User login
Stiff hands and feet, facial deformities
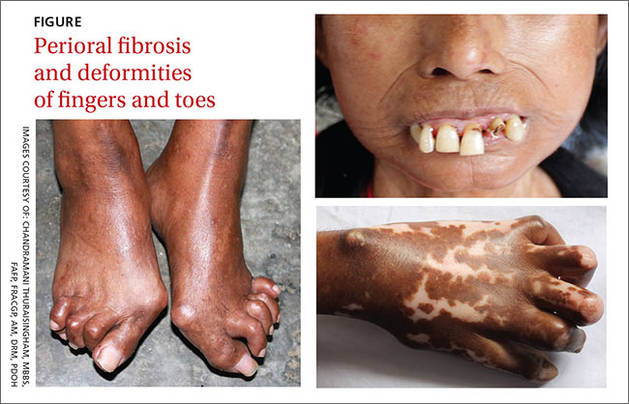
A 47-year-old Malaysian aboriginal woman presented to our clinic with stiffness in her fingers and feet that had been bothering her for about 10 years. The patient had multiple facial deformities, including perioral fibrosis, which gave her face a bird-like appearance; micrognathia (which affected the alignment of her teeth); a narrow mouth with pursed, puckered lips; bound-down skin of the nose; and a loss of wrinkling.
Ivory-colored plaques of hard, thickened skin caused a pigmented, “salt and pepper” appearance on our patient’s face. She also had deformities of all of her fingers and toes that severely restricted her ability to move them (FIGURE).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Limited systemic sclerosis
Systemic sclerosis (SSc) is a rare autoimmune disease that mainly affects women ages 30 to 50.1 SSc is classified according to the extent of skin involvement and includes limited SSc (lSSc), which is also called CREST syndrome, and diffuse SSc (dSSc).
CREST stands for:
- calcinosis, or subcutaneous calcium deposits,
- Raynaud’s phenomenon,
- esophageal dysfunction,
- sclerodactyly, which presents as tightening of the skin of the fingers or toes, and
- telangiectasia, which is characterized by dilated superficial capillaries, usually on the face, arms, and hands.2
Patients like ours who have lSSc experience a gradual progression of symptoms. Their skin is affected in limited areas, such as the fingers, hands, face, lower arms, and feet. There is no involvement of the chest, abdomen, or internal organs, with the exception of the esophagus. Esophageal smooth muscle becomes atrophied and is replaced by fibrous tissue, leading to esophageal motility dysfunction. This is in contrast to dSSc, in which you would see involvement of internal organs such as the lungs, kidneys, and gastrointestinal tract.
Raynaud’s phenomenon is commonly the first symptom of lSSc, and often precedes other manifestations of the disease by years. Raynaud’s phenomenon is triggered by cold conditions and emotional stress, which cause spasms and narrowing of the blood vessels of the skin. Telangiectasia and calcinosis often follow skin tightening and thickening on the face and hands.
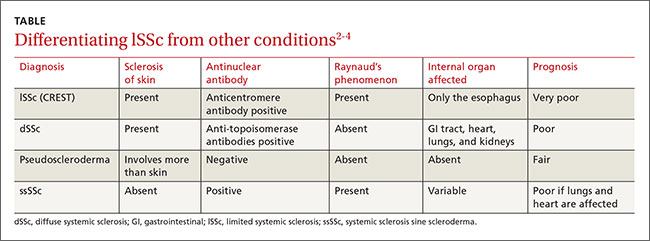
The differential diagnosis for lSSc includes dSSc, pseudoscleroderma, and systemic sclerosis sine scleroderma (ssSSc) (TABLE).2-4 In 2013, a joint committee of the American College of Rheumatology (ACR) and the European League Against Rheumatism published revised classification criteria for SSc based on a scoring systemto improve sensitivity because the 1980 ACR criteria did not classify some cases of limited cutaneous SSc.5 Based on these revised criteria, patients having a total score of 9 or more are classified as having SSc. Our patient’s score was 15.
Lab tests, imaging studies are used to diagnose SSc
Generally, blood testing in patients with SSc may show thrombocytopenia, hypergammaglobulinemia, or (in patients with renal involvement of SSc) elevated blood urea and creatinine levels. Creatine kinase, erythrocyte sedimentation rate, and C-reactive protein may be elevated due to myositis, vasculitis, malignancy, or an overlap of systemic sclerosis with another autoimmune disease.3
Serologic testing. Antinuclear antibodies (ANA) are positive in 60% to 80% of patients with SSc.2 Antibodies to topoisomerase-1 (Scl-70 antibodies) are present in 30% of cases of dSSc.2 The presence of either anticentromere antibodies (ACA) or anti-Scl-70 is highly specific (95%-99%) for the diagnosis of lSSc and dSSc, respectively.2 Anti-polymerase 1 and 3 antibodies (RNAP) are associated with dSSc and a significantly higher incidence of renal involvement.4
Capillary microscopy can be helpful in showing dilated, tortuous, and enlarged capillaries in the nail fold and adjacent areas. It is an effective method for distinguishing between primary and secondary Raynaud’s phenomenon.6
Chest x-ray. The lungs are involved in approximately 80% of all patients with SSc, and lung involvement is the leading cause of morbidity and mortality.7 The 2 most common types of direct pulmonary involvement are interstitial lung disease and pulmonary hypertension, which together account for 60% of SSc-related deaths.8
Key elements of sclerodermal lung disease are inflammation, lung scarring, and pulmonary hypertension due to progressive scarring of the inner lining of the small arteries. Inflammation and scarring of lung tissue causes interstitial lung disease. This is more common in dSSc, whereas pulmonary hypertension is more common in lSSc.8 In ssSSc, pulmonary disease can occur without any skin involvement. High-resolution computer tomography scans of the lungs (HRCT) can sensitively detect scarring and severity of lung inflammation, while a simple chest x-ray cannot.9 Interstitial abnormalities on HRCT have been found in 90% of patients.9
Spirometry. Interstitial lung disease leads to a reduction in forced vital capacity (FVC) and diffusing capacity of the lungs for carbon monoxide (DLCO). Significant pulmonary involvement is detectable in 25% of patients with SSc within 3 years of diagnosis.10 Interstitial fibrosis shows a restrictive pattern on spirometry. Forced expiratory volume in 1 second (FEV1) and FVC are reduced, and the FEV1/FVC ratio is normal or increased.
No cure, but patient-specific Tx can improve quality of life
SSc greatly reduces a patient’s self-esteem and quality of life. Because there is no cure, many patients develop depression.11,12 However, depending on the patient, some treatments can control symptoms and complications, and thus improve quality of life.
Because SSc is an autoimmune disease, immunosuppressive agents are a pillar of treatment. Recent studies have shown that low-dose pulse cyclophosphamide can stabilize pulmonary function.13
Raynaud’s phenomenon is treated with low-dose calcium-channel blockers, such as amlodipine 5 mg/d that is gradually increased to as much as 20 mg/d to increase blood flow to the fingers.14
Fibrosis of the skin is treated with daily doses of PUVA (photochemotherapy) 0.25 J/cm2 or 0.4 J/cm2 for 3 to 8 weeks (total doses between 3.5 J/cm2 and 9.6 J/cm2); this results in improvements in hand closure, skin sclerosis index, and flexion of fingers or knee joints.15 D-penicillamine, an anti-fibrotic drug that has the ability to not only destabilize tissue collagen but also reduce its production, is started early with a low dose and then carefully increased.16
Our patient declined hospital care or treatment and refused referral to a physiotherapist for hand exercises, paraffin baths, massages, splints, or water aerobics. Her condition remained stable and she has been able to manage on her own, despite her advanced stage of CREST syndrome.
CORRESPONDENCE
Chandramani Thuraisingham, MBBS, FAFP, FRACGP, AM, DRM, PDOH, International Medical University, Clinical School Seremban, Jalan Rasah, 70300 Seremban, Negeri Sembilan, Malaysia; [email protected].
1. Scleroderma. American College of Rheumatology Web site. Available at: http://www.rheumatology.org/I-Am-A/Patient-Caregiver/Diseases-Conditions/Scleroderma. Accessed January 7, 2016.
2. Chatterjee S. Systemic scleroderma. Cleveland Clinic Web site. August 2010. Available at: http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/rheumatology/systemic-sclerosis/. Accessed January 7, 2016.
3. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78:961-968.
4. Bunn CC, Denton CP, Shi-Wen X, et al. Anti-RNA polymerases and other autoantibody specificities in systemic sclerosis. Br J Rheumatol. 1998;37:15-20.
5. van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013;72:1747-1755.
6. Pavlov-Dolijanovic S, Damjanov NS, Stojanovic RM, et al. Scleroderma pattern of nailfold capillary changes as predictive value for the development of a connective tissue disease: a follow-up study of 3,029 patients with primary Raynaud’s phenomenon. Rheumatol Int. 2012;32:3039-3045.
7. Lung involvement. University of Michigan Health System, Scleroderma Program Web site. Available at: https://www.med.umich.edu/scleroderma/patients/lung.htm. Accessed January 7, 2016.
8. Morelli S, Barbieri C, Sgreccia A, et al. Relationship between cutaneous and pulmonary involvement in systemic sclerosis. J Rheumatol. 1997;24:81-85.
9. Schurawitzki H, Stiglbauer R, Graninger W, et al. Interstitial lung disease in progressive systemic sclerosis: high-resolution CT versus radiography. Radiology. 1990;176:755-759.
10. Wells AU, Steen V, Valentini G. Pulmonary complications: one of the most challenging complications of systemic sclerosis. Rheumatology (Oxford). 2009;48:iii40-iii44.
11. Thombs BD, Taillefer SS, Hudson M, et al. Depression in patients with systemic sclerosis: a systematic review of the evidence. Arthritis Rheum. 2007;57:1089-1097.
12. Benrud-Larson LM, Heinberg LJ, Boling C, et al. Body image dissatisfaction among women with scleroderma: extent and relationship to psychosocial function. Health Psychol. 2003;22:130-139.
13. Iudici M, Cuomo G, Vettori S, et al. Low-dose pulse cyclophosphamide in interstitial lung disease associated with systemic sclerosis (SSc-ILD): efficacy of maintenance immunosuppression in responders and non-responders. Semin Arthritis Rheum. 2015;44:437-444.
14. Treatment of the Raynaud phenomenon resistant to initial therapy. UpToDate Website. Available at: http://www.uptodate.com/contents/treatment-of-the-raynaud-phenomenon-resistant-to-initial-therapy. Accessed January 7, 2016.
15. Kanekura T, Fukumaru S, Matsushita S, et al. Successful treatment of scleroderma with PUVA therapy. J Dermatol. 1996;23:455-459.
16. Jayson MI, Lovell C, Black CM, et al. Penicillamine therapy in systemic sclerosis. Proc R Soc Med. 1977;70:82-88.
A 47-year-old Malaysian aboriginal woman presented to our clinic with stiffness in her fingers and feet that had been bothering her for about 10 years. The patient had multiple facial deformities, including perioral fibrosis, which gave her face a bird-like appearance; micrognathia (which affected the alignment of her teeth); a narrow mouth with pursed, puckered lips; bound-down skin of the nose; and a loss of wrinkling.
Ivory-colored plaques of hard, thickened skin caused a pigmented, “salt and pepper” appearance on our patient’s face. She also had deformities of all of her fingers and toes that severely restricted her ability to move them (FIGURE).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Limited systemic sclerosis
Systemic sclerosis (SSc) is a rare autoimmune disease that mainly affects women ages 30 to 50.1 SSc is classified according to the extent of skin involvement and includes limited SSc (lSSc), which is also called CREST syndrome, and diffuse SSc (dSSc).
CREST stands for:
- calcinosis, or subcutaneous calcium deposits,
- Raynaud’s phenomenon,
- esophageal dysfunction,
- sclerodactyly, which presents as tightening of the skin of the fingers or toes, and
- telangiectasia, which is characterized by dilated superficial capillaries, usually on the face, arms, and hands.2
Patients like ours who have lSSc experience a gradual progression of symptoms. Their skin is affected in limited areas, such as the fingers, hands, face, lower arms, and feet. There is no involvement of the chest, abdomen, or internal organs, with the exception of the esophagus. Esophageal smooth muscle becomes atrophied and is replaced by fibrous tissue, leading to esophageal motility dysfunction. This is in contrast to dSSc, in which you would see involvement of internal organs such as the lungs, kidneys, and gastrointestinal tract.
Raynaud’s phenomenon is commonly the first symptom of lSSc, and often precedes other manifestations of the disease by years. Raynaud’s phenomenon is triggered by cold conditions and emotional stress, which cause spasms and narrowing of the blood vessels of the skin. Telangiectasia and calcinosis often follow skin tightening and thickening on the face and hands.
The differential diagnosis for lSSc includes dSSc, pseudoscleroderma, and systemic sclerosis sine scleroderma (ssSSc) (TABLE).2-4 In 2013, a joint committee of the American College of Rheumatology (ACR) and the European League Against Rheumatism published revised classification criteria for SSc based on a scoring systemto improve sensitivity because the 1980 ACR criteria did not classify some cases of limited cutaneous SSc.5 Based on these revised criteria, patients having a total score of 9 or more are classified as having SSc. Our patient’s score was 15.
Lab tests, imaging studies are used to diagnose SSc
Generally, blood testing in patients with SSc may show thrombocytopenia, hypergammaglobulinemia, or (in patients with renal involvement of SSc) elevated blood urea and creatinine levels. Creatine kinase, erythrocyte sedimentation rate, and C-reactive protein may be elevated due to myositis, vasculitis, malignancy, or an overlap of systemic sclerosis with another autoimmune disease.3
Serologic testing. Antinuclear antibodies (ANA) are positive in 60% to 80% of patients with SSc.2 Antibodies to topoisomerase-1 (Scl-70 antibodies) are present in 30% of cases of dSSc.2 The presence of either anticentromere antibodies (ACA) or anti-Scl-70 is highly specific (95%-99%) for the diagnosis of lSSc and dSSc, respectively.2 Anti-polymerase 1 and 3 antibodies (RNAP) are associated with dSSc and a significantly higher incidence of renal involvement.4
Capillary microscopy can be helpful in showing dilated, tortuous, and enlarged capillaries in the nail fold and adjacent areas. It is an effective method for distinguishing between primary and secondary Raynaud’s phenomenon.6
Chest x-ray. The lungs are involved in approximately 80% of all patients with SSc, and lung involvement is the leading cause of morbidity and mortality.7 The 2 most common types of direct pulmonary involvement are interstitial lung disease and pulmonary hypertension, which together account for 60% of SSc-related deaths.8
Key elements of sclerodermal lung disease are inflammation, lung scarring, and pulmonary hypertension due to progressive scarring of the inner lining of the small arteries. Inflammation and scarring of lung tissue causes interstitial lung disease. This is more common in dSSc, whereas pulmonary hypertension is more common in lSSc.8 In ssSSc, pulmonary disease can occur without any skin involvement. High-resolution computer tomography scans of the lungs (HRCT) can sensitively detect scarring and severity of lung inflammation, while a simple chest x-ray cannot.9 Interstitial abnormalities on HRCT have been found in 90% of patients.9
Spirometry. Interstitial lung disease leads to a reduction in forced vital capacity (FVC) and diffusing capacity of the lungs for carbon monoxide (DLCO). Significant pulmonary involvement is detectable in 25% of patients with SSc within 3 years of diagnosis.10 Interstitial fibrosis shows a restrictive pattern on spirometry. Forced expiratory volume in 1 second (FEV1) and FVC are reduced, and the FEV1/FVC ratio is normal or increased.
No cure, but patient-specific Tx can improve quality of life
SSc greatly reduces a patient’s self-esteem and quality of life. Because there is no cure, many patients develop depression.11,12 However, depending on the patient, some treatments can control symptoms and complications, and thus improve quality of life.
Because SSc is an autoimmune disease, immunosuppressive agents are a pillar of treatment. Recent studies have shown that low-dose pulse cyclophosphamide can stabilize pulmonary function.13
Raynaud’s phenomenon is treated with low-dose calcium-channel blockers, such as amlodipine 5 mg/d that is gradually increased to as much as 20 mg/d to increase blood flow to the fingers.14
Fibrosis of the skin is treated with daily doses of PUVA (photochemotherapy) 0.25 J/cm2 or 0.4 J/cm2 for 3 to 8 weeks (total doses between 3.5 J/cm2 and 9.6 J/cm2); this results in improvements in hand closure, skin sclerosis index, and flexion of fingers or knee joints.15 D-penicillamine, an anti-fibrotic drug that has the ability to not only destabilize tissue collagen but also reduce its production, is started early with a low dose and then carefully increased.16
Our patient declined hospital care or treatment and refused referral to a physiotherapist for hand exercises, paraffin baths, massages, splints, or water aerobics. Her condition remained stable and she has been able to manage on her own, despite her advanced stage of CREST syndrome.
CORRESPONDENCE
Chandramani Thuraisingham, MBBS, FAFP, FRACGP, AM, DRM, PDOH, International Medical University, Clinical School Seremban, Jalan Rasah, 70300 Seremban, Negeri Sembilan, Malaysia; [email protected].
A 47-year-old Malaysian aboriginal woman presented to our clinic with stiffness in her fingers and feet that had been bothering her for about 10 years. The patient had multiple facial deformities, including perioral fibrosis, which gave her face a bird-like appearance; micrognathia (which affected the alignment of her teeth); a narrow mouth with pursed, puckered lips; bound-down skin of the nose; and a loss of wrinkling.
Ivory-colored plaques of hard, thickened skin caused a pigmented, “salt and pepper” appearance on our patient’s face. She also had deformities of all of her fingers and toes that severely restricted her ability to move them (FIGURE).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Limited systemic sclerosis
Systemic sclerosis (SSc) is a rare autoimmune disease that mainly affects women ages 30 to 50.1 SSc is classified according to the extent of skin involvement and includes limited SSc (lSSc), which is also called CREST syndrome, and diffuse SSc (dSSc).
CREST stands for:
- calcinosis, or subcutaneous calcium deposits,
- Raynaud’s phenomenon,
- esophageal dysfunction,
- sclerodactyly, which presents as tightening of the skin of the fingers or toes, and
- telangiectasia, which is characterized by dilated superficial capillaries, usually on the face, arms, and hands.2
Patients like ours who have lSSc experience a gradual progression of symptoms. Their skin is affected in limited areas, such as the fingers, hands, face, lower arms, and feet. There is no involvement of the chest, abdomen, or internal organs, with the exception of the esophagus. Esophageal smooth muscle becomes atrophied and is replaced by fibrous tissue, leading to esophageal motility dysfunction. This is in contrast to dSSc, in which you would see involvement of internal organs such as the lungs, kidneys, and gastrointestinal tract.
Raynaud’s phenomenon is commonly the first symptom of lSSc, and often precedes other manifestations of the disease by years. Raynaud’s phenomenon is triggered by cold conditions and emotional stress, which cause spasms and narrowing of the blood vessels of the skin. Telangiectasia and calcinosis often follow skin tightening and thickening on the face and hands.
The differential diagnosis for lSSc includes dSSc, pseudoscleroderma, and systemic sclerosis sine scleroderma (ssSSc) (TABLE).2-4 In 2013, a joint committee of the American College of Rheumatology (ACR) and the European League Against Rheumatism published revised classification criteria for SSc based on a scoring systemto improve sensitivity because the 1980 ACR criteria did not classify some cases of limited cutaneous SSc.5 Based on these revised criteria, patients having a total score of 9 or more are classified as having SSc. Our patient’s score was 15.
Lab tests, imaging studies are used to diagnose SSc
Generally, blood testing in patients with SSc may show thrombocytopenia, hypergammaglobulinemia, or (in patients with renal involvement of SSc) elevated blood urea and creatinine levels. Creatine kinase, erythrocyte sedimentation rate, and C-reactive protein may be elevated due to myositis, vasculitis, malignancy, or an overlap of systemic sclerosis with another autoimmune disease.3
Serologic testing. Antinuclear antibodies (ANA) are positive in 60% to 80% of patients with SSc.2 Antibodies to topoisomerase-1 (Scl-70 antibodies) are present in 30% of cases of dSSc.2 The presence of either anticentromere antibodies (ACA) or anti-Scl-70 is highly specific (95%-99%) for the diagnosis of lSSc and dSSc, respectively.2 Anti-polymerase 1 and 3 antibodies (RNAP) are associated with dSSc and a significantly higher incidence of renal involvement.4
Capillary microscopy can be helpful in showing dilated, tortuous, and enlarged capillaries in the nail fold and adjacent areas. It is an effective method for distinguishing between primary and secondary Raynaud’s phenomenon.6
Chest x-ray. The lungs are involved in approximately 80% of all patients with SSc, and lung involvement is the leading cause of morbidity and mortality.7 The 2 most common types of direct pulmonary involvement are interstitial lung disease and pulmonary hypertension, which together account for 60% of SSc-related deaths.8
Key elements of sclerodermal lung disease are inflammation, lung scarring, and pulmonary hypertension due to progressive scarring of the inner lining of the small arteries. Inflammation and scarring of lung tissue causes interstitial lung disease. This is more common in dSSc, whereas pulmonary hypertension is more common in lSSc.8 In ssSSc, pulmonary disease can occur without any skin involvement. High-resolution computer tomography scans of the lungs (HRCT) can sensitively detect scarring and severity of lung inflammation, while a simple chest x-ray cannot.9 Interstitial abnormalities on HRCT have been found in 90% of patients.9
Spirometry. Interstitial lung disease leads to a reduction in forced vital capacity (FVC) and diffusing capacity of the lungs for carbon monoxide (DLCO). Significant pulmonary involvement is detectable in 25% of patients with SSc within 3 years of diagnosis.10 Interstitial fibrosis shows a restrictive pattern on spirometry. Forced expiratory volume in 1 second (FEV1) and FVC are reduced, and the FEV1/FVC ratio is normal or increased.
No cure, but patient-specific Tx can improve quality of life
SSc greatly reduces a patient’s self-esteem and quality of life. Because there is no cure, many patients develop depression.11,12 However, depending on the patient, some treatments can control symptoms and complications, and thus improve quality of life.
Because SSc is an autoimmune disease, immunosuppressive agents are a pillar of treatment. Recent studies have shown that low-dose pulse cyclophosphamide can stabilize pulmonary function.13
Raynaud’s phenomenon is treated with low-dose calcium-channel blockers, such as amlodipine 5 mg/d that is gradually increased to as much as 20 mg/d to increase blood flow to the fingers.14
Fibrosis of the skin is treated with daily doses of PUVA (photochemotherapy) 0.25 J/cm2 or 0.4 J/cm2 for 3 to 8 weeks (total doses between 3.5 J/cm2 and 9.6 J/cm2); this results in improvements in hand closure, skin sclerosis index, and flexion of fingers or knee joints.15 D-penicillamine, an anti-fibrotic drug that has the ability to not only destabilize tissue collagen but also reduce its production, is started early with a low dose and then carefully increased.16
Our patient declined hospital care or treatment and refused referral to a physiotherapist for hand exercises, paraffin baths, massages, splints, or water aerobics. Her condition remained stable and she has been able to manage on her own, despite her advanced stage of CREST syndrome.
CORRESPONDENCE
Chandramani Thuraisingham, MBBS, FAFP, FRACGP, AM, DRM, PDOH, International Medical University, Clinical School Seremban, Jalan Rasah, 70300 Seremban, Negeri Sembilan, Malaysia; [email protected].
1. Scleroderma. American College of Rheumatology Web site. Available at: http://www.rheumatology.org/I-Am-A/Patient-Caregiver/Diseases-Conditions/Scleroderma. Accessed January 7, 2016.
2. Chatterjee S. Systemic scleroderma. Cleveland Clinic Web site. August 2010. Available at: http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/rheumatology/systemic-sclerosis/. Accessed January 7, 2016.
3. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78:961-968.
4. Bunn CC, Denton CP, Shi-Wen X, et al. Anti-RNA polymerases and other autoantibody specificities in systemic sclerosis. Br J Rheumatol. 1998;37:15-20.
5. van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013;72:1747-1755.
6. Pavlov-Dolijanovic S, Damjanov NS, Stojanovic RM, et al. Scleroderma pattern of nailfold capillary changes as predictive value for the development of a connective tissue disease: a follow-up study of 3,029 patients with primary Raynaud’s phenomenon. Rheumatol Int. 2012;32:3039-3045.
7. Lung involvement. University of Michigan Health System, Scleroderma Program Web site. Available at: https://www.med.umich.edu/scleroderma/patients/lung.htm. Accessed January 7, 2016.
8. Morelli S, Barbieri C, Sgreccia A, et al. Relationship between cutaneous and pulmonary involvement in systemic sclerosis. J Rheumatol. 1997;24:81-85.
9. Schurawitzki H, Stiglbauer R, Graninger W, et al. Interstitial lung disease in progressive systemic sclerosis: high-resolution CT versus radiography. Radiology. 1990;176:755-759.
10. Wells AU, Steen V, Valentini G. Pulmonary complications: one of the most challenging complications of systemic sclerosis. Rheumatology (Oxford). 2009;48:iii40-iii44.
11. Thombs BD, Taillefer SS, Hudson M, et al. Depression in patients with systemic sclerosis: a systematic review of the evidence. Arthritis Rheum. 2007;57:1089-1097.
12. Benrud-Larson LM, Heinberg LJ, Boling C, et al. Body image dissatisfaction among women with scleroderma: extent and relationship to psychosocial function. Health Psychol. 2003;22:130-139.
13. Iudici M, Cuomo G, Vettori S, et al. Low-dose pulse cyclophosphamide in interstitial lung disease associated with systemic sclerosis (SSc-ILD): efficacy of maintenance immunosuppression in responders and non-responders. Semin Arthritis Rheum. 2015;44:437-444.
14. Treatment of the Raynaud phenomenon resistant to initial therapy. UpToDate Website. Available at: http://www.uptodate.com/contents/treatment-of-the-raynaud-phenomenon-resistant-to-initial-therapy. Accessed January 7, 2016.
15. Kanekura T, Fukumaru S, Matsushita S, et al. Successful treatment of scleroderma with PUVA therapy. J Dermatol. 1996;23:455-459.
16. Jayson MI, Lovell C, Black CM, et al. Penicillamine therapy in systemic sclerosis. Proc R Soc Med. 1977;70:82-88.
1. Scleroderma. American College of Rheumatology Web site. Available at: http://www.rheumatology.org/I-Am-A/Patient-Caregiver/Diseases-Conditions/Scleroderma. Accessed January 7, 2016.
2. Chatterjee S. Systemic scleroderma. Cleveland Clinic Web site. August 2010. Available at: http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/rheumatology/systemic-sclerosis/. Accessed January 7, 2016.
3. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78:961-968.
4. Bunn CC, Denton CP, Shi-Wen X, et al. Anti-RNA polymerases and other autoantibody specificities in systemic sclerosis. Br J Rheumatol. 1998;37:15-20.
5. van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013;72:1747-1755.
6. Pavlov-Dolijanovic S, Damjanov NS, Stojanovic RM, et al. Scleroderma pattern of nailfold capillary changes as predictive value for the development of a connective tissue disease: a follow-up study of 3,029 patients with primary Raynaud’s phenomenon. Rheumatol Int. 2012;32:3039-3045.
7. Lung involvement. University of Michigan Health System, Scleroderma Program Web site. Available at: https://www.med.umich.edu/scleroderma/patients/lung.htm. Accessed January 7, 2016.
8. Morelli S, Barbieri C, Sgreccia A, et al. Relationship between cutaneous and pulmonary involvement in systemic sclerosis. J Rheumatol. 1997;24:81-85.
9. Schurawitzki H, Stiglbauer R, Graninger W, et al. Interstitial lung disease in progressive systemic sclerosis: high-resolution CT versus radiography. Radiology. 1990;176:755-759.
10. Wells AU, Steen V, Valentini G. Pulmonary complications: one of the most challenging complications of systemic sclerosis. Rheumatology (Oxford). 2009;48:iii40-iii44.
11. Thombs BD, Taillefer SS, Hudson M, et al. Depression in patients with systemic sclerosis: a systematic review of the evidence. Arthritis Rheum. 2007;57:1089-1097.
12. Benrud-Larson LM, Heinberg LJ, Boling C, et al. Body image dissatisfaction among women with scleroderma: extent and relationship to psychosocial function. Health Psychol. 2003;22:130-139.
13. Iudici M, Cuomo G, Vettori S, et al. Low-dose pulse cyclophosphamide in interstitial lung disease associated with systemic sclerosis (SSc-ILD): efficacy of maintenance immunosuppression in responders and non-responders. Semin Arthritis Rheum. 2015;44:437-444.
14. Treatment of the Raynaud phenomenon resistant to initial therapy. UpToDate Website. Available at: http://www.uptodate.com/contents/treatment-of-the-raynaud-phenomenon-resistant-to-initial-therapy. Accessed January 7, 2016.
15. Kanekura T, Fukumaru S, Matsushita S, et al. Successful treatment of scleroderma with PUVA therapy. J Dermatol. 1996;23:455-459.
16. Jayson MI, Lovell C, Black CM, et al. Penicillamine therapy in systemic sclerosis. Proc R Soc Med. 1977;70:82-88.
