User login
How to diagnose and repair congenital uterovaginal malformations
The author reports no financial relationships relevant to this article.
CASE 1 Fluid-filled abdominal mass
A 14-year-old girl complains of crampy, episodic, lower abdominal pain of 3 months’ duration and acute retention of urine. She also reports back pain and dyschezia. She has never menstruated, but her breasts began developing 18 months earlier. Examination reveals normal Tanner stage 3 breast development and female hair distribution and a large abdominal mass. No fetal heart tones are apparent. The external genitalia appear to be normal except for a bulging mass at the introitus.
Imaging (FIGURE 1) reveals that the mass is fluid-filled. The uterus, tubes, and ovaries are present at the dome of the mass. The kidneys are normal.
What is the mass?
FIGURE 1 No exit for menstrual products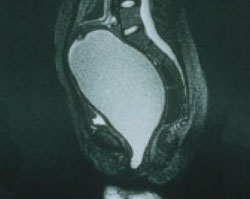
An MRI shows a large hematocolpos that develops after menarche in women who have outflow obstruction, such as imperforate hymen—as this patient had.A list of the processes involved in normal development of the female reproductive tract highlights its precarious complexity: cellular differentiation, migration, canalization, fusion, and programmed cell death. A failure in any of these processes can cause a malformation. When that malformation becomes apparent depends on the stage of life of the patient and the nature of the abnormality. As you might imagine, diagnosis and treatment are not always straightforward.
In the patient just described, the likely diagnosis is imperforate hymen or transverse vaginal septum due to failed canalization of the vaginal plate. The patient has been menstruating internally for many months and now has a large hematocolpos. Urinary retention develops when the amount of retained blood in the vagina causes acute angulation of the urethrovesical junction. Evacuation of the blood restores the physiologic angle, enabling the patient to void normally.
Most anomalies involving the external genitalia are apparent at birth (clitoromegaly, imperforate hymen), whereas obstructive and nonobstructive malformations may become evident at birth; during childhood, puberty, or adolescence; or with menarche or childbearing.
Treatment of these abnormalities has changed significantly over the past few years—largely due to refinements in diagnostic imaging, surgical and nonsurgical techniques, and instrumentation. These advances have improved reproductive function and enhanced the psychosexual attitude of these patients.
In this article, I review the most common abnormalities and describe their evaluation and management.
Vaginal canalization
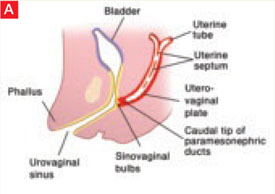
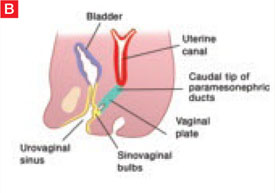
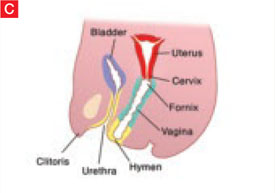
The sinovaginal bulbs are two solid evaginations originating from the urogenital sinus at the distal aspect of the müllerian tubercle, as shown at right (A). The sinovaginal bulbs proliferate into the caudal end of the uterovaginal canal to become the solid vaginal plate (B). The lumen of the lower vagina is formed by degeneration of the central cells of this vaginal plate, which occurs in a cephalad direction (C). This process of canalization is complete by 20 weeks’ gestation.
Hymen usually ruptures before birth
The vaginal hymen is separated from the urogenital sinus by the hymeneal membrane. The hymen usually ruptures before birth with the degeneration of central epithelial cells (i.e., canalization), leaving a thin fold of mucous membrane around the vaginal introitus.
Uterus, fallopian tubes develop from solid tissue
The müllerian ducts are first identifiable at approximately 6 weeks of gestation, when they begin to elongate caudally and cross the metanephric ducts medially to meet in the midline. By the seventh week, the urorectal septum has developed, separating the rectum from the urogenital sinus. Around 12 weeks’ gestation, the caudal portion of the müllerian ducts fuses to form the uterovaginal canal, which inserts into the dorsal wall of the urogenital sinus at Müller’s tubercle.
The two müllerian ducts are initially solid tissue and lie side by side. Subsequently, internal canalization of each duct produces two channels divided by a septum that is reabsorbed in a cephalad direction by 20 weeks. The cranial unfused portions of the müllerian ducts develop into the fimbria and fallopian tubes, whereas the caudal, fused portions form the uterus and upper vagina.
Impaired canalization
Imperforate hymen
The most common obstructive lesion of the female reproductive tract is an imperforate hymen. It can be diagnosed at birth with careful examination to ensure patency of both the rectum and vagina. Not infrequently, the infant presents with a bulging introitus due to mucocolpos from vaginal secretions stimulated by maternal estrogen (FIGURE 2). If diagnosis is delayed, the mucus usually resorbs and the child remains asymptomatic until menarche, when she presents with a bulging hymen and a large hematocolpos, as in the opening case. Management of imperforate hymen is feasible for the generalist. Repair can be performed in a patient of any age, but is facilitated if the tissue has undergone estrogen stimulation. Surgery is, therefore, ideal in the neonatal, postpubertal, and premenarchal periods.
Repair involves incision of the membrane near the hymeneal ring, followed by evacuation of accumulated blood. The cervix may appear to be flush with the vaginal apex, owing to compression, but this condition usually corrects itself within 2 or 3 weeks. Extra hymeneal tissue is excised to create an orifice of normal size, and the vaginal mucosa is intermittently sutured to the hymeneal ring using absorbable material (FIGURE 3).
FIGURE 2 Imperforate hymen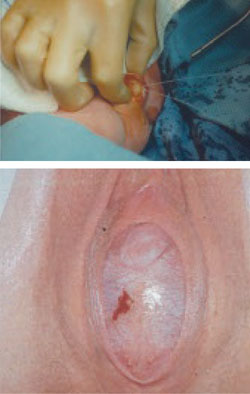
Bulging hymen due to mucocolpos in a newborn (top) and hematocolpos in an adolescent (bottom).
FIGURE 3 Hymenectomy restores vaginal orifice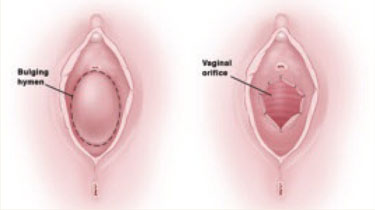
Extra hymeneal tissue is excised to create an orifice of normal size. The vaginal mucosa is intermittently sutured to the hymeneal ring.
Incomplete hymeneal fenestration
This condition often goes undiagnosed until the patient seeks evaluation for an inability to insert tampons or medication or because of coital difficulty. Young girls with microperforate hymen may present with postmenstrual spotting or malodorous discharge due to partial obstruction and poor drainage.
Treatment of microperforate, cribriform, or septate hymen entails resection of excess hymeneal tissue to create a functional hymeneal ring.
Transverse vaginal septum
A transverse septum represents failed canalization of the vaginal plate. This condition occurs in approximately one of every 18,000 to 30,000 women.1,2 The septum can be located at any of various levels in the vagina; approximately 46% are found in the upper vagina, 35% to 40% in the middle portion, and 15% to 20% in the lower vagina.1,2 Lower septa appear to be thinner than those in the proximal vagina; thinness may be due to the pressure of accumulated fluid. If a small perforation is present, the patient will experience irregular spotting.
Examination. The external genitalia appear to be normal, but the vagina is shortened. Children may present with mucocolpos, whereas adolescents develop hematocolpos. If a small perforation is present, pyohematocolpos may result from ascending infection. A mass can be palpated on rectal examination.
Ultrasonography (US) or magnetic resonance imaging (MRI) helps define the location and thickness of the septum and distinguish a high septum from congenital absence of the cervix.
Treatment. Resect a small septum and perform end-to-end anastomosis of the upper and lower edges of the vaginal wall (FIGURE 4). Only an experienced surgeon should excise a thick septum. Reanastomosis is easier if the upper vagina is distended with menstrual blood. To ensure healing without stricture, mobilize the vaginal edge circumferentially before suturing the raw edges.
Outflow obstruction caused by a transverse septum in the distal vagina almost never leads to hematometra or hematosalpinx. However, septa that arise in the proximal vagina are usually thicker and, if left untreated, can cause reflux that damages the upper reproductive tract. These cases are almost always associated with severe dysmenorrhea and should be referred to an experienced vaginal surgeon. Pelvic MRI is recommended to assess the thickness and precise anatomy of the defect before repair.
FIGURE 4 Removing a transverse vaginal septum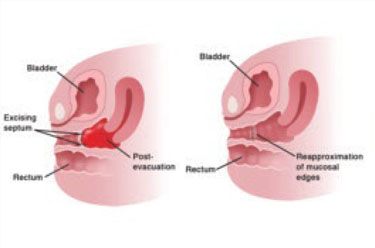
The septum is resected from the distal vagina. This is followed by end-to-end anastomosis of mucosal edges.
Uterine septum
A septate uterus has a normal external surface but two endometrial cavities. It is caused by a defect in fusion or resorption of the midline septum between the two müllerian ducts. The degree of septation varies from a small midline septum to a total failure of resorption that produces a septate uterus with a longitudinal vaginal septum. A slight midline septum with minimal fundal indentation is classified by some as a septate uterus; by others, as a bicornuate uterus.
A higher risk of recurrent miscarriage is associated with longer septa.
Surgical repair with hysteroscopic resection of the septum yields excellent results.
CASE 2 The problem of agenesis
An 18-year-old patient consults a gynecologist because she has never menstruated and is unable to have sexual intercourse. She appears to be phenotypically normal, with normal labia upon examination, but no hymeneal opening is found. Pelvic US reveals absence of the uterus with ovaries that appear to be normal.
How should her condition be managed?
Uterovaginal agenesis, also known as müllerian aplasia or Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome, refers to congenital absence of the vagina with variable uterine development (FIGURE 5). It arises from agenesis of the
müllerian duct system, although the underlying cause is unknown. Vaginal agenesis is usually accompanied by cervical and uterine agenesis. However, 7% to 12% of patients with this syndrome have a normal but obstructed or rudimentary uterus with functional endometrium. This condition occurs in roughly one of every 5,000 females.1,3
In addition, these girls often exhibit extragenital anomalies.1,4 Approximately 25% to 50% have a urologic anomaly, such as unilateral renal agenesis, pelvic or horseshoe kidneys, or irregularities of the collecting systems; 10% to 15% have another type of anomaly, such as congenital heart disease, abnormalities of the hands, congenital deafness, cleft palate, and inguinal or femoral hernia.
The genetic basis of MRKH syndrome is unknown. These patients have normal female karyotypes and phenotypes with normal ovaries and ovarian function—although there is evidence that they are prone to early menopause. They develop secondary sexual characteristics. They usually present with primary amenorrhea at 15 to 17 years of age.
Vaginal agenesis is the second most common cause of primary amenorrhea after gonadal dysgenesis. Most patients have a rudimentary, nonfunctioning uterus, but 2% to 7% have a uterus with functioning endometrium and present with cyclic or chronic abdominopelvic pain secondary to obstruction or endometriosis, hematocolpos, or hematometra.
Imaging. US examination facilitates evaluation of the kidneys and confirms the presence of ovaries and absence of the uterus. MRI may be useful to confirm the presence of functioning endometrium.
The differential diagnosis of vaginal agenesis includes androgen insensitivity and low-lying transverse vaginal septum. Treatment includes psychosocial support to reassure the patient that a normal sex life will be possible after treatment.5 Construction of a neovagina, using vaginal dilators (the first-line strategy) or surgical placement of split-thickness skin graft or artificial skin (Abbe–McIndoe procedure), is the treatment. In one case series, more than 90% of patients with vaginal agenesis were successfully treated using vaginal dilation.6
The American College of Obstetricians and Gynecologists (ACOG) recommends that patients be referred to centers with expertise in surgical creation of a neovagina.5
FIGURE 5 Uterine agenesis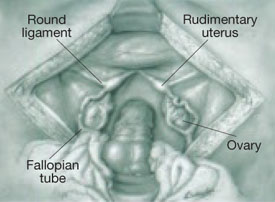
This depiction of uterine agenesis shows small, nonfunctioning müllerian remnants and normal tubes and ovaries.
Agenesis of the lower vagina
This malformation is caused by a failure of canalization of the sinovaginal bulbs and vaginal plate. The tubes, ovaries, cervix, and upper vagina are normal, but the lower vagina is replaced by fibrous tissue. Adolescents present with primary amenorrhea accompanied by cyclic pelvic pain. A vaginal dimple may be apparent on external inspection.
Because these girls have normal cyclic pituitary–ovarian–endometrial function at menarche, the upper vagina will distend with blood and secretions, and a mass can be palpated on rectoabdominal examination. US confirms the presence of a fluid-filled mass, and MRI further defines the length of obstruction.
Treatment. Surgery (by an experienced vaginal surgeon) is best performed when a large hematocolpos is present because distention increases the amount of tissue available for pull-through.
CASE 3 Fusion failure
A 17-year-old woman complains of lower abdominal pain and an enlarging abdomen. Menarche occurred at 13 years. The patient began to notice right-sided lower abdominal pain and abdominal swelling about 2 years ago, but denies any other symptoms.
Examination reveals a mass arising from the right lateral vaginal wall. The cervix appears to be normal. US shows a large, predominantly right-sided, fluid-filled mass arising from the pelvis. The uterus appears to be didelphic, with normal ovaries and an absent right kidney.
What is the mass?
This is a classic description of uterovaginal duplication that arises when the müllerian ducts fail to fuse (FIGURE 6).
In this case, the right-sided vagina failed to canalize as well, causing a large hematocolpos. Generally, duplication is limited to the uterus and cervix (uterus didelphys bicollis), although duplication of the vulva, bladder, urethra, vagina, and anus have been described.
Women with a didelphic uterus often have a good reproductive outcome. A septated vagina occurs in 75% of cases and may cause difficulty with sexual intercourse or vaginal delivery. Symptomatic women usually opt for resection of the vaginal septum.
Women who have an obstructed hemivagina and ipsilateral renal agenesis have regular menses because menstrual blood from one uterus can exit through its unobstructed side.
Treatment involves resection of the wall of the obstructed vagina, creating a single vaginal vault. If both vaginas have failed to canalize, the patient will present much earlier, with primary amenorrhea and cyclic pelvic pain—a presentation similar to that of transverse septum. Reconstructive surgery of the uterus is not recommended for complete duplication of the uterus.1,7
FIGURE 6 Uterus didelphys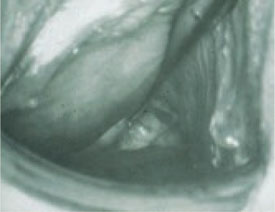
This patient has uterus didelphys with an obstructed hemivagina.
Unicornuate and bicornuate uterus
A unicornuate uterus represents an asymmetric lateral fusion defect. One cavity is usually normal, with a fallopian tube and cervix, but the failed müllerian duct can have various configurations. The affected müllerian duct may fail to develop, or it may develop partially as a horn on the uterus or a separate structure that may or may not communicate with the uterus.
Most rudimentary horns are asymptomatic, but others contain functional endometrium that is shed cyclically. If the rudimentary horn is obstructed, as is usually the case, the woman develops cyclic or chronic abdominopelvic pain that necessitates surgical excision of the horn. Endometriosis, a result of reflux, is common.
In a bicornuate uterus, the fundus is indented and the vagina is normal. This anomaly occurs when the müllerian ducts fuse only partially. The result is varying degrees of separation of the uterine horns.
Pregnancy outcomes appear to be similar to those of the general population. However, some women develop early pregnancy loss, preterm labor, incompetent cervix, or malpresentation.
Treatment is open surgical reunification.
CASE 4 Embryonic rest cells
A 55-year-old woman complains of “fullness” in the vagina, especially during intercourse. Upon examination, a 4- to 5-cm fluid-filled cyst is visible at the right posterior fornix.
What type of cyst is it?
Gartner’s duct cysts from embryonic rest cells arise from remnants of the metanephric duct system. They usually develop on the lateral vaginal walls or fornix (FIGURE 7), unlike epidermal inclusion cysts, which are generally located on the posterior wall or at the cervical–vesical junction. Although they sometimes cause dyspareunia or make it difficult for the patient to insert a tampon, they are usually asymptomatic unless they are large. Intervention is not indicated unless the patient is symptomatic.
Treatment. Marsupialization rather than excision is the treatment of choice because these embryonic rest cells may extend deep into the retroperitoneum.
FIGURE 7 Gartner’s duct cysts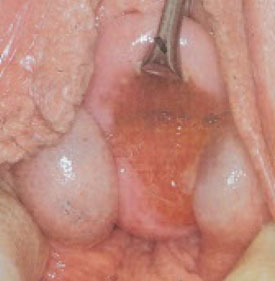
These bilateral cysts developed from embryonic rest cells.
1. Emans SJ, Laufer MR, Goldstein DP. Pediatric and Adolescent Gynecology. 5th ed. Chapter 10. Philadelphia: Lippincott Williams & Wilkins;. 2005:334-416.
2. Acién P. Incidence of Müllerian defects in fertile and infertile women. Hum Reprod. 1997;12:1372-1376.
3. Evans TN, Poland ML, Boving RL. Vaginal malformations. Am J Obstet Gynecol. 1981;141:910-920.
4. Fedele L, Bianchi S, Agnoli B, Tozzi L, Vignali M. Urinary tract anomalies associated with unicornuate uterus. J Urol. 1996;155:847-858.
5. Vaginal agenesis: diagnosis, management, and routine care. Committee Opinion No. 355. Washington, DC: ACOG; December 2006.
6. Roberts CP, Haber MJ, Rock JA. Vaginal creation for müllerian agenesis. Am J Obstet Gynecol. 2001;185:1349-1353.
7. Stassart JP, Nagel TC, Prem KA, Phipps WR. Uterus didelphys, obstructed hemivagina, and ipsilateral renal agenesis: the University of Minnesota experience. Fertil Steril. 1992;57:756-761.
The author reports no financial relationships relevant to this article.
CASE 1 Fluid-filled abdominal mass
A 14-year-old girl complains of crampy, episodic, lower abdominal pain of 3 months’ duration and acute retention of urine. She also reports back pain and dyschezia. She has never menstruated, but her breasts began developing 18 months earlier. Examination reveals normal Tanner stage 3 breast development and female hair distribution and a large abdominal mass. No fetal heart tones are apparent. The external genitalia appear to be normal except for a bulging mass at the introitus.
Imaging (FIGURE 1) reveals that the mass is fluid-filled. The uterus, tubes, and ovaries are present at the dome of the mass. The kidneys are normal.
What is the mass?
FIGURE 1 No exit for menstrual products
An MRI shows a large hematocolpos that develops after menarche in women who have outflow obstruction, such as imperforate hymen—as this patient had.A list of the processes involved in normal development of the female reproductive tract highlights its precarious complexity: cellular differentiation, migration, canalization, fusion, and programmed cell death. A failure in any of these processes can cause a malformation. When that malformation becomes apparent depends on the stage of life of the patient and the nature of the abnormality. As you might imagine, diagnosis and treatment are not always straightforward.
In the patient just described, the likely diagnosis is imperforate hymen or transverse vaginal septum due to failed canalization of the vaginal plate. The patient has been menstruating internally for many months and now has a large hematocolpos. Urinary retention develops when the amount of retained blood in the vagina causes acute angulation of the urethrovesical junction. Evacuation of the blood restores the physiologic angle, enabling the patient to void normally.
Most anomalies involving the external genitalia are apparent at birth (clitoromegaly, imperforate hymen), whereas obstructive and nonobstructive malformations may become evident at birth; during childhood, puberty, or adolescence; or with menarche or childbearing.
Treatment of these abnormalities has changed significantly over the past few years—largely due to refinements in diagnostic imaging, surgical and nonsurgical techniques, and instrumentation. These advances have improved reproductive function and enhanced the psychosexual attitude of these patients.
In this article, I review the most common abnormalities and describe their evaluation and management.
Vaginal canalization
The sinovaginal bulbs are two solid evaginations originating from the urogenital sinus at the distal aspect of the müllerian tubercle, as shown at right (A). The sinovaginal bulbs proliferate into the caudal end of the uterovaginal canal to become the solid vaginal plate (B). The lumen of the lower vagina is formed by degeneration of the central cells of this vaginal plate, which occurs in a cephalad direction (C). This process of canalization is complete by 20 weeks’ gestation.
Hymen usually ruptures before birth
The vaginal hymen is separated from the urogenital sinus by the hymeneal membrane. The hymen usually ruptures before birth with the degeneration of central epithelial cells (i.e., canalization), leaving a thin fold of mucous membrane around the vaginal introitus.
Uterus, fallopian tubes develop from solid tissue
The müllerian ducts are first identifiable at approximately 6 weeks of gestation, when they begin to elongate caudally and cross the metanephric ducts medially to meet in the midline. By the seventh week, the urorectal septum has developed, separating the rectum from the urogenital sinus. Around 12 weeks’ gestation, the caudal portion of the müllerian ducts fuses to form the uterovaginal canal, which inserts into the dorsal wall of the urogenital sinus at Müller’s tubercle.
The two müllerian ducts are initially solid tissue and lie side by side. Subsequently, internal canalization of each duct produces two channels divided by a septum that is reabsorbed in a cephalad direction by 20 weeks. The cranial unfused portions of the müllerian ducts develop into the fimbria and fallopian tubes, whereas the caudal, fused portions form the uterus and upper vagina.
Impaired canalization
Imperforate hymen
The most common obstructive lesion of the female reproductive tract is an imperforate hymen. It can be diagnosed at birth with careful examination to ensure patency of both the rectum and vagina. Not infrequently, the infant presents with a bulging introitus due to mucocolpos from vaginal secretions stimulated by maternal estrogen (FIGURE 2). If diagnosis is delayed, the mucus usually resorbs and the child remains asymptomatic until menarche, when she presents with a bulging hymen and a large hematocolpos, as in the opening case. Management of imperforate hymen is feasible for the generalist. Repair can be performed in a patient of any age, but is facilitated if the tissue has undergone estrogen stimulation. Surgery is, therefore, ideal in the neonatal, postpubertal, and premenarchal periods.
Repair involves incision of the membrane near the hymeneal ring, followed by evacuation of accumulated blood. The cervix may appear to be flush with the vaginal apex, owing to compression, but this condition usually corrects itself within 2 or 3 weeks. Extra hymeneal tissue is excised to create an orifice of normal size, and the vaginal mucosa is intermittently sutured to the hymeneal ring using absorbable material (FIGURE 3).
FIGURE 2 Imperforate hymen
Bulging hymen due to mucocolpos in a newborn (top) and hematocolpos in an adolescent (bottom).
FIGURE 3 Hymenectomy restores vaginal orifice
Extra hymeneal tissue is excised to create an orifice of normal size. The vaginal mucosa is intermittently sutured to the hymeneal ring.
Incomplete hymeneal fenestration
This condition often goes undiagnosed until the patient seeks evaluation for an inability to insert tampons or medication or because of coital difficulty. Young girls with microperforate hymen may present with postmenstrual spotting or malodorous discharge due to partial obstruction and poor drainage.
Treatment of microperforate, cribriform, or septate hymen entails resection of excess hymeneal tissue to create a functional hymeneal ring.
Transverse vaginal septum
A transverse septum represents failed canalization of the vaginal plate. This condition occurs in approximately one of every 18,000 to 30,000 women.1,2 The septum can be located at any of various levels in the vagina; approximately 46% are found in the upper vagina, 35% to 40% in the middle portion, and 15% to 20% in the lower vagina.1,2 Lower septa appear to be thinner than those in the proximal vagina; thinness may be due to the pressure of accumulated fluid. If a small perforation is present, the patient will experience irregular spotting.
Examination. The external genitalia appear to be normal, but the vagina is shortened. Children may present with mucocolpos, whereas adolescents develop hematocolpos. If a small perforation is present, pyohematocolpos may result from ascending infection. A mass can be palpated on rectal examination.
Ultrasonography (US) or magnetic resonance imaging (MRI) helps define the location and thickness of the septum and distinguish a high septum from congenital absence of the cervix.
Treatment. Resect a small septum and perform end-to-end anastomosis of the upper and lower edges of the vaginal wall (FIGURE 4). Only an experienced surgeon should excise a thick septum. Reanastomosis is easier if the upper vagina is distended with menstrual blood. To ensure healing without stricture, mobilize the vaginal edge circumferentially before suturing the raw edges.
Outflow obstruction caused by a transverse septum in the distal vagina almost never leads to hematometra or hematosalpinx. However, septa that arise in the proximal vagina are usually thicker and, if left untreated, can cause reflux that damages the upper reproductive tract. These cases are almost always associated with severe dysmenorrhea and should be referred to an experienced vaginal surgeon. Pelvic MRI is recommended to assess the thickness and precise anatomy of the defect before repair.
FIGURE 4 Removing a transverse vaginal septum
The septum is resected from the distal vagina. This is followed by end-to-end anastomosis of mucosal edges.
Uterine septum
A septate uterus has a normal external surface but two endometrial cavities. It is caused by a defect in fusion or resorption of the midline septum between the two müllerian ducts. The degree of septation varies from a small midline septum to a total failure of resorption that produces a septate uterus with a longitudinal vaginal septum. A slight midline septum with minimal fundal indentation is classified by some as a septate uterus; by others, as a bicornuate uterus.
A higher risk of recurrent miscarriage is associated with longer septa.
Surgical repair with hysteroscopic resection of the septum yields excellent results.
CASE 2 The problem of agenesis
An 18-year-old patient consults a gynecologist because she has never menstruated and is unable to have sexual intercourse. She appears to be phenotypically normal, with normal labia upon examination, but no hymeneal opening is found. Pelvic US reveals absence of the uterus with ovaries that appear to be normal.
How should her condition be managed?
Uterovaginal agenesis, also known as müllerian aplasia or Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome, refers to congenital absence of the vagina with variable uterine development (FIGURE 5). It arises from agenesis of the
müllerian duct system, although the underlying cause is unknown. Vaginal agenesis is usually accompanied by cervical and uterine agenesis. However, 7% to 12% of patients with this syndrome have a normal but obstructed or rudimentary uterus with functional endometrium. This condition occurs in roughly one of every 5,000 females.1,3
In addition, these girls often exhibit extragenital anomalies.1,4 Approximately 25% to 50% have a urologic anomaly, such as unilateral renal agenesis, pelvic or horseshoe kidneys, or irregularities of the collecting systems; 10% to 15% have another type of anomaly, such as congenital heart disease, abnormalities of the hands, congenital deafness, cleft palate, and inguinal or femoral hernia.
The genetic basis of MRKH syndrome is unknown. These patients have normal female karyotypes and phenotypes with normal ovaries and ovarian function—although there is evidence that they are prone to early menopause. They develop secondary sexual characteristics. They usually present with primary amenorrhea at 15 to 17 years of age.
Vaginal agenesis is the second most common cause of primary amenorrhea after gonadal dysgenesis. Most patients have a rudimentary, nonfunctioning uterus, but 2% to 7% have a uterus with functioning endometrium and present with cyclic or chronic abdominopelvic pain secondary to obstruction or endometriosis, hematocolpos, or hematometra.
Imaging. US examination facilitates evaluation of the kidneys and confirms the presence of ovaries and absence of the uterus. MRI may be useful to confirm the presence of functioning endometrium.
The differential diagnosis of vaginal agenesis includes androgen insensitivity and low-lying transverse vaginal septum. Treatment includes psychosocial support to reassure the patient that a normal sex life will be possible after treatment.5 Construction of a neovagina, using vaginal dilators (the first-line strategy) or surgical placement of split-thickness skin graft or artificial skin (Abbe–McIndoe procedure), is the treatment. In one case series, more than 90% of patients with vaginal agenesis were successfully treated using vaginal dilation.6
The American College of Obstetricians and Gynecologists (ACOG) recommends that patients be referred to centers with expertise in surgical creation of a neovagina.5
FIGURE 5 Uterine agenesis
This depiction of uterine agenesis shows small, nonfunctioning müllerian remnants and normal tubes and ovaries.
Agenesis of the lower vagina
This malformation is caused by a failure of canalization of the sinovaginal bulbs and vaginal plate. The tubes, ovaries, cervix, and upper vagina are normal, but the lower vagina is replaced by fibrous tissue. Adolescents present with primary amenorrhea accompanied by cyclic pelvic pain. A vaginal dimple may be apparent on external inspection.
Because these girls have normal cyclic pituitary–ovarian–endometrial function at menarche, the upper vagina will distend with blood and secretions, and a mass can be palpated on rectoabdominal examination. US confirms the presence of a fluid-filled mass, and MRI further defines the length of obstruction.
Treatment. Surgery (by an experienced vaginal surgeon) is best performed when a large hematocolpos is present because distention increases the amount of tissue available for pull-through.
CASE 3 Fusion failure
A 17-year-old woman complains of lower abdominal pain and an enlarging abdomen. Menarche occurred at 13 years. The patient began to notice right-sided lower abdominal pain and abdominal swelling about 2 years ago, but denies any other symptoms.
Examination reveals a mass arising from the right lateral vaginal wall. The cervix appears to be normal. US shows a large, predominantly right-sided, fluid-filled mass arising from the pelvis. The uterus appears to be didelphic, with normal ovaries and an absent right kidney.
What is the mass?
This is a classic description of uterovaginal duplication that arises when the müllerian ducts fail to fuse (FIGURE 6).
In this case, the right-sided vagina failed to canalize as well, causing a large hematocolpos. Generally, duplication is limited to the uterus and cervix (uterus didelphys bicollis), although duplication of the vulva, bladder, urethra, vagina, and anus have been described.
Women with a didelphic uterus often have a good reproductive outcome. A septated vagina occurs in 75% of cases and may cause difficulty with sexual intercourse or vaginal delivery. Symptomatic women usually opt for resection of the vaginal septum.
Women who have an obstructed hemivagina and ipsilateral renal agenesis have regular menses because menstrual blood from one uterus can exit through its unobstructed side.
Treatment involves resection of the wall of the obstructed vagina, creating a single vaginal vault. If both vaginas have failed to canalize, the patient will present much earlier, with primary amenorrhea and cyclic pelvic pain—a presentation similar to that of transverse septum. Reconstructive surgery of the uterus is not recommended for complete duplication of the uterus.1,7
FIGURE 6 Uterus didelphys
This patient has uterus didelphys with an obstructed hemivagina.
Unicornuate and bicornuate uterus
A unicornuate uterus represents an asymmetric lateral fusion defect. One cavity is usually normal, with a fallopian tube and cervix, but the failed müllerian duct can have various configurations. The affected müllerian duct may fail to develop, or it may develop partially as a horn on the uterus or a separate structure that may or may not communicate with the uterus.
Most rudimentary horns are asymptomatic, but others contain functional endometrium that is shed cyclically. If the rudimentary horn is obstructed, as is usually the case, the woman develops cyclic or chronic abdominopelvic pain that necessitates surgical excision of the horn. Endometriosis, a result of reflux, is common.
In a bicornuate uterus, the fundus is indented and the vagina is normal. This anomaly occurs when the müllerian ducts fuse only partially. The result is varying degrees of separation of the uterine horns.
Pregnancy outcomes appear to be similar to those of the general population. However, some women develop early pregnancy loss, preterm labor, incompetent cervix, or malpresentation.
Treatment is open surgical reunification.
CASE 4 Embryonic rest cells
A 55-year-old woman complains of “fullness” in the vagina, especially during intercourse. Upon examination, a 4- to 5-cm fluid-filled cyst is visible at the right posterior fornix.
What type of cyst is it?
Gartner’s duct cysts from embryonic rest cells arise from remnants of the metanephric duct system. They usually develop on the lateral vaginal walls or fornix (FIGURE 7), unlike epidermal inclusion cysts, which are generally located on the posterior wall or at the cervical–vesical junction. Although they sometimes cause dyspareunia or make it difficult for the patient to insert a tampon, they are usually asymptomatic unless they are large. Intervention is not indicated unless the patient is symptomatic.
Treatment. Marsupialization rather than excision is the treatment of choice because these embryonic rest cells may extend deep into the retroperitoneum.
FIGURE 7 Gartner’s duct cysts
These bilateral cysts developed from embryonic rest cells.
The author reports no financial relationships relevant to this article.
CASE 1 Fluid-filled abdominal mass
A 14-year-old girl complains of crampy, episodic, lower abdominal pain of 3 months’ duration and acute retention of urine. She also reports back pain and dyschezia. She has never menstruated, but her breasts began developing 18 months earlier. Examination reveals normal Tanner stage 3 breast development and female hair distribution and a large abdominal mass. No fetal heart tones are apparent. The external genitalia appear to be normal except for a bulging mass at the introitus.
Imaging (FIGURE 1) reveals that the mass is fluid-filled. The uterus, tubes, and ovaries are present at the dome of the mass. The kidneys are normal.
What is the mass?
FIGURE 1 No exit for menstrual products
An MRI shows a large hematocolpos that develops after menarche in women who have outflow obstruction, such as imperforate hymen—as this patient had.A list of the processes involved in normal development of the female reproductive tract highlights its precarious complexity: cellular differentiation, migration, canalization, fusion, and programmed cell death. A failure in any of these processes can cause a malformation. When that malformation becomes apparent depends on the stage of life of the patient and the nature of the abnormality. As you might imagine, diagnosis and treatment are not always straightforward.
In the patient just described, the likely diagnosis is imperforate hymen or transverse vaginal septum due to failed canalization of the vaginal plate. The patient has been menstruating internally for many months and now has a large hematocolpos. Urinary retention develops when the amount of retained blood in the vagina causes acute angulation of the urethrovesical junction. Evacuation of the blood restores the physiologic angle, enabling the patient to void normally.
Most anomalies involving the external genitalia are apparent at birth (clitoromegaly, imperforate hymen), whereas obstructive and nonobstructive malformations may become evident at birth; during childhood, puberty, or adolescence; or with menarche or childbearing.
Treatment of these abnormalities has changed significantly over the past few years—largely due to refinements in diagnostic imaging, surgical and nonsurgical techniques, and instrumentation. These advances have improved reproductive function and enhanced the psychosexual attitude of these patients.
In this article, I review the most common abnormalities and describe their evaluation and management.
Vaginal canalization
The sinovaginal bulbs are two solid evaginations originating from the urogenital sinus at the distal aspect of the müllerian tubercle, as shown at right (A). The sinovaginal bulbs proliferate into the caudal end of the uterovaginal canal to become the solid vaginal plate (B). The lumen of the lower vagina is formed by degeneration of the central cells of this vaginal plate, which occurs in a cephalad direction (C). This process of canalization is complete by 20 weeks’ gestation.
Hymen usually ruptures before birth
The vaginal hymen is separated from the urogenital sinus by the hymeneal membrane. The hymen usually ruptures before birth with the degeneration of central epithelial cells (i.e., canalization), leaving a thin fold of mucous membrane around the vaginal introitus.
Uterus, fallopian tubes develop from solid tissue
The müllerian ducts are first identifiable at approximately 6 weeks of gestation, when they begin to elongate caudally and cross the metanephric ducts medially to meet in the midline. By the seventh week, the urorectal septum has developed, separating the rectum from the urogenital sinus. Around 12 weeks’ gestation, the caudal portion of the müllerian ducts fuses to form the uterovaginal canal, which inserts into the dorsal wall of the urogenital sinus at Müller’s tubercle.
The two müllerian ducts are initially solid tissue and lie side by side. Subsequently, internal canalization of each duct produces two channels divided by a septum that is reabsorbed in a cephalad direction by 20 weeks. The cranial unfused portions of the müllerian ducts develop into the fimbria and fallopian tubes, whereas the caudal, fused portions form the uterus and upper vagina.
Impaired canalization
Imperforate hymen
The most common obstructive lesion of the female reproductive tract is an imperforate hymen. It can be diagnosed at birth with careful examination to ensure patency of both the rectum and vagina. Not infrequently, the infant presents with a bulging introitus due to mucocolpos from vaginal secretions stimulated by maternal estrogen (FIGURE 2). If diagnosis is delayed, the mucus usually resorbs and the child remains asymptomatic until menarche, when she presents with a bulging hymen and a large hematocolpos, as in the opening case. Management of imperforate hymen is feasible for the generalist. Repair can be performed in a patient of any age, but is facilitated if the tissue has undergone estrogen stimulation. Surgery is, therefore, ideal in the neonatal, postpubertal, and premenarchal periods.
Repair involves incision of the membrane near the hymeneal ring, followed by evacuation of accumulated blood. The cervix may appear to be flush with the vaginal apex, owing to compression, but this condition usually corrects itself within 2 or 3 weeks. Extra hymeneal tissue is excised to create an orifice of normal size, and the vaginal mucosa is intermittently sutured to the hymeneal ring using absorbable material (FIGURE 3).
FIGURE 2 Imperforate hymen
Bulging hymen due to mucocolpos in a newborn (top) and hematocolpos in an adolescent (bottom).
FIGURE 3 Hymenectomy restores vaginal orifice
Extra hymeneal tissue is excised to create an orifice of normal size. The vaginal mucosa is intermittently sutured to the hymeneal ring.
Incomplete hymeneal fenestration
This condition often goes undiagnosed until the patient seeks evaluation for an inability to insert tampons or medication or because of coital difficulty. Young girls with microperforate hymen may present with postmenstrual spotting or malodorous discharge due to partial obstruction and poor drainage.
Treatment of microperforate, cribriform, or septate hymen entails resection of excess hymeneal tissue to create a functional hymeneal ring.
Transverse vaginal septum
A transverse septum represents failed canalization of the vaginal plate. This condition occurs in approximately one of every 18,000 to 30,000 women.1,2 The septum can be located at any of various levels in the vagina; approximately 46% are found in the upper vagina, 35% to 40% in the middle portion, and 15% to 20% in the lower vagina.1,2 Lower septa appear to be thinner than those in the proximal vagina; thinness may be due to the pressure of accumulated fluid. If a small perforation is present, the patient will experience irregular spotting.
Examination. The external genitalia appear to be normal, but the vagina is shortened. Children may present with mucocolpos, whereas adolescents develop hematocolpos. If a small perforation is present, pyohematocolpos may result from ascending infection. A mass can be palpated on rectal examination.
Ultrasonography (US) or magnetic resonance imaging (MRI) helps define the location and thickness of the septum and distinguish a high septum from congenital absence of the cervix.
Treatment. Resect a small septum and perform end-to-end anastomosis of the upper and lower edges of the vaginal wall (FIGURE 4). Only an experienced surgeon should excise a thick septum. Reanastomosis is easier if the upper vagina is distended with menstrual blood. To ensure healing without stricture, mobilize the vaginal edge circumferentially before suturing the raw edges.
Outflow obstruction caused by a transverse septum in the distal vagina almost never leads to hematometra or hematosalpinx. However, septa that arise in the proximal vagina are usually thicker and, if left untreated, can cause reflux that damages the upper reproductive tract. These cases are almost always associated with severe dysmenorrhea and should be referred to an experienced vaginal surgeon. Pelvic MRI is recommended to assess the thickness and precise anatomy of the defect before repair.
FIGURE 4 Removing a transverse vaginal septum
The septum is resected from the distal vagina. This is followed by end-to-end anastomosis of mucosal edges.
Uterine septum
A septate uterus has a normal external surface but two endometrial cavities. It is caused by a defect in fusion or resorption of the midline septum between the two müllerian ducts. The degree of septation varies from a small midline septum to a total failure of resorption that produces a septate uterus with a longitudinal vaginal septum. A slight midline septum with minimal fundal indentation is classified by some as a septate uterus; by others, as a bicornuate uterus.
A higher risk of recurrent miscarriage is associated with longer septa.
Surgical repair with hysteroscopic resection of the septum yields excellent results.
CASE 2 The problem of agenesis
An 18-year-old patient consults a gynecologist because she has never menstruated and is unable to have sexual intercourse. She appears to be phenotypically normal, with normal labia upon examination, but no hymeneal opening is found. Pelvic US reveals absence of the uterus with ovaries that appear to be normal.
How should her condition be managed?
Uterovaginal agenesis, also known as müllerian aplasia or Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome, refers to congenital absence of the vagina with variable uterine development (FIGURE 5). It arises from agenesis of the
müllerian duct system, although the underlying cause is unknown. Vaginal agenesis is usually accompanied by cervical and uterine agenesis. However, 7% to 12% of patients with this syndrome have a normal but obstructed or rudimentary uterus with functional endometrium. This condition occurs in roughly one of every 5,000 females.1,3
In addition, these girls often exhibit extragenital anomalies.1,4 Approximately 25% to 50% have a urologic anomaly, such as unilateral renal agenesis, pelvic or horseshoe kidneys, or irregularities of the collecting systems; 10% to 15% have another type of anomaly, such as congenital heart disease, abnormalities of the hands, congenital deafness, cleft palate, and inguinal or femoral hernia.
The genetic basis of MRKH syndrome is unknown. These patients have normal female karyotypes and phenotypes with normal ovaries and ovarian function—although there is evidence that they are prone to early menopause. They develop secondary sexual characteristics. They usually present with primary amenorrhea at 15 to 17 years of age.
Vaginal agenesis is the second most common cause of primary amenorrhea after gonadal dysgenesis. Most patients have a rudimentary, nonfunctioning uterus, but 2% to 7% have a uterus with functioning endometrium and present with cyclic or chronic abdominopelvic pain secondary to obstruction or endometriosis, hematocolpos, or hematometra.
Imaging. US examination facilitates evaluation of the kidneys and confirms the presence of ovaries and absence of the uterus. MRI may be useful to confirm the presence of functioning endometrium.
The differential diagnosis of vaginal agenesis includes androgen insensitivity and low-lying transverse vaginal septum. Treatment includes psychosocial support to reassure the patient that a normal sex life will be possible after treatment.5 Construction of a neovagina, using vaginal dilators (the first-line strategy) or surgical placement of split-thickness skin graft or artificial skin (Abbe–McIndoe procedure), is the treatment. In one case series, more than 90% of patients with vaginal agenesis were successfully treated using vaginal dilation.6
The American College of Obstetricians and Gynecologists (ACOG) recommends that patients be referred to centers with expertise in surgical creation of a neovagina.5
FIGURE 5 Uterine agenesis
This depiction of uterine agenesis shows small, nonfunctioning müllerian remnants and normal tubes and ovaries.
Agenesis of the lower vagina
This malformation is caused by a failure of canalization of the sinovaginal bulbs and vaginal plate. The tubes, ovaries, cervix, and upper vagina are normal, but the lower vagina is replaced by fibrous tissue. Adolescents present with primary amenorrhea accompanied by cyclic pelvic pain. A vaginal dimple may be apparent on external inspection.
Because these girls have normal cyclic pituitary–ovarian–endometrial function at menarche, the upper vagina will distend with blood and secretions, and a mass can be palpated on rectoabdominal examination. US confirms the presence of a fluid-filled mass, and MRI further defines the length of obstruction.
Treatment. Surgery (by an experienced vaginal surgeon) is best performed when a large hematocolpos is present because distention increases the amount of tissue available for pull-through.
CASE 3 Fusion failure
A 17-year-old woman complains of lower abdominal pain and an enlarging abdomen. Menarche occurred at 13 years. The patient began to notice right-sided lower abdominal pain and abdominal swelling about 2 years ago, but denies any other symptoms.
Examination reveals a mass arising from the right lateral vaginal wall. The cervix appears to be normal. US shows a large, predominantly right-sided, fluid-filled mass arising from the pelvis. The uterus appears to be didelphic, with normal ovaries and an absent right kidney.
What is the mass?
This is a classic description of uterovaginal duplication that arises when the müllerian ducts fail to fuse (FIGURE 6).
In this case, the right-sided vagina failed to canalize as well, causing a large hematocolpos. Generally, duplication is limited to the uterus and cervix (uterus didelphys bicollis), although duplication of the vulva, bladder, urethra, vagina, and anus have been described.
Women with a didelphic uterus often have a good reproductive outcome. A septated vagina occurs in 75% of cases and may cause difficulty with sexual intercourse or vaginal delivery. Symptomatic women usually opt for resection of the vaginal septum.
Women who have an obstructed hemivagina and ipsilateral renal agenesis have regular menses because menstrual blood from one uterus can exit through its unobstructed side.
Treatment involves resection of the wall of the obstructed vagina, creating a single vaginal vault. If both vaginas have failed to canalize, the patient will present much earlier, with primary amenorrhea and cyclic pelvic pain—a presentation similar to that of transverse septum. Reconstructive surgery of the uterus is not recommended for complete duplication of the uterus.1,7
FIGURE 6 Uterus didelphys
This patient has uterus didelphys with an obstructed hemivagina.
Unicornuate and bicornuate uterus
A unicornuate uterus represents an asymmetric lateral fusion defect. One cavity is usually normal, with a fallopian tube and cervix, but the failed müllerian duct can have various configurations. The affected müllerian duct may fail to develop, or it may develop partially as a horn on the uterus or a separate structure that may or may not communicate with the uterus.
Most rudimentary horns are asymptomatic, but others contain functional endometrium that is shed cyclically. If the rudimentary horn is obstructed, as is usually the case, the woman develops cyclic or chronic abdominopelvic pain that necessitates surgical excision of the horn. Endometriosis, a result of reflux, is common.
In a bicornuate uterus, the fundus is indented and the vagina is normal. This anomaly occurs when the müllerian ducts fuse only partially. The result is varying degrees of separation of the uterine horns.
Pregnancy outcomes appear to be similar to those of the general population. However, some women develop early pregnancy loss, preterm labor, incompetent cervix, or malpresentation.
Treatment is open surgical reunification.
CASE 4 Embryonic rest cells
A 55-year-old woman complains of “fullness” in the vagina, especially during intercourse. Upon examination, a 4- to 5-cm fluid-filled cyst is visible at the right posterior fornix.
What type of cyst is it?
Gartner’s duct cysts from embryonic rest cells arise from remnants of the metanephric duct system. They usually develop on the lateral vaginal walls or fornix (FIGURE 7), unlike epidermal inclusion cysts, which are generally located on the posterior wall or at the cervical–vesical junction. Although they sometimes cause dyspareunia or make it difficult for the patient to insert a tampon, they are usually asymptomatic unless they are large. Intervention is not indicated unless the patient is symptomatic.
Treatment. Marsupialization rather than excision is the treatment of choice because these embryonic rest cells may extend deep into the retroperitoneum.
FIGURE 7 Gartner’s duct cysts
These bilateral cysts developed from embryonic rest cells.
1. Emans SJ, Laufer MR, Goldstein DP. Pediatric and Adolescent Gynecology. 5th ed. Chapter 10. Philadelphia: Lippincott Williams & Wilkins;. 2005:334-416.
2. Acién P. Incidence of Müllerian defects in fertile and infertile women. Hum Reprod. 1997;12:1372-1376.
3. Evans TN, Poland ML, Boving RL. Vaginal malformations. Am J Obstet Gynecol. 1981;141:910-920.
4. Fedele L, Bianchi S, Agnoli B, Tozzi L, Vignali M. Urinary tract anomalies associated with unicornuate uterus. J Urol. 1996;155:847-858.
5. Vaginal agenesis: diagnosis, management, and routine care. Committee Opinion No. 355. Washington, DC: ACOG; December 2006.
6. Roberts CP, Haber MJ, Rock JA. Vaginal creation for müllerian agenesis. Am J Obstet Gynecol. 2001;185:1349-1353.
7. Stassart JP, Nagel TC, Prem KA, Phipps WR. Uterus didelphys, obstructed hemivagina, and ipsilateral renal agenesis: the University of Minnesota experience. Fertil Steril. 1992;57:756-761.
1. Emans SJ, Laufer MR, Goldstein DP. Pediatric and Adolescent Gynecology. 5th ed. Chapter 10. Philadelphia: Lippincott Williams & Wilkins;. 2005:334-416.
2. Acién P. Incidence of Müllerian defects in fertile and infertile women. Hum Reprod. 1997;12:1372-1376.
3. Evans TN, Poland ML, Boving RL. Vaginal malformations. Am J Obstet Gynecol. 1981;141:910-920.
4. Fedele L, Bianchi S, Agnoli B, Tozzi L, Vignali M. Urinary tract anomalies associated with unicornuate uterus. J Urol. 1996;155:847-858.
5. Vaginal agenesis: diagnosis, management, and routine care. Committee Opinion No. 355. Washington, DC: ACOG; December 2006.
6. Roberts CP, Haber MJ, Rock JA. Vaginal creation for müllerian agenesis. Am J Obstet Gynecol. 2001;185:1349-1353.
7. Stassart JP, Nagel TC, Prem KA, Phipps WR. Uterus didelphys, obstructed hemivagina, and ipsilateral renal agenesis: the University of Minnesota experience. Fertil Steril. 1992;57:756-761.