User login
ASH preview: Key themes include tackling CAR T obstacles, sickle cell advances, VTE
Chimeric antigen receptor (CAR) T-cell therapies have garnered a great deal of attention given their “incredible efficacy” in treating B-cell malignancies, and new findings are taking aim at the drawbacks of therapy, such as the time, expense, and toxicity involved, according to Robert A. Brodsky, MD.

One example, from a study slated for presentation during a plenary session at the upcoming annual meeting of the American Society of Hematology involves the investigational T-cell bispecific antibody mosunetuzumab, which targets both CD20 on the surface of malignant B cells, and CD3 on cytotoxic T cells, engaging the T cells and directing their cytotoxicity against B cells.
In a study (Abstract 6) of 218 non-Hodgkin lymphoma patients, including 23 who had already received CAR T-cell therapy and had relapsed or were refractory to the treatment, 64% responded, 42% had a complete response, and the median duration of response is now out to 9 months, Dr. Brodsky, ASH secretary and director of the division of hematology at Johns Hopkins University, Baltimore, said during a premeeting press conference.
“It’s basically an antibody using the patient’s own T cell to do what a CAR-T cell would do – [a] very exciting study and large study,” he said. “It is an off-the-shelf product, it completely gets around the problem of the time to generate the CAR T-cell product, and because it’s going to be much simpler and faster to produce, it’s likely going to be much cheaper than CAR T cells.”
The preliminary results also suggest it is less toxic than CAR T-cell therapy, he added.
Two other CAR T-cell therapy–related studies highlighted during the press conference address its use for multiple myeloma. One, the phase 1b/2 CARTITUDE study (Abstract 577) uses CAR T cells against the B-cell maturation antigen (BCMA) in the relapsed/refractory setting.
Of 25 patients treated with chemotherapy followed by CAR T-cell infusion and followed for a median of 3 months, 91% responded, two achieved a complete remission, and “many other responses were very deep responses,” Dr. Brodsky said, noting that the second featured multiple myeloma trial (Abstract 930) looked at bispecific CAR T-cell therapy targeting BCMA and CD38 in an effort to reduce resistance to the therapy.
“Again, very interesting preliminary results,” he said, noting that of 16 patients followed for a median of 36 weeks, 87.5% responded, the treatment was well tolerated, and progression-free survival at 9 months was 75%.
In addition to the “key theme” of overcoming CAR T-cell therapy obstacles, three other themes have emerged from among the thousands of abstracts submitted for presentation at ASH. These, as presented during the press conference, include new venous thromboembolism (VTE) therapies and approaches to research; inclusive medicine, with abstracts focused on age- and race-related issues in clinical trials; and new advances in the treatment of sickle cell disease. All of these have potentially practice-changing implications, as do the six late-breaking abstracts selected from 93 abstracts submitted for consideration for oral presentation at ASH, Dr. Brodsky said.
One of the “truly practice-changing” late-breakers is a randomized phase 3 trial (Abstract LBA-1) comparing the bispecific antibody blinatumomab to chemotherapy for post-re-induction therapy in high- and intermediate-risk acute lymphoblastic leukemia (ALL) at first relapse in children, adolescents and young adults.
The study demonstrated the superiority of blinatumomab for efficacy and tolerability, which is particularly encouraging given the challenges in getting relapsed ALL patients back into remission so they can undergo bone marrow transplant, Dr. Brodsky said.
Of 208 patients randomized, 73% vs. 45% in the blinatumomab vs. chemotherapy arms were able to get to transplant – and therefore to potential cure, he said.
“Of note, the blinatumomab arm was less toxic and there was marked improvement in disease-free and overall survival, so this is clearly going to become a new standard of care for relapsed and refractory ALL,” he added.
Chimeric antigen receptor (CAR) T-cell therapies have garnered a great deal of attention given their “incredible efficacy” in treating B-cell malignancies, and new findings are taking aim at the drawbacks of therapy, such as the time, expense, and toxicity involved, according to Robert A. Brodsky, MD.
One example, from a study slated for presentation during a plenary session at the upcoming annual meeting of the American Society of Hematology involves the investigational T-cell bispecific antibody mosunetuzumab, which targets both CD20 on the surface of malignant B cells, and CD3 on cytotoxic T cells, engaging the T cells and directing their cytotoxicity against B cells.
In a study (Abstract 6) of 218 non-Hodgkin lymphoma patients, including 23 who had already received CAR T-cell therapy and had relapsed or were refractory to the treatment, 64% responded, 42% had a complete response, and the median duration of response is now out to 9 months, Dr. Brodsky, ASH secretary and director of the division of hematology at Johns Hopkins University, Baltimore, said during a premeeting press conference.
“It’s basically an antibody using the patient’s own T cell to do what a CAR-T cell would do – [a] very exciting study and large study,” he said. “It is an off-the-shelf product, it completely gets around the problem of the time to generate the CAR T-cell product, and because it’s going to be much simpler and faster to produce, it’s likely going to be much cheaper than CAR T cells.”
The preliminary results also suggest it is less toxic than CAR T-cell therapy, he added.
Two other CAR T-cell therapy–related studies highlighted during the press conference address its use for multiple myeloma. One, the phase 1b/2 CARTITUDE study (Abstract 577) uses CAR T cells against the B-cell maturation antigen (BCMA) in the relapsed/refractory setting.
Of 25 patients treated with chemotherapy followed by CAR T-cell infusion and followed for a median of 3 months, 91% responded, two achieved a complete remission, and “many other responses were very deep responses,” Dr. Brodsky said, noting that the second featured multiple myeloma trial (Abstract 930) looked at bispecific CAR T-cell therapy targeting BCMA and CD38 in an effort to reduce resistance to the therapy.
“Again, very interesting preliminary results,” he said, noting that of 16 patients followed for a median of 36 weeks, 87.5% responded, the treatment was well tolerated, and progression-free survival at 9 months was 75%.
In addition to the “key theme” of overcoming CAR T-cell therapy obstacles, three other themes have emerged from among the thousands of abstracts submitted for presentation at ASH. These, as presented during the press conference, include new venous thromboembolism (VTE) therapies and approaches to research; inclusive medicine, with abstracts focused on age- and race-related issues in clinical trials; and new advances in the treatment of sickle cell disease. All of these have potentially practice-changing implications, as do the six late-breaking abstracts selected from 93 abstracts submitted for consideration for oral presentation at ASH, Dr. Brodsky said.
One of the “truly practice-changing” late-breakers is a randomized phase 3 trial (Abstract LBA-1) comparing the bispecific antibody blinatumomab to chemotherapy for post-re-induction therapy in high- and intermediate-risk acute lymphoblastic leukemia (ALL) at first relapse in children, adolescents and young adults.
The study demonstrated the superiority of blinatumomab for efficacy and tolerability, which is particularly encouraging given the challenges in getting relapsed ALL patients back into remission so they can undergo bone marrow transplant, Dr. Brodsky said.
Of 208 patients randomized, 73% vs. 45% in the blinatumomab vs. chemotherapy arms were able to get to transplant – and therefore to potential cure, he said.
“Of note, the blinatumomab arm was less toxic and there was marked improvement in disease-free and overall survival, so this is clearly going to become a new standard of care for relapsed and refractory ALL,” he added.
Chimeric antigen receptor (CAR) T-cell therapies have garnered a great deal of attention given their “incredible efficacy” in treating B-cell malignancies, and new findings are taking aim at the drawbacks of therapy, such as the time, expense, and toxicity involved, according to Robert A. Brodsky, MD.
One example, from a study slated for presentation during a plenary session at the upcoming annual meeting of the American Society of Hematology involves the investigational T-cell bispecific antibody mosunetuzumab, which targets both CD20 on the surface of malignant B cells, and CD3 on cytotoxic T cells, engaging the T cells and directing their cytotoxicity against B cells.
In a study (Abstract 6) of 218 non-Hodgkin lymphoma patients, including 23 who had already received CAR T-cell therapy and had relapsed or were refractory to the treatment, 64% responded, 42% had a complete response, and the median duration of response is now out to 9 months, Dr. Brodsky, ASH secretary and director of the division of hematology at Johns Hopkins University, Baltimore, said during a premeeting press conference.
“It’s basically an antibody using the patient’s own T cell to do what a CAR-T cell would do – [a] very exciting study and large study,” he said. “It is an off-the-shelf product, it completely gets around the problem of the time to generate the CAR T-cell product, and because it’s going to be much simpler and faster to produce, it’s likely going to be much cheaper than CAR T cells.”
The preliminary results also suggest it is less toxic than CAR T-cell therapy, he added.
Two other CAR T-cell therapy–related studies highlighted during the press conference address its use for multiple myeloma. One, the phase 1b/2 CARTITUDE study (Abstract 577) uses CAR T cells against the B-cell maturation antigen (BCMA) in the relapsed/refractory setting.
Of 25 patients treated with chemotherapy followed by CAR T-cell infusion and followed for a median of 3 months, 91% responded, two achieved a complete remission, and “many other responses were very deep responses,” Dr. Brodsky said, noting that the second featured multiple myeloma trial (Abstract 930) looked at bispecific CAR T-cell therapy targeting BCMA and CD38 in an effort to reduce resistance to the therapy.
“Again, very interesting preliminary results,” he said, noting that of 16 patients followed for a median of 36 weeks, 87.5% responded, the treatment was well tolerated, and progression-free survival at 9 months was 75%.
In addition to the “key theme” of overcoming CAR T-cell therapy obstacles, three other themes have emerged from among the thousands of abstracts submitted for presentation at ASH. These, as presented during the press conference, include new venous thromboembolism (VTE) therapies and approaches to research; inclusive medicine, with abstracts focused on age- and race-related issues in clinical trials; and new advances in the treatment of sickle cell disease. All of these have potentially practice-changing implications, as do the six late-breaking abstracts selected from 93 abstracts submitted for consideration for oral presentation at ASH, Dr. Brodsky said.
One of the “truly practice-changing” late-breakers is a randomized phase 3 trial (Abstract LBA-1) comparing the bispecific antibody blinatumomab to chemotherapy for post-re-induction therapy in high- and intermediate-risk acute lymphoblastic leukemia (ALL) at first relapse in children, adolescents and young adults.
The study demonstrated the superiority of blinatumomab for efficacy and tolerability, which is particularly encouraging given the challenges in getting relapsed ALL patients back into remission so they can undergo bone marrow transplant, Dr. Brodsky said.
Of 208 patients randomized, 73% vs. 45% in the blinatumomab vs. chemotherapy arms were able to get to transplant – and therefore to potential cure, he said.
“Of note, the blinatumomab arm was less toxic and there was marked improvement in disease-free and overall survival, so this is clearly going to become a new standard of care for relapsed and refractory ALL,” he added.
Early lenalidomide may delay progression of smoldering myeloma
Early treatment with lenalidomide may delay disease progression and prevent end-organ damage in patients with high-risk smoldering multiple myeloma (SMM), according to findings from a phase 3 trial.

While observation is the current standard of care in SMM, early therapy may represent a new standard for patients with high-risk disease, explained Sagar Lonial, MD, of Winship Cancer Institute, Emory University, Atlanta, and colleagues. Their findings were published in the Journal of Clinical Oncology.
The randomized, open-label, phase 3 study included 182 patients with intermediate- or high-risk SMM. Study patients were randomly allocated to receive either oral lenalidomide at 25 mg daily on days 1-21 of a 28-day cycle or observation.
Study subjects were stratified based on time since SMM diagnosis – 1 year or less vs. more than 1 year, and all patients in the lenalidomide arm received aspirin at 325 mg on days 1-28. Both interventions were maintained until unacceptable toxicity, disease progression, or withdrawal for other reasons.
The primary outcome was progression-free survival (PFS), measured from baseline to the development of symptomatic multiple myeloma (MM). The criteria for progression included evidence of end-organ damage in relation to MM and biochemical disease progression.
The researchers found that at 1 year PFS was 98% in the lenalidomide group and 89% in the observation group. At 2 years, PFS was 93% in the lenalidomide group and 76% in the observation group. PFS was 91% in the lenalidomide group and 66% in the observation group at 3 years (hazard ratio, 0.28; P = .002).
Among lenalidomide-treated patients, grade 3 or 4 hematologic and nonhematologic adverse events occurred in 36 patients (41%). Nonhematologic adverse events occurred in 25 patients (28%).
Frequent AEs among lenalidomide-treated patients included grade 4 decreased neutrophil count (4.5%), as well as grade 3 infections (20.5%), hypertension (9.1%), fatigue (6.8%), skin problems (5.7%), dyspnea (5.7%), and hypokalemia (3.4%). “In most cases, [adverse events] could be managed with dose modifications,” they wrote.
To reduce long-term toxicity, the researchers recommended a 2-year duration of therapy for patients at highest risk.
“Our results support the use of early intervention in patients with high-risk SMM – as defined by the 20/2/20 criteria where our magnitude of benefit was the greatest – rather than continued observation,” the researchers wrote.
The trial was funded by the National Cancer Institute. The authors reported financial affiliations with AbbVie, Aduro Biotech, Amgen, Bristol-Myers Squibb, Celgene, Juno Therapeutics, Kite Pharma, Sanofi, Takeda, and several other companies.
SOURCE: Lonial S et al. J Clin Oncol. 2019 Oct 25. doi: 10.1200/JCO.19.01740.
Early treatment with lenalidomide may delay disease progression and prevent end-organ damage in patients with high-risk smoldering multiple myeloma (SMM), according to findings from a phase 3 trial.
While observation is the current standard of care in SMM, early therapy may represent a new standard for patients with high-risk disease, explained Sagar Lonial, MD, of Winship Cancer Institute, Emory University, Atlanta, and colleagues. Their findings were published in the Journal of Clinical Oncology.
The randomized, open-label, phase 3 study included 182 patients with intermediate- or high-risk SMM. Study patients were randomly allocated to receive either oral lenalidomide at 25 mg daily on days 1-21 of a 28-day cycle or observation.
Study subjects were stratified based on time since SMM diagnosis – 1 year or less vs. more than 1 year, and all patients in the lenalidomide arm received aspirin at 325 mg on days 1-28. Both interventions were maintained until unacceptable toxicity, disease progression, or withdrawal for other reasons.
The primary outcome was progression-free survival (PFS), measured from baseline to the development of symptomatic multiple myeloma (MM). The criteria for progression included evidence of end-organ damage in relation to MM and biochemical disease progression.
The researchers found that at 1 year PFS was 98% in the lenalidomide group and 89% in the observation group. At 2 years, PFS was 93% in the lenalidomide group and 76% in the observation group. PFS was 91% in the lenalidomide group and 66% in the observation group at 3 years (hazard ratio, 0.28; P = .002).
Among lenalidomide-treated patients, grade 3 or 4 hematologic and nonhematologic adverse events occurred in 36 patients (41%). Nonhematologic adverse events occurred in 25 patients (28%).
Frequent AEs among lenalidomide-treated patients included grade 4 decreased neutrophil count (4.5%), as well as grade 3 infections (20.5%), hypertension (9.1%), fatigue (6.8%), skin problems (5.7%), dyspnea (5.7%), and hypokalemia (3.4%). “In most cases, [adverse events] could be managed with dose modifications,” they wrote.
To reduce long-term toxicity, the researchers recommended a 2-year duration of therapy for patients at highest risk.
“Our results support the use of early intervention in patients with high-risk SMM – as defined by the 20/2/20 criteria where our magnitude of benefit was the greatest – rather than continued observation,” the researchers wrote.
The trial was funded by the National Cancer Institute. The authors reported financial affiliations with AbbVie, Aduro Biotech, Amgen, Bristol-Myers Squibb, Celgene, Juno Therapeutics, Kite Pharma, Sanofi, Takeda, and several other companies.
SOURCE: Lonial S et al. J Clin Oncol. 2019 Oct 25. doi: 10.1200/JCO.19.01740.
Early treatment with lenalidomide may delay disease progression and prevent end-organ damage in patients with high-risk smoldering multiple myeloma (SMM), according to findings from a phase 3 trial.
While observation is the current standard of care in SMM, early therapy may represent a new standard for patients with high-risk disease, explained Sagar Lonial, MD, of Winship Cancer Institute, Emory University, Atlanta, and colleagues. Their findings were published in the Journal of Clinical Oncology.
The randomized, open-label, phase 3 study included 182 patients with intermediate- or high-risk SMM. Study patients were randomly allocated to receive either oral lenalidomide at 25 mg daily on days 1-21 of a 28-day cycle or observation.
Study subjects were stratified based on time since SMM diagnosis – 1 year or less vs. more than 1 year, and all patients in the lenalidomide arm received aspirin at 325 mg on days 1-28. Both interventions were maintained until unacceptable toxicity, disease progression, or withdrawal for other reasons.
The primary outcome was progression-free survival (PFS), measured from baseline to the development of symptomatic multiple myeloma (MM). The criteria for progression included evidence of end-organ damage in relation to MM and biochemical disease progression.
The researchers found that at 1 year PFS was 98% in the lenalidomide group and 89% in the observation group. At 2 years, PFS was 93% in the lenalidomide group and 76% in the observation group. PFS was 91% in the lenalidomide group and 66% in the observation group at 3 years (hazard ratio, 0.28; P = .002).
Among lenalidomide-treated patients, grade 3 or 4 hematologic and nonhematologic adverse events occurred in 36 patients (41%). Nonhematologic adverse events occurred in 25 patients (28%).
Frequent AEs among lenalidomide-treated patients included grade 4 decreased neutrophil count (4.5%), as well as grade 3 infections (20.5%), hypertension (9.1%), fatigue (6.8%), skin problems (5.7%), dyspnea (5.7%), and hypokalemia (3.4%). “In most cases, [adverse events] could be managed with dose modifications,” they wrote.
To reduce long-term toxicity, the researchers recommended a 2-year duration of therapy for patients at highest risk.
“Our results support the use of early intervention in patients with high-risk SMM – as defined by the 20/2/20 criteria where our magnitude of benefit was the greatest – rather than continued observation,” the researchers wrote.
The trial was funded by the National Cancer Institute. The authors reported financial affiliations with AbbVie, Aduro Biotech, Amgen, Bristol-Myers Squibb, Celgene, Juno Therapeutics, Kite Pharma, Sanofi, Takeda, and several other companies.
SOURCE: Lonial S et al. J Clin Oncol. 2019 Oct 25. doi: 10.1200/JCO.19.01740.
FROM JOURNAL OF CLINICAL ONCOLOGY
Rituximab maintenance has a durable benefit in follicular lymphoma
The benefit of rituximab maintenance therapy in follicular lymphoma is long lasting and evident out to at least 9 years, according to the final efficacy analysis of the PRIMA phase 3, randomized, controlled trial.
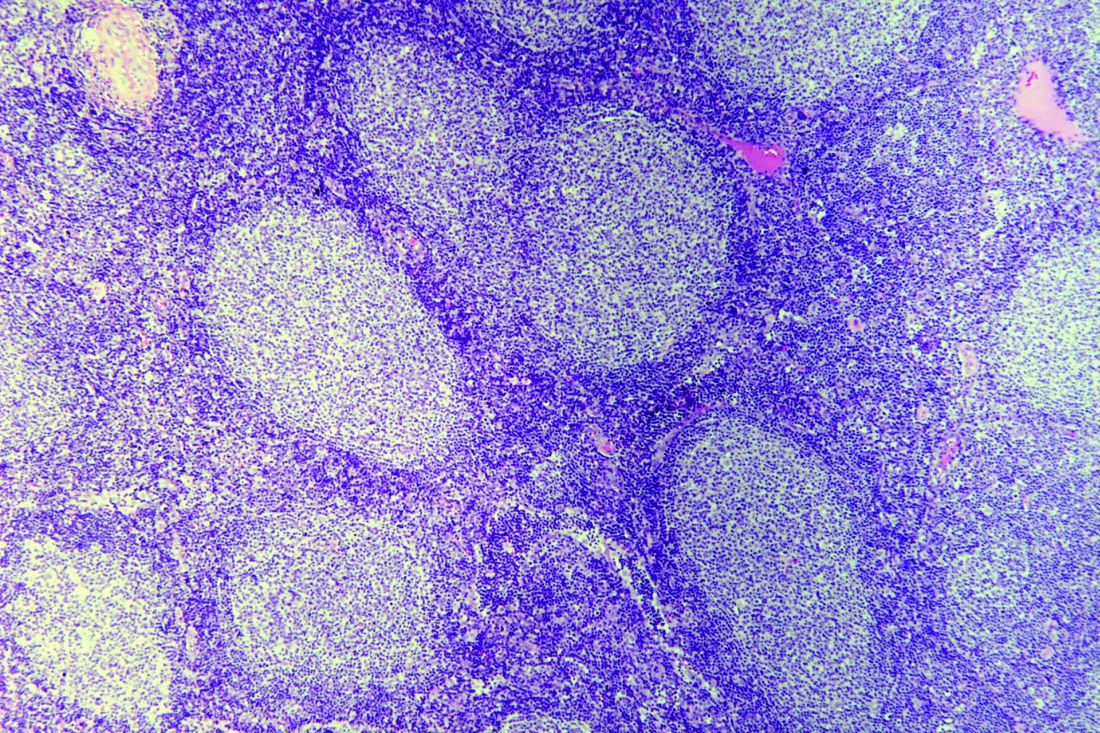
Previously reported results of this pivotal trial showed prolongation of progression-free survival and time to next treatment with rituximab maintenance at a follow-up of 3 years and 6 years.
“Rituximab maintenance is now widely recommended for patients with follicular lymphoma responding to first-line rituximab-based immunochemotherapy,” Emmanuel Bachy, MD, PhD, of Institut National de la Santé et de la Recherche Médicale in Pierre-Bénite, France, and colleagues reported in the Journal of Clinical Oncology.
In PRIMA, patients with previously untreated high–tumor burden follicular lymphoma received any of three immunochemotherapy induction regimens. The 1,018 patients having a response were then randomly assigned to receive 2 years of rituximab (Rituxan) maintenance or observation only.
Dr. Bachy and colleagues performed the trial’s final efficacy analyses, now at a median follow-up of 9 years.
Among the 607 patients consenting to the extended follow-up, median progression-free survival was 10.5 years in the rituximab-maintenance group, compared with 4.1 years in the observation group (hazard ratio, 0.61; P less than .001).
“Subgroup analyses showed the substantial progression-free survival improvement associated with rituximab maintenance was independent of age, sex, induction immunochemotherapy regimen, response to induction, or [Follicular Lymphoma International Prognostic Index] risk score,” the investigators wrote.
Rituximab maintenance also prolonged the median time to next antilymphoma treatment (not reached vs. 6.1 years; hazard ratio, 0.66; P less than .001) and time to next chemotherapy treatment (not reached vs. 9.3 years; hazard ratio, 0.71; P less than .001).
But there was no significant difference in overall survival. Median overall survival was not reached in either group (hazard ratio, 1.04; P = .7948). The estimated 10-year overall survival rate was about 80% in both groups.
“This 9-year follow-up of the PRIMA study demonstrates that rituximab maintenance after induction immunochemotherapy provides a significant long-term [progression-free survival] benefit over observation,” the investigators wrote. “Despite the lack of [overall survival] advantage, it is noteworthy that more than half of the patients in the rituximab maintenance arm remain free of disease progression and have not required new antilymphoma treatment beyond 10 years.”
The trial was sponsored by the Lymphoma Study Association and supported by F. Hoffmann–La Roche and Biogen. Dr. Bachy reported financial disclosures related to Roche and other companies.
SOURCE: Bachy E et al. J Clin Oncol. 2019 Nov 1;37(31):2815-24.
The benefit of rituximab maintenance therapy in follicular lymphoma is long lasting and evident out to at least 9 years, according to the final efficacy analysis of the PRIMA phase 3, randomized, controlled trial.
Previously reported results of this pivotal trial showed prolongation of progression-free survival and time to next treatment with rituximab maintenance at a follow-up of 3 years and 6 years.
“Rituximab maintenance is now widely recommended for patients with follicular lymphoma responding to first-line rituximab-based immunochemotherapy,” Emmanuel Bachy, MD, PhD, of Institut National de la Santé et de la Recherche Médicale in Pierre-Bénite, France, and colleagues reported in the Journal of Clinical Oncology.
In PRIMA, patients with previously untreated high–tumor burden follicular lymphoma received any of three immunochemotherapy induction regimens. The 1,018 patients having a response were then randomly assigned to receive 2 years of rituximab (Rituxan) maintenance or observation only.
Dr. Bachy and colleagues performed the trial’s final efficacy analyses, now at a median follow-up of 9 years.
Among the 607 patients consenting to the extended follow-up, median progression-free survival was 10.5 years in the rituximab-maintenance group, compared with 4.1 years in the observation group (hazard ratio, 0.61; P less than .001).
“Subgroup analyses showed the substantial progression-free survival improvement associated with rituximab maintenance was independent of age, sex, induction immunochemotherapy regimen, response to induction, or [Follicular Lymphoma International Prognostic Index] risk score,” the investigators wrote.
Rituximab maintenance also prolonged the median time to next antilymphoma treatment (not reached vs. 6.1 years; hazard ratio, 0.66; P less than .001) and time to next chemotherapy treatment (not reached vs. 9.3 years; hazard ratio, 0.71; P less than .001).
But there was no significant difference in overall survival. Median overall survival was not reached in either group (hazard ratio, 1.04; P = .7948). The estimated 10-year overall survival rate was about 80% in both groups.
“This 9-year follow-up of the PRIMA study demonstrates that rituximab maintenance after induction immunochemotherapy provides a significant long-term [progression-free survival] benefit over observation,” the investigators wrote. “Despite the lack of [overall survival] advantage, it is noteworthy that more than half of the patients in the rituximab maintenance arm remain free of disease progression and have not required new antilymphoma treatment beyond 10 years.”
The trial was sponsored by the Lymphoma Study Association and supported by F. Hoffmann–La Roche and Biogen. Dr. Bachy reported financial disclosures related to Roche and other companies.
SOURCE: Bachy E et al. J Clin Oncol. 2019 Nov 1;37(31):2815-24.
The benefit of rituximab maintenance therapy in follicular lymphoma is long lasting and evident out to at least 9 years, according to the final efficacy analysis of the PRIMA phase 3, randomized, controlled trial.
Previously reported results of this pivotal trial showed prolongation of progression-free survival and time to next treatment with rituximab maintenance at a follow-up of 3 years and 6 years.
“Rituximab maintenance is now widely recommended for patients with follicular lymphoma responding to first-line rituximab-based immunochemotherapy,” Emmanuel Bachy, MD, PhD, of Institut National de la Santé et de la Recherche Médicale in Pierre-Bénite, France, and colleagues reported in the Journal of Clinical Oncology.
In PRIMA, patients with previously untreated high–tumor burden follicular lymphoma received any of three immunochemotherapy induction regimens. The 1,018 patients having a response were then randomly assigned to receive 2 years of rituximab (Rituxan) maintenance or observation only.
Dr. Bachy and colleagues performed the trial’s final efficacy analyses, now at a median follow-up of 9 years.
Among the 607 patients consenting to the extended follow-up, median progression-free survival was 10.5 years in the rituximab-maintenance group, compared with 4.1 years in the observation group (hazard ratio, 0.61; P less than .001).
“Subgroup analyses showed the substantial progression-free survival improvement associated with rituximab maintenance was independent of age, sex, induction immunochemotherapy regimen, response to induction, or [Follicular Lymphoma International Prognostic Index] risk score,” the investigators wrote.
Rituximab maintenance also prolonged the median time to next antilymphoma treatment (not reached vs. 6.1 years; hazard ratio, 0.66; P less than .001) and time to next chemotherapy treatment (not reached vs. 9.3 years; hazard ratio, 0.71; P less than .001).
But there was no significant difference in overall survival. Median overall survival was not reached in either group (hazard ratio, 1.04; P = .7948). The estimated 10-year overall survival rate was about 80% in both groups.
“This 9-year follow-up of the PRIMA study demonstrates that rituximab maintenance after induction immunochemotherapy provides a significant long-term [progression-free survival] benefit over observation,” the investigators wrote. “Despite the lack of [overall survival] advantage, it is noteworthy that more than half of the patients in the rituximab maintenance arm remain free of disease progression and have not required new antilymphoma treatment beyond 10 years.”
The trial was sponsored by the Lymphoma Study Association and supported by F. Hoffmann–La Roche and Biogen. Dr. Bachy reported financial disclosures related to Roche and other companies.
SOURCE: Bachy E et al. J Clin Oncol. 2019 Nov 1;37(31):2815-24.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
CAR T-cell ‘cocktail’ may overcome antigen escape relapse
A chimeric antigen receptor (CAR) T-cell “cocktail” targeting both CD19 and CD22 could improve outcomes for patients with refractory or relapsed B-cell malignancies, according to investigators.
This dual approach, which appeared safe and effective, may be able to overcome antigen escape relapse, reported Na Wang, MD, of Huazhong University of Science and Technology in China, and colleagues.
The investigators tested this method in an open-label, single-arm pilot study involving 89 patients with refractory/relapsed B cell malignancies. Of these, 51 patients had B-cell acute lymphoblastic leukemia (B-ALL), while the remaining 38 had non-Hodgkin lymphoma (NHL). All patients had dual expression of CD19 and CD22 on malignant B cells, good performance status, and “essentially” normal organ function, the investigators reported in Blood.
Following lymphodepletion, patients were infused with CAR19 and CAR22 T cells, then evaluated for responses with imaging or bone marrow aspiration on a monthly basis for 6 months, then every 3 months thereafter.
After 30 days, most patients with ALL (96%) achieved a minimal residual disease-negative complete response or complete response with incomplete count recovery. After a median follow-up of 16.7 months, almost half of these responders relapsed (49%), median progression-free survival was 13.6 months, and overall survival was 31 months.
With a minimum follow-up of 3 months, half of the patients with NHL (50%) achieved complete responses, with the caveat that two patients who died of septic shock and severe cytokine release syndrome were excluded from this efficacy analysis. After a median follow-up of 14.4 months, in the NHL group, median progression-free survival was 9.9 months and overall survival was 18 months.
Across disease types, almost all patients (95.5%) experienced cytokine release syndrome, with more than three-quarters (77.6%) categorized as grade 1 or 2. CAR T cell-related encephalopathy syndrome (CRES) occurred in 13.5% of patients; most were low grade, apart from one case that was grade 4. In total, 12 patients died due to adverse events.
“The severe [adverse events] were mostly cytopenias and the most frequent fatal [adverse event] was lung infection, which was attributable in part to the high disease burden and heavy pretreatment of the enrolled patients,” the investigators wrote. “Nearly all the high-grade CRS and CRES were reversible and occurred in similar incidences as previously reported. Thus, the sequential infusion of CAR19/22 T-cell “cocktail” was an efficient and well-tolerated approach to circumvent antigen loss of CD19 or CD22.”
The investigators reported having no conflicts of interest.
SOURCE: Wang N et al. 2019 Oct 29. doi: 10.1182/blood.2019000017.
A chimeric antigen receptor (CAR) T-cell “cocktail” targeting both CD19 and CD22 could improve outcomes for patients with refractory or relapsed B-cell malignancies, according to investigators.
This dual approach, which appeared safe and effective, may be able to overcome antigen escape relapse, reported Na Wang, MD, of Huazhong University of Science and Technology in China, and colleagues.
The investigators tested this method in an open-label, single-arm pilot study involving 89 patients with refractory/relapsed B cell malignancies. Of these, 51 patients had B-cell acute lymphoblastic leukemia (B-ALL), while the remaining 38 had non-Hodgkin lymphoma (NHL). All patients had dual expression of CD19 and CD22 on malignant B cells, good performance status, and “essentially” normal organ function, the investigators reported in Blood.
Following lymphodepletion, patients were infused with CAR19 and CAR22 T cells, then evaluated for responses with imaging or bone marrow aspiration on a monthly basis for 6 months, then every 3 months thereafter.
After 30 days, most patients with ALL (96%) achieved a minimal residual disease-negative complete response or complete response with incomplete count recovery. After a median follow-up of 16.7 months, almost half of these responders relapsed (49%), median progression-free survival was 13.6 months, and overall survival was 31 months.
With a minimum follow-up of 3 months, half of the patients with NHL (50%) achieved complete responses, with the caveat that two patients who died of septic shock and severe cytokine release syndrome were excluded from this efficacy analysis. After a median follow-up of 14.4 months, in the NHL group, median progression-free survival was 9.9 months and overall survival was 18 months.
Across disease types, almost all patients (95.5%) experienced cytokine release syndrome, with more than three-quarters (77.6%) categorized as grade 1 or 2. CAR T cell-related encephalopathy syndrome (CRES) occurred in 13.5% of patients; most were low grade, apart from one case that was grade 4. In total, 12 patients died due to adverse events.
“The severe [adverse events] were mostly cytopenias and the most frequent fatal [adverse event] was lung infection, which was attributable in part to the high disease burden and heavy pretreatment of the enrolled patients,” the investigators wrote. “Nearly all the high-grade CRS and CRES were reversible and occurred in similar incidences as previously reported. Thus, the sequential infusion of CAR19/22 T-cell “cocktail” was an efficient and well-tolerated approach to circumvent antigen loss of CD19 or CD22.”
The investigators reported having no conflicts of interest.
SOURCE: Wang N et al. 2019 Oct 29. doi: 10.1182/blood.2019000017.
A chimeric antigen receptor (CAR) T-cell “cocktail” targeting both CD19 and CD22 could improve outcomes for patients with refractory or relapsed B-cell malignancies, according to investigators.
This dual approach, which appeared safe and effective, may be able to overcome antigen escape relapse, reported Na Wang, MD, of Huazhong University of Science and Technology in China, and colleagues.
The investigators tested this method in an open-label, single-arm pilot study involving 89 patients with refractory/relapsed B cell malignancies. Of these, 51 patients had B-cell acute lymphoblastic leukemia (B-ALL), while the remaining 38 had non-Hodgkin lymphoma (NHL). All patients had dual expression of CD19 and CD22 on malignant B cells, good performance status, and “essentially” normal organ function, the investigators reported in Blood.
Following lymphodepletion, patients were infused with CAR19 and CAR22 T cells, then evaluated for responses with imaging or bone marrow aspiration on a monthly basis for 6 months, then every 3 months thereafter.
After 30 days, most patients with ALL (96%) achieved a minimal residual disease-negative complete response or complete response with incomplete count recovery. After a median follow-up of 16.7 months, almost half of these responders relapsed (49%), median progression-free survival was 13.6 months, and overall survival was 31 months.
With a minimum follow-up of 3 months, half of the patients with NHL (50%) achieved complete responses, with the caveat that two patients who died of septic shock and severe cytokine release syndrome were excluded from this efficacy analysis. After a median follow-up of 14.4 months, in the NHL group, median progression-free survival was 9.9 months and overall survival was 18 months.
Across disease types, almost all patients (95.5%) experienced cytokine release syndrome, with more than three-quarters (77.6%) categorized as grade 1 or 2. CAR T cell-related encephalopathy syndrome (CRES) occurred in 13.5% of patients; most were low grade, apart from one case that was grade 4. In total, 12 patients died due to adverse events.
“The severe [adverse events] were mostly cytopenias and the most frequent fatal [adverse event] was lung infection, which was attributable in part to the high disease burden and heavy pretreatment of the enrolled patients,” the investigators wrote. “Nearly all the high-grade CRS and CRES were reversible and occurred in similar incidences as previously reported. Thus, the sequential infusion of CAR19/22 T-cell “cocktail” was an efficient and well-tolerated approach to circumvent antigen loss of CD19 or CD22.”
The investigators reported having no conflicts of interest.
SOURCE: Wang N et al. 2019 Oct 29. doi: 10.1182/blood.2019000017.
FROM BLOOD
Adding polatuzumab extends survival in relapsed/refractory DLBCL
For patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), adding polatuzumab vedotin to bendamustine and rituximab can improve complete response rates and extend overall survival, according to findings from a phase 1b/2 trial.
Adding polatuzumab decreased mortality risk by 58%, reported lead author Laurie H. Sehn, MD, of the University of British Columbia, Vancouver, and colleagues.

“Patients with transplantation-ineligible [relapsed/refractory] DLBCL, including those who experienced treatment failure with [autologous stem cell transplant], have dismal outcomes with limited therapeutic options,” the investigators wrote in the Journal of Clinical Oncology. “To our knowledge, this is the first randomized trial demonstrating an [overall survival] benefit in patients with transplantation-ineligible [relapsed/refractory] DLBCL.”
In the first part of the study, 27 patients were treated with polatuzumab vedotin, bendamustine, and obinutuzumab. After a median follow-up of 27 months, this regimen returned a complete response rate of 29.6%, median progression-free survival of 6.3 months, and median overall survival of 10.8 months.
In the primary analysis, 80 patients were randomized to receive bendamustine and rituximab, with or without polatuzumab. Adding polatuzumab had a significant benefit, as 40.0% of these patients achieved a complete response, compared with 17.5% of patients who did not receive polatuzumab. After a median follow-up of 22.3 months, outcomes also were significantly improved with the addition of polatuzumab for both median progression-free survival (9.5 vs. 3.7 months) and overall survival (12.4 vs. 4.7 months).
Adding polatuzumab did come with some safety trade-offs. Rates of certain grade 3 or 4 adverse events were higher, including thrombocytopenia (41% vs. 23.1%), neutropenia (46.2% vs. 33.3%), and anemia (28.2% vs. 17.9%), while infection rates were comparable. Almost half of the patients treated with polatuzumab (43.6%) developed grade 1 or 2 peripheral neuropathy, but most cases resolved.
Combination therapy with polatuzumab, bendamustine, and rituximab “represents a novel, effective therapeutic regimen to address the unmet need of patients with transplantation-ineligible [relapsed/refractory] DLBCL,” the investigators wrote. Since just 25% of polatuzumab combination–treated patients had received prior autologous stem cell transplant, the investigators said they could not make definitive conclusions on this combination’s efficacy in the post-ASCT setting.
Additional trials involving polatuzumab in the relapsed/refractory setting are ongoing. For patients with treatment-naive DLBCL, a phase 3 trial (NCT03274492) is evaluating substitution of polatuzumab for vincristine in the R-CHOP regimen.
The study was funded by F. Hoffmann-La Roche and Genentech. The investigators reported additional relationships with AbbVie, Kite Pharma, Lundbeck, and others.
SOURCE: Sehn LH et al. J Clin Oncol. 2019 Nov 6. doi: 10.1200/JCO.19.00172.
For patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), adding polatuzumab vedotin to bendamustine and rituximab can improve complete response rates and extend overall survival, according to findings from a phase 1b/2 trial.
Adding polatuzumab decreased mortality risk by 58%, reported lead author Laurie H. Sehn, MD, of the University of British Columbia, Vancouver, and colleagues.
“Patients with transplantation-ineligible [relapsed/refractory] DLBCL, including those who experienced treatment failure with [autologous stem cell transplant], have dismal outcomes with limited therapeutic options,” the investigators wrote in the Journal of Clinical Oncology. “To our knowledge, this is the first randomized trial demonstrating an [overall survival] benefit in patients with transplantation-ineligible [relapsed/refractory] DLBCL.”
In the first part of the study, 27 patients were treated with polatuzumab vedotin, bendamustine, and obinutuzumab. After a median follow-up of 27 months, this regimen returned a complete response rate of 29.6%, median progression-free survival of 6.3 months, and median overall survival of 10.8 months.
In the primary analysis, 80 patients were randomized to receive bendamustine and rituximab, with or without polatuzumab. Adding polatuzumab had a significant benefit, as 40.0% of these patients achieved a complete response, compared with 17.5% of patients who did not receive polatuzumab. After a median follow-up of 22.3 months, outcomes also were significantly improved with the addition of polatuzumab for both median progression-free survival (9.5 vs. 3.7 months) and overall survival (12.4 vs. 4.7 months).
Adding polatuzumab did come with some safety trade-offs. Rates of certain grade 3 or 4 adverse events were higher, including thrombocytopenia (41% vs. 23.1%), neutropenia (46.2% vs. 33.3%), and anemia (28.2% vs. 17.9%), while infection rates were comparable. Almost half of the patients treated with polatuzumab (43.6%) developed grade 1 or 2 peripheral neuropathy, but most cases resolved.
Combination therapy with polatuzumab, bendamustine, and rituximab “represents a novel, effective therapeutic regimen to address the unmet need of patients with transplantation-ineligible [relapsed/refractory] DLBCL,” the investigators wrote. Since just 25% of polatuzumab combination–treated patients had received prior autologous stem cell transplant, the investigators said they could not make definitive conclusions on this combination’s efficacy in the post-ASCT setting.
Additional trials involving polatuzumab in the relapsed/refractory setting are ongoing. For patients with treatment-naive DLBCL, a phase 3 trial (NCT03274492) is evaluating substitution of polatuzumab for vincristine in the R-CHOP regimen.
The study was funded by F. Hoffmann-La Roche and Genentech. The investigators reported additional relationships with AbbVie, Kite Pharma, Lundbeck, and others.
SOURCE: Sehn LH et al. J Clin Oncol. 2019 Nov 6. doi: 10.1200/JCO.19.00172.
For patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), adding polatuzumab vedotin to bendamustine and rituximab can improve complete response rates and extend overall survival, according to findings from a phase 1b/2 trial.
Adding polatuzumab decreased mortality risk by 58%, reported lead author Laurie H. Sehn, MD, of the University of British Columbia, Vancouver, and colleagues.
“Patients with transplantation-ineligible [relapsed/refractory] DLBCL, including those who experienced treatment failure with [autologous stem cell transplant], have dismal outcomes with limited therapeutic options,” the investigators wrote in the Journal of Clinical Oncology. “To our knowledge, this is the first randomized trial demonstrating an [overall survival] benefit in patients with transplantation-ineligible [relapsed/refractory] DLBCL.”
In the first part of the study, 27 patients were treated with polatuzumab vedotin, bendamustine, and obinutuzumab. After a median follow-up of 27 months, this regimen returned a complete response rate of 29.6%, median progression-free survival of 6.3 months, and median overall survival of 10.8 months.
In the primary analysis, 80 patients were randomized to receive bendamustine and rituximab, with or without polatuzumab. Adding polatuzumab had a significant benefit, as 40.0% of these patients achieved a complete response, compared with 17.5% of patients who did not receive polatuzumab. After a median follow-up of 22.3 months, outcomes also were significantly improved with the addition of polatuzumab for both median progression-free survival (9.5 vs. 3.7 months) and overall survival (12.4 vs. 4.7 months).
Adding polatuzumab did come with some safety trade-offs. Rates of certain grade 3 or 4 adverse events were higher, including thrombocytopenia (41% vs. 23.1%), neutropenia (46.2% vs. 33.3%), and anemia (28.2% vs. 17.9%), while infection rates were comparable. Almost half of the patients treated with polatuzumab (43.6%) developed grade 1 or 2 peripheral neuropathy, but most cases resolved.
Combination therapy with polatuzumab, bendamustine, and rituximab “represents a novel, effective therapeutic regimen to address the unmet need of patients with transplantation-ineligible [relapsed/refractory] DLBCL,” the investigators wrote. Since just 25% of polatuzumab combination–treated patients had received prior autologous stem cell transplant, the investigators said they could not make definitive conclusions on this combination’s efficacy in the post-ASCT setting.
Additional trials involving polatuzumab in the relapsed/refractory setting are ongoing. For patients with treatment-naive DLBCL, a phase 3 trial (NCT03274492) is evaluating substitution of polatuzumab for vincristine in the R-CHOP regimen.
The study was funded by F. Hoffmann-La Roche and Genentech. The investigators reported additional relationships with AbbVie, Kite Pharma, Lundbeck, and others.
SOURCE: Sehn LH et al. J Clin Oncol. 2019 Nov 6. doi: 10.1200/JCO.19.00172.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
FDA approves Brukinsa for relapsed, refractory MCL
The Food and Drug Administration has approved zanubrutinib (Brukinsa) for the treatment of mantle cell lymphoma (MCL) in adult patients who have received at least one prior therapy.

The approval is based on results from two separate studies; in a global phase 1/2 trial, patients with relapsed or refractory MCL who received zanubrutinib had an overall response rate of 84%, with 22% experiencing a complete response and 62% experiencing partial response. Median duration of response was 18.5 months. The ORR in the second study – a multicenter phase 2 trial – was also 84%, but with 59% experiencing a complete response and 24% experiencing partial response; duration of response was 19.5 months.
The most common adverse events reported during the trials were decreased neutrophil count, decreased platelet count, upper respiratory tract infection, decreased white blood cell count, decreased hemoglobin, rash, bruising, diarrhea, cough, musculoskeletal pain, pneumonia, urinary tract infection, hematuria, fatigue, constipation, and hemorrhage. The most common serious adverse events were pneumonia and hemorrhage.
Of the 118 patients with MCL treated with zanubrutinib over the two trials, 8 had to be discontinued because of adverse events.
The recommended dose of zanubrutinib is 320 mg, taken orally 160 mg twice daily or 320 mg once daily, with or without food.
“BTK [Bruton kinase] inhibition is an established mode of treatment for patients with MCL, but many patients treated with previously approved BTK inhibitors do not fully respond to BTK therapy or are forced to discontinue treatment early due to side effects. Today we have a new option for our adult patients who have received one prior systemic or targeted therapy and are living with MCL,” Luhua (Michael) Wang, MD, clinical trial investigator and professor in the department of lymphoma and myeloma at the University of Texas MD Anderson Cancer Center, Houston, said in a statement.
The Food and Drug Administration has approved zanubrutinib (Brukinsa) for the treatment of mantle cell lymphoma (MCL) in adult patients who have received at least one prior therapy.
The approval is based on results from two separate studies; in a global phase 1/2 trial, patients with relapsed or refractory MCL who received zanubrutinib had an overall response rate of 84%, with 22% experiencing a complete response and 62% experiencing partial response. Median duration of response was 18.5 months. The ORR in the second study – a multicenter phase 2 trial – was also 84%, but with 59% experiencing a complete response and 24% experiencing partial response; duration of response was 19.5 months.
The most common adverse events reported during the trials were decreased neutrophil count, decreased platelet count, upper respiratory tract infection, decreased white blood cell count, decreased hemoglobin, rash, bruising, diarrhea, cough, musculoskeletal pain, pneumonia, urinary tract infection, hematuria, fatigue, constipation, and hemorrhage. The most common serious adverse events were pneumonia and hemorrhage.
Of the 118 patients with MCL treated with zanubrutinib over the two trials, 8 had to be discontinued because of adverse events.
The recommended dose of zanubrutinib is 320 mg, taken orally 160 mg twice daily or 320 mg once daily, with or without food.
“BTK [Bruton kinase] inhibition is an established mode of treatment for patients with MCL, but many patients treated with previously approved BTK inhibitors do not fully respond to BTK therapy or are forced to discontinue treatment early due to side effects. Today we have a new option for our adult patients who have received one prior systemic or targeted therapy and are living with MCL,” Luhua (Michael) Wang, MD, clinical trial investigator and professor in the department of lymphoma and myeloma at the University of Texas MD Anderson Cancer Center, Houston, said in a statement.
The Food and Drug Administration has approved zanubrutinib (Brukinsa) for the treatment of mantle cell lymphoma (MCL) in adult patients who have received at least one prior therapy.
The approval is based on results from two separate studies; in a global phase 1/2 trial, patients with relapsed or refractory MCL who received zanubrutinib had an overall response rate of 84%, with 22% experiencing a complete response and 62% experiencing partial response. Median duration of response was 18.5 months. The ORR in the second study – a multicenter phase 2 trial – was also 84%, but with 59% experiencing a complete response and 24% experiencing partial response; duration of response was 19.5 months.
The most common adverse events reported during the trials were decreased neutrophil count, decreased platelet count, upper respiratory tract infection, decreased white blood cell count, decreased hemoglobin, rash, bruising, diarrhea, cough, musculoskeletal pain, pneumonia, urinary tract infection, hematuria, fatigue, constipation, and hemorrhage. The most common serious adverse events were pneumonia and hemorrhage.
Of the 118 patients with MCL treated with zanubrutinib over the two trials, 8 had to be discontinued because of adverse events.
The recommended dose of zanubrutinib is 320 mg, taken orally 160 mg twice daily or 320 mg once daily, with or without food.
“BTK [Bruton kinase] inhibition is an established mode of treatment for patients with MCL, but many patients treated with previously approved BTK inhibitors do not fully respond to BTK therapy or are forced to discontinue treatment early due to side effects. Today we have a new option for our adult patients who have received one prior systemic or targeted therapy and are living with MCL,” Luhua (Michael) Wang, MD, clinical trial investigator and professor in the department of lymphoma and myeloma at the University of Texas MD Anderson Cancer Center, Houston, said in a statement.
Gene signature may help guide initial CLL treatment choice
A novel 17-gene expression signature may help guide the choice of initial treatment in patients with IGHV-unmutated chronic lymphocytic leukemia (CLL), according to findings of a retrospective dual cohort study.

“[Fludarabine, cyclophosphamide, and rituximab] was the first regimen to improve progression-free survival and overall survival in patients with chronic lymphocytic leukaemia, and has become a gold-standard chemoimmunotherapy regimen in physically fit patients,” wrote the investigators, who were led by Carmen D. Herling, MD, of the Center for Integrated Oncology, Cologne, Germany; and Kevin R. Coombes, PhD, of Ohio State University, Columbus.
While several studies demonstrate that young, fit patients with mutated IGHV gene and no high-risk cytogenetic abnormalities achieve durable remission with the FCR (fludarabine, cyclophosphamide, and rituximab) regimen, there have been no studies to identify if this is true for patients with unmutated IGHV gene, they reported in the Lancet Oncology.
The investigators performed transcriptional profiling using peripheral blood samples collected from two cohorts of patients with CLL who were treated with frontline FCR.
The discovery and training cohort consisted of 101 patients (65% with IGHV-unmutated disease) treated at the MD Anderson Cancer Center who had a median follow-up of about 12 years. The validation cohort consisted of 109 patients with IGHV-unmutated disease treated on the German CLL8 single-arm trial who had a median follow-up of about 6 years.
A total of 1,136 genes showed a significant univariate association with time to progression. Ultimately, 17 of these genes – most of them involved in purine metabolism and oxidative phosphorylation – were included in the expression signature.
Among patients with IGHV-unmutated CLL, the 17-gene signature discriminated between two groups having differing time to progression after their frontline FCR chemoimmunotherapy: an unfavorable prognosis group and an intermediate prognosis group.
The unfavorable prognosis group had a significantly higher relative risk of progression in both the discovery/training cohort (hazard ratio, 3.83; P less than .0001) and the validation cohort (HR, 1.90; P = .008). In the validation cohort, the median time to progression was 39 months among patients with a signature-defined unfavorable prognosis, compared with 59 months among patients with a signature-defined intermediate prognosis.
“We would recommend testing the value of the 17-gene signature in a prospective study that compares FCR treatment with alternative therapies, such as ibrutinib, as part of a randomised clinical trial,” the investigators wrote.
Dr. Herling reported financial disclosures related to Hoffmann-La Roche, and Dr. Coombes reported grants from the National Institutes of Health. The study was funded by the Chronic Lymphocytic Leukaemia Global Research Foundation and the National Institutes of Health/National Cancer Institute.
SOURCE: Herling CD et al. Lancet Oncol. 2019;20(11):1576-86.
A novel 17-gene expression signature may help guide the choice of initial treatment in patients with IGHV-unmutated chronic lymphocytic leukemia (CLL), according to findings of a retrospective dual cohort study.
“[Fludarabine, cyclophosphamide, and rituximab] was the first regimen to improve progression-free survival and overall survival in patients with chronic lymphocytic leukaemia, and has become a gold-standard chemoimmunotherapy regimen in physically fit patients,” wrote the investigators, who were led by Carmen D. Herling, MD, of the Center for Integrated Oncology, Cologne, Germany; and Kevin R. Coombes, PhD, of Ohio State University, Columbus.
While several studies demonstrate that young, fit patients with mutated IGHV gene and no high-risk cytogenetic abnormalities achieve durable remission with the FCR (fludarabine, cyclophosphamide, and rituximab) regimen, there have been no studies to identify if this is true for patients with unmutated IGHV gene, they reported in the Lancet Oncology.
The investigators performed transcriptional profiling using peripheral blood samples collected from two cohorts of patients with CLL who were treated with frontline FCR.
The discovery and training cohort consisted of 101 patients (65% with IGHV-unmutated disease) treated at the MD Anderson Cancer Center who had a median follow-up of about 12 years. The validation cohort consisted of 109 patients with IGHV-unmutated disease treated on the German CLL8 single-arm trial who had a median follow-up of about 6 years.
A total of 1,136 genes showed a significant univariate association with time to progression. Ultimately, 17 of these genes – most of them involved in purine metabolism and oxidative phosphorylation – were included in the expression signature.
Among patients with IGHV-unmutated CLL, the 17-gene signature discriminated between two groups having differing time to progression after their frontline FCR chemoimmunotherapy: an unfavorable prognosis group and an intermediate prognosis group.
The unfavorable prognosis group had a significantly higher relative risk of progression in both the discovery/training cohort (hazard ratio, 3.83; P less than .0001) and the validation cohort (HR, 1.90; P = .008). In the validation cohort, the median time to progression was 39 months among patients with a signature-defined unfavorable prognosis, compared with 59 months among patients with a signature-defined intermediate prognosis.
“We would recommend testing the value of the 17-gene signature in a prospective study that compares FCR treatment with alternative therapies, such as ibrutinib, as part of a randomised clinical trial,” the investigators wrote.
Dr. Herling reported financial disclosures related to Hoffmann-La Roche, and Dr. Coombes reported grants from the National Institutes of Health. The study was funded by the Chronic Lymphocytic Leukaemia Global Research Foundation and the National Institutes of Health/National Cancer Institute.
SOURCE: Herling CD et al. Lancet Oncol. 2019;20(11):1576-86.
A novel 17-gene expression signature may help guide the choice of initial treatment in patients with IGHV-unmutated chronic lymphocytic leukemia (CLL), according to findings of a retrospective dual cohort study.
“[Fludarabine, cyclophosphamide, and rituximab] was the first regimen to improve progression-free survival and overall survival in patients with chronic lymphocytic leukaemia, and has become a gold-standard chemoimmunotherapy regimen in physically fit patients,” wrote the investigators, who were led by Carmen D. Herling, MD, of the Center for Integrated Oncology, Cologne, Germany; and Kevin R. Coombes, PhD, of Ohio State University, Columbus.
While several studies demonstrate that young, fit patients with mutated IGHV gene and no high-risk cytogenetic abnormalities achieve durable remission with the FCR (fludarabine, cyclophosphamide, and rituximab) regimen, there have been no studies to identify if this is true for patients with unmutated IGHV gene, they reported in the Lancet Oncology.
The investigators performed transcriptional profiling using peripheral blood samples collected from two cohorts of patients with CLL who were treated with frontline FCR.
The discovery and training cohort consisted of 101 patients (65% with IGHV-unmutated disease) treated at the MD Anderson Cancer Center who had a median follow-up of about 12 years. The validation cohort consisted of 109 patients with IGHV-unmutated disease treated on the German CLL8 single-arm trial who had a median follow-up of about 6 years.
A total of 1,136 genes showed a significant univariate association with time to progression. Ultimately, 17 of these genes – most of them involved in purine metabolism and oxidative phosphorylation – were included in the expression signature.
Among patients with IGHV-unmutated CLL, the 17-gene signature discriminated between two groups having differing time to progression after their frontline FCR chemoimmunotherapy: an unfavorable prognosis group and an intermediate prognosis group.
The unfavorable prognosis group had a significantly higher relative risk of progression in both the discovery/training cohort (hazard ratio, 3.83; P less than .0001) and the validation cohort (HR, 1.90; P = .008). In the validation cohort, the median time to progression was 39 months among patients with a signature-defined unfavorable prognosis, compared with 59 months among patients with a signature-defined intermediate prognosis.
“We would recommend testing the value of the 17-gene signature in a prospective study that compares FCR treatment with alternative therapies, such as ibrutinib, as part of a randomised clinical trial,” the investigators wrote.
Dr. Herling reported financial disclosures related to Hoffmann-La Roche, and Dr. Coombes reported grants from the National Institutes of Health. The study was funded by the Chronic Lymphocytic Leukaemia Global Research Foundation and the National Institutes of Health/National Cancer Institute.
SOURCE: Herling CD et al. Lancet Oncol. 2019;20(11):1576-86.
FROM LANCET ONCOLOGY
Foundation launches direct-to-patient registry in multiple myeloma
The Multiple Myeloma Research Foundation (MMRF) recently launched its Direct-to-Patient registry, in what the organization’s leaders are describing as a “disruptive” step toward improving outcomes for patients with multiple myeloma.
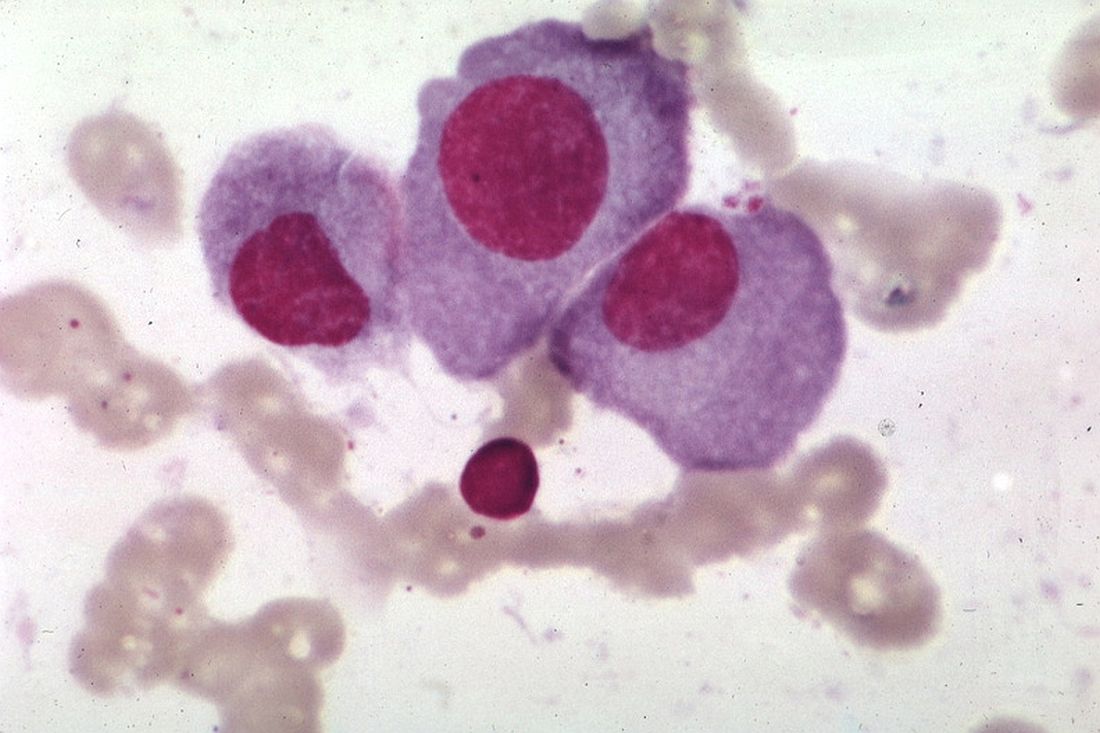
The new registry is intended to build upon CoMMpass, a program started 8 years ago that now represents the largest genomic database of any type of cancer. Although CoMMpass includes data from about 1,150 patients with myeloma, it’s not enough information, according to the chief marketing and development officer at the MMRF, Anne Quinn Young.

“For a disease as heterogenous as myeloma is, we need more, particularly because we don’t have all the samples for later-stage disease,” Ms. Quinn Young said in an interview. “And even with the clinical data, given the patient population, both [in terms of] demographics and the nature of the disease, the numbers of patients still living after multiple relapses is rather small.”
In an earlier effort to gather more data, the MMRF first turned to other organizations for help, but this approach fell short because of scarcity of data, and in some cases, unwillingness to share. Steven Labkoff, MD, chief data officer at the MMRF, described this experience in an interview.

“When the MMRF was looking around for different data sources for myeloma data, it was always the claim that, ‘Sure, we have plenty of patients, we have plenty of data, and it’s rich and really complete.’ However, as we approached an array of organizations – big organizations – as we dug into the details and reviewed patient counts or data completeness, they either didn’t have a sufficient number of patients, they didn’t have sufficiently complete data for our needs, and in the case where some did have sufficient numbers and complete data sets, they simply weren’t in a position to share that data outside their institution,” Dr. Labkoff said.
Undeterred, the MMRF switched tactics to the current, patient-centric approach.
“We’re leveraging one aspect of the HIPAA legislation,” Dr. Labkoff said, referring to patients’ rights to request their own medical records and an institution’s legal obligation to provide those records.
In the short-term, the registry will collect three types of data: patient donated data (answers from a patient survey), electronic medical records abstracted from all relevant past providers, and genomic test results. Participating patients will have blood drawn at home by a phlebotomist for the genomic assay. Additional tubes of blood will be concurrently collected and biobanked. This will eventually allow for immune profiling, Dr. Labkoff said.
Future goals include a patient-reported outcomes module and the ability to link data with medical claims.
So far, 79 patients have participated in the pilot program, according to the MMRF. As the database builds, Ms. Quinn Young and Dr. Labkoff anticipate that it will yield answers to a variety of real-world questions.
Dr. Labkoff offered two examples. “Of all the patients who have been exposed to ‘name your drug,’ what were the costs of their therapy, and what were the outcomes?” he said. In addition, researchers will be able to query clinical trial inclusion criteria to search for data on a specific patient profile, such as patients with a 4:14 translocation, who have had a bone marrow transplant in the last 2 years, and have been exposed to a certain drug regimen.
Ms. Quinn Young noted that doctors may be able to use the database to reliably identify high-risk patients and guide agent selection. Common patient questions also will be addressed, she said, including best treatment regimens for certain types of patients.
“For patients who may have run out of all commercially available options, or for patients who are perhaps seen at a community center, where certainly this type of profiling is not standard, it’s opening up a whole new set of options for them,” Ms. Quinn Young said. “And if their physician doesn’t pursue those options, they have the report that they can use to seek a second opinion.”
The Direct-to-Patient registry is unique because it aims to empower patients in a way that hasn’t been done before, Ms. Quinn Young said. “We are committed ... ever since we conceived of this project, to giving results back to patients. That is disruptive because right now that doesn’t exist.”
But the cost of implementing the registry, which has an approximate budget of $20 million, stands in the way of a completely free flow of anonymized data. MMRF leaders are exploring different strategies to sustain funding for the program.
Another MMRF program, CoMMpass, uses a precompetitive consortium model, in which several pharmaceutical companies pay for a preview of the data 6 months in advance of nonprofit researchers. A similar model may be used with the Direct-to-Patient registry, but this has yet to be determined, according to Dr. Labkoff and Ms. Quinn Young.
For now, Ms. Quinn Young said she hopes that physicians will be receptive to the program. “[The short term goal is that] when patients come to their doctors asking about this, that there is support and open-mindedness,” she said.
Looking to the future, Dr. Labkoff described how the registry could accelerate myeloma research, ultimately toward a cure.
“It is generally accepted that it can take 17 years to get something – a therapy, a new drug, or a guideline – from the bench to the bedside,” he said. “It’s my hope that we can take next generation sequencing and the results of this registry and bend that curve, maybe ... to 10 [years], or very aggressively, to 7 or 5 [years], where doctors are able to use the information in these reports for the patients that have literally given themselves, and use this to help guide the choices of their therapy or the trials they apply for, to help them get a better outcome in general.”
The Direct-to-Patient registry is a collaborative effort between the MMRF and multiple organizations, including the health care technology company COTA, the Broad Institute of Harvard and MIT, Prometheus Research, Tempus, and the Dana Farber Cancer Institute.
The Multiple Myeloma Research Foundation (MMRF) recently launched its Direct-to-Patient registry, in what the organization’s leaders are describing as a “disruptive” step toward improving outcomes for patients with multiple myeloma.
The new registry is intended to build upon CoMMpass, a program started 8 years ago that now represents the largest genomic database of any type of cancer. Although CoMMpass includes data from about 1,150 patients with myeloma, it’s not enough information, according to the chief marketing and development officer at the MMRF, Anne Quinn Young.
“For a disease as heterogenous as myeloma is, we need more, particularly because we don’t have all the samples for later-stage disease,” Ms. Quinn Young said in an interview. “And even with the clinical data, given the patient population, both [in terms of] demographics and the nature of the disease, the numbers of patients still living after multiple relapses is rather small.”
In an earlier effort to gather more data, the MMRF first turned to other organizations for help, but this approach fell short because of scarcity of data, and in some cases, unwillingness to share. Steven Labkoff, MD, chief data officer at the MMRF, described this experience in an interview.
“When the MMRF was looking around for different data sources for myeloma data, it was always the claim that, ‘Sure, we have plenty of patients, we have plenty of data, and it’s rich and really complete.’ However, as we approached an array of organizations – big organizations – as we dug into the details and reviewed patient counts or data completeness, they either didn’t have a sufficient number of patients, they didn’t have sufficiently complete data for our needs, and in the case where some did have sufficient numbers and complete data sets, they simply weren’t in a position to share that data outside their institution,” Dr. Labkoff said.
Undeterred, the MMRF switched tactics to the current, patient-centric approach.
“We’re leveraging one aspect of the HIPAA legislation,” Dr. Labkoff said, referring to patients’ rights to request their own medical records and an institution’s legal obligation to provide those records.
In the short-term, the registry will collect three types of data: patient donated data (answers from a patient survey), electronic medical records abstracted from all relevant past providers, and genomic test results. Participating patients will have blood drawn at home by a phlebotomist for the genomic assay. Additional tubes of blood will be concurrently collected and biobanked. This will eventually allow for immune profiling, Dr. Labkoff said.
Future goals include a patient-reported outcomes module and the ability to link data with medical claims.
So far, 79 patients have participated in the pilot program, according to the MMRF. As the database builds, Ms. Quinn Young and Dr. Labkoff anticipate that it will yield answers to a variety of real-world questions.
Dr. Labkoff offered two examples. “Of all the patients who have been exposed to ‘name your drug,’ what were the costs of their therapy, and what were the outcomes?” he said. In addition, researchers will be able to query clinical trial inclusion criteria to search for data on a specific patient profile, such as patients with a 4:14 translocation, who have had a bone marrow transplant in the last 2 years, and have been exposed to a certain drug regimen.
Ms. Quinn Young noted that doctors may be able to use the database to reliably identify high-risk patients and guide agent selection. Common patient questions also will be addressed, she said, including best treatment regimens for certain types of patients.
“For patients who may have run out of all commercially available options, or for patients who are perhaps seen at a community center, where certainly this type of profiling is not standard, it’s opening up a whole new set of options for them,” Ms. Quinn Young said. “And if their physician doesn’t pursue those options, they have the report that they can use to seek a second opinion.”
The Direct-to-Patient registry is unique because it aims to empower patients in a way that hasn’t been done before, Ms. Quinn Young said. “We are committed ... ever since we conceived of this project, to giving results back to patients. That is disruptive because right now that doesn’t exist.”
But the cost of implementing the registry, which has an approximate budget of $20 million, stands in the way of a completely free flow of anonymized data. MMRF leaders are exploring different strategies to sustain funding for the program.
Another MMRF program, CoMMpass, uses a precompetitive consortium model, in which several pharmaceutical companies pay for a preview of the data 6 months in advance of nonprofit researchers. A similar model may be used with the Direct-to-Patient registry, but this has yet to be determined, according to Dr. Labkoff and Ms. Quinn Young.
For now, Ms. Quinn Young said she hopes that physicians will be receptive to the program. “[The short term goal is that] when patients come to their doctors asking about this, that there is support and open-mindedness,” she said.
Looking to the future, Dr. Labkoff described how the registry could accelerate myeloma research, ultimately toward a cure.
“It is generally accepted that it can take 17 years to get something – a therapy, a new drug, or a guideline – from the bench to the bedside,” he said. “It’s my hope that we can take next generation sequencing and the results of this registry and bend that curve, maybe ... to 10 [years], or very aggressively, to 7 or 5 [years], where doctors are able to use the information in these reports for the patients that have literally given themselves, and use this to help guide the choices of their therapy or the trials they apply for, to help them get a better outcome in general.”
The Direct-to-Patient registry is a collaborative effort between the MMRF and multiple organizations, including the health care technology company COTA, the Broad Institute of Harvard and MIT, Prometheus Research, Tempus, and the Dana Farber Cancer Institute.
The Multiple Myeloma Research Foundation (MMRF) recently launched its Direct-to-Patient registry, in what the organization’s leaders are describing as a “disruptive” step toward improving outcomes for patients with multiple myeloma.
The new registry is intended to build upon CoMMpass, a program started 8 years ago that now represents the largest genomic database of any type of cancer. Although CoMMpass includes data from about 1,150 patients with myeloma, it’s not enough information, according to the chief marketing and development officer at the MMRF, Anne Quinn Young.
“For a disease as heterogenous as myeloma is, we need more, particularly because we don’t have all the samples for later-stage disease,” Ms. Quinn Young said in an interview. “And even with the clinical data, given the patient population, both [in terms of] demographics and the nature of the disease, the numbers of patients still living after multiple relapses is rather small.”
In an earlier effort to gather more data, the MMRF first turned to other organizations for help, but this approach fell short because of scarcity of data, and in some cases, unwillingness to share. Steven Labkoff, MD, chief data officer at the MMRF, described this experience in an interview.
“When the MMRF was looking around for different data sources for myeloma data, it was always the claim that, ‘Sure, we have plenty of patients, we have plenty of data, and it’s rich and really complete.’ However, as we approached an array of organizations – big organizations – as we dug into the details and reviewed patient counts or data completeness, they either didn’t have a sufficient number of patients, they didn’t have sufficiently complete data for our needs, and in the case where some did have sufficient numbers and complete data sets, they simply weren’t in a position to share that data outside their institution,” Dr. Labkoff said.
Undeterred, the MMRF switched tactics to the current, patient-centric approach.
“We’re leveraging one aspect of the HIPAA legislation,” Dr. Labkoff said, referring to patients’ rights to request their own medical records and an institution’s legal obligation to provide those records.
In the short-term, the registry will collect three types of data: patient donated data (answers from a patient survey), electronic medical records abstracted from all relevant past providers, and genomic test results. Participating patients will have blood drawn at home by a phlebotomist for the genomic assay. Additional tubes of blood will be concurrently collected and biobanked. This will eventually allow for immune profiling, Dr. Labkoff said.
Future goals include a patient-reported outcomes module and the ability to link data with medical claims.
So far, 79 patients have participated in the pilot program, according to the MMRF. As the database builds, Ms. Quinn Young and Dr. Labkoff anticipate that it will yield answers to a variety of real-world questions.
Dr. Labkoff offered two examples. “Of all the patients who have been exposed to ‘name your drug,’ what were the costs of their therapy, and what were the outcomes?” he said. In addition, researchers will be able to query clinical trial inclusion criteria to search for data on a specific patient profile, such as patients with a 4:14 translocation, who have had a bone marrow transplant in the last 2 years, and have been exposed to a certain drug regimen.
Ms. Quinn Young noted that doctors may be able to use the database to reliably identify high-risk patients and guide agent selection. Common patient questions also will be addressed, she said, including best treatment regimens for certain types of patients.
“For patients who may have run out of all commercially available options, or for patients who are perhaps seen at a community center, where certainly this type of profiling is not standard, it’s opening up a whole new set of options for them,” Ms. Quinn Young said. “And if their physician doesn’t pursue those options, they have the report that they can use to seek a second opinion.”
The Direct-to-Patient registry is unique because it aims to empower patients in a way that hasn’t been done before, Ms. Quinn Young said. “We are committed ... ever since we conceived of this project, to giving results back to patients. That is disruptive because right now that doesn’t exist.”
But the cost of implementing the registry, which has an approximate budget of $20 million, stands in the way of a completely free flow of anonymized data. MMRF leaders are exploring different strategies to sustain funding for the program.
Another MMRF program, CoMMpass, uses a precompetitive consortium model, in which several pharmaceutical companies pay for a preview of the data 6 months in advance of nonprofit researchers. A similar model may be used with the Direct-to-Patient registry, but this has yet to be determined, according to Dr. Labkoff and Ms. Quinn Young.
For now, Ms. Quinn Young said she hopes that physicians will be receptive to the program. “[The short term goal is that] when patients come to their doctors asking about this, that there is support and open-mindedness,” she said.
Looking to the future, Dr. Labkoff described how the registry could accelerate myeloma research, ultimately toward a cure.
“It is generally accepted that it can take 17 years to get something – a therapy, a new drug, or a guideline – from the bench to the bedside,” he said. “It’s my hope that we can take next generation sequencing and the results of this registry and bend that curve, maybe ... to 10 [years], or very aggressively, to 7 or 5 [years], where doctors are able to use the information in these reports for the patients that have literally given themselves, and use this to help guide the choices of their therapy or the trials they apply for, to help them get a better outcome in general.”
The Direct-to-Patient registry is a collaborative effort between the MMRF and multiple organizations, including the health care technology company COTA, the Broad Institute of Harvard and MIT, Prometheus Research, Tempus, and the Dana Farber Cancer Institute.
Armored CAR T cells elicit responses in NHL patients
NATIONAL HARBOR, MD – An armored chimeric antigen receptor (CAR) T-cell therapy has demonstrated efficacy in vitro and in patients with relapsed or refractory non-Hodgkin lymphoma (NHL), according to findings presented at the annual meeting of the Society for Immunotherapy of Cancer.
ICTCAR014, a dominant negative PD-1 armored CAR T-cell therapy, proved more cytotoxic than traditional CAR T-cell therapy in vitro and produced responses in 12 of 13 NHL patients who received it.
Xiaobin Victor Lu, PhD, of Innovative Cellular Therapeutics, Shanghai, China, presented results with ICTCAR014 at the meeting.
Dr. Lu explained that ICTCAR014 consists of CD19-targeted CAR T cells genetically engineered to overexpress a PD-1 dominant negative protein with an altered intracellular signaling domain. The dominant negative protein can act as a “decoy receptor” to bind and block the PD-L1/2 inhibitory signal, thereby enhancing the efficacy of CAR T cells.
Innovative Cellular Therapeutics is developing ICTCAR014 because there is “some room to improve” with commercially available CAR T-cell products, Dr. Lu said. Specifically, tisagenlecleucel produced a 52% response rate in the JULIET trial (N Engl J Med. 2019;380:45-56), and axicabtagene ciloleucel produced an 82% response rate in the ZUMA-1 trial (N Engl J Med. 2017;377:2531-44).
There is also evidence to suggest that PD-1 blockade can modulate and “refuel” CAR T cells in relapsed/refractory NHL patients who fail or relapse after traditional anti-CD19 CAR T-cell therapy (Blood. 2017 Feb 23;129[8]:1039-41). This finding has prompted researchers to conduct trials of PD-1 inhibitors in combination with CAR T-cell therapies. But this combination approach may be expensive and cause more side effects than the armored CAR T-cell approach, Dr. Lu said.
In preclinical studies, Dr. Lu and colleagues found that ICTCAR014 was more effective than traditional anti-CD19 CAR T cells in killing Nalm6-PDL1 cells. In addition, the PD-1 dominant negative protein protected CAR T cells from exhaustion.
Dr. Lu also presented results in 13 NHL patients who have received ICTCAR014 in a phase 1 trial in China. Eleven patients had diffuse large B-cell lymphoma (DLBCL), and two had follicular lymphoma.
The objective response rate was 92.3% (12/13), which included five partial responses (38.5%) and seven complete responses (53.8%). Both follicular lymphoma patients and five DLBCL patients achieved a complete response. Five DLBCL patients achieved a partial response, and the remaining DLBCL patient did not respond.
Dr. Lu did not present safety data. However, he reported that there was no increased incidence of cytokine release syndrome or neurotoxicity in these patients, compared with patients receiving traditional CAR T-cell therapy.
Dr. Lu is employed by Innovative Cellular Therapeutics, which funded the research and is developing ICTCAR014.
SOURCE: Lu V et al. SITC 2019, Abstract O25.
NATIONAL HARBOR, MD – An armored chimeric antigen receptor (CAR) T-cell therapy has demonstrated efficacy in vitro and in patients with relapsed or refractory non-Hodgkin lymphoma (NHL), according to findings presented at the annual meeting of the Society for Immunotherapy of Cancer.
ICTCAR014, a dominant negative PD-1 armored CAR T-cell therapy, proved more cytotoxic than traditional CAR T-cell therapy in vitro and produced responses in 12 of 13 NHL patients who received it.
Xiaobin Victor Lu, PhD, of Innovative Cellular Therapeutics, Shanghai, China, presented results with ICTCAR014 at the meeting.
Dr. Lu explained that ICTCAR014 consists of CD19-targeted CAR T cells genetically engineered to overexpress a PD-1 dominant negative protein with an altered intracellular signaling domain. The dominant negative protein can act as a “decoy receptor” to bind and block the PD-L1/2 inhibitory signal, thereby enhancing the efficacy of CAR T cells.
Innovative Cellular Therapeutics is developing ICTCAR014 because there is “some room to improve” with commercially available CAR T-cell products, Dr. Lu said. Specifically, tisagenlecleucel produced a 52% response rate in the JULIET trial (N Engl J Med. 2019;380:45-56), and axicabtagene ciloleucel produced an 82% response rate in the ZUMA-1 trial (N Engl J Med. 2017;377:2531-44).
There is also evidence to suggest that PD-1 blockade can modulate and “refuel” CAR T cells in relapsed/refractory NHL patients who fail or relapse after traditional anti-CD19 CAR T-cell therapy (Blood. 2017 Feb 23;129[8]:1039-41). This finding has prompted researchers to conduct trials of PD-1 inhibitors in combination with CAR T-cell therapies. But this combination approach may be expensive and cause more side effects than the armored CAR T-cell approach, Dr. Lu said.
In preclinical studies, Dr. Lu and colleagues found that ICTCAR014 was more effective than traditional anti-CD19 CAR T cells in killing Nalm6-PDL1 cells. In addition, the PD-1 dominant negative protein protected CAR T cells from exhaustion.
Dr. Lu also presented results in 13 NHL patients who have received ICTCAR014 in a phase 1 trial in China. Eleven patients had diffuse large B-cell lymphoma (DLBCL), and two had follicular lymphoma.
The objective response rate was 92.3% (12/13), which included five partial responses (38.5%) and seven complete responses (53.8%). Both follicular lymphoma patients and five DLBCL patients achieved a complete response. Five DLBCL patients achieved a partial response, and the remaining DLBCL patient did not respond.
Dr. Lu did not present safety data. However, he reported that there was no increased incidence of cytokine release syndrome or neurotoxicity in these patients, compared with patients receiving traditional CAR T-cell therapy.
Dr. Lu is employed by Innovative Cellular Therapeutics, which funded the research and is developing ICTCAR014.
SOURCE: Lu V et al. SITC 2019, Abstract O25.
NATIONAL HARBOR, MD – An armored chimeric antigen receptor (CAR) T-cell therapy has demonstrated efficacy in vitro and in patients with relapsed or refractory non-Hodgkin lymphoma (NHL), according to findings presented at the annual meeting of the Society for Immunotherapy of Cancer.
ICTCAR014, a dominant negative PD-1 armored CAR T-cell therapy, proved more cytotoxic than traditional CAR T-cell therapy in vitro and produced responses in 12 of 13 NHL patients who received it.
Xiaobin Victor Lu, PhD, of Innovative Cellular Therapeutics, Shanghai, China, presented results with ICTCAR014 at the meeting.
Dr. Lu explained that ICTCAR014 consists of CD19-targeted CAR T cells genetically engineered to overexpress a PD-1 dominant negative protein with an altered intracellular signaling domain. The dominant negative protein can act as a “decoy receptor” to bind and block the PD-L1/2 inhibitory signal, thereby enhancing the efficacy of CAR T cells.
Innovative Cellular Therapeutics is developing ICTCAR014 because there is “some room to improve” with commercially available CAR T-cell products, Dr. Lu said. Specifically, tisagenlecleucel produced a 52% response rate in the JULIET trial (N Engl J Med. 2019;380:45-56), and axicabtagene ciloleucel produced an 82% response rate in the ZUMA-1 trial (N Engl J Med. 2017;377:2531-44).
There is also evidence to suggest that PD-1 blockade can modulate and “refuel” CAR T cells in relapsed/refractory NHL patients who fail or relapse after traditional anti-CD19 CAR T-cell therapy (Blood. 2017 Feb 23;129[8]:1039-41). This finding has prompted researchers to conduct trials of PD-1 inhibitors in combination with CAR T-cell therapies. But this combination approach may be expensive and cause more side effects than the armored CAR T-cell approach, Dr. Lu said.
In preclinical studies, Dr. Lu and colleagues found that ICTCAR014 was more effective than traditional anti-CD19 CAR T cells in killing Nalm6-PDL1 cells. In addition, the PD-1 dominant negative protein protected CAR T cells from exhaustion.
Dr. Lu also presented results in 13 NHL patients who have received ICTCAR014 in a phase 1 trial in China. Eleven patients had diffuse large B-cell lymphoma (DLBCL), and two had follicular lymphoma.
The objective response rate was 92.3% (12/13), which included five partial responses (38.5%) and seven complete responses (53.8%). Both follicular lymphoma patients and five DLBCL patients achieved a complete response. Five DLBCL patients achieved a partial response, and the remaining DLBCL patient did not respond.
Dr. Lu did not present safety data. However, he reported that there was no increased incidence of cytokine release syndrome or neurotoxicity in these patients, compared with patients receiving traditional CAR T-cell therapy.
Dr. Lu is employed by Innovative Cellular Therapeutics, which funded the research and is developing ICTCAR014.
SOURCE: Lu V et al. SITC 2019, Abstract O25.
REPORTING FROM SITC 2019
Survival ‘excellent’ after rituximab-bendamustine induction in transplant-eligible MCL
The combination of rituximab and bendamustine (RB) provided “excellent” survival with less toxicity, compared with a cytarabine-based induction regimen, in transplant-eligible patients with mantle cell lymphoma, according to a long-term follow-up report from randomized phase 2 trial.

The 5-year survival rates for RB were “provocatively similar” to what was achieved with the standard, intensive R-hyperCVAD regimen, investigators said in this update on the Southwest Oncology Group (SWOG) S1106 study.
By contrast, the R-hyperCVAD regimen was associated with more toxicity and higher failure rates for stem cell mobilization, according to the report’s lead author, Manali Kamdar, MD, of the University of Colorado, Denver, and coauthors.
“Overall, S1106 demonstrated that an outpatient-based, less intensive induction therapy of bendamustine plus rituximab is highly effective, safe, and durable in untreated transplant-eligible MCL patients,” Dr. Kamdar and her colleagues reported in Blood Advances.
The results have guided the design of an upcoming study, EA4181, in which patients with mantle cell lymphoma will be treated with an RB backbone plus cytarabine, the BTK inhibitor acalabrutinib, or both, according to the authors.
In the present study, S1106, patients with mantle cell lymphoma were randomized to receive RB or the R-hyperCVAD regimen, which consisted of rituximab with hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone, alternating with high-dose cytarabine and methotrexate. Both regimens were followed by autologous hematopoietic stem cell transplant.
The stem cell mobilization failure rate was 29% in the R-hyperCVAD arm in an interim analysis conducted after 53 of a planned 160 patients had been enrolled, including 35 in the RB arm and 17 in the R-hyperCVAD arm, according to a report published in the British Journal of Haematology (2016 Dec 19. doi: 10.1111/bjh.14480). That analysis triggered a shutdown of the study, based on a rule stating that either arm would be deemed “unacceptably toxic” if the mobilization rate exceeded 10%.
Accordingly, R-hyperCVAD is “not an ideal platform” for future trials, the investigators said. At that time, the estimated 2-year progression-free survival (PFS) was 81% versus 82% for RB and R-hyperCVAD, respectively, while overall survival (OS) was 87% versus 88%.
With additional follow-up, the 5-year PFS is 66% and 62% in the RB and R-hyperCVAD arms, respectively, while 5-year OS is 80% and 74%, according to the investigators.
The RB regimen also results in “excellent” minimal residual disease (MRD) negativity, they added.
MRD status was evaluated in 12 paired pre- and postinduction therapy specimens, of which 2 pairs were from patients in the R-hyperCVAD arm, and 10 pairs were from patients in the RB arm.
In the R-hyperCVAD arm, both patients were MRD positive at baseline, and MRD negative after induction, according to the investigators. Similarly, 9 of 10 patients in the RB arm were MRD positive at baseline, and of those, 7 converted to MRD negative following induction.
The research was supported by the National Cancer Institute, and in part by Sequenta (Adaptive Biotechnologies). Dr. Kamdar reported being on the speakers bureau of Seattle Genetics and receiving consultancy fees from AstraZeneca, Celgene, and Genentech. Co-authors of the study provided disclosures related to Millennium Pharmaceuticals, Affimed, Seattle Genetics, Pharmacyclics, and Merck, among others.
SOURCE: Kamdar M et al. Blood Adv. 2019 Oct 22;3(20):3132-5.
The combination of rituximab and bendamustine (RB) provided “excellent” survival with less toxicity, compared with a cytarabine-based induction regimen, in transplant-eligible patients with mantle cell lymphoma, according to a long-term follow-up report from randomized phase 2 trial.
The 5-year survival rates for RB were “provocatively similar” to what was achieved with the standard, intensive R-hyperCVAD regimen, investigators said in this update on the Southwest Oncology Group (SWOG) S1106 study.
By contrast, the R-hyperCVAD regimen was associated with more toxicity and higher failure rates for stem cell mobilization, according to the report’s lead author, Manali Kamdar, MD, of the University of Colorado, Denver, and coauthors.
“Overall, S1106 demonstrated that an outpatient-based, less intensive induction therapy of bendamustine plus rituximab is highly effective, safe, and durable in untreated transplant-eligible MCL patients,” Dr. Kamdar and her colleagues reported in Blood Advances.
The results have guided the design of an upcoming study, EA4181, in which patients with mantle cell lymphoma will be treated with an RB backbone plus cytarabine, the BTK inhibitor acalabrutinib, or both, according to the authors.
In the present study, S1106, patients with mantle cell lymphoma were randomized to receive RB or the R-hyperCVAD regimen, which consisted of rituximab with hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone, alternating with high-dose cytarabine and methotrexate. Both regimens were followed by autologous hematopoietic stem cell transplant.
The stem cell mobilization failure rate was 29% in the R-hyperCVAD arm in an interim analysis conducted after 53 of a planned 160 patients had been enrolled, including 35 in the RB arm and 17 in the R-hyperCVAD arm, according to a report published in the British Journal of Haematology (2016 Dec 19. doi: 10.1111/bjh.14480). That analysis triggered a shutdown of the study, based on a rule stating that either arm would be deemed “unacceptably toxic” if the mobilization rate exceeded 10%.
Accordingly, R-hyperCVAD is “not an ideal platform” for future trials, the investigators said. At that time, the estimated 2-year progression-free survival (PFS) was 81% versus 82% for RB and R-hyperCVAD, respectively, while overall survival (OS) was 87% versus 88%.
With additional follow-up, the 5-year PFS is 66% and 62% in the RB and R-hyperCVAD arms, respectively, while 5-year OS is 80% and 74%, according to the investigators.
The RB regimen also results in “excellent” minimal residual disease (MRD) negativity, they added.
MRD status was evaluated in 12 paired pre- and postinduction therapy specimens, of which 2 pairs were from patients in the R-hyperCVAD arm, and 10 pairs were from patients in the RB arm.
In the R-hyperCVAD arm, both patients were MRD positive at baseline, and MRD negative after induction, according to the investigators. Similarly, 9 of 10 patients in the RB arm were MRD positive at baseline, and of those, 7 converted to MRD negative following induction.
The research was supported by the National Cancer Institute, and in part by Sequenta (Adaptive Biotechnologies). Dr. Kamdar reported being on the speakers bureau of Seattle Genetics and receiving consultancy fees from AstraZeneca, Celgene, and Genentech. Co-authors of the study provided disclosures related to Millennium Pharmaceuticals, Affimed, Seattle Genetics, Pharmacyclics, and Merck, among others.
SOURCE: Kamdar M et al. Blood Adv. 2019 Oct 22;3(20):3132-5.
The combination of rituximab and bendamustine (RB) provided “excellent” survival with less toxicity, compared with a cytarabine-based induction regimen, in transplant-eligible patients with mantle cell lymphoma, according to a long-term follow-up report from randomized phase 2 trial.
The 5-year survival rates for RB were “provocatively similar” to what was achieved with the standard, intensive R-hyperCVAD regimen, investigators said in this update on the Southwest Oncology Group (SWOG) S1106 study.
By contrast, the R-hyperCVAD regimen was associated with more toxicity and higher failure rates for stem cell mobilization, according to the report’s lead author, Manali Kamdar, MD, of the University of Colorado, Denver, and coauthors.
“Overall, S1106 demonstrated that an outpatient-based, less intensive induction therapy of bendamustine plus rituximab is highly effective, safe, and durable in untreated transplant-eligible MCL patients,” Dr. Kamdar and her colleagues reported in Blood Advances.
The results have guided the design of an upcoming study, EA4181, in which patients with mantle cell lymphoma will be treated with an RB backbone plus cytarabine, the BTK inhibitor acalabrutinib, or both, according to the authors.
In the present study, S1106, patients with mantle cell lymphoma were randomized to receive RB or the R-hyperCVAD regimen, which consisted of rituximab with hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone, alternating with high-dose cytarabine and methotrexate. Both regimens were followed by autologous hematopoietic stem cell transplant.
The stem cell mobilization failure rate was 29% in the R-hyperCVAD arm in an interim analysis conducted after 53 of a planned 160 patients had been enrolled, including 35 in the RB arm and 17 in the R-hyperCVAD arm, according to a report published in the British Journal of Haematology (2016 Dec 19. doi: 10.1111/bjh.14480). That analysis triggered a shutdown of the study, based on a rule stating that either arm would be deemed “unacceptably toxic” if the mobilization rate exceeded 10%.
Accordingly, R-hyperCVAD is “not an ideal platform” for future trials, the investigators said. At that time, the estimated 2-year progression-free survival (PFS) was 81% versus 82% for RB and R-hyperCVAD, respectively, while overall survival (OS) was 87% versus 88%.
With additional follow-up, the 5-year PFS is 66% and 62% in the RB and R-hyperCVAD arms, respectively, while 5-year OS is 80% and 74%, according to the investigators.
The RB regimen also results in “excellent” minimal residual disease (MRD) negativity, they added.
MRD status was evaluated in 12 paired pre- and postinduction therapy specimens, of which 2 pairs were from patients in the R-hyperCVAD arm, and 10 pairs were from patients in the RB arm.
In the R-hyperCVAD arm, both patients were MRD positive at baseline, and MRD negative after induction, according to the investigators. Similarly, 9 of 10 patients in the RB arm were MRD positive at baseline, and of those, 7 converted to MRD negative following induction.
The research was supported by the National Cancer Institute, and in part by Sequenta (Adaptive Biotechnologies). Dr. Kamdar reported being on the speakers bureau of Seattle Genetics and receiving consultancy fees from AstraZeneca, Celgene, and Genentech. Co-authors of the study provided disclosures related to Millennium Pharmaceuticals, Affimed, Seattle Genetics, Pharmacyclics, and Merck, among others.
SOURCE: Kamdar M et al. Blood Adv. 2019 Oct 22;3(20):3132-5.
FROM BLOOD ADVANCES