enrolled 302 patients and demonstrated a 3-month improvement in progression-free survival (PFS) for olaparib compared with the control arm (7.0 vs 4.2 months, respectively; P = .0009). The patient population for this BRCA-mutated trial was relatively young, with a median age of 45 years, and 50% of the women were hormone positive and 30%, platinum resistant.
The CDK4/6 inhibitors continue to be impressive, with the recently reported results from the MONARCH 2 trial showing encouraging PFS and overall response rate results with the addition of the CDK4/6 inhibitor abemaciclib to fulvestrant, a selective estrogen-receptor degrader. In this study, hormone-positive, HER2-negative women who had progressed on previous endocrine therapy were randomized 2:1 to abemaciclib plus fulvestrant or placebo plus fulvestrant. A total of 669 patients were accrued, and after a median follow-up of 19 months, a highly significant PFS difference of 7 months between the abemaciclib–fulvestrant and fulvestrant–only groups was observed (16.4 vs 9.3 months, respectively; P < .0000001) along with an overall response rate of 48.1 months, compared with 21.3 months. Previous findings have demonstrated monotherapy activity for abemaciclib, and the comparisons with palbociclib and ribociclib will be forthcoming, although no comparative trials are underway. These agents will be extensively assessed in a variety of settings, including adjuvantly.
The results of the much anticipated APHINITY study, which evaluated the addition of pertuzumab to trastuzumab in the adjuvant HER2-positive setting, were met with mixed reviews. Patients were included if they had node-positive invasive breast cancer or node-negative tumors of >1.0 cm. A total of 4,804 patients (37% node negative) were enrolled in the study. The intent-to-treat primary endpoint of invasive disease-free survival (DFS) was statistically positive (P = .045), although the 3-year absolute percentages for the pertuzumab–trastuzumab and trastuzumab-only groups were 94.1% and 93.2%, respectively. It should be noted that the planned statistical assumption was for a delta of 2.6% – 91.8% and 89.2%, respectively. Thus, both arms actually did better than had been planned, which was based on historical comparisons, and the node-positive and hormone-negative subgroups trended toward a greater benefit with the addition of pertuzumab. There was, and will continue to be, much debate around the cost–benefit ratio and which patients should be offered the combination. The outstanding results with the addition of pertuzumab in the neoadjuvant setting will continue to be the setting in which the greatest absolute clinical benefit will be seen. It is unusual in this era to see trials this large planned to identify a small difference, and it is likely that resource constraints will make such studies a thing of the past.
The very active hormonal therapies, abiraterone and enzalutimide, for castrate-resistant prostate cancer remain of high interest in the area of clinical trials. The LATITUDE study evaluated a straightforward design that compared abiraterone with placebo in patients who were newly diagnosed with high-risk, metastatic hormone-naïve prostate cancer. Patients in both arms received androgen-deprivation therapy and high risk was defined by having 2 of 3 criteria: a Gleason score of ≥8; 3 or more bone lesions; or visceral disease. Of note is that 1,199 patients were enrolled before publication of the CHAARTED or STAMPEDE results, which established docetaxel as a standard for these patients. The median age in the LATITUDE trial was 68 years, with 17% of patients having visceral disease and 48% having nodal disease, making it a similar patient population to those in the docetaxel studies. The results favoring abiraterone were strikingly positive, with a 38% reduction in the risk of death (P < .0001) and a 53% reduction in the risk of radiographic progression or death (P < .0001). The regimen was well tolerated overall, and it is clear that this option will be widely considered by physicians and their patients.
Two studies addressing the importance of managing symptoms and improving outcomes were also part of the plenary session. The IDEA Collaboration conducted a prospective pooled analysis of 6 phase 3 studies that assessed 3 and 6 months of oxaliplatin-based regimens for stage 3 colon cancer. FOLFOX and CAPOX given to 12,834 patients in 6 studies from the United States, European Union, Canada, Australia, New Zealand, and Japan were evaluated for DFS, treatment compliance, and adverse events. As would be anticipated, fewer side effects, particularly neurotoxicity, and greater compliance were observed in the 3-month group. Although DFS noninferiority for 3 months of therapy was not established statistically, the overall data led the investigators to issue a consensus statement advocating for a risk-based approach in deciding the duration of therapy and recommending 3 months of therapy for patients with stage 3, T1-3N1 disease, and consideration of 6 months therapy for T4 and/ or N2 disease. The investigators also acknowledged the leader and creator of IDEA, the late Daniel Sargent, PhD, of the Mayo Clinic, who passed away far too young after a brief illness last fall (1970-2016).
The second symptom-based study was performed at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and designed by a group of investigators from the Dana-Farber Cancer Institute in Boston; the Mayo Clinic in Rochester, Minnesota; the University of North Carolina in Chapel Hill; and MSKCC (p. e236). The hypothesis was simply that proactive symptom monitoring during chemotherapy would improve symptom management and lead to better outcomes. For the study, 766 patients with advanced solid tumors who were receiving outpatient chemotherapy were randomized to a control arm with standard follow-up or to the intervention arm, on which patients self-reported on 12 common symptoms before and between visits using a web-based tool and received weekly e-mail reminders and nursing alerts. At 6 months, and compared with baseline, the self-reporting patients in the intervention arm experienced an improved quality of life (P < .001). In addition, 7% fewer of the self-reporting patients visited the emergency department (P = .02), and they experienced longer survival by 5 months compared with the standard follow-up group (31.2 vs 26.0 months, respectively; P = .03). Although there are limitations to such a study, the growth in technological advances should create the opportunity to expand on this strategy in further trials and in practice. With such an emphasis in the Medicare Oncology Home Model on decreasing hospital admissions and visits to the emergency department, there should great motivation for all involved to consider incorporating self-reporting into their patterns of care.
A continued emphasis on molecular profiling, personalized and/or precision medicine, and identifying or matching the patient to the best possible therapy or the most appropriate clinical trial remains vital to improving outcomes. Just before the ASCO meeting, the US Food and Drug Administration approved pembrolizumab for the treatment of patients with high-level microsatellite instability (MSI-H) and mismatch-repair deficient (dMMR) cancers, regardless of the site of origin. The approval was based on data from 149 patients with MSI-H or dMMR cancers, which showed a 40% response rate in this group of patients, two-thirds of whom had previously treated colon cancer. This landmark approval of a cancer therapy for a specific molecular profile and not the site of the disease, will certainly shape the future of oncology drug development. One of the highlighted stories at ASCO was the success of the larotrectinib (LOXO 101) tropomyosin receptor kinase inhibitor in patients with the TRK fusion mutations (p. e237). The data, including waterfall charts, swimmer plots, and computed-tomography scans, were impressive in this targeted population with a 76% response rate and a 91% duration of response at 6 months with a mild side effect profile.
In summary, across a variety of cancers, with treatment strategies of an equally diverse nature, we saw practice-changing data from the ASCO meeting that will benefit our patients. Continuing to seek out clinical trial options for patients will be critical in answering the many questions that have emerged and the substantial number of studies that are ongoing with combination immunotherapies, targeted small molecules, and a growing armamentarium of monoclonal antibodies.
Cartilage Restoration in the Patellofemoral Joint
Take-Home Points
- Careful evaluation is key in attributing knee pain to patellofemoral cartilage lesions-that is, in making a "diagnosis by exclusion".
- Initial treatment is nonoperative management focused on weight loss and extensive "core-to-floor" rehabilitation.
- Optimization of anatomy and biomechanics is crucial.
- Factors important in surgical decision-making incude defect location and size, subchondral bone status, unipolar vs bipolar lesions, and previous cartilage procedure.
- The most commonly used surgical procedures-autologous chondrocyte implantation, osteochondral autograft transfer, and osteochondral allograft-have demonstrated improved intermediate-term outcomes.
Patellofemoral (PF) pain is often a component of more general anterior knee pain. One source of PF pain is chondral lesions. As these lesions are commonly seen on magnetic resonance imaging (MRI) and during arthroscopy, it is necessary to differentiate incidental and symptomatic lesions.1 In addition, the correlation between symptoms and lesion presence and severity is poor.
PF pain is multifactorial (structural lesions, malalignment, deconditioning, muscle imbalance and overuse) and can coexist with other lesions in the knee (ligament tears, meniscal injuries, and cartilage lesions in other compartments). Therefore, careful evaluation is key in attributing knee pain to PF cartilage lesions—that is, in making a "diagnosis by exclusion."
From the start, it must be appreciated that the vast majority of patients will not require surgery, and many who require surgery for pain will not require cartilage restoration. One key to success with PF patients is a good working relationship with an experienced physical therapist.
Etiology
The primary causes of PF cartilage lesions are patellar instability, chronic maltracking without instability, direct trauma, repetitive microtrauma, and idiopathic.
Patellar Instability
Patients with patellar instability often present with underlying anatomical risk factors (eg, trochlear dysplasia, increased Q-angle/tibial tubercle-trochlear groove [TT-TG] distance, patella alta, and unbalanced medial and lateral soft tissues2). These factors should be addressed before surgery.
Patellar instability can cause cartilage damage during the dislocation event or by chronic subluxation. Cartilage becomes damaged in up to 96% of patellar dislocations.3 Most commonly, the damage consists of fissuring and/or fibrillation, but chondral and osteochondral fractures can occur as well. During dislocation, the medial patella strikes the lateral aspect of the femur, and, as the knee collapses into flexion, the lateral aspect of the proximal lateral femoral condyle (weight-bearing area) can sustain damage. In the patella, typically the injury is distal-medial (occasionally crossing the median ridge). A shear lesion may involve the chondral surface or be osteochondral (Figure 1A).
Chronic Maltracking Without Instability
Chronic maltracking is usually related to anatomical abnormalities, which include the same factors that can cause patellar instability. A common combination is trochlear dysplasia, increased TT-TG or TT-posterior cruciate ligament distance, and lateral soft-tissue contracture. These are often seen in PF joints that progress to lateral PF arthritis. As lateral PF arthritis progresses, lateral soft-tissue contracture worsens, compounding symptoms of laterally based pain. With respect to cartilage repair, these joints can be treated if recognized early; however, once osteoarthritis is fully established in the joint, facetectomy or PF replacement may be necessary.
Direct Trauma
With the knee in flexion during a direct trauma over the patella (eg, fall or dashboard trauma), all zones of cartilage and subchondral bone in both patella and trochlea can be injured, leading to macrostructural damage, chondral/osteochondral fracture, or, with a subcritical force, microstructural damage and chondrocyte death, subsequently causing cartilage degeneration (cartilage may look normal initially; the matrix takes months to years to deteriorate). Direct trauma usually occurs with the knee flexed. Therefore, these lesions typically are located in the distal trochlea and superior pole of the patella.
Repetitive Microtrauma
Minor injuries, which by themselves do not immediately cause apparent chondral or osteochondral fractures, may eventually exceed the capacity of natural cartilage homeostasis and result in repetitive microtrauma. Common causes are repeated jumping (as in basketball and volleyball) and prolonged flexed-knee position (eg, what a baseball catcher experiences), which may also be associated with other lesions caused by extensor apparatus overload (eg, quadriceps tendon or patellar tendon tendinitis, and fat pad impingement syndrome).
Idiopathic
In a subset of patients with osteochondritis dissecans, the patella is the lesion site. In another subset, idiopathic lesions may be related to a genetic predisposition to osteoarthritis and may not be restricted to the PF joint. In some cases, the PF joint is the first compartment to degenerate and is the most symptomatic in a setting of truly tricompartmental disease. In these cases, treating only the PF lesion can result in functional failure, owing to disease progression in other compartments. Even mild disease in other compartments should be carefully evaluated.
History and Physical Examination
Patients often report a history of anterior knee pain that worsens with stair use, prolonged sitting, and flexed-knee activities (eg, squatting). Compared with pain alone, swelling, though not specific to cartilage disease, is more suspicious for a cartilage etiology. Identifying the cartilage defect as the sole source of pain is particularly difficult in patients with recurrent patellar instability. In these patients, pain and swelling, even between instability episodes, suggest that cartilage damage is at least a component of the symptomology.
Important diagnostic components of physical examination are gait analysis, tibiofemoral alignment, and patellar alignment in all 3 planes, both static and functional. Patella-specific measurements include medial-lateral position and quadrants of excursion, lateral tilt, and patella alta, as well as J-sign and subluxation with quadriceps contraction in extension.
It is also important to document effusion; crepitus; active and passive range of motion (spine, hips, knees); site of pain or tenderness to palpation (medial, lateral, distal, retropatellar) and whether it matches the complaints and the location of the cartilage lesion; results of the grind test (placing downward force on the patella during flexion and extension) and whether they match the flexion angle of the tenderness and the flexion angle in which the cartilage lesion has increased PF contact; ligamentous and soft-tissue stability or imbalance (tibiofemoral and patellar; apprehension test, glide test, tilt test); and muscle strength, flexibility, and atrophy of the core (abdomen, dorsal and hip muscles) and lower extremities (quadriceps, hamstrings, gastrocnemius).
Imaging
Imaging should be used to evaluate both PF alignment and the cartilage lesions. For alignment, standard radiographs (weight-bearing knee sequence and axial view; full limb length when needed), computed tomography, and MRI can be used.
Meaningful evaluation requires MRI with cartilage-specific sequences, including standard spin-echo (SE) and gradient-recalled echo (GRE), fast SE, and, for cartilage morphology, T2-weighted fat suppression (FS) and 3-dimensional SE and GRE.5 For evaluation of cartilage function and metabolism, the collagen network, and proteoglycan content in the knee cartilage matrix, consideration should be given to compositional assessment techniques, such as T2 mapping, delayed gadolinium-enhanced MRI of cartilage, T1ρ imaging, sodium imaging, and diffusion-weighted sequences.5 Use of the latter functional sequences is still debatable, and these sequences are not widely available.
Treatment
In general, the initial approach is nonoperative management focused on weight loss and extensive core-to-floor rehabilitation, unless surgery is specifically indicated (eg, for loose body removal or osteochondral fracture reattachment). Rehabilitation focuses on achieving adequate range of motion of the spine, hips, and knees along with muscle strength and flexibility of the core (abdomen, dorsal and hip muscles) and lower limbs (quadriceps, hamstrings, gastrocnemius). Rehabilitation is not defined by time but rather by development of an optimized soft-tissue envelope that decreases joint reactive forces. The full process can take 6 to 9 months, but there should be some improvement by 3 months.
Corticosteroid, hyaluronic acid,6 or platelet-rich plasma7 injections can provide temporary relief and facilitate rehabilitation in the setting of pain inhibition. As stand-alone treatment, injections are more suitable for more diffuse degenerative lesions in older and low-demand patients than for focal traumatic lesions in young and high-demand patients.
Surgery is indicated for full-thickness or nearly full-thickness lesions (International Cartilage Repair Society grade 3a or higher) >1 cm2 after failed conservative treatment.
Optimization of anatomy and biomechanics is crucial, as persistent abnormalities lead to high rates of failure of cartilage procedures, and correction of those factors results in outcomes similar to those of patients without such abnormal anatomy.8 The procedures most commonly used to improve patellar tracking or unloading in the PF compartment are lateral retinacular lengthening and TT transfer: medialization and/or distalization for correction of malalignment, and straight anteriorization or anteromedialization for unloading. These procedures can improve symptoms and function in lateral and distal patellar and trochlear lesions even without the addition of a cartilage restoration procedure.
Factors that are important in surgical decision-making include defect location and size, subchondral bone status, unipolar vs bipolar lesions, and previous cartilage procedure.
Location. The shapes of the patella and trochlea vary much more than the shapes of the condyles and plateaus. This variability complicates morphology matching, particularly with involvement of the central TG and median patellar ridge. Therefore, focal contained lesions of the patella and trochlea may be more technically amenable to cell therapy techniques than to osteochondral procedures, which require contour matching between donor and recipient
Size. Although small lesions in the femoral condyles can be considered for microfracture (MFx) or osteochondral autograft transfer (OAT), MFx is less suitable because of poor results in the PF joint, and OAT because of donor-site morbidity in the trochlea.
Subchondral bone status. When subchondral bone is compromised, such as with bone loss, cysts, or significant bone edema, the entire osteochondral unit should be treated. Here, OAT and osteochondral allograft (OCA) are the preferred treatments, depending on lesion size.
Unipolar vs bipolar lesions. Compared with unipolar lesions, bipolar lesions tend to have worse outcomes. Therefore, an associated unloading procedure (TT osteotomy) should be given special consideration. Autologous chondrocyte implantation (ACI) appears to have better outcomes than OCA for bipolar PF lesions.9,10
Previous surgery. Although a failed cartilage procedure can negatively affect ACI outcomes, particularly in the presence of intralesional osteophytes,11 it does not affect OCA outcomes.12 Therefore, after previous MFx, OCA instead of ACI may be considered.
Fragment Fixation
Viable fragments from traumatic lesions (direct trauma or patellar dislocation) or osteochondritis dissecans should be repaired if possible, particularly in young patients. In a fragment that contains a substantial amount of bone, compression screws provide stable fixation. More recently, it has been recognized that fixation of predominantly cartilaginous fragments can be successful13 (Figure 1B). Débridement of soft tissue in the lesion bed and on the fragment is important in facilitating healing, as is removal of sclerotic bone.
MFx
Although MFx can have good outcomes in small contained femoral condyle lesions, in the PF joint treatment has been more challenging, and clinical outcomes have been poor (increased subchondral edema, increased effusion).14 In addition, deterioration becomes significant after 36 months. Therefore, MFx should be restricted to small (<2 cm2), well-contained trochlear defects, particularly in low-demand patients.
ACI and Matrix-Induced ACI
As stated, ACI (Figure 2) is suitable for PF joints because it intrinsically respects the complex anatomy.
OAT
As mentioned, donor-site morbidity may compromise final outcomes of harvest and implantation in the PF joint. Nonetheless, in carefully selected patients with small lesions that are limited to 1 facet (not including the patellar ridge or the TG) and that require only 1 plug (Figure 3), OAT can have good clinical results.16
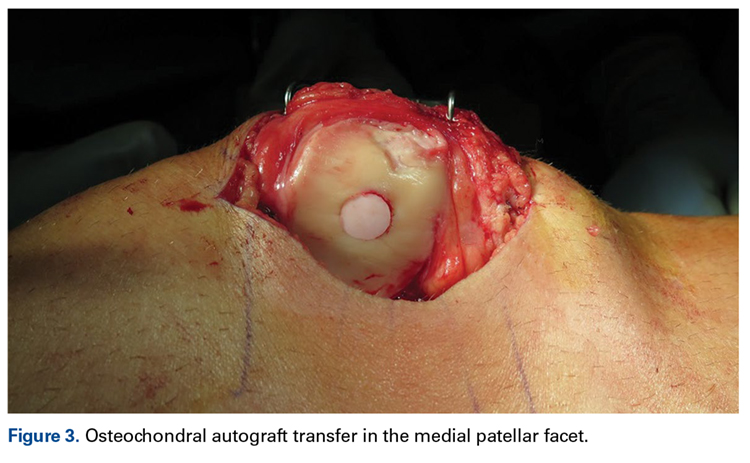
OCA
Two techniques can be used with OCA in the PF joint. The dowel technique, in which circular plugs are implanted, is predominantly used for defects that do not cross the midline (those located in their entirety on the medial or lateral aspect of the patella or trochlea). Central defects, which can be treated with the dowel technique as well, are technically more challenging to match perfectly, because of the complex geometry of the median ridge and the TG (Figure 4).
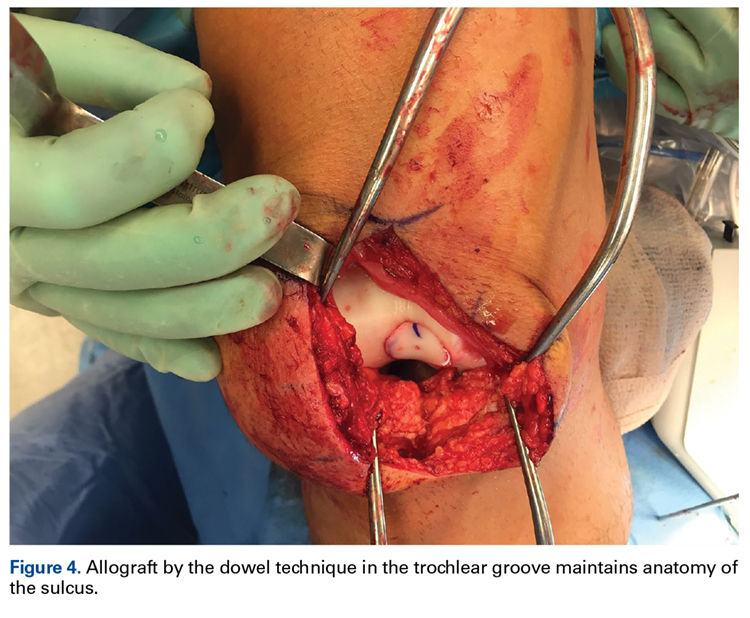
Experimental and Emerging Technologies
Biocartilage
Biocartilage, a dehydrated, micronized allogeneic cartilage scaffold implanted with platelet-rich plasma and fibrin glue added over a contained MFx-treated defect, can be used in the patella and trochlea and has the same indications as MFx (small lesions, contained lesions). There are limited clinical studies of short- or long-term outcomes.
Fresh and Viable OCA
Fresh OCA (ProChondrix; AlloSource) and viable/cryopreserved OCA (Cartiform; Arthrex) are thin osteochondral scaffolds that contain viable chondrocytes and growth factors. They can be implanted alone or used with MFx, and are indicated for lesions measuring 1 cm2 to 3 cm2. Aside from a case report,17 there are no clinical studies on outcomes.
Bone Marrow Aspirate Concentrate Implantation
Bone marrow aspirate concentrate from centrifuged iliac crest–harvested aspirate containing mesenchymal stem cells with chondrogenic potential is applied under a synthetic scaffold. Indications are the same as for ACI. Medium-term follow-up studies in the PF joint have shown good results, similar to those obtained with matrix-induced ACI.18
Particulated Juvenile Allograft Cartilage
Particulated juvenile allograft cartilage (DeNovo NT Graft; Zimmer Biomet) is minced cartilage allograft (from juvenile donors) that has been cut into cubes (~1 mm3). Indications are for patellar and trochlear lesions 1 cm2 to 6 cm2. For both the trochlea and the patella, short-term outcomes have been good.19,20
Rehabilitation After Surgery
Isolated PF cartilage restoration generally does not require prolonged weight-bearing restrictions, and ambulation with the knee locked in full extension is permitted as tolerated. Concurrent TT osteotomy, however, requires protection with 4 to 6 weeks of toe-touch weight-bearing to minimize the risk of tibial fracture.
Conclusion
Comprehensive preoperative assessment is essential and should include a thorough core-to-floor physical examination as well as PF-specific imaging. Treatment of symptomatic chondral lesions in the PF joint requires specific technical and postoperative management, which differs significantly from management involving the condyles. Attending to all these details makes the outcomes of PF cartilage treatment reproducible. These outcomes may rival those of condylar treatment.
1. Curl WW, Krome J, Gordon ES, Rushing J, Smith BP, Poehling GG. Cartilage injuries: a review of 31,516 knee arthroscopies. Arthroscopy. 1997;13(4):456-460.
2. Steensen RN, Bentley JC, Trinh TQ, Backes JR, Wiltfong RE. The prevalence and combined prevalences of anatomic factors associated with recurrent patellar dislocation: a magnetic resonance imaging study. Am J Sports Med. 2015;43(4):921-927.
3. Nomura E, Inoue M. Cartilage lesions of the patella in recurrent patellar dislocation. Am J Sports Med. 2004;32(2):498-502.
4. Vollnberg B, Koehlitz T, Jung T, et al. Prevalence of cartilage lesions and early osteoarthritis in patients with patellar dislocation. Eur Radiol. 2012;22(11):2347-2356.
5. Crema MD, Roemer FW, Marra MD, et al. Articular cartilage in the knee: current MR imaging techniques and applications in clinical practice and research. Radiographics. 2011;31(1):37-61.
6. Campbell KA, Erickson BJ, Saltzman BM, et al. Is local viscosupplementation injection clinically superior to other therapies in the treatment of osteoarthritis of the knee: a systematic review of overlapping meta-analyses. Arthroscopy. 2015;31(10):2036-2045.e14.
7. Saltzman BM, Jain A, Campbell KA, et al. Does the use of platelet-rich plasma at the time of surgery improve clinical outcomes in arthroscopic rotator cuff repair when compared with control cohorts? A systematic review of meta-analyses. Arthroscopy. 2016;32(5):906-918.
8. Gomoll AH, Gillogly SD, Cole BJ, et al. Autologous chondrocyte implantation in the patella: a multicenter experience. Am J Sports Med. 2014;42(5):1074-1081.
9. Meric G, Gracitelli GC, Gortz S, De Young AJ, Bugbee WD. Fresh osteochondral allograft transplantation for bipolar reciprocal osteochondral lesions of the knee. Am J Sports Med. 2015;43(3):709-714.
10. Peterson L, Vasiliadis HS, Brittberg M, Lindahl A. Autologous chondrocyte implantation: a long-term follow-up. Am J Sports Med. 2010;38(6):1117-1124.
11. Minas T, Gomoll AH, Rosenberger R, Royce RO, Bryant T. Increased failure rate of autologous chondrocyte implantation after previous treatment with marrow stimulation techniques. Am J Sports Med. 2009;37(5):902-908.
12. Gracitelli GC, Meric G, Briggs DT, et al. Fresh osteochondral allografts in the knee: comparison of primary transplantation versus transplantation after failure of previous subchondral marrow stimulation. Am J Sports Med. 2015;43(4):885-891.
13. Anderson CN, Magnussen RA, Block JJ, Anderson AF, Spindler KP. Operative fixation of chondral loose bodies in osteochondritis dissecans in the knee: a report of 5 cases. Orthop J Sports Med. 2013;1(2):2325967113496546.
14. Kreuz PC, Steinwachs MR, Erggelet C, et al. Results after microfracture of full-thickness chondral defects in different compartments in the knee. Osteoarthritis Cartilage. 2006;14(11):1119-1125.
15. Vasiliadis HS, Lindahl A, Georgoulis AD, Peterson L. Malalignment and cartilage lesions in the patellofemoral joint treated with autologous chondrocyte implantation. Knee Surg Sports Traumatol Arthrosc. 2011;19(3):452-457.
16. Astur DC, Arliani GG, Binz M, et al. Autologous osteochondral transplantation for treating patellar chondral injuries: evaluation, treatment, and outcomes of a two-year follow-up study. J Bone Joint Surg Am. 2014;96(10):816-823.
17. Hoffman JK, Geraghty S, Protzman NM. Articular cartilage repair using marrow simulation augmented with a viable chondral allograft: 9-month postoperative histological evaluation. Case Rep Orthop. 2015;2015:617365.
18. Gobbi A, Chaurasia S, Karnatzikos G, Nakamura N. Matrix-induced autologous chondrocyte implantation versus multipotent stem cells for the treatment of large patellofemoral chondral lesions: a nonrandomized prospective trial. Cartilage. 2015;6(2):82-97.
19. Farr J, Tabet SK, Margerrison E, Cole BJ. Clinical, radiographic, and histological outcomes after cartilage repair with particulated juvenile articular cartilage: a 2-year prospective study. Am J Sports Med. 2014;42(6):1417-1425.
20. Tompkins M, Hamann JC, Diduch DR, et al. Preliminary results of a novel single-stage cartilage restoration technique: particulated juvenile articular cartilage allograft for chondral defects of the patella. Arthroscopy. 2013;29(10):1661-1670.
Take-Home Points
- Careful evaluation is key in attributing knee pain to patellofemoral cartilage lesions-that is, in making a "diagnosis by exclusion".
- Initial treatment is nonoperative management focused on weight loss and extensive "core-to-floor" rehabilitation.
- Optimization of anatomy and biomechanics is crucial.
- Factors important in surgical decision-making incude defect location and size, subchondral bone status, unipolar vs bipolar lesions, and previous cartilage procedure.
- The most commonly used surgical procedures-autologous chondrocyte implantation, osteochondral autograft transfer, and osteochondral allograft-have demonstrated improved intermediate-term outcomes.
Patellofemoral (PF) pain is often a component of more general anterior knee pain. One source of PF pain is chondral lesions. As these lesions are commonly seen on magnetic resonance imaging (MRI) and during arthroscopy, it is necessary to differentiate incidental and symptomatic lesions.1 In addition, the correlation between symptoms and lesion presence and severity is poor.
PF pain is multifactorial (structural lesions, malalignment, deconditioning, muscle imbalance and overuse) and can coexist with other lesions in the knee (ligament tears, meniscal injuries, and cartilage lesions in other compartments). Therefore, careful evaluation is key in attributing knee pain to PF cartilage lesions—that is, in making a "diagnosis by exclusion."
From the start, it must be appreciated that the vast majority of patients will not require surgery, and many who require surgery for pain will not require cartilage restoration. One key to success with PF patients is a good working relationship with an experienced physical therapist.
Etiology
The primary causes of PF cartilage lesions are patellar instability, chronic maltracking without instability, direct trauma, repetitive microtrauma, and idiopathic.
Patellar Instability
Patients with patellar instability often present with underlying anatomical risk factors (eg, trochlear dysplasia, increased Q-angle/tibial tubercle-trochlear groove [TT-TG] distance, patella alta, and unbalanced medial and lateral soft tissues2). These factors should be addressed before surgery.
Patellar instability can cause cartilage damage during the dislocation event or by chronic subluxation. Cartilage becomes damaged in up to 96% of patellar dislocations.3 Most commonly, the damage consists of fissuring and/or fibrillation, but chondral and osteochondral fractures can occur as well. During dislocation, the medial patella strikes the lateral aspect of the femur, and, as the knee collapses into flexion, the lateral aspect of the proximal lateral femoral condyle (weight-bearing area) can sustain damage. In the patella, typically the injury is distal-medial (occasionally crossing the median ridge). A shear lesion may involve the chondral surface or be osteochondral (Figure 1A).
Chronic Maltracking Without Instability
Chronic maltracking is usually related to anatomical abnormalities, which include the same factors that can cause patellar instability. A common combination is trochlear dysplasia, increased TT-TG or TT-posterior cruciate ligament distance, and lateral soft-tissue contracture. These are often seen in PF joints that progress to lateral PF arthritis. As lateral PF arthritis progresses, lateral soft-tissue contracture worsens, compounding symptoms of laterally based pain. With respect to cartilage repair, these joints can be treated if recognized early; however, once osteoarthritis is fully established in the joint, facetectomy or PF replacement may be necessary.
Direct Trauma
With the knee in flexion during a direct trauma over the patella (eg, fall or dashboard trauma), all zones of cartilage and subchondral bone in both patella and trochlea can be injured, leading to macrostructural damage, chondral/osteochondral fracture, or, with a subcritical force, microstructural damage and chondrocyte death, subsequently causing cartilage degeneration (cartilage may look normal initially; the matrix takes months to years to deteriorate). Direct trauma usually occurs with the knee flexed. Therefore, these lesions typically are located in the distal trochlea and superior pole of the patella.
Repetitive Microtrauma
Minor injuries, which by themselves do not immediately cause apparent chondral or osteochondral fractures, may eventually exceed the capacity of natural cartilage homeostasis and result in repetitive microtrauma. Common causes are repeated jumping (as in basketball and volleyball) and prolonged flexed-knee position (eg, what a baseball catcher experiences), which may also be associated with other lesions caused by extensor apparatus overload (eg, quadriceps tendon or patellar tendon tendinitis, and fat pad impingement syndrome).
Idiopathic
In a subset of patients with osteochondritis dissecans, the patella is the lesion site. In another subset, idiopathic lesions may be related to a genetic predisposition to osteoarthritis and may not be restricted to the PF joint. In some cases, the PF joint is the first compartment to degenerate and is the most symptomatic in a setting of truly tricompartmental disease. In these cases, treating only the PF lesion can result in functional failure, owing to disease progression in other compartments. Even mild disease in other compartments should be carefully evaluated.
History and Physical Examination
Patients often report a history of anterior knee pain that worsens with stair use, prolonged sitting, and flexed-knee activities (eg, squatting). Compared with pain alone, swelling, though not specific to cartilage disease, is more suspicious for a cartilage etiology. Identifying the cartilage defect as the sole source of pain is particularly difficult in patients with recurrent patellar instability. In these patients, pain and swelling, even between instability episodes, suggest that cartilage damage is at least a component of the symptomology.
Important diagnostic components of physical examination are gait analysis, tibiofemoral alignment, and patellar alignment in all 3 planes, both static and functional. Patella-specific measurements include medial-lateral position and quadrants of excursion, lateral tilt, and patella alta, as well as J-sign and subluxation with quadriceps contraction in extension.
It is also important to document effusion; crepitus; active and passive range of motion (spine, hips, knees); site of pain or tenderness to palpation (medial, lateral, distal, retropatellar) and whether it matches the complaints and the location of the cartilage lesion; results of the grind test (placing downward force on the patella during flexion and extension) and whether they match the flexion angle of the tenderness and the flexion angle in which the cartilage lesion has increased PF contact; ligamentous and soft-tissue stability or imbalance (tibiofemoral and patellar; apprehension test, glide test, tilt test); and muscle strength, flexibility, and atrophy of the core (abdomen, dorsal and hip muscles) and lower extremities (quadriceps, hamstrings, gastrocnemius).
Imaging
Imaging should be used to evaluate both PF alignment and the cartilage lesions. For alignment, standard radiographs (weight-bearing knee sequence and axial view; full limb length when needed), computed tomography, and MRI can be used.
Meaningful evaluation requires MRI with cartilage-specific sequences, including standard spin-echo (SE) and gradient-recalled echo (GRE), fast SE, and, for cartilage morphology, T2-weighted fat suppression (FS) and 3-dimensional SE and GRE.5 For evaluation of cartilage function and metabolism, the collagen network, and proteoglycan content in the knee cartilage matrix, consideration should be given to compositional assessment techniques, such as T2 mapping, delayed gadolinium-enhanced MRI of cartilage, T1ρ imaging, sodium imaging, and diffusion-weighted sequences.5 Use of the latter functional sequences is still debatable, and these sequences are not widely available.
Treatment
In general, the initial approach is nonoperative management focused on weight loss and extensive core-to-floor rehabilitation, unless surgery is specifically indicated (eg, for loose body removal or osteochondral fracture reattachment). Rehabilitation focuses on achieving adequate range of motion of the spine, hips, and knees along with muscle strength and flexibility of the core (abdomen, dorsal and hip muscles) and lower limbs (quadriceps, hamstrings, gastrocnemius). Rehabilitation is not defined by time but rather by development of an optimized soft-tissue envelope that decreases joint reactive forces. The full process can take 6 to 9 months, but there should be some improvement by 3 months.
Corticosteroid, hyaluronic acid,6 or platelet-rich plasma7 injections can provide temporary relief and facilitate rehabilitation in the setting of pain inhibition. As stand-alone treatment, injections are more suitable for more diffuse degenerative lesions in older and low-demand patients than for focal traumatic lesions in young and high-demand patients.
Surgery is indicated for full-thickness or nearly full-thickness lesions (International Cartilage Repair Society grade 3a or higher) >1 cm2 after failed conservative treatment.
Optimization of anatomy and biomechanics is crucial, as persistent abnormalities lead to high rates of failure of cartilage procedures, and correction of those factors results in outcomes similar to those of patients without such abnormal anatomy.8 The procedures most commonly used to improve patellar tracking or unloading in the PF compartment are lateral retinacular lengthening and TT transfer: medialization and/or distalization for correction of malalignment, and straight anteriorization or anteromedialization for unloading. These procedures can improve symptoms and function in lateral and distal patellar and trochlear lesions even without the addition of a cartilage restoration procedure.
Factors that are important in surgical decision-making include defect location and size, subchondral bone status, unipolar vs bipolar lesions, and previous cartilage procedure.
Location. The shapes of the patella and trochlea vary much more than the shapes of the condyles and plateaus. This variability complicates morphology matching, particularly with involvement of the central TG and median patellar ridge. Therefore, focal contained lesions of the patella and trochlea may be more technically amenable to cell therapy techniques than to osteochondral procedures, which require contour matching between donor and recipient
Size. Although small lesions in the femoral condyles can be considered for microfracture (MFx) or osteochondral autograft transfer (OAT), MFx is less suitable because of poor results in the PF joint, and OAT because of donor-site morbidity in the trochlea.
Subchondral bone status. When subchondral bone is compromised, such as with bone loss, cysts, or significant bone edema, the entire osteochondral unit should be treated. Here, OAT and osteochondral allograft (OCA) are the preferred treatments, depending on lesion size.
Unipolar vs bipolar lesions. Compared with unipolar lesions, bipolar lesions tend to have worse outcomes. Therefore, an associated unloading procedure (TT osteotomy) should be given special consideration. Autologous chondrocyte implantation (ACI) appears to have better outcomes than OCA for bipolar PF lesions.9,10
Previous surgery. Although a failed cartilage procedure can negatively affect ACI outcomes, particularly in the presence of intralesional osteophytes,11 it does not affect OCA outcomes.12 Therefore, after previous MFx, OCA instead of ACI may be considered.
Fragment Fixation
Viable fragments from traumatic lesions (direct trauma or patellar dislocation) or osteochondritis dissecans should be repaired if possible, particularly in young patients. In a fragment that contains a substantial amount of bone, compression screws provide stable fixation. More recently, it has been recognized that fixation of predominantly cartilaginous fragments can be successful13 (Figure 1B). Débridement of soft tissue in the lesion bed and on the fragment is important in facilitating healing, as is removal of sclerotic bone.
MFx
Although MFx can have good outcomes in small contained femoral condyle lesions, in the PF joint treatment has been more challenging, and clinical outcomes have been poor (increased subchondral edema, increased effusion).14 In addition, deterioration becomes significant after 36 months. Therefore, MFx should be restricted to small (<2 cm2), well-contained trochlear defects, particularly in low-demand patients.
ACI and Matrix-Induced ACI
As stated, ACI (Figure 2) is suitable for PF joints because it intrinsically respects the complex anatomy.
OAT
As mentioned, donor-site morbidity may compromise final outcomes of harvest and implantation in the PF joint. Nonetheless, in carefully selected patients with small lesions that are limited to 1 facet (not including the patellar ridge or the TG) and that require only 1 plug (Figure 3), OAT can have good clinical results.16
OCA
Two techniques can be used with OCA in the PF joint. The dowel technique, in which circular plugs are implanted, is predominantly used for defects that do not cross the midline (those located in their entirety on the medial or lateral aspect of the patella or trochlea). Central defects, which can be treated with the dowel technique as well, are technically more challenging to match perfectly, because of the complex geometry of the median ridge and the TG (Figure 4).
Experimental and Emerging Technologies
Biocartilage
Biocartilage, a dehydrated, micronized allogeneic cartilage scaffold implanted with platelet-rich plasma and fibrin glue added over a contained MFx-treated defect, can be used in the patella and trochlea and has the same indications as MFx (small lesions, contained lesions). There are limited clinical studies of short- or long-term outcomes.
Fresh and Viable OCA
Fresh OCA (ProChondrix; AlloSource) and viable/cryopreserved OCA (Cartiform; Arthrex) are thin osteochondral scaffolds that contain viable chondrocytes and growth factors. They can be implanted alone or used with MFx, and are indicated for lesions measuring 1 cm2 to 3 cm2. Aside from a case report,17 there are no clinical studies on outcomes.
Bone Marrow Aspirate Concentrate Implantation
Bone marrow aspirate concentrate from centrifuged iliac crest–harvested aspirate containing mesenchymal stem cells with chondrogenic potential is applied under a synthetic scaffold. Indications are the same as for ACI. Medium-term follow-up studies in the PF joint have shown good results, similar to those obtained with matrix-induced ACI.18
Particulated Juvenile Allograft Cartilage
Particulated juvenile allograft cartilage (DeNovo NT Graft; Zimmer Biomet) is minced cartilage allograft (from juvenile donors) that has been cut into cubes (~1 mm3). Indications are for patellar and trochlear lesions 1 cm2 to 6 cm2. For both the trochlea and the patella, short-term outcomes have been good.19,20
Rehabilitation After Surgery
Isolated PF cartilage restoration generally does not require prolonged weight-bearing restrictions, and ambulation with the knee locked in full extension is permitted as tolerated. Concurrent TT osteotomy, however, requires protection with 4 to 6 weeks of toe-touch weight-bearing to minimize the risk of tibial fracture.
Conclusion
Comprehensive preoperative assessment is essential and should include a thorough core-to-floor physical examination as well as PF-specific imaging. Treatment of symptomatic chondral lesions in the PF joint requires specific technical and postoperative management, which differs significantly from management involving the condyles. Attending to all these details makes the outcomes of PF cartilage treatment reproducible. These outcomes may rival those of condylar treatment.
Take-Home Points
- Careful evaluation is key in attributing knee pain to patellofemoral cartilage lesions-that is, in making a "diagnosis by exclusion".
- Initial treatment is nonoperative management focused on weight loss and extensive "core-to-floor" rehabilitation.
- Optimization of anatomy and biomechanics is crucial.
- Factors important in surgical decision-making incude defect location and size, subchondral bone status, unipolar vs bipolar lesions, and previous cartilage procedure.
- The most commonly used surgical procedures-autologous chondrocyte implantation, osteochondral autograft transfer, and osteochondral allograft-have demonstrated improved intermediate-term outcomes.
Patellofemoral (PF) pain is often a component of more general anterior knee pain. One source of PF pain is chondral lesions. As these lesions are commonly seen on magnetic resonance imaging (MRI) and during arthroscopy, it is necessary to differentiate incidental and symptomatic lesions.1 In addition, the correlation between symptoms and lesion presence and severity is poor.
PF pain is multifactorial (structural lesions, malalignment, deconditioning, muscle imbalance and overuse) and can coexist with other lesions in the knee (ligament tears, meniscal injuries, and cartilage lesions in other compartments). Therefore, careful evaluation is key in attributing knee pain to PF cartilage lesions—that is, in making a "diagnosis by exclusion."
From the start, it must be appreciated that the vast majority of patients will not require surgery, and many who require surgery for pain will not require cartilage restoration. One key to success with PF patients is a good working relationship with an experienced physical therapist.
Etiology
The primary causes of PF cartilage lesions are patellar instability, chronic maltracking without instability, direct trauma, repetitive microtrauma, and idiopathic.
Patellar Instability
Patients with patellar instability often present with underlying anatomical risk factors (eg, trochlear dysplasia, increased Q-angle/tibial tubercle-trochlear groove [TT-TG] distance, patella alta, and unbalanced medial and lateral soft tissues2). These factors should be addressed before surgery.
Patellar instability can cause cartilage damage during the dislocation event or by chronic subluxation. Cartilage becomes damaged in up to 96% of patellar dislocations.3 Most commonly, the damage consists of fissuring and/or fibrillation, but chondral and osteochondral fractures can occur as well. During dislocation, the medial patella strikes the lateral aspect of the femur, and, as the knee collapses into flexion, the lateral aspect of the proximal lateral femoral condyle (weight-bearing area) can sustain damage. In the patella, typically the injury is distal-medial (occasionally crossing the median ridge). A shear lesion may involve the chondral surface or be osteochondral (Figure 1A).
Chronic Maltracking Without Instability
Chronic maltracking is usually related to anatomical abnormalities, which include the same factors that can cause patellar instability. A common combination is trochlear dysplasia, increased TT-TG or TT-posterior cruciate ligament distance, and lateral soft-tissue contracture. These are often seen in PF joints that progress to lateral PF arthritis. As lateral PF arthritis progresses, lateral soft-tissue contracture worsens, compounding symptoms of laterally based pain. With respect to cartilage repair, these joints can be treated if recognized early; however, once osteoarthritis is fully established in the joint, facetectomy or PF replacement may be necessary.
Direct Trauma
With the knee in flexion during a direct trauma over the patella (eg, fall or dashboard trauma), all zones of cartilage and subchondral bone in both patella and trochlea can be injured, leading to macrostructural damage, chondral/osteochondral fracture, or, with a subcritical force, microstructural damage and chondrocyte death, subsequently causing cartilage degeneration (cartilage may look normal initially; the matrix takes months to years to deteriorate). Direct trauma usually occurs with the knee flexed. Therefore, these lesions typically are located in the distal trochlea and superior pole of the patella.
Repetitive Microtrauma
Minor injuries, which by themselves do not immediately cause apparent chondral or osteochondral fractures, may eventually exceed the capacity of natural cartilage homeostasis and result in repetitive microtrauma. Common causes are repeated jumping (as in basketball and volleyball) and prolonged flexed-knee position (eg, what a baseball catcher experiences), which may also be associated with other lesions caused by extensor apparatus overload (eg, quadriceps tendon or patellar tendon tendinitis, and fat pad impingement syndrome).
Idiopathic
In a subset of patients with osteochondritis dissecans, the patella is the lesion site. In another subset, idiopathic lesions may be related to a genetic predisposition to osteoarthritis and may not be restricted to the PF joint. In some cases, the PF joint is the first compartment to degenerate and is the most symptomatic in a setting of truly tricompartmental disease. In these cases, treating only the PF lesion can result in functional failure, owing to disease progression in other compartments. Even mild disease in other compartments should be carefully evaluated.
History and Physical Examination
Patients often report a history of anterior knee pain that worsens with stair use, prolonged sitting, and flexed-knee activities (eg, squatting). Compared with pain alone, swelling, though not specific to cartilage disease, is more suspicious for a cartilage etiology. Identifying the cartilage defect as the sole source of pain is particularly difficult in patients with recurrent patellar instability. In these patients, pain and swelling, even between instability episodes, suggest that cartilage damage is at least a component of the symptomology.
Important diagnostic components of physical examination are gait analysis, tibiofemoral alignment, and patellar alignment in all 3 planes, both static and functional. Patella-specific measurements include medial-lateral position and quadrants of excursion, lateral tilt, and patella alta, as well as J-sign and subluxation with quadriceps contraction in extension.
It is also important to document effusion; crepitus; active and passive range of motion (spine, hips, knees); site of pain or tenderness to palpation (medial, lateral, distal, retropatellar) and whether it matches the complaints and the location of the cartilage lesion; results of the grind test (placing downward force on the patella during flexion and extension) and whether they match the flexion angle of the tenderness and the flexion angle in which the cartilage lesion has increased PF contact; ligamentous and soft-tissue stability or imbalance (tibiofemoral and patellar; apprehension test, glide test, tilt test); and muscle strength, flexibility, and atrophy of the core (abdomen, dorsal and hip muscles) and lower extremities (quadriceps, hamstrings, gastrocnemius).
Imaging
Imaging should be used to evaluate both PF alignment and the cartilage lesions. For alignment, standard radiographs (weight-bearing knee sequence and axial view; full limb length when needed), computed tomography, and MRI can be used.
Meaningful evaluation requires MRI with cartilage-specific sequences, including standard spin-echo (SE) and gradient-recalled echo (GRE), fast SE, and, for cartilage morphology, T2-weighted fat suppression (FS) and 3-dimensional SE and GRE.5 For evaluation of cartilage function and metabolism, the collagen network, and proteoglycan content in the knee cartilage matrix, consideration should be given to compositional assessment techniques, such as T2 mapping, delayed gadolinium-enhanced MRI of cartilage, T1ρ imaging, sodium imaging, and diffusion-weighted sequences.5 Use of the latter functional sequences is still debatable, and these sequences are not widely available.
Treatment
In general, the initial approach is nonoperative management focused on weight loss and extensive core-to-floor rehabilitation, unless surgery is specifically indicated (eg, for loose body removal or osteochondral fracture reattachment). Rehabilitation focuses on achieving adequate range of motion of the spine, hips, and knees along with muscle strength and flexibility of the core (abdomen, dorsal and hip muscles) and lower limbs (quadriceps, hamstrings, gastrocnemius). Rehabilitation is not defined by time but rather by development of an optimized soft-tissue envelope that decreases joint reactive forces. The full process can take 6 to 9 months, but there should be some improvement by 3 months.
Corticosteroid, hyaluronic acid,6 or platelet-rich plasma7 injections can provide temporary relief and facilitate rehabilitation in the setting of pain inhibition. As stand-alone treatment, injections are more suitable for more diffuse degenerative lesions in older and low-demand patients than for focal traumatic lesions in young and high-demand patients.
Surgery is indicated for full-thickness or nearly full-thickness lesions (International Cartilage Repair Society grade 3a or higher) >1 cm2 after failed conservative treatment.
Optimization of anatomy and biomechanics is crucial, as persistent abnormalities lead to high rates of failure of cartilage procedures, and correction of those factors results in outcomes similar to those of patients without such abnormal anatomy.8 The procedures most commonly used to improve patellar tracking or unloading in the PF compartment are lateral retinacular lengthening and TT transfer: medialization and/or distalization for correction of malalignment, and straight anteriorization or anteromedialization for unloading. These procedures can improve symptoms and function in lateral and distal patellar and trochlear lesions even without the addition of a cartilage restoration procedure.
Factors that are important in surgical decision-making include defect location and size, subchondral bone status, unipolar vs bipolar lesions, and previous cartilage procedure.
Location. The shapes of the patella and trochlea vary much more than the shapes of the condyles and plateaus. This variability complicates morphology matching, particularly with involvement of the central TG and median patellar ridge. Therefore, focal contained lesions of the patella and trochlea may be more technically amenable to cell therapy techniques than to osteochondral procedures, which require contour matching between donor and recipient
Size. Although small lesions in the femoral condyles can be considered for microfracture (MFx) or osteochondral autograft transfer (OAT), MFx is less suitable because of poor results in the PF joint, and OAT because of donor-site morbidity in the trochlea.
Subchondral bone status. When subchondral bone is compromised, such as with bone loss, cysts, or significant bone edema, the entire osteochondral unit should be treated. Here, OAT and osteochondral allograft (OCA) are the preferred treatments, depending on lesion size.
Unipolar vs bipolar lesions. Compared with unipolar lesions, bipolar lesions tend to have worse outcomes. Therefore, an associated unloading procedure (TT osteotomy) should be given special consideration. Autologous chondrocyte implantation (ACI) appears to have better outcomes than OCA for bipolar PF lesions.9,10
Previous surgery. Although a failed cartilage procedure can negatively affect ACI outcomes, particularly in the presence of intralesional osteophytes,11 it does not affect OCA outcomes.12 Therefore, after previous MFx, OCA instead of ACI may be considered.
Fragment Fixation
Viable fragments from traumatic lesions (direct trauma or patellar dislocation) or osteochondritis dissecans should be repaired if possible, particularly in young patients. In a fragment that contains a substantial amount of bone, compression screws provide stable fixation. More recently, it has been recognized that fixation of predominantly cartilaginous fragments can be successful13 (Figure 1B). Débridement of soft tissue in the lesion bed and on the fragment is important in facilitating healing, as is removal of sclerotic bone.
MFx
Although MFx can have good outcomes in small contained femoral condyle lesions, in the PF joint treatment has been more challenging, and clinical outcomes have been poor (increased subchondral edema, increased effusion).14 In addition, deterioration becomes significant after 36 months. Therefore, MFx should be restricted to small (<2 cm2), well-contained trochlear defects, particularly in low-demand patients.
ACI and Matrix-Induced ACI
As stated, ACI (Figure 2) is suitable for PF joints because it intrinsically respects the complex anatomy.
OAT
As mentioned, donor-site morbidity may compromise final outcomes of harvest and implantation in the PF joint. Nonetheless, in carefully selected patients with small lesions that are limited to 1 facet (not including the patellar ridge or the TG) and that require only 1 plug (Figure 3), OAT can have good clinical results.16
OCA
Two techniques can be used with OCA in the PF joint. The dowel technique, in which circular plugs are implanted, is predominantly used for defects that do not cross the midline (those located in their entirety on the medial or lateral aspect of the patella or trochlea). Central defects, which can be treated with the dowel technique as well, are technically more challenging to match perfectly, because of the complex geometry of the median ridge and the TG (Figure 4).
Experimental and Emerging Technologies
Biocartilage
Biocartilage, a dehydrated, micronized allogeneic cartilage scaffold implanted with platelet-rich plasma and fibrin glue added over a contained MFx-treated defect, can be used in the patella and trochlea and has the same indications as MFx (small lesions, contained lesions). There are limited clinical studies of short- or long-term outcomes.
Fresh and Viable OCA
Fresh OCA (ProChondrix; AlloSource) and viable/cryopreserved OCA (Cartiform; Arthrex) are thin osteochondral scaffolds that contain viable chondrocytes and growth factors. They can be implanted alone or used with MFx, and are indicated for lesions measuring 1 cm2 to 3 cm2. Aside from a case report,17 there are no clinical studies on outcomes.
Bone Marrow Aspirate Concentrate Implantation
Bone marrow aspirate concentrate from centrifuged iliac crest–harvested aspirate containing mesenchymal stem cells with chondrogenic potential is applied under a synthetic scaffold. Indications are the same as for ACI. Medium-term follow-up studies in the PF joint have shown good results, similar to those obtained with matrix-induced ACI.18
Particulated Juvenile Allograft Cartilage
Particulated juvenile allograft cartilage (DeNovo NT Graft; Zimmer Biomet) is minced cartilage allograft (from juvenile donors) that has been cut into cubes (~1 mm3). Indications are for patellar and trochlear lesions 1 cm2 to 6 cm2. For both the trochlea and the patella, short-term outcomes have been good.19,20
Rehabilitation After Surgery
Isolated PF cartilage restoration generally does not require prolonged weight-bearing restrictions, and ambulation with the knee locked in full extension is permitted as tolerated. Concurrent TT osteotomy, however, requires protection with 4 to 6 weeks of toe-touch weight-bearing to minimize the risk of tibial fracture.
Conclusion
Comprehensive preoperative assessment is essential and should include a thorough core-to-floor physical examination as well as PF-specific imaging. Treatment of symptomatic chondral lesions in the PF joint requires specific technical and postoperative management, which differs significantly from management involving the condyles. Attending to all these details makes the outcomes of PF cartilage treatment reproducible. These outcomes may rival those of condylar treatment.
1. Curl WW, Krome J, Gordon ES, Rushing J, Smith BP, Poehling GG. Cartilage injuries: a review of 31,516 knee arthroscopies. Arthroscopy. 1997;13(4):456-460.
2. Steensen RN, Bentley JC, Trinh TQ, Backes JR, Wiltfong RE. The prevalence and combined prevalences of anatomic factors associated with recurrent patellar dislocation: a magnetic resonance imaging study. Am J Sports Med. 2015;43(4):921-927.
3. Nomura E, Inoue M. Cartilage lesions of the patella in recurrent patellar dislocation. Am J Sports Med. 2004;32(2):498-502.
4. Vollnberg B, Koehlitz T, Jung T, et al. Prevalence of cartilage lesions and early osteoarthritis in patients with patellar dislocation. Eur Radiol. 2012;22(11):2347-2356.
5. Crema MD, Roemer FW, Marra MD, et al. Articular cartilage in the knee: current MR imaging techniques and applications in clinical practice and research. Radiographics. 2011;31(1):37-61.
6. Campbell KA, Erickson BJ, Saltzman BM, et al. Is local viscosupplementation injection clinically superior to other therapies in the treatment of osteoarthritis of the knee: a systematic review of overlapping meta-analyses. Arthroscopy. 2015;31(10):2036-2045.e14.
7. Saltzman BM, Jain A, Campbell KA, et al. Does the use of platelet-rich plasma at the time of surgery improve clinical outcomes in arthroscopic rotator cuff repair when compared with control cohorts? A systematic review of meta-analyses. Arthroscopy. 2016;32(5):906-918.
8. Gomoll AH, Gillogly SD, Cole BJ, et al. Autologous chondrocyte implantation in the patella: a multicenter experience. Am J Sports Med. 2014;42(5):1074-1081.
9. Meric G, Gracitelli GC, Gortz S, De Young AJ, Bugbee WD. Fresh osteochondral allograft transplantation for bipolar reciprocal osteochondral lesions of the knee. Am J Sports Med. 2015;43(3):709-714.
10. Peterson L, Vasiliadis HS, Brittberg M, Lindahl A. Autologous chondrocyte implantation: a long-term follow-up. Am J Sports Med. 2010;38(6):1117-1124.
11. Minas T, Gomoll AH, Rosenberger R, Royce RO, Bryant T. Increased failure rate of autologous chondrocyte implantation after previous treatment with marrow stimulation techniques. Am J Sports Med. 2009;37(5):902-908.
12. Gracitelli GC, Meric G, Briggs DT, et al. Fresh osteochondral allografts in the knee: comparison of primary transplantation versus transplantation after failure of previous subchondral marrow stimulation. Am J Sports Med. 2015;43(4):885-891.
13. Anderson CN, Magnussen RA, Block JJ, Anderson AF, Spindler KP. Operative fixation of chondral loose bodies in osteochondritis dissecans in the knee: a report of 5 cases. Orthop J Sports Med. 2013;1(2):2325967113496546.
14. Kreuz PC, Steinwachs MR, Erggelet C, et al. Results after microfracture of full-thickness chondral defects in different compartments in the knee. Osteoarthritis Cartilage. 2006;14(11):1119-1125.
15. Vasiliadis HS, Lindahl A, Georgoulis AD, Peterson L. Malalignment and cartilage lesions in the patellofemoral joint treated with autologous chondrocyte implantation. Knee Surg Sports Traumatol Arthrosc. 2011;19(3):452-457.
16. Astur DC, Arliani GG, Binz M, et al. Autologous osteochondral transplantation for treating patellar chondral injuries: evaluation, treatment, and outcomes of a two-year follow-up study. J Bone Joint Surg Am. 2014;96(10):816-823.
17. Hoffman JK, Geraghty S, Protzman NM. Articular cartilage repair using marrow simulation augmented with a viable chondral allograft: 9-month postoperative histological evaluation. Case Rep Orthop. 2015;2015:617365.
18. Gobbi A, Chaurasia S, Karnatzikos G, Nakamura N. Matrix-induced autologous chondrocyte implantation versus multipotent stem cells for the treatment of large patellofemoral chondral lesions: a nonrandomized prospective trial. Cartilage. 2015;6(2):82-97.
19. Farr J, Tabet SK, Margerrison E, Cole BJ. Clinical, radiographic, and histological outcomes after cartilage repair with particulated juvenile articular cartilage: a 2-year prospective study. Am J Sports Med. 2014;42(6):1417-1425.
20. Tompkins M, Hamann JC, Diduch DR, et al. Preliminary results of a novel single-stage cartilage restoration technique: particulated juvenile articular cartilage allograft for chondral defects of the patella. Arthroscopy. 2013;29(10):1661-1670.
1. Curl WW, Krome J, Gordon ES, Rushing J, Smith BP, Poehling GG. Cartilage injuries: a review of 31,516 knee arthroscopies. Arthroscopy. 1997;13(4):456-460.
2. Steensen RN, Bentley JC, Trinh TQ, Backes JR, Wiltfong RE. The prevalence and combined prevalences of anatomic factors associated with recurrent patellar dislocation: a magnetic resonance imaging study. Am J Sports Med. 2015;43(4):921-927.
3. Nomura E, Inoue M. Cartilage lesions of the patella in recurrent patellar dislocation. Am J Sports Med. 2004;32(2):498-502.
4. Vollnberg B, Koehlitz T, Jung T, et al. Prevalence of cartilage lesions and early osteoarthritis in patients with patellar dislocation. Eur Radiol. 2012;22(11):2347-2356.
5. Crema MD, Roemer FW, Marra MD, et al. Articular cartilage in the knee: current MR imaging techniques and applications in clinical practice and research. Radiographics. 2011;31(1):37-61.
6. Campbell KA, Erickson BJ, Saltzman BM, et al. Is local viscosupplementation injection clinically superior to other therapies in the treatment of osteoarthritis of the knee: a systematic review of overlapping meta-analyses. Arthroscopy. 2015;31(10):2036-2045.e14.
7. Saltzman BM, Jain A, Campbell KA, et al. Does the use of platelet-rich plasma at the time of surgery improve clinical outcomes in arthroscopic rotator cuff repair when compared with control cohorts? A systematic review of meta-analyses. Arthroscopy. 2016;32(5):906-918.
8. Gomoll AH, Gillogly SD, Cole BJ, et al. Autologous chondrocyte implantation in the patella: a multicenter experience. Am J Sports Med. 2014;42(5):1074-1081.
9. Meric G, Gracitelli GC, Gortz S, De Young AJ, Bugbee WD. Fresh osteochondral allograft transplantation for bipolar reciprocal osteochondral lesions of the knee. Am J Sports Med. 2015;43(3):709-714.
10. Peterson L, Vasiliadis HS, Brittberg M, Lindahl A. Autologous chondrocyte implantation: a long-term follow-up. Am J Sports Med. 2010;38(6):1117-1124.
11. Minas T, Gomoll AH, Rosenberger R, Royce RO, Bryant T. Increased failure rate of autologous chondrocyte implantation after previous treatment with marrow stimulation techniques. Am J Sports Med. 2009;37(5):902-908.
12. Gracitelli GC, Meric G, Briggs DT, et al. Fresh osteochondral allografts in the knee: comparison of primary transplantation versus transplantation after failure of previous subchondral marrow stimulation. Am J Sports Med. 2015;43(4):885-891.
13. Anderson CN, Magnussen RA, Block JJ, Anderson AF, Spindler KP. Operative fixation of chondral loose bodies in osteochondritis dissecans in the knee: a report of 5 cases. Orthop J Sports Med. 2013;1(2):2325967113496546.
14. Kreuz PC, Steinwachs MR, Erggelet C, et al. Results after microfracture of full-thickness chondral defects in different compartments in the knee. Osteoarthritis Cartilage. 2006;14(11):1119-1125.
15. Vasiliadis HS, Lindahl A, Georgoulis AD, Peterson L. Malalignment and cartilage lesions in the patellofemoral joint treated with autologous chondrocyte implantation. Knee Surg Sports Traumatol Arthrosc. 2011;19(3):452-457.
16. Astur DC, Arliani GG, Binz M, et al. Autologous osteochondral transplantation for treating patellar chondral injuries: evaluation, treatment, and outcomes of a two-year follow-up study. J Bone Joint Surg Am. 2014;96(10):816-823.
17. Hoffman JK, Geraghty S, Protzman NM. Articular cartilage repair using marrow simulation augmented with a viable chondral allograft: 9-month postoperative histological evaluation. Case Rep Orthop. 2015;2015:617365.
18. Gobbi A, Chaurasia S, Karnatzikos G, Nakamura N. Matrix-induced autologous chondrocyte implantation versus multipotent stem cells for the treatment of large patellofemoral chondral lesions: a nonrandomized prospective trial. Cartilage. 2015;6(2):82-97.
19. Farr J, Tabet SK, Margerrison E, Cole BJ. Clinical, radiographic, and histological outcomes after cartilage repair with particulated juvenile articular cartilage: a 2-year prospective study. Am J Sports Med. 2014;42(6):1417-1425.
20. Tompkins M, Hamann JC, Diduch DR, et al. Preliminary results of a novel single-stage cartilage restoration technique: particulated juvenile articular cartilage allograft for chondral defects of the patella. Arthroscopy. 2013;29(10):1661-1670.
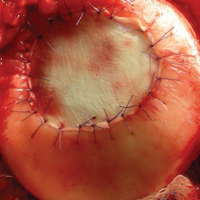
Ribociclib: another CDK inhibitor hits the mark in breast cancer
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
Approval makes olaratumab the first first-line treatment option for soft tissue sarcoma in more than 40 years
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.