User login
Portable hematology analyzer gets FDA nod
The Food and Drug Administration has granted .

HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100 (J Clin Pathol. 2016 Aug;69[8]:720-5).
“The HemoScreen delivers lab accurate results,” Avishay Bransky, PhD, CEO of PixCell, said in a statement.
HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations, he added.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
The Food and Drug Administration has granted .
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100 (J Clin Pathol. 2016 Aug;69[8]:720-5).
“The HemoScreen delivers lab accurate results,” Avishay Bransky, PhD, CEO of PixCell, said in a statement.
HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations, he added.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
The Food and Drug Administration has granted .
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100 (J Clin Pathol. 2016 Aug;69[8]:720-5).
“The HemoScreen delivers lab accurate results,” Avishay Bransky, PhD, CEO of PixCell, said in a statement.
HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations, he added.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
Antigen profiling may help prevent transfusion complications
BETHESDA, MD. – according to one researcher.

“We strongly feel that you should get an extended red cell type on the first encounter” for patients facing long-term transfusion support, Connie Westhoff, PhD, of the New York Blood Center, said at Sickle Cell in Focus, a conference held by the National Institutes of Health. “This can be a one-time test. It doesn’t have to be repeated.”
She also stressed the importance of ensuring that this information travels with patients, who may be seen at various hospitals. “One of the challenges here is making this part of the patient’s electronic medical record.”
Alloimmunization has been a major concern for chronically transfused patients, so there’s a real advantage to knowing a patient’s extended red cell antigen profile before the patient develops alloantibodies following transfusion, Dr. Westhoff said. Hemolytic transfusion reactions can sometimes destroy patients’ own RBCs in addition to the transfused RBCs. Having the patient’s profile to identify the potential cause of the incompatibility and guide transfusion support in an emergency can be lifesaving.
The implementation of genotyping by DNA-based methods has brought down the cost of this screening, making it no longer a barrier in care.
Dr. Westhoff cited a study she and her colleagues published on a study typing patients who have sickle cell disease (Transfusion. 2015 Jun;55[6 Pt 2]:1388-93). In that paper, they found that DNA-based RBC typing provided improved accuracy and expanded information on RBC antigens, compared with hemagglutination methods. This led to its implementation as the primary method for extended RBC typing for patients with sickle cell disease at the Children’s Hospital of Philadelphia.
About 65%-70% of antibodies drop to levels that are not detectable by routine assays, further demonstrating the need for DNA-based methods. The drugs used to dampen the immune response in many of these patients may also hinder or impact detection of antibodies that remain at levels that continue to cause in vivo hemolysis, Dr. Westhoff said in an interview.
For patients with sickle cell disease in many Western countries, antigen matching for CEK, at a minimum, is routine. The United States is now moving in this direction.
“Modern transfusion practice is moving to knowing what antigens the patient is at risk to become immunized against. ... What antigens does the patient lack and what antibodies could the patient make,” Dr. Westhoff said in an interview. “It’s a major advantage in your transfusion service to expedite work-ups and patient care by having that extended antigen profile.”
Dr. Westhoff reported no relevant financial disclosures.
BETHESDA, MD. – according to one researcher.
“We strongly feel that you should get an extended red cell type on the first encounter” for patients facing long-term transfusion support, Connie Westhoff, PhD, of the New York Blood Center, said at Sickle Cell in Focus, a conference held by the National Institutes of Health. “This can be a one-time test. It doesn’t have to be repeated.”
She also stressed the importance of ensuring that this information travels with patients, who may be seen at various hospitals. “One of the challenges here is making this part of the patient’s electronic medical record.”
Alloimmunization has been a major concern for chronically transfused patients, so there’s a real advantage to knowing a patient’s extended red cell antigen profile before the patient develops alloantibodies following transfusion, Dr. Westhoff said. Hemolytic transfusion reactions can sometimes destroy patients’ own RBCs in addition to the transfused RBCs. Having the patient’s profile to identify the potential cause of the incompatibility and guide transfusion support in an emergency can be lifesaving.
The implementation of genotyping by DNA-based methods has brought down the cost of this screening, making it no longer a barrier in care.
Dr. Westhoff cited a study she and her colleagues published on a study typing patients who have sickle cell disease (Transfusion. 2015 Jun;55[6 Pt 2]:1388-93). In that paper, they found that DNA-based RBC typing provided improved accuracy and expanded information on RBC antigens, compared with hemagglutination methods. This led to its implementation as the primary method for extended RBC typing for patients with sickle cell disease at the Children’s Hospital of Philadelphia.
About 65%-70% of antibodies drop to levels that are not detectable by routine assays, further demonstrating the need for DNA-based methods. The drugs used to dampen the immune response in many of these patients may also hinder or impact detection of antibodies that remain at levels that continue to cause in vivo hemolysis, Dr. Westhoff said in an interview.
For patients with sickle cell disease in many Western countries, antigen matching for CEK, at a minimum, is routine. The United States is now moving in this direction.
“Modern transfusion practice is moving to knowing what antigens the patient is at risk to become immunized against. ... What antigens does the patient lack and what antibodies could the patient make,” Dr. Westhoff said in an interview. “It’s a major advantage in your transfusion service to expedite work-ups and patient care by having that extended antigen profile.”
Dr. Westhoff reported no relevant financial disclosures.
BETHESDA, MD. – according to one researcher.
“We strongly feel that you should get an extended red cell type on the first encounter” for patients facing long-term transfusion support, Connie Westhoff, PhD, of the New York Blood Center, said at Sickle Cell in Focus, a conference held by the National Institutes of Health. “This can be a one-time test. It doesn’t have to be repeated.”
She also stressed the importance of ensuring that this information travels with patients, who may be seen at various hospitals. “One of the challenges here is making this part of the patient’s electronic medical record.”
Alloimmunization has been a major concern for chronically transfused patients, so there’s a real advantage to knowing a patient’s extended red cell antigen profile before the patient develops alloantibodies following transfusion, Dr. Westhoff said. Hemolytic transfusion reactions can sometimes destroy patients’ own RBCs in addition to the transfused RBCs. Having the patient’s profile to identify the potential cause of the incompatibility and guide transfusion support in an emergency can be lifesaving.
The implementation of genotyping by DNA-based methods has brought down the cost of this screening, making it no longer a barrier in care.
Dr. Westhoff cited a study she and her colleagues published on a study typing patients who have sickle cell disease (Transfusion. 2015 Jun;55[6 Pt 2]:1388-93). In that paper, they found that DNA-based RBC typing provided improved accuracy and expanded information on RBC antigens, compared with hemagglutination methods. This led to its implementation as the primary method for extended RBC typing for patients with sickle cell disease at the Children’s Hospital of Philadelphia.
About 65%-70% of antibodies drop to levels that are not detectable by routine assays, further demonstrating the need for DNA-based methods. The drugs used to dampen the immune response in many of these patients may also hinder or impact detection of antibodies that remain at levels that continue to cause in vivo hemolysis, Dr. Westhoff said in an interview.
For patients with sickle cell disease in many Western countries, antigen matching for CEK, at a minimum, is routine. The United States is now moving in this direction.
“Modern transfusion practice is moving to knowing what antigens the patient is at risk to become immunized against. ... What antigens does the patient lack and what antibodies could the patient make,” Dr. Westhoff said in an interview. “It’s a major advantage in your transfusion service to expedite work-ups and patient care by having that extended antigen profile.”
Dr. Westhoff reported no relevant financial disclosures.
REPORTING FROM SICKLE CELL IN FOCUS
Platelet transfusion threshold matters for preterm infants
A lower threshold for platelet transfusions in preterm infants with severe thrombocytopenia is associated with significantly lower incidence of death and major bleeding, compared with a higher threshold, a new study suggests.
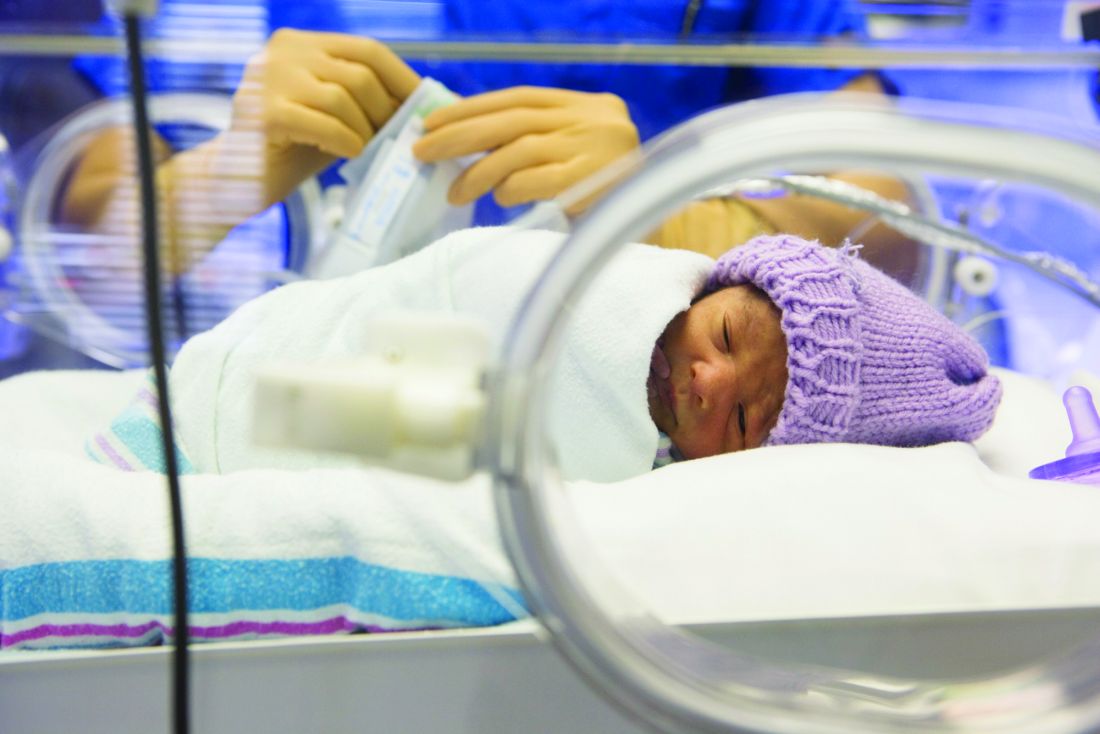
A new major bleeding episode or death occurred in 26% of infants in the high-threshold group, compared with 19% in the low-threshold group, representing a 57% higher risk of poor outcomes even after researchers adjusted for gestational age and intrauterine growth restriction (odds ratio, 1.57; P = .02).
Researchers reported the results of a trial in 660 infants with a mean gestational age of 26.6 weeks, who were randomized to a platelet infusion either at a high platelet–count threshold of 50,000/mm3 or a low threshold of 25,000/mm3.
“Although retrospective studies have suggested that platelet transfusions may cause harm in neonates independently of the disease process, data from randomized controlled trials to support this are lacking,” Anna Curley, MD, of the National Maternity Hospital in Dublin and her coauthors reported in the New England Journal of Medicine.
The rates of minor or worse bleeding were similar between the two groups, and the percentage of infants surviving with bronchopulmonary dysplasia at 36 weeks of corrected age was higher in the high-threshold group (63% vs. 54%; OR, 1.54).
The rates of serious adverse events, not including major bleeding, were similar between the high- and low-threshold groups.
The outcomes of transfusions were not influenced by other factors such as intrauterine growth restriction, gestational age, or postnatal age at randomization.
“Our trial highlights the importance of trials of platelet transfusion involving patients with conditions other than haematological malignancies,” the authors wrote.
However they acknowledged that the reasons for the differences in mortality and outcomes between the two study groups were unknown.
“Platelets have recognized immunological and inflammatory effects, outside of effects on hemostasis,” they wrote. “The effect of transfusing adult platelets to a delicately balance neonatal hemostatic system with relatively hypofunctional platelets is also poorly understood.”
The study was supported by the National Health Service Blood and Transplant Research and Development Committee and other foundations. Authors reported financial disclosures related to Sanquin and Cerus.
SOURCE: Curley A et al. N Engl J Med. 2018 Nov 2. doi: 10.1056/NEJMoa1807320.
A lower threshold for platelet transfusions in preterm infants with severe thrombocytopenia is associated with significantly lower incidence of death and major bleeding, compared with a higher threshold, a new study suggests.
A new major bleeding episode or death occurred in 26% of infants in the high-threshold group, compared with 19% in the low-threshold group, representing a 57% higher risk of poor outcomes even after researchers adjusted for gestational age and intrauterine growth restriction (odds ratio, 1.57; P = .02).
Researchers reported the results of a trial in 660 infants with a mean gestational age of 26.6 weeks, who were randomized to a platelet infusion either at a high platelet–count threshold of 50,000/mm3 or a low threshold of 25,000/mm3.
“Although retrospective studies have suggested that platelet transfusions may cause harm in neonates independently of the disease process, data from randomized controlled trials to support this are lacking,” Anna Curley, MD, of the National Maternity Hospital in Dublin and her coauthors reported in the New England Journal of Medicine.
The rates of minor or worse bleeding were similar between the two groups, and the percentage of infants surviving with bronchopulmonary dysplasia at 36 weeks of corrected age was higher in the high-threshold group (63% vs. 54%; OR, 1.54).
The rates of serious adverse events, not including major bleeding, were similar between the high- and low-threshold groups.
The outcomes of transfusions were not influenced by other factors such as intrauterine growth restriction, gestational age, or postnatal age at randomization.
“Our trial highlights the importance of trials of platelet transfusion involving patients with conditions other than haematological malignancies,” the authors wrote.
However they acknowledged that the reasons for the differences in mortality and outcomes between the two study groups were unknown.
“Platelets have recognized immunological and inflammatory effects, outside of effects on hemostasis,” they wrote. “The effect of transfusing adult platelets to a delicately balance neonatal hemostatic system with relatively hypofunctional platelets is also poorly understood.”
The study was supported by the National Health Service Blood and Transplant Research and Development Committee and other foundations. Authors reported financial disclosures related to Sanquin and Cerus.
SOURCE: Curley A et al. N Engl J Med. 2018 Nov 2. doi: 10.1056/NEJMoa1807320.
A lower threshold for platelet transfusions in preterm infants with severe thrombocytopenia is associated with significantly lower incidence of death and major bleeding, compared with a higher threshold, a new study suggests.
A new major bleeding episode or death occurred in 26% of infants in the high-threshold group, compared with 19% in the low-threshold group, representing a 57% higher risk of poor outcomes even after researchers adjusted for gestational age and intrauterine growth restriction (odds ratio, 1.57; P = .02).
Researchers reported the results of a trial in 660 infants with a mean gestational age of 26.6 weeks, who were randomized to a platelet infusion either at a high platelet–count threshold of 50,000/mm3 or a low threshold of 25,000/mm3.
“Although retrospective studies have suggested that platelet transfusions may cause harm in neonates independently of the disease process, data from randomized controlled trials to support this are lacking,” Anna Curley, MD, of the National Maternity Hospital in Dublin and her coauthors reported in the New England Journal of Medicine.
The rates of minor or worse bleeding were similar between the two groups, and the percentage of infants surviving with bronchopulmonary dysplasia at 36 weeks of corrected age was higher in the high-threshold group (63% vs. 54%; OR, 1.54).
The rates of serious adverse events, not including major bleeding, were similar between the high- and low-threshold groups.
The outcomes of transfusions were not influenced by other factors such as intrauterine growth restriction, gestational age, or postnatal age at randomization.
“Our trial highlights the importance of trials of platelet transfusion involving patients with conditions other than haematological malignancies,” the authors wrote.
However they acknowledged that the reasons for the differences in mortality and outcomes between the two study groups were unknown.
“Platelets have recognized immunological and inflammatory effects, outside of effects on hemostasis,” they wrote. “The effect of transfusing adult platelets to a delicately balance neonatal hemostatic system with relatively hypofunctional platelets is also poorly understood.”
The study was supported by the National Health Service Blood and Transplant Research and Development Committee and other foundations. Authors reported financial disclosures related to Sanquin and Cerus.
SOURCE: Curley A et al. N Engl J Med. 2018 Nov 2. doi: 10.1056/NEJMoa1807320.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: The odds of new major bleeding or death were 57% higher in preterm infants who received a platelet transfusion at a higher threshold of 50,000 per mm3 than at a lower threshold of 25,000 mm3 (P = .02).Study details: Randomized study in 660 preterm infants with severe thrombocytopenia.
Disclosures: The study was supported by the National Health Service Blood and Transplant Research and Development Committee, and other foundations. Authors reported financial disclosures related to Sanquin and Cerus.
Source: Curley A et al. N Engl J Med. 2018 Nov 2. doi: 10.1056/NEJMoa1807320.
Sickle cell disease gene therapy seen advancing
BETHESDA, MD. – Experimental gene therapies for sickle cell disease and thalassemia appear to be advancing, with BCL11A among the most promising targets in this field, researchers said at Sickle Cell in Focus, a conference held by the National Institutes of Health.

Several highly anticipated presentations on the topic are expected for December meeting of the American Society of Hematology.
Alexis A. Thompson, MD, of Northwestern University, Chicago, reviewed highlights from a study in which the majority of patients given a gene therapy for transfusion dependent beta-thalassemia didn’t need subsequent blood transfusions. The New England Journal of Medicine in April published the results of this work done with Bluebird Bio’s LentiGlobin gene therapy (N Engl J Med. 2018; 378:1479-93).
Of the 22 patients in this trial, 15 have become transfusion independent, Dr. Thompson said in her presentation. Those patients that did not have this positive outcome still appear to have been helped by the gene therapy, she said. They had a median of 60% reduction in their transfusion volumes and nearly 60% in their number of transfusions.
“Whether it was transfusion independence or reduction in their transfusion volume or number, the vast majority of individuals in this first large-scale study had clinical benefit” from the therapy, said Dr. Thompson, who was the lead author of the study.
Dr. Thompson, the current president of the American Society of Hematology (ASH), said she’s looking forward to presentations on some of the most advanced gene therapies for sickle cell disease and thalassemia at the group’s annual meeting in December. The ASH presentations include those of John F. Tisdale, MD, who will report the latest data on LentiGlobin gene therapy in sickle cell disease, and Punam Malik, MD, of Cincinnati Children’s Hospital, who has developed a gamma globin lentivirus vector. There also will be a first readout on a particularly novel approach taken by researchers at Boston Children’s Hospital, led by David Williams, MD.
The development of CRISPR-Cas9 “has really opened up the field” of gene therapy, aiding researchers at Boston Children’s in their efforts to develop a treatment to maintain fetal hemoglobin production, Daniel E. Bauer, MD, PhD, of Boston Children’s Hospital, said during his presentation at the NIH conference.

Dr. Bauer provided an update on the BCL11A research that seeks to block what amounts to a genetic “off switch” for production of fetal hemoglobin. It’s long been known that erythrocytes of newborns with sickle cell disease are protected from sickling by high levels of fetal hemoglobin. Clinical manifestations of sickle cell disease then emerge in the first year of life as fetal hemoglobin levels decline.
“A main goal in hematology has been to understand how is it that these alternative fetal hemoglobin genes get silenced and how can we turn them back on,” said Dr. Bauer, a staff physician in pediatric hematology/oncology.
The gene BCL11A also plays key roles in the development of the central nervous system, B lymphatic lymphocyte maturation, and hematopoietic stem cell self-renewal. That led the researchers to hone in on targeting sequences around the BCL11A gene that act as erythroid enhancers, intending to limit potential complications by creating a very specific therapy for sickle cell disease.
In a related clinical trial, using lentiviral gene therapy rather than gene editing, researchers at Boston Children’s began an open-label, nonrandomized, single-center pilot study that involves a single infusion of autologous bone marrow derived CD34+ HSC cells transduced by a vector containing a short-hairpin segment of RNA targeting the gene BCL11A.
The study has a maximum accrual of seven evaluable patients, according to the NIH’s clinical trials website. The protocol is similar to bone marrow transplant, in that native blood stem cells are eliminated by myeloablative conditioning therapy. In this gene therapy, the patient’s own blood stem cells are then infused after the new genetic material has been added to counter the normal BCL11A off switch.
Swee Lay Thein, MBBS, an organizer of the NIH’s sickle cell conference, said in an interview that the “gene therapy side is really looking very optimistic.”
Dr. Thein, a senior investigator for sickle-cell genetics at the National Heart, Lung, and Blood Institute, earlier in her career discovered segments of DNA, including the BCL11A gene. She said that gaining greater understanding about genomic variation might someday aid in determining which people need more intense intervention for their sickle cell disease.
“You would be able to predict who will have more severe disease; we could monitor them more closely and perhaps even advocate for gene therapy or bone marrow transplant before complications have occurred rather than waiting for them to occur,” Dr. Thein said.
She referred to this as her “dream” for care of people with sickle cell disease. “This is still far off in the horizon.”
The NHLBI in September 2018 launched its Cure Sickle Cell Initiative. The agency estimates that it spends about $100 million on sickle cell disease research each year. The inherited blood disorder affects about 100,000 people in the United States and 20 million individuals worldwide. In sickle cell disease, a single genetic mutation causes red blood cells to form abnormal, sickle shapes that can clog the blood vessels and deprive cells of oxygen.
Dr. Thompson reported research funding and consulting agreements with Biomarin, Bluebird Bio, Celgene, Novartis, and Shire. Dr. Bauer reported patents related to BCL11A enhancer editing, consulting agreements with Merck and Pfizer, and research support from Bioverativ.
BETHESDA, MD. – Experimental gene therapies for sickle cell disease and thalassemia appear to be advancing, with BCL11A among the most promising targets in this field, researchers said at Sickle Cell in Focus, a conference held by the National Institutes of Health.
Several highly anticipated presentations on the topic are expected for December meeting of the American Society of Hematology.
Alexis A. Thompson, MD, of Northwestern University, Chicago, reviewed highlights from a study in which the majority of patients given a gene therapy for transfusion dependent beta-thalassemia didn’t need subsequent blood transfusions. The New England Journal of Medicine in April published the results of this work done with Bluebird Bio’s LentiGlobin gene therapy (N Engl J Med. 2018; 378:1479-93).
Of the 22 patients in this trial, 15 have become transfusion independent, Dr. Thompson said in her presentation. Those patients that did not have this positive outcome still appear to have been helped by the gene therapy, she said. They had a median of 60% reduction in their transfusion volumes and nearly 60% in their number of transfusions.
“Whether it was transfusion independence or reduction in their transfusion volume or number, the vast majority of individuals in this first large-scale study had clinical benefit” from the therapy, said Dr. Thompson, who was the lead author of the study.
Dr. Thompson, the current president of the American Society of Hematology (ASH), said she’s looking forward to presentations on some of the most advanced gene therapies for sickle cell disease and thalassemia at the group’s annual meeting in December. The ASH presentations include those of John F. Tisdale, MD, who will report the latest data on LentiGlobin gene therapy in sickle cell disease, and Punam Malik, MD, of Cincinnati Children’s Hospital, who has developed a gamma globin lentivirus vector. There also will be a first readout on a particularly novel approach taken by researchers at Boston Children’s Hospital, led by David Williams, MD.
The development of CRISPR-Cas9 “has really opened up the field” of gene therapy, aiding researchers at Boston Children’s in their efforts to develop a treatment to maintain fetal hemoglobin production, Daniel E. Bauer, MD, PhD, of Boston Children’s Hospital, said during his presentation at the NIH conference.
Dr. Bauer provided an update on the BCL11A research that seeks to block what amounts to a genetic “off switch” for production of fetal hemoglobin. It’s long been known that erythrocytes of newborns with sickle cell disease are protected from sickling by high levels of fetal hemoglobin. Clinical manifestations of sickle cell disease then emerge in the first year of life as fetal hemoglobin levels decline.
“A main goal in hematology has been to understand how is it that these alternative fetal hemoglobin genes get silenced and how can we turn them back on,” said Dr. Bauer, a staff physician in pediatric hematology/oncology.
The gene BCL11A also plays key roles in the development of the central nervous system, B lymphatic lymphocyte maturation, and hematopoietic stem cell self-renewal. That led the researchers to hone in on targeting sequences around the BCL11A gene that act as erythroid enhancers, intending to limit potential complications by creating a very specific therapy for sickle cell disease.
In a related clinical trial, using lentiviral gene therapy rather than gene editing, researchers at Boston Children’s began an open-label, nonrandomized, single-center pilot study that involves a single infusion of autologous bone marrow derived CD34+ HSC cells transduced by a vector containing a short-hairpin segment of RNA targeting the gene BCL11A.
The study has a maximum accrual of seven evaluable patients, according to the NIH’s clinical trials website. The protocol is similar to bone marrow transplant, in that native blood stem cells are eliminated by myeloablative conditioning therapy. In this gene therapy, the patient’s own blood stem cells are then infused after the new genetic material has been added to counter the normal BCL11A off switch.
Swee Lay Thein, MBBS, an organizer of the NIH’s sickle cell conference, said in an interview that the “gene therapy side is really looking very optimistic.”
Dr. Thein, a senior investigator for sickle-cell genetics at the National Heart, Lung, and Blood Institute, earlier in her career discovered segments of DNA, including the BCL11A gene. She said that gaining greater understanding about genomic variation might someday aid in determining which people need more intense intervention for their sickle cell disease.
“You would be able to predict who will have more severe disease; we could monitor them more closely and perhaps even advocate for gene therapy or bone marrow transplant before complications have occurred rather than waiting for them to occur,” Dr. Thein said.
She referred to this as her “dream” for care of people with sickle cell disease. “This is still far off in the horizon.”
The NHLBI in September 2018 launched its Cure Sickle Cell Initiative. The agency estimates that it spends about $100 million on sickle cell disease research each year. The inherited blood disorder affects about 100,000 people in the United States and 20 million individuals worldwide. In sickle cell disease, a single genetic mutation causes red blood cells to form abnormal, sickle shapes that can clog the blood vessels and deprive cells of oxygen.
Dr. Thompson reported research funding and consulting agreements with Biomarin, Bluebird Bio, Celgene, Novartis, and Shire. Dr. Bauer reported patents related to BCL11A enhancer editing, consulting agreements with Merck and Pfizer, and research support from Bioverativ.
BETHESDA, MD. – Experimental gene therapies for sickle cell disease and thalassemia appear to be advancing, with BCL11A among the most promising targets in this field, researchers said at Sickle Cell in Focus, a conference held by the National Institutes of Health.
Several highly anticipated presentations on the topic are expected for December meeting of the American Society of Hematology.
Alexis A. Thompson, MD, of Northwestern University, Chicago, reviewed highlights from a study in which the majority of patients given a gene therapy for transfusion dependent beta-thalassemia didn’t need subsequent blood transfusions. The New England Journal of Medicine in April published the results of this work done with Bluebird Bio’s LentiGlobin gene therapy (N Engl J Med. 2018; 378:1479-93).
Of the 22 patients in this trial, 15 have become transfusion independent, Dr. Thompson said in her presentation. Those patients that did not have this positive outcome still appear to have been helped by the gene therapy, she said. They had a median of 60% reduction in their transfusion volumes and nearly 60% in their number of transfusions.
“Whether it was transfusion independence or reduction in their transfusion volume or number, the vast majority of individuals in this first large-scale study had clinical benefit” from the therapy, said Dr. Thompson, who was the lead author of the study.
Dr. Thompson, the current president of the American Society of Hematology (ASH), said she’s looking forward to presentations on some of the most advanced gene therapies for sickle cell disease and thalassemia at the group’s annual meeting in December. The ASH presentations include those of John F. Tisdale, MD, who will report the latest data on LentiGlobin gene therapy in sickle cell disease, and Punam Malik, MD, of Cincinnati Children’s Hospital, who has developed a gamma globin lentivirus vector. There also will be a first readout on a particularly novel approach taken by researchers at Boston Children’s Hospital, led by David Williams, MD.
The development of CRISPR-Cas9 “has really opened up the field” of gene therapy, aiding researchers at Boston Children’s in their efforts to develop a treatment to maintain fetal hemoglobin production, Daniel E. Bauer, MD, PhD, of Boston Children’s Hospital, said during his presentation at the NIH conference.
Dr. Bauer provided an update on the BCL11A research that seeks to block what amounts to a genetic “off switch” for production of fetal hemoglobin. It’s long been known that erythrocytes of newborns with sickle cell disease are protected from sickling by high levels of fetal hemoglobin. Clinical manifestations of sickle cell disease then emerge in the first year of life as fetal hemoglobin levels decline.
“A main goal in hematology has been to understand how is it that these alternative fetal hemoglobin genes get silenced and how can we turn them back on,” said Dr. Bauer, a staff physician in pediatric hematology/oncology.
The gene BCL11A also plays key roles in the development of the central nervous system, B lymphatic lymphocyte maturation, and hematopoietic stem cell self-renewal. That led the researchers to hone in on targeting sequences around the BCL11A gene that act as erythroid enhancers, intending to limit potential complications by creating a very specific therapy for sickle cell disease.
In a related clinical trial, using lentiviral gene therapy rather than gene editing, researchers at Boston Children’s began an open-label, nonrandomized, single-center pilot study that involves a single infusion of autologous bone marrow derived CD34+ HSC cells transduced by a vector containing a short-hairpin segment of RNA targeting the gene BCL11A.
The study has a maximum accrual of seven evaluable patients, according to the NIH’s clinical trials website. The protocol is similar to bone marrow transplant, in that native blood stem cells are eliminated by myeloablative conditioning therapy. In this gene therapy, the patient’s own blood stem cells are then infused after the new genetic material has been added to counter the normal BCL11A off switch.
Swee Lay Thein, MBBS, an organizer of the NIH’s sickle cell conference, said in an interview that the “gene therapy side is really looking very optimistic.”
Dr. Thein, a senior investigator for sickle-cell genetics at the National Heart, Lung, and Blood Institute, earlier in her career discovered segments of DNA, including the BCL11A gene. She said that gaining greater understanding about genomic variation might someday aid in determining which people need more intense intervention for their sickle cell disease.
“You would be able to predict who will have more severe disease; we could monitor them more closely and perhaps even advocate for gene therapy or bone marrow transplant before complications have occurred rather than waiting for them to occur,” Dr. Thein said.
She referred to this as her “dream” for care of people with sickle cell disease. “This is still far off in the horizon.”
The NHLBI in September 2018 launched its Cure Sickle Cell Initiative. The agency estimates that it spends about $100 million on sickle cell disease research each year. The inherited blood disorder affects about 100,000 people in the United States and 20 million individuals worldwide. In sickle cell disease, a single genetic mutation causes red blood cells to form abnormal, sickle shapes that can clog the blood vessels and deprive cells of oxygen.
Dr. Thompson reported research funding and consulting agreements with Biomarin, Bluebird Bio, Celgene, Novartis, and Shire. Dr. Bauer reported patents related to BCL11A enhancer editing, consulting agreements with Merck and Pfizer, and research support from Bioverativ.
REPORTING FROM SICKLE CELL IN FOCUS
Bleeding score could help identify hemophilia
Bleeding scores may be helpful in identifying hemophilia patients, regardless of whether or not clotting factor levels are known, results of a recent investigation suggest.

Both hemophilia A and B patients had significantly higher bleeding scores as assessed by the ISTH-BAT (International Society on Thrombosis and Hemostasis–Bleeding Assessment Tool), compared with control subjects, according to results of the study.
Moreover, hemophilia patients classified as severe had significantly higher ISTH-BAT scores compared with those classified as mild, reported by Munira Borhany, MD, of the National Institute of Blood Disease and Bone Marrow Transplantation, Karachi, Pakistan, and her colleagues.
“The ISTH-BAT can be easily used in the clinics by physicians and can help to identify those patients who should be further investigated,” Dr. Borhany and her coauthors reported in the journal Transfusion and Apheresis Science.
The ISTH-BAT, established to standardize the reporting of bleeding symptoms, scores symptoms from 0, which indicates absent or trivial, to 4, meaning a symptom that requires medical intervention. Total scores considered abnormal are 4 or greater in men, 6 and greater in women, and 3 and greater in children, according to previously published reports.
In the present cross-sectional study, Dr. Borhany and her colleagues evaluated bleeding scores for 115 adult and pediatric patients – 78 with hemophilia A and 37 with hemophilia B – who were treated in Pakistan between 2014 and 2016.
Bleeding scores were a mean of 13.5 and 13.2 for hemophilia A and B patients, respectively, and 0.8 for 100 healthy male controls also included in the study. Scores were significantly higher in hemophilia patients versus controls (P less than .001), but not different between hemophilia A and B patients, the investigators reported.
Further results suggested a correlation between factor levels and clinical presentation of bleeding symptoms, according to the investigators. Statistically significant differences in bleeding scores also were seen between patients with severe and mild disease, and between severe and moderate disease, but not between the mild and moderate groups, they added.
Most studies of bleeding questionnaires to date have been in patients with von Willebrand disease or platelet disorders, with very little data on hemophilia.
“Apart from one recent study using ISTH-BAT in hemophilia carriers as part of assessing quality of life, we are unaware of other studies examining this assessment tool in hemophilia,” the researchers wrote.
This study cohort was unique, according to the investigators, because it included a substantial number of adults who were new patients with bleeding symptoms who had no previous diagnosis of hemophilia. “This allowed assessing whether investigators may tend to apply a higher score when knowing very low factor levels in hemophilia patients,” they said.
In fact, there was no major difference in bleeding scores for those newly diagnosed patients versus already diagnosed patients.
Results of an ongoing study will determine whether the ISTH BAT bleeding score can predict risk of bleeding in hemophilia patients, according to Dr. Borhany and her coauthors.
They reported having no conflicts of interest.
SOURCE: Borhany M et al. Transfus Apher Sci. 2018 Aug;57(4):556-60.
Bleeding scores may be helpful in identifying hemophilia patients, regardless of whether or not clotting factor levels are known, results of a recent investigation suggest.
Both hemophilia A and B patients had significantly higher bleeding scores as assessed by the ISTH-BAT (International Society on Thrombosis and Hemostasis–Bleeding Assessment Tool), compared with control subjects, according to results of the study.
Moreover, hemophilia patients classified as severe had significantly higher ISTH-BAT scores compared with those classified as mild, reported by Munira Borhany, MD, of the National Institute of Blood Disease and Bone Marrow Transplantation, Karachi, Pakistan, and her colleagues.
“The ISTH-BAT can be easily used in the clinics by physicians and can help to identify those patients who should be further investigated,” Dr. Borhany and her coauthors reported in the journal Transfusion and Apheresis Science.
The ISTH-BAT, established to standardize the reporting of bleeding symptoms, scores symptoms from 0, which indicates absent or trivial, to 4, meaning a symptom that requires medical intervention. Total scores considered abnormal are 4 or greater in men, 6 and greater in women, and 3 and greater in children, according to previously published reports.
In the present cross-sectional study, Dr. Borhany and her colleagues evaluated bleeding scores for 115 adult and pediatric patients – 78 with hemophilia A and 37 with hemophilia B – who were treated in Pakistan between 2014 and 2016.
Bleeding scores were a mean of 13.5 and 13.2 for hemophilia A and B patients, respectively, and 0.8 for 100 healthy male controls also included in the study. Scores were significantly higher in hemophilia patients versus controls (P less than .001), but not different between hemophilia A and B patients, the investigators reported.
Further results suggested a correlation between factor levels and clinical presentation of bleeding symptoms, according to the investigators. Statistically significant differences in bleeding scores also were seen between patients with severe and mild disease, and between severe and moderate disease, but not between the mild and moderate groups, they added.
Most studies of bleeding questionnaires to date have been in patients with von Willebrand disease or platelet disorders, with very little data on hemophilia.
“Apart from one recent study using ISTH-BAT in hemophilia carriers as part of assessing quality of life, we are unaware of other studies examining this assessment tool in hemophilia,” the researchers wrote.
This study cohort was unique, according to the investigators, because it included a substantial number of adults who were new patients with bleeding symptoms who had no previous diagnosis of hemophilia. “This allowed assessing whether investigators may tend to apply a higher score when knowing very low factor levels in hemophilia patients,” they said.
In fact, there was no major difference in bleeding scores for those newly diagnosed patients versus already diagnosed patients.
Results of an ongoing study will determine whether the ISTH BAT bleeding score can predict risk of bleeding in hemophilia patients, according to Dr. Borhany and her coauthors.
They reported having no conflicts of interest.
SOURCE: Borhany M et al. Transfus Apher Sci. 2018 Aug;57(4):556-60.
Bleeding scores may be helpful in identifying hemophilia patients, regardless of whether or not clotting factor levels are known, results of a recent investigation suggest.
Both hemophilia A and B patients had significantly higher bleeding scores as assessed by the ISTH-BAT (International Society on Thrombosis and Hemostasis–Bleeding Assessment Tool), compared with control subjects, according to results of the study.
Moreover, hemophilia patients classified as severe had significantly higher ISTH-BAT scores compared with those classified as mild, reported by Munira Borhany, MD, of the National Institute of Blood Disease and Bone Marrow Transplantation, Karachi, Pakistan, and her colleagues.
“The ISTH-BAT can be easily used in the clinics by physicians and can help to identify those patients who should be further investigated,” Dr. Borhany and her coauthors reported in the journal Transfusion and Apheresis Science.
The ISTH-BAT, established to standardize the reporting of bleeding symptoms, scores symptoms from 0, which indicates absent or trivial, to 4, meaning a symptom that requires medical intervention. Total scores considered abnormal are 4 or greater in men, 6 and greater in women, and 3 and greater in children, according to previously published reports.
In the present cross-sectional study, Dr. Borhany and her colleagues evaluated bleeding scores for 115 adult and pediatric patients – 78 with hemophilia A and 37 with hemophilia B – who were treated in Pakistan between 2014 and 2016.
Bleeding scores were a mean of 13.5 and 13.2 for hemophilia A and B patients, respectively, and 0.8 for 100 healthy male controls also included in the study. Scores were significantly higher in hemophilia patients versus controls (P less than .001), but not different between hemophilia A and B patients, the investigators reported.
Further results suggested a correlation between factor levels and clinical presentation of bleeding symptoms, according to the investigators. Statistically significant differences in bleeding scores also were seen between patients with severe and mild disease, and between severe and moderate disease, but not between the mild and moderate groups, they added.
Most studies of bleeding questionnaires to date have been in patients with von Willebrand disease or platelet disorders, with very little data on hemophilia.
“Apart from one recent study using ISTH-BAT in hemophilia carriers as part of assessing quality of life, we are unaware of other studies examining this assessment tool in hemophilia,” the researchers wrote.
This study cohort was unique, according to the investigators, because it included a substantial number of adults who were new patients with bleeding symptoms who had no previous diagnosis of hemophilia. “This allowed assessing whether investigators may tend to apply a higher score when knowing very low factor levels in hemophilia patients,” they said.
In fact, there was no major difference in bleeding scores for those newly diagnosed patients versus already diagnosed patients.
Results of an ongoing study will determine whether the ISTH BAT bleeding score can predict risk of bleeding in hemophilia patients, according to Dr. Borhany and her coauthors.
They reported having no conflicts of interest.
SOURCE: Borhany M et al. Transfus Apher Sci. 2018 Aug;57(4):556-60.
FROM TRANSFUSION AND APHERESIS SCIENCE
Key clinical point:
Major finding: Bleeding scores were a mean of 13.5 and 13.2 for hemophilia A and B patients, respectively, and 0.8 for healthy male controls (P less than .001).
Study details: A cross-sectional study included 115 adult and pediatric patients with hemophilia A or B treated in Pakistan between 2014 and 2016.
Disclosures: The authors reported having no conflicts of interest.
Source: Borhany M et al. Transfus Apher Sci. 2018 Aug;57(4):556-60.
Rapid bacterial testing of platelets saves money, reduces waste
BOSTON – Rapid bacterial testing of platelets in a hospital blood bank can result in both significant cost savings and reduced wastage of blood products, investigators said.

Rapid bacterial testing of 6- or 7-day-old apheresis platelets resulted in projected annual cost savings of nearly $89,000 per year and cut the rate of platelet wastage from expiration by more than half, reported Adam L. Booth, MD, chief resident in the department of pathology at the University of Texas, Galveston, and his colleagues.
“When a person takes all this time to come in and donate, they do it under the impression that they’re going to help somebody, or several people, and you hate to see those platelets wasted. You want them to be used,” he said in an interview at AABB 2018, the annual meeting of the group formerly known as the American Association of Blood Banks.
Platelets typically have a shelf life of just 5 days because longer storage increases the risk for bacterial growth and the potential for transfusion-transmitted infections, Dr. Booth and his colleagues noted in a poster presentation.
A recently published Food and Drug Administration draft guidance for blood banks and transfusion services proposed changing regulations regarding bacterial control of blood products to allow for extended dating if the platelets are collected in an FDA-approved 7-day storage container with labeling that requires testing every product with a bacterial detection device, or if the platelets are individually tested for bacterial detection using an approved device.
To see what effect the regulations, if implemented as expected, might have on acquisition costs and wastage of apheresis platelets, the investigators reviewed their center’s platelet acquisition costs and wastage from expiration 12 months before and 6 months after implementation of a rapid bacterial testing protocol, with 6-month results projected out to 1 year for comparison purposes.
They looked at data on bacterial testing of 6-day and 7-day-old apheresis platelets, and excluded data on platelet units that were due to expire on day 5 because they were not stored in FDA-approved containers.
Prior to testing, 332 units at a mean per-unit cost of $516.96 were wasted, for an annual cost of more than $171,000. After the start of testing, however, the annualized rate of waste dropped to 117 units, for an annualized cost of more than $60,000. The difference – minus the cost of rapid bacterial testing – resulted in an annual savings for the institution of nearly $89,000.
Prior to rapid testing, the annual wastage rate was 24%; after testing, it dropped to an annualized 10% rate, the investigators reported.
The number of units transfused and the associated costs of transfusions were similar between the time periods studied.
“Our findings suggest that rapid bacterial testing can simultaneously enhance the safety of apheresis platelet transfusions and contribute to significant cost savings,” Dr. Booth and his colleagues wrote.
The study was internally funded. The authors reported having no conflicts of interest.
SOURCE: Booth AL et al. AABB18, Abstract INV4.
BOSTON – Rapid bacterial testing of platelets in a hospital blood bank can result in both significant cost savings and reduced wastage of blood products, investigators said.
Rapid bacterial testing of 6- or 7-day-old apheresis platelets resulted in projected annual cost savings of nearly $89,000 per year and cut the rate of platelet wastage from expiration by more than half, reported Adam L. Booth, MD, chief resident in the department of pathology at the University of Texas, Galveston, and his colleagues.
“When a person takes all this time to come in and donate, they do it under the impression that they’re going to help somebody, or several people, and you hate to see those platelets wasted. You want them to be used,” he said in an interview at AABB 2018, the annual meeting of the group formerly known as the American Association of Blood Banks.
Platelets typically have a shelf life of just 5 days because longer storage increases the risk for bacterial growth and the potential for transfusion-transmitted infections, Dr. Booth and his colleagues noted in a poster presentation.
A recently published Food and Drug Administration draft guidance for blood banks and transfusion services proposed changing regulations regarding bacterial control of blood products to allow for extended dating if the platelets are collected in an FDA-approved 7-day storage container with labeling that requires testing every product with a bacterial detection device, or if the platelets are individually tested for bacterial detection using an approved device.
To see what effect the regulations, if implemented as expected, might have on acquisition costs and wastage of apheresis platelets, the investigators reviewed their center’s platelet acquisition costs and wastage from expiration 12 months before and 6 months after implementation of a rapid bacterial testing protocol, with 6-month results projected out to 1 year for comparison purposes.
They looked at data on bacterial testing of 6-day and 7-day-old apheresis platelets, and excluded data on platelet units that were due to expire on day 5 because they were not stored in FDA-approved containers.
Prior to testing, 332 units at a mean per-unit cost of $516.96 were wasted, for an annual cost of more than $171,000. After the start of testing, however, the annualized rate of waste dropped to 117 units, for an annualized cost of more than $60,000. The difference – minus the cost of rapid bacterial testing – resulted in an annual savings for the institution of nearly $89,000.
Prior to rapid testing, the annual wastage rate was 24%; after testing, it dropped to an annualized 10% rate, the investigators reported.
The number of units transfused and the associated costs of transfusions were similar between the time periods studied.
“Our findings suggest that rapid bacterial testing can simultaneously enhance the safety of apheresis platelet transfusions and contribute to significant cost savings,” Dr. Booth and his colleagues wrote.
The study was internally funded. The authors reported having no conflicts of interest.
SOURCE: Booth AL et al. AABB18, Abstract INV4.
BOSTON – Rapid bacterial testing of platelets in a hospital blood bank can result in both significant cost savings and reduced wastage of blood products, investigators said.
Rapid bacterial testing of 6- or 7-day-old apheresis platelets resulted in projected annual cost savings of nearly $89,000 per year and cut the rate of platelet wastage from expiration by more than half, reported Adam L. Booth, MD, chief resident in the department of pathology at the University of Texas, Galveston, and his colleagues.
“When a person takes all this time to come in and donate, they do it under the impression that they’re going to help somebody, or several people, and you hate to see those platelets wasted. You want them to be used,” he said in an interview at AABB 2018, the annual meeting of the group formerly known as the American Association of Blood Banks.
Platelets typically have a shelf life of just 5 days because longer storage increases the risk for bacterial growth and the potential for transfusion-transmitted infections, Dr. Booth and his colleagues noted in a poster presentation.
A recently published Food and Drug Administration draft guidance for blood banks and transfusion services proposed changing regulations regarding bacterial control of blood products to allow for extended dating if the platelets are collected in an FDA-approved 7-day storage container with labeling that requires testing every product with a bacterial detection device, or if the platelets are individually tested for bacterial detection using an approved device.
To see what effect the regulations, if implemented as expected, might have on acquisition costs and wastage of apheresis platelets, the investigators reviewed their center’s platelet acquisition costs and wastage from expiration 12 months before and 6 months after implementation of a rapid bacterial testing protocol, with 6-month results projected out to 1 year for comparison purposes.
They looked at data on bacterial testing of 6-day and 7-day-old apheresis platelets, and excluded data on platelet units that were due to expire on day 5 because they were not stored in FDA-approved containers.
Prior to testing, 332 units at a mean per-unit cost of $516.96 were wasted, for an annual cost of more than $171,000. After the start of testing, however, the annualized rate of waste dropped to 117 units, for an annualized cost of more than $60,000. The difference – minus the cost of rapid bacterial testing – resulted in an annual savings for the institution of nearly $89,000.
Prior to rapid testing, the annual wastage rate was 24%; after testing, it dropped to an annualized 10% rate, the investigators reported.
The number of units transfused and the associated costs of transfusions were similar between the time periods studied.
“Our findings suggest that rapid bacterial testing can simultaneously enhance the safety of apheresis platelet transfusions and contribute to significant cost savings,” Dr. Booth and his colleagues wrote.
The study was internally funded. The authors reported having no conflicts of interest.
SOURCE: Booth AL et al. AABB18, Abstract INV4.
REPORTING FROM AABB18
Key clinical point:
Major finding: Annualized cost savings with rapid bacterial testing were nearly $89,000; platelet wastage decreased from 24% to 10%.
Study details: A retrospective analysis of costs and product wastage before and after implementation of rapid bacterial testing.
Disclosures: The study was internally funded. The authors reported having no conflicts of interest.
Source: Booth AL et al. AABB18, Abstract INV4.
Few clinical outcomes convincingly linked to sickle cell trait
Although sickle cell trait (SCT) has been linked to numerous adverse clinical outcomes in multiple studies, only a handful of those associations have strong supporting evidence, results of a systematic review suggest.
Venous and renal complications had the strongest evidence supporting an association with SCT, while exertion-related sudden death – perhaps the highest-profile potential complication of SCT – had moderate-strength evidence supporting a link, according to the review.
By contrast, most other associations between SCT and clinical outcomes had either low-strength evidence or insufficient data to support a link, according to Rakhi P. Naik, MD, of Johns Hopkins University, Baltimore, and coauthors of the review.
“Future rigorous studies are needed to address potential complications of SCT and to determine modifiers of risk,” they wrote. The report in the Annals of Internal Medicine.
The systematic review by Dr. Naik and colleagues focused on 41 studies, most of which were population-based cohort or case-control studies. They rated the evidence quality of each study and grouped 24 clinical outcomes of interest into six categories: exertion-related injury, renal, vascular, pediatric, surgery- and trauma-related outcomes, and mortality.
Exercise-related injury has received considerable attention, particularly in relation to the military and athletics.
The strength of evidence for a link between SCT and exertion-related death was low in their analysis, which included two studies evaluating the outcome. However, Dr. Naik and coauthors did note that SCT may be associated with a small absolute risk of exertion-related death in extreme conditions such a highly strenuous athletic training or the military.
“We do concur with the American Society of Hematology statement recommending against routine SCT screening in athletics and supporting the consistent use of universal precautions to mitigate exertion-related risk in all persons, regardless of SCT status,” they wrote.
Similarly, the absolute risk of exertional rhabdomyolysis in SCT is small and probably occurs only in high-intensity settings, with risk modified by other genetic and environmental factors, Dr. Naik and coauthors said, based on their analysis of two studies looking at this outcome.
Venous complications had a stronger body of evidence, including several studies showing high levels of procoagulants, which makes elevated venous thromboembolism risk plausible in individuals with SCT.
High-strength evidence linked pulmonary embolism, with or without deep-vein thrombosis, to SCT. In contrast, there was no increased risk of isolated deep-vein thrombosis in these individuals.
“The cause of this paradoxical observation is unknown but may be an increased risk for clot embolization in SCT,” Dr. Naik and colleagues wrote in a discussion of the results.
Renal outcomes were often attributed to SCT, and in this review, the authors said there was evidence to support SCT as a risk factor for both proteinuria and chronic kidney disease.
Out of six studies looking at proteinuria, the one high-quality study found a 1.86-fold increased risk for baseline albuminuria in African Americans with SCT versus those without, according to the review.
Out of four studies looking at chronic kidney disease, the two high-quality studies found 1.57- to 1.89-fold increased risk of those outcomes in African Americans with SCT.
Support for the study came in part from the National Human Genome Research Institute and the National Heart, Lung, and Blood Institute. The authors reported disclosures related to Novartis, Addmedica, and Global Blood Therapeutics, among others.
SOURCE: Naik RP et al. Ann Intern Med. 2018 Oct 30. doi:10.7326/M18-1161.
Although sickle cell trait (SCT) has been linked to numerous adverse clinical outcomes in multiple studies, only a handful of those associations have strong supporting evidence, results of a systematic review suggest.
Venous and renal complications had the strongest evidence supporting an association with SCT, while exertion-related sudden death – perhaps the highest-profile potential complication of SCT – had moderate-strength evidence supporting a link, according to the review.
By contrast, most other associations between SCT and clinical outcomes had either low-strength evidence or insufficient data to support a link, according to Rakhi P. Naik, MD, of Johns Hopkins University, Baltimore, and coauthors of the review.
“Future rigorous studies are needed to address potential complications of SCT and to determine modifiers of risk,” they wrote. The report in the Annals of Internal Medicine.
The systematic review by Dr. Naik and colleagues focused on 41 studies, most of which were population-based cohort or case-control studies. They rated the evidence quality of each study and grouped 24 clinical outcomes of interest into six categories: exertion-related injury, renal, vascular, pediatric, surgery- and trauma-related outcomes, and mortality.
Exercise-related injury has received considerable attention, particularly in relation to the military and athletics.
The strength of evidence for a link between SCT and exertion-related death was low in their analysis, which included two studies evaluating the outcome. However, Dr. Naik and coauthors did note that SCT may be associated with a small absolute risk of exertion-related death in extreme conditions such a highly strenuous athletic training or the military.
“We do concur with the American Society of Hematology statement recommending against routine SCT screening in athletics and supporting the consistent use of universal precautions to mitigate exertion-related risk in all persons, regardless of SCT status,” they wrote.
Similarly, the absolute risk of exertional rhabdomyolysis in SCT is small and probably occurs only in high-intensity settings, with risk modified by other genetic and environmental factors, Dr. Naik and coauthors said, based on their analysis of two studies looking at this outcome.
Venous complications had a stronger body of evidence, including several studies showing high levels of procoagulants, which makes elevated venous thromboembolism risk plausible in individuals with SCT.
High-strength evidence linked pulmonary embolism, with or without deep-vein thrombosis, to SCT. In contrast, there was no increased risk of isolated deep-vein thrombosis in these individuals.
“The cause of this paradoxical observation is unknown but may be an increased risk for clot embolization in SCT,” Dr. Naik and colleagues wrote in a discussion of the results.
Renal outcomes were often attributed to SCT, and in this review, the authors said there was evidence to support SCT as a risk factor for both proteinuria and chronic kidney disease.
Out of six studies looking at proteinuria, the one high-quality study found a 1.86-fold increased risk for baseline albuminuria in African Americans with SCT versus those without, according to the review.
Out of four studies looking at chronic kidney disease, the two high-quality studies found 1.57- to 1.89-fold increased risk of those outcomes in African Americans with SCT.
Support for the study came in part from the National Human Genome Research Institute and the National Heart, Lung, and Blood Institute. The authors reported disclosures related to Novartis, Addmedica, and Global Blood Therapeutics, among others.
SOURCE: Naik RP et al. Ann Intern Med. 2018 Oct 30. doi:10.7326/M18-1161.
Although sickle cell trait (SCT) has been linked to numerous adverse clinical outcomes in multiple studies, only a handful of those associations have strong supporting evidence, results of a systematic review suggest.
Venous and renal complications had the strongest evidence supporting an association with SCT, while exertion-related sudden death – perhaps the highest-profile potential complication of SCT – had moderate-strength evidence supporting a link, according to the review.
By contrast, most other associations between SCT and clinical outcomes had either low-strength evidence or insufficient data to support a link, according to Rakhi P. Naik, MD, of Johns Hopkins University, Baltimore, and coauthors of the review.
“Future rigorous studies are needed to address potential complications of SCT and to determine modifiers of risk,” they wrote. The report in the Annals of Internal Medicine.
The systematic review by Dr. Naik and colleagues focused on 41 studies, most of which were population-based cohort or case-control studies. They rated the evidence quality of each study and grouped 24 clinical outcomes of interest into six categories: exertion-related injury, renal, vascular, pediatric, surgery- and trauma-related outcomes, and mortality.
Exercise-related injury has received considerable attention, particularly in relation to the military and athletics.
The strength of evidence for a link between SCT and exertion-related death was low in their analysis, which included two studies evaluating the outcome. However, Dr. Naik and coauthors did note that SCT may be associated with a small absolute risk of exertion-related death in extreme conditions such a highly strenuous athletic training or the military.
“We do concur with the American Society of Hematology statement recommending against routine SCT screening in athletics and supporting the consistent use of universal precautions to mitigate exertion-related risk in all persons, regardless of SCT status,” they wrote.
Similarly, the absolute risk of exertional rhabdomyolysis in SCT is small and probably occurs only in high-intensity settings, with risk modified by other genetic and environmental factors, Dr. Naik and coauthors said, based on their analysis of two studies looking at this outcome.
Venous complications had a stronger body of evidence, including several studies showing high levels of procoagulants, which makes elevated venous thromboembolism risk plausible in individuals with SCT.
High-strength evidence linked pulmonary embolism, with or without deep-vein thrombosis, to SCT. In contrast, there was no increased risk of isolated deep-vein thrombosis in these individuals.
“The cause of this paradoxical observation is unknown but may be an increased risk for clot embolization in SCT,” Dr. Naik and colleagues wrote in a discussion of the results.
Renal outcomes were often attributed to SCT, and in this review, the authors said there was evidence to support SCT as a risk factor for both proteinuria and chronic kidney disease.
Out of six studies looking at proteinuria, the one high-quality study found a 1.86-fold increased risk for baseline albuminuria in African Americans with SCT versus those without, according to the review.
Out of four studies looking at chronic kidney disease, the two high-quality studies found 1.57- to 1.89-fold increased risk of those outcomes in African Americans with SCT.
Support for the study came in part from the National Human Genome Research Institute and the National Heart, Lung, and Blood Institute. The authors reported disclosures related to Novartis, Addmedica, and Global Blood Therapeutics, among others.
SOURCE: Naik RP et al. Ann Intern Med. 2018 Oct 30. doi:10.7326/M18-1161.
FROM ANNALS OF INTERNAL MEDICINE
Key clinical point:
Major finding: Risks of 1.57-fold and higher were seen in high-quality studies linking SCT to venous and renal complications, while studies of moderate quality suggested small absolute risks of exertion-related mortality or rhabdomyolysis.
Study details: A systematic review including 41 mostly population-based cohort or case-control studies looking at 24 clinical outcomes of interest.
Disclosures: Support for the study came in part from the National Human Genome Research Institute and the National Heart, Lung, and Blood Institute. The authors reported disclosures related to Novartis, Addmedica, and Global Blood Therapeutics, among others.
Source: Naik RP et al. Ann Intern Med. 2018 Oct 30. doi:10.7326/M18-1161.
Improved treatments emerge for hemophilia patients with high-titer inhibitors
For patients with hemophilia and high-titer inhibitors, a new era of treatment has begun with the development of improved variants of traditional bypassing agents and novel, nonfactor-based, prophylactic agents, say authors of a recent review article.

“These new agents may transform the treatment of inhibitor patients and inhibitor-related bleeds, potentially decreasing morbidity and mortality and improving patients’ quality of life,” Amy D. Shapiro, MD, and coauthors wrote in the Journal of Thrombosis and Haemostasis.
Until recently, the only two bypassing agents available were activated prothrombin complex concentrates and recombinant factor VIIa, noted Dr. Shapiro, who is CEO and co-medical director of the Indiana Hemophilia and Thrombosis Center, Indianapolis, and her coauthors.
The first of the novel targeted agents, emicizumab, is a humanized, bispecific monoclonal antibody that was approved for prophylactic use in hemophilia A in the United States in November 2017.
Emicizumab could transform the treatment of patients with inhibitors, but it also requires reconsideration of how to treat breakthrough bleeds and eliminate the underlying inhibitor, the authors said.
In a phase 3 study, this biologic significantly decreased the annualized bleeding rate by 87% versus on-demand bypassing agent therapy, and by 79% versus a prophylactic bypassing agent regimen, they said, noting that 63% of patients had no bleeding events during the study.
Serious adverse events were seen in patients receiving emicizumab prophylaxis, including thrombosis in 2 out of 103 subjects and thrombotic microangiopathy in 3. In all cases, the patients were treating breakthrough bleeds with activated prothrombin complex concentrate, Dr. Shapiro and coauthors wrote.
Other novel agents in clinical development include fitusiran and tissue factor pathway inhibitors, which each target a different natural anticoagulant and could result in new prophylactic options, according to the study coauthors.
“The possibility of multiple therapeutic targets may allow for a highly personalized approach to prophylaxis therapy, with traditional bypassing agents providing options when breakthrough bleeds occur,” they wrote.
In the meantime, eptacog alfa is the “de facto standard” for recombinant factor VIIa, though a new variant under development, eptacog beta, has been accepted for regulatory review in the United States. In a phase 3 clinical trial, eptacog beta appeared to provide improved efficacy and decreased dosing requirements, possibly due to increased binding affinity to endothelial protein C receptor (EPCR), the authors said.
“The addition of improved rFVIIa variants with unique pharmacological and pharmacokinetic profiles will provide new tools to treat bleeding events in inhibitor patients,” Dr. Shapiro and her colleagues wrote.
The use of traditional bypassing agents is expected to decrease over time as new and improved therapeutics are developed. Traditional agents, however, will “remain a necessity” for breakthrough bleeds in hemophilia A patients with inhibitors, and until novel agents become available for hemophilia B, the authors said.
The review article was supported by an unrestricted educational grant from HEMA Biologics, LLC, which had no involvement or editorial control in research, writing or submission. Dr. Shapiro reported disclosures related to HEMA Biologics, Shire, Novo Nordisk, Kedrion Biopharma, Bioverativ, and Genentech.
SOURCE: Shapiro AD, et al. J Thromb Haemost. 2018 Sep 28.
For patients with hemophilia and high-titer inhibitors, a new era of treatment has begun with the development of improved variants of traditional bypassing agents and novel, nonfactor-based, prophylactic agents, say authors of a recent review article.
“These new agents may transform the treatment of inhibitor patients and inhibitor-related bleeds, potentially decreasing morbidity and mortality and improving patients’ quality of life,” Amy D. Shapiro, MD, and coauthors wrote in the Journal of Thrombosis and Haemostasis.
Until recently, the only two bypassing agents available were activated prothrombin complex concentrates and recombinant factor VIIa, noted Dr. Shapiro, who is CEO and co-medical director of the Indiana Hemophilia and Thrombosis Center, Indianapolis, and her coauthors.
The first of the novel targeted agents, emicizumab, is a humanized, bispecific monoclonal antibody that was approved for prophylactic use in hemophilia A in the United States in November 2017.
Emicizumab could transform the treatment of patients with inhibitors, but it also requires reconsideration of how to treat breakthrough bleeds and eliminate the underlying inhibitor, the authors said.
In a phase 3 study, this biologic significantly decreased the annualized bleeding rate by 87% versus on-demand bypassing agent therapy, and by 79% versus a prophylactic bypassing agent regimen, they said, noting that 63% of patients had no bleeding events during the study.
Serious adverse events were seen in patients receiving emicizumab prophylaxis, including thrombosis in 2 out of 103 subjects and thrombotic microangiopathy in 3. In all cases, the patients were treating breakthrough bleeds with activated prothrombin complex concentrate, Dr. Shapiro and coauthors wrote.
Other novel agents in clinical development include fitusiran and tissue factor pathway inhibitors, which each target a different natural anticoagulant and could result in new prophylactic options, according to the study coauthors.
“The possibility of multiple therapeutic targets may allow for a highly personalized approach to prophylaxis therapy, with traditional bypassing agents providing options when breakthrough bleeds occur,” they wrote.
In the meantime, eptacog alfa is the “de facto standard” for recombinant factor VIIa, though a new variant under development, eptacog beta, has been accepted for regulatory review in the United States. In a phase 3 clinical trial, eptacog beta appeared to provide improved efficacy and decreased dosing requirements, possibly due to increased binding affinity to endothelial protein C receptor (EPCR), the authors said.
“The addition of improved rFVIIa variants with unique pharmacological and pharmacokinetic profiles will provide new tools to treat bleeding events in inhibitor patients,” Dr. Shapiro and her colleagues wrote.
The use of traditional bypassing agents is expected to decrease over time as new and improved therapeutics are developed. Traditional agents, however, will “remain a necessity” for breakthrough bleeds in hemophilia A patients with inhibitors, and until novel agents become available for hemophilia B, the authors said.
The review article was supported by an unrestricted educational grant from HEMA Biologics, LLC, which had no involvement or editorial control in research, writing or submission. Dr. Shapiro reported disclosures related to HEMA Biologics, Shire, Novo Nordisk, Kedrion Biopharma, Bioverativ, and Genentech.
SOURCE: Shapiro AD, et al. J Thromb Haemost. 2018 Sep 28.
For patients with hemophilia and high-titer inhibitors, a new era of treatment has begun with the development of improved variants of traditional bypassing agents and novel, nonfactor-based, prophylactic agents, say authors of a recent review article.
“These new agents may transform the treatment of inhibitor patients and inhibitor-related bleeds, potentially decreasing morbidity and mortality and improving patients’ quality of life,” Amy D. Shapiro, MD, and coauthors wrote in the Journal of Thrombosis and Haemostasis.
Until recently, the only two bypassing agents available were activated prothrombin complex concentrates and recombinant factor VIIa, noted Dr. Shapiro, who is CEO and co-medical director of the Indiana Hemophilia and Thrombosis Center, Indianapolis, and her coauthors.
The first of the novel targeted agents, emicizumab, is a humanized, bispecific monoclonal antibody that was approved for prophylactic use in hemophilia A in the United States in November 2017.
Emicizumab could transform the treatment of patients with inhibitors, but it also requires reconsideration of how to treat breakthrough bleeds and eliminate the underlying inhibitor, the authors said.
In a phase 3 study, this biologic significantly decreased the annualized bleeding rate by 87% versus on-demand bypassing agent therapy, and by 79% versus a prophylactic bypassing agent regimen, they said, noting that 63% of patients had no bleeding events during the study.
Serious adverse events were seen in patients receiving emicizumab prophylaxis, including thrombosis in 2 out of 103 subjects and thrombotic microangiopathy in 3. In all cases, the patients were treating breakthrough bleeds with activated prothrombin complex concentrate, Dr. Shapiro and coauthors wrote.
Other novel agents in clinical development include fitusiran and tissue factor pathway inhibitors, which each target a different natural anticoagulant and could result in new prophylactic options, according to the study coauthors.
“The possibility of multiple therapeutic targets may allow for a highly personalized approach to prophylaxis therapy, with traditional bypassing agents providing options when breakthrough bleeds occur,” they wrote.
In the meantime, eptacog alfa is the “de facto standard” for recombinant factor VIIa, though a new variant under development, eptacog beta, has been accepted for regulatory review in the United States. In a phase 3 clinical trial, eptacog beta appeared to provide improved efficacy and decreased dosing requirements, possibly due to increased binding affinity to endothelial protein C receptor (EPCR), the authors said.
“The addition of improved rFVIIa variants with unique pharmacological and pharmacokinetic profiles will provide new tools to treat bleeding events in inhibitor patients,” Dr. Shapiro and her colleagues wrote.
The use of traditional bypassing agents is expected to decrease over time as new and improved therapeutics are developed. Traditional agents, however, will “remain a necessity” for breakthrough bleeds in hemophilia A patients with inhibitors, and until novel agents become available for hemophilia B, the authors said.
The review article was supported by an unrestricted educational grant from HEMA Biologics, LLC, which had no involvement or editorial control in research, writing or submission. Dr. Shapiro reported disclosures related to HEMA Biologics, Shire, Novo Nordisk, Kedrion Biopharma, Bioverativ, and Genentech.
SOURCE: Shapiro AD, et al. J Thromb Haemost. 2018 Sep 28.
FROM THE JOURNAL OF THROMBOSIS AND HAEMOSTASIS
Cardiovascular disease risk unchanged in men with hemophilia A
Concerns may be unfounded for risks of earlier-onset cardiovascular disease in men with hemophilia A, according to investigators.
Cardiovascular comorbidities between groups were generally comparable, regardless of hemophilia A status, reported lead author Thomas J. Humphries, MD, of Bayer, and his colleagues.
“To date, there have been conflicting data in the literature regarding the risks of [cardiovascular] comorbidities in patients with hemophilia A, compared with the general population,” the investigators wrote in Advances in Medical Sciences. “Some studies have reported lower mortality from [cardiovascular] diseases and/or decreased atherogenesis in patients with hemophilia … conversely, other reports indicate comparable or higher [cardiovascular] comorbidities in patients with hemophilia, compared with the general population.”
In two previous commercial database reviews conducted by Dr. Humphries and his colleagues, cardiovascular disease appeared to occur more commonly and at a younger age in men with hemophilia A. More concerning, patients aged under 40 years showed elevated incidence of stroke and thrombosis. The authors sought to clarify these findings in the present study.
The retrospective chart review involved 74 men with hemophilia A and 222 men without the condition, matched by study year, payer type, race, and age. Patients presented at any of 31 medical facilities within the Henry Ford Health System in Detroit. Diagnoses were made between Jan. 1, 1995, and Dec. 31, 2014.
For the most part there were no significant differences in cardiovascular disease prevalence between the two cohorts. Rates of hypertension, obesity, coronary artery disease, heart failure, stroke, venous and arterial thrombosis, ventricular arrhythmias, atrial fibrillation, and chronic renal disease were numerically higher in the control group, but those differences were not statistically significant. There were significantly higher prevalence rates for diabetes (P = .0108) and hyperlipidemia (P = .0001) in the control group versus patients with hemophilia A.
The investigators pointed out that meaningful statistical differences using standardized differences were not reached for venous and arterial thrombosis and atrial fibrillation.
“It is worth noting that in the hemophilia A group, hypertension appeared first in the 18- to 29-year age group, as did venous thrombosis,” the investigators wrote, suggesting that monitoring, starting in the late teens, may be warranted.
The investigators also noted multiple study limitations, notably the small sample size, compared with commercial databases that were reviewed in previous studies. Additionally, the severity of disease was unknown for some of the hemophilia A patients and the study only followed patients for 1 year.
“The results of this retrospective chart review did not confirm diffuse statistically significant differences in [cardiovascular] comorbidities and their earlier onset in hemophilia A versus controls,” the investigators concluded.
The study was funded by Bayer. Three of the authors were employed by Bayer when the study was conducted. Other authors reported employment with Xcenda and the Henry Ford Health System and research funding from Xcenda.
SOURCE: Humphries TJ et al. Adv Med Sci. 2018;63(2):329-33.
Concerns may be unfounded for risks of earlier-onset cardiovascular disease in men with hemophilia A, according to investigators.
Cardiovascular comorbidities between groups were generally comparable, regardless of hemophilia A status, reported lead author Thomas J. Humphries, MD, of Bayer, and his colleagues.
“To date, there have been conflicting data in the literature regarding the risks of [cardiovascular] comorbidities in patients with hemophilia A, compared with the general population,” the investigators wrote in Advances in Medical Sciences. “Some studies have reported lower mortality from [cardiovascular] diseases and/or decreased atherogenesis in patients with hemophilia … conversely, other reports indicate comparable or higher [cardiovascular] comorbidities in patients with hemophilia, compared with the general population.”
In two previous commercial database reviews conducted by Dr. Humphries and his colleagues, cardiovascular disease appeared to occur more commonly and at a younger age in men with hemophilia A. More concerning, patients aged under 40 years showed elevated incidence of stroke and thrombosis. The authors sought to clarify these findings in the present study.
The retrospective chart review involved 74 men with hemophilia A and 222 men without the condition, matched by study year, payer type, race, and age. Patients presented at any of 31 medical facilities within the Henry Ford Health System in Detroit. Diagnoses were made between Jan. 1, 1995, and Dec. 31, 2014.
For the most part there were no significant differences in cardiovascular disease prevalence between the two cohorts. Rates of hypertension, obesity, coronary artery disease, heart failure, stroke, venous and arterial thrombosis, ventricular arrhythmias, atrial fibrillation, and chronic renal disease were numerically higher in the control group, but those differences were not statistically significant. There were significantly higher prevalence rates for diabetes (P = .0108) and hyperlipidemia (P = .0001) in the control group versus patients with hemophilia A.
The investigators pointed out that meaningful statistical differences using standardized differences were not reached for venous and arterial thrombosis and atrial fibrillation.
“It is worth noting that in the hemophilia A group, hypertension appeared first in the 18- to 29-year age group, as did venous thrombosis,” the investigators wrote, suggesting that monitoring, starting in the late teens, may be warranted.
The investigators also noted multiple study limitations, notably the small sample size, compared with commercial databases that were reviewed in previous studies. Additionally, the severity of disease was unknown for some of the hemophilia A patients and the study only followed patients for 1 year.
“The results of this retrospective chart review did not confirm diffuse statistically significant differences in [cardiovascular] comorbidities and their earlier onset in hemophilia A versus controls,” the investigators concluded.
The study was funded by Bayer. Three of the authors were employed by Bayer when the study was conducted. Other authors reported employment with Xcenda and the Henry Ford Health System and research funding from Xcenda.
SOURCE: Humphries TJ et al. Adv Med Sci. 2018;63(2):329-33.
Concerns may be unfounded for risks of earlier-onset cardiovascular disease in men with hemophilia A, according to investigators.
Cardiovascular comorbidities between groups were generally comparable, regardless of hemophilia A status, reported lead author Thomas J. Humphries, MD, of Bayer, and his colleagues.
“To date, there have been conflicting data in the literature regarding the risks of [cardiovascular] comorbidities in patients with hemophilia A, compared with the general population,” the investigators wrote in Advances in Medical Sciences. “Some studies have reported lower mortality from [cardiovascular] diseases and/or decreased atherogenesis in patients with hemophilia … conversely, other reports indicate comparable or higher [cardiovascular] comorbidities in patients with hemophilia, compared with the general population.”
In two previous commercial database reviews conducted by Dr. Humphries and his colleagues, cardiovascular disease appeared to occur more commonly and at a younger age in men with hemophilia A. More concerning, patients aged under 40 years showed elevated incidence of stroke and thrombosis. The authors sought to clarify these findings in the present study.
The retrospective chart review involved 74 men with hemophilia A and 222 men without the condition, matched by study year, payer type, race, and age. Patients presented at any of 31 medical facilities within the Henry Ford Health System in Detroit. Diagnoses were made between Jan. 1, 1995, and Dec. 31, 2014.
For the most part there were no significant differences in cardiovascular disease prevalence between the two cohorts. Rates of hypertension, obesity, coronary artery disease, heart failure, stroke, venous and arterial thrombosis, ventricular arrhythmias, atrial fibrillation, and chronic renal disease were numerically higher in the control group, but those differences were not statistically significant. There were significantly higher prevalence rates for diabetes (P = .0108) and hyperlipidemia (P = .0001) in the control group versus patients with hemophilia A.
The investigators pointed out that meaningful statistical differences using standardized differences were not reached for venous and arterial thrombosis and atrial fibrillation.
“It is worth noting that in the hemophilia A group, hypertension appeared first in the 18- to 29-year age group, as did venous thrombosis,” the investigators wrote, suggesting that monitoring, starting in the late teens, may be warranted.
The investigators also noted multiple study limitations, notably the small sample size, compared with commercial databases that were reviewed in previous studies. Additionally, the severity of disease was unknown for some of the hemophilia A patients and the study only followed patients for 1 year.
“The results of this retrospective chart review did not confirm diffuse statistically significant differences in [cardiovascular] comorbidities and their earlier onset in hemophilia A versus controls,” the investigators concluded.
The study was funded by Bayer. Three of the authors were employed by Bayer when the study was conducted. Other authors reported employment with Xcenda and the Henry Ford Health System and research funding from Xcenda.
SOURCE: Humphries TJ et al. Adv Med Sci. 2018;63(2):329-33.
FROM ADVANCES IN MEDICAL SCIENCES
Key clinical point:
Major finding: Prevalence rates of diabetes (P = .0108) and hyperlipidemia (P = .0001) were higher in the control group, compared with patients with hemophilia A.
Study details: A retrospective chart review involving 74 men with hemophilia A and 222 men without the condition, matched by study year, payer type, race, and age.
Disclosures: The study was funded by Bayer. Three authors were employed by Bayer when the study was conducted. Other authors reported employment by Xcenda and the Henry Ford Health System and research funding from Xcenda.
Source: Humphries TJ et al. Adv Med Sci. 2018;63(2):329-33.
Canada approves Jivi for hemophilia A
Health Canada has approved Jivi® (antihemophilic factor [recombinant, B-domain deleted, PEGylated]) for use in patients with hemophilia A.
Jivi (formerly BAY94-9027) is a DNA-derived, factor VIII concentrate developed by Bayer.
Health Canada has approved Jivi for use as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in previously treated hemophilia A patients age 12 and older.
Jivi is also approved for the control and prevention of episodic bleeding and for perioperative management of bleeding.
The recommended initial dosing for Jivi as prophylaxis is twice weekly, with the ability to dose every 5 days and further adjust dosing based on bleeding episodes.
Health Canada’s approval of Jivi is based on the PROTECT VIII trial. Results from this trial are available in the U.S. prescribing information for Jivi.
PROTECT VIII enrolled previously treated adults and adolescents (ages 12 to 65) with severe hemophilia A.
In part A, researchers evaluated different dosing regimens for Jivi used as prophylaxis and on-demand treatment. An optional extension study was available to patients who completed part A. In part B, Jivi was used for perioperative management of bleeding.
Efficacy
In part A, there were 132 patients in the intent‐to‐treat population—112 in the prophylaxis group and 20 in the on-demand group.
Patients received Jivi for 36 weeks. For the first 10 weeks, patients in the prophylaxis group received twice-weekly dosing at 25 IU/kg.
Patients with more than one bleed during this time went on to receive 30–40 IU/kg twice weekly. Patients with one or fewer bleeds were eligible for randomization to dosing every 5 days (45–60 IU/kg) or every 7 days (60 IU/kg).
The median annualized bleeding rate (ABR) was 4.1 for the patients who were treated twice weekly and were not eligible for randomization (n=13) and 1.9 for patients who were eligible for randomization but continued on twice-weekly treatment (n=11).
The median ABR was 1.9 for patients who were randomized to treatment every 5 days (n=43) and 0.96 for patients who completed prophylaxis with dosing every 7 days (32/43).
The median ABR for patients treated on demand was 24.1.
There were 388 treated bleeds in the on-demand group and 317 treated bleeds in the prophylaxis group. Overall, 73.3% of responses to treatment were considered “excellent” or “good,” 23.3% were considered “moderate,” and 3.3% were considered “poor.”
There were 17 patients who underwent 20 major surgeries in part B or the extension study and 10 patients who underwent minor surgeries in part A. Jivi provided “good” or “excellent” hemostatic control during all surgeries.
Safety
Safety data are available for 148 patients age 12 and older.
Adverse events in these patients included abdominal pain (3%), nausea (5%), vomiting (3%), injection site reactions (1%), pyrexia (5%), hypersensitivity (2%), dizziness (2%), headache (14%), insomnia (3%), cough (7%), erythema (1%), pruritus (1%), rash (2%), and flushing (1%).
A factor VIII inhibitor was reported in one adult patient, but repeat testing did not confirm the report.
One adult with asthma had a clinical hypersensitivity reaction and a transient increase of IgM anti-PEG antibody titer, which was negative upon retesting.
Health Canada has approved Jivi® (antihemophilic factor [recombinant, B-domain deleted, PEGylated]) for use in patients with hemophilia A.
Jivi (formerly BAY94-9027) is a DNA-derived, factor VIII concentrate developed by Bayer.
Health Canada has approved Jivi for use as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in previously treated hemophilia A patients age 12 and older.
Jivi is also approved for the control and prevention of episodic bleeding and for perioperative management of bleeding.
The recommended initial dosing for Jivi as prophylaxis is twice weekly, with the ability to dose every 5 days and further adjust dosing based on bleeding episodes.
Health Canada’s approval of Jivi is based on the PROTECT VIII trial. Results from this trial are available in the U.S. prescribing information for Jivi.
PROTECT VIII enrolled previously treated adults and adolescents (ages 12 to 65) with severe hemophilia A.
In part A, researchers evaluated different dosing regimens for Jivi used as prophylaxis and on-demand treatment. An optional extension study was available to patients who completed part A. In part B, Jivi was used for perioperative management of bleeding.
Efficacy
In part A, there were 132 patients in the intent‐to‐treat population—112 in the prophylaxis group and 20 in the on-demand group.
Patients received Jivi for 36 weeks. For the first 10 weeks, patients in the prophylaxis group received twice-weekly dosing at 25 IU/kg.
Patients with more than one bleed during this time went on to receive 30–40 IU/kg twice weekly. Patients with one or fewer bleeds were eligible for randomization to dosing every 5 days (45–60 IU/kg) or every 7 days (60 IU/kg).
The median annualized bleeding rate (ABR) was 4.1 for the patients who were treated twice weekly and were not eligible for randomization (n=13) and 1.9 for patients who were eligible for randomization but continued on twice-weekly treatment (n=11).
The median ABR was 1.9 for patients who were randomized to treatment every 5 days (n=43) and 0.96 for patients who completed prophylaxis with dosing every 7 days (32/43).
The median ABR for patients treated on demand was 24.1.
There were 388 treated bleeds in the on-demand group and 317 treated bleeds in the prophylaxis group. Overall, 73.3% of responses to treatment were considered “excellent” or “good,” 23.3% were considered “moderate,” and 3.3% were considered “poor.”
There were 17 patients who underwent 20 major surgeries in part B or the extension study and 10 patients who underwent minor surgeries in part A. Jivi provided “good” or “excellent” hemostatic control during all surgeries.
Safety
Safety data are available for 148 patients age 12 and older.
Adverse events in these patients included abdominal pain (3%), nausea (5%), vomiting (3%), injection site reactions (1%), pyrexia (5%), hypersensitivity (2%), dizziness (2%), headache (14%), insomnia (3%), cough (7%), erythema (1%), pruritus (1%), rash (2%), and flushing (1%).
A factor VIII inhibitor was reported in one adult patient, but repeat testing did not confirm the report.
One adult with asthma had a clinical hypersensitivity reaction and a transient increase of IgM anti-PEG antibody titer, which was negative upon retesting.
Health Canada has approved Jivi® (antihemophilic factor [recombinant, B-domain deleted, PEGylated]) for use in patients with hemophilia A.
Jivi (formerly BAY94-9027) is a DNA-derived, factor VIII concentrate developed by Bayer.
Health Canada has approved Jivi for use as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in previously treated hemophilia A patients age 12 and older.
Jivi is also approved for the control and prevention of episodic bleeding and for perioperative management of bleeding.
The recommended initial dosing for Jivi as prophylaxis is twice weekly, with the ability to dose every 5 days and further adjust dosing based on bleeding episodes.
Health Canada’s approval of Jivi is based on the PROTECT VIII trial. Results from this trial are available in the U.S. prescribing information for Jivi.
PROTECT VIII enrolled previously treated adults and adolescents (ages 12 to 65) with severe hemophilia A.
In part A, researchers evaluated different dosing regimens for Jivi used as prophylaxis and on-demand treatment. An optional extension study was available to patients who completed part A. In part B, Jivi was used for perioperative management of bleeding.
Efficacy
In part A, there were 132 patients in the intent‐to‐treat population—112 in the prophylaxis group and 20 in the on-demand group.
Patients received Jivi for 36 weeks. For the first 10 weeks, patients in the prophylaxis group received twice-weekly dosing at 25 IU/kg.
Patients with more than one bleed during this time went on to receive 30–40 IU/kg twice weekly. Patients with one or fewer bleeds were eligible for randomization to dosing every 5 days (45–60 IU/kg) or every 7 days (60 IU/kg).
The median annualized bleeding rate (ABR) was 4.1 for the patients who were treated twice weekly and were not eligible for randomization (n=13) and 1.9 for patients who were eligible for randomization but continued on twice-weekly treatment (n=11).
The median ABR was 1.9 for patients who were randomized to treatment every 5 days (n=43) and 0.96 for patients who completed prophylaxis with dosing every 7 days (32/43).
The median ABR for patients treated on demand was 24.1.
There were 388 treated bleeds in the on-demand group and 317 treated bleeds in the prophylaxis group. Overall, 73.3% of responses to treatment were considered “excellent” or “good,” 23.3% were considered “moderate,” and 3.3% were considered “poor.”
There were 17 patients who underwent 20 major surgeries in part B or the extension study and 10 patients who underwent minor surgeries in part A. Jivi provided “good” or “excellent” hemostatic control during all surgeries.
Safety
Safety data are available for 148 patients age 12 and older.
Adverse events in these patients included abdominal pain (3%), nausea (5%), vomiting (3%), injection site reactions (1%), pyrexia (5%), hypersensitivity (2%), dizziness (2%), headache (14%), insomnia (3%), cough (7%), erythema (1%), pruritus (1%), rash (2%), and flushing (1%).
A factor VIII inhibitor was reported in one adult patient, but repeat testing did not confirm the report.
One adult with asthma had a clinical hypersensitivity reaction and a transient increase of IgM anti-PEG antibody titer, which was negative upon retesting.