User login
Rapid Deterioration and Death Caused by Bilateral Phlegmasia Cerulea Dolens
Phlegmasia cerulea dolens (PCD), a life-threatening complication of deep venous thrombosis (DVT), is characterized by massive iliofemoral thrombus that extends to the collateral veins, leading to fluid sequestration and elevated compartment pressures that ultimately compromise arterial flow. Phlegmasia cerulea dolens can rapidly progress to compartment syndrome and gangrene.1,2 The affected limbs of patients with PCD can be hypoxic and appear purple in color due to substantial lack of blood flow, with diminished or absent pulses. Risk factors for PCD include malignancy, hypercoagulable states, venous stasis, contraceptive agents, inferior vena cava (IVC) filter, aneurysm, history of DVT, trauma, heparin-induced thrombocytopenia, femoral vein catheterization, antiphospholipid syndrome, or pregnancy.3-6 Failure to treat PCD early and aggressively carries an amputation rate of up to 50% and a mortality rate of up to 40%.4
We present the case of a patient with PCD, whose condition rapidly deteriorated despite prompt diagnosis and treatment.
Case
A 58-year-old woman presented to the ED with a 1-day history of back and leg pain and difficulty walking. When asked about the severity of her pain, she rated her leg pain at 10 on a scale of 0 to 10. The patient’s history was significant for DVT and pulmonary embolism (PE), for which a Greenfield IVC had been placed and for which she was on prophylactic warfarin therapy. The patient stated that she had been taken off warfarin several weeks prior to presentation in preparation for an elective colonoscopy and dental procedure, but had restarted the warfarin therapy 2 days prior to presentation. She had no history of diabetes mellitus or renal disease.
Initial vital signs at presentation were: blood pressure, 120/91 mm Hg; heart rate, 110 beats/min; respiratory rate, 24 breaths/min; and temperature, 96.6°F. Oxygen saturation was 100% on a nonrebreather mask.
On examination, the patient was alert and oriented to person, time, and place, but appeared dyspneic. An electrocardiogram revealed sinus tachycardia. On physical examination, lung sounds were clear to auscultation bilaterally with good air movement, and the abdomen was soft and nontender with normal bowel sounds. The dorsalis pedis and posterior tibial pulses were absent bilaterally, lower extremity capillary refill was 3 seconds, and the legs appeared mildly erythematous and cool to touch. No speech or neurological deficits were present.
Laboratory evaluation was remarkable for metabolic acidosis, venous pH, 7.11; bicarbonate, 11.7; partial pressure of carbon dioxide, 37.6; lactic acid, 8.8 mEq/L leukocytosis, 24,900 u/L; glucose, 296 mg/dL; creatinine, 2.41 mg/dL; and international normalized ratio, 1.36.
Before additional laboratory studies and imaging could be obtained, the patient developed altered mental status, hypotension, and paralysis of the lower extremities. She was orally intubated for airway protection and was given a total of 4 L of normal saline intravenously (IV) for hypotension and acidosis; sodium bicarbonate for metabolic acidosis; norepinephrine for hypotension; fentanyl for pain; and ondansetron for nausea. A central line and arterial line were placed for administering medication and hemodynamic monitoring.
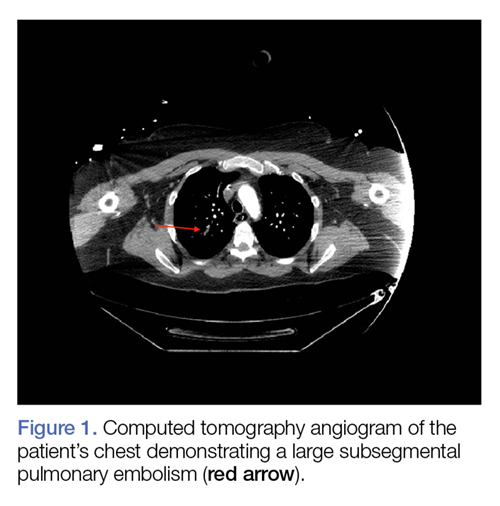
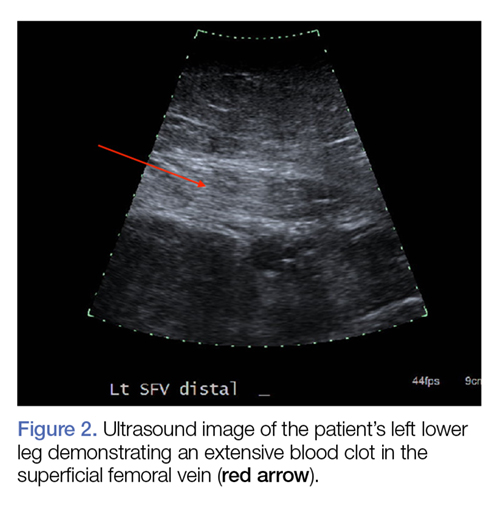
Computed tomography (CT) angiography of the chest, abdomen, and pelvis demonstrated multiple subsegmental bilateral PE with no arterial pathology (Figure 1). Beside ultrasound revealed extensive bilateral DVTs involving the superficial and common femoral veins (Figure 2). The patient’s bilateral DVTs, arterial compromise, and leg cyanosis led to the diagnosis of PCD.
Critical care and vascular surgery services were consulted, and the patient was admitted to the intensive care unit. Since the patient was too unstable to undergo thrombectomy, she was given IV tissue plasminogen activator. Despite aggressive pharmacological treatment, the patient’s condition continued to deteriorate. On hospital day 2, the patient’s family changed the patient’s code status to do-not-resuscitate/comfort-care only; she died shortly thereafter.
Discussion
This case illustrates the severity and complications of PCD and the rapidity with which this condition can deteriorate. At the time of ED presentation, the patient had already developed bilateral PCD, metabolic acidosis, and bilateral PE. Unfortunately, due to decreased venous return, decreased cardiac output, and severe shock, she quickly became unstable and progressed rapidly to multisystem organ failure leading to death.
Risk Factors
A prior patient history DVT and an IVC filter are both significant risk factors for the progression of DVT to PCD;3,6 however, in this case, IVC filter failed to prevent emboli from reaching the lungs. Extensive thrombi led to severely decreased venous return and cardiac output, causing life-threatening shock, ischemia, and metabolic acidosis. A lactic acid level taken on hospital day 2 was elevated at 19 mEq/L, demonstrating the severity, morbidity, and progression of PCD.
Signs and Symptoms
The three cardinal signs that lead to a clinical diagnosis of PCD are edema, pain, and violaceous discoloration or skin mottling.3 Although most commonly found in the lower extremity, PCD can occur in any limb due to occlusion of venous outflow.7 Unfortunately, a clinical diagnosis of PCD is not often made until the venous occlusion becomes severe enough to impair arterial flow and cause venous gangrene, tissue ischemia, shock, and death.8
Although IVC filters are designed to prevent life-threatening PE, there are risk factors associated with their use. Whether placed recently or decades prior, urgent investigation, such as immediate CT scan, should be undertaken in patients presenting with DVT-like symptoms who have a history of an IVC filter, to ensure the filter has not shifted from its original placement and is not occluding the IVC.
Conclusion
Phlegmasia cerulea dolens is an uncommon vascular emergency, but one that has a high-morbidity and high-mortality rate. This case demonstrates the importance of early diagnosis, aggressive treatment, and the severe complications that can develop in PCD.
There are cases in the literature where patients diagnosed with PCD had a successful outcome with pharmacological or surgical intervention such as thrombectomy. Treatment for PCD is most effective when instituted early in onset. As seen in our patient, the tendency for rapid deterioration in PCD can limit potentially lifesaving therapeutic options, decreasing the chances of a successful outcome. Emergency physicians, therefore, must be aware of the high-mortality rate associated with this disorder and the possibility of rapid progression from stable to critical condition.
1. Kesieme E, Kesieme C, Jebbin N, Irekpita E, Dongo A. Deep vein thrombosis: a clinical review. J Blood Med. 2011;2:59-69. doi:10.2147/JBM.S19009.
2. Bhatt S, Wehbe C, Dogra VS. Phlegmasia cerulea dolens. J Clin Ultrasound. 2007;35(7):401-404. doi:10.1002/jcu.20317.
3. Maiti A, Das A, Smith DT. Phlegmasia cerulean dolens. Postgrad Med J. 2016;pii: postgradmedj-2016-134185. doi:10.1136/postgradmedj-2016-134185.
4. Abdul W, Hickey B, Wilson C. Lower extremity compartment syndrome in the setting of iliofemoral deep vein thrombosis, phlegmasia cerulea dolens and factor VII deficiency. BMJ Case Rep. 2016;2016:pii:bcr2016215078. doi:10.1136/bcr-2016-215078.
5. Onuoha CU. Phlegmasia cerulea dolens: A rare clinical presentation. Am J Med. 2015;128(9):e27-e28. doi:10.1016/j.amjmed.2015.04.009.
6. Chinsakchai K, Ten Duis K, Moll FL, de Borst GJ. Trends in management of phlegmasia cerulea dolens. Vasc Endovascular Surg. 2011;45(1):5-14. doi:10.1177/1538574410388309.
7. Bagenal JD, Nasralla D. Bilateral phlegmasia cerulea dolens in an occluded inferior vena cava filter. BMJ Case Rep. 2013;pii: bcr2013009302. doi:10.1136/bcr-2013-009302.
8. Kiefer CS, Colletti JE. Phlegmasia cerulea dolens in a patient with an inferior vena cava filter. J Emerg Med. 2013;44(1):e95-e97. doi:10.1016/j.jemermed.2012.01.018.
Phlegmasia cerulea dolens (PCD), a life-threatening complication of deep venous thrombosis (DVT), is characterized by massive iliofemoral thrombus that extends to the collateral veins, leading to fluid sequestration and elevated compartment pressures that ultimately compromise arterial flow. Phlegmasia cerulea dolens can rapidly progress to compartment syndrome and gangrene.1,2 The affected limbs of patients with PCD can be hypoxic and appear purple in color due to substantial lack of blood flow, with diminished or absent pulses. Risk factors for PCD include malignancy, hypercoagulable states, venous stasis, contraceptive agents, inferior vena cava (IVC) filter, aneurysm, history of DVT, trauma, heparin-induced thrombocytopenia, femoral vein catheterization, antiphospholipid syndrome, or pregnancy.3-6 Failure to treat PCD early and aggressively carries an amputation rate of up to 50% and a mortality rate of up to 40%.4
We present the case of a patient with PCD, whose condition rapidly deteriorated despite prompt diagnosis and treatment.
Case
A 58-year-old woman presented to the ED with a 1-day history of back and leg pain and difficulty walking. When asked about the severity of her pain, she rated her leg pain at 10 on a scale of 0 to 10. The patient’s history was significant for DVT and pulmonary embolism (PE), for which a Greenfield IVC had been placed and for which she was on prophylactic warfarin therapy. The patient stated that she had been taken off warfarin several weeks prior to presentation in preparation for an elective colonoscopy and dental procedure, but had restarted the warfarin therapy 2 days prior to presentation. She had no history of diabetes mellitus or renal disease.
Initial vital signs at presentation were: blood pressure, 120/91 mm Hg; heart rate, 110 beats/min; respiratory rate, 24 breaths/min; and temperature, 96.6°F. Oxygen saturation was 100% on a nonrebreather mask.
On examination, the patient was alert and oriented to person, time, and place, but appeared dyspneic. An electrocardiogram revealed sinus tachycardia. On physical examination, lung sounds were clear to auscultation bilaterally with good air movement, and the abdomen was soft and nontender with normal bowel sounds. The dorsalis pedis and posterior tibial pulses were absent bilaterally, lower extremity capillary refill was 3 seconds, and the legs appeared mildly erythematous and cool to touch. No speech or neurological deficits were present.
Laboratory evaluation was remarkable for metabolic acidosis, venous pH, 7.11; bicarbonate, 11.7; partial pressure of carbon dioxide, 37.6; lactic acid, 8.8 mEq/L leukocytosis, 24,900 u/L; glucose, 296 mg/dL; creatinine, 2.41 mg/dL; and international normalized ratio, 1.36.
Before additional laboratory studies and imaging could be obtained, the patient developed altered mental status, hypotension, and paralysis of the lower extremities. She was orally intubated for airway protection and was given a total of 4 L of normal saline intravenously (IV) for hypotension and acidosis; sodium bicarbonate for metabolic acidosis; norepinephrine for hypotension; fentanyl for pain; and ondansetron for nausea. A central line and arterial line were placed for administering medication and hemodynamic monitoring.
Computed tomography (CT) angiography of the chest, abdomen, and pelvis demonstrated multiple subsegmental bilateral PE with no arterial pathology (Figure 1). Beside ultrasound revealed extensive bilateral DVTs involving the superficial and common femoral veins (Figure 2). The patient’s bilateral DVTs, arterial compromise, and leg cyanosis led to the diagnosis of PCD.
Critical care and vascular surgery services were consulted, and the patient was admitted to the intensive care unit. Since the patient was too unstable to undergo thrombectomy, she was given IV tissue plasminogen activator. Despite aggressive pharmacological treatment, the patient’s condition continued to deteriorate. On hospital day 2, the patient’s family changed the patient’s code status to do-not-resuscitate/comfort-care only; she died shortly thereafter.
Discussion
This case illustrates the severity and complications of PCD and the rapidity with which this condition can deteriorate. At the time of ED presentation, the patient had already developed bilateral PCD, metabolic acidosis, and bilateral PE. Unfortunately, due to decreased venous return, decreased cardiac output, and severe shock, she quickly became unstable and progressed rapidly to multisystem organ failure leading to death.
Risk Factors
A prior patient history DVT and an IVC filter are both significant risk factors for the progression of DVT to PCD;3,6 however, in this case, IVC filter failed to prevent emboli from reaching the lungs. Extensive thrombi led to severely decreased venous return and cardiac output, causing life-threatening shock, ischemia, and metabolic acidosis. A lactic acid level taken on hospital day 2 was elevated at 19 mEq/L, demonstrating the severity, morbidity, and progression of PCD.
Signs and Symptoms
The three cardinal signs that lead to a clinical diagnosis of PCD are edema, pain, and violaceous discoloration or skin mottling.3 Although most commonly found in the lower extremity, PCD can occur in any limb due to occlusion of venous outflow.7 Unfortunately, a clinical diagnosis of PCD is not often made until the venous occlusion becomes severe enough to impair arterial flow and cause venous gangrene, tissue ischemia, shock, and death.8
Although IVC filters are designed to prevent life-threatening PE, there are risk factors associated with their use. Whether placed recently or decades prior, urgent investigation, such as immediate CT scan, should be undertaken in patients presenting with DVT-like symptoms who have a history of an IVC filter, to ensure the filter has not shifted from its original placement and is not occluding the IVC.
Conclusion
Phlegmasia cerulea dolens is an uncommon vascular emergency, but one that has a high-morbidity and high-mortality rate. This case demonstrates the importance of early diagnosis, aggressive treatment, and the severe complications that can develop in PCD.
There are cases in the literature where patients diagnosed with PCD had a successful outcome with pharmacological or surgical intervention such as thrombectomy. Treatment for PCD is most effective when instituted early in onset. As seen in our patient, the tendency for rapid deterioration in PCD can limit potentially lifesaving therapeutic options, decreasing the chances of a successful outcome. Emergency physicians, therefore, must be aware of the high-mortality rate associated with this disorder and the possibility of rapid progression from stable to critical condition.
Phlegmasia cerulea dolens (PCD), a life-threatening complication of deep venous thrombosis (DVT), is characterized by massive iliofemoral thrombus that extends to the collateral veins, leading to fluid sequestration and elevated compartment pressures that ultimately compromise arterial flow. Phlegmasia cerulea dolens can rapidly progress to compartment syndrome and gangrene.1,2 The affected limbs of patients with PCD can be hypoxic and appear purple in color due to substantial lack of blood flow, with diminished or absent pulses. Risk factors for PCD include malignancy, hypercoagulable states, venous stasis, contraceptive agents, inferior vena cava (IVC) filter, aneurysm, history of DVT, trauma, heparin-induced thrombocytopenia, femoral vein catheterization, antiphospholipid syndrome, or pregnancy.3-6 Failure to treat PCD early and aggressively carries an amputation rate of up to 50% and a mortality rate of up to 40%.4
We present the case of a patient with PCD, whose condition rapidly deteriorated despite prompt diagnosis and treatment.
Case
A 58-year-old woman presented to the ED with a 1-day history of back and leg pain and difficulty walking. When asked about the severity of her pain, she rated her leg pain at 10 on a scale of 0 to 10. The patient’s history was significant for DVT and pulmonary embolism (PE), for which a Greenfield IVC had been placed and for which she was on prophylactic warfarin therapy. The patient stated that she had been taken off warfarin several weeks prior to presentation in preparation for an elective colonoscopy and dental procedure, but had restarted the warfarin therapy 2 days prior to presentation. She had no history of diabetes mellitus or renal disease.
Initial vital signs at presentation were: blood pressure, 120/91 mm Hg; heart rate, 110 beats/min; respiratory rate, 24 breaths/min; and temperature, 96.6°F. Oxygen saturation was 100% on a nonrebreather mask.
On examination, the patient was alert and oriented to person, time, and place, but appeared dyspneic. An electrocardiogram revealed sinus tachycardia. On physical examination, lung sounds were clear to auscultation bilaterally with good air movement, and the abdomen was soft and nontender with normal bowel sounds. The dorsalis pedis and posterior tibial pulses were absent bilaterally, lower extremity capillary refill was 3 seconds, and the legs appeared mildly erythematous and cool to touch. No speech or neurological deficits were present.
Laboratory evaluation was remarkable for metabolic acidosis, venous pH, 7.11; bicarbonate, 11.7; partial pressure of carbon dioxide, 37.6; lactic acid, 8.8 mEq/L leukocytosis, 24,900 u/L; glucose, 296 mg/dL; creatinine, 2.41 mg/dL; and international normalized ratio, 1.36.
Before additional laboratory studies and imaging could be obtained, the patient developed altered mental status, hypotension, and paralysis of the lower extremities. She was orally intubated for airway protection and was given a total of 4 L of normal saline intravenously (IV) for hypotension and acidosis; sodium bicarbonate for metabolic acidosis; norepinephrine for hypotension; fentanyl for pain; and ondansetron for nausea. A central line and arterial line were placed for administering medication and hemodynamic monitoring.
Computed tomography (CT) angiography of the chest, abdomen, and pelvis demonstrated multiple subsegmental bilateral PE with no arterial pathology (Figure 1). Beside ultrasound revealed extensive bilateral DVTs involving the superficial and common femoral veins (Figure 2). The patient’s bilateral DVTs, arterial compromise, and leg cyanosis led to the diagnosis of PCD.
Critical care and vascular surgery services were consulted, and the patient was admitted to the intensive care unit. Since the patient was too unstable to undergo thrombectomy, she was given IV tissue plasminogen activator. Despite aggressive pharmacological treatment, the patient’s condition continued to deteriorate. On hospital day 2, the patient’s family changed the patient’s code status to do-not-resuscitate/comfort-care only; she died shortly thereafter.
Discussion
This case illustrates the severity and complications of PCD and the rapidity with which this condition can deteriorate. At the time of ED presentation, the patient had already developed bilateral PCD, metabolic acidosis, and bilateral PE. Unfortunately, due to decreased venous return, decreased cardiac output, and severe shock, she quickly became unstable and progressed rapidly to multisystem organ failure leading to death.
Risk Factors
A prior patient history DVT and an IVC filter are both significant risk factors for the progression of DVT to PCD;3,6 however, in this case, IVC filter failed to prevent emboli from reaching the lungs. Extensive thrombi led to severely decreased venous return and cardiac output, causing life-threatening shock, ischemia, and metabolic acidosis. A lactic acid level taken on hospital day 2 was elevated at 19 mEq/L, demonstrating the severity, morbidity, and progression of PCD.
Signs and Symptoms
The three cardinal signs that lead to a clinical diagnosis of PCD are edema, pain, and violaceous discoloration or skin mottling.3 Although most commonly found in the lower extremity, PCD can occur in any limb due to occlusion of venous outflow.7 Unfortunately, a clinical diagnosis of PCD is not often made until the venous occlusion becomes severe enough to impair arterial flow and cause venous gangrene, tissue ischemia, shock, and death.8
Although IVC filters are designed to prevent life-threatening PE, there are risk factors associated with their use. Whether placed recently or decades prior, urgent investigation, such as immediate CT scan, should be undertaken in patients presenting with DVT-like symptoms who have a history of an IVC filter, to ensure the filter has not shifted from its original placement and is not occluding the IVC.
Conclusion
Phlegmasia cerulea dolens is an uncommon vascular emergency, but one that has a high-morbidity and high-mortality rate. This case demonstrates the importance of early diagnosis, aggressive treatment, and the severe complications that can develop in PCD.
There are cases in the literature where patients diagnosed with PCD had a successful outcome with pharmacological or surgical intervention such as thrombectomy. Treatment for PCD is most effective when instituted early in onset. As seen in our patient, the tendency for rapid deterioration in PCD can limit potentially lifesaving therapeutic options, decreasing the chances of a successful outcome. Emergency physicians, therefore, must be aware of the high-mortality rate associated with this disorder and the possibility of rapid progression from stable to critical condition.
1. Kesieme E, Kesieme C, Jebbin N, Irekpita E, Dongo A. Deep vein thrombosis: a clinical review. J Blood Med. 2011;2:59-69. doi:10.2147/JBM.S19009.
2. Bhatt S, Wehbe C, Dogra VS. Phlegmasia cerulea dolens. J Clin Ultrasound. 2007;35(7):401-404. doi:10.1002/jcu.20317.
3. Maiti A, Das A, Smith DT. Phlegmasia cerulean dolens. Postgrad Med J. 2016;pii: postgradmedj-2016-134185. doi:10.1136/postgradmedj-2016-134185.
4. Abdul W, Hickey B, Wilson C. Lower extremity compartment syndrome in the setting of iliofemoral deep vein thrombosis, phlegmasia cerulea dolens and factor VII deficiency. BMJ Case Rep. 2016;2016:pii:bcr2016215078. doi:10.1136/bcr-2016-215078.
5. Onuoha CU. Phlegmasia cerulea dolens: A rare clinical presentation. Am J Med. 2015;128(9):e27-e28. doi:10.1016/j.amjmed.2015.04.009.
6. Chinsakchai K, Ten Duis K, Moll FL, de Borst GJ. Trends in management of phlegmasia cerulea dolens. Vasc Endovascular Surg. 2011;45(1):5-14. doi:10.1177/1538574410388309.
7. Bagenal JD, Nasralla D. Bilateral phlegmasia cerulea dolens in an occluded inferior vena cava filter. BMJ Case Rep. 2013;pii: bcr2013009302. doi:10.1136/bcr-2013-009302.
8. Kiefer CS, Colletti JE. Phlegmasia cerulea dolens in a patient with an inferior vena cava filter. J Emerg Med. 2013;44(1):e95-e97. doi:10.1016/j.jemermed.2012.01.018.
1. Kesieme E, Kesieme C, Jebbin N, Irekpita E, Dongo A. Deep vein thrombosis: a clinical review. J Blood Med. 2011;2:59-69. doi:10.2147/JBM.S19009.
2. Bhatt S, Wehbe C, Dogra VS. Phlegmasia cerulea dolens. J Clin Ultrasound. 2007;35(7):401-404. doi:10.1002/jcu.20317.
3. Maiti A, Das A, Smith DT. Phlegmasia cerulean dolens. Postgrad Med J. 2016;pii: postgradmedj-2016-134185. doi:10.1136/postgradmedj-2016-134185.
4. Abdul W, Hickey B, Wilson C. Lower extremity compartment syndrome in the setting of iliofemoral deep vein thrombosis, phlegmasia cerulea dolens and factor VII deficiency. BMJ Case Rep. 2016;2016:pii:bcr2016215078. doi:10.1136/bcr-2016-215078.
5. Onuoha CU. Phlegmasia cerulea dolens: A rare clinical presentation. Am J Med. 2015;128(9):e27-e28. doi:10.1016/j.amjmed.2015.04.009.
6. Chinsakchai K, Ten Duis K, Moll FL, de Borst GJ. Trends in management of phlegmasia cerulea dolens. Vasc Endovascular Surg. 2011;45(1):5-14. doi:10.1177/1538574410388309.
7. Bagenal JD, Nasralla D. Bilateral phlegmasia cerulea dolens in an occluded inferior vena cava filter. BMJ Case Rep. 2013;pii: bcr2013009302. doi:10.1136/bcr-2013-009302.
8. Kiefer CS, Colletti JE. Phlegmasia cerulea dolens in a patient with an inferior vena cava filter. J Emerg Med. 2013;44(1):e95-e97. doi:10.1016/j.jemermed.2012.01.018.
Case Report: A 48-Year-Old Woman With Acute Abdomen
Case
A 48-year-old woman presented to the ED with significant periumbilical abdominal pain and left lower extremity pain, which she rated an “8” on a scale of 1 to 10. She stated that the pain worsened with movement and change in position. The claudication in the patient’s left lower extremity began a few weeks prior to presentation, at which time she had received medical attention, including ankle brachial index testing that showed an abnormal value in the left lower extremity. The patient noted that when the abdominal pain began, the pain in her leg became more frequent and of higher intensity, with intermittent numbness. She reported some nausea, paresthesia, and sensory changes to the left lower extremity; however, she denied diarrhea, headache, fever, back pain, urinary symptoms, chest pain, and shortness of breath.
Regarding social history, the patient admitted to smoking half a pack of cigarettes a day and drinking alcohol socially. She denied any significant family history of disease. The patient’s own medical history included colon cancer, claudication, and multiple abdominal surgeries. The patient had been diagnosed with stage II colon cancer 4 years earlier, for which she had undergone a colon resection.
During the physical examination, the patient was diaphoretic, uncomfortable, and in severe distress. Her vital signs were: blood pressure, 146/77 mm Hg; respiratory rate, 18 breaths/minute; heart rate, 129 beats/minute; and temperature within normal limits. Oxygen saturation was 94% on room air.
The abdominal examination revealed a distended abdomen that was severely tender to palpation, with rigidity, guarding, and rebound tenderness. Examination of the lower extremities revealed an absent palpable dorsalis pedis pulse to the left lower extremity, but dorsalis pedis pulse and posterior tibial pulse in the left lower extremity were appreciated by Doppler. The right lower extremity had palpable 2+ dorsalis pedis and posterior tibial pulses.
The patient was immediately started on fentanyl and intravenous (IV) fluids; she was also given IV ondansetron and promethazine for nausea. Her pain was refractory to treatment, and required multiple doses of hydromorphone. Laboratory evaluation revealed leukocytosis with a white blood cell (WBC) count of 15.1 thou/cmm.
Computed tomography angiography (CTA) with runoff was ordered to evaluate lower extremity vasculature and perfusion, as well as abdominal vasculature and intra-abdominal organ pathology. The CTA revealed 99% stenosis in the left iliac artery; multiple areas of stenosis within the abdominal vasculature, including the superior mesenteric artery (SMA) and inferior mesenteric artery (IMA); and a small ventral hernia slightly left of the umbilicus but without evidence of obstruction. The patient remained stable while in the ED, and an emergent vascular surgery consultation was ordered. She was transferred to surgical services.
Mesenteric Ischemia
Mesenteric ischemia is a condition in which the intestine does not receive adequate blood supply, resulting in inflammation and injury. Cases of the disease may be acute or chronic. Acute mesenteric ischemia (AMI) may be occlusive or nonocclusive. Occlusive AMI is most commonly caused by embolic or thrombotic occlusion of one or more mesenteric arteries. Nonocclusive AMI (NMI) is most commonly due to primary splanchnic vasoconstriction.1 It can also be seen in patients on high-dose vasopressor agents. Chronic mesenteric ischemia indicates continuous intestinal hypoperfusion that is often associated with meals and referred to as postprandial or intestinal angina.
Mesenteric ischemia is associated with poor outcomes, having a mortality rate ranging from 40% to 70%.2 It is imperative that diagnosis and treatment commence rapidly to avoid potentially catastrophic complications such as transmural bowel infarction. Although visceral ischemia is rare, occurring in only 2 to 3 per 100,000, the high mortality rate makes prompt and accurate diagnosis essential to decreasing morbidity and mortality.3
Symptoms and Signs
The classical presentation of mesenteric ischemia is sudden onset of abdominal pain out of proportion to physical examination findings; however, peritoneal signs are also not uncommon later in the disease process. The most common presenting symptoms are abdominal pain, nausea, and diarrhea. Laboratory findings associated with mesenteric ischemia include leukocytosis, metabolic acidosis, elevated lactate, and an elevated D-dimer.2
Early recognition is crucial given the significant risk of bowel necrosis. Signs of peritonitis are frequently present late in the disease course; signs such as nausea, vomiting, and constipation are more frequent. Patients may also have complications such as ileus, gastrointestinal bleeding, and pancreatitis, which may mask the diagnosis of AMI.4
Prompt diagnosis and treatment are paramount. Acute AMI should especially be considered in patients who are over age 60 years, have a history of atrial fibrillation, claudication, hypercoagulable states or a previous history of atherosclerotic disease, myocardial infarction, and a history of postprandial abdominal pain and weight loss.
Laboratory Evaluation
The most common laboratory abnormalities in AMI are hemoconcentration, leukocytosis, elevated lactic acid, metabolic acidosis, and a high anion gap. Elevated amylase and creatinine phosphokinase are also frequently observed but are not specific for AMI. Hyperphosphatemia and hyperkalemia are frequently late signs and are associated with bowel infarction. Findings on plain abdominal radiographs are nonspecific and should not be utilized in the workups. Barium enemas also have no place in diagnosis, as this may reduce perfusion to the bowel wall and cause perforation.5 Leukocytosis and high lactate levels appear to be present in the majority of patients, though these are not specific for acute mesenteric ischemia.4
Imaging Studies
In the past, catheter-based angiography was considered the gold standard for diagnosis. However, the more readily available CTA is emerging as the primary imaging modality to diagnose mesenteric ischemia.3 Both CT and contrast angiography play a major role in the diagnosis. In addition to mesenteric ischemia, CT also allows for identification of nonvascular causes of abdominal pain. Contrast angiography has an important role in early diagnosis and is helpful in treatment planning as well as operative interventions.4
While CTA is the most frequently used technique in suspected AMI, contrast-enhanced three-dimensional magnetic resonance angiography (MRA) is also widely used. However, the inferior mesenteric artery and other splanchnic vessel periphery are currently better assessed with CTA due to the higher special and temporal resolution of the former. Both CTA and MRA are excellent screening techniques for AMI due to various causes.6
Duplex Doppler sonography has also been suggested as a screening tool in patients with suspected mesenteric ischemia, but this modality has multiple limitations, including failure to obtain adequate Doppler signal due to bowel gas or vessel wall calcification. Since significant disease is often common in the SMA and the celiac arteries of asymptomatic elderly patients, this modality should be considered when examining patients with suspected mesenteric ischemia.7
Treatment
Endovascular intervention or catheter-directed vasodilator therapy can be started immediately postangiography. The role of endovascular therapy in AMI is controversial. In NMI, a catheter-directed vasodilator infusion continues to be the treatment of choice in patients without peritonitis. Catheter-directed thrombolysis and percutaneous angioplasty have also been investigated in the treatment of AMI.4
The goal of surgical care is the removal of necrotic and nonsalvageable bowel and the prevention of further infarction. Stenting of the affected arteries may be utilized. An exploratory laparotomy remains the gold standard for assessment of bowel viability. Multiorgan failure poses a great risk in patients with AMI and mortality remains high.4 The most preferred surgical revascularization technique in embolic AMI remains the balloon catheter thromboembolectomy—with or without patch angioplasty of the superior mesenteric artery.
Prevention therapy should be utilized aggressively for AMI; patients with atrial fibrillation should be started on anticoagulants. Elective and timely revascularization may be undertaken in patients with chronic claudication and AMI secondary to atherosclerotic disease. In addition, patients should be advised not to smoke.4
Upon diagnosis of AMI, aggressive IV fluid resuscitation with crystalloids should be administered starting with volumes as high as 100 mL/kg to correct any metabolic derangements. A broad-spectrum antibiotic should also be started as early as possible. If no contraindications to anticoagulation exist, therapeutic IV heparin sodium should be administered to maintain an activated partial thromboplastin time at twice the normal value.5 The patient in this case was started on IV heparin and broad-spectrum antibiotics. In an optimized hemodynamic status, attempts to reduce acute vasospasm in AMI can be made with an IV glucagon infusion, starting at 1 mcg/kg/minute. The presence of peritoneal signs indicates bowel infarction and mandates an emergency laparotomy.5 As noted in the patient’s history, she was not on any anticoagulants on presentation and was a smoker.
Conclusion
The causes of abdominal pain range from benign to life threatening; therefore, it is imperative for clinicians to obtain a thorough history and physical examination of patients presenting with abdominal pain, and to consider a vascular etiology in the differential diagnosis. This case is unique in that the patient had multiple areas of stenosis within the abdomen, including the SMA and IMA, and either an acute or chronic occlusion, and claudication of her left lower extremity.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Bosman is an undergraduate research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
- Tendler DA, Lamont JT. Nonocclusive mesenteric ischemia. UpToDate. http://www.uptodate.com/contents/nonocclusive-mesenteric-ischemia?source=search_result&search=Acute+Mesenteric+Ischemia&selectedTitle=2~72. Accessed March 27, 2015.
- Bobadilla JL. Mesenteric ischemia. Surg Clin North Am. 2013;93(4):925-940, ix.
- van den Heijkant TC, Aerts BA, Teijink JA, Buurman WA, Luyer MD. Challenges in diagnosing mesenteric ischemia. World J Gastroenterol. 2013;19(9):1338-1341.
- Park WM, Gloviczki P, Cherry KJ jR, et al. Contemporary management of acute mesenteric ischemia: Factors associated with survival. J Vasc Surg. 2002;35(3):445-452.
- Oldenburg AW, Lau LL, Rodenberg TJ, Edmonds HJ, Burger CD. Acute mesenteric ischemia: a clinical review. Arch Intern Med. 2004;164(10):1054-1062.
- Shih MC, Hagspiel, KD. CTA and MRA in mesenteric ischemia: part 1, Role in diagnosis and differential diagnosis. AJR Am J Roentgenol. 2007;188(2):452-461.
- Roobottom CA, Dubbins PA. Significant disease of the celiac and superior mesenteric arteries in asymptomatic patients: predictive value of Doppler sonography. AJR
Case
A 48-year-old woman presented to the ED with significant periumbilical abdominal pain and left lower extremity pain, which she rated an “8” on a scale of 1 to 10. She stated that the pain worsened with movement and change in position. The claudication in the patient’s left lower extremity began a few weeks prior to presentation, at which time she had received medical attention, including ankle brachial index testing that showed an abnormal value in the left lower extremity. The patient noted that when the abdominal pain began, the pain in her leg became more frequent and of higher intensity, with intermittent numbness. She reported some nausea, paresthesia, and sensory changes to the left lower extremity; however, she denied diarrhea, headache, fever, back pain, urinary symptoms, chest pain, and shortness of breath.
Regarding social history, the patient admitted to smoking half a pack of cigarettes a day and drinking alcohol socially. She denied any significant family history of disease. The patient’s own medical history included colon cancer, claudication, and multiple abdominal surgeries. The patient had been diagnosed with stage II colon cancer 4 years earlier, for which she had undergone a colon resection.
During the physical examination, the patient was diaphoretic, uncomfortable, and in severe distress. Her vital signs were: blood pressure, 146/77 mm Hg; respiratory rate, 18 breaths/minute; heart rate, 129 beats/minute; and temperature within normal limits. Oxygen saturation was 94% on room air.
The abdominal examination revealed a distended abdomen that was severely tender to palpation, with rigidity, guarding, and rebound tenderness. Examination of the lower extremities revealed an absent palpable dorsalis pedis pulse to the left lower extremity, but dorsalis pedis pulse and posterior tibial pulse in the left lower extremity were appreciated by Doppler. The right lower extremity had palpable 2+ dorsalis pedis and posterior tibial pulses.
The patient was immediately started on fentanyl and intravenous (IV) fluids; she was also given IV ondansetron and promethazine for nausea. Her pain was refractory to treatment, and required multiple doses of hydromorphone. Laboratory evaluation revealed leukocytosis with a white blood cell (WBC) count of 15.1 thou/cmm.
Computed tomography angiography (CTA) with runoff was ordered to evaluate lower extremity vasculature and perfusion, as well as abdominal vasculature and intra-abdominal organ pathology. The CTA revealed 99% stenosis in the left iliac artery; multiple areas of stenosis within the abdominal vasculature, including the superior mesenteric artery (SMA) and inferior mesenteric artery (IMA); and a small ventral hernia slightly left of the umbilicus but without evidence of obstruction. The patient remained stable while in the ED, and an emergent vascular surgery consultation was ordered. She was transferred to surgical services.
Mesenteric Ischemia
Mesenteric ischemia is a condition in which the intestine does not receive adequate blood supply, resulting in inflammation and injury. Cases of the disease may be acute or chronic. Acute mesenteric ischemia (AMI) may be occlusive or nonocclusive. Occlusive AMI is most commonly caused by embolic or thrombotic occlusion of one or more mesenteric arteries. Nonocclusive AMI (NMI) is most commonly due to primary splanchnic vasoconstriction.1 It can also be seen in patients on high-dose vasopressor agents. Chronic mesenteric ischemia indicates continuous intestinal hypoperfusion that is often associated with meals and referred to as postprandial or intestinal angina.
Mesenteric ischemia is associated with poor outcomes, having a mortality rate ranging from 40% to 70%.2 It is imperative that diagnosis and treatment commence rapidly to avoid potentially catastrophic complications such as transmural bowel infarction. Although visceral ischemia is rare, occurring in only 2 to 3 per 100,000, the high mortality rate makes prompt and accurate diagnosis essential to decreasing morbidity and mortality.3
Symptoms and Signs
The classical presentation of mesenteric ischemia is sudden onset of abdominal pain out of proportion to physical examination findings; however, peritoneal signs are also not uncommon later in the disease process. The most common presenting symptoms are abdominal pain, nausea, and diarrhea. Laboratory findings associated with mesenteric ischemia include leukocytosis, metabolic acidosis, elevated lactate, and an elevated D-dimer.2
Early recognition is crucial given the significant risk of bowel necrosis. Signs of peritonitis are frequently present late in the disease course; signs such as nausea, vomiting, and constipation are more frequent. Patients may also have complications such as ileus, gastrointestinal bleeding, and pancreatitis, which may mask the diagnosis of AMI.4
Prompt diagnosis and treatment are paramount. Acute AMI should especially be considered in patients who are over age 60 years, have a history of atrial fibrillation, claudication, hypercoagulable states or a previous history of atherosclerotic disease, myocardial infarction, and a history of postprandial abdominal pain and weight loss.
Laboratory Evaluation
The most common laboratory abnormalities in AMI are hemoconcentration, leukocytosis, elevated lactic acid, metabolic acidosis, and a high anion gap. Elevated amylase and creatinine phosphokinase are also frequently observed but are not specific for AMI. Hyperphosphatemia and hyperkalemia are frequently late signs and are associated with bowel infarction. Findings on plain abdominal radiographs are nonspecific and should not be utilized in the workups. Barium enemas also have no place in diagnosis, as this may reduce perfusion to the bowel wall and cause perforation.5 Leukocytosis and high lactate levels appear to be present in the majority of patients, though these are not specific for acute mesenteric ischemia.4
Imaging Studies
In the past, catheter-based angiography was considered the gold standard for diagnosis. However, the more readily available CTA is emerging as the primary imaging modality to diagnose mesenteric ischemia.3 Both CT and contrast angiography play a major role in the diagnosis. In addition to mesenteric ischemia, CT also allows for identification of nonvascular causes of abdominal pain. Contrast angiography has an important role in early diagnosis and is helpful in treatment planning as well as operative interventions.4
While CTA is the most frequently used technique in suspected AMI, contrast-enhanced three-dimensional magnetic resonance angiography (MRA) is also widely used. However, the inferior mesenteric artery and other splanchnic vessel periphery are currently better assessed with CTA due to the higher special and temporal resolution of the former. Both CTA and MRA are excellent screening techniques for AMI due to various causes.6
Duplex Doppler sonography has also been suggested as a screening tool in patients with suspected mesenteric ischemia, but this modality has multiple limitations, including failure to obtain adequate Doppler signal due to bowel gas or vessel wall calcification. Since significant disease is often common in the SMA and the celiac arteries of asymptomatic elderly patients, this modality should be considered when examining patients with suspected mesenteric ischemia.7
Treatment
Endovascular intervention or catheter-directed vasodilator therapy can be started immediately postangiography. The role of endovascular therapy in AMI is controversial. In NMI, a catheter-directed vasodilator infusion continues to be the treatment of choice in patients without peritonitis. Catheter-directed thrombolysis and percutaneous angioplasty have also been investigated in the treatment of AMI.4
The goal of surgical care is the removal of necrotic and nonsalvageable bowel and the prevention of further infarction. Stenting of the affected arteries may be utilized. An exploratory laparotomy remains the gold standard for assessment of bowel viability. Multiorgan failure poses a great risk in patients with AMI and mortality remains high.4 The most preferred surgical revascularization technique in embolic AMI remains the balloon catheter thromboembolectomy—with or without patch angioplasty of the superior mesenteric artery.
Prevention therapy should be utilized aggressively for AMI; patients with atrial fibrillation should be started on anticoagulants. Elective and timely revascularization may be undertaken in patients with chronic claudication and AMI secondary to atherosclerotic disease. In addition, patients should be advised not to smoke.4
Upon diagnosis of AMI, aggressive IV fluid resuscitation with crystalloids should be administered starting with volumes as high as 100 mL/kg to correct any metabolic derangements. A broad-spectrum antibiotic should also be started as early as possible. If no contraindications to anticoagulation exist, therapeutic IV heparin sodium should be administered to maintain an activated partial thromboplastin time at twice the normal value.5 The patient in this case was started on IV heparin and broad-spectrum antibiotics. In an optimized hemodynamic status, attempts to reduce acute vasospasm in AMI can be made with an IV glucagon infusion, starting at 1 mcg/kg/minute. The presence of peritoneal signs indicates bowel infarction and mandates an emergency laparotomy.5 As noted in the patient’s history, she was not on any anticoagulants on presentation and was a smoker.
Conclusion
The causes of abdominal pain range from benign to life threatening; therefore, it is imperative for clinicians to obtain a thorough history and physical examination of patients presenting with abdominal pain, and to consider a vascular etiology in the differential diagnosis. This case is unique in that the patient had multiple areas of stenosis within the abdomen, including the SMA and IMA, and either an acute or chronic occlusion, and claudication of her left lower extremity.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Bosman is an undergraduate research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
Case
A 48-year-old woman presented to the ED with significant periumbilical abdominal pain and left lower extremity pain, which she rated an “8” on a scale of 1 to 10. She stated that the pain worsened with movement and change in position. The claudication in the patient’s left lower extremity began a few weeks prior to presentation, at which time she had received medical attention, including ankle brachial index testing that showed an abnormal value in the left lower extremity. The patient noted that when the abdominal pain began, the pain in her leg became more frequent and of higher intensity, with intermittent numbness. She reported some nausea, paresthesia, and sensory changes to the left lower extremity; however, she denied diarrhea, headache, fever, back pain, urinary symptoms, chest pain, and shortness of breath.
Regarding social history, the patient admitted to smoking half a pack of cigarettes a day and drinking alcohol socially. She denied any significant family history of disease. The patient’s own medical history included colon cancer, claudication, and multiple abdominal surgeries. The patient had been diagnosed with stage II colon cancer 4 years earlier, for which she had undergone a colon resection.
During the physical examination, the patient was diaphoretic, uncomfortable, and in severe distress. Her vital signs were: blood pressure, 146/77 mm Hg; respiratory rate, 18 breaths/minute; heart rate, 129 beats/minute; and temperature within normal limits. Oxygen saturation was 94% on room air.
The abdominal examination revealed a distended abdomen that was severely tender to palpation, with rigidity, guarding, and rebound tenderness. Examination of the lower extremities revealed an absent palpable dorsalis pedis pulse to the left lower extremity, but dorsalis pedis pulse and posterior tibial pulse in the left lower extremity were appreciated by Doppler. The right lower extremity had palpable 2+ dorsalis pedis and posterior tibial pulses.
The patient was immediately started on fentanyl and intravenous (IV) fluids; she was also given IV ondansetron and promethazine for nausea. Her pain was refractory to treatment, and required multiple doses of hydromorphone. Laboratory evaluation revealed leukocytosis with a white blood cell (WBC) count of 15.1 thou/cmm.
Computed tomography angiography (CTA) with runoff was ordered to evaluate lower extremity vasculature and perfusion, as well as abdominal vasculature and intra-abdominal organ pathology. The CTA revealed 99% stenosis in the left iliac artery; multiple areas of stenosis within the abdominal vasculature, including the superior mesenteric artery (SMA) and inferior mesenteric artery (IMA); and a small ventral hernia slightly left of the umbilicus but without evidence of obstruction. The patient remained stable while in the ED, and an emergent vascular surgery consultation was ordered. She was transferred to surgical services.
Mesenteric Ischemia
Mesenteric ischemia is a condition in which the intestine does not receive adequate blood supply, resulting in inflammation and injury. Cases of the disease may be acute or chronic. Acute mesenteric ischemia (AMI) may be occlusive or nonocclusive. Occlusive AMI is most commonly caused by embolic or thrombotic occlusion of one or more mesenteric arteries. Nonocclusive AMI (NMI) is most commonly due to primary splanchnic vasoconstriction.1 It can also be seen in patients on high-dose vasopressor agents. Chronic mesenteric ischemia indicates continuous intestinal hypoperfusion that is often associated with meals and referred to as postprandial or intestinal angina.
Mesenteric ischemia is associated with poor outcomes, having a mortality rate ranging from 40% to 70%.2 It is imperative that diagnosis and treatment commence rapidly to avoid potentially catastrophic complications such as transmural bowel infarction. Although visceral ischemia is rare, occurring in only 2 to 3 per 100,000, the high mortality rate makes prompt and accurate diagnosis essential to decreasing morbidity and mortality.3
Symptoms and Signs
The classical presentation of mesenteric ischemia is sudden onset of abdominal pain out of proportion to physical examination findings; however, peritoneal signs are also not uncommon later in the disease process. The most common presenting symptoms are abdominal pain, nausea, and diarrhea. Laboratory findings associated with mesenteric ischemia include leukocytosis, metabolic acidosis, elevated lactate, and an elevated D-dimer.2
Early recognition is crucial given the significant risk of bowel necrosis. Signs of peritonitis are frequently present late in the disease course; signs such as nausea, vomiting, and constipation are more frequent. Patients may also have complications such as ileus, gastrointestinal bleeding, and pancreatitis, which may mask the diagnosis of AMI.4
Prompt diagnosis and treatment are paramount. Acute AMI should especially be considered in patients who are over age 60 years, have a history of atrial fibrillation, claudication, hypercoagulable states or a previous history of atherosclerotic disease, myocardial infarction, and a history of postprandial abdominal pain and weight loss.
Laboratory Evaluation
The most common laboratory abnormalities in AMI are hemoconcentration, leukocytosis, elevated lactic acid, metabolic acidosis, and a high anion gap. Elevated amylase and creatinine phosphokinase are also frequently observed but are not specific for AMI. Hyperphosphatemia and hyperkalemia are frequently late signs and are associated with bowel infarction. Findings on plain abdominal radiographs are nonspecific and should not be utilized in the workups. Barium enemas also have no place in diagnosis, as this may reduce perfusion to the bowel wall and cause perforation.5 Leukocytosis and high lactate levels appear to be present in the majority of patients, though these are not specific for acute mesenteric ischemia.4
Imaging Studies
In the past, catheter-based angiography was considered the gold standard for diagnosis. However, the more readily available CTA is emerging as the primary imaging modality to diagnose mesenteric ischemia.3 Both CT and contrast angiography play a major role in the diagnosis. In addition to mesenteric ischemia, CT also allows for identification of nonvascular causes of abdominal pain. Contrast angiography has an important role in early diagnosis and is helpful in treatment planning as well as operative interventions.4
While CTA is the most frequently used technique in suspected AMI, contrast-enhanced three-dimensional magnetic resonance angiography (MRA) is also widely used. However, the inferior mesenteric artery and other splanchnic vessel periphery are currently better assessed with CTA due to the higher special and temporal resolution of the former. Both CTA and MRA are excellent screening techniques for AMI due to various causes.6
Duplex Doppler sonography has also been suggested as a screening tool in patients with suspected mesenteric ischemia, but this modality has multiple limitations, including failure to obtain adequate Doppler signal due to bowel gas or vessel wall calcification. Since significant disease is often common in the SMA and the celiac arteries of asymptomatic elderly patients, this modality should be considered when examining patients with suspected mesenteric ischemia.7
Treatment
Endovascular intervention or catheter-directed vasodilator therapy can be started immediately postangiography. The role of endovascular therapy in AMI is controversial. In NMI, a catheter-directed vasodilator infusion continues to be the treatment of choice in patients without peritonitis. Catheter-directed thrombolysis and percutaneous angioplasty have also been investigated in the treatment of AMI.4
The goal of surgical care is the removal of necrotic and nonsalvageable bowel and the prevention of further infarction. Stenting of the affected arteries may be utilized. An exploratory laparotomy remains the gold standard for assessment of bowel viability. Multiorgan failure poses a great risk in patients with AMI and mortality remains high.4 The most preferred surgical revascularization technique in embolic AMI remains the balloon catheter thromboembolectomy—with or without patch angioplasty of the superior mesenteric artery.
Prevention therapy should be utilized aggressively for AMI; patients with atrial fibrillation should be started on anticoagulants. Elective and timely revascularization may be undertaken in patients with chronic claudication and AMI secondary to atherosclerotic disease. In addition, patients should be advised not to smoke.4
Upon diagnosis of AMI, aggressive IV fluid resuscitation with crystalloids should be administered starting with volumes as high as 100 mL/kg to correct any metabolic derangements. A broad-spectrum antibiotic should also be started as early as possible. If no contraindications to anticoagulation exist, therapeutic IV heparin sodium should be administered to maintain an activated partial thromboplastin time at twice the normal value.5 The patient in this case was started on IV heparin and broad-spectrum antibiotics. In an optimized hemodynamic status, attempts to reduce acute vasospasm in AMI can be made with an IV glucagon infusion, starting at 1 mcg/kg/minute. The presence of peritoneal signs indicates bowel infarction and mandates an emergency laparotomy.5 As noted in the patient’s history, she was not on any anticoagulants on presentation and was a smoker.
Conclusion
The causes of abdominal pain range from benign to life threatening; therefore, it is imperative for clinicians to obtain a thorough history and physical examination of patients presenting with abdominal pain, and to consider a vascular etiology in the differential diagnosis. This case is unique in that the patient had multiple areas of stenosis within the abdomen, including the SMA and IMA, and either an acute or chronic occlusion, and claudication of her left lower extremity.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Bosman is an undergraduate research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
- Tendler DA, Lamont JT. Nonocclusive mesenteric ischemia. UpToDate. http://www.uptodate.com/contents/nonocclusive-mesenteric-ischemia?source=search_result&search=Acute+Mesenteric+Ischemia&selectedTitle=2~72. Accessed March 27, 2015.
- Bobadilla JL. Mesenteric ischemia. Surg Clin North Am. 2013;93(4):925-940, ix.
- van den Heijkant TC, Aerts BA, Teijink JA, Buurman WA, Luyer MD. Challenges in diagnosing mesenteric ischemia. World J Gastroenterol. 2013;19(9):1338-1341.
- Park WM, Gloviczki P, Cherry KJ jR, et al. Contemporary management of acute mesenteric ischemia: Factors associated with survival. J Vasc Surg. 2002;35(3):445-452.
- Oldenburg AW, Lau LL, Rodenberg TJ, Edmonds HJ, Burger CD. Acute mesenteric ischemia: a clinical review. Arch Intern Med. 2004;164(10):1054-1062.
- Shih MC, Hagspiel, KD. CTA and MRA in mesenteric ischemia: part 1, Role in diagnosis and differential diagnosis. AJR Am J Roentgenol. 2007;188(2):452-461.
- Roobottom CA, Dubbins PA. Significant disease of the celiac and superior mesenteric arteries in asymptomatic patients: predictive value of Doppler sonography. AJR
- Tendler DA, Lamont JT. Nonocclusive mesenteric ischemia. UpToDate. http://www.uptodate.com/contents/nonocclusive-mesenteric-ischemia?source=search_result&search=Acute+Mesenteric+Ischemia&selectedTitle=2~72. Accessed March 27, 2015.
- Bobadilla JL. Mesenteric ischemia. Surg Clin North Am. 2013;93(4):925-940, ix.
- van den Heijkant TC, Aerts BA, Teijink JA, Buurman WA, Luyer MD. Challenges in diagnosing mesenteric ischemia. World J Gastroenterol. 2013;19(9):1338-1341.
- Park WM, Gloviczki P, Cherry KJ jR, et al. Contemporary management of acute mesenteric ischemia: Factors associated with survival. J Vasc Surg. 2002;35(3):445-452.
- Oldenburg AW, Lau LL, Rodenberg TJ, Edmonds HJ, Burger CD. Acute mesenteric ischemia: a clinical review. Arch Intern Med. 2004;164(10):1054-1062.
- Shih MC, Hagspiel, KD. CTA and MRA in mesenteric ischemia: part 1, Role in diagnosis and differential diagnosis. AJR Am J Roentgenol. 2007;188(2):452-461.
- Roobottom CA, Dubbins PA. Significant disease of the celiac and superior mesenteric arteries in asymptomatic patients: predictive value of Doppler sonography. AJR

Case Report: Recurrent Sagittal Sinus Thrombosis
Case

The patient’s past medical history included one miscarriage, as well as a papillary thyroid carcinoma with resection, which was discovered a few months before her presentation to the ED and after diagnosis of the initial SSS thrombosis.
Physical examination revealed a well-developed, mildly obese female. On arrival at the ED, the patient’s National Institutes of Health Stroke Scale score was 0. Her vital signs and ocular, neurological, and psychiatric examinations were all normal. The social history was negative for tobacco or alcohol use, and she had no family history of deep vein thrombosis (DVT) or pulmonary embolism.
A noncontrast computed tomography (CT) of the head demonstrated a hemorrhagic venous infarction involving the posterior right parietal lobe. Intracranial magnetic resonance venography (MRV) and brain magnetic resonance imaging (MRI) revealed thrombosis of the posterior third of the SSS as the source of the infarction. This sinus had been patent during the patient’s previous hospital admissions.
The patient’s international normalized ratio (INR) was therapeutic on presentation. Warfarin was discontinued, and she was started on an intravenous (IV) heparin drip. For anticoagulation, she was prescribed 20 mg rivaroxaban daily and 2,000 mg levetiracetam daily.
One week after discharge, the patient again presented to the ED with a recurrence of symptoms, including confusion, slurred speech, and headache, which she rated a “5” on a pain scale of 0 to 10. Similar to the previous ED visit, the slurred speech had resolved by the time of examination. The patient did not exhibit facial asymmetry but did complain of bilateral numbness and tingling in both hands. A noncontrast CT of the head showed no changes in the right parietal hemorrhagic venous infarct and intraparenchymal hemorrhage; however, there was an interval increase in edema compared to the prior CT. Rivaroxaban and levetiracetam were continued, and 20 mg simvastatin daily was prescribed.
Overview
Cerebral venous sinus thrombosis is a rare condition with an often varied clinical presentation—the symptoms of which can take hours to weeks to evolve, thus making the diagnosis challenging. In 70% of cases, the SSS and lateral sinuses are individually involved, and in 30% of cases, both regions are affected simultaneously.1 Only recently have clinicians been able to diagnose this condition antemortem.
Risk Factors and Etiology
Inherited and Acquired hypercoagulable states
Cerebral venous sinus thrombosis (CVST) and cerebrovascular accident (CVA) often result from a hypercoagulable state (HCS), and both acquired and inherited factors place patients at risk. Inherited factors are the most common cause of venous thromboembolism in patients younger than age 40 years. Acquired factors have a combined effect with inherited ones, leading to increased risk of CVST or CVA.2
The patient in this case possessed both acquired and inherited factors of an HCS. Inherited factors can be found through a thrombophilia evaluation. In general, acquired factors of thrombophilia include obesity, a prior history of thrombosis, pregnancy, and cancer and its treatment. A thrombophilia evaluation revealed the patient was homozygous for the 4G allele, which has been shown to increase concentration of plasminogen activator inhibitor (PAI-1) by 30%. An inhibitor to the pathway of fibrinolysis, PAI-1 is a major factor preventing the excessive presence and magnitude of blood clots.3
Pregnancy and the Puerperium
Cerebral vascular sinus thrombosis is most commonly seen in young to middle-aged women. High risk factors include pregnancy and the puerperium due to increased HCS during these periods.4 The incidence of CVST in this population is approximately 10 per 100,000 women.4
Oral Hormonal Contraceptives
In approximately 10% of CVST cases, oral hormonal contraceptive use in the presence of a coagulation disorder are frequently the cause—as observed in the incidence of DVT in this patient population.
Septic Cerebral Venous Sinus Thrombosis
Septic CVST occurs mainly in children and up to 18% of adult cases in developing countries. It is associated with localized infections (eg, mastoiditis, otitis media, sinusitis, meningitis).
Other Causes
Although rare, other causes of CVST include intracranial hypotension, hydrocephalus, and the use of certain drugs and supplements (eg, corticosteroids, high doses of vitamin A). Each of these potential causes also should be considered when evaluating for CVST.4
Symptoms and Signs
Common symptoms and signs of CVST include headache, nausea, vomiting, seizure, and focal neurological deficit. Papilledema is present in 40% of cases, primarily in patients with delayed diagnosis or a chronic course.
Neurological Deficits
Cerebral venous sinus thrombosis may not necessarily cause focal neurological deficits due to numerous pathways of venous drainage and the possibility of reversal of venous blood flow. However, the condition can lead to impaired resorption of CSF causing intracranial hypertension.
Headache
In 70% of cases, headache is the initial symptom of CVST, and it is the only symptom in 16% of cases. With respect to headache presentation, it is important to remember that thunderclap headache is not exclusive to the diagnosis of subarachnoid hemorrhage (SAH). The absence of findings on workup to support the diagnosis of SAH should prompt investigation with MRV and evaluation of CVST.4
Seizure
Focal or generalized seizure on initial presentation occurs in 30% to 40% of cases of CVST. When smaller cerebral veins are involved, this can lead to focal edema, neurological deficits, venous infarction, and seizure. Focal deficits are determined by the localization of CVST and associated lesions. Other symptoms may include migraine headache, transient ischemic attack, cranial nerve palsies, and subarachnoid hemorrhage.4
Complications
Cerebral venous sinus thrombosis is a rare condition with an often varied clinical presentation—the symptoms of which can take hours to weeks to evolve, thus making the diagnosis challenging.
Complications in patients with CVST occur when venous congestion increases and raises dural venous sinus and cerebral spinal fluid (CSF) pressure. Parenchymal edema with venous infarction and hemorrhage complicates up to 50% of venous sinus thromboses (as seen in this patient).1 Unfortunately, little is known about long-term risk outcomes or recurrence of CVST.1
As previously noted, the patient presented with transient slurred speech, mild headache, and bilateral hand tingling. On workup, she was found to have an SSS thrombosis with an associated right intraparenchymal hemorrhage that occurred despite therapeutic INR levels and the initiation of coumadin therapy prior to admission.
Evaluation and Diagnosis
D-Dimer Evaluation
There is a strong association between D-dimer levels above 500 ng/mL and acute CVST. Nevertheless, lower levels do not rule out the diagnosis in a patient presenting with headache.4
Imaging Techniques
Important imaging techniques in the evaluation of CVST include CT, MRV, MRI, and magnetic resonance angiography (MRA). The first imaging modality in evaluating a patient with neurological symptoms and headache in the ED is CT, which can show evidence of an infarction that does not respond to an arterial distribution. In the absence of a hemorrhagic component, however, infarct demonstration may be delayed for up to 72 hours.5 On contrast CT, an empty delta sign may be apparent due to enhancement of the collateral veins in the SSS walls surrounding a nonenhanced thrombus. The delta sign is not frequently present and may be false due to early division of the SSS.5
Computed tomography venography, CT angiography, and MRI can also be utilized to evaluate for CVST. Computed tomography venography is especially useful in identifying the cerebral veins and dural sinuses,6 and MRV is an excellent method for visualizing the dural venous sinuses and larger cerebral veins. Single-slice phase-contrast angiography is also a rapid and reliable test for CVST.7 Conventional angiography and direct venography should be considered if MR studies are nondiagnostic; however, this test is invasive with associated risks.5
Treatment
Heparin therapy should be initiated in patients presenting with dural sinus thrombosis even if pre-existing hemorrhage exists. Patients failing to respond to therapy with worsening neurological deficits may warrant local thrombolysis with tissue plasminogen activator. Identifying those in the acute state of disease is essential as they may have poor prognostic outcomes that may warrant more invasive intervention.1
Conclusion
Cerebral venous sinus thrombosis is a rare condition with a diverse clinical presentation. As demonstrated in this case, some patients present with stroke-like symptoms of nontraumatic headache, slurred speech, and bilateral hand tingling, which, on workup, reveal SSS thrombosis associated right intraparenchymal hemorrhage.
This case draws attention to the importance of risk stratification in patients with a history of HCS and neurological complaints presenting to the ED. Dural sinus thrombosis may have a vague initial neurological presentation; therefore, early recognition and initiation of therapy will assist in reducing morbidity and mortality.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
- Kimber J. Cerebral venous sinus thrombosis. QJM. 2002;95(3):137-142.
- Anderson JA, Weitz JI. Hypercoagulable states. In: Hoffman R, Benz EJ, Jr, Silberstein LE, Heslop HE, Weitz JI, Anastasi J, eds. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:2013-2024.
- Humphries SE, Panahloo A, Montgomery HE, Green F, Yudkin J. Gene-environment interaction in the determination of levels of haemostatic variables involved in thrombosis and fibrinolysis. Thromb Haemost. 1997;78(1):457-461.
- Weimar C. Diagnosis and treatment of cerebral venous and sinus thrombosis. Curr Neurol Neurosci Rep. 2014;14(1):417.
- Masuhr F, Mehraein S, Einhäupl K. Cerebral venous and sinus thrombosis. J Neurol. 2004;251(1):11-23.
- Ozsvath RR, Casey SO, Lustrin ES, et al. Cerebral venography: comparison of CT and MR projection venography. AJR Am J Roentgenol. 1997;169(6):1699-1707.
- Adams WM, Laitt RD, Beards SC, Kassner A, Jackson A. Use of single-slice thick slab phase-contrast angiography for the diagnosis of dural venous sinus thrombosis. Eur Radiol. 1999;9(8):1614-1619.
Case
The patient’s past medical history included one miscarriage, as well as a papillary thyroid carcinoma with resection, which was discovered a few months before her presentation to the ED and after diagnosis of the initial SSS thrombosis.
Physical examination revealed a well-developed, mildly obese female. On arrival at the ED, the patient’s National Institutes of Health Stroke Scale score was 0. Her vital signs and ocular, neurological, and psychiatric examinations were all normal. The social history was negative for tobacco or alcohol use, and she had no family history of deep vein thrombosis (DVT) or pulmonary embolism.
A noncontrast computed tomography (CT) of the head demonstrated a hemorrhagic venous infarction involving the posterior right parietal lobe. Intracranial magnetic resonance venography (MRV) and brain magnetic resonance imaging (MRI) revealed thrombosis of the posterior third of the SSS as the source of the infarction. This sinus had been patent during the patient’s previous hospital admissions.
The patient’s international normalized ratio (INR) was therapeutic on presentation. Warfarin was discontinued, and she was started on an intravenous (IV) heparin drip. For anticoagulation, she was prescribed 20 mg rivaroxaban daily and 2,000 mg levetiracetam daily.
One week after discharge, the patient again presented to the ED with a recurrence of symptoms, including confusion, slurred speech, and headache, which she rated a “5” on a pain scale of 0 to 10. Similar to the previous ED visit, the slurred speech had resolved by the time of examination. The patient did not exhibit facial asymmetry but did complain of bilateral numbness and tingling in both hands. A noncontrast CT of the head showed no changes in the right parietal hemorrhagic venous infarct and intraparenchymal hemorrhage; however, there was an interval increase in edema compared to the prior CT. Rivaroxaban and levetiracetam were continued, and 20 mg simvastatin daily was prescribed.
Overview
Cerebral venous sinus thrombosis is a rare condition with an often varied clinical presentation—the symptoms of which can take hours to weeks to evolve, thus making the diagnosis challenging. In 70% of cases, the SSS and lateral sinuses are individually involved, and in 30% of cases, both regions are affected simultaneously.1 Only recently have clinicians been able to diagnose this condition antemortem.
Risk Factors and Etiology
Inherited and Acquired hypercoagulable states
Cerebral venous sinus thrombosis (CVST) and cerebrovascular accident (CVA) often result from a hypercoagulable state (HCS), and both acquired and inherited factors place patients at risk. Inherited factors are the most common cause of venous thromboembolism in patients younger than age 40 years. Acquired factors have a combined effect with inherited ones, leading to increased risk of CVST or CVA.2
The patient in this case possessed both acquired and inherited factors of an HCS. Inherited factors can be found through a thrombophilia evaluation. In general, acquired factors of thrombophilia include obesity, a prior history of thrombosis, pregnancy, and cancer and its treatment. A thrombophilia evaluation revealed the patient was homozygous for the 4G allele, which has been shown to increase concentration of plasminogen activator inhibitor (PAI-1) by 30%. An inhibitor to the pathway of fibrinolysis, PAI-1 is a major factor preventing the excessive presence and magnitude of blood clots.3
Pregnancy and the Puerperium
Cerebral vascular sinus thrombosis is most commonly seen in young to middle-aged women. High risk factors include pregnancy and the puerperium due to increased HCS during these periods.4 The incidence of CVST in this population is approximately 10 per 100,000 women.4
Oral Hormonal Contraceptives
In approximately 10% of CVST cases, oral hormonal contraceptive use in the presence of a coagulation disorder are frequently the cause—as observed in the incidence of DVT in this patient population.
Septic Cerebral Venous Sinus Thrombosis
Septic CVST occurs mainly in children and up to 18% of adult cases in developing countries. It is associated with localized infections (eg, mastoiditis, otitis media, sinusitis, meningitis).
Other Causes
Although rare, other causes of CVST include intracranial hypotension, hydrocephalus, and the use of certain drugs and supplements (eg, corticosteroids, high doses of vitamin A). Each of these potential causes also should be considered when evaluating for CVST.4
Symptoms and Signs
Common symptoms and signs of CVST include headache, nausea, vomiting, seizure, and focal neurological deficit. Papilledema is present in 40% of cases, primarily in patients with delayed diagnosis or a chronic course.
Neurological Deficits
Cerebral venous sinus thrombosis may not necessarily cause focal neurological deficits due to numerous pathways of venous drainage and the possibility of reversal of venous blood flow. However, the condition can lead to impaired resorption of CSF causing intracranial hypertension.
Headache
In 70% of cases, headache is the initial symptom of CVST, and it is the only symptom in 16% of cases. With respect to headache presentation, it is important to remember that thunderclap headache is not exclusive to the diagnosis of subarachnoid hemorrhage (SAH). The absence of findings on workup to support the diagnosis of SAH should prompt investigation with MRV and evaluation of CVST.4
Seizure
Focal or generalized seizure on initial presentation occurs in 30% to 40% of cases of CVST. When smaller cerebral veins are involved, this can lead to focal edema, neurological deficits, venous infarction, and seizure. Focal deficits are determined by the localization of CVST and associated lesions. Other symptoms may include migraine headache, transient ischemic attack, cranial nerve palsies, and subarachnoid hemorrhage.4
Complications
Cerebral venous sinus thrombosis is a rare condition with an often varied clinical presentation—the symptoms of which can take hours to weeks to evolve, thus making the diagnosis challenging.
Complications in patients with CVST occur when venous congestion increases and raises dural venous sinus and cerebral spinal fluid (CSF) pressure. Parenchymal edema with venous infarction and hemorrhage complicates up to 50% of venous sinus thromboses (as seen in this patient).1 Unfortunately, little is known about long-term risk outcomes or recurrence of CVST.1
As previously noted, the patient presented with transient slurred speech, mild headache, and bilateral hand tingling. On workup, she was found to have an SSS thrombosis with an associated right intraparenchymal hemorrhage that occurred despite therapeutic INR levels and the initiation of coumadin therapy prior to admission.
Evaluation and Diagnosis
D-Dimer Evaluation
There is a strong association between D-dimer levels above 500 ng/mL and acute CVST. Nevertheless, lower levels do not rule out the diagnosis in a patient presenting with headache.4
Imaging Techniques
Important imaging techniques in the evaluation of CVST include CT, MRV, MRI, and magnetic resonance angiography (MRA). The first imaging modality in evaluating a patient with neurological symptoms and headache in the ED is CT, which can show evidence of an infarction that does not respond to an arterial distribution. In the absence of a hemorrhagic component, however, infarct demonstration may be delayed for up to 72 hours.5 On contrast CT, an empty delta sign may be apparent due to enhancement of the collateral veins in the SSS walls surrounding a nonenhanced thrombus. The delta sign is not frequently present and may be false due to early division of the SSS.5
Computed tomography venography, CT angiography, and MRI can also be utilized to evaluate for CVST. Computed tomography venography is especially useful in identifying the cerebral veins and dural sinuses,6 and MRV is an excellent method for visualizing the dural venous sinuses and larger cerebral veins. Single-slice phase-contrast angiography is also a rapid and reliable test for CVST.7 Conventional angiography and direct venography should be considered if MR studies are nondiagnostic; however, this test is invasive with associated risks.5
Treatment
Heparin therapy should be initiated in patients presenting with dural sinus thrombosis even if pre-existing hemorrhage exists. Patients failing to respond to therapy with worsening neurological deficits may warrant local thrombolysis with tissue plasminogen activator. Identifying those in the acute state of disease is essential as they may have poor prognostic outcomes that may warrant more invasive intervention.1
Conclusion
Cerebral venous sinus thrombosis is a rare condition with a diverse clinical presentation. As demonstrated in this case, some patients present with stroke-like symptoms of nontraumatic headache, slurred speech, and bilateral hand tingling, which, on workup, reveal SSS thrombosis associated right intraparenchymal hemorrhage.
This case draws attention to the importance of risk stratification in patients with a history of HCS and neurological complaints presenting to the ED. Dural sinus thrombosis may have a vague initial neurological presentation; therefore, early recognition and initiation of therapy will assist in reducing morbidity and mortality.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
Case
The patient’s past medical history included one miscarriage, as well as a papillary thyroid carcinoma with resection, which was discovered a few months before her presentation to the ED and after diagnosis of the initial SSS thrombosis.
Physical examination revealed a well-developed, mildly obese female. On arrival at the ED, the patient’s National Institutes of Health Stroke Scale score was 0. Her vital signs and ocular, neurological, and psychiatric examinations were all normal. The social history was negative for tobacco or alcohol use, and she had no family history of deep vein thrombosis (DVT) or pulmonary embolism.
A noncontrast computed tomography (CT) of the head demonstrated a hemorrhagic venous infarction involving the posterior right parietal lobe. Intracranial magnetic resonance venography (MRV) and brain magnetic resonance imaging (MRI) revealed thrombosis of the posterior third of the SSS as the source of the infarction. This sinus had been patent during the patient’s previous hospital admissions.
The patient’s international normalized ratio (INR) was therapeutic on presentation. Warfarin was discontinued, and she was started on an intravenous (IV) heparin drip. For anticoagulation, she was prescribed 20 mg rivaroxaban daily and 2,000 mg levetiracetam daily.
One week after discharge, the patient again presented to the ED with a recurrence of symptoms, including confusion, slurred speech, and headache, which she rated a “5” on a pain scale of 0 to 10. Similar to the previous ED visit, the slurred speech had resolved by the time of examination. The patient did not exhibit facial asymmetry but did complain of bilateral numbness and tingling in both hands. A noncontrast CT of the head showed no changes in the right parietal hemorrhagic venous infarct and intraparenchymal hemorrhage; however, there was an interval increase in edema compared to the prior CT. Rivaroxaban and levetiracetam were continued, and 20 mg simvastatin daily was prescribed.
Overview
Cerebral venous sinus thrombosis is a rare condition with an often varied clinical presentation—the symptoms of which can take hours to weeks to evolve, thus making the diagnosis challenging. In 70% of cases, the SSS and lateral sinuses are individually involved, and in 30% of cases, both regions are affected simultaneously.1 Only recently have clinicians been able to diagnose this condition antemortem.
Risk Factors and Etiology
Inherited and Acquired hypercoagulable states
Cerebral venous sinus thrombosis (CVST) and cerebrovascular accident (CVA) often result from a hypercoagulable state (HCS), and both acquired and inherited factors place patients at risk. Inherited factors are the most common cause of venous thromboembolism in patients younger than age 40 years. Acquired factors have a combined effect with inherited ones, leading to increased risk of CVST or CVA.2
The patient in this case possessed both acquired and inherited factors of an HCS. Inherited factors can be found through a thrombophilia evaluation. In general, acquired factors of thrombophilia include obesity, a prior history of thrombosis, pregnancy, and cancer and its treatment. A thrombophilia evaluation revealed the patient was homozygous for the 4G allele, which has been shown to increase concentration of plasminogen activator inhibitor (PAI-1) by 30%. An inhibitor to the pathway of fibrinolysis, PAI-1 is a major factor preventing the excessive presence and magnitude of blood clots.3
Pregnancy and the Puerperium
Cerebral vascular sinus thrombosis is most commonly seen in young to middle-aged women. High risk factors include pregnancy and the puerperium due to increased HCS during these periods.4 The incidence of CVST in this population is approximately 10 per 100,000 women.4
Oral Hormonal Contraceptives
In approximately 10% of CVST cases, oral hormonal contraceptive use in the presence of a coagulation disorder are frequently the cause—as observed in the incidence of DVT in this patient population.
Septic Cerebral Venous Sinus Thrombosis
Septic CVST occurs mainly in children and up to 18% of adult cases in developing countries. It is associated with localized infections (eg, mastoiditis, otitis media, sinusitis, meningitis).
Other Causes
Although rare, other causes of CVST include intracranial hypotension, hydrocephalus, and the use of certain drugs and supplements (eg, corticosteroids, high doses of vitamin A). Each of these potential causes also should be considered when evaluating for CVST.4
Symptoms and Signs
Common symptoms and signs of CVST include headache, nausea, vomiting, seizure, and focal neurological deficit. Papilledema is present in 40% of cases, primarily in patients with delayed diagnosis or a chronic course.
Neurological Deficits
Cerebral venous sinus thrombosis may not necessarily cause focal neurological deficits due to numerous pathways of venous drainage and the possibility of reversal of venous blood flow. However, the condition can lead to impaired resorption of CSF causing intracranial hypertension.
Headache
In 70% of cases, headache is the initial symptom of CVST, and it is the only symptom in 16% of cases. With respect to headache presentation, it is important to remember that thunderclap headache is not exclusive to the diagnosis of subarachnoid hemorrhage (SAH). The absence of findings on workup to support the diagnosis of SAH should prompt investigation with MRV and evaluation of CVST.4
Seizure
Focal or generalized seizure on initial presentation occurs in 30% to 40% of cases of CVST. When smaller cerebral veins are involved, this can lead to focal edema, neurological deficits, venous infarction, and seizure. Focal deficits are determined by the localization of CVST and associated lesions. Other symptoms may include migraine headache, transient ischemic attack, cranial nerve palsies, and subarachnoid hemorrhage.4
Complications
Cerebral venous sinus thrombosis is a rare condition with an often varied clinical presentation—the symptoms of which can take hours to weeks to evolve, thus making the diagnosis challenging.
Complications in patients with CVST occur when venous congestion increases and raises dural venous sinus and cerebral spinal fluid (CSF) pressure. Parenchymal edema with venous infarction and hemorrhage complicates up to 50% of venous sinus thromboses (as seen in this patient).1 Unfortunately, little is known about long-term risk outcomes or recurrence of CVST.1
As previously noted, the patient presented with transient slurred speech, mild headache, and bilateral hand tingling. On workup, she was found to have an SSS thrombosis with an associated right intraparenchymal hemorrhage that occurred despite therapeutic INR levels and the initiation of coumadin therapy prior to admission.
Evaluation and Diagnosis
D-Dimer Evaluation
There is a strong association between D-dimer levels above 500 ng/mL and acute CVST. Nevertheless, lower levels do not rule out the diagnosis in a patient presenting with headache.4
Imaging Techniques
Important imaging techniques in the evaluation of CVST include CT, MRV, MRI, and magnetic resonance angiography (MRA). The first imaging modality in evaluating a patient with neurological symptoms and headache in the ED is CT, which can show evidence of an infarction that does not respond to an arterial distribution. In the absence of a hemorrhagic component, however, infarct demonstration may be delayed for up to 72 hours.5 On contrast CT, an empty delta sign may be apparent due to enhancement of the collateral veins in the SSS walls surrounding a nonenhanced thrombus. The delta sign is not frequently present and may be false due to early division of the SSS.5
Computed tomography venography, CT angiography, and MRI can also be utilized to evaluate for CVST. Computed tomography venography is especially useful in identifying the cerebral veins and dural sinuses,6 and MRV is an excellent method for visualizing the dural venous sinuses and larger cerebral veins. Single-slice phase-contrast angiography is also a rapid and reliable test for CVST.7 Conventional angiography and direct venography should be considered if MR studies are nondiagnostic; however, this test is invasive with associated risks.5
Treatment
Heparin therapy should be initiated in patients presenting with dural sinus thrombosis even if pre-existing hemorrhage exists. Patients failing to respond to therapy with worsening neurological deficits may warrant local thrombolysis with tissue plasminogen activator. Identifying those in the acute state of disease is essential as they may have poor prognostic outcomes that may warrant more invasive intervention.1
Conclusion
Cerebral venous sinus thrombosis is a rare condition with a diverse clinical presentation. As demonstrated in this case, some patients present with stroke-like symptoms of nontraumatic headache, slurred speech, and bilateral hand tingling, which, on workup, reveal SSS thrombosis associated right intraparenchymal hemorrhage.
This case draws attention to the importance of risk stratification in patients with a history of HCS and neurological complaints presenting to the ED. Dural sinus thrombosis may have a vague initial neurological presentation; therefore, early recognition and initiation of therapy will assist in reducing morbidity and mortality.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
- Kimber J. Cerebral venous sinus thrombosis. QJM. 2002;95(3):137-142.
- Anderson JA, Weitz JI. Hypercoagulable states. In: Hoffman R, Benz EJ, Jr, Silberstein LE, Heslop HE, Weitz JI, Anastasi J, eds. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:2013-2024.
- Humphries SE, Panahloo A, Montgomery HE, Green F, Yudkin J. Gene-environment interaction in the determination of levels of haemostatic variables involved in thrombosis and fibrinolysis. Thromb Haemost. 1997;78(1):457-461.
- Weimar C. Diagnosis and treatment of cerebral venous and sinus thrombosis. Curr Neurol Neurosci Rep. 2014;14(1):417.
- Masuhr F, Mehraein S, Einhäupl K. Cerebral venous and sinus thrombosis. J Neurol. 2004;251(1):11-23.
- Ozsvath RR, Casey SO, Lustrin ES, et al. Cerebral venography: comparison of CT and MR projection venography. AJR Am J Roentgenol. 1997;169(6):1699-1707.
- Adams WM, Laitt RD, Beards SC, Kassner A, Jackson A. Use of single-slice thick slab phase-contrast angiography for the diagnosis of dural venous sinus thrombosis. Eur Radiol. 1999;9(8):1614-1619.
- Kimber J. Cerebral venous sinus thrombosis. QJM. 2002;95(3):137-142.
- Anderson JA, Weitz JI. Hypercoagulable states. In: Hoffman R, Benz EJ, Jr, Silberstein LE, Heslop HE, Weitz JI, Anastasi J, eds. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:2013-2024.
- Humphries SE, Panahloo A, Montgomery HE, Green F, Yudkin J. Gene-environment interaction in the determination of levels of haemostatic variables involved in thrombosis and fibrinolysis. Thromb Haemost. 1997;78(1):457-461.
- Weimar C. Diagnosis and treatment of cerebral venous and sinus thrombosis. Curr Neurol Neurosci Rep. 2014;14(1):417.
- Masuhr F, Mehraein S, Einhäupl K. Cerebral venous and sinus thrombosis. J Neurol. 2004;251(1):11-23.
- Ozsvath RR, Casey SO, Lustrin ES, et al. Cerebral venography: comparison of CT and MR projection venography. AJR Am J Roentgenol. 1997;169(6):1699-1707.
- Adams WM, Laitt RD, Beards SC, Kassner A, Jackson A. Use of single-slice thick slab phase-contrast angiography for the diagnosis of dural venous sinus thrombosis. Eur Radiol. 1999;9(8):1614-1619.

Case Report: Rapidly Ascending Weakness in a 22-year-old Man
Case
A 22-year-old white man presented to the ED after waking from sleep and experiencing painless bilateral lower extremity weakness that ascended to the right upper extremity. This weakness left the patient unable to walk. He denied incontinence and was otherwise healthy with no prior history of similar symptoms. A family history of the presenting symptomatology was denied, and there was no family history of diabetes or kidney disease. The patient did state he had upper respiratory symptoms with a sore throat and runny nose 2 weeks earlier. He denied use of tobacco, alcohol, and illicit drugs.
Physical examination revealed abnormal deep tendon reflexes, absent patellar and achilles reflexes on the right side, reduced patellar and achilles reflexes of the left side, and diminished bilateral brachioradialis reflexes. Examination of the lower extremities revealed motor strength at 2/5 on the left and 1/5 on the right. His upper extremities demonstrated bilateral 4/5 motor strength. No ptosis was observed. All pulses of the extremities were normal with good capillary refill. His vital signs were: blood pressure, 131/75 mm Hg; pulse, 75 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 97.2°F
Brain and cervical spine magnetic resonance imaging, computed tomography of the head, and blood cultures were ordered; the results of each were unremarkable. A lumbar puncture (LP) was attempted in the ED, but was unsuccessful due to difficulty in patient positioning secondary to profound weakness. An LP was later succesfully performed under fluoroscopy and revealed no abnormal findings. An electrocardiogram (ECG) demonstrated normal sinus rhythm with ST-segment flattening. The differential diagnosis for rapidly ascending weakness included Guillain-Barré syndrome, transverse myelitis, toxic metabolic syndromes, and myelopathies.
A basic metabolic panel showed that the patient was profoundly hypokalemic, with a potassium level of 1.9 mEq/L. After further blood analysis, mild hypomagnesemia was observed, with a level of 1.4 mEq/L. Whle in the ED, he was given 40 mEq potassium chloride intravenously (IV) and orally, and 2 g IV magnesium sulfate in the ED. The patient was evaluated by a neurologist in the ED. No respiratory compromise was present and the patient was admitted to neurology service with consultations from both nephrology and endocrinology.
Within 12 hours of starting potassium and magnesium supplementation, the patient’s potassium and magnesium levels returned to normal and his weakness concurrently subsided. The final diagnosis of the patient was hypokalemic periodic paralysis.
Discussion
Hypokalemic periodic paralysis (HPP), a rare autosomal dominant disorder with a prevalence of one in 100,000, is characterized by muscle weakness with accompanying hypokalemia. The first onset of symptoms is usually in the first or second decade of life, with a quarter of cases presenting before the age 10 years, and half before age 16 years. Being a typically-inherited autosomal dominant disorder, approximately two-thirds of cases present with a family history—unlike this patient case in which there was no family history of the disease. It is three to four times more common in men than women.
Hypokalemic periodic paralysis typically occurs upon waking from sleep or during rest following exercise. It may also be triggered by high-carbohydrate or high-salt meals, or from alcohol consumption. The muscles of the extremeties are usually affected, while those of the eyes, face, and sphincters are typically spared. If untreated, an attack can last from several hours to several days. Tendon and cutaneous reflexes can also be reduced or absent.1 The manifestation of HPP can, at first impression, lead the differential diagnosis in the direction of the many other conditions that cause weakness of the extremities. Guillain-Barré syndrome and transverse myelitis are the more common diagnoses for which HPP can be mistaken. It is therefore important that the EP consider HPP in the differential so that an unusual presentation such as the one in this case may be diagnosed.
When presented with extraneous complications (eg, when the condition is encountered perioperatively or suffered comorbidly with similar conditions such as Guillain-Barré syndrome), HPP can be especially difficult to recognize.2,3 Although a relatively uncommon condition, it can be potentially life-threatening if not recognized and treated appropriately. Severe hypokalemia may cause sequela such as respiratory failure and cardiac arhythmia. Thus, ECG and cardiorespiratory monitoring are essential in patients with HPP and hypokalemia as very severe hypokalemia may also cause paralysis of bulbar and cranial nerve musculature. Electocardiographic changes are common, and may include ST-segment sagging and flattening, U waves, and T-wave inversion. Other causes in patients presenting with HPP must also be considered, as hypokalemic paralysis of a sporadic nature may be associated with conditions including barium poisoning, hyperthyroidism, renal disorders, certain endocrinopathies, and gastrointestinal potassium losses.4
The pathogenesis of HPP is not completely understood; however, alteration in potassium regulation are well documented. Treatment of HPP includes both oral and IV potassium supplementation. Prophylaxis against recurrent attacks has been successful with various modalities including spiranolactone daily and acetazolamide.4 Care must be taken to consider thyrotoxic periodic paralysis, which most commonly presents in Asian men, as hyperthyroid symptoms may be subtle. Treatment focuses on reversal of the thyrotoxic state. β-Adrenergic blocking agents reduce the frequency and severity of attacks and should be started while measures to control thyrotoxicosis are being instituted.4
Before a diagnosis of HPP is made, other causes of hypokalemic paralysis must first be excluded.
Conclusion
This case is important because it demonstrates an unusual presentation of HPP in an emergency setting. This perspective of HPP can help the EP in recognizing and differentiating the condsition from similar disorders.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
- Ropper AH, Samuels MA. The myotonias, periodic paralyses, cramps, spasms, and states of persistent muscle fiber activity. In: Ropper AH, Samuels MA, Klein JP, eds. Adams & Victor’s Principles of Neurology. 10th ed. New York, NY: McGraw-Hill Education; 2014:1490-1508.
- Abbas H, Kothari N, Bogra J. Hypokalemic periodic paralysis. Natl J Maxillofac Surg. 2012;3(2):220-221.
- Saroja AO, Naik KR, Khanpet MS. Uncommon dyselectrolytemia complicating Guillain-Barré syndrome. J Neurosci Rural Pract. 2013;4(3):328-330.
- Ahlawat SK, Sachdev A. Hypokalemic paralysis. Postgrad Med J. 1999;75(882):193-197.
Case
A 22-year-old white man presented to the ED after waking from sleep and experiencing painless bilateral lower extremity weakness that ascended to the right upper extremity. This weakness left the patient unable to walk. He denied incontinence and was otherwise healthy with no prior history of similar symptoms. A family history of the presenting symptomatology was denied, and there was no family history of diabetes or kidney disease. The patient did state he had upper respiratory symptoms with a sore throat and runny nose 2 weeks earlier. He denied use of tobacco, alcohol, and illicit drugs.
Physical examination revealed abnormal deep tendon reflexes, absent patellar and achilles reflexes on the right side, reduced patellar and achilles reflexes of the left side, and diminished bilateral brachioradialis reflexes. Examination of the lower extremities revealed motor strength at 2/5 on the left and 1/5 on the right. His upper extremities demonstrated bilateral 4/5 motor strength. No ptosis was observed. All pulses of the extremities were normal with good capillary refill. His vital signs were: blood pressure, 131/75 mm Hg; pulse, 75 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 97.2°F
Brain and cervical spine magnetic resonance imaging, computed tomography of the head, and blood cultures were ordered; the results of each were unremarkable. A lumbar puncture (LP) was attempted in the ED, but was unsuccessful due to difficulty in patient positioning secondary to profound weakness. An LP was later succesfully performed under fluoroscopy and revealed no abnormal findings. An electrocardiogram (ECG) demonstrated normal sinus rhythm with ST-segment flattening. The differential diagnosis for rapidly ascending weakness included Guillain-Barré syndrome, transverse myelitis, toxic metabolic syndromes, and myelopathies.
A basic metabolic panel showed that the patient was profoundly hypokalemic, with a potassium level of 1.9 mEq/L. After further blood analysis, mild hypomagnesemia was observed, with a level of 1.4 mEq/L. Whle in the ED, he was given 40 mEq potassium chloride intravenously (IV) and orally, and 2 g IV magnesium sulfate in the ED. The patient was evaluated by a neurologist in the ED. No respiratory compromise was present and the patient was admitted to neurology service with consultations from both nephrology and endocrinology.
Within 12 hours of starting potassium and magnesium supplementation, the patient’s potassium and magnesium levels returned to normal and his weakness concurrently subsided. The final diagnosis of the patient was hypokalemic periodic paralysis.
Discussion
Hypokalemic periodic paralysis (HPP), a rare autosomal dominant disorder with a prevalence of one in 100,000, is characterized by muscle weakness with accompanying hypokalemia. The first onset of symptoms is usually in the first or second decade of life, with a quarter of cases presenting before the age 10 years, and half before age 16 years. Being a typically-inherited autosomal dominant disorder, approximately two-thirds of cases present with a family history—unlike this patient case in which there was no family history of the disease. It is three to four times more common in men than women.
Hypokalemic periodic paralysis typically occurs upon waking from sleep or during rest following exercise. It may also be triggered by high-carbohydrate or high-salt meals, or from alcohol consumption. The muscles of the extremeties are usually affected, while those of the eyes, face, and sphincters are typically spared. If untreated, an attack can last from several hours to several days. Tendon and cutaneous reflexes can also be reduced or absent.1 The manifestation of HPP can, at first impression, lead the differential diagnosis in the direction of the many other conditions that cause weakness of the extremities. Guillain-Barré syndrome and transverse myelitis are the more common diagnoses for which HPP can be mistaken. It is therefore important that the EP consider HPP in the differential so that an unusual presentation such as the one in this case may be diagnosed.
When presented with extraneous complications (eg, when the condition is encountered perioperatively or suffered comorbidly with similar conditions such as Guillain-Barré syndrome), HPP can be especially difficult to recognize.2,3 Although a relatively uncommon condition, it can be potentially life-threatening if not recognized and treated appropriately. Severe hypokalemia may cause sequela such as respiratory failure and cardiac arhythmia. Thus, ECG and cardiorespiratory monitoring are essential in patients with HPP and hypokalemia as very severe hypokalemia may also cause paralysis of bulbar and cranial nerve musculature. Electocardiographic changes are common, and may include ST-segment sagging and flattening, U waves, and T-wave inversion. Other causes in patients presenting with HPP must also be considered, as hypokalemic paralysis of a sporadic nature may be associated with conditions including barium poisoning, hyperthyroidism, renal disorders, certain endocrinopathies, and gastrointestinal potassium losses.4
The pathogenesis of HPP is not completely understood; however, alteration in potassium regulation are well documented. Treatment of HPP includes both oral and IV potassium supplementation. Prophylaxis against recurrent attacks has been successful with various modalities including spiranolactone daily and acetazolamide.4 Care must be taken to consider thyrotoxic periodic paralysis, which most commonly presents in Asian men, as hyperthyroid symptoms may be subtle. Treatment focuses on reversal of the thyrotoxic state. β-Adrenergic blocking agents reduce the frequency and severity of attacks and should be started while measures to control thyrotoxicosis are being instituted.4
Before a diagnosis of HPP is made, other causes of hypokalemic paralysis must first be excluded.
Conclusion
This case is important because it demonstrates an unusual presentation of HPP in an emergency setting. This perspective of HPP can help the EP in recognizing and differentiating the condsition from similar disorders.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
Case
A 22-year-old white man presented to the ED after waking from sleep and experiencing painless bilateral lower extremity weakness that ascended to the right upper extremity. This weakness left the patient unable to walk. He denied incontinence and was otherwise healthy with no prior history of similar symptoms. A family history of the presenting symptomatology was denied, and there was no family history of diabetes or kidney disease. The patient did state he had upper respiratory symptoms with a sore throat and runny nose 2 weeks earlier. He denied use of tobacco, alcohol, and illicit drugs.
Physical examination revealed abnormal deep tendon reflexes, absent patellar and achilles reflexes on the right side, reduced patellar and achilles reflexes of the left side, and diminished bilateral brachioradialis reflexes. Examination of the lower extremities revealed motor strength at 2/5 on the left and 1/5 on the right. His upper extremities demonstrated bilateral 4/5 motor strength. No ptosis was observed. All pulses of the extremities were normal with good capillary refill. His vital signs were: blood pressure, 131/75 mm Hg; pulse, 75 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 97.2°F
Brain and cervical spine magnetic resonance imaging, computed tomography of the head, and blood cultures were ordered; the results of each were unremarkable. A lumbar puncture (LP) was attempted in the ED, but was unsuccessful due to difficulty in patient positioning secondary to profound weakness. An LP was later succesfully performed under fluoroscopy and revealed no abnormal findings. An electrocardiogram (ECG) demonstrated normal sinus rhythm with ST-segment flattening. The differential diagnosis for rapidly ascending weakness included Guillain-Barré syndrome, transverse myelitis, toxic metabolic syndromes, and myelopathies.
A basic metabolic panel showed that the patient was profoundly hypokalemic, with a potassium level of 1.9 mEq/L. After further blood analysis, mild hypomagnesemia was observed, with a level of 1.4 mEq/L. Whle in the ED, he was given 40 mEq potassium chloride intravenously (IV) and orally, and 2 g IV magnesium sulfate in the ED. The patient was evaluated by a neurologist in the ED. No respiratory compromise was present and the patient was admitted to neurology service with consultations from both nephrology and endocrinology.
Within 12 hours of starting potassium and magnesium supplementation, the patient’s potassium and magnesium levels returned to normal and his weakness concurrently subsided. The final diagnosis of the patient was hypokalemic periodic paralysis.
Discussion
Hypokalemic periodic paralysis (HPP), a rare autosomal dominant disorder with a prevalence of one in 100,000, is characterized by muscle weakness with accompanying hypokalemia. The first onset of symptoms is usually in the first or second decade of life, with a quarter of cases presenting before the age 10 years, and half before age 16 years. Being a typically-inherited autosomal dominant disorder, approximately two-thirds of cases present with a family history—unlike this patient case in which there was no family history of the disease. It is three to four times more common in men than women.
Hypokalemic periodic paralysis typically occurs upon waking from sleep or during rest following exercise. It may also be triggered by high-carbohydrate or high-salt meals, or from alcohol consumption. The muscles of the extremeties are usually affected, while those of the eyes, face, and sphincters are typically spared. If untreated, an attack can last from several hours to several days. Tendon and cutaneous reflexes can also be reduced or absent.1 The manifestation of HPP can, at first impression, lead the differential diagnosis in the direction of the many other conditions that cause weakness of the extremities. Guillain-Barré syndrome and transverse myelitis are the more common diagnoses for which HPP can be mistaken. It is therefore important that the EP consider HPP in the differential so that an unusual presentation such as the one in this case may be diagnosed.
When presented with extraneous complications (eg, when the condition is encountered perioperatively or suffered comorbidly with similar conditions such as Guillain-Barré syndrome), HPP can be especially difficult to recognize.2,3 Although a relatively uncommon condition, it can be potentially life-threatening if not recognized and treated appropriately. Severe hypokalemia may cause sequela such as respiratory failure and cardiac arhythmia. Thus, ECG and cardiorespiratory monitoring are essential in patients with HPP and hypokalemia as very severe hypokalemia may also cause paralysis of bulbar and cranial nerve musculature. Electocardiographic changes are common, and may include ST-segment sagging and flattening, U waves, and T-wave inversion. Other causes in patients presenting with HPP must also be considered, as hypokalemic paralysis of a sporadic nature may be associated with conditions including barium poisoning, hyperthyroidism, renal disorders, certain endocrinopathies, and gastrointestinal potassium losses.4
The pathogenesis of HPP is not completely understood; however, alteration in potassium regulation are well documented. Treatment of HPP includes both oral and IV potassium supplementation. Prophylaxis against recurrent attacks has been successful with various modalities including spiranolactone daily and acetazolamide.4 Care must be taken to consider thyrotoxic periodic paralysis, which most commonly presents in Asian men, as hyperthyroid symptoms may be subtle. Treatment focuses on reversal of the thyrotoxic state. β-Adrenergic blocking agents reduce the frequency and severity of attacks and should be started while measures to control thyrotoxicosis are being instituted.4
Before a diagnosis of HPP is made, other causes of hypokalemic paralysis must first be excluded.
Conclusion
This case is important because it demonstrates an unusual presentation of HPP in an emergency setting. This perspective of HPP can help the EP in recognizing and differentiating the condsition from similar disorders.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
- Ropper AH, Samuels MA. The myotonias, periodic paralyses, cramps, spasms, and states of persistent muscle fiber activity. In: Ropper AH, Samuels MA, Klein JP, eds. Adams & Victor’s Principles of Neurology. 10th ed. New York, NY: McGraw-Hill Education; 2014:1490-1508.
- Abbas H, Kothari N, Bogra J. Hypokalemic periodic paralysis. Natl J Maxillofac Surg. 2012;3(2):220-221.
- Saroja AO, Naik KR, Khanpet MS. Uncommon dyselectrolytemia complicating Guillain-Barré syndrome. J Neurosci Rural Pract. 2013;4(3):328-330.
- Ahlawat SK, Sachdev A. Hypokalemic paralysis. Postgrad Med J. 1999;75(882):193-197.
- Ropper AH, Samuels MA. The myotonias, periodic paralyses, cramps, spasms, and states of persistent muscle fiber activity. In: Ropper AH, Samuels MA, Klein JP, eds. Adams & Victor’s Principles of Neurology. 10th ed. New York, NY: McGraw-Hill Education; 2014:1490-1508.
- Abbas H, Kothari N, Bogra J. Hypokalemic periodic paralysis. Natl J Maxillofac Surg. 2012;3(2):220-221.
- Saroja AO, Naik KR, Khanpet MS. Uncommon dyselectrolytemia complicating Guillain-Barré syndrome. J Neurosci Rural Pract. 2013;4(3):328-330.
- Ahlawat SK, Sachdev A. Hypokalemic paralysis. Postgrad Med J. 1999;75(882):193-197.