User login
A progressive scalp lesion
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 47-YEAR-OLD AFRICAN AMERICAN WOMAN sought care at our clinic for multiple progressive scalp lesions. She said that she first noticed the lesions 13 years ago when her children were diagnosed with ringworm on the scalp. At that time, her physician thought that she, too, had tinea capitis, and she was treated with 6 weeks of griseofulvin. The lesions persisted, however.
She told us that the lesions were nonpruritic and that she didn’t have any other symptoms. The patient did not have a history of trauma or exposure of chemicals to the scalp, and she was not taking any prescription or over-the-counter medications.
Examination of her scalp revealed scattered irregularly shaped, nontender lesions that were centrally hypopigmented and peripherally hyperpigmented. She also had scarring and hair loss (FIGURE 1). She had no other lesions on her body.
FIGURE 1
Irregularly shaped scalp lesions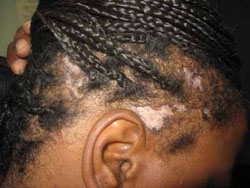
Our 47-year-old patient had multiple scattered scalp lesions that were nontender and centrally hypopigmented. scarring, alopecia, and surrounding areas of hyperpigmentation were also visible.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU MANAGE THIS CONDITION?
Diagnosis: Discoid lupus erythematosus
A punch biopsy and tissue pathology confirmed that our patient had discoid lupus erythematosus (DLE).
DLE is a common type of cutaneous lupus that is chronic and is typically associated with atrophy and scarring of the skin. The primary discoid lesion is a discrete erythematous papule or plaque with adherent scaling, follicular plugging, atrophic scarring, central hypopigmentation, and hyperpigmented borders.
DLE is a dermatosis that is localized in 80% of patients and occurs mainly on sun-exposed areas of the skin, such as the scalp, face, and ears. In 20% of cases it occurs on the extremities and upper trunk. Women are affected more than men, and it can affect any age group, although it is more common in individuals between the ages of 20 and 40.1
The etiology of DLE is unknown. What we do know is that less than 5% of patients with DLE eventually end up with systemic lupus erythematosus (SLE), while up to 25% of patients with SLE go on to develop chronic discoid lesions.2
Abnormalities in serology are not common in DLE. About 20% of patients with DLE show positive antinuclear antibody titers when tested. This is the case in the presence of widespread disease more so than in localized DLE. The presence of antinative (antidouble-stranded) or anti-Smith antibodies is usually suggestive of systemic symptoms and occurs in 5% of cases.3
Several conditions mimic DLE
The differential diagnosis for DLE includes the following:
Lichen plano pilaris. This condition has a predilection to the crown of the head and the frontal central region of the scalp; it is also associated with bilateral eyebrow hair loss. Patients may complain of pruritus, localized tenderness, and a burning sensation. The etiology is unknown; it is most often seen in middle-aged women with a chronic, progressive clinical course.4
Alopecia areata. Patients suddenly lose hair in patches. The hair grows back, and then falls out again. This asymptomatic condition is a tissue-restricted autoimmune disease of the hair follicle that most commonly occurs among children and young adults.5
Dissecting scalp cellulitis. This is a suppurating and cicatrizing disease of the scalp of unknown etiology. It typically affects the scalp vertex and occipital region, and is most common among young black men. The erythematous papules eventually discharge seropurulent material and form underlying intercommunicating sinuses with eventual scarring.6 Patients are likely to complain of pain and pruritus.
Tinea capitis. As noted earlier, this is commonly referred to as “ringworm” and is of a fungal, infectious etiology that typically affects children. Permanent scarring and alopecia are common in affected areas, and patients complain of itching and burning.
Diagnosis can be made on clinical grounds
While a clinical diagnosis of DLE can be made, a tissue biopsy of a new inflamed site is confirmatory. Histopathologic findings show hyperkeratosis, follicular plugging, thickening of the basement membrane, atrophic epidermis, and dermal perifollicular and periappendageal lymphocytic inflammatory infiltrate.
Direct immunofluorescence of lesions shows granular immunoglobulin and complement deposition at the dermal-epidermal junction.7,8
Early treatment is key
Early treatment may be helpful in preventing permanent scarring. Therapeutic options commonly used are oral antimalarials (strength of recommendation [SOR]: A) and topical (SOR: A) or intralesional (SOR: B) corticosteroids.9 Other topical agents, such as calcineurin inhibitors, retinoids, and imiquimod, have been found to be helpful in some cases.
Alternative systemic agents that appear to be useful include methotrexate, azathioprine, thalidomide, dapsone, and mycophenolate mofetil. Patients should be advised to avoid the sun and wear broad-spectrum sunscreen.7,10
Finally, the patient sees some improvement
We discussed the risks and benefits of the various treatments, and our patient elected to start hydroxychloroquine (Plaquenil), 200 to 400 mg/day orally (not to exceed 6.5 mg/kg per day). We referred her for a baseline ophthalmological exam and stressed that she needed a repeat exam every 6 to 12 months while she remained on the hydroxychloroquine. We also referred her to a rheumatologist.
After 4 months of treatment, she showed some improvement (FIGURE 2), with no side effects from the medication. The patient was subsequently lost to follow-up.
FIGURE 2
A visible improvement
After 4 months of treatment with hydroxychloroquine, the patient’s scalp lesions improved and there was evidence of hair growth.
CORRESPONDENCE: Ahunna Ahiarah, MD, UB Family Medicine, 1315 Jefferson Avenue, Buffalo, NY 14208; [email protected]
1. Tlacuilo-Parra A, Guevara-Gutierrez E, Gutierrez-Murillo F, et al. Pimecrolimus 1% cream for the treatment of discoid lupus erythematosus. Rheumatology. 2005;44:1564-1568.
2. Wouters CH, Diegenant C, Ceuppens JL, et al. The circulating lymphocyte profiles in patients with discoid lupus erythematosus and systemic lupus erythematosus suggest a pathogenetic relationship. Br J Dermatol. 2004;150:693-700.
3. Callen JP. Chronic cutaneous lupus erythematosus. Clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416.
4. Tandon YK, Somani N, Cevasco NC, et al. A histologic review of 27 patients with lichen planopilaris. J Am Acad Dermatol. 2008;59:91-98.
5. Gilhar A, Kalish RS. Alopecia areata: a tissue specific autoimmune disease of the hair follicle. Autoimmun Rev. 2006;5:64-69.
6. Monroe M, Crutchfield C. Dissecting cellulitis of the scalp. Dermatol Nurs. 2005;17:208.-
7. Callen JP. Collagen vascular diseases. J Am Acad Dermatol. 2004;51:427-439.
8. Lee LA. Lupus erythematosus. In: Sams WM Jr, Lynch PJ, eds. Principles and Practice of Dermatology. New York: Churchill Livingstone; 1996:581–598.
9. Callen JP. Update on the management of cutaneous lupus erythematosus. Br J Dermatol. 2004;151:731-736.
10. Jessop S, Whitelaw D, Jordaan F. Drugs for discoid lupus erythematosus. Cochrane Database Syst Rev. 2000;(2):CD002954.-
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 47-YEAR-OLD AFRICAN AMERICAN WOMAN sought care at our clinic for multiple progressive scalp lesions. She said that she first noticed the lesions 13 years ago when her children were diagnosed with ringworm on the scalp. At that time, her physician thought that she, too, had tinea capitis, and she was treated with 6 weeks of griseofulvin. The lesions persisted, however.
She told us that the lesions were nonpruritic and that she didn’t have any other symptoms. The patient did not have a history of trauma or exposure of chemicals to the scalp, and she was not taking any prescription or over-the-counter medications.
Examination of her scalp revealed scattered irregularly shaped, nontender lesions that were centrally hypopigmented and peripherally hyperpigmented. She also had scarring and hair loss (FIGURE 1). She had no other lesions on her body.
FIGURE 1
Irregularly shaped scalp lesions
Our 47-year-old patient had multiple scattered scalp lesions that were nontender and centrally hypopigmented. scarring, alopecia, and surrounding areas of hyperpigmentation were also visible.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU MANAGE THIS CONDITION?
Diagnosis: Discoid lupus erythematosus
A punch biopsy and tissue pathology confirmed that our patient had discoid lupus erythematosus (DLE).
DLE is a common type of cutaneous lupus that is chronic and is typically associated with atrophy and scarring of the skin. The primary discoid lesion is a discrete erythematous papule or plaque with adherent scaling, follicular plugging, atrophic scarring, central hypopigmentation, and hyperpigmented borders.
DLE is a dermatosis that is localized in 80% of patients and occurs mainly on sun-exposed areas of the skin, such as the scalp, face, and ears. In 20% of cases it occurs on the extremities and upper trunk. Women are affected more than men, and it can affect any age group, although it is more common in individuals between the ages of 20 and 40.1
The etiology of DLE is unknown. What we do know is that less than 5% of patients with DLE eventually end up with systemic lupus erythematosus (SLE), while up to 25% of patients with SLE go on to develop chronic discoid lesions.2
Abnormalities in serology are not common in DLE. About 20% of patients with DLE show positive antinuclear antibody titers when tested. This is the case in the presence of widespread disease more so than in localized DLE. The presence of antinative (antidouble-stranded) or anti-Smith antibodies is usually suggestive of systemic symptoms and occurs in 5% of cases.3
Several conditions mimic DLE
The differential diagnosis for DLE includes the following:
Lichen plano pilaris. This condition has a predilection to the crown of the head and the frontal central region of the scalp; it is also associated with bilateral eyebrow hair loss. Patients may complain of pruritus, localized tenderness, and a burning sensation. The etiology is unknown; it is most often seen in middle-aged women with a chronic, progressive clinical course.4
Alopecia areata. Patients suddenly lose hair in patches. The hair grows back, and then falls out again. This asymptomatic condition is a tissue-restricted autoimmune disease of the hair follicle that most commonly occurs among children and young adults.5
Dissecting scalp cellulitis. This is a suppurating and cicatrizing disease of the scalp of unknown etiology. It typically affects the scalp vertex and occipital region, and is most common among young black men. The erythematous papules eventually discharge seropurulent material and form underlying intercommunicating sinuses with eventual scarring.6 Patients are likely to complain of pain and pruritus.
Tinea capitis. As noted earlier, this is commonly referred to as “ringworm” and is of a fungal, infectious etiology that typically affects children. Permanent scarring and alopecia are common in affected areas, and patients complain of itching and burning.
Diagnosis can be made on clinical grounds
While a clinical diagnosis of DLE can be made, a tissue biopsy of a new inflamed site is confirmatory. Histopathologic findings show hyperkeratosis, follicular plugging, thickening of the basement membrane, atrophic epidermis, and dermal perifollicular and periappendageal lymphocytic inflammatory infiltrate.
Direct immunofluorescence of lesions shows granular immunoglobulin and complement deposition at the dermal-epidermal junction.7,8
Early treatment is key
Early treatment may be helpful in preventing permanent scarring. Therapeutic options commonly used are oral antimalarials (strength of recommendation [SOR]: A) and topical (SOR: A) or intralesional (SOR: B) corticosteroids.9 Other topical agents, such as calcineurin inhibitors, retinoids, and imiquimod, have been found to be helpful in some cases.
Alternative systemic agents that appear to be useful include methotrexate, azathioprine, thalidomide, dapsone, and mycophenolate mofetil. Patients should be advised to avoid the sun and wear broad-spectrum sunscreen.7,10
Finally, the patient sees some improvement
We discussed the risks and benefits of the various treatments, and our patient elected to start hydroxychloroquine (Plaquenil), 200 to 400 mg/day orally (not to exceed 6.5 mg/kg per day). We referred her for a baseline ophthalmological exam and stressed that she needed a repeat exam every 6 to 12 months while she remained on the hydroxychloroquine. We also referred her to a rheumatologist.
After 4 months of treatment, she showed some improvement (FIGURE 2), with no side effects from the medication. The patient was subsequently lost to follow-up.
FIGURE 2
A visible improvement
After 4 months of treatment with hydroxychloroquine, the patient’s scalp lesions improved and there was evidence of hair growth.
CORRESPONDENCE: Ahunna Ahiarah, MD, UB Family Medicine, 1315 Jefferson Avenue, Buffalo, NY 14208; [email protected]
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 47-YEAR-OLD AFRICAN AMERICAN WOMAN sought care at our clinic for multiple progressive scalp lesions. She said that she first noticed the lesions 13 years ago when her children were diagnosed with ringworm on the scalp. At that time, her physician thought that she, too, had tinea capitis, and she was treated with 6 weeks of griseofulvin. The lesions persisted, however.
She told us that the lesions were nonpruritic and that she didn’t have any other symptoms. The patient did not have a history of trauma or exposure of chemicals to the scalp, and she was not taking any prescription or over-the-counter medications.
Examination of her scalp revealed scattered irregularly shaped, nontender lesions that were centrally hypopigmented and peripherally hyperpigmented. She also had scarring and hair loss (FIGURE 1). She had no other lesions on her body.
FIGURE 1
Irregularly shaped scalp lesions
Our 47-year-old patient had multiple scattered scalp lesions that were nontender and centrally hypopigmented. scarring, alopecia, and surrounding areas of hyperpigmentation were also visible.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU MANAGE THIS CONDITION?
Diagnosis: Discoid lupus erythematosus
A punch biopsy and tissue pathology confirmed that our patient had discoid lupus erythematosus (DLE).
DLE is a common type of cutaneous lupus that is chronic and is typically associated with atrophy and scarring of the skin. The primary discoid lesion is a discrete erythematous papule or plaque with adherent scaling, follicular plugging, atrophic scarring, central hypopigmentation, and hyperpigmented borders.
DLE is a dermatosis that is localized in 80% of patients and occurs mainly on sun-exposed areas of the skin, such as the scalp, face, and ears. In 20% of cases it occurs on the extremities and upper trunk. Women are affected more than men, and it can affect any age group, although it is more common in individuals between the ages of 20 and 40.1
The etiology of DLE is unknown. What we do know is that less than 5% of patients with DLE eventually end up with systemic lupus erythematosus (SLE), while up to 25% of patients with SLE go on to develop chronic discoid lesions.2
Abnormalities in serology are not common in DLE. About 20% of patients with DLE show positive antinuclear antibody titers when tested. This is the case in the presence of widespread disease more so than in localized DLE. The presence of antinative (antidouble-stranded) or anti-Smith antibodies is usually suggestive of systemic symptoms and occurs in 5% of cases.3
Several conditions mimic DLE
The differential diagnosis for DLE includes the following:
Lichen plano pilaris. This condition has a predilection to the crown of the head and the frontal central region of the scalp; it is also associated with bilateral eyebrow hair loss. Patients may complain of pruritus, localized tenderness, and a burning sensation. The etiology is unknown; it is most often seen in middle-aged women with a chronic, progressive clinical course.4
Alopecia areata. Patients suddenly lose hair in patches. The hair grows back, and then falls out again. This asymptomatic condition is a tissue-restricted autoimmune disease of the hair follicle that most commonly occurs among children and young adults.5
Dissecting scalp cellulitis. This is a suppurating and cicatrizing disease of the scalp of unknown etiology. It typically affects the scalp vertex and occipital region, and is most common among young black men. The erythematous papules eventually discharge seropurulent material and form underlying intercommunicating sinuses with eventual scarring.6 Patients are likely to complain of pain and pruritus.
Tinea capitis. As noted earlier, this is commonly referred to as “ringworm” and is of a fungal, infectious etiology that typically affects children. Permanent scarring and alopecia are common in affected areas, and patients complain of itching and burning.
Diagnosis can be made on clinical grounds
While a clinical diagnosis of DLE can be made, a tissue biopsy of a new inflamed site is confirmatory. Histopathologic findings show hyperkeratosis, follicular plugging, thickening of the basement membrane, atrophic epidermis, and dermal perifollicular and periappendageal lymphocytic inflammatory infiltrate.
Direct immunofluorescence of lesions shows granular immunoglobulin and complement deposition at the dermal-epidermal junction.7,8
Early treatment is key
Early treatment may be helpful in preventing permanent scarring. Therapeutic options commonly used are oral antimalarials (strength of recommendation [SOR]: A) and topical (SOR: A) or intralesional (SOR: B) corticosteroids.9 Other topical agents, such as calcineurin inhibitors, retinoids, and imiquimod, have been found to be helpful in some cases.
Alternative systemic agents that appear to be useful include methotrexate, azathioprine, thalidomide, dapsone, and mycophenolate mofetil. Patients should be advised to avoid the sun and wear broad-spectrum sunscreen.7,10
Finally, the patient sees some improvement
We discussed the risks and benefits of the various treatments, and our patient elected to start hydroxychloroquine (Plaquenil), 200 to 400 mg/day orally (not to exceed 6.5 mg/kg per day). We referred her for a baseline ophthalmological exam and stressed that she needed a repeat exam every 6 to 12 months while she remained on the hydroxychloroquine. We also referred her to a rheumatologist.
After 4 months of treatment, she showed some improvement (FIGURE 2), with no side effects from the medication. The patient was subsequently lost to follow-up.
FIGURE 2
A visible improvement
After 4 months of treatment with hydroxychloroquine, the patient’s scalp lesions improved and there was evidence of hair growth.
CORRESPONDENCE: Ahunna Ahiarah, MD, UB Family Medicine, 1315 Jefferson Avenue, Buffalo, NY 14208; [email protected]
1. Tlacuilo-Parra A, Guevara-Gutierrez E, Gutierrez-Murillo F, et al. Pimecrolimus 1% cream for the treatment of discoid lupus erythematosus. Rheumatology. 2005;44:1564-1568.
2. Wouters CH, Diegenant C, Ceuppens JL, et al. The circulating lymphocyte profiles in patients with discoid lupus erythematosus and systemic lupus erythematosus suggest a pathogenetic relationship. Br J Dermatol. 2004;150:693-700.
3. Callen JP. Chronic cutaneous lupus erythematosus. Clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416.
4. Tandon YK, Somani N, Cevasco NC, et al. A histologic review of 27 patients with lichen planopilaris. J Am Acad Dermatol. 2008;59:91-98.
5. Gilhar A, Kalish RS. Alopecia areata: a tissue specific autoimmune disease of the hair follicle. Autoimmun Rev. 2006;5:64-69.
6. Monroe M, Crutchfield C. Dissecting cellulitis of the scalp. Dermatol Nurs. 2005;17:208.-
7. Callen JP. Collagen vascular diseases. J Am Acad Dermatol. 2004;51:427-439.
8. Lee LA. Lupus erythematosus. In: Sams WM Jr, Lynch PJ, eds. Principles and Practice of Dermatology. New York: Churchill Livingstone; 1996:581–598.
9. Callen JP. Update on the management of cutaneous lupus erythematosus. Br J Dermatol. 2004;151:731-736.
10. Jessop S, Whitelaw D, Jordaan F. Drugs for discoid lupus erythematosus. Cochrane Database Syst Rev. 2000;(2):CD002954.-
1. Tlacuilo-Parra A, Guevara-Gutierrez E, Gutierrez-Murillo F, et al. Pimecrolimus 1% cream for the treatment of discoid lupus erythematosus. Rheumatology. 2005;44:1564-1568.
2. Wouters CH, Diegenant C, Ceuppens JL, et al. The circulating lymphocyte profiles in patients with discoid lupus erythematosus and systemic lupus erythematosus suggest a pathogenetic relationship. Br J Dermatol. 2004;150:693-700.
3. Callen JP. Chronic cutaneous lupus erythematosus. Clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416.
4. Tandon YK, Somani N, Cevasco NC, et al. A histologic review of 27 patients with lichen planopilaris. J Am Acad Dermatol. 2008;59:91-98.
5. Gilhar A, Kalish RS. Alopecia areata: a tissue specific autoimmune disease of the hair follicle. Autoimmun Rev. 2006;5:64-69.
6. Monroe M, Crutchfield C. Dissecting cellulitis of the scalp. Dermatol Nurs. 2005;17:208.-
7. Callen JP. Collagen vascular diseases. J Am Acad Dermatol. 2004;51:427-439.
8. Lee LA. Lupus erythematosus. In: Sams WM Jr, Lynch PJ, eds. Principles and Practice of Dermatology. New York: Churchill Livingstone; 1996:581–598.
9. Callen JP. Update on the management of cutaneous lupus erythematosus. Br J Dermatol. 2004;151:731-736.
10. Jessop S, Whitelaw D, Jordaan F. Drugs for discoid lupus erythematosus. Cochrane Database Syst Rev. 2000;(2):CD002954.-
The Journal of Family Practice ©2010 Dowden Health Media
Rupturing bullae not responding to antibiotics
A 33-year-old African American woman came to the office with a 2-week history of skin lesions and itching. The lesions started with a single blister on her left elbow; numerous other blisters subsequently appeared on her forearm and hands. One week before this visit, she had been given a presumptive diagnosis of bullous impetigo and was treated with cephalexin.
Despite the antibiotics, other lesions soon appeared in the nuchal and breast folds, axillae, and scalp areas. Several had ruptured, producing purulent, malodorous material. She had no known allergies, no medical problems aside from obesity, and no significant family history or recent travels. She denied any illicit drug use and had not been on any medications.
On physical exam, 1 large bulla was seen on the fourth digit of her left hand (Figure 1). The patient was obese, and inspection of the skin folds of her abdomen showed multiple suppurative lesions and erosions where previous bullae were found (Figure 2). No oral or gingival erosions were seen. Labs showed a white blood cell (WBC) count of 10.5 x109L], hemoglobin of 11.0 g/dL, and hemoglobin A1cof 5.5; liver function tests were normal. Gram stain showed no WBC and had rare Gram-positive bacilli. Potassium hydroxide prep of a skin lesion scraping showed no fungal elements. A herpes culture was performed along with a punch biopsy.
FIGURE 1
Bulla on the index finger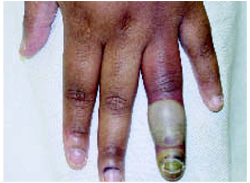
FIGURE 2
Multiple bullae on the trunk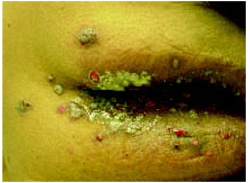
What is the diagnosis?
Differential diagnosis
Many diseases manifest with bullae/vesicles. Workup should begin with a complete history and physical exam. A skin biopsy may be needed to make a definitive diagnosis.
Herpes zoster typically manifests with clustered pruritic vesicular lesions on a red base that follow a dermatomal distribution. Pemphigus vulgaris appears with flaccid blisters, erosions, and tend to have oral mucosal lesions. A positive Nikolsky’s sign is characteristic of pemphigus vulgaris. Bullous impetigo appears with scattered lesions of erythema and macules, progressing to thin roofed bullae and subsequently to “honey-crusted” lesions. In toxic epidermal necrolysis, the bullae are widespread and lead to sloughing of the skin. Pyoderma gangrenosum has ulcer formation preceded by pustules that typically expand rapidly to approximately 20 cm. These ulcers have necrotic bluish edges.
Diagnostic test results
The patient’s herpes culture was negative. Fortunately, the punch biopsy was sent for direct immunofluorescence. Direct immunofluorescence showed positive staining with immunoglobulin (Ig) G in the intercellular regions of the epidermis and no staining with IgA, IgM, C3, or fibrinogen. Hematoxylin and eosin-stained sections showed suprabasal blistering containing neutrophils and a few eosinophils. These results are consistent with pemphigus vulgaris.
Diagnosis: pemphigus vulgaris
Pemphigus vulgaris is blistering disease involving the skin and mucous membranes, with severe morbidity and occasional mortality. Prior to the development of effective treatment, the disease was 75% fatal within 5 years.1
Its prevalence is equal among men and women, with a rate of occurrence of 0.5/100,000 people per year. The average age of onset is in the fifth and sixth decades of life, but there is wide variation in age. It has multiple causes and risk factors (Table).
The clinical manifestation of pemphigus typically features mucocutaneous blisters followed by erosions. Often they appear first in mucous membranes and may not appear cutaneously until several months later.2 The skin lesions are painful flaccid blisters that may appear anywhere. A characteristic finding of pemphigus vulgaris is the Nikolsky sign, in which lateral stress applied to perilesional skin causes an expansion of the blistering.
There are 2 major subtypes of pemphigus. Pemphigus vulgaris has blisters extending to the deep epidermis, and pemphigus foliaceus has more superficial involvement of the epidermis. Pemphigus can also be seen in paraneoplastics syndromes.
In pemphigus, the epidermal cells lose normal cell contacts and form a blister. Electron microscopy shows desmosomal abnormalities at desmosomal junctions. It is these junctions that guarantee the integrity of the epithelium. Direct immunofluorescence shows IgG deposition in intercellular spaces.
TABLECauses and risk factors for pemphigus vulgaris
| Penicillamine |
| Captopril |
| Rifampin |
| Phenol-based drugs |
| Amide-based drugs |
| Foods: garlic, leek, onion |
| Pregnancy |
| Pesticide exposure |
| Herpes virus infection |
| Cytomegalovirus infection |
| Epstein-Barr virus infectyion |
| Adapted from: Benner et al 2003.4 |
Treatment: Steroids and adjuvant drugs
Inducing remission is the main goal of therapy for pemphigus vulgaris. Epidemiological studies have shown up to a 75% remission rate 10 years after initial diagnosis.3 Corticosteroids are the preferred therapy for the management of pemphigus vulgaris (based on expert opinion). Also, adjuvant drugs such as azathioprine and cyclophosphamide are commonly used in combination with corticosteroids, with the aim of increasing efficacy and of having a steroid-sparing action (level of evidence: 5, expert opinion).
A tailored dosing schedule of steroids has been advocated according to the severity of the disease. Mild disease can be treated with an initial prednisone dose of 40 to 60 mg/d; in severe cases, 60 to 100 mg/d. Other agents that have been used to treat pemphigus vulgaris are intramuscular gold, dapsone, and intravenous immunoglobulin.
Patient outcome
The patient was treated with oral prednisone starting at 60 mg/d and her skin began to clear. A full course of oral prednisone was continued and tapered over 1 month. Currently, she remains in remission off all medications.
Conclusion
Pemphigus vulgaris is a potentially life-threatening condition that must be recognized and treated promptly. With a lack of large-scale controlled studies, the diagnosis and management of pemphigus vulgaris has based on expert opinion.3 Complications such as superimposed infection of the lesions, cellulitis, and sepsis can occur. Its association with underlying neoplasm, thymomas, myasthenia gravis, and other autoimmune disorders warrants consideration for additional workup when indicated.
Correspondence
John Sauret, MD, Department of Family Medicine, State University of New York at Buffalo, 150 Family Medical Modular Complex, Buffalo, NY 14214-3013. E-mail: [email protected].
1. Sami N, Ahmed AR. Dual diagnosis of pemphigus and pemphigoid. Retrospective review of 30 cases in the literature. Dermatol 2001;202:293-301.
2. Ahmed AR, Graham J. Pemphigus: current concepts. Ann Intern Med 1980;92:396-405.
3. Harman KE, Albert S, Black MM. Guidelines for the management of pemphigus vulgaris. Br J Dermatol 2003;149:926-937.
4. Benner S, Mashiah J, Tamir E, Goldberg I, Wohl Y. PEMPHIGUS: an acronym for a disease with multiple etiologies. Skinmed 2003;2:163-167.
A 33-year-old African American woman came to the office with a 2-week history of skin lesions and itching. The lesions started with a single blister on her left elbow; numerous other blisters subsequently appeared on her forearm and hands. One week before this visit, she had been given a presumptive diagnosis of bullous impetigo and was treated with cephalexin.
Despite the antibiotics, other lesions soon appeared in the nuchal and breast folds, axillae, and scalp areas. Several had ruptured, producing purulent, malodorous material. She had no known allergies, no medical problems aside from obesity, and no significant family history or recent travels. She denied any illicit drug use and had not been on any medications.
On physical exam, 1 large bulla was seen on the fourth digit of her left hand (Figure 1). The patient was obese, and inspection of the skin folds of her abdomen showed multiple suppurative lesions and erosions where previous bullae were found (Figure 2). No oral or gingival erosions were seen. Labs showed a white blood cell (WBC) count of 10.5 x109L], hemoglobin of 11.0 g/dL, and hemoglobin A1cof 5.5; liver function tests were normal. Gram stain showed no WBC and had rare Gram-positive bacilli. Potassium hydroxide prep of a skin lesion scraping showed no fungal elements. A herpes culture was performed along with a punch biopsy.
FIGURE 1
Bulla on the index finger
FIGURE 2
Multiple bullae on the trunk
What is the diagnosis?
Differential diagnosis
Many diseases manifest with bullae/vesicles. Workup should begin with a complete history and physical exam. A skin biopsy may be needed to make a definitive diagnosis.
Herpes zoster typically manifests with clustered pruritic vesicular lesions on a red base that follow a dermatomal distribution. Pemphigus vulgaris appears with flaccid blisters, erosions, and tend to have oral mucosal lesions. A positive Nikolsky’s sign is characteristic of pemphigus vulgaris. Bullous impetigo appears with scattered lesions of erythema and macules, progressing to thin roofed bullae and subsequently to “honey-crusted” lesions. In toxic epidermal necrolysis, the bullae are widespread and lead to sloughing of the skin. Pyoderma gangrenosum has ulcer formation preceded by pustules that typically expand rapidly to approximately 20 cm. These ulcers have necrotic bluish edges.
Diagnostic test results
The patient’s herpes culture was negative. Fortunately, the punch biopsy was sent for direct immunofluorescence. Direct immunofluorescence showed positive staining with immunoglobulin (Ig) G in the intercellular regions of the epidermis and no staining with IgA, IgM, C3, or fibrinogen. Hematoxylin and eosin-stained sections showed suprabasal blistering containing neutrophils and a few eosinophils. These results are consistent with pemphigus vulgaris.
Diagnosis: pemphigus vulgaris
Pemphigus vulgaris is blistering disease involving the skin and mucous membranes, with severe morbidity and occasional mortality. Prior to the development of effective treatment, the disease was 75% fatal within 5 years.1
Its prevalence is equal among men and women, with a rate of occurrence of 0.5/100,000 people per year. The average age of onset is in the fifth and sixth decades of life, but there is wide variation in age. It has multiple causes and risk factors (Table).
The clinical manifestation of pemphigus typically features mucocutaneous blisters followed by erosions. Often they appear first in mucous membranes and may not appear cutaneously until several months later.2 The skin lesions are painful flaccid blisters that may appear anywhere. A characteristic finding of pemphigus vulgaris is the Nikolsky sign, in which lateral stress applied to perilesional skin causes an expansion of the blistering.
There are 2 major subtypes of pemphigus. Pemphigus vulgaris has blisters extending to the deep epidermis, and pemphigus foliaceus has more superficial involvement of the epidermis. Pemphigus can also be seen in paraneoplastics syndromes.
In pemphigus, the epidermal cells lose normal cell contacts and form a blister. Electron microscopy shows desmosomal abnormalities at desmosomal junctions. It is these junctions that guarantee the integrity of the epithelium. Direct immunofluorescence shows IgG deposition in intercellular spaces.
TABLECauses and risk factors for pemphigus vulgaris
| Penicillamine |
| Captopril |
| Rifampin |
| Phenol-based drugs |
| Amide-based drugs |
| Foods: garlic, leek, onion |
| Pregnancy |
| Pesticide exposure |
| Herpes virus infection |
| Cytomegalovirus infection |
| Epstein-Barr virus infectyion |
| Adapted from: Benner et al 2003.4 |
Treatment: Steroids and adjuvant drugs
Inducing remission is the main goal of therapy for pemphigus vulgaris. Epidemiological studies have shown up to a 75% remission rate 10 years after initial diagnosis.3 Corticosteroids are the preferred therapy for the management of pemphigus vulgaris (based on expert opinion). Also, adjuvant drugs such as azathioprine and cyclophosphamide are commonly used in combination with corticosteroids, with the aim of increasing efficacy and of having a steroid-sparing action (level of evidence: 5, expert opinion).
A tailored dosing schedule of steroids has been advocated according to the severity of the disease. Mild disease can be treated with an initial prednisone dose of 40 to 60 mg/d; in severe cases, 60 to 100 mg/d. Other agents that have been used to treat pemphigus vulgaris are intramuscular gold, dapsone, and intravenous immunoglobulin.
Patient outcome
The patient was treated with oral prednisone starting at 60 mg/d and her skin began to clear. A full course of oral prednisone was continued and tapered over 1 month. Currently, she remains in remission off all medications.
Conclusion
Pemphigus vulgaris is a potentially life-threatening condition that must be recognized and treated promptly. With a lack of large-scale controlled studies, the diagnosis and management of pemphigus vulgaris has based on expert opinion.3 Complications such as superimposed infection of the lesions, cellulitis, and sepsis can occur. Its association with underlying neoplasm, thymomas, myasthenia gravis, and other autoimmune disorders warrants consideration for additional workup when indicated.
Correspondence
John Sauret, MD, Department of Family Medicine, State University of New York at Buffalo, 150 Family Medical Modular Complex, Buffalo, NY 14214-3013. E-mail: [email protected].
A 33-year-old African American woman came to the office with a 2-week history of skin lesions and itching. The lesions started with a single blister on her left elbow; numerous other blisters subsequently appeared on her forearm and hands. One week before this visit, she had been given a presumptive diagnosis of bullous impetigo and was treated with cephalexin.
Despite the antibiotics, other lesions soon appeared in the nuchal and breast folds, axillae, and scalp areas. Several had ruptured, producing purulent, malodorous material. She had no known allergies, no medical problems aside from obesity, and no significant family history or recent travels. She denied any illicit drug use and had not been on any medications.
On physical exam, 1 large bulla was seen on the fourth digit of her left hand (Figure 1). The patient was obese, and inspection of the skin folds of her abdomen showed multiple suppurative lesions and erosions where previous bullae were found (Figure 2). No oral or gingival erosions were seen. Labs showed a white blood cell (WBC) count of 10.5 x109L], hemoglobin of 11.0 g/dL, and hemoglobin A1cof 5.5; liver function tests were normal. Gram stain showed no WBC and had rare Gram-positive bacilli. Potassium hydroxide prep of a skin lesion scraping showed no fungal elements. A herpes culture was performed along with a punch biopsy.
FIGURE 1
Bulla on the index finger
FIGURE 2
Multiple bullae on the trunk
What is the diagnosis?
Differential diagnosis
Many diseases manifest with bullae/vesicles. Workup should begin with a complete history and physical exam. A skin biopsy may be needed to make a definitive diagnosis.
Herpes zoster typically manifests with clustered pruritic vesicular lesions on a red base that follow a dermatomal distribution. Pemphigus vulgaris appears with flaccid blisters, erosions, and tend to have oral mucosal lesions. A positive Nikolsky’s sign is characteristic of pemphigus vulgaris. Bullous impetigo appears with scattered lesions of erythema and macules, progressing to thin roofed bullae and subsequently to “honey-crusted” lesions. In toxic epidermal necrolysis, the bullae are widespread and lead to sloughing of the skin. Pyoderma gangrenosum has ulcer formation preceded by pustules that typically expand rapidly to approximately 20 cm. These ulcers have necrotic bluish edges.
Diagnostic test results
The patient’s herpes culture was negative. Fortunately, the punch biopsy was sent for direct immunofluorescence. Direct immunofluorescence showed positive staining with immunoglobulin (Ig) G in the intercellular regions of the epidermis and no staining with IgA, IgM, C3, or fibrinogen. Hematoxylin and eosin-stained sections showed suprabasal blistering containing neutrophils and a few eosinophils. These results are consistent with pemphigus vulgaris.
Diagnosis: pemphigus vulgaris
Pemphigus vulgaris is blistering disease involving the skin and mucous membranes, with severe morbidity and occasional mortality. Prior to the development of effective treatment, the disease was 75% fatal within 5 years.1
Its prevalence is equal among men and women, with a rate of occurrence of 0.5/100,000 people per year. The average age of onset is in the fifth and sixth decades of life, but there is wide variation in age. It has multiple causes and risk factors (Table).
The clinical manifestation of pemphigus typically features mucocutaneous blisters followed by erosions. Often they appear first in mucous membranes and may not appear cutaneously until several months later.2 The skin lesions are painful flaccid blisters that may appear anywhere. A characteristic finding of pemphigus vulgaris is the Nikolsky sign, in which lateral stress applied to perilesional skin causes an expansion of the blistering.
There are 2 major subtypes of pemphigus. Pemphigus vulgaris has blisters extending to the deep epidermis, and pemphigus foliaceus has more superficial involvement of the epidermis. Pemphigus can also be seen in paraneoplastics syndromes.
In pemphigus, the epidermal cells lose normal cell contacts and form a blister. Electron microscopy shows desmosomal abnormalities at desmosomal junctions. It is these junctions that guarantee the integrity of the epithelium. Direct immunofluorescence shows IgG deposition in intercellular spaces.
TABLECauses and risk factors for pemphigus vulgaris
| Penicillamine |
| Captopril |
| Rifampin |
| Phenol-based drugs |
| Amide-based drugs |
| Foods: garlic, leek, onion |
| Pregnancy |
| Pesticide exposure |
| Herpes virus infection |
| Cytomegalovirus infection |
| Epstein-Barr virus infectyion |
| Adapted from: Benner et al 2003.4 |
Treatment: Steroids and adjuvant drugs
Inducing remission is the main goal of therapy for pemphigus vulgaris. Epidemiological studies have shown up to a 75% remission rate 10 years after initial diagnosis.3 Corticosteroids are the preferred therapy for the management of pemphigus vulgaris (based on expert opinion). Also, adjuvant drugs such as azathioprine and cyclophosphamide are commonly used in combination with corticosteroids, with the aim of increasing efficacy and of having a steroid-sparing action (level of evidence: 5, expert opinion).
A tailored dosing schedule of steroids has been advocated according to the severity of the disease. Mild disease can be treated with an initial prednisone dose of 40 to 60 mg/d; in severe cases, 60 to 100 mg/d. Other agents that have been used to treat pemphigus vulgaris are intramuscular gold, dapsone, and intravenous immunoglobulin.
Patient outcome
The patient was treated with oral prednisone starting at 60 mg/d and her skin began to clear. A full course of oral prednisone was continued and tapered over 1 month. Currently, she remains in remission off all medications.
Conclusion
Pemphigus vulgaris is a potentially life-threatening condition that must be recognized and treated promptly. With a lack of large-scale controlled studies, the diagnosis and management of pemphigus vulgaris has based on expert opinion.3 Complications such as superimposed infection of the lesions, cellulitis, and sepsis can occur. Its association with underlying neoplasm, thymomas, myasthenia gravis, and other autoimmune disorders warrants consideration for additional workup when indicated.
Correspondence
John Sauret, MD, Department of Family Medicine, State University of New York at Buffalo, 150 Family Medical Modular Complex, Buffalo, NY 14214-3013. E-mail: [email protected].
1. Sami N, Ahmed AR. Dual diagnosis of pemphigus and pemphigoid. Retrospective review of 30 cases in the literature. Dermatol 2001;202:293-301.
2. Ahmed AR, Graham J. Pemphigus: current concepts. Ann Intern Med 1980;92:396-405.
3. Harman KE, Albert S, Black MM. Guidelines for the management of pemphigus vulgaris. Br J Dermatol 2003;149:926-937.
4. Benner S, Mashiah J, Tamir E, Goldberg I, Wohl Y. PEMPHIGUS: an acronym for a disease with multiple etiologies. Skinmed 2003;2:163-167.
1. Sami N, Ahmed AR. Dual diagnosis of pemphigus and pemphigoid. Retrospective review of 30 cases in the literature. Dermatol 2001;202:293-301.
2. Ahmed AR, Graham J. Pemphigus: current concepts. Ann Intern Med 1980;92:396-405.
3. Harman KE, Albert S, Black MM. Guidelines for the management of pemphigus vulgaris. Br J Dermatol 2003;149:926-937.
4. Benner S, Mashiah J, Tamir E, Goldberg I, Wohl Y. PEMPHIGUS: an acronym for a disease with multiple etiologies. Skinmed 2003;2:163-167.