User login
Factor V Leiden: How great is the risk of venous thromboembolism?
A 29-year-old white man with no chronic medical problems presents to the emergency department with shortness of breath, left-sided pleuritic chest pain, cough, and hemoptysis. These symptoms began abruptly 1 day ago and have persisted. He also has mild pain and swelling in both calves. He denies having any fever, night sweats, or chills. On further questioning, he reports having taken a long, nonstop driving trip that lasted 8 hours 1 week ago.
His medical history is negative, and he specifically reports no history of deep venous thrombosis or pulmonary embolism. He underwent appendectomy 10 years ago but has had no other operations. He does not take any medications. His family history is noncontributory and is negative for venous thromboembolism. He smokes and uses alcohol occasionally but not illicit drugs.
Examination. He appears to be in considerable distress because of his chest pain. His temperature is 100.4°F (38.0°C), blood pressure 125/70 mm Hg, heart rate 125 beats per minute, respiratory rate 26 breaths per minute, oxygen saturation 92% on room air, and body mass index 19 kg/m2.
Chest examination reveals diminished vesicular breathing in the left base, which is normal to percussion without added sounds. Both calves are swollen and tender to palpation without skin discoloration. The rest of his examination is normal.
Laboratory values:
- White blood cell count 9.3 × 109/L (reference range 4.5–11.0)
- Hemoglobin 15.9 g/dL (14.0–17.5)
- Platelets 205 × 109/L (150–350)
- Sodium 140 mEq/L (136–142)
- Potassium 3.9 mEq/L (3.5–5.0)
- Chloride 108 mEq/L (96–106)
- Bicarbonate 23 mEq/L (21–28)
- Blood urea nitrogen 14 mg/dL (8–23)
- Creatinine 0.9 mg/dL (0.6–1.2)
- Glucose 95 mg/dL (70–110)
- International normalized ratio (INR) 0.90 (0.00–1.2)
- Partial thromboplastin time 27.5 seconds (24.6–31.8)
- Creatine phosphokinase 205 U/L (39–308)
- Troponin T < 0.015 ng/mL (0.01–0.045).
Pulmonary embolism is diagnosed
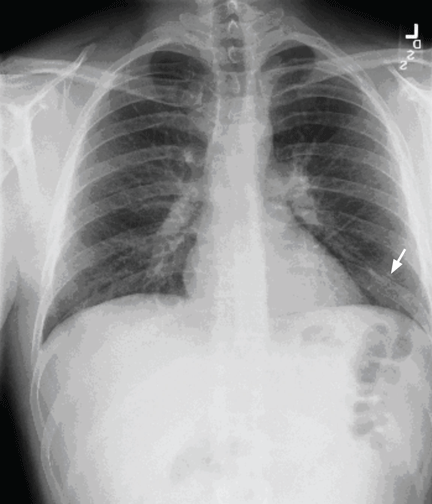
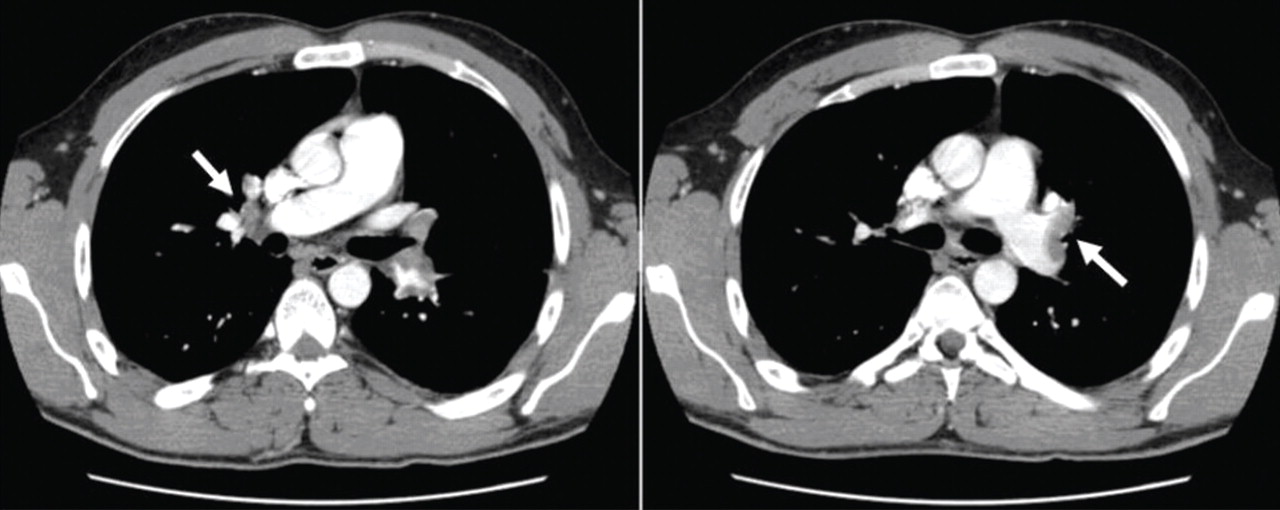
Factor V Leiden is diagnosed, and the patient recovers with treatment
Anticoagulation is started in the emergency department.
Given this patient’s young age and clot burden, a hypercoagulable state is suspected. Thrombophilia screening is performed, with tests for the factor V Leiden mutation, the prothrombin G20210A mutation, and antiphospholipid and lupus anticoagulant antibodies. The rest of the thrombophilia panel, including antithrombin III, factor VIII, protein C, and protein S, is deferred because the levels of these substances would be expected to change during the acute thrombosis.
The direct test for factor V Leiden mutation is positive for the heterozygous type. The test for the prothrombin G20210A mutation is negative, and his antiphospholipid antibody levels, including the lupus anticoagulant titer, are within normal limits.
The patient is kept on a standard regimen of unfractionated heparin, overlapped with warfarin (Coumadin) until his INR is 2.0 to 3.0 on 2 consecutive days. His hospital course is uneventful and his condition gradually improves.
He is discharged home to continue on oral anticoagulation for 6 months with a target INR of 2.0 to 3.0. Two weeks after completing his anticoagulation therapy, his levels of antithrombin III, factor VIII, protein C, and protein S are all within normal limits.
FACTOR V LEIDEN IS COMMON
Factor V Leiden is the most common inherited thrombophilia, with a prevalence of 3% to 7% in the general US population,1 approximately 5% in whites, 2.2% in Hispanics, and 1.2% in blacks.2 Its prevalence in patients with venous thromboembolism, however, is 50%.1,3 The annual incidence of venous thromboembolism in patients with factor V Leiden is 0.5%.4,5
MORE COAGULATION, LESS ANTICOAGULATION
Factor V has a critical position in both the coagulant and anticoagulant pathways. Factor V Leiden results in a hypercoagulable state by both increasing coagulation and decreasing anticoagulation.
This mutation causes factor V to be resistant to being cleaved and inactivated by activated protein C, a condition known as APC resistance. As a result, more factor Va is available within the prothrombinase complex, increasing coagulation by increased generation of thrombin.6–8
Furthermore, a cofactor formed by cleavage of factor V at position 506 is thought to support activated protein C in degrading factor VIIIa (in the tenase complex), along with protein S. People with factor V Leiden lack this cleavage product and thus have less anticoagulant activity from activated protein C. The increased coagulation and decreased anticoagulation appear to contribute equally to the hypercoagulable state in factor V Leiden-associated APC resistance.9–11
Heterozygosity for the factor V Leiden mutation accounts for 90% to 95% of cases of APC resistance. A much smaller number of people are homozygous for it.1
People who are homozygous for factor V Leiden are at higher risk of venous thromboembolism than those who are heterozygous for it, since the latter group’s blood contains both factor V Leiden and normal factor V. The normal factor V allows anticoagulation via the second pathway of inactivation of factor VIIIa by activated protein C, giving some protection against thrombosis. In people who are homozygous for factor V Leiden, the lack of normal factor V acting as an anticoagulant protein results in a higher thrombotic risk.9–11
Other factor V mutations may also cause APC resistance
Although factor V Leiden is the only genetic defect for which a causal relationship with APC resistance has been clearly determined, other, rarer hereditary factor V mutations or polymorphisms have been described, such as factor V Cambridge (Arg306Thr)12 and factor V Hong Kong (Arg306Gly).13 These mutations may result in APC resistance, but their clinical association with thrombosis is less clear.14 Factor V Liverpool (Ile359Thr) is associated with a higher risk of thrombosis, apparently because of reduced APC-mediated inactivation of factor Va and because it is a poor cofactor with activated protein C for the inactivation of factor VIIIa.15
An R2 haplotype has also been described in association with APC resistance.16,17 The phenomenon may be due to a reduction in activated protein C cofactor activity.9 However, not all studies have been convincing regarding the role of this haplotype in clinical disease.18 Coinheritance of this haplotype with factor V Leiden may increase the risk of venous thromboembolism above that associated with factor V Leiden alone.19
Although factor V Leiden is the most common cause of inherited APC resistance, other changes in hemostasis cause acquired APC resistance and may contribute to the thrombotic tendency in these patients.20–22 The most common causes of acquired APC resistance include elevated factor VIII levels,23–25 pregnancy,26–28 use of oral contraceptives,29,30 and antiphospholipid antibodies.31
USUALLY MANIFESTS AS DEEP VEIN THROMBOSIS
Factor V Leiden usually manifests as deep vein thrombosis with or without pulmonary embolism, but thrombosis in unusual locations also occurs.32
The risk of a first episode of venous thromboembolism is two to five times higher with heterozygous factor V Leiden. However, even though the relative risk is high, the absolute risk is low. Furthermore, despite the higher risk of venous thrombosis, there is no evidence that heterozygosity for factor V Leiden increases the overall mortality rate.4,33–36
In people with homozygous factor V Leiden or with combined inherited thrombophilias, the risk of venous thromboembolism is increased to a greater degree: it is 20 to 50 times higher.7,8,37–39 However, whether the risk of death is higher is not clear.
VENOUS THROMBOEMBOLISM IS MULTIFACTORIAL
The pathogenesis of venous thromboembolism is multifactorial and involves an interaction between inherited and acquired factors. Very often, people with factor V Leiden have additional risk factors that contribute to the development of venous clots, and it is very unusual for them to have thrombosis in the absence of these additional factors.
These factors include older age, surgery, obesity, prolonged travel, immobility, hospitalization, oral contraceptive use, hormonal replacement therapy, pregnancy, and malignancy. They increase the risk of venous thrombosis in normal individuals as well, but more so in people with factor V Leiden.40–43
Testing for other known causes of thrombophilia may also be pursued. These include elevated homocysteine levels, the factor II (prothrombin) G20210A mutation, anticardiolipin antibody, lupus anticoagulant, and deficiencies of antithrombin III, protein C, and protein S.
Factor V Leiden by itself does not appear to increase the risk of arterial thrombosis, ie, heart attack and stroke.33,38,44–46
Family history: A risk indicator for venous thrombosis
Family history is an important indicator of risk for a first venous thromboembolic event, regardless of other risk factors identified. The risk of a first event is two to three times higher in people with a family history of thrombosis in a first-degree relative. The risk is four times higher when multiple family members are affected, at least one of them before age 50.47
In people with genetic thrombophilia, the risk of thrombosis (especially unprovoked thrombosis at a young age) is also higher in those with a strong family history than in those without a family history. In those with factor V Leiden, the risk of venous thromboembolism is three to four times higher if there is a positive family history. The risk is five times higher in carriers of factor V Leiden with a family history of venous thromboembolism before age 50, and 13 times higher in those with more than one affected family member.47
Possible shared environmental factors or coinheritance of other unidentified genetic factors may also contribute to the higher susceptibility in thrombosis-prone families.
TESTING FOR APC RESISTANCE AND FACTOR V LEIDEN
The factor V Leiden mutation can be detected directly by genetic testing of peripheral blood mononuclear cells. This method is relatively time-consuming and expensive, however.
At present, the most cost-effective approach is to test first for APC resistance using a second-generation coagulation assay—the modified APC sensitivity test. In this clot-based method, the patient’s sample is prediluted with factor V-deficient plasma to eliminate the effect of lupus anticoagulants and factor deficiencies that could prolong the baseline clotting time, and heparin is inactivated by polybrene. Then either an augmented partial-thromboplastin-time-based assay or a tissue-factor-dependent factor V assay is performed.
This test is nearly 100% sensitive and specific for factor V Leiden, in contrast to the first-generation, or classic, APC sensitivity test, which lacked specificity and sensitivity for it.9–11,48–60 This modification also permits testing of patients receiving anticoagulants or who have abnormal augmented partial thromboplastin times due to coagulation factor deficiencies.
A positive result on the modified APC sensitivity test should be confirmed by a direct genetic test for the factor V Leiden mutation. An APC resistance assay is unnecessary if a direct genetic test is used initially.
HOW LONG TO GIVE ANTICOAGULATION AFTER VENOUS THROMBOEMBOLISM?
Patients who have had an episode of venous thromboembolism have to be treated with anticoagulants.
In general, the initial management of venous thromboembolism in patients with heritable thrombophilias is no different from that in any other patient with a clot. Anticoagulants such as warfarin are given at a target INR of 2.5 (range 2.0–3.0).32 The duration of treatment is based on the risk factors that resulted in the thrombotic event.
After a first event, some authorities recommend anticoagulant therapy for 6 months.32 A shorter period (3 months) is recommended if there is a transient risk factor (eg, surgery, oral contraceptive use, travel, pregnancy, the puerperium) and the thrombosis is confined to distal veins (eg, the calf veins).32
Factor V Leiden does not necessarily increase the risk of recurrent events in patients who have a transient risk factor. Therefore, people who are heterozygous for this mutation do not usually need to be treated lifelong with anticoagulants if they have had only one episode of deep vein thrombosis or pulmonary embolism, given the risk of bleeding associated with anticoagulation, unless they have additional risk factors.
Conditions in which indefinite anticoagulation may be required after careful consideration of the risks and benefits are:
- Life-threatening events such as near-fatal pulmonary embolism
- Cerebral or visceral vein thrombosis
- Recurrent thrombotic events
- Additional persistent risk factors (eg, active malignant neoplasm, extremity paresis, and antiphospholipid antibodies)
- Combined thrombophilias (eg, combined heterozygosity for factor V Leiden and the prothrombin G20210A mutation)
- Homozygosity for factor V Leiden.32,46,48
Factor V Leiden by itself or combined with other thrombophilic abnormalities is not associated with a higher risk of recurrent venous thromboembolism during warfarin therapy (a possible exception is the combination of factor V Leiden plus antiphospholipid antibodies).32,34 Furthermore, current evidence suggests that no thrombophilic defect is a clinically important risk factor for recurrent venous thromboembolism after anticoagulant therapy is stopped. All these facts indicate that clinical factors are probably more important than laboratory abnormalities in determining the duration of anticoagulation therapy.32,35,36,61–63
PRIMARY PROPHYLAXIS IN PATIENTS WITH FACTOR V LEIDEN
Factor V Leiden is only one of many risk factors for deep vein thrombosis or pulmonary embolism. If carriers of factor V Leiden have never had a blood clot, then they are not routinely treated with an anticoagulant. Rather, they should be counseled about reducing or eliminating other factors that may add to their risk of developing a clot in the future.
Usually, the effect of risk factors is additive: the more risk factors present, the higher the risk.46,50 Sometimes, however, the effect of multiple risk factors is more than additive.
Some risk factors, such as genetics or age, are not alterable, but many can be controlled by medications or lifestyle modifications. Therefore, general measures and precautions are recommended to minimize the risk of thrombosis. For example:
Losing weight (if the patient is overweight) is an important intervention for risk reduction, since obesity is probably the most common modifiable risk factor for developing blood clots.
Avoiding long periods of immobility is recommended. For example, if the patient is taking a long car ride (more than 2 hours), then stopping every few hours and walking around for a few minutes is a good way to keep the blood circulating. If the patient has a desk job, getting up and walking around the office periodically is advised. On long airplane trips, a walk in the aisle every so often and preventing dehydration by drinking plenty of fluids and avoiding alcohol are recommended.
Wearing elastic stockings with a graduated elastic pressure may prevent deep venous thrombosis from developing on long flights.63–65
Staying active and getting regular exercise through such activities as walking, bicycling, or swimming are helpful.
Avoiding smoking is critical.50,63
Thromboprophylaxis is recommended for most acutely ill hospitalized patients, especially those confined to bed with additional risk factors. Guidelines for prophylaxis are based on an individualized risk assessment and not on thrombophilia status. Prophylactic anticoagulation is routinely recommended for patients undergoing major high-risk surgery, such as an orthopedic, urologic, gynecologic, or bariatric procedure. Any excess thrombotic risk conferred by thrombophilia is likely small compared with the risk of surgery, and recommendations on the duration and intensity of thromboprophylaxis are not based on thrombophilic status.46,48
Education. Pain, swelling, redness of a limb, unexplained shortness of breath, and chest pain are the most common symptoms of deep vein thrombosis and pulmonary embolism.46,50 It is crucial to teach patients with factor V Leiden to recognize these symptoms and to seek early medical attention in case they experience any of them.
SCREENING FAMILY MEMBERS FOR THE FACTOR V LEIDEN MUTATION
Factor V Leiden by itself is a relatively mild thrombophilic defect that does not cause thrombosis in all carriers, and there is no evidence that early diagnosis reduces rates of morbidity or mortality. Therefore, routine screening of all asymptomatic relatives of affected patients with venous thrombosis is not recommended. Rather, the decision to screen should be made on an individual basis.50,66
Screening may be beneficial in selected cases, especially when patients have a strong family history of recurrent venous thrombosis at a young age (younger than 50 years) and the family member has additional risk factors for venous thromboembolism such as oral contraception or is planning for pregnancy.32,48,49,66
- Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. Lancet 1995; 346:1133–1134.
- Ridker PM, Miletich JP, Hennekens CH, Buring JE. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA 1997; 277:1305–1307.
- Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995; 85:1504–1508.
- Stolz E, Kemkes-Matthes B, Pötzsch B, et al. Screening for thrombophilic risk factors among 25 German patients with cerebral venous thrombosis. Acta Neurol Scand 2000; 102:31–36.
- Langlois NJ, Wells PS. Risk of venous thromboembolism in relatives of symptomatic probands with thrombophilia: a systematic review. Thromb Haemost 2003; 90:17–26.
- Juul K, Tybjaerg-Hansen A, Mortensen J, Lange P, Vestbo J, Nordestgaard BG. Factor V leiden homozygosity, dyspnea, and reduced pulmonary function. Arch Intern Med 2005; 165:2032–2036.
- Bertina RM, Koeleman BP, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369:64–67.
- Dahlbäck B. New molecular insights into the genetics of thrombophilia. Resistance to activated protein C caused by Arg506 to Gln mutation in factor V as a pathogenic risk factor for venous thrombosis. Thromb Haemost 1995; 74:139–148.
- Castoldi E, Brugge JM, Nicolaes GA, Girelli D, Tans G, Rosing J. Impaired APC cofactor activity of factor V plays a major role in the APC resistance associated with the factor V Leiden (R506Q) and R2 (H1299R) mutations. Blood 2004; 103:4173–4179.
- Dahlback B. Anticoagulant factor V and thrombosis risk (editorial). Blood 2004; 103:3995.
- Simioni P, Castoldi E, Lunghi B, Tormene D, Rosing J, Bernardi F. An underestimated combination of opposites resulting in enhanced thrombotic tendency. Blood 2005; 106:2363–2365.
- Williamson D, Brown K, Luddington R, Baglin C, Baglin T. Factor V Cambridge: a new mutation (Arg306-->Thr) associated with resistance to activated protein C. Blood 1998; 91:1140–1144.
- Chan WP, Lee CK, Kwong YL, Lam CK, Liang R. A novel mutation of Arg306 of factor V gene in Hong Kong Chinese. Blood 1998; 91:1135–1139.
- Liang R, Lee CK, Wat MS, Kwong YL, Lam CK, Liu HW. Clinical significance of Arg306 mutations of factor V gene. Blood 1998; 92:2599–2600.
- Steen M, Norstrøm EA, Tholander AL, et al. Functional characterization of factor V-Ile359Thr: a novel mutation associated with thrombosis. Blood 2004; 103:3381–3387.
- Bernardi F, Faioni EM, Castoldi E, et al. A factor V genetic component differing from factor V R506Q contributes to the activated protein C resistance phenotype. Blood 1997; 90:1552–1557.
- Lunghi B, Castoldi E, Mingozzi F, Bernardi F. A new factor V gene polymorphism (His 1254 Arg) present in subjects of African origin mimics the R2 polymorphism (His 1299 Arg). Blood 1998; 91:364–365.
- Luddington R, Jackson A, Pannerselvam S, Brown K, Baglin T. The factor V R2 allele: risk of venous thromboembolism, factor V levels and resistance to activated protein C. Thromb Haemost 2000; 83:204–208.
- Faioni EM, Franchi F, Bucciarelli P, et al. Coinheritance of the HR2 haplotype in the factor V gene confers an increased risk of venous thromboembolism to carriers of factor V R506Q (factor V Leiden). Blood 1999; 94:3062–3066.
- Clark P, Walker ID. The phenomenon known as acquired activated protein C resistance. Br J Haematol 2001; 115:767–773.
- Tosetto A, Simioni M, Madeo D, Rodeghiero F. Intraindividual consistency of the activated protein C resistance phenotype. Br J Haematol 2004; 126:405–409.
- de Visser MC, Rosendaal FR, Bertina RM. A reduced sensitivity for activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood 1999; 93:1271–1276.
- Kraaijenhagen RA, in’t Anker PS, Koopman MM, et al. High plasma concentration of factor VIIIc is a major risk factor for venous thromboembolism. Thromb Haemost 2000; 83:5–9.
- Kamphuisen PW, Eikenboom JC, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol 2001; 21:731–738.
- Koster T, Blann AD, Briët E, Vandenbroucke JP, Rosendaal FR. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995; 345:152–155.
- Clark P, Brennand J, Conkie JA, McCall F, Greer IA, Walker ID. Activated protein C sensitivity, protein C, protein S and coagulation in normal pregnancy. Thromb Haemost 1998; 79:1166–1170.
- Cumming AM, Tait RC, Fildes S, Yoong A, Keeney S, Hay CR. Development of resistance to activated protein C during pregnancy. Br J Haematol 1995; 90:725–727.
- Mathonnet F, de Mazancourt P, Bastenaire B, et al. Activated protein C sensitivity ratio in pregnant women at delivery. Br J Haematol 1996; 92:244–246.
- Post MS, Rosing J, Van Der Mooren MJ, et al; Ageing Women’ and the Institute for Cardiovascular Research-Vrije Universiteit (ICaRVU). Increased resistance to activated protein C after short-term oral hormone replacement therapy in healthy post-menopausal women. Br J Haematol 2002; 119:1017–1023.
- Olivieri O, Friso S, Manzato F, et al. Resistance to activated protein C in healthy women taking oral contraceptives. Br J Haematol 1995; 91:465–470.
- Bokarewa MI, Blombäck M, Egberg N, Rosén S. A new variant of interaction between phospholipid antibodies and the protein C system. Blood Coagul Fibrinolysis 1994; 5:37–41.
- Baglin T, Gray E, Greaves M, et al; British Committee for Standards in Haematology. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149:209–220.
- van Stralen KJ, Doggen CJ, Bezemer ID, Pomp ER, Lisman T, Rosendaal FR. Mechanisms of the factor V Leiden paradox. Arterioscler Thromb Vasc Biol 2008; 28:1872–1877.
- Agaoglu N, Mustafa NA, Turkyilmaz S. Prothrombotic disorders in patients with mesenteric vein thrombosis. J Invest Surg 2003; 16:299–304.
- El-Karaksy H, El-Koofy N, El-Hawary M, et al. Prevalence of factor V Leiden mutation and other hereditary thrombophilic factors in Egyptian children with portal vein thrombosis: results of a single-center case-control study. Ann Hematol 2004; 83:712–715.
- Heijmans BT, Westendorp RG, Knook DL, Kluft C, Slagboom PE. The risk of mortality and the factor V Leiden mutation in a population-based cohort. Thromb Haemost 1998; 80:607–609.
- Turkstra F, Karemaker R, Kuijer PM, Prins MH, Büller HR. Is the prevalence of the factor V Leiden mutation in patients with pulmonary embolism and deep vein thrombosis really different? Thromb Haemost 1999; 81:345–348.
- Ridker PM, Hennekens CH, Lindpaintner K, Stampfer MJ, Eisenberg PR, Miletich JP. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995; 332:912–917.
- Manten B, Westendorp RG, Koster T, Reitsma PH, Rosendaal FR. Risk factor profiles in patients with different clinical manifestations of venous thromboembolism: a focus on the factor V Leiden mutation. Thromb Haemost 1996; 76:510–513.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Bloemenkamp KW, Rosendaal FR, Helmerhorst FM, Büller HR, Vandenbroucke JP. Enhancement by factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing a third-generation progestagen. Lancet 1995; 346:1593–1596.
- Murphy PT. Factor V Leiden and venous thromboembolism. Ann Intern Med 2004; 141:483–484.
- Nizankowska-Mogilnicka E, Adamek L, Grzanka P, et al. Genetic polymorphisms associated with acute pulmonary embolism and deep venous thrombosis. Eur Respir J 2003; 21:25–30.
- Arsov T, Miladinova D, Spiroski M. Factor V Leiden is associated with higher risk of deep venous thrombosis of large blood vessels. Croat Med J 2006; 47:433–439.
- Simioni P, Prandoni P, Lensing AW, et al. Risk for subsequent venous thromboembolic complications in carriers of the prothrombin or the factor V gene mutation with a first episode of deep-vein thrombosis. Blood 2000; 96:3329–3333.
- Ornstein DL, Cushman M. Cardiology patient page. Factor V Leiden. Circulation 2003; 107:e94–e97.
- Bezemer ID, van der Meer FJ, Eikenboom JC, Rosendaal FR, Doggen CJ. The value of family history as a risk indicator for venous thrombosis. Arch Intern Med 2009; 169:610–615.
- Press RD, Bauer KA, Kujovich JL, Heit JA. Clinical utility of factor V leiden (R506Q) testing for the diagnosis and management of thromboembolic disorders. Arch Pathol Lab Med 2002; 126:1304–1318.
- Gadelha T, Roldán V, Lecumberri R, et al; RIETE Investigators. Clinical characteristics of patients with factor V Leiden or prothrombin G20210A and a first episode of venous thromboembolism. Findings from the RIETE Registry. Thromb Res 2010; 126:283–286.
- Severinsen MT, Overvad K, Johnsen SP, et al. Genetic susceptibility, smoking, obesity and risk of venous thromboembolism. Br J Haematol 2010; 149:273–279.
- Kujovich JL. Factor V Leiden thrombophilia. Genet Med 2011; 13:1–16.
- Lijfering WM, Brouwer JL, Veeger NJ, et al. Selective testing for thrombophilia in patients with first venous thrombosis: results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood 2009; 113:5314–5322.
- Kearon C, Julian JA, Kovacs MJ, et al; ELATE Investigators. Influence of thrombophilia on risk of recurrent venous thromboembolism while on warfarin: results from a randomized trial. Blood 2008; 112:4432–4436.
- Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med 2006; 166:729–736.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Strobl FJ, Hoffman S, Huber S, Williams EC, Voelkerding KV. Activated protein C resistance assay performance: improvement by sample dilution with factor V-deficient plasma. Arch Pathol Lab Med 1998; 122:430–433.
- Legnani C, Palareti G, Biagi R, et al. Activated protein C resistance: a comparison between two clotting assays and their relationship to the presence of the factor V Leiden mutation. Br J Haematol 1996; 93:694–699.
- Gouault-Heilmann M, Leroy-Matheron C. Factor V Leiden-dependent APC resistance: improved sensitivity and specificity of the APC resistance test by plasma dilution in factor V-depleted plasma. Thromb Res 1996; 82:281–283.
- Svensson PJ, Zöller B, Dahlbäck B. Evaluation of original and modified APC-resistance tests in unselected outpatients with clinically suspected thrombosis and in healthy controls. Thromb Haemost 1997; 77:332–335.
- Tripodi A, Negri B, Bertina RM, Mannucci PM. Screening for the FV:Q506 mutation—evaluation of thirteen plasma-based methods for their diagnostic efficacy in comparison with DNA analysis. Thromb Haemost 1997; 77:436–439.
- Wåhlander K, Larson G, Lindahl TL, et al. Factor V Leiden (G1691A) and prothrombin gene G20210A mutations as potential risk factors for venous thromboembolism after total hip or total knee replacement surgery. Thromb Haemost 2002; 87:580–585.
- Joseph JE, Low J, Courtenay B, Neil MJ, McGrath M, Ma D. A single-centre prospective study of clinical and haemostatic risk factors for venous thromboembolism following lower limb arthroplasty. Br J Haematol 2005; 129:87–92.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):381S–453S.
- Brenner B. Prophylaxis for travel-related thrombosis? Yes. J Thromb Haemost 2004; 2:2089–2091.
- Gavish I, Brenner B. Air travel and the risk of thromboembolism. Intern Emerg Med 2011; 6:113–116.
- Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA; ACMG Factor V Leiden Working Group. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med 2001; 3:139–148.
A 29-year-old white man with no chronic medical problems presents to the emergency department with shortness of breath, left-sided pleuritic chest pain, cough, and hemoptysis. These symptoms began abruptly 1 day ago and have persisted. He also has mild pain and swelling in both calves. He denies having any fever, night sweats, or chills. On further questioning, he reports having taken a long, nonstop driving trip that lasted 8 hours 1 week ago.
His medical history is negative, and he specifically reports no history of deep venous thrombosis or pulmonary embolism. He underwent appendectomy 10 years ago but has had no other operations. He does not take any medications. His family history is noncontributory and is negative for venous thromboembolism. He smokes and uses alcohol occasionally but not illicit drugs.
Examination. He appears to be in considerable distress because of his chest pain. His temperature is 100.4°F (38.0°C), blood pressure 125/70 mm Hg, heart rate 125 beats per minute, respiratory rate 26 breaths per minute, oxygen saturation 92% on room air, and body mass index 19 kg/m2.
Chest examination reveals diminished vesicular breathing in the left base, which is normal to percussion without added sounds. Both calves are swollen and tender to palpation without skin discoloration. The rest of his examination is normal.
Laboratory values:
- White blood cell count 9.3 × 109/L (reference range 4.5–11.0)
- Hemoglobin 15.9 g/dL (14.0–17.5)
- Platelets 205 × 109/L (150–350)
- Sodium 140 mEq/L (136–142)
- Potassium 3.9 mEq/L (3.5–5.0)
- Chloride 108 mEq/L (96–106)
- Bicarbonate 23 mEq/L (21–28)
- Blood urea nitrogen 14 mg/dL (8–23)
- Creatinine 0.9 mg/dL (0.6–1.2)
- Glucose 95 mg/dL (70–110)
- International normalized ratio (INR) 0.90 (0.00–1.2)
- Partial thromboplastin time 27.5 seconds (24.6–31.8)
- Creatine phosphokinase 205 U/L (39–308)
- Troponin T < 0.015 ng/mL (0.01–0.045).
Pulmonary embolism is diagnosed
Factor V Leiden is diagnosed, and the patient recovers with treatment
Anticoagulation is started in the emergency department.
Given this patient’s young age and clot burden, a hypercoagulable state is suspected. Thrombophilia screening is performed, with tests for the factor V Leiden mutation, the prothrombin G20210A mutation, and antiphospholipid and lupus anticoagulant antibodies. The rest of the thrombophilia panel, including antithrombin III, factor VIII, protein C, and protein S, is deferred because the levels of these substances would be expected to change during the acute thrombosis.
The direct test for factor V Leiden mutation is positive for the heterozygous type. The test for the prothrombin G20210A mutation is negative, and his antiphospholipid antibody levels, including the lupus anticoagulant titer, are within normal limits.
The patient is kept on a standard regimen of unfractionated heparin, overlapped with warfarin (Coumadin) until his INR is 2.0 to 3.0 on 2 consecutive days. His hospital course is uneventful and his condition gradually improves.
He is discharged home to continue on oral anticoagulation for 6 months with a target INR of 2.0 to 3.0. Two weeks after completing his anticoagulation therapy, his levels of antithrombin III, factor VIII, protein C, and protein S are all within normal limits.
FACTOR V LEIDEN IS COMMON
Factor V Leiden is the most common inherited thrombophilia, with a prevalence of 3% to 7% in the general US population,1 approximately 5% in whites, 2.2% in Hispanics, and 1.2% in blacks.2 Its prevalence in patients with venous thromboembolism, however, is 50%.1,3 The annual incidence of venous thromboembolism in patients with factor V Leiden is 0.5%.4,5
MORE COAGULATION, LESS ANTICOAGULATION
Factor V has a critical position in both the coagulant and anticoagulant pathways. Factor V Leiden results in a hypercoagulable state by both increasing coagulation and decreasing anticoagulation.
This mutation causes factor V to be resistant to being cleaved and inactivated by activated protein C, a condition known as APC resistance. As a result, more factor Va is available within the prothrombinase complex, increasing coagulation by increased generation of thrombin.6–8
Furthermore, a cofactor formed by cleavage of factor V at position 506 is thought to support activated protein C in degrading factor VIIIa (in the tenase complex), along with protein S. People with factor V Leiden lack this cleavage product and thus have less anticoagulant activity from activated protein C. The increased coagulation and decreased anticoagulation appear to contribute equally to the hypercoagulable state in factor V Leiden-associated APC resistance.9–11
Heterozygosity for the factor V Leiden mutation accounts for 90% to 95% of cases of APC resistance. A much smaller number of people are homozygous for it.1
People who are homozygous for factor V Leiden are at higher risk of venous thromboembolism than those who are heterozygous for it, since the latter group’s blood contains both factor V Leiden and normal factor V. The normal factor V allows anticoagulation via the second pathway of inactivation of factor VIIIa by activated protein C, giving some protection against thrombosis. In people who are homozygous for factor V Leiden, the lack of normal factor V acting as an anticoagulant protein results in a higher thrombotic risk.9–11
Other factor V mutations may also cause APC resistance
Although factor V Leiden is the only genetic defect for which a causal relationship with APC resistance has been clearly determined, other, rarer hereditary factor V mutations or polymorphisms have been described, such as factor V Cambridge (Arg306Thr)12 and factor V Hong Kong (Arg306Gly).13 These mutations may result in APC resistance, but their clinical association with thrombosis is less clear.14 Factor V Liverpool (Ile359Thr) is associated with a higher risk of thrombosis, apparently because of reduced APC-mediated inactivation of factor Va and because it is a poor cofactor with activated protein C for the inactivation of factor VIIIa.15
An R2 haplotype has also been described in association with APC resistance.16,17 The phenomenon may be due to a reduction in activated protein C cofactor activity.9 However, not all studies have been convincing regarding the role of this haplotype in clinical disease.18 Coinheritance of this haplotype with factor V Leiden may increase the risk of venous thromboembolism above that associated with factor V Leiden alone.19
Although factor V Leiden is the most common cause of inherited APC resistance, other changes in hemostasis cause acquired APC resistance and may contribute to the thrombotic tendency in these patients.20–22 The most common causes of acquired APC resistance include elevated factor VIII levels,23–25 pregnancy,26–28 use of oral contraceptives,29,30 and antiphospholipid antibodies.31
USUALLY MANIFESTS AS DEEP VEIN THROMBOSIS
Factor V Leiden usually manifests as deep vein thrombosis with or without pulmonary embolism, but thrombosis in unusual locations also occurs.32
The risk of a first episode of venous thromboembolism is two to five times higher with heterozygous factor V Leiden. However, even though the relative risk is high, the absolute risk is low. Furthermore, despite the higher risk of venous thrombosis, there is no evidence that heterozygosity for factor V Leiden increases the overall mortality rate.4,33–36
In people with homozygous factor V Leiden or with combined inherited thrombophilias, the risk of venous thromboembolism is increased to a greater degree: it is 20 to 50 times higher.7,8,37–39 However, whether the risk of death is higher is not clear.
VENOUS THROMBOEMBOLISM IS MULTIFACTORIAL
The pathogenesis of venous thromboembolism is multifactorial and involves an interaction between inherited and acquired factors. Very often, people with factor V Leiden have additional risk factors that contribute to the development of venous clots, and it is very unusual for them to have thrombosis in the absence of these additional factors.
These factors include older age, surgery, obesity, prolonged travel, immobility, hospitalization, oral contraceptive use, hormonal replacement therapy, pregnancy, and malignancy. They increase the risk of venous thrombosis in normal individuals as well, but more so in people with factor V Leiden.40–43
Testing for other known causes of thrombophilia may also be pursued. These include elevated homocysteine levels, the factor II (prothrombin) G20210A mutation, anticardiolipin antibody, lupus anticoagulant, and deficiencies of antithrombin III, protein C, and protein S.
Factor V Leiden by itself does not appear to increase the risk of arterial thrombosis, ie, heart attack and stroke.33,38,44–46
Family history: A risk indicator for venous thrombosis
Family history is an important indicator of risk for a first venous thromboembolic event, regardless of other risk factors identified. The risk of a first event is two to three times higher in people with a family history of thrombosis in a first-degree relative. The risk is four times higher when multiple family members are affected, at least one of them before age 50.47
In people with genetic thrombophilia, the risk of thrombosis (especially unprovoked thrombosis at a young age) is also higher in those with a strong family history than in those without a family history. In those with factor V Leiden, the risk of venous thromboembolism is three to four times higher if there is a positive family history. The risk is five times higher in carriers of factor V Leiden with a family history of venous thromboembolism before age 50, and 13 times higher in those with more than one affected family member.47
Possible shared environmental factors or coinheritance of other unidentified genetic factors may also contribute to the higher susceptibility in thrombosis-prone families.
TESTING FOR APC RESISTANCE AND FACTOR V LEIDEN
The factor V Leiden mutation can be detected directly by genetic testing of peripheral blood mononuclear cells. This method is relatively time-consuming and expensive, however.
At present, the most cost-effective approach is to test first for APC resistance using a second-generation coagulation assay—the modified APC sensitivity test. In this clot-based method, the patient’s sample is prediluted with factor V-deficient plasma to eliminate the effect of lupus anticoagulants and factor deficiencies that could prolong the baseline clotting time, and heparin is inactivated by polybrene. Then either an augmented partial-thromboplastin-time-based assay or a tissue-factor-dependent factor V assay is performed.
This test is nearly 100% sensitive and specific for factor V Leiden, in contrast to the first-generation, or classic, APC sensitivity test, which lacked specificity and sensitivity for it.9–11,48–60 This modification also permits testing of patients receiving anticoagulants or who have abnormal augmented partial thromboplastin times due to coagulation factor deficiencies.
A positive result on the modified APC sensitivity test should be confirmed by a direct genetic test for the factor V Leiden mutation. An APC resistance assay is unnecessary if a direct genetic test is used initially.
HOW LONG TO GIVE ANTICOAGULATION AFTER VENOUS THROMBOEMBOLISM?
Patients who have had an episode of venous thromboembolism have to be treated with anticoagulants.
In general, the initial management of venous thromboembolism in patients with heritable thrombophilias is no different from that in any other patient with a clot. Anticoagulants such as warfarin are given at a target INR of 2.5 (range 2.0–3.0).32 The duration of treatment is based on the risk factors that resulted in the thrombotic event.
After a first event, some authorities recommend anticoagulant therapy for 6 months.32 A shorter period (3 months) is recommended if there is a transient risk factor (eg, surgery, oral contraceptive use, travel, pregnancy, the puerperium) and the thrombosis is confined to distal veins (eg, the calf veins).32
Factor V Leiden does not necessarily increase the risk of recurrent events in patients who have a transient risk factor. Therefore, people who are heterozygous for this mutation do not usually need to be treated lifelong with anticoagulants if they have had only one episode of deep vein thrombosis or pulmonary embolism, given the risk of bleeding associated with anticoagulation, unless they have additional risk factors.
Conditions in which indefinite anticoagulation may be required after careful consideration of the risks and benefits are:
- Life-threatening events such as near-fatal pulmonary embolism
- Cerebral or visceral vein thrombosis
- Recurrent thrombotic events
- Additional persistent risk factors (eg, active malignant neoplasm, extremity paresis, and antiphospholipid antibodies)
- Combined thrombophilias (eg, combined heterozygosity for factor V Leiden and the prothrombin G20210A mutation)
- Homozygosity for factor V Leiden.32,46,48
Factor V Leiden by itself or combined with other thrombophilic abnormalities is not associated with a higher risk of recurrent venous thromboembolism during warfarin therapy (a possible exception is the combination of factor V Leiden plus antiphospholipid antibodies).32,34 Furthermore, current evidence suggests that no thrombophilic defect is a clinically important risk factor for recurrent venous thromboembolism after anticoagulant therapy is stopped. All these facts indicate that clinical factors are probably more important than laboratory abnormalities in determining the duration of anticoagulation therapy.32,35,36,61–63
PRIMARY PROPHYLAXIS IN PATIENTS WITH FACTOR V LEIDEN
Factor V Leiden is only one of many risk factors for deep vein thrombosis or pulmonary embolism. If carriers of factor V Leiden have never had a blood clot, then they are not routinely treated with an anticoagulant. Rather, they should be counseled about reducing or eliminating other factors that may add to their risk of developing a clot in the future.
Usually, the effect of risk factors is additive: the more risk factors present, the higher the risk.46,50 Sometimes, however, the effect of multiple risk factors is more than additive.
Some risk factors, such as genetics or age, are not alterable, but many can be controlled by medications or lifestyle modifications. Therefore, general measures and precautions are recommended to minimize the risk of thrombosis. For example:
Losing weight (if the patient is overweight) is an important intervention for risk reduction, since obesity is probably the most common modifiable risk factor for developing blood clots.
Avoiding long periods of immobility is recommended. For example, if the patient is taking a long car ride (more than 2 hours), then stopping every few hours and walking around for a few minutes is a good way to keep the blood circulating. If the patient has a desk job, getting up and walking around the office periodically is advised. On long airplane trips, a walk in the aisle every so often and preventing dehydration by drinking plenty of fluids and avoiding alcohol are recommended.
Wearing elastic stockings with a graduated elastic pressure may prevent deep venous thrombosis from developing on long flights.63–65
Staying active and getting regular exercise through such activities as walking, bicycling, or swimming are helpful.
Avoiding smoking is critical.50,63
Thromboprophylaxis is recommended for most acutely ill hospitalized patients, especially those confined to bed with additional risk factors. Guidelines for prophylaxis are based on an individualized risk assessment and not on thrombophilia status. Prophylactic anticoagulation is routinely recommended for patients undergoing major high-risk surgery, such as an orthopedic, urologic, gynecologic, or bariatric procedure. Any excess thrombotic risk conferred by thrombophilia is likely small compared with the risk of surgery, and recommendations on the duration and intensity of thromboprophylaxis are not based on thrombophilic status.46,48
Education. Pain, swelling, redness of a limb, unexplained shortness of breath, and chest pain are the most common symptoms of deep vein thrombosis and pulmonary embolism.46,50 It is crucial to teach patients with factor V Leiden to recognize these symptoms and to seek early medical attention in case they experience any of them.
SCREENING FAMILY MEMBERS FOR THE FACTOR V LEIDEN MUTATION
Factor V Leiden by itself is a relatively mild thrombophilic defect that does not cause thrombosis in all carriers, and there is no evidence that early diagnosis reduces rates of morbidity or mortality. Therefore, routine screening of all asymptomatic relatives of affected patients with venous thrombosis is not recommended. Rather, the decision to screen should be made on an individual basis.50,66
Screening may be beneficial in selected cases, especially when patients have a strong family history of recurrent venous thrombosis at a young age (younger than 50 years) and the family member has additional risk factors for venous thromboembolism such as oral contraception or is planning for pregnancy.32,48,49,66
A 29-year-old white man with no chronic medical problems presents to the emergency department with shortness of breath, left-sided pleuritic chest pain, cough, and hemoptysis. These symptoms began abruptly 1 day ago and have persisted. He also has mild pain and swelling in both calves. He denies having any fever, night sweats, or chills. On further questioning, he reports having taken a long, nonstop driving trip that lasted 8 hours 1 week ago.
His medical history is negative, and he specifically reports no history of deep venous thrombosis or pulmonary embolism. He underwent appendectomy 10 years ago but has had no other operations. He does not take any medications. His family history is noncontributory and is negative for venous thromboembolism. He smokes and uses alcohol occasionally but not illicit drugs.
Examination. He appears to be in considerable distress because of his chest pain. His temperature is 100.4°F (38.0°C), blood pressure 125/70 mm Hg, heart rate 125 beats per minute, respiratory rate 26 breaths per minute, oxygen saturation 92% on room air, and body mass index 19 kg/m2.
Chest examination reveals diminished vesicular breathing in the left base, which is normal to percussion without added sounds. Both calves are swollen and tender to palpation without skin discoloration. The rest of his examination is normal.
Laboratory values:
- White blood cell count 9.3 × 109/L (reference range 4.5–11.0)
- Hemoglobin 15.9 g/dL (14.0–17.5)
- Platelets 205 × 109/L (150–350)
- Sodium 140 mEq/L (136–142)
- Potassium 3.9 mEq/L (3.5–5.0)
- Chloride 108 mEq/L (96–106)
- Bicarbonate 23 mEq/L (21–28)
- Blood urea nitrogen 14 mg/dL (8–23)
- Creatinine 0.9 mg/dL (0.6–1.2)
- Glucose 95 mg/dL (70–110)
- International normalized ratio (INR) 0.90 (0.00–1.2)
- Partial thromboplastin time 27.5 seconds (24.6–31.8)
- Creatine phosphokinase 205 U/L (39–308)
- Troponin T < 0.015 ng/mL (0.01–0.045).
Pulmonary embolism is diagnosed
Factor V Leiden is diagnosed, and the patient recovers with treatment
Anticoagulation is started in the emergency department.
Given this patient’s young age and clot burden, a hypercoagulable state is suspected. Thrombophilia screening is performed, with tests for the factor V Leiden mutation, the prothrombin G20210A mutation, and antiphospholipid and lupus anticoagulant antibodies. The rest of the thrombophilia panel, including antithrombin III, factor VIII, protein C, and protein S, is deferred because the levels of these substances would be expected to change during the acute thrombosis.
The direct test for factor V Leiden mutation is positive for the heterozygous type. The test for the prothrombin G20210A mutation is negative, and his antiphospholipid antibody levels, including the lupus anticoagulant titer, are within normal limits.
The patient is kept on a standard regimen of unfractionated heparin, overlapped with warfarin (Coumadin) until his INR is 2.0 to 3.0 on 2 consecutive days. His hospital course is uneventful and his condition gradually improves.
He is discharged home to continue on oral anticoagulation for 6 months with a target INR of 2.0 to 3.0. Two weeks after completing his anticoagulation therapy, his levels of antithrombin III, factor VIII, protein C, and protein S are all within normal limits.
FACTOR V LEIDEN IS COMMON
Factor V Leiden is the most common inherited thrombophilia, with a prevalence of 3% to 7% in the general US population,1 approximately 5% in whites, 2.2% in Hispanics, and 1.2% in blacks.2 Its prevalence in patients with venous thromboembolism, however, is 50%.1,3 The annual incidence of venous thromboembolism in patients with factor V Leiden is 0.5%.4,5
MORE COAGULATION, LESS ANTICOAGULATION
Factor V has a critical position in both the coagulant and anticoagulant pathways. Factor V Leiden results in a hypercoagulable state by both increasing coagulation and decreasing anticoagulation.
This mutation causes factor V to be resistant to being cleaved and inactivated by activated protein C, a condition known as APC resistance. As a result, more factor Va is available within the prothrombinase complex, increasing coagulation by increased generation of thrombin.6–8
Furthermore, a cofactor formed by cleavage of factor V at position 506 is thought to support activated protein C in degrading factor VIIIa (in the tenase complex), along with protein S. People with factor V Leiden lack this cleavage product and thus have less anticoagulant activity from activated protein C. The increased coagulation and decreased anticoagulation appear to contribute equally to the hypercoagulable state in factor V Leiden-associated APC resistance.9–11
Heterozygosity for the factor V Leiden mutation accounts for 90% to 95% of cases of APC resistance. A much smaller number of people are homozygous for it.1
People who are homozygous for factor V Leiden are at higher risk of venous thromboembolism than those who are heterozygous for it, since the latter group’s blood contains both factor V Leiden and normal factor V. The normal factor V allows anticoagulation via the second pathway of inactivation of factor VIIIa by activated protein C, giving some protection against thrombosis. In people who are homozygous for factor V Leiden, the lack of normal factor V acting as an anticoagulant protein results in a higher thrombotic risk.9–11
Other factor V mutations may also cause APC resistance
Although factor V Leiden is the only genetic defect for which a causal relationship with APC resistance has been clearly determined, other, rarer hereditary factor V mutations or polymorphisms have been described, such as factor V Cambridge (Arg306Thr)12 and factor V Hong Kong (Arg306Gly).13 These mutations may result in APC resistance, but their clinical association with thrombosis is less clear.14 Factor V Liverpool (Ile359Thr) is associated with a higher risk of thrombosis, apparently because of reduced APC-mediated inactivation of factor Va and because it is a poor cofactor with activated protein C for the inactivation of factor VIIIa.15
An R2 haplotype has also been described in association with APC resistance.16,17 The phenomenon may be due to a reduction in activated protein C cofactor activity.9 However, not all studies have been convincing regarding the role of this haplotype in clinical disease.18 Coinheritance of this haplotype with factor V Leiden may increase the risk of venous thromboembolism above that associated with factor V Leiden alone.19
Although factor V Leiden is the most common cause of inherited APC resistance, other changes in hemostasis cause acquired APC resistance and may contribute to the thrombotic tendency in these patients.20–22 The most common causes of acquired APC resistance include elevated factor VIII levels,23–25 pregnancy,26–28 use of oral contraceptives,29,30 and antiphospholipid antibodies.31
USUALLY MANIFESTS AS DEEP VEIN THROMBOSIS
Factor V Leiden usually manifests as deep vein thrombosis with or without pulmonary embolism, but thrombosis in unusual locations also occurs.32
The risk of a first episode of venous thromboembolism is two to five times higher with heterozygous factor V Leiden. However, even though the relative risk is high, the absolute risk is low. Furthermore, despite the higher risk of venous thrombosis, there is no evidence that heterozygosity for factor V Leiden increases the overall mortality rate.4,33–36
In people with homozygous factor V Leiden or with combined inherited thrombophilias, the risk of venous thromboembolism is increased to a greater degree: it is 20 to 50 times higher.7,8,37–39 However, whether the risk of death is higher is not clear.
VENOUS THROMBOEMBOLISM IS MULTIFACTORIAL
The pathogenesis of venous thromboembolism is multifactorial and involves an interaction between inherited and acquired factors. Very often, people with factor V Leiden have additional risk factors that contribute to the development of venous clots, and it is very unusual for them to have thrombosis in the absence of these additional factors.
These factors include older age, surgery, obesity, prolonged travel, immobility, hospitalization, oral contraceptive use, hormonal replacement therapy, pregnancy, and malignancy. They increase the risk of venous thrombosis in normal individuals as well, but more so in people with factor V Leiden.40–43
Testing for other known causes of thrombophilia may also be pursued. These include elevated homocysteine levels, the factor II (prothrombin) G20210A mutation, anticardiolipin antibody, lupus anticoagulant, and deficiencies of antithrombin III, protein C, and protein S.
Factor V Leiden by itself does not appear to increase the risk of arterial thrombosis, ie, heart attack and stroke.33,38,44–46
Family history: A risk indicator for venous thrombosis
Family history is an important indicator of risk for a first venous thromboembolic event, regardless of other risk factors identified. The risk of a first event is two to three times higher in people with a family history of thrombosis in a first-degree relative. The risk is four times higher when multiple family members are affected, at least one of them before age 50.47
In people with genetic thrombophilia, the risk of thrombosis (especially unprovoked thrombosis at a young age) is also higher in those with a strong family history than in those without a family history. In those with factor V Leiden, the risk of venous thromboembolism is three to four times higher if there is a positive family history. The risk is five times higher in carriers of factor V Leiden with a family history of venous thromboembolism before age 50, and 13 times higher in those with more than one affected family member.47
Possible shared environmental factors or coinheritance of other unidentified genetic factors may also contribute to the higher susceptibility in thrombosis-prone families.
TESTING FOR APC RESISTANCE AND FACTOR V LEIDEN
The factor V Leiden mutation can be detected directly by genetic testing of peripheral blood mononuclear cells. This method is relatively time-consuming and expensive, however.
At present, the most cost-effective approach is to test first for APC resistance using a second-generation coagulation assay—the modified APC sensitivity test. In this clot-based method, the patient’s sample is prediluted with factor V-deficient plasma to eliminate the effect of lupus anticoagulants and factor deficiencies that could prolong the baseline clotting time, and heparin is inactivated by polybrene. Then either an augmented partial-thromboplastin-time-based assay or a tissue-factor-dependent factor V assay is performed.
This test is nearly 100% sensitive and specific for factor V Leiden, in contrast to the first-generation, or classic, APC sensitivity test, which lacked specificity and sensitivity for it.9–11,48–60 This modification also permits testing of patients receiving anticoagulants or who have abnormal augmented partial thromboplastin times due to coagulation factor deficiencies.
A positive result on the modified APC sensitivity test should be confirmed by a direct genetic test for the factor V Leiden mutation. An APC resistance assay is unnecessary if a direct genetic test is used initially.
HOW LONG TO GIVE ANTICOAGULATION AFTER VENOUS THROMBOEMBOLISM?
Patients who have had an episode of venous thromboembolism have to be treated with anticoagulants.
In general, the initial management of venous thromboembolism in patients with heritable thrombophilias is no different from that in any other patient with a clot. Anticoagulants such as warfarin are given at a target INR of 2.5 (range 2.0–3.0).32 The duration of treatment is based on the risk factors that resulted in the thrombotic event.
After a first event, some authorities recommend anticoagulant therapy for 6 months.32 A shorter period (3 months) is recommended if there is a transient risk factor (eg, surgery, oral contraceptive use, travel, pregnancy, the puerperium) and the thrombosis is confined to distal veins (eg, the calf veins).32
Factor V Leiden does not necessarily increase the risk of recurrent events in patients who have a transient risk factor. Therefore, people who are heterozygous for this mutation do not usually need to be treated lifelong with anticoagulants if they have had only one episode of deep vein thrombosis or pulmonary embolism, given the risk of bleeding associated with anticoagulation, unless they have additional risk factors.
Conditions in which indefinite anticoagulation may be required after careful consideration of the risks and benefits are:
- Life-threatening events such as near-fatal pulmonary embolism
- Cerebral or visceral vein thrombosis
- Recurrent thrombotic events
- Additional persistent risk factors (eg, active malignant neoplasm, extremity paresis, and antiphospholipid antibodies)
- Combined thrombophilias (eg, combined heterozygosity for factor V Leiden and the prothrombin G20210A mutation)
- Homozygosity for factor V Leiden.32,46,48
Factor V Leiden by itself or combined with other thrombophilic abnormalities is not associated with a higher risk of recurrent venous thromboembolism during warfarin therapy (a possible exception is the combination of factor V Leiden plus antiphospholipid antibodies).32,34 Furthermore, current evidence suggests that no thrombophilic defect is a clinically important risk factor for recurrent venous thromboembolism after anticoagulant therapy is stopped. All these facts indicate that clinical factors are probably more important than laboratory abnormalities in determining the duration of anticoagulation therapy.32,35,36,61–63
PRIMARY PROPHYLAXIS IN PATIENTS WITH FACTOR V LEIDEN
Factor V Leiden is only one of many risk factors for deep vein thrombosis or pulmonary embolism. If carriers of factor V Leiden have never had a blood clot, then they are not routinely treated with an anticoagulant. Rather, they should be counseled about reducing or eliminating other factors that may add to their risk of developing a clot in the future.
Usually, the effect of risk factors is additive: the more risk factors present, the higher the risk.46,50 Sometimes, however, the effect of multiple risk factors is more than additive.
Some risk factors, such as genetics or age, are not alterable, but many can be controlled by medications or lifestyle modifications. Therefore, general measures and precautions are recommended to minimize the risk of thrombosis. For example:
Losing weight (if the patient is overweight) is an important intervention for risk reduction, since obesity is probably the most common modifiable risk factor for developing blood clots.
Avoiding long periods of immobility is recommended. For example, if the patient is taking a long car ride (more than 2 hours), then stopping every few hours and walking around for a few minutes is a good way to keep the blood circulating. If the patient has a desk job, getting up and walking around the office periodically is advised. On long airplane trips, a walk in the aisle every so often and preventing dehydration by drinking plenty of fluids and avoiding alcohol are recommended.
Wearing elastic stockings with a graduated elastic pressure may prevent deep venous thrombosis from developing on long flights.63–65
Staying active and getting regular exercise through such activities as walking, bicycling, or swimming are helpful.
Avoiding smoking is critical.50,63
Thromboprophylaxis is recommended for most acutely ill hospitalized patients, especially those confined to bed with additional risk factors. Guidelines for prophylaxis are based on an individualized risk assessment and not on thrombophilia status. Prophylactic anticoagulation is routinely recommended for patients undergoing major high-risk surgery, such as an orthopedic, urologic, gynecologic, or bariatric procedure. Any excess thrombotic risk conferred by thrombophilia is likely small compared with the risk of surgery, and recommendations on the duration and intensity of thromboprophylaxis are not based on thrombophilic status.46,48
Education. Pain, swelling, redness of a limb, unexplained shortness of breath, and chest pain are the most common symptoms of deep vein thrombosis and pulmonary embolism.46,50 It is crucial to teach patients with factor V Leiden to recognize these symptoms and to seek early medical attention in case they experience any of them.
SCREENING FAMILY MEMBERS FOR THE FACTOR V LEIDEN MUTATION
Factor V Leiden by itself is a relatively mild thrombophilic defect that does not cause thrombosis in all carriers, and there is no evidence that early diagnosis reduces rates of morbidity or mortality. Therefore, routine screening of all asymptomatic relatives of affected patients with venous thrombosis is not recommended. Rather, the decision to screen should be made on an individual basis.50,66
Screening may be beneficial in selected cases, especially when patients have a strong family history of recurrent venous thrombosis at a young age (younger than 50 years) and the family member has additional risk factors for venous thromboembolism such as oral contraception or is planning for pregnancy.32,48,49,66
- Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. Lancet 1995; 346:1133–1134.
- Ridker PM, Miletich JP, Hennekens CH, Buring JE. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA 1997; 277:1305–1307.
- Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995; 85:1504–1508.
- Stolz E, Kemkes-Matthes B, Pötzsch B, et al. Screening for thrombophilic risk factors among 25 German patients with cerebral venous thrombosis. Acta Neurol Scand 2000; 102:31–36.
- Langlois NJ, Wells PS. Risk of venous thromboembolism in relatives of symptomatic probands with thrombophilia: a systematic review. Thromb Haemost 2003; 90:17–26.
- Juul K, Tybjaerg-Hansen A, Mortensen J, Lange P, Vestbo J, Nordestgaard BG. Factor V leiden homozygosity, dyspnea, and reduced pulmonary function. Arch Intern Med 2005; 165:2032–2036.
- Bertina RM, Koeleman BP, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369:64–67.
- Dahlbäck B. New molecular insights into the genetics of thrombophilia. Resistance to activated protein C caused by Arg506 to Gln mutation in factor V as a pathogenic risk factor for venous thrombosis. Thromb Haemost 1995; 74:139–148.
- Castoldi E, Brugge JM, Nicolaes GA, Girelli D, Tans G, Rosing J. Impaired APC cofactor activity of factor V plays a major role in the APC resistance associated with the factor V Leiden (R506Q) and R2 (H1299R) mutations. Blood 2004; 103:4173–4179.
- Dahlback B. Anticoagulant factor V and thrombosis risk (editorial). Blood 2004; 103:3995.
- Simioni P, Castoldi E, Lunghi B, Tormene D, Rosing J, Bernardi F. An underestimated combination of opposites resulting in enhanced thrombotic tendency. Blood 2005; 106:2363–2365.
- Williamson D, Brown K, Luddington R, Baglin C, Baglin T. Factor V Cambridge: a new mutation (Arg306-->Thr) associated with resistance to activated protein C. Blood 1998; 91:1140–1144.
- Chan WP, Lee CK, Kwong YL, Lam CK, Liang R. A novel mutation of Arg306 of factor V gene in Hong Kong Chinese. Blood 1998; 91:1135–1139.
- Liang R, Lee CK, Wat MS, Kwong YL, Lam CK, Liu HW. Clinical significance of Arg306 mutations of factor V gene. Blood 1998; 92:2599–2600.
- Steen M, Norstrøm EA, Tholander AL, et al. Functional characterization of factor V-Ile359Thr: a novel mutation associated with thrombosis. Blood 2004; 103:3381–3387.
- Bernardi F, Faioni EM, Castoldi E, et al. A factor V genetic component differing from factor V R506Q contributes to the activated protein C resistance phenotype. Blood 1997; 90:1552–1557.
- Lunghi B, Castoldi E, Mingozzi F, Bernardi F. A new factor V gene polymorphism (His 1254 Arg) present in subjects of African origin mimics the R2 polymorphism (His 1299 Arg). Blood 1998; 91:364–365.
- Luddington R, Jackson A, Pannerselvam S, Brown K, Baglin T. The factor V R2 allele: risk of venous thromboembolism, factor V levels and resistance to activated protein C. Thromb Haemost 2000; 83:204–208.
- Faioni EM, Franchi F, Bucciarelli P, et al. Coinheritance of the HR2 haplotype in the factor V gene confers an increased risk of venous thromboembolism to carriers of factor V R506Q (factor V Leiden). Blood 1999; 94:3062–3066.
- Clark P, Walker ID. The phenomenon known as acquired activated protein C resistance. Br J Haematol 2001; 115:767–773.
- Tosetto A, Simioni M, Madeo D, Rodeghiero F. Intraindividual consistency of the activated protein C resistance phenotype. Br J Haematol 2004; 126:405–409.
- de Visser MC, Rosendaal FR, Bertina RM. A reduced sensitivity for activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood 1999; 93:1271–1276.
- Kraaijenhagen RA, in’t Anker PS, Koopman MM, et al. High plasma concentration of factor VIIIc is a major risk factor for venous thromboembolism. Thromb Haemost 2000; 83:5–9.
- Kamphuisen PW, Eikenboom JC, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol 2001; 21:731–738.
- Koster T, Blann AD, Briët E, Vandenbroucke JP, Rosendaal FR. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995; 345:152–155.
- Clark P, Brennand J, Conkie JA, McCall F, Greer IA, Walker ID. Activated protein C sensitivity, protein C, protein S and coagulation in normal pregnancy. Thromb Haemost 1998; 79:1166–1170.
- Cumming AM, Tait RC, Fildes S, Yoong A, Keeney S, Hay CR. Development of resistance to activated protein C during pregnancy. Br J Haematol 1995; 90:725–727.
- Mathonnet F, de Mazancourt P, Bastenaire B, et al. Activated protein C sensitivity ratio in pregnant women at delivery. Br J Haematol 1996; 92:244–246.
- Post MS, Rosing J, Van Der Mooren MJ, et al; Ageing Women’ and the Institute for Cardiovascular Research-Vrije Universiteit (ICaRVU). Increased resistance to activated protein C after short-term oral hormone replacement therapy in healthy post-menopausal women. Br J Haematol 2002; 119:1017–1023.
- Olivieri O, Friso S, Manzato F, et al. Resistance to activated protein C in healthy women taking oral contraceptives. Br J Haematol 1995; 91:465–470.
- Bokarewa MI, Blombäck M, Egberg N, Rosén S. A new variant of interaction between phospholipid antibodies and the protein C system. Blood Coagul Fibrinolysis 1994; 5:37–41.
- Baglin T, Gray E, Greaves M, et al; British Committee for Standards in Haematology. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149:209–220.
- van Stralen KJ, Doggen CJ, Bezemer ID, Pomp ER, Lisman T, Rosendaal FR. Mechanisms of the factor V Leiden paradox. Arterioscler Thromb Vasc Biol 2008; 28:1872–1877.
- Agaoglu N, Mustafa NA, Turkyilmaz S. Prothrombotic disorders in patients with mesenteric vein thrombosis. J Invest Surg 2003; 16:299–304.
- El-Karaksy H, El-Koofy N, El-Hawary M, et al. Prevalence of factor V Leiden mutation and other hereditary thrombophilic factors in Egyptian children with portal vein thrombosis: results of a single-center case-control study. Ann Hematol 2004; 83:712–715.
- Heijmans BT, Westendorp RG, Knook DL, Kluft C, Slagboom PE. The risk of mortality and the factor V Leiden mutation in a population-based cohort. Thromb Haemost 1998; 80:607–609.
- Turkstra F, Karemaker R, Kuijer PM, Prins MH, Büller HR. Is the prevalence of the factor V Leiden mutation in patients with pulmonary embolism and deep vein thrombosis really different? Thromb Haemost 1999; 81:345–348.
- Ridker PM, Hennekens CH, Lindpaintner K, Stampfer MJ, Eisenberg PR, Miletich JP. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995; 332:912–917.
- Manten B, Westendorp RG, Koster T, Reitsma PH, Rosendaal FR. Risk factor profiles in patients with different clinical manifestations of venous thromboembolism: a focus on the factor V Leiden mutation. Thromb Haemost 1996; 76:510–513.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Bloemenkamp KW, Rosendaal FR, Helmerhorst FM, Büller HR, Vandenbroucke JP. Enhancement by factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing a third-generation progestagen. Lancet 1995; 346:1593–1596.
- Murphy PT. Factor V Leiden and venous thromboembolism. Ann Intern Med 2004; 141:483–484.
- Nizankowska-Mogilnicka E, Adamek L, Grzanka P, et al. Genetic polymorphisms associated with acute pulmonary embolism and deep venous thrombosis. Eur Respir J 2003; 21:25–30.
- Arsov T, Miladinova D, Spiroski M. Factor V Leiden is associated with higher risk of deep venous thrombosis of large blood vessels. Croat Med J 2006; 47:433–439.
- Simioni P, Prandoni P, Lensing AW, et al. Risk for subsequent venous thromboembolic complications in carriers of the prothrombin or the factor V gene mutation with a first episode of deep-vein thrombosis. Blood 2000; 96:3329–3333.
- Ornstein DL, Cushman M. Cardiology patient page. Factor V Leiden. Circulation 2003; 107:e94–e97.
- Bezemer ID, van der Meer FJ, Eikenboom JC, Rosendaal FR, Doggen CJ. The value of family history as a risk indicator for venous thrombosis. Arch Intern Med 2009; 169:610–615.
- Press RD, Bauer KA, Kujovich JL, Heit JA. Clinical utility of factor V leiden (R506Q) testing for the diagnosis and management of thromboembolic disorders. Arch Pathol Lab Med 2002; 126:1304–1318.
- Gadelha T, Roldán V, Lecumberri R, et al; RIETE Investigators. Clinical characteristics of patients with factor V Leiden or prothrombin G20210A and a first episode of venous thromboembolism. Findings from the RIETE Registry. Thromb Res 2010; 126:283–286.
- Severinsen MT, Overvad K, Johnsen SP, et al. Genetic susceptibility, smoking, obesity and risk of venous thromboembolism. Br J Haematol 2010; 149:273–279.
- Kujovich JL. Factor V Leiden thrombophilia. Genet Med 2011; 13:1–16.
- Lijfering WM, Brouwer JL, Veeger NJ, et al. Selective testing for thrombophilia in patients with first venous thrombosis: results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood 2009; 113:5314–5322.
- Kearon C, Julian JA, Kovacs MJ, et al; ELATE Investigators. Influence of thrombophilia on risk of recurrent venous thromboembolism while on warfarin: results from a randomized trial. Blood 2008; 112:4432–4436.
- Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med 2006; 166:729–736.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Strobl FJ, Hoffman S, Huber S, Williams EC, Voelkerding KV. Activated protein C resistance assay performance: improvement by sample dilution with factor V-deficient plasma. Arch Pathol Lab Med 1998; 122:430–433.
- Legnani C, Palareti G, Biagi R, et al. Activated protein C resistance: a comparison between two clotting assays and their relationship to the presence of the factor V Leiden mutation. Br J Haematol 1996; 93:694–699.
- Gouault-Heilmann M, Leroy-Matheron C. Factor V Leiden-dependent APC resistance: improved sensitivity and specificity of the APC resistance test by plasma dilution in factor V-depleted plasma. Thromb Res 1996; 82:281–283.
- Svensson PJ, Zöller B, Dahlbäck B. Evaluation of original and modified APC-resistance tests in unselected outpatients with clinically suspected thrombosis and in healthy controls. Thromb Haemost 1997; 77:332–335.
- Tripodi A, Negri B, Bertina RM, Mannucci PM. Screening for the FV:Q506 mutation—evaluation of thirteen plasma-based methods for their diagnostic efficacy in comparison with DNA analysis. Thromb Haemost 1997; 77:436–439.
- Wåhlander K, Larson G, Lindahl TL, et al. Factor V Leiden (G1691A) and prothrombin gene G20210A mutations as potential risk factors for venous thromboembolism after total hip or total knee replacement surgery. Thromb Haemost 2002; 87:580–585.
- Joseph JE, Low J, Courtenay B, Neil MJ, McGrath M, Ma D. A single-centre prospective study of clinical and haemostatic risk factors for venous thromboembolism following lower limb arthroplasty. Br J Haematol 2005; 129:87–92.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):381S–453S.
- Brenner B. Prophylaxis for travel-related thrombosis? Yes. J Thromb Haemost 2004; 2:2089–2091.
- Gavish I, Brenner B. Air travel and the risk of thromboembolism. Intern Emerg Med 2011; 6:113–116.
- Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA; ACMG Factor V Leiden Working Group. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med 2001; 3:139–148.
- Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. Lancet 1995; 346:1133–1134.
- Ridker PM, Miletich JP, Hennekens CH, Buring JE. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA 1997; 277:1305–1307.
- Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995; 85:1504–1508.
- Stolz E, Kemkes-Matthes B, Pötzsch B, et al. Screening for thrombophilic risk factors among 25 German patients with cerebral venous thrombosis. Acta Neurol Scand 2000; 102:31–36.
- Langlois NJ, Wells PS. Risk of venous thromboembolism in relatives of symptomatic probands with thrombophilia: a systematic review. Thromb Haemost 2003; 90:17–26.
- Juul K, Tybjaerg-Hansen A, Mortensen J, Lange P, Vestbo J, Nordestgaard BG. Factor V leiden homozygosity, dyspnea, and reduced pulmonary function. Arch Intern Med 2005; 165:2032–2036.
- Bertina RM, Koeleman BP, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369:64–67.
- Dahlbäck B. New molecular insights into the genetics of thrombophilia. Resistance to activated protein C caused by Arg506 to Gln mutation in factor V as a pathogenic risk factor for venous thrombosis. Thromb Haemost 1995; 74:139–148.
- Castoldi E, Brugge JM, Nicolaes GA, Girelli D, Tans G, Rosing J. Impaired APC cofactor activity of factor V plays a major role in the APC resistance associated with the factor V Leiden (R506Q) and R2 (H1299R) mutations. Blood 2004; 103:4173–4179.
- Dahlback B. Anticoagulant factor V and thrombosis risk (editorial). Blood 2004; 103:3995.
- Simioni P, Castoldi E, Lunghi B, Tormene D, Rosing J, Bernardi F. An underestimated combination of opposites resulting in enhanced thrombotic tendency. Blood 2005; 106:2363–2365.
- Williamson D, Brown K, Luddington R, Baglin C, Baglin T. Factor V Cambridge: a new mutation (Arg306-->Thr) associated with resistance to activated protein C. Blood 1998; 91:1140–1144.
- Chan WP, Lee CK, Kwong YL, Lam CK, Liang R. A novel mutation of Arg306 of factor V gene in Hong Kong Chinese. Blood 1998; 91:1135–1139.
- Liang R, Lee CK, Wat MS, Kwong YL, Lam CK, Liu HW. Clinical significance of Arg306 mutations of factor V gene. Blood 1998; 92:2599–2600.
- Steen M, Norstrøm EA, Tholander AL, et al. Functional characterization of factor V-Ile359Thr: a novel mutation associated with thrombosis. Blood 2004; 103:3381–3387.
- Bernardi F, Faioni EM, Castoldi E, et al. A factor V genetic component differing from factor V R506Q contributes to the activated protein C resistance phenotype. Blood 1997; 90:1552–1557.
- Lunghi B, Castoldi E, Mingozzi F, Bernardi F. A new factor V gene polymorphism (His 1254 Arg) present in subjects of African origin mimics the R2 polymorphism (His 1299 Arg). Blood 1998; 91:364–365.
- Luddington R, Jackson A, Pannerselvam S, Brown K, Baglin T. The factor V R2 allele: risk of venous thromboembolism, factor V levels and resistance to activated protein C. Thromb Haemost 2000; 83:204–208.
- Faioni EM, Franchi F, Bucciarelli P, et al. Coinheritance of the HR2 haplotype in the factor V gene confers an increased risk of venous thromboembolism to carriers of factor V R506Q (factor V Leiden). Blood 1999; 94:3062–3066.
- Clark P, Walker ID. The phenomenon known as acquired activated protein C resistance. Br J Haematol 2001; 115:767–773.
- Tosetto A, Simioni M, Madeo D, Rodeghiero F. Intraindividual consistency of the activated protein C resistance phenotype. Br J Haematol 2004; 126:405–409.
- de Visser MC, Rosendaal FR, Bertina RM. A reduced sensitivity for activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood 1999; 93:1271–1276.
- Kraaijenhagen RA, in’t Anker PS, Koopman MM, et al. High plasma concentration of factor VIIIc is a major risk factor for venous thromboembolism. Thromb Haemost 2000; 83:5–9.
- Kamphuisen PW, Eikenboom JC, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol 2001; 21:731–738.
- Koster T, Blann AD, Briët E, Vandenbroucke JP, Rosendaal FR. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995; 345:152–155.
- Clark P, Brennand J, Conkie JA, McCall F, Greer IA, Walker ID. Activated protein C sensitivity, protein C, protein S and coagulation in normal pregnancy. Thromb Haemost 1998; 79:1166–1170.
- Cumming AM, Tait RC, Fildes S, Yoong A, Keeney S, Hay CR. Development of resistance to activated protein C during pregnancy. Br J Haematol 1995; 90:725–727.
- Mathonnet F, de Mazancourt P, Bastenaire B, et al. Activated protein C sensitivity ratio in pregnant women at delivery. Br J Haematol 1996; 92:244–246.
- Post MS, Rosing J, Van Der Mooren MJ, et al; Ageing Women’ and the Institute for Cardiovascular Research-Vrije Universiteit (ICaRVU). Increased resistance to activated protein C after short-term oral hormone replacement therapy in healthy post-menopausal women. Br J Haematol 2002; 119:1017–1023.
- Olivieri O, Friso S, Manzato F, et al. Resistance to activated protein C in healthy women taking oral contraceptives. Br J Haematol 1995; 91:465–470.
- Bokarewa MI, Blombäck M, Egberg N, Rosén S. A new variant of interaction between phospholipid antibodies and the protein C system. Blood Coagul Fibrinolysis 1994; 5:37–41.
- Baglin T, Gray E, Greaves M, et al; British Committee for Standards in Haematology. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149:209–220.
- van Stralen KJ, Doggen CJ, Bezemer ID, Pomp ER, Lisman T, Rosendaal FR. Mechanisms of the factor V Leiden paradox. Arterioscler Thromb Vasc Biol 2008; 28:1872–1877.
- Agaoglu N, Mustafa NA, Turkyilmaz S. Prothrombotic disorders in patients with mesenteric vein thrombosis. J Invest Surg 2003; 16:299–304.
- El-Karaksy H, El-Koofy N, El-Hawary M, et al. Prevalence of factor V Leiden mutation and other hereditary thrombophilic factors in Egyptian children with portal vein thrombosis: results of a single-center case-control study. Ann Hematol 2004; 83:712–715.
- Heijmans BT, Westendorp RG, Knook DL, Kluft C, Slagboom PE. The risk of mortality and the factor V Leiden mutation in a population-based cohort. Thromb Haemost 1998; 80:607–609.
- Turkstra F, Karemaker R, Kuijer PM, Prins MH, Büller HR. Is the prevalence of the factor V Leiden mutation in patients with pulmonary embolism and deep vein thrombosis really different? Thromb Haemost 1999; 81:345–348.
- Ridker PM, Hennekens CH, Lindpaintner K, Stampfer MJ, Eisenberg PR, Miletich JP. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995; 332:912–917.
- Manten B, Westendorp RG, Koster T, Reitsma PH, Rosendaal FR. Risk factor profiles in patients with different clinical manifestations of venous thromboembolism: a focus on the factor V Leiden mutation. Thromb Haemost 1996; 76:510–513.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Bloemenkamp KW, Rosendaal FR, Helmerhorst FM, Büller HR, Vandenbroucke JP. Enhancement by factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing a third-generation progestagen. Lancet 1995; 346:1593–1596.
- Murphy PT. Factor V Leiden and venous thromboembolism. Ann Intern Med 2004; 141:483–484.
- Nizankowska-Mogilnicka E, Adamek L, Grzanka P, et al. Genetic polymorphisms associated with acute pulmonary embolism and deep venous thrombosis. Eur Respir J 2003; 21:25–30.
- Arsov T, Miladinova D, Spiroski M. Factor V Leiden is associated with higher risk of deep venous thrombosis of large blood vessels. Croat Med J 2006; 47:433–439.
- Simioni P, Prandoni P, Lensing AW, et al. Risk for subsequent venous thromboembolic complications in carriers of the prothrombin or the factor V gene mutation with a first episode of deep-vein thrombosis. Blood 2000; 96:3329–3333.
- Ornstein DL, Cushman M. Cardiology patient page. Factor V Leiden. Circulation 2003; 107:e94–e97.
- Bezemer ID, van der Meer FJ, Eikenboom JC, Rosendaal FR, Doggen CJ. The value of family history as a risk indicator for venous thrombosis. Arch Intern Med 2009; 169:610–615.
- Press RD, Bauer KA, Kujovich JL, Heit JA. Clinical utility of factor V leiden (R506Q) testing for the diagnosis and management of thromboembolic disorders. Arch Pathol Lab Med 2002; 126:1304–1318.
- Gadelha T, Roldán V, Lecumberri R, et al; RIETE Investigators. Clinical characteristics of patients with factor V Leiden or prothrombin G20210A and a first episode of venous thromboembolism. Findings from the RIETE Registry. Thromb Res 2010; 126:283–286.
- Severinsen MT, Overvad K, Johnsen SP, et al. Genetic susceptibility, smoking, obesity and risk of venous thromboembolism. Br J Haematol 2010; 149:273–279.
- Kujovich JL. Factor V Leiden thrombophilia. Genet Med 2011; 13:1–16.
- Lijfering WM, Brouwer JL, Veeger NJ, et al. Selective testing for thrombophilia in patients with first venous thrombosis: results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood 2009; 113:5314–5322.
- Kearon C, Julian JA, Kovacs MJ, et al; ELATE Investigators. Influence of thrombophilia on risk of recurrent venous thromboembolism while on warfarin: results from a randomized trial. Blood 2008; 112:4432–4436.
- Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med 2006; 166:729–736.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Strobl FJ, Hoffman S, Huber S, Williams EC, Voelkerding KV. Activated protein C resistance assay performance: improvement by sample dilution with factor V-deficient plasma. Arch Pathol Lab Med 1998; 122:430–433.
- Legnani C, Palareti G, Biagi R, et al. Activated protein C resistance: a comparison between two clotting assays and their relationship to the presence of the factor V Leiden mutation. Br J Haematol 1996; 93:694–699.
- Gouault-Heilmann M, Leroy-Matheron C. Factor V Leiden-dependent APC resistance: improved sensitivity and specificity of the APC resistance test by plasma dilution in factor V-depleted plasma. Thromb Res 1996; 82:281–283.
- Svensson PJ, Zöller B, Dahlbäck B. Evaluation of original and modified APC-resistance tests in unselected outpatients with clinically suspected thrombosis and in healthy controls. Thromb Haemost 1997; 77:332–335.
- Tripodi A, Negri B, Bertina RM, Mannucci PM. Screening for the FV:Q506 mutation—evaluation of thirteen plasma-based methods for their diagnostic efficacy in comparison with DNA analysis. Thromb Haemost 1997; 77:436–439.
- Wåhlander K, Larson G, Lindahl TL, et al. Factor V Leiden (G1691A) and prothrombin gene G20210A mutations as potential risk factors for venous thromboembolism after total hip or total knee replacement surgery. Thromb Haemost 2002; 87:580–585.
- Joseph JE, Low J, Courtenay B, Neil MJ, McGrath M, Ma D. A single-centre prospective study of clinical and haemostatic risk factors for venous thromboembolism following lower limb arthroplasty. Br J Haematol 2005; 129:87–92.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):381S–453S.
- Brenner B. Prophylaxis for travel-related thrombosis? Yes. J Thromb Haemost 2004; 2:2089–2091.
- Gavish I, Brenner B. Air travel and the risk of thromboembolism. Intern Emerg Med 2011; 6:113–116.
- Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA; ACMG Factor V Leiden Working Group. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med 2001; 3:139–148.
KEY POINTS
- The pathogenesis of venous thromboembolism is complex and multifactorial, often reflecting the interplay between environmental, clinical, and genetic factors.
- Factor V Leiden increases the risk of venous thromboembolism but by itself does not appear to increase the risk of arterial thrombosis.
- Often, people with factor V Leiden may have additional risk factors that increase the rate of venous clots, such as older age, surgery, obesity, immobility, prolonged travel, hospitalization, oral contraceptive use, hormonal replacement therapy, pregnancy, and malignancy.
- General measures and precautions are needed to minimize the risk of venous thromboembolism in people with the factor V Leiden mutation, especially when modifiable factors are present, such as obesity and long periods of immobilization.
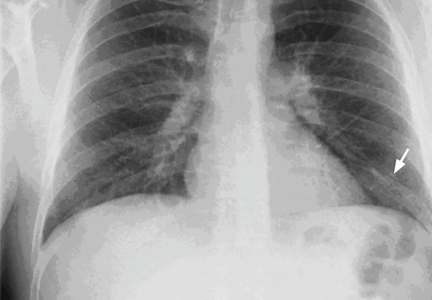
Synthetic legal intoxicating drugs: The emerging ‘incense’ and ‘bath salt’ phenomenon
Over the past year, it has been hard to avoid news reports involving people getting high on “bath salts” and “incense” (also known as “Spice” or “K2”). Addiction treatment professionals have been overwhelmed by questions regarding why one would want to “snort bath salts” or “smoke incense.”
These substances are not what they appear to be. They are sold as bath salts and incense and are labeled “not for human consumption” simply to avoid regulation by the US Food and Drug Administration (FDA). In reality, they are powerful psychoactive drugs, with effects that mimic those of more commonly abused drugs such as amphetamines and marijuana. Until recently, they were legally available over the counter at quick-marts, head shops, and on the Internet. Because they are relatively new, they may not be detectable on routine urine drug screens, and users may be unaware of the specific chemicals contained in them.
These drugs, which we have collectively termed synthetic legal intoxicating drugs (SLIDs), are increasing dramatically in use.1–3 A survey of youths at a rave party indicated that 21% had used one of them on at least one occasion.4 The general impression held by the drug-using public is that SLIDs are relatively cheap, are not detected on standard urine drug screens, can produce a powerful high, and, until recently, were readily available through legitimate sources.
Physicians need to be aware of SLIDs in order to recognize and manage the intoxication syndromes associated with these substances when encountered in clinical practice, and in order to educate patients about their potential dangers.
SYNTHETIC CANNABINOIDS MARKETED AS INCENSE
Herbal incense products that could be smoked as an alternative to marijuana started appearing on the Internet in Europe in 2004. By 2008, when such products first appeared in the United States, their use in Europe was already widespread.
Initially, consumers were led to believe that such herbal smoking blends were safe, legal alternatives to marijuana, and that it was the proprietary blend of herbs that was responsible for the “natural” high. Spice, a specific brand name, was originally trademarked in England as incense and also as an herbal smoking product.5
Legal authorities, however, suspected that these herbal blends were adulterated with synthetic substances. In December 2008, the first such substance was found when Austrian authorities isolated a synthetic cannabinoid, JWH-018, from an herbal incense product.6 By the end of 2009, five other synthetic cannabinoids—CP-47,497, HU-210, JWH-073, JWH-250, and JWH-398—had been isolated from various herbal incense samples around the world.7
The synthetic cannabinoids in herbal incense products are not derived from the hemp plant (Cannabis sativa), but are synthesized in laboratories and are formulated to interact with the endogenous cannabinoid receptors in the brain to produce psychoactive effects.
Synthetic cannabinoids are full agonists; natural THC is only a partial agonist
Two types of cannabinoid receptors have been discovered in humans: CB1 and CB2. Both types are found in the central nervous system, and CB2 is also found extensively in the periphery. CB1 is the receptor responsible for the psychoactive effects of cannabinoids, including altered consciousness, euphoria, relaxation, perceptual disturbances, intensified sensory experiences, cognitive impairment, and increased reaction time.6 The physiologic role of CB2 remains uncertain.
The major psychoactive cannabinoid in naturally occurring marijuana is delta-9-tetrahydrocannabinol (THC). The so-called classic cannabinoids, such as HU-210, are analogues of THC and are based on its chemical structure. The rest of the synthetic cannabinoids commonly found in incense products differ in chemical structure from naturally occurring cannabinoids such as THC, but have activity at the CB1 receptor and are thus psychoactive.
Of clinical relevance is that THC is only a partial agonist at the CB1 receptor, while all synthetic cannabinoids commonly found in incense products are full agonists at CB1.7 This difference is important because partial agonists bind to receptors but stimulate them only partially and therefore exhibit a plateau effect in terms of dose vs clinical response. In contrast, full agonists have no ceiling on the dose-response relationship and therefore have a greater potential for overdose and severe toxic effects.
Despite uncertainties, use is widespread
Most of the synthetic cannabinoids in herbal incense products were developed for research purposes, and there are almost no reliable scientific data on their effects in humans. Of additional concern is that no research has been conducted on their pyrolytic effects, ie, how these chemicals are transformed when they are burned, such as when consumers smoke them. Furthermore, herbal incense products often vary in their active substances and concentrations, so consumers really do not know what they are getting.
Despite the many uncertainties, the use of these products is widespread. Data submitted to the US Drug Enforcement Administration (DEA) from a major toxicology laboratory indicated that from July through November of 2010, 3,700 samples tested positive for either JWH-018 or JWH-073. This report also indicated that 30% to 35% of specimens submitted by juvenile probation departments were positive for synthetic cannabinoids.8
MEDICAL CONCERNS OVER SYNTHETIC CANNABINOIDS
Amid the mysteries surrounding synthetic cannabinoids, one thing is clear: users are increasingly seeking medical attention. In 2010, there were 2,906 calls to poison control centers across the United States pertaining to “synthetic marijuana”; in 2011 there were 6,959 calls, and in January 2012, 639 such calls had been placed.9

The duration of the intoxicating effects of synthetic cannabinoids is generally longer than that of THC, but this seems to be variable. JWH-018, for instance, seems to have a shorter duration of action, at around 1 to 2 hours, while a longer, 5- to 6-hour intoxicating effect has been observed with CP-47,497.7,12
Serious adverse effects
Although the prevalence of serious adverse effects associated with the use of synthetic cannabinoids is not known, a number of serious complications have been recognized.
Seizures. One case of seizure has been reported in association with the use of synthetic cannabinoids, specifically JWH-018.12 This case involved a previously healthy 48-year-old man who had ingested a powder that was subsequently confirmed to be JWH-018, which he mixed with alcohol. Of further concern in this case is that this individual developed a refractory supraventricular tachycardia that required cardioversion on the first hospital day.
The authors speculated that the seizure may have been due to a dose-response mechanism that resulted in either the release of presynaptic excitatory neurotransmitters or the decreased release of inhibitory neurotransmitters. They further postulated that the supraventricular tachycardia could have been caused by one of two mechanisms previously reported in association with CB1 agonists: an increase in circulating catecholamines or heightened oxidative demands on the myocardium.12
Psychosis. The occurrence of psychotic symptoms such as hallucinations and paranoid delusions in association with synthetic cannabinoids is not surprising, given the well-documented link between marijuana use and psychosis.13,14
A case report of a 25-year-old patient with a 7-year history of recurrent psychosis that was initially triggered by cannabis use indicated that the use of 3 g of herbal incense on three occasions was associated with worsening of previous psychotic symptoms and the emergence of command and paranoid types of auditory hallucination.10
Semistructured interviews of 15 patients in a forensic rehabilitative service, all of whom had a history of psychotic illness, showed that 69% experienced symptoms consistent with psychotic relapse after smoking an herbal incense product containing JWH-018.15
It is possible that psychotic symptoms may be more prominent with synthetic cannabinoids than with natural marijuana because not only are synthetic cannabinoids more potent and work as full agonists, but, unlike marijuana, they do not contain cannabidiol, which is thought to have antipsychotic efficacy.10,16 However, the risk of psychotic symptoms in association with synthetic cannabinoid usage in otherwise healthy people is unknown.
Regulation lags behind
Growing concern over the perceived dangers posed by synthetic cannabinoids has led to a ban on some of the more common ones contained in herbal incense preparations. On March 1, 2011, the US DEA temporarily placed five synthetic cannabinoids (JWH-018, JWH-073, JWH-200, CP-47,497, and cannabicyclohexanol) under schedule I (banned substances).
Such a ban, however, may be futile because there are an estimated 100 synthetic cannabinoids that have yet to enter the market, and when one is banned, a new one is likely to be introduced immediately as a replacement.8
SYNTHETIC STIMULANTS MARKETED AS BATH SALTS
Like the herbal incense products, “bath salts” may likewise not be what they appear to be. They too may be labeled “not for human consumption” in an effort to bypass laws governing mind-altering substances.
Several pharmacologically active substances have been marketed as bath salts. Two of the more common ingredients are 3,4-methylenedioxypyrovalerone (MDPV) and 4-methylcathinone (mephedrone).
MDPV is a dopamine and norepineph-rine reuptake inhibitor that acts as a powerful stimulant. It has no FDA-approved medical use, but it is an analogue of the stimulant pyrovalerone, which was once used to treat chronic fatigue.17
MDPV seems to be the most common substance found in bath salt products in the United States. A sample of this substance was first seized on the streets by German authorities in 2007. A study in Finland conducted from August 2009 to September 2010 estimated that 5.7% of all arrests for driving under the influence (DUI) unrelated to alcohol consumption involved MDPV intoxication.17 In 2009, the National Forensic Laboratory Information System of the US DEA had seized only two samples of MDPV, but by 2010 that had increased to 161.18
Mephedrone is derived from phenethylamine and is closely related to cathinone, the active ingredient in the African khat plant (Catha edulis).19 Khat has a history of abuse, and the chemical structure of cathinone and its derivatives is similar to that of amphetamine.
Mephedrone, a powerful stimulant, is suspected of working as a monoamine reuptake inhibitor, and it may also directly induce the presynaptic release of monoamines.20 The net effect is an increase in serotonin, norepineph-rine, and dopamine levels at neuronal synapses.
Mephedrone was first described in 1929 by chemist Saem de Burnaga Sanchez, and it remained an obscure research chemical for many years.21 It was formally recognized as a drug of abuse in Europe in 2007, and by 2009 it was the sixth most frequently used such drug in Europe.8,22
Although MDPV and mephedrone are the most common psychoactive ingredients in bath salts, many other synthetic drugs have been found on the market.
A temporary ban
On September 7, 2011, the US government made it illegal to possess or sell any substance containing MDPV, mephedrone, or methy-lone. This temporary restriction was to remain in effect for 1 year to give the DEA time to collect data to support a move to permanently control these substances.3
Like synthetic cannabinoids, however, synthetic stimulants are very difficult to regulate because they are a large group of substances. As soon as one substance is outlawed, another synthetic stimulant will likely take its place.
MEDICAL CONCERNS REGARDING SYNTHETIC STIMULANTS
The medical and psychiatric sequelae that are associated with the use of bath salts have sent an increasing number of people to emergency rooms. The number of bath-salt-related calls to US poison control centers increased dramatically from 303 in 2010 to 4,720 by August 31, 2011. Most of these calls were related to tachycardia, agitation, hallucinations, extreme paranoia, delusions, and elevations in blood pressure.3
A report of 35 cases of people who had used bath salts and who had reported to Michigan emergency rooms between November 13, 2010, and March 31, 2011, indicated that agitation was present in 66%, tachycardia in 63%, delusions and hallucinations in 40%, seizure or tremor in 29%, hypertension in 23%, drowsiness in 23%, paranoia in 20%, and mydriasis in 20%; one patient was dead on arrival. Of the 34 patients who were alive on arrival, 17 (50%) were hospitalized, 15 were released, and 2 left against medical advice. In the patients in this study, 63% had injected the drug, 26% snorted it, and 11% ingested it orally.2 Toxicology results obtained during an autopsy on the one person who died revealed a high level of MDPV, and the coroner ruled that MDPV toxicity was the primary cause of death.2

Though the pharmacokinetic properties of mephedrone are unknown, James et al24 noted that an interesting feature is that its clinical effects seem to persist for more than 24 hours after the last exposure to the drug, which would not be expected based on the rapid elimination of other similar cathinones.
Sympathomimetic toxicity. Many of the symptoms listed in Table 2 are consistent with a sympathomimetic syndrome. In a case series reported by Regan et al,26 most of the 57 patients exhibited cardiovascular findings consistent with sympathomimetic toxicity.
In the study by James et al,24 one of the patients with chest pain had electrocardiographic changes consistent with acute myocardial infarction. Though it is not possible to conclude from a single case that mephedrone poses a risk of myocardial infarction, such a risk has been reported with khat.28 More research is needed to determine whether mephedrone poses a risk of cardiac events when used by people with or without an underlying cardiac condition.
Seizure also seems to be a relatively common feature associated with mephedrone use in case series of emergency room presentations. The US Centers for Disease Control and Prevention l2 reported that of 35 patients who had used bath salts, 40% experienced seizures or “tremors.” A recent case series27 of 15 patients presenting to an emergency department after mephedrone use reported that 20% had experienced seizures. In the study by James et al,24 four patients (3% of the total group) experienced seizures after using mephedrone. It should be noted that, aside from people presenting to emergency rooms, seizures are rarely reported in the wider population of mephedrone users.
Psychotic symptoms are also quite common in users of synthetic stimulants who present to emergency rooms, occurring, as previously stated, in 14% to 40% of cases.2,24
In a small case series, Penders and Gestring29 pointed out some common features in three patients who had used MDPV and had presented with psychosis: sleep problems, inattention, vivid hallucinations of intruders, fearfulness, and inability to remember many of the events surrounding their drug use. The authors concluded that the psychotic syndrome present in their three patients was indicative of a short-term delirium rather than a substance-induced psychosis based on the presence of attention deficits and memory problems. The patients in this series responded well to brief hospitalization and antipsychotic medications.
As with seizure, extreme presentations such as psychosis are infrequently mentioned except in people requiring treatment at a hospital. There are simply no data regarding the prevalence of psychotic symptoms in the larger group of all synthetic stimulant users.
SUSPECT SLID INTOXICATION IN ‘PSYCHIATRIC’ PATIENTS
Despite the temporary ban on the more common substances found in Spice and bath salts, it is premature for the medical community to breath a sigh of relief. Producers of these products are already likely bringing to market new ones containing similar but as yet nonbanned substances. Furthermore, such bans will do little to affect Internet commerce; rather than go to a head shop, consumers will order the products online.
Doctors in urgent care centers, emergency rooms, and on general medical floors should pay close attention to any patient without a known psychiatric history who is acting in a bizarre fashion. Most SLID-intoxicated patients will present with anxiety, agitation, and psychosis. Rather than assume that they are psychiatric patients, one should consider the possibility of SLID intoxication and pay close attention to the possible medical sequelae associated with SLID use, such as elevated blood pressure, tachycardia, and seizure.
Benzodiazepines, especially lorazepam (Ativan), have been the agents most commonly used to treat both agitation and seizures associated with SLID intoxication.
Antipsychotics should be used judiciously because of their propensity to lower the seizure threshold, and patients with synthetic stimulant toxicity are already at increased risk of seizure.
A psychiatric consult should be considered in the event of any suspected toxicity or for any patient whose behavior is difficult to manage.
Restraints may be needed in some circumstances when agitation cannot be controlled with benzodiazepines alone, to ensure safety for the patient as well as that of others in the emergency department.

Routine laboratory tests should be part of the workup of patients suspected of being under the influence of SLIDs. These include a complete blood cell count, complete metabolic panel, and urine toxicology (Table 3).23,25 A routine urine toxicology study will likely be negative, but either the patient or collateral information may give you a general idea of what the patient used, in which case the sample could be sent out for special tests for the more common substances found in herbal incense or bath salt products.
Electroencephalography may be indicated if there is any question as to whether the patient may have suffered a seizure. There should be a low threshold to order electrocardiography, especially in the case of synthetic stimulant intoxication.
Serial cardiac enzymes may be warranted if a patient with synthetic-stimulant intoxicated has chest pain.
Education, addiction treatment. Much is unknown about the risk of SLIDs, but given the adverse events reported in the literature, it seems likely that those with underlying cardiac or psychiatric issues may be at higher risk for the most serious drug-related consequences. With regard to synthetic stimulants, Winstock et al20 recommend a harm-reduction approach involving educating patients about avoiding the development of tolerance, not engaging in polydrug use, not injecting, and paying special attention to remaining cool and well hydrated.
Experience shows that once SLID patients get through their acute crisis and are no longer psychotic, they tend to be forthright in divulging what they used to get high. At that point, consideration should be given to consulting an addiction treatment specialist for further evaluation of the patient’s drug use history and for formulation of a treatment plan to help ensure that the patient doesn’t return to using these drugs.
SLIDs POSE A REAL CHALLENGE
SLIDs present a real challenge to law enforcement, governments, the public, and the addiction treatment community. There is currently no way to routinely test for these substances. Furthermore, any tests that are developed or laws that are enacted will be easily evaded, as there are many more synthetic substances waiting in the wings to be released.
Don’t be lulled into thinking that SLIDs are gone with the recent bans against some of the more common substances. More SLIDs are coming, and more morbidity should be expected in medical settings.
Doctors in emergency departments and other settings need to be prepared for the agitated and often psychotic presentation of SLID-intoxicated patients and should be ready with benzodiazepines, restraints, and a calm and reassuring manner. And for patients who present with psychotic symptoms, medical staff should also be ready to consider involuntary short-term commitment to an inpatient psychiatric unit.
Once they recover, patients need to be educated about the dangers of substances such as SLIDs that, because of their novelty, may be perceived as less dangerous alternatives to traditional illicit drugs.
- Wehrman J. Fake marijuana spurs more than 4,500 calls to US poison centers. American Association of Poison Control Centers (AAPCC), May 12, 2011. http://www.aapcc.org/dnn/Portals/0/prrel/updatedk2-may112011.pdf. Accessed February 20, 2012.
- Centers for Disease Control and Prevention. Emergency department visits after use of a drug sold as “bath salts”—Michigan, November 13, 2010–March 31, 2011. MMWR Morb Mortal Wkly Rep 2011; 60( 19):624–627.
- Canton L. Poison control centers applaud DEA’s ban of bath salts. American Association of Poison Control Centers (AAPCC). September 8, 2011. http://www.mc.vanderbilt.edu/root/vumc.php?site=poisoncenter&doc=36028. Accessed February 20, 2012.
- Banta-Green C. “Club drug” use patterns and related behaviors in Seattle, King County. Survey data collected for STEPS (Stemming the Tide of Ecstasy through Prevention Strategies). Report to public health-Seattle, King County, Feb. 9, 2004.
- Erowid EF, Erowid F. Spice & spin-offs: prohibition’s high-tech cannabis substitutes. June 2009. http://www.erowid.org/chemicals/spice_product/spice_product_article1.shtml. Accessed February 20, 2012.
- Cary P. Spice, K2 and the problem of synthetic cannabinoids. Drug Court Practitioner Fact Sheet 2010; 6:2–3.
- European Monitoring Centre for Drugs and Drug Addiction. EMCDDA 2009 thematic paper—understanding the ‘Spice’ phenomenon. Luxembourg: Office for Official Publications of the European Communities, 2009.
- Rannazzi T. The dangers of synthetic cannabinoids and stimulants. Testimony before the Senate Caucus on International Narcotics Control, United States Senate. April 6, 2011. http://www.justice.gov/dea/speeches/110412_testimony.pdf. Accessed February 20, 2012.
- American Association of Poison Control Centers. Poison centers report calls about synthetic marijuana. www.AAPCC.org. Accessed February 22, 2012.
- Müller H, Sperling W, Körhrmann M, Huttner HB, Kornhuber J, Maler JM. The synthetic cannabinoid Spice as a trigger for an acute exacerbation of cannabis induced recurrent psychotic episodes. Schizophr Res 2010; 118:309–310.
- Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, Moran JH. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol (Phila) 2011: 49;760–764.
- Vardakou I, Pistos C, Spiliopoulou CH. Spice drugs as a new trend: mode of action, identification and legislation. Toxicol Lett 2010; 197:157–162.
- Fergusson DM, Poulton R, Smith PF, Boden JM. Cannabis and psychosis. BMJ 2006; 332:172–175.
- Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 2007; 370:319–328.
- Every-Palmer S. Synthetic cannabinoid JWH-018 and psychosis: an explorative study. Drug Alcohol Depend 2011; 117:152–157.
- Huffman JW, Thompson AL, Wilety JL, Martin BR. Synthesis and pharmacology of 1-deoxy analogs of CP-47,497 and CP-55,940. Bioorg Med Chem 2008; 16:322–335.
- Kriikku P, Wilhelm L, Schwarz O, Rintatalo J. New designer drug of abuse: 3,4-methylenedioxypyrovalerone (MDPV). Findings from apprehended drivers in Finland. Forensic Sci Int 2011; 210:195–200.
- Drug Enforcement Administration. 3,4-Methylenedioxypyrovalerone (MDPV). (Street names: “bath salts,” Ivory Wave,” “plant fertilizer,” “Vanilla Sky,” “Energy-1”). October 2011. www.deadiversion.usdoj.gov/drugs_concern/mdpv.pdf. Accessed February 20, 2012.
- Kalix P. Cathinone, a natural amphetamine. Pharmacol Toxicol 1992; 70:77–86.
- Winstock AR, Marsen J, Mitcheson L. What should be done about mephedrone? BMJ 2010; 340:c1605.
- Saem de Burnaga Sanchez J. Sur un homologue de l’ éphédrine. Bulletin de la Societé Chimique de France 1929; 45:284–286.
- Winstock A, Mitcheson L, Ramsey J, Davies S, Puchnarewicz M, Marsden J. Mephedrone: use, subjective effects and health risks. Addiction 2011; 106:1991–1996.
- Winstock AR, Mitcheson LR, Deluca P, Davey Z, Corazza O, Schifano F. Mephedrone, new kid for the chop? Addiction 2011; 106:154–161.
- James D, Adams RD, Spears R, et al; National Poisons Information Service. Clinical characteristics of mephedrone toxicity reported to the UK National Poisons Information Service. Emerg Med J 2011; 28:686–689.
- Wood DM, Davies S, Puchnarewicz M, et al. Recreational use of 4-methylmethcathinone (4-MMC) with associated sympathomimetic toxicity. J Med Toxicol 2010; 6:327–330.
- Regan L, Mitchelson M, Macdonald C. Mephedrone toxicity in a Scottish emergency department. Emerg Med J 2011; 28:1055–1058.
- Wood DM, Greene SL, Dargan PI. Clinical pattern of toxicity associated with the novel synthetic cathinone mephedrone. Emerg Med J 2011; 28:280–282.
- Al-Motarreb A, Briancon S, Al-Jaber N, et al. Khat chewing is a risk factor for acute myocardial infarction: a case-control study. Br J Clin Pharmacol 2005; 59:574–581.
- Penders TM, Gestring R. Hallucinatory delirium following use of MDPV: “bath salts.” Gen Hosp Psychiatry 2011; 33:525–526.
Over the past year, it has been hard to avoid news reports involving people getting high on “bath salts” and “incense” (also known as “Spice” or “K2”). Addiction treatment professionals have been overwhelmed by questions regarding why one would want to “snort bath salts” or “smoke incense.”
These substances are not what they appear to be. They are sold as bath salts and incense and are labeled “not for human consumption” simply to avoid regulation by the US Food and Drug Administration (FDA). In reality, they are powerful psychoactive drugs, with effects that mimic those of more commonly abused drugs such as amphetamines and marijuana. Until recently, they were legally available over the counter at quick-marts, head shops, and on the Internet. Because they are relatively new, they may not be detectable on routine urine drug screens, and users may be unaware of the specific chemicals contained in them.
These drugs, which we have collectively termed synthetic legal intoxicating drugs (SLIDs), are increasing dramatically in use.1–3 A survey of youths at a rave party indicated that 21% had used one of them on at least one occasion.4 The general impression held by the drug-using public is that SLIDs are relatively cheap, are not detected on standard urine drug screens, can produce a powerful high, and, until recently, were readily available through legitimate sources.
Physicians need to be aware of SLIDs in order to recognize and manage the intoxication syndromes associated with these substances when encountered in clinical practice, and in order to educate patients about their potential dangers.
SYNTHETIC CANNABINOIDS MARKETED AS INCENSE
Herbal incense products that could be smoked as an alternative to marijuana started appearing on the Internet in Europe in 2004. By 2008, when such products first appeared in the United States, their use in Europe was already widespread.
Initially, consumers were led to believe that such herbal smoking blends were safe, legal alternatives to marijuana, and that it was the proprietary blend of herbs that was responsible for the “natural” high. Spice, a specific brand name, was originally trademarked in England as incense and also as an herbal smoking product.5
Legal authorities, however, suspected that these herbal blends were adulterated with synthetic substances. In December 2008, the first such substance was found when Austrian authorities isolated a synthetic cannabinoid, JWH-018, from an herbal incense product.6 By the end of 2009, five other synthetic cannabinoids—CP-47,497, HU-210, JWH-073, JWH-250, and JWH-398—had been isolated from various herbal incense samples around the world.7
The synthetic cannabinoids in herbal incense products are not derived from the hemp plant (Cannabis sativa), but are synthesized in laboratories and are formulated to interact with the endogenous cannabinoid receptors in the brain to produce psychoactive effects.
Synthetic cannabinoids are full agonists; natural THC is only a partial agonist
Two types of cannabinoid receptors have been discovered in humans: CB1 and CB2. Both types are found in the central nervous system, and CB2 is also found extensively in the periphery. CB1 is the receptor responsible for the psychoactive effects of cannabinoids, including altered consciousness, euphoria, relaxation, perceptual disturbances, intensified sensory experiences, cognitive impairment, and increased reaction time.6 The physiologic role of CB2 remains uncertain.
The major psychoactive cannabinoid in naturally occurring marijuana is delta-9-tetrahydrocannabinol (THC). The so-called classic cannabinoids, such as HU-210, are analogues of THC and are based on its chemical structure. The rest of the synthetic cannabinoids commonly found in incense products differ in chemical structure from naturally occurring cannabinoids such as THC, but have activity at the CB1 receptor and are thus psychoactive.
Of clinical relevance is that THC is only a partial agonist at the CB1 receptor, while all synthetic cannabinoids commonly found in incense products are full agonists at CB1.7 This difference is important because partial agonists bind to receptors but stimulate them only partially and therefore exhibit a plateau effect in terms of dose vs clinical response. In contrast, full agonists have no ceiling on the dose-response relationship and therefore have a greater potential for overdose and severe toxic effects.
Despite uncertainties, use is widespread
Most of the synthetic cannabinoids in herbal incense products were developed for research purposes, and there are almost no reliable scientific data on their effects in humans. Of additional concern is that no research has been conducted on their pyrolytic effects, ie, how these chemicals are transformed when they are burned, such as when consumers smoke them. Furthermore, herbal incense products often vary in their active substances and concentrations, so consumers really do not know what they are getting.
Despite the many uncertainties, the use of these products is widespread. Data submitted to the US Drug Enforcement Administration (DEA) from a major toxicology laboratory indicated that from July through November of 2010, 3,700 samples tested positive for either JWH-018 or JWH-073. This report also indicated that 30% to 35% of specimens submitted by juvenile probation departments were positive for synthetic cannabinoids.8
MEDICAL CONCERNS OVER SYNTHETIC CANNABINOIDS
Amid the mysteries surrounding synthetic cannabinoids, one thing is clear: users are increasingly seeking medical attention. In 2010, there were 2,906 calls to poison control centers across the United States pertaining to “synthetic marijuana”; in 2011 there were 6,959 calls, and in January 2012, 639 such calls had been placed.9
The duration of the intoxicating effects of synthetic cannabinoids is generally longer than that of THC, but this seems to be variable. JWH-018, for instance, seems to have a shorter duration of action, at around 1 to 2 hours, while a longer, 5- to 6-hour intoxicating effect has been observed with CP-47,497.7,12
Serious adverse effects
Although the prevalence of serious adverse effects associated with the use of synthetic cannabinoids is not known, a number of serious complications have been recognized.
Seizures. One case of seizure has been reported in association with the use of synthetic cannabinoids, specifically JWH-018.12 This case involved a previously healthy 48-year-old man who had ingested a powder that was subsequently confirmed to be JWH-018, which he mixed with alcohol. Of further concern in this case is that this individual developed a refractory supraventricular tachycardia that required cardioversion on the first hospital day.
The authors speculated that the seizure may have been due to a dose-response mechanism that resulted in either the release of presynaptic excitatory neurotransmitters or the decreased release of inhibitory neurotransmitters. They further postulated that the supraventricular tachycardia could have been caused by one of two mechanisms previously reported in association with CB1 agonists: an increase in circulating catecholamines or heightened oxidative demands on the myocardium.12
Psychosis. The occurrence of psychotic symptoms such as hallucinations and paranoid delusions in association with synthetic cannabinoids is not surprising, given the well-documented link between marijuana use and psychosis.13,14
A case report of a 25-year-old patient with a 7-year history of recurrent psychosis that was initially triggered by cannabis use indicated that the use of 3 g of herbal incense on three occasions was associated with worsening of previous psychotic symptoms and the emergence of command and paranoid types of auditory hallucination.10
Semistructured interviews of 15 patients in a forensic rehabilitative service, all of whom had a history of psychotic illness, showed that 69% experienced symptoms consistent with psychotic relapse after smoking an herbal incense product containing JWH-018.15
It is possible that psychotic symptoms may be more prominent with synthetic cannabinoids than with natural marijuana because not only are synthetic cannabinoids more potent and work as full agonists, but, unlike marijuana, they do not contain cannabidiol, which is thought to have antipsychotic efficacy.10,16 However, the risk of psychotic symptoms in association with synthetic cannabinoid usage in otherwise healthy people is unknown.
Regulation lags behind
Growing concern over the perceived dangers posed by synthetic cannabinoids has led to a ban on some of the more common ones contained in herbal incense preparations. On March 1, 2011, the US DEA temporarily placed five synthetic cannabinoids (JWH-018, JWH-073, JWH-200, CP-47,497, and cannabicyclohexanol) under schedule I (banned substances).
Such a ban, however, may be futile because there are an estimated 100 synthetic cannabinoids that have yet to enter the market, and when one is banned, a new one is likely to be introduced immediately as a replacement.8
SYNTHETIC STIMULANTS MARKETED AS BATH SALTS
Like the herbal incense products, “bath salts” may likewise not be what they appear to be. They too may be labeled “not for human consumption” in an effort to bypass laws governing mind-altering substances.
Several pharmacologically active substances have been marketed as bath salts. Two of the more common ingredients are 3,4-methylenedioxypyrovalerone (MDPV) and 4-methylcathinone (mephedrone).
MDPV is a dopamine and norepineph-rine reuptake inhibitor that acts as a powerful stimulant. It has no FDA-approved medical use, but it is an analogue of the stimulant pyrovalerone, which was once used to treat chronic fatigue.17
MDPV seems to be the most common substance found in bath salt products in the United States. A sample of this substance was first seized on the streets by German authorities in 2007. A study in Finland conducted from August 2009 to September 2010 estimated that 5.7% of all arrests for driving under the influence (DUI) unrelated to alcohol consumption involved MDPV intoxication.17 In 2009, the National Forensic Laboratory Information System of the US DEA had seized only two samples of MDPV, but by 2010 that had increased to 161.18
Mephedrone is derived from phenethylamine and is closely related to cathinone, the active ingredient in the African khat plant (Catha edulis).19 Khat has a history of abuse, and the chemical structure of cathinone and its derivatives is similar to that of amphetamine.
Mephedrone, a powerful stimulant, is suspected of working as a monoamine reuptake inhibitor, and it may also directly induce the presynaptic release of monoamines.20 The net effect is an increase in serotonin, norepineph-rine, and dopamine levels at neuronal synapses.
Mephedrone was first described in 1929 by chemist Saem de Burnaga Sanchez, and it remained an obscure research chemical for many years.21 It was formally recognized as a drug of abuse in Europe in 2007, and by 2009 it was the sixth most frequently used such drug in Europe.8,22
Although MDPV and mephedrone are the most common psychoactive ingredients in bath salts, many other synthetic drugs have been found on the market.
A temporary ban
On September 7, 2011, the US government made it illegal to possess or sell any substance containing MDPV, mephedrone, or methy-lone. This temporary restriction was to remain in effect for 1 year to give the DEA time to collect data to support a move to permanently control these substances.3
Like synthetic cannabinoids, however, synthetic stimulants are very difficult to regulate because they are a large group of substances. As soon as one substance is outlawed, another synthetic stimulant will likely take its place.
MEDICAL CONCERNS REGARDING SYNTHETIC STIMULANTS
The medical and psychiatric sequelae that are associated with the use of bath salts have sent an increasing number of people to emergency rooms. The number of bath-salt-related calls to US poison control centers increased dramatically from 303 in 2010 to 4,720 by August 31, 2011. Most of these calls were related to tachycardia, agitation, hallucinations, extreme paranoia, delusions, and elevations in blood pressure.3
A report of 35 cases of people who had used bath salts and who had reported to Michigan emergency rooms between November 13, 2010, and March 31, 2011, indicated that agitation was present in 66%, tachycardia in 63%, delusions and hallucinations in 40%, seizure or tremor in 29%, hypertension in 23%, drowsiness in 23%, paranoia in 20%, and mydriasis in 20%; one patient was dead on arrival. Of the 34 patients who were alive on arrival, 17 (50%) were hospitalized, 15 were released, and 2 left against medical advice. In the patients in this study, 63% had injected the drug, 26% snorted it, and 11% ingested it orally.2 Toxicology results obtained during an autopsy on the one person who died revealed a high level of MDPV, and the coroner ruled that MDPV toxicity was the primary cause of death.2
Though the pharmacokinetic properties of mephedrone are unknown, James et al24 noted that an interesting feature is that its clinical effects seem to persist for more than 24 hours after the last exposure to the drug, which would not be expected based on the rapid elimination of other similar cathinones.
Sympathomimetic toxicity. Many of the symptoms listed in Table 2 are consistent with a sympathomimetic syndrome. In a case series reported by Regan et al,26 most of the 57 patients exhibited cardiovascular findings consistent with sympathomimetic toxicity.
In the study by James et al,24 one of the patients with chest pain had electrocardiographic changes consistent with acute myocardial infarction. Though it is not possible to conclude from a single case that mephedrone poses a risk of myocardial infarction, such a risk has been reported with khat.28 More research is needed to determine whether mephedrone poses a risk of cardiac events when used by people with or without an underlying cardiac condition.
Seizure also seems to be a relatively common feature associated with mephedrone use in case series of emergency room presentations. The US Centers for Disease Control and Prevention l2 reported that of 35 patients who had used bath salts, 40% experienced seizures or “tremors.” A recent case series27 of 15 patients presenting to an emergency department after mephedrone use reported that 20% had experienced seizures. In the study by James et al,24 four patients (3% of the total group) experienced seizures after using mephedrone. It should be noted that, aside from people presenting to emergency rooms, seizures are rarely reported in the wider population of mephedrone users.
Psychotic symptoms are also quite common in users of synthetic stimulants who present to emergency rooms, occurring, as previously stated, in 14% to 40% of cases.2,24
In a small case series, Penders and Gestring29 pointed out some common features in three patients who had used MDPV and had presented with psychosis: sleep problems, inattention, vivid hallucinations of intruders, fearfulness, and inability to remember many of the events surrounding their drug use. The authors concluded that the psychotic syndrome present in their three patients was indicative of a short-term delirium rather than a substance-induced psychosis based on the presence of attention deficits and memory problems. The patients in this series responded well to brief hospitalization and antipsychotic medications.
As with seizure, extreme presentations such as psychosis are infrequently mentioned except in people requiring treatment at a hospital. There are simply no data regarding the prevalence of psychotic symptoms in the larger group of all synthetic stimulant users.
SUSPECT SLID INTOXICATION IN ‘PSYCHIATRIC’ PATIENTS
Despite the temporary ban on the more common substances found in Spice and bath salts, it is premature for the medical community to breath a sigh of relief. Producers of these products are already likely bringing to market new ones containing similar but as yet nonbanned substances. Furthermore, such bans will do little to affect Internet commerce; rather than go to a head shop, consumers will order the products online.
Doctors in urgent care centers, emergency rooms, and on general medical floors should pay close attention to any patient without a known psychiatric history who is acting in a bizarre fashion. Most SLID-intoxicated patients will present with anxiety, agitation, and psychosis. Rather than assume that they are psychiatric patients, one should consider the possibility of SLID intoxication and pay close attention to the possible medical sequelae associated with SLID use, such as elevated blood pressure, tachycardia, and seizure.
Benzodiazepines, especially lorazepam (Ativan), have been the agents most commonly used to treat both agitation and seizures associated with SLID intoxication.
Antipsychotics should be used judiciously because of their propensity to lower the seizure threshold, and patients with synthetic stimulant toxicity are already at increased risk of seizure.
A psychiatric consult should be considered in the event of any suspected toxicity or for any patient whose behavior is difficult to manage.
Restraints may be needed in some circumstances when agitation cannot be controlled with benzodiazepines alone, to ensure safety for the patient as well as that of others in the emergency department.
Routine laboratory tests should be part of the workup of patients suspected of being under the influence of SLIDs. These include a complete blood cell count, complete metabolic panel, and urine toxicology (Table 3).23,25 A routine urine toxicology study will likely be negative, but either the patient or collateral information may give you a general idea of what the patient used, in which case the sample could be sent out for special tests for the more common substances found in herbal incense or bath salt products.
Electroencephalography may be indicated if there is any question as to whether the patient may have suffered a seizure. There should be a low threshold to order electrocardiography, especially in the case of synthetic stimulant intoxication.
Serial cardiac enzymes may be warranted if a patient with synthetic-stimulant intoxicated has chest pain.
Education, addiction treatment. Much is unknown about the risk of SLIDs, but given the adverse events reported in the literature, it seems likely that those with underlying cardiac or psychiatric issues may be at higher risk for the most serious drug-related consequences. With regard to synthetic stimulants, Winstock et al20 recommend a harm-reduction approach involving educating patients about avoiding the development of tolerance, not engaging in polydrug use, not injecting, and paying special attention to remaining cool and well hydrated.
Experience shows that once SLID patients get through their acute crisis and are no longer psychotic, they tend to be forthright in divulging what they used to get high. At that point, consideration should be given to consulting an addiction treatment specialist for further evaluation of the patient’s drug use history and for formulation of a treatment plan to help ensure that the patient doesn’t return to using these drugs.
SLIDs POSE A REAL CHALLENGE
SLIDs present a real challenge to law enforcement, governments, the public, and the addiction treatment community. There is currently no way to routinely test for these substances. Furthermore, any tests that are developed or laws that are enacted will be easily evaded, as there are many more synthetic substances waiting in the wings to be released.
Don’t be lulled into thinking that SLIDs are gone with the recent bans against some of the more common substances. More SLIDs are coming, and more morbidity should be expected in medical settings.
Doctors in emergency departments and other settings need to be prepared for the agitated and often psychotic presentation of SLID-intoxicated patients and should be ready with benzodiazepines, restraints, and a calm and reassuring manner. And for patients who present with psychotic symptoms, medical staff should also be ready to consider involuntary short-term commitment to an inpatient psychiatric unit.
Once they recover, patients need to be educated about the dangers of substances such as SLIDs that, because of their novelty, may be perceived as less dangerous alternatives to traditional illicit drugs.
Over the past year, it has been hard to avoid news reports involving people getting high on “bath salts” and “incense” (also known as “Spice” or “K2”). Addiction treatment professionals have been overwhelmed by questions regarding why one would want to “snort bath salts” or “smoke incense.”
These substances are not what they appear to be. They are sold as bath salts and incense and are labeled “not for human consumption” simply to avoid regulation by the US Food and Drug Administration (FDA). In reality, they are powerful psychoactive drugs, with effects that mimic those of more commonly abused drugs such as amphetamines and marijuana. Until recently, they were legally available over the counter at quick-marts, head shops, and on the Internet. Because they are relatively new, they may not be detectable on routine urine drug screens, and users may be unaware of the specific chemicals contained in them.
These drugs, which we have collectively termed synthetic legal intoxicating drugs (SLIDs), are increasing dramatically in use.1–3 A survey of youths at a rave party indicated that 21% had used one of them on at least one occasion.4 The general impression held by the drug-using public is that SLIDs are relatively cheap, are not detected on standard urine drug screens, can produce a powerful high, and, until recently, were readily available through legitimate sources.
Physicians need to be aware of SLIDs in order to recognize and manage the intoxication syndromes associated with these substances when encountered in clinical practice, and in order to educate patients about their potential dangers.
SYNTHETIC CANNABINOIDS MARKETED AS INCENSE
Herbal incense products that could be smoked as an alternative to marijuana started appearing on the Internet in Europe in 2004. By 2008, when such products first appeared in the United States, their use in Europe was already widespread.
Initially, consumers were led to believe that such herbal smoking blends were safe, legal alternatives to marijuana, and that it was the proprietary blend of herbs that was responsible for the “natural” high. Spice, a specific brand name, was originally trademarked in England as incense and also as an herbal smoking product.5
Legal authorities, however, suspected that these herbal blends were adulterated with synthetic substances. In December 2008, the first such substance was found when Austrian authorities isolated a synthetic cannabinoid, JWH-018, from an herbal incense product.6 By the end of 2009, five other synthetic cannabinoids—CP-47,497, HU-210, JWH-073, JWH-250, and JWH-398—had been isolated from various herbal incense samples around the world.7
The synthetic cannabinoids in herbal incense products are not derived from the hemp plant (Cannabis sativa), but are synthesized in laboratories and are formulated to interact with the endogenous cannabinoid receptors in the brain to produce psychoactive effects.
Synthetic cannabinoids are full agonists; natural THC is only a partial agonist
Two types of cannabinoid receptors have been discovered in humans: CB1 and CB2. Both types are found in the central nervous system, and CB2 is also found extensively in the periphery. CB1 is the receptor responsible for the psychoactive effects of cannabinoids, including altered consciousness, euphoria, relaxation, perceptual disturbances, intensified sensory experiences, cognitive impairment, and increased reaction time.6 The physiologic role of CB2 remains uncertain.
The major psychoactive cannabinoid in naturally occurring marijuana is delta-9-tetrahydrocannabinol (THC). The so-called classic cannabinoids, such as HU-210, are analogues of THC and are based on its chemical structure. The rest of the synthetic cannabinoids commonly found in incense products differ in chemical structure from naturally occurring cannabinoids such as THC, but have activity at the CB1 receptor and are thus psychoactive.
Of clinical relevance is that THC is only a partial agonist at the CB1 receptor, while all synthetic cannabinoids commonly found in incense products are full agonists at CB1.7 This difference is important because partial agonists bind to receptors but stimulate them only partially and therefore exhibit a plateau effect in terms of dose vs clinical response. In contrast, full agonists have no ceiling on the dose-response relationship and therefore have a greater potential for overdose and severe toxic effects.
Despite uncertainties, use is widespread
Most of the synthetic cannabinoids in herbal incense products were developed for research purposes, and there are almost no reliable scientific data on their effects in humans. Of additional concern is that no research has been conducted on their pyrolytic effects, ie, how these chemicals are transformed when they are burned, such as when consumers smoke them. Furthermore, herbal incense products often vary in their active substances and concentrations, so consumers really do not know what they are getting.
Despite the many uncertainties, the use of these products is widespread. Data submitted to the US Drug Enforcement Administration (DEA) from a major toxicology laboratory indicated that from July through November of 2010, 3,700 samples tested positive for either JWH-018 or JWH-073. This report also indicated that 30% to 35% of specimens submitted by juvenile probation departments were positive for synthetic cannabinoids.8
MEDICAL CONCERNS OVER SYNTHETIC CANNABINOIDS
Amid the mysteries surrounding synthetic cannabinoids, one thing is clear: users are increasingly seeking medical attention. In 2010, there were 2,906 calls to poison control centers across the United States pertaining to “synthetic marijuana”; in 2011 there were 6,959 calls, and in January 2012, 639 such calls had been placed.9
The duration of the intoxicating effects of synthetic cannabinoids is generally longer than that of THC, but this seems to be variable. JWH-018, for instance, seems to have a shorter duration of action, at around 1 to 2 hours, while a longer, 5- to 6-hour intoxicating effect has been observed with CP-47,497.7,12
Serious adverse effects
Although the prevalence of serious adverse effects associated with the use of synthetic cannabinoids is not known, a number of serious complications have been recognized.
Seizures. One case of seizure has been reported in association with the use of synthetic cannabinoids, specifically JWH-018.12 This case involved a previously healthy 48-year-old man who had ingested a powder that was subsequently confirmed to be JWH-018, which he mixed with alcohol. Of further concern in this case is that this individual developed a refractory supraventricular tachycardia that required cardioversion on the first hospital day.
The authors speculated that the seizure may have been due to a dose-response mechanism that resulted in either the release of presynaptic excitatory neurotransmitters or the decreased release of inhibitory neurotransmitters. They further postulated that the supraventricular tachycardia could have been caused by one of two mechanisms previously reported in association with CB1 agonists: an increase in circulating catecholamines or heightened oxidative demands on the myocardium.12
Psychosis. The occurrence of psychotic symptoms such as hallucinations and paranoid delusions in association with synthetic cannabinoids is not surprising, given the well-documented link between marijuana use and psychosis.13,14
A case report of a 25-year-old patient with a 7-year history of recurrent psychosis that was initially triggered by cannabis use indicated that the use of 3 g of herbal incense on three occasions was associated with worsening of previous psychotic symptoms and the emergence of command and paranoid types of auditory hallucination.10
Semistructured interviews of 15 patients in a forensic rehabilitative service, all of whom had a history of psychotic illness, showed that 69% experienced symptoms consistent with psychotic relapse after smoking an herbal incense product containing JWH-018.15
It is possible that psychotic symptoms may be more prominent with synthetic cannabinoids than with natural marijuana because not only are synthetic cannabinoids more potent and work as full agonists, but, unlike marijuana, they do not contain cannabidiol, which is thought to have antipsychotic efficacy.10,16 However, the risk of psychotic symptoms in association with synthetic cannabinoid usage in otherwise healthy people is unknown.
Regulation lags behind
Growing concern over the perceived dangers posed by synthetic cannabinoids has led to a ban on some of the more common ones contained in herbal incense preparations. On March 1, 2011, the US DEA temporarily placed five synthetic cannabinoids (JWH-018, JWH-073, JWH-200, CP-47,497, and cannabicyclohexanol) under schedule I (banned substances).
Such a ban, however, may be futile because there are an estimated 100 synthetic cannabinoids that have yet to enter the market, and when one is banned, a new one is likely to be introduced immediately as a replacement.8
SYNTHETIC STIMULANTS MARKETED AS BATH SALTS
Like the herbal incense products, “bath salts” may likewise not be what they appear to be. They too may be labeled “not for human consumption” in an effort to bypass laws governing mind-altering substances.
Several pharmacologically active substances have been marketed as bath salts. Two of the more common ingredients are 3,4-methylenedioxypyrovalerone (MDPV) and 4-methylcathinone (mephedrone).
MDPV is a dopamine and norepineph-rine reuptake inhibitor that acts as a powerful stimulant. It has no FDA-approved medical use, but it is an analogue of the stimulant pyrovalerone, which was once used to treat chronic fatigue.17
MDPV seems to be the most common substance found in bath salt products in the United States. A sample of this substance was first seized on the streets by German authorities in 2007. A study in Finland conducted from August 2009 to September 2010 estimated that 5.7% of all arrests for driving under the influence (DUI) unrelated to alcohol consumption involved MDPV intoxication.17 In 2009, the National Forensic Laboratory Information System of the US DEA had seized only two samples of MDPV, but by 2010 that had increased to 161.18
Mephedrone is derived from phenethylamine and is closely related to cathinone, the active ingredient in the African khat plant (Catha edulis).19 Khat has a history of abuse, and the chemical structure of cathinone and its derivatives is similar to that of amphetamine.
Mephedrone, a powerful stimulant, is suspected of working as a monoamine reuptake inhibitor, and it may also directly induce the presynaptic release of monoamines.20 The net effect is an increase in serotonin, norepineph-rine, and dopamine levels at neuronal synapses.
Mephedrone was first described in 1929 by chemist Saem de Burnaga Sanchez, and it remained an obscure research chemical for many years.21 It was formally recognized as a drug of abuse in Europe in 2007, and by 2009 it was the sixth most frequently used such drug in Europe.8,22
Although MDPV and mephedrone are the most common psychoactive ingredients in bath salts, many other synthetic drugs have been found on the market.
A temporary ban
On September 7, 2011, the US government made it illegal to possess or sell any substance containing MDPV, mephedrone, or methy-lone. This temporary restriction was to remain in effect for 1 year to give the DEA time to collect data to support a move to permanently control these substances.3
Like synthetic cannabinoids, however, synthetic stimulants are very difficult to regulate because they are a large group of substances. As soon as one substance is outlawed, another synthetic stimulant will likely take its place.
MEDICAL CONCERNS REGARDING SYNTHETIC STIMULANTS
The medical and psychiatric sequelae that are associated with the use of bath salts have sent an increasing number of people to emergency rooms. The number of bath-salt-related calls to US poison control centers increased dramatically from 303 in 2010 to 4,720 by August 31, 2011. Most of these calls were related to tachycardia, agitation, hallucinations, extreme paranoia, delusions, and elevations in blood pressure.3
A report of 35 cases of people who had used bath salts and who had reported to Michigan emergency rooms between November 13, 2010, and March 31, 2011, indicated that agitation was present in 66%, tachycardia in 63%, delusions and hallucinations in 40%, seizure or tremor in 29%, hypertension in 23%, drowsiness in 23%, paranoia in 20%, and mydriasis in 20%; one patient was dead on arrival. Of the 34 patients who were alive on arrival, 17 (50%) were hospitalized, 15 were released, and 2 left against medical advice. In the patients in this study, 63% had injected the drug, 26% snorted it, and 11% ingested it orally.2 Toxicology results obtained during an autopsy on the one person who died revealed a high level of MDPV, and the coroner ruled that MDPV toxicity was the primary cause of death.2
Though the pharmacokinetic properties of mephedrone are unknown, James et al24 noted that an interesting feature is that its clinical effects seem to persist for more than 24 hours after the last exposure to the drug, which would not be expected based on the rapid elimination of other similar cathinones.
Sympathomimetic toxicity. Many of the symptoms listed in Table 2 are consistent with a sympathomimetic syndrome. In a case series reported by Regan et al,26 most of the 57 patients exhibited cardiovascular findings consistent with sympathomimetic toxicity.
In the study by James et al,24 one of the patients with chest pain had electrocardiographic changes consistent with acute myocardial infarction. Though it is not possible to conclude from a single case that mephedrone poses a risk of myocardial infarction, such a risk has been reported with khat.28 More research is needed to determine whether mephedrone poses a risk of cardiac events when used by people with or without an underlying cardiac condition.
Seizure also seems to be a relatively common feature associated with mephedrone use in case series of emergency room presentations. The US Centers for Disease Control and Prevention l2 reported that of 35 patients who had used bath salts, 40% experienced seizures or “tremors.” A recent case series27 of 15 patients presenting to an emergency department after mephedrone use reported that 20% had experienced seizures. In the study by James et al,24 four patients (3% of the total group) experienced seizures after using mephedrone. It should be noted that, aside from people presenting to emergency rooms, seizures are rarely reported in the wider population of mephedrone users.
Psychotic symptoms are also quite common in users of synthetic stimulants who present to emergency rooms, occurring, as previously stated, in 14% to 40% of cases.2,24
In a small case series, Penders and Gestring29 pointed out some common features in three patients who had used MDPV and had presented with psychosis: sleep problems, inattention, vivid hallucinations of intruders, fearfulness, and inability to remember many of the events surrounding their drug use. The authors concluded that the psychotic syndrome present in their three patients was indicative of a short-term delirium rather than a substance-induced psychosis based on the presence of attention deficits and memory problems. The patients in this series responded well to brief hospitalization and antipsychotic medications.
As with seizure, extreme presentations such as psychosis are infrequently mentioned except in people requiring treatment at a hospital. There are simply no data regarding the prevalence of psychotic symptoms in the larger group of all synthetic stimulant users.
SUSPECT SLID INTOXICATION IN ‘PSYCHIATRIC’ PATIENTS
Despite the temporary ban on the more common substances found in Spice and bath salts, it is premature for the medical community to breath a sigh of relief. Producers of these products are already likely bringing to market new ones containing similar but as yet nonbanned substances. Furthermore, such bans will do little to affect Internet commerce; rather than go to a head shop, consumers will order the products online.
Doctors in urgent care centers, emergency rooms, and on general medical floors should pay close attention to any patient without a known psychiatric history who is acting in a bizarre fashion. Most SLID-intoxicated patients will present with anxiety, agitation, and psychosis. Rather than assume that they are psychiatric patients, one should consider the possibility of SLID intoxication and pay close attention to the possible medical sequelae associated with SLID use, such as elevated blood pressure, tachycardia, and seizure.
Benzodiazepines, especially lorazepam (Ativan), have been the agents most commonly used to treat both agitation and seizures associated with SLID intoxication.
Antipsychotics should be used judiciously because of their propensity to lower the seizure threshold, and patients with synthetic stimulant toxicity are already at increased risk of seizure.
A psychiatric consult should be considered in the event of any suspected toxicity or for any patient whose behavior is difficult to manage.
Restraints may be needed in some circumstances when agitation cannot be controlled with benzodiazepines alone, to ensure safety for the patient as well as that of others in the emergency department.
Routine laboratory tests should be part of the workup of patients suspected of being under the influence of SLIDs. These include a complete blood cell count, complete metabolic panel, and urine toxicology (Table 3).23,25 A routine urine toxicology study will likely be negative, but either the patient or collateral information may give you a general idea of what the patient used, in which case the sample could be sent out for special tests for the more common substances found in herbal incense or bath salt products.
Electroencephalography may be indicated if there is any question as to whether the patient may have suffered a seizure. There should be a low threshold to order electrocardiography, especially in the case of synthetic stimulant intoxication.
Serial cardiac enzymes may be warranted if a patient with synthetic-stimulant intoxicated has chest pain.
Education, addiction treatment. Much is unknown about the risk of SLIDs, but given the adverse events reported in the literature, it seems likely that those with underlying cardiac or psychiatric issues may be at higher risk for the most serious drug-related consequences. With regard to synthetic stimulants, Winstock et al20 recommend a harm-reduction approach involving educating patients about avoiding the development of tolerance, not engaging in polydrug use, not injecting, and paying special attention to remaining cool and well hydrated.
Experience shows that once SLID patients get through their acute crisis and are no longer psychotic, they tend to be forthright in divulging what they used to get high. At that point, consideration should be given to consulting an addiction treatment specialist for further evaluation of the patient’s drug use history and for formulation of a treatment plan to help ensure that the patient doesn’t return to using these drugs.
SLIDs POSE A REAL CHALLENGE
SLIDs present a real challenge to law enforcement, governments, the public, and the addiction treatment community. There is currently no way to routinely test for these substances. Furthermore, any tests that are developed or laws that are enacted will be easily evaded, as there are many more synthetic substances waiting in the wings to be released.
Don’t be lulled into thinking that SLIDs are gone with the recent bans against some of the more common substances. More SLIDs are coming, and more morbidity should be expected in medical settings.
Doctors in emergency departments and other settings need to be prepared for the agitated and often psychotic presentation of SLID-intoxicated patients and should be ready with benzodiazepines, restraints, and a calm and reassuring manner. And for patients who present with psychotic symptoms, medical staff should also be ready to consider involuntary short-term commitment to an inpatient psychiatric unit.
Once they recover, patients need to be educated about the dangers of substances such as SLIDs that, because of their novelty, may be perceived as less dangerous alternatives to traditional illicit drugs.
- Wehrman J. Fake marijuana spurs more than 4,500 calls to US poison centers. American Association of Poison Control Centers (AAPCC), May 12, 2011. http://www.aapcc.org/dnn/Portals/0/prrel/updatedk2-may112011.pdf. Accessed February 20, 2012.
- Centers for Disease Control and Prevention. Emergency department visits after use of a drug sold as “bath salts”—Michigan, November 13, 2010–March 31, 2011. MMWR Morb Mortal Wkly Rep 2011; 60( 19):624–627.
- Canton L. Poison control centers applaud DEA’s ban of bath salts. American Association of Poison Control Centers (AAPCC). September 8, 2011. http://www.mc.vanderbilt.edu/root/vumc.php?site=poisoncenter&doc=36028. Accessed February 20, 2012.
- Banta-Green C. “Club drug” use patterns and related behaviors in Seattle, King County. Survey data collected for STEPS (Stemming the Tide of Ecstasy through Prevention Strategies). Report to public health-Seattle, King County, Feb. 9, 2004.
- Erowid EF, Erowid F. Spice & spin-offs: prohibition’s high-tech cannabis substitutes. June 2009. http://www.erowid.org/chemicals/spice_product/spice_product_article1.shtml. Accessed February 20, 2012.
- Cary P. Spice, K2 and the problem of synthetic cannabinoids. Drug Court Practitioner Fact Sheet 2010; 6:2–3.
- European Monitoring Centre for Drugs and Drug Addiction. EMCDDA 2009 thematic paper—understanding the ‘Spice’ phenomenon. Luxembourg: Office for Official Publications of the European Communities, 2009.
- Rannazzi T. The dangers of synthetic cannabinoids and stimulants. Testimony before the Senate Caucus on International Narcotics Control, United States Senate. April 6, 2011. http://www.justice.gov/dea/speeches/110412_testimony.pdf. Accessed February 20, 2012.
- American Association of Poison Control Centers. Poison centers report calls about synthetic marijuana. www.AAPCC.org. Accessed February 22, 2012.
- Müller H, Sperling W, Körhrmann M, Huttner HB, Kornhuber J, Maler JM. The synthetic cannabinoid Spice as a trigger for an acute exacerbation of cannabis induced recurrent psychotic episodes. Schizophr Res 2010; 118:309–310.
- Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, Moran JH. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol (Phila) 2011: 49;760–764.
- Vardakou I, Pistos C, Spiliopoulou CH. Spice drugs as a new trend: mode of action, identification and legislation. Toxicol Lett 2010; 197:157–162.
- Fergusson DM, Poulton R, Smith PF, Boden JM. Cannabis and psychosis. BMJ 2006; 332:172–175.
- Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 2007; 370:319–328.
- Every-Palmer S. Synthetic cannabinoid JWH-018 and psychosis: an explorative study. Drug Alcohol Depend 2011; 117:152–157.
- Huffman JW, Thompson AL, Wilety JL, Martin BR. Synthesis and pharmacology of 1-deoxy analogs of CP-47,497 and CP-55,940. Bioorg Med Chem 2008; 16:322–335.
- Kriikku P, Wilhelm L, Schwarz O, Rintatalo J. New designer drug of abuse: 3,4-methylenedioxypyrovalerone (MDPV). Findings from apprehended drivers in Finland. Forensic Sci Int 2011; 210:195–200.
- Drug Enforcement Administration. 3,4-Methylenedioxypyrovalerone (MDPV). (Street names: “bath salts,” Ivory Wave,” “plant fertilizer,” “Vanilla Sky,” “Energy-1”). October 2011. www.deadiversion.usdoj.gov/drugs_concern/mdpv.pdf. Accessed February 20, 2012.
- Kalix P. Cathinone, a natural amphetamine. Pharmacol Toxicol 1992; 70:77–86.
- Winstock AR, Marsen J, Mitcheson L. What should be done about mephedrone? BMJ 2010; 340:c1605.
- Saem de Burnaga Sanchez J. Sur un homologue de l’ éphédrine. Bulletin de la Societé Chimique de France 1929; 45:284–286.
- Winstock A, Mitcheson L, Ramsey J, Davies S, Puchnarewicz M, Marsden J. Mephedrone: use, subjective effects and health risks. Addiction 2011; 106:1991–1996.
- Winstock AR, Mitcheson LR, Deluca P, Davey Z, Corazza O, Schifano F. Mephedrone, new kid for the chop? Addiction 2011; 106:154–161.
- James D, Adams RD, Spears R, et al; National Poisons Information Service. Clinical characteristics of mephedrone toxicity reported to the UK National Poisons Information Service. Emerg Med J 2011; 28:686–689.
- Wood DM, Davies S, Puchnarewicz M, et al. Recreational use of 4-methylmethcathinone (4-MMC) with associated sympathomimetic toxicity. J Med Toxicol 2010; 6:327–330.
- Regan L, Mitchelson M, Macdonald C. Mephedrone toxicity in a Scottish emergency department. Emerg Med J 2011; 28:1055–1058.
- Wood DM, Greene SL, Dargan PI. Clinical pattern of toxicity associated with the novel synthetic cathinone mephedrone. Emerg Med J 2011; 28:280–282.
- Al-Motarreb A, Briancon S, Al-Jaber N, et al. Khat chewing is a risk factor for acute myocardial infarction: a case-control study. Br J Clin Pharmacol 2005; 59:574–581.
- Penders TM, Gestring R. Hallucinatory delirium following use of MDPV: “bath salts.” Gen Hosp Psychiatry 2011; 33:525–526.
- Wehrman J. Fake marijuana spurs more than 4,500 calls to US poison centers. American Association of Poison Control Centers (AAPCC), May 12, 2011. http://www.aapcc.org/dnn/Portals/0/prrel/updatedk2-may112011.pdf. Accessed February 20, 2012.
- Centers for Disease Control and Prevention. Emergency department visits after use of a drug sold as “bath salts”—Michigan, November 13, 2010–March 31, 2011. MMWR Morb Mortal Wkly Rep 2011; 60( 19):624–627.
- Canton L. Poison control centers applaud DEA’s ban of bath salts. American Association of Poison Control Centers (AAPCC). September 8, 2011. http://www.mc.vanderbilt.edu/root/vumc.php?site=poisoncenter&doc=36028. Accessed February 20, 2012.
- Banta-Green C. “Club drug” use patterns and related behaviors in Seattle, King County. Survey data collected for STEPS (Stemming the Tide of Ecstasy through Prevention Strategies). Report to public health-Seattle, King County, Feb. 9, 2004.
- Erowid EF, Erowid F. Spice & spin-offs: prohibition’s high-tech cannabis substitutes. June 2009. http://www.erowid.org/chemicals/spice_product/spice_product_article1.shtml. Accessed February 20, 2012.
- Cary P. Spice, K2 and the problem of synthetic cannabinoids. Drug Court Practitioner Fact Sheet 2010; 6:2–3.
- European Monitoring Centre for Drugs and Drug Addiction. EMCDDA 2009 thematic paper—understanding the ‘Spice’ phenomenon. Luxembourg: Office for Official Publications of the European Communities, 2009.
- Rannazzi T. The dangers of synthetic cannabinoids and stimulants. Testimony before the Senate Caucus on International Narcotics Control, United States Senate. April 6, 2011. http://www.justice.gov/dea/speeches/110412_testimony.pdf. Accessed February 20, 2012.
- American Association of Poison Control Centers. Poison centers report calls about synthetic marijuana. www.AAPCC.org. Accessed February 22, 2012.
- Müller H, Sperling W, Körhrmann M, Huttner HB, Kornhuber J, Maler JM. The synthetic cannabinoid Spice as a trigger for an acute exacerbation of cannabis induced recurrent psychotic episodes. Schizophr Res 2010; 118:309–310.
- Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, Moran JH. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol (Phila) 2011: 49;760–764.
- Vardakou I, Pistos C, Spiliopoulou CH. Spice drugs as a new trend: mode of action, identification and legislation. Toxicol Lett 2010; 197:157–162.
- Fergusson DM, Poulton R, Smith PF, Boden JM. Cannabis and psychosis. BMJ 2006; 332:172–175.
- Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 2007; 370:319–328.
- Every-Palmer S. Synthetic cannabinoid JWH-018 and psychosis: an explorative study. Drug Alcohol Depend 2011; 117:152–157.
- Huffman JW, Thompson AL, Wilety JL, Martin BR. Synthesis and pharmacology of 1-deoxy analogs of CP-47,497 and CP-55,940. Bioorg Med Chem 2008; 16:322–335.
- Kriikku P, Wilhelm L, Schwarz O, Rintatalo J. New designer drug of abuse: 3,4-methylenedioxypyrovalerone (MDPV). Findings from apprehended drivers in Finland. Forensic Sci Int 2011; 210:195–200.
- Drug Enforcement Administration. 3,4-Methylenedioxypyrovalerone (MDPV). (Street names: “bath salts,” Ivory Wave,” “plant fertilizer,” “Vanilla Sky,” “Energy-1”). October 2011. www.deadiversion.usdoj.gov/drugs_concern/mdpv.pdf. Accessed February 20, 2012.
- Kalix P. Cathinone, a natural amphetamine. Pharmacol Toxicol 1992; 70:77–86.
- Winstock AR, Marsen J, Mitcheson L. What should be done about mephedrone? BMJ 2010; 340:c1605.
- Saem de Burnaga Sanchez J. Sur un homologue de l’ éphédrine. Bulletin de la Societé Chimique de France 1929; 45:284–286.
- Winstock A, Mitcheson L, Ramsey J, Davies S, Puchnarewicz M, Marsden J. Mephedrone: use, subjective effects and health risks. Addiction 2011; 106:1991–1996.
- Winstock AR, Mitcheson LR, Deluca P, Davey Z, Corazza O, Schifano F. Mephedrone, new kid for the chop? Addiction 2011; 106:154–161.
- James D, Adams RD, Spears R, et al; National Poisons Information Service. Clinical characteristics of mephedrone toxicity reported to the UK National Poisons Information Service. Emerg Med J 2011; 28:686–689.
- Wood DM, Davies S, Puchnarewicz M, et al. Recreational use of 4-methylmethcathinone (4-MMC) with associated sympathomimetic toxicity. J Med Toxicol 2010; 6:327–330.
- Regan L, Mitchelson M, Macdonald C. Mephedrone toxicity in a Scottish emergency department. Emerg Med J 2011; 28:1055–1058.
- Wood DM, Greene SL, Dargan PI. Clinical pattern of toxicity associated with the novel synthetic cathinone mephedrone. Emerg Med J 2011; 28:280–282.
- Al-Motarreb A, Briancon S, Al-Jaber N, et al. Khat chewing is a risk factor for acute myocardial infarction: a case-control study. Br J Clin Pharmacol 2005; 59:574–581.
- Penders TM, Gestring R. Hallucinatory delirium following use of MDPV: “bath salts.” Gen Hosp Psychiatry 2011; 33:525–526.
KEY POINTS
- These products are sold under misleading names and deceptive labels to avoid regulation. Although several have recently been banned, many more are waiting to be brought to the market in a similar fashion.
- “Incense” products often contain synthetic cannabinoids; scientific research into their potential long-term effects in humans has been very limited.
- The potential for medical and psychiatric adverse events from synthetic cannabinoids may be heightened because of their full-agonist mechanism of action and because of the variable concentration and unregulated potency of these compounds in incense products.
- Bath salt intoxication, when encountered in the emergency department, may present as a psychiatric disorder or as a range of medical problems including cardiovascular issues, seizures, and hyperthermia.
Child has congenital disorder after negative prenatal testing … and more
WHEN A POSSIBLE FETAL ABNORMALITY WAS SEEN on ultrasonography, the ObGyn suggested both parents have DNA testing for a hormonal disorder. Blood samples were taken in the hospital laboratory and sent to an outside lab. The parents were told that the results were negative.
The child was born with congenital adrenal hyperplasia, causing hormonal imbalance and development of ambiguous genitalia. She underwent genital reconstruction surgery at 4 months, and is expected to require additional surgery and lifelong hormone replacement therapy and monitoring.
PATIENTS’ CLAIM The hospital lab technician ordered the wrong test. The ObGyn was at fault for not confirming the test’s name. The parents would have terminated the pregnancy if they had been correctly informed of the child’s condition.
DEFENDANTS’ DEFENSE The test requested by the lab technician was similar in name to that ordered by the ObGyn. The ObGyn denied negligence; she relied on the lab to order the test she requested. The hospital claimed the error had been the fault of other entities involved in the handling and testing of the blood samples.
VERDICT A New Jersey jury found the hospital 75% and the lab technician 25% liable. The $1 million verdict included $625,000 for the child and $375,000 for her parents. A defense verdict was returned for the ObGyn.
Decision-to-delivery time challenged
A WOMAN WAS ADMITTED to the hospital for induction of labor for vaginal birth after cesarean delivery (VBAC). Because of fetal distress, the child was delivered by cesarean and later given a diagnosis of cerebral palsy. He has deficits involving grip, writing, and gait, and developmental delays.
PATIENT’S CLAIM Cesarean delivery should have been performed earlier because of a non-reassuring fetal heart rate.
PHYSICIAN’S DEFENSE The child’s heart rate was properly monitored in utero, and there were no contraindications to VBAC. As soon as the fetal tracings were disturbing, the physician converted to cesarean delivery. Only 18 minutes elapsed from the time of that decision until delivery. The child’s injury was mild and he has no cognitive impairment.
VERDICT A Mississippi defense verdict was returned.
Hematoma following vaginal hysterectomy
A 32-YEAR-OLD WOMAN underwent a vaginal hysterectomy. She developed a hematoma and was readmitted a week later for emergency surgery that included a bilateral salpingo-oophorectomy. She was scheduled for drainage of an abscess using interventional radiology, but the abscess ruptured during the preprocedure physical examination. The patient was discharged but returned the next day with serious pulmonary problems.
PATIENT’S CLAIM She chose vaginal hysterectomy to avoid scarring; now her abdomen was scarred from emergency surgery. The drainage procedure should have been performed despite the rupture. She was discharged prematurely after emergency surgery. A different antibiotic should have been prescribed.
PHYSICIAN’S DEFENSE A hematoma is a known complication of surgery. The drainage procedure was unnecessary after the rupture; the patient appeared to improve before she was discharged. Appropriate antibiotics were prescribed.
VERDICT A Ohio defense verdict was returned.
Oxygen deprivation blamed for fetal brain damage
LABOR WAS INDUCED after a mother reported a decrease in fetal movement. The child, age 9 at time of trial, has the developmental, motor, and language skills of a toddler.
PATIENT’S CLAIM The child’s grandparents, his legal guardians, claimed the doctors and nurses failed to properly monitor the oxytocin medication given to the mother, leading to oxygen deprivation that caused traumatic brain and neurological injuries.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT An Illinois settlement of $7.5 million was reached with the medical center before trial. Claims against the delivering ObGyn are still pending.
A 38-YEAR-OLD WOMAN underwent diagnostic hysteroscopy. During the procedure, visualization was poor and the gynecologist inadvertently perforated the uterus and rectum. Massive infection developed. Surgery to treat the infection and repair the injury included hysterectomy.
PATIENT’S CLAIM The gynecologist did not properly perform the hysteroscopy, and did not investigate for perforations at the end of the procedure. A small hole in the rectum allowed fecal contents to spill into the abdomen and pelvis, and caused the infection. The patient is now incapable of bearing children.
PHYSICIAN’S DEFENSE The infection that developed came solely from the perforation of the uterus, a known complication of hysteroscopy. The rectal perforation occurred during diagnostic laparoscopy and hysterectomy that was performed to treat the infection.
VERDICT A $650,000 Virginia settlement was reached.
12 lb, 7 oz baby, brachial plexus injury
A DIABETIC MOTHER GAINED 62 LBS during pregnancy. The baby, delivered vaginally, weighed 12 lbs, 7 oz. He suffered a brachial plexus injury, with avulsion injuries at C5, C6, and C7. The child’s right hand is in a pronated position; he cannot supinate without using his other hand to assist, despite three operations.
PATIENT’S CLAIM The ObGyn never discussed the risk of a large baby. Three weeks before delivery, ultrasonography estimated fetal weight at 9 lbs, 2 oz. The mother asked if cesarean delivery would be safer; the ObGyn responded that he believed the child weighed less than 10 lbs, and that a vaginal delivery would be appropriate.
PHYSICIAN’S DEFENSE The ObGyn did not offer cesarean delivery because he believed there was no medical necessity for that discussion.
VERDICT A $1,174,365 Ohio verdict was returned.
Despite gastroschisis, neonatal team called after birth
ULTRASONOGRAPHY showed fetal gastroschisis with a moderate amount of exposed bowel. The mother went into labor at 38 weeks. Electronic external fetal heart-rate tracing showed fetal bradycardia at 60–70 beats per minute (bpm). When the membranes were artificially ruptured, the amniotic fluid was full of thick meconium. A fetal scalp electrode showed a heart rate of 30–120 bpm; a second electrode confirmed the range.
The baby was delivered vaginally with Apgar scores of 2, 2, and 4 at 1, 5, and 10 minutes, respectively. The newborn was depressed, flaccid, blue, and unresponsive, with thick meconium below the vocal cords.
When the neonatal intensive care unit (NICU) team arrived, the baby was making no respiratory effort, and had a heart rate of 60 bpm. Meconium blocked the airway; he was intubated at 4 minutes of life. Arterial blood
sampling showed severe metabolic acidosis from hypoxia. Gastroschisis ruled out fetal cooling, which might have ameliorated the brain injury. The child suffered hypoxic ischemic encephalopathy from intrapartum asyphyxia that led to microcephaly. He requires a feeding tube and lifetime care.
PATIENT’S CLAIM Knowing that gastroschisis was present, the NICU team should have been called to the patient’s bedside before her membranes were ruptured. A cesarean delivery should have been performed when fetal distress was evident.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT A $2.8 million Virginia settlement was reached: $1.8 million for the child; $1 million for the mother.
Twin-to-twin transfusion syndrome
A WOMAN EXPECTING TWINS had multiple ultrasonographic studies during pregnancy; all were read as normal. The babies were born prematurely and both died shortly after birth.
PATIENT’S CLAIM The radiologist and two ObGyns failed to correctly analyze the sonograms and diagnose and treat twin-to-twin transfusion syndrome.
PHYSICIANS’ DEFENSE The case was settled before trial.
VERDICT A $375,000 Virginia settlement was reached.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
WHEN A POSSIBLE FETAL ABNORMALITY WAS SEEN on ultrasonography, the ObGyn suggested both parents have DNA testing for a hormonal disorder. Blood samples were taken in the hospital laboratory and sent to an outside lab. The parents were told that the results were negative.
The child was born with congenital adrenal hyperplasia, causing hormonal imbalance and development of ambiguous genitalia. She underwent genital reconstruction surgery at 4 months, and is expected to require additional surgery and lifelong hormone replacement therapy and monitoring.
PATIENTS’ CLAIM The hospital lab technician ordered the wrong test. The ObGyn was at fault for not confirming the test’s name. The parents would have terminated the pregnancy if they had been correctly informed of the child’s condition.
DEFENDANTS’ DEFENSE The test requested by the lab technician was similar in name to that ordered by the ObGyn. The ObGyn denied negligence; she relied on the lab to order the test she requested. The hospital claimed the error had been the fault of other entities involved in the handling and testing of the blood samples.
VERDICT A New Jersey jury found the hospital 75% and the lab technician 25% liable. The $1 million verdict included $625,000 for the child and $375,000 for her parents. A defense verdict was returned for the ObGyn.
Decision-to-delivery time challenged
A WOMAN WAS ADMITTED to the hospital for induction of labor for vaginal birth after cesarean delivery (VBAC). Because of fetal distress, the child was delivered by cesarean and later given a diagnosis of cerebral palsy. He has deficits involving grip, writing, and gait, and developmental delays.
PATIENT’S CLAIM Cesarean delivery should have been performed earlier because of a non-reassuring fetal heart rate.
PHYSICIAN’S DEFENSE The child’s heart rate was properly monitored in utero, and there were no contraindications to VBAC. As soon as the fetal tracings were disturbing, the physician converted to cesarean delivery. Only 18 minutes elapsed from the time of that decision until delivery. The child’s injury was mild and he has no cognitive impairment.
VERDICT A Mississippi defense verdict was returned.
Hematoma following vaginal hysterectomy
A 32-YEAR-OLD WOMAN underwent a vaginal hysterectomy. She developed a hematoma and was readmitted a week later for emergency surgery that included a bilateral salpingo-oophorectomy. She was scheduled for drainage of an abscess using interventional radiology, but the abscess ruptured during the preprocedure physical examination. The patient was discharged but returned the next day with serious pulmonary problems.
PATIENT’S CLAIM She chose vaginal hysterectomy to avoid scarring; now her abdomen was scarred from emergency surgery. The drainage procedure should have been performed despite the rupture. She was discharged prematurely after emergency surgery. A different antibiotic should have been prescribed.
PHYSICIAN’S DEFENSE A hematoma is a known complication of surgery. The drainage procedure was unnecessary after the rupture; the patient appeared to improve before she was discharged. Appropriate antibiotics were prescribed.
VERDICT A Ohio defense verdict was returned.
Oxygen deprivation blamed for fetal brain damage
LABOR WAS INDUCED after a mother reported a decrease in fetal movement. The child, age 9 at time of trial, has the developmental, motor, and language skills of a toddler.
PATIENT’S CLAIM The child’s grandparents, his legal guardians, claimed the doctors and nurses failed to properly monitor the oxytocin medication given to the mother, leading to oxygen deprivation that caused traumatic brain and neurological injuries.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT An Illinois settlement of $7.5 million was reached with the medical center before trial. Claims against the delivering ObGyn are still pending.
A 38-YEAR-OLD WOMAN underwent diagnostic hysteroscopy. During the procedure, visualization was poor and the gynecologist inadvertently perforated the uterus and rectum. Massive infection developed. Surgery to treat the infection and repair the injury included hysterectomy.
PATIENT’S CLAIM The gynecologist did not properly perform the hysteroscopy, and did not investigate for perforations at the end of the procedure. A small hole in the rectum allowed fecal contents to spill into the abdomen and pelvis, and caused the infection. The patient is now incapable of bearing children.
PHYSICIAN’S DEFENSE The infection that developed came solely from the perforation of the uterus, a known complication of hysteroscopy. The rectal perforation occurred during diagnostic laparoscopy and hysterectomy that was performed to treat the infection.
VERDICT A $650,000 Virginia settlement was reached.
12 lb, 7 oz baby, brachial plexus injury
A DIABETIC MOTHER GAINED 62 LBS during pregnancy. The baby, delivered vaginally, weighed 12 lbs, 7 oz. He suffered a brachial plexus injury, with avulsion injuries at C5, C6, and C7. The child’s right hand is in a pronated position; he cannot supinate without using his other hand to assist, despite three operations.
PATIENT’S CLAIM The ObGyn never discussed the risk of a large baby. Three weeks before delivery, ultrasonography estimated fetal weight at 9 lbs, 2 oz. The mother asked if cesarean delivery would be safer; the ObGyn responded that he believed the child weighed less than 10 lbs, and that a vaginal delivery would be appropriate.
PHYSICIAN’S DEFENSE The ObGyn did not offer cesarean delivery because he believed there was no medical necessity for that discussion.
VERDICT A $1,174,365 Ohio verdict was returned.
Despite gastroschisis, neonatal team called after birth
ULTRASONOGRAPHY showed fetal gastroschisis with a moderate amount of exposed bowel. The mother went into labor at 38 weeks. Electronic external fetal heart-rate tracing showed fetal bradycardia at 60–70 beats per minute (bpm). When the membranes were artificially ruptured, the amniotic fluid was full of thick meconium. A fetal scalp electrode showed a heart rate of 30–120 bpm; a second electrode confirmed the range.
The baby was delivered vaginally with Apgar scores of 2, 2, and 4 at 1, 5, and 10 minutes, respectively. The newborn was depressed, flaccid, blue, and unresponsive, with thick meconium below the vocal cords.
When the neonatal intensive care unit (NICU) team arrived, the baby was making no respiratory effort, and had a heart rate of 60 bpm. Meconium blocked the airway; he was intubated at 4 minutes of life. Arterial blood
sampling showed severe metabolic acidosis from hypoxia. Gastroschisis ruled out fetal cooling, which might have ameliorated the brain injury. The child suffered hypoxic ischemic encephalopathy from intrapartum asyphyxia that led to microcephaly. He requires a feeding tube and lifetime care.
PATIENT’S CLAIM Knowing that gastroschisis was present, the NICU team should have been called to the patient’s bedside before her membranes were ruptured. A cesarean delivery should have been performed when fetal distress was evident.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT A $2.8 million Virginia settlement was reached: $1.8 million for the child; $1 million for the mother.
Twin-to-twin transfusion syndrome
A WOMAN EXPECTING TWINS had multiple ultrasonographic studies during pregnancy; all were read as normal. The babies were born prematurely and both died shortly after birth.
PATIENT’S CLAIM The radiologist and two ObGyns failed to correctly analyze the sonograms and diagnose and treat twin-to-twin transfusion syndrome.
PHYSICIANS’ DEFENSE The case was settled before trial.
VERDICT A $375,000 Virginia settlement was reached.
WHEN A POSSIBLE FETAL ABNORMALITY WAS SEEN on ultrasonography, the ObGyn suggested both parents have DNA testing for a hormonal disorder. Blood samples were taken in the hospital laboratory and sent to an outside lab. The parents were told that the results were negative.
The child was born with congenital adrenal hyperplasia, causing hormonal imbalance and development of ambiguous genitalia. She underwent genital reconstruction surgery at 4 months, and is expected to require additional surgery and lifelong hormone replacement therapy and monitoring.
PATIENTS’ CLAIM The hospital lab technician ordered the wrong test. The ObGyn was at fault for not confirming the test’s name. The parents would have terminated the pregnancy if they had been correctly informed of the child’s condition.
DEFENDANTS’ DEFENSE The test requested by the lab technician was similar in name to that ordered by the ObGyn. The ObGyn denied negligence; she relied on the lab to order the test she requested. The hospital claimed the error had been the fault of other entities involved in the handling and testing of the blood samples.
VERDICT A New Jersey jury found the hospital 75% and the lab technician 25% liable. The $1 million verdict included $625,000 for the child and $375,000 for her parents. A defense verdict was returned for the ObGyn.
Decision-to-delivery time challenged
A WOMAN WAS ADMITTED to the hospital for induction of labor for vaginal birth after cesarean delivery (VBAC). Because of fetal distress, the child was delivered by cesarean and later given a diagnosis of cerebral palsy. He has deficits involving grip, writing, and gait, and developmental delays.
PATIENT’S CLAIM Cesarean delivery should have been performed earlier because of a non-reassuring fetal heart rate.
PHYSICIAN’S DEFENSE The child’s heart rate was properly monitored in utero, and there were no contraindications to VBAC. As soon as the fetal tracings were disturbing, the physician converted to cesarean delivery. Only 18 minutes elapsed from the time of that decision until delivery. The child’s injury was mild and he has no cognitive impairment.
VERDICT A Mississippi defense verdict was returned.
Hematoma following vaginal hysterectomy
A 32-YEAR-OLD WOMAN underwent a vaginal hysterectomy. She developed a hematoma and was readmitted a week later for emergency surgery that included a bilateral salpingo-oophorectomy. She was scheduled for drainage of an abscess using interventional radiology, but the abscess ruptured during the preprocedure physical examination. The patient was discharged but returned the next day with serious pulmonary problems.
PATIENT’S CLAIM She chose vaginal hysterectomy to avoid scarring; now her abdomen was scarred from emergency surgery. The drainage procedure should have been performed despite the rupture. She was discharged prematurely after emergency surgery. A different antibiotic should have been prescribed.
PHYSICIAN’S DEFENSE A hematoma is a known complication of surgery. The drainage procedure was unnecessary after the rupture; the patient appeared to improve before she was discharged. Appropriate antibiotics were prescribed.
VERDICT A Ohio defense verdict was returned.
Oxygen deprivation blamed for fetal brain damage
LABOR WAS INDUCED after a mother reported a decrease in fetal movement. The child, age 9 at time of trial, has the developmental, motor, and language skills of a toddler.
PATIENT’S CLAIM The child’s grandparents, his legal guardians, claimed the doctors and nurses failed to properly monitor the oxytocin medication given to the mother, leading to oxygen deprivation that caused traumatic brain and neurological injuries.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT An Illinois settlement of $7.5 million was reached with the medical center before trial. Claims against the delivering ObGyn are still pending.
A 38-YEAR-OLD WOMAN underwent diagnostic hysteroscopy. During the procedure, visualization was poor and the gynecologist inadvertently perforated the uterus and rectum. Massive infection developed. Surgery to treat the infection and repair the injury included hysterectomy.
PATIENT’S CLAIM The gynecologist did not properly perform the hysteroscopy, and did not investigate for perforations at the end of the procedure. A small hole in the rectum allowed fecal contents to spill into the abdomen and pelvis, and caused the infection. The patient is now incapable of bearing children.
PHYSICIAN’S DEFENSE The infection that developed came solely from the perforation of the uterus, a known complication of hysteroscopy. The rectal perforation occurred during diagnostic laparoscopy and hysterectomy that was performed to treat the infection.
VERDICT A $650,000 Virginia settlement was reached.
12 lb, 7 oz baby, brachial plexus injury
A DIABETIC MOTHER GAINED 62 LBS during pregnancy. The baby, delivered vaginally, weighed 12 lbs, 7 oz. He suffered a brachial plexus injury, with avulsion injuries at C5, C6, and C7. The child’s right hand is in a pronated position; he cannot supinate without using his other hand to assist, despite three operations.
PATIENT’S CLAIM The ObGyn never discussed the risk of a large baby. Three weeks before delivery, ultrasonography estimated fetal weight at 9 lbs, 2 oz. The mother asked if cesarean delivery would be safer; the ObGyn responded that he believed the child weighed less than 10 lbs, and that a vaginal delivery would be appropriate.
PHYSICIAN’S DEFENSE The ObGyn did not offer cesarean delivery because he believed there was no medical necessity for that discussion.
VERDICT A $1,174,365 Ohio verdict was returned.
Despite gastroschisis, neonatal team called after birth
ULTRASONOGRAPHY showed fetal gastroschisis with a moderate amount of exposed bowel. The mother went into labor at 38 weeks. Electronic external fetal heart-rate tracing showed fetal bradycardia at 60–70 beats per minute (bpm). When the membranes were artificially ruptured, the amniotic fluid was full of thick meconium. A fetal scalp electrode showed a heart rate of 30–120 bpm; a second electrode confirmed the range.
The baby was delivered vaginally with Apgar scores of 2, 2, and 4 at 1, 5, and 10 minutes, respectively. The newborn was depressed, flaccid, blue, and unresponsive, with thick meconium below the vocal cords.
When the neonatal intensive care unit (NICU) team arrived, the baby was making no respiratory effort, and had a heart rate of 60 bpm. Meconium blocked the airway; he was intubated at 4 minutes of life. Arterial blood
sampling showed severe metabolic acidosis from hypoxia. Gastroschisis ruled out fetal cooling, which might have ameliorated the brain injury. The child suffered hypoxic ischemic encephalopathy from intrapartum asyphyxia that led to microcephaly. He requires a feeding tube and lifetime care.
PATIENT’S CLAIM Knowing that gastroschisis was present, the NICU team should have been called to the patient’s bedside before her membranes were ruptured. A cesarean delivery should have been performed when fetal distress was evident.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT A $2.8 million Virginia settlement was reached: $1.8 million for the child; $1 million for the mother.
Twin-to-twin transfusion syndrome
A WOMAN EXPECTING TWINS had multiple ultrasonographic studies during pregnancy; all were read as normal. The babies were born prematurely and both died shortly after birth.
PATIENT’S CLAIM The radiologist and two ObGyns failed to correctly analyze the sonograms and diagnose and treat twin-to-twin transfusion syndrome.
PHYSICIANS’ DEFENSE The case was settled before trial.
VERDICT A $375,000 Virginia settlement was reached.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
Evaluating older adults’ capacity and need for guardianship
Discuss this article at www.facebook.com/CurrentPsychiatry
Although forensic psychiatrists typically are consulted in complex legal matters, geriatric, consultation-liaison, and general psychiatrists are on the front lines of assessing capacity to give informed consent and need for guardianship. Psychiatrists often find such consultations daunting because residency training usually includes little to no formal training in performing psycho-legal assessments. Evaluating issues such as decision-making capacity, guardianship, and capacity to give informed consent requires a delicate balance between autonomy and beneficence. This article reviews 4 common legal issues in geriatric consultation—capacity evaluations, informed consent, guardianship, and elder abuse—and suggests a systematic approach to psycho-legal consultations in older adults.
Confidentiality and dual agency
Every psychiatrist should be familiar with basic principles of medical ethics as well as key aspects of local mental health law. Relevant ethical principles include autonomy, beneficence, confidentiality, and dual agency. A review of all these ethical issues is beyond the scope of this article, so here I highlight confidentiality and dual agency.
Confidentiality—the clinician’s obligation not to disclose private medical information—is a legal as well as an ethical requirement. A psychiatrist who performs a psycho-legal evaluation must disclose to the patient the purpose of the evaluation, that a report will be prepared, and to whom it will be submitted. Exceptions to confidentiality include medical emergencies, mandatory reporting of abuse and infectious diseases, and the duty to protect (warning police and the intended victim when a patient makes a threat).
Dual agency or dual role refers to serving as both a treating physician and a forensic evaluator. Although it is ideal to avoid serving in a dual role, sometimes it is impractical or impossible to avoid doing so, such as in guardianship or civil commitment evaluations, or in state forensic hospitals. In such cases, the psychiatrist must be aware of potential conflicts between clinical and forensic evaluations. A treating psychiatrist primarily serves his or her patient’s best interest, whereas a forensic psychiatrist primarily seeks truth.1 A treating psychiatrist is at risk of consciously or unconsciously biasing his or her psycho-legal evaluation in favor of or against the patient/litigant, depending upon the psychiatrist’s countertransference. Further, performing a psycho-legal evaluation can cause problems in ongoing treatment. A psychiatrist who testifies that his or her fiercely independent patient needs a guardian or nursing home placement will experience significant challenges in continuing to work with that patient.
4 common issues for older adults
Decision-making capacity. Although “capacity” and “competence” often are used interchangeably, “capacity” broadly refers to the ability to perform a specific task, whereas “competence” refers to the legally defined standard for performing a specific task such as making a will. “Competence” is legally determined, whereas “capacity” may be determined clinically.
Capacity usually is task-specific rather than a general construct. The existence of physical or mental illness per se does not mean that a patient lacks capacity. Rather, capacity is determined by whether an individual has specific abilities, regardless of diagnosis. Specific capacities include the ability to give informed consent, manage finances, make a will, or enter into contracts (Table 1).2-4 Appelbaum and Gutheil describe 4 components for assessing specific capacity:
- communication of a choice
- factual understanding of the issues
- appreciation of the situation and its consequences
- rational manipulation of information.5,6
Table 1
Criteria of 3 specific capacities
| Capacity | Criteria |
|---|---|
| Capacity to give informed consent | Understand nature of illness and treatment Understand risks and benefits of treatment Understand treatment alternatives Understand risk of refusing treatment |
| Testamentary capacity | Understand that he/she is making a will Know the nature and extent of their property Understand the “natural objects” of their bounty and their claims upon them |
| Contractual capacity | Understand the transaction Act in a reasonable manner |
| Source: References 2-4 | |
Ability to communicate a choice refers to a patient’s ability to express his or her wishes in a reasonably stable manner. Factual understanding of the issues refers to an individual’s ability to understand the relevant facts before making a decision. Appreciation of the situation and its consequences refers to a person’s ability to rationally understand the effect of decisions. Appreciation is a higher level of understanding than mere factual understanding—eg, a delusional patient who believes himself immortal may intellectually understand that a surgical procedure carries a 50% mortality risk, but may be unable to appreciate the information as it relates to him because he believes he is immortal. Rational manipulation of information refers to a patient’s reasoning process and how the patient integrates data into his or her decision-making process.5
Informed consent. In my experience, capacity to give informed consent is the most commonly requested specific capacity assessment in general medical settings. Informed consent must be knowing, voluntary, and competent. All material information—information that would cause a reasonable person to accept or reject a proposed treatment—should be communicated to the patient. Informed consent requires an understanding of the patient’s condition and indication for treatment, risks and benefits of and alternatives to treatment, and risks of declining treatment.2,3 Exceptions to informed consent include incompetence, medical emergencies, patient waiver of informed consent, and a limited therapeutic privilege (when a physician determines that the information would harm the patient).3
Several instruments can help clinicians assess patients’ capacity to give informed consent. The benefits of using a structured instrument include:
- ensuring that specific information is covered during each evaluation
- systematically recording a patient’s response.5
Disadvantages of using instruments include the fact that no instrument can take into account all aspects of a particular case, and some instruments are time-consuming and require training. Structured instruments can be a useful adjunct to the clinical interview in some cases, but should not substitute for it. In a review of 23 instruments for assessing decisional capacity to consent to treatment or clinical research, the MacArthur Competence Assessment Tool for Clinical Research and the MacArthur Competence Assessment Tool for Treatment had the most empirical support, although the authors noted that other instruments might be better suited to specific situations.7
Psychiatrists may be consulted when a patient refuses treatment or decides to leave the hospital against medical advice. The key issue in both situations is whether the patient has capacity to refuse treatment.8 If there is evidence that the patient is mentally ill and poses an imminent risk of suicide or violence or is unable to provide for his or her basic needs, the psychiatrist should assess whether the patient meets criteria for civil commitment.
Many clinicians employ a “sliding scale” approach to competence, requiring a lower degree of competence for consenting to low-risk, high-benefit interventions and a greater degree of competence for higher-risk procedures.5,9 Family members often serve as informal surrogate decision makers for incapacitated patients, except when there is significant family discord or no family members are available.5
Guardianship. Guardians are appointed by courts to make decisions for individuals who have been found incompetent (wards). Although its purposes are beneficent, the guardianship system could do significant harm.10 Determining that an individual is incompetent is tantamount to depriving him or her of basic personhood. In many cases, the ward loses the ability to consent to or refuse medical care, manage his or her finances, enter into contracts, marry, and determine where he or she will live. On the other hand, failing to recognize incompetence can leave a vulnerable person in danger of physical deterioration, abuse, neglect, or exploitation.
It is critical that guardianship evaluations be conducted carefully. In a review of 298 guardianship cases from 3 states, Moye and colleagues11 found that the quality of the reports was significantly better in Colorado, a state with guardianship reforms, but documentation of functional strengths and weaknesses was “particularly rare” in guardianship evaluations and prognosis often was not included. This information is relevant to judges, who need to determine which areas of function are impaired and how long the impairment is likely to last.
Guardianship evaluations often focus on general rather than specific capacity. In other words, often there is not a specific task such as consenting to surgery that the alleged incompetent person needs to perform. Rather, the question is whether an individual can manage his or her finances or make treatment decisions in general. Appelbaum and Gutheil suggest considering 6 factors when assessing general capacity:
- awareness of the situation
- factual understanding of the issues
- appreciation of the likely consequences
- rational manipulation of information
- functioning in one’s environment
- extent of demands on patient.5
The first 4 are closely related to the elements of specific capacity described above. Functioning in one’s environment and extent of demands on the patient attempt to anticipate the tasks that an individual will need to perform. A patient with mild dementia may be unable to manage a complex estate but can handle a bank account and a fixed income. Similarly, it is important to consider the patient’s support system. An impaired patient may function adequately with his wife’s help but may lose the capacity to live independently if his wife dies or becomes impaired.
Traditionally, guardianship has resulted in a complete loss of decision-making ability. Several state legislatures have passed laws allowing for limited guardianship, although orders for limited guardianship remain underutilized.10 Limited guardianship delineates specific areas of incompetence and limits the guardian’s decision-making authority to those areas while leaving intact the ward’s ability to make all other decisions for himself or herself.
The use of less-restrictive alternatives to guardianship—such as powers of attorney, durable powers of attorney, living wills, payees, and trusts—is increasing. A power of attorney allows a patient to authorize a specific individual to act on his or her behalf. The scope of the power of attorney can be limited, such as to manage finances or even to a specific transaction, such as selling a home or car. A durable power of attorney also allows an agent to make decisions on the patient’s behalf but becomes active only when the patient becomes incompetent. It often is used to appoint an individual to make medical decisions on behalf of an incompetent patient. Living wills allow patients to determine what treatment they would like in the event they become incompetent.
Elder abuse. An estimated 1 to 2 million adults age >65 have been abused, exploited, or neglected.12 Elder abuse includes physical abuse, neglect, emotional abuse, sexual abuse, and financial exploitation (including undue influence). Most states have mandatory reporting of elder abuse, although they vary regarding who must report and what the report must entail. Psychiatrists should be vigilant in looking for signs of elder abuse (Table 2),13 regardless of the reason for the consult.
Table 2
Signs of elder abuse: What to wlook for
| Type of abuse | Signs |
|---|---|
| Physical | Bruises, burns (especially circular, suggesting cigarette burns), slap marks |
| Sexual | Unexplained sexually transmitted diseases, bruises in genital area, breasts, or anal area |
| Emotional | Withdrawal, new-onset depression |
| Financial | Sudden loss of property, unusual increase in spending, checks paid in large, round numbers, checks marked as gifts or loans |
| Neglect | Malnutrition, lack of medical care, poor hygiene, pressure ulcers |
| Source: Reference 13 | |
10 tips for thorough evaluations
1. Consider the context of the consultation. This includes medical factors (such as the patient’s condition, prognosis, relationship with the treatment team, and recommended course of treatment), legal factors (eg, pending litigation and relevant legal standards for issues such as guardianship), and psychosocial issues (eg, the patient’s current support structure and family conflicts).
2. Identify the legal issue and any relevant legal standards. The legal standard will inform you of the issues you need to address in the evaluation. If an attorney has consulted you, ask him or her to provide the legal standard.
3. Gather relevant collateral information, which may include interviews with family members or a review of financial or medical records.
4. Explain the purpose of the examination and the limits of confidentiality.
5. Perform a focused psychiatric evaluation, paying special attention to cognitive functioning, reasoning, and unusual thought content such as delusional beliefs.
6. Perform an interview specific to the referral issue.
7. Consider using a relevant assessment instrument.
8. Consider psychological testing, laboratory testing, imaging, or further medical evaluation. These assessments can help determine the diagnosis, the cause of any deficits in capacity, and whether any deficits are reversible.
9. Determine what opinions you are able to render. Limit opinions and remember that it may be appropriate to decline to address certain issues if there is insufficient information or if the issue is outside your area of expertise.
10. Prepare a written report. Consider the audience. Minimize the use of medical jargon and define all medical terms. State your opinions clearly and with reasonable medical certainty (in most jurisdictions, this means more likely than not). State the basis for all opinions.
For a case study that provides an example of a psycho-legal evaluation of a geriatric patient, see the Box.
Mr. A, age 75, recently started taking a dopaminergic agonist to treat Parkinson’s disease. He says he wants to divorce his wife of 35 years because of “scandalous affairs” she allegedly engaged in. His wife reports that he has been accusing her of having affairs with various men, including a man who recently painted their house.
On evaluation, Mr. A’s Mini-Mental State Examination score is 30/30. He has no signs of depression and his sleep patterns have not changed. There have been no changes in his spending patterns, although he no longer gives his wife money for grocery shopping, telling her to get money from her “boyfriends.” He is adamant about this decision, saying, “It’s my money and I can do with it as I please. This is still a free country, isn’t it?”
He says he has $70,000 in his individual retirement account, $20,000 in his bank account, and receives a pension of $1,785 per month. He estimates that his home is worth $200,000. His financial records essentially are consistent with his reports. He is able to perform basic calculations without difficulty and is aware of his monthly expenses. He describes his relationship with his wife by saying, “It was fine until she started screwing around.”
When asked about the likely consequences of his decision, he shrugs and says, “I guess she’ll have to get money from her boyfriends. I don’t really care who she sees as long as they stay away from me.” He denies having thoughts of harming his wife or her alleged “boyfriends.” He recognizes that his wife might divorce him, leaving him alone.
When I ask Mr. A if it is possible he is mistaken in his belief that his wife is having affairs, he says, “No, doctor. You don’t know her.” When I ask how he knows she is having affairs, he says that the painter started looking at him “funny” and that the busboy at a restaurant they frequent called his wife “dear.” He believes his wife is having sexual relations with both of these men.
Does Mr. A require a guardian? I opine that Mr. A requires a guardian of estate (to manage his property) but not a guardian of person because he is capable of making decisions about his medical care and other personal decisions. He is failing to care for his wife because of his delusional jealousy. Although cognitively intact, he is unable to appreciate the consequences of his actions or rationally manipulate information because of his delusional thinking. He believes he is “cutting off” an unfaithful spouse when, in fact, there is no evidence that she has been unfaithful. His inability to rationally manipulate information is demonstrated by the fact that he uses innocuous facts such as a busboy calling his elderly wife “dear” to support his delusion that she was having affairs.
I note that his psychosis is reversible because it is likely due to his antiparkinsonian regimen. However, he declines both a dose reduction in his medication and antipsychotic treatment. I note that should his psychosis resolve, he may regain financial decision-making capacity.
Related Resources
- American Academy of Psychiatry and the Law. www.aapl.org.
- American Bar Association Commission on Law and Aging. www.americanbar.org/groups/law_aging.html.
- National Center on Elder Abuse. www.ncea.aoa.gov.
Disclosure
Dr. Soliman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
The author thanks forensic psychiatry fellow Abhishek Jain, MD for reviewing the article and offering editorial suggestions.
1. American Academy of Psychiatry and the Law. Ethics guidelines for the practice of forensic psychiatry. http://www.aapl.org/ethics.htm. Adopted May 2005. Accessed February 24 2012.
2. Blank K. Legal and ethical issues. In: Sadvoy J Jarvik LF, Grossberg GT, et al, eds. Comprehensive textbook of geriatric psychiatry. 3rd ed. New York, NY: American Association of Geriatric Psychiatry; 2004:1183-1206.
3. Schwartz HI, Mack DM. Informed consent and competency. In: Rosner R ed. Principles and practice of forensic psychiatry. 2nd ed. New York, NY: Oxford University Press; 2003:97-106.
4. Ciccone RJ. Civil competencies. In: Rosner R ed. Principles and practice of forensic psychiatry. 2nd ed. New York, NY: Oxford University Press; 2003:308-315.
5. Appelbaum PS, Gutheil TG. Competence and substitute decision making. In: Appelbaum PS Gutheil TG, eds. Clinical handbook of psychiatry and the law. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:177-213.
6. Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
7. Dunn LB, Nowrangi MA, Palmer BW, et al. Assessing decisional capacity for clinical research or treatment: a review of instruments. Am J Psychiatry. 2006;163(8):1323-1334.
8. Resnick PJ, Sorrentino R. Forensic issues in consultation-liaison psychiatry. Psychiatric Times. 2005;23(14).-
9. Drane JF. The many faces of competency. Hastings Cent Rep. 1985;15(2):17-21.
10. Wright JL. Guardianship for your own good: improving the well being of respondents and wards in the USA. Int J Law Psychiatry. 2010;33(5-6):350-368.
11. Moye J, Wood S, Edelstein B, et al. Clinical evidence in guardianship of older adults is inadequate: findings from a tri-state study. Gerontologist. 2007;47(5):604-612.
12. National Center on Elder Abuse. Fact sheet: elder abuse prevalence and incidence. http://www.ncea.aoa.gov/NCEAroot/Main_Site/pdf/publication/FinalStatistics050331.pdf. Accessed February 24, 2012.
13. National Center on Elder Abuse. Why should I care about elder abuse? http://www.ncea.aoa.gov/Main_Site/pdf/publication/NCEA_WhatIsAbuse-2010.pdf. Published March 3 2010. Accessed February 24, 2012.
Discuss this article at www.facebook.com/CurrentPsychiatry
Although forensic psychiatrists typically are consulted in complex legal matters, geriatric, consultation-liaison, and general psychiatrists are on the front lines of assessing capacity to give informed consent and need for guardianship. Psychiatrists often find such consultations daunting because residency training usually includes little to no formal training in performing psycho-legal assessments. Evaluating issues such as decision-making capacity, guardianship, and capacity to give informed consent requires a delicate balance between autonomy and beneficence. This article reviews 4 common legal issues in geriatric consultation—capacity evaluations, informed consent, guardianship, and elder abuse—and suggests a systematic approach to psycho-legal consultations in older adults.
Confidentiality and dual agency
Every psychiatrist should be familiar with basic principles of medical ethics as well as key aspects of local mental health law. Relevant ethical principles include autonomy, beneficence, confidentiality, and dual agency. A review of all these ethical issues is beyond the scope of this article, so here I highlight confidentiality and dual agency.
Confidentiality—the clinician’s obligation not to disclose private medical information—is a legal as well as an ethical requirement. A psychiatrist who performs a psycho-legal evaluation must disclose to the patient the purpose of the evaluation, that a report will be prepared, and to whom it will be submitted. Exceptions to confidentiality include medical emergencies, mandatory reporting of abuse and infectious diseases, and the duty to protect (warning police and the intended victim when a patient makes a threat).
Dual agency or dual role refers to serving as both a treating physician and a forensic evaluator. Although it is ideal to avoid serving in a dual role, sometimes it is impractical or impossible to avoid doing so, such as in guardianship or civil commitment evaluations, or in state forensic hospitals. In such cases, the psychiatrist must be aware of potential conflicts between clinical and forensic evaluations. A treating psychiatrist primarily serves his or her patient’s best interest, whereas a forensic psychiatrist primarily seeks truth.1 A treating psychiatrist is at risk of consciously or unconsciously biasing his or her psycho-legal evaluation in favor of or against the patient/litigant, depending upon the psychiatrist’s countertransference. Further, performing a psycho-legal evaluation can cause problems in ongoing treatment. A psychiatrist who testifies that his or her fiercely independent patient needs a guardian or nursing home placement will experience significant challenges in continuing to work with that patient.
4 common issues for older adults
Decision-making capacity. Although “capacity” and “competence” often are used interchangeably, “capacity” broadly refers to the ability to perform a specific task, whereas “competence” refers to the legally defined standard for performing a specific task such as making a will. “Competence” is legally determined, whereas “capacity” may be determined clinically.
Capacity usually is task-specific rather than a general construct. The existence of physical or mental illness per se does not mean that a patient lacks capacity. Rather, capacity is determined by whether an individual has specific abilities, regardless of diagnosis. Specific capacities include the ability to give informed consent, manage finances, make a will, or enter into contracts (Table 1).2-4 Appelbaum and Gutheil describe 4 components for assessing specific capacity:
- communication of a choice
- factual understanding of the issues
- appreciation of the situation and its consequences
- rational manipulation of information.5,6
Table 1
Criteria of 3 specific capacities
| Capacity | Criteria |
|---|---|
| Capacity to give informed consent | Understand nature of illness and treatment Understand risks and benefits of treatment Understand treatment alternatives Understand risk of refusing treatment |
| Testamentary capacity | Understand that he/she is making a will Know the nature and extent of their property Understand the “natural objects” of their bounty and their claims upon them |
| Contractual capacity | Understand the transaction Act in a reasonable manner |
| Source: References 2-4 | |
Ability to communicate a choice refers to a patient’s ability to express his or her wishes in a reasonably stable manner. Factual understanding of the issues refers to an individual’s ability to understand the relevant facts before making a decision. Appreciation of the situation and its consequences refers to a person’s ability to rationally understand the effect of decisions. Appreciation is a higher level of understanding than mere factual understanding—eg, a delusional patient who believes himself immortal may intellectually understand that a surgical procedure carries a 50% mortality risk, but may be unable to appreciate the information as it relates to him because he believes he is immortal. Rational manipulation of information refers to a patient’s reasoning process and how the patient integrates data into his or her decision-making process.5
Informed consent. In my experience, capacity to give informed consent is the most commonly requested specific capacity assessment in general medical settings. Informed consent must be knowing, voluntary, and competent. All material information—information that would cause a reasonable person to accept or reject a proposed treatment—should be communicated to the patient. Informed consent requires an understanding of the patient’s condition and indication for treatment, risks and benefits of and alternatives to treatment, and risks of declining treatment.2,3 Exceptions to informed consent include incompetence, medical emergencies, patient waiver of informed consent, and a limited therapeutic privilege (when a physician determines that the information would harm the patient).3
Several instruments can help clinicians assess patients’ capacity to give informed consent. The benefits of using a structured instrument include:
- ensuring that specific information is covered during each evaluation
- systematically recording a patient’s response.5
Disadvantages of using instruments include the fact that no instrument can take into account all aspects of a particular case, and some instruments are time-consuming and require training. Structured instruments can be a useful adjunct to the clinical interview in some cases, but should not substitute for it. In a review of 23 instruments for assessing decisional capacity to consent to treatment or clinical research, the MacArthur Competence Assessment Tool for Clinical Research and the MacArthur Competence Assessment Tool for Treatment had the most empirical support, although the authors noted that other instruments might be better suited to specific situations.7
Psychiatrists may be consulted when a patient refuses treatment or decides to leave the hospital against medical advice. The key issue in both situations is whether the patient has capacity to refuse treatment.8 If there is evidence that the patient is mentally ill and poses an imminent risk of suicide or violence or is unable to provide for his or her basic needs, the psychiatrist should assess whether the patient meets criteria for civil commitment.
Many clinicians employ a “sliding scale” approach to competence, requiring a lower degree of competence for consenting to low-risk, high-benefit interventions and a greater degree of competence for higher-risk procedures.5,9 Family members often serve as informal surrogate decision makers for incapacitated patients, except when there is significant family discord or no family members are available.5
Guardianship. Guardians are appointed by courts to make decisions for individuals who have been found incompetent (wards). Although its purposes are beneficent, the guardianship system could do significant harm.10 Determining that an individual is incompetent is tantamount to depriving him or her of basic personhood. In many cases, the ward loses the ability to consent to or refuse medical care, manage his or her finances, enter into contracts, marry, and determine where he or she will live. On the other hand, failing to recognize incompetence can leave a vulnerable person in danger of physical deterioration, abuse, neglect, or exploitation.
It is critical that guardianship evaluations be conducted carefully. In a review of 298 guardianship cases from 3 states, Moye and colleagues11 found that the quality of the reports was significantly better in Colorado, a state with guardianship reforms, but documentation of functional strengths and weaknesses was “particularly rare” in guardianship evaluations and prognosis often was not included. This information is relevant to judges, who need to determine which areas of function are impaired and how long the impairment is likely to last.
Guardianship evaluations often focus on general rather than specific capacity. In other words, often there is not a specific task such as consenting to surgery that the alleged incompetent person needs to perform. Rather, the question is whether an individual can manage his or her finances or make treatment decisions in general. Appelbaum and Gutheil suggest considering 6 factors when assessing general capacity:
- awareness of the situation
- factual understanding of the issues
- appreciation of the likely consequences
- rational manipulation of information
- functioning in one’s environment
- extent of demands on patient.5
The first 4 are closely related to the elements of specific capacity described above. Functioning in one’s environment and extent of demands on the patient attempt to anticipate the tasks that an individual will need to perform. A patient with mild dementia may be unable to manage a complex estate but can handle a bank account and a fixed income. Similarly, it is important to consider the patient’s support system. An impaired patient may function adequately with his wife’s help but may lose the capacity to live independently if his wife dies or becomes impaired.
Traditionally, guardianship has resulted in a complete loss of decision-making ability. Several state legislatures have passed laws allowing for limited guardianship, although orders for limited guardianship remain underutilized.10 Limited guardianship delineates specific areas of incompetence and limits the guardian’s decision-making authority to those areas while leaving intact the ward’s ability to make all other decisions for himself or herself.
The use of less-restrictive alternatives to guardianship—such as powers of attorney, durable powers of attorney, living wills, payees, and trusts—is increasing. A power of attorney allows a patient to authorize a specific individual to act on his or her behalf. The scope of the power of attorney can be limited, such as to manage finances or even to a specific transaction, such as selling a home or car. A durable power of attorney also allows an agent to make decisions on the patient’s behalf but becomes active only when the patient becomes incompetent. It often is used to appoint an individual to make medical decisions on behalf of an incompetent patient. Living wills allow patients to determine what treatment they would like in the event they become incompetent.
Elder abuse. An estimated 1 to 2 million adults age >65 have been abused, exploited, or neglected.12 Elder abuse includes physical abuse, neglect, emotional abuse, sexual abuse, and financial exploitation (including undue influence). Most states have mandatory reporting of elder abuse, although they vary regarding who must report and what the report must entail. Psychiatrists should be vigilant in looking for signs of elder abuse (Table 2),13 regardless of the reason for the consult.
Table 2
Signs of elder abuse: What to wlook for
| Type of abuse | Signs |
|---|---|
| Physical | Bruises, burns (especially circular, suggesting cigarette burns), slap marks |
| Sexual | Unexplained sexually transmitted diseases, bruises in genital area, breasts, or anal area |
| Emotional | Withdrawal, new-onset depression |
| Financial | Sudden loss of property, unusual increase in spending, checks paid in large, round numbers, checks marked as gifts or loans |
| Neglect | Malnutrition, lack of medical care, poor hygiene, pressure ulcers |
| Source: Reference 13 | |
10 tips for thorough evaluations
1. Consider the context of the consultation. This includes medical factors (such as the patient’s condition, prognosis, relationship with the treatment team, and recommended course of treatment), legal factors (eg, pending litigation and relevant legal standards for issues such as guardianship), and psychosocial issues (eg, the patient’s current support structure and family conflicts).
2. Identify the legal issue and any relevant legal standards. The legal standard will inform you of the issues you need to address in the evaluation. If an attorney has consulted you, ask him or her to provide the legal standard.
3. Gather relevant collateral information, which may include interviews with family members or a review of financial or medical records.
4. Explain the purpose of the examination and the limits of confidentiality.
5. Perform a focused psychiatric evaluation, paying special attention to cognitive functioning, reasoning, and unusual thought content such as delusional beliefs.
6. Perform an interview specific to the referral issue.
7. Consider using a relevant assessment instrument.
8. Consider psychological testing, laboratory testing, imaging, or further medical evaluation. These assessments can help determine the diagnosis, the cause of any deficits in capacity, and whether any deficits are reversible.
9. Determine what opinions you are able to render. Limit opinions and remember that it may be appropriate to decline to address certain issues if there is insufficient information or if the issue is outside your area of expertise.
10. Prepare a written report. Consider the audience. Minimize the use of medical jargon and define all medical terms. State your opinions clearly and with reasonable medical certainty (in most jurisdictions, this means more likely than not). State the basis for all opinions.
For a case study that provides an example of a psycho-legal evaluation of a geriatric patient, see the Box.
Mr. A, age 75, recently started taking a dopaminergic agonist to treat Parkinson’s disease. He says he wants to divorce his wife of 35 years because of “scandalous affairs” she allegedly engaged in. His wife reports that he has been accusing her of having affairs with various men, including a man who recently painted their house.
On evaluation, Mr. A’s Mini-Mental State Examination score is 30/30. He has no signs of depression and his sleep patterns have not changed. There have been no changes in his spending patterns, although he no longer gives his wife money for grocery shopping, telling her to get money from her “boyfriends.” He is adamant about this decision, saying, “It’s my money and I can do with it as I please. This is still a free country, isn’t it?”
He says he has $70,000 in his individual retirement account, $20,000 in his bank account, and receives a pension of $1,785 per month. He estimates that his home is worth $200,000. His financial records essentially are consistent with his reports. He is able to perform basic calculations without difficulty and is aware of his monthly expenses. He describes his relationship with his wife by saying, “It was fine until she started screwing around.”
When asked about the likely consequences of his decision, he shrugs and says, “I guess she’ll have to get money from her boyfriends. I don’t really care who she sees as long as they stay away from me.” He denies having thoughts of harming his wife or her alleged “boyfriends.” He recognizes that his wife might divorce him, leaving him alone.
When I ask Mr. A if it is possible he is mistaken in his belief that his wife is having affairs, he says, “No, doctor. You don’t know her.” When I ask how he knows she is having affairs, he says that the painter started looking at him “funny” and that the busboy at a restaurant they frequent called his wife “dear.” He believes his wife is having sexual relations with both of these men.
Does Mr. A require a guardian? I opine that Mr. A requires a guardian of estate (to manage his property) but not a guardian of person because he is capable of making decisions about his medical care and other personal decisions. He is failing to care for his wife because of his delusional jealousy. Although cognitively intact, he is unable to appreciate the consequences of his actions or rationally manipulate information because of his delusional thinking. He believes he is “cutting off” an unfaithful spouse when, in fact, there is no evidence that she has been unfaithful. His inability to rationally manipulate information is demonstrated by the fact that he uses innocuous facts such as a busboy calling his elderly wife “dear” to support his delusion that she was having affairs.
I note that his psychosis is reversible because it is likely due to his antiparkinsonian regimen. However, he declines both a dose reduction in his medication and antipsychotic treatment. I note that should his psychosis resolve, he may regain financial decision-making capacity.
Related Resources
- American Academy of Psychiatry and the Law. www.aapl.org.
- American Bar Association Commission on Law and Aging. www.americanbar.org/groups/law_aging.html.
- National Center on Elder Abuse. www.ncea.aoa.gov.
Disclosure
Dr. Soliman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
The author thanks forensic psychiatry fellow Abhishek Jain, MD for reviewing the article and offering editorial suggestions.
Discuss this article at www.facebook.com/CurrentPsychiatry
Although forensic psychiatrists typically are consulted in complex legal matters, geriatric, consultation-liaison, and general psychiatrists are on the front lines of assessing capacity to give informed consent and need for guardianship. Psychiatrists often find such consultations daunting because residency training usually includes little to no formal training in performing psycho-legal assessments. Evaluating issues such as decision-making capacity, guardianship, and capacity to give informed consent requires a delicate balance between autonomy and beneficence. This article reviews 4 common legal issues in geriatric consultation—capacity evaluations, informed consent, guardianship, and elder abuse—and suggests a systematic approach to psycho-legal consultations in older adults.
Confidentiality and dual agency
Every psychiatrist should be familiar with basic principles of medical ethics as well as key aspects of local mental health law. Relevant ethical principles include autonomy, beneficence, confidentiality, and dual agency. A review of all these ethical issues is beyond the scope of this article, so here I highlight confidentiality and dual agency.
Confidentiality—the clinician’s obligation not to disclose private medical information—is a legal as well as an ethical requirement. A psychiatrist who performs a psycho-legal evaluation must disclose to the patient the purpose of the evaluation, that a report will be prepared, and to whom it will be submitted. Exceptions to confidentiality include medical emergencies, mandatory reporting of abuse and infectious diseases, and the duty to protect (warning police and the intended victim when a patient makes a threat).
Dual agency or dual role refers to serving as both a treating physician and a forensic evaluator. Although it is ideal to avoid serving in a dual role, sometimes it is impractical or impossible to avoid doing so, such as in guardianship or civil commitment evaluations, or in state forensic hospitals. In such cases, the psychiatrist must be aware of potential conflicts between clinical and forensic evaluations. A treating psychiatrist primarily serves his or her patient’s best interest, whereas a forensic psychiatrist primarily seeks truth.1 A treating psychiatrist is at risk of consciously or unconsciously biasing his or her psycho-legal evaluation in favor of or against the patient/litigant, depending upon the psychiatrist’s countertransference. Further, performing a psycho-legal evaluation can cause problems in ongoing treatment. A psychiatrist who testifies that his or her fiercely independent patient needs a guardian or nursing home placement will experience significant challenges in continuing to work with that patient.
4 common issues for older adults
Decision-making capacity. Although “capacity” and “competence” often are used interchangeably, “capacity” broadly refers to the ability to perform a specific task, whereas “competence” refers to the legally defined standard for performing a specific task such as making a will. “Competence” is legally determined, whereas “capacity” may be determined clinically.
Capacity usually is task-specific rather than a general construct. The existence of physical or mental illness per se does not mean that a patient lacks capacity. Rather, capacity is determined by whether an individual has specific abilities, regardless of diagnosis. Specific capacities include the ability to give informed consent, manage finances, make a will, or enter into contracts (Table 1).2-4 Appelbaum and Gutheil describe 4 components for assessing specific capacity:
- communication of a choice
- factual understanding of the issues
- appreciation of the situation and its consequences
- rational manipulation of information.5,6
Table 1
Criteria of 3 specific capacities
| Capacity | Criteria |
|---|---|
| Capacity to give informed consent | Understand nature of illness and treatment Understand risks and benefits of treatment Understand treatment alternatives Understand risk of refusing treatment |
| Testamentary capacity | Understand that he/she is making a will Know the nature and extent of their property Understand the “natural objects” of their bounty and their claims upon them |
| Contractual capacity | Understand the transaction Act in a reasonable manner |
| Source: References 2-4 | |
Ability to communicate a choice refers to a patient’s ability to express his or her wishes in a reasonably stable manner. Factual understanding of the issues refers to an individual’s ability to understand the relevant facts before making a decision. Appreciation of the situation and its consequences refers to a person’s ability to rationally understand the effect of decisions. Appreciation is a higher level of understanding than mere factual understanding—eg, a delusional patient who believes himself immortal may intellectually understand that a surgical procedure carries a 50% mortality risk, but may be unable to appreciate the information as it relates to him because he believes he is immortal. Rational manipulation of information refers to a patient’s reasoning process and how the patient integrates data into his or her decision-making process.5
Informed consent. In my experience, capacity to give informed consent is the most commonly requested specific capacity assessment in general medical settings. Informed consent must be knowing, voluntary, and competent. All material information—information that would cause a reasonable person to accept or reject a proposed treatment—should be communicated to the patient. Informed consent requires an understanding of the patient’s condition and indication for treatment, risks and benefits of and alternatives to treatment, and risks of declining treatment.2,3 Exceptions to informed consent include incompetence, medical emergencies, patient waiver of informed consent, and a limited therapeutic privilege (when a physician determines that the information would harm the patient).3
Several instruments can help clinicians assess patients’ capacity to give informed consent. The benefits of using a structured instrument include:
- ensuring that specific information is covered during each evaluation
- systematically recording a patient’s response.5
Disadvantages of using instruments include the fact that no instrument can take into account all aspects of a particular case, and some instruments are time-consuming and require training. Structured instruments can be a useful adjunct to the clinical interview in some cases, but should not substitute for it. In a review of 23 instruments for assessing decisional capacity to consent to treatment or clinical research, the MacArthur Competence Assessment Tool for Clinical Research and the MacArthur Competence Assessment Tool for Treatment had the most empirical support, although the authors noted that other instruments might be better suited to specific situations.7
Psychiatrists may be consulted when a patient refuses treatment or decides to leave the hospital against medical advice. The key issue in both situations is whether the patient has capacity to refuse treatment.8 If there is evidence that the patient is mentally ill and poses an imminent risk of suicide or violence or is unable to provide for his or her basic needs, the psychiatrist should assess whether the patient meets criteria for civil commitment.
Many clinicians employ a “sliding scale” approach to competence, requiring a lower degree of competence for consenting to low-risk, high-benefit interventions and a greater degree of competence for higher-risk procedures.5,9 Family members often serve as informal surrogate decision makers for incapacitated patients, except when there is significant family discord or no family members are available.5
Guardianship. Guardians are appointed by courts to make decisions for individuals who have been found incompetent (wards). Although its purposes are beneficent, the guardianship system could do significant harm.10 Determining that an individual is incompetent is tantamount to depriving him or her of basic personhood. In many cases, the ward loses the ability to consent to or refuse medical care, manage his or her finances, enter into contracts, marry, and determine where he or she will live. On the other hand, failing to recognize incompetence can leave a vulnerable person in danger of physical deterioration, abuse, neglect, or exploitation.
It is critical that guardianship evaluations be conducted carefully. In a review of 298 guardianship cases from 3 states, Moye and colleagues11 found that the quality of the reports was significantly better in Colorado, a state with guardianship reforms, but documentation of functional strengths and weaknesses was “particularly rare” in guardianship evaluations and prognosis often was not included. This information is relevant to judges, who need to determine which areas of function are impaired and how long the impairment is likely to last.
Guardianship evaluations often focus on general rather than specific capacity. In other words, often there is not a specific task such as consenting to surgery that the alleged incompetent person needs to perform. Rather, the question is whether an individual can manage his or her finances or make treatment decisions in general. Appelbaum and Gutheil suggest considering 6 factors when assessing general capacity:
- awareness of the situation
- factual understanding of the issues
- appreciation of the likely consequences
- rational manipulation of information
- functioning in one’s environment
- extent of demands on patient.5
The first 4 are closely related to the elements of specific capacity described above. Functioning in one’s environment and extent of demands on the patient attempt to anticipate the tasks that an individual will need to perform. A patient with mild dementia may be unable to manage a complex estate but can handle a bank account and a fixed income. Similarly, it is important to consider the patient’s support system. An impaired patient may function adequately with his wife’s help but may lose the capacity to live independently if his wife dies or becomes impaired.
Traditionally, guardianship has resulted in a complete loss of decision-making ability. Several state legislatures have passed laws allowing for limited guardianship, although orders for limited guardianship remain underutilized.10 Limited guardianship delineates specific areas of incompetence and limits the guardian’s decision-making authority to those areas while leaving intact the ward’s ability to make all other decisions for himself or herself.
The use of less-restrictive alternatives to guardianship—such as powers of attorney, durable powers of attorney, living wills, payees, and trusts—is increasing. A power of attorney allows a patient to authorize a specific individual to act on his or her behalf. The scope of the power of attorney can be limited, such as to manage finances or even to a specific transaction, such as selling a home or car. A durable power of attorney also allows an agent to make decisions on the patient’s behalf but becomes active only when the patient becomes incompetent. It often is used to appoint an individual to make medical decisions on behalf of an incompetent patient. Living wills allow patients to determine what treatment they would like in the event they become incompetent.
Elder abuse. An estimated 1 to 2 million adults age >65 have been abused, exploited, or neglected.12 Elder abuse includes physical abuse, neglect, emotional abuse, sexual abuse, and financial exploitation (including undue influence). Most states have mandatory reporting of elder abuse, although they vary regarding who must report and what the report must entail. Psychiatrists should be vigilant in looking for signs of elder abuse (Table 2),13 regardless of the reason for the consult.
Table 2
Signs of elder abuse: What to wlook for
| Type of abuse | Signs |
|---|---|
| Physical | Bruises, burns (especially circular, suggesting cigarette burns), slap marks |
| Sexual | Unexplained sexually transmitted diseases, bruises in genital area, breasts, or anal area |
| Emotional | Withdrawal, new-onset depression |
| Financial | Sudden loss of property, unusual increase in spending, checks paid in large, round numbers, checks marked as gifts or loans |
| Neglect | Malnutrition, lack of medical care, poor hygiene, pressure ulcers |
| Source: Reference 13 | |
10 tips for thorough evaluations
1. Consider the context of the consultation. This includes medical factors (such as the patient’s condition, prognosis, relationship with the treatment team, and recommended course of treatment), legal factors (eg, pending litigation and relevant legal standards for issues such as guardianship), and psychosocial issues (eg, the patient’s current support structure and family conflicts).
2. Identify the legal issue and any relevant legal standards. The legal standard will inform you of the issues you need to address in the evaluation. If an attorney has consulted you, ask him or her to provide the legal standard.
3. Gather relevant collateral information, which may include interviews with family members or a review of financial or medical records.
4. Explain the purpose of the examination and the limits of confidentiality.
5. Perform a focused psychiatric evaluation, paying special attention to cognitive functioning, reasoning, and unusual thought content such as delusional beliefs.
6. Perform an interview specific to the referral issue.
7. Consider using a relevant assessment instrument.
8. Consider psychological testing, laboratory testing, imaging, or further medical evaluation. These assessments can help determine the diagnosis, the cause of any deficits in capacity, and whether any deficits are reversible.
9. Determine what opinions you are able to render. Limit opinions and remember that it may be appropriate to decline to address certain issues if there is insufficient information or if the issue is outside your area of expertise.
10. Prepare a written report. Consider the audience. Minimize the use of medical jargon and define all medical terms. State your opinions clearly and with reasonable medical certainty (in most jurisdictions, this means more likely than not). State the basis for all opinions.
For a case study that provides an example of a psycho-legal evaluation of a geriatric patient, see the Box.
Mr. A, age 75, recently started taking a dopaminergic agonist to treat Parkinson’s disease. He says he wants to divorce his wife of 35 years because of “scandalous affairs” she allegedly engaged in. His wife reports that he has been accusing her of having affairs with various men, including a man who recently painted their house.
On evaluation, Mr. A’s Mini-Mental State Examination score is 30/30. He has no signs of depression and his sleep patterns have not changed. There have been no changes in his spending patterns, although he no longer gives his wife money for grocery shopping, telling her to get money from her “boyfriends.” He is adamant about this decision, saying, “It’s my money and I can do with it as I please. This is still a free country, isn’t it?”
He says he has $70,000 in his individual retirement account, $20,000 in his bank account, and receives a pension of $1,785 per month. He estimates that his home is worth $200,000. His financial records essentially are consistent with his reports. He is able to perform basic calculations without difficulty and is aware of his monthly expenses. He describes his relationship with his wife by saying, “It was fine until she started screwing around.”
When asked about the likely consequences of his decision, he shrugs and says, “I guess she’ll have to get money from her boyfriends. I don’t really care who she sees as long as they stay away from me.” He denies having thoughts of harming his wife or her alleged “boyfriends.” He recognizes that his wife might divorce him, leaving him alone.
When I ask Mr. A if it is possible he is mistaken in his belief that his wife is having affairs, he says, “No, doctor. You don’t know her.” When I ask how he knows she is having affairs, he says that the painter started looking at him “funny” and that the busboy at a restaurant they frequent called his wife “dear.” He believes his wife is having sexual relations with both of these men.
Does Mr. A require a guardian? I opine that Mr. A requires a guardian of estate (to manage his property) but not a guardian of person because he is capable of making decisions about his medical care and other personal decisions. He is failing to care for his wife because of his delusional jealousy. Although cognitively intact, he is unable to appreciate the consequences of his actions or rationally manipulate information because of his delusional thinking. He believes he is “cutting off” an unfaithful spouse when, in fact, there is no evidence that she has been unfaithful. His inability to rationally manipulate information is demonstrated by the fact that he uses innocuous facts such as a busboy calling his elderly wife “dear” to support his delusion that she was having affairs.
I note that his psychosis is reversible because it is likely due to his antiparkinsonian regimen. However, he declines both a dose reduction in his medication and antipsychotic treatment. I note that should his psychosis resolve, he may regain financial decision-making capacity.
Related Resources
- American Academy of Psychiatry and the Law. www.aapl.org.
- American Bar Association Commission on Law and Aging. www.americanbar.org/groups/law_aging.html.
- National Center on Elder Abuse. www.ncea.aoa.gov.
Disclosure
Dr. Soliman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
The author thanks forensic psychiatry fellow Abhishek Jain, MD for reviewing the article and offering editorial suggestions.
1. American Academy of Psychiatry and the Law. Ethics guidelines for the practice of forensic psychiatry. http://www.aapl.org/ethics.htm. Adopted May 2005. Accessed February 24 2012.
2. Blank K. Legal and ethical issues. In: Sadvoy J Jarvik LF, Grossberg GT, et al, eds. Comprehensive textbook of geriatric psychiatry. 3rd ed. New York, NY: American Association of Geriatric Psychiatry; 2004:1183-1206.
3. Schwartz HI, Mack DM. Informed consent and competency. In: Rosner R ed. Principles and practice of forensic psychiatry. 2nd ed. New York, NY: Oxford University Press; 2003:97-106.
4. Ciccone RJ. Civil competencies. In: Rosner R ed. Principles and practice of forensic psychiatry. 2nd ed. New York, NY: Oxford University Press; 2003:308-315.
5. Appelbaum PS, Gutheil TG. Competence and substitute decision making. In: Appelbaum PS Gutheil TG, eds. Clinical handbook of psychiatry and the law. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:177-213.
6. Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
7. Dunn LB, Nowrangi MA, Palmer BW, et al. Assessing decisional capacity for clinical research or treatment: a review of instruments. Am J Psychiatry. 2006;163(8):1323-1334.
8. Resnick PJ, Sorrentino R. Forensic issues in consultation-liaison psychiatry. Psychiatric Times. 2005;23(14).-
9. Drane JF. The many faces of competency. Hastings Cent Rep. 1985;15(2):17-21.
10. Wright JL. Guardianship for your own good: improving the well being of respondents and wards in the USA. Int J Law Psychiatry. 2010;33(5-6):350-368.
11. Moye J, Wood S, Edelstein B, et al. Clinical evidence in guardianship of older adults is inadequate: findings from a tri-state study. Gerontologist. 2007;47(5):604-612.
12. National Center on Elder Abuse. Fact sheet: elder abuse prevalence and incidence. http://www.ncea.aoa.gov/NCEAroot/Main_Site/pdf/publication/FinalStatistics050331.pdf. Accessed February 24, 2012.
13. National Center on Elder Abuse. Why should I care about elder abuse? http://www.ncea.aoa.gov/Main_Site/pdf/publication/NCEA_WhatIsAbuse-2010.pdf. Published March 3 2010. Accessed February 24, 2012.
1. American Academy of Psychiatry and the Law. Ethics guidelines for the practice of forensic psychiatry. http://www.aapl.org/ethics.htm. Adopted May 2005. Accessed February 24 2012.
2. Blank K. Legal and ethical issues. In: Sadvoy J Jarvik LF, Grossberg GT, et al, eds. Comprehensive textbook of geriatric psychiatry. 3rd ed. New York, NY: American Association of Geriatric Psychiatry; 2004:1183-1206.
3. Schwartz HI, Mack DM. Informed consent and competency. In: Rosner R ed. Principles and practice of forensic psychiatry. 2nd ed. New York, NY: Oxford University Press; 2003:97-106.
4. Ciccone RJ. Civil competencies. In: Rosner R ed. Principles and practice of forensic psychiatry. 2nd ed. New York, NY: Oxford University Press; 2003:308-315.
5. Appelbaum PS, Gutheil TG. Competence and substitute decision making. In: Appelbaum PS Gutheil TG, eds. Clinical handbook of psychiatry and the law. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:177-213.
6. Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
7. Dunn LB, Nowrangi MA, Palmer BW, et al. Assessing decisional capacity for clinical research or treatment: a review of instruments. Am J Psychiatry. 2006;163(8):1323-1334.
8. Resnick PJ, Sorrentino R. Forensic issues in consultation-liaison psychiatry. Psychiatric Times. 2005;23(14).-
9. Drane JF. The many faces of competency. Hastings Cent Rep. 1985;15(2):17-21.
10. Wright JL. Guardianship for your own good: improving the well being of respondents and wards in the USA. Int J Law Psychiatry. 2010;33(5-6):350-368.
11. Moye J, Wood S, Edelstein B, et al. Clinical evidence in guardianship of older adults is inadequate: findings from a tri-state study. Gerontologist. 2007;47(5):604-612.
12. National Center on Elder Abuse. Fact sheet: elder abuse prevalence and incidence. http://www.ncea.aoa.gov/NCEAroot/Main_Site/pdf/publication/FinalStatistics050331.pdf. Accessed February 24, 2012.
13. National Center on Elder Abuse. Why should I care about elder abuse? http://www.ncea.aoa.gov/Main_Site/pdf/publication/NCEA_WhatIsAbuse-2010.pdf. Published March 3 2010. Accessed February 24, 2012.
The hidden danger of hand sanitizer
Discuss this article at www.facebook.com/CurrentPsychiatry
The Centers for Disease Control and Prevention recommends that all health care professionals use an ethanol-based hand sanitizer to decontaminate their hands before and after direct contact with patients to prevent infection.1,2 As a result, many psychiatric hospitals use alcohol-based hand sanitizers as a primary infection control measure.
Patient misuse of these products as intoxicants has been reported in prisons, emergency rooms, and medical units.3-7 We report 2 cases of psychiatric inpatients who intentionally ingested alcohol-based hand sanitizers to become intoxicated; there were no permanent toxic effects in either case.
Case 1
Mr. F, age 52, is diagnosed with polysubstance dependence and bipolar disorder and hospitalized for acute exacerbation of mania characterized by unrestrained buying sprees, racing thoughts, grandiosity, and a persistently irritable mood. On day 3 of admission, he presents as stuporous and disorganized, with a strong odor of alcohol on his breath. He admits drinking an alcohol-based hand sanitizer foaming solution, an empty bottle of which is found in his room. His serum alcohol level is 176 mg/dL; the threshold concentration above which a person is considered legally drunk when operating a motor vehicle is 100 mg/dL. Other laboratory values, including urine toxicology, were negative.
Case 2
Mr. V, age 47, has schizophrenia, cocaine dependence, and antisocial personality disorder. He is admitted for command auditory hallucinations and a suicide attempt by overdose. On day 6 of hospitalization, staff members find him delirious and confused. Mr. V confesses to drinking an alcohol-based hand sanitizer solution for the past 3 days. His vital signs are stable, and his serum alcohol level is 142 mg/dL.
Limiting access
Hand sanitizer has a much higher alcohol concentration than several common alcoholic drinks Table8,9 Ethyl alcohol, the active ingredient in hand sanitizers, is responsible for the adverse effects seen in our patients; the inactive ingredients—glycerin, propylene glycol, tocopherol acetate, isopropyl myristate, and aminomethyl propanol—generally are recognized as safe by the FDA and the Cosmetic Ingredient Review Expert Panel.10,11 Although hospitals routinely restrict patients’ access to traditional forms of alcohol, hand sanitizer is easily accessible in many facilities. In our cases, having the alcohol-based sanitizer placed throughout the unit and readily available to patients made it easy for at-risk patients to become intoxicated. As suggested by Weiner,7 replacing bottles of hand sanitizer with self-contained, wall-mounted dispensers that are difficult for patients to remove might decrease the likelihood of ingestion.
Table
Alcohol content of hand sanitizers and beverages
| Product | Percentage of alcohol by volume |
|---|---|
| Beer | 5% alcohol8 |
| Wine | 12% alcohol8 |
| Distilled spirits | 40% alcohol8 |
| Purell Foaming Hand Sanitizer | 62% ethyl alcohol9 |
| Purell Instant Hand Sanitizer | 62% ethyl alcohol, 5% isopropanol by volume9 |
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. The Joint Commission National patient safety goals. http://www.jointcommission.org/standards_information/npsgs.aspx. Accessed December 8 2011.
2. Emadi A, Coberly L. Intoxication of a hospitalized patient with an isopropanol-based hand sanitizer. N Engl J Med. 2007;356(5):530-531.
3. Boyce JM, Pittet D. Guideline for hand hygiene in health-care settings: recommendations of the Healthcare Infection Control Practices Advisory Committee and the HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force. Infect Control Hosp Epidemiol. 2002;23(12 suppl):S3-S40.
4. Bookstaver PB, Norris LB, Michels JE. Ingestion of hand sanitizer by a hospitalized patient with a history of alcohol abuse. Am J Health Syst Pharm. 2008;65(23):2203-2204.
5. Doyon S, Welsh C. Intoxication of a prison inmate with an ethyl alcohol-based hand sanitizer. N Engl J Med. 2007;356(5):529-530.
6. Thanarajasingam G, Diedrich DA, Mueller PS. Intentional ingestion of ethanol-based hand sanitizer by a hospitalized patient with alcoholism. Mayo Clin Proc. 2007;82(10):1288-1289.
7. Weiner SG. Changing dispensers may prevent intoxication from isopropanol and ethyl alcohol-based hand sanitizers. Ann Emerg Med. 2007;50(4):486.-
8. National Institute on Alcohol Abuse and Alcoholism What’s a “standard” drink? http://rethinkingdrinking.niaaa.nih.gov/WhatCountsDrink/WhatsAStandardDrink.asp. Accessed February 17, 2012.
9. GOJO Industries. Purell instant hand sanitizier. http://www.gojo.com/United-States/Brands/PURELL/MSDS.aspx. Accessed December 8, 2011.
10. U.S. Food and Drug Administration. Generally Recognized as Safe (GRAS) notice inventory. http://www.fda.gov/Food/FoodIngredientsPackaging/GenerallyRecognizedasSafeGRAS/
GRASListings/default.htm. Accessed February 21, 2012.
11. The Cosmetic Ingredient Review Find ingredient reviews and documents. http://www.cir-safety.org/ingredients/glossary/all. Accessed February 21, 2012.
Discuss this article at www.facebook.com/CurrentPsychiatry
The Centers for Disease Control and Prevention recommends that all health care professionals use an ethanol-based hand sanitizer to decontaminate their hands before and after direct contact with patients to prevent infection.1,2 As a result, many psychiatric hospitals use alcohol-based hand sanitizers as a primary infection control measure.
Patient misuse of these products as intoxicants has been reported in prisons, emergency rooms, and medical units.3-7 We report 2 cases of psychiatric inpatients who intentionally ingested alcohol-based hand sanitizers to become intoxicated; there were no permanent toxic effects in either case.
Case 1
Mr. F, age 52, is diagnosed with polysubstance dependence and bipolar disorder and hospitalized for acute exacerbation of mania characterized by unrestrained buying sprees, racing thoughts, grandiosity, and a persistently irritable mood. On day 3 of admission, he presents as stuporous and disorganized, with a strong odor of alcohol on his breath. He admits drinking an alcohol-based hand sanitizer foaming solution, an empty bottle of which is found in his room. His serum alcohol level is 176 mg/dL; the threshold concentration above which a person is considered legally drunk when operating a motor vehicle is 100 mg/dL. Other laboratory values, including urine toxicology, were negative.
Case 2
Mr. V, age 47, has schizophrenia, cocaine dependence, and antisocial personality disorder. He is admitted for command auditory hallucinations and a suicide attempt by overdose. On day 6 of hospitalization, staff members find him delirious and confused. Mr. V confesses to drinking an alcohol-based hand sanitizer solution for the past 3 days. His vital signs are stable, and his serum alcohol level is 142 mg/dL.
Limiting access
Hand sanitizer has a much higher alcohol concentration than several common alcoholic drinks Table8,9 Ethyl alcohol, the active ingredient in hand sanitizers, is responsible for the adverse effects seen in our patients; the inactive ingredients—glycerin, propylene glycol, tocopherol acetate, isopropyl myristate, and aminomethyl propanol—generally are recognized as safe by the FDA and the Cosmetic Ingredient Review Expert Panel.10,11 Although hospitals routinely restrict patients’ access to traditional forms of alcohol, hand sanitizer is easily accessible in many facilities. In our cases, having the alcohol-based sanitizer placed throughout the unit and readily available to patients made it easy for at-risk patients to become intoxicated. As suggested by Weiner,7 replacing bottles of hand sanitizer with self-contained, wall-mounted dispensers that are difficult for patients to remove might decrease the likelihood of ingestion.
Table
Alcohol content of hand sanitizers and beverages
| Product | Percentage of alcohol by volume |
|---|---|
| Beer | 5% alcohol8 |
| Wine | 12% alcohol8 |
| Distilled spirits | 40% alcohol8 |
| Purell Foaming Hand Sanitizer | 62% ethyl alcohol9 |
| Purell Instant Hand Sanitizer | 62% ethyl alcohol, 5% isopropanol by volume9 |
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
The Centers for Disease Control and Prevention recommends that all health care professionals use an ethanol-based hand sanitizer to decontaminate their hands before and after direct contact with patients to prevent infection.1,2 As a result, many psychiatric hospitals use alcohol-based hand sanitizers as a primary infection control measure.
Patient misuse of these products as intoxicants has been reported in prisons, emergency rooms, and medical units.3-7 We report 2 cases of psychiatric inpatients who intentionally ingested alcohol-based hand sanitizers to become intoxicated; there were no permanent toxic effects in either case.
Case 1
Mr. F, age 52, is diagnosed with polysubstance dependence and bipolar disorder and hospitalized for acute exacerbation of mania characterized by unrestrained buying sprees, racing thoughts, grandiosity, and a persistently irritable mood. On day 3 of admission, he presents as stuporous and disorganized, with a strong odor of alcohol on his breath. He admits drinking an alcohol-based hand sanitizer foaming solution, an empty bottle of which is found in his room. His serum alcohol level is 176 mg/dL; the threshold concentration above which a person is considered legally drunk when operating a motor vehicle is 100 mg/dL. Other laboratory values, including urine toxicology, were negative.
Case 2
Mr. V, age 47, has schizophrenia, cocaine dependence, and antisocial personality disorder. He is admitted for command auditory hallucinations and a suicide attempt by overdose. On day 6 of hospitalization, staff members find him delirious and confused. Mr. V confesses to drinking an alcohol-based hand sanitizer solution for the past 3 days. His vital signs are stable, and his serum alcohol level is 142 mg/dL.
Limiting access
Hand sanitizer has a much higher alcohol concentration than several common alcoholic drinks Table8,9 Ethyl alcohol, the active ingredient in hand sanitizers, is responsible for the adverse effects seen in our patients; the inactive ingredients—glycerin, propylene glycol, tocopherol acetate, isopropyl myristate, and aminomethyl propanol—generally are recognized as safe by the FDA and the Cosmetic Ingredient Review Expert Panel.10,11 Although hospitals routinely restrict patients’ access to traditional forms of alcohol, hand sanitizer is easily accessible in many facilities. In our cases, having the alcohol-based sanitizer placed throughout the unit and readily available to patients made it easy for at-risk patients to become intoxicated. As suggested by Weiner,7 replacing bottles of hand sanitizer with self-contained, wall-mounted dispensers that are difficult for patients to remove might decrease the likelihood of ingestion.
Table
Alcohol content of hand sanitizers and beverages
| Product | Percentage of alcohol by volume |
|---|---|
| Beer | 5% alcohol8 |
| Wine | 12% alcohol8 |
| Distilled spirits | 40% alcohol8 |
| Purell Foaming Hand Sanitizer | 62% ethyl alcohol9 |
| Purell Instant Hand Sanitizer | 62% ethyl alcohol, 5% isopropanol by volume9 |
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. The Joint Commission National patient safety goals. http://www.jointcommission.org/standards_information/npsgs.aspx. Accessed December 8 2011.
2. Emadi A, Coberly L. Intoxication of a hospitalized patient with an isopropanol-based hand sanitizer. N Engl J Med. 2007;356(5):530-531.
3. Boyce JM, Pittet D. Guideline for hand hygiene in health-care settings: recommendations of the Healthcare Infection Control Practices Advisory Committee and the HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force. Infect Control Hosp Epidemiol. 2002;23(12 suppl):S3-S40.
4. Bookstaver PB, Norris LB, Michels JE. Ingestion of hand sanitizer by a hospitalized patient with a history of alcohol abuse. Am J Health Syst Pharm. 2008;65(23):2203-2204.
5. Doyon S, Welsh C. Intoxication of a prison inmate with an ethyl alcohol-based hand sanitizer. N Engl J Med. 2007;356(5):529-530.
6. Thanarajasingam G, Diedrich DA, Mueller PS. Intentional ingestion of ethanol-based hand sanitizer by a hospitalized patient with alcoholism. Mayo Clin Proc. 2007;82(10):1288-1289.
7. Weiner SG. Changing dispensers may prevent intoxication from isopropanol and ethyl alcohol-based hand sanitizers. Ann Emerg Med. 2007;50(4):486.-
8. National Institute on Alcohol Abuse and Alcoholism What’s a “standard” drink? http://rethinkingdrinking.niaaa.nih.gov/WhatCountsDrink/WhatsAStandardDrink.asp. Accessed February 17, 2012.
9. GOJO Industries. Purell instant hand sanitizier. http://www.gojo.com/United-States/Brands/PURELL/MSDS.aspx. Accessed December 8, 2011.
10. U.S. Food and Drug Administration. Generally Recognized as Safe (GRAS) notice inventory. http://www.fda.gov/Food/FoodIngredientsPackaging/GenerallyRecognizedasSafeGRAS/
GRASListings/default.htm. Accessed February 21, 2012.
11. The Cosmetic Ingredient Review Find ingredient reviews and documents. http://www.cir-safety.org/ingredients/glossary/all. Accessed February 21, 2012.
1. The Joint Commission National patient safety goals. http://www.jointcommission.org/standards_information/npsgs.aspx. Accessed December 8 2011.
2. Emadi A, Coberly L. Intoxication of a hospitalized patient with an isopropanol-based hand sanitizer. N Engl J Med. 2007;356(5):530-531.
3. Boyce JM, Pittet D. Guideline for hand hygiene in health-care settings: recommendations of the Healthcare Infection Control Practices Advisory Committee and the HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force. Infect Control Hosp Epidemiol. 2002;23(12 suppl):S3-S40.
4. Bookstaver PB, Norris LB, Michels JE. Ingestion of hand sanitizer by a hospitalized patient with a history of alcohol abuse. Am J Health Syst Pharm. 2008;65(23):2203-2204.
5. Doyon S, Welsh C. Intoxication of a prison inmate with an ethyl alcohol-based hand sanitizer. N Engl J Med. 2007;356(5):529-530.
6. Thanarajasingam G, Diedrich DA, Mueller PS. Intentional ingestion of ethanol-based hand sanitizer by a hospitalized patient with alcoholism. Mayo Clin Proc. 2007;82(10):1288-1289.
7. Weiner SG. Changing dispensers may prevent intoxication from isopropanol and ethyl alcohol-based hand sanitizers. Ann Emerg Med. 2007;50(4):486.-
8. National Institute on Alcohol Abuse and Alcoholism What’s a “standard” drink? http://rethinkingdrinking.niaaa.nih.gov/WhatCountsDrink/WhatsAStandardDrink.asp. Accessed February 17, 2012.
9. GOJO Industries. Purell instant hand sanitizier. http://www.gojo.com/United-States/Brands/PURELL/MSDS.aspx. Accessed December 8, 2011.
10. U.S. Food and Drug Administration. Generally Recognized as Safe (GRAS) notice inventory. http://www.fda.gov/Food/FoodIngredientsPackaging/GenerallyRecognizedasSafeGRAS/
GRASListings/default.htm. Accessed February 21, 2012.
11. The Cosmetic Ingredient Review Find ingredient reviews and documents. http://www.cir-safety.org/ingredients/glossary/all. Accessed February 21, 2012.
Becoming PARTNERS in recovery-oriented care
Discuss this article at www.facebook.com/CurrentPsychiatry
Over the past 20 years, the recovery movement in mental health care has transformed the way in which clinicians and behavioral health agencies view and treat persons with serious mental illnesses. The New Freedom Commission on Mental Health defined recovery as “the process in which people are able to live, work, learn, and participate in their communities.” This process supports a clinical approach to psychiatric care that promotes opportunity, hope, and social inclusion.1 As professionals, “our primary interest should be to take the principles and concepts of [r]ecovery and to look at ways in which our practices and services could be orientated to facilitate [r]ecovery in the people who use them.”2
Drawing upon the Substance Abuse and Mental Health Services Administration’s core principles of a recovery-oriented approach,3 the mnemonic PARTNERS may help you recall key concepts to delivering and assessing recovery-oriented care.
Person-centered approaches recognize the patient’s unique gifts, strengths, needs, and cultural perspectives. Treatment is based upon the patient’s resiliencies, deficits, and personal goals, not just algorithms.
Autonomy emphasizes the patient’s right to determine his or her own destiny. It serves as the justification for the recovery maxim of “no decision about me, without me.”
Responsibility recognizes that the choices, decisions, and consequences about the type, amount, and frequency of care are a shared responsibility between physician and patient.
Transformational interactions and attitudes occur between the physician, the patient, and the system in a recovery-oriented model of care. The physician, patient, and agency are transformed into partners in the healing process.
Nonlinear concepts highlight the expectation that a patient’s pathway to recovery inevitably will be punctuated by personal gains, uneventful plateaus, and the occasional setback.
Empowerment is patients’ growing sense that they can speak openly and freely about their needs, hopes, and life goals, individually or through support and advocacy groups.
Respect is the bedrock value that defines the physician/patient relationship and ensures the absence of discrimination and stigmatization within behavioral health systems.
Strength-based approaches remind the physician that valuing and building upon the patient’s core strengths, talents, and positive attributes, rather than narrowly focusing on personal deficits, fuels the recovery process.
Disclosure
Dr. Christensen reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. President’s New Freedom Commission on Mental Health. Achieving the promise: transforming mental health care in America. Final report. Rockville, MD: Substance Abuse and Mental Health Administration; 2003.
2. South London and Maudsley NHS Foundation Trust and South West London and St. George’s Mental Health NHS Trust. Recovery is for all: hope, agency and opportunity in psychiatry. A position statement by consultant psychiatrists. London, United Kingdom: SLAM/SWLSTG; 2010.
3. Substance Abuse and Mental Health Services Administration. National consensus statement on mental health recovery. http://store.samhsa.gov/shin/content//SMA05-4129/SMA05-4129.pdf. Accessed February 24, 2012.
Discuss this article at www.facebook.com/CurrentPsychiatry
Over the past 20 years, the recovery movement in mental health care has transformed the way in which clinicians and behavioral health agencies view and treat persons with serious mental illnesses. The New Freedom Commission on Mental Health defined recovery as “the process in which people are able to live, work, learn, and participate in their communities.” This process supports a clinical approach to psychiatric care that promotes opportunity, hope, and social inclusion.1 As professionals, “our primary interest should be to take the principles and concepts of [r]ecovery and to look at ways in which our practices and services could be orientated to facilitate [r]ecovery in the people who use them.”2
Drawing upon the Substance Abuse and Mental Health Services Administration’s core principles of a recovery-oriented approach,3 the mnemonic PARTNERS may help you recall key concepts to delivering and assessing recovery-oriented care.
Person-centered approaches recognize the patient’s unique gifts, strengths, needs, and cultural perspectives. Treatment is based upon the patient’s resiliencies, deficits, and personal goals, not just algorithms.
Autonomy emphasizes the patient’s right to determine his or her own destiny. It serves as the justification for the recovery maxim of “no decision about me, without me.”
Responsibility recognizes that the choices, decisions, and consequences about the type, amount, and frequency of care are a shared responsibility between physician and patient.
Transformational interactions and attitudes occur between the physician, the patient, and the system in a recovery-oriented model of care. The physician, patient, and agency are transformed into partners in the healing process.
Nonlinear concepts highlight the expectation that a patient’s pathway to recovery inevitably will be punctuated by personal gains, uneventful plateaus, and the occasional setback.
Empowerment is patients’ growing sense that they can speak openly and freely about their needs, hopes, and life goals, individually or through support and advocacy groups.
Respect is the bedrock value that defines the physician/patient relationship and ensures the absence of discrimination and stigmatization within behavioral health systems.
Strength-based approaches remind the physician that valuing and building upon the patient’s core strengths, talents, and positive attributes, rather than narrowly focusing on personal deficits, fuels the recovery process.
Disclosure
Dr. Christensen reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Over the past 20 years, the recovery movement in mental health care has transformed the way in which clinicians and behavioral health agencies view and treat persons with serious mental illnesses. The New Freedom Commission on Mental Health defined recovery as “the process in which people are able to live, work, learn, and participate in their communities.” This process supports a clinical approach to psychiatric care that promotes opportunity, hope, and social inclusion.1 As professionals, “our primary interest should be to take the principles and concepts of [r]ecovery and to look at ways in which our practices and services could be orientated to facilitate [r]ecovery in the people who use them.”2
Drawing upon the Substance Abuse and Mental Health Services Administration’s core principles of a recovery-oriented approach,3 the mnemonic PARTNERS may help you recall key concepts to delivering and assessing recovery-oriented care.
Person-centered approaches recognize the patient’s unique gifts, strengths, needs, and cultural perspectives. Treatment is based upon the patient’s resiliencies, deficits, and personal goals, not just algorithms.
Autonomy emphasizes the patient’s right to determine his or her own destiny. It serves as the justification for the recovery maxim of “no decision about me, without me.”
Responsibility recognizes that the choices, decisions, and consequences about the type, amount, and frequency of care are a shared responsibility between physician and patient.
Transformational interactions and attitudes occur between the physician, the patient, and the system in a recovery-oriented model of care. The physician, patient, and agency are transformed into partners in the healing process.
Nonlinear concepts highlight the expectation that a patient’s pathway to recovery inevitably will be punctuated by personal gains, uneventful plateaus, and the occasional setback.
Empowerment is patients’ growing sense that they can speak openly and freely about their needs, hopes, and life goals, individually or through support and advocacy groups.
Respect is the bedrock value that defines the physician/patient relationship and ensures the absence of discrimination and stigmatization within behavioral health systems.
Strength-based approaches remind the physician that valuing and building upon the patient’s core strengths, talents, and positive attributes, rather than narrowly focusing on personal deficits, fuels the recovery process.
Disclosure
Dr. Christensen reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. President’s New Freedom Commission on Mental Health. Achieving the promise: transforming mental health care in America. Final report. Rockville, MD: Substance Abuse and Mental Health Administration; 2003.
2. South London and Maudsley NHS Foundation Trust and South West London and St. George’s Mental Health NHS Trust. Recovery is for all: hope, agency and opportunity in psychiatry. A position statement by consultant psychiatrists. London, United Kingdom: SLAM/SWLSTG; 2010.
3. Substance Abuse and Mental Health Services Administration. National consensus statement on mental health recovery. http://store.samhsa.gov/shin/content//SMA05-4129/SMA05-4129.pdf. Accessed February 24, 2012.
1. President’s New Freedom Commission on Mental Health. Achieving the promise: transforming mental health care in America. Final report. Rockville, MD: Substance Abuse and Mental Health Administration; 2003.
2. South London and Maudsley NHS Foundation Trust and South West London and St. George’s Mental Health NHS Trust. Recovery is for all: hope, agency and opportunity in psychiatry. A position statement by consultant psychiatrists. London, United Kingdom: SLAM/SWLSTG; 2010.
3. Substance Abuse and Mental Health Services Administration. National consensus statement on mental health recovery. http://store.samhsa.gov/shin/content//SMA05-4129/SMA05-4129.pdf. Accessed February 24, 2012.
Using melatonin to reset the clock of hospitalized older patients
Helping hospitalized geriatric patients maintain an appropriate sleep-wake cycle can be a challenge. Older patients’ circadian rhythm may be affected by several factors—eg, obstructive sleep apnea and restless leg syndrome—that contribute to disrupted sleep and daytime fatigue. Some patients may have dementing illnesses that could dysregulate sleep. Many older patients experience delirium during hospitalization, of which sleep-wake cycle disturbances are a hallmark. Finally, geriatric patients’ natural sleep pattern often does not mimic a hospital’s typical schedule.
Sleep medication side effects
Medications used to promote sleep can cause side effects in geriatric patients. Benzodiazepine use by older adults is discouraged because these medications could cause falls or contribute to delirium. Non-benzodiazepine hypnotics such as zolpidem, zaleplon, and eszopiclone pose a similar risk. Medications containing diphenhydramine predispose patients to deliriogenic effects via their anticholinergic properties. Tricyclic antidepressants carry risks, such as delirium secondary to anticholinergic effects, orthostatic hypotension, falls from α-1 blockade, and cardiac arrythmias.
Atypical antipsychotics sometimes are used off-label to help initiate sleep, but they carry a “black-box” warning regarding sudden death from cardiovascular events in geriatric patients with dementia. Hydroxyzine and trazodone also are associated with side effects such as orthostatic hypotension and daytime sedation, and are not always effective.
Melatonin is a hormone secreted by the pineal gland in response to darkness, under the control of the suprachiasmatic nucleus (SCN), and is thought to promote sleep via synchronizing effects on the SCN.1 Melatonin is available as an over-the-counter dietary supplement and via prescription in dosages of 1 or 3 mg. The typical effective dose is 3 to 9 mg.1 Patients should take melatonin in the mid-evening, ideally between 7 pm and 8 pm, and effects become evident after a few days. Side effects are rare; the most common are headache and nausea. Daytime sedation and vivid dreams also have been reported. Melatonin can be used safely in conjunction with other sleep aids and its major drug-drug interactions involve enhancing the effects of other sedatives.2
We have found melatonin to be effective for treating sleep disturbances in older hospitalized patients. Its effectiveness may stem from the high incidence of dysregulated or calcified pineal glands in geriatric patients, which leads to a marked reduction in melatonin secretion.3 Recent evidence also suggests melatonin may reduce the incidence of delirium in older adults, and it has been proposed as a delirium treatment in post-operative and intensive care unit settings.4
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. de Jonghe A, Korevaar JC, van Munster BC, et al. Effectiveness of melatonin treatment on circadian rhythm disturbances in dementia. Are there implications for delirium? A systematic review. Int J Geriatr Psychiatry. 2010;25(12):1201-1208.
2. Werneke U, Turner T, Priebe S. Complementary medicines in psychiatry: review of effectiveness and safety. Br J Psychiatry. 2006;188:109-121.
3. Schmid HA. Decreased melatonin biosynthesis calcium flux, pineal gland calcification and aging: a hypothetical framework. Gerontology. 1993;39(4):189-199.
4. Al-Aama T, Brymer C, Gutmanis I, et al. Melatonin decreases delirium in elderly patients: a randomized, placebo-controlled trial. Int J Geriatr Psychiatry. 2011;26(7):687-694.
Helping hospitalized geriatric patients maintain an appropriate sleep-wake cycle can be a challenge. Older patients’ circadian rhythm may be affected by several factors—eg, obstructive sleep apnea and restless leg syndrome—that contribute to disrupted sleep and daytime fatigue. Some patients may have dementing illnesses that could dysregulate sleep. Many older patients experience delirium during hospitalization, of which sleep-wake cycle disturbances are a hallmark. Finally, geriatric patients’ natural sleep pattern often does not mimic a hospital’s typical schedule.
Sleep medication side effects
Medications used to promote sleep can cause side effects in geriatric patients. Benzodiazepine use by older adults is discouraged because these medications could cause falls or contribute to delirium. Non-benzodiazepine hypnotics such as zolpidem, zaleplon, and eszopiclone pose a similar risk. Medications containing diphenhydramine predispose patients to deliriogenic effects via their anticholinergic properties. Tricyclic antidepressants carry risks, such as delirium secondary to anticholinergic effects, orthostatic hypotension, falls from α-1 blockade, and cardiac arrythmias.
Atypical antipsychotics sometimes are used off-label to help initiate sleep, but they carry a “black-box” warning regarding sudden death from cardiovascular events in geriatric patients with dementia. Hydroxyzine and trazodone also are associated with side effects such as orthostatic hypotension and daytime sedation, and are not always effective.
Melatonin is a hormone secreted by the pineal gland in response to darkness, under the control of the suprachiasmatic nucleus (SCN), and is thought to promote sleep via synchronizing effects on the SCN.1 Melatonin is available as an over-the-counter dietary supplement and via prescription in dosages of 1 or 3 mg. The typical effective dose is 3 to 9 mg.1 Patients should take melatonin in the mid-evening, ideally between 7 pm and 8 pm, and effects become evident after a few days. Side effects are rare; the most common are headache and nausea. Daytime sedation and vivid dreams also have been reported. Melatonin can be used safely in conjunction with other sleep aids and its major drug-drug interactions involve enhancing the effects of other sedatives.2
We have found melatonin to be effective for treating sleep disturbances in older hospitalized patients. Its effectiveness may stem from the high incidence of dysregulated or calcified pineal glands in geriatric patients, which leads to a marked reduction in melatonin secretion.3 Recent evidence also suggests melatonin may reduce the incidence of delirium in older adults, and it has been proposed as a delirium treatment in post-operative and intensive care unit settings.4
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Helping hospitalized geriatric patients maintain an appropriate sleep-wake cycle can be a challenge. Older patients’ circadian rhythm may be affected by several factors—eg, obstructive sleep apnea and restless leg syndrome—that contribute to disrupted sleep and daytime fatigue. Some patients may have dementing illnesses that could dysregulate sleep. Many older patients experience delirium during hospitalization, of which sleep-wake cycle disturbances are a hallmark. Finally, geriatric patients’ natural sleep pattern often does not mimic a hospital’s typical schedule.
Sleep medication side effects
Medications used to promote sleep can cause side effects in geriatric patients. Benzodiazepine use by older adults is discouraged because these medications could cause falls or contribute to delirium. Non-benzodiazepine hypnotics such as zolpidem, zaleplon, and eszopiclone pose a similar risk. Medications containing diphenhydramine predispose patients to deliriogenic effects via their anticholinergic properties. Tricyclic antidepressants carry risks, such as delirium secondary to anticholinergic effects, orthostatic hypotension, falls from α-1 blockade, and cardiac arrythmias.
Atypical antipsychotics sometimes are used off-label to help initiate sleep, but they carry a “black-box” warning regarding sudden death from cardiovascular events in geriatric patients with dementia. Hydroxyzine and trazodone also are associated with side effects such as orthostatic hypotension and daytime sedation, and are not always effective.
Melatonin is a hormone secreted by the pineal gland in response to darkness, under the control of the suprachiasmatic nucleus (SCN), and is thought to promote sleep via synchronizing effects on the SCN.1 Melatonin is available as an over-the-counter dietary supplement and via prescription in dosages of 1 or 3 mg. The typical effective dose is 3 to 9 mg.1 Patients should take melatonin in the mid-evening, ideally between 7 pm and 8 pm, and effects become evident after a few days. Side effects are rare; the most common are headache and nausea. Daytime sedation and vivid dreams also have been reported. Melatonin can be used safely in conjunction with other sleep aids and its major drug-drug interactions involve enhancing the effects of other sedatives.2
We have found melatonin to be effective for treating sleep disturbances in older hospitalized patients. Its effectiveness may stem from the high incidence of dysregulated or calcified pineal glands in geriatric patients, which leads to a marked reduction in melatonin secretion.3 Recent evidence also suggests melatonin may reduce the incidence of delirium in older adults, and it has been proposed as a delirium treatment in post-operative and intensive care unit settings.4
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. de Jonghe A, Korevaar JC, van Munster BC, et al. Effectiveness of melatonin treatment on circadian rhythm disturbances in dementia. Are there implications for delirium? A systematic review. Int J Geriatr Psychiatry. 2010;25(12):1201-1208.
2. Werneke U, Turner T, Priebe S. Complementary medicines in psychiatry: review of effectiveness and safety. Br J Psychiatry. 2006;188:109-121.
3. Schmid HA. Decreased melatonin biosynthesis calcium flux, pineal gland calcification and aging: a hypothetical framework. Gerontology. 1993;39(4):189-199.
4. Al-Aama T, Brymer C, Gutmanis I, et al. Melatonin decreases delirium in elderly patients: a randomized, placebo-controlled trial. Int J Geriatr Psychiatry. 2011;26(7):687-694.
1. de Jonghe A, Korevaar JC, van Munster BC, et al. Effectiveness of melatonin treatment on circadian rhythm disturbances in dementia. Are there implications for delirium? A systematic review. Int J Geriatr Psychiatry. 2010;25(12):1201-1208.
2. Werneke U, Turner T, Priebe S. Complementary medicines in psychiatry: review of effectiveness and safety. Br J Psychiatry. 2006;188:109-121.
3. Schmid HA. Decreased melatonin biosynthesis calcium flux, pineal gland calcification and aging: a hypothetical framework. Gerontology. 1993;39(4):189-199.
4. Al-Aama T, Brymer C, Gutmanis I, et al. Melatonin decreases delirium in elderly patients: a randomized, placebo-controlled trial. Int J Geriatr Psychiatry. 2011;26(7):687-694.
Premenstrual dysphoric disorder: How to alleviate her suffering
Approximately 75% of women experience a premenstrual change in emotional or physical symptoms commonly referred to as premenstrual syndrome (PMS). These symptoms—including increased irritability, tension, depressed mood, and somatic complaints such as breast tenderness and bloating—often are mild to moderate and cause minimal distress.1 However, approximately 3% to 9% of women experience moderate to severe premenstrual mood symptoms that meet criteria for premenstrual dysphoric disorder (PMDD).2
PMDD includes depressed or labile mood, anxiety, irritability, anger, insomnia, difficulty concentrating, and other symptoms that occur exclusively during the 2 weeks before menses and cause significant deterioration in daily functioning. Women with PMDD use general and mental health services more often than women without the condition.3 They may experience impairment in marital and parental relationships as severe as that experienced by women with recurrent or chronic major depression.2
PMDD often responds to treatment. Unfortunately, many women with PMDD do not seek treatment, and up to 90% may go undiagnosed.4 In this article, we review the prevalence, etiology, diagnosis, and treatment of PMDD.
A complex disorder
A distinguishing characteristic of PMDD is the timing of symptom onset. In women with PMDD, mood symptoms occur only during the luteal phase of the menstrual cycle (ovulation until onset of menses) and resolve after menstruation onset. Women with PMDD report normal mood and functioning during the follicular phase of the menstrual cycle (first day of the menstrual cycle until ovulation).
Although PMS and PMDD criteria share affective and somatic symptoms, more symptoms are required for a PMDD diagnosis, and symptoms often are more severe.5 As defined in DSM-IV-TR (Table),6 PMDD has a broader range of symptoms than PMS and includes symptoms not included in the American College of Obstetrics and Gynecology criteria for PMS,7 such as impaired concentration, appetite, and sleep (hypersomnia or insomnia); and mood lability. PMDD symptoms must occur only during the 2 weeks preceding menses, although on average symptoms last 6 days and severity usually peaks in the 2 days before menses.1 The prevalence of subthreshold PMDD is fairly common; approximately 19% of women will meet some—but not all—DSM-IV-TR criteria for PMDD.3
In a revision proposed for DSM-5, PMDD would be included as a mood disorder, which represents a significant change from DSM-IV-TR, where it is listed in the appendix as “research criteria.”8 In addition, in oral contraceptive users, a PMDD diagnosis should not be made unless the premenstrual symptoms are reported to be present and as severe when the woman is not taking the oral contraceptive.8
Comorbidity with other axis I disorders such as major depressive disorder (MDD), bipolar disorder (BD), and anxiety disorders is high.9-11 Women with an MDD history have the highest correlation with PMDD,9 and worsening premenstrual mood symptoms are more common in women with BD.12 Payne et al11 found that premenstrual symptoms were reported by twice as many women diagnosed with mood disorders (68%) than women without a psychiatric diagnosis (34%). Moreover, 38% to 46% of women with PMDD have comorbid seasonal affective disorder, and 11% to 38% report a comorbid anxiety disorder.12 Women with PMDD and a history of MDD have lower cortisol concentrations than non-PMDD women.10 Although interventions for PMDD and a comorbid axis I disorder may be similar, it is important to consider both when planning treatment.
Abuse, trauma, and PMDD. An association between PMS/PMDD and a history of sexual and physical abuse is well-documented.13 Studies have reported abuse histories among almost 60% of women with PMDD,14 although studies comparing abuse and trauma in PMDD vs non-PMDD women have been small. A recent study found that trauma and posttraumatic stress disorder are independently associated with PMDD and premenstrual symptoms.15
Evidence suggests that a history of abuse is associated with specific biological sequelae in PMDD women, particularly with respect to hypothalamic-pituitary-thyroid axis measures and noradrenergic activity.16-18 Women with PMDD and a history of sexual abuse show:
- markedly elevated triiodothyronine (T3) concentrations (the more biologically potent thyroid hormone) that appear to result from increased conversion of thyroxine (T4) to T316
- lower circulating plasma norepinephrine concentrations17
- greater resting and stress-induced heart rates and systolic blood pressure compared with non-abused PMDD women, an effect that is eliminated by clonidine (an α-2 adrenergic receptor agonist).18
One study showed that PMDD women with abuse histories had higher blood pressure measurements at rest and during stress and exhibited greater vascular tone than non-abused women; these effects were not seen in non-PMDD women with similar abuse histories.14 This body of evidence is consistent with the concept that PMDD is a stress-related disorder,19 and that a history of abuse is prevalent and may identify a clinically distinct subgroup of PMDD women with respect to thyroid axis and adrenergic physiology. Screening PMDD patients for abuse histories may help manage the disorder.
For a discussion of the etiology of PMDD, see Box 1.
Table
DSM-IV-TR research criteria for PMDD
|
| Source: Reference 6 |
Although questions remain about the pathogenesis of premenstrual dysphoric disorder (PMDD), literature documents the role of gonadal steroids (estrogen and progesterone) in the etiology of premenstrual syndrome (PMS)/PMDD and suggests that women with PMDD are differentially sensitive to the normal physiologic fluctuations of gonadal hormones throughout the menstrual cycle.a
The first half of the menstrual cycle—the follicular phase—begins with increasing levels of follicular stimulating hormone (FSH) leading to maturity of the ovarian follicle. Once the follicle is ripe, the luteal phase of the menstrual cycle begins with a surge in luteinizing hormone (LH), which results in ovulation of the mature follicle, followed by increased secretion of progesterone, followed by increased estrogen secretion. The system is regulated via negative feedback, and high levels of progesterone decrease gonadotropin-releasing hormone (GnRH) pulse frequency, which leads to decreased secretion of FSH and LH, and subsequent decline of estrogen and progesterone. If the ovarian follicle is not fertilized, menstruation begins and FSH levels rise again, initiating the follicular phase of the menstrual cycle.
Fluctuations in reproductive steroid levels have been implicated in the etiology of PMDD from studies showing that oophorectomy and ovulation inhibitors (GnRH agonists) relieve symptoms.b Some researchers proposed that symptoms are related to the drop of progesterone in the late luteal phase; however, many women have symptoms that start at ovulation or during the early luteal phase before the fall in progesterone concentrations.c PMS symptoms may occur independently of the mid-to-late luteal phase.d Because production of gonadal steroids does not differ between women with or without PMS or PMDD,e it may be that follicular or periovulatory changes in levels of estradiol or progesterone secretion trigger symptoms of PMDD in susceptible women, while women without PMDD appear to be immune to these effects of gonadal steroids. This idea is supported by a study showing that pharmacologic induction of a hypogonadal state eliminates symptoms in most women with severe PMS, while “adding back” estrogen or progesterone within the context of hypogonadism elicits return of PMS symptoms in those with PMS but not in controls.a
Abnormalities in serotonin levels also may contribute to PMDD.f In 1 study, a serotonin receptor antagonist precipitated return of symptoms within 24 hours of administration in women with PMDD but not in controls.g PMDD symptoms also can be evoked by depleting the serotonin precursor tryptophan.h When women with PMDD received paroxetine at different phases of their menstrual cycle, they showed fluctuations in serotonergic function across their cycles; these fluctuations were not seen in controls.i Other neurotransmitters implicated in PMDD include γ-aminobutyric acid (GABA),j glutamate,k lower levels of cortisol and beta-endorphins,l and an abnormal stress response.m
Other studies have focused on differing concentrations of luteal phase hormonesn and gene associations. Two studies suggested that PMDD is heritableo,p and other studies have looked at the association between specific psychological traits that are more prominent in PMDD and single nucleotide polymorphisms in the estrogen receptor alpha gene.q,r
Thyroid hormones also may play a role in the etiology of PMS/PMDD. Thyroid function tests have shown greater variability in women with PMS vs controls,s although this variability appears to be limited to women with a sexual abuse history.t Other studies have evaluated hormones regulated across the circadian and sleep-wake cycles, including melatonin, cortisol, thyroid-stimulating hormone, and prolactin, which suggests that although levels of these hormones may not differ between women with PMDD and controls, the timing of their excretion may vary.s Additionally, women with PMDD are characterized by prefrontal brain asymmetry on electroencephalography that also is evident in patients with major depressive disorder.u
There also may be dysregulation of allopregnanolone (ALLO) in women with PMDD.v,w ALLO is a metabolite of progesterone that is a neurosteroid produced in the brain as well as in the ovary and adrenals.v It produces anxiolytic effects by acting as a modulator of GABA receptors.x In PMDD, ALLO levels may influence the severity of premenstrual symptoms.w
References
- Schmidt PJ, Nieman LK, Danaceau MA, et al. Differential behavioral effects of gonadal steroids in women with and in those without premenstrual syndrome. N Engl J Med. 1998;338(4):209-216.
- Muse KN, Cetel NS, Futterman LA, et al. The premenstrual syndrome. Effects of “medical ovariectomy.” N Engl J Med. 1984;311(21):1345-1349.
- Yonkers KA, O’Brien PM, Eriksson E. Premenstrual syndrome. Lancet. 2008;371(9619):1200-1210.
- Schmidt PJ, Nieman LK, Grover GN, et al. Lack of effect of induced menses on symptoms in women with premenstrual syndrome. N Engl J Med. 1991;324(17):1174-1179.
- Rubinow DR, Schmidt PJ. The neuroendocrinology of menstrual cycle mood disorders. Ann N Y Acad Sci. 1995;771:648-659.
- Steiner M, Pearlstein T. Premenstrual dysphoria and the serotonin system: pathophysiology and treatment. J Clin Psychiatry. 2000;61(suppl 12):17-21.
- Roca CA, Schmidt PJ, Smith MJ, et al. Effects of metergoline on symptoms in women with premenstrual dysphoric disorder. Am J Psychiatry. 2002;159(11):1876-1881.
- Menkes DB, Coates DC, Fawcett JP. Acute tryptophan depletion aggravates premenstrual syndrome. J Affect Disord. 1994;32(1):37-44.
- Inoue Y, Terao T, Iwata N, et al. Fluctuating serotonergic function in premenstrual dysphoric disorder and premenstrual syndrome: findings from neuroendocrine challenge tests. Psychopharmacology (Berl). 2007;190(2):213-219.
- Epperson CN, Haga K, Mason GF, et al. Cortical gamma-aminobutyric acid levels across the menstrual cycle in healthy women and those with premenstrual dysphoric disorder: a proton magnetic resonance spectroscopy study. Arch Gen Psychiatry. 2002;59(9):851-858.
- Batra NA, Seres-Mailo J, Hanstock C, et al. Proton magnetic resonance spectroscopy measurement of brain glutamate levels in premenstrual dysphoric disorder. Biol Psychiatry. 2008;63(12):1178-1184.
- Straneva PA, Maixner W, Light KC, et al. Menstrual cycle, beta-endorphins, and pain sensitivity in premenstrual dysphoric disorder. Health Psychol. 2002;21(4):358-367.
- Epperson CN, Pittman B, Czarkowski KA, et al. Luteal-phase accentuation of acoustic startle response in women with premenstrual dysphoric disorder. Neuropsychopharmacology. 2007;32(10):2190-2198.
- Thys-Jacobs S, McMahon D, Bilezikian JP. Differences in free estradiol and sex hormone-binding globulin in women with and without premenstrual dysphoric disorder. J Clin Endocrinol Metab. 2008;93(1):96-102.
- Payne JL, Klein SR, Zamoiski RB, et al. Premenstrual mood symptoms: study of familiality and personality correlates in mood disorder pedigrees. Arch Womens Ment Health. 2009;12(1):27-34.
- Kendler KS, Karkowski LM, Corey LA, et al. Longitudinal population-based twin study of retrospectively reported premenstrual symptoms and lifetime major depression. Am J Psychiatry. 1998;155(9):1234-1240.
- Miller A, Vo H, Huo L, et al. Estrogen receptor alpha (ESR-1) associations with psychological traits in women with PMDD and controls. J Psychiatr Res. 2010;44(12):788-794.
- Huo L, Straub RE, Roca C, et al. Risk for premenstrual dysphoric disorder is associated with genetic variation in ESR1, the estrogen receptor alpha gene. Biol Psychiatry. 2007;62(8):925-933.
- Girdler SS, Pedersen CA, Light KC. Thyroid axis function during the menstrual cycle in women with premenstrual syndrome. Psychoneuroendocrinology. 1995;20(4):395-403.
- Girdler SS, Thompson KS, Light KC, et al. Historical sexual abuse and current thyroid axis profiles in women with premenstrual dysphoric disorder. Psychosom Med. 2004;66(3):403-410.
- Accortt EE, Stewart JL, Coan JA, et al. Prefrontal brain asymmetry and pre-menstrual dysphoric disorder symptomatology. J Affect Disord. 2011;128(1-2):178-183.
- Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6(6):2311-2322.
- Girdler SS, Straneva PA, Light KC, et al. Allopregnanolone levels and reactivity to mental stress in premenstrual dysphoric disorder. Biol Psychiatry. 2001;49(9):788-797.
- Brot MD, Akwa Y, Purdy RH, et al. The anxiolytic-like effects of the neurosteroid allopregnanolone: interactions with GABA(A) receptors. Eur J Pharmacol. 1997;325(1):1-7.
Mood charting aids diagnosis
A PMDD diagnosis requires prospective daily monitoring of symptoms for ≥2 consecutive months. Because only 25% to 35% of women who present with PMDD meet diagnostic criteria when prospective daily monitoring is used,20 it is important for patients to keep a daily diary of PMDD symptoms to distinguish the disorder from PMS (Box 2). The Prospective Record of the Impact and Severity of Premenstrual Symptoms calendar and the Daily Record of Severity of Problems (DRSPP)21 may help make the diagnosis.
The widely used DRSPP allows clinicians to quantify the severity of physical, emotional, and behavioral symptoms and may be the easiest to use in clinical practice because it creates a graphic representation of cyclical symptom changes. The DRSPP includes all PMDD symptoms and severity ratings21 and is recognized as a valid instrument for diagnosing PMDD. Another option is a revised visual analogue scale. Lastly, a new revised Premenstrual Tension Syndrome (PMTS) rating scale, which combines the PMTS Observer rating scale plus multiple visual analogue scales, shows promise as a tool to assess PMDD symptoms.
To distinguish premenstrual syndrome (PMS) from premenstrual dysphoric disorder (PMDD), premenstrual exacerbation of an underlying psychiatric disorder, general medical conditions, or other disorders with no association to the menstrual cycle, it is necessary to have patients conduct daily symptom charting over 2 menstrual cycles. This charting should include documentation of emotional, behavioral, and physical symptoms. PMDD can be differentiated from PMS by the severity and number of symptoms. In PMDD, 1 of the symptoms must be a mood disturbance (depressed, anxious, labile, and/or irritable). For a sample form used for PMDD charting, the Daily Record of Severity of Problems, see http://pmdd.factsforhealth.org/drsp/drsp_month.pdf.
Treatment options
Hormonal interventions. Attempts to treat PMS with progesterone during the luteal phase have been largely unsuccessful, although progesterone is approved to treat PMS in the United Kingdom. Long-acting gonadotropin-releasing hormone (GnRH) agonists are effective but result in medical menopause with its accompanying symptoms, which puts women at risk for osteoporosis.22 Approximately 60% to 70% of women with PMDD respond to leuprolide (a GnRH agonist), but it is difficult to predict who will respond; daily mood self-ratings of sadness, anxiety, and irritability predict a positive response to leuprolide with high probability.23 Side effects of GnRH agonists (hot flashes, night sweats, vaginal dryness, etc.) can be tempered by “adding back” some estrogen with a hormonal agent with progestational activity to reduce the risks of unopposed estrogen (ie, endometrial hyperplasia).24
Surgical bilateral oophorectomy is effective but extremely invasive, especially in younger women in whom removal of ovaries generally is inadvisable. Patients should receive a trial of a GnRH agonist before a surgical intervention, because oophorectomy may not reduce symptoms and is irreversible. Oophorectomy also would require hormone replacement therapy.
High-dose estrogen as transdermal patches or subcutaneous implants to inhibit ovulation is effective, but because of the risks of unopposed estrogen, a progestin would be needed. Risks of estrogen therapy (alone and in combination with progestins) include increased risk of endometrial cancer, coronary heart disease, breast cancer, stroke, and pulmonary embolism.25 Danazol, a synthetic androgen and gonadotropin inhibitor, effectively blocks ovulation, but side effects include hirsutism and possible teratogenicity.26 Although these hormonal manipulations may effectively treat PMDD, none are considered practical.
The use of combined oral contraceptives (estrogen and progestin) is common. Although continuous cycle oral contraceptives often are recommended for PMDD, limited evidence supports their use; studies have been mostly negative.27,28 A recent review of 4 studies of a continuous oral contraceptive (levonorgestrel 90 mcg/ethinyl estradiol 20 mcg) for PMDD and PMS had more promising results, although the results were highly variable among studies and a large placebo effect was observed.29
A combination oral contraceptive, drospirenone/ethinyl estradiol, is FDA-approved for treating PMDD in women seeking hormonal contraception because it has shown efficacy compared with placebo, with reported improvements in perceived productivity, social activities, and interpersonal relationships.30 The nature of hormone delivery (ie, a reduction in the pill-free interval from 7 to 4 days) in drospirenone/ethinyl estradiol may contribute to its efficacy because a reduced pill-free interval minimizes the degree of follicular recruitment and subsequent estrogen production and cyclicity seen with standard oral contraceptive.31
Antidepressants have been shown to effectively ameliorate affective and physical symptoms and improve quality of life and psychosocial function in patients with PMS and PMDD. The response rates for selective serotonin reuptake inhibitors (SSRIs) in PMDD treatment vary from 60% to 90%, vs 30% to 40% for placebo.32 A 2009 Cochrane review found SSRIs reduced premenstrual symptoms compared with placebo.33 However, a literature review suggested that the percentage of women with PMDD who respond to SSRIs or continuous oral contraceptives is lower than the percentage of women who do not respond at all, once the placebo effect is taken into account, and that approximately 40% of women with PMDD do not respond to SSRIs.34 A small study found that citalopram may be effective for women with PMDD who did not respond to a prior SSRI.35
However, only antidepressants that affect serotonergic—not noradrenergic—transmission are effective in PMDD.22 These include:
- the tricyclic antidepressant clomipramine
- the SSRIs citalopram, escitalopram, fluoxetine, paroxetine, and sertraline
- the serotonin-noradrenergic reuptake inhibitor venlafaxine.
It appears that in PMDD, serotonergic agents play a role other than their antidepressant effect.36 The effect of these agents is rapid in PMS/PMDD; women with PMDD who take antidepressants often experience reduced symptoms within the first menstrual cycle, whereas in MDD the onset of action can take weeks or months.37
Although why onset of antidepressant action is quick in PMDD is unclear, rapid onset allows for several dosing options. Some women prefer continuous dosing throughout the month because they do not have to keep track of ovulation. Dosing antidepressants only in the luteal phase (taking the antidepressant from ovulation onset to the start of menses) is an effective treatment strategy.38 Many women prefer to take medication for only 2 weeks per month, which can decrease side effects and lower treatment costs. Alternatively, symptom-onset dosing—initiating the antidepressant when PMDD symptoms begin and stopping at menses onset or within 3 days thereafter—has shown promising results.39,40 Paroxetine, sertraline, and fluoxetine are FDA-approved for PMDD as continuous or intermittent regimens, although using fluoxetine intermittently may not make sense because its biologically active metabolite has an extended half-life.37
Other treatments. Dietary interventions, psychotherapy, vitamins, bright light treatment, and spironolactone have been assessed for PMS/PMDD, although for many evidence-based findings are lacking (Box 3).
See the Bibliography below for studies that support using antidepressants to treat PMDD
Two reviews of 10 randomized controlled trials (RCTs) that evaluated 62 herbs, vitamins, and mineral treatments for premenstrual symptoms found efficacy for chasteberry (Vitex agnus-castus), calcium, and vitamin B6 but not for primrose oil, magnesium oxide, or St. John’s wort.a,b A study comparing fluoxetine with chasteberry found a similar percentage of patients responded to either agent (68% vs 58%, respectively).c Another study showed calcium resulted in a 48% reduction in premenstrual symptoms from baseline, compared with a 30% reduction with placebo.d Bright light treatment significantly reduced depression ratings in women with premenstrual dysphoric disorder (PMDD).e Compared with placebo, the aldosterone antagonist spironolactone improved irritability, depression, feelings of swelling, breast tenderness, and food craving in women with premenstrual syndrome (PMS).f
A recent systematic review of 7 trials of cognitive-behavioral therapy (CBT) for PMDD, including 3 RCTs, showed a lack of a statistically significant effect.g However, a separate review of RCTs of alternative treatments for PMDD—5 of which included CBT—suggested that CBT may be beneficial in reducing premenstrual symptoms, but the evidence was low quality.h
References
- Dante G, Facchinetti F. Herbal treatments for alleviating premenstrual symptoms: a systematic review. J Psychosom Obstet Gynaecol. 2011;32(1):42-51.
- Whelan AM, Jurgens TM, Naylor H. Herbs, vitamins and minerals in the treatment of premenstrual syndrome: a systematic review. Can J Clin Pharmacol. 2009;16(3):e407-e429.
- Atmaca M, Kumru S, Tezcan E. Fluoxetine versus Vitex agnus castus extract in the treatment of premenstrual dysphoric disorder. Hum Psychopharmacol. 2003;18(3):191-195.
- Thys-Jacobs S, Starkey P, Bernstein D, et al. Calcium carbonate and the premenstrual syndrome: effects on premenstrual and menstrual symptoms. Premenstrual Syndrome Study Group. Am J Obstet Gynecol. 1998;179(2):444-452.
- Parry BL, Berga SL, Mostofi N, et al. Morning versus evening bright light treatment of late luteal phase dysphoric disorder. Am J Psychiatry. 1989;146(9):1215-1217.
- Wang M, Hammarbäck S, Lindhe BA, et al. Treatment of premenstrual syndrome by spironolactone: a double-blind, placebo-controlled study. Acta Obstet Gynecol Scand. 1995;74(10):803-808.
- Lustyk MK, Gerrish WG, Shaver S, et al. Cognitive-behavioral therapy for premenstrual syndrome and premenstrual dysphoric disorder: a systematic review. Arch Womens Ment Health. 2009;12(2):85-96.
- Busse JW, Montori VM, Krasnik C, et al. Psychological intervention for premenstrual syndrome: a meta-analysis of randomized controlled trials. Psychother Psychosom. 2009;78(1):6-15.
Cohen LS, Miner C, Brown EW, et al. Premenstrual daily fluoxetine for premenstrual dysphoric disorder: a placebo-controlled, clinical trial using computerized diaries. Obstet Gynecol. 2002;100(3):435-444.
Eriksson E, Ekman A, Sinclair S, et al. Escitalopram administered in the luteal phase exerts a marked and dose-dependent effect in premenstrual dysphoric disorder. J Clin Psychopharmacol. 2008;28(2):195-202.
Eriksson E, Hedberg MA, Andersch B, et al. The serotonin reuptake inhibitor paroxetin is superior to the noradrenaline reuptake inhibitor maprotiline in the treatment of premenstrual syndrome. Neuropsychopharmacology. 1995;12(2):167-176.
Menkes DB, Taghavi E, Mason PA, et al. Fluoxetine treatment of severe premenstrual syndrome. BMJ. 1992;305(6849):346-347.
Miner C, Brown E, McCray S, et al. Weekly luteal-phase dosing with enteric-coated fluoxetine 90 mg in premenstrual dysphoric disorder: a randomized, double-blind, placebo-controlled clinical trial. Clin Ther. 2002;24(3):417-433.
Ozeren S, Corakçi A, Yücesoy I, et al. Fluoxetine in the treatment of premenstrual syndrome. Eur J Obstet Gynecol Reprod Biol. 1997;73(2):167-170.
Pearlstein TB, Stone AB, Lund SA, et al. Comparison of fluoxetine, bupropion, and placebo in the treatment of premenstrual dysphoric disorder. J Clin Psychopharmacol. 1997;17(4):261-266.
Ravindran LN, Woods SA, Steiner M, et al. Symptom-onset dosing with citalopram in the treatment of premenstrual dysphoric disorder (PMDD): a case series. Arch Womens Ment Health. 2007;10(3):125-127.
Steiner M, Brown E, Trzepacz P, et al. Fluoxetine improves functional work capacity in women with premenstrual dysphoric disorder. Arch Womens Ment Health. 2003;6(1):71-77.
Stone AB, Pearlstein TB, Brown WA. Fluoxetine in the treatment of late luteal phase dysphoric disorder. J Clin Psychiatry. 1991;52(7):290-293.
Sundblad C, Hedberg MA, Eriksson E. Clomipramine administered during the luteal phase reduces the symptoms of premenstrual syndrome: a placebo-controlled trial. Neuropsychopharmacology. 1993;9(2):133-145.
Sundblad C, Modigh K, Andersch B, et al. Clomipramine effectively reduces premenstrual irritability and dysphoria: a placebo-controlled trial. Acta Psychiatr Scand. 1992;85(1):39-47.
Su TP, Schmidt PJ, Danaceau MA, et al. Fluoxetine in the treatment of premenstrual dysphoria. Neuropsychopharmacology. 1997; 16(5):346-356.
Wikander I, Sundblad C, Andersch B, et al. Citalopram in premenstrual dysphoria: is intermittent treatment during luteal phases more effective than continuous medication throughout the menstrual cycle? J Clin Psychopharmacol. 1998;18(5):390-398.
Treatment selection
To enhance compliance and improve the likelihood of successful treatment, tailor treatment decisions to your patient’s needs. Carefully discuss with your patient the evidence-based literature to select the best option for her. Factors to consider when counseling patients include:
- the patient’s age, cigarette smoking habits, and body mass index, which may contraindicate oral contraceptives
- does the patient have regular cycle lengths?
- can she adhere to an on-off schedule? If so, intermittent SSRI dosing may be a good treatment option
- does the patient have irregular cycles?
- is there evidence that symptoms persist into the follicular phase, albeit at a lower level? If so, continuous SSRI dosing may be the best option.
Related Resources
- American Psychiatric Association. DSM-5 development: D 04 Premenstrual dysphoric disorder. www.dsm5.org/proposedrevision/pages/proposedrevision.aspx?rid=484.
Drug Brand Names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clonidine • Catapres, others
- Danazol • Danocrine
- Drospirenone/ethinyl estradiol • Yaz
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Leuprolide • Lupron
- Levonorgestrel/ethinyl estradiol • Seasonale, others
- Paroxetine • Paxil
- Progesterone • Prometrium
- Sertraline • Zoloft
- Spironolactone • Aldactone
- Venlafaxine • Effexor
Disclosures
Drs. Wakil and Girdler report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products
Dr. Meltzer-Brody receives research/grant support from AstraZeneca, The Foundation of Hope, and the National Institutes of Health.
1. Yonkers KA, O’Brien PM, Eriksson E. Premenstrual syndrome. Lancet. 2008;371(9619):1200-1210.
2. Halbreich U, Borenstein J, Pearlstein T, et al. The prevalence, impairment, impact and burden of premenstrual dysphoric disorder (PMS/PMDD). Psychoneuroendocrinology. 2008;28(suppl 3):1-23.
3. Wittchen HU, Becker E, Lieb R, et al. Prevalence, incidence and stability of premenstrual dysphoric disorder in the community. Psychol Med. 2002;32(1):119-132.
4. Hylan TR, Sundell K, Judge R. The impact of premenstrual symptomatology on functioning and treatment-seeking behavior: experience from the United States United Kingdom, and France. J Womens Health Gend Based Med. 1999;8(8):1043-1052.
5. Biggs WS, Demuth RH. Premenstrual syndrome and premenstrual dysphoric disorder. Am Fam Physician. 2011;84(8):918-924.
6. Diagnostic and statistical manual of mental disorders. 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
7. ACOG Practice Bulletin. Clinical management guidelines for obstetrician-gynecologists. Number 15 April 2000. Premenstrual syndrome. Obstet Gynecol. 2000;95:1-9.
8. American Psychiatric Association. Proposed draft revisions to DSM disorders and criteria. DSM-5 development. http://www.dsm5.org/proposedrevision/Pages/Default.aspx. Accessed February 23 2012.
9. Yonkers KA. The association between premenstrual dysphoric disorder and other mood disorders. J Clin Psychiatry. 1997;58(suppl 15):19-25.
10. Klatzkin RR, Lindgren ME, Forneris CA, et al. Histories of major depression and premenstrual dysphoric disorder: evidence for phenotypic differences. Biol Psychol. 2010;84(2):235-247.
11. Payne JL, Roy PS, Murphy-Eberenz K, et al. Reproductive cycle-associated mood symptoms in women with major depression and bipolar disorder. J Affect Disord. 2007;99(1-3):221-229.
12. Kim DR, Gyulai L, Freeman EW, et al. Premenstrual dysphoric disorder and psychiatric co-morbidity. Arch Womens Ment Health. 2004;7(1):37-47.
13. Golding JM, Taylor DL, Menard L, et al. Prevalence of sexual abuse history in a sample of women seeking treatment for premenstrual syndrome. J Psychosom Obstet Gynaecol. 2000;21(2):69-80.
14. Girdler SS, Leserman J, Bunevicius R, et al. Persistent alterations in biological profiles in women with abuse histories: influence of premenstrual dysphoric disorder. Health Psychol. 2007;26(2):201-213.
15. Pilver CE, Levy BR, Libby DJ, et al. Posttraumatic stress disorder and trauma characteristics are correlates of premenstrual dysphoric disorder. Arch Womens Ment Health. 2011;14(5):383-393.
16. Girdler SS, Thompson KS, Light KC, et al. Historical sexual abuse and current thyroid axis profiles in women with premenstrual dysphoric disorder. Psychosom Med. 2004;66(3):403-410.
17. Girdler SS, Sherwood A, Hinderliter AL, et al. Biological correlates of abuse in women with premenstrual dysphoric disorder and healthy controls. Psychosom Med. 2003;65(5):849-856.
18. Bunevicius R, Hinderliter AL, Light KC, et al. Histories of sexual abuse are associated with differential effects of clonidine on autonomic function in women with premenstrual dysphoric disorder. Biol Psychol. 2005;69(3):281-296.
19. Perkonigg A, Yonkers KA, Pfister H, et al. Risk factors for premenstrual dysphoric disorder in a community sample of young women: the role of traumatic events and posttraumatic stress disorder. J Clin Psychiatry. 2004;65(10):1314-1322.
20. Girdler SS, Pedersen CA, Stern RA, et al. Menstrual cycle and premenstrual syndrome: modifiers of cardiovascular reactivity in women. Health Psychol. 1993;12(3):180-192.
21. Endicott J, Nee J, Harrison W. Daily Record of Severity of Problems (DRSP): reliability and validity. Arch Womens Ment Health. 2006;9(1):41-49.
22. Cunningham J, Yonkers KA, O’Brien S, et al. Update on research and treatment of premenstrual dysphoric disorder. Harv Rev Psychiatry. 2009;17(2):120-137.
23. Pincus SM, Alam S, Rubinow DR, et al. Predicting response to leuprolide of women with premenstrual dysphoric disorder by daily mood rating dynamics. J Psychiatr Res. 2011;45(3):386-394.
24. Wyatt KM, Dimmock PW, Ismail KM, et al. The effectiveness of GnRHa with and without ‘add-back’ therapy in treating premenstrual syndrome: a meta analysis. BJOG. 2004;111(6):585-593.
25. Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321-333.
26. Hahn PM, Van Vugt DA, Reid RL. A randomized placebo-controlled, crossover trial of danazol for the treatment of premenstrual syndrome. Psychoneuroendocrinology. 1995;20(2):193-209.
27. Bancroft J, Rennie D. The impact of oral contraceptives on the experience of perimenstrual mood clumsiness, food craving and other symptoms. J Psychosom Res. 1993;37(2):195-202.
28. Graham CA, Sherwin BB. A prospective treatment study of premenstrual symptoms using a triphasic oral contraceptive. J Psychosom Res. 1992;36(3):257-266.
29. Freeman EW, Halbreich U, Grubb GS, et al. An overview of four studies of a continuous oral contraceptive (levonorgestrel 90 mcg/ethinyl estradiol 20 mcg) on premenstrual dysphoric disorder and premenstrual syndrome [published online ahead of print December 5, 2011]. Contraception. doi: 10.1016/j.contraception.2011.09.010.
30. Lopez LM, Kaptein AA, Helmerhorst FM. Oral contraceptives containing drospirenone for premenstrual syndrome. Cochrane Database Syst Rev. 2009;(2):CD006586.
31. Sullivan H, Furniss H, Spona J, et al. Effect of 21-day and 24-day oral contraceptive regimens containing gestodene (60 microg) andethinyl estradiol (15 microg) on ovarian activity. Fertil Steril. 1999;72(1):115-120.
32. Yonkers KA, Clark RH, Trivedi MH. The psychopharmacological treatment of nonmajor mood disorders. Mod Probl Pharmacopsychiatry. 1997;25:146-166.
33. Brown J, O’Brien PM, Marjoribanks J, et al. Selective serotonin reuptake inhibitors for premenstrual syndrome. Cochrane Database Syst Rev. 2009;(2):CD001396.
34. Halbreich U. Selective serotonin reuptake inhibitors and initial oral contraceptives for the treatment of PMDD: effective but not enough. CNS Spectr. 2008;13(7):566-572.
35. Freeman EW, Jabara S, Sondheimer SJ, et al. Citalopram in PMS patients with prior SSRI treatment failure: a preliminary study. J Womens Health Gend Based Med. 2002;11(5):
459-464.
36. Freeman EW, Rickels K, Sondheimer SJ, et al. Differential response to antidepressants in women with premenstrual syndrome/premenstrual dysphoric disorder: a randomized controlled trial. Arch Gen Psychiatry. 1999;56(10):932-939.
37. Steiner M, Pearlstein T, Cohen LS, et al. Expert guidelines for the treatment of severe PMS, PMDD, and comorbidities: the role of SSRIs. J Womens Health (Larchmt). 2006;15(1):57-69.
38. Freeman EW. Luteal phase administration of agents for the treatment of premenstrual dysphoric disorder. CNS Drugs. 2004;18(7):453-468.
39. Yonkers KA, Holthausen GA, Poschman K, et al. Symptom-onset treatment for women with premenstrual dysphoric disorder. J Clin Psychopharmacol. 2006;26(2):198-202.
40. Freeman EW, Sondheimer SJ, Sammel MD, et al. A preliminary study of luteal phase versus symptom-onset dosing with escitalopram for premenstrual dysphoric disorder. J Clin Psychiatry. 2005;66(6):769-773.
Approximately 75% of women experience a premenstrual change in emotional or physical symptoms commonly referred to as premenstrual syndrome (PMS). These symptoms—including increased irritability, tension, depressed mood, and somatic complaints such as breast tenderness and bloating—often are mild to moderate and cause minimal distress.1 However, approximately 3% to 9% of women experience moderate to severe premenstrual mood symptoms that meet criteria for premenstrual dysphoric disorder (PMDD).2
PMDD includes depressed or labile mood, anxiety, irritability, anger, insomnia, difficulty concentrating, and other symptoms that occur exclusively during the 2 weeks before menses and cause significant deterioration in daily functioning. Women with PMDD use general and mental health services more often than women without the condition.3 They may experience impairment in marital and parental relationships as severe as that experienced by women with recurrent or chronic major depression.2
PMDD often responds to treatment. Unfortunately, many women with PMDD do not seek treatment, and up to 90% may go undiagnosed.4 In this article, we review the prevalence, etiology, diagnosis, and treatment of PMDD.
A complex disorder
A distinguishing characteristic of PMDD is the timing of symptom onset. In women with PMDD, mood symptoms occur only during the luteal phase of the menstrual cycle (ovulation until onset of menses) and resolve after menstruation onset. Women with PMDD report normal mood and functioning during the follicular phase of the menstrual cycle (first day of the menstrual cycle until ovulation).
Although PMS and PMDD criteria share affective and somatic symptoms, more symptoms are required for a PMDD diagnosis, and symptoms often are more severe.5 As defined in DSM-IV-TR (Table),6 PMDD has a broader range of symptoms than PMS and includes symptoms not included in the American College of Obstetrics and Gynecology criteria for PMS,7 such as impaired concentration, appetite, and sleep (hypersomnia or insomnia); and mood lability. PMDD symptoms must occur only during the 2 weeks preceding menses, although on average symptoms last 6 days and severity usually peaks in the 2 days before menses.1 The prevalence of subthreshold PMDD is fairly common; approximately 19% of women will meet some—but not all—DSM-IV-TR criteria for PMDD.3
In a revision proposed for DSM-5, PMDD would be included as a mood disorder, which represents a significant change from DSM-IV-TR, where it is listed in the appendix as “research criteria.”8 In addition, in oral contraceptive users, a PMDD diagnosis should not be made unless the premenstrual symptoms are reported to be present and as severe when the woman is not taking the oral contraceptive.8
Comorbidity with other axis I disorders such as major depressive disorder (MDD), bipolar disorder (BD), and anxiety disorders is high.9-11 Women with an MDD history have the highest correlation with PMDD,9 and worsening premenstrual mood symptoms are more common in women with BD.12 Payne et al11 found that premenstrual symptoms were reported by twice as many women diagnosed with mood disorders (68%) than women without a psychiatric diagnosis (34%). Moreover, 38% to 46% of women with PMDD have comorbid seasonal affective disorder, and 11% to 38% report a comorbid anxiety disorder.12 Women with PMDD and a history of MDD have lower cortisol concentrations than non-PMDD women.10 Although interventions for PMDD and a comorbid axis I disorder may be similar, it is important to consider both when planning treatment.
Abuse, trauma, and PMDD. An association between PMS/PMDD and a history of sexual and physical abuse is well-documented.13 Studies have reported abuse histories among almost 60% of women with PMDD,14 although studies comparing abuse and trauma in PMDD vs non-PMDD women have been small. A recent study found that trauma and posttraumatic stress disorder are independently associated with PMDD and premenstrual symptoms.15
Evidence suggests that a history of abuse is associated with specific biological sequelae in PMDD women, particularly with respect to hypothalamic-pituitary-thyroid axis measures and noradrenergic activity.16-18 Women with PMDD and a history of sexual abuse show:
- markedly elevated triiodothyronine (T3) concentrations (the more biologically potent thyroid hormone) that appear to result from increased conversion of thyroxine (T4) to T316
- lower circulating plasma norepinephrine concentrations17
- greater resting and stress-induced heart rates and systolic blood pressure compared with non-abused PMDD women, an effect that is eliminated by clonidine (an α-2 adrenergic receptor agonist).18
One study showed that PMDD women with abuse histories had higher blood pressure measurements at rest and during stress and exhibited greater vascular tone than non-abused women; these effects were not seen in non-PMDD women with similar abuse histories.14 This body of evidence is consistent with the concept that PMDD is a stress-related disorder,19 and that a history of abuse is prevalent and may identify a clinically distinct subgroup of PMDD women with respect to thyroid axis and adrenergic physiology. Screening PMDD patients for abuse histories may help manage the disorder.
For a discussion of the etiology of PMDD, see Box 1.
Table
DSM-IV-TR research criteria for PMDD
|
| Source: Reference 6 |
Although questions remain about the pathogenesis of premenstrual dysphoric disorder (PMDD), literature documents the role of gonadal steroids (estrogen and progesterone) in the etiology of premenstrual syndrome (PMS)/PMDD and suggests that women with PMDD are differentially sensitive to the normal physiologic fluctuations of gonadal hormones throughout the menstrual cycle.a
The first half of the menstrual cycle—the follicular phase—begins with increasing levels of follicular stimulating hormone (FSH) leading to maturity of the ovarian follicle. Once the follicle is ripe, the luteal phase of the menstrual cycle begins with a surge in luteinizing hormone (LH), which results in ovulation of the mature follicle, followed by increased secretion of progesterone, followed by increased estrogen secretion. The system is regulated via negative feedback, and high levels of progesterone decrease gonadotropin-releasing hormone (GnRH) pulse frequency, which leads to decreased secretion of FSH and LH, and subsequent decline of estrogen and progesterone. If the ovarian follicle is not fertilized, menstruation begins and FSH levels rise again, initiating the follicular phase of the menstrual cycle.
Fluctuations in reproductive steroid levels have been implicated in the etiology of PMDD from studies showing that oophorectomy and ovulation inhibitors (GnRH agonists) relieve symptoms.b Some researchers proposed that symptoms are related to the drop of progesterone in the late luteal phase; however, many women have symptoms that start at ovulation or during the early luteal phase before the fall in progesterone concentrations.c PMS symptoms may occur independently of the mid-to-late luteal phase.d Because production of gonadal steroids does not differ between women with or without PMS or PMDD,e it may be that follicular or periovulatory changes in levels of estradiol or progesterone secretion trigger symptoms of PMDD in susceptible women, while women without PMDD appear to be immune to these effects of gonadal steroids. This idea is supported by a study showing that pharmacologic induction of a hypogonadal state eliminates symptoms in most women with severe PMS, while “adding back” estrogen or progesterone within the context of hypogonadism elicits return of PMS symptoms in those with PMS but not in controls.a
Abnormalities in serotonin levels also may contribute to PMDD.f In 1 study, a serotonin receptor antagonist precipitated return of symptoms within 24 hours of administration in women with PMDD but not in controls.g PMDD symptoms also can be evoked by depleting the serotonin precursor tryptophan.h When women with PMDD received paroxetine at different phases of their menstrual cycle, they showed fluctuations in serotonergic function across their cycles; these fluctuations were not seen in controls.i Other neurotransmitters implicated in PMDD include γ-aminobutyric acid (GABA),j glutamate,k lower levels of cortisol and beta-endorphins,l and an abnormal stress response.m
Other studies have focused on differing concentrations of luteal phase hormonesn and gene associations. Two studies suggested that PMDD is heritableo,p and other studies have looked at the association between specific psychological traits that are more prominent in PMDD and single nucleotide polymorphisms in the estrogen receptor alpha gene.q,r
Thyroid hormones also may play a role in the etiology of PMS/PMDD. Thyroid function tests have shown greater variability in women with PMS vs controls,s although this variability appears to be limited to women with a sexual abuse history.t Other studies have evaluated hormones regulated across the circadian and sleep-wake cycles, including melatonin, cortisol, thyroid-stimulating hormone, and prolactin, which suggests that although levels of these hormones may not differ between women with PMDD and controls, the timing of their excretion may vary.s Additionally, women with PMDD are characterized by prefrontal brain asymmetry on electroencephalography that also is evident in patients with major depressive disorder.u
There also may be dysregulation of allopregnanolone (ALLO) in women with PMDD.v,w ALLO is a metabolite of progesterone that is a neurosteroid produced in the brain as well as in the ovary and adrenals.v It produces anxiolytic effects by acting as a modulator of GABA receptors.x In PMDD, ALLO levels may influence the severity of premenstrual symptoms.w
References
- Schmidt PJ, Nieman LK, Danaceau MA, et al. Differential behavioral effects of gonadal steroids in women with and in those without premenstrual syndrome. N Engl J Med. 1998;338(4):209-216.
- Muse KN, Cetel NS, Futterman LA, et al. The premenstrual syndrome. Effects of “medical ovariectomy.” N Engl J Med. 1984;311(21):1345-1349.
- Yonkers KA, O’Brien PM, Eriksson E. Premenstrual syndrome. Lancet. 2008;371(9619):1200-1210.
- Schmidt PJ, Nieman LK, Grover GN, et al. Lack of effect of induced menses on symptoms in women with premenstrual syndrome. N Engl J Med. 1991;324(17):1174-1179.
- Rubinow DR, Schmidt PJ. The neuroendocrinology of menstrual cycle mood disorders. Ann N Y Acad Sci. 1995;771:648-659.
- Steiner M, Pearlstein T. Premenstrual dysphoria and the serotonin system: pathophysiology and treatment. J Clin Psychiatry. 2000;61(suppl 12):17-21.
- Roca CA, Schmidt PJ, Smith MJ, et al. Effects of metergoline on symptoms in women with premenstrual dysphoric disorder. Am J Psychiatry. 2002;159(11):1876-1881.
- Menkes DB, Coates DC, Fawcett JP. Acute tryptophan depletion aggravates premenstrual syndrome. J Affect Disord. 1994;32(1):37-44.
- Inoue Y, Terao T, Iwata N, et al. Fluctuating serotonergic function in premenstrual dysphoric disorder and premenstrual syndrome: findings from neuroendocrine challenge tests. Psychopharmacology (Berl). 2007;190(2):213-219.
- Epperson CN, Haga K, Mason GF, et al. Cortical gamma-aminobutyric acid levels across the menstrual cycle in healthy women and those with premenstrual dysphoric disorder: a proton magnetic resonance spectroscopy study. Arch Gen Psychiatry. 2002;59(9):851-858.
- Batra NA, Seres-Mailo J, Hanstock C, et al. Proton magnetic resonance spectroscopy measurement of brain glutamate levels in premenstrual dysphoric disorder. Biol Psychiatry. 2008;63(12):1178-1184.
- Straneva PA, Maixner W, Light KC, et al. Menstrual cycle, beta-endorphins, and pain sensitivity in premenstrual dysphoric disorder. Health Psychol. 2002;21(4):358-367.
- Epperson CN, Pittman B, Czarkowski KA, et al. Luteal-phase accentuation of acoustic startle response in women with premenstrual dysphoric disorder. Neuropsychopharmacology. 2007;32(10):2190-2198.
- Thys-Jacobs S, McMahon D, Bilezikian JP. Differences in free estradiol and sex hormone-binding globulin in women with and without premenstrual dysphoric disorder. J Clin Endocrinol Metab. 2008;93(1):96-102.
- Payne JL, Klein SR, Zamoiski RB, et al. Premenstrual mood symptoms: study of familiality and personality correlates in mood disorder pedigrees. Arch Womens Ment Health. 2009;12(1):27-34.
- Kendler KS, Karkowski LM, Corey LA, et al. Longitudinal population-based twin study of retrospectively reported premenstrual symptoms and lifetime major depression. Am J Psychiatry. 1998;155(9):1234-1240.
- Miller A, Vo H, Huo L, et al. Estrogen receptor alpha (ESR-1) associations with psychological traits in women with PMDD and controls. J Psychiatr Res. 2010;44(12):788-794.
- Huo L, Straub RE, Roca C, et al. Risk for premenstrual dysphoric disorder is associated with genetic variation in ESR1, the estrogen receptor alpha gene. Biol Psychiatry. 2007;62(8):925-933.
- Girdler SS, Pedersen CA, Light KC. Thyroid axis function during the menstrual cycle in women with premenstrual syndrome. Psychoneuroendocrinology. 1995;20(4):395-403.
- Girdler SS, Thompson KS, Light KC, et al. Historical sexual abuse and current thyroid axis profiles in women with premenstrual dysphoric disorder. Psychosom Med. 2004;66(3):403-410.
- Accortt EE, Stewart JL, Coan JA, et al. Prefrontal brain asymmetry and pre-menstrual dysphoric disorder symptomatology. J Affect Disord. 2011;128(1-2):178-183.
- Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6(6):2311-2322.
- Girdler SS, Straneva PA, Light KC, et al. Allopregnanolone levels and reactivity to mental stress in premenstrual dysphoric disorder. Biol Psychiatry. 2001;49(9):788-797.
- Brot MD, Akwa Y, Purdy RH, et al. The anxiolytic-like effects of the neurosteroid allopregnanolone: interactions with GABA(A) receptors. Eur J Pharmacol. 1997;325(1):1-7.
Mood charting aids diagnosis
A PMDD diagnosis requires prospective daily monitoring of symptoms for ≥2 consecutive months. Because only 25% to 35% of women who present with PMDD meet diagnostic criteria when prospective daily monitoring is used,20 it is important for patients to keep a daily diary of PMDD symptoms to distinguish the disorder from PMS (Box 2). The Prospective Record of the Impact and Severity of Premenstrual Symptoms calendar and the Daily Record of Severity of Problems (DRSPP)21 may help make the diagnosis.
The widely used DRSPP allows clinicians to quantify the severity of physical, emotional, and behavioral symptoms and may be the easiest to use in clinical practice because it creates a graphic representation of cyclical symptom changes. The DRSPP includes all PMDD symptoms and severity ratings21 and is recognized as a valid instrument for diagnosing PMDD. Another option is a revised visual analogue scale. Lastly, a new revised Premenstrual Tension Syndrome (PMTS) rating scale, which combines the PMTS Observer rating scale plus multiple visual analogue scales, shows promise as a tool to assess PMDD symptoms.
To distinguish premenstrual syndrome (PMS) from premenstrual dysphoric disorder (PMDD), premenstrual exacerbation of an underlying psychiatric disorder, general medical conditions, or other disorders with no association to the menstrual cycle, it is necessary to have patients conduct daily symptom charting over 2 menstrual cycles. This charting should include documentation of emotional, behavioral, and physical symptoms. PMDD can be differentiated from PMS by the severity and number of symptoms. In PMDD, 1 of the symptoms must be a mood disturbance (depressed, anxious, labile, and/or irritable). For a sample form used for PMDD charting, the Daily Record of Severity of Problems, see http://pmdd.factsforhealth.org/drsp/drsp_month.pdf.
Treatment options
Hormonal interventions. Attempts to treat PMS with progesterone during the luteal phase have been largely unsuccessful, although progesterone is approved to treat PMS in the United Kingdom. Long-acting gonadotropin-releasing hormone (GnRH) agonists are effective but result in medical menopause with its accompanying symptoms, which puts women at risk for osteoporosis.22 Approximately 60% to 70% of women with PMDD respond to leuprolide (a GnRH agonist), but it is difficult to predict who will respond; daily mood self-ratings of sadness, anxiety, and irritability predict a positive response to leuprolide with high probability.23 Side effects of GnRH agonists (hot flashes, night sweats, vaginal dryness, etc.) can be tempered by “adding back” some estrogen with a hormonal agent with progestational activity to reduce the risks of unopposed estrogen (ie, endometrial hyperplasia).24
Surgical bilateral oophorectomy is effective but extremely invasive, especially in younger women in whom removal of ovaries generally is inadvisable. Patients should receive a trial of a GnRH agonist before a surgical intervention, because oophorectomy may not reduce symptoms and is irreversible. Oophorectomy also would require hormone replacement therapy.
High-dose estrogen as transdermal patches or subcutaneous implants to inhibit ovulation is effective, but because of the risks of unopposed estrogen, a progestin would be needed. Risks of estrogen therapy (alone and in combination with progestins) include increased risk of endometrial cancer, coronary heart disease, breast cancer, stroke, and pulmonary embolism.25 Danazol, a synthetic androgen and gonadotropin inhibitor, effectively blocks ovulation, but side effects include hirsutism and possible teratogenicity.26 Although these hormonal manipulations may effectively treat PMDD, none are considered practical.
The use of combined oral contraceptives (estrogen and progestin) is common. Although continuous cycle oral contraceptives often are recommended for PMDD, limited evidence supports their use; studies have been mostly negative.27,28 A recent review of 4 studies of a continuous oral contraceptive (levonorgestrel 90 mcg/ethinyl estradiol 20 mcg) for PMDD and PMS had more promising results, although the results were highly variable among studies and a large placebo effect was observed.29
A combination oral contraceptive, drospirenone/ethinyl estradiol, is FDA-approved for treating PMDD in women seeking hormonal contraception because it has shown efficacy compared with placebo, with reported improvements in perceived productivity, social activities, and interpersonal relationships.30 The nature of hormone delivery (ie, a reduction in the pill-free interval from 7 to 4 days) in drospirenone/ethinyl estradiol may contribute to its efficacy because a reduced pill-free interval minimizes the degree of follicular recruitment and subsequent estrogen production and cyclicity seen with standard oral contraceptive.31
Antidepressants have been shown to effectively ameliorate affective and physical symptoms and improve quality of life and psychosocial function in patients with PMS and PMDD. The response rates for selective serotonin reuptake inhibitors (SSRIs) in PMDD treatment vary from 60% to 90%, vs 30% to 40% for placebo.32 A 2009 Cochrane review found SSRIs reduced premenstrual symptoms compared with placebo.33 However, a literature review suggested that the percentage of women with PMDD who respond to SSRIs or continuous oral contraceptives is lower than the percentage of women who do not respond at all, once the placebo effect is taken into account, and that approximately 40% of women with PMDD do not respond to SSRIs.34 A small study found that citalopram may be effective for women with PMDD who did not respond to a prior SSRI.35
However, only antidepressants that affect serotonergic—not noradrenergic—transmission are effective in PMDD.22 These include:
- the tricyclic antidepressant clomipramine
- the SSRIs citalopram, escitalopram, fluoxetine, paroxetine, and sertraline
- the serotonin-noradrenergic reuptake inhibitor venlafaxine.
It appears that in PMDD, serotonergic agents play a role other than their antidepressant effect.36 The effect of these agents is rapid in PMS/PMDD; women with PMDD who take antidepressants often experience reduced symptoms within the first menstrual cycle, whereas in MDD the onset of action can take weeks or months.37
Although why onset of antidepressant action is quick in PMDD is unclear, rapid onset allows for several dosing options. Some women prefer continuous dosing throughout the month because they do not have to keep track of ovulation. Dosing antidepressants only in the luteal phase (taking the antidepressant from ovulation onset to the start of menses) is an effective treatment strategy.38 Many women prefer to take medication for only 2 weeks per month, which can decrease side effects and lower treatment costs. Alternatively, symptom-onset dosing—initiating the antidepressant when PMDD symptoms begin and stopping at menses onset or within 3 days thereafter—has shown promising results.39,40 Paroxetine, sertraline, and fluoxetine are FDA-approved for PMDD as continuous or intermittent regimens, although using fluoxetine intermittently may not make sense because its biologically active metabolite has an extended half-life.37
Other treatments. Dietary interventions, psychotherapy, vitamins, bright light treatment, and spironolactone have been assessed for PMS/PMDD, although for many evidence-based findings are lacking (Box 3).
See the Bibliography below for studies that support using antidepressants to treat PMDD
Two reviews of 10 randomized controlled trials (RCTs) that evaluated 62 herbs, vitamins, and mineral treatments for premenstrual symptoms found efficacy for chasteberry (Vitex agnus-castus), calcium, and vitamin B6 but not for primrose oil, magnesium oxide, or St. John’s wort.a,b A study comparing fluoxetine with chasteberry found a similar percentage of patients responded to either agent (68% vs 58%, respectively).c Another study showed calcium resulted in a 48% reduction in premenstrual symptoms from baseline, compared with a 30% reduction with placebo.d Bright light treatment significantly reduced depression ratings in women with premenstrual dysphoric disorder (PMDD).e Compared with placebo, the aldosterone antagonist spironolactone improved irritability, depression, feelings of swelling, breast tenderness, and food craving in women with premenstrual syndrome (PMS).f
A recent systematic review of 7 trials of cognitive-behavioral therapy (CBT) for PMDD, including 3 RCTs, showed a lack of a statistically significant effect.g However, a separate review of RCTs of alternative treatments for PMDD—5 of which included CBT—suggested that CBT may be beneficial in reducing premenstrual symptoms, but the evidence was low quality.h
References
- Dante G, Facchinetti F. Herbal treatments for alleviating premenstrual symptoms: a systematic review. J Psychosom Obstet Gynaecol. 2011;32(1):42-51.
- Whelan AM, Jurgens TM, Naylor H. Herbs, vitamins and minerals in the treatment of premenstrual syndrome: a systematic review. Can J Clin Pharmacol. 2009;16(3):e407-e429.
- Atmaca M, Kumru S, Tezcan E. Fluoxetine versus Vitex agnus castus extract in the treatment of premenstrual dysphoric disorder. Hum Psychopharmacol. 2003;18(3):191-195.
- Thys-Jacobs S, Starkey P, Bernstein D, et al. Calcium carbonate and the premenstrual syndrome: effects on premenstrual and menstrual symptoms. Premenstrual Syndrome Study Group. Am J Obstet Gynecol. 1998;179(2):444-452.
- Parry BL, Berga SL, Mostofi N, et al. Morning versus evening bright light treatment of late luteal phase dysphoric disorder. Am J Psychiatry. 1989;146(9):1215-1217.
- Wang M, Hammarbäck S, Lindhe BA, et al. Treatment of premenstrual syndrome by spironolactone: a double-blind, placebo-controlled study. Acta Obstet Gynecol Scand. 1995;74(10):803-808.
- Lustyk MK, Gerrish WG, Shaver S, et al. Cognitive-behavioral therapy for premenstrual syndrome and premenstrual dysphoric disorder: a systematic review. Arch Womens Ment Health. 2009;12(2):85-96.
- Busse JW, Montori VM, Krasnik C, et al. Psychological intervention for premenstrual syndrome: a meta-analysis of randomized controlled trials. Psychother Psychosom. 2009;78(1):6-15.
Cohen LS, Miner C, Brown EW, et al. Premenstrual daily fluoxetine for premenstrual dysphoric disorder: a placebo-controlled, clinical trial using computerized diaries. Obstet Gynecol. 2002;100(3):435-444.
Eriksson E, Ekman A, Sinclair S, et al. Escitalopram administered in the luteal phase exerts a marked and dose-dependent effect in premenstrual dysphoric disorder. J Clin Psychopharmacol. 2008;28(2):195-202.
Eriksson E, Hedberg MA, Andersch B, et al. The serotonin reuptake inhibitor paroxetin is superior to the noradrenaline reuptake inhibitor maprotiline in the treatment of premenstrual syndrome. Neuropsychopharmacology. 1995;12(2):167-176.
Menkes DB, Taghavi E, Mason PA, et al. Fluoxetine treatment of severe premenstrual syndrome. BMJ. 1992;305(6849):346-347.
Miner C, Brown E, McCray S, et al. Weekly luteal-phase dosing with enteric-coated fluoxetine 90 mg in premenstrual dysphoric disorder: a randomized, double-blind, placebo-controlled clinical trial. Clin Ther. 2002;24(3):417-433.
Ozeren S, Corakçi A, Yücesoy I, et al. Fluoxetine in the treatment of premenstrual syndrome. Eur J Obstet Gynecol Reprod Biol. 1997;73(2):167-170.
Pearlstein TB, Stone AB, Lund SA, et al. Comparison of fluoxetine, bupropion, and placebo in the treatment of premenstrual dysphoric disorder. J Clin Psychopharmacol. 1997;17(4):261-266.
Ravindran LN, Woods SA, Steiner M, et al. Symptom-onset dosing with citalopram in the treatment of premenstrual dysphoric disorder (PMDD): a case series. Arch Womens Ment Health. 2007;10(3):125-127.
Steiner M, Brown E, Trzepacz P, et al. Fluoxetine improves functional work capacity in women with premenstrual dysphoric disorder. Arch Womens Ment Health. 2003;6(1):71-77.
Stone AB, Pearlstein TB, Brown WA. Fluoxetine in the treatment of late luteal phase dysphoric disorder. J Clin Psychiatry. 1991;52(7):290-293.
Sundblad C, Hedberg MA, Eriksson E. Clomipramine administered during the luteal phase reduces the symptoms of premenstrual syndrome: a placebo-controlled trial. Neuropsychopharmacology. 1993;9(2):133-145.
Sundblad C, Modigh K, Andersch B, et al. Clomipramine effectively reduces premenstrual irritability and dysphoria: a placebo-controlled trial. Acta Psychiatr Scand. 1992;85(1):39-47.
Su TP, Schmidt PJ, Danaceau MA, et al. Fluoxetine in the treatment of premenstrual dysphoria. Neuropsychopharmacology. 1997; 16(5):346-356.
Wikander I, Sundblad C, Andersch B, et al. Citalopram in premenstrual dysphoria: is intermittent treatment during luteal phases more effective than continuous medication throughout the menstrual cycle? J Clin Psychopharmacol. 1998;18(5):390-398.
Treatment selection
To enhance compliance and improve the likelihood of successful treatment, tailor treatment decisions to your patient’s needs. Carefully discuss with your patient the evidence-based literature to select the best option for her. Factors to consider when counseling patients include:
- the patient’s age, cigarette smoking habits, and body mass index, which may contraindicate oral contraceptives
- does the patient have regular cycle lengths?
- can she adhere to an on-off schedule? If so, intermittent SSRI dosing may be a good treatment option
- does the patient have irregular cycles?
- is there evidence that symptoms persist into the follicular phase, albeit at a lower level? If so, continuous SSRI dosing may be the best option.
Related Resources
- American Psychiatric Association. DSM-5 development: D 04 Premenstrual dysphoric disorder. www.dsm5.org/proposedrevision/pages/proposedrevision.aspx?rid=484.
Drug Brand Names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clonidine • Catapres, others
- Danazol • Danocrine
- Drospirenone/ethinyl estradiol • Yaz
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Leuprolide • Lupron
- Levonorgestrel/ethinyl estradiol • Seasonale, others
- Paroxetine • Paxil
- Progesterone • Prometrium
- Sertraline • Zoloft
- Spironolactone • Aldactone
- Venlafaxine • Effexor
Disclosures
Drs. Wakil and Girdler report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products
Dr. Meltzer-Brody receives research/grant support from AstraZeneca, The Foundation of Hope, and the National Institutes of Health.
Approximately 75% of women experience a premenstrual change in emotional or physical symptoms commonly referred to as premenstrual syndrome (PMS). These symptoms—including increased irritability, tension, depressed mood, and somatic complaints such as breast tenderness and bloating—often are mild to moderate and cause minimal distress.1 However, approximately 3% to 9% of women experience moderate to severe premenstrual mood symptoms that meet criteria for premenstrual dysphoric disorder (PMDD).2
PMDD includes depressed or labile mood, anxiety, irritability, anger, insomnia, difficulty concentrating, and other symptoms that occur exclusively during the 2 weeks before menses and cause significant deterioration in daily functioning. Women with PMDD use general and mental health services more often than women without the condition.3 They may experience impairment in marital and parental relationships as severe as that experienced by women with recurrent or chronic major depression.2
PMDD often responds to treatment. Unfortunately, many women with PMDD do not seek treatment, and up to 90% may go undiagnosed.4 In this article, we review the prevalence, etiology, diagnosis, and treatment of PMDD.
A complex disorder
A distinguishing characteristic of PMDD is the timing of symptom onset. In women with PMDD, mood symptoms occur only during the luteal phase of the menstrual cycle (ovulation until onset of menses) and resolve after menstruation onset. Women with PMDD report normal mood and functioning during the follicular phase of the menstrual cycle (first day of the menstrual cycle until ovulation).
Although PMS and PMDD criteria share affective and somatic symptoms, more symptoms are required for a PMDD diagnosis, and symptoms often are more severe.5 As defined in DSM-IV-TR (Table),6 PMDD has a broader range of symptoms than PMS and includes symptoms not included in the American College of Obstetrics and Gynecology criteria for PMS,7 such as impaired concentration, appetite, and sleep (hypersomnia or insomnia); and mood lability. PMDD symptoms must occur only during the 2 weeks preceding menses, although on average symptoms last 6 days and severity usually peaks in the 2 days before menses.1 The prevalence of subthreshold PMDD is fairly common; approximately 19% of women will meet some—but not all—DSM-IV-TR criteria for PMDD.3
In a revision proposed for DSM-5, PMDD would be included as a mood disorder, which represents a significant change from DSM-IV-TR, where it is listed in the appendix as “research criteria.”8 In addition, in oral contraceptive users, a PMDD diagnosis should not be made unless the premenstrual symptoms are reported to be present and as severe when the woman is not taking the oral contraceptive.8
Comorbidity with other axis I disorders such as major depressive disorder (MDD), bipolar disorder (BD), and anxiety disorders is high.9-11 Women with an MDD history have the highest correlation with PMDD,9 and worsening premenstrual mood symptoms are more common in women with BD.12 Payne et al11 found that premenstrual symptoms were reported by twice as many women diagnosed with mood disorders (68%) than women without a psychiatric diagnosis (34%). Moreover, 38% to 46% of women with PMDD have comorbid seasonal affective disorder, and 11% to 38% report a comorbid anxiety disorder.12 Women with PMDD and a history of MDD have lower cortisol concentrations than non-PMDD women.10 Although interventions for PMDD and a comorbid axis I disorder may be similar, it is important to consider both when planning treatment.
Abuse, trauma, and PMDD. An association between PMS/PMDD and a history of sexual and physical abuse is well-documented.13 Studies have reported abuse histories among almost 60% of women with PMDD,14 although studies comparing abuse and trauma in PMDD vs non-PMDD women have been small. A recent study found that trauma and posttraumatic stress disorder are independently associated with PMDD and premenstrual symptoms.15
Evidence suggests that a history of abuse is associated with specific biological sequelae in PMDD women, particularly with respect to hypothalamic-pituitary-thyroid axis measures and noradrenergic activity.16-18 Women with PMDD and a history of sexual abuse show:
- markedly elevated triiodothyronine (T3) concentrations (the more biologically potent thyroid hormone) that appear to result from increased conversion of thyroxine (T4) to T316
- lower circulating plasma norepinephrine concentrations17
- greater resting and stress-induced heart rates and systolic blood pressure compared with non-abused PMDD women, an effect that is eliminated by clonidine (an α-2 adrenergic receptor agonist).18
One study showed that PMDD women with abuse histories had higher blood pressure measurements at rest and during stress and exhibited greater vascular tone than non-abused women; these effects were not seen in non-PMDD women with similar abuse histories.14 This body of evidence is consistent with the concept that PMDD is a stress-related disorder,19 and that a history of abuse is prevalent and may identify a clinically distinct subgroup of PMDD women with respect to thyroid axis and adrenergic physiology. Screening PMDD patients for abuse histories may help manage the disorder.
For a discussion of the etiology of PMDD, see Box 1.
Table
DSM-IV-TR research criteria for PMDD
|
| Source: Reference 6 |
Although questions remain about the pathogenesis of premenstrual dysphoric disorder (PMDD), literature documents the role of gonadal steroids (estrogen and progesterone) in the etiology of premenstrual syndrome (PMS)/PMDD and suggests that women with PMDD are differentially sensitive to the normal physiologic fluctuations of gonadal hormones throughout the menstrual cycle.a
The first half of the menstrual cycle—the follicular phase—begins with increasing levels of follicular stimulating hormone (FSH) leading to maturity of the ovarian follicle. Once the follicle is ripe, the luteal phase of the menstrual cycle begins with a surge in luteinizing hormone (LH), which results in ovulation of the mature follicle, followed by increased secretion of progesterone, followed by increased estrogen secretion. The system is regulated via negative feedback, and high levels of progesterone decrease gonadotropin-releasing hormone (GnRH) pulse frequency, which leads to decreased secretion of FSH and LH, and subsequent decline of estrogen and progesterone. If the ovarian follicle is not fertilized, menstruation begins and FSH levels rise again, initiating the follicular phase of the menstrual cycle.
Fluctuations in reproductive steroid levels have been implicated in the etiology of PMDD from studies showing that oophorectomy and ovulation inhibitors (GnRH agonists) relieve symptoms.b Some researchers proposed that symptoms are related to the drop of progesterone in the late luteal phase; however, many women have symptoms that start at ovulation or during the early luteal phase before the fall in progesterone concentrations.c PMS symptoms may occur independently of the mid-to-late luteal phase.d Because production of gonadal steroids does not differ between women with or without PMS or PMDD,e it may be that follicular or periovulatory changes in levels of estradiol or progesterone secretion trigger symptoms of PMDD in susceptible women, while women without PMDD appear to be immune to these effects of gonadal steroids. This idea is supported by a study showing that pharmacologic induction of a hypogonadal state eliminates symptoms in most women with severe PMS, while “adding back” estrogen or progesterone within the context of hypogonadism elicits return of PMS symptoms in those with PMS but not in controls.a
Abnormalities in serotonin levels also may contribute to PMDD.f In 1 study, a serotonin receptor antagonist precipitated return of symptoms within 24 hours of administration in women with PMDD but not in controls.g PMDD symptoms also can be evoked by depleting the serotonin precursor tryptophan.h When women with PMDD received paroxetine at different phases of their menstrual cycle, they showed fluctuations in serotonergic function across their cycles; these fluctuations were not seen in controls.i Other neurotransmitters implicated in PMDD include γ-aminobutyric acid (GABA),j glutamate,k lower levels of cortisol and beta-endorphins,l and an abnormal stress response.m
Other studies have focused on differing concentrations of luteal phase hormonesn and gene associations. Two studies suggested that PMDD is heritableo,p and other studies have looked at the association between specific psychological traits that are more prominent in PMDD and single nucleotide polymorphisms in the estrogen receptor alpha gene.q,r
Thyroid hormones also may play a role in the etiology of PMS/PMDD. Thyroid function tests have shown greater variability in women with PMS vs controls,s although this variability appears to be limited to women with a sexual abuse history.t Other studies have evaluated hormones regulated across the circadian and sleep-wake cycles, including melatonin, cortisol, thyroid-stimulating hormone, and prolactin, which suggests that although levels of these hormones may not differ between women with PMDD and controls, the timing of their excretion may vary.s Additionally, women with PMDD are characterized by prefrontal brain asymmetry on electroencephalography that also is evident in patients with major depressive disorder.u
There also may be dysregulation of allopregnanolone (ALLO) in women with PMDD.v,w ALLO is a metabolite of progesterone that is a neurosteroid produced in the brain as well as in the ovary and adrenals.v It produces anxiolytic effects by acting as a modulator of GABA receptors.x In PMDD, ALLO levels may influence the severity of premenstrual symptoms.w
References
- Schmidt PJ, Nieman LK, Danaceau MA, et al. Differential behavioral effects of gonadal steroids in women with and in those without premenstrual syndrome. N Engl J Med. 1998;338(4):209-216.
- Muse KN, Cetel NS, Futterman LA, et al. The premenstrual syndrome. Effects of “medical ovariectomy.” N Engl J Med. 1984;311(21):1345-1349.
- Yonkers KA, O’Brien PM, Eriksson E. Premenstrual syndrome. Lancet. 2008;371(9619):1200-1210.
- Schmidt PJ, Nieman LK, Grover GN, et al. Lack of effect of induced menses on symptoms in women with premenstrual syndrome. N Engl J Med. 1991;324(17):1174-1179.
- Rubinow DR, Schmidt PJ. The neuroendocrinology of menstrual cycle mood disorders. Ann N Y Acad Sci. 1995;771:648-659.
- Steiner M, Pearlstein T. Premenstrual dysphoria and the serotonin system: pathophysiology and treatment. J Clin Psychiatry. 2000;61(suppl 12):17-21.
- Roca CA, Schmidt PJ, Smith MJ, et al. Effects of metergoline on symptoms in women with premenstrual dysphoric disorder. Am J Psychiatry. 2002;159(11):1876-1881.
- Menkes DB, Coates DC, Fawcett JP. Acute tryptophan depletion aggravates premenstrual syndrome. J Affect Disord. 1994;32(1):37-44.
- Inoue Y, Terao T, Iwata N, et al. Fluctuating serotonergic function in premenstrual dysphoric disorder and premenstrual syndrome: findings from neuroendocrine challenge tests. Psychopharmacology (Berl). 2007;190(2):213-219.
- Epperson CN, Haga K, Mason GF, et al. Cortical gamma-aminobutyric acid levels across the menstrual cycle in healthy women and those with premenstrual dysphoric disorder: a proton magnetic resonance spectroscopy study. Arch Gen Psychiatry. 2002;59(9):851-858.
- Batra NA, Seres-Mailo J, Hanstock C, et al. Proton magnetic resonance spectroscopy measurement of brain glutamate levels in premenstrual dysphoric disorder. Biol Psychiatry. 2008;63(12):1178-1184.
- Straneva PA, Maixner W, Light KC, et al. Menstrual cycle, beta-endorphins, and pain sensitivity in premenstrual dysphoric disorder. Health Psychol. 2002;21(4):358-367.
- Epperson CN, Pittman B, Czarkowski KA, et al. Luteal-phase accentuation of acoustic startle response in women with premenstrual dysphoric disorder. Neuropsychopharmacology. 2007;32(10):2190-2198.
- Thys-Jacobs S, McMahon D, Bilezikian JP. Differences in free estradiol and sex hormone-binding globulin in women with and without premenstrual dysphoric disorder. J Clin Endocrinol Metab. 2008;93(1):96-102.
- Payne JL, Klein SR, Zamoiski RB, et al. Premenstrual mood symptoms: study of familiality and personality correlates in mood disorder pedigrees. Arch Womens Ment Health. 2009;12(1):27-34.
- Kendler KS, Karkowski LM, Corey LA, et al. Longitudinal population-based twin study of retrospectively reported premenstrual symptoms and lifetime major depression. Am J Psychiatry. 1998;155(9):1234-1240.
- Miller A, Vo H, Huo L, et al. Estrogen receptor alpha (ESR-1) associations with psychological traits in women with PMDD and controls. J Psychiatr Res. 2010;44(12):788-794.
- Huo L, Straub RE, Roca C, et al. Risk for premenstrual dysphoric disorder is associated with genetic variation in ESR1, the estrogen receptor alpha gene. Biol Psychiatry. 2007;62(8):925-933.
- Girdler SS, Pedersen CA, Light KC. Thyroid axis function during the menstrual cycle in women with premenstrual syndrome. Psychoneuroendocrinology. 1995;20(4):395-403.
- Girdler SS, Thompson KS, Light KC, et al. Historical sexual abuse and current thyroid axis profiles in women with premenstrual dysphoric disorder. Psychosom Med. 2004;66(3):403-410.
- Accortt EE, Stewart JL, Coan JA, et al. Prefrontal brain asymmetry and pre-menstrual dysphoric disorder symptomatology. J Affect Disord. 2011;128(1-2):178-183.
- Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6(6):2311-2322.
- Girdler SS, Straneva PA, Light KC, et al. Allopregnanolone levels and reactivity to mental stress in premenstrual dysphoric disorder. Biol Psychiatry. 2001;49(9):788-797.
- Brot MD, Akwa Y, Purdy RH, et al. The anxiolytic-like effects of the neurosteroid allopregnanolone: interactions with GABA(A) receptors. Eur J Pharmacol. 1997;325(1):1-7.
Mood charting aids diagnosis
A PMDD diagnosis requires prospective daily monitoring of symptoms for ≥2 consecutive months. Because only 25% to 35% of women who present with PMDD meet diagnostic criteria when prospective daily monitoring is used,20 it is important for patients to keep a daily diary of PMDD symptoms to distinguish the disorder from PMS (Box 2). The Prospective Record of the Impact and Severity of Premenstrual Symptoms calendar and the Daily Record of Severity of Problems (DRSPP)21 may help make the diagnosis.
The widely used DRSPP allows clinicians to quantify the severity of physical, emotional, and behavioral symptoms and may be the easiest to use in clinical practice because it creates a graphic representation of cyclical symptom changes. The DRSPP includes all PMDD symptoms and severity ratings21 and is recognized as a valid instrument for diagnosing PMDD. Another option is a revised visual analogue scale. Lastly, a new revised Premenstrual Tension Syndrome (PMTS) rating scale, which combines the PMTS Observer rating scale plus multiple visual analogue scales, shows promise as a tool to assess PMDD symptoms.
To distinguish premenstrual syndrome (PMS) from premenstrual dysphoric disorder (PMDD), premenstrual exacerbation of an underlying psychiatric disorder, general medical conditions, or other disorders with no association to the menstrual cycle, it is necessary to have patients conduct daily symptom charting over 2 menstrual cycles. This charting should include documentation of emotional, behavioral, and physical symptoms. PMDD can be differentiated from PMS by the severity and number of symptoms. In PMDD, 1 of the symptoms must be a mood disturbance (depressed, anxious, labile, and/or irritable). For a sample form used for PMDD charting, the Daily Record of Severity of Problems, see http://pmdd.factsforhealth.org/drsp/drsp_month.pdf.
Treatment options
Hormonal interventions. Attempts to treat PMS with progesterone during the luteal phase have been largely unsuccessful, although progesterone is approved to treat PMS in the United Kingdom. Long-acting gonadotropin-releasing hormone (GnRH) agonists are effective but result in medical menopause with its accompanying symptoms, which puts women at risk for osteoporosis.22 Approximately 60% to 70% of women with PMDD respond to leuprolide (a GnRH agonist), but it is difficult to predict who will respond; daily mood self-ratings of sadness, anxiety, and irritability predict a positive response to leuprolide with high probability.23 Side effects of GnRH agonists (hot flashes, night sweats, vaginal dryness, etc.) can be tempered by “adding back” some estrogen with a hormonal agent with progestational activity to reduce the risks of unopposed estrogen (ie, endometrial hyperplasia).24
Surgical bilateral oophorectomy is effective but extremely invasive, especially in younger women in whom removal of ovaries generally is inadvisable. Patients should receive a trial of a GnRH agonist before a surgical intervention, because oophorectomy may not reduce symptoms and is irreversible. Oophorectomy also would require hormone replacement therapy.
High-dose estrogen as transdermal patches or subcutaneous implants to inhibit ovulation is effective, but because of the risks of unopposed estrogen, a progestin would be needed. Risks of estrogen therapy (alone and in combination with progestins) include increased risk of endometrial cancer, coronary heart disease, breast cancer, stroke, and pulmonary embolism.25 Danazol, a synthetic androgen and gonadotropin inhibitor, effectively blocks ovulation, but side effects include hirsutism and possible teratogenicity.26 Although these hormonal manipulations may effectively treat PMDD, none are considered practical.
The use of combined oral contraceptives (estrogen and progestin) is common. Although continuous cycle oral contraceptives often are recommended for PMDD, limited evidence supports their use; studies have been mostly negative.27,28 A recent review of 4 studies of a continuous oral contraceptive (levonorgestrel 90 mcg/ethinyl estradiol 20 mcg) for PMDD and PMS had more promising results, although the results were highly variable among studies and a large placebo effect was observed.29
A combination oral contraceptive, drospirenone/ethinyl estradiol, is FDA-approved for treating PMDD in women seeking hormonal contraception because it has shown efficacy compared with placebo, with reported improvements in perceived productivity, social activities, and interpersonal relationships.30 The nature of hormone delivery (ie, a reduction in the pill-free interval from 7 to 4 days) in drospirenone/ethinyl estradiol may contribute to its efficacy because a reduced pill-free interval minimizes the degree of follicular recruitment and subsequent estrogen production and cyclicity seen with standard oral contraceptive.31
Antidepressants have been shown to effectively ameliorate affective and physical symptoms and improve quality of life and psychosocial function in patients with PMS and PMDD. The response rates for selective serotonin reuptake inhibitors (SSRIs) in PMDD treatment vary from 60% to 90%, vs 30% to 40% for placebo.32 A 2009 Cochrane review found SSRIs reduced premenstrual symptoms compared with placebo.33 However, a literature review suggested that the percentage of women with PMDD who respond to SSRIs or continuous oral contraceptives is lower than the percentage of women who do not respond at all, once the placebo effect is taken into account, and that approximately 40% of women with PMDD do not respond to SSRIs.34 A small study found that citalopram may be effective for women with PMDD who did not respond to a prior SSRI.35
However, only antidepressants that affect serotonergic—not noradrenergic—transmission are effective in PMDD.22 These include:
- the tricyclic antidepressant clomipramine
- the SSRIs citalopram, escitalopram, fluoxetine, paroxetine, and sertraline
- the serotonin-noradrenergic reuptake inhibitor venlafaxine.
It appears that in PMDD, serotonergic agents play a role other than their antidepressant effect.36 The effect of these agents is rapid in PMS/PMDD; women with PMDD who take antidepressants often experience reduced symptoms within the first menstrual cycle, whereas in MDD the onset of action can take weeks or months.37
Although why onset of antidepressant action is quick in PMDD is unclear, rapid onset allows for several dosing options. Some women prefer continuous dosing throughout the month because they do not have to keep track of ovulation. Dosing antidepressants only in the luteal phase (taking the antidepressant from ovulation onset to the start of menses) is an effective treatment strategy.38 Many women prefer to take medication for only 2 weeks per month, which can decrease side effects and lower treatment costs. Alternatively, symptom-onset dosing—initiating the antidepressant when PMDD symptoms begin and stopping at menses onset or within 3 days thereafter—has shown promising results.39,40 Paroxetine, sertraline, and fluoxetine are FDA-approved for PMDD as continuous or intermittent regimens, although using fluoxetine intermittently may not make sense because its biologically active metabolite has an extended half-life.37
Other treatments. Dietary interventions, psychotherapy, vitamins, bright light treatment, and spironolactone have been assessed for PMS/PMDD, although for many evidence-based findings are lacking (Box 3).
See the Bibliography below for studies that support using antidepressants to treat PMDD
Two reviews of 10 randomized controlled trials (RCTs) that evaluated 62 herbs, vitamins, and mineral treatments for premenstrual symptoms found efficacy for chasteberry (Vitex agnus-castus), calcium, and vitamin B6 but not for primrose oil, magnesium oxide, or St. John’s wort.a,b A study comparing fluoxetine with chasteberry found a similar percentage of patients responded to either agent (68% vs 58%, respectively).c Another study showed calcium resulted in a 48% reduction in premenstrual symptoms from baseline, compared with a 30% reduction with placebo.d Bright light treatment significantly reduced depression ratings in women with premenstrual dysphoric disorder (PMDD).e Compared with placebo, the aldosterone antagonist spironolactone improved irritability, depression, feelings of swelling, breast tenderness, and food craving in women with premenstrual syndrome (PMS).f
A recent systematic review of 7 trials of cognitive-behavioral therapy (CBT) for PMDD, including 3 RCTs, showed a lack of a statistically significant effect.g However, a separate review of RCTs of alternative treatments for PMDD—5 of which included CBT—suggested that CBT may be beneficial in reducing premenstrual symptoms, but the evidence was low quality.h
References
- Dante G, Facchinetti F. Herbal treatments for alleviating premenstrual symptoms: a systematic review. J Psychosom Obstet Gynaecol. 2011;32(1):42-51.
- Whelan AM, Jurgens TM, Naylor H. Herbs, vitamins and minerals in the treatment of premenstrual syndrome: a systematic review. Can J Clin Pharmacol. 2009;16(3):e407-e429.
- Atmaca M, Kumru S, Tezcan E. Fluoxetine versus Vitex agnus castus extract in the treatment of premenstrual dysphoric disorder. Hum Psychopharmacol. 2003;18(3):191-195.
- Thys-Jacobs S, Starkey P, Bernstein D, et al. Calcium carbonate and the premenstrual syndrome: effects on premenstrual and menstrual symptoms. Premenstrual Syndrome Study Group. Am J Obstet Gynecol. 1998;179(2):444-452.
- Parry BL, Berga SL, Mostofi N, et al. Morning versus evening bright light treatment of late luteal phase dysphoric disorder. Am J Psychiatry. 1989;146(9):1215-1217.
- Wang M, Hammarbäck S, Lindhe BA, et al. Treatment of premenstrual syndrome by spironolactone: a double-blind, placebo-controlled study. Acta Obstet Gynecol Scand. 1995;74(10):803-808.
- Lustyk MK, Gerrish WG, Shaver S, et al. Cognitive-behavioral therapy for premenstrual syndrome and premenstrual dysphoric disorder: a systematic review. Arch Womens Ment Health. 2009;12(2):85-96.
- Busse JW, Montori VM, Krasnik C, et al. Psychological intervention for premenstrual syndrome: a meta-analysis of randomized controlled trials. Psychother Psychosom. 2009;78(1):6-15.
Cohen LS, Miner C, Brown EW, et al. Premenstrual daily fluoxetine for premenstrual dysphoric disorder: a placebo-controlled, clinical trial using computerized diaries. Obstet Gynecol. 2002;100(3):435-444.
Eriksson E, Ekman A, Sinclair S, et al. Escitalopram administered in the luteal phase exerts a marked and dose-dependent effect in premenstrual dysphoric disorder. J Clin Psychopharmacol. 2008;28(2):195-202.
Eriksson E, Hedberg MA, Andersch B, et al. The serotonin reuptake inhibitor paroxetin is superior to the noradrenaline reuptake inhibitor maprotiline in the treatment of premenstrual syndrome. Neuropsychopharmacology. 1995;12(2):167-176.
Menkes DB, Taghavi E, Mason PA, et al. Fluoxetine treatment of severe premenstrual syndrome. BMJ. 1992;305(6849):346-347.
Miner C, Brown E, McCray S, et al. Weekly luteal-phase dosing with enteric-coated fluoxetine 90 mg in premenstrual dysphoric disorder: a randomized, double-blind, placebo-controlled clinical trial. Clin Ther. 2002;24(3):417-433.
Ozeren S, Corakçi A, Yücesoy I, et al. Fluoxetine in the treatment of premenstrual syndrome. Eur J Obstet Gynecol Reprod Biol. 1997;73(2):167-170.
Pearlstein TB, Stone AB, Lund SA, et al. Comparison of fluoxetine, bupropion, and placebo in the treatment of premenstrual dysphoric disorder. J Clin Psychopharmacol. 1997;17(4):261-266.
Ravindran LN, Woods SA, Steiner M, et al. Symptom-onset dosing with citalopram in the treatment of premenstrual dysphoric disorder (PMDD): a case series. Arch Womens Ment Health. 2007;10(3):125-127.
Steiner M, Brown E, Trzepacz P, et al. Fluoxetine improves functional work capacity in women with premenstrual dysphoric disorder. Arch Womens Ment Health. 2003;6(1):71-77.
Stone AB, Pearlstein TB, Brown WA. Fluoxetine in the treatment of late luteal phase dysphoric disorder. J Clin Psychiatry. 1991;52(7):290-293.
Sundblad C, Hedberg MA, Eriksson E. Clomipramine administered during the luteal phase reduces the symptoms of premenstrual syndrome: a placebo-controlled trial. Neuropsychopharmacology. 1993;9(2):133-145.
Sundblad C, Modigh K, Andersch B, et al. Clomipramine effectively reduces premenstrual irritability and dysphoria: a placebo-controlled trial. Acta Psychiatr Scand. 1992;85(1):39-47.
Su TP, Schmidt PJ, Danaceau MA, et al. Fluoxetine in the treatment of premenstrual dysphoria. Neuropsychopharmacology. 1997; 16(5):346-356.
Wikander I, Sundblad C, Andersch B, et al. Citalopram in premenstrual dysphoria: is intermittent treatment during luteal phases more effective than continuous medication throughout the menstrual cycle? J Clin Psychopharmacol. 1998;18(5):390-398.
Treatment selection
To enhance compliance and improve the likelihood of successful treatment, tailor treatment decisions to your patient’s needs. Carefully discuss with your patient the evidence-based literature to select the best option for her. Factors to consider when counseling patients include:
- the patient’s age, cigarette smoking habits, and body mass index, which may contraindicate oral contraceptives
- does the patient have regular cycle lengths?
- can she adhere to an on-off schedule? If so, intermittent SSRI dosing may be a good treatment option
- does the patient have irregular cycles?
- is there evidence that symptoms persist into the follicular phase, albeit at a lower level? If so, continuous SSRI dosing may be the best option.
Related Resources
- American Psychiatric Association. DSM-5 development: D 04 Premenstrual dysphoric disorder. www.dsm5.org/proposedrevision/pages/proposedrevision.aspx?rid=484.
Drug Brand Names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clonidine • Catapres, others
- Danazol • Danocrine
- Drospirenone/ethinyl estradiol • Yaz
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Leuprolide • Lupron
- Levonorgestrel/ethinyl estradiol • Seasonale, others
- Paroxetine • Paxil
- Progesterone • Prometrium
- Sertraline • Zoloft
- Spironolactone • Aldactone
- Venlafaxine • Effexor
Disclosures
Drs. Wakil and Girdler report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products
Dr. Meltzer-Brody receives research/grant support from AstraZeneca, The Foundation of Hope, and the National Institutes of Health.
1. Yonkers KA, O’Brien PM, Eriksson E. Premenstrual syndrome. Lancet. 2008;371(9619):1200-1210.
2. Halbreich U, Borenstein J, Pearlstein T, et al. The prevalence, impairment, impact and burden of premenstrual dysphoric disorder (PMS/PMDD). Psychoneuroendocrinology. 2008;28(suppl 3):1-23.
3. Wittchen HU, Becker E, Lieb R, et al. Prevalence, incidence and stability of premenstrual dysphoric disorder in the community. Psychol Med. 2002;32(1):119-132.
4. Hylan TR, Sundell K, Judge R. The impact of premenstrual symptomatology on functioning and treatment-seeking behavior: experience from the United States United Kingdom, and France. J Womens Health Gend Based Med. 1999;8(8):1043-1052.
5. Biggs WS, Demuth RH. Premenstrual syndrome and premenstrual dysphoric disorder. Am Fam Physician. 2011;84(8):918-924.
6. Diagnostic and statistical manual of mental disorders. 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
7. ACOG Practice Bulletin. Clinical management guidelines for obstetrician-gynecologists. Number 15 April 2000. Premenstrual syndrome. Obstet Gynecol. 2000;95:1-9.
8. American Psychiatric Association. Proposed draft revisions to DSM disorders and criteria. DSM-5 development. http://www.dsm5.org/proposedrevision/Pages/Default.aspx. Accessed February 23 2012.
9. Yonkers KA. The association between premenstrual dysphoric disorder and other mood disorders. J Clin Psychiatry. 1997;58(suppl 15):19-25.
10. Klatzkin RR, Lindgren ME, Forneris CA, et al. Histories of major depression and premenstrual dysphoric disorder: evidence for phenotypic differences. Biol Psychol. 2010;84(2):235-247.
11. Payne JL, Roy PS, Murphy-Eberenz K, et al. Reproductive cycle-associated mood symptoms in women with major depression and bipolar disorder. J Affect Disord. 2007;99(1-3):221-229.
12. Kim DR, Gyulai L, Freeman EW, et al. Premenstrual dysphoric disorder and psychiatric co-morbidity. Arch Womens Ment Health. 2004;7(1):37-47.
13. Golding JM, Taylor DL, Menard L, et al. Prevalence of sexual abuse history in a sample of women seeking treatment for premenstrual syndrome. J Psychosom Obstet Gynaecol. 2000;21(2):69-80.
14. Girdler SS, Leserman J, Bunevicius R, et al. Persistent alterations in biological profiles in women with abuse histories: influence of premenstrual dysphoric disorder. Health Psychol. 2007;26(2):201-213.
15. Pilver CE, Levy BR, Libby DJ, et al. Posttraumatic stress disorder and trauma characteristics are correlates of premenstrual dysphoric disorder. Arch Womens Ment Health. 2011;14(5):383-393.
16. Girdler SS, Thompson KS, Light KC, et al. Historical sexual abuse and current thyroid axis profiles in women with premenstrual dysphoric disorder. Psychosom Med. 2004;66(3):403-410.
17. Girdler SS, Sherwood A, Hinderliter AL, et al. Biological correlates of abuse in women with premenstrual dysphoric disorder and healthy controls. Psychosom Med. 2003;65(5):849-856.
18. Bunevicius R, Hinderliter AL, Light KC, et al. Histories of sexual abuse are associated with differential effects of clonidine on autonomic function in women with premenstrual dysphoric disorder. Biol Psychol. 2005;69(3):281-296.
19. Perkonigg A, Yonkers KA, Pfister H, et al. Risk factors for premenstrual dysphoric disorder in a community sample of young women: the role of traumatic events and posttraumatic stress disorder. J Clin Psychiatry. 2004;65(10):1314-1322.
20. Girdler SS, Pedersen CA, Stern RA, et al. Menstrual cycle and premenstrual syndrome: modifiers of cardiovascular reactivity in women. Health Psychol. 1993;12(3):180-192.
21. Endicott J, Nee J, Harrison W. Daily Record of Severity of Problems (DRSP): reliability and validity. Arch Womens Ment Health. 2006;9(1):41-49.
22. Cunningham J, Yonkers KA, O’Brien S, et al. Update on research and treatment of premenstrual dysphoric disorder. Harv Rev Psychiatry. 2009;17(2):120-137.
23. Pincus SM, Alam S, Rubinow DR, et al. Predicting response to leuprolide of women with premenstrual dysphoric disorder by daily mood rating dynamics. J Psychiatr Res. 2011;45(3):386-394.
24. Wyatt KM, Dimmock PW, Ismail KM, et al. The effectiveness of GnRHa with and without ‘add-back’ therapy in treating premenstrual syndrome: a meta analysis. BJOG. 2004;111(6):585-593.
25. Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321-333.
26. Hahn PM, Van Vugt DA, Reid RL. A randomized placebo-controlled, crossover trial of danazol for the treatment of premenstrual syndrome. Psychoneuroendocrinology. 1995;20(2):193-209.
27. Bancroft J, Rennie D. The impact of oral contraceptives on the experience of perimenstrual mood clumsiness, food craving and other symptoms. J Psychosom Res. 1993;37(2):195-202.
28. Graham CA, Sherwin BB. A prospective treatment study of premenstrual symptoms using a triphasic oral contraceptive. J Psychosom Res. 1992;36(3):257-266.
29. Freeman EW, Halbreich U, Grubb GS, et al. An overview of four studies of a continuous oral contraceptive (levonorgestrel 90 mcg/ethinyl estradiol 20 mcg) on premenstrual dysphoric disorder and premenstrual syndrome [published online ahead of print December 5, 2011]. Contraception. doi: 10.1016/j.contraception.2011.09.010.
30. Lopez LM, Kaptein AA, Helmerhorst FM. Oral contraceptives containing drospirenone for premenstrual syndrome. Cochrane Database Syst Rev. 2009;(2):CD006586.
31. Sullivan H, Furniss H, Spona J, et al. Effect of 21-day and 24-day oral contraceptive regimens containing gestodene (60 microg) andethinyl estradiol (15 microg) on ovarian activity. Fertil Steril. 1999;72(1):115-120.
32. Yonkers KA, Clark RH, Trivedi MH. The psychopharmacological treatment of nonmajor mood disorders. Mod Probl Pharmacopsychiatry. 1997;25:146-166.
33. Brown J, O’Brien PM, Marjoribanks J, et al. Selective serotonin reuptake inhibitors for premenstrual syndrome. Cochrane Database Syst Rev. 2009;(2):CD001396.
34. Halbreich U. Selective serotonin reuptake inhibitors and initial oral contraceptives for the treatment of PMDD: effective but not enough. CNS Spectr. 2008;13(7):566-572.
35. Freeman EW, Jabara S, Sondheimer SJ, et al. Citalopram in PMS patients with prior SSRI treatment failure: a preliminary study. J Womens Health Gend Based Med. 2002;11(5):
459-464.
36. Freeman EW, Rickels K, Sondheimer SJ, et al. Differential response to antidepressants in women with premenstrual syndrome/premenstrual dysphoric disorder: a randomized controlled trial. Arch Gen Psychiatry. 1999;56(10):932-939.
37. Steiner M, Pearlstein T, Cohen LS, et al. Expert guidelines for the treatment of severe PMS, PMDD, and comorbidities: the role of SSRIs. J Womens Health (Larchmt). 2006;15(1):57-69.
38. Freeman EW. Luteal phase administration of agents for the treatment of premenstrual dysphoric disorder. CNS Drugs. 2004;18(7):453-468.
39. Yonkers KA, Holthausen GA, Poschman K, et al. Symptom-onset treatment for women with premenstrual dysphoric disorder. J Clin Psychopharmacol. 2006;26(2):198-202.
40. Freeman EW, Sondheimer SJ, Sammel MD, et al. A preliminary study of luteal phase versus symptom-onset dosing with escitalopram for premenstrual dysphoric disorder. J Clin Psychiatry. 2005;66(6):769-773.
1. Yonkers KA, O’Brien PM, Eriksson E. Premenstrual syndrome. Lancet. 2008;371(9619):1200-1210.
2. Halbreich U, Borenstein J, Pearlstein T, et al. The prevalence, impairment, impact and burden of premenstrual dysphoric disorder (PMS/PMDD). Psychoneuroendocrinology. 2008;28(suppl 3):1-23.
3. Wittchen HU, Becker E, Lieb R, et al. Prevalence, incidence and stability of premenstrual dysphoric disorder in the community. Psychol Med. 2002;32(1):119-132.
4. Hylan TR, Sundell K, Judge R. The impact of premenstrual symptomatology on functioning and treatment-seeking behavior: experience from the United States United Kingdom, and France. J Womens Health Gend Based Med. 1999;8(8):1043-1052.
5. Biggs WS, Demuth RH. Premenstrual syndrome and premenstrual dysphoric disorder. Am Fam Physician. 2011;84(8):918-924.
6. Diagnostic and statistical manual of mental disorders. 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
7. ACOG Practice Bulletin. Clinical management guidelines for obstetrician-gynecologists. Number 15 April 2000. Premenstrual syndrome. Obstet Gynecol. 2000;95:1-9.
8. American Psychiatric Association. Proposed draft revisions to DSM disorders and criteria. DSM-5 development. http://www.dsm5.org/proposedrevision/Pages/Default.aspx. Accessed February 23 2012.
9. Yonkers KA. The association between premenstrual dysphoric disorder and other mood disorders. J Clin Psychiatry. 1997;58(suppl 15):19-25.
10. Klatzkin RR, Lindgren ME, Forneris CA, et al. Histories of major depression and premenstrual dysphoric disorder: evidence for phenotypic differences. Biol Psychol. 2010;84(2):235-247.
11. Payne JL, Roy PS, Murphy-Eberenz K, et al. Reproductive cycle-associated mood symptoms in women with major depression and bipolar disorder. J Affect Disord. 2007;99(1-3):221-229.
12. Kim DR, Gyulai L, Freeman EW, et al. Premenstrual dysphoric disorder and psychiatric co-morbidity. Arch Womens Ment Health. 2004;7(1):37-47.
13. Golding JM, Taylor DL, Menard L, et al. Prevalence of sexual abuse history in a sample of women seeking treatment for premenstrual syndrome. J Psychosom Obstet Gynaecol. 2000;21(2):69-80.
14. Girdler SS, Leserman J, Bunevicius R, et al. Persistent alterations in biological profiles in women with abuse histories: influence of premenstrual dysphoric disorder. Health Psychol. 2007;26(2):201-213.
15. Pilver CE, Levy BR, Libby DJ, et al. Posttraumatic stress disorder and trauma characteristics are correlates of premenstrual dysphoric disorder. Arch Womens Ment Health. 2011;14(5):383-393.
16. Girdler SS, Thompson KS, Light KC, et al. Historical sexual abuse and current thyroid axis profiles in women with premenstrual dysphoric disorder. Psychosom Med. 2004;66(3):403-410.
17. Girdler SS, Sherwood A, Hinderliter AL, et al. Biological correlates of abuse in women with premenstrual dysphoric disorder and healthy controls. Psychosom Med. 2003;65(5):849-856.
18. Bunevicius R, Hinderliter AL, Light KC, et al. Histories of sexual abuse are associated with differential effects of clonidine on autonomic function in women with premenstrual dysphoric disorder. Biol Psychol. 2005;69(3):281-296.
19. Perkonigg A, Yonkers KA, Pfister H, et al. Risk factors for premenstrual dysphoric disorder in a community sample of young women: the role of traumatic events and posttraumatic stress disorder. J Clin Psychiatry. 2004;65(10):1314-1322.
20. Girdler SS, Pedersen CA, Stern RA, et al. Menstrual cycle and premenstrual syndrome: modifiers of cardiovascular reactivity in women. Health Psychol. 1993;12(3):180-192.
21. Endicott J, Nee J, Harrison W. Daily Record of Severity of Problems (DRSP): reliability and validity. Arch Womens Ment Health. 2006;9(1):41-49.
22. Cunningham J, Yonkers KA, O’Brien S, et al. Update on research and treatment of premenstrual dysphoric disorder. Harv Rev Psychiatry. 2009;17(2):120-137.
23. Pincus SM, Alam S, Rubinow DR, et al. Predicting response to leuprolide of women with premenstrual dysphoric disorder by daily mood rating dynamics. J Psychiatr Res. 2011;45(3):386-394.
24. Wyatt KM, Dimmock PW, Ismail KM, et al. The effectiveness of GnRHa with and without ‘add-back’ therapy in treating premenstrual syndrome: a meta analysis. BJOG. 2004;111(6):585-593.
25. Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321-333.
26. Hahn PM, Van Vugt DA, Reid RL. A randomized placebo-controlled, crossover trial of danazol for the treatment of premenstrual syndrome. Psychoneuroendocrinology. 1995;20(2):193-209.
27. Bancroft J, Rennie D. The impact of oral contraceptives on the experience of perimenstrual mood clumsiness, food craving and other symptoms. J Psychosom Res. 1993;37(2):195-202.
28. Graham CA, Sherwin BB. A prospective treatment study of premenstrual symptoms using a triphasic oral contraceptive. J Psychosom Res. 1992;36(3):257-266.
29. Freeman EW, Halbreich U, Grubb GS, et al. An overview of four studies of a continuous oral contraceptive (levonorgestrel 90 mcg/ethinyl estradiol 20 mcg) on premenstrual dysphoric disorder and premenstrual syndrome [published online ahead of print December 5, 2011]. Contraception. doi: 10.1016/j.contraception.2011.09.010.
30. Lopez LM, Kaptein AA, Helmerhorst FM. Oral contraceptives containing drospirenone for premenstrual syndrome. Cochrane Database Syst Rev. 2009;(2):CD006586.
31. Sullivan H, Furniss H, Spona J, et al. Effect of 21-day and 24-day oral contraceptive regimens containing gestodene (60 microg) andethinyl estradiol (15 microg) on ovarian activity. Fertil Steril. 1999;72(1):115-120.
32. Yonkers KA, Clark RH, Trivedi MH. The psychopharmacological treatment of nonmajor mood disorders. Mod Probl Pharmacopsychiatry. 1997;25:146-166.
33. Brown J, O’Brien PM, Marjoribanks J, et al. Selective serotonin reuptake inhibitors for premenstrual syndrome. Cochrane Database Syst Rev. 2009;(2):CD001396.
34. Halbreich U. Selective serotonin reuptake inhibitors and initial oral contraceptives for the treatment of PMDD: effective but not enough. CNS Spectr. 2008;13(7):566-572.
35. Freeman EW, Jabara S, Sondheimer SJ, et al. Citalopram in PMS patients with prior SSRI treatment failure: a preliminary study. J Womens Health Gend Based Med. 2002;11(5):
459-464.
36. Freeman EW, Rickels K, Sondheimer SJ, et al. Differential response to antidepressants in women with premenstrual syndrome/premenstrual dysphoric disorder: a randomized controlled trial. Arch Gen Psychiatry. 1999;56(10):932-939.
37. Steiner M, Pearlstein T, Cohen LS, et al. Expert guidelines for the treatment of severe PMS, PMDD, and comorbidities: the role of SSRIs. J Womens Health (Larchmt). 2006;15(1):57-69.
38. Freeman EW. Luteal phase administration of agents for the treatment of premenstrual dysphoric disorder. CNS Drugs. 2004;18(7):453-468.
39. Yonkers KA, Holthausen GA, Poschman K, et al. Symptom-onset treatment for women with premenstrual dysphoric disorder. J Clin Psychopharmacol. 2006;26(2):198-202.
40. Freeman EW, Sondheimer SJ, Sammel MD, et al. A preliminary study of luteal phase versus symptom-onset dosing with escitalopram for premenstrual dysphoric disorder. J Clin Psychiatry. 2005;66(6):769-773.
Getting ready for DSM-5: Psychotic disorders
Discuss this article at www.facebook.com/CurrentPsychiatry
In DSM-IV,1 the section on schizophrenia and other psychotic disorders includes schizophrenia (with 5 subtypes), schizophreniform disorder, schizoaffective disorder, delusional disorder, shared psychotic disorder, brief psychotic disorder, substance-induced psychotic disorder, psychotic disorder due to a general medical condition, and psychotic disorder not otherwise specified. As we consider proposed changes to DSM-5 (Table 1),2 it is useful to consider limitations in our current construct of schizophrenia.
Table 1
Psychotic disorders in DSM-5: Summary of proposed changes
| Replace existing subtypes with dimensions |
| Include diagnosis of attenuated psychosis syndrome |
| Modify criteria for schizoaffective disorder |
| ‘Delink’ catatonia from schizophrenia |
| Source: Reference 2 |
First, many etiological factors and pathophysiological processes appear relevant to what we consider schizophrenia and it is almost certain that our construct of schizophrenia encompasses not one but numerous diseases with a shared phenotype.3-5
Second, the boundary between schizophrenia and schizoaffective disorder is imprecisely defined, and a proportion of patients with schizophrenia with some mood symptoms may inappropriately receive a schizoaffective disorder diagnosis. This is compounded by the poor reliability and low diagnostic stability of a schizoaffective disorder diagnosis.6-8
Third, the current classic schizophrenia subtypes provide an inadequate description of the enormous heterogeneity of this condition. Additionally, subtype stability is low, and only the paranoid and undifferentiated subtypes are used frequently in clinical practice.
Fourth, the prominence given to Schneiderian first-rank symptoms (“bizarre” delusions or “special” hallucinations) appears misplaced.
Fifth, the current construct of schizophrenia inadequately describes the major psychopathological dimensions of the condition and stages of its evolution.8,9
Finally, the current clinical construct of schizophrenia does not match neurobiological markers and genetic findings or specific pharmacological treatment provided.5,10 Proposed DSM-5 revisions2 to the definition of schizophrenia to address these limitations are summarized below.
Schizophrenia syndrome
Proposed changes to the diagnostic criteria for schizophrenia are modest and continuity with DSM-IV is broadly maintained. Two modest changes to criterion A (active phase symptoms) are proposed:
- Eliminate special treatment of bizarre delusions and other Schneiderian first-rank symptoms. In DSM-IV, only 1 criterion A is required if it is a bizarre delusion or hallucination. Because Schneiderian first-rank symptoms do not have diagnostic specificity and diagnosing “bizarreness” of delusions and hallucinations has low reliability, it is proposed that these positive symptoms be treated like any other with regard to their diagnostic implications.
- Require that at least 1 of the 2 symptoms required to meet criterion A be delusions, hallucinations, or disorganized thinking. These are core positive symptoms diagnosed with high reliability and might reasonably be considered necessary for a reliable schizophrenia diagnosis.
Subtypes
The DSM-5 proposal for describing schizophrenia advocates eliminating DSM-IV schizophrenia subtypes. These subtypes have limited diagnostic stability, low reliability, and poor validity. Furthermore, except for the paranoid and undifferentiated subtypes, other subtypes rarely are used in most mental health care systems.
Schizoaffective disorder
Characterizing patients with both psychotic and mood symptoms either concurrently or at different points during their illness always has been controversial. In DSM-I and DSM-II, a diagnosis of schizophrenia, schizoaffective subtype, generally was recommended for such patients. DSM-III reversed this recommendation and specified that schizophrenia was to be diagnosed only in the absence of prominent mood symptoms. Furthermore, in DSM-III, diagnosing schizoaffective disorder was strongly discouraged, and it was the only condition in DSM-III without operational criteria. Schizoaffective disorder saw a revival in DSM-III-R that has continued through DSM-IV. In fact, in many mental health care systems, almost one-third of patients with psychotic symptoms receive a schizoaffective disorder diagnosis. One of the insidious changes to the definition of schizoaffective disorder from DSM-III to DSM-IV is that it moved from being a lifetime diagnosis to a cross-sectional diagnosis—ie, in DSM-IV, only mood/psychotic symptoms in the current episode are considered, and the longitudinal course of these symptoms in the patient’s life are ignored. The current DSM-5 proposal attempts to improve reliability of this diagnosis by providing more specific criteria and is reconceptualizing schizoaffective disorder as a longitudinal diagnosis. To this end, the most significant proposed change is to criterion C of schizoaffective disorder, which attempts to demarcate schizoaffective disorder from schizophrenia with prominent mood symptoms. Criterion C will be revised to state “symptoms that meet criteria for a mood episode are present for a majority (>50%) of the total duration of the active and residual periods of the illness.”2
Psychopathological dimensions
Schizophrenic illness is characterized by several psychopathological domains, with a distinctive course, patterns of treatment-response, and prognostic implications. The relative severity of symptom dimensions—positive, negative, mood, disorganization, motor, and cognitive—vary among patients and also within patients at different stages of their illness. Measuring the relative severity of these symptom dimensions throughout the illness course can provide clinicians with useful information about the nature of a patient’s schizophrenic illness and the specific impact of treatment on different aspects of his or her illness (Table 2). In addition to being clinically useful, dimensional measurement also should improve schizophrenia research because having dimensional information will permit studies on etiology and pathogenesis that cut across current diagnostic categories. Although field trials are evaluating 9 dimensions—delusions, hallucinations, disorganization, depression, mania, cognitive impairment, restricted emotional expression, avolition, and psychomotor—it is likely that fewer dimensions will be recommended for DSM-5, based on reliability results of these trials, clinical utility, and logistic feasibility in routine clinical settings.
Table 2
Goals of a dimensional approach to schizophrenia
| Better understanding of schizophrenia |
| Distinct dimensions of illness |
| Distinct stages of illness |
| Elucidation of neurobiology |
| More precise delineation of etiology |
| More refined treatment development |
| Direction at specific dimension-endophenotype |
| Stage-specific treatment |
| Novel treatment targets |
Attenuated psychosis syndrome
Some clinicians and researchers believe that many patients with schizophrenia experience unsatisfactory outcomes because we identify the illness and initiate treatment after substantial brain tissue damage has occurred. Introducing attenuated psychosis syndrome will support clinicians’ efforts to recognize mild psychotic symptoms early in their evolution and to monitor—and if necessary, intervene—during these crucial early stages. Risks include possible stigma and inappropriate use of medications and other treatments. This controversial proposal is being field tested. It is unclear if this category will be included in DSM-5 and if it does, whether it will be in the main text or the appendix.
Catatonia
Catatonia will be used as a specifier for various psychotic disorders, major mood disorders, and associated with a general medical condition. Additionally, the same criteria will be used to diagnose catatonia across DSM-5. Catatonia Not Elsewhere Classified might be added as a residual category for other conditions in which a clear catatonic syndrome is present and the parent disorder has not yet been identified.2
Other psychotic disorders
Relatively minor changes are proposed in criteria for other disorders in this section. There are likely to be changes in the text, however, that incorporate new information about these conditions generated since publication of DSM-IV-TR in 2000. Some proposed changes include:
- deleting shared delusional disorder (folie à deux) as a separate diagnosis and instead characterizing it as a specifier for delusional disorder
- clarifying the distinction between substance-induced psychotic disorder and other psychotic disorders accompanied by comorbid substance use.
Current status of DSM-5
Field trials are being completed and their results remain to be analyzed. Major changes being evaluated in the field trials include:
- the impact of the change in concept and criteria for schizoaffective disorder
- the addition of a series of psychopathology dimensions
- the impact of adding attenuated psychosis syndrome as a new class.
Changes proposed by the Psychosis Disorders Work Group are intended to increase clinical utility (fewer diagnoses, better demarcation between disorders, greater treatment relevance [dimensions]) and modestly improve validity (more consistent with current information about the nature of various psychotic disorders), while retaining reliability in diagnosing various psychotic disorders (and improving it for schizoaffective disorder). Proposed changes are modest by and large but hope to set a better stage for a future etiopathophysiological classification.
The Psychosis Disorders Work Group’s recommendations are posted on the DSM-5 Web site2 at www.dsm5.org and are being reviewed by 2 expert committees established by the American Psychiatric Association Board of Trustees: a Scientific Review Committee and a Clinical and Public Health Implications Committee. Based on the results of the field trials, ongoing reviews, and other emerging data and discussions, additional changes to the current DSM-5 proposals may occur. DSM-5 is likely to be finalized in early 2013 and the published manual will be released in May 2013.
Related Resources
- American Psychiatric Association. DSM-5 Development. www.dsm5.org.
- Woods SW, McGlashan TH. The risk-benefit ratio of the proposed DSM-5 attenuated psychosis syndrome. Am J Psychiatry. 2011;168(12):1338.
Disclosure
Dr. Tandon is a member of the DSM-5 Psychotic Disorders Work Group. He is solely responsible for the content of this article.
1. Diagnostic and statistical manual of mental disorders, 4th ed. Washington, DC: American Psychiatric Association; 1994.
2. American Psychiatric Association. DSM-5 development. http://www.dsm5.org. Accessed March 19, 2011.
3. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia “just the facts”: what we know in 2008. Part 1: overview. Schizophr Res. 2008;100(1-3):4-19.
4. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia “just the facts” what we know in 2008. 2: epidemiology and etiology. Schizophr Res. 2008;102(1-3):1-18.
5. Keshavan MS, Tandon R, Boutros N, et al. Schizophrenia, “just the facts” what we know in 2008. Part 3: neurobiology. Schizophr Res. 2008;106(2-3):89-107.
6. Tandon R, Maj M. Nosological status and definition of schizophrenia. Some considerations for DSM-V and ICD-11. Asian Journal of Psychiatry. 2008;1(2):22-27.
7. Fiedorowicz JG, Epping EA, Flaum M. Toward defining schizophrenia as a more useful clinical construct. Curr Psychiatry Rep. 2008;10(4):344-351.
8. Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia “just the facts” 4. Clinical features and conceptualization. Schizophr Res. 2009;110(1-3):1-23.
9. McGorry PD. Risk syndromes clinical staging, and DSM V: new diagnostic infrastructure for early intervention in psychiatry. Schizophr Res. 2010;120(1-3):49-53.
10. Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia “just the facts” 5. Treatment and prevention. Past, present, and future. Schizophr Res. 2010;122(1-3):1-23.

Rajiv Tandon, MD
Professor of Psychiatry, University of Florida, Gainesville, FL
Rajiv Tandon, MD
Professor of Psychiatry, University of Florida, Gainesville, FL
Rajiv Tandon, MD
Professor of Psychiatry, University of Florida, Gainesville, FL
Discuss this article at www.facebook.com/CurrentPsychiatry
In DSM-IV,1 the section on schizophrenia and other psychotic disorders includes schizophrenia (with 5 subtypes), schizophreniform disorder, schizoaffective disorder, delusional disorder, shared psychotic disorder, brief psychotic disorder, substance-induced psychotic disorder, psychotic disorder due to a general medical condition, and psychotic disorder not otherwise specified. As we consider proposed changes to DSM-5 (Table 1),2 it is useful to consider limitations in our current construct of schizophrenia.
Table 1
Psychotic disorders in DSM-5: Summary of proposed changes
| Replace existing subtypes with dimensions |
| Include diagnosis of attenuated psychosis syndrome |
| Modify criteria for schizoaffective disorder |
| ‘Delink’ catatonia from schizophrenia |
| Source: Reference 2 |
First, many etiological factors and pathophysiological processes appear relevant to what we consider schizophrenia and it is almost certain that our construct of schizophrenia encompasses not one but numerous diseases with a shared phenotype.3-5
Second, the boundary between schizophrenia and schizoaffective disorder is imprecisely defined, and a proportion of patients with schizophrenia with some mood symptoms may inappropriately receive a schizoaffective disorder diagnosis. This is compounded by the poor reliability and low diagnostic stability of a schizoaffective disorder diagnosis.6-8
Third, the current classic schizophrenia subtypes provide an inadequate description of the enormous heterogeneity of this condition. Additionally, subtype stability is low, and only the paranoid and undifferentiated subtypes are used frequently in clinical practice.
Fourth, the prominence given to Schneiderian first-rank symptoms (“bizarre” delusions or “special” hallucinations) appears misplaced.
Fifth, the current construct of schizophrenia inadequately describes the major psychopathological dimensions of the condition and stages of its evolution.8,9
Finally, the current clinical construct of schizophrenia does not match neurobiological markers and genetic findings or specific pharmacological treatment provided.5,10 Proposed DSM-5 revisions2 to the definition of schizophrenia to address these limitations are summarized below.
Schizophrenia syndrome
Proposed changes to the diagnostic criteria for schizophrenia are modest and continuity with DSM-IV is broadly maintained. Two modest changes to criterion A (active phase symptoms) are proposed:
- Eliminate special treatment of bizarre delusions and other Schneiderian first-rank symptoms. In DSM-IV, only 1 criterion A is required if it is a bizarre delusion or hallucination. Because Schneiderian first-rank symptoms do not have diagnostic specificity and diagnosing “bizarreness” of delusions and hallucinations has low reliability, it is proposed that these positive symptoms be treated like any other with regard to their diagnostic implications.
- Require that at least 1 of the 2 symptoms required to meet criterion A be delusions, hallucinations, or disorganized thinking. These are core positive symptoms diagnosed with high reliability and might reasonably be considered necessary for a reliable schizophrenia diagnosis.
Subtypes
The DSM-5 proposal for describing schizophrenia advocates eliminating DSM-IV schizophrenia subtypes. These subtypes have limited diagnostic stability, low reliability, and poor validity. Furthermore, except for the paranoid and undifferentiated subtypes, other subtypes rarely are used in most mental health care systems.
Schizoaffective disorder
Characterizing patients with both psychotic and mood symptoms either concurrently or at different points during their illness always has been controversial. In DSM-I and DSM-II, a diagnosis of schizophrenia, schizoaffective subtype, generally was recommended for such patients. DSM-III reversed this recommendation and specified that schizophrenia was to be diagnosed only in the absence of prominent mood symptoms. Furthermore, in DSM-III, diagnosing schizoaffective disorder was strongly discouraged, and it was the only condition in DSM-III without operational criteria. Schizoaffective disorder saw a revival in DSM-III-R that has continued through DSM-IV. In fact, in many mental health care systems, almost one-third of patients with psychotic symptoms receive a schizoaffective disorder diagnosis. One of the insidious changes to the definition of schizoaffective disorder from DSM-III to DSM-IV is that it moved from being a lifetime diagnosis to a cross-sectional diagnosis—ie, in DSM-IV, only mood/psychotic symptoms in the current episode are considered, and the longitudinal course of these symptoms in the patient’s life are ignored. The current DSM-5 proposal attempts to improve reliability of this diagnosis by providing more specific criteria and is reconceptualizing schizoaffective disorder as a longitudinal diagnosis. To this end, the most significant proposed change is to criterion C of schizoaffective disorder, which attempts to demarcate schizoaffective disorder from schizophrenia with prominent mood symptoms. Criterion C will be revised to state “symptoms that meet criteria for a mood episode are present for a majority (>50%) of the total duration of the active and residual periods of the illness.”2
Psychopathological dimensions
Schizophrenic illness is characterized by several psychopathological domains, with a distinctive course, patterns of treatment-response, and prognostic implications. The relative severity of symptom dimensions—positive, negative, mood, disorganization, motor, and cognitive—vary among patients and also within patients at different stages of their illness. Measuring the relative severity of these symptom dimensions throughout the illness course can provide clinicians with useful information about the nature of a patient’s schizophrenic illness and the specific impact of treatment on different aspects of his or her illness (Table 2). In addition to being clinically useful, dimensional measurement also should improve schizophrenia research because having dimensional information will permit studies on etiology and pathogenesis that cut across current diagnostic categories. Although field trials are evaluating 9 dimensions—delusions, hallucinations, disorganization, depression, mania, cognitive impairment, restricted emotional expression, avolition, and psychomotor—it is likely that fewer dimensions will be recommended for DSM-5, based on reliability results of these trials, clinical utility, and logistic feasibility in routine clinical settings.
Table 2
Goals of a dimensional approach to schizophrenia
| Better understanding of schizophrenia |
| Distinct dimensions of illness |
| Distinct stages of illness |
| Elucidation of neurobiology |
| More precise delineation of etiology |
| More refined treatment development |
| Direction at specific dimension-endophenotype |
| Stage-specific treatment |
| Novel treatment targets |
Attenuated psychosis syndrome
Some clinicians and researchers believe that many patients with schizophrenia experience unsatisfactory outcomes because we identify the illness and initiate treatment after substantial brain tissue damage has occurred. Introducing attenuated psychosis syndrome will support clinicians’ efforts to recognize mild psychotic symptoms early in their evolution and to monitor—and if necessary, intervene—during these crucial early stages. Risks include possible stigma and inappropriate use of medications and other treatments. This controversial proposal is being field tested. It is unclear if this category will be included in DSM-5 and if it does, whether it will be in the main text or the appendix.
Catatonia
Catatonia will be used as a specifier for various psychotic disorders, major mood disorders, and associated with a general medical condition. Additionally, the same criteria will be used to diagnose catatonia across DSM-5. Catatonia Not Elsewhere Classified might be added as a residual category for other conditions in which a clear catatonic syndrome is present and the parent disorder has not yet been identified.2
Other psychotic disorders
Relatively minor changes are proposed in criteria for other disorders in this section. There are likely to be changes in the text, however, that incorporate new information about these conditions generated since publication of DSM-IV-TR in 2000. Some proposed changes include:
- deleting shared delusional disorder (folie à deux) as a separate diagnosis and instead characterizing it as a specifier for delusional disorder
- clarifying the distinction between substance-induced psychotic disorder and other psychotic disorders accompanied by comorbid substance use.
Current status of DSM-5
Field trials are being completed and their results remain to be analyzed. Major changes being evaluated in the field trials include:
- the impact of the change in concept and criteria for schizoaffective disorder
- the addition of a series of psychopathology dimensions
- the impact of adding attenuated psychosis syndrome as a new class.
Changes proposed by the Psychosis Disorders Work Group are intended to increase clinical utility (fewer diagnoses, better demarcation between disorders, greater treatment relevance [dimensions]) and modestly improve validity (more consistent with current information about the nature of various psychotic disorders), while retaining reliability in diagnosing various psychotic disorders (and improving it for schizoaffective disorder). Proposed changes are modest by and large but hope to set a better stage for a future etiopathophysiological classification.
The Psychosis Disorders Work Group’s recommendations are posted on the DSM-5 Web site2 at www.dsm5.org and are being reviewed by 2 expert committees established by the American Psychiatric Association Board of Trustees: a Scientific Review Committee and a Clinical and Public Health Implications Committee. Based on the results of the field trials, ongoing reviews, and other emerging data and discussions, additional changes to the current DSM-5 proposals may occur. DSM-5 is likely to be finalized in early 2013 and the published manual will be released in May 2013.
Related Resources
- American Psychiatric Association. DSM-5 Development. www.dsm5.org.
- Woods SW, McGlashan TH. The risk-benefit ratio of the proposed DSM-5 attenuated psychosis syndrome. Am J Psychiatry. 2011;168(12):1338.
Disclosure
Dr. Tandon is a member of the DSM-5 Psychotic Disorders Work Group. He is solely responsible for the content of this article.
Discuss this article at www.facebook.com/CurrentPsychiatry
In DSM-IV,1 the section on schizophrenia and other psychotic disorders includes schizophrenia (with 5 subtypes), schizophreniform disorder, schizoaffective disorder, delusional disorder, shared psychotic disorder, brief psychotic disorder, substance-induced psychotic disorder, psychotic disorder due to a general medical condition, and psychotic disorder not otherwise specified. As we consider proposed changes to DSM-5 (Table 1),2 it is useful to consider limitations in our current construct of schizophrenia.
Table 1
Psychotic disorders in DSM-5: Summary of proposed changes
| Replace existing subtypes with dimensions |
| Include diagnosis of attenuated psychosis syndrome |
| Modify criteria for schizoaffective disorder |
| ‘Delink’ catatonia from schizophrenia |
| Source: Reference 2 |
First, many etiological factors and pathophysiological processes appear relevant to what we consider schizophrenia and it is almost certain that our construct of schizophrenia encompasses not one but numerous diseases with a shared phenotype.3-5
Second, the boundary between schizophrenia and schizoaffective disorder is imprecisely defined, and a proportion of patients with schizophrenia with some mood symptoms may inappropriately receive a schizoaffective disorder diagnosis. This is compounded by the poor reliability and low diagnostic stability of a schizoaffective disorder diagnosis.6-8
Third, the current classic schizophrenia subtypes provide an inadequate description of the enormous heterogeneity of this condition. Additionally, subtype stability is low, and only the paranoid and undifferentiated subtypes are used frequently in clinical practice.
Fourth, the prominence given to Schneiderian first-rank symptoms (“bizarre” delusions or “special” hallucinations) appears misplaced.
Fifth, the current construct of schizophrenia inadequately describes the major psychopathological dimensions of the condition and stages of its evolution.8,9
Finally, the current clinical construct of schizophrenia does not match neurobiological markers and genetic findings or specific pharmacological treatment provided.5,10 Proposed DSM-5 revisions2 to the definition of schizophrenia to address these limitations are summarized below.
Schizophrenia syndrome
Proposed changes to the diagnostic criteria for schizophrenia are modest and continuity with DSM-IV is broadly maintained. Two modest changes to criterion A (active phase symptoms) are proposed:
- Eliminate special treatment of bizarre delusions and other Schneiderian first-rank symptoms. In DSM-IV, only 1 criterion A is required if it is a bizarre delusion or hallucination. Because Schneiderian first-rank symptoms do not have diagnostic specificity and diagnosing “bizarreness” of delusions and hallucinations has low reliability, it is proposed that these positive symptoms be treated like any other with regard to their diagnostic implications.
- Require that at least 1 of the 2 symptoms required to meet criterion A be delusions, hallucinations, or disorganized thinking. These are core positive symptoms diagnosed with high reliability and might reasonably be considered necessary for a reliable schizophrenia diagnosis.
Subtypes
The DSM-5 proposal for describing schizophrenia advocates eliminating DSM-IV schizophrenia subtypes. These subtypes have limited diagnostic stability, low reliability, and poor validity. Furthermore, except for the paranoid and undifferentiated subtypes, other subtypes rarely are used in most mental health care systems.
Schizoaffective disorder
Characterizing patients with both psychotic and mood symptoms either concurrently or at different points during their illness always has been controversial. In DSM-I and DSM-II, a diagnosis of schizophrenia, schizoaffective subtype, generally was recommended for such patients. DSM-III reversed this recommendation and specified that schizophrenia was to be diagnosed only in the absence of prominent mood symptoms. Furthermore, in DSM-III, diagnosing schizoaffective disorder was strongly discouraged, and it was the only condition in DSM-III without operational criteria. Schizoaffective disorder saw a revival in DSM-III-R that has continued through DSM-IV. In fact, in many mental health care systems, almost one-third of patients with psychotic symptoms receive a schizoaffective disorder diagnosis. One of the insidious changes to the definition of schizoaffective disorder from DSM-III to DSM-IV is that it moved from being a lifetime diagnosis to a cross-sectional diagnosis—ie, in DSM-IV, only mood/psychotic symptoms in the current episode are considered, and the longitudinal course of these symptoms in the patient’s life are ignored. The current DSM-5 proposal attempts to improve reliability of this diagnosis by providing more specific criteria and is reconceptualizing schizoaffective disorder as a longitudinal diagnosis. To this end, the most significant proposed change is to criterion C of schizoaffective disorder, which attempts to demarcate schizoaffective disorder from schizophrenia with prominent mood symptoms. Criterion C will be revised to state “symptoms that meet criteria for a mood episode are present for a majority (>50%) of the total duration of the active and residual periods of the illness.”2
Psychopathological dimensions
Schizophrenic illness is characterized by several psychopathological domains, with a distinctive course, patterns of treatment-response, and prognostic implications. The relative severity of symptom dimensions—positive, negative, mood, disorganization, motor, and cognitive—vary among patients and also within patients at different stages of their illness. Measuring the relative severity of these symptom dimensions throughout the illness course can provide clinicians with useful information about the nature of a patient’s schizophrenic illness and the specific impact of treatment on different aspects of his or her illness (Table 2). In addition to being clinically useful, dimensional measurement also should improve schizophrenia research because having dimensional information will permit studies on etiology and pathogenesis that cut across current diagnostic categories. Although field trials are evaluating 9 dimensions—delusions, hallucinations, disorganization, depression, mania, cognitive impairment, restricted emotional expression, avolition, and psychomotor—it is likely that fewer dimensions will be recommended for DSM-5, based on reliability results of these trials, clinical utility, and logistic feasibility in routine clinical settings.
Table 2
Goals of a dimensional approach to schizophrenia
| Better understanding of schizophrenia |
| Distinct dimensions of illness |
| Distinct stages of illness |
| Elucidation of neurobiology |
| More precise delineation of etiology |
| More refined treatment development |
| Direction at specific dimension-endophenotype |
| Stage-specific treatment |
| Novel treatment targets |
Attenuated psychosis syndrome
Some clinicians and researchers believe that many patients with schizophrenia experience unsatisfactory outcomes because we identify the illness and initiate treatment after substantial brain tissue damage has occurred. Introducing attenuated psychosis syndrome will support clinicians’ efforts to recognize mild psychotic symptoms early in their evolution and to monitor—and if necessary, intervene—during these crucial early stages. Risks include possible stigma and inappropriate use of medications and other treatments. This controversial proposal is being field tested. It is unclear if this category will be included in DSM-5 and if it does, whether it will be in the main text or the appendix.
Catatonia
Catatonia will be used as a specifier for various psychotic disorders, major mood disorders, and associated with a general medical condition. Additionally, the same criteria will be used to diagnose catatonia across DSM-5. Catatonia Not Elsewhere Classified might be added as a residual category for other conditions in which a clear catatonic syndrome is present and the parent disorder has not yet been identified.2
Other psychotic disorders
Relatively minor changes are proposed in criteria for other disorders in this section. There are likely to be changes in the text, however, that incorporate new information about these conditions generated since publication of DSM-IV-TR in 2000. Some proposed changes include:
- deleting shared delusional disorder (folie à deux) as a separate diagnosis and instead characterizing it as a specifier for delusional disorder
- clarifying the distinction between substance-induced psychotic disorder and other psychotic disorders accompanied by comorbid substance use.
Current status of DSM-5
Field trials are being completed and their results remain to be analyzed. Major changes being evaluated in the field trials include:
- the impact of the change in concept and criteria for schizoaffective disorder
- the addition of a series of psychopathology dimensions
- the impact of adding attenuated psychosis syndrome as a new class.
Changes proposed by the Psychosis Disorders Work Group are intended to increase clinical utility (fewer diagnoses, better demarcation between disorders, greater treatment relevance [dimensions]) and modestly improve validity (more consistent with current information about the nature of various psychotic disorders), while retaining reliability in diagnosing various psychotic disorders (and improving it for schizoaffective disorder). Proposed changes are modest by and large but hope to set a better stage for a future etiopathophysiological classification.
The Psychosis Disorders Work Group’s recommendations are posted on the DSM-5 Web site2 at www.dsm5.org and are being reviewed by 2 expert committees established by the American Psychiatric Association Board of Trustees: a Scientific Review Committee and a Clinical and Public Health Implications Committee. Based on the results of the field trials, ongoing reviews, and other emerging data and discussions, additional changes to the current DSM-5 proposals may occur. DSM-5 is likely to be finalized in early 2013 and the published manual will be released in May 2013.
Related Resources
- American Psychiatric Association. DSM-5 Development. www.dsm5.org.
- Woods SW, McGlashan TH. The risk-benefit ratio of the proposed DSM-5 attenuated psychosis syndrome. Am J Psychiatry. 2011;168(12):1338.
Disclosure
Dr. Tandon is a member of the DSM-5 Psychotic Disorders Work Group. He is solely responsible for the content of this article.
1. Diagnostic and statistical manual of mental disorders, 4th ed. Washington, DC: American Psychiatric Association; 1994.
2. American Psychiatric Association. DSM-5 development. http://www.dsm5.org. Accessed March 19, 2011.
3. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia “just the facts”: what we know in 2008. Part 1: overview. Schizophr Res. 2008;100(1-3):4-19.
4. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia “just the facts” what we know in 2008. 2: epidemiology and etiology. Schizophr Res. 2008;102(1-3):1-18.
5. Keshavan MS, Tandon R, Boutros N, et al. Schizophrenia, “just the facts” what we know in 2008. Part 3: neurobiology. Schizophr Res. 2008;106(2-3):89-107.
6. Tandon R, Maj M. Nosological status and definition of schizophrenia. Some considerations for DSM-V and ICD-11. Asian Journal of Psychiatry. 2008;1(2):22-27.
7. Fiedorowicz JG, Epping EA, Flaum M. Toward defining schizophrenia as a more useful clinical construct. Curr Psychiatry Rep. 2008;10(4):344-351.
8. Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia “just the facts” 4. Clinical features and conceptualization. Schizophr Res. 2009;110(1-3):1-23.
9. McGorry PD. Risk syndromes clinical staging, and DSM V: new diagnostic infrastructure for early intervention in psychiatry. Schizophr Res. 2010;120(1-3):49-53.
10. Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia “just the facts” 5. Treatment and prevention. Past, present, and future. Schizophr Res. 2010;122(1-3):1-23.
1. Diagnostic and statistical manual of mental disorders, 4th ed. Washington, DC: American Psychiatric Association; 1994.
2. American Psychiatric Association. DSM-5 development. http://www.dsm5.org. Accessed March 19, 2011.
3. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia “just the facts”: what we know in 2008. Part 1: overview. Schizophr Res. 2008;100(1-3):4-19.
4. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia “just the facts” what we know in 2008. 2: epidemiology and etiology. Schizophr Res. 2008;102(1-3):1-18.
5. Keshavan MS, Tandon R, Boutros N, et al. Schizophrenia, “just the facts” what we know in 2008. Part 3: neurobiology. Schizophr Res. 2008;106(2-3):89-107.
6. Tandon R, Maj M. Nosological status and definition of schizophrenia. Some considerations for DSM-V and ICD-11. Asian Journal of Psychiatry. 2008;1(2):22-27.
7. Fiedorowicz JG, Epping EA, Flaum M. Toward defining schizophrenia as a more useful clinical construct. Curr Psychiatry Rep. 2008;10(4):344-351.
8. Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia “just the facts” 4. Clinical features and conceptualization. Schizophr Res. 2009;110(1-3):1-23.
9. McGorry PD. Risk syndromes clinical staging, and DSM V: new diagnostic infrastructure for early intervention in psychiatry. Schizophr Res. 2010;120(1-3):49-53.
10. Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia “just the facts” 5. Treatment and prevention. Past, present, and future. Schizophr Res. 2010;122(1-3):1-23.
Benzodiazepines: A versatile clinical tool
Since the discovery of chlordiazepoxide in the 1950s, benzodiazepines have revolutionized the treatment of anxiety and insomnia, largely because of their improved safety profile compared with barbiturates, formerly the preferred sedative-hypnotic.1 In addition to their anxiolytic and sedative-hypnotic effects, benzodiazepines exhibit anterograde amnesia, anticonvulsant, and muscle relaxant properties.1 Psychiatrists use benzodiazepines to treat anxiety and sleep disorders, acute agitation, alcohol withdrawal, catatonia, and psychotropic side effects such as akathisia. This article highlights the evidence for using benzodiazepines in anxiety and other disorders and why they generally should not be used for obsessive-compulsive disorder and posttraumatic stress disorder (Box 1).
Current evidence indicates little support for using benzodiazepines for obsessive-compulsive disorder (OCD). The American Psychiatric Association (APA) and the World Federation of Biological Psychiatry do not recommend benzodiazepines for treating OCD because of a lack of evidence for efficacy.a,b An earlier study suggested clonazepam monotherapy was effective for OCDc; however, a more recent study did not show a benefit on rate of response or degree of symptom improvement.d Augmentation strategies with benzodiazepines also do not appear to be beneficial for OCD management. A recent double-blind, placebo-controlled study failed to demonstrate faster symptom improvement by augmenting sertraline with clonazepam, although the study had a small sample size and high drop-out rate.e
Because benzodiazepines have negligible action on core posttraumatic stress disorder (PTSD) symptoms (re-experiencing, avoidance, and hyperarousal), selective serotonin reuptake inhibitors and other agents largely have supplanted them for PTSD treatment.f Use of benzodiazepines for PTSD is associated with withdrawal symptoms, more severe symptoms after discontinuation, and possible disinhibition, and may interfere with patients’ efforts to integrate trauma experiences. Although benzodiazepines may reduce distress associated with acute trauma, there is evidence—in clinical studies and animal models—that early benzodiazepine administration fails to prevent PTSD and may increase its incidence.g The International Consensus Group on Depression and Anxiety, the APA, and the British Association for Psychopharmacology all highlight the limited role, if any, for benzodiazepines in PTSD.h-j
References
- Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
- American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Arlington, VA: American Psychiatric Publishing, Inc.; 2007.
- Hewlett WA, Vinogradov S, Agras WS. Clomipramine, clonazepam, and clonidine treatment of obsessive compulsive disorder. J Clin Psychopharmacol. 1992;12(6):420-430.
- Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
- Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam and sertraline in OCD. Ann Clin Psychiatry. 2004;16(3):127-132.
- Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs. Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
- Matar MA, Zohar J, Kaplan Z, et al. Alprazolam treatment immediately after stress exposure interferes with the normal HPA-stress response and increases vulnerability to subsequent stress in an animal model of PTSD. Eur Neuropsychopharmacol. 2009;19(4):283-295.
- Ballenger JC, Davidson JR, Lecrubier Y, et al. Consensus statement update on posttraumatic stress disorder from the international consensus group on depression and anxiety. J Clin Psychiatry. 2004;65(suppl 1):55-62.
- Ursano RJ, Bell C, Eth S, et al. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Am J Psychiatry. 2004;161(11 suppl):3-31.
- Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based guidelines for the pharmacological treatment of anxiety disorders: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2005;19(6):567-596.
Pharmacokinetic properties
Most benzodiazepines are considered to have similar efficacy; therefore, selection is based on pharmacokinetic considerations. Table 1 compares the indication, onset, and half-life of 12 commonly used benzodiazepines.2-6 Although Table 1 lists approximate equivalent doses, studies report inconsistent data. These are approximations only and should not be used independently to make therapy decisions.
Table 1
Oral benzodiazepines: Indications, onset, half-life, and equivalent doses
| Drug | FDA-approved indication(s) | Onset of action | Approximate half-life (hours) in healthy adults | Approximate equivalent dose (mg)a | Comments |
|---|---|---|---|---|---|
| Alprazolam | Anxiety disorders, panic disorder | Intermediate | 6.3 to 26.9 (IR), 10.7 to 15.8 (XR) | 0.5 | Increased risk for abuse because of greater lipid solubility |
| Chlordiazepoxide | Anxiety disorders, acute alcohol withdrawal, preoperative apprehension and anxiety | Intermediate | 24 to 48 | 10 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Clonazepam | Seizure disorders, panic disorder | Intermediate | 18 to 50 | 0.25 to 0.5 | Use caution in patients with liver disease |
| Clorazepate | Anxiety, seizures, acute alcohol withdrawal | Fast | 40 to 50 | 7.5 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Diazepam | Anxiety disorders, acute alcohol withdrawal, muscle spasms, convulsive disorders | Very fast | 20 to 100 | 5 | Risk for accumulation because of long-acting metabolites (temazepam, desmethyldiazepam, oxazepam). Increased risk for abuse because of quick onset |
| Estazolam | Insomnia | Intermediate | 10 to 24 | 0.3 to 2 | None |
| Flurazepam | Insomnia | Intermediate | 47 to 100 | 30 | Avoid in geriatric patients or patients with liver impairment |
| Lorazepam | Anxiety | Intermediate | 10 to 20 | 1 | Preferred for patients with liver impairment and geriatric patients |
| Oxazepam | Anxiety, acute alcohol withdrawal | Slow to intermediate | 5 to 20 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Quazepam | Insomnia | Intermediate | 39 to 73 | 5 to 15 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Temazepam | Insomnia | Intermediate | 3.5 to 18.4 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Triazolam | Insomnia | Fast | 1.5 to 5.5 | 0.25 | Lacks active metabolites |
| IR: immediate release; XR: extended release aInterpret with caution, conflicting data exist Source: References 2-6 | |||||
A diverse range of indications
Alcohol withdrawal. Benzodiazepines are the treatment of choice for alcohol withdrawal syndrome, particularly to prevent seizures.7 Research supports symptom-triggered therapy using the revised Clinical Institute Withdrawal Assessment for Alcohol. Benzodiazepines reduce CNS sympathetic hyperactivity to mitigate withdrawal from alcohol by decreasing tachycardia, tremor, insomnia, agitation, and anxiety. Furthermore, these agents provide prophylaxis against serious sequelae such as seizures and delirium.
Insomnia. The American Academy of Sleep Medicine considers benzodiazepine receptor agonists (BzRAs, which include benzodiazepines and non-benzodiazepines) and ramelteon first-line pharmacotherapy for primary insomnia.8 However, pharmacologic treatment should be short-term. Agents with short to intermediate half-lives and rapid onset, such as triazolam, can aid sleep initiation. Those with longer half-lives, such as temazepam, could address sleep maintenance. If a patient does not respond to the initial agent, try another medication within the same class, because patients may respond differently. Use lower starting doses in geriatric patients.9 Closely monitor for adverse effects, rebound insomnia, and potential abuse or tolerance. Identify comorbid conditions and medications that may impair sleep, and address them accordingly.
Psychological and behavioral treatments given over 4 to 8 weeks can yield stable sleep improvements for up to 2 years. If available, these interventions may be considered first-line for treating insomnia because of their lasting effects compared with BzRAs.10
Generalized anxiety disorder (GAD). Benzodiazepines effectively treat GAD because they work quickly and are well tolerated. However, there are better first-line treatment options when considering efficacy studies and dependence and tolerance concerns. One effect-size comparison of 21 double-blind, placebo-controlled trials showed that the efficacy of selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), and pregabalin are comparable to benzodiazepines.11 Benzodiazepines can be used in the first 2 to 3 weeks after initiating antidepressants to alleviate and prevent worsening of anxiety that may occur at the start of antidepressant therapy. Recent treatment guidelines recommend benzodiazepines as a second-line treatment or for treatment-resistant GAD in patients who do not have a substance abuse history.12,13
Panic disorder. Efficacy of benzodiazepines for panic disorder is comparable to SSRIs, SNRIs, and tricyclic antidepressants (TCAs). SSRIs and SNRIs are considered first-line treatments for panic disorder because of their favorable side effect profile.14 In practice, benzodiazepines often are combined with SSRIs, SNRIs, or TCAs. A randomized controlled trial demonstrated that paroxetine and clonazepam (mean dose 1.6 mg/d at 5 weeks) resulted in a more rapid response compared with paroxetine alone, although this difference lasted only a few weeks.15 Furthermore, this study suggested that brief treatment with clonazepam followed by a taper is as effective as sustained treatment with paroxetine and clonazepam.15
There is a lack of high-quality data on combining cognitive-behavioral therapy (CBT) and benzodiazepines for panic disorder, although a Cochrane Review found that adding a benzodiazepine to CBT did not lead to a significant difference in response compared with psychotherapy alone.16 A recent randomized controlled trial demonstrated that tapering benzodiazepines combined with CBT was associated with successful discontinuation of the drug and prevented return of panic symptoms.17
Social anxiety. A meta-analysis found that for treating social anxiety, benzodiazepines have better efficacy than SSRIs, monoamine oxidase inhibitors, and anticonvulsants.18 Longer-acting benzodiazepines may be more effective than shorter-acting agents. One study of patients with social anxiety showed a 38% response rate for alprazolam vs 20% for placebo over 12 weeks, and a similar 10-week study demonstrated a 73% recovery rate with clonazepam vs 22% for placebo.19 In addition, studies have observed that patients can be maintained on clonazepam for up to 2 years without symptom relapse and will tolerate slow-taper discontinuation.18,20 Sedation and drowsiness can be lessened by limiting clonazepam doses to 2 to 3 mg/d.
Akathisia and tremor. Akathisia, a syndrome of motor restlessness and inner turmoil, is associated with antipsychotics but can occur with SSRIs. Reducing the dosage or switching to another, usually less potent agent often can relieve akathisia. When these remedies are not tenable, consider benzodiazepines along with other medications—including beta blockers and anticholinergic agents—with demonstrated efficacy in reducing akathisia symptoms. Lorazepam, diazepam, and clonazepam have demonstrated efficacy for relieving akathisia in comparison studies with placebo, propranolol, and diphenhydramine.21,22
Drug-induced postural tremor can occur with several psychotropics, including lithium, valproic acid, antidepressants, and antipsychotics. A tremor is considered mild if a patient can drink a glass of water with 1 hand without spilling and severe if holding a glass with 2 hands is difficult. Propranolol is most commonly prescribed for these tremors, but alprazolam and clonazepam have demonstrated efficacy, either as monotherapy or coadministered with a beta blocker.23
Acute agitation. Agitated patients often have acute psychosis and/or mania or dyscontrol secondary to axis II disorders.24 Patients may be paranoid, hostile, disruptive, and combative. Rapidly initiating medication can prevent the need for more restrictive measures, such as seclusion or restraint. Antipsychotics—especially high-potency agents such as haloperidol—and benzodiazepines, as monotherapy or in combination, are a mainstay treatment. Although treatment protocols favor atypical antipsychotics over typical antipsychotics, benzodiazepines are a viable option because of their anxiolytic and sedative effects. Advantages of benzodiazepine monotherapy include decreased extrapyramidal symptoms, greater patient acceptance/preference, and increased sedation compared with antipsychotics. Lorazepam, 1 to 2 mg intramuscularly (IM) or orally, is well tolerated because of its favorable drug-drug interaction profile and lack of significant cardiac side effects. Benzodiazepines can cause respiratory depression in patients with chronic lung disease and additive sedation secondary to opiates, other sedatives/hypnotics, or alcohol. Behavioral disinhibition is rare and is associated with preexisting CNS pathology or mental retardation.25 The IM olanzapine package insert warns against coadministering IM lorazepam because of additive cardiorespiratory depressive effects and excessive somnolence.26
Catatonia. The characteristic symptoms of catatonia are immobility, negativism, muteness, and failure to eat or drink. Benzodiazepines improve these symptoms in approximately 70% to 80% of catatonic patients with affective disorders. Response rates are lower in catatonia in patients with schizophrenia.27 If catatonia in a patient with psychosis is missed, giving antipsychotics before benzodiazepines may worsen catatonic symptoms or precipitate neuroleptic malignant syndrome in some cases. When you suspect a patient has catatonia, start with lorazepam, 1 to 2 mg IV or IM, and examine the patient for diminishing catatonic signs within 1 to 2 hours. If catatonia signs lessen, begin regularly scheduled lorazepam, with dosing varying by age—be more cautious in geriatric patients—and symptom severity. Titrate benzodiazepines for stuporous patients more slowly (eg, 1 mg 3 times a day as a starting dose) than for excited catatonic patients. Lorazepam can be increased gradually as tolerated; it is not unusual for patients to require up to 8 to 12 mg/d. Electroconvulsive therapy (ECT) is the treatment of choice when catatonic patients respond poorly or partially to high-dose benzodiazepines.28,29
Benzodiazepine reversal for ECT
Benzodiazepines have anticonvulsant properties that may interfere with the therapeutic efficacy of ECT.30 A multi-center study demonstrated that lorazepam (up to 4 mg/d as needed) in the 48 hours before the first ECT session was not associated with effects on seizure threshold or duration; however, larger lorazepam dosages were associated with briefer EEG seizure duration.31 Some patients may not tolerate withholding or tapering benzodiazepines in preparation for ECT. Studies investigating flumazenil for pre-ECT benzodiazepine reversal are lacking. One retrospective analysis showed that flumazenil administration immediately before and after ECT resulted in adequate seizures with no difference in clinical outcome compared with patients who were not receiving benzodiazepines or flumazenil.32
Tapering benzodiazepines
Slow discontinuation of benzodiazepines is recommended to avoid withdrawal symptoms, such as rebound anxiety, agitation, insomnia, or seizures, particularly when use exceeds 8 weeks. The onset of withdrawal symptoms varies, depending on the medication used. Withdrawal symptoms may appear in 1 to 2 days for agents with shorter half-lives, but may not appear until 3 to 7 days for agents with longer half-lives.33Table 2 lists recommended durations for tapering benzodiazepines.33,34 In general, decrease the total daily dose by 25% the first week, another 25% the second week, then 10% a week until discontinuation. When benzodiazepine use exceeds 1 year, a slower taper is recommended; for example, decrease 10% every 1 to 2 weeks. When 20% of the dosage remains, begin a 5% dose reduction every 2 to 4 weeks. Monitor patients for withdrawal symptoms or symptom exacerbation. If either occur, consider maintaining the current benzodiazepine dose or increasing the dose for 1 to 2 weeks or longer, if necessary, then continue to taper at a slower rate.34
Table 2
Recommendations for tapering benzodiazepines
| Duration of use | Recommended taper length | Comments |
| <6 to 8 weeks | Taper may not be required | Depending on clinical judgment and patient stability/preference, consider implementing a taper, particularly if using a high-dose benzodiazepine or an agent with a short or intermediate half-life, such as alprazolam or triazolam |
| 8 weeks to 6 months | Slowly over 2 to 3 weeks | Go slower during latter half of taper. Tapering will reduce, not eliminate, withdrawal symptoms. Patients should avoid alcohol and stimulants during benzodiazepine withdrawal |
| 6 months to 1 year | Slowly over 4 to 8 weeks | |
| >1 year | Slowly over 2 to 4 months | |
| Source: References 33,34 | ||
Risks of benzodiazepine use
For most indications, benzodiazepine therapy should be short-term.35 Use exceeding 2 to 4 weeks increases the risk for dependence and withdrawal. Tell patients to avoid alcohol while taking a benzodiazepine because this combination is potentially lethal. Benzodiazepines are commonly abused and abuse can lead to unintentional drug overdose. Benzodiazepines accounted for 37% of unintentional drug overdose deaths in West Virginia in 2006; in 46% of these cases, benzodiazepines were used for nonmedical purposes. Clinicians can help reduce the risk of diversion by limiting prescriptions to 30 days with no refills.36
Older patients taking benzodiazepines are at increased risk of falls and hip fractures.37 Lorazepam, oxazepam, and temazepam—agents with shorter half-lives that are not greatly affected by pharmacokinetic changes associated with aging—are preferred for these patients.34 Patients with dementia or other CNS-compromising conditions may become confused or delirious with regular benzodiazepine dosing. Educate patients to whom you prescribe benzodiazepines about the importance of gauging their level of sedation before driving or engaging in other tasks for which sedation could compromise their safety. Benzodiazepine use during pregnancy requires a careful discussion of risks and benefits (Box 2).38
Benzodiazepine use during pregnancy has been associated with cleft palate and urogenital and neurologic malformations in the fetus.38 Although data are conflicting—particularly among recent meta-analyses that fail to demonstrate an association—some experts advise against benzodiazepine use in the first trimester. Participate in shared decision making with your patients and educate them about the potential risks and benefits of benzodiazepine use during the first trimester and throughout pregnancy. After delivery, newborns may develop “floppy baby syndrome”—which is associated with lethargy, difficulty eating, and respiratory depression—or withdrawal.38 To minimize this risk, consider tapering the benzodiazepine as the patient approaches delivery.
Related Resources
- Substance Abuse and Mental Health Services Administration. www.samhsa.gov.
- National Institute on Drug Abuse resources for medical and health professionals. www.drugabuse.gov/medical-health-professionals.
- American Academy of Sleep Medicine. www.aasmnet.org.
Drug Brand Names
- Alprazolam • Xanax
- Chlordiazepoxide • Librium, Limbitrol
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Diazepam • Valium
- Diphenhydramine • Benadryl, others
- Estazolam • ProSom
- Flumazenil • Romazicon
- Flurazepam • Dalmane
- Haloperidol • Haldol
- Lithium • Lithobid
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Oxazepam • Serax
- Paroxetine • Paxil
- Pregabalin • Lyrica
- Propranolol • Inderal, InnoPran XL, others
- Quazepam • Doral
- Ramelteon • Rozerem
- Sertraline • Zoloft
- Temazepam • Restoril
- Triazolam • Halcion
- Valproic acid • Depakene, Stavzor, others
Disclosures
Drs. Bostwick and Yasugi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Casher is a speaker for AstraZeneca and Sunovion Pharmaceuticals.
1. Mihic SJ, Harris RA. Hypnotics and sedatives. In: Brunton LL Chabner BA, Knollmann BC, eds. Goodman & Gilman’s the pharmacological basis of therapeutics. New York, NY: McGraw Hill and Company; 2011:457-480.
2. Facts and comparisons Web site. 2011 Wolters Kluwer Health Inc. http://online.factsandcomparisons.com. Accessed August 16, 2011.
3. DuPont RL, Greene W, Lydiard RB. Sedatives and hypnotics: pharmacology and epidemiology. In: Gold MS Hermann R, eds. UpToDate. http://www.uptodate.com/contents/sedatives-and-hypnotics-abuse-and-dependence-pharmacology-and-epidemiology. Accessed August 16, 2011.
4. U.S. Food and Drug Administration. Orange book: approved drug products with therapeutic equivalence evaluations. http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm. Accessed August 16, 2011.
5. Chouinard G. Issues in the clinical use of benzodiazepines: potency withdrawal, and rebound. J Clin Psychiatry. 2004;65(suppl 5):7-12.
6. Shader RI, Greenblatt DJ. Can you provide a table of equivalencies for benzodiazepines and other marketed benzodiazepine receptor agonists? J Clin Psychopharmacol. 1997;17(4):331.-
7. Amato L, Minozzi S, Davoli M. Efficacy and safety of pharmacologic interventions for the treatment of the alcohol withdrawal syndrome. Cochrane Database Syst Rev. 2011;15(6):CD008537.-
8. Schutte-Rodin S, Broch L, Buysse D, et al. Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med. 2008;4(5):487-504.
9. Foral P, Dewan N, Malesker M. Insomnia: a therapeutic review for pharmacists. Consult Pharm. 2011;26(5):332-341.
10. Riemann D, Perlis ML. The treatments of chronic insomnia: a review of benzodiazepine receptor agonists and psychological and behavioral therapies. Sleep Med Rev. 2009;13(3):205-214.
11. Hidalgo RB, Tupler LA, Davidson JR. An effect-size analysis of pharmacologic treatments of generalized anxiety disorder. J Psychopharmacol. 2007;21(8):864-872.
12. Davidson JR, Zhang W, Connor KM, et al. A psychopharmacological treatment algorithm for generalised anxiety disorder (GAD). J Psychopharmacol. 2010;24(1):3-26.
13. Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
14. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Arlington VA: American Psychiatric Publishing, Inc.; 2009.
15. Pollack MH, Simon NM, Worthington JJ, et al. Combined paroxetine and clonazepam treatment strategies compared to paroxetine monotherapy for panic disorder. J Psychopharmacol. 2003;17(3):276-282.
16. Watanabe N, Churchill R, Furukawa TA. Combined psychotherapy plus benzodiazepines for panic disorder. Cochrane Database Syst Rev. 2009;(1):CD005335.-
17. Otto MW, McHugh RK, Simon NM, et al. Efficacy of CBT for benzodiazepine discontinuation in patients with panic disorder: further evaluation. Behav Res Ther. 2010;48(8):720-727.
18. Davidson JR. Use of benzodiazepines in social anxiety disorder generalized anxiety disorder, and posttraumatic stress disorder. J Clin Psychiatry. 2004;65(suppl 5):29-33.
19. Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
20. Connor KM, Davidson JR, Potts NL, et al. Discontinuation of clonazepam in the treatment of social phobia. J Clin Psychopharmacol. 1998;18(5):373-378.
21. Miller CH, Fleischhacker WW. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
22. Rodnitzky RL. Drug-induced movement disorders. Clin Neuropharmacol. 2002;25(3):142-151.
23. Arbaizar B, Gómez-Acebo I, Llorca J. Postural induced tremor in psychiatry. Psychiatry Clin Neurosci. 2008;62(6):638-645.
24. Casher MI, Bess JD. Manual of inpatient psychiatry. New York NY: Cambridge University Press; 2010.
25. Battaglia J. Pharmacological management of acute agitation. Drugs. 2005;65(9):1207-1222.
26. Physicians’ desk reference. Montvale NJ: PDR Network, LLC; 2010.
27. Rosebush PI, Mazurek MF. Catatonia and its treatment. Schizophr Bull. 2010;36(2):239-242.
28. Ungvari GS, Kau LS, Wai-Kwong T, et al. The pharmacological treatment of catatonia: an overview. Eur Arch Psychiatry Clin Neurosci. 2001;251(suppl 1):I31-I34.
29. Fink M, Taylor MA. Catatonia: a clinician’s guide to diagnosis and treatment. New York NY: Cambridge University Press; 2003.
30. Naguib N, Koorn R. Interactions between psychotropics anaesthetics and electroconvulsive therapy: implications for drug choice and patient management. CNS Drugs. 2002;16(4):229-247.
31. Boylan LS, Haskett RF, Mulsant BH, et al. Determinants of seizure threshold in ECT: benzodiazepine use, anesthetic dosage, and other factors. J ECT. 2000;16(1):3-18.
32. Krystal AD, Watts BV, Weiner RD, et al. The use of flumazenil in the anxious and benzodiazepine-dependent ECT patient. J ECT. 1998;14(1):5-14.
33. Melton ST, Kirkwood CK. Anxiety disorders I: generalized anxiety panic, and social anxiety disorders. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. New York, NY: McGraw-Hill Companies; 2011:1209-1228.
34. Benzodiazepine toolkit. The Pharmacist’s Letter/Prescriber’s Letter. 2011;27(4):270406.-
35. Lader M. Benzodiazepines revisited – will we ever learn? Addiction. 2011;106(12):2086-2109.
36. Toblin RL, Paulozzi LJ, Logan JE, et al. Mental illness and psychotropic drug use among prescription drug overdose deaths: a medical examiner chart review. J Clin Psychiatry. 2010;71(4):491-496.
37. Ashton H. The diagnosis and management of benzodiazepine dependence. Curr Opin Psychiatry. 2005;18(3):249-255.
38. Menon SJ. Psychotropic medication during pregnancy and lactation. Arch Gynecol Obstet. 2008;277(1):1-13.
Since the discovery of chlordiazepoxide in the 1950s, benzodiazepines have revolutionized the treatment of anxiety and insomnia, largely because of their improved safety profile compared with barbiturates, formerly the preferred sedative-hypnotic.1 In addition to their anxiolytic and sedative-hypnotic effects, benzodiazepines exhibit anterograde amnesia, anticonvulsant, and muscle relaxant properties.1 Psychiatrists use benzodiazepines to treat anxiety and sleep disorders, acute agitation, alcohol withdrawal, catatonia, and psychotropic side effects such as akathisia. This article highlights the evidence for using benzodiazepines in anxiety and other disorders and why they generally should not be used for obsessive-compulsive disorder and posttraumatic stress disorder (Box 1).
Current evidence indicates little support for using benzodiazepines for obsessive-compulsive disorder (OCD). The American Psychiatric Association (APA) and the World Federation of Biological Psychiatry do not recommend benzodiazepines for treating OCD because of a lack of evidence for efficacy.a,b An earlier study suggested clonazepam monotherapy was effective for OCDc; however, a more recent study did not show a benefit on rate of response or degree of symptom improvement.d Augmentation strategies with benzodiazepines also do not appear to be beneficial for OCD management. A recent double-blind, placebo-controlled study failed to demonstrate faster symptom improvement by augmenting sertraline with clonazepam, although the study had a small sample size and high drop-out rate.e
Because benzodiazepines have negligible action on core posttraumatic stress disorder (PTSD) symptoms (re-experiencing, avoidance, and hyperarousal), selective serotonin reuptake inhibitors and other agents largely have supplanted them for PTSD treatment.f Use of benzodiazepines for PTSD is associated with withdrawal symptoms, more severe symptoms after discontinuation, and possible disinhibition, and may interfere with patients’ efforts to integrate trauma experiences. Although benzodiazepines may reduce distress associated with acute trauma, there is evidence—in clinical studies and animal models—that early benzodiazepine administration fails to prevent PTSD and may increase its incidence.g The International Consensus Group on Depression and Anxiety, the APA, and the British Association for Psychopharmacology all highlight the limited role, if any, for benzodiazepines in PTSD.h-j
References
- Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
- American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Arlington, VA: American Psychiatric Publishing, Inc.; 2007.
- Hewlett WA, Vinogradov S, Agras WS. Clomipramine, clonazepam, and clonidine treatment of obsessive compulsive disorder. J Clin Psychopharmacol. 1992;12(6):420-430.
- Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
- Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam and sertraline in OCD. Ann Clin Psychiatry. 2004;16(3):127-132.
- Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs. Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
- Matar MA, Zohar J, Kaplan Z, et al. Alprazolam treatment immediately after stress exposure interferes with the normal HPA-stress response and increases vulnerability to subsequent stress in an animal model of PTSD. Eur Neuropsychopharmacol. 2009;19(4):283-295.
- Ballenger JC, Davidson JR, Lecrubier Y, et al. Consensus statement update on posttraumatic stress disorder from the international consensus group on depression and anxiety. J Clin Psychiatry. 2004;65(suppl 1):55-62.
- Ursano RJ, Bell C, Eth S, et al. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Am J Psychiatry. 2004;161(11 suppl):3-31.
- Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based guidelines for the pharmacological treatment of anxiety disorders: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2005;19(6):567-596.
Pharmacokinetic properties
Most benzodiazepines are considered to have similar efficacy; therefore, selection is based on pharmacokinetic considerations. Table 1 compares the indication, onset, and half-life of 12 commonly used benzodiazepines.2-6 Although Table 1 lists approximate equivalent doses, studies report inconsistent data. These are approximations only and should not be used independently to make therapy decisions.
Table 1
Oral benzodiazepines: Indications, onset, half-life, and equivalent doses
| Drug | FDA-approved indication(s) | Onset of action | Approximate half-life (hours) in healthy adults | Approximate equivalent dose (mg)a | Comments |
|---|---|---|---|---|---|
| Alprazolam | Anxiety disorders, panic disorder | Intermediate | 6.3 to 26.9 (IR), 10.7 to 15.8 (XR) | 0.5 | Increased risk for abuse because of greater lipid solubility |
| Chlordiazepoxide | Anxiety disorders, acute alcohol withdrawal, preoperative apprehension and anxiety | Intermediate | 24 to 48 | 10 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Clonazepam | Seizure disorders, panic disorder | Intermediate | 18 to 50 | 0.25 to 0.5 | Use caution in patients with liver disease |
| Clorazepate | Anxiety, seizures, acute alcohol withdrawal | Fast | 40 to 50 | 7.5 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Diazepam | Anxiety disorders, acute alcohol withdrawal, muscle spasms, convulsive disorders | Very fast | 20 to 100 | 5 | Risk for accumulation because of long-acting metabolites (temazepam, desmethyldiazepam, oxazepam). Increased risk for abuse because of quick onset |
| Estazolam | Insomnia | Intermediate | 10 to 24 | 0.3 to 2 | None |
| Flurazepam | Insomnia | Intermediate | 47 to 100 | 30 | Avoid in geriatric patients or patients with liver impairment |
| Lorazepam | Anxiety | Intermediate | 10 to 20 | 1 | Preferred for patients with liver impairment and geriatric patients |
| Oxazepam | Anxiety, acute alcohol withdrawal | Slow to intermediate | 5 to 20 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Quazepam | Insomnia | Intermediate | 39 to 73 | 5 to 15 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Temazepam | Insomnia | Intermediate | 3.5 to 18.4 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Triazolam | Insomnia | Fast | 1.5 to 5.5 | 0.25 | Lacks active metabolites |
| IR: immediate release; XR: extended release aInterpret with caution, conflicting data exist Source: References 2-6 | |||||
A diverse range of indications
Alcohol withdrawal. Benzodiazepines are the treatment of choice for alcohol withdrawal syndrome, particularly to prevent seizures.7 Research supports symptom-triggered therapy using the revised Clinical Institute Withdrawal Assessment for Alcohol. Benzodiazepines reduce CNS sympathetic hyperactivity to mitigate withdrawal from alcohol by decreasing tachycardia, tremor, insomnia, agitation, and anxiety. Furthermore, these agents provide prophylaxis against serious sequelae such as seizures and delirium.
Insomnia. The American Academy of Sleep Medicine considers benzodiazepine receptor agonists (BzRAs, which include benzodiazepines and non-benzodiazepines) and ramelteon first-line pharmacotherapy for primary insomnia.8 However, pharmacologic treatment should be short-term. Agents with short to intermediate half-lives and rapid onset, such as triazolam, can aid sleep initiation. Those with longer half-lives, such as temazepam, could address sleep maintenance. If a patient does not respond to the initial agent, try another medication within the same class, because patients may respond differently. Use lower starting doses in geriatric patients.9 Closely monitor for adverse effects, rebound insomnia, and potential abuse or tolerance. Identify comorbid conditions and medications that may impair sleep, and address them accordingly.
Psychological and behavioral treatments given over 4 to 8 weeks can yield stable sleep improvements for up to 2 years. If available, these interventions may be considered first-line for treating insomnia because of their lasting effects compared with BzRAs.10
Generalized anxiety disorder (GAD). Benzodiazepines effectively treat GAD because they work quickly and are well tolerated. However, there are better first-line treatment options when considering efficacy studies and dependence and tolerance concerns. One effect-size comparison of 21 double-blind, placebo-controlled trials showed that the efficacy of selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), and pregabalin are comparable to benzodiazepines.11 Benzodiazepines can be used in the first 2 to 3 weeks after initiating antidepressants to alleviate and prevent worsening of anxiety that may occur at the start of antidepressant therapy. Recent treatment guidelines recommend benzodiazepines as a second-line treatment or for treatment-resistant GAD in patients who do not have a substance abuse history.12,13
Panic disorder. Efficacy of benzodiazepines for panic disorder is comparable to SSRIs, SNRIs, and tricyclic antidepressants (TCAs). SSRIs and SNRIs are considered first-line treatments for panic disorder because of their favorable side effect profile.14 In practice, benzodiazepines often are combined with SSRIs, SNRIs, or TCAs. A randomized controlled trial demonstrated that paroxetine and clonazepam (mean dose 1.6 mg/d at 5 weeks) resulted in a more rapid response compared with paroxetine alone, although this difference lasted only a few weeks.15 Furthermore, this study suggested that brief treatment with clonazepam followed by a taper is as effective as sustained treatment with paroxetine and clonazepam.15
There is a lack of high-quality data on combining cognitive-behavioral therapy (CBT) and benzodiazepines for panic disorder, although a Cochrane Review found that adding a benzodiazepine to CBT did not lead to a significant difference in response compared with psychotherapy alone.16 A recent randomized controlled trial demonstrated that tapering benzodiazepines combined with CBT was associated with successful discontinuation of the drug and prevented return of panic symptoms.17
Social anxiety. A meta-analysis found that for treating social anxiety, benzodiazepines have better efficacy than SSRIs, monoamine oxidase inhibitors, and anticonvulsants.18 Longer-acting benzodiazepines may be more effective than shorter-acting agents. One study of patients with social anxiety showed a 38% response rate for alprazolam vs 20% for placebo over 12 weeks, and a similar 10-week study demonstrated a 73% recovery rate with clonazepam vs 22% for placebo.19 In addition, studies have observed that patients can be maintained on clonazepam for up to 2 years without symptom relapse and will tolerate slow-taper discontinuation.18,20 Sedation and drowsiness can be lessened by limiting clonazepam doses to 2 to 3 mg/d.
Akathisia and tremor. Akathisia, a syndrome of motor restlessness and inner turmoil, is associated with antipsychotics but can occur with SSRIs. Reducing the dosage or switching to another, usually less potent agent often can relieve akathisia. When these remedies are not tenable, consider benzodiazepines along with other medications—including beta blockers and anticholinergic agents—with demonstrated efficacy in reducing akathisia symptoms. Lorazepam, diazepam, and clonazepam have demonstrated efficacy for relieving akathisia in comparison studies with placebo, propranolol, and diphenhydramine.21,22
Drug-induced postural tremor can occur with several psychotropics, including lithium, valproic acid, antidepressants, and antipsychotics. A tremor is considered mild if a patient can drink a glass of water with 1 hand without spilling and severe if holding a glass with 2 hands is difficult. Propranolol is most commonly prescribed for these tremors, but alprazolam and clonazepam have demonstrated efficacy, either as monotherapy or coadministered with a beta blocker.23
Acute agitation. Agitated patients often have acute psychosis and/or mania or dyscontrol secondary to axis II disorders.24 Patients may be paranoid, hostile, disruptive, and combative. Rapidly initiating medication can prevent the need for more restrictive measures, such as seclusion or restraint. Antipsychotics—especially high-potency agents such as haloperidol—and benzodiazepines, as monotherapy or in combination, are a mainstay treatment. Although treatment protocols favor atypical antipsychotics over typical antipsychotics, benzodiazepines are a viable option because of their anxiolytic and sedative effects. Advantages of benzodiazepine monotherapy include decreased extrapyramidal symptoms, greater patient acceptance/preference, and increased sedation compared with antipsychotics. Lorazepam, 1 to 2 mg intramuscularly (IM) or orally, is well tolerated because of its favorable drug-drug interaction profile and lack of significant cardiac side effects. Benzodiazepines can cause respiratory depression in patients with chronic lung disease and additive sedation secondary to opiates, other sedatives/hypnotics, or alcohol. Behavioral disinhibition is rare and is associated with preexisting CNS pathology or mental retardation.25 The IM olanzapine package insert warns against coadministering IM lorazepam because of additive cardiorespiratory depressive effects and excessive somnolence.26
Catatonia. The characteristic symptoms of catatonia are immobility, negativism, muteness, and failure to eat or drink. Benzodiazepines improve these symptoms in approximately 70% to 80% of catatonic patients with affective disorders. Response rates are lower in catatonia in patients with schizophrenia.27 If catatonia in a patient with psychosis is missed, giving antipsychotics before benzodiazepines may worsen catatonic symptoms or precipitate neuroleptic malignant syndrome in some cases. When you suspect a patient has catatonia, start with lorazepam, 1 to 2 mg IV or IM, and examine the patient for diminishing catatonic signs within 1 to 2 hours. If catatonia signs lessen, begin regularly scheduled lorazepam, with dosing varying by age—be more cautious in geriatric patients—and symptom severity. Titrate benzodiazepines for stuporous patients more slowly (eg, 1 mg 3 times a day as a starting dose) than for excited catatonic patients. Lorazepam can be increased gradually as tolerated; it is not unusual for patients to require up to 8 to 12 mg/d. Electroconvulsive therapy (ECT) is the treatment of choice when catatonic patients respond poorly or partially to high-dose benzodiazepines.28,29
Benzodiazepine reversal for ECT
Benzodiazepines have anticonvulsant properties that may interfere with the therapeutic efficacy of ECT.30 A multi-center study demonstrated that lorazepam (up to 4 mg/d as needed) in the 48 hours before the first ECT session was not associated with effects on seizure threshold or duration; however, larger lorazepam dosages were associated with briefer EEG seizure duration.31 Some patients may not tolerate withholding or tapering benzodiazepines in preparation for ECT. Studies investigating flumazenil for pre-ECT benzodiazepine reversal are lacking. One retrospective analysis showed that flumazenil administration immediately before and after ECT resulted in adequate seizures with no difference in clinical outcome compared with patients who were not receiving benzodiazepines or flumazenil.32
Tapering benzodiazepines
Slow discontinuation of benzodiazepines is recommended to avoid withdrawal symptoms, such as rebound anxiety, agitation, insomnia, or seizures, particularly when use exceeds 8 weeks. The onset of withdrawal symptoms varies, depending on the medication used. Withdrawal symptoms may appear in 1 to 2 days for agents with shorter half-lives, but may not appear until 3 to 7 days for agents with longer half-lives.33Table 2 lists recommended durations for tapering benzodiazepines.33,34 In general, decrease the total daily dose by 25% the first week, another 25% the second week, then 10% a week until discontinuation. When benzodiazepine use exceeds 1 year, a slower taper is recommended; for example, decrease 10% every 1 to 2 weeks. When 20% of the dosage remains, begin a 5% dose reduction every 2 to 4 weeks. Monitor patients for withdrawal symptoms or symptom exacerbation. If either occur, consider maintaining the current benzodiazepine dose or increasing the dose for 1 to 2 weeks or longer, if necessary, then continue to taper at a slower rate.34
Table 2
Recommendations for tapering benzodiazepines
| Duration of use | Recommended taper length | Comments |
| <6 to 8 weeks | Taper may not be required | Depending on clinical judgment and patient stability/preference, consider implementing a taper, particularly if using a high-dose benzodiazepine or an agent with a short or intermediate half-life, such as alprazolam or triazolam |
| 8 weeks to 6 months | Slowly over 2 to 3 weeks | Go slower during latter half of taper. Tapering will reduce, not eliminate, withdrawal symptoms. Patients should avoid alcohol and stimulants during benzodiazepine withdrawal |
| 6 months to 1 year | Slowly over 4 to 8 weeks | |
| >1 year | Slowly over 2 to 4 months | |
| Source: References 33,34 | ||
Risks of benzodiazepine use
For most indications, benzodiazepine therapy should be short-term.35 Use exceeding 2 to 4 weeks increases the risk for dependence and withdrawal. Tell patients to avoid alcohol while taking a benzodiazepine because this combination is potentially lethal. Benzodiazepines are commonly abused and abuse can lead to unintentional drug overdose. Benzodiazepines accounted for 37% of unintentional drug overdose deaths in West Virginia in 2006; in 46% of these cases, benzodiazepines were used for nonmedical purposes. Clinicians can help reduce the risk of diversion by limiting prescriptions to 30 days with no refills.36
Older patients taking benzodiazepines are at increased risk of falls and hip fractures.37 Lorazepam, oxazepam, and temazepam—agents with shorter half-lives that are not greatly affected by pharmacokinetic changes associated with aging—are preferred for these patients.34 Patients with dementia or other CNS-compromising conditions may become confused or delirious with regular benzodiazepine dosing. Educate patients to whom you prescribe benzodiazepines about the importance of gauging their level of sedation before driving or engaging in other tasks for which sedation could compromise their safety. Benzodiazepine use during pregnancy requires a careful discussion of risks and benefits (Box 2).38
Benzodiazepine use during pregnancy has been associated with cleft palate and urogenital and neurologic malformations in the fetus.38 Although data are conflicting—particularly among recent meta-analyses that fail to demonstrate an association—some experts advise against benzodiazepine use in the first trimester. Participate in shared decision making with your patients and educate them about the potential risks and benefits of benzodiazepine use during the first trimester and throughout pregnancy. After delivery, newborns may develop “floppy baby syndrome”—which is associated with lethargy, difficulty eating, and respiratory depression—or withdrawal.38 To minimize this risk, consider tapering the benzodiazepine as the patient approaches delivery.
Related Resources
- Substance Abuse and Mental Health Services Administration. www.samhsa.gov.
- National Institute on Drug Abuse resources for medical and health professionals. www.drugabuse.gov/medical-health-professionals.
- American Academy of Sleep Medicine. www.aasmnet.org.
Drug Brand Names
- Alprazolam • Xanax
- Chlordiazepoxide • Librium, Limbitrol
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Diazepam • Valium
- Diphenhydramine • Benadryl, others
- Estazolam • ProSom
- Flumazenil • Romazicon
- Flurazepam • Dalmane
- Haloperidol • Haldol
- Lithium • Lithobid
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Oxazepam • Serax
- Paroxetine • Paxil
- Pregabalin • Lyrica
- Propranolol • Inderal, InnoPran XL, others
- Quazepam • Doral
- Ramelteon • Rozerem
- Sertraline • Zoloft
- Temazepam • Restoril
- Triazolam • Halcion
- Valproic acid • Depakene, Stavzor, others
Disclosures
Drs. Bostwick and Yasugi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Casher is a speaker for AstraZeneca and Sunovion Pharmaceuticals.
Since the discovery of chlordiazepoxide in the 1950s, benzodiazepines have revolutionized the treatment of anxiety and insomnia, largely because of their improved safety profile compared with barbiturates, formerly the preferred sedative-hypnotic.1 In addition to their anxiolytic and sedative-hypnotic effects, benzodiazepines exhibit anterograde amnesia, anticonvulsant, and muscle relaxant properties.1 Psychiatrists use benzodiazepines to treat anxiety and sleep disorders, acute agitation, alcohol withdrawal, catatonia, and psychotropic side effects such as akathisia. This article highlights the evidence for using benzodiazepines in anxiety and other disorders and why they generally should not be used for obsessive-compulsive disorder and posttraumatic stress disorder (Box 1).
Current evidence indicates little support for using benzodiazepines for obsessive-compulsive disorder (OCD). The American Psychiatric Association (APA) and the World Federation of Biological Psychiatry do not recommend benzodiazepines for treating OCD because of a lack of evidence for efficacy.a,b An earlier study suggested clonazepam monotherapy was effective for OCDc; however, a more recent study did not show a benefit on rate of response or degree of symptom improvement.d Augmentation strategies with benzodiazepines also do not appear to be beneficial for OCD management. A recent double-blind, placebo-controlled study failed to demonstrate faster symptom improvement by augmenting sertraline with clonazepam, although the study had a small sample size and high drop-out rate.e
Because benzodiazepines have negligible action on core posttraumatic stress disorder (PTSD) symptoms (re-experiencing, avoidance, and hyperarousal), selective serotonin reuptake inhibitors and other agents largely have supplanted them for PTSD treatment.f Use of benzodiazepines for PTSD is associated with withdrawal symptoms, more severe symptoms after discontinuation, and possible disinhibition, and may interfere with patients’ efforts to integrate trauma experiences. Although benzodiazepines may reduce distress associated with acute trauma, there is evidence—in clinical studies and animal models—that early benzodiazepine administration fails to prevent PTSD and may increase its incidence.g The International Consensus Group on Depression and Anxiety, the APA, and the British Association for Psychopharmacology all highlight the limited role, if any, for benzodiazepines in PTSD.h-j
References
- Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
- American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Arlington, VA: American Psychiatric Publishing, Inc.; 2007.
- Hewlett WA, Vinogradov S, Agras WS. Clomipramine, clonazepam, and clonidine treatment of obsessive compulsive disorder. J Clin Psychopharmacol. 1992;12(6):420-430.
- Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
- Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam and sertraline in OCD. Ann Clin Psychiatry. 2004;16(3):127-132.
- Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs. Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
- Matar MA, Zohar J, Kaplan Z, et al. Alprazolam treatment immediately after stress exposure interferes with the normal HPA-stress response and increases vulnerability to subsequent stress in an animal model of PTSD. Eur Neuropsychopharmacol. 2009;19(4):283-295.
- Ballenger JC, Davidson JR, Lecrubier Y, et al. Consensus statement update on posttraumatic stress disorder from the international consensus group on depression and anxiety. J Clin Psychiatry. 2004;65(suppl 1):55-62.
- Ursano RJ, Bell C, Eth S, et al. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Am J Psychiatry. 2004;161(11 suppl):3-31.
- Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based guidelines for the pharmacological treatment of anxiety disorders: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2005;19(6):567-596.
Pharmacokinetic properties
Most benzodiazepines are considered to have similar efficacy; therefore, selection is based on pharmacokinetic considerations. Table 1 compares the indication, onset, and half-life of 12 commonly used benzodiazepines.2-6 Although Table 1 lists approximate equivalent doses, studies report inconsistent data. These are approximations only and should not be used independently to make therapy decisions.
Table 1
Oral benzodiazepines: Indications, onset, half-life, and equivalent doses
| Drug | FDA-approved indication(s) | Onset of action | Approximate half-life (hours) in healthy adults | Approximate equivalent dose (mg)a | Comments |
|---|---|---|---|---|---|
| Alprazolam | Anxiety disorders, panic disorder | Intermediate | 6.3 to 26.9 (IR), 10.7 to 15.8 (XR) | 0.5 | Increased risk for abuse because of greater lipid solubility |
| Chlordiazepoxide | Anxiety disorders, acute alcohol withdrawal, preoperative apprehension and anxiety | Intermediate | 24 to 48 | 10 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Clonazepam | Seizure disorders, panic disorder | Intermediate | 18 to 50 | 0.25 to 0.5 | Use caution in patients with liver disease |
| Clorazepate | Anxiety, seizures, acute alcohol withdrawal | Fast | 40 to 50 | 7.5 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Diazepam | Anxiety disorders, acute alcohol withdrawal, muscle spasms, convulsive disorders | Very fast | 20 to 100 | 5 | Risk for accumulation because of long-acting metabolites (temazepam, desmethyldiazepam, oxazepam). Increased risk for abuse because of quick onset |
| Estazolam | Insomnia | Intermediate | 10 to 24 | 0.3 to 2 | None |
| Flurazepam | Insomnia | Intermediate | 47 to 100 | 30 | Avoid in geriatric patients or patients with liver impairment |
| Lorazepam | Anxiety | Intermediate | 10 to 20 | 1 | Preferred for patients with liver impairment and geriatric patients |
| Oxazepam | Anxiety, acute alcohol withdrawal | Slow to intermediate | 5 to 20 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Quazepam | Insomnia | Intermediate | 39 to 73 | 5 to 15 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Temazepam | Insomnia | Intermediate | 3.5 to 18.4 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Triazolam | Insomnia | Fast | 1.5 to 5.5 | 0.25 | Lacks active metabolites |
| IR: immediate release; XR: extended release aInterpret with caution, conflicting data exist Source: References 2-6 | |||||
A diverse range of indications
Alcohol withdrawal. Benzodiazepines are the treatment of choice for alcohol withdrawal syndrome, particularly to prevent seizures.7 Research supports symptom-triggered therapy using the revised Clinical Institute Withdrawal Assessment for Alcohol. Benzodiazepines reduce CNS sympathetic hyperactivity to mitigate withdrawal from alcohol by decreasing tachycardia, tremor, insomnia, agitation, and anxiety. Furthermore, these agents provide prophylaxis against serious sequelae such as seizures and delirium.
Insomnia. The American Academy of Sleep Medicine considers benzodiazepine receptor agonists (BzRAs, which include benzodiazepines and non-benzodiazepines) and ramelteon first-line pharmacotherapy for primary insomnia.8 However, pharmacologic treatment should be short-term. Agents with short to intermediate half-lives and rapid onset, such as triazolam, can aid sleep initiation. Those with longer half-lives, such as temazepam, could address sleep maintenance. If a patient does not respond to the initial agent, try another medication within the same class, because patients may respond differently. Use lower starting doses in geriatric patients.9 Closely monitor for adverse effects, rebound insomnia, and potential abuse or tolerance. Identify comorbid conditions and medications that may impair sleep, and address them accordingly.
Psychological and behavioral treatments given over 4 to 8 weeks can yield stable sleep improvements for up to 2 years. If available, these interventions may be considered first-line for treating insomnia because of their lasting effects compared with BzRAs.10
Generalized anxiety disorder (GAD). Benzodiazepines effectively treat GAD because they work quickly and are well tolerated. However, there are better first-line treatment options when considering efficacy studies and dependence and tolerance concerns. One effect-size comparison of 21 double-blind, placebo-controlled trials showed that the efficacy of selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), and pregabalin are comparable to benzodiazepines.11 Benzodiazepines can be used in the first 2 to 3 weeks after initiating antidepressants to alleviate and prevent worsening of anxiety that may occur at the start of antidepressant therapy. Recent treatment guidelines recommend benzodiazepines as a second-line treatment or for treatment-resistant GAD in patients who do not have a substance abuse history.12,13
Panic disorder. Efficacy of benzodiazepines for panic disorder is comparable to SSRIs, SNRIs, and tricyclic antidepressants (TCAs). SSRIs and SNRIs are considered first-line treatments for panic disorder because of their favorable side effect profile.14 In practice, benzodiazepines often are combined with SSRIs, SNRIs, or TCAs. A randomized controlled trial demonstrated that paroxetine and clonazepam (mean dose 1.6 mg/d at 5 weeks) resulted in a more rapid response compared with paroxetine alone, although this difference lasted only a few weeks.15 Furthermore, this study suggested that brief treatment with clonazepam followed by a taper is as effective as sustained treatment with paroxetine and clonazepam.15
There is a lack of high-quality data on combining cognitive-behavioral therapy (CBT) and benzodiazepines for panic disorder, although a Cochrane Review found that adding a benzodiazepine to CBT did not lead to a significant difference in response compared with psychotherapy alone.16 A recent randomized controlled trial demonstrated that tapering benzodiazepines combined with CBT was associated with successful discontinuation of the drug and prevented return of panic symptoms.17
Social anxiety. A meta-analysis found that for treating social anxiety, benzodiazepines have better efficacy than SSRIs, monoamine oxidase inhibitors, and anticonvulsants.18 Longer-acting benzodiazepines may be more effective than shorter-acting agents. One study of patients with social anxiety showed a 38% response rate for alprazolam vs 20% for placebo over 12 weeks, and a similar 10-week study demonstrated a 73% recovery rate with clonazepam vs 22% for placebo.19 In addition, studies have observed that patients can be maintained on clonazepam for up to 2 years without symptom relapse and will tolerate slow-taper discontinuation.18,20 Sedation and drowsiness can be lessened by limiting clonazepam doses to 2 to 3 mg/d.
Akathisia and tremor. Akathisia, a syndrome of motor restlessness and inner turmoil, is associated with antipsychotics but can occur with SSRIs. Reducing the dosage or switching to another, usually less potent agent often can relieve akathisia. When these remedies are not tenable, consider benzodiazepines along with other medications—including beta blockers and anticholinergic agents—with demonstrated efficacy in reducing akathisia symptoms. Lorazepam, diazepam, and clonazepam have demonstrated efficacy for relieving akathisia in comparison studies with placebo, propranolol, and diphenhydramine.21,22
Drug-induced postural tremor can occur with several psychotropics, including lithium, valproic acid, antidepressants, and antipsychotics. A tremor is considered mild if a patient can drink a glass of water with 1 hand without spilling and severe if holding a glass with 2 hands is difficult. Propranolol is most commonly prescribed for these tremors, but alprazolam and clonazepam have demonstrated efficacy, either as monotherapy or coadministered with a beta blocker.23
Acute agitation. Agitated patients often have acute psychosis and/or mania or dyscontrol secondary to axis II disorders.24 Patients may be paranoid, hostile, disruptive, and combative. Rapidly initiating medication can prevent the need for more restrictive measures, such as seclusion or restraint. Antipsychotics—especially high-potency agents such as haloperidol—and benzodiazepines, as monotherapy or in combination, are a mainstay treatment. Although treatment protocols favor atypical antipsychotics over typical antipsychotics, benzodiazepines are a viable option because of their anxiolytic and sedative effects. Advantages of benzodiazepine monotherapy include decreased extrapyramidal symptoms, greater patient acceptance/preference, and increased sedation compared with antipsychotics. Lorazepam, 1 to 2 mg intramuscularly (IM) or orally, is well tolerated because of its favorable drug-drug interaction profile and lack of significant cardiac side effects. Benzodiazepines can cause respiratory depression in patients with chronic lung disease and additive sedation secondary to opiates, other sedatives/hypnotics, or alcohol. Behavioral disinhibition is rare and is associated with preexisting CNS pathology or mental retardation.25 The IM olanzapine package insert warns against coadministering IM lorazepam because of additive cardiorespiratory depressive effects and excessive somnolence.26
Catatonia. The characteristic symptoms of catatonia are immobility, negativism, muteness, and failure to eat or drink. Benzodiazepines improve these symptoms in approximately 70% to 80% of catatonic patients with affective disorders. Response rates are lower in catatonia in patients with schizophrenia.27 If catatonia in a patient with psychosis is missed, giving antipsychotics before benzodiazepines may worsen catatonic symptoms or precipitate neuroleptic malignant syndrome in some cases. When you suspect a patient has catatonia, start with lorazepam, 1 to 2 mg IV or IM, and examine the patient for diminishing catatonic signs within 1 to 2 hours. If catatonia signs lessen, begin regularly scheduled lorazepam, with dosing varying by age—be more cautious in geriatric patients—and symptom severity. Titrate benzodiazepines for stuporous patients more slowly (eg, 1 mg 3 times a day as a starting dose) than for excited catatonic patients. Lorazepam can be increased gradually as tolerated; it is not unusual for patients to require up to 8 to 12 mg/d. Electroconvulsive therapy (ECT) is the treatment of choice when catatonic patients respond poorly or partially to high-dose benzodiazepines.28,29
Benzodiazepine reversal for ECT
Benzodiazepines have anticonvulsant properties that may interfere with the therapeutic efficacy of ECT.30 A multi-center study demonstrated that lorazepam (up to 4 mg/d as needed) in the 48 hours before the first ECT session was not associated with effects on seizure threshold or duration; however, larger lorazepam dosages were associated with briefer EEG seizure duration.31 Some patients may not tolerate withholding or tapering benzodiazepines in preparation for ECT. Studies investigating flumazenil for pre-ECT benzodiazepine reversal are lacking. One retrospective analysis showed that flumazenil administration immediately before and after ECT resulted in adequate seizures with no difference in clinical outcome compared with patients who were not receiving benzodiazepines or flumazenil.32
Tapering benzodiazepines
Slow discontinuation of benzodiazepines is recommended to avoid withdrawal symptoms, such as rebound anxiety, agitation, insomnia, or seizures, particularly when use exceeds 8 weeks. The onset of withdrawal symptoms varies, depending on the medication used. Withdrawal symptoms may appear in 1 to 2 days for agents with shorter half-lives, but may not appear until 3 to 7 days for agents with longer half-lives.33Table 2 lists recommended durations for tapering benzodiazepines.33,34 In general, decrease the total daily dose by 25% the first week, another 25% the second week, then 10% a week until discontinuation. When benzodiazepine use exceeds 1 year, a slower taper is recommended; for example, decrease 10% every 1 to 2 weeks. When 20% of the dosage remains, begin a 5% dose reduction every 2 to 4 weeks. Monitor patients for withdrawal symptoms or symptom exacerbation. If either occur, consider maintaining the current benzodiazepine dose or increasing the dose for 1 to 2 weeks or longer, if necessary, then continue to taper at a slower rate.34
Table 2
Recommendations for tapering benzodiazepines
| Duration of use | Recommended taper length | Comments |
| <6 to 8 weeks | Taper may not be required | Depending on clinical judgment and patient stability/preference, consider implementing a taper, particularly if using a high-dose benzodiazepine or an agent with a short or intermediate half-life, such as alprazolam or triazolam |
| 8 weeks to 6 months | Slowly over 2 to 3 weeks | Go slower during latter half of taper. Tapering will reduce, not eliminate, withdrawal symptoms. Patients should avoid alcohol and stimulants during benzodiazepine withdrawal |
| 6 months to 1 year | Slowly over 4 to 8 weeks | |
| >1 year | Slowly over 2 to 4 months | |
| Source: References 33,34 | ||
Risks of benzodiazepine use
For most indications, benzodiazepine therapy should be short-term.35 Use exceeding 2 to 4 weeks increases the risk for dependence and withdrawal. Tell patients to avoid alcohol while taking a benzodiazepine because this combination is potentially lethal. Benzodiazepines are commonly abused and abuse can lead to unintentional drug overdose. Benzodiazepines accounted for 37% of unintentional drug overdose deaths in West Virginia in 2006; in 46% of these cases, benzodiazepines were used for nonmedical purposes. Clinicians can help reduce the risk of diversion by limiting prescriptions to 30 days with no refills.36
Older patients taking benzodiazepines are at increased risk of falls and hip fractures.37 Lorazepam, oxazepam, and temazepam—agents with shorter half-lives that are not greatly affected by pharmacokinetic changes associated with aging—are preferred for these patients.34 Patients with dementia or other CNS-compromising conditions may become confused or delirious with regular benzodiazepine dosing. Educate patients to whom you prescribe benzodiazepines about the importance of gauging their level of sedation before driving or engaging in other tasks for which sedation could compromise their safety. Benzodiazepine use during pregnancy requires a careful discussion of risks and benefits (Box 2).38
Benzodiazepine use during pregnancy has been associated with cleft palate and urogenital and neurologic malformations in the fetus.38 Although data are conflicting—particularly among recent meta-analyses that fail to demonstrate an association—some experts advise against benzodiazepine use in the first trimester. Participate in shared decision making with your patients and educate them about the potential risks and benefits of benzodiazepine use during the first trimester and throughout pregnancy. After delivery, newborns may develop “floppy baby syndrome”—which is associated with lethargy, difficulty eating, and respiratory depression—or withdrawal.38 To minimize this risk, consider tapering the benzodiazepine as the patient approaches delivery.
Related Resources
- Substance Abuse and Mental Health Services Administration. www.samhsa.gov.
- National Institute on Drug Abuse resources for medical and health professionals. www.drugabuse.gov/medical-health-professionals.
- American Academy of Sleep Medicine. www.aasmnet.org.
Drug Brand Names
- Alprazolam • Xanax
- Chlordiazepoxide • Librium, Limbitrol
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Diazepam • Valium
- Diphenhydramine • Benadryl, others
- Estazolam • ProSom
- Flumazenil • Romazicon
- Flurazepam • Dalmane
- Haloperidol • Haldol
- Lithium • Lithobid
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Oxazepam • Serax
- Paroxetine • Paxil
- Pregabalin • Lyrica
- Propranolol • Inderal, InnoPran XL, others
- Quazepam • Doral
- Ramelteon • Rozerem
- Sertraline • Zoloft
- Temazepam • Restoril
- Triazolam • Halcion
- Valproic acid • Depakene, Stavzor, others
Disclosures
Drs. Bostwick and Yasugi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Casher is a speaker for AstraZeneca and Sunovion Pharmaceuticals.
1. Mihic SJ, Harris RA. Hypnotics and sedatives. In: Brunton LL Chabner BA, Knollmann BC, eds. Goodman & Gilman’s the pharmacological basis of therapeutics. New York, NY: McGraw Hill and Company; 2011:457-480.
2. Facts and comparisons Web site. 2011 Wolters Kluwer Health Inc. http://online.factsandcomparisons.com. Accessed August 16, 2011.
3. DuPont RL, Greene W, Lydiard RB. Sedatives and hypnotics: pharmacology and epidemiology. In: Gold MS Hermann R, eds. UpToDate. http://www.uptodate.com/contents/sedatives-and-hypnotics-abuse-and-dependence-pharmacology-and-epidemiology. Accessed August 16, 2011.
4. U.S. Food and Drug Administration. Orange book: approved drug products with therapeutic equivalence evaluations. http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm. Accessed August 16, 2011.
5. Chouinard G. Issues in the clinical use of benzodiazepines: potency withdrawal, and rebound. J Clin Psychiatry. 2004;65(suppl 5):7-12.
6. Shader RI, Greenblatt DJ. Can you provide a table of equivalencies for benzodiazepines and other marketed benzodiazepine receptor agonists? J Clin Psychopharmacol. 1997;17(4):331.-
7. Amato L, Minozzi S, Davoli M. Efficacy and safety of pharmacologic interventions for the treatment of the alcohol withdrawal syndrome. Cochrane Database Syst Rev. 2011;15(6):CD008537.-
8. Schutte-Rodin S, Broch L, Buysse D, et al. Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med. 2008;4(5):487-504.
9. Foral P, Dewan N, Malesker M. Insomnia: a therapeutic review for pharmacists. Consult Pharm. 2011;26(5):332-341.
10. Riemann D, Perlis ML. The treatments of chronic insomnia: a review of benzodiazepine receptor agonists and psychological and behavioral therapies. Sleep Med Rev. 2009;13(3):205-214.
11. Hidalgo RB, Tupler LA, Davidson JR. An effect-size analysis of pharmacologic treatments of generalized anxiety disorder. J Psychopharmacol. 2007;21(8):864-872.
12. Davidson JR, Zhang W, Connor KM, et al. A psychopharmacological treatment algorithm for generalised anxiety disorder (GAD). J Psychopharmacol. 2010;24(1):3-26.
13. Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
14. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Arlington VA: American Psychiatric Publishing, Inc.; 2009.
15. Pollack MH, Simon NM, Worthington JJ, et al. Combined paroxetine and clonazepam treatment strategies compared to paroxetine monotherapy for panic disorder. J Psychopharmacol. 2003;17(3):276-282.
16. Watanabe N, Churchill R, Furukawa TA. Combined psychotherapy plus benzodiazepines for panic disorder. Cochrane Database Syst Rev. 2009;(1):CD005335.-
17. Otto MW, McHugh RK, Simon NM, et al. Efficacy of CBT for benzodiazepine discontinuation in patients with panic disorder: further evaluation. Behav Res Ther. 2010;48(8):720-727.
18. Davidson JR. Use of benzodiazepines in social anxiety disorder generalized anxiety disorder, and posttraumatic stress disorder. J Clin Psychiatry. 2004;65(suppl 5):29-33.
19. Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
20. Connor KM, Davidson JR, Potts NL, et al. Discontinuation of clonazepam in the treatment of social phobia. J Clin Psychopharmacol. 1998;18(5):373-378.
21. Miller CH, Fleischhacker WW. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
22. Rodnitzky RL. Drug-induced movement disorders. Clin Neuropharmacol. 2002;25(3):142-151.
23. Arbaizar B, Gómez-Acebo I, Llorca J. Postural induced tremor in psychiatry. Psychiatry Clin Neurosci. 2008;62(6):638-645.
24. Casher MI, Bess JD. Manual of inpatient psychiatry. New York NY: Cambridge University Press; 2010.
25. Battaglia J. Pharmacological management of acute agitation. Drugs. 2005;65(9):1207-1222.
26. Physicians’ desk reference. Montvale NJ: PDR Network, LLC; 2010.
27. Rosebush PI, Mazurek MF. Catatonia and its treatment. Schizophr Bull. 2010;36(2):239-242.
28. Ungvari GS, Kau LS, Wai-Kwong T, et al. The pharmacological treatment of catatonia: an overview. Eur Arch Psychiatry Clin Neurosci. 2001;251(suppl 1):I31-I34.
29. Fink M, Taylor MA. Catatonia: a clinician’s guide to diagnosis and treatment. New York NY: Cambridge University Press; 2003.
30. Naguib N, Koorn R. Interactions between psychotropics anaesthetics and electroconvulsive therapy: implications for drug choice and patient management. CNS Drugs. 2002;16(4):229-247.
31. Boylan LS, Haskett RF, Mulsant BH, et al. Determinants of seizure threshold in ECT: benzodiazepine use, anesthetic dosage, and other factors. J ECT. 2000;16(1):3-18.
32. Krystal AD, Watts BV, Weiner RD, et al. The use of flumazenil in the anxious and benzodiazepine-dependent ECT patient. J ECT. 1998;14(1):5-14.
33. Melton ST, Kirkwood CK. Anxiety disorders I: generalized anxiety panic, and social anxiety disorders. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. New York, NY: McGraw-Hill Companies; 2011:1209-1228.
34. Benzodiazepine toolkit. The Pharmacist’s Letter/Prescriber’s Letter. 2011;27(4):270406.-
35. Lader M. Benzodiazepines revisited – will we ever learn? Addiction. 2011;106(12):2086-2109.
36. Toblin RL, Paulozzi LJ, Logan JE, et al. Mental illness and psychotropic drug use among prescription drug overdose deaths: a medical examiner chart review. J Clin Psychiatry. 2010;71(4):491-496.
37. Ashton H. The diagnosis and management of benzodiazepine dependence. Curr Opin Psychiatry. 2005;18(3):249-255.
38. Menon SJ. Psychotropic medication during pregnancy and lactation. Arch Gynecol Obstet. 2008;277(1):1-13.
1. Mihic SJ, Harris RA. Hypnotics and sedatives. In: Brunton LL Chabner BA, Knollmann BC, eds. Goodman & Gilman’s the pharmacological basis of therapeutics. New York, NY: McGraw Hill and Company; 2011:457-480.
2. Facts and comparisons Web site. 2011 Wolters Kluwer Health Inc. http://online.factsandcomparisons.com. Accessed August 16, 2011.
3. DuPont RL, Greene W, Lydiard RB. Sedatives and hypnotics: pharmacology and epidemiology. In: Gold MS Hermann R, eds. UpToDate. http://www.uptodate.com/contents/sedatives-and-hypnotics-abuse-and-dependence-pharmacology-and-epidemiology. Accessed August 16, 2011.
4. U.S. Food and Drug Administration. Orange book: approved drug products with therapeutic equivalence evaluations. http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm. Accessed August 16, 2011.
5. Chouinard G. Issues in the clinical use of benzodiazepines: potency withdrawal, and rebound. J Clin Psychiatry. 2004;65(suppl 5):7-12.
6. Shader RI, Greenblatt DJ. Can you provide a table of equivalencies for benzodiazepines and other marketed benzodiazepine receptor agonists? J Clin Psychopharmacol. 1997;17(4):331.-
7. Amato L, Minozzi S, Davoli M. Efficacy and safety of pharmacologic interventions for the treatment of the alcohol withdrawal syndrome. Cochrane Database Syst Rev. 2011;15(6):CD008537.-
8. Schutte-Rodin S, Broch L, Buysse D, et al. Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med. 2008;4(5):487-504.
9. Foral P, Dewan N, Malesker M. Insomnia: a therapeutic review for pharmacists. Consult Pharm. 2011;26(5):332-341.
10. Riemann D, Perlis ML. The treatments of chronic insomnia: a review of benzodiazepine receptor agonists and psychological and behavioral therapies. Sleep Med Rev. 2009;13(3):205-214.
11. Hidalgo RB, Tupler LA, Davidson JR. An effect-size analysis of pharmacologic treatments of generalized anxiety disorder. J Psychopharmacol. 2007;21(8):864-872.
12. Davidson JR, Zhang W, Connor KM, et al. A psychopharmacological treatment algorithm for generalised anxiety disorder (GAD). J Psychopharmacol. 2010;24(1):3-26.
13. Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
14. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Arlington VA: American Psychiatric Publishing, Inc.; 2009.
15. Pollack MH, Simon NM, Worthington JJ, et al. Combined paroxetine and clonazepam treatment strategies compared to paroxetine monotherapy for panic disorder. J Psychopharmacol. 2003;17(3):276-282.
16. Watanabe N, Churchill R, Furukawa TA. Combined psychotherapy plus benzodiazepines for panic disorder. Cochrane Database Syst Rev. 2009;(1):CD005335.-
17. Otto MW, McHugh RK, Simon NM, et al. Efficacy of CBT for benzodiazepine discontinuation in patients with panic disorder: further evaluation. Behav Res Ther. 2010;48(8):720-727.
18. Davidson JR. Use of benzodiazepines in social anxiety disorder generalized anxiety disorder, and posttraumatic stress disorder. J Clin Psychiatry. 2004;65(suppl 5):29-33.
19. Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
20. Connor KM, Davidson JR, Potts NL, et al. Discontinuation of clonazepam in the treatment of social phobia. J Clin Psychopharmacol. 1998;18(5):373-378.
21. Miller CH, Fleischhacker WW. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
22. Rodnitzky RL. Drug-induced movement disorders. Clin Neuropharmacol. 2002;25(3):142-151.
23. Arbaizar B, Gómez-Acebo I, Llorca J. Postural induced tremor in psychiatry. Psychiatry Clin Neurosci. 2008;62(6):638-645.
24. Casher MI, Bess JD. Manual of inpatient psychiatry. New York NY: Cambridge University Press; 2010.
25. Battaglia J. Pharmacological management of acute agitation. Drugs. 2005;65(9):1207-1222.
26. Physicians’ desk reference. Montvale NJ: PDR Network, LLC; 2010.
27. Rosebush PI, Mazurek MF. Catatonia and its treatment. Schizophr Bull. 2010;36(2):239-242.
28. Ungvari GS, Kau LS, Wai-Kwong T, et al. The pharmacological treatment of catatonia: an overview. Eur Arch Psychiatry Clin Neurosci. 2001;251(suppl 1):I31-I34.
29. Fink M, Taylor MA. Catatonia: a clinician’s guide to diagnosis and treatment. New York NY: Cambridge University Press; 2003.
30. Naguib N, Koorn R. Interactions between psychotropics anaesthetics and electroconvulsive therapy: implications for drug choice and patient management. CNS Drugs. 2002;16(4):229-247.
31. Boylan LS, Haskett RF, Mulsant BH, et al. Determinants of seizure threshold in ECT: benzodiazepine use, anesthetic dosage, and other factors. J ECT. 2000;16(1):3-18.
32. Krystal AD, Watts BV, Weiner RD, et al. The use of flumazenil in the anxious and benzodiazepine-dependent ECT patient. J ECT. 1998;14(1):5-14.
33. Melton ST, Kirkwood CK. Anxiety disorders I: generalized anxiety panic, and social anxiety disorders. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. New York, NY: McGraw-Hill Companies; 2011:1209-1228.
34. Benzodiazepine toolkit. The Pharmacist’s Letter/Prescriber’s Letter. 2011;27(4):270406.-
35. Lader M. Benzodiazepines revisited – will we ever learn? Addiction. 2011;106(12):2086-2109.
36. Toblin RL, Paulozzi LJ, Logan JE, et al. Mental illness and psychotropic drug use among prescription drug overdose deaths: a medical examiner chart review. J Clin Psychiatry. 2010;71(4):491-496.
37. Ashton H. The diagnosis and management of benzodiazepine dependence. Curr Opin Psychiatry. 2005;18(3):249-255.
38. Menon SJ. Psychotropic medication during pregnancy and lactation. Arch Gynecol Obstet. 2008;277(1):1-13.