User login
How plasma donation can affect your patient’s pharmacotherapy
Discuss this article at www.facebook.com/CurrentPsychiatry
Many economically disadvantaged psychiatric patients donate plasma for financial incentive. However, plasmapheresis (PP)—separation of plasma and cellular components of blood—can increase drug clearance, which may affect how you manage patients who donate plasma frequently.
Donated plasma is used to help patients with hemophilia and other blood disorders and burn victims. It’s also valuable for medical research. Typically, donors cannot be taking lithium, experiencing active hallucinations, receiving ≥3 psychotropic medications, or have had a psychiatric hospitalization in the past 12 months. Patients can donate while taking antidepressants, mood stabilizers, antipsychotics, and anticonvulsants.1
Pharmacotherapeutic concerns
During PP, solutes in plasma such as drugs can be removed, increasing drug clearance by 30%.2,3 PP affects both protein-bound and free drug concentrations. PP effectively clears drugs that are highly protein bound and have a small volume of distribution. As a result, serum levels of psychotropics are lowered. Most psychotropics except lithium are bound to plasma protein. Because of high protein binding, plasma concentrations of psychotropics may rebound after PP. Antipsychotics are highly protein bound—85% to 90%—and highly lipophilic. For a list of protein binding percentages of commonly used psychotropics, see the Table.4
Table
Protein binding percentages of common psychotropics
| Drug(s) | Percentage of protein binding |
|---|---|
| Lamotrigine; topiramate | Minimal |
| Desvenlafaxine | 30% |
| Carbamazepine | 40% to 90% |
| Venlafaxine | 40% to 50% |
| Oxcarbazepine | 40% to 60% |
| Escitalopram | 56% |
| All other SSRIs | 75% |
| Bupropion | 84% |
| Mirtazapine | 85% |
| Duloxetine; divalproex | 90% |
| Tricyclic antidepressants | 98% |
| SSRIs: selective serotonin reuptake inhibitors Source: Reference 4 | |
Plasma is regenerated 24 to 48 hours after PP; therefore, the clinical effect on daily psychotropic dosing should be small unless the donations are frequent. Long-term and regular plasma donation may result in hypoalbuminemia and hypocholesterolemia5; however, the effects of hypoalbuminemia on psychotropics routinely bound to serum proteins are unknown. Patients with an acute infection or malnourishment could have further decreased albumin production or increased catabolism, resulting in a significant decrease in serum albumin concentration, which may affect psychotropic pharmacokinetics.5
Other concerns
Beware of financial incentives because economically disadvantaged psychiatric patients are vulnerable to coercion. Some plasma donor centers will pay donors a specific amount—ranging from $20 to $30— for their first 2 donations and offer monthly bonuses if a patient donates 8 times a month.
The amount of plasma a patient can donate is based on their weight; patients who weigh more get paid more. This may conflict with your attempts to motivate patients to lose weight.
Disclosures
Dr. Selvaraj receives an internal grant from Creighton University.
Drs. Gabel and Ramaswamy report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
The authors would like to thank Darrel E. Willoughby, Librarian, Omaha Veterans Affairs Medical Center, Omaha, NE, for his assistance with this article.
1. American Red Cross. Eligibility criteria by topic. http://www.redcrossblood.org/donating-blood/eligibility-requirements/eligibility-criteria-topic#meds_vaccinations. Accessed April 11 2012.
2. Kale-Pradhan PB, Woo MH. A review of the effects of plasmapheresis on drug clearance. Pharmacotherapy. 1997;17(4):684-695.
3. Ibrahim RB, Liu C, Cronin SM, et al. Drug removal by plasmapheresis: an evidence-based review. Pharmacotherapy. 2007;27(11):1529-1549.
4. Clinical pharmacology online. http://www.clinicalpharmacology.com. Accessed April 11 2012.
5. Jones DK, Dunn MI. ‘Vampire syndrome’: serum protein and lipid abnormalities related to frequent sale of plasma. J Fam Pract. 1995;40(3):288-290.
Discuss this article at www.facebook.com/CurrentPsychiatry
Many economically disadvantaged psychiatric patients donate plasma for financial incentive. However, plasmapheresis (PP)—separation of plasma and cellular components of blood—can increase drug clearance, which may affect how you manage patients who donate plasma frequently.
Donated plasma is used to help patients with hemophilia and other blood disorders and burn victims. It’s also valuable for medical research. Typically, donors cannot be taking lithium, experiencing active hallucinations, receiving ≥3 psychotropic medications, or have had a psychiatric hospitalization in the past 12 months. Patients can donate while taking antidepressants, mood stabilizers, antipsychotics, and anticonvulsants.1
Pharmacotherapeutic concerns
During PP, solutes in plasma such as drugs can be removed, increasing drug clearance by 30%.2,3 PP affects both protein-bound and free drug concentrations. PP effectively clears drugs that are highly protein bound and have a small volume of distribution. As a result, serum levels of psychotropics are lowered. Most psychotropics except lithium are bound to plasma protein. Because of high protein binding, plasma concentrations of psychotropics may rebound after PP. Antipsychotics are highly protein bound—85% to 90%—and highly lipophilic. For a list of protein binding percentages of commonly used psychotropics, see the Table.4
Table
Protein binding percentages of common psychotropics
| Drug(s) | Percentage of protein binding |
|---|---|
| Lamotrigine; topiramate | Minimal |
| Desvenlafaxine | 30% |
| Carbamazepine | 40% to 90% |
| Venlafaxine | 40% to 50% |
| Oxcarbazepine | 40% to 60% |
| Escitalopram | 56% |
| All other SSRIs | 75% |
| Bupropion | 84% |
| Mirtazapine | 85% |
| Duloxetine; divalproex | 90% |
| Tricyclic antidepressants | 98% |
| SSRIs: selective serotonin reuptake inhibitors Source: Reference 4 | |
Plasma is regenerated 24 to 48 hours after PP; therefore, the clinical effect on daily psychotropic dosing should be small unless the donations are frequent. Long-term and regular plasma donation may result in hypoalbuminemia and hypocholesterolemia5; however, the effects of hypoalbuminemia on psychotropics routinely bound to serum proteins are unknown. Patients with an acute infection or malnourishment could have further decreased albumin production or increased catabolism, resulting in a significant decrease in serum albumin concentration, which may affect psychotropic pharmacokinetics.5
Other concerns
Beware of financial incentives because economically disadvantaged psychiatric patients are vulnerable to coercion. Some plasma donor centers will pay donors a specific amount—ranging from $20 to $30— for their first 2 donations and offer monthly bonuses if a patient donates 8 times a month.
The amount of plasma a patient can donate is based on their weight; patients who weigh more get paid more. This may conflict with your attempts to motivate patients to lose weight.
Disclosures
Dr. Selvaraj receives an internal grant from Creighton University.
Drs. Gabel and Ramaswamy report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
The authors would like to thank Darrel E. Willoughby, Librarian, Omaha Veterans Affairs Medical Center, Omaha, NE, for his assistance with this article.
Discuss this article at www.facebook.com/CurrentPsychiatry
Many economically disadvantaged psychiatric patients donate plasma for financial incentive. However, plasmapheresis (PP)—separation of plasma and cellular components of blood—can increase drug clearance, which may affect how you manage patients who donate plasma frequently.
Donated plasma is used to help patients with hemophilia and other blood disorders and burn victims. It’s also valuable for medical research. Typically, donors cannot be taking lithium, experiencing active hallucinations, receiving ≥3 psychotropic medications, or have had a psychiatric hospitalization in the past 12 months. Patients can donate while taking antidepressants, mood stabilizers, antipsychotics, and anticonvulsants.1
Pharmacotherapeutic concerns
During PP, solutes in plasma such as drugs can be removed, increasing drug clearance by 30%.2,3 PP affects both protein-bound and free drug concentrations. PP effectively clears drugs that are highly protein bound and have a small volume of distribution. As a result, serum levels of psychotropics are lowered. Most psychotropics except lithium are bound to plasma protein. Because of high protein binding, plasma concentrations of psychotropics may rebound after PP. Antipsychotics are highly protein bound—85% to 90%—and highly lipophilic. For a list of protein binding percentages of commonly used psychotropics, see the Table.4
Table
Protein binding percentages of common psychotropics
| Drug(s) | Percentage of protein binding |
|---|---|
| Lamotrigine; topiramate | Minimal |
| Desvenlafaxine | 30% |
| Carbamazepine | 40% to 90% |
| Venlafaxine | 40% to 50% |
| Oxcarbazepine | 40% to 60% |
| Escitalopram | 56% |
| All other SSRIs | 75% |
| Bupropion | 84% |
| Mirtazapine | 85% |
| Duloxetine; divalproex | 90% |
| Tricyclic antidepressants | 98% |
| SSRIs: selective serotonin reuptake inhibitors Source: Reference 4 | |
Plasma is regenerated 24 to 48 hours after PP; therefore, the clinical effect on daily psychotropic dosing should be small unless the donations are frequent. Long-term and regular plasma donation may result in hypoalbuminemia and hypocholesterolemia5; however, the effects of hypoalbuminemia on psychotropics routinely bound to serum proteins are unknown. Patients with an acute infection or malnourishment could have further decreased albumin production or increased catabolism, resulting in a significant decrease in serum albumin concentration, which may affect psychotropic pharmacokinetics.5
Other concerns
Beware of financial incentives because economically disadvantaged psychiatric patients are vulnerable to coercion. Some plasma donor centers will pay donors a specific amount—ranging from $20 to $30— for their first 2 donations and offer monthly bonuses if a patient donates 8 times a month.
The amount of plasma a patient can donate is based on their weight; patients who weigh more get paid more. This may conflict with your attempts to motivate patients to lose weight.
Disclosures
Dr. Selvaraj receives an internal grant from Creighton University.
Drs. Gabel and Ramaswamy report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
The authors would like to thank Darrel E. Willoughby, Librarian, Omaha Veterans Affairs Medical Center, Omaha, NE, for his assistance with this article.
1. American Red Cross. Eligibility criteria by topic. http://www.redcrossblood.org/donating-blood/eligibility-requirements/eligibility-criteria-topic#meds_vaccinations. Accessed April 11 2012.
2. Kale-Pradhan PB, Woo MH. A review of the effects of plasmapheresis on drug clearance. Pharmacotherapy. 1997;17(4):684-695.
3. Ibrahim RB, Liu C, Cronin SM, et al. Drug removal by plasmapheresis: an evidence-based review. Pharmacotherapy. 2007;27(11):1529-1549.
4. Clinical pharmacology online. http://www.clinicalpharmacology.com. Accessed April 11 2012.
5. Jones DK, Dunn MI. ‘Vampire syndrome’: serum protein and lipid abnormalities related to frequent sale of plasma. J Fam Pract. 1995;40(3):288-290.
1. American Red Cross. Eligibility criteria by topic. http://www.redcrossblood.org/donating-blood/eligibility-requirements/eligibility-criteria-topic#meds_vaccinations. Accessed April 11 2012.
2. Kale-Pradhan PB, Woo MH. A review of the effects of plasmapheresis on drug clearance. Pharmacotherapy. 1997;17(4):684-695.
3. Ibrahim RB, Liu C, Cronin SM, et al. Drug removal by plasmapheresis: an evidence-based review. Pharmacotherapy. 2007;27(11):1529-1549.
4. Clinical pharmacology online. http://www.clinicalpharmacology.com. Accessed April 11 2012.
5. Jones DK, Dunn MI. ‘Vampire syndrome’: serum protein and lipid abnormalities related to frequent sale of plasma. J Fam Pract. 1995;40(3):288-290.
How to lower suicide risk in depressed children and adolescents
Discuss this article at www.facebook.com/CurrentPsychiatry
Although depression affects nearly 2% of children (age ≤12) and up to 10% of adolescents (age 13 to 18),1 the disorder often is underdiagnosed and undertreated in pediatric patients.2 Treating depression in young patients is challenging. Only 30% to 40% of depressed children and adolescents who receive evidence-based treatment achieve remission.3 In addition, 50% to 70% of those who initially achieve remission will experience recurrence within 5 years.4 Suicide is the third leading cause of death among children and adolescents, and depression greatly increases the likelihood of suicide.5,6
This article reviews assessing and treating depression in children and adolescents, and how to lower suicide risk in pediatric patients.
Symptoms vary with age
Depressive symptoms vary as a function of the child’s cognitive development and social functioning. Hopelessness and vegetative and motivational symptoms may be more frequent in adolescents than in children.7
In preschool-age children, depression manifests indirectly through somatic symptoms and behavioral disturbances. In this age group, sadness or irritability are sensitive and predominant symptoms of depression.8 In older children, sadness and loss of interest in social activities may indicate depression. In adolescents, feelings of mental and physical weariness, aloneness, disconnectedness, uncertainty, vulnerability, anger, irritability, and ambivalence toward friends suggest a depressive disorder.9
Genetic predisposition to depression, poor family support, dysfunctional parenting, and individual vulnerabilities such as poor self-esteem or emotional dysregulation may increase young patients’ risk for depression.10 Peer and family support may protect against depression. Personal competence stemming from social acceptance and body image satisfaction also may be protective factors. A sense of religious and existential well-being (finding meaning and purpose in life) are significantly associated with lower rates of depression among adolescents.11
A persistent illness
The mean duration of a depressive episode in children and adolescents is 7 to 8 months.12 However, subsyndromal depressive symptoms—as well as relapse and recurrence—are common. Long-term studies indicate that many depressed adolescents experience depressive episodes into adulthood.12 Factors that may predict recurrence in adulthood include:
- severity of depressive episodes
- concurrent psychotic symptoms
- suicidal thoughts
- history of recurrent depressive episodes
- threshold residual symptoms
- recent stressful life events
- adverse family environment
- family history of depression.12
Early symptom onset, greater depression severity, suicidality, presence of comorbid anxiety, disruptive disorders, and an adverse family environment also predict longer recovery time.12 A study of depressed adolescents found that a history of recurrent depression, family history of recurrent depression, personality disorder traits, and (for girls only) conflict with parents predicted recurrence of depression in young adulthood.4Table 1 summarizes factors that affect depression outcomes in children and adolescents.
Table 1
What affects depression outcomes in children and adolescents?
| Factor | Outcomes |
|---|---|
| Age | Pharmacotherapy and CBT are equally effective in younger and older adolescents.a Although age does not affect long-term treatment outcomes, older adolescents (age 18 to 19) with treatment-resistant depression may respond better to a combination of CBT and medicationb |
| Sex | Females are more likely to experience relapse.c However, sex does not influence response to initial treatmentc |
| Socioeconomic status | Adolescents with high socioeconomic status are more likely to respond to CBT |
| Illness characteristics | Severity of depression is the strongest predictor of poor outcome.d-f Patients with moderate depression are more likely to benefit from CBT added to medication.g However, adding CBT to medication did not affect outcomes in adolescents with self-injurious behavior.b,f Suicidal behaviors during treatment are less frequent when CBT is combined with medicationh,i |
| Substance abuse | Patients with substance use disorders are less likely to respond to depression treatmentf and those who continued to abuse substances during treatment are less likely to achieve remission than those who abstainb |
| Cognitive measures | Higher levels of hopelessness are associated with poor outcomes. For adolescents with treatment-resistant depression who experience hopelessness, adding CBT to pharmacotherapy did not provide additional benefit. Some studies have noted that adolescents with cognitive distortions are more likely to benefit from CBT plus pharmacotherapyb |
| Family characteristics/environment | High family stress is associated with poor treatment outcomes.f Experiencing loss and physically dangerous events does not affect depression outcomes. Trauma and history of abuse adversely effect depression treatment outcomes |
CBT: cognitive-behavioral therapy
| |
Assessment strategies
Semi-structured interviews such as the Child and Adolescent Psychiatric Assessment, the Diagnostic Interview for Children and Adolescents, and the Kiddie Schedule for Affective Disorder and Schizophrenia are useful for assessing depression in pediatric patients (Table 2).13-16 These tools can be used to assess depression criteria based on information gathered from several sources. Many instruments can be used to assess and monitor pediatric depression, including the Children’s Depression Inventory, the Reynolds Child Depression Scales and Adolescent Depression Scales, and the Child Depression Rating Scale.
To assess suicide risk in depressed younger patients:
- ask about emotional difficulties
- identify lack of developmental progress
- estimate their level of distress
- detect impairment in functioning
- estimate the level of danger to themselves and others.17
The best way to assess for suicidal ideation is to ask about it directly while interviewing the patient and his or her parents. Simple questions such as “Have you ever thought about killing yourself or wish you were dead?” and “Have you ever done anything on purpose to hurt or kill yourself?” can be effective.10 These questions are best placed in the middle or toward the end of a list of questions about depressive symptoms.
Adolescents may be more likely than adults to disclose information about suicidality on self-reports.6 However, self-assessment suicide scales are not a substitute for clinical assessment because they tend to be oversensitive and non-specific and lack predictive value. A positive response to either of these questions should prompt a more detailed clinical investigation. There is no evidence that asking about suicide risk increases suicidal behavior, even in high-risk youths.
Table 2
Assessing children and adolescents: 3 semi-structured interviews
| Interview | Features |
|---|---|
| Child and Adolescent Psychiatric Assessment13 | For patients age 9 to 17. Assesses symptoms from the past 3 months. Administration time: 1 to 2 hours. Requires minimal interviewer experience. Assesses impairment in multiple areas (family, peers, school, leisure activities) |
| Diagnostic Interview for Children and Adolescents14 | Separate versions for children (age 6 to 12) and adolescents (age 13 to 17). Assesses lifetime psychopathology. Administration time: 1 to 2 hours. Interrater reliability varies (poor to good) |
| Kiddie Schedule for Affective Disorders and Schizophrenia15 | Assesses lifetime and current psychopathology. Administration time: 35 minutes to 2.5 hours. Interrater reliability: fair to excellent16 |
Treatment options
Psychotherapy. Several controlled studies and meta-analyses support the efficacy of cognitive-behavioral therapy (CBT) for mild depression in pediatric patients.18-20 Two recent meta-analyses of CBT studies in depressed adolescents found the mean effect size of CBT was 0.34 to 0.35.19,21 However, a separate analysis found CBT did not have long-term benefits for depressed adolescents, particularly patients with a history of abuse.22
Interpersonal therapy also can be effective in adolescent outpatients with mild to moderate depression. One study found the effect size of psychotherapy was modest (0.36).19
Pharmacotherapy. Two meta-analyses support selective serotonin reuptake inhibitors (SSRIs) for treating mild to moderate depression in children and adolescents. One found 61% of depressed patients age <19 who received an SSRI were “much improved” or “very much improved.”23 Another meta-analysis that compared SSRIs and placebo found fluoxetine was more effective than sertraline or citalopram for depressed adolescents.24 Other studies have shown that for severe depression, the effect size of antidepressants (0.69) is higher than that of placebo (0.39).25 Antidepressants are more effective in adolescents than in children.25
Fluoxetine is the only FDA-approved medication for treating depression in children age ≥8. In 2007 the FDA extended to all antidepressants its “black-box” warning about increased risk of suicidality in patients up to age 24. The results of studies that analyzed data about the safety of antidepressants in children and adolescents have been mixed—some found evidence of increased suicidality with antidepressant use,26,27 whereas others showed no increased risk.28,29Table 3 summarizes steps to minimize the risk of antidepressant-induced suicidality.17
Psychotherapy plus pharmacotherapy. Researchers who compared fluoxetine to CBT and to a combination of the 2 in adolescents with moderate to severe depression found that fluoxetine was most effective in the first 12 weeks of treatment.30 Surprisingly, CBT’s effectiveness was not different from placebo.30 However, studies have shown that combining psychotherapy and medication results in greater symptom improvement,30 faster clinical response,31 improvement of global functioning and quality of life,32 and reduced suicidality.33 At 6 months, the difference in response between medication and psychotherapy was small.25 The Treatment of Resistant Depression in Adolescents study found that for chronic adolescent depression, pharmacotherapy (fluoxetine and venlafaxine) combined with CBT produced a higher response rate than pharmacotherapy alone (54% vs 41%).34
Table 3
Protecting against antidepressant-induced suicidality
| Before initiating antidepressant treatment |
|---|
| Review the patient’s psychiatric history |
| Assess for past suicidal behavior |
| Assess for a family history of mental illness or mood disorders and suicide attempts |
| Screen for unrecognized bipolar spectrum disorders |
| Educate patients and their families to watch for signs of worsening depression or suicidality, and to report such symptoms immediately |
| During antidepressant treatment |
| Pay attention to abrupt changes in symptoms, particularly symptoms that were not part of the patient’s initial presentation |
| Watch for deterioration of symptoms |
| Monitor for emergence of ‘activating’ symptoms (ie, irritability, impulsivity, anxiety, insomnia, agitation, hostility, akathisia, hypomania, or mania) |
| Evaluate the patient’s suicide risk factors, including having a specific plan and/or access to lethal means |
| Consider hospitalization if the patient is at high risk for suicide |
| Source: Reference 17 |
Lowering suicide risk
Up to 60% of adolescents who commit suicide had a depressive disorder. Risk factors for child and adolescent suicide attempts include:
- self-harm behaviors
- psychiatric disorders
- family disturbances
- substance abuse
- physical/sexual abuse.17
How to best manage suicidal youths depends on an adequate assessment of the severity of the patient’s current problems and conflicts and the degree of suicidal intent. Assessment of coping resources, access to support systems, and the attitude of the patient and family toward intervention and follow-up also is important.
Children and adolescents at high risk for suicide—those with a plan or recent suicide attempt with high probability of lethality, stated current intent to kill themselves, or recent suicidal ideation or behavior—may need inpatient psychiatric admission. Although no studies have shown that admitting high-risk suicidal patients prevents suicide, hospitalization often is the safest course of action. Develop ing a comprehensive outpatient treatment plan before discharge is essential. Patients with fewer risk factors, especially those who want help and have social support, hope for the future, and a desire to resolve conflicts, may require only a brief crisis-oriented intervention.
The following recommendations for managing suicidality in children and adolescents are based on clinical experience and have not been empirically validated.
Develop a safety plan to direct the patient’s behavior under various situations. For example, the patient would agree in writing that “If I feel depressed, I will do X, Y, and Z to address it,” or “If I find myself having suicidal thoughts, I will contact ABC.” Having a safety plan lowers the risk of a suicide attempt more than having a suicide contract, which does not give the patient any tools.35
Create a ‘hope box.’ This is a box in which the patient collects mementos and other objects that remind him or her of hope and reasons to live. The patient should be able to access it at all times, so he or she can tap into it during crisis periods to avert suicidal acts.35
Counteract alienation. A sense of social isolation and burdensomeness may be “tipping factors” for suicidal acts when adolescents feel depressed.35 Clinicians should try to help connect patients to meaningful social activities, even in small doses.
Manage overarousal. Overarousal in depressed children and adolescents is manifested as agitation. Insomnia is a clinically modifiable risk factor. Insomnia initially responds well to behavioral interventions such as sleep hygiene, sleep restriction, and stimulus control techniques.35
Related Resources
- National Suicide Prevention Lifeline. 800-273-TALK (8255). www.suicidepreventionlifeline.org.
- Suicide Prevention Resource Center. www.sprc.org.
Drug Brand Names
- Citalopram • Celexa
- Fluoxetine • Prozac
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Drs. Shailesh Jain and Islam report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rakesh Jain is a consultant to or has received research/grant support from Eli Lilly and Company, Merck, Pfizer Inc., Shionogi Pharmaceuticals, and Shire.
1. Birmaher B, Brent D. AACAP Work Group on Quality Issues, et al. Practice parameter for the assessment and treatment of children and adolescents with depressive disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(11):1503-1526.
2. Lewinsohn PM, Clarke GN, Seeley JR, et al. Major depression in community adolescents: age at onset, episode duration, and time to recurrence. J Am Acad Child Adolesc Psychiatry. 1994;33(6):809-818.
3. Emslie GJ, Kennard BD, Mayes TL. Predictors of treatment response in adolescent depression. Pediatr Ann. 2011;40(6):300-306.
4. Lewinsohn PM, Rohde P, Seeley JR, et al. Natural course of adolescent major depressive disorder in a community sample: predictors of recurrence in young adults. Am J Psychiatry. 2000;157(10):1584-1591.
5. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
6. Gould MS, Greenberg T, Velting DM, et al. Youth suicide risk and preventive interventions: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry. 2003;42(4):386-405.
7. Weiss B, Garber J. Developmental differences in the phenomenology of depression. Dev Psychopathol. 2003;15(2):403-430.
8. Calles JL, Jr. Depression in children and adolescents. Prim Care. 2007;34(2):243-258abstract vi.
9. Farmer TJ. The experience of major depression: adolescents’ perspectives. Issues Ment Health Nurs. 2002;23(6):567-585.
10. Zalsman G, Brent DA, Weersing VR. Depressive disorders in childhood and adolescence: an overview: epidemiology clinical manifestation and risk factors. Child Adolesc Psychiatr Clin N Am. 2006;15(4):827-841, vii.
11. Cotton S, Larkin E, Hoopes A, et al. The impact of adolescent spirituality on depressive symptoms and health risk behaviors. J Adolesc Health. 2005;36(6):529.-
12. Birmaher B, Arbelaez C, Brent D. Course and outcome of child and adolescent major depressive disorder. Child Adolesc Psychiatr Clin N Am. 2002;11(3):619-637, x.
13. Angold A, Costello EJ. A test-retest reliability study of child-reported psychiatric symptoms and diagnoses using the Child and Adolescent Psychiatric Assessment (CAPA-C). Psychol Med. 1995;25(4):755-762.
14. Reich W. Diagnostic interview for children and adolescents (DICA). J Am Acad Child Adolesc Psychiatry. 2000;39(1):59-66.
15. Puig-Antich J, Lukens E, Brent D. Psychosocial schedule for school age children - revised. Pittsburgh PA: Western Psychiatric Institute and Clinic; 1986.
16. Ambrosini PJ. Historical development and present status of the schedule for affective disorders and schizophrenia for school-age children (K-SADS). J Am Acad Child Adolesc Psychiatry. 2000;39(1):49-58.
17. Dodig-Curković K, Curković M, Radić J, et al. Suicidal behavior and suicide among children and adolescents-risk factors and epidemiological characteristics. Coll Antropol. 2010;34(2):771-777.
18. Harrington R, Whittaker J, Shoebridge P, et al. Systematic review of efficacy of cognitive behaviour therapies in childhood and adolescent depressive disorder. BMJ. 1998;316(7144):1559-1563.
19. Weisz JR, McCarty CA, Valeri SM. Effects of psychotherapy for depression in children and adolescents: a meta-analysis. Psychol Bull. 2006;132(1):132-149.
20. Mufson L, Dorta KP, Wickramaratne P, et al. A randomized effectiveness trial of interpersonal psychotherapy for depressed adolescents. Arch Gen Psychiatry. 2004;61(6):577-584.
21. Klein JB, Jacobs RH, Reinecke MA. Cognitive-behavioral therapy for adolescent depression: a meta-analytic investigation of changes in effect-size estimates. J Am Acad Child Adolesc Psychiatry. 2007;46(11):1403-1413.
22. Vitiello B, Emslie G, Clarke G, et al. Long-term outcome of adolescent depression initially resistant to selective serotonin reuptake inhibitor treatment: a follow-up study of the TORDIA sample. J Clin Psychiatry. 2011;72(3):388-396.
23. Bridge JA, Iyengar S, Salary CB, et al. Clinical response and risk for reported suicidal ideation and suicide attempts in pediatric antidepressant treatment: a meta-analysis of randomized controlled trials. JAMA. 2007;297(15):1683-1696.
24. Usala T, Clavenna A, Zuddas A, et al. Randomised controlled trials of selective serotonin reuptake inhibitors in treating depression in children and adolescents: a systematic review and meta-analysis. Eur Neuropsychopharmacol. 2008;18(1):62-73.
25. March JS, Silva S, Petrycki S, et al. The Treatment for Adolescents With Depression Study (TADS): long-term effectiveness and safety outcomes. Arch Gen Psychiatry. 2007;64(10):1132-1143.
26. Bridge JA, Iyengar S, Salary CB, et al. Clinical response and risk for reported suicidal ideation and suicide attempts in pediatric antidepressant treatment: a meta-analysis of randomized controlled trials. JAMA. 2007;297(15):1683-1696.
27. Stone M, Laughren T, Jones ML, et al. Risk of suicidality in clinical trials of antidepressants in adults: analysis of proprietary data submitted to US Food and Drug Administration. BMJ. 2009;339:b2880.-
28. Khan A, Khan S, Kolts R, et al. Suicide rates in clinical trials of SSRIs, other antidepressants, and placebo: analysis of FDA reports. Am J Psychiatry. 2003;160(4):790-792.
29. Simon GE, Savarino J. Suicide attempts among patients starting depression treatment with medications or psychotherapy. Am J Psychiatry. 2007;164(7):1029-1034.
30. March J, Silva S, Petrycki S, et al. Fluoxetine, cognitive-behavioral therapy, and their combination for adolescents with depression: Treatment for Adolescents With Depression Study (TADS) randomized controlled trial. JAMA. 2004;292(7):807-820.
31. Kratochvil C, Emslie G, Silva S, et al. Acute time to response in the Treatment for Adolescents with Depression Study (TADS). J Am Acad Child Adolesc Psychiatry. 2006;45(12):1412-1418.
32. Vitiello B, Rohde P, Silva S, et al. Functioning and quality of life in the Treatment for Adolescents with Depression Study (TADS). J Am Acad Child Adolesc Psychiatry. 2006;45(12):1419-1426.
33. Emslie G, Kratochvil C, Vitiello B, et al. Treatment for Adolescents with Depression Study (TADS): safety results. J Am Acad Child Adolesc Psychiatry. 2006;45(12):1440-1455.
34. Brent D, Emslie G, Clarke G, et al. Switching to another SSRI or to venlafaxine with or without cognitive behavioral therapy for adolescents with SSRI-resistant depression: the TORDIA randomized controlled trial. JAMA. 2008;299(8):901-913.
35. Joiner TE, Ribeiro JD. Assessment and management of suicidal behavior in teens. Psychiatr Ann. 2011;41(4):220-225.
Discuss this article at www.facebook.com/CurrentPsychiatry
Although depression affects nearly 2% of children (age ≤12) and up to 10% of adolescents (age 13 to 18),1 the disorder often is underdiagnosed and undertreated in pediatric patients.2 Treating depression in young patients is challenging. Only 30% to 40% of depressed children and adolescents who receive evidence-based treatment achieve remission.3 In addition, 50% to 70% of those who initially achieve remission will experience recurrence within 5 years.4 Suicide is the third leading cause of death among children and adolescents, and depression greatly increases the likelihood of suicide.5,6
This article reviews assessing and treating depression in children and adolescents, and how to lower suicide risk in pediatric patients.
Symptoms vary with age
Depressive symptoms vary as a function of the child’s cognitive development and social functioning. Hopelessness and vegetative and motivational symptoms may be more frequent in adolescents than in children.7
In preschool-age children, depression manifests indirectly through somatic symptoms and behavioral disturbances. In this age group, sadness or irritability are sensitive and predominant symptoms of depression.8 In older children, sadness and loss of interest in social activities may indicate depression. In adolescents, feelings of mental and physical weariness, aloneness, disconnectedness, uncertainty, vulnerability, anger, irritability, and ambivalence toward friends suggest a depressive disorder.9
Genetic predisposition to depression, poor family support, dysfunctional parenting, and individual vulnerabilities such as poor self-esteem or emotional dysregulation may increase young patients’ risk for depression.10 Peer and family support may protect against depression. Personal competence stemming from social acceptance and body image satisfaction also may be protective factors. A sense of religious and existential well-being (finding meaning and purpose in life) are significantly associated with lower rates of depression among adolescents.11
A persistent illness
The mean duration of a depressive episode in children and adolescents is 7 to 8 months.12 However, subsyndromal depressive symptoms—as well as relapse and recurrence—are common. Long-term studies indicate that many depressed adolescents experience depressive episodes into adulthood.12 Factors that may predict recurrence in adulthood include:
- severity of depressive episodes
- concurrent psychotic symptoms
- suicidal thoughts
- history of recurrent depressive episodes
- threshold residual symptoms
- recent stressful life events
- adverse family environment
- family history of depression.12
Early symptom onset, greater depression severity, suicidality, presence of comorbid anxiety, disruptive disorders, and an adverse family environment also predict longer recovery time.12 A study of depressed adolescents found that a history of recurrent depression, family history of recurrent depression, personality disorder traits, and (for girls only) conflict with parents predicted recurrence of depression in young adulthood.4Table 1 summarizes factors that affect depression outcomes in children and adolescents.
Table 1
What affects depression outcomes in children and adolescents?
| Factor | Outcomes |
|---|---|
| Age | Pharmacotherapy and CBT are equally effective in younger and older adolescents.a Although age does not affect long-term treatment outcomes, older adolescents (age 18 to 19) with treatment-resistant depression may respond better to a combination of CBT and medicationb |
| Sex | Females are more likely to experience relapse.c However, sex does not influence response to initial treatmentc |
| Socioeconomic status | Adolescents with high socioeconomic status are more likely to respond to CBT |
| Illness characteristics | Severity of depression is the strongest predictor of poor outcome.d-f Patients with moderate depression are more likely to benefit from CBT added to medication.g However, adding CBT to medication did not affect outcomes in adolescents with self-injurious behavior.b,f Suicidal behaviors during treatment are less frequent when CBT is combined with medicationh,i |
| Substance abuse | Patients with substance use disorders are less likely to respond to depression treatmentf and those who continued to abuse substances during treatment are less likely to achieve remission than those who abstainb |
| Cognitive measures | Higher levels of hopelessness are associated with poor outcomes. For adolescents with treatment-resistant depression who experience hopelessness, adding CBT to pharmacotherapy did not provide additional benefit. Some studies have noted that adolescents with cognitive distortions are more likely to benefit from CBT plus pharmacotherapyb |
| Family characteristics/environment | High family stress is associated with poor treatment outcomes.f Experiencing loss and physically dangerous events does not affect depression outcomes. Trauma and history of abuse adversely effect depression treatment outcomes |
CBT: cognitive-behavioral therapy
| |
Assessment strategies
Semi-structured interviews such as the Child and Adolescent Psychiatric Assessment, the Diagnostic Interview for Children and Adolescents, and the Kiddie Schedule for Affective Disorder and Schizophrenia are useful for assessing depression in pediatric patients (Table 2).13-16 These tools can be used to assess depression criteria based on information gathered from several sources. Many instruments can be used to assess and monitor pediatric depression, including the Children’s Depression Inventory, the Reynolds Child Depression Scales and Adolescent Depression Scales, and the Child Depression Rating Scale.
To assess suicide risk in depressed younger patients:
- ask about emotional difficulties
- identify lack of developmental progress
- estimate their level of distress
- detect impairment in functioning
- estimate the level of danger to themselves and others.17
The best way to assess for suicidal ideation is to ask about it directly while interviewing the patient and his or her parents. Simple questions such as “Have you ever thought about killing yourself or wish you were dead?” and “Have you ever done anything on purpose to hurt or kill yourself?” can be effective.10 These questions are best placed in the middle or toward the end of a list of questions about depressive symptoms.
Adolescents may be more likely than adults to disclose information about suicidality on self-reports.6 However, self-assessment suicide scales are not a substitute for clinical assessment because they tend to be oversensitive and non-specific and lack predictive value. A positive response to either of these questions should prompt a more detailed clinical investigation. There is no evidence that asking about suicide risk increases suicidal behavior, even in high-risk youths.
Table 2
Assessing children and adolescents: 3 semi-structured interviews
| Interview | Features |
|---|---|
| Child and Adolescent Psychiatric Assessment13 | For patients age 9 to 17. Assesses symptoms from the past 3 months. Administration time: 1 to 2 hours. Requires minimal interviewer experience. Assesses impairment in multiple areas (family, peers, school, leisure activities) |
| Diagnostic Interview for Children and Adolescents14 | Separate versions for children (age 6 to 12) and adolescents (age 13 to 17). Assesses lifetime psychopathology. Administration time: 1 to 2 hours. Interrater reliability varies (poor to good) |
| Kiddie Schedule for Affective Disorders and Schizophrenia15 | Assesses lifetime and current psychopathology. Administration time: 35 minutes to 2.5 hours. Interrater reliability: fair to excellent16 |
Treatment options
Psychotherapy. Several controlled studies and meta-analyses support the efficacy of cognitive-behavioral therapy (CBT) for mild depression in pediatric patients.18-20 Two recent meta-analyses of CBT studies in depressed adolescents found the mean effect size of CBT was 0.34 to 0.35.19,21 However, a separate analysis found CBT did not have long-term benefits for depressed adolescents, particularly patients with a history of abuse.22
Interpersonal therapy also can be effective in adolescent outpatients with mild to moderate depression. One study found the effect size of psychotherapy was modest (0.36).19
Pharmacotherapy. Two meta-analyses support selective serotonin reuptake inhibitors (SSRIs) for treating mild to moderate depression in children and adolescents. One found 61% of depressed patients age <19 who received an SSRI were “much improved” or “very much improved.”23 Another meta-analysis that compared SSRIs and placebo found fluoxetine was more effective than sertraline or citalopram for depressed adolescents.24 Other studies have shown that for severe depression, the effect size of antidepressants (0.69) is higher than that of placebo (0.39).25 Antidepressants are more effective in adolescents than in children.25
Fluoxetine is the only FDA-approved medication for treating depression in children age ≥8. In 2007 the FDA extended to all antidepressants its “black-box” warning about increased risk of suicidality in patients up to age 24. The results of studies that analyzed data about the safety of antidepressants in children and adolescents have been mixed—some found evidence of increased suicidality with antidepressant use,26,27 whereas others showed no increased risk.28,29Table 3 summarizes steps to minimize the risk of antidepressant-induced suicidality.17
Psychotherapy plus pharmacotherapy. Researchers who compared fluoxetine to CBT and to a combination of the 2 in adolescents with moderate to severe depression found that fluoxetine was most effective in the first 12 weeks of treatment.30 Surprisingly, CBT’s effectiveness was not different from placebo.30 However, studies have shown that combining psychotherapy and medication results in greater symptom improvement,30 faster clinical response,31 improvement of global functioning and quality of life,32 and reduced suicidality.33 At 6 months, the difference in response between medication and psychotherapy was small.25 The Treatment of Resistant Depression in Adolescents study found that for chronic adolescent depression, pharmacotherapy (fluoxetine and venlafaxine) combined with CBT produced a higher response rate than pharmacotherapy alone (54% vs 41%).34
Table 3
Protecting against antidepressant-induced suicidality
| Before initiating antidepressant treatment |
|---|
| Review the patient’s psychiatric history |
| Assess for past suicidal behavior |
| Assess for a family history of mental illness or mood disorders and suicide attempts |
| Screen for unrecognized bipolar spectrum disorders |
| Educate patients and their families to watch for signs of worsening depression or suicidality, and to report such symptoms immediately |
| During antidepressant treatment |
| Pay attention to abrupt changes in symptoms, particularly symptoms that were not part of the patient’s initial presentation |
| Watch for deterioration of symptoms |
| Monitor for emergence of ‘activating’ symptoms (ie, irritability, impulsivity, anxiety, insomnia, agitation, hostility, akathisia, hypomania, or mania) |
| Evaluate the patient’s suicide risk factors, including having a specific plan and/or access to lethal means |
| Consider hospitalization if the patient is at high risk for suicide |
| Source: Reference 17 |
Lowering suicide risk
Up to 60% of adolescents who commit suicide had a depressive disorder. Risk factors for child and adolescent suicide attempts include:
- self-harm behaviors
- psychiatric disorders
- family disturbances
- substance abuse
- physical/sexual abuse.17
How to best manage suicidal youths depends on an adequate assessment of the severity of the patient’s current problems and conflicts and the degree of suicidal intent. Assessment of coping resources, access to support systems, and the attitude of the patient and family toward intervention and follow-up also is important.
Children and adolescents at high risk for suicide—those with a plan or recent suicide attempt with high probability of lethality, stated current intent to kill themselves, or recent suicidal ideation or behavior—may need inpatient psychiatric admission. Although no studies have shown that admitting high-risk suicidal patients prevents suicide, hospitalization often is the safest course of action. Develop ing a comprehensive outpatient treatment plan before discharge is essential. Patients with fewer risk factors, especially those who want help and have social support, hope for the future, and a desire to resolve conflicts, may require only a brief crisis-oriented intervention.
The following recommendations for managing suicidality in children and adolescents are based on clinical experience and have not been empirically validated.
Develop a safety plan to direct the patient’s behavior under various situations. For example, the patient would agree in writing that “If I feel depressed, I will do X, Y, and Z to address it,” or “If I find myself having suicidal thoughts, I will contact ABC.” Having a safety plan lowers the risk of a suicide attempt more than having a suicide contract, which does not give the patient any tools.35
Create a ‘hope box.’ This is a box in which the patient collects mementos and other objects that remind him or her of hope and reasons to live. The patient should be able to access it at all times, so he or she can tap into it during crisis periods to avert suicidal acts.35
Counteract alienation. A sense of social isolation and burdensomeness may be “tipping factors” for suicidal acts when adolescents feel depressed.35 Clinicians should try to help connect patients to meaningful social activities, even in small doses.
Manage overarousal. Overarousal in depressed children and adolescents is manifested as agitation. Insomnia is a clinically modifiable risk factor. Insomnia initially responds well to behavioral interventions such as sleep hygiene, sleep restriction, and stimulus control techniques.35
Related Resources
- National Suicide Prevention Lifeline. 800-273-TALK (8255). www.suicidepreventionlifeline.org.
- Suicide Prevention Resource Center. www.sprc.org.
Drug Brand Names
- Citalopram • Celexa
- Fluoxetine • Prozac
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Drs. Shailesh Jain and Islam report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rakesh Jain is a consultant to or has received research/grant support from Eli Lilly and Company, Merck, Pfizer Inc., Shionogi Pharmaceuticals, and Shire.
Discuss this article at www.facebook.com/CurrentPsychiatry
Although depression affects nearly 2% of children (age ≤12) and up to 10% of adolescents (age 13 to 18),1 the disorder often is underdiagnosed and undertreated in pediatric patients.2 Treating depression in young patients is challenging. Only 30% to 40% of depressed children and adolescents who receive evidence-based treatment achieve remission.3 In addition, 50% to 70% of those who initially achieve remission will experience recurrence within 5 years.4 Suicide is the third leading cause of death among children and adolescents, and depression greatly increases the likelihood of suicide.5,6
This article reviews assessing and treating depression in children and adolescents, and how to lower suicide risk in pediatric patients.
Symptoms vary with age
Depressive symptoms vary as a function of the child’s cognitive development and social functioning. Hopelessness and vegetative and motivational symptoms may be more frequent in adolescents than in children.7
In preschool-age children, depression manifests indirectly through somatic symptoms and behavioral disturbances. In this age group, sadness or irritability are sensitive and predominant symptoms of depression.8 In older children, sadness and loss of interest in social activities may indicate depression. In adolescents, feelings of mental and physical weariness, aloneness, disconnectedness, uncertainty, vulnerability, anger, irritability, and ambivalence toward friends suggest a depressive disorder.9
Genetic predisposition to depression, poor family support, dysfunctional parenting, and individual vulnerabilities such as poor self-esteem or emotional dysregulation may increase young patients’ risk for depression.10 Peer and family support may protect against depression. Personal competence stemming from social acceptance and body image satisfaction also may be protective factors. A sense of religious and existential well-being (finding meaning and purpose in life) are significantly associated with lower rates of depression among adolescents.11
A persistent illness
The mean duration of a depressive episode in children and adolescents is 7 to 8 months.12 However, subsyndromal depressive symptoms—as well as relapse and recurrence—are common. Long-term studies indicate that many depressed adolescents experience depressive episodes into adulthood.12 Factors that may predict recurrence in adulthood include:
- severity of depressive episodes
- concurrent psychotic symptoms
- suicidal thoughts
- history of recurrent depressive episodes
- threshold residual symptoms
- recent stressful life events
- adverse family environment
- family history of depression.12
Early symptom onset, greater depression severity, suicidality, presence of comorbid anxiety, disruptive disorders, and an adverse family environment also predict longer recovery time.12 A study of depressed adolescents found that a history of recurrent depression, family history of recurrent depression, personality disorder traits, and (for girls only) conflict with parents predicted recurrence of depression in young adulthood.4Table 1 summarizes factors that affect depression outcomes in children and adolescents.
Table 1
What affects depression outcomes in children and adolescents?
| Factor | Outcomes |
|---|---|
| Age | Pharmacotherapy and CBT are equally effective in younger and older adolescents.a Although age does not affect long-term treatment outcomes, older adolescents (age 18 to 19) with treatment-resistant depression may respond better to a combination of CBT and medicationb |
| Sex | Females are more likely to experience relapse.c However, sex does not influence response to initial treatmentc |
| Socioeconomic status | Adolescents with high socioeconomic status are more likely to respond to CBT |
| Illness characteristics | Severity of depression is the strongest predictor of poor outcome.d-f Patients with moderate depression are more likely to benefit from CBT added to medication.g However, adding CBT to medication did not affect outcomes in adolescents with self-injurious behavior.b,f Suicidal behaviors during treatment are less frequent when CBT is combined with medicationh,i |
| Substance abuse | Patients with substance use disorders are less likely to respond to depression treatmentf and those who continued to abuse substances during treatment are less likely to achieve remission than those who abstainb |
| Cognitive measures | Higher levels of hopelessness are associated with poor outcomes. For adolescents with treatment-resistant depression who experience hopelessness, adding CBT to pharmacotherapy did not provide additional benefit. Some studies have noted that adolescents with cognitive distortions are more likely to benefit from CBT plus pharmacotherapyb |
| Family characteristics/environment | High family stress is associated with poor treatment outcomes.f Experiencing loss and physically dangerous events does not affect depression outcomes. Trauma and history of abuse adversely effect depression treatment outcomes |
CBT: cognitive-behavioral therapy
| |
Assessment strategies
Semi-structured interviews such as the Child and Adolescent Psychiatric Assessment, the Diagnostic Interview for Children and Adolescents, and the Kiddie Schedule for Affective Disorder and Schizophrenia are useful for assessing depression in pediatric patients (Table 2).13-16 These tools can be used to assess depression criteria based on information gathered from several sources. Many instruments can be used to assess and monitor pediatric depression, including the Children’s Depression Inventory, the Reynolds Child Depression Scales and Adolescent Depression Scales, and the Child Depression Rating Scale.
To assess suicide risk in depressed younger patients:
- ask about emotional difficulties
- identify lack of developmental progress
- estimate their level of distress
- detect impairment in functioning
- estimate the level of danger to themselves and others.17
The best way to assess for suicidal ideation is to ask about it directly while interviewing the patient and his or her parents. Simple questions such as “Have you ever thought about killing yourself or wish you were dead?” and “Have you ever done anything on purpose to hurt or kill yourself?” can be effective.10 These questions are best placed in the middle or toward the end of a list of questions about depressive symptoms.
Adolescents may be more likely than adults to disclose information about suicidality on self-reports.6 However, self-assessment suicide scales are not a substitute for clinical assessment because they tend to be oversensitive and non-specific and lack predictive value. A positive response to either of these questions should prompt a more detailed clinical investigation. There is no evidence that asking about suicide risk increases suicidal behavior, even in high-risk youths.
Table 2
Assessing children and adolescents: 3 semi-structured interviews
| Interview | Features |
|---|---|
| Child and Adolescent Psychiatric Assessment13 | For patients age 9 to 17. Assesses symptoms from the past 3 months. Administration time: 1 to 2 hours. Requires minimal interviewer experience. Assesses impairment in multiple areas (family, peers, school, leisure activities) |
| Diagnostic Interview for Children and Adolescents14 | Separate versions for children (age 6 to 12) and adolescents (age 13 to 17). Assesses lifetime psychopathology. Administration time: 1 to 2 hours. Interrater reliability varies (poor to good) |
| Kiddie Schedule for Affective Disorders and Schizophrenia15 | Assesses lifetime and current psychopathology. Administration time: 35 minutes to 2.5 hours. Interrater reliability: fair to excellent16 |
Treatment options
Psychotherapy. Several controlled studies and meta-analyses support the efficacy of cognitive-behavioral therapy (CBT) for mild depression in pediatric patients.18-20 Two recent meta-analyses of CBT studies in depressed adolescents found the mean effect size of CBT was 0.34 to 0.35.19,21 However, a separate analysis found CBT did not have long-term benefits for depressed adolescents, particularly patients with a history of abuse.22
Interpersonal therapy also can be effective in adolescent outpatients with mild to moderate depression. One study found the effect size of psychotherapy was modest (0.36).19
Pharmacotherapy. Two meta-analyses support selective serotonin reuptake inhibitors (SSRIs) for treating mild to moderate depression in children and adolescents. One found 61% of depressed patients age <19 who received an SSRI were “much improved” or “very much improved.”23 Another meta-analysis that compared SSRIs and placebo found fluoxetine was more effective than sertraline or citalopram for depressed adolescents.24 Other studies have shown that for severe depression, the effect size of antidepressants (0.69) is higher than that of placebo (0.39).25 Antidepressants are more effective in adolescents than in children.25
Fluoxetine is the only FDA-approved medication for treating depression in children age ≥8. In 2007 the FDA extended to all antidepressants its “black-box” warning about increased risk of suicidality in patients up to age 24. The results of studies that analyzed data about the safety of antidepressants in children and adolescents have been mixed—some found evidence of increased suicidality with antidepressant use,26,27 whereas others showed no increased risk.28,29Table 3 summarizes steps to minimize the risk of antidepressant-induced suicidality.17
Psychotherapy plus pharmacotherapy. Researchers who compared fluoxetine to CBT and to a combination of the 2 in adolescents with moderate to severe depression found that fluoxetine was most effective in the first 12 weeks of treatment.30 Surprisingly, CBT’s effectiveness was not different from placebo.30 However, studies have shown that combining psychotherapy and medication results in greater symptom improvement,30 faster clinical response,31 improvement of global functioning and quality of life,32 and reduced suicidality.33 At 6 months, the difference in response between medication and psychotherapy was small.25 The Treatment of Resistant Depression in Adolescents study found that for chronic adolescent depression, pharmacotherapy (fluoxetine and venlafaxine) combined with CBT produced a higher response rate than pharmacotherapy alone (54% vs 41%).34
Table 3
Protecting against antidepressant-induced suicidality
| Before initiating antidepressant treatment |
|---|
| Review the patient’s psychiatric history |
| Assess for past suicidal behavior |
| Assess for a family history of mental illness or mood disorders and suicide attempts |
| Screen for unrecognized bipolar spectrum disorders |
| Educate patients and their families to watch for signs of worsening depression or suicidality, and to report such symptoms immediately |
| During antidepressant treatment |
| Pay attention to abrupt changes in symptoms, particularly symptoms that were not part of the patient’s initial presentation |
| Watch for deterioration of symptoms |
| Monitor for emergence of ‘activating’ symptoms (ie, irritability, impulsivity, anxiety, insomnia, agitation, hostility, akathisia, hypomania, or mania) |
| Evaluate the patient’s suicide risk factors, including having a specific plan and/or access to lethal means |
| Consider hospitalization if the patient is at high risk for suicide |
| Source: Reference 17 |
Lowering suicide risk
Up to 60% of adolescents who commit suicide had a depressive disorder. Risk factors for child and adolescent suicide attempts include:
- self-harm behaviors
- psychiatric disorders
- family disturbances
- substance abuse
- physical/sexual abuse.17
How to best manage suicidal youths depends on an adequate assessment of the severity of the patient’s current problems and conflicts and the degree of suicidal intent. Assessment of coping resources, access to support systems, and the attitude of the patient and family toward intervention and follow-up also is important.
Children and adolescents at high risk for suicide—those with a plan or recent suicide attempt with high probability of lethality, stated current intent to kill themselves, or recent suicidal ideation or behavior—may need inpatient psychiatric admission. Although no studies have shown that admitting high-risk suicidal patients prevents suicide, hospitalization often is the safest course of action. Develop ing a comprehensive outpatient treatment plan before discharge is essential. Patients with fewer risk factors, especially those who want help and have social support, hope for the future, and a desire to resolve conflicts, may require only a brief crisis-oriented intervention.
The following recommendations for managing suicidality in children and adolescents are based on clinical experience and have not been empirically validated.
Develop a safety plan to direct the patient’s behavior under various situations. For example, the patient would agree in writing that “If I feel depressed, I will do X, Y, and Z to address it,” or “If I find myself having suicidal thoughts, I will contact ABC.” Having a safety plan lowers the risk of a suicide attempt more than having a suicide contract, which does not give the patient any tools.35
Create a ‘hope box.’ This is a box in which the patient collects mementos and other objects that remind him or her of hope and reasons to live. The patient should be able to access it at all times, so he or she can tap into it during crisis periods to avert suicidal acts.35
Counteract alienation. A sense of social isolation and burdensomeness may be “tipping factors” for suicidal acts when adolescents feel depressed.35 Clinicians should try to help connect patients to meaningful social activities, even in small doses.
Manage overarousal. Overarousal in depressed children and adolescents is manifested as agitation. Insomnia is a clinically modifiable risk factor. Insomnia initially responds well to behavioral interventions such as sleep hygiene, sleep restriction, and stimulus control techniques.35
Related Resources
- National Suicide Prevention Lifeline. 800-273-TALK (8255). www.suicidepreventionlifeline.org.
- Suicide Prevention Resource Center. www.sprc.org.
Drug Brand Names
- Citalopram • Celexa
- Fluoxetine • Prozac
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Drs. Shailesh Jain and Islam report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rakesh Jain is a consultant to or has received research/grant support from Eli Lilly and Company, Merck, Pfizer Inc., Shionogi Pharmaceuticals, and Shire.
1. Birmaher B, Brent D. AACAP Work Group on Quality Issues, et al. Practice parameter for the assessment and treatment of children and adolescents with depressive disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(11):1503-1526.
2. Lewinsohn PM, Clarke GN, Seeley JR, et al. Major depression in community adolescents: age at onset, episode duration, and time to recurrence. J Am Acad Child Adolesc Psychiatry. 1994;33(6):809-818.
3. Emslie GJ, Kennard BD, Mayes TL. Predictors of treatment response in adolescent depression. Pediatr Ann. 2011;40(6):300-306.
4. Lewinsohn PM, Rohde P, Seeley JR, et al. Natural course of adolescent major depressive disorder in a community sample: predictors of recurrence in young adults. Am J Psychiatry. 2000;157(10):1584-1591.
5. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
6. Gould MS, Greenberg T, Velting DM, et al. Youth suicide risk and preventive interventions: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry. 2003;42(4):386-405.
7. Weiss B, Garber J. Developmental differences in the phenomenology of depression. Dev Psychopathol. 2003;15(2):403-430.
8. Calles JL, Jr. Depression in children and adolescents. Prim Care. 2007;34(2):243-258abstract vi.
9. Farmer TJ. The experience of major depression: adolescents’ perspectives. Issues Ment Health Nurs. 2002;23(6):567-585.
10. Zalsman G, Brent DA, Weersing VR. Depressive disorders in childhood and adolescence: an overview: epidemiology clinical manifestation and risk factors. Child Adolesc Psychiatr Clin N Am. 2006;15(4):827-841, vii.
11. Cotton S, Larkin E, Hoopes A, et al. The impact of adolescent spirituality on depressive symptoms and health risk behaviors. J Adolesc Health. 2005;36(6):529.-
12. Birmaher B, Arbelaez C, Brent D. Course and outcome of child and adolescent major depressive disorder. Child Adolesc Psychiatr Clin N Am. 2002;11(3):619-637, x.
13. Angold A, Costello EJ. A test-retest reliability study of child-reported psychiatric symptoms and diagnoses using the Child and Adolescent Psychiatric Assessment (CAPA-C). Psychol Med. 1995;25(4):755-762.
14. Reich W. Diagnostic interview for children and adolescents (DICA). J Am Acad Child Adolesc Psychiatry. 2000;39(1):59-66.
15. Puig-Antich J, Lukens E, Brent D. Psychosocial schedule for school age children - revised. Pittsburgh PA: Western Psychiatric Institute and Clinic; 1986.
16. Ambrosini PJ. Historical development and present status of the schedule for affective disorders and schizophrenia for school-age children (K-SADS). J Am Acad Child Adolesc Psychiatry. 2000;39(1):49-58.
17. Dodig-Curković K, Curković M, Radić J, et al. Suicidal behavior and suicide among children and adolescents-risk factors and epidemiological characteristics. Coll Antropol. 2010;34(2):771-777.
18. Harrington R, Whittaker J, Shoebridge P, et al. Systematic review of efficacy of cognitive behaviour therapies in childhood and adolescent depressive disorder. BMJ. 1998;316(7144):1559-1563.
19. Weisz JR, McCarty CA, Valeri SM. Effects of psychotherapy for depression in children and adolescents: a meta-analysis. Psychol Bull. 2006;132(1):132-149.
20. Mufson L, Dorta KP, Wickramaratne P, et al. A randomized effectiveness trial of interpersonal psychotherapy for depressed adolescents. Arch Gen Psychiatry. 2004;61(6):577-584.
21. Klein JB, Jacobs RH, Reinecke MA. Cognitive-behavioral therapy for adolescent depression: a meta-analytic investigation of changes in effect-size estimates. J Am Acad Child Adolesc Psychiatry. 2007;46(11):1403-1413.
22. Vitiello B, Emslie G, Clarke G, et al. Long-term outcome of adolescent depression initially resistant to selective serotonin reuptake inhibitor treatment: a follow-up study of the TORDIA sample. J Clin Psychiatry. 2011;72(3):388-396.
23. Bridge JA, Iyengar S, Salary CB, et al. Clinical response and risk for reported suicidal ideation and suicide attempts in pediatric antidepressant treatment: a meta-analysis of randomized controlled trials. JAMA. 2007;297(15):1683-1696.
24. Usala T, Clavenna A, Zuddas A, et al. Randomised controlled trials of selective serotonin reuptake inhibitors in treating depression in children and adolescents: a systematic review and meta-analysis. Eur Neuropsychopharmacol. 2008;18(1):62-73.
25. March JS, Silva S, Petrycki S, et al. The Treatment for Adolescents With Depression Study (TADS): long-term effectiveness and safety outcomes. Arch Gen Psychiatry. 2007;64(10):1132-1143.
26. Bridge JA, Iyengar S, Salary CB, et al. Clinical response and risk for reported suicidal ideation and suicide attempts in pediatric antidepressant treatment: a meta-analysis of randomized controlled trials. JAMA. 2007;297(15):1683-1696.
27. Stone M, Laughren T, Jones ML, et al. Risk of suicidality in clinical trials of antidepressants in adults: analysis of proprietary data submitted to US Food and Drug Administration. BMJ. 2009;339:b2880.-
28. Khan A, Khan S, Kolts R, et al. Suicide rates in clinical trials of SSRIs, other antidepressants, and placebo: analysis of FDA reports. Am J Psychiatry. 2003;160(4):790-792.
29. Simon GE, Savarino J. Suicide attempts among patients starting depression treatment with medications or psychotherapy. Am J Psychiatry. 2007;164(7):1029-1034.
30. March J, Silva S, Petrycki S, et al. Fluoxetine, cognitive-behavioral therapy, and their combination for adolescents with depression: Treatment for Adolescents With Depression Study (TADS) randomized controlled trial. JAMA. 2004;292(7):807-820.
31. Kratochvil C, Emslie G, Silva S, et al. Acute time to response in the Treatment for Adolescents with Depression Study (TADS). J Am Acad Child Adolesc Psychiatry. 2006;45(12):1412-1418.
32. Vitiello B, Rohde P, Silva S, et al. Functioning and quality of life in the Treatment for Adolescents with Depression Study (TADS). J Am Acad Child Adolesc Psychiatry. 2006;45(12):1419-1426.
33. Emslie G, Kratochvil C, Vitiello B, et al. Treatment for Adolescents with Depression Study (TADS): safety results. J Am Acad Child Adolesc Psychiatry. 2006;45(12):1440-1455.
34. Brent D, Emslie G, Clarke G, et al. Switching to another SSRI or to venlafaxine with or without cognitive behavioral therapy for adolescents with SSRI-resistant depression: the TORDIA randomized controlled trial. JAMA. 2008;299(8):901-913.
35. Joiner TE, Ribeiro JD. Assessment and management of suicidal behavior in teens. Psychiatr Ann. 2011;41(4):220-225.
1. Birmaher B, Brent D. AACAP Work Group on Quality Issues, et al. Practice parameter for the assessment and treatment of children and adolescents with depressive disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(11):1503-1526.
2. Lewinsohn PM, Clarke GN, Seeley JR, et al. Major depression in community adolescents: age at onset, episode duration, and time to recurrence. J Am Acad Child Adolesc Psychiatry. 1994;33(6):809-818.
3. Emslie GJ, Kennard BD, Mayes TL. Predictors of treatment response in adolescent depression. Pediatr Ann. 2011;40(6):300-306.
4. Lewinsohn PM, Rohde P, Seeley JR, et al. Natural course of adolescent major depressive disorder in a community sample: predictors of recurrence in young adults. Am J Psychiatry. 2000;157(10):1584-1591.
5. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
6. Gould MS, Greenberg T, Velting DM, et al. Youth suicide risk and preventive interventions: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry. 2003;42(4):386-405.
7. Weiss B, Garber J. Developmental differences in the phenomenology of depression. Dev Psychopathol. 2003;15(2):403-430.
8. Calles JL, Jr. Depression in children and adolescents. Prim Care. 2007;34(2):243-258abstract vi.
9. Farmer TJ. The experience of major depression: adolescents’ perspectives. Issues Ment Health Nurs. 2002;23(6):567-585.
10. Zalsman G, Brent DA, Weersing VR. Depressive disorders in childhood and adolescence: an overview: epidemiology clinical manifestation and risk factors. Child Adolesc Psychiatr Clin N Am. 2006;15(4):827-841, vii.
11. Cotton S, Larkin E, Hoopes A, et al. The impact of adolescent spirituality on depressive symptoms and health risk behaviors. J Adolesc Health. 2005;36(6):529.-
12. Birmaher B, Arbelaez C, Brent D. Course and outcome of child and adolescent major depressive disorder. Child Adolesc Psychiatr Clin N Am. 2002;11(3):619-637, x.
13. Angold A, Costello EJ. A test-retest reliability study of child-reported psychiatric symptoms and diagnoses using the Child and Adolescent Psychiatric Assessment (CAPA-C). Psychol Med. 1995;25(4):755-762.
14. Reich W. Diagnostic interview for children and adolescents (DICA). J Am Acad Child Adolesc Psychiatry. 2000;39(1):59-66.
15. Puig-Antich J, Lukens E, Brent D. Psychosocial schedule for school age children - revised. Pittsburgh PA: Western Psychiatric Institute and Clinic; 1986.
16. Ambrosini PJ. Historical development and present status of the schedule for affective disorders and schizophrenia for school-age children (K-SADS). J Am Acad Child Adolesc Psychiatry. 2000;39(1):49-58.
17. Dodig-Curković K, Curković M, Radić J, et al. Suicidal behavior and suicide among children and adolescents-risk factors and epidemiological characteristics. Coll Antropol. 2010;34(2):771-777.
18. Harrington R, Whittaker J, Shoebridge P, et al. Systematic review of efficacy of cognitive behaviour therapies in childhood and adolescent depressive disorder. BMJ. 1998;316(7144):1559-1563.
19. Weisz JR, McCarty CA, Valeri SM. Effects of psychotherapy for depression in children and adolescents: a meta-analysis. Psychol Bull. 2006;132(1):132-149.
20. Mufson L, Dorta KP, Wickramaratne P, et al. A randomized effectiveness trial of interpersonal psychotherapy for depressed adolescents. Arch Gen Psychiatry. 2004;61(6):577-584.
21. Klein JB, Jacobs RH, Reinecke MA. Cognitive-behavioral therapy for adolescent depression: a meta-analytic investigation of changes in effect-size estimates. J Am Acad Child Adolesc Psychiatry. 2007;46(11):1403-1413.
22. Vitiello B, Emslie G, Clarke G, et al. Long-term outcome of adolescent depression initially resistant to selective serotonin reuptake inhibitor treatment: a follow-up study of the TORDIA sample. J Clin Psychiatry. 2011;72(3):388-396.
23. Bridge JA, Iyengar S, Salary CB, et al. Clinical response and risk for reported suicidal ideation and suicide attempts in pediatric antidepressant treatment: a meta-analysis of randomized controlled trials. JAMA. 2007;297(15):1683-1696.
24. Usala T, Clavenna A, Zuddas A, et al. Randomised controlled trials of selective serotonin reuptake inhibitors in treating depression in children and adolescents: a systematic review and meta-analysis. Eur Neuropsychopharmacol. 2008;18(1):62-73.
25. March JS, Silva S, Petrycki S, et al. The Treatment for Adolescents With Depression Study (TADS): long-term effectiveness and safety outcomes. Arch Gen Psychiatry. 2007;64(10):1132-1143.
26. Bridge JA, Iyengar S, Salary CB, et al. Clinical response and risk for reported suicidal ideation and suicide attempts in pediatric antidepressant treatment: a meta-analysis of randomized controlled trials. JAMA. 2007;297(15):1683-1696.
27. Stone M, Laughren T, Jones ML, et al. Risk of suicidality in clinical trials of antidepressants in adults: analysis of proprietary data submitted to US Food and Drug Administration. BMJ. 2009;339:b2880.-
28. Khan A, Khan S, Kolts R, et al. Suicide rates in clinical trials of SSRIs, other antidepressants, and placebo: analysis of FDA reports. Am J Psychiatry. 2003;160(4):790-792.
29. Simon GE, Savarino J. Suicide attempts among patients starting depression treatment with medications or psychotherapy. Am J Psychiatry. 2007;164(7):1029-1034.
30. March J, Silva S, Petrycki S, et al. Fluoxetine, cognitive-behavioral therapy, and their combination for adolescents with depression: Treatment for Adolescents With Depression Study (TADS) randomized controlled trial. JAMA. 2004;292(7):807-820.
31. Kratochvil C, Emslie G, Silva S, et al. Acute time to response in the Treatment for Adolescents with Depression Study (TADS). J Am Acad Child Adolesc Psychiatry. 2006;45(12):1412-1418.
32. Vitiello B, Rohde P, Silva S, et al. Functioning and quality of life in the Treatment for Adolescents with Depression Study (TADS). J Am Acad Child Adolesc Psychiatry. 2006;45(12):1419-1426.
33. Emslie G, Kratochvil C, Vitiello B, et al. Treatment for Adolescents with Depression Study (TADS): safety results. J Am Acad Child Adolesc Psychiatry. 2006;45(12):1440-1455.
34. Brent D, Emslie G, Clarke G, et al. Switching to another SSRI or to venlafaxine with or without cognitive behavioral therapy for adolescents with SSRI-resistant depression: the TORDIA randomized controlled trial. JAMA. 2008;299(8):901-913.
35. Joiner TE, Ribeiro JD. Assessment and management of suicidal behavior in teens. Psychiatr Ann. 2011;41(4):220-225.
Binge eating disorder: Evidence-based treatments
Binge eating is consumption of an unusually large amount of food coupled with a feeling of loss of control over eating. Binge eating disorder (BED) is characterized by recurrent episodes of binge eating without inappropriate compensatory behaviors (eg, self-induced vomiting, misuse of laxatives, diuretics, or other agents, excessive exercise).1 It is the most common eating disorder in the United States, with a lifetime prevalence of approximately 3.5% in women and 2% in men.2 The diagnosis falls within the DSM-IV-TR category of eating disorders not otherwise specified,1 but clinicians often view it as a distinct clinical phenomenon. In DSM-IV-TR, an individual would meet criteria for BED if he or she engages in regular binge eating behavior in the absence of recurrent compensatory behaviors ≥2 days per week over 6 months.1 Proposed changes for DSM-5 recognize a distinct BED diagnosis, reduce the frequency criterion to once per week and the duration criterion to the past 3 months, and shift the focus from binge days to binge episodes (Table 1).3
Table 1
Proposed DSM-5 criteria for binge eating disorder
|
| Source:Reference 3 |
BED can occur in individuals of all body mass indices (BMI), but is common among individuals who are overweight or obese as well as those with depression or type 2 diabetes; BED can complicate treatment of these conditions.2,4,5 Primary treatment goals are:
- abstinence from binge eating
- improved psychological functioning
- appropriate weight regulation in overweight patients.
We report on 3 approaches to BED treatment: medication only, behavioral intervention only, and medication plus behavioral intervention. This article provides insights about emerging changes in diagnostic criteria for BED as well as evidence-informed treatment options and recommendations.
The evidence base
We conducted a review of 23 BED studies: 7 medication only, 5 medication plus behavioral, and 11 behavioral only. We focused on studies conducted since September 2005 that included binge frequency, weight, and depression as primary outcomes (see Berkman et al6 for a review of BED treatment studies before 2005). The studies included 2,527 participants (2,216 women and 311 men). Although the sex distribution of BED in the general population tends to slightly favor women,2 the proportion of women presenting for treatment generally is considerably higher than that of men. In studies that reported on race and/or ethnicity, 1,639 participants were identified as white, 191 as African American, 25 as Hispanic, 2 as Asian, 1 as Native American, and 25 as “other.” Ages ranged from 18 to 77.
Several medications are effective
In placebo-controlled studies, a high-dose selective serotonin reuptake inhibitor (escitalopram7), 2 anticonvulsants (zonisamide8 and topiramate9), a selective norepinephrine reuptake inhibitor (atomoxetine10), and an appetite suppressant (sibutramine11) were associated with significant decreases in binge eating frequency, weight, and BMI in overweight/obese patients diagnosed with BED (Table 2). In an open-label trial, memantine—a N-methyl-D-aspartate receptor antagonist often used to treat symptoms of Alzheimer’s disease—was associated with a significant reduction in binge eating but no change in weight.12 Lamotrigine was not significantly different from placebo in reducing binge eating or weight, but showed promise in reducing metabolic parameters such as glucose and triglyceride levels commonly associated with obesity and type 2 diabetes.13 Because BED often is comorbid with obesity and type 2 diabetes, lamotrigine augmentation when treating obese individuals with BED warrants further investigation. As with any pharmacologic agent, carefully consider potential side effects and interactions with other drugs before prescribing medications for BED. Informing patients of potential side effects is crucial for patient safety and accuracy of the data collected in well-controlled treatment studies.
Table 2
Pharmacotherapy for binge eating disorder
| Study | Drug/dosage | Comments |
|---|---|---|
| Guerdjikova et al, 20087 | Escitalopram, 10 to 30 mg/d, vs placebo for 12 weeks | Escitalopram was significantly better than placebo in reducing weight, BMI, and illness severity |
| McElroy et al, 20068 | Zonisamide, 100 to 600 mg/d, vs placebo for 16 weeks | Zonisamide was significantly better than placebo in reducing BE, weight, BMI, and various aspects of unhealthy eating behavior |
| McElroy et al, 20079 | Topiramate, 25 to 400 mg/d, vs placebo for 16 weeks | Topiramate was significantly better than placebo in reducing BE, weight, BMI, and related psychological features of BE |
| McElroy et al, 200710 | Atomoxetine, 40 to 120 mg/d, vs placebo for 10 weeks | Atomoxetine was significantly better than placebo in reducing BE, weight, BMI, and obsessive-compulsive features of BE, and in achieving remission |
| Wilfley et al, 200811 | Sibutramine, 15 mg/d, vs placebo for 24 weeks | Sibutramine was significantly better than placebo in reducing BE, weight, BMI, and related psychological features of BE |
| Brennan et al, 200812 | Open-label memantine, 5 to 20 mg/d, for 12 weeks | Memantine was associated with decreased binge frequency and related psychological features of BE |
| Guerdjikova et al, 200913 | Lamotrigine, 50 to 400 mg/d, vs placebo for 16 weeks | Lamotrigine was not significantly different from placebo |
| BE: binge eating; BMI: body mass index | ||
CBT vs other behavioral approaches
Cognitive-behavioral therapy (CBT), which focuses on identifying and modifying unhealthy thoughts that maintain disordered eating behaviors, is the most widely studied behavioral intervention for BED. Other studied treatments include interpersonal psychotherapy (IPT), motivational interviewing (MI), and structured behavioral weight loss (BWL) (Table 3).14-24 IPT is a psychodynamically based, time-limited treatment that focuses on the interpersonal context of the disorder and on building interpersonal skills. MI emphasizes exploring and resolving ambivalence about treatment, and works to facilitate change through motivational processes. BWL is centered on making dietary and physical activity changes to achieve weight loss. Behavioral treatments have been delivered in various formats, such as an individual or group setting, by electronic interface, and via self-help approaches. Most studies compared active treatment to a control group, but some compared active treatments head-to-head.
Table 3
CBT and other behavioral interventions for BED
| Study | Intervention | Comments |
|---|---|---|
| Annunziato et al, 200914 | 2 groups received CBT and hypocaloric diet for 8 weeks followed by 14 weeks of enhanced nutritional program (ie, reduced consumption of high energy density foods and once-daily liquid meal replacement) or control (normal diet) | Enhanced nutritional program was not significantly different from the control in reducing weight, BE, or psychological features of BE; variability in adherence to the enhanced nutritional program was identified as a significant effect modifier |
| Ashton et al, 200915 | 4 sessions of group CBT in an open trial | CBT was associated with significant reductions in BE and psychological features of BE in post-bariatric surgery patients |
| Dingemans et al, 200716 | CBT vs wait-list control | CBT significantly better than the wait-list control in reducing BE and psychological features of BE, and in achieving abstinence from BE |
| Friederich et al, 200717 | 15-session CBT blended with elements of interpersonal therapy (IPT), nutritional counseling, and supervised walking program; no control group | Treatment significantly reduced weight, BE, and related psychological features of BE in patients meeting sub-threshold and full criteria for BED |
| Grilo et al, 200518 | Guided self-help CBT (CBTgsh) vs guided self-help behavioral weight loss (BWLgsh) vs non-specific attention control for 12 weeks | CBTgsh significantly better than BWLgsh and control in BE remission; CBTgsh significantly better than BWLgsh, which was significantly better than control in reducing cognitive restraint; CBTgsh significantly better than control in reducing depression and eating-related psychopathology; no differences between groups in BMI change |
| Ricca et al, 201019 | Individual (I-CBT) vs group CBT (G-CBT) for 24 weeks in patients meeting subthreshold and full criteria for BED | BE and BMI were significantly reduced in both groups at 24 weeks and 3-year follow-up. I-CBT was not better than G-CBT in reducing BE or weight at 24 weeks or 3-year follow-up; I-CBT was significantly better than G-CBT in reducing eating-related psychopathology at 24 weeks and 3-year follow-up; I-CBT was significantly better than G-CBT in recovery (ie, no longer meeting full BED criteria) at 24 weeks but not at 3-year follow-up |
| Schlup et al, 200920 | 8 weekly sessions of group CBT vs wait-list control | CBT was significantly better than wait-list control in reducing BE and eating concerns and in achieving abstinence at end of treatment; CBT was not different than control in reducing BMI; treatment-related reductions in BE and eating concerns were maintained at 12-month follow-up |
| Shapiro et al, 200721 | 10 weekly sessions of group CBT (G-CBT) vs CD-ROM delivered CBT (CD-CBT) vs wait-list control | G-CBT and CD-CBT were not different from each other but both were significantly better than wait-list control in reducing BE |
| Tasca et al, 200622 | Group CBT (G-CBT) vs group psychodynamic interpersonal therapy (G-IPT) vs wait-list control for 16 weeks | G-CBT and G-IPT were not different from each other; G-CBT and G-IPT were significantly better than wait list in reducing BE and interpersonal problems (but not BMI) and increasing cognitive restraint post-treatment; depression was reduced in both groups at 6 months but only in G-IPT at 12 months; reductions in BE maintained at 12 months |
| Wilson et al, 201023 | 10 sessions of guided self-help CBT (CBTgsh) vs 19 sessions of IPT vs 20 sessions of behavioral weight loss (BWL) over 6 months | BWL was significantly better than IPT and CBTgsh in reducing BMI and in the number of patients achieving 5% weight loss at post-treatment but effects were not sustained over time; BWL was significantly better than CBTgsh in increasing dietary restraint |
| Cassin et al, 200824 | Self-help book + motivational interviewing (SH-MI) vs self-help book alone (SH) for 16 weeks | SH-MI was significantly better than SH in reducing BE and depression |
| BE: binge eating; BED: binge eating disorder; BMI: body mass index; CBT: cognitive-behavioral therapy | ||
Studies found that CBT and IPT are effective in reducing the frequency of binge eating, whether measured by the number of binge eating episodes or days a patient reports having engaged in binge eating.14-23 However, some studies suggested that CBT can help a substantial number of patients achieve abstinence from binge eating.16,20 Adding MI to a self-help approach may improve binge eating outcomes,24 and binge eating can be successfully reduced using individual, group, and CD-ROM delivery formats.21 In direct comparisons, individual CBT outperformed group CBT in helping patients recover from BED (ie, no longer meeting diagnostic criteria),19 and CBT delivered via guided self-help outperformed BWL in helping patients achieve remission.18
Psychological features of BED typically include low levels of cognitive restraint and high levels of disinhibition, hunger, and shape and weight concerns. Improvements in these psychological measures were observed with CBT,15-20,22 IPT,22 and MI.24 In direct comparisons, self-help CBT demonstrated greater reductions in perceived hunger and disinhibition than self-help BWL,18 and individual CBT outperformed group CBT in reducing shape and weight concerns.19 Isolated studies reported improvements in depression after self-help CBT18 and MI,24 and sustained improvements22 after group CBT (6 months) and group IPT (12 months). Additional research is needed to determine whether CBT crafted specifically for BED improves self-rated depression or if enhancements targeting depressive symptoms are required.
The impact of behavioral interventions on weight in overweight patients has been mixed. Although some CBT studies reported a substantial decrease in weight,17,19 others suggested that weight loss among patients treated with CBT is not superior to those in a wait-list control group16 or is not significant over the course of treatment.20,21 The impact of BWL on weight outcomes in BED also has been unimpressive: after 12 weeks, self-help BWL was no better than self-help CBT in reducing BMI18; after 16 weeks, BWL was better than CBT and IPT in achieving clinically significant (≥5%) weight loss, but this advantage was not sustained at 1- and 2-year follow-up.23 It is difficult to determine why successfully treated BED patients fail to lose weight because one would expect decreases in binge eating to lead to weight loss. It is possible that calories previously consumed during binge eating episodes are distributed over non-binge meals or that patients label binges and non-binge meals differently as a result of treatment.
Combining treatments
BED patients often are treated with a combination of psychotherapy and pharmacotherapy (Table 4).25-29 When added to CBT, topiramate was associated with improvements in weight and some psychological outcomes,25,26 but fluoxetine was not.27,28 Direct comparisons also showed that CBT, alone or combined with fluoxetine, was better than fluoxetine alone in reducing binge eating.27 When combined with an individualized hypocaloric diet, the anti-obesity medication orlistat reduced weight in obese BED patients but had no appreciable effect on binge eating.29 Collectively, the studies we reviewed suggested that combining medication and CBT may improve binge eating and weight loss outcomes; however, additional trials are necessary to determine more definitively which medications combined with CBT are best at producing sustained weight loss while reducing binge eating frequency.
Table 4
Combining medication with behavioral interventions for BED
| Study | Drug/dosage | Comments |
|---|---|---|
| Brambilla et al, 200925 | 3 groups treated for 6 months: Group 1: CBT + setraline (50 to 150 mg/d) + topiramate (25 to 150 mg/d) + reduced calorie diet Group 2: CBT + sertraline (50 to 150 mg/d) + reduced calorie diet Group 3: CBT + nutritional counseling | Weight, BMI, and psychological features of BE reduced significantly only in group 1 |
| Claudino et al, 200726 | Group 1: CBT + topiramate (25 to 300 mg/d) Group 2: CBT + placebo 19 sessions over 21 weeks | Significant reductions in BE and depression in both groups; topiramate significantly better than placebo in reducing weight and in achieving BE remission |
| Devlin et al, 200527 | 4 groups, all received behavioral weight control intervention for 5 months (20 weeks) plus either: Group 1: CBT + fluoxetine Group 2: CBT + placebo Group 3: fluoxetineGroup 4 placebo (fluoxetine dose, 20 to 60 mg/d) | CBT groups (1 and 2) significantly better than non-CBT groups (3 and 4) in reducing BE and achieving abstinence from BE; fluoxetine significantly better than placebo in reducing depression |
| Molinari et al, 200528 | 3 groups, all received nutritional and diet counseling for 54 weeks (4 were inpatient) plus: Group 1: CBT Group 2: fluoxetine (20 to 60 mg/d) Group 3: CBT + fluoxetine | At 12 months, CBT (groups 1 and 3) associated with lower BE frequency and greater percentage of weight loss than fluoxetine |
| Golay et al, 200529 | Hypocaloric diet + orlistat (120 mg/d) vs hypocaloric diet + placebo for 24 weeks | Orlistat not different from placebo in reducing the number of patients classified with BED; orlistat significantly better than placebo in reducing weight and body fat |
| BE: binge eating; BED: binge eating disorder; BMI: body mass index; CBT: cognitive-behavioral therapy | ||
Recommendations
Evidence suggests that pharmacotherapy and CBT—alone or in combination—are effective in reducing binge eating, and pharmacotherapy is effective in reducing weight in overweight individuals with BED. More research is needed for IPT and MI. It is unclear which medications provide the greatest benefit in terms of binge eating remission; however, pharmacotherapy has a clear advantage in facilitating short-term weight loss. Also, all BED patients should receive medical management to address possible complications such as hypertension or type 2 diabetes. In addition to reducing binge eating, CBT can improve related psychological comorbidities (eg, eating-related psychopathology and depression) and may have additional benefit when combined with pharmacotherapy.
In light of these findings, we recommend augmenting psychotherapeutic care with pharmacotherapy and medical management to address all relevant psychological and medical domains. Future investigations should address the benefits of coordinated psychological and medical care and evaluate how to maintain treatment gains.
Related Resources
- Binge Eating Disorder Association. www.bedaonline.com.
- Brownley KA, Berkman ND, Sedway JA, et al. Binge eating disorder treatment: a systematic review of randomized controlled trials. Int J Eat Disord. 2007;40(4):337-348.
Drug Brand Names
- Atomoxetine • Strattera
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Lamotrigine • Lamictal
- Memantine • Namenda
- Orlistat • Alli, Xenical
- Sertraline • Zoloft
- Sibutramine • Meridia
- Topiramate • Topamax, Topiragen
- Zonisamide • Zonegram
Disclosures
Dr. Peat receives a post-doctoral trainee grant from the National Institutes of Health.
Drs. Brownley, Berkman, and Bulik report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Hudson JI, Hiripi E, Pope HG, et al. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry. 2007;61(3):348-358.
3. American Psychiatric Association. Proposed revision to DSM-5: K-05 Feeding and eating disorders. http://www.dsm5.org/ProposedRevision/Pages/proposedrevision.aspx?rid=372. Updated January 31 2011. Accessed March 26, 2012.
4. Grucza RA, Pryzbeck TR, Cloninger CR. Prevalence and correlates of binge eating disorder in a community sample. Compr Psychiatry. 2007;48(2):124-131.
5. Meneghini LF, Spadola J, Florez H. Prevalence and associations of binge eating disorder in a multiethnic population with type 2 diabetes. Diabetes Care. 2006;29(12):2760.-
6. Berkman ND, Bulik CM, Brownley KA, et al. Management of eating disorders. Evidence report/technology assessment No. 135. Rockville, MD: Agency for Healthcare Research and Quality; 2006. AHRQ Publication No. 06-E010.
7. Guerdjikova AI, McElroy SL, Kotwal R, et al. High-dose escitalopram in the treatment of binge-eating disorder with obesity: a placebo-controlled monotherapy trial. Hum Psychopharmacol. 2008;23(1):1-11.
8. McElroy SL, Kotwal R, Guerdjikova AL, et al. Zonisamide in the treatment of binge eating disorder with obesity: a randomized controlled trial. J Clin Psychiatry. 2006;67(12):1897-1906.
9. McElroy SL, Hudson JI, Capece JA, et al. Topiramate for the treatment of binge eating disorder associated with obesity: a placebo-controlled study. Biol Psychiatry. 2007;61(9):1039-1048.
10. McElroy SL, Guerdjikova A, Kotwal R, et al. Atomoxetine in the treatment of binge-eating disorder: a randomized placebo-controlled trial. J Clin Psychiatry. 2007;68(3):390-398.
11. Wilfley DE, Crow SJ, Hudson JI, et al. Efficacy of sibutramine for the treatment of binge eating disorder: a randomized multicenter placebo-controlled double-blind study. Am J Psychiatry. 2008;165(1):51-58.
12. Brennan BP, Roberts JL, Fogarty KV, et al. Memantine in the treatment of binge eating disorder: an open-label, prospective trial. Int J Eat Disord. 2008;41(6):520-526.
13. Guerdjikova AI, McElroy SL, Welge JA, et al. Lamotrigine in the treatment of binge-eating disorder with obesity: a randomized, placebo-controlled monotherapy trial. Int Clin Psychopharmacol. 2009;24(3):150-158.
14. Annunziato RA, Timko CA, Crerand CE, et al. A randomized trial examining differential meal replacement adherence in a weight loss maintenance program after one-year follow-up. Eat Behav. 2009;10(3):176-183.
15. Ashton K, Drerup M, Windover A, et al. Brief, four-session group CBT reduces binge eating behaviors among bariatric surgery candidates. Surg Obes Relat Dis. 2009;5(2):257-262.
16. Dingemans AE, Spinhoven P, van Furth EF. Predictors and mediators of treatment outcome in patients with binge eating disorder. Behav Res Ther. 2007;45(11):2551-2562.
17. Friederich HC, Schild S, Wild B, et al. Treatment outcome in people with subthreshold compared with full-syndrome binge eating disorder. Obesity. 2007;15(2):283-287.
18. Grilo CM, Masheb RM. A randomized controlled comparison of guided self-help cognitive behavioral therapy and behavioral weight loss for binge eating disorder. Behav Res Ther. 2005;43(11):1509-1525.
19. Ricca V, Castellini G, Mannucci E, et al. Comparison of individual and group cognitive behavioral therapy for binge eating disorder. A randomized, three-year follow-up study. Appetite. 2010;55(3):656-665.
20. Schlup B, Munsch S, Meyer AH, et al. The efficacy of a short version of a cognitive-behavioral treatment followed by booster sessions for binge eating disorder. Behav Res Ther. 2009;47(7):628-635.
21. Shapiro JR, Reba-Harrelson L, Dymek-Valentine M, et al. Feasibility and acceptability of CD-ROM-based cognitive-behavioral treatment for binge-eating disorder. Eur Eat Disord Rev. 2007;15(3):175-184.
22. Tasca GA, Ritchie K, Conrad G, et al. Attachment scales predict outcome in a randomized controlled trial of two group therapies for binge eating disorder: an aptitude by treatment interaction. Psychother Res. 2006;16(1):106-121.
23. Wilson GT, Wilfley DE, Agras WS, et al. Psychological treatments of binge eating disorder. Arch Gen Psychiatry. 2010;67(1):94-101.
24. Cassin SE, von Ranson KM, Heng K, et al. Adapted motivational interviewing for women with binge eating disorder: a randomized controlled trial. Psychol Addict Behav. 2008;22(3):417-425.
25. Brambilla F, Samek L, Company M, et al. Multivariate therapeutic approach to binge-eating disorder: combined nutritional, psychological and pharmacological treatment. Int Clin Psychopharmacol. 2009;24(6):312-317.
26. Claudino AM, de Oliveira IR, Appolinario JC, et al. Double-blind, randomized, placebo-controlled trial of topiramate plus cognitive-behavior therapy in binge-eating disorder. J Clin Psychiatry. 2007;68(9):1324-1332.
27. Devlin MJ, Goldfein JA, Petkova E, et al. Cognitive behavioral therapy and fluoxetine as adjuncts to group behavioral therapy for binge eating disorder. Obes Res. 2005;13(6):1077-1088.
28. Molinari E, Baruffi M, Croci M, et al. Binge eating disorder in obesity: comparison of different therapeutic strategies. Eat Weight Disord. 2005;10(3):154-161.
29. Golay A, Laurent-Jaccard A, Habicht F, et al. Effect of orlistat in obese patients with binge eating disorder. Obes Res. 2005;13(10):1701-1708.
Binge eating is consumption of an unusually large amount of food coupled with a feeling of loss of control over eating. Binge eating disorder (BED) is characterized by recurrent episodes of binge eating without inappropriate compensatory behaviors (eg, self-induced vomiting, misuse of laxatives, diuretics, or other agents, excessive exercise).1 It is the most common eating disorder in the United States, with a lifetime prevalence of approximately 3.5% in women and 2% in men.2 The diagnosis falls within the DSM-IV-TR category of eating disorders not otherwise specified,1 but clinicians often view it as a distinct clinical phenomenon. In DSM-IV-TR, an individual would meet criteria for BED if he or she engages in regular binge eating behavior in the absence of recurrent compensatory behaviors ≥2 days per week over 6 months.1 Proposed changes for DSM-5 recognize a distinct BED diagnosis, reduce the frequency criterion to once per week and the duration criterion to the past 3 months, and shift the focus from binge days to binge episodes (Table 1).3
Table 1
Proposed DSM-5 criteria for binge eating disorder
|
| Source:Reference 3 |
BED can occur in individuals of all body mass indices (BMI), but is common among individuals who are overweight or obese as well as those with depression or type 2 diabetes; BED can complicate treatment of these conditions.2,4,5 Primary treatment goals are:
- abstinence from binge eating
- improved psychological functioning
- appropriate weight regulation in overweight patients.
We report on 3 approaches to BED treatment: medication only, behavioral intervention only, and medication plus behavioral intervention. This article provides insights about emerging changes in diagnostic criteria for BED as well as evidence-informed treatment options and recommendations.
The evidence base
We conducted a review of 23 BED studies: 7 medication only, 5 medication plus behavioral, and 11 behavioral only. We focused on studies conducted since September 2005 that included binge frequency, weight, and depression as primary outcomes (see Berkman et al6 for a review of BED treatment studies before 2005). The studies included 2,527 participants (2,216 women and 311 men). Although the sex distribution of BED in the general population tends to slightly favor women,2 the proportion of women presenting for treatment generally is considerably higher than that of men. In studies that reported on race and/or ethnicity, 1,639 participants were identified as white, 191 as African American, 25 as Hispanic, 2 as Asian, 1 as Native American, and 25 as “other.” Ages ranged from 18 to 77.
Several medications are effective
In placebo-controlled studies, a high-dose selective serotonin reuptake inhibitor (escitalopram7), 2 anticonvulsants (zonisamide8 and topiramate9), a selective norepinephrine reuptake inhibitor (atomoxetine10), and an appetite suppressant (sibutramine11) were associated with significant decreases in binge eating frequency, weight, and BMI in overweight/obese patients diagnosed with BED (Table 2). In an open-label trial, memantine—a N-methyl-D-aspartate receptor antagonist often used to treat symptoms of Alzheimer’s disease—was associated with a significant reduction in binge eating but no change in weight.12 Lamotrigine was not significantly different from placebo in reducing binge eating or weight, but showed promise in reducing metabolic parameters such as glucose and triglyceride levels commonly associated with obesity and type 2 diabetes.13 Because BED often is comorbid with obesity and type 2 diabetes, lamotrigine augmentation when treating obese individuals with BED warrants further investigation. As with any pharmacologic agent, carefully consider potential side effects and interactions with other drugs before prescribing medications for BED. Informing patients of potential side effects is crucial for patient safety and accuracy of the data collected in well-controlled treatment studies.
Table 2
Pharmacotherapy for binge eating disorder
| Study | Drug/dosage | Comments |
|---|---|---|
| Guerdjikova et al, 20087 | Escitalopram, 10 to 30 mg/d, vs placebo for 12 weeks | Escitalopram was significantly better than placebo in reducing weight, BMI, and illness severity |
| McElroy et al, 20068 | Zonisamide, 100 to 600 mg/d, vs placebo for 16 weeks | Zonisamide was significantly better than placebo in reducing BE, weight, BMI, and various aspects of unhealthy eating behavior |
| McElroy et al, 20079 | Topiramate, 25 to 400 mg/d, vs placebo for 16 weeks | Topiramate was significantly better than placebo in reducing BE, weight, BMI, and related psychological features of BE |
| McElroy et al, 200710 | Atomoxetine, 40 to 120 mg/d, vs placebo for 10 weeks | Atomoxetine was significantly better than placebo in reducing BE, weight, BMI, and obsessive-compulsive features of BE, and in achieving remission |
| Wilfley et al, 200811 | Sibutramine, 15 mg/d, vs placebo for 24 weeks | Sibutramine was significantly better than placebo in reducing BE, weight, BMI, and related psychological features of BE |
| Brennan et al, 200812 | Open-label memantine, 5 to 20 mg/d, for 12 weeks | Memantine was associated with decreased binge frequency and related psychological features of BE |
| Guerdjikova et al, 200913 | Lamotrigine, 50 to 400 mg/d, vs placebo for 16 weeks | Lamotrigine was not significantly different from placebo |
| BE: binge eating; BMI: body mass index | ||
CBT vs other behavioral approaches
Cognitive-behavioral therapy (CBT), which focuses on identifying and modifying unhealthy thoughts that maintain disordered eating behaviors, is the most widely studied behavioral intervention for BED. Other studied treatments include interpersonal psychotherapy (IPT), motivational interviewing (MI), and structured behavioral weight loss (BWL) (Table 3).14-24 IPT is a psychodynamically based, time-limited treatment that focuses on the interpersonal context of the disorder and on building interpersonal skills. MI emphasizes exploring and resolving ambivalence about treatment, and works to facilitate change through motivational processes. BWL is centered on making dietary and physical activity changes to achieve weight loss. Behavioral treatments have been delivered in various formats, such as an individual or group setting, by electronic interface, and via self-help approaches. Most studies compared active treatment to a control group, but some compared active treatments head-to-head.
Table 3
CBT and other behavioral interventions for BED
| Study | Intervention | Comments |
|---|---|---|
| Annunziato et al, 200914 | 2 groups received CBT and hypocaloric diet for 8 weeks followed by 14 weeks of enhanced nutritional program (ie, reduced consumption of high energy density foods and once-daily liquid meal replacement) or control (normal diet) | Enhanced nutritional program was not significantly different from the control in reducing weight, BE, or psychological features of BE; variability in adherence to the enhanced nutritional program was identified as a significant effect modifier |
| Ashton et al, 200915 | 4 sessions of group CBT in an open trial | CBT was associated with significant reductions in BE and psychological features of BE in post-bariatric surgery patients |
| Dingemans et al, 200716 | CBT vs wait-list control | CBT significantly better than the wait-list control in reducing BE and psychological features of BE, and in achieving abstinence from BE |
| Friederich et al, 200717 | 15-session CBT blended with elements of interpersonal therapy (IPT), nutritional counseling, and supervised walking program; no control group | Treatment significantly reduced weight, BE, and related psychological features of BE in patients meeting sub-threshold and full criteria for BED |
| Grilo et al, 200518 | Guided self-help CBT (CBTgsh) vs guided self-help behavioral weight loss (BWLgsh) vs non-specific attention control for 12 weeks | CBTgsh significantly better than BWLgsh and control in BE remission; CBTgsh significantly better than BWLgsh, which was significantly better than control in reducing cognitive restraint; CBTgsh significantly better than control in reducing depression and eating-related psychopathology; no differences between groups in BMI change |
| Ricca et al, 201019 | Individual (I-CBT) vs group CBT (G-CBT) for 24 weeks in patients meeting subthreshold and full criteria for BED | BE and BMI were significantly reduced in both groups at 24 weeks and 3-year follow-up. I-CBT was not better than G-CBT in reducing BE or weight at 24 weeks or 3-year follow-up; I-CBT was significantly better than G-CBT in reducing eating-related psychopathology at 24 weeks and 3-year follow-up; I-CBT was significantly better than G-CBT in recovery (ie, no longer meeting full BED criteria) at 24 weeks but not at 3-year follow-up |
| Schlup et al, 200920 | 8 weekly sessions of group CBT vs wait-list control | CBT was significantly better than wait-list control in reducing BE and eating concerns and in achieving abstinence at end of treatment; CBT was not different than control in reducing BMI; treatment-related reductions in BE and eating concerns were maintained at 12-month follow-up |
| Shapiro et al, 200721 | 10 weekly sessions of group CBT (G-CBT) vs CD-ROM delivered CBT (CD-CBT) vs wait-list control | G-CBT and CD-CBT were not different from each other but both were significantly better than wait-list control in reducing BE |
| Tasca et al, 200622 | Group CBT (G-CBT) vs group psychodynamic interpersonal therapy (G-IPT) vs wait-list control for 16 weeks | G-CBT and G-IPT were not different from each other; G-CBT and G-IPT were significantly better than wait list in reducing BE and interpersonal problems (but not BMI) and increasing cognitive restraint post-treatment; depression was reduced in both groups at 6 months but only in G-IPT at 12 months; reductions in BE maintained at 12 months |
| Wilson et al, 201023 | 10 sessions of guided self-help CBT (CBTgsh) vs 19 sessions of IPT vs 20 sessions of behavioral weight loss (BWL) over 6 months | BWL was significantly better than IPT and CBTgsh in reducing BMI and in the number of patients achieving 5% weight loss at post-treatment but effects were not sustained over time; BWL was significantly better than CBTgsh in increasing dietary restraint |
| Cassin et al, 200824 | Self-help book + motivational interviewing (SH-MI) vs self-help book alone (SH) for 16 weeks | SH-MI was significantly better than SH in reducing BE and depression |
| BE: binge eating; BED: binge eating disorder; BMI: body mass index; CBT: cognitive-behavioral therapy | ||
Studies found that CBT and IPT are effective in reducing the frequency of binge eating, whether measured by the number of binge eating episodes or days a patient reports having engaged in binge eating.14-23 However, some studies suggested that CBT can help a substantial number of patients achieve abstinence from binge eating.16,20 Adding MI to a self-help approach may improve binge eating outcomes,24 and binge eating can be successfully reduced using individual, group, and CD-ROM delivery formats.21 In direct comparisons, individual CBT outperformed group CBT in helping patients recover from BED (ie, no longer meeting diagnostic criteria),19 and CBT delivered via guided self-help outperformed BWL in helping patients achieve remission.18
Psychological features of BED typically include low levels of cognitive restraint and high levels of disinhibition, hunger, and shape and weight concerns. Improvements in these psychological measures were observed with CBT,15-20,22 IPT,22 and MI.24 In direct comparisons, self-help CBT demonstrated greater reductions in perceived hunger and disinhibition than self-help BWL,18 and individual CBT outperformed group CBT in reducing shape and weight concerns.19 Isolated studies reported improvements in depression after self-help CBT18 and MI,24 and sustained improvements22 after group CBT (6 months) and group IPT (12 months). Additional research is needed to determine whether CBT crafted specifically for BED improves self-rated depression or if enhancements targeting depressive symptoms are required.
The impact of behavioral interventions on weight in overweight patients has been mixed. Although some CBT studies reported a substantial decrease in weight,17,19 others suggested that weight loss among patients treated with CBT is not superior to those in a wait-list control group16 or is not significant over the course of treatment.20,21 The impact of BWL on weight outcomes in BED also has been unimpressive: after 12 weeks, self-help BWL was no better than self-help CBT in reducing BMI18; after 16 weeks, BWL was better than CBT and IPT in achieving clinically significant (≥5%) weight loss, but this advantage was not sustained at 1- and 2-year follow-up.23 It is difficult to determine why successfully treated BED patients fail to lose weight because one would expect decreases in binge eating to lead to weight loss. It is possible that calories previously consumed during binge eating episodes are distributed over non-binge meals or that patients label binges and non-binge meals differently as a result of treatment.
Combining treatments
BED patients often are treated with a combination of psychotherapy and pharmacotherapy (Table 4).25-29 When added to CBT, topiramate was associated with improvements in weight and some psychological outcomes,25,26 but fluoxetine was not.27,28 Direct comparisons also showed that CBT, alone or combined with fluoxetine, was better than fluoxetine alone in reducing binge eating.27 When combined with an individualized hypocaloric diet, the anti-obesity medication orlistat reduced weight in obese BED patients but had no appreciable effect on binge eating.29 Collectively, the studies we reviewed suggested that combining medication and CBT may improve binge eating and weight loss outcomes; however, additional trials are necessary to determine more definitively which medications combined with CBT are best at producing sustained weight loss while reducing binge eating frequency.
Table 4
Combining medication with behavioral interventions for BED
| Study | Drug/dosage | Comments |
|---|---|---|
| Brambilla et al, 200925 | 3 groups treated for 6 months: Group 1: CBT + setraline (50 to 150 mg/d) + topiramate (25 to 150 mg/d) + reduced calorie diet Group 2: CBT + sertraline (50 to 150 mg/d) + reduced calorie diet Group 3: CBT + nutritional counseling | Weight, BMI, and psychological features of BE reduced significantly only in group 1 |
| Claudino et al, 200726 | Group 1: CBT + topiramate (25 to 300 mg/d) Group 2: CBT + placebo 19 sessions over 21 weeks | Significant reductions in BE and depression in both groups; topiramate significantly better than placebo in reducing weight and in achieving BE remission |
| Devlin et al, 200527 | 4 groups, all received behavioral weight control intervention for 5 months (20 weeks) plus either: Group 1: CBT + fluoxetine Group 2: CBT + placebo Group 3: fluoxetineGroup 4 placebo (fluoxetine dose, 20 to 60 mg/d) | CBT groups (1 and 2) significantly better than non-CBT groups (3 and 4) in reducing BE and achieving abstinence from BE; fluoxetine significantly better than placebo in reducing depression |
| Molinari et al, 200528 | 3 groups, all received nutritional and diet counseling for 54 weeks (4 were inpatient) plus: Group 1: CBT Group 2: fluoxetine (20 to 60 mg/d) Group 3: CBT + fluoxetine | At 12 months, CBT (groups 1 and 3) associated with lower BE frequency and greater percentage of weight loss than fluoxetine |
| Golay et al, 200529 | Hypocaloric diet + orlistat (120 mg/d) vs hypocaloric diet + placebo for 24 weeks | Orlistat not different from placebo in reducing the number of patients classified with BED; orlistat significantly better than placebo in reducing weight and body fat |
| BE: binge eating; BED: binge eating disorder; BMI: body mass index; CBT: cognitive-behavioral therapy | ||
Recommendations
Evidence suggests that pharmacotherapy and CBT—alone or in combination—are effective in reducing binge eating, and pharmacotherapy is effective in reducing weight in overweight individuals with BED. More research is needed for IPT and MI. It is unclear which medications provide the greatest benefit in terms of binge eating remission; however, pharmacotherapy has a clear advantage in facilitating short-term weight loss. Also, all BED patients should receive medical management to address possible complications such as hypertension or type 2 diabetes. In addition to reducing binge eating, CBT can improve related psychological comorbidities (eg, eating-related psychopathology and depression) and may have additional benefit when combined with pharmacotherapy.
In light of these findings, we recommend augmenting psychotherapeutic care with pharmacotherapy and medical management to address all relevant psychological and medical domains. Future investigations should address the benefits of coordinated psychological and medical care and evaluate how to maintain treatment gains.
Related Resources
- Binge Eating Disorder Association. www.bedaonline.com.
- Brownley KA, Berkman ND, Sedway JA, et al. Binge eating disorder treatment: a systematic review of randomized controlled trials. Int J Eat Disord. 2007;40(4):337-348.
Drug Brand Names
- Atomoxetine • Strattera
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Lamotrigine • Lamictal
- Memantine • Namenda
- Orlistat • Alli, Xenical
- Sertraline • Zoloft
- Sibutramine • Meridia
- Topiramate • Topamax, Topiragen
- Zonisamide • Zonegram
Disclosures
Dr. Peat receives a post-doctoral trainee grant from the National Institutes of Health.
Drs. Brownley, Berkman, and Bulik report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Binge eating is consumption of an unusually large amount of food coupled with a feeling of loss of control over eating. Binge eating disorder (BED) is characterized by recurrent episodes of binge eating without inappropriate compensatory behaviors (eg, self-induced vomiting, misuse of laxatives, diuretics, or other agents, excessive exercise).1 It is the most common eating disorder in the United States, with a lifetime prevalence of approximately 3.5% in women and 2% in men.2 The diagnosis falls within the DSM-IV-TR category of eating disorders not otherwise specified,1 but clinicians often view it as a distinct clinical phenomenon. In DSM-IV-TR, an individual would meet criteria for BED if he or she engages in regular binge eating behavior in the absence of recurrent compensatory behaviors ≥2 days per week over 6 months.1 Proposed changes for DSM-5 recognize a distinct BED diagnosis, reduce the frequency criterion to once per week and the duration criterion to the past 3 months, and shift the focus from binge days to binge episodes (Table 1).3
Table 1
Proposed DSM-5 criteria for binge eating disorder
|
| Source:Reference 3 |
BED can occur in individuals of all body mass indices (BMI), but is common among individuals who are overweight or obese as well as those with depression or type 2 diabetes; BED can complicate treatment of these conditions.2,4,5 Primary treatment goals are:
- abstinence from binge eating
- improved psychological functioning
- appropriate weight regulation in overweight patients.
We report on 3 approaches to BED treatment: medication only, behavioral intervention only, and medication plus behavioral intervention. This article provides insights about emerging changes in diagnostic criteria for BED as well as evidence-informed treatment options and recommendations.
The evidence base
We conducted a review of 23 BED studies: 7 medication only, 5 medication plus behavioral, and 11 behavioral only. We focused on studies conducted since September 2005 that included binge frequency, weight, and depression as primary outcomes (see Berkman et al6 for a review of BED treatment studies before 2005). The studies included 2,527 participants (2,216 women and 311 men). Although the sex distribution of BED in the general population tends to slightly favor women,2 the proportion of women presenting for treatment generally is considerably higher than that of men. In studies that reported on race and/or ethnicity, 1,639 participants were identified as white, 191 as African American, 25 as Hispanic, 2 as Asian, 1 as Native American, and 25 as “other.” Ages ranged from 18 to 77.
Several medications are effective
In placebo-controlled studies, a high-dose selective serotonin reuptake inhibitor (escitalopram7), 2 anticonvulsants (zonisamide8 and topiramate9), a selective norepinephrine reuptake inhibitor (atomoxetine10), and an appetite suppressant (sibutramine11) were associated with significant decreases in binge eating frequency, weight, and BMI in overweight/obese patients diagnosed with BED (Table 2). In an open-label trial, memantine—a N-methyl-D-aspartate receptor antagonist often used to treat symptoms of Alzheimer’s disease—was associated with a significant reduction in binge eating but no change in weight.12 Lamotrigine was not significantly different from placebo in reducing binge eating or weight, but showed promise in reducing metabolic parameters such as glucose and triglyceride levels commonly associated with obesity and type 2 diabetes.13 Because BED often is comorbid with obesity and type 2 diabetes, lamotrigine augmentation when treating obese individuals with BED warrants further investigation. As with any pharmacologic agent, carefully consider potential side effects and interactions with other drugs before prescribing medications for BED. Informing patients of potential side effects is crucial for patient safety and accuracy of the data collected in well-controlled treatment studies.
Table 2
Pharmacotherapy for binge eating disorder
| Study | Drug/dosage | Comments |
|---|---|---|
| Guerdjikova et al, 20087 | Escitalopram, 10 to 30 mg/d, vs placebo for 12 weeks | Escitalopram was significantly better than placebo in reducing weight, BMI, and illness severity |
| McElroy et al, 20068 | Zonisamide, 100 to 600 mg/d, vs placebo for 16 weeks | Zonisamide was significantly better than placebo in reducing BE, weight, BMI, and various aspects of unhealthy eating behavior |
| McElroy et al, 20079 | Topiramate, 25 to 400 mg/d, vs placebo for 16 weeks | Topiramate was significantly better than placebo in reducing BE, weight, BMI, and related psychological features of BE |
| McElroy et al, 200710 | Atomoxetine, 40 to 120 mg/d, vs placebo for 10 weeks | Atomoxetine was significantly better than placebo in reducing BE, weight, BMI, and obsessive-compulsive features of BE, and in achieving remission |
| Wilfley et al, 200811 | Sibutramine, 15 mg/d, vs placebo for 24 weeks | Sibutramine was significantly better than placebo in reducing BE, weight, BMI, and related psychological features of BE |
| Brennan et al, 200812 | Open-label memantine, 5 to 20 mg/d, for 12 weeks | Memantine was associated with decreased binge frequency and related psychological features of BE |
| Guerdjikova et al, 200913 | Lamotrigine, 50 to 400 mg/d, vs placebo for 16 weeks | Lamotrigine was not significantly different from placebo |
| BE: binge eating; BMI: body mass index | ||
CBT vs other behavioral approaches
Cognitive-behavioral therapy (CBT), which focuses on identifying and modifying unhealthy thoughts that maintain disordered eating behaviors, is the most widely studied behavioral intervention for BED. Other studied treatments include interpersonal psychotherapy (IPT), motivational interviewing (MI), and structured behavioral weight loss (BWL) (Table 3).14-24 IPT is a psychodynamically based, time-limited treatment that focuses on the interpersonal context of the disorder and on building interpersonal skills. MI emphasizes exploring and resolving ambivalence about treatment, and works to facilitate change through motivational processes. BWL is centered on making dietary and physical activity changes to achieve weight loss. Behavioral treatments have been delivered in various formats, such as an individual or group setting, by electronic interface, and via self-help approaches. Most studies compared active treatment to a control group, but some compared active treatments head-to-head.
Table 3
CBT and other behavioral interventions for BED
| Study | Intervention | Comments |
|---|---|---|
| Annunziato et al, 200914 | 2 groups received CBT and hypocaloric diet for 8 weeks followed by 14 weeks of enhanced nutritional program (ie, reduced consumption of high energy density foods and once-daily liquid meal replacement) or control (normal diet) | Enhanced nutritional program was not significantly different from the control in reducing weight, BE, or psychological features of BE; variability in adherence to the enhanced nutritional program was identified as a significant effect modifier |
| Ashton et al, 200915 | 4 sessions of group CBT in an open trial | CBT was associated with significant reductions in BE and psychological features of BE in post-bariatric surgery patients |
| Dingemans et al, 200716 | CBT vs wait-list control | CBT significantly better than the wait-list control in reducing BE and psychological features of BE, and in achieving abstinence from BE |
| Friederich et al, 200717 | 15-session CBT blended with elements of interpersonal therapy (IPT), nutritional counseling, and supervised walking program; no control group | Treatment significantly reduced weight, BE, and related psychological features of BE in patients meeting sub-threshold and full criteria for BED |
| Grilo et al, 200518 | Guided self-help CBT (CBTgsh) vs guided self-help behavioral weight loss (BWLgsh) vs non-specific attention control for 12 weeks | CBTgsh significantly better than BWLgsh and control in BE remission; CBTgsh significantly better than BWLgsh, which was significantly better than control in reducing cognitive restraint; CBTgsh significantly better than control in reducing depression and eating-related psychopathology; no differences between groups in BMI change |
| Ricca et al, 201019 | Individual (I-CBT) vs group CBT (G-CBT) for 24 weeks in patients meeting subthreshold and full criteria for BED | BE and BMI were significantly reduced in both groups at 24 weeks and 3-year follow-up. I-CBT was not better than G-CBT in reducing BE or weight at 24 weeks or 3-year follow-up; I-CBT was significantly better than G-CBT in reducing eating-related psychopathology at 24 weeks and 3-year follow-up; I-CBT was significantly better than G-CBT in recovery (ie, no longer meeting full BED criteria) at 24 weeks but not at 3-year follow-up |
| Schlup et al, 200920 | 8 weekly sessions of group CBT vs wait-list control | CBT was significantly better than wait-list control in reducing BE and eating concerns and in achieving abstinence at end of treatment; CBT was not different than control in reducing BMI; treatment-related reductions in BE and eating concerns were maintained at 12-month follow-up |
| Shapiro et al, 200721 | 10 weekly sessions of group CBT (G-CBT) vs CD-ROM delivered CBT (CD-CBT) vs wait-list control | G-CBT and CD-CBT were not different from each other but both were significantly better than wait-list control in reducing BE |
| Tasca et al, 200622 | Group CBT (G-CBT) vs group psychodynamic interpersonal therapy (G-IPT) vs wait-list control for 16 weeks | G-CBT and G-IPT were not different from each other; G-CBT and G-IPT were significantly better than wait list in reducing BE and interpersonal problems (but not BMI) and increasing cognitive restraint post-treatment; depression was reduced in both groups at 6 months but only in G-IPT at 12 months; reductions in BE maintained at 12 months |
| Wilson et al, 201023 | 10 sessions of guided self-help CBT (CBTgsh) vs 19 sessions of IPT vs 20 sessions of behavioral weight loss (BWL) over 6 months | BWL was significantly better than IPT and CBTgsh in reducing BMI and in the number of patients achieving 5% weight loss at post-treatment but effects were not sustained over time; BWL was significantly better than CBTgsh in increasing dietary restraint |
| Cassin et al, 200824 | Self-help book + motivational interviewing (SH-MI) vs self-help book alone (SH) for 16 weeks | SH-MI was significantly better than SH in reducing BE and depression |
| BE: binge eating; BED: binge eating disorder; BMI: body mass index; CBT: cognitive-behavioral therapy | ||
Studies found that CBT and IPT are effective in reducing the frequency of binge eating, whether measured by the number of binge eating episodes or days a patient reports having engaged in binge eating.14-23 However, some studies suggested that CBT can help a substantial number of patients achieve abstinence from binge eating.16,20 Adding MI to a self-help approach may improve binge eating outcomes,24 and binge eating can be successfully reduced using individual, group, and CD-ROM delivery formats.21 In direct comparisons, individual CBT outperformed group CBT in helping patients recover from BED (ie, no longer meeting diagnostic criteria),19 and CBT delivered via guided self-help outperformed BWL in helping patients achieve remission.18
Psychological features of BED typically include low levels of cognitive restraint and high levels of disinhibition, hunger, and shape and weight concerns. Improvements in these psychological measures were observed with CBT,15-20,22 IPT,22 and MI.24 In direct comparisons, self-help CBT demonstrated greater reductions in perceived hunger and disinhibition than self-help BWL,18 and individual CBT outperformed group CBT in reducing shape and weight concerns.19 Isolated studies reported improvements in depression after self-help CBT18 and MI,24 and sustained improvements22 after group CBT (6 months) and group IPT (12 months). Additional research is needed to determine whether CBT crafted specifically for BED improves self-rated depression or if enhancements targeting depressive symptoms are required.
The impact of behavioral interventions on weight in overweight patients has been mixed. Although some CBT studies reported a substantial decrease in weight,17,19 others suggested that weight loss among patients treated with CBT is not superior to those in a wait-list control group16 or is not significant over the course of treatment.20,21 The impact of BWL on weight outcomes in BED also has been unimpressive: after 12 weeks, self-help BWL was no better than self-help CBT in reducing BMI18; after 16 weeks, BWL was better than CBT and IPT in achieving clinically significant (≥5%) weight loss, but this advantage was not sustained at 1- and 2-year follow-up.23 It is difficult to determine why successfully treated BED patients fail to lose weight because one would expect decreases in binge eating to lead to weight loss. It is possible that calories previously consumed during binge eating episodes are distributed over non-binge meals or that patients label binges and non-binge meals differently as a result of treatment.
Combining treatments
BED patients often are treated with a combination of psychotherapy and pharmacotherapy (Table 4).25-29 When added to CBT, topiramate was associated with improvements in weight and some psychological outcomes,25,26 but fluoxetine was not.27,28 Direct comparisons also showed that CBT, alone or combined with fluoxetine, was better than fluoxetine alone in reducing binge eating.27 When combined with an individualized hypocaloric diet, the anti-obesity medication orlistat reduced weight in obese BED patients but had no appreciable effect on binge eating.29 Collectively, the studies we reviewed suggested that combining medication and CBT may improve binge eating and weight loss outcomes; however, additional trials are necessary to determine more definitively which medications combined with CBT are best at producing sustained weight loss while reducing binge eating frequency.
Table 4
Combining medication with behavioral interventions for BED
| Study | Drug/dosage | Comments |
|---|---|---|
| Brambilla et al, 200925 | 3 groups treated for 6 months: Group 1: CBT + setraline (50 to 150 mg/d) + topiramate (25 to 150 mg/d) + reduced calorie diet Group 2: CBT + sertraline (50 to 150 mg/d) + reduced calorie diet Group 3: CBT + nutritional counseling | Weight, BMI, and psychological features of BE reduced significantly only in group 1 |
| Claudino et al, 200726 | Group 1: CBT + topiramate (25 to 300 mg/d) Group 2: CBT + placebo 19 sessions over 21 weeks | Significant reductions in BE and depression in both groups; topiramate significantly better than placebo in reducing weight and in achieving BE remission |
| Devlin et al, 200527 | 4 groups, all received behavioral weight control intervention for 5 months (20 weeks) plus either: Group 1: CBT + fluoxetine Group 2: CBT + placebo Group 3: fluoxetineGroup 4 placebo (fluoxetine dose, 20 to 60 mg/d) | CBT groups (1 and 2) significantly better than non-CBT groups (3 and 4) in reducing BE and achieving abstinence from BE; fluoxetine significantly better than placebo in reducing depression |
| Molinari et al, 200528 | 3 groups, all received nutritional and diet counseling for 54 weeks (4 were inpatient) plus: Group 1: CBT Group 2: fluoxetine (20 to 60 mg/d) Group 3: CBT + fluoxetine | At 12 months, CBT (groups 1 and 3) associated with lower BE frequency and greater percentage of weight loss than fluoxetine |
| Golay et al, 200529 | Hypocaloric diet + orlistat (120 mg/d) vs hypocaloric diet + placebo for 24 weeks | Orlistat not different from placebo in reducing the number of patients classified with BED; orlistat significantly better than placebo in reducing weight and body fat |
| BE: binge eating; BED: binge eating disorder; BMI: body mass index; CBT: cognitive-behavioral therapy | ||
Recommendations
Evidence suggests that pharmacotherapy and CBT—alone or in combination—are effective in reducing binge eating, and pharmacotherapy is effective in reducing weight in overweight individuals with BED. More research is needed for IPT and MI. It is unclear which medications provide the greatest benefit in terms of binge eating remission; however, pharmacotherapy has a clear advantage in facilitating short-term weight loss. Also, all BED patients should receive medical management to address possible complications such as hypertension or type 2 diabetes. In addition to reducing binge eating, CBT can improve related psychological comorbidities (eg, eating-related psychopathology and depression) and may have additional benefit when combined with pharmacotherapy.
In light of these findings, we recommend augmenting psychotherapeutic care with pharmacotherapy and medical management to address all relevant psychological and medical domains. Future investigations should address the benefits of coordinated psychological and medical care and evaluate how to maintain treatment gains.
Related Resources
- Binge Eating Disorder Association. www.bedaonline.com.
- Brownley KA, Berkman ND, Sedway JA, et al. Binge eating disorder treatment: a systematic review of randomized controlled trials. Int J Eat Disord. 2007;40(4):337-348.
Drug Brand Names
- Atomoxetine • Strattera
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Lamotrigine • Lamictal
- Memantine • Namenda
- Orlistat • Alli, Xenical
- Sertraline • Zoloft
- Sibutramine • Meridia
- Topiramate • Topamax, Topiragen
- Zonisamide • Zonegram
Disclosures
Dr. Peat receives a post-doctoral trainee grant from the National Institutes of Health.
Drs. Brownley, Berkman, and Bulik report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Hudson JI, Hiripi E, Pope HG, et al. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry. 2007;61(3):348-358.
3. American Psychiatric Association. Proposed revision to DSM-5: K-05 Feeding and eating disorders. http://www.dsm5.org/ProposedRevision/Pages/proposedrevision.aspx?rid=372. Updated January 31 2011. Accessed March 26, 2012.
4. Grucza RA, Pryzbeck TR, Cloninger CR. Prevalence and correlates of binge eating disorder in a community sample. Compr Psychiatry. 2007;48(2):124-131.
5. Meneghini LF, Spadola J, Florez H. Prevalence and associations of binge eating disorder in a multiethnic population with type 2 diabetes. Diabetes Care. 2006;29(12):2760.-
6. Berkman ND, Bulik CM, Brownley KA, et al. Management of eating disorders. Evidence report/technology assessment No. 135. Rockville, MD: Agency for Healthcare Research and Quality; 2006. AHRQ Publication No. 06-E010.
7. Guerdjikova AI, McElroy SL, Kotwal R, et al. High-dose escitalopram in the treatment of binge-eating disorder with obesity: a placebo-controlled monotherapy trial. Hum Psychopharmacol. 2008;23(1):1-11.
8. McElroy SL, Kotwal R, Guerdjikova AL, et al. Zonisamide in the treatment of binge eating disorder with obesity: a randomized controlled trial. J Clin Psychiatry. 2006;67(12):1897-1906.
9. McElroy SL, Hudson JI, Capece JA, et al. Topiramate for the treatment of binge eating disorder associated with obesity: a placebo-controlled study. Biol Psychiatry. 2007;61(9):1039-1048.
10. McElroy SL, Guerdjikova A, Kotwal R, et al. Atomoxetine in the treatment of binge-eating disorder: a randomized placebo-controlled trial. J Clin Psychiatry. 2007;68(3):390-398.
11. Wilfley DE, Crow SJ, Hudson JI, et al. Efficacy of sibutramine for the treatment of binge eating disorder: a randomized multicenter placebo-controlled double-blind study. Am J Psychiatry. 2008;165(1):51-58.
12. Brennan BP, Roberts JL, Fogarty KV, et al. Memantine in the treatment of binge eating disorder: an open-label, prospective trial. Int J Eat Disord. 2008;41(6):520-526.
13. Guerdjikova AI, McElroy SL, Welge JA, et al. Lamotrigine in the treatment of binge-eating disorder with obesity: a randomized, placebo-controlled monotherapy trial. Int Clin Psychopharmacol. 2009;24(3):150-158.
14. Annunziato RA, Timko CA, Crerand CE, et al. A randomized trial examining differential meal replacement adherence in a weight loss maintenance program after one-year follow-up. Eat Behav. 2009;10(3):176-183.
15. Ashton K, Drerup M, Windover A, et al. Brief, four-session group CBT reduces binge eating behaviors among bariatric surgery candidates. Surg Obes Relat Dis. 2009;5(2):257-262.
16. Dingemans AE, Spinhoven P, van Furth EF. Predictors and mediators of treatment outcome in patients with binge eating disorder. Behav Res Ther. 2007;45(11):2551-2562.
17. Friederich HC, Schild S, Wild B, et al. Treatment outcome in people with subthreshold compared with full-syndrome binge eating disorder. Obesity. 2007;15(2):283-287.
18. Grilo CM, Masheb RM. A randomized controlled comparison of guided self-help cognitive behavioral therapy and behavioral weight loss for binge eating disorder. Behav Res Ther. 2005;43(11):1509-1525.
19. Ricca V, Castellini G, Mannucci E, et al. Comparison of individual and group cognitive behavioral therapy for binge eating disorder. A randomized, three-year follow-up study. Appetite. 2010;55(3):656-665.
20. Schlup B, Munsch S, Meyer AH, et al. The efficacy of a short version of a cognitive-behavioral treatment followed by booster sessions for binge eating disorder. Behav Res Ther. 2009;47(7):628-635.
21. Shapiro JR, Reba-Harrelson L, Dymek-Valentine M, et al. Feasibility and acceptability of CD-ROM-based cognitive-behavioral treatment for binge-eating disorder. Eur Eat Disord Rev. 2007;15(3):175-184.
22. Tasca GA, Ritchie K, Conrad G, et al. Attachment scales predict outcome in a randomized controlled trial of two group therapies for binge eating disorder: an aptitude by treatment interaction. Psychother Res. 2006;16(1):106-121.
23. Wilson GT, Wilfley DE, Agras WS, et al. Psychological treatments of binge eating disorder. Arch Gen Psychiatry. 2010;67(1):94-101.
24. Cassin SE, von Ranson KM, Heng K, et al. Adapted motivational interviewing for women with binge eating disorder: a randomized controlled trial. Psychol Addict Behav. 2008;22(3):417-425.
25. Brambilla F, Samek L, Company M, et al. Multivariate therapeutic approach to binge-eating disorder: combined nutritional, psychological and pharmacological treatment. Int Clin Psychopharmacol. 2009;24(6):312-317.
26. Claudino AM, de Oliveira IR, Appolinario JC, et al. Double-blind, randomized, placebo-controlled trial of topiramate plus cognitive-behavior therapy in binge-eating disorder. J Clin Psychiatry. 2007;68(9):1324-1332.
27. Devlin MJ, Goldfein JA, Petkova E, et al. Cognitive behavioral therapy and fluoxetine as adjuncts to group behavioral therapy for binge eating disorder. Obes Res. 2005;13(6):1077-1088.
28. Molinari E, Baruffi M, Croci M, et al. Binge eating disorder in obesity: comparison of different therapeutic strategies. Eat Weight Disord. 2005;10(3):154-161.
29. Golay A, Laurent-Jaccard A, Habicht F, et al. Effect of orlistat in obese patients with binge eating disorder. Obes Res. 2005;13(10):1701-1708.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Hudson JI, Hiripi E, Pope HG, et al. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry. 2007;61(3):348-358.
3. American Psychiatric Association. Proposed revision to DSM-5: K-05 Feeding and eating disorders. http://www.dsm5.org/ProposedRevision/Pages/proposedrevision.aspx?rid=372. Updated January 31 2011. Accessed March 26, 2012.
4. Grucza RA, Pryzbeck TR, Cloninger CR. Prevalence and correlates of binge eating disorder in a community sample. Compr Psychiatry. 2007;48(2):124-131.
5. Meneghini LF, Spadola J, Florez H. Prevalence and associations of binge eating disorder in a multiethnic population with type 2 diabetes. Diabetes Care. 2006;29(12):2760.-
6. Berkman ND, Bulik CM, Brownley KA, et al. Management of eating disorders. Evidence report/technology assessment No. 135. Rockville, MD: Agency for Healthcare Research and Quality; 2006. AHRQ Publication No. 06-E010.
7. Guerdjikova AI, McElroy SL, Kotwal R, et al. High-dose escitalopram in the treatment of binge-eating disorder with obesity: a placebo-controlled monotherapy trial. Hum Psychopharmacol. 2008;23(1):1-11.
8. McElroy SL, Kotwal R, Guerdjikova AL, et al. Zonisamide in the treatment of binge eating disorder with obesity: a randomized controlled trial. J Clin Psychiatry. 2006;67(12):1897-1906.
9. McElroy SL, Hudson JI, Capece JA, et al. Topiramate for the treatment of binge eating disorder associated with obesity: a placebo-controlled study. Biol Psychiatry. 2007;61(9):1039-1048.
10. McElroy SL, Guerdjikova A, Kotwal R, et al. Atomoxetine in the treatment of binge-eating disorder: a randomized placebo-controlled trial. J Clin Psychiatry. 2007;68(3):390-398.
11. Wilfley DE, Crow SJ, Hudson JI, et al. Efficacy of sibutramine for the treatment of binge eating disorder: a randomized multicenter placebo-controlled double-blind study. Am J Psychiatry. 2008;165(1):51-58.
12. Brennan BP, Roberts JL, Fogarty KV, et al. Memantine in the treatment of binge eating disorder: an open-label, prospective trial. Int J Eat Disord. 2008;41(6):520-526.
13. Guerdjikova AI, McElroy SL, Welge JA, et al. Lamotrigine in the treatment of binge-eating disorder with obesity: a randomized, placebo-controlled monotherapy trial. Int Clin Psychopharmacol. 2009;24(3):150-158.
14. Annunziato RA, Timko CA, Crerand CE, et al. A randomized trial examining differential meal replacement adherence in a weight loss maintenance program after one-year follow-up. Eat Behav. 2009;10(3):176-183.
15. Ashton K, Drerup M, Windover A, et al. Brief, four-session group CBT reduces binge eating behaviors among bariatric surgery candidates. Surg Obes Relat Dis. 2009;5(2):257-262.
16. Dingemans AE, Spinhoven P, van Furth EF. Predictors and mediators of treatment outcome in patients with binge eating disorder. Behav Res Ther. 2007;45(11):2551-2562.
17. Friederich HC, Schild S, Wild B, et al. Treatment outcome in people with subthreshold compared with full-syndrome binge eating disorder. Obesity. 2007;15(2):283-287.
18. Grilo CM, Masheb RM. A randomized controlled comparison of guided self-help cognitive behavioral therapy and behavioral weight loss for binge eating disorder. Behav Res Ther. 2005;43(11):1509-1525.
19. Ricca V, Castellini G, Mannucci E, et al. Comparison of individual and group cognitive behavioral therapy for binge eating disorder. A randomized, three-year follow-up study. Appetite. 2010;55(3):656-665.
20. Schlup B, Munsch S, Meyer AH, et al. The efficacy of a short version of a cognitive-behavioral treatment followed by booster sessions for binge eating disorder. Behav Res Ther. 2009;47(7):628-635.
21. Shapiro JR, Reba-Harrelson L, Dymek-Valentine M, et al. Feasibility and acceptability of CD-ROM-based cognitive-behavioral treatment for binge-eating disorder. Eur Eat Disord Rev. 2007;15(3):175-184.
22. Tasca GA, Ritchie K, Conrad G, et al. Attachment scales predict outcome in a randomized controlled trial of two group therapies for binge eating disorder: an aptitude by treatment interaction. Psychother Res. 2006;16(1):106-121.
23. Wilson GT, Wilfley DE, Agras WS, et al. Psychological treatments of binge eating disorder. Arch Gen Psychiatry. 2010;67(1):94-101.
24. Cassin SE, von Ranson KM, Heng K, et al. Adapted motivational interviewing for women with binge eating disorder: a randomized controlled trial. Psychol Addict Behav. 2008;22(3):417-425.
25. Brambilla F, Samek L, Company M, et al. Multivariate therapeutic approach to binge-eating disorder: combined nutritional, psychological and pharmacological treatment. Int Clin Psychopharmacol. 2009;24(6):312-317.
26. Claudino AM, de Oliveira IR, Appolinario JC, et al. Double-blind, randomized, placebo-controlled trial of topiramate plus cognitive-behavior therapy in binge-eating disorder. J Clin Psychiatry. 2007;68(9):1324-1332.
27. Devlin MJ, Goldfein JA, Petkova E, et al. Cognitive behavioral therapy and fluoxetine as adjuncts to group behavioral therapy for binge eating disorder. Obes Res. 2005;13(6):1077-1088.
28. Molinari E, Baruffi M, Croci M, et al. Binge eating disorder in obesity: comparison of different therapeutic strategies. Eat Weight Disord. 2005;10(3):154-161.
29. Golay A, Laurent-Jaccard A, Habicht F, et al. Effect of orlistat in obese patients with binge eating disorder. Obes Res. 2005;13(10):1701-1708.
Generalized anxiety disorder: Helping patients overcome worry
Discuss this article at www.facebook.com/CurrentPsychiatry
Mrs. M, age 44, is a married mother of 2 who presents to the psychiatric clinic with increased anxiety that recently has become intolerable, stating “I can’t stop my head.” She has experienced anxiety for “as long as I can remember.” She was a shy, anxious child who worried about her parents’ health. Her anxiety worsened at college, where she first sought care. She was prescribed diazepam as needed. The next semester, she had a depressive episode, treated with imipramine, 75 mg/d, which she tolerated poorly.
Mrs. M has received episodic supportive therapy since college. She has been plagued by bouts of anxiety and worry, with insomnia, tension, and fatigue. She worries about financial, career, family, and safety issues and has a phobia of spiders. Her family and friends often comment about her excessive worry, and it has strained her marriage and career; she was passed over for a promotion in part because of her anxiousness. Mrs. M also has experienced several depressive episodes.
Mrs. M has sought medical care for various non-specific somatic complaints; all laboratory tests were normal. Approximately 10 years ago, Mrs. M’s primary care physician prescribed fluoxetine, 20 mg/d, but Mrs. M stopped taking it after a few days, stating she felt “more anxious and jittery.”
To meet DSM-IV-TR diagnostic criteria for generalized anxiety disorder (GAD), patients must experience anxiety and worry that they find difficult to control. The worry and anxiety occur more days than not for at least 6 months and cause clinically significant distress and impairment (Table 1).1 These diagnostic criteria are being reevaluated—the DSM-5 Anxiety Work Group has proposed renaming the condition generalized worry disorder, specifying that only 2 domains need to be impacted by worry, shortening the required time frame of impairment from 6 months to 3 months, and including at least 1 behavioral change spawned by excessive worry.2 Although provisional, these recommendations suggest DSM-5 will include changes to GAD when it is published in 2013.
Table 1
DSM-IV-TR diagnostic criteria for generalized anxiety disorder
|
| OCD: obsessive-compulsive disorder; PTSD: posttraumatic stress disorder Source: Reference 1 |
A common, chronic condition
In the United States, the lifetime prevalence of GAD is 5.7%.3 It is twice as common in women. Although GAD can occur at any age, 75% of patients develop it before age 47; the median age is 31.3,4 Patients who present with GAD later in life have a better prognosis.3,4
Approximately 90% of GAD patients will meet criteria for another axis I disorder.3 When GAD patients present for treatment, social phobia and panic disorder are the most common comorbid psychiatric disorders. The lifetime prevalence of a mood disorder among GAD patients is 62%, but as few as 6% of GAD patients will meet criteria for a mood disorder at presentation.3 The onset of GAD usually precedes depression.5,6
Patients with GAD often first seek treatment from their primary care provider.7 A useful screening tool is the GAD-7 (Table 2).8 This instrument has a specificity of 92% and sensitivity of 76% for GAD for patients who score ≥8.7 Although the GAD-7 cannot confirm a GAD diagnosis, it can prompt clinicians to conduct a more structured interview. Because higher scores correlate with more severe symptoms, the GAD-7 can be used to measure progress.
Table 2
Screening for generalized anxiety disorder: The GAD-7
| Over the last 2 weeks, how often have you been bothered by any of the following problems? | Not at all | Several days | More than half the days | Nearly every day | |
|---|---|---|---|---|---|
| 1. | Feeling nervous, anxious, or on edge | 0 | 1 | 2 | 3 |
| 2. | Not being able to stop or control worrying | 0 | 1 | 2 | 3 |
| 3. | Worrying too much about different things | 0 | 1 | 2 | 3 |
| 4. | Trouble relaxing | 0 | 1 | 2 | 3 |
| 5. | Being so restless that it is hard to sit still | 0 | 1 | 2 | 3 |
| 6. | Becoming easily annoyed or irritable | 0 | 1 | 2 | 3 |
| 7. | Feeling afraid as if something awful might happen | 0 | 1 | 2 | 3 |
| GAD: generalized anxiety disorder Source: Reference 8 | |||||
Differential diagnosis
GAD typically has a chronic course with fluctuating symptom severity over the patient’s lifespan. Assess patients who present with anxiety for medical conditions that mimic GAD. These include:
- endocrine (hyperthyroidism), metabolic (electrolyte abnormalities), respiratory (asthma), neurologic (seizure disorder), or cardiac (arrhythmia) conditions
- nutritional deficiencies, especially of B vitamins and folate
- ingestion of substances or medications that may cause anxiety, such as caffeine or amphetamines.
A thorough history, medication review, and physical examination—as well as routine tests such as metabolic panel, complete blood count, thyroid function tests, urine drug screen, and electrocardiography—will capture most of these potential etiologies. In addition to ruling out medical causes, also assess for comorbid psychiatric conditions before reaching a diagnosis.
Evidence-based treatments
The treatment armamentarium for GAD includes psychotherapy and pharmacotherapy; complementary and alternative medicine (CAM) modalities may be useful adjunctive treatments. Which approach to use is determined by clinical judgment and the patient’s symptom severity and preferences. Combination therapy consisting of psychotherapy and medication often is appropriate.
Psychotherapy. Cognitive-behavioral therapy (CBT) is the preferred form of psychotherapy for GAD because it results in sustained improvements for patients with anxiety.5,9 Other modalities that may be effective include psychodynamic psychotherapy, mindfulness-based therapy, and interpersonal psychotherapy.10,11
Pharmacotherapy. As few as one-quarter of patients with GAD receive medications at appropriate dose and duration.12 Antidepressants are a first-line pharmacotherapy.5 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are highly effective for treating GAD.5 Paroxetine, escitalopram, duloxetine, and venlafaxine are FDA-approved for GAD, but other SSRIs also are used as primary treatment.5 Assuming the selected agent is tolerable and efficacious, a treatment course of 12 months is recommended.13
Benzodiazepines promote binding of the neuroinhibitory transmitter γ-aminobutyric acid and enhance chloride ion influx, thus reducing anxiety. Benzodiazepines have been widely used because of their rapid onset of action and effectiveness in managing anxiety, but their role in long-term management of GAD is unclear because these medications increase the risk of addiction, cognitive dulling, memory impairment, psychomotor retardation, and respiratory depression when combined with other CNS depressants such as alcohol and opiates. Before prescribing a benzodiazepine, conduct a thorough risk-benefit analysis and obtain informed consent. Long-term benzodiazepine monotherapy is not recommended.5,14
Hydroxyzine is an alternative to benzodiazepines.15 It works as an antihistamine and is FDA-approved for psychogenic neurosis, a Freudian distinction encompassing anxiety derived from psychological rather than physiological factors.
The azapirone buspirone is a nonaddictive, generally non-sedating 5-HT1A agonist. Although anecdotally some psychiatrists may report limited clinical utility, many analyses found azapirones, including buspirone, were effective for GAD,16,17 particularly for patients with comorbid depression.18
Tricyclic antidepressants are not a first-line choice because of their side effect profile and potential for drug-drug interactions. Nonetheless, some research suggests imipramine may be a reasonable option for GAD.14,19
Although not FDA-approved for GAD, anticonvulsant and antipsychotic medications may be reasonable adjunctive agents for patients with refractory GAD. Studies have suggested gabapentin20 and quetiapine21 as options.
Investigational treatments. Glutaminergic transmission is being investigated as a target for pharmacotherapy for GAD. In an 8-week, open-label trial, 12 of 15 GAD patients responded to the antiglutamatergic agent riluzole, 100 mg/d, and 8 patients achieved remission.22 In another study, pregabalin, which promotes calcium channel blockade, significantly reduced patients’ scores on the Hamilton Anxiety Rating Scale.23 However, this medication is a schedule V controlled substance and little is known about its long-term effects. Researchers had proposed that inhibition of corticotropin-releasing factor (CRF) may help reduce anxiety, but in a double-blind, placebo-controlled trial, they found that the CRF antagonist pexacerfont was no more effective than placebo.24
CAM treatments. A meta-analysis found that compared with placebo, kava extract (Piper methysticum) effectively reduced anxiety symptoms.25 However, considering its risk for hepatotoxicity, kava is not a recommended treatment.26 Although valerian, St. John’s wort, and passionflower have been used to manage GAD, there is insufficient evidence of their effectiveness and safety.26 No strong evidence supports nutritional supplements such as ginger, amino acids, and omega-3 fatty acids (fish oils) as treatment for GAD. Although there’s limited research on resistance training, aromatherapy, yoga, meditation, or acupuncture for treating anxiety, consider these treatments if your patient finds them helpful, because generally they are not contraindicated.
CASE CONTINUED: An antidepressant and CBT
Mrs. M reluctantly agrees to a trial of sertraline, 50 mg/d. She refuses a prescription for clonazepam because she is afraid of drug dependence but accepts a referral for CBT. Two days later, she calls the clinic and says she is more anxious and wants to stop the sertraline. The psychiatrist reassures her and reduces the dosage to 25 mg/d.
Mrs. M’s spike of anxiety resolves by her 2-week follow-up appointment and sertraline is titrated to 200 mg/d. Her irritability, anxiety, and mood improve within 2 months. The worry does not completely resolve, but she is much improved at 6 months, and the focus of her therapy shifts to her marriage.
Related Resources
- Goldberg D, Kendler KS, Sirvatka PJ, et al, eds. Diagnostic issues in depression and generalized anxiety disorder—refining the research agenda for DSM-V. Arlington, VA: American Psychiatric Publishing; 2010.
- Fricchione G. Generalized anxiety disorder. N Engl J Med. 2004;351(7):675-682.
Drug Brand Names
- Buspirone • Buspar
- Clonazepam •Klonopin
- Diazepam • Valium
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Gabapentin• Neurontin
- Hydroxyzine • Vistaril, Atarax
- Imipramine • Tofranil
- Paroxetine • Paxil
- Pregabalin • Lyrica
- Quetiapine • Seroquel
- Riluzole • Rilutek
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Barry reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Andrews G, Hobbs MJ, Borkovec TD, et al. Generalized worry disorder: a review of DSM-IV generalized anxiety disorder and options for DSM-V. Depress Anxiety. 2010;27(2):134-147.
3. Turk CL, Mennin DS. Phenomenology of generalized anxiety disorder. Psychiatr Ann. 2011;41(2):72-78.
4. Kessler RC, Chiu WT, Demler O, et al. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
5. Ballenger JC, Davidson JR, Lecrubier Y, et al. Consensus statement on generalized anxiety disorder from the International Consensus Group on Depression and Anxiety. J Clin Psychiatry. 2001;6(suppl 11):53-58.
6. Kessler RC, Keller MB, Wittchen HU. The epidemiology of generalized anxiety disorder. Psychiatr Clin North Am. 2001;24(1):19-39.
7. Kavan MG, Elsasser G, Barone EJ. Generalized anxiety disorder: practical assessment and management. Am Fam Physician. 2009;79(9):785-791.
8. Spitzer RL, Kroenke K, Williams JBW, et al. A brief measure for assessing generalized anxiety disorder. Arch Intern Med. 2006;166(10):1092-1097.
9. Newman MG, Borkovec TD. Cognitive-behavioral treatment of generalized anxiety disorder. Clin Psychol. 1995;48(4):5-7.
10. Leichsenring F, Salzer S, Jaeger U, et al. Short-term psychodynamic psychotherapy and cognitive-behavioral therapy in generalized anxiety disorder: a randomized, controlled trial. Am J Psychiatry. 2009;166(8):875-881.
11. Evans S, Ferrando S, Findler M, et al. Mindfulness-based cognitive therapy for generalized anxiety disorder. J Anxiety Disord. 2008;22(4):716-721.
12. Stein MB, Sherbourne CD, Craske MG, et al. Quality of care for primary care patients with anxiety disorders. Am J Psychiatry. 2004;161(12):2230-2237.
13. Fricchione G. Generalized anxiety disorder. N Engl J Med. 2004;351(7):675-682.
14. Rickels K, Downing R, Schweizer E, et al. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry. 1993;50(11):884-895.
15. Guaiana G, Barbui C, Cipriani A. Hydroxyzine for generalised anxiety disorder. Cochrane Database Syst Rev. 2010;(12):CD006815.-
16. Gammans RE, Stringfellow JC, Hvizdos AJ, et al. Use of buspirone in patients with generalized anxiety disorder and coexisting depressive symptoms. A meta-analysis of eight randomized, controlled studies. Neuropsychobiology. 1992;25(4):193-201.
17. Chessick CA, Allen MH, Thase M, et al. Azapirones for generalized anxiety disorder. Cochrane Database Syst Rev. 2006;(3):CD006115.-
18. Sramek JJ, Tansman M, Suri A, et al. Efficacy of buspirone in generalized anxiety disorder with coexisting mild depressive symptoms. J Clin Psychiatry. 1996;57(7):287-291.
19. Rickels K, DeMartinis N, García-España F, et al. Imipramine and buspirone in treatment of patients with generalized anxiety disorder who are discontinuing long-term benzodiazepine therapy. Am J Psychiatry. 2000;157(12):1973-1979.
20. Pollack MH, Matthews J, Scott EL. Gabapentin as a potential treatment for anxiety disorders. Am J Psychiatry. 1998;155(7):992-993.
21. Khan A, Joyce M, Atkinson S, et al. A randomized, double-blind study of once-daily extended release quetiapine fumarate (quetiapine XR) monotherapy in patients with generalized anxiety disorder. J Clin Psychopharmacol. 2011;31(4):418-428.
22. Mathew SJ, Amiel JM, Coplan JD, et al. Open-label trial of riluzole in generalized anxiety disorder. Am J Psychiatry. 2005;162(12):2379-2381.
23. Pande AC, Crockatt JG, Feltner DE, et al. Pregabalin in generalized anxiety disorder: a placebo-controlled trial. Am J Psychiatry. 2003;160(3):533-540.
24. Coric V, Feldman HH, Oren DA, et al. Multicenter, randomized, double-blind, active comparator and placebo-controlled trial of a corticotropin-releasing factor receptor-1 antagonist in generalized anxiety disorder. Depress Anxiety. 2010;27(5):417-425.
25. Pittler MH, Ernst E. Kava extract for treating anxiety. Cochrane Database Syst Rev. 2003;(1):CD003383.-
26. Zoberi K, Pollard CA. Treating anxiety without SSRIs. J Fam Pract. 2010;59(3):148-154.
Discuss this article at www.facebook.com/CurrentPsychiatry
Mrs. M, age 44, is a married mother of 2 who presents to the psychiatric clinic with increased anxiety that recently has become intolerable, stating “I can’t stop my head.” She has experienced anxiety for “as long as I can remember.” She was a shy, anxious child who worried about her parents’ health. Her anxiety worsened at college, where she first sought care. She was prescribed diazepam as needed. The next semester, she had a depressive episode, treated with imipramine, 75 mg/d, which she tolerated poorly.
Mrs. M has received episodic supportive therapy since college. She has been plagued by bouts of anxiety and worry, with insomnia, tension, and fatigue. She worries about financial, career, family, and safety issues and has a phobia of spiders. Her family and friends often comment about her excessive worry, and it has strained her marriage and career; she was passed over for a promotion in part because of her anxiousness. Mrs. M also has experienced several depressive episodes.
Mrs. M has sought medical care for various non-specific somatic complaints; all laboratory tests were normal. Approximately 10 years ago, Mrs. M’s primary care physician prescribed fluoxetine, 20 mg/d, but Mrs. M stopped taking it after a few days, stating she felt “more anxious and jittery.”
To meet DSM-IV-TR diagnostic criteria for generalized anxiety disorder (GAD), patients must experience anxiety and worry that they find difficult to control. The worry and anxiety occur more days than not for at least 6 months and cause clinically significant distress and impairment (Table 1).1 These diagnostic criteria are being reevaluated—the DSM-5 Anxiety Work Group has proposed renaming the condition generalized worry disorder, specifying that only 2 domains need to be impacted by worry, shortening the required time frame of impairment from 6 months to 3 months, and including at least 1 behavioral change spawned by excessive worry.2 Although provisional, these recommendations suggest DSM-5 will include changes to GAD when it is published in 2013.
Table 1
DSM-IV-TR diagnostic criteria for generalized anxiety disorder
|
| OCD: obsessive-compulsive disorder; PTSD: posttraumatic stress disorder Source: Reference 1 |
A common, chronic condition
In the United States, the lifetime prevalence of GAD is 5.7%.3 It is twice as common in women. Although GAD can occur at any age, 75% of patients develop it before age 47; the median age is 31.3,4 Patients who present with GAD later in life have a better prognosis.3,4
Approximately 90% of GAD patients will meet criteria for another axis I disorder.3 When GAD patients present for treatment, social phobia and panic disorder are the most common comorbid psychiatric disorders. The lifetime prevalence of a mood disorder among GAD patients is 62%, but as few as 6% of GAD patients will meet criteria for a mood disorder at presentation.3 The onset of GAD usually precedes depression.5,6
Patients with GAD often first seek treatment from their primary care provider.7 A useful screening tool is the GAD-7 (Table 2).8 This instrument has a specificity of 92% and sensitivity of 76% for GAD for patients who score ≥8.7 Although the GAD-7 cannot confirm a GAD diagnosis, it can prompt clinicians to conduct a more structured interview. Because higher scores correlate with more severe symptoms, the GAD-7 can be used to measure progress.
Table 2
Screening for generalized anxiety disorder: The GAD-7
| Over the last 2 weeks, how often have you been bothered by any of the following problems? | Not at all | Several days | More than half the days | Nearly every day | |
|---|---|---|---|---|---|
| 1. | Feeling nervous, anxious, or on edge | 0 | 1 | 2 | 3 |
| 2. | Not being able to stop or control worrying | 0 | 1 | 2 | 3 |
| 3. | Worrying too much about different things | 0 | 1 | 2 | 3 |
| 4. | Trouble relaxing | 0 | 1 | 2 | 3 |
| 5. | Being so restless that it is hard to sit still | 0 | 1 | 2 | 3 |
| 6. | Becoming easily annoyed or irritable | 0 | 1 | 2 | 3 |
| 7. | Feeling afraid as if something awful might happen | 0 | 1 | 2 | 3 |
| GAD: generalized anxiety disorder Source: Reference 8 | |||||
Differential diagnosis
GAD typically has a chronic course with fluctuating symptom severity over the patient’s lifespan. Assess patients who present with anxiety for medical conditions that mimic GAD. These include:
- endocrine (hyperthyroidism), metabolic (electrolyte abnormalities), respiratory (asthma), neurologic (seizure disorder), or cardiac (arrhythmia) conditions
- nutritional deficiencies, especially of B vitamins and folate
- ingestion of substances or medications that may cause anxiety, such as caffeine or amphetamines.
A thorough history, medication review, and physical examination—as well as routine tests such as metabolic panel, complete blood count, thyroid function tests, urine drug screen, and electrocardiography—will capture most of these potential etiologies. In addition to ruling out medical causes, also assess for comorbid psychiatric conditions before reaching a diagnosis.
Evidence-based treatments
The treatment armamentarium for GAD includes psychotherapy and pharmacotherapy; complementary and alternative medicine (CAM) modalities may be useful adjunctive treatments. Which approach to use is determined by clinical judgment and the patient’s symptom severity and preferences. Combination therapy consisting of psychotherapy and medication often is appropriate.
Psychotherapy. Cognitive-behavioral therapy (CBT) is the preferred form of psychotherapy for GAD because it results in sustained improvements for patients with anxiety.5,9 Other modalities that may be effective include psychodynamic psychotherapy, mindfulness-based therapy, and interpersonal psychotherapy.10,11
Pharmacotherapy. As few as one-quarter of patients with GAD receive medications at appropriate dose and duration.12 Antidepressants are a first-line pharmacotherapy.5 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are highly effective for treating GAD.5 Paroxetine, escitalopram, duloxetine, and venlafaxine are FDA-approved for GAD, but other SSRIs also are used as primary treatment.5 Assuming the selected agent is tolerable and efficacious, a treatment course of 12 months is recommended.13
Benzodiazepines promote binding of the neuroinhibitory transmitter γ-aminobutyric acid and enhance chloride ion influx, thus reducing anxiety. Benzodiazepines have been widely used because of their rapid onset of action and effectiveness in managing anxiety, but their role in long-term management of GAD is unclear because these medications increase the risk of addiction, cognitive dulling, memory impairment, psychomotor retardation, and respiratory depression when combined with other CNS depressants such as alcohol and opiates. Before prescribing a benzodiazepine, conduct a thorough risk-benefit analysis and obtain informed consent. Long-term benzodiazepine monotherapy is not recommended.5,14
Hydroxyzine is an alternative to benzodiazepines.15 It works as an antihistamine and is FDA-approved for psychogenic neurosis, a Freudian distinction encompassing anxiety derived from psychological rather than physiological factors.
The azapirone buspirone is a nonaddictive, generally non-sedating 5-HT1A agonist. Although anecdotally some psychiatrists may report limited clinical utility, many analyses found azapirones, including buspirone, were effective for GAD,16,17 particularly for patients with comorbid depression.18
Tricyclic antidepressants are not a first-line choice because of their side effect profile and potential for drug-drug interactions. Nonetheless, some research suggests imipramine may be a reasonable option for GAD.14,19
Although not FDA-approved for GAD, anticonvulsant and antipsychotic medications may be reasonable adjunctive agents for patients with refractory GAD. Studies have suggested gabapentin20 and quetiapine21 as options.
Investigational treatments. Glutaminergic transmission is being investigated as a target for pharmacotherapy for GAD. In an 8-week, open-label trial, 12 of 15 GAD patients responded to the antiglutamatergic agent riluzole, 100 mg/d, and 8 patients achieved remission.22 In another study, pregabalin, which promotes calcium channel blockade, significantly reduced patients’ scores on the Hamilton Anxiety Rating Scale.23 However, this medication is a schedule V controlled substance and little is known about its long-term effects. Researchers had proposed that inhibition of corticotropin-releasing factor (CRF) may help reduce anxiety, but in a double-blind, placebo-controlled trial, they found that the CRF antagonist pexacerfont was no more effective than placebo.24
CAM treatments. A meta-analysis found that compared with placebo, kava extract (Piper methysticum) effectively reduced anxiety symptoms.25 However, considering its risk for hepatotoxicity, kava is not a recommended treatment.26 Although valerian, St. John’s wort, and passionflower have been used to manage GAD, there is insufficient evidence of their effectiveness and safety.26 No strong evidence supports nutritional supplements such as ginger, amino acids, and omega-3 fatty acids (fish oils) as treatment for GAD. Although there’s limited research on resistance training, aromatherapy, yoga, meditation, or acupuncture for treating anxiety, consider these treatments if your patient finds them helpful, because generally they are not contraindicated.
CASE CONTINUED: An antidepressant and CBT
Mrs. M reluctantly agrees to a trial of sertraline, 50 mg/d. She refuses a prescription for clonazepam because she is afraid of drug dependence but accepts a referral for CBT. Two days later, she calls the clinic and says she is more anxious and wants to stop the sertraline. The psychiatrist reassures her and reduces the dosage to 25 mg/d.
Mrs. M’s spike of anxiety resolves by her 2-week follow-up appointment and sertraline is titrated to 200 mg/d. Her irritability, anxiety, and mood improve within 2 months. The worry does not completely resolve, but she is much improved at 6 months, and the focus of her therapy shifts to her marriage.
Related Resources
- Goldberg D, Kendler KS, Sirvatka PJ, et al, eds. Diagnostic issues in depression and generalized anxiety disorder—refining the research agenda for DSM-V. Arlington, VA: American Psychiatric Publishing; 2010.
- Fricchione G. Generalized anxiety disorder. N Engl J Med. 2004;351(7):675-682.
Drug Brand Names
- Buspirone • Buspar
- Clonazepam •Klonopin
- Diazepam • Valium
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Gabapentin• Neurontin
- Hydroxyzine • Vistaril, Atarax
- Imipramine • Tofranil
- Paroxetine • Paxil
- Pregabalin • Lyrica
- Quetiapine • Seroquel
- Riluzole • Rilutek
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Barry reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Mrs. M, age 44, is a married mother of 2 who presents to the psychiatric clinic with increased anxiety that recently has become intolerable, stating “I can’t stop my head.” She has experienced anxiety for “as long as I can remember.” She was a shy, anxious child who worried about her parents’ health. Her anxiety worsened at college, where she first sought care. She was prescribed diazepam as needed. The next semester, she had a depressive episode, treated with imipramine, 75 mg/d, which she tolerated poorly.
Mrs. M has received episodic supportive therapy since college. She has been plagued by bouts of anxiety and worry, with insomnia, tension, and fatigue. She worries about financial, career, family, and safety issues and has a phobia of spiders. Her family and friends often comment about her excessive worry, and it has strained her marriage and career; she was passed over for a promotion in part because of her anxiousness. Mrs. M also has experienced several depressive episodes.
Mrs. M has sought medical care for various non-specific somatic complaints; all laboratory tests were normal. Approximately 10 years ago, Mrs. M’s primary care physician prescribed fluoxetine, 20 mg/d, but Mrs. M stopped taking it after a few days, stating she felt “more anxious and jittery.”
To meet DSM-IV-TR diagnostic criteria for generalized anxiety disorder (GAD), patients must experience anxiety and worry that they find difficult to control. The worry and anxiety occur more days than not for at least 6 months and cause clinically significant distress and impairment (Table 1).1 These diagnostic criteria are being reevaluated—the DSM-5 Anxiety Work Group has proposed renaming the condition generalized worry disorder, specifying that only 2 domains need to be impacted by worry, shortening the required time frame of impairment from 6 months to 3 months, and including at least 1 behavioral change spawned by excessive worry.2 Although provisional, these recommendations suggest DSM-5 will include changes to GAD when it is published in 2013.
Table 1
DSM-IV-TR diagnostic criteria for generalized anxiety disorder
|
| OCD: obsessive-compulsive disorder; PTSD: posttraumatic stress disorder Source: Reference 1 |
A common, chronic condition
In the United States, the lifetime prevalence of GAD is 5.7%.3 It is twice as common in women. Although GAD can occur at any age, 75% of patients develop it before age 47; the median age is 31.3,4 Patients who present with GAD later in life have a better prognosis.3,4
Approximately 90% of GAD patients will meet criteria for another axis I disorder.3 When GAD patients present for treatment, social phobia and panic disorder are the most common comorbid psychiatric disorders. The lifetime prevalence of a mood disorder among GAD patients is 62%, but as few as 6% of GAD patients will meet criteria for a mood disorder at presentation.3 The onset of GAD usually precedes depression.5,6
Patients with GAD often first seek treatment from their primary care provider.7 A useful screening tool is the GAD-7 (Table 2).8 This instrument has a specificity of 92% and sensitivity of 76% for GAD for patients who score ≥8.7 Although the GAD-7 cannot confirm a GAD diagnosis, it can prompt clinicians to conduct a more structured interview. Because higher scores correlate with more severe symptoms, the GAD-7 can be used to measure progress.
Table 2
Screening for generalized anxiety disorder: The GAD-7
| Over the last 2 weeks, how often have you been bothered by any of the following problems? | Not at all | Several days | More than half the days | Nearly every day | |
|---|---|---|---|---|---|
| 1. | Feeling nervous, anxious, or on edge | 0 | 1 | 2 | 3 |
| 2. | Not being able to stop or control worrying | 0 | 1 | 2 | 3 |
| 3. | Worrying too much about different things | 0 | 1 | 2 | 3 |
| 4. | Trouble relaxing | 0 | 1 | 2 | 3 |
| 5. | Being so restless that it is hard to sit still | 0 | 1 | 2 | 3 |
| 6. | Becoming easily annoyed or irritable | 0 | 1 | 2 | 3 |
| 7. | Feeling afraid as if something awful might happen | 0 | 1 | 2 | 3 |
| GAD: generalized anxiety disorder Source: Reference 8 | |||||
Differential diagnosis
GAD typically has a chronic course with fluctuating symptom severity over the patient’s lifespan. Assess patients who present with anxiety for medical conditions that mimic GAD. These include:
- endocrine (hyperthyroidism), metabolic (electrolyte abnormalities), respiratory (asthma), neurologic (seizure disorder), or cardiac (arrhythmia) conditions
- nutritional deficiencies, especially of B vitamins and folate
- ingestion of substances or medications that may cause anxiety, such as caffeine or amphetamines.
A thorough history, medication review, and physical examination—as well as routine tests such as metabolic panel, complete blood count, thyroid function tests, urine drug screen, and electrocardiography—will capture most of these potential etiologies. In addition to ruling out medical causes, also assess for comorbid psychiatric conditions before reaching a diagnosis.
Evidence-based treatments
The treatment armamentarium for GAD includes psychotherapy and pharmacotherapy; complementary and alternative medicine (CAM) modalities may be useful adjunctive treatments. Which approach to use is determined by clinical judgment and the patient’s symptom severity and preferences. Combination therapy consisting of psychotherapy and medication often is appropriate.
Psychotherapy. Cognitive-behavioral therapy (CBT) is the preferred form of psychotherapy for GAD because it results in sustained improvements for patients with anxiety.5,9 Other modalities that may be effective include psychodynamic psychotherapy, mindfulness-based therapy, and interpersonal psychotherapy.10,11
Pharmacotherapy. As few as one-quarter of patients with GAD receive medications at appropriate dose and duration.12 Antidepressants are a first-line pharmacotherapy.5 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are highly effective for treating GAD.5 Paroxetine, escitalopram, duloxetine, and venlafaxine are FDA-approved for GAD, but other SSRIs also are used as primary treatment.5 Assuming the selected agent is tolerable and efficacious, a treatment course of 12 months is recommended.13
Benzodiazepines promote binding of the neuroinhibitory transmitter γ-aminobutyric acid and enhance chloride ion influx, thus reducing anxiety. Benzodiazepines have been widely used because of their rapid onset of action and effectiveness in managing anxiety, but their role in long-term management of GAD is unclear because these medications increase the risk of addiction, cognitive dulling, memory impairment, psychomotor retardation, and respiratory depression when combined with other CNS depressants such as alcohol and opiates. Before prescribing a benzodiazepine, conduct a thorough risk-benefit analysis and obtain informed consent. Long-term benzodiazepine monotherapy is not recommended.5,14
Hydroxyzine is an alternative to benzodiazepines.15 It works as an antihistamine and is FDA-approved for psychogenic neurosis, a Freudian distinction encompassing anxiety derived from psychological rather than physiological factors.
The azapirone buspirone is a nonaddictive, generally non-sedating 5-HT1A agonist. Although anecdotally some psychiatrists may report limited clinical utility, many analyses found azapirones, including buspirone, were effective for GAD,16,17 particularly for patients with comorbid depression.18
Tricyclic antidepressants are not a first-line choice because of their side effect profile and potential for drug-drug interactions. Nonetheless, some research suggests imipramine may be a reasonable option for GAD.14,19
Although not FDA-approved for GAD, anticonvulsant and antipsychotic medications may be reasonable adjunctive agents for patients with refractory GAD. Studies have suggested gabapentin20 and quetiapine21 as options.
Investigational treatments. Glutaminergic transmission is being investigated as a target for pharmacotherapy for GAD. In an 8-week, open-label trial, 12 of 15 GAD patients responded to the antiglutamatergic agent riluzole, 100 mg/d, and 8 patients achieved remission.22 In another study, pregabalin, which promotes calcium channel blockade, significantly reduced patients’ scores on the Hamilton Anxiety Rating Scale.23 However, this medication is a schedule V controlled substance and little is known about its long-term effects. Researchers had proposed that inhibition of corticotropin-releasing factor (CRF) may help reduce anxiety, but in a double-blind, placebo-controlled trial, they found that the CRF antagonist pexacerfont was no more effective than placebo.24
CAM treatments. A meta-analysis found that compared with placebo, kava extract (Piper methysticum) effectively reduced anxiety symptoms.25 However, considering its risk for hepatotoxicity, kava is not a recommended treatment.26 Although valerian, St. John’s wort, and passionflower have been used to manage GAD, there is insufficient evidence of their effectiveness and safety.26 No strong evidence supports nutritional supplements such as ginger, amino acids, and omega-3 fatty acids (fish oils) as treatment for GAD. Although there’s limited research on resistance training, aromatherapy, yoga, meditation, or acupuncture for treating anxiety, consider these treatments if your patient finds them helpful, because generally they are not contraindicated.
CASE CONTINUED: An antidepressant and CBT
Mrs. M reluctantly agrees to a trial of sertraline, 50 mg/d. She refuses a prescription for clonazepam because she is afraid of drug dependence but accepts a referral for CBT. Two days later, she calls the clinic and says she is more anxious and wants to stop the sertraline. The psychiatrist reassures her and reduces the dosage to 25 mg/d.
Mrs. M’s spike of anxiety resolves by her 2-week follow-up appointment and sertraline is titrated to 200 mg/d. Her irritability, anxiety, and mood improve within 2 months. The worry does not completely resolve, but she is much improved at 6 months, and the focus of her therapy shifts to her marriage.
Related Resources
- Goldberg D, Kendler KS, Sirvatka PJ, et al, eds. Diagnostic issues in depression and generalized anxiety disorder—refining the research agenda for DSM-V. Arlington, VA: American Psychiatric Publishing; 2010.
- Fricchione G. Generalized anxiety disorder. N Engl J Med. 2004;351(7):675-682.
Drug Brand Names
- Buspirone • Buspar
- Clonazepam •Klonopin
- Diazepam • Valium
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Gabapentin• Neurontin
- Hydroxyzine • Vistaril, Atarax
- Imipramine • Tofranil
- Paroxetine • Paxil
- Pregabalin • Lyrica
- Quetiapine • Seroquel
- Riluzole • Rilutek
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Barry reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Andrews G, Hobbs MJ, Borkovec TD, et al. Generalized worry disorder: a review of DSM-IV generalized anxiety disorder and options for DSM-V. Depress Anxiety. 2010;27(2):134-147.
3. Turk CL, Mennin DS. Phenomenology of generalized anxiety disorder. Psychiatr Ann. 2011;41(2):72-78.
4. Kessler RC, Chiu WT, Demler O, et al. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
5. Ballenger JC, Davidson JR, Lecrubier Y, et al. Consensus statement on generalized anxiety disorder from the International Consensus Group on Depression and Anxiety. J Clin Psychiatry. 2001;6(suppl 11):53-58.
6. Kessler RC, Keller MB, Wittchen HU. The epidemiology of generalized anxiety disorder. Psychiatr Clin North Am. 2001;24(1):19-39.
7. Kavan MG, Elsasser G, Barone EJ. Generalized anxiety disorder: practical assessment and management. Am Fam Physician. 2009;79(9):785-791.
8. Spitzer RL, Kroenke K, Williams JBW, et al. A brief measure for assessing generalized anxiety disorder. Arch Intern Med. 2006;166(10):1092-1097.
9. Newman MG, Borkovec TD. Cognitive-behavioral treatment of generalized anxiety disorder. Clin Psychol. 1995;48(4):5-7.
10. Leichsenring F, Salzer S, Jaeger U, et al. Short-term psychodynamic psychotherapy and cognitive-behavioral therapy in generalized anxiety disorder: a randomized, controlled trial. Am J Psychiatry. 2009;166(8):875-881.
11. Evans S, Ferrando S, Findler M, et al. Mindfulness-based cognitive therapy for generalized anxiety disorder. J Anxiety Disord. 2008;22(4):716-721.
12. Stein MB, Sherbourne CD, Craske MG, et al. Quality of care for primary care patients with anxiety disorders. Am J Psychiatry. 2004;161(12):2230-2237.
13. Fricchione G. Generalized anxiety disorder. N Engl J Med. 2004;351(7):675-682.
14. Rickels K, Downing R, Schweizer E, et al. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry. 1993;50(11):884-895.
15. Guaiana G, Barbui C, Cipriani A. Hydroxyzine for generalised anxiety disorder. Cochrane Database Syst Rev. 2010;(12):CD006815.-
16. Gammans RE, Stringfellow JC, Hvizdos AJ, et al. Use of buspirone in patients with generalized anxiety disorder and coexisting depressive symptoms. A meta-analysis of eight randomized, controlled studies. Neuropsychobiology. 1992;25(4):193-201.
17. Chessick CA, Allen MH, Thase M, et al. Azapirones for generalized anxiety disorder. Cochrane Database Syst Rev. 2006;(3):CD006115.-
18. Sramek JJ, Tansman M, Suri A, et al. Efficacy of buspirone in generalized anxiety disorder with coexisting mild depressive symptoms. J Clin Psychiatry. 1996;57(7):287-291.
19. Rickels K, DeMartinis N, García-España F, et al. Imipramine and buspirone in treatment of patients with generalized anxiety disorder who are discontinuing long-term benzodiazepine therapy. Am J Psychiatry. 2000;157(12):1973-1979.
20. Pollack MH, Matthews J, Scott EL. Gabapentin as a potential treatment for anxiety disorders. Am J Psychiatry. 1998;155(7):992-993.
21. Khan A, Joyce M, Atkinson S, et al. A randomized, double-blind study of once-daily extended release quetiapine fumarate (quetiapine XR) monotherapy in patients with generalized anxiety disorder. J Clin Psychopharmacol. 2011;31(4):418-428.
22. Mathew SJ, Amiel JM, Coplan JD, et al. Open-label trial of riluzole in generalized anxiety disorder. Am J Psychiatry. 2005;162(12):2379-2381.
23. Pande AC, Crockatt JG, Feltner DE, et al. Pregabalin in generalized anxiety disorder: a placebo-controlled trial. Am J Psychiatry. 2003;160(3):533-540.
24. Coric V, Feldman HH, Oren DA, et al. Multicenter, randomized, double-blind, active comparator and placebo-controlled trial of a corticotropin-releasing factor receptor-1 antagonist in generalized anxiety disorder. Depress Anxiety. 2010;27(5):417-425.
25. Pittler MH, Ernst E. Kava extract for treating anxiety. Cochrane Database Syst Rev. 2003;(1):CD003383.-
26. Zoberi K, Pollard CA. Treating anxiety without SSRIs. J Fam Pract. 2010;59(3):148-154.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Andrews G, Hobbs MJ, Borkovec TD, et al. Generalized worry disorder: a review of DSM-IV generalized anxiety disorder and options for DSM-V. Depress Anxiety. 2010;27(2):134-147.
3. Turk CL, Mennin DS. Phenomenology of generalized anxiety disorder. Psychiatr Ann. 2011;41(2):72-78.
4. Kessler RC, Chiu WT, Demler O, et al. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
5. Ballenger JC, Davidson JR, Lecrubier Y, et al. Consensus statement on generalized anxiety disorder from the International Consensus Group on Depression and Anxiety. J Clin Psychiatry. 2001;6(suppl 11):53-58.
6. Kessler RC, Keller MB, Wittchen HU. The epidemiology of generalized anxiety disorder. Psychiatr Clin North Am. 2001;24(1):19-39.
7. Kavan MG, Elsasser G, Barone EJ. Generalized anxiety disorder: practical assessment and management. Am Fam Physician. 2009;79(9):785-791.
8. Spitzer RL, Kroenke K, Williams JBW, et al. A brief measure for assessing generalized anxiety disorder. Arch Intern Med. 2006;166(10):1092-1097.
9. Newman MG, Borkovec TD. Cognitive-behavioral treatment of generalized anxiety disorder. Clin Psychol. 1995;48(4):5-7.
10. Leichsenring F, Salzer S, Jaeger U, et al. Short-term psychodynamic psychotherapy and cognitive-behavioral therapy in generalized anxiety disorder: a randomized, controlled trial. Am J Psychiatry. 2009;166(8):875-881.
11. Evans S, Ferrando S, Findler M, et al. Mindfulness-based cognitive therapy for generalized anxiety disorder. J Anxiety Disord. 2008;22(4):716-721.
12. Stein MB, Sherbourne CD, Craske MG, et al. Quality of care for primary care patients with anxiety disorders. Am J Psychiatry. 2004;161(12):2230-2237.
13. Fricchione G. Generalized anxiety disorder. N Engl J Med. 2004;351(7):675-682.
14. Rickels K, Downing R, Schweizer E, et al. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry. 1993;50(11):884-895.
15. Guaiana G, Barbui C, Cipriani A. Hydroxyzine for generalised anxiety disorder. Cochrane Database Syst Rev. 2010;(12):CD006815.-
16. Gammans RE, Stringfellow JC, Hvizdos AJ, et al. Use of buspirone in patients with generalized anxiety disorder and coexisting depressive symptoms. A meta-analysis of eight randomized, controlled studies. Neuropsychobiology. 1992;25(4):193-201.
17. Chessick CA, Allen MH, Thase M, et al. Azapirones for generalized anxiety disorder. Cochrane Database Syst Rev. 2006;(3):CD006115.-
18. Sramek JJ, Tansman M, Suri A, et al. Efficacy of buspirone in generalized anxiety disorder with coexisting mild depressive symptoms. J Clin Psychiatry. 1996;57(7):287-291.
19. Rickels K, DeMartinis N, García-España F, et al. Imipramine and buspirone in treatment of patients with generalized anxiety disorder who are discontinuing long-term benzodiazepine therapy. Am J Psychiatry. 2000;157(12):1973-1979.
20. Pollack MH, Matthews J, Scott EL. Gabapentin as a potential treatment for anxiety disorders. Am J Psychiatry. 1998;155(7):992-993.
21. Khan A, Joyce M, Atkinson S, et al. A randomized, double-blind study of once-daily extended release quetiapine fumarate (quetiapine XR) monotherapy in patients with generalized anxiety disorder. J Clin Psychopharmacol. 2011;31(4):418-428.
22. Mathew SJ, Amiel JM, Coplan JD, et al. Open-label trial of riluzole in generalized anxiety disorder. Am J Psychiatry. 2005;162(12):2379-2381.
23. Pande AC, Crockatt JG, Feltner DE, et al. Pregabalin in generalized anxiety disorder: a placebo-controlled trial. Am J Psychiatry. 2003;160(3):533-540.
24. Coric V, Feldman HH, Oren DA, et al. Multicenter, randomized, double-blind, active comparator and placebo-controlled trial of a corticotropin-releasing factor receptor-1 antagonist in generalized anxiety disorder. Depress Anxiety. 2010;27(5):417-425.
25. Pittler MH, Ernst E. Kava extract for treating anxiety. Cochrane Database Syst Rev. 2003;(1):CD003383.-
26. Zoberi K, Pollard CA. Treating anxiety without SSRIs. J Fam Pract. 2010;59(3):148-154.
Hyponatremia in Heart Failure
Hyponatremia, defined as a serum [Na+] 135 mEq/L, occurs in 2030% of patients with acute decompensated heart failure (HF)13 and has been independently associated with a poor prognosis. In clinical trials of acute decompensated HF, the reported mean serum sodium is often normal or near normal, but a significant proportion of study subjects can have serum sodium values that approach 130 mEq/L or lower.3 However, despite the association between hyponatremia and clinical outcomes like hospitalization and mortality, data from studies are sparse about the impact of drug or device interventions in the hyponatremic cohort, since patients are generally not stratified at the time of randomization by the value of baseline serum sodium.
HYPONATREMIA AND PROGNOSIS
Hyponatremia has long been recognized as a potential prognostic marker in heart failure, highlighted by Packer and Lee in 1986.4 Subsequently, a wealth of data derived from clinical trials, registries, and observational databases support the concept that hyponatremia is an independent predictor of both short‐ and long‐term outcomes.13, 511 As reviewed by Jao and Chiong,3 this relationship holds in patients on optimal evidence‐based medical therapy, including treatment with antagonists of the renin‐angiotensin system and beta blockers. In the Organized Program To Initiate Lifesaving Treatment In Hospitalized Patients With Heart Failure (OPTIMIZE)2 HF Registry of nearly 50,000 patients, in‐hospital and 60‐day mortality rates were higher in patients with lower serum sodium levels on admission (cut‐off point of 135 mEq/L). In‐hospital death and the combined endpoint of death or re‐hospitalization increased significantly for each 3 mEq/L decrease in serum [Na+] below 140 mEq/L. Patients with hyponatremia were more likely to have lower systolic blood pressures and receive intravenous inotropic agents; lengths of stay were also longer.
Similar findings were reported in the Evaluation Study of Congestive Heart Failure and Pulmonary Acute and Chronic Therapeutic Impact of a Vasopressin 2 Antagonist (Tolvaptan) in Congestive Heart Failure (ACTIV in CHF)10 trial.11 For example, in the former, Gheorghiade and colleagues tracked serum sodium levels in 433 hospitalized patients who had acute decompensated HF and examined the proportion free from a major event (defined as death and/or HF hospitalization).1 There was a clear association between the event rate and serum sodium level. Patients whose hyponatremia persisted from hospital admission to discharge were at higher risk relative to those whose hyponatremia was corrected during the hospital stay.
However, whether the way in which the serum sodium improvement is achieved has a bearing on outcomes is not known. In the studies comparing outcomes in patients with heart failure and hyponatremia versus normonatremia, no mention is made about how the patient arrived at either state. Despite this limitation, the findings are incontrovertibly consistent. Hyponatremia on discharge (prior to or after the adoption of renin‐angiotensin‐aldosterone system (RAAS) antagonists or beta blockers) is a marker for poorer outcomes, as is another laboratory abnormality frequently observed in patients hospitalized with heart failure: an elevated creatinine.
Additionally, serum sodium obtained shortly after hospitalization is a potent predictor of re‐hospitalization12 and persistently poor health‐related quality‐of‐life.13 The impact on longer‐term outcomes can also be demonstrated in multiple prognostic models6, 8, 9 in which serum sodium is a risk factor for adverse outcomes. For example, using the Seattle Heart Failure Model, overall prognosis worsens for each 1 mEq decline in serum sodium when all other variables are kept constant.8 This observation suggests that, in terms of prognosis, the value of serum sodium functions as a continuous not a binary variable.
HYPONATREMIA AND HF PATHOPHYSIOLOGY
The reasons underlying hyponatremia in heart failure are complex, but a key component is the non‐osmotic release of arginine vasopressin (AVP) in response to stimulation of carotid baroreceptors. This phenomenon occurs as a result of arterial underfilling (both lower blood pressure and lower cardiac output). AVP is one member of a family of neurohormones and cytokines that are upregulated in heart failure (eg, norepinephrine, renin, angiotensin, aldosterone, endothelin, and tumor necrosis factor‐alpha). Levels of AVP are increased most markedly in patients with advanced symptoms (ie, New York Heart Association Class III and IV),14 and this leads to impaired free water handling in the renal tubules and a hypervolemic form of hyponatremia. The reasons underlying the upregulation are debated, but likely reflect a short‐term hemodynamic adaptation that is designed to augment cardiac output by increasing circulating volume. In addition, multiple neurohormones have been shown to promote progressive ventricular dilation, referred to as remodeling. For example, chronic elevations of norepinephrine contribute to a multitude of genotypic and phenotypic changes at the level of the myocyte. The short‐term benefits of neurohormonal upregulation are offset by maladaptive responses in the long term, and this observation likely explains a major part of the clinical benefits seen with drugs such as angiotensin converting enzyme inhibitors, aldosterone antagonists, and beta blockers.
It is also clear that the development and management of patients with hyponatremia and heart failure are frequently complicated by the presence of other factors that impact sodium and water handling. Heart failure often occurs in older patients with renal dysfunction who are on medications that can exacerbate hyponatremia, such as diuretics, non‐steroidal anti‐inflammatory agents, antidepressants, and opiate derivatives. In addition, other conditions like hypothyroidism may coexist and contribute to the hyponatremic state. It is therefore crucial for the clinician to consider these possibilities when a patient with heart failure presents with or develops hyponatremia, and in particular to critically evaluate the potential role of concomitant medications that can cause a syndrome of inappropriate antidiuretic hormone secretion (SIADH)‐like picture.
HYPONATREMIA AND RESOURCE USE
As with other markers of poor outcome in heart failure, such as worsening renal insufficiency, chronic obstructive lung disease, and other comorbidities, hyponatremia is associated with longer lengths of stay (LOS) and cost. In an analysis of approximately 116,000 patients hospitalized with HF and grouped at admission by serum [Na+], risk‐adjusted mortality, LOS, and attributable cost were highest for patients with severe hyponatremia compared to patients with normonatremia.15 In addition, Amin and colleagues recently demonstrated that length of stay in the intensive care unit and associated costs were greater (21% and 23%, respectively) in patients who had an International Classification of Diseases, 9th revision, Clinical Modification (ICD‐9‐CM) code for hyponatremia compared to those that did not.16
CONSIDERATIONS FOR PATIENTS HOSPITALIZED WITH HEART FAILURE WITH AND WITHOUT HYPONATREMIA
A number of significant management challenges exist during the hospitalization phase of acute decompensated heart failure. Among other tasks, the clinician should evaluate the potential cause of the decompensation (eg, medication noncompliance, dietary noncompliance, increased metabolic demand from pneumonia or other infection, worsening renal failure, diuretic resistance, iatrogenic fluid overload) and decide whether the patient is fluid overloaded, in a low cardiac output state contributing to end‐organ perfusion, or both. Manifestations of worsening heart failure other than dyspnea may be present. For example, mental status changes in an elderly patient may reflect fluid overload with or without low cardiac output, but the differential diagnosis also includes impaired clearance of drugs due to liver congestion or worsening renal function (eg, digoxin toxicity), hyponatremia (potentially mediated through cerebral edema), low cardiac output, occult infection, cerebrovascular accident, and other complications of coronary heart disease.
Key components of the physical exam include the presence of jugular venous distention,17 a more sensitive and specific finding than pulmonary rales in chronic or acute‐on‐chronic heart failure. While the mainstay of therapy for fluid overload remains diuretic therapy, we have only recently learned in a definitive way from the Diuretic Optimization Strategies Evaluation (DOSE)18 study that the method of administration (bolus vs continuous intravenous infusion and high dose vs low dose) matters, albeit slightly. Patients who receive high doses of loop diuretic have greater dyspnea relief and weight loss but are at greater risk for developing worsening renal function.
Certain key clinical markers, when present on admission, place the patient in an at‐risk group for a longer length of stay (Table 1). In addition to new or established hyponatremia, these include a creatinine value above baseline, marked antecedent weight gain, and hypotension. During the hospitalization, development of new hyponatremia or worsening of established hyponatremia, worsening renal function (often simply defined by an increase in baseline creatinine by 0.3 mg/dL or more), lack of dyspnea relief, and lack of weight loss, increase the complexity of decision‐making. A proportion of these higher‐risk patients may benefit from the initiation of intravenous vasoactive therapy, mechanical fluid removal (eg, with ultrafiltration), or the use of a vaptan (or aquaretic), depending on the particular presentation and profile. Occasionally, mechanical support will be needed but this option only applies to a limited subgroup.19 However, aside from ventricular assist devices, none of these options have been associated with improved survival.
| Hyponatremia |
| Worsening renal failure |
| Advanced age |
| Comorbidities |
| Marked antecedent weight gain |
| Lack of (early) resolution of weight gain |
| Hypotension |
| Organ hypoperfusion |
Despite this limitation, the immediate goal of care in the acute setting is symptom relief. Thus, although neither intravenous dobutamine nor milrinone have been shown to decrease mortality, both are recognized as palliative options in patients with advanced or end‐stage symptoms20, 21; for example, milrinone, due to its inodilator characteristics, may improve symptoms and end‐organ perfusion while mitigating against an increase in pulmonary vascular resistance. However routine use in the management of acute decompensated heart failure is discouraged, based on the Outcome of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME‐CHF)2 Trial.22 Similarly, the routine use of nesiritide cannot be recommended, based on the neutral findings of the recently published Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND‐HF)23 study, though subsets of patients may still be candidates for this therapy.
Ultrafiltration appears to function well as an adjunct to fluid and salt removal as demonstrated in the Ultrafiltration versus Intravenous Diuretics for Patients Hospitalized for Acute Decompensated Congestive Heart Failure (UNLOAD)24 study, though a number of limitations have been cited.25 It should be strongly considered for patients who have developed refractory fluid overload and anasarca, especially if responsiveness to loop diuretics is blunted.
For hypervolemic hyponatremia, the standard approach has been fluid restriction, but this can require a prolonged and at times uncomfortable prescription for patients to follow. Hypertonic saline is contraindicated in most cases, given the salt load and risk of exacerbating fluid overload. Data for demeclocycline are sparse.26 The vaptan class is an interesting option, in large part because of the significant free water loss that can be achieved through the competitive antagonism of V2 receptors in renal tubules. Competitive binding to this receptor leads to a reduction in the deposition of new water channels (or aquaporins) on the luminal side of the tubule, resulting in a marked reduction in water reuptake from the urine.27 Indeed, data for tolvaptan, an orally available vaptan, suggest that short‐term treatment can increase urine output, weight loss, and serum sodium level.28 In both the Acute and Chronic Therapeutic Impact of a Vasopressin 2 Antagonist (Tolvaptan) in Congestive Heart Failure (ACTIV) and Efficacy of Vasopressin Antagonism in Heart Failure: Outcome Study With Tolvaptan (EVEREST)29 studies,28 a number of favorable short‐term effects were seen such as dyspnea relief and weight loss, but in the latter study, the trial did not meet 1 of its 2 prespecified co‐primary endpoints (change on a visual analog scale) in an embedded analysis of acute treatment effects. Further, EVEREST failed to show any meaningful impact on posthospitalization morbidity and mortality when tolvaptan was administered chronically.30 It is also noteworthy that in both trials, inclusion criteria required the presence of symptomatic heart failure rather than hyponatremia; in fact, in EVEREST only 11.5% of patients had a serum sodium level less than 135 mEq/L. To date, there are no long‐term prospectively collected data on the impact of the vaptan class in heart failure accompanied by hyponatremia.
Despite these caveats, the judicious use of vaptans may have a role in heart failure; at the very least, serum sodium increases by, on average, 5.2 mEq/L.31 Fluid restriction should be liberalized and serum sodium should be monitored frequently in the first few days of therapy to avoid rapid correction of serum sodium, which can lead to an unusual neurological complication (osmotic demyelination syndrome).32
OUTPATIENT MANAGEMENT CONSIDERATIONS
Patients who have chronic hyponatremia or who are at risk for worsening of preexisting hyponatremia should be closely monitored during the early postdischarge period, in part to detect further decreases in the serum sodium level and deterioration in overall clinical status. Worsening of hyponatremia may occur in the outpatient setting due to intentional or unintentional increased free water intake, initiation of new medications, exacerbation of the underlying condition, infection, or related conditions. Similar to the inpatient setting, the outpatient management of patients with fluid overload and hyponatremia can be difficult. Further study is required and clinical trials are needed to assess whether the chronic administration of a vaptan in this particular patient population will impact prognosis relative to fluid restriction alone.
Regardless of serum sodium, a frequently advocated intervention in long‐term management is daily weight monitoring which has become a gold standard, especially for patients with advanced symptoms. As shown in EVEREST, lean body weight increases prior to re‐hospitalization for HF were 1.96, 2.07, and 1.97 kg, compared with 0.74, 0.90, and 1.04 kg, respectively, in patients who were not re‐hospitalized (P < 0.001 for all groups).33 Recently, use of invasive hemodynamic monitoring, largely on the basis of the CardioMEMS Heart Sensor Allows Monitoring of Pressure to Improve Outcomes in NYHA Class III Heart Failure Patients (CHAMPION)34 trial, has been advocated as a potential breakthrough in outpatient management because increased right‐sided pressures, rather than weight gain, may precede a heart failure exacerbation.35, 36 It is, however, worthwhile to emphasize that routine hemodynamic monitoring with pulmonary artery catheterization has not been shown to be effective in the inpatient setting,37 despite the attractiveness of knowing the numbers. Additionally, the data supporting the use of serial measurements of biomarkers (in particular, brain natriuretic peptide or its precursor) as a surrogate for filling pressures are conflicting, and therefore this approach is not at present considered standard of care.38
Studies also suggest that postdischarge adherence and the intensity of follow‐up for patients recently admitted for HF may be critical to ensure optimal outcomes. From a practical standpoint, the presence of defined risk factors should lead clinicians to adopt a selective approach to postdischarge monitoring. For those patients deemed to be at risk, reasonable options include outpatient medication titration, more frequent nurse contact, and focused efforts at increasing patient self‐efficacy, all of which can be targeted in the context of a HF disease management program or HF clinic.39, 40 A recent consensus paper outlines the components that should be considered in the establishment of a clinic devoted to the care of patients with heart failure.40 Given increasing reimbursement pressures, these clinics may provide a mechanism to increase quality of care in the outpatient setting while decreasing risk of readmission for preventable heart failure exacerbations. However, other nonphysiological factors influence readmission rates, and not all of these factors can be easily addressed in a traditional medical model.41
SUMMARY
Hyponatremia, in addition to declining renal function, persistent dyspnea, and weight gain, is a major clinical concern during and following hospitalizations for acute decompensated heart failure. Low serum sodium (especially below 130 mEq/L) can contribute to symptoms, complicate diagnostic and therapeutic decision‐making, and significantly prolong length of stay and associated costs. Early recognition of the underlying etiologies, aggressive fluid restriction, and removal of medications that might exacerbate hyponatremia are key steps. The vaptan class is now a useful adjunct in select patients with hyponatremia and fluid overload who do not respond to standard approaches such as fluid restriction.
- ,,, et al.Characterization and prognostic value of persistent hyponatremia in patients with severe heart failure in the ESCAPE trial.Arch Intern Med.2007;167:1998–2005.
- ,,, et al,on behalf of the OPTIMIZE‐HF Investigators and Coordinators.Relationship between admission serum sodium concentration and clinical outcomes in patients hospitalized for heart failure: an analysis from the OPTIMIZE‐HF registry.Eur Heart J.2007;28:980–988.
- ,.Hyponatremia in acute decompensated heart failure: mechanisms, prognosis, and treatment options.Clin Cardiol.2010;33:666–671.
- ,.Prognostic importance of serum sodium concentration and its modification by converting enzyme inhibition in patients with severe chronic heart failure.Circulation.1986;73:257–267.
- ,,,,.Risk stratification of in‐hospital mortality in patients hospitalized for chronic congestive heart failure secondary to nonischemic cardiomyopathy.Cardiology.2003;100:136–142.
- ,,,,,.Predicting mortality among patients hospitalized for heart failure. Derivation and validation of a clinical model.JAMA.2003;290:2581–2587.
- ,,.Clinical relevance and management of the major electrolyte abnormalities in congestive heart failure: hyponatremia, hypokalemia, and hypomagnesemia.Am Heart J.1994;128:564–574.
- ,,, et al.The Seattle Heart Failure Model: prediction of survival in heart failure.Circulation.2006;113:1424–1433.
- ,,,,,.Development and prospective validation of a clinical index to predict survival in ambulatory patients referred for cardiac transplant evaluation.Circulation.1997;95:2660–2667.
- ,,, et alfor the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Congestive Heart Failure (ACTIV in CHF) Investigators.Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure.JAMA.2004;291:1963–1971.
- ,,, et al.Improvement in hyponatremia during hospitalization for worsening heart failure is associated with improved outcomes: insights from the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Chronic Heart Failure (ACTIV in CHF) trial.Acute Card Care.2007;9:82–86.
- ,,, et al.Critical elements of clinical follow‐up after hospital discharge for heart failure: insights from the EVEREST trial.Eur J Heart Fail.2010;12:367–374.
- ,,, et al.Identifying patients hospitalized with heart failure at risk for unfavorable future quality of life.Circ Cardiovasc Qual Outcomes.2011;4:389–398.
- ,,, et al.Possible vascular role of increased plasma arginine vasopressin in congestive heart failure.Int J Cardiol.2006;106:191–195.
- ,,,,,.Burden of sodium abnormalities in patients hospitalized for heart failure.Congest Heart Fail.2011;17:1–7.
- ,,, et al.Consequences of hyponatremia on cost and length of stay in heart failure patients.J Card Fail.2011;8:S72.
- ,,,.Prognostic importance of elevated jugular venous pressure and a third heart sound in patients with heart failure.N Engl J Med.2001;345:574–581.
- ,,, et alfor the NHLBI Heart Failure Clinical Research Network.Diuretic strategies in patients with acute decompensated heart failure.N Engl J Med.2011;364:797–805.
- ,,.Emerging ventricular assist devices for long‐term cardiac support.Nat Rev Cardiol.2010;7:71–76.
- ,,, et al.Chronic continuous home inotropic therapy in end‐stage heart failure.Am Heart J.2006;152:1096.e1–1096.e8.
- ,.Dobutamine for patients with end‐stage heart failure in a hospice program?J Palliat Med.2003;6:93–97.
- ,,, et al.Short‐term intravenous milrinone for acute exacerbation of chronic heart failure.JAMA.2002;287:1541–1547.
- ,,, et al.Effect of nesiritide in patients with acute decompensated heart failure.N Engl J Med.2011;365:32–43.
- ,,, et alfor the UNLOAD Trial Investigators.Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure.J Am Coll Cardiol.2007;49:675–683.
- ,,.The challenge of correcting volume overload in hospitalized patients with decompensated heart failure.J Am Coll Cardiol.2007;49:684–686.
- ,,.Demeclocycline treatment of water retention in congestive heart failure.Br Med J.1978;1:760.
- ,.Vasopressin antagonists.J Card Fail.2011;17:973–981.
- ,,, et al.A multicenter, randomized, double‐blind, placebo‐controlled study of tolvaptan monotherapy compared to furosemide and the combination of tolvaptan and furosemide in patients with heart failure and systolic dysfunction.JAMA.2004;291:1963–1971.
- ,,, et alfor the Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators.Short‐term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST Clinical Status Trials.JAMA.2007;297:1332–1343.
- ,,, et alfor the Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators.Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial.JAMA.2007;297:1319–1331.
- ,,,,,.Vasopressin receptor antagonists for the treatment of hyponatremia: systematic review and meta‐analysis.Am J Kidney Dis.2010;56:325–337.
- ,,,,.Central pontine myelinolysis and pontine lesions after rapid correction of hyponatremia: a prospective magnetic resonance imaging study.Ann Neurol.1990;27:61–66.
- ,,, et alfor the EVEREST Investigators.Weight changes after hospitalization for worsening heart failure and subsequent re‐hospitalization and mortality in the EVEREST trial.Eur Heart J.2009;30:1666–1673.
- ,,, et alfor the CHAMPION Trial Study Group.Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial.Lancet.2011;377:658–666.
- ,,.Sympathetically mediated changes in capacitance: redistribution of the venous reservoir as a cause of decompensation.Circ Heart Fail.2011;4:669–675.
- ,,, et al.Hemodynamic factors associated with acute decompensated heart failure: part 1—insights into pathophysiology.J Card Fail.2001;17:282–291.
- The ESCAPE Investigators.Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness.JAMA.2005;294:1625–1633.
- ,,, et al.B‐type natriuretic peptide‐guided heart failure therapy: a meta‐analysis.Arch Intern Med.2010;170:507–514.
- ,,,,,.A multidisciplinary intervention to prevent the readmission of elderly patients with congestive heart failure.N Engl J Med.1995;333:1190–1195.
- ,,, et al.The heart failure clinic: a consensus statement of the Heart Failure Society of America.J Card Fail.2008;14:801–815.
- ,,, et al.An automated model to identify heart failure patients at risk for 30‐day readmission or death using electronic medical record data.Med Care.2010;48:981–988.
Hyponatremia, defined as a serum [Na+] 135 mEq/L, occurs in 2030% of patients with acute decompensated heart failure (HF)13 and has been independently associated with a poor prognosis. In clinical trials of acute decompensated HF, the reported mean serum sodium is often normal or near normal, but a significant proportion of study subjects can have serum sodium values that approach 130 mEq/L or lower.3 However, despite the association between hyponatremia and clinical outcomes like hospitalization and mortality, data from studies are sparse about the impact of drug or device interventions in the hyponatremic cohort, since patients are generally not stratified at the time of randomization by the value of baseline serum sodium.
HYPONATREMIA AND PROGNOSIS
Hyponatremia has long been recognized as a potential prognostic marker in heart failure, highlighted by Packer and Lee in 1986.4 Subsequently, a wealth of data derived from clinical trials, registries, and observational databases support the concept that hyponatremia is an independent predictor of both short‐ and long‐term outcomes.13, 511 As reviewed by Jao and Chiong,3 this relationship holds in patients on optimal evidence‐based medical therapy, including treatment with antagonists of the renin‐angiotensin system and beta blockers. In the Organized Program To Initiate Lifesaving Treatment In Hospitalized Patients With Heart Failure (OPTIMIZE)2 HF Registry of nearly 50,000 patients, in‐hospital and 60‐day mortality rates were higher in patients with lower serum sodium levels on admission (cut‐off point of 135 mEq/L). In‐hospital death and the combined endpoint of death or re‐hospitalization increased significantly for each 3 mEq/L decrease in serum [Na+] below 140 mEq/L. Patients with hyponatremia were more likely to have lower systolic blood pressures and receive intravenous inotropic agents; lengths of stay were also longer.
Similar findings were reported in the Evaluation Study of Congestive Heart Failure and Pulmonary Acute and Chronic Therapeutic Impact of a Vasopressin 2 Antagonist (Tolvaptan) in Congestive Heart Failure (ACTIV in CHF)10 trial.11 For example, in the former, Gheorghiade and colleagues tracked serum sodium levels in 433 hospitalized patients who had acute decompensated HF and examined the proportion free from a major event (defined as death and/or HF hospitalization).1 There was a clear association between the event rate and serum sodium level. Patients whose hyponatremia persisted from hospital admission to discharge were at higher risk relative to those whose hyponatremia was corrected during the hospital stay.
However, whether the way in which the serum sodium improvement is achieved has a bearing on outcomes is not known. In the studies comparing outcomes in patients with heart failure and hyponatremia versus normonatremia, no mention is made about how the patient arrived at either state. Despite this limitation, the findings are incontrovertibly consistent. Hyponatremia on discharge (prior to or after the adoption of renin‐angiotensin‐aldosterone system (RAAS) antagonists or beta blockers) is a marker for poorer outcomes, as is another laboratory abnormality frequently observed in patients hospitalized with heart failure: an elevated creatinine.
Additionally, serum sodium obtained shortly after hospitalization is a potent predictor of re‐hospitalization12 and persistently poor health‐related quality‐of‐life.13 The impact on longer‐term outcomes can also be demonstrated in multiple prognostic models6, 8, 9 in which serum sodium is a risk factor for adverse outcomes. For example, using the Seattle Heart Failure Model, overall prognosis worsens for each 1 mEq decline in serum sodium when all other variables are kept constant.8 This observation suggests that, in terms of prognosis, the value of serum sodium functions as a continuous not a binary variable.
HYPONATREMIA AND HF PATHOPHYSIOLOGY
The reasons underlying hyponatremia in heart failure are complex, but a key component is the non‐osmotic release of arginine vasopressin (AVP) in response to stimulation of carotid baroreceptors. This phenomenon occurs as a result of arterial underfilling (both lower blood pressure and lower cardiac output). AVP is one member of a family of neurohormones and cytokines that are upregulated in heart failure (eg, norepinephrine, renin, angiotensin, aldosterone, endothelin, and tumor necrosis factor‐alpha). Levels of AVP are increased most markedly in patients with advanced symptoms (ie, New York Heart Association Class III and IV),14 and this leads to impaired free water handling in the renal tubules and a hypervolemic form of hyponatremia. The reasons underlying the upregulation are debated, but likely reflect a short‐term hemodynamic adaptation that is designed to augment cardiac output by increasing circulating volume. In addition, multiple neurohormones have been shown to promote progressive ventricular dilation, referred to as remodeling. For example, chronic elevations of norepinephrine contribute to a multitude of genotypic and phenotypic changes at the level of the myocyte. The short‐term benefits of neurohormonal upregulation are offset by maladaptive responses in the long term, and this observation likely explains a major part of the clinical benefits seen with drugs such as angiotensin converting enzyme inhibitors, aldosterone antagonists, and beta blockers.
It is also clear that the development and management of patients with hyponatremia and heart failure are frequently complicated by the presence of other factors that impact sodium and water handling. Heart failure often occurs in older patients with renal dysfunction who are on medications that can exacerbate hyponatremia, such as diuretics, non‐steroidal anti‐inflammatory agents, antidepressants, and opiate derivatives. In addition, other conditions like hypothyroidism may coexist and contribute to the hyponatremic state. It is therefore crucial for the clinician to consider these possibilities when a patient with heart failure presents with or develops hyponatremia, and in particular to critically evaluate the potential role of concomitant medications that can cause a syndrome of inappropriate antidiuretic hormone secretion (SIADH)‐like picture.
HYPONATREMIA AND RESOURCE USE
As with other markers of poor outcome in heart failure, such as worsening renal insufficiency, chronic obstructive lung disease, and other comorbidities, hyponatremia is associated with longer lengths of stay (LOS) and cost. In an analysis of approximately 116,000 patients hospitalized with HF and grouped at admission by serum [Na+], risk‐adjusted mortality, LOS, and attributable cost were highest for patients with severe hyponatremia compared to patients with normonatremia.15 In addition, Amin and colleagues recently demonstrated that length of stay in the intensive care unit and associated costs were greater (21% and 23%, respectively) in patients who had an International Classification of Diseases, 9th revision, Clinical Modification (ICD‐9‐CM) code for hyponatremia compared to those that did not.16
CONSIDERATIONS FOR PATIENTS HOSPITALIZED WITH HEART FAILURE WITH AND WITHOUT HYPONATREMIA
A number of significant management challenges exist during the hospitalization phase of acute decompensated heart failure. Among other tasks, the clinician should evaluate the potential cause of the decompensation (eg, medication noncompliance, dietary noncompliance, increased metabolic demand from pneumonia or other infection, worsening renal failure, diuretic resistance, iatrogenic fluid overload) and decide whether the patient is fluid overloaded, in a low cardiac output state contributing to end‐organ perfusion, or both. Manifestations of worsening heart failure other than dyspnea may be present. For example, mental status changes in an elderly patient may reflect fluid overload with or without low cardiac output, but the differential diagnosis also includes impaired clearance of drugs due to liver congestion or worsening renal function (eg, digoxin toxicity), hyponatremia (potentially mediated through cerebral edema), low cardiac output, occult infection, cerebrovascular accident, and other complications of coronary heart disease.
Key components of the physical exam include the presence of jugular venous distention,17 a more sensitive and specific finding than pulmonary rales in chronic or acute‐on‐chronic heart failure. While the mainstay of therapy for fluid overload remains diuretic therapy, we have only recently learned in a definitive way from the Diuretic Optimization Strategies Evaluation (DOSE)18 study that the method of administration (bolus vs continuous intravenous infusion and high dose vs low dose) matters, albeit slightly. Patients who receive high doses of loop diuretic have greater dyspnea relief and weight loss but are at greater risk for developing worsening renal function.
Certain key clinical markers, when present on admission, place the patient in an at‐risk group for a longer length of stay (Table 1). In addition to new or established hyponatremia, these include a creatinine value above baseline, marked antecedent weight gain, and hypotension. During the hospitalization, development of new hyponatremia or worsening of established hyponatremia, worsening renal function (often simply defined by an increase in baseline creatinine by 0.3 mg/dL or more), lack of dyspnea relief, and lack of weight loss, increase the complexity of decision‐making. A proportion of these higher‐risk patients may benefit from the initiation of intravenous vasoactive therapy, mechanical fluid removal (eg, with ultrafiltration), or the use of a vaptan (or aquaretic), depending on the particular presentation and profile. Occasionally, mechanical support will be needed but this option only applies to a limited subgroup.19 However, aside from ventricular assist devices, none of these options have been associated with improved survival.
| Hyponatremia |
| Worsening renal failure |
| Advanced age |
| Comorbidities |
| Marked antecedent weight gain |
| Lack of (early) resolution of weight gain |
| Hypotension |
| Organ hypoperfusion |
Despite this limitation, the immediate goal of care in the acute setting is symptom relief. Thus, although neither intravenous dobutamine nor milrinone have been shown to decrease mortality, both are recognized as palliative options in patients with advanced or end‐stage symptoms20, 21; for example, milrinone, due to its inodilator characteristics, may improve symptoms and end‐organ perfusion while mitigating against an increase in pulmonary vascular resistance. However routine use in the management of acute decompensated heart failure is discouraged, based on the Outcome of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME‐CHF)2 Trial.22 Similarly, the routine use of nesiritide cannot be recommended, based on the neutral findings of the recently published Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND‐HF)23 study, though subsets of patients may still be candidates for this therapy.
Ultrafiltration appears to function well as an adjunct to fluid and salt removal as demonstrated in the Ultrafiltration versus Intravenous Diuretics for Patients Hospitalized for Acute Decompensated Congestive Heart Failure (UNLOAD)24 study, though a number of limitations have been cited.25 It should be strongly considered for patients who have developed refractory fluid overload and anasarca, especially if responsiveness to loop diuretics is blunted.
For hypervolemic hyponatremia, the standard approach has been fluid restriction, but this can require a prolonged and at times uncomfortable prescription for patients to follow. Hypertonic saline is contraindicated in most cases, given the salt load and risk of exacerbating fluid overload. Data for demeclocycline are sparse.26 The vaptan class is an interesting option, in large part because of the significant free water loss that can be achieved through the competitive antagonism of V2 receptors in renal tubules. Competitive binding to this receptor leads to a reduction in the deposition of new water channels (or aquaporins) on the luminal side of the tubule, resulting in a marked reduction in water reuptake from the urine.27 Indeed, data for tolvaptan, an orally available vaptan, suggest that short‐term treatment can increase urine output, weight loss, and serum sodium level.28 In both the Acute and Chronic Therapeutic Impact of a Vasopressin 2 Antagonist (Tolvaptan) in Congestive Heart Failure (ACTIV) and Efficacy of Vasopressin Antagonism in Heart Failure: Outcome Study With Tolvaptan (EVEREST)29 studies,28 a number of favorable short‐term effects were seen such as dyspnea relief and weight loss, but in the latter study, the trial did not meet 1 of its 2 prespecified co‐primary endpoints (change on a visual analog scale) in an embedded analysis of acute treatment effects. Further, EVEREST failed to show any meaningful impact on posthospitalization morbidity and mortality when tolvaptan was administered chronically.30 It is also noteworthy that in both trials, inclusion criteria required the presence of symptomatic heart failure rather than hyponatremia; in fact, in EVEREST only 11.5% of patients had a serum sodium level less than 135 mEq/L. To date, there are no long‐term prospectively collected data on the impact of the vaptan class in heart failure accompanied by hyponatremia.
Despite these caveats, the judicious use of vaptans may have a role in heart failure; at the very least, serum sodium increases by, on average, 5.2 mEq/L.31 Fluid restriction should be liberalized and serum sodium should be monitored frequently in the first few days of therapy to avoid rapid correction of serum sodium, which can lead to an unusual neurological complication (osmotic demyelination syndrome).32
OUTPATIENT MANAGEMENT CONSIDERATIONS
Patients who have chronic hyponatremia or who are at risk for worsening of preexisting hyponatremia should be closely monitored during the early postdischarge period, in part to detect further decreases in the serum sodium level and deterioration in overall clinical status. Worsening of hyponatremia may occur in the outpatient setting due to intentional or unintentional increased free water intake, initiation of new medications, exacerbation of the underlying condition, infection, or related conditions. Similar to the inpatient setting, the outpatient management of patients with fluid overload and hyponatremia can be difficult. Further study is required and clinical trials are needed to assess whether the chronic administration of a vaptan in this particular patient population will impact prognosis relative to fluid restriction alone.
Regardless of serum sodium, a frequently advocated intervention in long‐term management is daily weight monitoring which has become a gold standard, especially for patients with advanced symptoms. As shown in EVEREST, lean body weight increases prior to re‐hospitalization for HF were 1.96, 2.07, and 1.97 kg, compared with 0.74, 0.90, and 1.04 kg, respectively, in patients who were not re‐hospitalized (P < 0.001 for all groups).33 Recently, use of invasive hemodynamic monitoring, largely on the basis of the CardioMEMS Heart Sensor Allows Monitoring of Pressure to Improve Outcomes in NYHA Class III Heart Failure Patients (CHAMPION)34 trial, has been advocated as a potential breakthrough in outpatient management because increased right‐sided pressures, rather than weight gain, may precede a heart failure exacerbation.35, 36 It is, however, worthwhile to emphasize that routine hemodynamic monitoring with pulmonary artery catheterization has not been shown to be effective in the inpatient setting,37 despite the attractiveness of knowing the numbers. Additionally, the data supporting the use of serial measurements of biomarkers (in particular, brain natriuretic peptide or its precursor) as a surrogate for filling pressures are conflicting, and therefore this approach is not at present considered standard of care.38
Studies also suggest that postdischarge adherence and the intensity of follow‐up for patients recently admitted for HF may be critical to ensure optimal outcomes. From a practical standpoint, the presence of defined risk factors should lead clinicians to adopt a selective approach to postdischarge monitoring. For those patients deemed to be at risk, reasonable options include outpatient medication titration, more frequent nurse contact, and focused efforts at increasing patient self‐efficacy, all of which can be targeted in the context of a HF disease management program or HF clinic.39, 40 A recent consensus paper outlines the components that should be considered in the establishment of a clinic devoted to the care of patients with heart failure.40 Given increasing reimbursement pressures, these clinics may provide a mechanism to increase quality of care in the outpatient setting while decreasing risk of readmission for preventable heart failure exacerbations. However, other nonphysiological factors influence readmission rates, and not all of these factors can be easily addressed in a traditional medical model.41
SUMMARY
Hyponatremia, in addition to declining renal function, persistent dyspnea, and weight gain, is a major clinical concern during and following hospitalizations for acute decompensated heart failure. Low serum sodium (especially below 130 mEq/L) can contribute to symptoms, complicate diagnostic and therapeutic decision‐making, and significantly prolong length of stay and associated costs. Early recognition of the underlying etiologies, aggressive fluid restriction, and removal of medications that might exacerbate hyponatremia are key steps. The vaptan class is now a useful adjunct in select patients with hyponatremia and fluid overload who do not respond to standard approaches such as fluid restriction.
Hyponatremia, defined as a serum [Na+] 135 mEq/L, occurs in 2030% of patients with acute decompensated heart failure (HF)13 and has been independently associated with a poor prognosis. In clinical trials of acute decompensated HF, the reported mean serum sodium is often normal or near normal, but a significant proportion of study subjects can have serum sodium values that approach 130 mEq/L or lower.3 However, despite the association between hyponatremia and clinical outcomes like hospitalization and mortality, data from studies are sparse about the impact of drug or device interventions in the hyponatremic cohort, since patients are generally not stratified at the time of randomization by the value of baseline serum sodium.
HYPONATREMIA AND PROGNOSIS
Hyponatremia has long been recognized as a potential prognostic marker in heart failure, highlighted by Packer and Lee in 1986.4 Subsequently, a wealth of data derived from clinical trials, registries, and observational databases support the concept that hyponatremia is an independent predictor of both short‐ and long‐term outcomes.13, 511 As reviewed by Jao and Chiong,3 this relationship holds in patients on optimal evidence‐based medical therapy, including treatment with antagonists of the renin‐angiotensin system and beta blockers. In the Organized Program To Initiate Lifesaving Treatment In Hospitalized Patients With Heart Failure (OPTIMIZE)2 HF Registry of nearly 50,000 patients, in‐hospital and 60‐day mortality rates were higher in patients with lower serum sodium levels on admission (cut‐off point of 135 mEq/L). In‐hospital death and the combined endpoint of death or re‐hospitalization increased significantly for each 3 mEq/L decrease in serum [Na+] below 140 mEq/L. Patients with hyponatremia were more likely to have lower systolic blood pressures and receive intravenous inotropic agents; lengths of stay were also longer.
Similar findings were reported in the Evaluation Study of Congestive Heart Failure and Pulmonary Acute and Chronic Therapeutic Impact of a Vasopressin 2 Antagonist (Tolvaptan) in Congestive Heart Failure (ACTIV in CHF)10 trial.11 For example, in the former, Gheorghiade and colleagues tracked serum sodium levels in 433 hospitalized patients who had acute decompensated HF and examined the proportion free from a major event (defined as death and/or HF hospitalization).1 There was a clear association between the event rate and serum sodium level. Patients whose hyponatremia persisted from hospital admission to discharge were at higher risk relative to those whose hyponatremia was corrected during the hospital stay.
However, whether the way in which the serum sodium improvement is achieved has a bearing on outcomes is not known. In the studies comparing outcomes in patients with heart failure and hyponatremia versus normonatremia, no mention is made about how the patient arrived at either state. Despite this limitation, the findings are incontrovertibly consistent. Hyponatremia on discharge (prior to or after the adoption of renin‐angiotensin‐aldosterone system (RAAS) antagonists or beta blockers) is a marker for poorer outcomes, as is another laboratory abnormality frequently observed in patients hospitalized with heart failure: an elevated creatinine.
Additionally, serum sodium obtained shortly after hospitalization is a potent predictor of re‐hospitalization12 and persistently poor health‐related quality‐of‐life.13 The impact on longer‐term outcomes can also be demonstrated in multiple prognostic models6, 8, 9 in which serum sodium is a risk factor for adverse outcomes. For example, using the Seattle Heart Failure Model, overall prognosis worsens for each 1 mEq decline in serum sodium when all other variables are kept constant.8 This observation suggests that, in terms of prognosis, the value of serum sodium functions as a continuous not a binary variable.
HYPONATREMIA AND HF PATHOPHYSIOLOGY
The reasons underlying hyponatremia in heart failure are complex, but a key component is the non‐osmotic release of arginine vasopressin (AVP) in response to stimulation of carotid baroreceptors. This phenomenon occurs as a result of arterial underfilling (both lower blood pressure and lower cardiac output). AVP is one member of a family of neurohormones and cytokines that are upregulated in heart failure (eg, norepinephrine, renin, angiotensin, aldosterone, endothelin, and tumor necrosis factor‐alpha). Levels of AVP are increased most markedly in patients with advanced symptoms (ie, New York Heart Association Class III and IV),14 and this leads to impaired free water handling in the renal tubules and a hypervolemic form of hyponatremia. The reasons underlying the upregulation are debated, but likely reflect a short‐term hemodynamic adaptation that is designed to augment cardiac output by increasing circulating volume. In addition, multiple neurohormones have been shown to promote progressive ventricular dilation, referred to as remodeling. For example, chronic elevations of norepinephrine contribute to a multitude of genotypic and phenotypic changes at the level of the myocyte. The short‐term benefits of neurohormonal upregulation are offset by maladaptive responses in the long term, and this observation likely explains a major part of the clinical benefits seen with drugs such as angiotensin converting enzyme inhibitors, aldosterone antagonists, and beta blockers.
It is also clear that the development and management of patients with hyponatremia and heart failure are frequently complicated by the presence of other factors that impact sodium and water handling. Heart failure often occurs in older patients with renal dysfunction who are on medications that can exacerbate hyponatremia, such as diuretics, non‐steroidal anti‐inflammatory agents, antidepressants, and opiate derivatives. In addition, other conditions like hypothyroidism may coexist and contribute to the hyponatremic state. It is therefore crucial for the clinician to consider these possibilities when a patient with heart failure presents with or develops hyponatremia, and in particular to critically evaluate the potential role of concomitant medications that can cause a syndrome of inappropriate antidiuretic hormone secretion (SIADH)‐like picture.
HYPONATREMIA AND RESOURCE USE
As with other markers of poor outcome in heart failure, such as worsening renal insufficiency, chronic obstructive lung disease, and other comorbidities, hyponatremia is associated with longer lengths of stay (LOS) and cost. In an analysis of approximately 116,000 patients hospitalized with HF and grouped at admission by serum [Na+], risk‐adjusted mortality, LOS, and attributable cost were highest for patients with severe hyponatremia compared to patients with normonatremia.15 In addition, Amin and colleagues recently demonstrated that length of stay in the intensive care unit and associated costs were greater (21% and 23%, respectively) in patients who had an International Classification of Diseases, 9th revision, Clinical Modification (ICD‐9‐CM) code for hyponatremia compared to those that did not.16
CONSIDERATIONS FOR PATIENTS HOSPITALIZED WITH HEART FAILURE WITH AND WITHOUT HYPONATREMIA
A number of significant management challenges exist during the hospitalization phase of acute decompensated heart failure. Among other tasks, the clinician should evaluate the potential cause of the decompensation (eg, medication noncompliance, dietary noncompliance, increased metabolic demand from pneumonia or other infection, worsening renal failure, diuretic resistance, iatrogenic fluid overload) and decide whether the patient is fluid overloaded, in a low cardiac output state contributing to end‐organ perfusion, or both. Manifestations of worsening heart failure other than dyspnea may be present. For example, mental status changes in an elderly patient may reflect fluid overload with or without low cardiac output, but the differential diagnosis also includes impaired clearance of drugs due to liver congestion or worsening renal function (eg, digoxin toxicity), hyponatremia (potentially mediated through cerebral edema), low cardiac output, occult infection, cerebrovascular accident, and other complications of coronary heart disease.
Key components of the physical exam include the presence of jugular venous distention,17 a more sensitive and specific finding than pulmonary rales in chronic or acute‐on‐chronic heart failure. While the mainstay of therapy for fluid overload remains diuretic therapy, we have only recently learned in a definitive way from the Diuretic Optimization Strategies Evaluation (DOSE)18 study that the method of administration (bolus vs continuous intravenous infusion and high dose vs low dose) matters, albeit slightly. Patients who receive high doses of loop diuretic have greater dyspnea relief and weight loss but are at greater risk for developing worsening renal function.
Certain key clinical markers, when present on admission, place the patient in an at‐risk group for a longer length of stay (Table 1). In addition to new or established hyponatremia, these include a creatinine value above baseline, marked antecedent weight gain, and hypotension. During the hospitalization, development of new hyponatremia or worsening of established hyponatremia, worsening renal function (often simply defined by an increase in baseline creatinine by 0.3 mg/dL or more), lack of dyspnea relief, and lack of weight loss, increase the complexity of decision‐making. A proportion of these higher‐risk patients may benefit from the initiation of intravenous vasoactive therapy, mechanical fluid removal (eg, with ultrafiltration), or the use of a vaptan (or aquaretic), depending on the particular presentation and profile. Occasionally, mechanical support will be needed but this option only applies to a limited subgroup.19 However, aside from ventricular assist devices, none of these options have been associated with improved survival.
| Hyponatremia |
| Worsening renal failure |
| Advanced age |
| Comorbidities |
| Marked antecedent weight gain |
| Lack of (early) resolution of weight gain |
| Hypotension |
| Organ hypoperfusion |
Despite this limitation, the immediate goal of care in the acute setting is symptom relief. Thus, although neither intravenous dobutamine nor milrinone have been shown to decrease mortality, both are recognized as palliative options in patients with advanced or end‐stage symptoms20, 21; for example, milrinone, due to its inodilator characteristics, may improve symptoms and end‐organ perfusion while mitigating against an increase in pulmonary vascular resistance. However routine use in the management of acute decompensated heart failure is discouraged, based on the Outcome of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME‐CHF)2 Trial.22 Similarly, the routine use of nesiritide cannot be recommended, based on the neutral findings of the recently published Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND‐HF)23 study, though subsets of patients may still be candidates for this therapy.
Ultrafiltration appears to function well as an adjunct to fluid and salt removal as demonstrated in the Ultrafiltration versus Intravenous Diuretics for Patients Hospitalized for Acute Decompensated Congestive Heart Failure (UNLOAD)24 study, though a number of limitations have been cited.25 It should be strongly considered for patients who have developed refractory fluid overload and anasarca, especially if responsiveness to loop diuretics is blunted.
For hypervolemic hyponatremia, the standard approach has been fluid restriction, but this can require a prolonged and at times uncomfortable prescription for patients to follow. Hypertonic saline is contraindicated in most cases, given the salt load and risk of exacerbating fluid overload. Data for demeclocycline are sparse.26 The vaptan class is an interesting option, in large part because of the significant free water loss that can be achieved through the competitive antagonism of V2 receptors in renal tubules. Competitive binding to this receptor leads to a reduction in the deposition of new water channels (or aquaporins) on the luminal side of the tubule, resulting in a marked reduction in water reuptake from the urine.27 Indeed, data for tolvaptan, an orally available vaptan, suggest that short‐term treatment can increase urine output, weight loss, and serum sodium level.28 In both the Acute and Chronic Therapeutic Impact of a Vasopressin 2 Antagonist (Tolvaptan) in Congestive Heart Failure (ACTIV) and Efficacy of Vasopressin Antagonism in Heart Failure: Outcome Study With Tolvaptan (EVEREST)29 studies,28 a number of favorable short‐term effects were seen such as dyspnea relief and weight loss, but in the latter study, the trial did not meet 1 of its 2 prespecified co‐primary endpoints (change on a visual analog scale) in an embedded analysis of acute treatment effects. Further, EVEREST failed to show any meaningful impact on posthospitalization morbidity and mortality when tolvaptan was administered chronically.30 It is also noteworthy that in both trials, inclusion criteria required the presence of symptomatic heart failure rather than hyponatremia; in fact, in EVEREST only 11.5% of patients had a serum sodium level less than 135 mEq/L. To date, there are no long‐term prospectively collected data on the impact of the vaptan class in heart failure accompanied by hyponatremia.
Despite these caveats, the judicious use of vaptans may have a role in heart failure; at the very least, serum sodium increases by, on average, 5.2 mEq/L.31 Fluid restriction should be liberalized and serum sodium should be monitored frequently in the first few days of therapy to avoid rapid correction of serum sodium, which can lead to an unusual neurological complication (osmotic demyelination syndrome).32
OUTPATIENT MANAGEMENT CONSIDERATIONS
Patients who have chronic hyponatremia or who are at risk for worsening of preexisting hyponatremia should be closely monitored during the early postdischarge period, in part to detect further decreases in the serum sodium level and deterioration in overall clinical status. Worsening of hyponatremia may occur in the outpatient setting due to intentional or unintentional increased free water intake, initiation of new medications, exacerbation of the underlying condition, infection, or related conditions. Similar to the inpatient setting, the outpatient management of patients with fluid overload and hyponatremia can be difficult. Further study is required and clinical trials are needed to assess whether the chronic administration of a vaptan in this particular patient population will impact prognosis relative to fluid restriction alone.
Regardless of serum sodium, a frequently advocated intervention in long‐term management is daily weight monitoring which has become a gold standard, especially for patients with advanced symptoms. As shown in EVEREST, lean body weight increases prior to re‐hospitalization for HF were 1.96, 2.07, and 1.97 kg, compared with 0.74, 0.90, and 1.04 kg, respectively, in patients who were not re‐hospitalized (P < 0.001 for all groups).33 Recently, use of invasive hemodynamic monitoring, largely on the basis of the CardioMEMS Heart Sensor Allows Monitoring of Pressure to Improve Outcomes in NYHA Class III Heart Failure Patients (CHAMPION)34 trial, has been advocated as a potential breakthrough in outpatient management because increased right‐sided pressures, rather than weight gain, may precede a heart failure exacerbation.35, 36 It is, however, worthwhile to emphasize that routine hemodynamic monitoring with pulmonary artery catheterization has not been shown to be effective in the inpatient setting,37 despite the attractiveness of knowing the numbers. Additionally, the data supporting the use of serial measurements of biomarkers (in particular, brain natriuretic peptide or its precursor) as a surrogate for filling pressures are conflicting, and therefore this approach is not at present considered standard of care.38
Studies also suggest that postdischarge adherence and the intensity of follow‐up for patients recently admitted for HF may be critical to ensure optimal outcomes. From a practical standpoint, the presence of defined risk factors should lead clinicians to adopt a selective approach to postdischarge monitoring. For those patients deemed to be at risk, reasonable options include outpatient medication titration, more frequent nurse contact, and focused efforts at increasing patient self‐efficacy, all of which can be targeted in the context of a HF disease management program or HF clinic.39, 40 A recent consensus paper outlines the components that should be considered in the establishment of a clinic devoted to the care of patients with heart failure.40 Given increasing reimbursement pressures, these clinics may provide a mechanism to increase quality of care in the outpatient setting while decreasing risk of readmission for preventable heart failure exacerbations. However, other nonphysiological factors influence readmission rates, and not all of these factors can be easily addressed in a traditional medical model.41
SUMMARY
Hyponatremia, in addition to declining renal function, persistent dyspnea, and weight gain, is a major clinical concern during and following hospitalizations for acute decompensated heart failure. Low serum sodium (especially below 130 mEq/L) can contribute to symptoms, complicate diagnostic and therapeutic decision‐making, and significantly prolong length of stay and associated costs. Early recognition of the underlying etiologies, aggressive fluid restriction, and removal of medications that might exacerbate hyponatremia are key steps. The vaptan class is now a useful adjunct in select patients with hyponatremia and fluid overload who do not respond to standard approaches such as fluid restriction.
- ,,, et al.Characterization and prognostic value of persistent hyponatremia in patients with severe heart failure in the ESCAPE trial.Arch Intern Med.2007;167:1998–2005.
- ,,, et al,on behalf of the OPTIMIZE‐HF Investigators and Coordinators.Relationship between admission serum sodium concentration and clinical outcomes in patients hospitalized for heart failure: an analysis from the OPTIMIZE‐HF registry.Eur Heart J.2007;28:980–988.
- ,.Hyponatremia in acute decompensated heart failure: mechanisms, prognosis, and treatment options.Clin Cardiol.2010;33:666–671.
- ,.Prognostic importance of serum sodium concentration and its modification by converting enzyme inhibition in patients with severe chronic heart failure.Circulation.1986;73:257–267.
- ,,,,.Risk stratification of in‐hospital mortality in patients hospitalized for chronic congestive heart failure secondary to nonischemic cardiomyopathy.Cardiology.2003;100:136–142.
- ,,,,,.Predicting mortality among patients hospitalized for heart failure. Derivation and validation of a clinical model.JAMA.2003;290:2581–2587.
- ,,.Clinical relevance and management of the major electrolyte abnormalities in congestive heart failure: hyponatremia, hypokalemia, and hypomagnesemia.Am Heart J.1994;128:564–574.
- ,,, et al.The Seattle Heart Failure Model: prediction of survival in heart failure.Circulation.2006;113:1424–1433.
- ,,,,,.Development and prospective validation of a clinical index to predict survival in ambulatory patients referred for cardiac transplant evaluation.Circulation.1997;95:2660–2667.
- ,,, et alfor the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Congestive Heart Failure (ACTIV in CHF) Investigators.Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure.JAMA.2004;291:1963–1971.
- ,,, et al.Improvement in hyponatremia during hospitalization for worsening heart failure is associated with improved outcomes: insights from the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Chronic Heart Failure (ACTIV in CHF) trial.Acute Card Care.2007;9:82–86.
- ,,, et al.Critical elements of clinical follow‐up after hospital discharge for heart failure: insights from the EVEREST trial.Eur J Heart Fail.2010;12:367–374.
- ,,, et al.Identifying patients hospitalized with heart failure at risk for unfavorable future quality of life.Circ Cardiovasc Qual Outcomes.2011;4:389–398.
- ,,, et al.Possible vascular role of increased plasma arginine vasopressin in congestive heart failure.Int J Cardiol.2006;106:191–195.
- ,,,,,.Burden of sodium abnormalities in patients hospitalized for heart failure.Congest Heart Fail.2011;17:1–7.
- ,,, et al.Consequences of hyponatremia on cost and length of stay in heart failure patients.J Card Fail.2011;8:S72.
- ,,,.Prognostic importance of elevated jugular venous pressure and a third heart sound in patients with heart failure.N Engl J Med.2001;345:574–581.
- ,,, et alfor the NHLBI Heart Failure Clinical Research Network.Diuretic strategies in patients with acute decompensated heart failure.N Engl J Med.2011;364:797–805.
- ,,.Emerging ventricular assist devices for long‐term cardiac support.Nat Rev Cardiol.2010;7:71–76.
- ,,, et al.Chronic continuous home inotropic therapy in end‐stage heart failure.Am Heart J.2006;152:1096.e1–1096.e8.
- ,.Dobutamine for patients with end‐stage heart failure in a hospice program?J Palliat Med.2003;6:93–97.
- ,,, et al.Short‐term intravenous milrinone for acute exacerbation of chronic heart failure.JAMA.2002;287:1541–1547.
- ,,, et al.Effect of nesiritide in patients with acute decompensated heart failure.N Engl J Med.2011;365:32–43.
- ,,, et alfor the UNLOAD Trial Investigators.Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure.J Am Coll Cardiol.2007;49:675–683.
- ,,.The challenge of correcting volume overload in hospitalized patients with decompensated heart failure.J Am Coll Cardiol.2007;49:684–686.
- ,,.Demeclocycline treatment of water retention in congestive heart failure.Br Med J.1978;1:760.
- ,.Vasopressin antagonists.J Card Fail.2011;17:973–981.
- ,,, et al.A multicenter, randomized, double‐blind, placebo‐controlled study of tolvaptan monotherapy compared to furosemide and the combination of tolvaptan and furosemide in patients with heart failure and systolic dysfunction.JAMA.2004;291:1963–1971.
- ,,, et alfor the Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators.Short‐term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST Clinical Status Trials.JAMA.2007;297:1332–1343.
- ,,, et alfor the Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators.Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial.JAMA.2007;297:1319–1331.
- ,,,,,.Vasopressin receptor antagonists for the treatment of hyponatremia: systematic review and meta‐analysis.Am J Kidney Dis.2010;56:325–337.
- ,,,,.Central pontine myelinolysis and pontine lesions after rapid correction of hyponatremia: a prospective magnetic resonance imaging study.Ann Neurol.1990;27:61–66.
- ,,, et alfor the EVEREST Investigators.Weight changes after hospitalization for worsening heart failure and subsequent re‐hospitalization and mortality in the EVEREST trial.Eur Heart J.2009;30:1666–1673.
- ,,, et alfor the CHAMPION Trial Study Group.Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial.Lancet.2011;377:658–666.
- ,,.Sympathetically mediated changes in capacitance: redistribution of the venous reservoir as a cause of decompensation.Circ Heart Fail.2011;4:669–675.
- ,,, et al.Hemodynamic factors associated with acute decompensated heart failure: part 1—insights into pathophysiology.J Card Fail.2001;17:282–291.
- The ESCAPE Investigators.Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness.JAMA.2005;294:1625–1633.
- ,,, et al.B‐type natriuretic peptide‐guided heart failure therapy: a meta‐analysis.Arch Intern Med.2010;170:507–514.
- ,,,,,.A multidisciplinary intervention to prevent the readmission of elderly patients with congestive heart failure.N Engl J Med.1995;333:1190–1195.
- ,,, et al.The heart failure clinic: a consensus statement of the Heart Failure Society of America.J Card Fail.2008;14:801–815.
- ,,, et al.An automated model to identify heart failure patients at risk for 30‐day readmission or death using electronic medical record data.Med Care.2010;48:981–988.
- ,,, et al.Characterization and prognostic value of persistent hyponatremia in patients with severe heart failure in the ESCAPE trial.Arch Intern Med.2007;167:1998–2005.
- ,,, et al,on behalf of the OPTIMIZE‐HF Investigators and Coordinators.Relationship between admission serum sodium concentration and clinical outcomes in patients hospitalized for heart failure: an analysis from the OPTIMIZE‐HF registry.Eur Heart J.2007;28:980–988.
- ,.Hyponatremia in acute decompensated heart failure: mechanisms, prognosis, and treatment options.Clin Cardiol.2010;33:666–671.
- ,.Prognostic importance of serum sodium concentration and its modification by converting enzyme inhibition in patients with severe chronic heart failure.Circulation.1986;73:257–267.
- ,,,,.Risk stratification of in‐hospital mortality in patients hospitalized for chronic congestive heart failure secondary to nonischemic cardiomyopathy.Cardiology.2003;100:136–142.
- ,,,,,.Predicting mortality among patients hospitalized for heart failure. Derivation and validation of a clinical model.JAMA.2003;290:2581–2587.
- ,,.Clinical relevance and management of the major electrolyte abnormalities in congestive heart failure: hyponatremia, hypokalemia, and hypomagnesemia.Am Heart J.1994;128:564–574.
- ,,, et al.The Seattle Heart Failure Model: prediction of survival in heart failure.Circulation.2006;113:1424–1433.
- ,,,,,.Development and prospective validation of a clinical index to predict survival in ambulatory patients referred for cardiac transplant evaluation.Circulation.1997;95:2660–2667.
- ,,, et alfor the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Congestive Heart Failure (ACTIV in CHF) Investigators.Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure.JAMA.2004;291:1963–1971.
- ,,, et al.Improvement in hyponatremia during hospitalization for worsening heart failure is associated with improved outcomes: insights from the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Chronic Heart Failure (ACTIV in CHF) trial.Acute Card Care.2007;9:82–86.
- ,,, et al.Critical elements of clinical follow‐up after hospital discharge for heart failure: insights from the EVEREST trial.Eur J Heart Fail.2010;12:367–374.
- ,,, et al.Identifying patients hospitalized with heart failure at risk for unfavorable future quality of life.Circ Cardiovasc Qual Outcomes.2011;4:389–398.
- ,,, et al.Possible vascular role of increased plasma arginine vasopressin in congestive heart failure.Int J Cardiol.2006;106:191–195.
- ,,,,,.Burden of sodium abnormalities in patients hospitalized for heart failure.Congest Heart Fail.2011;17:1–7.
- ,,, et al.Consequences of hyponatremia on cost and length of stay in heart failure patients.J Card Fail.2011;8:S72.
- ,,,.Prognostic importance of elevated jugular venous pressure and a third heart sound in patients with heart failure.N Engl J Med.2001;345:574–581.
- ,,, et alfor the NHLBI Heart Failure Clinical Research Network.Diuretic strategies in patients with acute decompensated heart failure.N Engl J Med.2011;364:797–805.
- ,,.Emerging ventricular assist devices for long‐term cardiac support.Nat Rev Cardiol.2010;7:71–76.
- ,,, et al.Chronic continuous home inotropic therapy in end‐stage heart failure.Am Heart J.2006;152:1096.e1–1096.e8.
- ,.Dobutamine for patients with end‐stage heart failure in a hospice program?J Palliat Med.2003;6:93–97.
- ,,, et al.Short‐term intravenous milrinone for acute exacerbation of chronic heart failure.JAMA.2002;287:1541–1547.
- ,,, et al.Effect of nesiritide in patients with acute decompensated heart failure.N Engl J Med.2011;365:32–43.
- ,,, et alfor the UNLOAD Trial Investigators.Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure.J Am Coll Cardiol.2007;49:675–683.
- ,,.The challenge of correcting volume overload in hospitalized patients with decompensated heart failure.J Am Coll Cardiol.2007;49:684–686.
- ,,.Demeclocycline treatment of water retention in congestive heart failure.Br Med J.1978;1:760.
- ,.Vasopressin antagonists.J Card Fail.2011;17:973–981.
- ,,, et al.A multicenter, randomized, double‐blind, placebo‐controlled study of tolvaptan monotherapy compared to furosemide and the combination of tolvaptan and furosemide in patients with heart failure and systolic dysfunction.JAMA.2004;291:1963–1971.
- ,,, et alfor the Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators.Short‐term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST Clinical Status Trials.JAMA.2007;297:1332–1343.
- ,,, et alfor the Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators.Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial.JAMA.2007;297:1319–1331.
- ,,,,,.Vasopressin receptor antagonists for the treatment of hyponatremia: systematic review and meta‐analysis.Am J Kidney Dis.2010;56:325–337.
- ,,,,.Central pontine myelinolysis and pontine lesions after rapid correction of hyponatremia: a prospective magnetic resonance imaging study.Ann Neurol.1990;27:61–66.
- ,,, et alfor the EVEREST Investigators.Weight changes after hospitalization for worsening heart failure and subsequent re‐hospitalization and mortality in the EVEREST trial.Eur Heart J.2009;30:1666–1673.
- ,,, et alfor the CHAMPION Trial Study Group.Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial.Lancet.2011;377:658–666.
- ,,.Sympathetically mediated changes in capacitance: redistribution of the venous reservoir as a cause of decompensation.Circ Heart Fail.2011;4:669–675.
- ,,, et al.Hemodynamic factors associated with acute decompensated heart failure: part 1—insights into pathophysiology.J Card Fail.2001;17:282–291.
- The ESCAPE Investigators.Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness.JAMA.2005;294:1625–1633.
- ,,, et al.B‐type natriuretic peptide‐guided heart failure therapy: a meta‐analysis.Arch Intern Med.2010;170:507–514.
- ,,,,,.A multidisciplinary intervention to prevent the readmission of elderly patients with congestive heart failure.N Engl J Med.1995;333:1190–1195.
- ,,, et al.The heart failure clinic: a consensus statement of the Heart Failure Society of America.J Card Fail.2008;14:801–815.
- ,,, et al.An automated model to identify heart failure patients at risk for 30‐day readmission or death using electronic medical record data.Med Care.2010;48:981–988.
Hyponatremia in Pneumonia
M.C. is an 82‐year‐old female resident of a skilled nursing facility with a past medical history of moderate dementia, hypertension, type 2 diabetes, and stage 3 chronic kidney disease (baseline creatinine, 1.4 mg/dL; creatinine clearance, 33 mL/min). Her serum sodium concentration ([Na+]) is normal (range, 136139 mEq/L) at baseline. She is brought to the emergency department with a 2‐day history of fever, productive cough, and altered mental status from baseline. She is febrile (38.7C), and has tachycardia (114 bpm), normal blood pressure (128/76 mmHg), and hypoxemia (89% on 2 L). Physical examination suggests euvolemia. Notable laboratory values include: serum [Na+], 127 mEq/L; serum potassium, 4.1 mEq/L; blood urea nitrogen, 14 mg/dL; serum creatinine, 1.5 mg/dL; glucose, 110 mg/dL; plasma osmolality, 253 mOsm/kg; urine [Na+], 92 mEq/L; and urine osmolality, 480 mOsm/kg. Chest radiography shows a right lower lobe infiltrate with prominent air‐bronchograms. The patient is started on intravenous (IV) antibiotics and normal saline (75 mL/hr), and is admitted to the medical service for management of healthcare‐associated pneumonia.
HYPONATREMIA AND PNEUMONIA
The association of hyponatremia with respiratory illness has been recognized for more than 70 years. Winkler and Crankshaw first reported low serum [Na+] in patients with pulmonary tuberculosis in 1938.1 Roughly 25 years later, reports of hyponatremia in patients with pneumonia began to surface in the literature.2 The prevalence of hyponatremia (serum [Na+] <135 mEq/L) is up to 29% of patients with pneumonia.3 Low serum [Na+] is associated with worse outcomes in such patients.36 In a large retrospective cohort (n = 7965), Zilberberg and colleagues found that pneumonia patients with hyponatremia (serum [Na+] <135 mEq/L) had statistically higher rates of intensive care unit (ICU) admission (10.0% vs 6.3%, P < 0.001), mechanical ventilation (3.9% vs 2.3%, P = 0.01), longer ICU (6.3 vs 5.3 days, P = 0.07) and hospital lengths of stay (7.6 vs 7.0 days, P < 0.001), and a trend toward higher hospital mortality (5.4% vs 4.0%, P = 0.1) as compared with those with normal serum [Na+].4 Hyponatremia is also associated with higher illness severity in a variety of other patient populations. The underlying nature of these associations, however, remains obscure.
The mechanism of hyponatremia in pneumonia is incompletely understood. Syndrome of inappropriate antidiuretic hormone secretion (SIADH) is most often implicated.7 Patients with pneumonia often present with several factors that are associated with nonosmotic stimulation of antidiuretic hormone (ADH), most notably inflammatory cytokines such as interleukin‐6,8 stress, nausea, and hypoxemia.9, 10 Others implicate a reset osmostat, citing evidence for this mechanism in other infectious conditions (ie, tuberculosis and malaria).11, 12 Patients with pneumonia may also have concomitant hypovolemia due to factors such as inadequate oral intake, systemic vasodilation, and extrarenal sodium losses from vomiting and diarrhea.13 In contrast to SIADH, hypovolemia is a potent stimulus for appropriate ADH secretion through activation of the carotid baroreceptors.
CASE STUDY REVISITED
M.C.'s initial laboratory assessment would suggest SIADH. Additional testing rules out endocrinopathy (thyroid‐stimulating hormone, 2.2 mIU/L; AM serum cortisol, 16 g/dL). After 3 days of normal saline infusion (75 mL/hr) and IV vancomycin, cefepime, and levofloxacin, her serum [Na+] has dropped to 125 mEq/L. Her vital signs have normalized and she is now saturating well on ambient air. She remains euvolemic. Notable laboratory values on hospital day 4 include serum [Na+], 125 mEq/L; serum creatinine, 1.3 mg/dL; plasma osmolality, 261 mOsm/kg; urine [Na+], 103 mEq/L; urine potassium, 58 mEq/L; and urine osmolality, 518 mOsm/kg. Her provider invokes a diagnosis of SIADH and appropriately discontinues the normal saline. A fluid restriction of 500 mL/day is then instituted based on her average daily urine volume (1.7 L) and urine/plasma electrolyte ratio (electrolyte‐free water clearance = urine volume {1 [(UNa + UK)/PNa].14 After 48 hours, her serum [Na+] has improved to 128 mEq/L, yet she notes extreme thirst. A trial of increased dietary salt is offered, but she refuses, stating that her primary care physician has advised her for years to avoid salt due to her blood pressure. At this point, the nephrology service is consulted for consideration of a vasopressin receptor antagonist.
MANAGEMENT OF HYPONATREMIA IN PATIENTS WITH PNEUMONIA
As mentioned above, hyponatremia has been identified as a marker of increased disease severity in patients with pneumonia, and as such should serve as a reminder to implement the appropriate level of monitoring and vigilance so as to minimize unfavorable outcomes.
Pneumonia patients with hyponatremia often have concomitant hypovolemia. Administering isotonic fluids at admission is appropriate to treat volume depletion, as well as reduce the risk of hyponatremia developing during hospitalization.3 Nair and colleagues reported that 10.5% of the pneumonia patients with normal serum [Na+] levels at admission developed hyponatremia during their hospital stay.3 The choice of initial IV fluid treatment influenced this risk significantly: 3.9% of patients given isotonic saline developed hyponatremia compared with 14.5% of those given hypotonic fluids and 13.5% given no IV fluids. Volume status must be followed closely in pneumonia patients who are given isotonic fluids such as normal saline. If hyponatremia persists once euvolemia is achieved, isotonic fluids should be discontinued or used with caution in patients with other indications for IV fluids. Although patients with SIADH have impaired free water excretion, their ability to excrete sodium remains intact.15 Therefore, giving normal saline to euvolemic patients with SIADH can lead to free water retention and downward pressure on the serum [Na+].
Once euvolemia is established in this patient group, treatment mirrors the general management principles for SIADH. Many approaches exist to managing this condition, yet the majority of options have significant drawbacks. Although fluid restriction has been promoted for years, the level of restriction must generally be significant and ongoing to be effective. A goal intake of <800 mL/day is usually required to maintain the negative water balance necessary to treat hyponatremia and maintain a normal serum [Na+].16 Patients on such a fluid restriction experience thirst, a fundamentally strong impulse that is difficult to manage. As a result, long‐term compliance is extremely challenging.1719 Diets high in solute (sodium and/or protein) have also been used to manage SIADH. Unfortunately, there are no guidelines to follow, and such diets are generally contraindicated in patients with comorbidities such as heart failure and kidney disease. Demeclocycline has been used successfully to treat hyponatremia, but its effects are variable and it can be nephrotoxic.20 Urea induces an osmotic diuresis and concomitant free water excretion. However, its use is very limited by an unpleasant bitter taste and the lack of availability in many countries.20 Vasopressin receptor antagonists (also known as vaptans) have a US Food and Drug Administration (FDA) indication for the treatment of clinically significant hypervolemic or euvolemic hyponatremia (associated with heart failure, cirrhosis or SIADH) with either a serum [Na+] level <125 mEq/L or less marked hyponatremia that is symptomatic and resistant to fluid restriction. The use of vaptans in patients with pneumonia has not been studied specifically or extensively (unlike patients with heart failure or cirrhosis), and therefore should be used with extra caution in this group, under the supervision of a nephrologist. Additional studies are needed to evaluate long‐term clinical outcomes and cost/benefit ratios for the use of vaptans in patients with SIADH.
SUMMARY
The presence of hyponatremia in patients admitted with pneumonia should be recognized and actively managed. Isotonic fluids are generally appropriate initially to address underlying volume depletion and reduce the risk of hyponatremia developing during hospitalization. If hyponatremia persists once euvolemia is achieved, patients are traditionally then managed with fluid restriction, increased dietary solute, or demeclocycline, each of which has significant limitations. Vasopressin receptor antagonists represent a new option for managing these patients, but must be used carefully under the supervision of a nephrologist.
- ,.Chloride depletion in conditions other than Addison's disease.J Clin Invest.1938;17(1):1–6.
- ,.Severe hyponatremia associated with pneumonia.Metabolism.1962;11:1181–1186.
- ,,,.Hyponatremia in community‐acquired pneumonia.Am J Nephrol.2007;27(2):184–190.
- ,,, et al.Hyponatremia and hospital outcomes among patients with pneumonia: a retrospective cohort study.BMC Pulm Med.2008;8:16.
- ,,, et al.Epidemiology and clinical outcomes of community‐acquired pneumonia in adult patients in Asian countries: a prospective study by the Asian network for surveillance of resistant pathogens.Int J Antimicrob Agents.2008;31:107–114.
- ,.Frequency and significance of electrolyte abnormalities in pneumonia.Indian Pediatr.1992;29(6):735–740.
- ,.Pneumonia and the syndrome of inappropriate antidiuretic hormone secretion: don't pour water on the fire.Am Rev Respir Dis.1988;138:512–513.
- ,,,,.Hypothalamic‐pituitary‐adrenal axis activation and stimulation of systemic vasopressin secretion by recombinant interleukin‐6 in humans: potential implications for the syndrome of inappropriate vasopressin secretion.J Clin Endocrinol Metab.1994;79(4):934–939.
- ,,,,,.Abnormalities of sodium and H2O handling in chronic obstructive lung disease.Arch Intern Med.1982;142(7):1326–1330.
- ,,, et al.Effect of hypoxemia on sodium and water excretion in chronic obstructive lung disease.Am J Med.1985;78(1):87–94.
- ,,,,.Hyponatraemia in malaria.Ann Trop Med Parasitol.1967;61:265–279.
- ,,,.Altered water metabolism in tuberculosis: role of vasopressin.Am J Med.1990;88(4):357–364.
- ,,, et al.Laboratory abnormalities in patients with bacterial pneumonia.Chest.1997;111(3):595–600.
- ,,, et al.The urine/plasma electrolyte ratio: a predictive guide to water restriction.Am J Med Sci.2000;319(4):240–244.
- ,,,,,.Postoperative hyponatremia despite near‐isotonic saline infusion: a phenomenon of desalination.Ann Intern Med.1997;126(1):20–25.
- ,.Hyponatremia.N Engl J Med.2000;342(21):1581–1589.
- ,.The syndrome of inappropriate antidiuresis.N Engl J Med.2007;356:2064–2072.
- ,.Managing hyponatremia in cirrhosis.J Hosp Med.2010;5:S8–S17.
- .Current treatments and novel pharmacologic treatments for hyponatremia in congestive heart failure.Am J Cardiol.2005;95(9A):14B–23B.
- ,.The syndrome of inappropriate antidiuretic hormone: current and future management options.Eur J Endocrinol.2010;162 (suppl 1):S13–S18.
M.C. is an 82‐year‐old female resident of a skilled nursing facility with a past medical history of moderate dementia, hypertension, type 2 diabetes, and stage 3 chronic kidney disease (baseline creatinine, 1.4 mg/dL; creatinine clearance, 33 mL/min). Her serum sodium concentration ([Na+]) is normal (range, 136139 mEq/L) at baseline. She is brought to the emergency department with a 2‐day history of fever, productive cough, and altered mental status from baseline. She is febrile (38.7C), and has tachycardia (114 bpm), normal blood pressure (128/76 mmHg), and hypoxemia (89% on 2 L). Physical examination suggests euvolemia. Notable laboratory values include: serum [Na+], 127 mEq/L; serum potassium, 4.1 mEq/L; blood urea nitrogen, 14 mg/dL; serum creatinine, 1.5 mg/dL; glucose, 110 mg/dL; plasma osmolality, 253 mOsm/kg; urine [Na+], 92 mEq/L; and urine osmolality, 480 mOsm/kg. Chest radiography shows a right lower lobe infiltrate with prominent air‐bronchograms. The patient is started on intravenous (IV) antibiotics and normal saline (75 mL/hr), and is admitted to the medical service for management of healthcare‐associated pneumonia.
HYPONATREMIA AND PNEUMONIA
The association of hyponatremia with respiratory illness has been recognized for more than 70 years. Winkler and Crankshaw first reported low serum [Na+] in patients with pulmonary tuberculosis in 1938.1 Roughly 25 years later, reports of hyponatremia in patients with pneumonia began to surface in the literature.2 The prevalence of hyponatremia (serum [Na+] <135 mEq/L) is up to 29% of patients with pneumonia.3 Low serum [Na+] is associated with worse outcomes in such patients.36 In a large retrospective cohort (n = 7965), Zilberberg and colleagues found that pneumonia patients with hyponatremia (serum [Na+] <135 mEq/L) had statistically higher rates of intensive care unit (ICU) admission (10.0% vs 6.3%, P < 0.001), mechanical ventilation (3.9% vs 2.3%, P = 0.01), longer ICU (6.3 vs 5.3 days, P = 0.07) and hospital lengths of stay (7.6 vs 7.0 days, P < 0.001), and a trend toward higher hospital mortality (5.4% vs 4.0%, P = 0.1) as compared with those with normal serum [Na+].4 Hyponatremia is also associated with higher illness severity in a variety of other patient populations. The underlying nature of these associations, however, remains obscure.
The mechanism of hyponatremia in pneumonia is incompletely understood. Syndrome of inappropriate antidiuretic hormone secretion (SIADH) is most often implicated.7 Patients with pneumonia often present with several factors that are associated with nonosmotic stimulation of antidiuretic hormone (ADH), most notably inflammatory cytokines such as interleukin‐6,8 stress, nausea, and hypoxemia.9, 10 Others implicate a reset osmostat, citing evidence for this mechanism in other infectious conditions (ie, tuberculosis and malaria).11, 12 Patients with pneumonia may also have concomitant hypovolemia due to factors such as inadequate oral intake, systemic vasodilation, and extrarenal sodium losses from vomiting and diarrhea.13 In contrast to SIADH, hypovolemia is a potent stimulus for appropriate ADH secretion through activation of the carotid baroreceptors.
CASE STUDY REVISITED
M.C.'s initial laboratory assessment would suggest SIADH. Additional testing rules out endocrinopathy (thyroid‐stimulating hormone, 2.2 mIU/L; AM serum cortisol, 16 g/dL). After 3 days of normal saline infusion (75 mL/hr) and IV vancomycin, cefepime, and levofloxacin, her serum [Na+] has dropped to 125 mEq/L. Her vital signs have normalized and she is now saturating well on ambient air. She remains euvolemic. Notable laboratory values on hospital day 4 include serum [Na+], 125 mEq/L; serum creatinine, 1.3 mg/dL; plasma osmolality, 261 mOsm/kg; urine [Na+], 103 mEq/L; urine potassium, 58 mEq/L; and urine osmolality, 518 mOsm/kg. Her provider invokes a diagnosis of SIADH and appropriately discontinues the normal saline. A fluid restriction of 500 mL/day is then instituted based on her average daily urine volume (1.7 L) and urine/plasma electrolyte ratio (electrolyte‐free water clearance = urine volume {1 [(UNa + UK)/PNa].14 After 48 hours, her serum [Na+] has improved to 128 mEq/L, yet she notes extreme thirst. A trial of increased dietary salt is offered, but she refuses, stating that her primary care physician has advised her for years to avoid salt due to her blood pressure. At this point, the nephrology service is consulted for consideration of a vasopressin receptor antagonist.
MANAGEMENT OF HYPONATREMIA IN PATIENTS WITH PNEUMONIA
As mentioned above, hyponatremia has been identified as a marker of increased disease severity in patients with pneumonia, and as such should serve as a reminder to implement the appropriate level of monitoring and vigilance so as to minimize unfavorable outcomes.
Pneumonia patients with hyponatremia often have concomitant hypovolemia. Administering isotonic fluids at admission is appropriate to treat volume depletion, as well as reduce the risk of hyponatremia developing during hospitalization.3 Nair and colleagues reported that 10.5% of the pneumonia patients with normal serum [Na+] levels at admission developed hyponatremia during their hospital stay.3 The choice of initial IV fluid treatment influenced this risk significantly: 3.9% of patients given isotonic saline developed hyponatremia compared with 14.5% of those given hypotonic fluids and 13.5% given no IV fluids. Volume status must be followed closely in pneumonia patients who are given isotonic fluids such as normal saline. If hyponatremia persists once euvolemia is achieved, isotonic fluids should be discontinued or used with caution in patients with other indications for IV fluids. Although patients with SIADH have impaired free water excretion, their ability to excrete sodium remains intact.15 Therefore, giving normal saline to euvolemic patients with SIADH can lead to free water retention and downward pressure on the serum [Na+].
Once euvolemia is established in this patient group, treatment mirrors the general management principles for SIADH. Many approaches exist to managing this condition, yet the majority of options have significant drawbacks. Although fluid restriction has been promoted for years, the level of restriction must generally be significant and ongoing to be effective. A goal intake of <800 mL/day is usually required to maintain the negative water balance necessary to treat hyponatremia and maintain a normal serum [Na+].16 Patients on such a fluid restriction experience thirst, a fundamentally strong impulse that is difficult to manage. As a result, long‐term compliance is extremely challenging.1719 Diets high in solute (sodium and/or protein) have also been used to manage SIADH. Unfortunately, there are no guidelines to follow, and such diets are generally contraindicated in patients with comorbidities such as heart failure and kidney disease. Demeclocycline has been used successfully to treat hyponatremia, but its effects are variable and it can be nephrotoxic.20 Urea induces an osmotic diuresis and concomitant free water excretion. However, its use is very limited by an unpleasant bitter taste and the lack of availability in many countries.20 Vasopressin receptor antagonists (also known as vaptans) have a US Food and Drug Administration (FDA) indication for the treatment of clinically significant hypervolemic or euvolemic hyponatremia (associated with heart failure, cirrhosis or SIADH) with either a serum [Na+] level <125 mEq/L or less marked hyponatremia that is symptomatic and resistant to fluid restriction. The use of vaptans in patients with pneumonia has not been studied specifically or extensively (unlike patients with heart failure or cirrhosis), and therefore should be used with extra caution in this group, under the supervision of a nephrologist. Additional studies are needed to evaluate long‐term clinical outcomes and cost/benefit ratios for the use of vaptans in patients with SIADH.
SUMMARY
The presence of hyponatremia in patients admitted with pneumonia should be recognized and actively managed. Isotonic fluids are generally appropriate initially to address underlying volume depletion and reduce the risk of hyponatremia developing during hospitalization. If hyponatremia persists once euvolemia is achieved, patients are traditionally then managed with fluid restriction, increased dietary solute, or demeclocycline, each of which has significant limitations. Vasopressin receptor antagonists represent a new option for managing these patients, but must be used carefully under the supervision of a nephrologist.
M.C. is an 82‐year‐old female resident of a skilled nursing facility with a past medical history of moderate dementia, hypertension, type 2 diabetes, and stage 3 chronic kidney disease (baseline creatinine, 1.4 mg/dL; creatinine clearance, 33 mL/min). Her serum sodium concentration ([Na+]) is normal (range, 136139 mEq/L) at baseline. She is brought to the emergency department with a 2‐day history of fever, productive cough, and altered mental status from baseline. She is febrile (38.7C), and has tachycardia (114 bpm), normal blood pressure (128/76 mmHg), and hypoxemia (89% on 2 L). Physical examination suggests euvolemia. Notable laboratory values include: serum [Na+], 127 mEq/L; serum potassium, 4.1 mEq/L; blood urea nitrogen, 14 mg/dL; serum creatinine, 1.5 mg/dL; glucose, 110 mg/dL; plasma osmolality, 253 mOsm/kg; urine [Na+], 92 mEq/L; and urine osmolality, 480 mOsm/kg. Chest radiography shows a right lower lobe infiltrate with prominent air‐bronchograms. The patient is started on intravenous (IV) antibiotics and normal saline (75 mL/hr), and is admitted to the medical service for management of healthcare‐associated pneumonia.
HYPONATREMIA AND PNEUMONIA
The association of hyponatremia with respiratory illness has been recognized for more than 70 years. Winkler and Crankshaw first reported low serum [Na+] in patients with pulmonary tuberculosis in 1938.1 Roughly 25 years later, reports of hyponatremia in patients with pneumonia began to surface in the literature.2 The prevalence of hyponatremia (serum [Na+] <135 mEq/L) is up to 29% of patients with pneumonia.3 Low serum [Na+] is associated with worse outcomes in such patients.36 In a large retrospective cohort (n = 7965), Zilberberg and colleagues found that pneumonia patients with hyponatremia (serum [Na+] <135 mEq/L) had statistically higher rates of intensive care unit (ICU) admission (10.0% vs 6.3%, P < 0.001), mechanical ventilation (3.9% vs 2.3%, P = 0.01), longer ICU (6.3 vs 5.3 days, P = 0.07) and hospital lengths of stay (7.6 vs 7.0 days, P < 0.001), and a trend toward higher hospital mortality (5.4% vs 4.0%, P = 0.1) as compared with those with normal serum [Na+].4 Hyponatremia is also associated with higher illness severity in a variety of other patient populations. The underlying nature of these associations, however, remains obscure.
The mechanism of hyponatremia in pneumonia is incompletely understood. Syndrome of inappropriate antidiuretic hormone secretion (SIADH) is most often implicated.7 Patients with pneumonia often present with several factors that are associated with nonosmotic stimulation of antidiuretic hormone (ADH), most notably inflammatory cytokines such as interleukin‐6,8 stress, nausea, and hypoxemia.9, 10 Others implicate a reset osmostat, citing evidence for this mechanism in other infectious conditions (ie, tuberculosis and malaria).11, 12 Patients with pneumonia may also have concomitant hypovolemia due to factors such as inadequate oral intake, systemic vasodilation, and extrarenal sodium losses from vomiting and diarrhea.13 In contrast to SIADH, hypovolemia is a potent stimulus for appropriate ADH secretion through activation of the carotid baroreceptors.
CASE STUDY REVISITED
M.C.'s initial laboratory assessment would suggest SIADH. Additional testing rules out endocrinopathy (thyroid‐stimulating hormone, 2.2 mIU/L; AM serum cortisol, 16 g/dL). After 3 days of normal saline infusion (75 mL/hr) and IV vancomycin, cefepime, and levofloxacin, her serum [Na+] has dropped to 125 mEq/L. Her vital signs have normalized and she is now saturating well on ambient air. She remains euvolemic. Notable laboratory values on hospital day 4 include serum [Na+], 125 mEq/L; serum creatinine, 1.3 mg/dL; plasma osmolality, 261 mOsm/kg; urine [Na+], 103 mEq/L; urine potassium, 58 mEq/L; and urine osmolality, 518 mOsm/kg. Her provider invokes a diagnosis of SIADH and appropriately discontinues the normal saline. A fluid restriction of 500 mL/day is then instituted based on her average daily urine volume (1.7 L) and urine/plasma electrolyte ratio (electrolyte‐free water clearance = urine volume {1 [(UNa + UK)/PNa].14 After 48 hours, her serum [Na+] has improved to 128 mEq/L, yet she notes extreme thirst. A trial of increased dietary salt is offered, but she refuses, stating that her primary care physician has advised her for years to avoid salt due to her blood pressure. At this point, the nephrology service is consulted for consideration of a vasopressin receptor antagonist.
MANAGEMENT OF HYPONATREMIA IN PATIENTS WITH PNEUMONIA
As mentioned above, hyponatremia has been identified as a marker of increased disease severity in patients with pneumonia, and as such should serve as a reminder to implement the appropriate level of monitoring and vigilance so as to minimize unfavorable outcomes.
Pneumonia patients with hyponatremia often have concomitant hypovolemia. Administering isotonic fluids at admission is appropriate to treat volume depletion, as well as reduce the risk of hyponatremia developing during hospitalization.3 Nair and colleagues reported that 10.5% of the pneumonia patients with normal serum [Na+] levels at admission developed hyponatremia during their hospital stay.3 The choice of initial IV fluid treatment influenced this risk significantly: 3.9% of patients given isotonic saline developed hyponatremia compared with 14.5% of those given hypotonic fluids and 13.5% given no IV fluids. Volume status must be followed closely in pneumonia patients who are given isotonic fluids such as normal saline. If hyponatremia persists once euvolemia is achieved, isotonic fluids should be discontinued or used with caution in patients with other indications for IV fluids. Although patients with SIADH have impaired free water excretion, their ability to excrete sodium remains intact.15 Therefore, giving normal saline to euvolemic patients with SIADH can lead to free water retention and downward pressure on the serum [Na+].
Once euvolemia is established in this patient group, treatment mirrors the general management principles for SIADH. Many approaches exist to managing this condition, yet the majority of options have significant drawbacks. Although fluid restriction has been promoted for years, the level of restriction must generally be significant and ongoing to be effective. A goal intake of <800 mL/day is usually required to maintain the negative water balance necessary to treat hyponatremia and maintain a normal serum [Na+].16 Patients on such a fluid restriction experience thirst, a fundamentally strong impulse that is difficult to manage. As a result, long‐term compliance is extremely challenging.1719 Diets high in solute (sodium and/or protein) have also been used to manage SIADH. Unfortunately, there are no guidelines to follow, and such diets are generally contraindicated in patients with comorbidities such as heart failure and kidney disease. Demeclocycline has been used successfully to treat hyponatremia, but its effects are variable and it can be nephrotoxic.20 Urea induces an osmotic diuresis and concomitant free water excretion. However, its use is very limited by an unpleasant bitter taste and the lack of availability in many countries.20 Vasopressin receptor antagonists (also known as vaptans) have a US Food and Drug Administration (FDA) indication for the treatment of clinically significant hypervolemic or euvolemic hyponatremia (associated with heart failure, cirrhosis or SIADH) with either a serum [Na+] level <125 mEq/L or less marked hyponatremia that is symptomatic and resistant to fluid restriction. The use of vaptans in patients with pneumonia has not been studied specifically or extensively (unlike patients with heart failure or cirrhosis), and therefore should be used with extra caution in this group, under the supervision of a nephrologist. Additional studies are needed to evaluate long‐term clinical outcomes and cost/benefit ratios for the use of vaptans in patients with SIADH.
SUMMARY
The presence of hyponatremia in patients admitted with pneumonia should be recognized and actively managed. Isotonic fluids are generally appropriate initially to address underlying volume depletion and reduce the risk of hyponatremia developing during hospitalization. If hyponatremia persists once euvolemia is achieved, patients are traditionally then managed with fluid restriction, increased dietary solute, or demeclocycline, each of which has significant limitations. Vasopressin receptor antagonists represent a new option for managing these patients, but must be used carefully under the supervision of a nephrologist.
- ,.Chloride depletion in conditions other than Addison's disease.J Clin Invest.1938;17(1):1–6.
- ,.Severe hyponatremia associated with pneumonia.Metabolism.1962;11:1181–1186.
- ,,,.Hyponatremia in community‐acquired pneumonia.Am J Nephrol.2007;27(2):184–190.
- ,,, et al.Hyponatremia and hospital outcomes among patients with pneumonia: a retrospective cohort study.BMC Pulm Med.2008;8:16.
- ,,, et al.Epidemiology and clinical outcomes of community‐acquired pneumonia in adult patients in Asian countries: a prospective study by the Asian network for surveillance of resistant pathogens.Int J Antimicrob Agents.2008;31:107–114.
- ,.Frequency and significance of electrolyte abnormalities in pneumonia.Indian Pediatr.1992;29(6):735–740.
- ,.Pneumonia and the syndrome of inappropriate antidiuretic hormone secretion: don't pour water on the fire.Am Rev Respir Dis.1988;138:512–513.
- ,,,,.Hypothalamic‐pituitary‐adrenal axis activation and stimulation of systemic vasopressin secretion by recombinant interleukin‐6 in humans: potential implications for the syndrome of inappropriate vasopressin secretion.J Clin Endocrinol Metab.1994;79(4):934–939.
- ,,,,,.Abnormalities of sodium and H2O handling in chronic obstructive lung disease.Arch Intern Med.1982;142(7):1326–1330.
- ,,, et al.Effect of hypoxemia on sodium and water excretion in chronic obstructive lung disease.Am J Med.1985;78(1):87–94.
- ,,,,.Hyponatraemia in malaria.Ann Trop Med Parasitol.1967;61:265–279.
- ,,,.Altered water metabolism in tuberculosis: role of vasopressin.Am J Med.1990;88(4):357–364.
- ,,, et al.Laboratory abnormalities in patients with bacterial pneumonia.Chest.1997;111(3):595–600.
- ,,, et al.The urine/plasma electrolyte ratio: a predictive guide to water restriction.Am J Med Sci.2000;319(4):240–244.
- ,,,,,.Postoperative hyponatremia despite near‐isotonic saline infusion: a phenomenon of desalination.Ann Intern Med.1997;126(1):20–25.
- ,.Hyponatremia.N Engl J Med.2000;342(21):1581–1589.
- ,.The syndrome of inappropriate antidiuresis.N Engl J Med.2007;356:2064–2072.
- ,.Managing hyponatremia in cirrhosis.J Hosp Med.2010;5:S8–S17.
- .Current treatments and novel pharmacologic treatments for hyponatremia in congestive heart failure.Am J Cardiol.2005;95(9A):14B–23B.
- ,.The syndrome of inappropriate antidiuretic hormone: current and future management options.Eur J Endocrinol.2010;162 (suppl 1):S13–S18.
- ,.Chloride depletion in conditions other than Addison's disease.J Clin Invest.1938;17(1):1–6.
- ,.Severe hyponatremia associated with pneumonia.Metabolism.1962;11:1181–1186.
- ,,,.Hyponatremia in community‐acquired pneumonia.Am J Nephrol.2007;27(2):184–190.
- ,,, et al.Hyponatremia and hospital outcomes among patients with pneumonia: a retrospective cohort study.BMC Pulm Med.2008;8:16.
- ,,, et al.Epidemiology and clinical outcomes of community‐acquired pneumonia in adult patients in Asian countries: a prospective study by the Asian network for surveillance of resistant pathogens.Int J Antimicrob Agents.2008;31:107–114.
- ,.Frequency and significance of electrolyte abnormalities in pneumonia.Indian Pediatr.1992;29(6):735–740.
- ,.Pneumonia and the syndrome of inappropriate antidiuretic hormone secretion: don't pour water on the fire.Am Rev Respir Dis.1988;138:512–513.
- ,,,,.Hypothalamic‐pituitary‐adrenal axis activation and stimulation of systemic vasopressin secretion by recombinant interleukin‐6 in humans: potential implications for the syndrome of inappropriate vasopressin secretion.J Clin Endocrinol Metab.1994;79(4):934–939.
- ,,,,,.Abnormalities of sodium and H2O handling in chronic obstructive lung disease.Arch Intern Med.1982;142(7):1326–1330.
- ,,, et al.Effect of hypoxemia on sodium and water excretion in chronic obstructive lung disease.Am J Med.1985;78(1):87–94.
- ,,,,.Hyponatraemia in malaria.Ann Trop Med Parasitol.1967;61:265–279.
- ,,,.Altered water metabolism in tuberculosis: role of vasopressin.Am J Med.1990;88(4):357–364.
- ,,, et al.Laboratory abnormalities in patients with bacterial pneumonia.Chest.1997;111(3):595–600.
- ,,, et al.The urine/plasma electrolyte ratio: a predictive guide to water restriction.Am J Med Sci.2000;319(4):240–244.
- ,,,,,.Postoperative hyponatremia despite near‐isotonic saline infusion: a phenomenon of desalination.Ann Intern Med.1997;126(1):20–25.
- ,.Hyponatremia.N Engl J Med.2000;342(21):1581–1589.
- ,.The syndrome of inappropriate antidiuresis.N Engl J Med.2007;356:2064–2072.
- ,.Managing hyponatremia in cirrhosis.J Hosp Med.2010;5:S8–S17.
- .Current treatments and novel pharmacologic treatments for hyponatremia in congestive heart failure.Am J Cardiol.2005;95(9A):14B–23B.
- ,.The syndrome of inappropriate antidiuretic hormone: current and future management options.Eur J Endocrinol.2010;162 (suppl 1):S13–S18.
Hyponatremia in Cirrhosis
Cirrhosis is one of the main causes of hypervolemic hyponatremia, a dilutional form of hyponatremia that occurs when there is an increase in total body water but a relatively smaller increase in total serum sodium. Portal hypertension is the main precipitating factor in fluid retention that leads to the development of cirrhotic hyponatremia. In cirrhosis, portal hypertension is determined by 2 main factors: increased intrahepatic resistance and increased spanchnic blood flow. The increased intrahepatic resistance is due to both structural (fibrosis, conversion of low resistance fenestrated sinusoids into capillaries) and dynamic (vasoconstriction due to endothelial cell dysfunction) changes.1
The hepatic circulation normally is able to accommodate an increase in portal blood flow associated with postprandial hyperemia. The elevated intrahepatic resistance in cirrhosis results in an inability to accommodate the normal increase in portal blood flow that occurs in the postprandial hyperemia state.3 As a result, portal pressure increases during postprandial hyperemia, leading to reflex vasoconstriction, which creates a shear stress and increases splanchnic nitric oxide (NO) production.4 NO, one of the most important vasodilators in the splanchnic circulation, increases splanchnic blood flow and portal pressures. When this happens repeatedly, it leads to a progressive dilation of preexisting portosystemic vascular channels and the development of varices.5 At the same time, levels of vascular endothelial growth factor rise; this is a very important mediator for angiogenesis because it increases NO, further increasing splanchnic vasodilation.6
Progressive splanchnic vasodilation and increased blood flow into the splanchnic circulation leads to central hypovolemia, arterial underfilling, and decreased blood flow in renal afferent arterioles. Vasoconstrictor norepinephrine and antinatriuretic mechanisms are subsequently activated in an attempt to normalize renal perfusion pressures. Baroreceptor‐mediated nonosmotic release of arginine vasopressin (AVP) is triggered and renin angiotensin‐aldosterone system activity is increased, which increases sodium reabsorption and activates the stellate cells, causing fibrosis, vasoconstriction, and increased portal pressures.6, 7
AVP acts at vasopression‐1A (V1A) receptors to increase arterial vasoconstriction, and at V2 receptors in renal tubule cells for solute‐free water retention.1 The increased sodium and water reabsorption leads to fluid retention, increased central blood volume, venous return to the heart, and an increase in cardiac output to maintain arterial perfusion and create the hyperdynamic circulation that is characteristic of cirrhosis with advanced portal hypertension. Dilutional hyponatremia develops when free water retention is more pronounced than that of sodium retention.
CLINICAL FACTORS ASSOCIATED WITH CIRRHOTIC HYPONATREMIA
Diuretics lead to hyponatremia through several mechanisms.8 First, they induce a contraction of the central blood volume, leading to the nonosmotic release of AVP. In advanced cirrhosis, there is activation of the renin‐angiotensin system in addition to the nonosmotic release of AVP, leading to sodium and free water reabsorption. Diuretics block the sodium reabsorption. However, the water‐retaining effects persist, further contributing to dilutional hyponatremia.8 This cycle is made worse by low sodium intake and frequent thirst experienced by these patients.8 Other medications (eg, non‐steroidal anti‐inflammatory drugs, proton pump inhibitors, and selective serotonin reuptake inhibitors) commonly prescribed for cirrhotic patients may also contribute to the development or worsening of dilutional hyponatremia.8
Increased intrathoracic pressure in patients with tense ascites can also contribute to dilutional hyponatremia by increasing baroreceptor‐mediated release of AVP.9 Large volume paracentesis without the oncotic influence of albumin, an intervention commonly required in patients with cirrhosis and recurrent ascites, may also lead to significant increases in plasma renin activity and plasma aldosterone, which further worsen these pathophysiologic mechanisms, resulting in reduced serum sodium concentration.10 Following removal of excess peritoneal fluid, blood flow to the kidneys is initially improved, but ascitic fluid reaccumulates and the patient becomes intravascularly depleted.10
Infection is an important clinical mediator for the development of both portal hypertension as well as hyponatremia. Bacterial translocation leads to endotoxemia and increased tumor necrosis factor (TNF)‐alpha, resulting in increased splanchnic NO and splanchnic arterial vasodilatation. This process reduces cardiac output, which leads to increased AVP secretion.11, 12 Endotoxin‐mediated splanchnic vasodilatation, especially with spontaneous bacterial peritonitis (SBP), can adversely affect central blood volume status, especially in the presence of severe ascites.1 Clinicians providing care for patients with cirrhosis should be aware of these factors and closely monitor at‐risk patients for the onset or worsening of hyponatremia.1
PROGNOSTIC SIGNIFICANCE OF HYPONATREMIA IN CIRRHOSIS
Hyponatremia has several important clinical implications for patients with cirrhosis.13 Hyponatremia is associated with refractory ascites, greater fluid accumulation, the need for paracentesis, and, importantly, impaired renal function. In patients with ascites and cirrhosis, approximately 50% have some degree of hyponatremia.2 Moreover, the severity of hyponatremia associated with advanced cirrhosis correlates with the degree of cirrhosis complications, especially hyponatremia associated with hepatorenal syndrome, encephalopathy, and SBP (Table 1).2
| Serum [Na+] mEq/L | |||
|---|---|---|---|
| 130 | 131‐135 | >135 | |
| |||
| Hepatorenal syndrome | 3.45 | 1.75 | 1 (reference value) |
| Hepatic encephalopathy | 3.40 | 1.69 | 1 (reference value) |
| Gastrointestinal bleeding | 1.48 | 0.93 | 1 (reference value) |
| Spontaneous bacterial peritonitis | 2.36 | 1.44 | 1 (reference value) |
Similarly, hyponatremia is strongly associated with increasing Child‐Pugh and Model for End‐Stage Liver Disease (MELD) scores.14 In an analysis of data among candidates for liver transplantation from the Organ Procurement and Transplantation Network, the combination of MELD score and serum sodium concentration was a better predictor of death than the MELD score alone.14 In addition, the effect of hyponatremia on clinical outcomes was greater in patients with a low MELD score than those with a relatively high MELD score.. These results suggest that combining serum sodium concentrations with MELD scores to assign transplantation priority might reduce mortality among patients on the waiting list.14
Hyponatremia is also a marker for the development of overt hepatic encephalopathy in patients with cirrhosis.13 One of the proposed mechanisms for encephalopathy is low‐grade cerebral edema. This leads to the conversion of glutamate to glutamine by ammonia, which accumulates within astrocytes, causing astrocyte swelling and dysfunction. Because hyponatremia complicates the management of fluid overload, it increases the risk of developing or exacerbating hepatic encephalopathy.13
Hyponatremia is intimately involved with the development of renal failure in the patient with cirrhosis. It is an earlier and more sensitive marker of renal impairment and/or circulatory dysfunction than serum creatinine.15 It is often the precursor to the development of hepatorenal syndrome.16, 17
Hyponatremia is more common in hospitalized versus ambulatory patients with cirrhosis.1 In a study of 126 patients with cirrhosis admitted to an intensive care unit, patients with serum [Na+] 135 mEq/L had a greater frequency of ascites, illness severity scores, hepatic encephalopathy, sepsis, renal failure, and in‐hospital mortality than normonatremic patients (73.1% vs 55.9%).18 Persistent ascites and low serum sodium identified cirrhotic patients with a high mortality risk, despite low MELD scores, in a study of 507 veterans in the United States with cirrhosis.19 In a retrospective review of 127 patients, hyponatremia was predictive of the development of acute renal failure during hospitalization; among patients with hyponatremia who developed renal failure in the hospital, 72% died.20
Clinical assessment of a patient with cirrhosis who has hyponatremia can be difficult.1 These patients have too much salt and water in the wrong spaces (ie, in the peritoneal cavity and peripheral tissue). As a result, it is possible to have fluid overload with intravascular depletion. A further complication is that dilutional hyponatremia is associated with hepatorenal syndrome. Because these patients have elevated blood urea nitrogen (BUN) and creatinine, and decreased urine output and urine sodium concentration, they appear to be indistinguishable from a patient with prerenal azotemia prior to volume expansion.1 Many of these factors and concerns are illustrated in the following case we handled several years ago.
A 70‐YEAR‐OLD WOMAN WITH CIRRHOSIS
K.R. is a 70‐year‐old white woman recently discharged from the hospital following treatment of recurrent cellulitis. Her past medical history is positive for cirrhosis secondary to active alcohol use, chronic autoimmune hepatitis, and iron overload. Her hospital course was notable for tense ascites, asterixis, and a serum [Na+] of 126 mEq/L at admission. K.R. was managed with large volume paracentesis with 25% salt‐poor albumin, elevation of her lower extremities, discontinuation of diuretics, and 1 L fluid restriction. Her serum [Na+] increased to 128 mEq/L. Although her cellulitis and edema both improved, both persisted. In addition, her mental status also improved, but asterixis persisted. At this point in the hospitalization, effective management of the cellulitis was hindered by the persistent edema, and its treatment with diuretics was limited by the hyponatremia and hepatic encephalopathy.
Today, we have better treatment options for managing this patient. To effectively correct the hyponatremia and facilitate treatment of the other complications of cirrhosis, we can now initiate therapy with one of the vaptans currently available.
TREATMENT OF MILD ASYMPTOMATIC HYPERVOLEMIC HYPONATREMIA
The initial approach to treatment of patients with mild asymptomatic, hypervolemic hyponatremia consists of fluid restriction and a sodium‐restricted diet.1 Fluid restriction, however, has limited efficacy and is often not well tolerated by patients. For patients with severe or progressive hyponatremia, diuretics should be minimized or discontinued to avoid intravascular volume depletion. If patients have severe dilutional hyponatremia and tense ascites, therapeutic paracentesis with plasma expanders is safe.1
The pharmacologic approach to treating hyponatremia has advanced with the discovery of vaptans, drugs that inhibit V2 receptors in cells of the collecting ducts.21 In contrast to conventional diuretics, vaptans do not increase natriuresis. Administration of a vaptan agent for 1 to 2 weeks has been shown to significantly improve low serum sodium levels in patients with hyponatremia, and promote aquaresis without significantly altering renal or circulatory function or activity of the renin‐angiotensin‐aldosterone system. The most frequent side effect of vaptan therapy is thirst.21
Two vaptan agents are currently approved for use in the United States: conivaptan and tolvaptan. Conivaptan is administered intravenously, and is a nonselective vasopressin inhibitor, blocking both V1A and V2 receptors. The course of therapy for conivaptan is 4 days. Tolvaptan, on the other hand, selectively blocks V2 receptors, and is a once‐daily oral vaptan that can be given long‐term.21
The efficacy of tolvaptan was evaluated in the Study of Ascending Levels of Tolvaptan in Hyponatremia 1 and 2 (SALT‐1 and SALT‐2).22 In these multicenter, prospective, randomized, placebo‐controlled trials, patients with dilutional hyponatremia (serum [Na+] <135 mEq/L) associated with cirrhosis (22.4% in SALT‐1, 30.5% in SALT‐2), heart failure, or syndrome of inappropriate antidiuretic hormone (ADH) hypersecretion, and who were hospitalized and clinically stable, received tolvaptan 15 mg daily or placebo. Repeat serum sodium levels were obtained at 8 hours, 2, 3, and 4 days, and then weekly at days 11, 18, 25, and 30. The study drug was discontinued on day 30, with follow‐up serum sodium levels taken 7 days later. (In patients with persistent hyponatremia, the tolvaptan dose was adjusted to 30 mg and then 60 mg with the goal of achieving a serum [Na+] <135 mEq/L.) Increases in serum sodium concentration were seen as early as 8 hours after the first administration of tolvaptan and persisted throughout the study period. After tolvaptan was discontinued, serum sodium levels decreased to baseline within 1 week.22 Tolvaptan was well tolerated, with the most common side effects being increased thirst, dry mouth, and increased urination.22
Longer‐term administration of tolvaptan was shown to maintain a higher serum sodium concentration with an acceptable safety profile in SALTWATER, the open‐label extension of the SALT‐1 and SALT‐2 trials.23 The study included 111 patients with hyponatremia who received oral tolvaptan for a mean follow‐up of 701 days. The most common adverse effects potentially related to tolvaptan were thirst, dry mouth, polydipsia, and polyuria.22, 23 Overall, there were 9 possible and 1 probable serious adverse events, which represents an acceptable safety profile over 77,369 patient‐days of exposure. Over time, 64 patients discontinued tolvaptan, 30 due to adverse reactions or death.22 The results of SALTWATER indicated that most patients received benefit from treatment with tolvaptan, with a decreased need for fluid restriction.23
PATIENT CHARACTERISTICS FOR TOLVAPTAN
In the SALT trials, tolvaptan was administered to clinically stable patients. Based on recommendations by the US Food and Drug Administration (FDA), tolvaptan should be initiated or reinitiated in a hospital setting.1 Patients with severe neurologic symptoms due to hyponatremia should be treated with normal saline instead of tolvaptan; combination therapy with tolvaptan and normal saline should be avoided due to the potential for a too‐rapid correction of hyponatremia and the potential for central pontine myelinolysis. Saline should be discontinued and persistent hyponatremia confirmed before beginning tolvaptan therapy.1
Several additional factors should be considered before patients begin tolvaptan. First, tolvaptan increases thirst, as well as the frequency and volume of urination. Therefore, patients must be able to respond appropriately to thirst with increased water intake. Patients should not be fluid‐restricted during the first day of tolvaptan therapy; instead, they should be instructed to respond to their thirst with increased water ingestion. Because of these factors, caution should be exercised in administering tolvaptan to a confused, restrained patient. In addition, patients should have adequate toileting aids, such as a bedside urinal or commode.1
As with most new drugs, acquisition costs for tolvaptan should be considered in light of the clinical benefits of treatment outcomes. In a retrospective review, median hospital costs for patients with moderate‐to‐severe ($16,606) and mild‐to‐moderate hyponatremia ($14,266) were higher than matched patients without hyponatremia ($13,066).24 In the Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) trial, in which patients with severe congestive heart failure (including those with and without hyponatremia) were randomized to tolvaptan or placebo, the adjusted mean length of hospital stay for those with hyponatremia at baseline who received tolvaptan was 1.72 days shorter than those who received placebo.25 Although tolvaptan is somewhat expensive, the cost compares favorably with the daily cost of hospitalization.
SUMMARY
Portal hypertension plays a pivotal role in the development of hyponatremia in patients with cirrhosis. Reflex vasodilation in the splanchnic circulation compromises the effective central blood volume, triggering compensatory vasoconstrictor and antinatriuretic mechanisms. The net effect is greater free water accumulation than sodium retention, creating dilutional hyponatremia.
The severity of hyponatremia correlates with the severity of cirrhosis complications, such as hepatorenal syndrome, encephalopathy, SBP, and renal failure. The presence of hyponatremia is a marker for poor outcomes and shortened survival, regardless of MELD scores.
In a hospitalized, acutely ill patient with cirrhosis, such as the person in this case, therapy may involve discontinuation of diuretics, evaluation and treatment of infection, volume expansion with salt‐poor albumin, and tolvaptan for treatment of hyponatremia. Regarding tolvaptan, early morning administration is recommended. At initiation of therapy, fluid restriction should be discontinued, and off‐floor testing should be avoided. Concomitant medications should be reviewed to avoid potentially harmful interactions.
- ,.Managing hyponatremia in cirrhosis.J Hosp Med.2010;5(suppl 3):S8–S17.
- ,,,; for the CAPPS Investigators.Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44:1535–1542.
- .Molecular mechanisms of increased intrahepatic resistance in portal hypertension.J Clin Gastroenterol.2007;41(suppl 3):S259–S261.
- ,,.The pathophysiology of portal hypertension.Dig Dis.2005;23:6–10.
- .The molecules: mechanisms of arterial vasodilatation observed in the splanchnic and systemic circulation in portal hypertension.J Clin Gastroenterol.2007;41(suppl 3):S288–S294.
- ,.Vascular endothelial dysfunction in cirrhosis.J Hepatol.2007;46:927–934.
- ,, et al.Management of cirrhosis and ascites.N Engl J Med.2004;350(16):1646–1654.
- ,,.A review of drug‐induced hyponatremia.Am J Kidney Dis.2008;52(1):144–153.
- ,,, et al.Effect of intrathoracic pressure on plasma arginine vasopressin levels.Gastroenterology.1991;101:607–617.
- ,,, et al.Randomized comparative study of therapeutic paracentesis with and without intravenous albumin in cirrhosis.Gastroenterol.1988;94:1493–1502.
- ,,, et al.Endogenous cannabinoids: a new system involved in the homeostasis of arterial pressure in experimental cirrhosis in the rat.Gastroenterology.2002;122:85–93.
- ,,, et al.Endocannabinoids acting at CB1 receptors mediate the cardiac contractile dysfunction in in vivo in cirrhotic rats.Am J Physiol Heart Circ Physiol.2007;293:H1689–H1695.
- ,.Pathogenetic mechanisms of hepatic encephalopathy.Gut.2008;57:1156–1165.
- ,,, et al.Hyponatremia and mortality among patients on the liver‐transplant waiting list.N Engl J Med.2008;359:1018–1026.
- ,,, et al.Addition of serum sodium into the MELD score predicts waiting list mortality better than MELD alone.Liver Transpl.2005;11:336–343.
- ,,, et al.Serum creatinine and bilirubin predict renal failure and mortality in patients with spontaneous bacterial peritonitis: a retrospective study.Liver Int.2009;29:415–419.
- ,,, et al.Natural history of patients hospitalized for management of cirrhotic ascites.Clin Gastroenterol Hepatol.2006;4:1385–1394.
- ,,, et al.Serum sodium predicts prognosis in critically ill cirrhotic patients.J Clin Gastroenterol.2010;44:220–226.
- ,,, et al.Persistent ascites and low serum sodium identify patients with cirrhosis and low MELD scores who are at high risk for early death.Hepatology.2004;40:802–810.
- ,,, et al.Incidence and factors predictive of acute renal failure in patients with advanced liver cirrhosis.Clin Nephrol.2006;65:28–33.
- ,.Hyponatremia in cirrhosis: pathogenesis, clinical significance, and management.Hepatology.2008;48(3):1002–1010.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.
- ,,, et al; for the SALTWATER Investigators.Oral tolvaptan is safe and effective in chronic hyponatremia.J Am Soc Nephrol.2010;21:705–712.
- ,,, et al.Economic impact of hyponatremia in hospitalized patients: a retrospective cohort study.Postgrad Med.2009;121(2):186–191.
- ,,, et al.Effect of serum sodium concentration and tolvaptan treatment on length of hospitalization in patients with heart failure.Am J Health Syst Pharm.2011;68(4):328–333.
Cirrhosis is one of the main causes of hypervolemic hyponatremia, a dilutional form of hyponatremia that occurs when there is an increase in total body water but a relatively smaller increase in total serum sodium. Portal hypertension is the main precipitating factor in fluid retention that leads to the development of cirrhotic hyponatremia. In cirrhosis, portal hypertension is determined by 2 main factors: increased intrahepatic resistance and increased spanchnic blood flow. The increased intrahepatic resistance is due to both structural (fibrosis, conversion of low resistance fenestrated sinusoids into capillaries) and dynamic (vasoconstriction due to endothelial cell dysfunction) changes.1
The hepatic circulation normally is able to accommodate an increase in portal blood flow associated with postprandial hyperemia. The elevated intrahepatic resistance in cirrhosis results in an inability to accommodate the normal increase in portal blood flow that occurs in the postprandial hyperemia state.3 As a result, portal pressure increases during postprandial hyperemia, leading to reflex vasoconstriction, which creates a shear stress and increases splanchnic nitric oxide (NO) production.4 NO, one of the most important vasodilators in the splanchnic circulation, increases splanchnic blood flow and portal pressures. When this happens repeatedly, it leads to a progressive dilation of preexisting portosystemic vascular channels and the development of varices.5 At the same time, levels of vascular endothelial growth factor rise; this is a very important mediator for angiogenesis because it increases NO, further increasing splanchnic vasodilation.6
Progressive splanchnic vasodilation and increased blood flow into the splanchnic circulation leads to central hypovolemia, arterial underfilling, and decreased blood flow in renal afferent arterioles. Vasoconstrictor norepinephrine and antinatriuretic mechanisms are subsequently activated in an attempt to normalize renal perfusion pressures. Baroreceptor‐mediated nonosmotic release of arginine vasopressin (AVP) is triggered and renin angiotensin‐aldosterone system activity is increased, which increases sodium reabsorption and activates the stellate cells, causing fibrosis, vasoconstriction, and increased portal pressures.6, 7
AVP acts at vasopression‐1A (V1A) receptors to increase arterial vasoconstriction, and at V2 receptors in renal tubule cells for solute‐free water retention.1 The increased sodium and water reabsorption leads to fluid retention, increased central blood volume, venous return to the heart, and an increase in cardiac output to maintain arterial perfusion and create the hyperdynamic circulation that is characteristic of cirrhosis with advanced portal hypertension. Dilutional hyponatremia develops when free water retention is more pronounced than that of sodium retention.
CLINICAL FACTORS ASSOCIATED WITH CIRRHOTIC HYPONATREMIA
Diuretics lead to hyponatremia through several mechanisms.8 First, they induce a contraction of the central blood volume, leading to the nonosmotic release of AVP. In advanced cirrhosis, there is activation of the renin‐angiotensin system in addition to the nonosmotic release of AVP, leading to sodium and free water reabsorption. Diuretics block the sodium reabsorption. However, the water‐retaining effects persist, further contributing to dilutional hyponatremia.8 This cycle is made worse by low sodium intake and frequent thirst experienced by these patients.8 Other medications (eg, non‐steroidal anti‐inflammatory drugs, proton pump inhibitors, and selective serotonin reuptake inhibitors) commonly prescribed for cirrhotic patients may also contribute to the development or worsening of dilutional hyponatremia.8
Increased intrathoracic pressure in patients with tense ascites can also contribute to dilutional hyponatremia by increasing baroreceptor‐mediated release of AVP.9 Large volume paracentesis without the oncotic influence of albumin, an intervention commonly required in patients with cirrhosis and recurrent ascites, may also lead to significant increases in plasma renin activity and plasma aldosterone, which further worsen these pathophysiologic mechanisms, resulting in reduced serum sodium concentration.10 Following removal of excess peritoneal fluid, blood flow to the kidneys is initially improved, but ascitic fluid reaccumulates and the patient becomes intravascularly depleted.10
Infection is an important clinical mediator for the development of both portal hypertension as well as hyponatremia. Bacterial translocation leads to endotoxemia and increased tumor necrosis factor (TNF)‐alpha, resulting in increased splanchnic NO and splanchnic arterial vasodilatation. This process reduces cardiac output, which leads to increased AVP secretion.11, 12 Endotoxin‐mediated splanchnic vasodilatation, especially with spontaneous bacterial peritonitis (SBP), can adversely affect central blood volume status, especially in the presence of severe ascites.1 Clinicians providing care for patients with cirrhosis should be aware of these factors and closely monitor at‐risk patients for the onset or worsening of hyponatremia.1
PROGNOSTIC SIGNIFICANCE OF HYPONATREMIA IN CIRRHOSIS
Hyponatremia has several important clinical implications for patients with cirrhosis.13 Hyponatremia is associated with refractory ascites, greater fluid accumulation, the need for paracentesis, and, importantly, impaired renal function. In patients with ascites and cirrhosis, approximately 50% have some degree of hyponatremia.2 Moreover, the severity of hyponatremia associated with advanced cirrhosis correlates with the degree of cirrhosis complications, especially hyponatremia associated with hepatorenal syndrome, encephalopathy, and SBP (Table 1).2
| Serum [Na+] mEq/L | |||
|---|---|---|---|
| 130 | 131‐135 | >135 | |
| |||
| Hepatorenal syndrome | 3.45 | 1.75 | 1 (reference value) |
| Hepatic encephalopathy | 3.40 | 1.69 | 1 (reference value) |
| Gastrointestinal bleeding | 1.48 | 0.93 | 1 (reference value) |
| Spontaneous bacterial peritonitis | 2.36 | 1.44 | 1 (reference value) |
Similarly, hyponatremia is strongly associated with increasing Child‐Pugh and Model for End‐Stage Liver Disease (MELD) scores.14 In an analysis of data among candidates for liver transplantation from the Organ Procurement and Transplantation Network, the combination of MELD score and serum sodium concentration was a better predictor of death than the MELD score alone.14 In addition, the effect of hyponatremia on clinical outcomes was greater in patients with a low MELD score than those with a relatively high MELD score.. These results suggest that combining serum sodium concentrations with MELD scores to assign transplantation priority might reduce mortality among patients on the waiting list.14
Hyponatremia is also a marker for the development of overt hepatic encephalopathy in patients with cirrhosis.13 One of the proposed mechanisms for encephalopathy is low‐grade cerebral edema. This leads to the conversion of glutamate to glutamine by ammonia, which accumulates within astrocytes, causing astrocyte swelling and dysfunction. Because hyponatremia complicates the management of fluid overload, it increases the risk of developing or exacerbating hepatic encephalopathy.13
Hyponatremia is intimately involved with the development of renal failure in the patient with cirrhosis. It is an earlier and more sensitive marker of renal impairment and/or circulatory dysfunction than serum creatinine.15 It is often the precursor to the development of hepatorenal syndrome.16, 17
Hyponatremia is more common in hospitalized versus ambulatory patients with cirrhosis.1 In a study of 126 patients with cirrhosis admitted to an intensive care unit, patients with serum [Na+] 135 mEq/L had a greater frequency of ascites, illness severity scores, hepatic encephalopathy, sepsis, renal failure, and in‐hospital mortality than normonatremic patients (73.1% vs 55.9%).18 Persistent ascites and low serum sodium identified cirrhotic patients with a high mortality risk, despite low MELD scores, in a study of 507 veterans in the United States with cirrhosis.19 In a retrospective review of 127 patients, hyponatremia was predictive of the development of acute renal failure during hospitalization; among patients with hyponatremia who developed renal failure in the hospital, 72% died.20
Clinical assessment of a patient with cirrhosis who has hyponatremia can be difficult.1 These patients have too much salt and water in the wrong spaces (ie, in the peritoneal cavity and peripheral tissue). As a result, it is possible to have fluid overload with intravascular depletion. A further complication is that dilutional hyponatremia is associated with hepatorenal syndrome. Because these patients have elevated blood urea nitrogen (BUN) and creatinine, and decreased urine output and urine sodium concentration, they appear to be indistinguishable from a patient with prerenal azotemia prior to volume expansion.1 Many of these factors and concerns are illustrated in the following case we handled several years ago.
A 70‐YEAR‐OLD WOMAN WITH CIRRHOSIS
K.R. is a 70‐year‐old white woman recently discharged from the hospital following treatment of recurrent cellulitis. Her past medical history is positive for cirrhosis secondary to active alcohol use, chronic autoimmune hepatitis, and iron overload. Her hospital course was notable for tense ascites, asterixis, and a serum [Na+] of 126 mEq/L at admission. K.R. was managed with large volume paracentesis with 25% salt‐poor albumin, elevation of her lower extremities, discontinuation of diuretics, and 1 L fluid restriction. Her serum [Na+] increased to 128 mEq/L. Although her cellulitis and edema both improved, both persisted. In addition, her mental status also improved, but asterixis persisted. At this point in the hospitalization, effective management of the cellulitis was hindered by the persistent edema, and its treatment with diuretics was limited by the hyponatremia and hepatic encephalopathy.
Today, we have better treatment options for managing this patient. To effectively correct the hyponatremia and facilitate treatment of the other complications of cirrhosis, we can now initiate therapy with one of the vaptans currently available.
TREATMENT OF MILD ASYMPTOMATIC HYPERVOLEMIC HYPONATREMIA
The initial approach to treatment of patients with mild asymptomatic, hypervolemic hyponatremia consists of fluid restriction and a sodium‐restricted diet.1 Fluid restriction, however, has limited efficacy and is often not well tolerated by patients. For patients with severe or progressive hyponatremia, diuretics should be minimized or discontinued to avoid intravascular volume depletion. If patients have severe dilutional hyponatremia and tense ascites, therapeutic paracentesis with plasma expanders is safe.1
The pharmacologic approach to treating hyponatremia has advanced with the discovery of vaptans, drugs that inhibit V2 receptors in cells of the collecting ducts.21 In contrast to conventional diuretics, vaptans do not increase natriuresis. Administration of a vaptan agent for 1 to 2 weeks has been shown to significantly improve low serum sodium levels in patients with hyponatremia, and promote aquaresis without significantly altering renal or circulatory function or activity of the renin‐angiotensin‐aldosterone system. The most frequent side effect of vaptan therapy is thirst.21
Two vaptan agents are currently approved for use in the United States: conivaptan and tolvaptan. Conivaptan is administered intravenously, and is a nonselective vasopressin inhibitor, blocking both V1A and V2 receptors. The course of therapy for conivaptan is 4 days. Tolvaptan, on the other hand, selectively blocks V2 receptors, and is a once‐daily oral vaptan that can be given long‐term.21
The efficacy of tolvaptan was evaluated in the Study of Ascending Levels of Tolvaptan in Hyponatremia 1 and 2 (SALT‐1 and SALT‐2).22 In these multicenter, prospective, randomized, placebo‐controlled trials, patients with dilutional hyponatremia (serum [Na+] <135 mEq/L) associated with cirrhosis (22.4% in SALT‐1, 30.5% in SALT‐2), heart failure, or syndrome of inappropriate antidiuretic hormone (ADH) hypersecretion, and who were hospitalized and clinically stable, received tolvaptan 15 mg daily or placebo. Repeat serum sodium levels were obtained at 8 hours, 2, 3, and 4 days, and then weekly at days 11, 18, 25, and 30. The study drug was discontinued on day 30, with follow‐up serum sodium levels taken 7 days later. (In patients with persistent hyponatremia, the tolvaptan dose was adjusted to 30 mg and then 60 mg with the goal of achieving a serum [Na+] <135 mEq/L.) Increases in serum sodium concentration were seen as early as 8 hours after the first administration of tolvaptan and persisted throughout the study period. After tolvaptan was discontinued, serum sodium levels decreased to baseline within 1 week.22 Tolvaptan was well tolerated, with the most common side effects being increased thirst, dry mouth, and increased urination.22
Longer‐term administration of tolvaptan was shown to maintain a higher serum sodium concentration with an acceptable safety profile in SALTWATER, the open‐label extension of the SALT‐1 and SALT‐2 trials.23 The study included 111 patients with hyponatremia who received oral tolvaptan for a mean follow‐up of 701 days. The most common adverse effects potentially related to tolvaptan were thirst, dry mouth, polydipsia, and polyuria.22, 23 Overall, there were 9 possible and 1 probable serious adverse events, which represents an acceptable safety profile over 77,369 patient‐days of exposure. Over time, 64 patients discontinued tolvaptan, 30 due to adverse reactions or death.22 The results of SALTWATER indicated that most patients received benefit from treatment with tolvaptan, with a decreased need for fluid restriction.23
PATIENT CHARACTERISTICS FOR TOLVAPTAN
In the SALT trials, tolvaptan was administered to clinically stable patients. Based on recommendations by the US Food and Drug Administration (FDA), tolvaptan should be initiated or reinitiated in a hospital setting.1 Patients with severe neurologic symptoms due to hyponatremia should be treated with normal saline instead of tolvaptan; combination therapy with tolvaptan and normal saline should be avoided due to the potential for a too‐rapid correction of hyponatremia and the potential for central pontine myelinolysis. Saline should be discontinued and persistent hyponatremia confirmed before beginning tolvaptan therapy.1
Several additional factors should be considered before patients begin tolvaptan. First, tolvaptan increases thirst, as well as the frequency and volume of urination. Therefore, patients must be able to respond appropriately to thirst with increased water intake. Patients should not be fluid‐restricted during the first day of tolvaptan therapy; instead, they should be instructed to respond to their thirst with increased water ingestion. Because of these factors, caution should be exercised in administering tolvaptan to a confused, restrained patient. In addition, patients should have adequate toileting aids, such as a bedside urinal or commode.1
As with most new drugs, acquisition costs for tolvaptan should be considered in light of the clinical benefits of treatment outcomes. In a retrospective review, median hospital costs for patients with moderate‐to‐severe ($16,606) and mild‐to‐moderate hyponatremia ($14,266) were higher than matched patients without hyponatremia ($13,066).24 In the Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) trial, in which patients with severe congestive heart failure (including those with and without hyponatremia) were randomized to tolvaptan or placebo, the adjusted mean length of hospital stay for those with hyponatremia at baseline who received tolvaptan was 1.72 days shorter than those who received placebo.25 Although tolvaptan is somewhat expensive, the cost compares favorably with the daily cost of hospitalization.
SUMMARY
Portal hypertension plays a pivotal role in the development of hyponatremia in patients with cirrhosis. Reflex vasodilation in the splanchnic circulation compromises the effective central blood volume, triggering compensatory vasoconstrictor and antinatriuretic mechanisms. The net effect is greater free water accumulation than sodium retention, creating dilutional hyponatremia.
The severity of hyponatremia correlates with the severity of cirrhosis complications, such as hepatorenal syndrome, encephalopathy, SBP, and renal failure. The presence of hyponatremia is a marker for poor outcomes and shortened survival, regardless of MELD scores.
In a hospitalized, acutely ill patient with cirrhosis, such as the person in this case, therapy may involve discontinuation of diuretics, evaluation and treatment of infection, volume expansion with salt‐poor albumin, and tolvaptan for treatment of hyponatremia. Regarding tolvaptan, early morning administration is recommended. At initiation of therapy, fluid restriction should be discontinued, and off‐floor testing should be avoided. Concomitant medications should be reviewed to avoid potentially harmful interactions.
Cirrhosis is one of the main causes of hypervolemic hyponatremia, a dilutional form of hyponatremia that occurs when there is an increase in total body water but a relatively smaller increase in total serum sodium. Portal hypertension is the main precipitating factor in fluid retention that leads to the development of cirrhotic hyponatremia. In cirrhosis, portal hypertension is determined by 2 main factors: increased intrahepatic resistance and increased spanchnic blood flow. The increased intrahepatic resistance is due to both structural (fibrosis, conversion of low resistance fenestrated sinusoids into capillaries) and dynamic (vasoconstriction due to endothelial cell dysfunction) changes.1
The hepatic circulation normally is able to accommodate an increase in portal blood flow associated with postprandial hyperemia. The elevated intrahepatic resistance in cirrhosis results in an inability to accommodate the normal increase in portal blood flow that occurs in the postprandial hyperemia state.3 As a result, portal pressure increases during postprandial hyperemia, leading to reflex vasoconstriction, which creates a shear stress and increases splanchnic nitric oxide (NO) production.4 NO, one of the most important vasodilators in the splanchnic circulation, increases splanchnic blood flow and portal pressures. When this happens repeatedly, it leads to a progressive dilation of preexisting portosystemic vascular channels and the development of varices.5 At the same time, levels of vascular endothelial growth factor rise; this is a very important mediator for angiogenesis because it increases NO, further increasing splanchnic vasodilation.6
Progressive splanchnic vasodilation and increased blood flow into the splanchnic circulation leads to central hypovolemia, arterial underfilling, and decreased blood flow in renal afferent arterioles. Vasoconstrictor norepinephrine and antinatriuretic mechanisms are subsequently activated in an attempt to normalize renal perfusion pressures. Baroreceptor‐mediated nonosmotic release of arginine vasopressin (AVP) is triggered and renin angiotensin‐aldosterone system activity is increased, which increases sodium reabsorption and activates the stellate cells, causing fibrosis, vasoconstriction, and increased portal pressures.6, 7
AVP acts at vasopression‐1A (V1A) receptors to increase arterial vasoconstriction, and at V2 receptors in renal tubule cells for solute‐free water retention.1 The increased sodium and water reabsorption leads to fluid retention, increased central blood volume, venous return to the heart, and an increase in cardiac output to maintain arterial perfusion and create the hyperdynamic circulation that is characteristic of cirrhosis with advanced portal hypertension. Dilutional hyponatremia develops when free water retention is more pronounced than that of sodium retention.
CLINICAL FACTORS ASSOCIATED WITH CIRRHOTIC HYPONATREMIA
Diuretics lead to hyponatremia through several mechanisms.8 First, they induce a contraction of the central blood volume, leading to the nonosmotic release of AVP. In advanced cirrhosis, there is activation of the renin‐angiotensin system in addition to the nonosmotic release of AVP, leading to sodium and free water reabsorption. Diuretics block the sodium reabsorption. However, the water‐retaining effects persist, further contributing to dilutional hyponatremia.8 This cycle is made worse by low sodium intake and frequent thirst experienced by these patients.8 Other medications (eg, non‐steroidal anti‐inflammatory drugs, proton pump inhibitors, and selective serotonin reuptake inhibitors) commonly prescribed for cirrhotic patients may also contribute to the development or worsening of dilutional hyponatremia.8
Increased intrathoracic pressure in patients with tense ascites can also contribute to dilutional hyponatremia by increasing baroreceptor‐mediated release of AVP.9 Large volume paracentesis without the oncotic influence of albumin, an intervention commonly required in patients with cirrhosis and recurrent ascites, may also lead to significant increases in plasma renin activity and plasma aldosterone, which further worsen these pathophysiologic mechanisms, resulting in reduced serum sodium concentration.10 Following removal of excess peritoneal fluid, blood flow to the kidneys is initially improved, but ascitic fluid reaccumulates and the patient becomes intravascularly depleted.10
Infection is an important clinical mediator for the development of both portal hypertension as well as hyponatremia. Bacterial translocation leads to endotoxemia and increased tumor necrosis factor (TNF)‐alpha, resulting in increased splanchnic NO and splanchnic arterial vasodilatation. This process reduces cardiac output, which leads to increased AVP secretion.11, 12 Endotoxin‐mediated splanchnic vasodilatation, especially with spontaneous bacterial peritonitis (SBP), can adversely affect central blood volume status, especially in the presence of severe ascites.1 Clinicians providing care for patients with cirrhosis should be aware of these factors and closely monitor at‐risk patients for the onset or worsening of hyponatremia.1
PROGNOSTIC SIGNIFICANCE OF HYPONATREMIA IN CIRRHOSIS
Hyponatremia has several important clinical implications for patients with cirrhosis.13 Hyponatremia is associated with refractory ascites, greater fluid accumulation, the need for paracentesis, and, importantly, impaired renal function. In patients with ascites and cirrhosis, approximately 50% have some degree of hyponatremia.2 Moreover, the severity of hyponatremia associated with advanced cirrhosis correlates with the degree of cirrhosis complications, especially hyponatremia associated with hepatorenal syndrome, encephalopathy, and SBP (Table 1).2
| Serum [Na+] mEq/L | |||
|---|---|---|---|
| 130 | 131‐135 | >135 | |
| |||
| Hepatorenal syndrome | 3.45 | 1.75 | 1 (reference value) |
| Hepatic encephalopathy | 3.40 | 1.69 | 1 (reference value) |
| Gastrointestinal bleeding | 1.48 | 0.93 | 1 (reference value) |
| Spontaneous bacterial peritonitis | 2.36 | 1.44 | 1 (reference value) |
Similarly, hyponatremia is strongly associated with increasing Child‐Pugh and Model for End‐Stage Liver Disease (MELD) scores.14 In an analysis of data among candidates for liver transplantation from the Organ Procurement and Transplantation Network, the combination of MELD score and serum sodium concentration was a better predictor of death than the MELD score alone.14 In addition, the effect of hyponatremia on clinical outcomes was greater in patients with a low MELD score than those with a relatively high MELD score.. These results suggest that combining serum sodium concentrations with MELD scores to assign transplantation priority might reduce mortality among patients on the waiting list.14
Hyponatremia is also a marker for the development of overt hepatic encephalopathy in patients with cirrhosis.13 One of the proposed mechanisms for encephalopathy is low‐grade cerebral edema. This leads to the conversion of glutamate to glutamine by ammonia, which accumulates within astrocytes, causing astrocyte swelling and dysfunction. Because hyponatremia complicates the management of fluid overload, it increases the risk of developing or exacerbating hepatic encephalopathy.13
Hyponatremia is intimately involved with the development of renal failure in the patient with cirrhosis. It is an earlier and more sensitive marker of renal impairment and/or circulatory dysfunction than serum creatinine.15 It is often the precursor to the development of hepatorenal syndrome.16, 17
Hyponatremia is more common in hospitalized versus ambulatory patients with cirrhosis.1 In a study of 126 patients with cirrhosis admitted to an intensive care unit, patients with serum [Na+] 135 mEq/L had a greater frequency of ascites, illness severity scores, hepatic encephalopathy, sepsis, renal failure, and in‐hospital mortality than normonatremic patients (73.1% vs 55.9%).18 Persistent ascites and low serum sodium identified cirrhotic patients with a high mortality risk, despite low MELD scores, in a study of 507 veterans in the United States with cirrhosis.19 In a retrospective review of 127 patients, hyponatremia was predictive of the development of acute renal failure during hospitalization; among patients with hyponatremia who developed renal failure in the hospital, 72% died.20
Clinical assessment of a patient with cirrhosis who has hyponatremia can be difficult.1 These patients have too much salt and water in the wrong spaces (ie, in the peritoneal cavity and peripheral tissue). As a result, it is possible to have fluid overload with intravascular depletion. A further complication is that dilutional hyponatremia is associated with hepatorenal syndrome. Because these patients have elevated blood urea nitrogen (BUN) and creatinine, and decreased urine output and urine sodium concentration, they appear to be indistinguishable from a patient with prerenal azotemia prior to volume expansion.1 Many of these factors and concerns are illustrated in the following case we handled several years ago.
A 70‐YEAR‐OLD WOMAN WITH CIRRHOSIS
K.R. is a 70‐year‐old white woman recently discharged from the hospital following treatment of recurrent cellulitis. Her past medical history is positive for cirrhosis secondary to active alcohol use, chronic autoimmune hepatitis, and iron overload. Her hospital course was notable for tense ascites, asterixis, and a serum [Na+] of 126 mEq/L at admission. K.R. was managed with large volume paracentesis with 25% salt‐poor albumin, elevation of her lower extremities, discontinuation of diuretics, and 1 L fluid restriction. Her serum [Na+] increased to 128 mEq/L. Although her cellulitis and edema both improved, both persisted. In addition, her mental status also improved, but asterixis persisted. At this point in the hospitalization, effective management of the cellulitis was hindered by the persistent edema, and its treatment with diuretics was limited by the hyponatremia and hepatic encephalopathy.
Today, we have better treatment options for managing this patient. To effectively correct the hyponatremia and facilitate treatment of the other complications of cirrhosis, we can now initiate therapy with one of the vaptans currently available.
TREATMENT OF MILD ASYMPTOMATIC HYPERVOLEMIC HYPONATREMIA
The initial approach to treatment of patients with mild asymptomatic, hypervolemic hyponatremia consists of fluid restriction and a sodium‐restricted diet.1 Fluid restriction, however, has limited efficacy and is often not well tolerated by patients. For patients with severe or progressive hyponatremia, diuretics should be minimized or discontinued to avoid intravascular volume depletion. If patients have severe dilutional hyponatremia and tense ascites, therapeutic paracentesis with plasma expanders is safe.1
The pharmacologic approach to treating hyponatremia has advanced with the discovery of vaptans, drugs that inhibit V2 receptors in cells of the collecting ducts.21 In contrast to conventional diuretics, vaptans do not increase natriuresis. Administration of a vaptan agent for 1 to 2 weeks has been shown to significantly improve low serum sodium levels in patients with hyponatremia, and promote aquaresis without significantly altering renal or circulatory function or activity of the renin‐angiotensin‐aldosterone system. The most frequent side effect of vaptan therapy is thirst.21
Two vaptan agents are currently approved for use in the United States: conivaptan and tolvaptan. Conivaptan is administered intravenously, and is a nonselective vasopressin inhibitor, blocking both V1A and V2 receptors. The course of therapy for conivaptan is 4 days. Tolvaptan, on the other hand, selectively blocks V2 receptors, and is a once‐daily oral vaptan that can be given long‐term.21
The efficacy of tolvaptan was evaluated in the Study of Ascending Levels of Tolvaptan in Hyponatremia 1 and 2 (SALT‐1 and SALT‐2).22 In these multicenter, prospective, randomized, placebo‐controlled trials, patients with dilutional hyponatremia (serum [Na+] <135 mEq/L) associated with cirrhosis (22.4% in SALT‐1, 30.5% in SALT‐2), heart failure, or syndrome of inappropriate antidiuretic hormone (ADH) hypersecretion, and who were hospitalized and clinically stable, received tolvaptan 15 mg daily or placebo. Repeat serum sodium levels were obtained at 8 hours, 2, 3, and 4 days, and then weekly at days 11, 18, 25, and 30. The study drug was discontinued on day 30, with follow‐up serum sodium levels taken 7 days later. (In patients with persistent hyponatremia, the tolvaptan dose was adjusted to 30 mg and then 60 mg with the goal of achieving a serum [Na+] <135 mEq/L.) Increases in serum sodium concentration were seen as early as 8 hours after the first administration of tolvaptan and persisted throughout the study period. After tolvaptan was discontinued, serum sodium levels decreased to baseline within 1 week.22 Tolvaptan was well tolerated, with the most common side effects being increased thirst, dry mouth, and increased urination.22
Longer‐term administration of tolvaptan was shown to maintain a higher serum sodium concentration with an acceptable safety profile in SALTWATER, the open‐label extension of the SALT‐1 and SALT‐2 trials.23 The study included 111 patients with hyponatremia who received oral tolvaptan for a mean follow‐up of 701 days. The most common adverse effects potentially related to tolvaptan were thirst, dry mouth, polydipsia, and polyuria.22, 23 Overall, there were 9 possible and 1 probable serious adverse events, which represents an acceptable safety profile over 77,369 patient‐days of exposure. Over time, 64 patients discontinued tolvaptan, 30 due to adverse reactions or death.22 The results of SALTWATER indicated that most patients received benefit from treatment with tolvaptan, with a decreased need for fluid restriction.23
PATIENT CHARACTERISTICS FOR TOLVAPTAN
In the SALT trials, tolvaptan was administered to clinically stable patients. Based on recommendations by the US Food and Drug Administration (FDA), tolvaptan should be initiated or reinitiated in a hospital setting.1 Patients with severe neurologic symptoms due to hyponatremia should be treated with normal saline instead of tolvaptan; combination therapy with tolvaptan and normal saline should be avoided due to the potential for a too‐rapid correction of hyponatremia and the potential for central pontine myelinolysis. Saline should be discontinued and persistent hyponatremia confirmed before beginning tolvaptan therapy.1
Several additional factors should be considered before patients begin tolvaptan. First, tolvaptan increases thirst, as well as the frequency and volume of urination. Therefore, patients must be able to respond appropriately to thirst with increased water intake. Patients should not be fluid‐restricted during the first day of tolvaptan therapy; instead, they should be instructed to respond to their thirst with increased water ingestion. Because of these factors, caution should be exercised in administering tolvaptan to a confused, restrained patient. In addition, patients should have adequate toileting aids, such as a bedside urinal or commode.1
As with most new drugs, acquisition costs for tolvaptan should be considered in light of the clinical benefits of treatment outcomes. In a retrospective review, median hospital costs for patients with moderate‐to‐severe ($16,606) and mild‐to‐moderate hyponatremia ($14,266) were higher than matched patients without hyponatremia ($13,066).24 In the Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) trial, in which patients with severe congestive heart failure (including those with and without hyponatremia) were randomized to tolvaptan or placebo, the adjusted mean length of hospital stay for those with hyponatremia at baseline who received tolvaptan was 1.72 days shorter than those who received placebo.25 Although tolvaptan is somewhat expensive, the cost compares favorably with the daily cost of hospitalization.
SUMMARY
Portal hypertension plays a pivotal role in the development of hyponatremia in patients with cirrhosis. Reflex vasodilation in the splanchnic circulation compromises the effective central blood volume, triggering compensatory vasoconstrictor and antinatriuretic mechanisms. The net effect is greater free water accumulation than sodium retention, creating dilutional hyponatremia.
The severity of hyponatremia correlates with the severity of cirrhosis complications, such as hepatorenal syndrome, encephalopathy, SBP, and renal failure. The presence of hyponatremia is a marker for poor outcomes and shortened survival, regardless of MELD scores.
In a hospitalized, acutely ill patient with cirrhosis, such as the person in this case, therapy may involve discontinuation of diuretics, evaluation and treatment of infection, volume expansion with salt‐poor albumin, and tolvaptan for treatment of hyponatremia. Regarding tolvaptan, early morning administration is recommended. At initiation of therapy, fluid restriction should be discontinued, and off‐floor testing should be avoided. Concomitant medications should be reviewed to avoid potentially harmful interactions.
- ,.Managing hyponatremia in cirrhosis.J Hosp Med.2010;5(suppl 3):S8–S17.
- ,,,; for the CAPPS Investigators.Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44:1535–1542.
- .Molecular mechanisms of increased intrahepatic resistance in portal hypertension.J Clin Gastroenterol.2007;41(suppl 3):S259–S261.
- ,,.The pathophysiology of portal hypertension.Dig Dis.2005;23:6–10.
- .The molecules: mechanisms of arterial vasodilatation observed in the splanchnic and systemic circulation in portal hypertension.J Clin Gastroenterol.2007;41(suppl 3):S288–S294.
- ,.Vascular endothelial dysfunction in cirrhosis.J Hepatol.2007;46:927–934.
- ,, et al.Management of cirrhosis and ascites.N Engl J Med.2004;350(16):1646–1654.
- ,,.A review of drug‐induced hyponatremia.Am J Kidney Dis.2008;52(1):144–153.
- ,,, et al.Effect of intrathoracic pressure on plasma arginine vasopressin levels.Gastroenterology.1991;101:607–617.
- ,,, et al.Randomized comparative study of therapeutic paracentesis with and without intravenous albumin in cirrhosis.Gastroenterol.1988;94:1493–1502.
- ,,, et al.Endogenous cannabinoids: a new system involved in the homeostasis of arterial pressure in experimental cirrhosis in the rat.Gastroenterology.2002;122:85–93.
- ,,, et al.Endocannabinoids acting at CB1 receptors mediate the cardiac contractile dysfunction in in vivo in cirrhotic rats.Am J Physiol Heart Circ Physiol.2007;293:H1689–H1695.
- ,.Pathogenetic mechanisms of hepatic encephalopathy.Gut.2008;57:1156–1165.
- ,,, et al.Hyponatremia and mortality among patients on the liver‐transplant waiting list.N Engl J Med.2008;359:1018–1026.
- ,,, et al.Addition of serum sodium into the MELD score predicts waiting list mortality better than MELD alone.Liver Transpl.2005;11:336–343.
- ,,, et al.Serum creatinine and bilirubin predict renal failure and mortality in patients with spontaneous bacterial peritonitis: a retrospective study.Liver Int.2009;29:415–419.
- ,,, et al.Natural history of patients hospitalized for management of cirrhotic ascites.Clin Gastroenterol Hepatol.2006;4:1385–1394.
- ,,, et al.Serum sodium predicts prognosis in critically ill cirrhotic patients.J Clin Gastroenterol.2010;44:220–226.
- ,,, et al.Persistent ascites and low serum sodium identify patients with cirrhosis and low MELD scores who are at high risk for early death.Hepatology.2004;40:802–810.
- ,,, et al.Incidence and factors predictive of acute renal failure in patients with advanced liver cirrhosis.Clin Nephrol.2006;65:28–33.
- ,.Hyponatremia in cirrhosis: pathogenesis, clinical significance, and management.Hepatology.2008;48(3):1002–1010.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.
- ,,, et al; for the SALTWATER Investigators.Oral tolvaptan is safe and effective in chronic hyponatremia.J Am Soc Nephrol.2010;21:705–712.
- ,,, et al.Economic impact of hyponatremia in hospitalized patients: a retrospective cohort study.Postgrad Med.2009;121(2):186–191.
- ,,, et al.Effect of serum sodium concentration and tolvaptan treatment on length of hospitalization in patients with heart failure.Am J Health Syst Pharm.2011;68(4):328–333.
- ,.Managing hyponatremia in cirrhosis.J Hosp Med.2010;5(suppl 3):S8–S17.
- ,,,; for the CAPPS Investigators.Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44:1535–1542.
- .Molecular mechanisms of increased intrahepatic resistance in portal hypertension.J Clin Gastroenterol.2007;41(suppl 3):S259–S261.
- ,,.The pathophysiology of portal hypertension.Dig Dis.2005;23:6–10.
- .The molecules: mechanisms of arterial vasodilatation observed in the splanchnic and systemic circulation in portal hypertension.J Clin Gastroenterol.2007;41(suppl 3):S288–S294.
- ,.Vascular endothelial dysfunction in cirrhosis.J Hepatol.2007;46:927–934.
- ,, et al.Management of cirrhosis and ascites.N Engl J Med.2004;350(16):1646–1654.
- ,,.A review of drug‐induced hyponatremia.Am J Kidney Dis.2008;52(1):144–153.
- ,,, et al.Effect of intrathoracic pressure on plasma arginine vasopressin levels.Gastroenterology.1991;101:607–617.
- ,,, et al.Randomized comparative study of therapeutic paracentesis with and without intravenous albumin in cirrhosis.Gastroenterol.1988;94:1493–1502.
- ,,, et al.Endogenous cannabinoids: a new system involved in the homeostasis of arterial pressure in experimental cirrhosis in the rat.Gastroenterology.2002;122:85–93.
- ,,, et al.Endocannabinoids acting at CB1 receptors mediate the cardiac contractile dysfunction in in vivo in cirrhotic rats.Am J Physiol Heart Circ Physiol.2007;293:H1689–H1695.
- ,.Pathogenetic mechanisms of hepatic encephalopathy.Gut.2008;57:1156–1165.
- ,,, et al.Hyponatremia and mortality among patients on the liver‐transplant waiting list.N Engl J Med.2008;359:1018–1026.
- ,,, et al.Addition of serum sodium into the MELD score predicts waiting list mortality better than MELD alone.Liver Transpl.2005;11:336–343.
- ,,, et al.Serum creatinine and bilirubin predict renal failure and mortality in patients with spontaneous bacterial peritonitis: a retrospective study.Liver Int.2009;29:415–419.
- ,,, et al.Natural history of patients hospitalized for management of cirrhotic ascites.Clin Gastroenterol Hepatol.2006;4:1385–1394.
- ,,, et al.Serum sodium predicts prognosis in critically ill cirrhotic patients.J Clin Gastroenterol.2010;44:220–226.
- ,,, et al.Persistent ascites and low serum sodium identify patients with cirrhosis and low MELD scores who are at high risk for early death.Hepatology.2004;40:802–810.
- ,,, et al.Incidence and factors predictive of acute renal failure in patients with advanced liver cirrhosis.Clin Nephrol.2006;65:28–33.
- ,.Hyponatremia in cirrhosis: pathogenesis, clinical significance, and management.Hepatology.2008;48(3):1002–1010.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.
- ,,, et al; for the SALTWATER Investigators.Oral tolvaptan is safe and effective in chronic hyponatremia.J Am Soc Nephrol.2010;21:705–712.
- ,,, et al.Economic impact of hyponatremia in hospitalized patients: a retrospective cohort study.Postgrad Med.2009;121(2):186–191.
- ,,, et al.Effect of serum sodium concentration and tolvaptan treatment on length of hospitalization in patients with heart failure.Am J Health Syst Pharm.2011;68(4):328–333.
Impact of Hyponatremia
The high prevalence of hyponatremia in hospitalized patients has been recognized for decades. Published reports dating back to the 1960s indicate that serum sodium concentrations ([Na+]) tend to be lower in hospitalized patients than in outpatients in the community.1 Current estimates for the prevalence of hyponatremia in hospitalized patients range from 15% to nearly 40%.2, 3 Several factors account for this wide range. While most studies estimate the presence of hyponatremia based on International Classification of Diseases, Ninth Revision (ICD‐9) codes, accurate reporting varies widely from institution to institution.4 Furthermore, the definition of hyponatremia depends entirely on the cut‐off value of [Na+] used (generally, <136 mEq/L).3 In addition to patients who have hyponatremia present on admission, a significant proportion develop the condition during their hospital stay.3 Deficits in water excretion can develop or worsen during hospitalization as a result of several factors, combined with intake of hypotonic fluid.3 In a study of hyponatremia in intensive care unit (ICU) patients, as many as 80% demonstrated impaired urinary dilution during their ICU course.5
The prevalence of hyponatremia is significant in patients hospitalized for heart failure (HF), cirrhosis, and pneumonia.6 The prevalence of hyponatremiadefined as serum sodium <135 mEq/Lranges from 18% to 25% in patients admitted for congestive heart failure.79 Rates of hyponatremia in patients admitted with cirrhosis are even higher on average, ranging between 18% and 49%.1012 Hyponatremia is also common in patients with community‐acquired pneumonia (CAP), with prevalence estimates ranging from 8% to 28%.1315
Overall, hyponatremia in each of these disease states portends worse outcome.16 In a retrospective study of 71 adults with pneumonia, admission serum [Na+] <135 mEq/L was a risk factor for in‐hospital mortality.13 In each of these conditions, hyponatremia is associated with the need for ICU care and mechanical ventilation, increased hospital length of stay (LOS), and higher costs of care.17, 18
PATHOPHYSIOLOGY OF HYPONATREMIA
There are 2 primary stimuli for the secretion of antidiuretic hormone (ADH), otherwise known as arginine vasopressin (AVP). Osmoreceptors in the hypothalamus measure the osmolality of the plasma.19 When osmolality increases, AVP is secreted; alternatively, when plasma osmolality drops, secretion of AVP under normal circumstances will diminish. The other stimulus results from baroreceptors throughout the body. Decreased intravascular volume (manifested by lower blood pressure) causes activation of the renin‐angiotensin‐aldosterone system, the sympathetic nervous system, as well as AVP secretion.16, 20 In turn, AVP acts on vasopressin V2 receptors in the kidney to encourage water reapsorption, therefore impairing the patient's ability to excrete dilute urine.6
The mechanism by which hyponatremia develops varies according to disease state. Whereas neurohormonal activation predominates in those with HF and cirrhosis, inappropriate AVP secretion (and in some cases, a resetting of the osmostat) occurs in patients with CAP.10, 13, 17 In both HF and cirrhosis, the degree of neurohormonal activation correlates with the degree of hyponatremia.17
In healthy individuals, the mechanism for free water excretion is AVP suppression caused by a fall in plasma osmolality. Patients with hyponatremia, however, are unable to suppress AVP due to true volume depletion (eg, as a result of inadequate oral intake, gastrointestinal fluids loss from vomiting/diarrhea, or use of thiazide diuretics), effective volume depletion (reduced cardiac output in HF patients vs vasodilation in patients with cirrhosis), or an inappropriate increase in AVP secretion.19, 21, 22
RISK FACTORS
The risk factors for hyponatremia are numerous.2, 22 The ability to excrete water declines with increasing age and is exacerbated by chronic illness. Other risk factors include low body weight, low sodium diets, and residence in a chronic care facility.22, 23 Patients with a low baseline serum sodium concentration also appear to be at increased risk of developing hyponatremia. Although the mechanisms by which such patients develop hyponatremia are not always clear, they generally involve an impaired ability to excrete free water due to an inability to appropriately suppress AVP secretion. Medications commonly associated with the syndrome of inappropriate ADH secretion (SIADH) include selective serotonin reuptake inhibitors (SSRIs), psychotropic drugs, non‐steroidal anti‐inflammatory drugs (NSAIDs), opiates, proton pump inhibitors, as well as certain chemotherapeutics.21, 22 Other risk factors associated with SIADH include major abdominal or thoracic surgery, pain, nausea, and excessive administration of hypotonic intravenous fluids. Finally, diuretic use (in particular thiazides) places patients at risk to develop hyponatremia by increasing total urine volume and solute excretion without an appreciable increase in free water excretion.24
MORBIDITY
The morbidities associated with hyponatremia vary widely in severity. Serious sequelae may occur as a result of hyponatremia itself, as well as from complications that occur due to the challenging nature of effective management. Much of the symptomatology relates to the central nervous system (CNS). Patients presenting with extremely low serum [Na+] levels (eg, <115 mEq/L) often have severe neurologic symptoms, while those with lesser degrees of hyponatremia may be asymptomatic, or present with milder nonspecific symptoms, such as confusion.25, 26 It is important to note that the clinical presentation of hyponatremia very much depends on whether it is acute (occurring over 2448 hours) or chronic (>48 hours).
Water shifts between the intracellular and extracellular fluid compartments are the primary means by which the body equalizes osmolality. When serum sodium changes, the ability of the brain to compensate is limited, and may result in various forms of neurologic impairment due to cerebral edema.25, 26 Such patients may become disoriented, restless, unable to attend, or unable to process information cognitively. There may also be peripheral neurologic dysfunction, such as muscle weakness, blunted neuromuscular reflexes, and impaired gait. Such impairments can lead to delirium, falls, and fractures.25, 27
HYPONATREMIA AND COGNITIVE IMPAIRMENT
Renneboog and colleagues performed a case‐control study to assess the impact of mild chronic asymptomatic hyponatremia (mean serum [Na+] 126 5 mEq/L) in 122 patients compared with 244 matched controls (mean age 72 13 years).28 Hyponatremic patients had significantly longer mean response times on concentration tests. Interestingly, the changes in cognitive function in hyponatremic patients were similar to healthy volunteers purposefully intoxicated with alcohol.28 Patients with hyponatremia have also been shown to score lower on the mental component summary of the 36‐item Short‐Form (SF‐36) survey.29 With treatment aimed at improving serum sodium, these same patients demonstrated improved cognitive function,29 suggesting that treating even mild forms of hyponatremia can improve patient outcomes.30
HYPONATREMIA AND FALLS/FRACTURES
Renneboog and colleagues also demonstrated a markedly increased risk of falls in their patients with chronic hyponatremia compared to controls.28 Hyponatremia increases not only the risk of falls, but also the risk of fracture following a fall. In another recent case‐control study of 513 patients, the adjusted odds ratio for fracture after a fall in a patient with hyponatremia was 4.16 compared with an age‐matched control with normal serum sodium who sustained a similar fall.27 Of note, hyponatremia was mild and asymptomatic in all patients studied. Medications (36% diuretics, 17% SSRIs) were the most common precipitating cause of hyponatremia in this study, which is notable because such risk factors should be recognized and addressed.
Although falls and fractures lead to obvious increases in morbidity and cost, delirium has also been identified as a risk factor for increased hospital LOS.18 Delirious patients are less likely and able to mobilize and participate in physical therapy. As such, they are more often bed‐bound and at increased risk for aspiration and other preventable issues, including deep vein thrombosis, bed sores, and debility, all of which may increase their LOS and cost of care.
MORTALITY
Hyponatremia is associated with a significantly increased mortality risk not only during hospitalization, but also at 1 and 5 years following discharge.31 In a prospective cohort study of approximately 100,000 patients, even those with mild hyponatremia ([Na+] 130134 mEq/L) had a significantly higher mortality at 5 years. The adjusted odds ratio for mortality in patients with serum sodium less than 135 mEq/L was 1.47 during hospitalization (95% CI, 1.331.62), 1.38 at 1 year post‐discharge (1.321.46), and 1.25 at 5 years (1.211.30). The significance of hyponatremia varied according to the underlying clinical condition, with the greatest risk observed in patients with metastatic cancer, heart disease, and those who had undergone orthopedic surgery.31 While the association between hyponatremia and mortality is profound, most experts do not believe that hyponatremia directly causes mortality per se. Instead, hyponatremia is felt to be a marker for increased illness severity.
It is difficult to isolate the direct costs of hyponatremia in the acute care setting because the condition is rarely treated in isolation. However, in a study of a managed‐care claims database of nearly 1,300 patients (excluding Medicare patients), hyponatremia was a predictor of higher medical costs at 6 months and at 1 year.32
DIAGNOSIS
The most common presentation of hyponatremia involves nonspecific symptoms or a total lack of symptoms.19 Many patients have comorbid diseases, and symptoms of these illnesses often predominate at hospital admission. Patients with mild to moderate hyponatremia may present with nausea, weakness, malaise, headache, and/or impaired mobility. With more severe hyponatremia, more dangerous neurologic symptoms appear, including generalized seizures, lethargy, and coma.19 Once hyponatremia is identified, the next step is to determine its acuity and classify it.
Although several classification systems exist to describe hyponatremia, the most common scheme begins with assessment of plasma osmolality and volume status.19 The majority of hyponatremic patients present with hypotonic or hypo‐osmolar serum (eg, plasma osmolality <275 mOsm/kg). The primary causes of hyponatremia in patients with normal or high serum osmolality are hyperglycemia, pseudohyponatremia, and advanced renal failure. Marked hyperglycemia increases plasma osmolality, and as a result, water moves out of cells into plasma and lowers serum sodium concentration in the process. Pseudohyponatremia arises from hyperlipidemia or hyperproteinemia, in which high concentrations of lipids/proteins reduce the free water component of plasma, therefore reducing the sodium concentration per liter of plasma. These patients do not have true hyponatremia since the physiologically important sodium concentration per liter of plasma water is normal. Finally, patients with advanced renal failure develop hyponatremia due to the inability to excrete water.
The first step in the diagnosis of hyponatremia is to assess the plasma osmolality and rule out the aforementioned conditions that cause normal or elevated serum osmolality. Patients with hypotonic serum must then be evaluated clinically to determine their volume status. Appropriate classification here has important implications for management.
In addition to clinical history and physical examination, additional laboratory assessments should be carried out. Thyroid dysfunction and adrenal insufficiency should be ruled out on the basis of thyroid stimulating hormone (TSH) and plasma cortisol levels. In addition, urine sodium and urine osmolality should be checked, as they can often help confirm the assessment of the patient's volume status and assist in the classification of the hyponatremia.
Hypovolemic hyponatremia commonly results from either renal or gastrointestinal losses of solute (sodium and potassium).19, 33 Such patients will typically have urine sodium values below 25 mEq/L. Hypervolemic hyponatremia occurs when both solute and water are increased, with water increases that are out of proportion to solute. It is seen in patients with HF, cirrhosis, and nephrotic syndrome.19, 33 These patients often also demonstrate low urine sodium levels. Although plasma and extracellular volumes are increased in these states, patients with HF and cirrhosis experience effective arterial blood volume depletion due to reduced cardiac output and arterial vasodilatation, respectively.
In euvolemic patients, hyponatremia is most often due to the syndrome of inappropriate antidiuretic hormone secretion. Such patients typically have urine sodium levels above 40 mEq/L. Free water excretion is impaired in SIADH, as evidenced by urine osmolality levels greater than 100 mOsm/kg (and often much higher). SIADH is the most common cause of hyponatremia in hospitalized patients.22 The heterogeneity of conditions that can lead to SIADH is striking, including pulmonary and CNS diseases, cancer, and various forms of endocrinopathy.22, 23 Consequently, SIADH is often a diagnosis of exclusion.
Other important causes of hyponatremia in euvolemic patients include primary polydipsia and low dietary solute intake. Primary polydipsia most commonly affects those with psychiatric illness.34 Increased thirst is a common side effect of antipsychotic medications. If water intake is excessive, the ability of the kidney to excrete water is overwhelmed and hyponatremia develops. These patients manifest with low urine osmolality (less than 100 mOsm/kg). In contrast, beer drinkers and other malnourished patients often have reduced ability to excrete free water based on low solute intake.35 In order to maximize the kidney's ability to excrete free water, a basic level of solute intake is required. Severe alcoholics (in particular beer drinkers) often do not meet this minimum solute level since beer is very low in solute. The result is markedly impaired free water excretion. Such patients develop hyponatremia with low urine omolality (less than 100 mOsm/kg).
MANAGEMENT
Although effective management of hyponatremia can be challenging, it is important to recognize that even modest improvements in serum [Na+] are associated with survival benefits.22, 36 The most important treatment factors relate to the severity of hyponatremia, its acuity, and the patient's volume status.33, 36 The first steps in effective management are to optimize treatment of any underlying disease(s) and to discontinue any medications that may be contributing to hyponatremia.
In the severe group are patients who present with either a documented acute drop in serum [Na+] or neurologic symptoms that are not attributable to another disease process. The mainstay of therapy for this group is prompt administration of hypertonic saline to rapidly address neurologic symptoms or prevent their development. Experts recommend correcting serum [Na+] at a rate of 2 mEq/L per hour in patients with documented severe acute hyponatremia, with the assistance of a nephrologist.22 Slower correction rates (0.51 mEq/L per hour) should be used in symptomatic patients who develop severe hyponatremia in a subacute or chronic timeframe, so as to reduce the risk of osmotic demyelination, which confers irreversible damage to neurons and serious CNS sequelae. In both cases, an initial correction of 46 mEq/L is generally sufficient to address neurologic symptoms.37 Correcting the sodium by more than 10 mEq/L in the first 24‐hour period is widely felt to place the patient at risk for iatrogenic brain injury, and should therefore be avoided. Serum sodium must be monitored very frequently (up to every 2 hours) in such patients to ensure appropriate management.22
Management of patients with hyponatremia of uncertain duration and nonspecific symptoms is more common, as well as more challenging. A recently published algorithm recommends looking for and promptly treating hypovolemia if it exists, and then beginning correction at a more gradual rate with normal saline ( furosemide).22 Appropriate management of these patients addresses the sequelae of hyponatremia while at the same time minimizing the risk of iatrogenic injury. Experts recommend therapeutic goals of 6 to 8 mEq/L in 24 hours, 12 to 14 mEq/L in 48 hours, and 14 to 16 mEq/L in 72 hours.37
In asymptomatic patients with chronic hyponatremia, the aim of treatment is gradual correction of serum [Na+]. A significant number of SIADH patients fall into this category. A common mistake seen in the management of such patients is inaccurate assessment of volume status and a blind trial of normal saline infusion. Administration of normal saline to such patients will not improve the serum sodium concentration, and may, in fact, drive it lower. While SIADH patients have a normal ability to excrete sodium, their ability to excrete water is impaired. Therefore, normal saline infusion will lead to free water retention.
For asymptomatic chronic hyponatremia patients, oral fluid restriction is the most simple and least toxic treatment. However, it is often difficult to calculate the actual fluid intake, since water present in food must be included. In addition, thirst often leads to patient nonadherence. Treatment with sodium chloride in the form of dietary salt or sodium chloride tablets is problematic in patients with hypertension, HF or cirrhosis.22 Demeclocycline is fairly well tolerated, but can cause nephrotoxicity and skin sensitivity. Urea, although effective, is available only as a powder that is bitter and difficult to tolerate.22
AVP‐receptor antagonists, commonly called vaptans, are the newest treatment option. Known as aquaretic drugs, they lead to free water excretion.38 Conivaptan and tolvaptan have been approved by the US Food and Drug Administration (FDA) for the treatment of hyponatremia. Conivaptan, available as an intravenous (IV) formulation only, is indicated for the acute treatment of euvolemic or hypervolemic hyponatremia in hospitalized patients for up to 4 days.21, 22, 38, 39 Due to its additional effects on the V1 receptor, this agent can cause vasodilation and resultant hypotension. In a randomized, placebo‐controlled study of patients with euvolemic or hypervolemic hyponatremia, a 4‐day IV infusion of conivaptan significantly increased serum [Na+] levels compared with placebo.40 Tolvaptan, available as an oral formulation, is more suitable for long‐term use, but must be started in the inpatient setting. Patients started on this agent must be followed closely after discharge. Based on the results of 2 multicenter, prospective, randomized, placebo‐controlled trials, tolvaptan is indicated for clinically significant euvolemic or hypervolemic hyponatremia (serum [Na+] <125 mEq/L, or less marked hyponatremia that is symptomatic and persistent, despite fluid restriction), in patients with HF, cirrhosis, and SIADH.22, 41, 42 The vaptans are contraindicated in hypovolemic patients because they can lead to hypotension and/or acute renal failure.38, 43 Fluid restrictions must also be relaxed in patients who are placed on a vaptan.
Long‐term clinical studies of these agents are needed to address their optimal duration of treatment, clinical outcomes, and comparative effectiveness to other treatment approaches. Although this is expected to change, vaptans are not included in current clinical practice guidelines for the management of hyponatremia.
SUMMARY
Hyponatremia is associated with significant morbidity and mortality in a variety of clinical scenarios. Prompt recognition and accurate diagnosis has the potential to improve patient outcomes, as even modest improvements in serum [Na+] are associated with survival benefits. The appropriate management of hyponatremia involves careful assessment of acuity, severity, and volume status. The recently approved vasopressin receptor antagonists show promise as a therapeutic option for this challenging clinical condition.
- ,.A comparison of plasma electrolyte and urea values in healthy persons and in hospital patients.Clin Chim Acta.1968;22:611–618.
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chim Acta.2003;337:169–172.
- ,,.Development of severe hyponatremia in hospitalized patients: treatment‐related risk factors and inadequate management.Nephrol Dial Transplant.2006;21:70–76.
- ,,,.Validity of hospital discharge International Classification of Diseases (ICD) codes for identifying patients with hyponatremia.J Clin Epidemiol.2003;56:530–535.
- ,,,.Incidence and etiology of hyponatremia in an intensive care unit.Clin Nephrol.1990;34:163–166.
- ,,.Epidemiology of hyponatremia.Semin Nephrol.2009;29:227–238.
- ,,, et al.Severe hyponatremia caused by an instrasellar carotid artery aneurysm.Med Health R I.2003;86(2):52–55.
- ,,, et al.Characterization and prognostic value of persistent hyponatremia in patients with severe heart failure in the ESCAPE trial.Arch Intern Med.2007;167:1998–2005.
- ,,, et al.Hyponatremia in a patient with a sellar mass.Chonnam Med J.2011;47(2):122–123.
- ,,, et al.Dilutional hyponatremia in patients with cirrhosis and ascites.Arch Intern Med.2002;162:323–328.
- ,,, et al.Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44(6):1535–1542.
- ,,, et al.Hyponatremia a valuable predictor of early mortality in patients with cirrhosis listed for liver transplantation.Clin Transplant.2011;25(4):638–645.
- ,,,.Streptococcus pneumoniae bacteremia in a community hospital.Chest.1998;113:387–390.
- ,,, et al.Hyponatremia in community‐acquired pneumonia.Am J Nephrol.2007;27(2):184–190.
- ,,, et al.Hyponatremia and hospital outcomes among patients with pneumonia: a retrospective cohort study.BMC Pulm Med.2008;8:16.
- .Consequences of inadequate management of hyponatremia.Am J Nephrol.2005;25:240–249.
- ,,.Incidence and prevalence of hyponatremia.Am J Med.2006;119:S30–S35.
- ,,,.The cost of delirium in the surgical patient.Psychosomatics.2001;42:68–73.
- ,.Diseases of water metabolism. In: Schrier RW, series ed;,, eds. Atlas of Diseases of the Kidney; vol 1. 1999;1–1.22. Available at: http://www.kidneyatlas.org/book1/ADK1_01.pdf. Accessed June 21,2011.
- .Vasopressin V2 receptor antagonists.J Mol Endocrinol.2002;29:1–9.
- ,.Managing hyponatremia in cirrhosis.J Hosp Med.2010;5:S8–S17.
- ,.The syndrome of inappropriate antidiuresis.N Engl J Med.2007;356:2064–2072.
- ,,,.Incidence and risk factors for hyponatremia following treatment with fluoxetine or paroxetine in elderly people.Br J Clin Pharmacol.1999;47:211–217.
- ,,.Diuretic‐associated hyponatremia.Semin Nephrol.2011;31(6):553–566.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- .Cerebral correlates of hyponatremia.Neurocrit Care.2007;6:72–78.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.Q J Med.2008;101:583–588.
- ,,, et al.Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits.Am J Med.2006;119:71.e1–71.e8.
- Advisory Committee of the Cardiovascular and Renal Drugs Division of the US Food and Drug Administration. Treatment of Hyponatremia: Medical Utility of Vasopressin V2 Receptor Antagonism. Briefing Document. June 25, 2008. Available at: http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008–4373b1–05.pdf. Accessed June 24,2011.
- ,.The syndrome of inappropriate antidiuretic hormone: current and future management options.Eur J Endocrinol.2010;162(suppl 1):S13–S18.
- ,,.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122:857–865.
- ,,,,.Medical costs of abnormal serum levels.J Am Soc Nephrol.2008;19:764–770.
- ,,,,.Hyponatremia treatment guidelines 2007: expert panel recommendations.Am J Med.2007;120:S1–S21.
- ,,, et al.Hyponatremia in psychogenic polydipsia.Arch Intern Med.1980;140(12):1639–1642.
- ,,.“Beer potomania” in non‐beer drinkers: effect of low dietary solute intake.Am J Kidney Dis.1998;31(6):1028–1031.
- ,.Hyponatremia: clinical diagnosis and management.Am J Med.2007;120:653–658.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29(3):282–299.
- ,,.Current and future treatment options in SIADH.NDT Plus.2009;2(suppl 3):iii12–iii19.
- Vaprisol (conivaptan hydrochloride injection). Prescribing information.Deerfield, IL:Astellas Pharma US, Inc; October2008.
- ,,, et al.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27:447–457.
- Samsca™ (oral selective vasopressin antagonist). Prescribing information.Rockville, MD:Otsuka America Pharmaceutical, Inc; November2009.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.
- ,,, et al.Vasopressin receptor antagonists for the treatment of hyponatremia: systematic review and meta‐analysis.Am J Kidney Dis.2010;56:325–337.
The high prevalence of hyponatremia in hospitalized patients has been recognized for decades. Published reports dating back to the 1960s indicate that serum sodium concentrations ([Na+]) tend to be lower in hospitalized patients than in outpatients in the community.1 Current estimates for the prevalence of hyponatremia in hospitalized patients range from 15% to nearly 40%.2, 3 Several factors account for this wide range. While most studies estimate the presence of hyponatremia based on International Classification of Diseases, Ninth Revision (ICD‐9) codes, accurate reporting varies widely from institution to institution.4 Furthermore, the definition of hyponatremia depends entirely on the cut‐off value of [Na+] used (generally, <136 mEq/L).3 In addition to patients who have hyponatremia present on admission, a significant proportion develop the condition during their hospital stay.3 Deficits in water excretion can develop or worsen during hospitalization as a result of several factors, combined with intake of hypotonic fluid.3 In a study of hyponatremia in intensive care unit (ICU) patients, as many as 80% demonstrated impaired urinary dilution during their ICU course.5
The prevalence of hyponatremia is significant in patients hospitalized for heart failure (HF), cirrhosis, and pneumonia.6 The prevalence of hyponatremiadefined as serum sodium <135 mEq/Lranges from 18% to 25% in patients admitted for congestive heart failure.79 Rates of hyponatremia in patients admitted with cirrhosis are even higher on average, ranging between 18% and 49%.1012 Hyponatremia is also common in patients with community‐acquired pneumonia (CAP), with prevalence estimates ranging from 8% to 28%.1315
Overall, hyponatremia in each of these disease states portends worse outcome.16 In a retrospective study of 71 adults with pneumonia, admission serum [Na+] <135 mEq/L was a risk factor for in‐hospital mortality.13 In each of these conditions, hyponatremia is associated with the need for ICU care and mechanical ventilation, increased hospital length of stay (LOS), and higher costs of care.17, 18
PATHOPHYSIOLOGY OF HYPONATREMIA
There are 2 primary stimuli for the secretion of antidiuretic hormone (ADH), otherwise known as arginine vasopressin (AVP). Osmoreceptors in the hypothalamus measure the osmolality of the plasma.19 When osmolality increases, AVP is secreted; alternatively, when plasma osmolality drops, secretion of AVP under normal circumstances will diminish. The other stimulus results from baroreceptors throughout the body. Decreased intravascular volume (manifested by lower blood pressure) causes activation of the renin‐angiotensin‐aldosterone system, the sympathetic nervous system, as well as AVP secretion.16, 20 In turn, AVP acts on vasopressin V2 receptors in the kidney to encourage water reapsorption, therefore impairing the patient's ability to excrete dilute urine.6
The mechanism by which hyponatremia develops varies according to disease state. Whereas neurohormonal activation predominates in those with HF and cirrhosis, inappropriate AVP secretion (and in some cases, a resetting of the osmostat) occurs in patients with CAP.10, 13, 17 In both HF and cirrhosis, the degree of neurohormonal activation correlates with the degree of hyponatremia.17
In healthy individuals, the mechanism for free water excretion is AVP suppression caused by a fall in plasma osmolality. Patients with hyponatremia, however, are unable to suppress AVP due to true volume depletion (eg, as a result of inadequate oral intake, gastrointestinal fluids loss from vomiting/diarrhea, or use of thiazide diuretics), effective volume depletion (reduced cardiac output in HF patients vs vasodilation in patients with cirrhosis), or an inappropriate increase in AVP secretion.19, 21, 22
RISK FACTORS
The risk factors for hyponatremia are numerous.2, 22 The ability to excrete water declines with increasing age and is exacerbated by chronic illness. Other risk factors include low body weight, low sodium diets, and residence in a chronic care facility.22, 23 Patients with a low baseline serum sodium concentration also appear to be at increased risk of developing hyponatremia. Although the mechanisms by which such patients develop hyponatremia are not always clear, they generally involve an impaired ability to excrete free water due to an inability to appropriately suppress AVP secretion. Medications commonly associated with the syndrome of inappropriate ADH secretion (SIADH) include selective serotonin reuptake inhibitors (SSRIs), psychotropic drugs, non‐steroidal anti‐inflammatory drugs (NSAIDs), opiates, proton pump inhibitors, as well as certain chemotherapeutics.21, 22 Other risk factors associated with SIADH include major abdominal or thoracic surgery, pain, nausea, and excessive administration of hypotonic intravenous fluids. Finally, diuretic use (in particular thiazides) places patients at risk to develop hyponatremia by increasing total urine volume and solute excretion without an appreciable increase in free water excretion.24
MORBIDITY
The morbidities associated with hyponatremia vary widely in severity. Serious sequelae may occur as a result of hyponatremia itself, as well as from complications that occur due to the challenging nature of effective management. Much of the symptomatology relates to the central nervous system (CNS). Patients presenting with extremely low serum [Na+] levels (eg, <115 mEq/L) often have severe neurologic symptoms, while those with lesser degrees of hyponatremia may be asymptomatic, or present with milder nonspecific symptoms, such as confusion.25, 26 It is important to note that the clinical presentation of hyponatremia very much depends on whether it is acute (occurring over 2448 hours) or chronic (>48 hours).
Water shifts between the intracellular and extracellular fluid compartments are the primary means by which the body equalizes osmolality. When serum sodium changes, the ability of the brain to compensate is limited, and may result in various forms of neurologic impairment due to cerebral edema.25, 26 Such patients may become disoriented, restless, unable to attend, or unable to process information cognitively. There may also be peripheral neurologic dysfunction, such as muscle weakness, blunted neuromuscular reflexes, and impaired gait. Such impairments can lead to delirium, falls, and fractures.25, 27
HYPONATREMIA AND COGNITIVE IMPAIRMENT
Renneboog and colleagues performed a case‐control study to assess the impact of mild chronic asymptomatic hyponatremia (mean serum [Na+] 126 5 mEq/L) in 122 patients compared with 244 matched controls (mean age 72 13 years).28 Hyponatremic patients had significantly longer mean response times on concentration tests. Interestingly, the changes in cognitive function in hyponatremic patients were similar to healthy volunteers purposefully intoxicated with alcohol.28 Patients with hyponatremia have also been shown to score lower on the mental component summary of the 36‐item Short‐Form (SF‐36) survey.29 With treatment aimed at improving serum sodium, these same patients demonstrated improved cognitive function,29 suggesting that treating even mild forms of hyponatremia can improve patient outcomes.30
HYPONATREMIA AND FALLS/FRACTURES
Renneboog and colleagues also demonstrated a markedly increased risk of falls in their patients with chronic hyponatremia compared to controls.28 Hyponatremia increases not only the risk of falls, but also the risk of fracture following a fall. In another recent case‐control study of 513 patients, the adjusted odds ratio for fracture after a fall in a patient with hyponatremia was 4.16 compared with an age‐matched control with normal serum sodium who sustained a similar fall.27 Of note, hyponatremia was mild and asymptomatic in all patients studied. Medications (36% diuretics, 17% SSRIs) were the most common precipitating cause of hyponatremia in this study, which is notable because such risk factors should be recognized and addressed.
Although falls and fractures lead to obvious increases in morbidity and cost, delirium has also been identified as a risk factor for increased hospital LOS.18 Delirious patients are less likely and able to mobilize and participate in physical therapy. As such, they are more often bed‐bound and at increased risk for aspiration and other preventable issues, including deep vein thrombosis, bed sores, and debility, all of which may increase their LOS and cost of care.
MORTALITY
Hyponatremia is associated with a significantly increased mortality risk not only during hospitalization, but also at 1 and 5 years following discharge.31 In a prospective cohort study of approximately 100,000 patients, even those with mild hyponatremia ([Na+] 130134 mEq/L) had a significantly higher mortality at 5 years. The adjusted odds ratio for mortality in patients with serum sodium less than 135 mEq/L was 1.47 during hospitalization (95% CI, 1.331.62), 1.38 at 1 year post‐discharge (1.321.46), and 1.25 at 5 years (1.211.30). The significance of hyponatremia varied according to the underlying clinical condition, with the greatest risk observed in patients with metastatic cancer, heart disease, and those who had undergone orthopedic surgery.31 While the association between hyponatremia and mortality is profound, most experts do not believe that hyponatremia directly causes mortality per se. Instead, hyponatremia is felt to be a marker for increased illness severity.
It is difficult to isolate the direct costs of hyponatremia in the acute care setting because the condition is rarely treated in isolation. However, in a study of a managed‐care claims database of nearly 1,300 patients (excluding Medicare patients), hyponatremia was a predictor of higher medical costs at 6 months and at 1 year.32
DIAGNOSIS
The most common presentation of hyponatremia involves nonspecific symptoms or a total lack of symptoms.19 Many patients have comorbid diseases, and symptoms of these illnesses often predominate at hospital admission. Patients with mild to moderate hyponatremia may present with nausea, weakness, malaise, headache, and/or impaired mobility. With more severe hyponatremia, more dangerous neurologic symptoms appear, including generalized seizures, lethargy, and coma.19 Once hyponatremia is identified, the next step is to determine its acuity and classify it.
Although several classification systems exist to describe hyponatremia, the most common scheme begins with assessment of plasma osmolality and volume status.19 The majority of hyponatremic patients present with hypotonic or hypo‐osmolar serum (eg, plasma osmolality <275 mOsm/kg). The primary causes of hyponatremia in patients with normal or high serum osmolality are hyperglycemia, pseudohyponatremia, and advanced renal failure. Marked hyperglycemia increases plasma osmolality, and as a result, water moves out of cells into plasma and lowers serum sodium concentration in the process. Pseudohyponatremia arises from hyperlipidemia or hyperproteinemia, in which high concentrations of lipids/proteins reduce the free water component of plasma, therefore reducing the sodium concentration per liter of plasma. These patients do not have true hyponatremia since the physiologically important sodium concentration per liter of plasma water is normal. Finally, patients with advanced renal failure develop hyponatremia due to the inability to excrete water.
The first step in the diagnosis of hyponatremia is to assess the plasma osmolality and rule out the aforementioned conditions that cause normal or elevated serum osmolality. Patients with hypotonic serum must then be evaluated clinically to determine their volume status. Appropriate classification here has important implications for management.
In addition to clinical history and physical examination, additional laboratory assessments should be carried out. Thyroid dysfunction and adrenal insufficiency should be ruled out on the basis of thyroid stimulating hormone (TSH) and plasma cortisol levels. In addition, urine sodium and urine osmolality should be checked, as they can often help confirm the assessment of the patient's volume status and assist in the classification of the hyponatremia.
Hypovolemic hyponatremia commonly results from either renal or gastrointestinal losses of solute (sodium and potassium).19, 33 Such patients will typically have urine sodium values below 25 mEq/L. Hypervolemic hyponatremia occurs when both solute and water are increased, with water increases that are out of proportion to solute. It is seen in patients with HF, cirrhosis, and nephrotic syndrome.19, 33 These patients often also demonstrate low urine sodium levels. Although plasma and extracellular volumes are increased in these states, patients with HF and cirrhosis experience effective arterial blood volume depletion due to reduced cardiac output and arterial vasodilatation, respectively.
In euvolemic patients, hyponatremia is most often due to the syndrome of inappropriate antidiuretic hormone secretion. Such patients typically have urine sodium levels above 40 mEq/L. Free water excretion is impaired in SIADH, as evidenced by urine osmolality levels greater than 100 mOsm/kg (and often much higher). SIADH is the most common cause of hyponatremia in hospitalized patients.22 The heterogeneity of conditions that can lead to SIADH is striking, including pulmonary and CNS diseases, cancer, and various forms of endocrinopathy.22, 23 Consequently, SIADH is often a diagnosis of exclusion.
Other important causes of hyponatremia in euvolemic patients include primary polydipsia and low dietary solute intake. Primary polydipsia most commonly affects those with psychiatric illness.34 Increased thirst is a common side effect of antipsychotic medications. If water intake is excessive, the ability of the kidney to excrete water is overwhelmed and hyponatremia develops. These patients manifest with low urine osmolality (less than 100 mOsm/kg). In contrast, beer drinkers and other malnourished patients often have reduced ability to excrete free water based on low solute intake.35 In order to maximize the kidney's ability to excrete free water, a basic level of solute intake is required. Severe alcoholics (in particular beer drinkers) often do not meet this minimum solute level since beer is very low in solute. The result is markedly impaired free water excretion. Such patients develop hyponatremia with low urine omolality (less than 100 mOsm/kg).
MANAGEMENT
Although effective management of hyponatremia can be challenging, it is important to recognize that even modest improvements in serum [Na+] are associated with survival benefits.22, 36 The most important treatment factors relate to the severity of hyponatremia, its acuity, and the patient's volume status.33, 36 The first steps in effective management are to optimize treatment of any underlying disease(s) and to discontinue any medications that may be contributing to hyponatremia.
In the severe group are patients who present with either a documented acute drop in serum [Na+] or neurologic symptoms that are not attributable to another disease process. The mainstay of therapy for this group is prompt administration of hypertonic saline to rapidly address neurologic symptoms or prevent their development. Experts recommend correcting serum [Na+] at a rate of 2 mEq/L per hour in patients with documented severe acute hyponatremia, with the assistance of a nephrologist.22 Slower correction rates (0.51 mEq/L per hour) should be used in symptomatic patients who develop severe hyponatremia in a subacute or chronic timeframe, so as to reduce the risk of osmotic demyelination, which confers irreversible damage to neurons and serious CNS sequelae. In both cases, an initial correction of 46 mEq/L is generally sufficient to address neurologic symptoms.37 Correcting the sodium by more than 10 mEq/L in the first 24‐hour period is widely felt to place the patient at risk for iatrogenic brain injury, and should therefore be avoided. Serum sodium must be monitored very frequently (up to every 2 hours) in such patients to ensure appropriate management.22
Management of patients with hyponatremia of uncertain duration and nonspecific symptoms is more common, as well as more challenging. A recently published algorithm recommends looking for and promptly treating hypovolemia if it exists, and then beginning correction at a more gradual rate with normal saline ( furosemide).22 Appropriate management of these patients addresses the sequelae of hyponatremia while at the same time minimizing the risk of iatrogenic injury. Experts recommend therapeutic goals of 6 to 8 mEq/L in 24 hours, 12 to 14 mEq/L in 48 hours, and 14 to 16 mEq/L in 72 hours.37
In asymptomatic patients with chronic hyponatremia, the aim of treatment is gradual correction of serum [Na+]. A significant number of SIADH patients fall into this category. A common mistake seen in the management of such patients is inaccurate assessment of volume status and a blind trial of normal saline infusion. Administration of normal saline to such patients will not improve the serum sodium concentration, and may, in fact, drive it lower. While SIADH patients have a normal ability to excrete sodium, their ability to excrete water is impaired. Therefore, normal saline infusion will lead to free water retention.
For asymptomatic chronic hyponatremia patients, oral fluid restriction is the most simple and least toxic treatment. However, it is often difficult to calculate the actual fluid intake, since water present in food must be included. In addition, thirst often leads to patient nonadherence. Treatment with sodium chloride in the form of dietary salt or sodium chloride tablets is problematic in patients with hypertension, HF or cirrhosis.22 Demeclocycline is fairly well tolerated, but can cause nephrotoxicity and skin sensitivity. Urea, although effective, is available only as a powder that is bitter and difficult to tolerate.22
AVP‐receptor antagonists, commonly called vaptans, are the newest treatment option. Known as aquaretic drugs, they lead to free water excretion.38 Conivaptan and tolvaptan have been approved by the US Food and Drug Administration (FDA) for the treatment of hyponatremia. Conivaptan, available as an intravenous (IV) formulation only, is indicated for the acute treatment of euvolemic or hypervolemic hyponatremia in hospitalized patients for up to 4 days.21, 22, 38, 39 Due to its additional effects on the V1 receptor, this agent can cause vasodilation and resultant hypotension. In a randomized, placebo‐controlled study of patients with euvolemic or hypervolemic hyponatremia, a 4‐day IV infusion of conivaptan significantly increased serum [Na+] levels compared with placebo.40 Tolvaptan, available as an oral formulation, is more suitable for long‐term use, but must be started in the inpatient setting. Patients started on this agent must be followed closely after discharge. Based on the results of 2 multicenter, prospective, randomized, placebo‐controlled trials, tolvaptan is indicated for clinically significant euvolemic or hypervolemic hyponatremia (serum [Na+] <125 mEq/L, or less marked hyponatremia that is symptomatic and persistent, despite fluid restriction), in patients with HF, cirrhosis, and SIADH.22, 41, 42 The vaptans are contraindicated in hypovolemic patients because they can lead to hypotension and/or acute renal failure.38, 43 Fluid restrictions must also be relaxed in patients who are placed on a vaptan.
Long‐term clinical studies of these agents are needed to address their optimal duration of treatment, clinical outcomes, and comparative effectiveness to other treatment approaches. Although this is expected to change, vaptans are not included in current clinical practice guidelines for the management of hyponatremia.
SUMMARY
Hyponatremia is associated with significant morbidity and mortality in a variety of clinical scenarios. Prompt recognition and accurate diagnosis has the potential to improve patient outcomes, as even modest improvements in serum [Na+] are associated with survival benefits. The appropriate management of hyponatremia involves careful assessment of acuity, severity, and volume status. The recently approved vasopressin receptor antagonists show promise as a therapeutic option for this challenging clinical condition.
The high prevalence of hyponatremia in hospitalized patients has been recognized for decades. Published reports dating back to the 1960s indicate that serum sodium concentrations ([Na+]) tend to be lower in hospitalized patients than in outpatients in the community.1 Current estimates for the prevalence of hyponatremia in hospitalized patients range from 15% to nearly 40%.2, 3 Several factors account for this wide range. While most studies estimate the presence of hyponatremia based on International Classification of Diseases, Ninth Revision (ICD‐9) codes, accurate reporting varies widely from institution to institution.4 Furthermore, the definition of hyponatremia depends entirely on the cut‐off value of [Na+] used (generally, <136 mEq/L).3 In addition to patients who have hyponatremia present on admission, a significant proportion develop the condition during their hospital stay.3 Deficits in water excretion can develop or worsen during hospitalization as a result of several factors, combined with intake of hypotonic fluid.3 In a study of hyponatremia in intensive care unit (ICU) patients, as many as 80% demonstrated impaired urinary dilution during their ICU course.5
The prevalence of hyponatremia is significant in patients hospitalized for heart failure (HF), cirrhosis, and pneumonia.6 The prevalence of hyponatremiadefined as serum sodium <135 mEq/Lranges from 18% to 25% in patients admitted for congestive heart failure.79 Rates of hyponatremia in patients admitted with cirrhosis are even higher on average, ranging between 18% and 49%.1012 Hyponatremia is also common in patients with community‐acquired pneumonia (CAP), with prevalence estimates ranging from 8% to 28%.1315
Overall, hyponatremia in each of these disease states portends worse outcome.16 In a retrospective study of 71 adults with pneumonia, admission serum [Na+] <135 mEq/L was a risk factor for in‐hospital mortality.13 In each of these conditions, hyponatremia is associated with the need for ICU care and mechanical ventilation, increased hospital length of stay (LOS), and higher costs of care.17, 18
PATHOPHYSIOLOGY OF HYPONATREMIA
There are 2 primary stimuli for the secretion of antidiuretic hormone (ADH), otherwise known as arginine vasopressin (AVP). Osmoreceptors in the hypothalamus measure the osmolality of the plasma.19 When osmolality increases, AVP is secreted; alternatively, when plasma osmolality drops, secretion of AVP under normal circumstances will diminish. The other stimulus results from baroreceptors throughout the body. Decreased intravascular volume (manifested by lower blood pressure) causes activation of the renin‐angiotensin‐aldosterone system, the sympathetic nervous system, as well as AVP secretion.16, 20 In turn, AVP acts on vasopressin V2 receptors in the kidney to encourage water reapsorption, therefore impairing the patient's ability to excrete dilute urine.6
The mechanism by which hyponatremia develops varies according to disease state. Whereas neurohormonal activation predominates in those with HF and cirrhosis, inappropriate AVP secretion (and in some cases, a resetting of the osmostat) occurs in patients with CAP.10, 13, 17 In both HF and cirrhosis, the degree of neurohormonal activation correlates with the degree of hyponatremia.17
In healthy individuals, the mechanism for free water excretion is AVP suppression caused by a fall in plasma osmolality. Patients with hyponatremia, however, are unable to suppress AVP due to true volume depletion (eg, as a result of inadequate oral intake, gastrointestinal fluids loss from vomiting/diarrhea, or use of thiazide diuretics), effective volume depletion (reduced cardiac output in HF patients vs vasodilation in patients with cirrhosis), or an inappropriate increase in AVP secretion.19, 21, 22
RISK FACTORS
The risk factors for hyponatremia are numerous.2, 22 The ability to excrete water declines with increasing age and is exacerbated by chronic illness. Other risk factors include low body weight, low sodium diets, and residence in a chronic care facility.22, 23 Patients with a low baseline serum sodium concentration also appear to be at increased risk of developing hyponatremia. Although the mechanisms by which such patients develop hyponatremia are not always clear, they generally involve an impaired ability to excrete free water due to an inability to appropriately suppress AVP secretion. Medications commonly associated with the syndrome of inappropriate ADH secretion (SIADH) include selective serotonin reuptake inhibitors (SSRIs), psychotropic drugs, non‐steroidal anti‐inflammatory drugs (NSAIDs), opiates, proton pump inhibitors, as well as certain chemotherapeutics.21, 22 Other risk factors associated with SIADH include major abdominal or thoracic surgery, pain, nausea, and excessive administration of hypotonic intravenous fluids. Finally, diuretic use (in particular thiazides) places patients at risk to develop hyponatremia by increasing total urine volume and solute excretion without an appreciable increase in free water excretion.24
MORBIDITY
The morbidities associated with hyponatremia vary widely in severity. Serious sequelae may occur as a result of hyponatremia itself, as well as from complications that occur due to the challenging nature of effective management. Much of the symptomatology relates to the central nervous system (CNS). Patients presenting with extremely low serum [Na+] levels (eg, <115 mEq/L) often have severe neurologic symptoms, while those with lesser degrees of hyponatremia may be asymptomatic, or present with milder nonspecific symptoms, such as confusion.25, 26 It is important to note that the clinical presentation of hyponatremia very much depends on whether it is acute (occurring over 2448 hours) or chronic (>48 hours).
Water shifts between the intracellular and extracellular fluid compartments are the primary means by which the body equalizes osmolality. When serum sodium changes, the ability of the brain to compensate is limited, and may result in various forms of neurologic impairment due to cerebral edema.25, 26 Such patients may become disoriented, restless, unable to attend, or unable to process information cognitively. There may also be peripheral neurologic dysfunction, such as muscle weakness, blunted neuromuscular reflexes, and impaired gait. Such impairments can lead to delirium, falls, and fractures.25, 27
HYPONATREMIA AND COGNITIVE IMPAIRMENT
Renneboog and colleagues performed a case‐control study to assess the impact of mild chronic asymptomatic hyponatremia (mean serum [Na+] 126 5 mEq/L) in 122 patients compared with 244 matched controls (mean age 72 13 years).28 Hyponatremic patients had significantly longer mean response times on concentration tests. Interestingly, the changes in cognitive function in hyponatremic patients were similar to healthy volunteers purposefully intoxicated with alcohol.28 Patients with hyponatremia have also been shown to score lower on the mental component summary of the 36‐item Short‐Form (SF‐36) survey.29 With treatment aimed at improving serum sodium, these same patients demonstrated improved cognitive function,29 suggesting that treating even mild forms of hyponatremia can improve patient outcomes.30
HYPONATREMIA AND FALLS/FRACTURES
Renneboog and colleagues also demonstrated a markedly increased risk of falls in their patients with chronic hyponatremia compared to controls.28 Hyponatremia increases not only the risk of falls, but also the risk of fracture following a fall. In another recent case‐control study of 513 patients, the adjusted odds ratio for fracture after a fall in a patient with hyponatremia was 4.16 compared with an age‐matched control with normal serum sodium who sustained a similar fall.27 Of note, hyponatremia was mild and asymptomatic in all patients studied. Medications (36% diuretics, 17% SSRIs) were the most common precipitating cause of hyponatremia in this study, which is notable because such risk factors should be recognized and addressed.
Although falls and fractures lead to obvious increases in morbidity and cost, delirium has also been identified as a risk factor for increased hospital LOS.18 Delirious patients are less likely and able to mobilize and participate in physical therapy. As such, they are more often bed‐bound and at increased risk for aspiration and other preventable issues, including deep vein thrombosis, bed sores, and debility, all of which may increase their LOS and cost of care.
MORTALITY
Hyponatremia is associated with a significantly increased mortality risk not only during hospitalization, but also at 1 and 5 years following discharge.31 In a prospective cohort study of approximately 100,000 patients, even those with mild hyponatremia ([Na+] 130134 mEq/L) had a significantly higher mortality at 5 years. The adjusted odds ratio for mortality in patients with serum sodium less than 135 mEq/L was 1.47 during hospitalization (95% CI, 1.331.62), 1.38 at 1 year post‐discharge (1.321.46), and 1.25 at 5 years (1.211.30). The significance of hyponatremia varied according to the underlying clinical condition, with the greatest risk observed in patients with metastatic cancer, heart disease, and those who had undergone orthopedic surgery.31 While the association between hyponatremia and mortality is profound, most experts do not believe that hyponatremia directly causes mortality per se. Instead, hyponatremia is felt to be a marker for increased illness severity.
It is difficult to isolate the direct costs of hyponatremia in the acute care setting because the condition is rarely treated in isolation. However, in a study of a managed‐care claims database of nearly 1,300 patients (excluding Medicare patients), hyponatremia was a predictor of higher medical costs at 6 months and at 1 year.32
DIAGNOSIS
The most common presentation of hyponatremia involves nonspecific symptoms or a total lack of symptoms.19 Many patients have comorbid diseases, and symptoms of these illnesses often predominate at hospital admission. Patients with mild to moderate hyponatremia may present with nausea, weakness, malaise, headache, and/or impaired mobility. With more severe hyponatremia, more dangerous neurologic symptoms appear, including generalized seizures, lethargy, and coma.19 Once hyponatremia is identified, the next step is to determine its acuity and classify it.
Although several classification systems exist to describe hyponatremia, the most common scheme begins with assessment of plasma osmolality and volume status.19 The majority of hyponatremic patients present with hypotonic or hypo‐osmolar serum (eg, plasma osmolality <275 mOsm/kg). The primary causes of hyponatremia in patients with normal or high serum osmolality are hyperglycemia, pseudohyponatremia, and advanced renal failure. Marked hyperglycemia increases plasma osmolality, and as a result, water moves out of cells into plasma and lowers serum sodium concentration in the process. Pseudohyponatremia arises from hyperlipidemia or hyperproteinemia, in which high concentrations of lipids/proteins reduce the free water component of plasma, therefore reducing the sodium concentration per liter of plasma. These patients do not have true hyponatremia since the physiologically important sodium concentration per liter of plasma water is normal. Finally, patients with advanced renal failure develop hyponatremia due to the inability to excrete water.
The first step in the diagnosis of hyponatremia is to assess the plasma osmolality and rule out the aforementioned conditions that cause normal or elevated serum osmolality. Patients with hypotonic serum must then be evaluated clinically to determine their volume status. Appropriate classification here has important implications for management.
In addition to clinical history and physical examination, additional laboratory assessments should be carried out. Thyroid dysfunction and adrenal insufficiency should be ruled out on the basis of thyroid stimulating hormone (TSH) and plasma cortisol levels. In addition, urine sodium and urine osmolality should be checked, as they can often help confirm the assessment of the patient's volume status and assist in the classification of the hyponatremia.
Hypovolemic hyponatremia commonly results from either renal or gastrointestinal losses of solute (sodium and potassium).19, 33 Such patients will typically have urine sodium values below 25 mEq/L. Hypervolemic hyponatremia occurs when both solute and water are increased, with water increases that are out of proportion to solute. It is seen in patients with HF, cirrhosis, and nephrotic syndrome.19, 33 These patients often also demonstrate low urine sodium levels. Although plasma and extracellular volumes are increased in these states, patients with HF and cirrhosis experience effective arterial blood volume depletion due to reduced cardiac output and arterial vasodilatation, respectively.
In euvolemic patients, hyponatremia is most often due to the syndrome of inappropriate antidiuretic hormone secretion. Such patients typically have urine sodium levels above 40 mEq/L. Free water excretion is impaired in SIADH, as evidenced by urine osmolality levels greater than 100 mOsm/kg (and often much higher). SIADH is the most common cause of hyponatremia in hospitalized patients.22 The heterogeneity of conditions that can lead to SIADH is striking, including pulmonary and CNS diseases, cancer, and various forms of endocrinopathy.22, 23 Consequently, SIADH is often a diagnosis of exclusion.
Other important causes of hyponatremia in euvolemic patients include primary polydipsia and low dietary solute intake. Primary polydipsia most commonly affects those with psychiatric illness.34 Increased thirst is a common side effect of antipsychotic medications. If water intake is excessive, the ability of the kidney to excrete water is overwhelmed and hyponatremia develops. These patients manifest with low urine osmolality (less than 100 mOsm/kg). In contrast, beer drinkers and other malnourished patients often have reduced ability to excrete free water based on low solute intake.35 In order to maximize the kidney's ability to excrete free water, a basic level of solute intake is required. Severe alcoholics (in particular beer drinkers) often do not meet this minimum solute level since beer is very low in solute. The result is markedly impaired free water excretion. Such patients develop hyponatremia with low urine omolality (less than 100 mOsm/kg).
MANAGEMENT
Although effective management of hyponatremia can be challenging, it is important to recognize that even modest improvements in serum [Na+] are associated with survival benefits.22, 36 The most important treatment factors relate to the severity of hyponatremia, its acuity, and the patient's volume status.33, 36 The first steps in effective management are to optimize treatment of any underlying disease(s) and to discontinue any medications that may be contributing to hyponatremia.
In the severe group are patients who present with either a documented acute drop in serum [Na+] or neurologic symptoms that are not attributable to another disease process. The mainstay of therapy for this group is prompt administration of hypertonic saline to rapidly address neurologic symptoms or prevent their development. Experts recommend correcting serum [Na+] at a rate of 2 mEq/L per hour in patients with documented severe acute hyponatremia, with the assistance of a nephrologist.22 Slower correction rates (0.51 mEq/L per hour) should be used in symptomatic patients who develop severe hyponatremia in a subacute or chronic timeframe, so as to reduce the risk of osmotic demyelination, which confers irreversible damage to neurons and serious CNS sequelae. In both cases, an initial correction of 46 mEq/L is generally sufficient to address neurologic symptoms.37 Correcting the sodium by more than 10 mEq/L in the first 24‐hour period is widely felt to place the patient at risk for iatrogenic brain injury, and should therefore be avoided. Serum sodium must be monitored very frequently (up to every 2 hours) in such patients to ensure appropriate management.22
Management of patients with hyponatremia of uncertain duration and nonspecific symptoms is more common, as well as more challenging. A recently published algorithm recommends looking for and promptly treating hypovolemia if it exists, and then beginning correction at a more gradual rate with normal saline ( furosemide).22 Appropriate management of these patients addresses the sequelae of hyponatremia while at the same time minimizing the risk of iatrogenic injury. Experts recommend therapeutic goals of 6 to 8 mEq/L in 24 hours, 12 to 14 mEq/L in 48 hours, and 14 to 16 mEq/L in 72 hours.37
In asymptomatic patients with chronic hyponatremia, the aim of treatment is gradual correction of serum [Na+]. A significant number of SIADH patients fall into this category. A common mistake seen in the management of such patients is inaccurate assessment of volume status and a blind trial of normal saline infusion. Administration of normal saline to such patients will not improve the serum sodium concentration, and may, in fact, drive it lower. While SIADH patients have a normal ability to excrete sodium, their ability to excrete water is impaired. Therefore, normal saline infusion will lead to free water retention.
For asymptomatic chronic hyponatremia patients, oral fluid restriction is the most simple and least toxic treatment. However, it is often difficult to calculate the actual fluid intake, since water present in food must be included. In addition, thirst often leads to patient nonadherence. Treatment with sodium chloride in the form of dietary salt or sodium chloride tablets is problematic in patients with hypertension, HF or cirrhosis.22 Demeclocycline is fairly well tolerated, but can cause nephrotoxicity and skin sensitivity. Urea, although effective, is available only as a powder that is bitter and difficult to tolerate.22
AVP‐receptor antagonists, commonly called vaptans, are the newest treatment option. Known as aquaretic drugs, they lead to free water excretion.38 Conivaptan and tolvaptan have been approved by the US Food and Drug Administration (FDA) for the treatment of hyponatremia. Conivaptan, available as an intravenous (IV) formulation only, is indicated for the acute treatment of euvolemic or hypervolemic hyponatremia in hospitalized patients for up to 4 days.21, 22, 38, 39 Due to its additional effects on the V1 receptor, this agent can cause vasodilation and resultant hypotension. In a randomized, placebo‐controlled study of patients with euvolemic or hypervolemic hyponatremia, a 4‐day IV infusion of conivaptan significantly increased serum [Na+] levels compared with placebo.40 Tolvaptan, available as an oral formulation, is more suitable for long‐term use, but must be started in the inpatient setting. Patients started on this agent must be followed closely after discharge. Based on the results of 2 multicenter, prospective, randomized, placebo‐controlled trials, tolvaptan is indicated for clinically significant euvolemic or hypervolemic hyponatremia (serum [Na+] <125 mEq/L, or less marked hyponatremia that is symptomatic and persistent, despite fluid restriction), in patients with HF, cirrhosis, and SIADH.22, 41, 42 The vaptans are contraindicated in hypovolemic patients because they can lead to hypotension and/or acute renal failure.38, 43 Fluid restrictions must also be relaxed in patients who are placed on a vaptan.
Long‐term clinical studies of these agents are needed to address their optimal duration of treatment, clinical outcomes, and comparative effectiveness to other treatment approaches. Although this is expected to change, vaptans are not included in current clinical practice guidelines for the management of hyponatremia.
SUMMARY
Hyponatremia is associated with significant morbidity and mortality in a variety of clinical scenarios. Prompt recognition and accurate diagnosis has the potential to improve patient outcomes, as even modest improvements in serum [Na+] are associated with survival benefits. The appropriate management of hyponatremia involves careful assessment of acuity, severity, and volume status. The recently approved vasopressin receptor antagonists show promise as a therapeutic option for this challenging clinical condition.
- ,.A comparison of plasma electrolyte and urea values in healthy persons and in hospital patients.Clin Chim Acta.1968;22:611–618.
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chim Acta.2003;337:169–172.
- ,,.Development of severe hyponatremia in hospitalized patients: treatment‐related risk factors and inadequate management.Nephrol Dial Transplant.2006;21:70–76.
- ,,,.Validity of hospital discharge International Classification of Diseases (ICD) codes for identifying patients with hyponatremia.J Clin Epidemiol.2003;56:530–535.
- ,,,.Incidence and etiology of hyponatremia in an intensive care unit.Clin Nephrol.1990;34:163–166.
- ,,.Epidemiology of hyponatremia.Semin Nephrol.2009;29:227–238.
- ,,, et al.Severe hyponatremia caused by an instrasellar carotid artery aneurysm.Med Health R I.2003;86(2):52–55.
- ,,, et al.Characterization and prognostic value of persistent hyponatremia in patients with severe heart failure in the ESCAPE trial.Arch Intern Med.2007;167:1998–2005.
- ,,, et al.Hyponatremia in a patient with a sellar mass.Chonnam Med J.2011;47(2):122–123.
- ,,, et al.Dilutional hyponatremia in patients with cirrhosis and ascites.Arch Intern Med.2002;162:323–328.
- ,,, et al.Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44(6):1535–1542.
- ,,, et al.Hyponatremia a valuable predictor of early mortality in patients with cirrhosis listed for liver transplantation.Clin Transplant.2011;25(4):638–645.
- ,,,.Streptococcus pneumoniae bacteremia in a community hospital.Chest.1998;113:387–390.
- ,,, et al.Hyponatremia in community‐acquired pneumonia.Am J Nephrol.2007;27(2):184–190.
- ,,, et al.Hyponatremia and hospital outcomes among patients with pneumonia: a retrospective cohort study.BMC Pulm Med.2008;8:16.
- .Consequences of inadequate management of hyponatremia.Am J Nephrol.2005;25:240–249.
- ,,.Incidence and prevalence of hyponatremia.Am J Med.2006;119:S30–S35.
- ,,,.The cost of delirium in the surgical patient.Psychosomatics.2001;42:68–73.
- ,.Diseases of water metabolism. In: Schrier RW, series ed;,, eds. Atlas of Diseases of the Kidney; vol 1. 1999;1–1.22. Available at: http://www.kidneyatlas.org/book1/ADK1_01.pdf. Accessed June 21,2011.
- .Vasopressin V2 receptor antagonists.J Mol Endocrinol.2002;29:1–9.
- ,.Managing hyponatremia in cirrhosis.J Hosp Med.2010;5:S8–S17.
- ,.The syndrome of inappropriate antidiuresis.N Engl J Med.2007;356:2064–2072.
- ,,,.Incidence and risk factors for hyponatremia following treatment with fluoxetine or paroxetine in elderly people.Br J Clin Pharmacol.1999;47:211–217.
- ,,.Diuretic‐associated hyponatremia.Semin Nephrol.2011;31(6):553–566.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- .Cerebral correlates of hyponatremia.Neurocrit Care.2007;6:72–78.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.Q J Med.2008;101:583–588.
- ,,, et al.Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits.Am J Med.2006;119:71.e1–71.e8.
- Advisory Committee of the Cardiovascular and Renal Drugs Division of the US Food and Drug Administration. Treatment of Hyponatremia: Medical Utility of Vasopressin V2 Receptor Antagonism. Briefing Document. June 25, 2008. Available at: http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008–4373b1–05.pdf. Accessed June 24,2011.
- ,.The syndrome of inappropriate antidiuretic hormone: current and future management options.Eur J Endocrinol.2010;162(suppl 1):S13–S18.
- ,,.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122:857–865.
- ,,,,.Medical costs of abnormal serum levels.J Am Soc Nephrol.2008;19:764–770.
- ,,,,.Hyponatremia treatment guidelines 2007: expert panel recommendations.Am J Med.2007;120:S1–S21.
- ,,, et al.Hyponatremia in psychogenic polydipsia.Arch Intern Med.1980;140(12):1639–1642.
- ,,.“Beer potomania” in non‐beer drinkers: effect of low dietary solute intake.Am J Kidney Dis.1998;31(6):1028–1031.
- ,.Hyponatremia: clinical diagnosis and management.Am J Med.2007;120:653–658.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29(3):282–299.
- ,,.Current and future treatment options in SIADH.NDT Plus.2009;2(suppl 3):iii12–iii19.
- Vaprisol (conivaptan hydrochloride injection). Prescribing information.Deerfield, IL:Astellas Pharma US, Inc; October2008.
- ,,, et al.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27:447–457.
- Samsca™ (oral selective vasopressin antagonist). Prescribing information.Rockville, MD:Otsuka America Pharmaceutical, Inc; November2009.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.
- ,,, et al.Vasopressin receptor antagonists for the treatment of hyponatremia: systematic review and meta‐analysis.Am J Kidney Dis.2010;56:325–337.
- ,.A comparison of plasma electrolyte and urea values in healthy persons and in hospital patients.Clin Chim Acta.1968;22:611–618.
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chim Acta.2003;337:169–172.
- ,,.Development of severe hyponatremia in hospitalized patients: treatment‐related risk factors and inadequate management.Nephrol Dial Transplant.2006;21:70–76.
- ,,,.Validity of hospital discharge International Classification of Diseases (ICD) codes for identifying patients with hyponatremia.J Clin Epidemiol.2003;56:530–535.
- ,,,.Incidence and etiology of hyponatremia in an intensive care unit.Clin Nephrol.1990;34:163–166.
- ,,.Epidemiology of hyponatremia.Semin Nephrol.2009;29:227–238.
- ,,, et al.Severe hyponatremia caused by an instrasellar carotid artery aneurysm.Med Health R I.2003;86(2):52–55.
- ,,, et al.Characterization and prognostic value of persistent hyponatremia in patients with severe heart failure in the ESCAPE trial.Arch Intern Med.2007;167:1998–2005.
- ,,, et al.Hyponatremia in a patient with a sellar mass.Chonnam Med J.2011;47(2):122–123.
- ,,, et al.Dilutional hyponatremia in patients with cirrhosis and ascites.Arch Intern Med.2002;162:323–328.
- ,,, et al.Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44(6):1535–1542.
- ,,, et al.Hyponatremia a valuable predictor of early mortality in patients with cirrhosis listed for liver transplantation.Clin Transplant.2011;25(4):638–645.
- ,,,.Streptococcus pneumoniae bacteremia in a community hospital.Chest.1998;113:387–390.
- ,,, et al.Hyponatremia in community‐acquired pneumonia.Am J Nephrol.2007;27(2):184–190.
- ,,, et al.Hyponatremia and hospital outcomes among patients with pneumonia: a retrospective cohort study.BMC Pulm Med.2008;8:16.
- .Consequences of inadequate management of hyponatremia.Am J Nephrol.2005;25:240–249.
- ,,.Incidence and prevalence of hyponatremia.Am J Med.2006;119:S30–S35.
- ,,,.The cost of delirium in the surgical patient.Psychosomatics.2001;42:68–73.
- ,.Diseases of water metabolism. In: Schrier RW, series ed;,, eds. Atlas of Diseases of the Kidney; vol 1. 1999;1–1.22. Available at: http://www.kidneyatlas.org/book1/ADK1_01.pdf. Accessed June 21,2011.
- .Vasopressin V2 receptor antagonists.J Mol Endocrinol.2002;29:1–9.
- ,.Managing hyponatremia in cirrhosis.J Hosp Med.2010;5:S8–S17.
- ,.The syndrome of inappropriate antidiuresis.N Engl J Med.2007;356:2064–2072.
- ,,,.Incidence and risk factors for hyponatremia following treatment with fluoxetine or paroxetine in elderly people.Br J Clin Pharmacol.1999;47:211–217.
- ,,.Diuretic‐associated hyponatremia.Semin Nephrol.2011;31(6):553–566.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- .Cerebral correlates of hyponatremia.Neurocrit Care.2007;6:72–78.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.Q J Med.2008;101:583–588.
- ,,, et al.Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits.Am J Med.2006;119:71.e1–71.e8.
- Advisory Committee of the Cardiovascular and Renal Drugs Division of the US Food and Drug Administration. Treatment of Hyponatremia: Medical Utility of Vasopressin V2 Receptor Antagonism. Briefing Document. June 25, 2008. Available at: http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008–4373b1–05.pdf. Accessed June 24,2011.
- ,.The syndrome of inappropriate antidiuretic hormone: current and future management options.Eur J Endocrinol.2010;162(suppl 1):S13–S18.
- ,,.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122:857–865.
- ,,,,.Medical costs of abnormal serum levels.J Am Soc Nephrol.2008;19:764–770.
- ,,,,.Hyponatremia treatment guidelines 2007: expert panel recommendations.Am J Med.2007;120:S1–S21.
- ,,, et al.Hyponatremia in psychogenic polydipsia.Arch Intern Med.1980;140(12):1639–1642.
- ,,.“Beer potomania” in non‐beer drinkers: effect of low dietary solute intake.Am J Kidney Dis.1998;31(6):1028–1031.
- ,.Hyponatremia: clinical diagnosis and management.Am J Med.2007;120:653–658.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29(3):282–299.
- ,,.Current and future treatment options in SIADH.NDT Plus.2009;2(suppl 3):iii12–iii19.
- Vaprisol (conivaptan hydrochloride injection). Prescribing information.Deerfield, IL:Astellas Pharma US, Inc; October2008.
- ,,, et al.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27:447–457.
- Samsca™ (oral selective vasopressin antagonist). Prescribing information.Rockville, MD:Otsuka America Pharmaceutical, Inc; November2009.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.
- ,,, et al.Vasopressin receptor antagonists for the treatment of hyponatremia: systematic review and meta‐analysis.Am J Kidney Dis.2010;56:325–337.
Nonallergic rhinitis: Common problem, chronic symptoms
A 55-year-old woman has come to the clinic because of clear rhinorrhea and nasal congestion, which occur year-round but are worse in the winter. She reports that at times her nose runs continuously. Nasal symptoms have been present for 4 to 5 years but are worsening. The clear discharge is not associated with sneezing or itching. Though she lives with a cat, her symptoms are not exacerbated by close contact with it.
One year ago, an allergist performed skin testing but found no evidence of allergies as a cause of her rhinitis. A short course of intranasal steroids did not seem to improve her nasal symptoms.
The patient also has hypertension, hypothyroidism, and hot flashes due to menopause; these conditions are well controlled with lisinopril (Zestril), levothyroxine (Synthroid), and estrogen replacement. She has no history of asthma and has had no allergies to drugs, including nonsteroidal anti-inflammatory drugs (NSAIDs.)
How should this patient be evaluated and treated?
COMMON, OFTEN OVERLOOKED
Many patients suffer from rhinitis, but this problem can be overshadowed by other chronic diseases seen in a medical clinic, especially during a brief office visit. When a patient presents with rhinitis, a key question is whether it is allergic or nonallergic.
This review will discuss the different forms of nonallergic rhinitis and their causes, and give recommendations about therapy.
RHINITIS: ALLERGIC OR NONALLERGIC?
While allergic rhinitis affects 30 and 60 million Americans annually, or between 10% to 30% of US adults,1 how many have nonallergic rhinitis has been difficult to determine.
In a study in allergy clinics, 23% of patients with rhinitis had the nonallergic form, 43% had the allergic form, and 34% had both forms (mixed rhinitis).2 Other studies have suggested that up to 52% of patients presenting to allergy clinics with rhinitis have nonallergic rhinitis.3
Over time, patients may not stay in the same category. One study found that 24% of patients originally diagnosed with nonallergic rhinitis developed positive allergy tests when retested 3 or more years after their initial evaluation.4
Regardless of the type, untreated or uncontrolled symptoms of rhinitis can significantly affect the quality of life.
All forms of rhinitis are characterized by one or more of the following symptoms: nasal congestion, clear rhinorrhea, sneezing, and itching. These symptoms can be episodic or chronic and can range from mild to debilitating. In addition, rhinitis can lead to systemic symptoms of fatigue, headache, sleep disturbance, and cognitive impairment and can be associated with respiratory symptoms such as sinusitis and asthma.1
Mechanisms are mostly unknown
While allergic rhinitis leads to symptoms when airborne allergens bind with specific immunoglobulin E (IgE) in the nose, the etiology of most forms of nonallergic rhinitis is unknown. However, several mechanisms have been proposed. These include entopy (local nasal IgE synthesis with negative skin tests),5 nocioceptive dysfunction (hyperactive sensory receptors),6 and autonomic nervous system abnormalities (hypoactive or hyperactive dysfunction of sympathetic or parasympathetic nerves in the nose).7
Does this patient have an allergic cause of rhinitis?
When considering a patient with rhinitis, the most important question is, “Does this patient have an allergic cause of rhinitis?” Allergic and nonallergic rhinitis have similar symptoms, making them difficult to distinguish. However, their mechanisms and treatment differ. By categorizing a patient’s type of rhinitis, the physician can make specific recommendations for avoidance and can initiate treatment with the most appropriate therapy. Misclassification can lead to treatment failure, multiple visits, poor adherence, and frustration for patients with uncontrolled symptoms.
Patients for whom an allergic cause cannot be found by allergy skin testing or serum specific IgE immunoassay (Immunocap/RAST) for environmental aeroallergens are classified as having nonallergic rhinitis.
CLUES POINTING TO NONALLERGIC VS ALLERGIC RHINITIS
Nonallergic rhinitis encompasses a range of syndromes with overlapping symptoms. While tools such as the Rhinitis Diagnostic Worksheet are available to help differentiate allergic from nonallergic rhinitis, debate continues about whether it is necessary to characterize different forms of rhinitis before initiating treatment.8
The diagnosis of nonallergic rhinitis depends on a thorough history and physical examination. Key questions relate to the triggers that bring on the rhinitis, which will assist the clinician in determining which subtype of rhinitis a patient may be experiencing and therefore how to manage it. Clues:
- Patients with nonallergic rhinitis more often report nasal congestion and rhinorrhea, rather than sneezing and itching, which are predominant symptoms of allergic rhinitis.
- Patients with nonallergic rhinitis tend to develop symptoms at a later age.
- Common triggers of nonallergic rhinitis are changes in weather and temperature, food, perfumes, odors, smoke, and fumes. Animal exposure does not lead to symptoms.
- Patients with nonallergic rhinitis have few complaints of concomitant symptoms of allergic conjunctivitis (itching, watering, redness, and swelling).
- Many patients with nonallergic rhinitis find that antihistamines have no benefit. Also, they do not have other atopic diseases such as eczema or food allergies and have no family history of atopy.
PHYSICAL FINDINGS
Some findings on physical examination may help distinguish allergic from nonallergic rhinitis.
- Patients with long-standing allergic rhinitis may have an “allergic crease,” ie, a horizontal wrinkle near the tip of the nose caused by frequent upward wiping. Another sign may be a gothic arch, which is a narrowing of the hard palate occurring as a child.
- In allergic rhinitis, the turbinates are often pale, moist, and boggy with a bluish tinge.


CASE CONTINUED
Our patient’s symptoms can be caused by many different factors. Allergic triggers for rhinitis include both indoor and outdoor sources. The most common allergens include cat, dog, dust mite, cockroach, mold, and pollen allergens. The absence of acute sneezing and itching when around her cat and her recent negative skin-prick tests confirm that the rhinitis symptoms are not allergic.
In this patient, who has symptoms throughout the year but no allergic triggers, consideration of the different subtypes of nonallergic rhinitis may help guide further therapy.
SUBTYPES OF NONALLERGIC RHINITIS
Vasomotor rhinitis
Vasomotor rhinitis is thought to be caused by a variety of neural and vascular triggers, often without an inflammatory cause. These triggers lead to symptoms involving nasal congestion and clear rhinorrhea more than sneezing and itching. The symptoms can be sporadic, with acute onset in relation to identifiable nonallergic triggers, or chronic, with no clear trigger.
Gustatory rhinitis, for example, is a form of vasomotor rhinitis in which clear rhinorrhea occurs suddenly while eating or while drinking alcohol. It may be prevented by using nasal ipratropium (Atrovent) before meals.
Irritant-sensitive vasomotor rhinitis. In some patients, acute vasomotor rhinitis symptoms are brought on by strong odors, cigarette smoke, air pollution, or perfume. When asked, most patients easily identify which of these irritant triggers cause symptoms.
Weather- or temperature-sensitive vasomotor rhinitis. In other patients, a change in temperature, humidity, or barometric pressure or exposure to cold or dry air can cause nasal symptoms.9 These triggers are often hard to identify. Weather- or temperature-sensitive vasomotor rhinitis is often mistaken for seasonal allergic rhinitis because weather changes occur in close relation to the peak allergy seasons in the spring and fall. However, this subtype does not respond as well to intranasal steroids.9
Other nonallergic triggers of vasomotor rhinitis may include exercise, emotion, and sexual arousal (honeymoon rhinitis).10
Some triggers, such as tobacco smoke and perfume, are easy to avoid. Other triggers, such as weather changes, are unavoidable. If avoidance measures fail or are inadequate, medications (described below) can be used for prophylaxis and symptomatic treatment.
Drug-induced rhinitis
Drugs of various classes are known to cause either acute or chronic rhinitis. Drug-induced rhinitis has been divided into different types based on the mechanism involved.11
The local inflammatory type occurs in aspirin-exacerbated respiratory disease, which is characterized by nasal polyposis with chronic rhinosinusitis, hyposmia, and moderate to severe persistent asthma. Aspirin and other NSAIDs induce an acute local inflammation, leading to severe rhinitis and asthma symptoms. Avoiding all NSAID products is recommended; aspirin desensitization may lead to improvement in rhinosinusitis and asthma control.
The neurogenic type of drug-induced rhinitis can occur with sympatholytic drugs such as alpha receptor agonists (eg, clonidine [Cat-apres]) and antagonists (eg, prazosin [Minipress]).11 Vasodilators, including phosphodiesterase-5 inhibitors such as sildenafil (Viagra), can lead to acute rhinitis symptoms (“anniversary rhinitis”).
Unknown mechanisms. Many other medications can lead to rhinitis by unknown mechanisms, usually with normal findings on physical examination. These include beta-blockers, angiotensin-converting enzyme inhibitors, calcium channel blockers, exogenous estrogens, oral contraceptives, antipsychotics, and gabapentin (Neurontin).
Correlating the initiation of a drug with the onset of rhinitis can help identify offending medications. Stopping the suspected medication, if feasible, is the first-line treatment.
Rhinitis medicamentosa, typically caused by overuse of over-the-counter topical nasal decongestants, is also classified under drug-induced rhinitis. Patients may not think of nasal decongestants as medications, and the physician may need to ask specifically about their use.
On examination, the nasal mucosa appears beefy red without mucous. Once a diagnosis is made, the physician should identify and treat the original etiology of the nasal congestion that led the patient to self-treat.
Patients with rhinitis medicamentosa often have difficulty discontinuing use of topical decongestants. They should be educated that the withdrawal symptoms can be severe and that more than one attempt at quitting may be needed. To break the cycle of rebound congestion, topical intranasal steroids should be used, though 5 to 7 days of oral steroids may be necessary.1
Cocaine is a potent vasoconstrictor. Its illicit use should be suspected, especially if the patient presents with symptoms of chronic irritation such as frequent nosebleeds, crusting, and scabbing.12
Infectious rhinitis
One of the most common causes of acute rhinitis is upper respiratory infection.
Acute viral upper respiratory infection often presents with thick nasal discharge, sneezing, and nasal obstruction that usually clears in 7 to 10 days but can last up to 3 weeks. Acute bacterial sinusitis can follow, typically in fewer than 2% of patients, with symptoms of persistent nasal congestion, discolored mucus, facial pain, cough, and sometimes fever.
Chronic rhinosinusitis is a syndrome with sinus mucosal inflammation with multiple causes. It is clinically defined as persistent nasal and sinus symptoms lasting longer than 12 weeks and confirmed with computed tomography (CT).13 The CT findings of chronic rhinosinusitis include thickening of the lining of the sinus cavities or complete opacification of the pneumatized sinuses.
Major symptoms to consider for diagnosis include facial pain, congestion, obstruction, purulent discharge on examination, and changes in olfaction. Minor symptoms are cough, fatigue, headache, halitosis, fever, ear symptoms, and dental pain.
Treatment may involve 3 or more weeks of an oral antibiotic and a short course of an oral steroid, a daily nasal steroid spray, or both oral and nasal steroids. Most patients can be managed in the primary care setting, but they can be referred to an ear, nose, and throat specialist, an allergist, or an immunologist if their symptoms do not respond to initial therapy.
Nonallergic rhinitis eosinophilic syndrome
Patients with nonallergic rhinitis eosinophilic syndrome (NARES) are typically middle-aged and have perennial symptoms of sneezing, itching, and rhinorrhea with intermittent exacerbations. They occasionally have associated hyposmia (impaired sense of smell).1 The diagnosis is made when eosinophils account for more than 5% of cells on a nasal smear and allergy testing is negative.
Patients may develop nasal polyposis and aspirin sensitivity.1 Entopy has been described in some.14
Because of the eosinophilic inflammation, this form of nonallergic rhinitis responds well to intranasal steroids.
Immunologic causes
Systemic diseases can affect the nose and cause variable nasal symptoms that can be mistaken for rhinitis. Wegener granulomatosis, sarcoidosis, relapsing polychondritis, midline granulomas, Churg-Strauss syndrome, and amyloidosis can all affect the structures in the nose even before manifesting systemic symptoms. Granulomatous infections in the nose may lead to crusting, bleeding, and nasal obstruction.1
A lack of a response to intranasal steroids or oral antibiotics should lead to consideration of these conditions, and treatment should be tailored to the specific disease.
Occupational rhinitis
Occupational exposure to chemicals, biologic aerosols, flour, and latex can lead to rhinitis, typically through an inflammatory mechanism. Many patients present with associated occupational asthma. The symptoms improve when the patient is away from work and worsen throughout the work week.
Avoiding the triggering agent is necessary to treat these symptoms.
Hormonal rhinitis
Hormonal rhinitis, ie, rhinitis related to metabolic and endocrine conditions, is most commonly associated with high estrogen states. Nasal congestion has been reported with pregnancy, menses, menarche, and the use of oral contraceptives.15 The mechanism for congestion in these conditions still needs clarification.
When considering drug therapy, only intranasal budesonide (Rhinocort) has a pregnancy category B rating.
While hypothyroidism and acromegaly have been mentioned in reviews of nonallergic rhinitis, evidence that these disorders cause nonallergic rhinitis is not strong.16,17
Structurally related rhinitis
Anatomic abnormalities that can cause persistent nasal congestion include nasal septal deviation, turbinate hypertrophy, enlarged adenoids, tumors, and foreign bodies. These can be visualized by simple anterior nasal examination, nasal endoscopy, or radiologic studies. If structural causes lead to impaired quality of life or chronic rhinosinusitis, then consider referral to a specialist for possible surgical treatment.
Clear spontaneous rhinorrhea, with or without trauma, can be caused by cerebrospinal fluid leaking into the nasal cavity.18 A salty, metallic taste in the mouth can be a clue that the fluid is cerebrospinal fluid. A definitive diagnosis of cerebrospinal fluid leak is made by finding beta-2-transferrin in nasal secretions.
Atrophic rhinitis
Atrophic rhinitis is categorized as primary or secondary.
Primary (idiopathic) atrophic rhinitis is characterized by atrophy of the nasal mucosa and mucosal colonization with Klebsiella ozaenae associated with a foul-smelling nasal discharge.19,20 This disorder has been primarily reported in young people who present with nasal obstruction, dryness, crusting, and epistaxis. They are from areas with warm climates, such as the Middle East, Southeast Asia, India, Africa, and the Mediterranean.
Secondary atrophic rhinitis can be a complication of nasal or sinus surgery, trauma, granulomatous disease, or exposure to radiation.21 This disorder is typically diagnosed with nasal endoscopy and treated with daily saline rinses with or without topical antibiotics.21
CASE CONTINUED
Questioned further, our patient says her symptoms are worse when her husband smokes, but that she continues to have congestion and rhinorrhea when he is away on business trips. She notes that her symptoms are often worse on airplanes (dry air with an acute change in barometric pressure), with weather changes, and in cold, dry environments. Symptoms are not induced by eating.
We note that she started taking lisinopril 2 years ago and conjugated equine estrogens 8 years ago. Review of systems reveals no history of facial or head trauma, polyps, or hyposmia.
The rhinitis and congestion are bilateral, and she denies headaches, acid reflux, and conjunctivitis. She has a mild throat-clearing cough that she attributes to postnasal drip.
On physical examination, her blood pressure is 118/76 mm Hg and her pulse is 64. Her turbinates are congested with clear rhinorrhea. The rest of the examination is normal.
AVOID TRIGGERS, PRETREAT BEFORE EXPOSURE
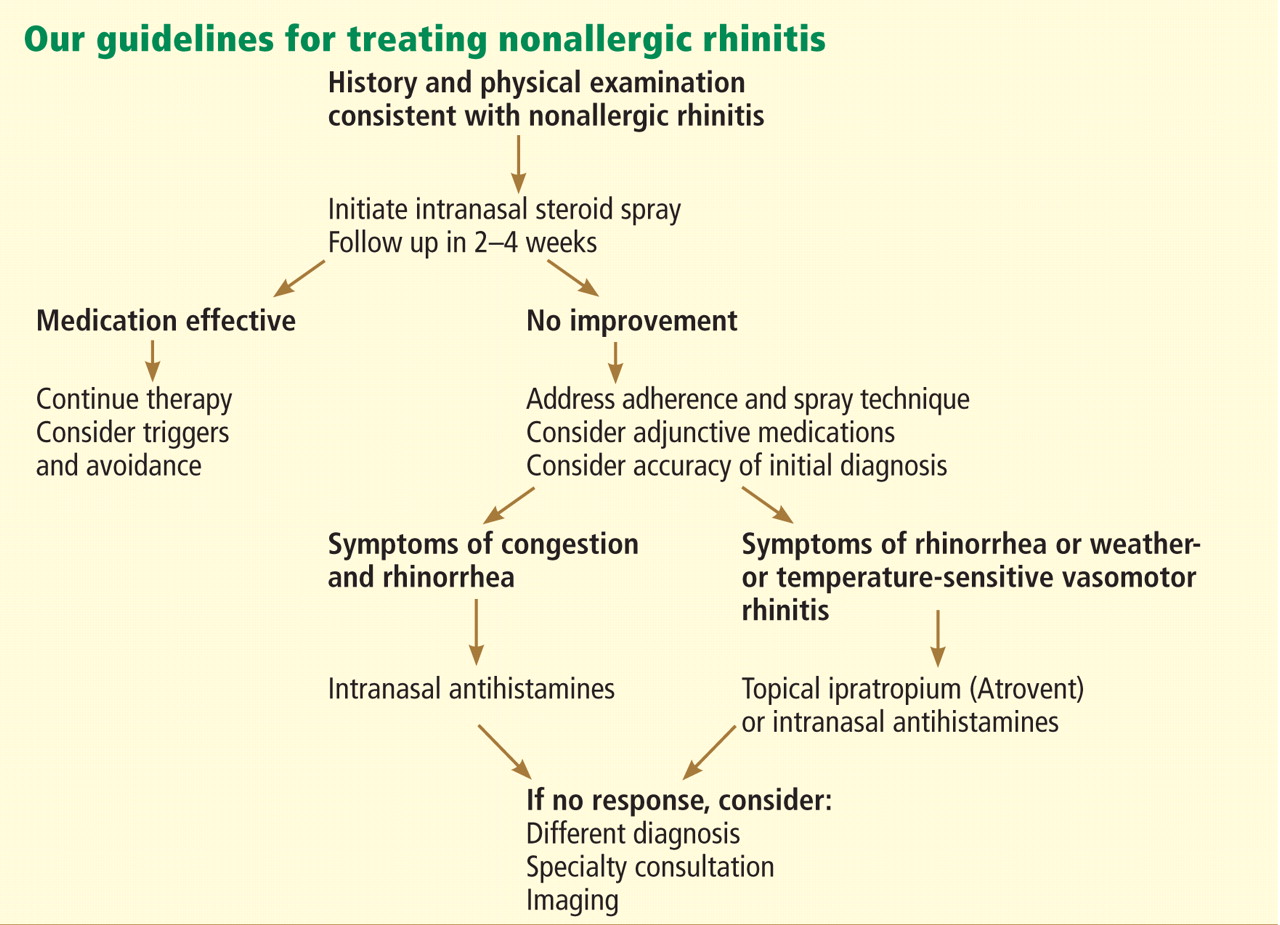
People with known environmental, non-immunologic, and irritant triggers should be reminded to avoid these exposures if possible.
If triggers are unavoidable, patients can pretreat themselves with topical nasal sprays before exposure. For example, if symptoms occur while on airplanes, then intranasal steroids or antihistamine sprays should be used before getting on the plane.
Many drugs available
Fortunately, many effective drugs are available to treat nonallergic rhinitis. These have few adverse effects or drug interactions.
Intranasal steroid sprays are considered first-line therapy, as there are studies demonstrating effectiveness in nonallergic rhinitis.22 Intranasal fluticasone propionate (Flonase) and beclomethasone dipropionate (Beconase AQ) are approved by the US Food and Drug Administration (FDA) for treating nonallergic rhinitis. Intranasal mometasone (Nasonex) is approved for treating nasal polyps.
Nasal steroid sprays are most effective if the dominant nasal symptom is congestion, but they have also shown benefit for rhinorrhea, sneezing, and itching.
Side effects of nasal steroid sprays include nasal irritation (dryness, burning, and stinging) and epistaxis, the latter occurring in 5% to 10% of patients.23
Intranasal antihistamines include azelastine (Astelin, Astepro) and olopatadine (Patanase). They are particularly useful for treating sneezing, congestion, and rhinorrhea.24 Astelin is the only intranasal antihistamine with FDA approval for nonallergic rhinitis.
Side effects of this drug class include bitter taste (with Astelin), sweet taste (with Astepro), headache, and somnolence.
Oral antihistamines such as loratadine (Claritin), cetirizine (Zyrtec), and fexofenadine (Allegra) are now available over the counter, and many patients try them before seeking medical care. These drugs may be helpful for those bothered by sneezing. However, no study has demonstrated their effectiveness for nonallergic rhinitis.25 First-generation antihistamines may help with rhinorrhea via their anticholinergic effects.
Ipratropium, an antimuscarinic agent, decreases secretions by inhibiting the nasal parasympathetic mucous glands. Intranasal ipratropium 0.03% (Atrovent 0.03%) should be considered first-line if the dominant symptom is rhinorrhea. Higher-dose ipratropium 0.06% is approved for rhinorrhea related to the common cold or allergic rhinitis. Because it is used topically, little is absorbed. Its major side effect is nasal dryness.
Decongestants, either oral or topical, can relieve the symptoms of congestion and rhinorrhea in nonallergic rhinitis. They should only be used short-term, as there is little evidence to support their chronic use.
Phenylpropanolamine, a decongestant previously found in over-the-counter cough medicines, was withdrawn from the market in 2000 owing to concern that the drug, especially when used for weight suppression, was linked to hemorrhagic stroke in young women.26,27 Other oral decongestants, ie, pseudoephedrine and phenylephrine, are still available, but there are no definitive guidelines for their use. Their side effects include tachycardia, increase in blood pressure, and insomnia.
Nasal saline irrigation has been used for centuries to treat rhinitis and sinusitis, despite limited evidence of benefit. A Cochrane review concluded that saline irrigation was well tolerated, had minor side effects, and could provide some relief of rhinosinusitis symptoms either as the sole therapeutic measure or as adjunctive treatment.28 Hypertonic saline solutions, while possibly more effective than isotonic saline in improving mucociliary clearance, are not as well tolerated since they can cause nasal burning and irritation. Presumed benefits of saline irrigation are clearance of nasal secretions, improvement of nasociliary function, and removal of irritants and pollen from the nose.
A strategy
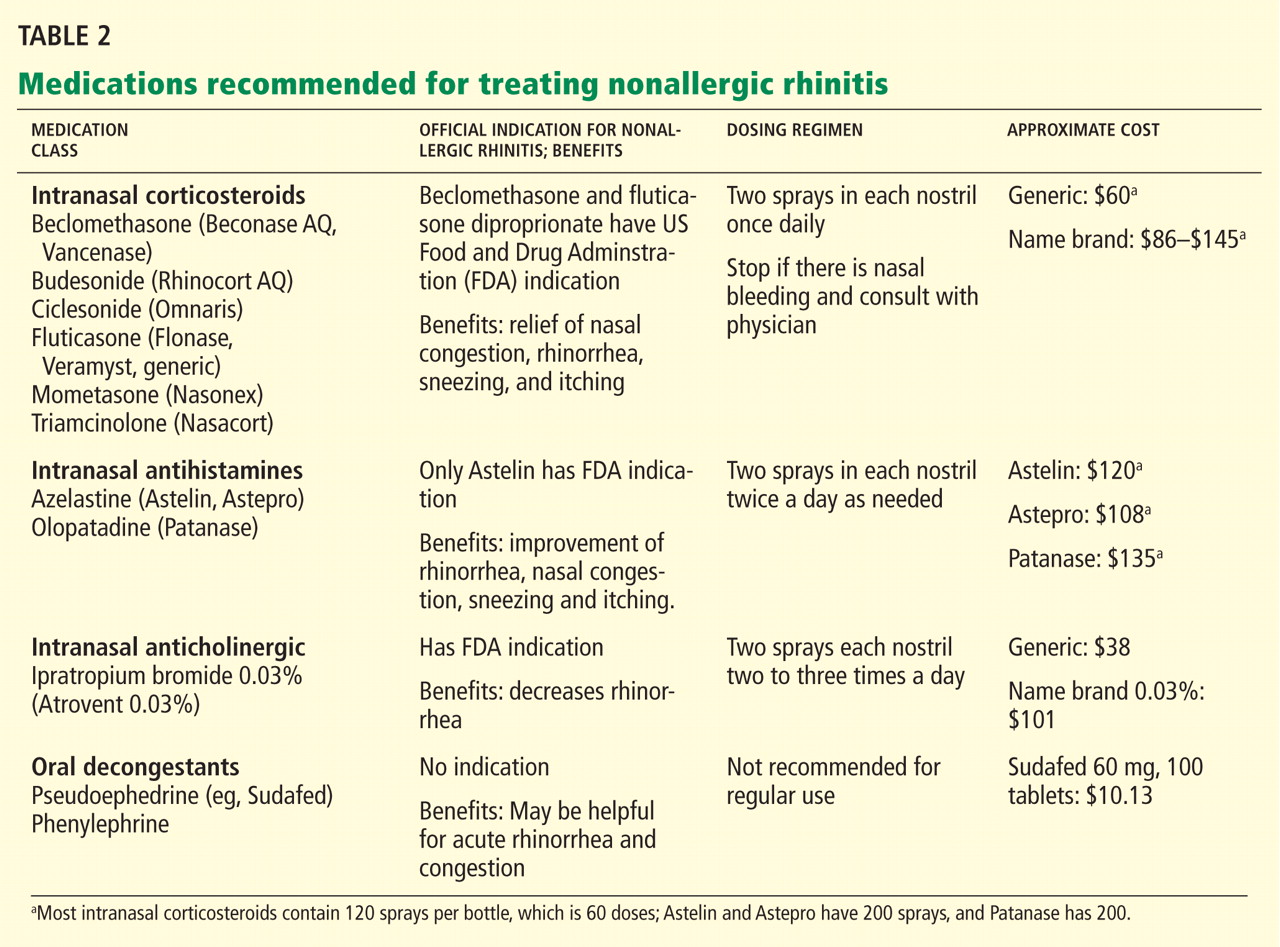
Imaging the sinuses with CT, which has replaced standard nasal radiography, may help if one is concerned about chronic rhinosinusitis, nasal polyps, or other anatomic condition that could contribute to persistent symptoms. Cost and radiation exposure should enter into the decision to obtain this study because a diagnosis based on the patient’s report of symptoms may be equally accurate.29,30
CASE CONTINUED
Our patient has a number of potential causes of her symptoms. Exposure to second-hand tobacco smoke at home and to the air in airplanes could be acute triggers. Weather and temperature changes could explain her chronic symptoms in the spring and fall. Use of an angiotensin-converting enzyme inhibitor (in her case, lisinopril) and estrogen replacement therapy may contribute to perennial symptoms, but the onset of her nonallergic rhinitis does not correlate with the use of these drugs. There are no symptoms to suggest chronic rhinosinusitis or anatomic causes of her symptoms.
This case is typical of vasomotor rhinitis of the weather- or temperature-sensitive type. This diagnosis may explain her lack of improvement with intranasal steroids, though adherence and spray technique should be assessed. At this point, we would recommend trying topical antihistamines daily when chronic symptoms are present or as needed for acute symptoms.
- Wallace DV, Dykewicz MS, Bernstein DI, et al. The diagnosis and management of rhinitis: an updated practice parameter. J Allergy Clin Immunol 2008; 122( suppl 2):S1–S84.
- Settipane RA, Charnock DR. Epidemiology of rhinitis: allergic and nonallergic. Clin Allergy Immunol 2007; 19:23–34.
- Settipane RA, Lieberman P. Update on nonallergic rhinitis. Ann Allergy Asthma Immunol 2001; 86:494–507.
- Rondón C, Doña I, Torres MJ, Campo P, Blanca M. Evolution of patients with nonallergic rhinitis supports conversion to allergic rhinitis. J Allergy Clin Immunol 2009; 123:1098–1102.
- Forester JP, Calabria CW. Local production of IgE in the respiratory mucosa and the concept of entopy: does allergy exist in nonallergic rhinitis? Ann Allergy Asthma Immunol 2010; 105:249–255.
- Silvers WS. The skier’s nose: a model of cold-induced rhinorrhea. Ann Allergy 1991; 67:32–36.
- Jaradeh SS, Smith TL, Torrico L, et al. Autonomic nervous system evaluation of patients with vasomotor rhinitis. Laryngoscope 2000; 110:1828–1831.
- Quan M, Casale TB, Blaiss MS. Should clinicians routinely determine rhinitis subtype on initial diagnosis and evaluation? A debate among experts. Clin Cornerstone 2009; 9:54–60.
- Jacobs R, Lieberman P, Kent E, Silvey M, Locantore N, Philpot EE. Weather/temperature-sensitive vasomotor rhinitis may be refractory to intranasal corticosteroid treatment. Allergy Asthma Proc 2009; 30:120–127.
- Monteseirin J, Camacho MJ, Bonilla I, Sanchez-Hernandez C, Hernandez M, Conde J. Honeymoon rhinitis. Allergy 2001; 56:353–354.
- Varghese M, Glaum MC, Lockey RF. Drug-induced rhinitis. Clin Exp Allergy 2010; 40:381–384.
- Schwartz RH, Estroff T, Fairbanks DN, Hoffmann NG. Nasal symptoms associated with cocaine abuse during adolescence. Arch Otolaryngol Head Neck Surg 1989; 115:63–64.
- Meltzer EO, Hamilos DL, Hadley JA, et al; American Academy of Allergy, Asthma and Immunology (AAAAI); American Academy of Otolaryngic Allergy (AAOA); American Academy of Otolaryngology--Head and Neck Surgery (AAO-HNS); American College of Allergy, Asthma and Immunology (ACAAI); American Rhinologic Society (ARS). Rhinosinusitis: establishing definitions for clinical research and patient care. J Allergy Clin Immunol 2004; 114( suppl 6):155–212.
- Powe DG, Huskisson RS, Carney AS, Jenkins D, Jones NS. Evidence for an inflammatory pathophysiology in idiopathic rhinitis. Clin Exp Allergy 2001; 31:864–872.
- Philpott CM, Robinson AM, Murty GE. Nasal pathophysiology and its relationship to the female ovarian hormones. J Otolaryngol Head Neck Surg 2008; 37:540–546.
- Dykewicz MS, Fineman S, Skoner DP, et al. Diagnosis and management of rhinitis: complete guidelines of the Joint Task Force on Practice Parameters in Allergy, Asthma and Immunology. American Academy of Allergy, Asthma, and Immunology. Ann Allergy Asthma Immunol 1998; 81:478–518.
- Ellegård EK, Karlsson NG, Ellegård LH. Rhinitis in the menstrual cycle, pregnancy, and some endocrine disorders. Clin Allergy Immunol 2007; 19:305–321.
- Dunn CJ, Alaani A, Johnson AP. Study on spontaneous cerebrospinal fluid rhinorrhoea: its aetiology and management. J Laryngol Otol 2005; 119:12–15.
- Bunnag C, Jareoncharsri P, Tansuriyawong P, Bhothisuwan W, Chantarakul N. Characteristics of atrophic rhinitis in Thai patients at the Siriraj Hospital. Rhinology 1999; 37:125–130.
- Dutt SN, Kameswaran M. The aetiology and management of atrophic rhinitis. J Laryngol Otol 2005; 119:843–852.
- deShazo RD, Stringer SP. Atrophic rhinosinusitis: progress toward explanation of an unsolved medical mystery. Curr Opin Allergy Clin Immunol 2011; 11:1–7.
- Greiner AN, Meltzer EO. Overview of the treatment of allergic rhinitis and nonallergic rhinopathy. Proc Am Thorac Soc 2011; 8:121–131.
- Corren J. Intranasal corticosteroids for allergic rhinitis: how do different agents compare? J Allergy Clin Immunol 1999; 104:S144–S149.
- Lieberman P, Meltzer EO, LaForce CF, Darter AL, Tort MJ. Two-week comparison study of olopatadine hydrochloride nasal spray 0.6% versus azelastine hydrochloride nasal spray 0.1% in patients with vasomotor rhinitis. Allergy Asthma Proc 2011; 32:151–158.
- Bousquet J, Khaltaev N, Cruz AA, et al; World Health Organization; GA(2)LEN. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008 update (in collaboration with the World Health Organization, GA(2)LEN and AllerGen). Allergy 2008; 63( suppl 86):8–160.
- SoRelle R. FDA warns of stroke risk associated with phenylpropanolamine; cold remedies and drugs removed from store shelves. Circulation 2000; 102:E9041–E9043.
- Kernan WN, Viscoli CM, Brass LM, et al. Phenylpropanolamine and the risk of hemorrhagic stroke. N Engl J Med 2000; 343:1826–1832.
- Harvey R, Hannan SA, Badia L, Scadding G. Nasal saline irrigations for the symptoms of chronic rhinosinusitis. Cochrane Database Syst Rev 2007;CD006394.
- Bhattacharyya N. The role of CT and MRI in the diagnosis of chronic rhinosinusitis. Curr Allergy Asthma Rep 2010; 10:171–174.
- Kenny TJ, Duncavage J, Bracikowski J, Yildirim A, Murray JJ, Tanner SB. Prospective analysis of sinus symptoms and correlation with paranasal computed tomography scan. Otolaryngol Head Neck Surg 2001; 125:40–43.
A 55-year-old woman has come to the clinic because of clear rhinorrhea and nasal congestion, which occur year-round but are worse in the winter. She reports that at times her nose runs continuously. Nasal symptoms have been present for 4 to 5 years but are worsening. The clear discharge is not associated with sneezing or itching. Though she lives with a cat, her symptoms are not exacerbated by close contact with it.
One year ago, an allergist performed skin testing but found no evidence of allergies as a cause of her rhinitis. A short course of intranasal steroids did not seem to improve her nasal symptoms.
The patient also has hypertension, hypothyroidism, and hot flashes due to menopause; these conditions are well controlled with lisinopril (Zestril), levothyroxine (Synthroid), and estrogen replacement. She has no history of asthma and has had no allergies to drugs, including nonsteroidal anti-inflammatory drugs (NSAIDs.)
How should this patient be evaluated and treated?
COMMON, OFTEN OVERLOOKED
Many patients suffer from rhinitis, but this problem can be overshadowed by other chronic diseases seen in a medical clinic, especially during a brief office visit. When a patient presents with rhinitis, a key question is whether it is allergic or nonallergic.
This review will discuss the different forms of nonallergic rhinitis and their causes, and give recommendations about therapy.
RHINITIS: ALLERGIC OR NONALLERGIC?
While allergic rhinitis affects 30 and 60 million Americans annually, or between 10% to 30% of US adults,1 how many have nonallergic rhinitis has been difficult to determine.
In a study in allergy clinics, 23% of patients with rhinitis had the nonallergic form, 43% had the allergic form, and 34% had both forms (mixed rhinitis).2 Other studies have suggested that up to 52% of patients presenting to allergy clinics with rhinitis have nonallergic rhinitis.3
Over time, patients may not stay in the same category. One study found that 24% of patients originally diagnosed with nonallergic rhinitis developed positive allergy tests when retested 3 or more years after their initial evaluation.4
Regardless of the type, untreated or uncontrolled symptoms of rhinitis can significantly affect the quality of life.
All forms of rhinitis are characterized by one or more of the following symptoms: nasal congestion, clear rhinorrhea, sneezing, and itching. These symptoms can be episodic or chronic and can range from mild to debilitating. In addition, rhinitis can lead to systemic symptoms of fatigue, headache, sleep disturbance, and cognitive impairment and can be associated with respiratory symptoms such as sinusitis and asthma.1
Mechanisms are mostly unknown
While allergic rhinitis leads to symptoms when airborne allergens bind with specific immunoglobulin E (IgE) in the nose, the etiology of most forms of nonallergic rhinitis is unknown. However, several mechanisms have been proposed. These include entopy (local nasal IgE synthesis with negative skin tests),5 nocioceptive dysfunction (hyperactive sensory receptors),6 and autonomic nervous system abnormalities (hypoactive or hyperactive dysfunction of sympathetic or parasympathetic nerves in the nose).7
Does this patient have an allergic cause of rhinitis?
When considering a patient with rhinitis, the most important question is, “Does this patient have an allergic cause of rhinitis?” Allergic and nonallergic rhinitis have similar symptoms, making them difficult to distinguish. However, their mechanisms and treatment differ. By categorizing a patient’s type of rhinitis, the physician can make specific recommendations for avoidance and can initiate treatment with the most appropriate therapy. Misclassification can lead to treatment failure, multiple visits, poor adherence, and frustration for patients with uncontrolled symptoms.
Patients for whom an allergic cause cannot be found by allergy skin testing or serum specific IgE immunoassay (Immunocap/RAST) for environmental aeroallergens are classified as having nonallergic rhinitis.
CLUES POINTING TO NONALLERGIC VS ALLERGIC RHINITIS
Nonallergic rhinitis encompasses a range of syndromes with overlapping symptoms. While tools such as the Rhinitis Diagnostic Worksheet are available to help differentiate allergic from nonallergic rhinitis, debate continues about whether it is necessary to characterize different forms of rhinitis before initiating treatment.8
The diagnosis of nonallergic rhinitis depends on a thorough history and physical examination. Key questions relate to the triggers that bring on the rhinitis, which will assist the clinician in determining which subtype of rhinitis a patient may be experiencing and therefore how to manage it. Clues:
- Patients with nonallergic rhinitis more often report nasal congestion and rhinorrhea, rather than sneezing and itching, which are predominant symptoms of allergic rhinitis.
- Patients with nonallergic rhinitis tend to develop symptoms at a later age.
- Common triggers of nonallergic rhinitis are changes in weather and temperature, food, perfumes, odors, smoke, and fumes. Animal exposure does not lead to symptoms.
- Patients with nonallergic rhinitis have few complaints of concomitant symptoms of allergic conjunctivitis (itching, watering, redness, and swelling).
- Many patients with nonallergic rhinitis find that antihistamines have no benefit. Also, they do not have other atopic diseases such as eczema or food allergies and have no family history of atopy.
PHYSICAL FINDINGS
Some findings on physical examination may help distinguish allergic from nonallergic rhinitis.
- Patients with long-standing allergic rhinitis may have an “allergic crease,” ie, a horizontal wrinkle near the tip of the nose caused by frequent upward wiping. Another sign may be a gothic arch, which is a narrowing of the hard palate occurring as a child.
- In allergic rhinitis, the turbinates are often pale, moist, and boggy with a bluish tinge.
CASE CONTINUED
Our patient’s symptoms can be caused by many different factors. Allergic triggers for rhinitis include both indoor and outdoor sources. The most common allergens include cat, dog, dust mite, cockroach, mold, and pollen allergens. The absence of acute sneezing and itching when around her cat and her recent negative skin-prick tests confirm that the rhinitis symptoms are not allergic.
In this patient, who has symptoms throughout the year but no allergic triggers, consideration of the different subtypes of nonallergic rhinitis may help guide further therapy.
SUBTYPES OF NONALLERGIC RHINITIS
Vasomotor rhinitis
Vasomotor rhinitis is thought to be caused by a variety of neural and vascular triggers, often without an inflammatory cause. These triggers lead to symptoms involving nasal congestion and clear rhinorrhea more than sneezing and itching. The symptoms can be sporadic, with acute onset in relation to identifiable nonallergic triggers, or chronic, with no clear trigger.
Gustatory rhinitis, for example, is a form of vasomotor rhinitis in which clear rhinorrhea occurs suddenly while eating or while drinking alcohol. It may be prevented by using nasal ipratropium (Atrovent) before meals.
Irritant-sensitive vasomotor rhinitis. In some patients, acute vasomotor rhinitis symptoms are brought on by strong odors, cigarette smoke, air pollution, or perfume. When asked, most patients easily identify which of these irritant triggers cause symptoms.
Weather- or temperature-sensitive vasomotor rhinitis. In other patients, a change in temperature, humidity, or barometric pressure or exposure to cold or dry air can cause nasal symptoms.9 These triggers are often hard to identify. Weather- or temperature-sensitive vasomotor rhinitis is often mistaken for seasonal allergic rhinitis because weather changes occur in close relation to the peak allergy seasons in the spring and fall. However, this subtype does not respond as well to intranasal steroids.9
Other nonallergic triggers of vasomotor rhinitis may include exercise, emotion, and sexual arousal (honeymoon rhinitis).10
Some triggers, such as tobacco smoke and perfume, are easy to avoid. Other triggers, such as weather changes, are unavoidable. If avoidance measures fail or are inadequate, medications (described below) can be used for prophylaxis and symptomatic treatment.
Drug-induced rhinitis
Drugs of various classes are known to cause either acute or chronic rhinitis. Drug-induced rhinitis has been divided into different types based on the mechanism involved.11
The local inflammatory type occurs in aspirin-exacerbated respiratory disease, which is characterized by nasal polyposis with chronic rhinosinusitis, hyposmia, and moderate to severe persistent asthma. Aspirin and other NSAIDs induce an acute local inflammation, leading to severe rhinitis and asthma symptoms. Avoiding all NSAID products is recommended; aspirin desensitization may lead to improvement in rhinosinusitis and asthma control.
The neurogenic type of drug-induced rhinitis can occur with sympatholytic drugs such as alpha receptor agonists (eg, clonidine [Cat-apres]) and antagonists (eg, prazosin [Minipress]).11 Vasodilators, including phosphodiesterase-5 inhibitors such as sildenafil (Viagra), can lead to acute rhinitis symptoms (“anniversary rhinitis”).
Unknown mechanisms. Many other medications can lead to rhinitis by unknown mechanisms, usually with normal findings on physical examination. These include beta-blockers, angiotensin-converting enzyme inhibitors, calcium channel blockers, exogenous estrogens, oral contraceptives, antipsychotics, and gabapentin (Neurontin).
Correlating the initiation of a drug with the onset of rhinitis can help identify offending medications. Stopping the suspected medication, if feasible, is the first-line treatment.
Rhinitis medicamentosa, typically caused by overuse of over-the-counter topical nasal decongestants, is also classified under drug-induced rhinitis. Patients may not think of nasal decongestants as medications, and the physician may need to ask specifically about their use.
On examination, the nasal mucosa appears beefy red without mucous. Once a diagnosis is made, the physician should identify and treat the original etiology of the nasal congestion that led the patient to self-treat.
Patients with rhinitis medicamentosa often have difficulty discontinuing use of topical decongestants. They should be educated that the withdrawal symptoms can be severe and that more than one attempt at quitting may be needed. To break the cycle of rebound congestion, topical intranasal steroids should be used, though 5 to 7 days of oral steroids may be necessary.1
Cocaine is a potent vasoconstrictor. Its illicit use should be suspected, especially if the patient presents with symptoms of chronic irritation such as frequent nosebleeds, crusting, and scabbing.12
Infectious rhinitis
One of the most common causes of acute rhinitis is upper respiratory infection.
Acute viral upper respiratory infection often presents with thick nasal discharge, sneezing, and nasal obstruction that usually clears in 7 to 10 days but can last up to 3 weeks. Acute bacterial sinusitis can follow, typically in fewer than 2% of patients, with symptoms of persistent nasal congestion, discolored mucus, facial pain, cough, and sometimes fever.
Chronic rhinosinusitis is a syndrome with sinus mucosal inflammation with multiple causes. It is clinically defined as persistent nasal and sinus symptoms lasting longer than 12 weeks and confirmed with computed tomography (CT).13 The CT findings of chronic rhinosinusitis include thickening of the lining of the sinus cavities or complete opacification of the pneumatized sinuses.
Major symptoms to consider for diagnosis include facial pain, congestion, obstruction, purulent discharge on examination, and changes in olfaction. Minor symptoms are cough, fatigue, headache, halitosis, fever, ear symptoms, and dental pain.
Treatment may involve 3 or more weeks of an oral antibiotic and a short course of an oral steroid, a daily nasal steroid spray, or both oral and nasal steroids. Most patients can be managed in the primary care setting, but they can be referred to an ear, nose, and throat specialist, an allergist, or an immunologist if their symptoms do not respond to initial therapy.
Nonallergic rhinitis eosinophilic syndrome
Patients with nonallergic rhinitis eosinophilic syndrome (NARES) are typically middle-aged and have perennial symptoms of sneezing, itching, and rhinorrhea with intermittent exacerbations. They occasionally have associated hyposmia (impaired sense of smell).1 The diagnosis is made when eosinophils account for more than 5% of cells on a nasal smear and allergy testing is negative.
Patients may develop nasal polyposis and aspirin sensitivity.1 Entopy has been described in some.14
Because of the eosinophilic inflammation, this form of nonallergic rhinitis responds well to intranasal steroids.
Immunologic causes
Systemic diseases can affect the nose and cause variable nasal symptoms that can be mistaken for rhinitis. Wegener granulomatosis, sarcoidosis, relapsing polychondritis, midline granulomas, Churg-Strauss syndrome, and amyloidosis can all affect the structures in the nose even before manifesting systemic symptoms. Granulomatous infections in the nose may lead to crusting, bleeding, and nasal obstruction.1
A lack of a response to intranasal steroids or oral antibiotics should lead to consideration of these conditions, and treatment should be tailored to the specific disease.
Occupational rhinitis
Occupational exposure to chemicals, biologic aerosols, flour, and latex can lead to rhinitis, typically through an inflammatory mechanism. Many patients present with associated occupational asthma. The symptoms improve when the patient is away from work and worsen throughout the work week.
Avoiding the triggering agent is necessary to treat these symptoms.
Hormonal rhinitis
Hormonal rhinitis, ie, rhinitis related to metabolic and endocrine conditions, is most commonly associated with high estrogen states. Nasal congestion has been reported with pregnancy, menses, menarche, and the use of oral contraceptives.15 The mechanism for congestion in these conditions still needs clarification.
When considering drug therapy, only intranasal budesonide (Rhinocort) has a pregnancy category B rating.
While hypothyroidism and acromegaly have been mentioned in reviews of nonallergic rhinitis, evidence that these disorders cause nonallergic rhinitis is not strong.16,17
Structurally related rhinitis
Anatomic abnormalities that can cause persistent nasal congestion include nasal septal deviation, turbinate hypertrophy, enlarged adenoids, tumors, and foreign bodies. These can be visualized by simple anterior nasal examination, nasal endoscopy, or radiologic studies. If structural causes lead to impaired quality of life or chronic rhinosinusitis, then consider referral to a specialist for possible surgical treatment.
Clear spontaneous rhinorrhea, with or without trauma, can be caused by cerebrospinal fluid leaking into the nasal cavity.18 A salty, metallic taste in the mouth can be a clue that the fluid is cerebrospinal fluid. A definitive diagnosis of cerebrospinal fluid leak is made by finding beta-2-transferrin in nasal secretions.
Atrophic rhinitis
Atrophic rhinitis is categorized as primary or secondary.
Primary (idiopathic) atrophic rhinitis is characterized by atrophy of the nasal mucosa and mucosal colonization with Klebsiella ozaenae associated with a foul-smelling nasal discharge.19,20 This disorder has been primarily reported in young people who present with nasal obstruction, dryness, crusting, and epistaxis. They are from areas with warm climates, such as the Middle East, Southeast Asia, India, Africa, and the Mediterranean.
Secondary atrophic rhinitis can be a complication of nasal or sinus surgery, trauma, granulomatous disease, or exposure to radiation.21 This disorder is typically diagnosed with nasal endoscopy and treated with daily saline rinses with or without topical antibiotics.21
CASE CONTINUED
Questioned further, our patient says her symptoms are worse when her husband smokes, but that she continues to have congestion and rhinorrhea when he is away on business trips. She notes that her symptoms are often worse on airplanes (dry air with an acute change in barometric pressure), with weather changes, and in cold, dry environments. Symptoms are not induced by eating.
We note that she started taking lisinopril 2 years ago and conjugated equine estrogens 8 years ago. Review of systems reveals no history of facial or head trauma, polyps, or hyposmia.
The rhinitis and congestion are bilateral, and she denies headaches, acid reflux, and conjunctivitis. She has a mild throat-clearing cough that she attributes to postnasal drip.
On physical examination, her blood pressure is 118/76 mm Hg and her pulse is 64. Her turbinates are congested with clear rhinorrhea. The rest of the examination is normal.
AVOID TRIGGERS, PRETREAT BEFORE EXPOSURE
People with known environmental, non-immunologic, and irritant triggers should be reminded to avoid these exposures if possible.
If triggers are unavoidable, patients can pretreat themselves with topical nasal sprays before exposure. For example, if symptoms occur while on airplanes, then intranasal steroids or antihistamine sprays should be used before getting on the plane.
Many drugs available
Fortunately, many effective drugs are available to treat nonallergic rhinitis. These have few adverse effects or drug interactions.
Intranasal steroid sprays are considered first-line therapy, as there are studies demonstrating effectiveness in nonallergic rhinitis.22 Intranasal fluticasone propionate (Flonase) and beclomethasone dipropionate (Beconase AQ) are approved by the US Food and Drug Administration (FDA) for treating nonallergic rhinitis. Intranasal mometasone (Nasonex) is approved for treating nasal polyps.
Nasal steroid sprays are most effective if the dominant nasal symptom is congestion, but they have also shown benefit for rhinorrhea, sneezing, and itching.
Side effects of nasal steroid sprays include nasal irritation (dryness, burning, and stinging) and epistaxis, the latter occurring in 5% to 10% of patients.23
Intranasal antihistamines include azelastine (Astelin, Astepro) and olopatadine (Patanase). They are particularly useful for treating sneezing, congestion, and rhinorrhea.24 Astelin is the only intranasal antihistamine with FDA approval for nonallergic rhinitis.
Side effects of this drug class include bitter taste (with Astelin), sweet taste (with Astepro), headache, and somnolence.
Oral antihistamines such as loratadine (Claritin), cetirizine (Zyrtec), and fexofenadine (Allegra) are now available over the counter, and many patients try them before seeking medical care. These drugs may be helpful for those bothered by sneezing. However, no study has demonstrated their effectiveness for nonallergic rhinitis.25 First-generation antihistamines may help with rhinorrhea via their anticholinergic effects.
Ipratropium, an antimuscarinic agent, decreases secretions by inhibiting the nasal parasympathetic mucous glands. Intranasal ipratropium 0.03% (Atrovent 0.03%) should be considered first-line if the dominant symptom is rhinorrhea. Higher-dose ipratropium 0.06% is approved for rhinorrhea related to the common cold or allergic rhinitis. Because it is used topically, little is absorbed. Its major side effect is nasal dryness.
Decongestants, either oral or topical, can relieve the symptoms of congestion and rhinorrhea in nonallergic rhinitis. They should only be used short-term, as there is little evidence to support their chronic use.
Phenylpropanolamine, a decongestant previously found in over-the-counter cough medicines, was withdrawn from the market in 2000 owing to concern that the drug, especially when used for weight suppression, was linked to hemorrhagic stroke in young women.26,27 Other oral decongestants, ie, pseudoephedrine and phenylephrine, are still available, but there are no definitive guidelines for their use. Their side effects include tachycardia, increase in blood pressure, and insomnia.
Nasal saline irrigation has been used for centuries to treat rhinitis and sinusitis, despite limited evidence of benefit. A Cochrane review concluded that saline irrigation was well tolerated, had minor side effects, and could provide some relief of rhinosinusitis symptoms either as the sole therapeutic measure or as adjunctive treatment.28 Hypertonic saline solutions, while possibly more effective than isotonic saline in improving mucociliary clearance, are not as well tolerated since they can cause nasal burning and irritation. Presumed benefits of saline irrigation are clearance of nasal secretions, improvement of nasociliary function, and removal of irritants and pollen from the nose.
A strategy
Imaging the sinuses with CT, which has replaced standard nasal radiography, may help if one is concerned about chronic rhinosinusitis, nasal polyps, or other anatomic condition that could contribute to persistent symptoms. Cost and radiation exposure should enter into the decision to obtain this study because a diagnosis based on the patient’s report of symptoms may be equally accurate.29,30
CASE CONTINUED
Our patient has a number of potential causes of her symptoms. Exposure to second-hand tobacco smoke at home and to the air in airplanes could be acute triggers. Weather and temperature changes could explain her chronic symptoms in the spring and fall. Use of an angiotensin-converting enzyme inhibitor (in her case, lisinopril) and estrogen replacement therapy may contribute to perennial symptoms, but the onset of her nonallergic rhinitis does not correlate with the use of these drugs. There are no symptoms to suggest chronic rhinosinusitis or anatomic causes of her symptoms.
This case is typical of vasomotor rhinitis of the weather- or temperature-sensitive type. This diagnosis may explain her lack of improvement with intranasal steroids, though adherence and spray technique should be assessed. At this point, we would recommend trying topical antihistamines daily when chronic symptoms are present or as needed for acute symptoms.
A 55-year-old woman has come to the clinic because of clear rhinorrhea and nasal congestion, which occur year-round but are worse in the winter. She reports that at times her nose runs continuously. Nasal symptoms have been present for 4 to 5 years but are worsening. The clear discharge is not associated with sneezing or itching. Though she lives with a cat, her symptoms are not exacerbated by close contact with it.
One year ago, an allergist performed skin testing but found no evidence of allergies as a cause of her rhinitis. A short course of intranasal steroids did not seem to improve her nasal symptoms.
The patient also has hypertension, hypothyroidism, and hot flashes due to menopause; these conditions are well controlled with lisinopril (Zestril), levothyroxine (Synthroid), and estrogen replacement. She has no history of asthma and has had no allergies to drugs, including nonsteroidal anti-inflammatory drugs (NSAIDs.)
How should this patient be evaluated and treated?
COMMON, OFTEN OVERLOOKED
Many patients suffer from rhinitis, but this problem can be overshadowed by other chronic diseases seen in a medical clinic, especially during a brief office visit. When a patient presents with rhinitis, a key question is whether it is allergic or nonallergic.
This review will discuss the different forms of nonallergic rhinitis and their causes, and give recommendations about therapy.
RHINITIS: ALLERGIC OR NONALLERGIC?
While allergic rhinitis affects 30 and 60 million Americans annually, or between 10% to 30% of US adults,1 how many have nonallergic rhinitis has been difficult to determine.
In a study in allergy clinics, 23% of patients with rhinitis had the nonallergic form, 43% had the allergic form, and 34% had both forms (mixed rhinitis).2 Other studies have suggested that up to 52% of patients presenting to allergy clinics with rhinitis have nonallergic rhinitis.3
Over time, patients may not stay in the same category. One study found that 24% of patients originally diagnosed with nonallergic rhinitis developed positive allergy tests when retested 3 or more years after their initial evaluation.4
Regardless of the type, untreated or uncontrolled symptoms of rhinitis can significantly affect the quality of life.
All forms of rhinitis are characterized by one or more of the following symptoms: nasal congestion, clear rhinorrhea, sneezing, and itching. These symptoms can be episodic or chronic and can range from mild to debilitating. In addition, rhinitis can lead to systemic symptoms of fatigue, headache, sleep disturbance, and cognitive impairment and can be associated with respiratory symptoms such as sinusitis and asthma.1
Mechanisms are mostly unknown
While allergic rhinitis leads to symptoms when airborne allergens bind with specific immunoglobulin E (IgE) in the nose, the etiology of most forms of nonallergic rhinitis is unknown. However, several mechanisms have been proposed. These include entopy (local nasal IgE synthesis with negative skin tests),5 nocioceptive dysfunction (hyperactive sensory receptors),6 and autonomic nervous system abnormalities (hypoactive or hyperactive dysfunction of sympathetic or parasympathetic nerves in the nose).7
Does this patient have an allergic cause of rhinitis?
When considering a patient with rhinitis, the most important question is, “Does this patient have an allergic cause of rhinitis?” Allergic and nonallergic rhinitis have similar symptoms, making them difficult to distinguish. However, their mechanisms and treatment differ. By categorizing a patient’s type of rhinitis, the physician can make specific recommendations for avoidance and can initiate treatment with the most appropriate therapy. Misclassification can lead to treatment failure, multiple visits, poor adherence, and frustration for patients with uncontrolled symptoms.
Patients for whom an allergic cause cannot be found by allergy skin testing or serum specific IgE immunoassay (Immunocap/RAST) for environmental aeroallergens are classified as having nonallergic rhinitis.
CLUES POINTING TO NONALLERGIC VS ALLERGIC RHINITIS
Nonallergic rhinitis encompasses a range of syndromes with overlapping symptoms. While tools such as the Rhinitis Diagnostic Worksheet are available to help differentiate allergic from nonallergic rhinitis, debate continues about whether it is necessary to characterize different forms of rhinitis before initiating treatment.8
The diagnosis of nonallergic rhinitis depends on a thorough history and physical examination. Key questions relate to the triggers that bring on the rhinitis, which will assist the clinician in determining which subtype of rhinitis a patient may be experiencing and therefore how to manage it. Clues:
- Patients with nonallergic rhinitis more often report nasal congestion and rhinorrhea, rather than sneezing and itching, which are predominant symptoms of allergic rhinitis.
- Patients with nonallergic rhinitis tend to develop symptoms at a later age.
- Common triggers of nonallergic rhinitis are changes in weather and temperature, food, perfumes, odors, smoke, and fumes. Animal exposure does not lead to symptoms.
- Patients with nonallergic rhinitis have few complaints of concomitant symptoms of allergic conjunctivitis (itching, watering, redness, and swelling).
- Many patients with nonallergic rhinitis find that antihistamines have no benefit. Also, they do not have other atopic diseases such as eczema or food allergies and have no family history of atopy.
PHYSICAL FINDINGS
Some findings on physical examination may help distinguish allergic from nonallergic rhinitis.
- Patients with long-standing allergic rhinitis may have an “allergic crease,” ie, a horizontal wrinkle near the tip of the nose caused by frequent upward wiping. Another sign may be a gothic arch, which is a narrowing of the hard palate occurring as a child.
- In allergic rhinitis, the turbinates are often pale, moist, and boggy with a bluish tinge.
CASE CONTINUED
Our patient’s symptoms can be caused by many different factors. Allergic triggers for rhinitis include both indoor and outdoor sources. The most common allergens include cat, dog, dust mite, cockroach, mold, and pollen allergens. The absence of acute sneezing and itching when around her cat and her recent negative skin-prick tests confirm that the rhinitis symptoms are not allergic.
In this patient, who has symptoms throughout the year but no allergic triggers, consideration of the different subtypes of nonallergic rhinitis may help guide further therapy.
SUBTYPES OF NONALLERGIC RHINITIS
Vasomotor rhinitis
Vasomotor rhinitis is thought to be caused by a variety of neural and vascular triggers, often without an inflammatory cause. These triggers lead to symptoms involving nasal congestion and clear rhinorrhea more than sneezing and itching. The symptoms can be sporadic, with acute onset in relation to identifiable nonallergic triggers, or chronic, with no clear trigger.
Gustatory rhinitis, for example, is a form of vasomotor rhinitis in which clear rhinorrhea occurs suddenly while eating or while drinking alcohol. It may be prevented by using nasal ipratropium (Atrovent) before meals.
Irritant-sensitive vasomotor rhinitis. In some patients, acute vasomotor rhinitis symptoms are brought on by strong odors, cigarette smoke, air pollution, or perfume. When asked, most patients easily identify which of these irritant triggers cause symptoms.
Weather- or temperature-sensitive vasomotor rhinitis. In other patients, a change in temperature, humidity, or barometric pressure or exposure to cold or dry air can cause nasal symptoms.9 These triggers are often hard to identify. Weather- or temperature-sensitive vasomotor rhinitis is often mistaken for seasonal allergic rhinitis because weather changes occur in close relation to the peak allergy seasons in the spring and fall. However, this subtype does not respond as well to intranasal steroids.9
Other nonallergic triggers of vasomotor rhinitis may include exercise, emotion, and sexual arousal (honeymoon rhinitis).10
Some triggers, such as tobacco smoke and perfume, are easy to avoid. Other triggers, such as weather changes, are unavoidable. If avoidance measures fail or are inadequate, medications (described below) can be used for prophylaxis and symptomatic treatment.
Drug-induced rhinitis
Drugs of various classes are known to cause either acute or chronic rhinitis. Drug-induced rhinitis has been divided into different types based on the mechanism involved.11
The local inflammatory type occurs in aspirin-exacerbated respiratory disease, which is characterized by nasal polyposis with chronic rhinosinusitis, hyposmia, and moderate to severe persistent asthma. Aspirin and other NSAIDs induce an acute local inflammation, leading to severe rhinitis and asthma symptoms. Avoiding all NSAID products is recommended; aspirin desensitization may lead to improvement in rhinosinusitis and asthma control.
The neurogenic type of drug-induced rhinitis can occur with sympatholytic drugs such as alpha receptor agonists (eg, clonidine [Cat-apres]) and antagonists (eg, prazosin [Minipress]).11 Vasodilators, including phosphodiesterase-5 inhibitors such as sildenafil (Viagra), can lead to acute rhinitis symptoms (“anniversary rhinitis”).
Unknown mechanisms. Many other medications can lead to rhinitis by unknown mechanisms, usually with normal findings on physical examination. These include beta-blockers, angiotensin-converting enzyme inhibitors, calcium channel blockers, exogenous estrogens, oral contraceptives, antipsychotics, and gabapentin (Neurontin).
Correlating the initiation of a drug with the onset of rhinitis can help identify offending medications. Stopping the suspected medication, if feasible, is the first-line treatment.
Rhinitis medicamentosa, typically caused by overuse of over-the-counter topical nasal decongestants, is also classified under drug-induced rhinitis. Patients may not think of nasal decongestants as medications, and the physician may need to ask specifically about their use.
On examination, the nasal mucosa appears beefy red without mucous. Once a diagnosis is made, the physician should identify and treat the original etiology of the nasal congestion that led the patient to self-treat.
Patients with rhinitis medicamentosa often have difficulty discontinuing use of topical decongestants. They should be educated that the withdrawal symptoms can be severe and that more than one attempt at quitting may be needed. To break the cycle of rebound congestion, topical intranasal steroids should be used, though 5 to 7 days of oral steroids may be necessary.1
Cocaine is a potent vasoconstrictor. Its illicit use should be suspected, especially if the patient presents with symptoms of chronic irritation such as frequent nosebleeds, crusting, and scabbing.12
Infectious rhinitis
One of the most common causes of acute rhinitis is upper respiratory infection.
Acute viral upper respiratory infection often presents with thick nasal discharge, sneezing, and nasal obstruction that usually clears in 7 to 10 days but can last up to 3 weeks. Acute bacterial sinusitis can follow, typically in fewer than 2% of patients, with symptoms of persistent nasal congestion, discolored mucus, facial pain, cough, and sometimes fever.
Chronic rhinosinusitis is a syndrome with sinus mucosal inflammation with multiple causes. It is clinically defined as persistent nasal and sinus symptoms lasting longer than 12 weeks and confirmed with computed tomography (CT).13 The CT findings of chronic rhinosinusitis include thickening of the lining of the sinus cavities or complete opacification of the pneumatized sinuses.
Major symptoms to consider for diagnosis include facial pain, congestion, obstruction, purulent discharge on examination, and changes in olfaction. Minor symptoms are cough, fatigue, headache, halitosis, fever, ear symptoms, and dental pain.
Treatment may involve 3 or more weeks of an oral antibiotic and a short course of an oral steroid, a daily nasal steroid spray, or both oral and nasal steroids. Most patients can be managed in the primary care setting, but they can be referred to an ear, nose, and throat specialist, an allergist, or an immunologist if their symptoms do not respond to initial therapy.
Nonallergic rhinitis eosinophilic syndrome
Patients with nonallergic rhinitis eosinophilic syndrome (NARES) are typically middle-aged and have perennial symptoms of sneezing, itching, and rhinorrhea with intermittent exacerbations. They occasionally have associated hyposmia (impaired sense of smell).1 The diagnosis is made when eosinophils account for more than 5% of cells on a nasal smear and allergy testing is negative.
Patients may develop nasal polyposis and aspirin sensitivity.1 Entopy has been described in some.14
Because of the eosinophilic inflammation, this form of nonallergic rhinitis responds well to intranasal steroids.
Immunologic causes
Systemic diseases can affect the nose and cause variable nasal symptoms that can be mistaken for rhinitis. Wegener granulomatosis, sarcoidosis, relapsing polychondritis, midline granulomas, Churg-Strauss syndrome, and amyloidosis can all affect the structures in the nose even before manifesting systemic symptoms. Granulomatous infections in the nose may lead to crusting, bleeding, and nasal obstruction.1
A lack of a response to intranasal steroids or oral antibiotics should lead to consideration of these conditions, and treatment should be tailored to the specific disease.
Occupational rhinitis
Occupational exposure to chemicals, biologic aerosols, flour, and latex can lead to rhinitis, typically through an inflammatory mechanism. Many patients present with associated occupational asthma. The symptoms improve when the patient is away from work and worsen throughout the work week.
Avoiding the triggering agent is necessary to treat these symptoms.
Hormonal rhinitis
Hormonal rhinitis, ie, rhinitis related to metabolic and endocrine conditions, is most commonly associated with high estrogen states. Nasal congestion has been reported with pregnancy, menses, menarche, and the use of oral contraceptives.15 The mechanism for congestion in these conditions still needs clarification.
When considering drug therapy, only intranasal budesonide (Rhinocort) has a pregnancy category B rating.
While hypothyroidism and acromegaly have been mentioned in reviews of nonallergic rhinitis, evidence that these disorders cause nonallergic rhinitis is not strong.16,17
Structurally related rhinitis
Anatomic abnormalities that can cause persistent nasal congestion include nasal septal deviation, turbinate hypertrophy, enlarged adenoids, tumors, and foreign bodies. These can be visualized by simple anterior nasal examination, nasal endoscopy, or radiologic studies. If structural causes lead to impaired quality of life or chronic rhinosinusitis, then consider referral to a specialist for possible surgical treatment.
Clear spontaneous rhinorrhea, with or without trauma, can be caused by cerebrospinal fluid leaking into the nasal cavity.18 A salty, metallic taste in the mouth can be a clue that the fluid is cerebrospinal fluid. A definitive diagnosis of cerebrospinal fluid leak is made by finding beta-2-transferrin in nasal secretions.
Atrophic rhinitis
Atrophic rhinitis is categorized as primary or secondary.
Primary (idiopathic) atrophic rhinitis is characterized by atrophy of the nasal mucosa and mucosal colonization with Klebsiella ozaenae associated with a foul-smelling nasal discharge.19,20 This disorder has been primarily reported in young people who present with nasal obstruction, dryness, crusting, and epistaxis. They are from areas with warm climates, such as the Middle East, Southeast Asia, India, Africa, and the Mediterranean.
Secondary atrophic rhinitis can be a complication of nasal or sinus surgery, trauma, granulomatous disease, or exposure to radiation.21 This disorder is typically diagnosed with nasal endoscopy and treated with daily saline rinses with or without topical antibiotics.21
CASE CONTINUED
Questioned further, our patient says her symptoms are worse when her husband smokes, but that she continues to have congestion and rhinorrhea when he is away on business trips. She notes that her symptoms are often worse on airplanes (dry air with an acute change in barometric pressure), with weather changes, and in cold, dry environments. Symptoms are not induced by eating.
We note that she started taking lisinopril 2 years ago and conjugated equine estrogens 8 years ago. Review of systems reveals no history of facial or head trauma, polyps, or hyposmia.
The rhinitis and congestion are bilateral, and she denies headaches, acid reflux, and conjunctivitis. She has a mild throat-clearing cough that she attributes to postnasal drip.
On physical examination, her blood pressure is 118/76 mm Hg and her pulse is 64. Her turbinates are congested with clear rhinorrhea. The rest of the examination is normal.
AVOID TRIGGERS, PRETREAT BEFORE EXPOSURE
People with known environmental, non-immunologic, and irritant triggers should be reminded to avoid these exposures if possible.
If triggers are unavoidable, patients can pretreat themselves with topical nasal sprays before exposure. For example, if symptoms occur while on airplanes, then intranasal steroids or antihistamine sprays should be used before getting on the plane.
Many drugs available
Fortunately, many effective drugs are available to treat nonallergic rhinitis. These have few adverse effects or drug interactions.
Intranasal steroid sprays are considered first-line therapy, as there are studies demonstrating effectiveness in nonallergic rhinitis.22 Intranasal fluticasone propionate (Flonase) and beclomethasone dipropionate (Beconase AQ) are approved by the US Food and Drug Administration (FDA) for treating nonallergic rhinitis. Intranasal mometasone (Nasonex) is approved for treating nasal polyps.
Nasal steroid sprays are most effective if the dominant nasal symptom is congestion, but they have also shown benefit for rhinorrhea, sneezing, and itching.
Side effects of nasal steroid sprays include nasal irritation (dryness, burning, and stinging) and epistaxis, the latter occurring in 5% to 10% of patients.23
Intranasal antihistamines include azelastine (Astelin, Astepro) and olopatadine (Patanase). They are particularly useful for treating sneezing, congestion, and rhinorrhea.24 Astelin is the only intranasal antihistamine with FDA approval for nonallergic rhinitis.
Side effects of this drug class include bitter taste (with Astelin), sweet taste (with Astepro), headache, and somnolence.
Oral antihistamines such as loratadine (Claritin), cetirizine (Zyrtec), and fexofenadine (Allegra) are now available over the counter, and many patients try them before seeking medical care. These drugs may be helpful for those bothered by sneezing. However, no study has demonstrated their effectiveness for nonallergic rhinitis.25 First-generation antihistamines may help with rhinorrhea via their anticholinergic effects.
Ipratropium, an antimuscarinic agent, decreases secretions by inhibiting the nasal parasympathetic mucous glands. Intranasal ipratropium 0.03% (Atrovent 0.03%) should be considered first-line if the dominant symptom is rhinorrhea. Higher-dose ipratropium 0.06% is approved for rhinorrhea related to the common cold or allergic rhinitis. Because it is used topically, little is absorbed. Its major side effect is nasal dryness.
Decongestants, either oral or topical, can relieve the symptoms of congestion and rhinorrhea in nonallergic rhinitis. They should only be used short-term, as there is little evidence to support their chronic use.
Phenylpropanolamine, a decongestant previously found in over-the-counter cough medicines, was withdrawn from the market in 2000 owing to concern that the drug, especially when used for weight suppression, was linked to hemorrhagic stroke in young women.26,27 Other oral decongestants, ie, pseudoephedrine and phenylephrine, are still available, but there are no definitive guidelines for their use. Their side effects include tachycardia, increase in blood pressure, and insomnia.
Nasal saline irrigation has been used for centuries to treat rhinitis and sinusitis, despite limited evidence of benefit. A Cochrane review concluded that saline irrigation was well tolerated, had minor side effects, and could provide some relief of rhinosinusitis symptoms either as the sole therapeutic measure or as adjunctive treatment.28 Hypertonic saline solutions, while possibly more effective than isotonic saline in improving mucociliary clearance, are not as well tolerated since they can cause nasal burning and irritation. Presumed benefits of saline irrigation are clearance of nasal secretions, improvement of nasociliary function, and removal of irritants and pollen from the nose.
A strategy
Imaging the sinuses with CT, which has replaced standard nasal radiography, may help if one is concerned about chronic rhinosinusitis, nasal polyps, or other anatomic condition that could contribute to persistent symptoms. Cost and radiation exposure should enter into the decision to obtain this study because a diagnosis based on the patient’s report of symptoms may be equally accurate.29,30
CASE CONTINUED
Our patient has a number of potential causes of her symptoms. Exposure to second-hand tobacco smoke at home and to the air in airplanes could be acute triggers. Weather and temperature changes could explain her chronic symptoms in the spring and fall. Use of an angiotensin-converting enzyme inhibitor (in her case, lisinopril) and estrogen replacement therapy may contribute to perennial symptoms, but the onset of her nonallergic rhinitis does not correlate with the use of these drugs. There are no symptoms to suggest chronic rhinosinusitis or anatomic causes of her symptoms.
This case is typical of vasomotor rhinitis of the weather- or temperature-sensitive type. This diagnosis may explain her lack of improvement with intranasal steroids, though adherence and spray technique should be assessed. At this point, we would recommend trying topical antihistamines daily when chronic symptoms are present or as needed for acute symptoms.
- Wallace DV, Dykewicz MS, Bernstein DI, et al. The diagnosis and management of rhinitis: an updated practice parameter. J Allergy Clin Immunol 2008; 122( suppl 2):S1–S84.
- Settipane RA, Charnock DR. Epidemiology of rhinitis: allergic and nonallergic. Clin Allergy Immunol 2007; 19:23–34.
- Settipane RA, Lieberman P. Update on nonallergic rhinitis. Ann Allergy Asthma Immunol 2001; 86:494–507.
- Rondón C, Doña I, Torres MJ, Campo P, Blanca M. Evolution of patients with nonallergic rhinitis supports conversion to allergic rhinitis. J Allergy Clin Immunol 2009; 123:1098–1102.
- Forester JP, Calabria CW. Local production of IgE in the respiratory mucosa and the concept of entopy: does allergy exist in nonallergic rhinitis? Ann Allergy Asthma Immunol 2010; 105:249–255.
- Silvers WS. The skier’s nose: a model of cold-induced rhinorrhea. Ann Allergy 1991; 67:32–36.
- Jaradeh SS, Smith TL, Torrico L, et al. Autonomic nervous system evaluation of patients with vasomotor rhinitis. Laryngoscope 2000; 110:1828–1831.
- Quan M, Casale TB, Blaiss MS. Should clinicians routinely determine rhinitis subtype on initial diagnosis and evaluation? A debate among experts. Clin Cornerstone 2009; 9:54–60.
- Jacobs R, Lieberman P, Kent E, Silvey M, Locantore N, Philpot EE. Weather/temperature-sensitive vasomotor rhinitis may be refractory to intranasal corticosteroid treatment. Allergy Asthma Proc 2009; 30:120–127.
- Monteseirin J, Camacho MJ, Bonilla I, Sanchez-Hernandez C, Hernandez M, Conde J. Honeymoon rhinitis. Allergy 2001; 56:353–354.
- Varghese M, Glaum MC, Lockey RF. Drug-induced rhinitis. Clin Exp Allergy 2010; 40:381–384.
- Schwartz RH, Estroff T, Fairbanks DN, Hoffmann NG. Nasal symptoms associated with cocaine abuse during adolescence. Arch Otolaryngol Head Neck Surg 1989; 115:63–64.
- Meltzer EO, Hamilos DL, Hadley JA, et al; American Academy of Allergy, Asthma and Immunology (AAAAI); American Academy of Otolaryngic Allergy (AAOA); American Academy of Otolaryngology--Head and Neck Surgery (AAO-HNS); American College of Allergy, Asthma and Immunology (ACAAI); American Rhinologic Society (ARS). Rhinosinusitis: establishing definitions for clinical research and patient care. J Allergy Clin Immunol 2004; 114( suppl 6):155–212.
- Powe DG, Huskisson RS, Carney AS, Jenkins D, Jones NS. Evidence for an inflammatory pathophysiology in idiopathic rhinitis. Clin Exp Allergy 2001; 31:864–872.
- Philpott CM, Robinson AM, Murty GE. Nasal pathophysiology and its relationship to the female ovarian hormones. J Otolaryngol Head Neck Surg 2008; 37:540–546.
- Dykewicz MS, Fineman S, Skoner DP, et al. Diagnosis and management of rhinitis: complete guidelines of the Joint Task Force on Practice Parameters in Allergy, Asthma and Immunology. American Academy of Allergy, Asthma, and Immunology. Ann Allergy Asthma Immunol 1998; 81:478–518.
- Ellegård EK, Karlsson NG, Ellegård LH. Rhinitis in the menstrual cycle, pregnancy, and some endocrine disorders. Clin Allergy Immunol 2007; 19:305–321.
- Dunn CJ, Alaani A, Johnson AP. Study on spontaneous cerebrospinal fluid rhinorrhoea: its aetiology and management. J Laryngol Otol 2005; 119:12–15.
- Bunnag C, Jareoncharsri P, Tansuriyawong P, Bhothisuwan W, Chantarakul N. Characteristics of atrophic rhinitis in Thai patients at the Siriraj Hospital. Rhinology 1999; 37:125–130.
- Dutt SN, Kameswaran M. The aetiology and management of atrophic rhinitis. J Laryngol Otol 2005; 119:843–852.
- deShazo RD, Stringer SP. Atrophic rhinosinusitis: progress toward explanation of an unsolved medical mystery. Curr Opin Allergy Clin Immunol 2011; 11:1–7.
- Greiner AN, Meltzer EO. Overview of the treatment of allergic rhinitis and nonallergic rhinopathy. Proc Am Thorac Soc 2011; 8:121–131.
- Corren J. Intranasal corticosteroids for allergic rhinitis: how do different agents compare? J Allergy Clin Immunol 1999; 104:S144–S149.
- Lieberman P, Meltzer EO, LaForce CF, Darter AL, Tort MJ. Two-week comparison study of olopatadine hydrochloride nasal spray 0.6% versus azelastine hydrochloride nasal spray 0.1% in patients with vasomotor rhinitis. Allergy Asthma Proc 2011; 32:151–158.
- Bousquet J, Khaltaev N, Cruz AA, et al; World Health Organization; GA(2)LEN. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008 update (in collaboration with the World Health Organization, GA(2)LEN and AllerGen). Allergy 2008; 63( suppl 86):8–160.
- SoRelle R. FDA warns of stroke risk associated with phenylpropanolamine; cold remedies and drugs removed from store shelves. Circulation 2000; 102:E9041–E9043.
- Kernan WN, Viscoli CM, Brass LM, et al. Phenylpropanolamine and the risk of hemorrhagic stroke. N Engl J Med 2000; 343:1826–1832.
- Harvey R, Hannan SA, Badia L, Scadding G. Nasal saline irrigations for the symptoms of chronic rhinosinusitis. Cochrane Database Syst Rev 2007;CD006394.
- Bhattacharyya N. The role of CT and MRI in the diagnosis of chronic rhinosinusitis. Curr Allergy Asthma Rep 2010; 10:171–174.
- Kenny TJ, Duncavage J, Bracikowski J, Yildirim A, Murray JJ, Tanner SB. Prospective analysis of sinus symptoms and correlation with paranasal computed tomography scan. Otolaryngol Head Neck Surg 2001; 125:40–43.
- Wallace DV, Dykewicz MS, Bernstein DI, et al. The diagnosis and management of rhinitis: an updated practice parameter. J Allergy Clin Immunol 2008; 122( suppl 2):S1–S84.
- Settipane RA, Charnock DR. Epidemiology of rhinitis: allergic and nonallergic. Clin Allergy Immunol 2007; 19:23–34.
- Settipane RA, Lieberman P. Update on nonallergic rhinitis. Ann Allergy Asthma Immunol 2001; 86:494–507.
- Rondón C, Doña I, Torres MJ, Campo P, Blanca M. Evolution of patients with nonallergic rhinitis supports conversion to allergic rhinitis. J Allergy Clin Immunol 2009; 123:1098–1102.
- Forester JP, Calabria CW. Local production of IgE in the respiratory mucosa and the concept of entopy: does allergy exist in nonallergic rhinitis? Ann Allergy Asthma Immunol 2010; 105:249–255.
- Silvers WS. The skier’s nose: a model of cold-induced rhinorrhea. Ann Allergy 1991; 67:32–36.
- Jaradeh SS, Smith TL, Torrico L, et al. Autonomic nervous system evaluation of patients with vasomotor rhinitis. Laryngoscope 2000; 110:1828–1831.
- Quan M, Casale TB, Blaiss MS. Should clinicians routinely determine rhinitis subtype on initial diagnosis and evaluation? A debate among experts. Clin Cornerstone 2009; 9:54–60.
- Jacobs R, Lieberman P, Kent E, Silvey M, Locantore N, Philpot EE. Weather/temperature-sensitive vasomotor rhinitis may be refractory to intranasal corticosteroid treatment. Allergy Asthma Proc 2009; 30:120–127.
- Monteseirin J, Camacho MJ, Bonilla I, Sanchez-Hernandez C, Hernandez M, Conde J. Honeymoon rhinitis. Allergy 2001; 56:353–354.
- Varghese M, Glaum MC, Lockey RF. Drug-induced rhinitis. Clin Exp Allergy 2010; 40:381–384.
- Schwartz RH, Estroff T, Fairbanks DN, Hoffmann NG. Nasal symptoms associated with cocaine abuse during adolescence. Arch Otolaryngol Head Neck Surg 1989; 115:63–64.
- Meltzer EO, Hamilos DL, Hadley JA, et al; American Academy of Allergy, Asthma and Immunology (AAAAI); American Academy of Otolaryngic Allergy (AAOA); American Academy of Otolaryngology--Head and Neck Surgery (AAO-HNS); American College of Allergy, Asthma and Immunology (ACAAI); American Rhinologic Society (ARS). Rhinosinusitis: establishing definitions for clinical research and patient care. J Allergy Clin Immunol 2004; 114( suppl 6):155–212.
- Powe DG, Huskisson RS, Carney AS, Jenkins D, Jones NS. Evidence for an inflammatory pathophysiology in idiopathic rhinitis. Clin Exp Allergy 2001; 31:864–872.
- Philpott CM, Robinson AM, Murty GE. Nasal pathophysiology and its relationship to the female ovarian hormones. J Otolaryngol Head Neck Surg 2008; 37:540–546.
- Dykewicz MS, Fineman S, Skoner DP, et al. Diagnosis and management of rhinitis: complete guidelines of the Joint Task Force on Practice Parameters in Allergy, Asthma and Immunology. American Academy of Allergy, Asthma, and Immunology. Ann Allergy Asthma Immunol 1998; 81:478–518.
- Ellegård EK, Karlsson NG, Ellegård LH. Rhinitis in the menstrual cycle, pregnancy, and some endocrine disorders. Clin Allergy Immunol 2007; 19:305–321.
- Dunn CJ, Alaani A, Johnson AP. Study on spontaneous cerebrospinal fluid rhinorrhoea: its aetiology and management. J Laryngol Otol 2005; 119:12–15.
- Bunnag C, Jareoncharsri P, Tansuriyawong P, Bhothisuwan W, Chantarakul N. Characteristics of atrophic rhinitis in Thai patients at the Siriraj Hospital. Rhinology 1999; 37:125–130.
- Dutt SN, Kameswaran M. The aetiology and management of atrophic rhinitis. J Laryngol Otol 2005; 119:843–852.
- deShazo RD, Stringer SP. Atrophic rhinosinusitis: progress toward explanation of an unsolved medical mystery. Curr Opin Allergy Clin Immunol 2011; 11:1–7.
- Greiner AN, Meltzer EO. Overview of the treatment of allergic rhinitis and nonallergic rhinopathy. Proc Am Thorac Soc 2011; 8:121–131.
- Corren J. Intranasal corticosteroids for allergic rhinitis: how do different agents compare? J Allergy Clin Immunol 1999; 104:S144–S149.
- Lieberman P, Meltzer EO, LaForce CF, Darter AL, Tort MJ. Two-week comparison study of olopatadine hydrochloride nasal spray 0.6% versus azelastine hydrochloride nasal spray 0.1% in patients with vasomotor rhinitis. Allergy Asthma Proc 2011; 32:151–158.
- Bousquet J, Khaltaev N, Cruz AA, et al; World Health Organization; GA(2)LEN. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008 update (in collaboration with the World Health Organization, GA(2)LEN and AllerGen). Allergy 2008; 63( suppl 86):8–160.
- SoRelle R. FDA warns of stroke risk associated with phenylpropanolamine; cold remedies and drugs removed from store shelves. Circulation 2000; 102:E9041–E9043.
- Kernan WN, Viscoli CM, Brass LM, et al. Phenylpropanolamine and the risk of hemorrhagic stroke. N Engl J Med 2000; 343:1826–1832.
- Harvey R, Hannan SA, Badia L, Scadding G. Nasal saline irrigations for the symptoms of chronic rhinosinusitis. Cochrane Database Syst Rev 2007;CD006394.
- Bhattacharyya N. The role of CT and MRI in the diagnosis of chronic rhinosinusitis. Curr Allergy Asthma Rep 2010; 10:171–174.
- Kenny TJ, Duncavage J, Bracikowski J, Yildirim A, Murray JJ, Tanner SB. Prospective analysis of sinus symptoms and correlation with paranasal computed tomography scan. Otolaryngol Head Neck Surg 2001; 125:40–43.
KEY POINTS
- When evaluating a patient with rhinitis, a key question is whether it is allergic or nonallergic.
- Identifying triggers that should be avoided is important for controlling symptoms.
- If symptoms continue, then the first-line treatment for nonallergic rhinitis is intranasal steroids.
- Failure of intranasal steroids to control symptoms should prompt a consideration of the many potential causes of rhinitis, and further evaluation and treatment can be tailored accordingly.
Antireflux surgery in the proton pump inhibitor era
For most patients with gastroesophageal reflux disease (GERD), a proton pump inhibitor (PPI) is the first choice for treatment.1 But some patients have symptoms that persist despite PPI therapy, some desire surgery despite successful PPI therapy, and some have persistent extraesophageal symptoms or other complications of reflux. For these patients, surgery is an option.2
In this article, we review the management of GERD and clarify the indications for antireflux surgery based on evidence of safety and efficacy.
GERD DEFINED: SYMPTOMS OR COMPLICATIONS
Defining the role of antireflux surgery is difficult, given the variety of presentations and the absence of a gold standard for diagnosing GERD. Most adults experience several episodes of physiologic reflux daily without symptoms.3 But a broad array of symptoms have been attributed to GERD, including chest pain, cough, and sore throat, and some conditions caused by acid reflux (eg, Barrett esophagus) can be asymptomatic.4,5

HEARTBURN ISN’T ALWAYS GERD
Typical GERD presents with the classic symptoms of pyrosis (heartburn) or acid regurgitation, or both.
Although these symptoms are often thought to be specific for GERD, other causes of esophageal injury— eg, eosinophilic esophagitis, infection (Candida, cytomegalovirus, herpes simplex virus), pill-induced esophagitis, or radiation therapy—can produce similar symptoms. Other causes, including coronary artery disease, biliary colic, foregut malignancy, or peptic ulcer disease, should also be considered in patients with supposedly typical GERD. Life-threatening mimics of GERD, such as unstable angina, should be excluded if they are likely, before proceeding with evaluating for possible GERD. Therefore, the initial history and examination should focus on appropriate diagnosis, with careful delineation of symptom quality.
Alarm features for advanced pathology6–8 include involuntary weight loss, dysphagia, vomiting, evidence of gastrointestinal blood loss, anemia, chest pain, and an epigastric mass.7 Admittedly, these features are only mediocre for detecting or excluding gastric or esophageal cancer, with a sensitivity of 67% and a specificity 66%.9 Nevertheless, they should prompt an endoscopic examination. In patients who have alarm features but have not yet been treated for GERD, upper endoscopy can identify an abnormality in about 60% of patients.10–12
PPIs HAVE REPLACED ANTACIDS AND HISTAMINE-2 RECEPTOR ANTAGONISTS
When the symptoms suggest GERD and no alarm features are present, an initial trial of the following lifestyle changes is reasonable:
- Avoiding acidic or refluxogenic foods (coffee, alcohol, chocolate, peppermint, fatty foods, citrus foods)
- Avoiding certain medications (anticholinergics, estrogens, calcium-channel blockers, nitroglycerine, benzodiazepines)
- Losing weight
- Quitting smoking
- Raising the head of the bed
- Staying upright for 2 to 3 hours after meals.
For someone with mild symptoms, these changes pose minimal risk. Unfortunately, they are unlikely to provide adequate symptom control for most patients.13–17
Before PPIs were invented, drug therapy for GERD symptoms that did not resolve with lifestyle changes consisted of antacids and, later, histamine-2 receptor antagonists. When maximal therapy failed to control symptoms, fundoplication surgery was considered an appropriate next step.
PPIs substantially changed the management of GERD, suppressing acid secretion much better than histamine-2 receptor antagonists. Taken 30 minutes before breakfast, a single daily dose of a PPI normalizes esophageal acid exposure in 67% of patients.18 Adding a second dose 30 minutes before dinner raises the number to more than 90%.19
PPIs have consistently outperformed histamine-2 blockers in the healing of esophagitis and in improving heartburn symptoms and are now the first-line medical therapy for uncomplicated GERD.6,8,20–25
WHEN PPIs WORK, SURGERY OFFERS NO ADVANTAGE
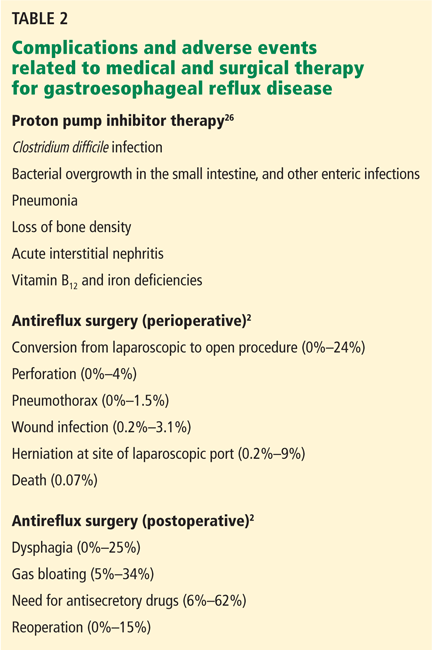
The LOTUS trial (Long-Term Usage of Esomeprazole vs Surgery for Treatment of Chronic GERD) compared long-term drug therapy with surgery to maintain remission of symptoms in GERD.27 In this trial, 554 patients whose symptoms initially responded to the PPI esomeprazole (Nexium) were randomized to continue to receive esomeprazole (n = 266) or to undergo laparoscopic antireflux surgery (288 were randomly assigned, and 248 had the operation). Dose adjustment of the esomeprazole was allowed (20–40 mg/day). A total of 372 patients completed 5 years of follow-up (192 esomeprazole, 180 surgery).
Symptoms stayed in remission in 92% of the esomeprazole group and 85% of the surgery group (P = .048). However, the difference was no longer statistically significant after modeling the effects of study dropout. The rate of severe adverse events was similar in both groups: 24.1% with esomeprazole and 28.6% with surgery.
These findings indicate that if symptoms fully abate with medical therapy, surgery offers no advantage. In addition, patients who desire surgery in the hope of avoiding lifelong drug therapy should be made aware that drug therapy and reoperation are often necessary after surgery.28 In most cases, antireflux surgery is unnecessary for patients whose GERD fully responds to PPI therapy.
IF PPIs FAIL, FURTHER TESTING NEEDED
But many patients who take PPIs still have symptoms, even though these drugs suppress acid secretion and heal esophagitis. In fact, symptoms completely resolve in only about one-half of patients with erosive disease and one-third of those without erosive disease.21
Reasons for an incomplete symptomatic response to PPIs are various. Acid reflux can persist, but this accounts for only 10% of cases.29 About one-third of patients have persistent reflux that is weakly acidic, with a pH higher than 4.29. However, most patients with persistent typical GERD symptoms do not have significant, persistent reflux, or their symptoms are not related to reflux events. In these cases, an alternative cause of the refractory symptoms should be sought.
Further diagnostic testing is indicated when symptoms persist despite PPI therapy. Upper endoscopy will reveal an abnormality such as persistent erosive esophagitis, eosinophilic esophagitis, esophageal stricture, Barrett esophagus, or esophageal cancer in roughly 10% of patients in whom empiric therapy fails.10
Although patients with persistent symptoms have not been enrolled in many randomized controlled trials, a multivariate analysis showed that failure of medical therapy heralds a poor response to surgery.30 Data such as these have led most experts to discourage fundoplication for such patients now, unlike in the pre-PPI era.
pH and intraluminal impedance testing
However, this recommendation against surgery is not a hard-and-fast rule.
In patients with esophageal regurgitation, most will not achieve adequate relief of symptoms with PPI therapy alone.34 The therapeutic gain of PPI therapy vs placebo averaged just 17% in seven randomized, controlled trials, more than 20% less than the response rate for heartburn.34 This is likely because of structural abnormalities such as reduced lower esophageal sphincter pressure, hiatal hernia, or delayed gastric emptying. Antireflux surgery can correct these structural abnormalities or prevent them from causing so much trouble; however, the presence of true regurgitation should first be confirmed by MII testing. If regurgitation is confirmed, antireflux surgery is warranted, particularly in patients with nocturnal symptoms who may be at high risk of aspiration. With careful patient selection, regurgitation symptoms improve in about 90% after surgery.2
In patients with heartburn, if esophageal acid exposure continues to be abnormal on MII-pH testing, then an escalation of therapy may improve symptoms, particularly if symptoms occur during reflux or if they partially responded to PPI therapy. Options in this scenario include alteration or intensification of acid-suppressive therapy, treatment with baclofen (Lioresal), and antireflux surgery.18,35,36 In randomized controlled trials of patients whose symptoms partially responded to PPIs, antireflux surgery has performed similarly to PPIs in terms of improving typical GERD symptoms, particularly regurgitation.27,37–41 Although this scenario is a reasonable indication for antireflux surgery, recommendations should be made with appropriate restraint since it is not easily reversible, some patients experience complications, and up to one-third will have no therapeutic benefit.30
Nonacid reflux. In some cases, MII-pH testing during PPI therapy will reveal reflux of weakly acidic (pH > 4) or alkaline stomach contents, often called “nonacid reflux.”29 Nonacid reflux is often present in patients with esophagitis that persists despite PPI therapy, indicating that even weakly acidic stomach contents can injure the mucosa.42 Since intensifying the acid-suppressive therapy is unlikely to improve these symptoms, antireflux surgery may have a role.
In one study,43 nonacid reflux was well controlled by laparoscopic Nissen fundoplication, although 15 (48%) of 31 patients had persistent symptoms of GERD after surgery. No patient had a strong symptom correlation with postoperative reflux events, suggesting an alternative cause of the persistent symptoms. Therefore, antireflux surgery for nonacid reflux should be limited exclusively to patients with strong symptom correlation, and even then it should be considered with restraint, given the limited evidence for benefit and the potential for harm.
If testing is negative. In studies investigating the diagnostic yield of MII-pH testing, more than 50% of patients who had refractory symptoms had a negative MII-pH test.29 In such situations, when the symptoms are strongly correlated with reflux events, the patient is said to have “esophageal hypersensitivity.” A few small studies have suggested that such patients may benefit from surgery, but these data have not been replicated in randomized controlled trials.32
When the testing is negative and there is no correlation between the patient’s symptoms and reflux events, the patient is unlikely to benefit from antireflux surgery. Care of these patients is beyond the scope of this review.
SURGERY RARELY IMPROVES COUGH, ASTHMA, OR LARYNGITIS
GERD has been implicated as a cause of chronic cough, asthma, and laryngitis, although each of these has many potential causes.44–46 Despite these associations, the evidence for therapeutic benefit from antireflux therapy is weak.
PPI therapy shows no benefit over placebo for chronic cough of uncertain etiology, but some benefit if GERD is objectively demonstrated.47 Laryngitis resolved in just 15% of patients on esomeprazole vs 16% of patients on placebo after excluding patients with moderate to severe heartburn.48
In a large randomized controlled trial in patients with asthma, there was no overall improvement in peak flow for the PPI group vs the placebo group, although significant improvement occurred in patients with heartburn and nocturnal respiratory symptoms.46
Potent antisecretory therapy seems to improve extraesophageal symptoms when typical GERD symptoms are also present, but it otherwise has shown little evidence of benefit.
The evidence for a benefit from antireflux surgery in patients with extraesophageal GERD syndromes is even more limited. Although one systematic review49 found that cough and other laryngeal symptoms improved in 60% to 100% of patients with objective evidence of GERD who underwent fundoplication, virtually all of the studies were uncontrolled case series.49
The lone randomized controlled trial in the systematic review compared Nissen fundoplication with ranitidine (Zantac) or antacids only for patients with asthma and GERD, and found no significant difference in peak expiratory flow among the three groups after 2 years. However, asthma symptom scores improved in 75% of the surgical group, 9% of the medical group, and 4% of the control group.50
In a study that was not included in the prior systematic review, patients with laryngopharyngeal reflux unresponsive to aggressive acid suppression who subsequently underwent fundoplication fared no better than those who did not.51
Thus, based on the available data, antireflux surgery is only rarely indicated for extraesophageal symptoms, especially in patients who have no typical GERD symptoms or in patients whose symptoms are refractory to medical therapy.
SURGERY FOR EROSIVE ESOPHAGITIS OR BARRETT ESOPHAGUS IF PPI FAILS
Lifelong antireflux therapy is indicated for patients with severe erosive esophagitis or Barrett esophagus. Erosive esophagitis recurs in more than 80% within 12 months of discontinuing antisecretory therapy.52 Both Barrett esophagus and esophageal adenocarcinoma are strongly associated with GERD, and nearly 10% of patients with chronic reflux have Barrett esophagus.53,54 It is suspected that suppressing reflux reduces the rate of progression of Barrett esophagus to esophageal adenocarcinoma, but this remains to be proven.
Perhaps the strongest indication for surgery in the PPI era is for patients who have persistent symptoms and severe erosive esophagitis (Los Angeles grade C or D) despite high-dose PPI therapy. If other causes of persistent esophagitis have been ruled out, fundoplication can induce healing and improve symptoms.55,56 In these cases, surgery is done to induce remission of the disease when maximal medical therapy has been truly unsuccessful.
Randomized controlled trials suggest that medical and surgical therapies are equally effective for preventing the recurrence of erosive esophagitis or the progression of Barrett esophagus. In a study of 225 patients, at 7 years of follow-up, esophagitis had recurred in 10.4% of patients on omeprazole vs 11.8% of those who had undergone antireflux surgery.40 Similarly, open Nissen fundoplication was no different from drug therapy (histamine-2 receptor antagonist or PPI) for progression of Barrett esophagus over a median of 5 years.57 A meta-analysis with nearly 5,000 person-years each in the medical and surgical groups also found no significant difference in rates of cancer progression.58
Notably, symptoms such as dysphagia, flatulence, and the inability to burp occurred significantly more often in the surgical groups in these studies.
In view of these data, antireflux surgery has no significant advantage over medical therapy for maintaining healing of erosive esophagitis or preventing progression of Barrett esophagus. Thus, it should be reserved for patients who do not desire lifelong drug therapy, provided they understand that there is no therapeutic advantage for their esophagitis or for Barrett esophagus.
SPECIFIC INDICATIONS FOR ANTIREFLUX SURGERY
Now that we have PPIs, several situations remain in which surgery for GERD is either indicated or worth considering.
Antireflux surgery is clearly indicated for:
- Patients with erosive esophagitis that does not heal with maximal drug therapy
- Patients with volume regurgitation, particularly if it occurs at night or if there is evidence of aspiration
- Patients who require lifelong treatment for reflux but who have had a serious adverse event related to PPI therapy, such as refractory Clostridium difficile infection.
Antireflux surgery is also worth considering in patients who for personal reasons wish to avoid long-term or lifelong drug therapy.
Patients should be informed, however, that antireflux surgery has not been shown to be better than medical therapy for maintaining remission of symptoms, for preventing progression of Barrett esophagus, or for maintaining healing of erosive esophagitis. Medical therapy is still the first option for these patients.
Surgery may also be considered in patients with persistent symptoms who have a partial response to medical therapy, who show persistent acidic or weakly acidic reflux on MII-pH testing, and whose symptoms have been correlated with reflux events. Although surgery is not sure to improve their symptoms, benefit is more likely in this patient population compared with those without these characteristics.
Extraesophageal GERD
In patients suspected of having extraesophageal GERD, surgery should be considered if typical GERD symptoms are present and improve with PPI therapy, if the extraesophageal syndrome partially responds to PPI therapy, and if MII-pH testing demonstrates a correlation between symptoms and reflux. Surgery may have a stronger indication in this setting if the patient has nocturnal reflux or extraesophageal symptoms.
When is surgery not an option?
In general, surgery should not be considered in patients who do not have a partial response to PPI therapy or who do not have a strong symptom-reflux correlation on MII-pH testing. In all cases of failed medical therapy without persistent severe erosive disease, the threshold for opting for surgery should be high, given the uncertain response of these patients to surgery.
Peristaltic dysfunction is a relative but not an absolute contraindication to surgery.59
RISKS, BENEFITS OF SURGERY FOR GERD
The patient’s preference for surgery over drug therapy should always be balanced against the risks of surgery, including both short-term and long-term adverse events, to allow the patient to make an adequately informed decision (Table 2).2,26
Adverse events associated with PPI therapy are rare and in many cases the association is debatable.26 Nonetheless, long-term PPI therapy has been most strongly associated with an increased risk of C difficile infection and other enteric infections, although the absolute risk of these events remains low.
Complication rates after antireflux surgery depend on the surgeon’s experience and technique. Death is exceedingly rare. In most high-volume centers, the need to convert from laparoscopic to open fundoplication occurs in fewer than 2.4% of patients.2
Potential perioperative complications include perforation (4%), wound infection (3%), and pneumothorax (2%).2
Antireflux surgery is also associated with a significant risk of dysphagia, bloating, an inability to burp, and excessive flatulence, all of which can markedly impair the quality of life.
A major consideration is that fundoplication is generally irreversible. Reoperation rates have been reported to range from 0% to 15%.2 Furthermore, up to 50% of patients still need medical therapy after surgery.60,61 Of note, only about 25% of patients on medical therapy after surgery will actually have an abnormal pH study.61
MORE STUDY NEEDED
Future studies directly comparing medical and surgical therapy for carefully selected patients with extraesophageal manifestations of GERD and refractory symptoms should help further delineate outcome in this difficult group of patients.
Under development are new drugs that may inhibit transient relaxation of the lower esophageal sphincter, as well as minimally invasive procedures, which may alter the indications for surgery in coming years.36
Acknowledgment: The research for this article was supported in part by a grant from the National Institutes of Health (T32 DK07634).
- Finks JF, Wei Y, Birkmeyer JD. The rise and fall of antireflux surgery in the United States. Surg Endosc 2006; 20:1698–1701.
- Stefanidis D, Hope WW, Kohn GP, Reardon PR, Richardson WS, Fanelli RD; SAGES Guidelines Committee. Guidelines for surgical treatment of gastroesophageal reflux disease. Surg Endosc 2010; 24:2647–2669.
- Richter JE. Typical and atypical presentations of gastroesophageal reflux disease. The role of esophageal testing in diagnosis and management. Gastroenterol Clin North Am 1996; 25:75–102.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Dickman R, Kim JL, Camargo L, et al. Correlation of gastroesophageal reflux disease symptoms characteristics with long-segment Barrett’s esophagus. Dis Esophagus 2006; 19:360–365.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Armstrong D, Marshall JK, Chiba N, et al; Canadian Association of Gastroenterology GERD Consensus Group. Canadian Consensus Conference on the management of gastroesophageal reflux disease in adults - update 2004. Can J Gastroenterol 2005; 19:15–35.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.
- Vakil N, Moayyedi P, Fennerty MB, Talley NJ. Limited value of alarm features in the diagnosis of upper gastrointestinal malignancy: systematic review and meta-analysis. Gastroenterology 2006; 131:390–401.
- Poh CH, Gasiorowska A, Navarro-Rodriguez T, et al. Upper GI tract findings in patients with heartburn in whom proton pump inhibitor treatment failed versus those not receiving antireflux treatment. Gastrointest Endosc 2010; 71:28–34.
- Dickman R, Mattek N, Holub J, Peters D, Fass R. Prevalence of upper gastrointestinal tract findings in patients with noncardiac chest pain versus those with gastroesophageal reflux disease (GERD)-related symptoms: results from a national endoscopic database. Am J Gastroenterol 2007; 102:1173–1179.
- Voutilainen M, Sipponen P, Mecklin JP, Juhola M, Färkkilä M. Gastroesophageal reflux disease: prevalence, clinical, endoscopic and histopathological findings in 1,128 consecutive patients referred for endoscopy due to dyspeptic and reflux symptoms. Digestion 2000; 61:6–13.
- Fraser-Moodie CA, Norton B, Gornall C, Magnago S, Weale AR, Holmes GK. Weight loss has an independent beneficial effect on symptoms of gastro-oesophageal reflux in patients who are overweight. Scand J Gastroenterol 1999; 34:337–340.
- Jacobson BC, Somers SC, Fuchs CS, Kelly CP, Camargo CA. Bodymass index and symptoms of gastroesophageal reflux in women. N Engl J Med 2006; 354:2340–2348.
- Kjellin A, Ramel S, Rössner S, Thor K. Gastroesophageal reflux in obese patients is not reduced by weight reduction. Scand J Gastroenterol 1996; 31:1047–1051.
- Waring JP, Eastwood TF, Austin JM, Sanowski RA. The immediate effects of cessation of cigarette smoking on gastroesophageal reflux. Am J Gastroenterol 1989; 84:1076–1078.
- Pehl C, Waizenhoefer A, Wendl B, Schmidt T, Schepp W, Pfeiffer A. Effect of low and high fat meals on lower esophageal sphincter motility and gastroesophageal reflux in healthy subjects. Am J Gastroenterol 1999; 94:1192–1196.
- Bajbouj M, Becker V, Phillip V, Wilhelm D, Schmid RM, Meining A. High-dose esomeprazole for treatment of symptomatic refractory gastroesophageal reflux disease—a prospective pH-metry/impedance-controlled study. Digestion 2009; 80:112–118.
- Charbel S, Khandwala F, Vaezi MF. The role of esophageal pH monitoring in symptomatic patients on PPI therapy. Am J Gastroenterol 2005; 100:283–289.
- Khan M, Santana J, Donnellan C, Preston C, Moayyedi P. Medical treatments in the short term management of reflux oesophagitis. Cochrane Database Syst Rev 2007;CD003244.
- Dean BB, Gano AD, Knight K, Ofman JJ, Fass R. Effectiveness of proton pump inhibitors in nonerosive reflux disease. Clin Gastroenterol Hepatol 2004; 2:656–664.
- Sabesin SM, Berlin RG, Humphries TJ, Bradstreet DC, Walton-Bowen KL, Zaidi S. Famotidine relieves symptoms of gastroesophageal reflux disease and heals erosions and ulcerations. Results of a multicenter, placebo-controlled, dose-ranging study. USA Merck Gastroesophageal Reflux Disease Study Group. Arch Intern Med 1991; 151:2394–2400.
- van Pinxteren B, Numans ME, Bonis PA, Lau J. Short-term treatment with proton pump inhibitors, H2-receptor antagonists and prokinetics for gastro-oesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Database Syst Rev 2004;CD002095.
- Chiba N, De Gara CJ, Wilkinson JM, Hunt RH. Speed of healing and symptom relief in grade II to IV gastroesophageal reflux disease: a meta-analysis. Gastroenterology 1997; 112:1798–1810.
- Venables TL, Newland RD, Patel AC, Hole J, Wilcock C, Turbitt ML. Omeprazole 10 milligrams once daily, omeprazole 20 milligrams once daily, or ranitidine 150 milligrams twice daily, evaluated as initial therapy for the relief of symptoms of gastro-oesophageal reflux disease in general practice. Scand J Gastroenterol 1997; 32:965–973.
- Madanick RD. Proton pump inhibitor side effects and drug interactions: much ado about nothing? Cleve Clin J Med 2011; 78:39–49.
- Galmiche JP, Hatlebakk J, Attwood S, et al; LOTUS Trial Collaborators. Laparoscopic antireflux surgery vs esomeprazole treatment for chronic GERD: the LOTUS randomized clinical trial. JAMA 2011; 305:1969–1977.
- Spechler SJ, Lee E, Ahnen D, et al. Long-term outcome of medical and surgical therapies for gastroesophageal reflux disease: followup of a randomized controlled trial. JAMA 2001; 285:2331–2338.
- Mainie I, Tutuian R, Shay S, et al. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: a multicentre study using combined ambulatory impedance-pH monitoring. Gut 2006; 55:1398–1402.
- Campos GM, Peters JH, DeMeester TR, et al. Multivariate analysis of factors predicting outcome after laparoscopic Nissen fundoplication. J Gastrointest Surg 1999; 3:292–300.
- Becker V, Bajbouj M, Waller K, Schmid RM, Meining A. Clinical trial: persistent gastro-oesophageal reflux symptoms despite standard therapy with proton pump inhibitors - a follow-up study of intraluminal-impedance guided therapy. Aliment Pharmacol Ther 2007; 26:1355–1360.
- Mainie I, Tutuian R, Agrawal A, Adams D, Castell DO. Combined multichannel intraluminal impedance-pH monitoring to select patients with persistent gastro-oesophageal reflux for laparoscopic Nissen fundoplication. Br J Surg 2006; 93:1483–1487.
- del Genio G, Tolone S, del Genio F, et al. Prospective assessment of patient selection for antireflux surgery by combined multichannel intraluminal impedance pH monitoring. J Gastrointest Surg 2008; 12:1491–1496.
- Kahrilas PJ, Howden CW, Hughes N. Response of regurgitation to proton pump inhibitor therapy in clinical trials of gastroesophageal reflux disease. Am J Gastroenterol 2011; 106:1419–1425.
- Koek GH, Sifrim D, Lerut T, Janssens J, Tack J. Effect of the GABA(B) agonist baclofen in patients with symptoms and duodeno-gastro-oesophageal reflux refractory to proton pump inhibitors. Gut 2003; 52:1397–1402.
- Boeckxstaens GE. Reflux inhibitors: a new approach for GERD? Curr Opin Pharmacol 2008; 8:685–689.
- Anvari M, Allen C, Marshall J, et al. A randomized controlled trial of laparoscopic nissen fundoplication versus proton pump inhibitors for treatment of patients with chronic gastroesophageal reflux disease: One-year follow-up. Surg Innov 2006; 13:238–249.
- Mahon D, Rhodes M, Decadt B, et al. Randomized clinical trial of laparoscopic Nissen fundoplication compared with proton-pump inhibitors for treatment of chronic gastro-oesophageal reflux. Br J Surg 2005; 92:695–699.
- Mehta S, Bennett J, Mahon D, Rhodes M. Prospective trial of laparoscopic nissen fundoplication versus proton pump inhibitor therapy for gastroesophageal reflux disease: Seven-year follow-up. J Gastrointest Surg 2006; 10:1312–1316.
- Lundell L, Miettinen P, Myrvold HE, et al; Nordic GORD Study Group. Seven-year follow-up of a randomized clinical trial comparing proton-pump inhibition with surgical therapy for reflux oesophagitis. Br J Surg 2007; 94:198–203.
- Lundell L, Attwood S, Ell C, et al; LOTUS trial collaborators. Comparing laparoscopic antireflux surgery with esomeprazole in the management of patients with chronic gastro-oesophageal reflux disease: a 3-year interim analysis of the LOTUS trial. Gut 2008; 57:1207–1213.
- Frazzoni M, Conigliaro R, Melotti G. Weakly acidic refluxes have a major role in the pathogenesis of proton pump inhibitor-resistant reflux oesophagitis. Aliment Pharmacol Ther 2011; 33:601–606.
- Broeders JA, Bredenoord AJ, Hazebroek EJ, Broeders IA, Gooszen HG, Smout AJ. Effects of anti-reflux surgery on weakly acidic reflux and belching. Gut 2011; 60:435–441.
- American Gastroenterological Association medical position statement: guidelines on the use of esophageal pH recording. Gastroenterology 1996; 110:1981.
- el-Serag HB, Sonnenberg A. Comorbid occurrence of laryngeal or pulmonary disease with esophagitis in United States military veterans. Gastroenterology 1997; 113:755–760.
- Kiljander TO, Laitinen JO. The prevalence of gastroesophageal reflux disease in adult asthmatics. Chest 2004; 126:1490–1494.
- Chang AB, Lasserson TJ, Kiljander TO, Connor FL, Gaffney JT, Garske LA. Systematic review and meta-analysis of randomised controlled trials of gastro-oesophageal reflux interventions for chronic cough associated with gastro-oesophageal reflux. BMJ 2006; 332:11–17.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116:254–260.
- Iqbal M, Batch AJ, Spychal RT, Cooper BT. Outcome of surgical fundoplication for extraesophageal (atypical) manifestations of gastroesophageal reflux disease in adults: a systematic review. J Laparoendosc Adv Surg Tech A 2008; 18:789–796.
- Sontag SJ, O’Connell S, Khandelwal S, et al. Asthmatics with gastroesophageal reflux: long term results of a randomized trial of medical and surgical antireflux therapies. Am J Gastroenterol 2003; 98:987–999.
- Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngopharyngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol 2006; 4:433–441.
- Johnson DA, Benjamin SB, Vakil NB, et al. Esomeprazole once daily for 6 months is effective therapy for maintaining healed erosive esophagitis and for controlling gastroesophageal reflux disease symptoms: a randomized, double-blind, placebo-controlled study of efficacy and safety. Am J Gastroenterol 2001; 96:27–34.
- Winters C, Spurling TJ, Chobanian SJ, et al. Barrett’s esophagus. A prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology 1987; 92:118–124.
- Westhoff B, Brotze S, Weston A, et al. The frequency of Barrett’s esophagus in high-risk patients with chronic GERD. Gastrointest Endosc 2005; 61:226–231.
- Rosenthal R, Peterli R, Guenin MO, von Flüe M, Ackermann C. Laparoscopic antireflux surgery: long-term outcomes and quality of life. J Laparoendosc Adv Surg Tech A 2006; 16:557–561.
- Broeders JA, Draaisma WA, Bredenoord AJ, Smout AJ, Broeders IA, Gooszen HG. Long-term outcome of Nissen fundoplication in non-erosive and erosive gastro-oesophageal reflux disease. Br J Surg 2010; 97:845–352.
- Parrilla P, Martínez de Haro LF, Ortiz A, et al. Long-term results of a randomized prospective study comparing medical and surgical treatment of Barrett’s esophagus. Ann Surg 2003; 237:291–298.
- Corey KE, Schmitz SM, Shaheen NJ. Does a surgical antireflux procedure decrease the incidence of esophageal adenocarcinoma in Barrett’s esophagus? A meta-analysis. Am J Gastroenterol 2003; 98:2390–2394.
- Pandolfino JE, Kahrilas PJ; American Gastroenterological Association. AGA technical review on the clinical use of esophageal manometry. Gastroenterology 2005; 128:209–224.
- Dominitz JA, Dire CA, Billingsley KG, Todd-Stenberg JA. Complications and antireflux medication use after antireflux surgery. Clin Gastroenterol Hepatol 2006; 4:299–305.
- Lord RV, Kaminski A, Oberg S, et al. Absence of gastroesophageal reflux disease in a majority of patients taking acid suppression medications after Nissen fundoplication. J Gastrointest Surg 2002; 6:3–9.
For most patients with gastroesophageal reflux disease (GERD), a proton pump inhibitor (PPI) is the first choice for treatment.1 But some patients have symptoms that persist despite PPI therapy, some desire surgery despite successful PPI therapy, and some have persistent extraesophageal symptoms or other complications of reflux. For these patients, surgery is an option.2
In this article, we review the management of GERD and clarify the indications for antireflux surgery based on evidence of safety and efficacy.
GERD DEFINED: SYMPTOMS OR COMPLICATIONS
Defining the role of antireflux surgery is difficult, given the variety of presentations and the absence of a gold standard for diagnosing GERD. Most adults experience several episodes of physiologic reflux daily without symptoms.3 But a broad array of symptoms have been attributed to GERD, including chest pain, cough, and sore throat, and some conditions caused by acid reflux (eg, Barrett esophagus) can be asymptomatic.4,5
HEARTBURN ISN’T ALWAYS GERD
Typical GERD presents with the classic symptoms of pyrosis (heartburn) or acid regurgitation, or both.
Although these symptoms are often thought to be specific for GERD, other causes of esophageal injury— eg, eosinophilic esophagitis, infection (Candida, cytomegalovirus, herpes simplex virus), pill-induced esophagitis, or radiation therapy—can produce similar symptoms. Other causes, including coronary artery disease, biliary colic, foregut malignancy, or peptic ulcer disease, should also be considered in patients with supposedly typical GERD. Life-threatening mimics of GERD, such as unstable angina, should be excluded if they are likely, before proceeding with evaluating for possible GERD. Therefore, the initial history and examination should focus on appropriate diagnosis, with careful delineation of symptom quality.
Alarm features for advanced pathology6–8 include involuntary weight loss, dysphagia, vomiting, evidence of gastrointestinal blood loss, anemia, chest pain, and an epigastric mass.7 Admittedly, these features are only mediocre for detecting or excluding gastric or esophageal cancer, with a sensitivity of 67% and a specificity 66%.9 Nevertheless, they should prompt an endoscopic examination. In patients who have alarm features but have not yet been treated for GERD, upper endoscopy can identify an abnormality in about 60% of patients.10–12
PPIs HAVE REPLACED ANTACIDS AND HISTAMINE-2 RECEPTOR ANTAGONISTS
When the symptoms suggest GERD and no alarm features are present, an initial trial of the following lifestyle changes is reasonable:
- Avoiding acidic or refluxogenic foods (coffee, alcohol, chocolate, peppermint, fatty foods, citrus foods)
- Avoiding certain medications (anticholinergics, estrogens, calcium-channel blockers, nitroglycerine, benzodiazepines)
- Losing weight
- Quitting smoking
- Raising the head of the bed
- Staying upright for 2 to 3 hours after meals.
For someone with mild symptoms, these changes pose minimal risk. Unfortunately, they are unlikely to provide adequate symptom control for most patients.13–17
Before PPIs were invented, drug therapy for GERD symptoms that did not resolve with lifestyle changes consisted of antacids and, later, histamine-2 receptor antagonists. When maximal therapy failed to control symptoms, fundoplication surgery was considered an appropriate next step.
PPIs substantially changed the management of GERD, suppressing acid secretion much better than histamine-2 receptor antagonists. Taken 30 minutes before breakfast, a single daily dose of a PPI normalizes esophageal acid exposure in 67% of patients.18 Adding a second dose 30 minutes before dinner raises the number to more than 90%.19
PPIs have consistently outperformed histamine-2 blockers in the healing of esophagitis and in improving heartburn symptoms and are now the first-line medical therapy for uncomplicated GERD.6,8,20–25
WHEN PPIs WORK, SURGERY OFFERS NO ADVANTAGE
The LOTUS trial (Long-Term Usage of Esomeprazole vs Surgery for Treatment of Chronic GERD) compared long-term drug therapy with surgery to maintain remission of symptoms in GERD.27 In this trial, 554 patients whose symptoms initially responded to the PPI esomeprazole (Nexium) were randomized to continue to receive esomeprazole (n = 266) or to undergo laparoscopic antireflux surgery (288 were randomly assigned, and 248 had the operation). Dose adjustment of the esomeprazole was allowed (20–40 mg/day). A total of 372 patients completed 5 years of follow-up (192 esomeprazole, 180 surgery).
Symptoms stayed in remission in 92% of the esomeprazole group and 85% of the surgery group (P = .048). However, the difference was no longer statistically significant after modeling the effects of study dropout. The rate of severe adverse events was similar in both groups: 24.1% with esomeprazole and 28.6% with surgery.
These findings indicate that if symptoms fully abate with medical therapy, surgery offers no advantage. In addition, patients who desire surgery in the hope of avoiding lifelong drug therapy should be made aware that drug therapy and reoperation are often necessary after surgery.28 In most cases, antireflux surgery is unnecessary for patients whose GERD fully responds to PPI therapy.
IF PPIs FAIL, FURTHER TESTING NEEDED
But many patients who take PPIs still have symptoms, even though these drugs suppress acid secretion and heal esophagitis. In fact, symptoms completely resolve in only about one-half of patients with erosive disease and one-third of those without erosive disease.21
Reasons for an incomplete symptomatic response to PPIs are various. Acid reflux can persist, but this accounts for only 10% of cases.29 About one-third of patients have persistent reflux that is weakly acidic, with a pH higher than 4.29. However, most patients with persistent typical GERD symptoms do not have significant, persistent reflux, or their symptoms are not related to reflux events. In these cases, an alternative cause of the refractory symptoms should be sought.
Further diagnostic testing is indicated when symptoms persist despite PPI therapy. Upper endoscopy will reveal an abnormality such as persistent erosive esophagitis, eosinophilic esophagitis, esophageal stricture, Barrett esophagus, or esophageal cancer in roughly 10% of patients in whom empiric therapy fails.10
Although patients with persistent symptoms have not been enrolled in many randomized controlled trials, a multivariate analysis showed that failure of medical therapy heralds a poor response to surgery.30 Data such as these have led most experts to discourage fundoplication for such patients now, unlike in the pre-PPI era.
pH and intraluminal impedance testing
However, this recommendation against surgery is not a hard-and-fast rule.
In patients with esophageal regurgitation, most will not achieve adequate relief of symptoms with PPI therapy alone.34 The therapeutic gain of PPI therapy vs placebo averaged just 17% in seven randomized, controlled trials, more than 20% less than the response rate for heartburn.34 This is likely because of structural abnormalities such as reduced lower esophageal sphincter pressure, hiatal hernia, or delayed gastric emptying. Antireflux surgery can correct these structural abnormalities or prevent them from causing so much trouble; however, the presence of true regurgitation should first be confirmed by MII testing. If regurgitation is confirmed, antireflux surgery is warranted, particularly in patients with nocturnal symptoms who may be at high risk of aspiration. With careful patient selection, regurgitation symptoms improve in about 90% after surgery.2
In patients with heartburn, if esophageal acid exposure continues to be abnormal on MII-pH testing, then an escalation of therapy may improve symptoms, particularly if symptoms occur during reflux or if they partially responded to PPI therapy. Options in this scenario include alteration or intensification of acid-suppressive therapy, treatment with baclofen (Lioresal), and antireflux surgery.18,35,36 In randomized controlled trials of patients whose symptoms partially responded to PPIs, antireflux surgery has performed similarly to PPIs in terms of improving typical GERD symptoms, particularly regurgitation.27,37–41 Although this scenario is a reasonable indication for antireflux surgery, recommendations should be made with appropriate restraint since it is not easily reversible, some patients experience complications, and up to one-third will have no therapeutic benefit.30
Nonacid reflux. In some cases, MII-pH testing during PPI therapy will reveal reflux of weakly acidic (pH > 4) or alkaline stomach contents, often called “nonacid reflux.”29 Nonacid reflux is often present in patients with esophagitis that persists despite PPI therapy, indicating that even weakly acidic stomach contents can injure the mucosa.42 Since intensifying the acid-suppressive therapy is unlikely to improve these symptoms, antireflux surgery may have a role.
In one study,43 nonacid reflux was well controlled by laparoscopic Nissen fundoplication, although 15 (48%) of 31 patients had persistent symptoms of GERD after surgery. No patient had a strong symptom correlation with postoperative reflux events, suggesting an alternative cause of the persistent symptoms. Therefore, antireflux surgery for nonacid reflux should be limited exclusively to patients with strong symptom correlation, and even then it should be considered with restraint, given the limited evidence for benefit and the potential for harm.
If testing is negative. In studies investigating the diagnostic yield of MII-pH testing, more than 50% of patients who had refractory symptoms had a negative MII-pH test.29 In such situations, when the symptoms are strongly correlated with reflux events, the patient is said to have “esophageal hypersensitivity.” A few small studies have suggested that such patients may benefit from surgery, but these data have not been replicated in randomized controlled trials.32
When the testing is negative and there is no correlation between the patient’s symptoms and reflux events, the patient is unlikely to benefit from antireflux surgery. Care of these patients is beyond the scope of this review.
SURGERY RARELY IMPROVES COUGH, ASTHMA, OR LARYNGITIS
GERD has been implicated as a cause of chronic cough, asthma, and laryngitis, although each of these has many potential causes.44–46 Despite these associations, the evidence for therapeutic benefit from antireflux therapy is weak.
PPI therapy shows no benefit over placebo for chronic cough of uncertain etiology, but some benefit if GERD is objectively demonstrated.47 Laryngitis resolved in just 15% of patients on esomeprazole vs 16% of patients on placebo after excluding patients with moderate to severe heartburn.48
In a large randomized controlled trial in patients with asthma, there was no overall improvement in peak flow for the PPI group vs the placebo group, although significant improvement occurred in patients with heartburn and nocturnal respiratory symptoms.46
Potent antisecretory therapy seems to improve extraesophageal symptoms when typical GERD symptoms are also present, but it otherwise has shown little evidence of benefit.
The evidence for a benefit from antireflux surgery in patients with extraesophageal GERD syndromes is even more limited. Although one systematic review49 found that cough and other laryngeal symptoms improved in 60% to 100% of patients with objective evidence of GERD who underwent fundoplication, virtually all of the studies were uncontrolled case series.49
The lone randomized controlled trial in the systematic review compared Nissen fundoplication with ranitidine (Zantac) or antacids only for patients with asthma and GERD, and found no significant difference in peak expiratory flow among the three groups after 2 years. However, asthma symptom scores improved in 75% of the surgical group, 9% of the medical group, and 4% of the control group.50
In a study that was not included in the prior systematic review, patients with laryngopharyngeal reflux unresponsive to aggressive acid suppression who subsequently underwent fundoplication fared no better than those who did not.51
Thus, based on the available data, antireflux surgery is only rarely indicated for extraesophageal symptoms, especially in patients who have no typical GERD symptoms or in patients whose symptoms are refractory to medical therapy.
SURGERY FOR EROSIVE ESOPHAGITIS OR BARRETT ESOPHAGUS IF PPI FAILS
Lifelong antireflux therapy is indicated for patients with severe erosive esophagitis or Barrett esophagus. Erosive esophagitis recurs in more than 80% within 12 months of discontinuing antisecretory therapy.52 Both Barrett esophagus and esophageal adenocarcinoma are strongly associated with GERD, and nearly 10% of patients with chronic reflux have Barrett esophagus.53,54 It is suspected that suppressing reflux reduces the rate of progression of Barrett esophagus to esophageal adenocarcinoma, but this remains to be proven.
Perhaps the strongest indication for surgery in the PPI era is for patients who have persistent symptoms and severe erosive esophagitis (Los Angeles grade C or D) despite high-dose PPI therapy. If other causes of persistent esophagitis have been ruled out, fundoplication can induce healing and improve symptoms.55,56 In these cases, surgery is done to induce remission of the disease when maximal medical therapy has been truly unsuccessful.
Randomized controlled trials suggest that medical and surgical therapies are equally effective for preventing the recurrence of erosive esophagitis or the progression of Barrett esophagus. In a study of 225 patients, at 7 years of follow-up, esophagitis had recurred in 10.4% of patients on omeprazole vs 11.8% of those who had undergone antireflux surgery.40 Similarly, open Nissen fundoplication was no different from drug therapy (histamine-2 receptor antagonist or PPI) for progression of Barrett esophagus over a median of 5 years.57 A meta-analysis with nearly 5,000 person-years each in the medical and surgical groups also found no significant difference in rates of cancer progression.58
Notably, symptoms such as dysphagia, flatulence, and the inability to burp occurred significantly more often in the surgical groups in these studies.
In view of these data, antireflux surgery has no significant advantage over medical therapy for maintaining healing of erosive esophagitis or preventing progression of Barrett esophagus. Thus, it should be reserved for patients who do not desire lifelong drug therapy, provided they understand that there is no therapeutic advantage for their esophagitis or for Barrett esophagus.
SPECIFIC INDICATIONS FOR ANTIREFLUX SURGERY
Now that we have PPIs, several situations remain in which surgery for GERD is either indicated or worth considering.
Antireflux surgery is clearly indicated for:
- Patients with erosive esophagitis that does not heal with maximal drug therapy
- Patients with volume regurgitation, particularly if it occurs at night or if there is evidence of aspiration
- Patients who require lifelong treatment for reflux but who have had a serious adverse event related to PPI therapy, such as refractory Clostridium difficile infection.
Antireflux surgery is also worth considering in patients who for personal reasons wish to avoid long-term or lifelong drug therapy.
Patients should be informed, however, that antireflux surgery has not been shown to be better than medical therapy for maintaining remission of symptoms, for preventing progression of Barrett esophagus, or for maintaining healing of erosive esophagitis. Medical therapy is still the first option for these patients.
Surgery may also be considered in patients with persistent symptoms who have a partial response to medical therapy, who show persistent acidic or weakly acidic reflux on MII-pH testing, and whose symptoms have been correlated with reflux events. Although surgery is not sure to improve their symptoms, benefit is more likely in this patient population compared with those without these characteristics.
Extraesophageal GERD
In patients suspected of having extraesophageal GERD, surgery should be considered if typical GERD symptoms are present and improve with PPI therapy, if the extraesophageal syndrome partially responds to PPI therapy, and if MII-pH testing demonstrates a correlation between symptoms and reflux. Surgery may have a stronger indication in this setting if the patient has nocturnal reflux or extraesophageal symptoms.
When is surgery not an option?
In general, surgery should not be considered in patients who do not have a partial response to PPI therapy or who do not have a strong symptom-reflux correlation on MII-pH testing. In all cases of failed medical therapy without persistent severe erosive disease, the threshold for opting for surgery should be high, given the uncertain response of these patients to surgery.
Peristaltic dysfunction is a relative but not an absolute contraindication to surgery.59
RISKS, BENEFITS OF SURGERY FOR GERD
The patient’s preference for surgery over drug therapy should always be balanced against the risks of surgery, including both short-term and long-term adverse events, to allow the patient to make an adequately informed decision (Table 2).2,26
Adverse events associated with PPI therapy are rare and in many cases the association is debatable.26 Nonetheless, long-term PPI therapy has been most strongly associated with an increased risk of C difficile infection and other enteric infections, although the absolute risk of these events remains low.
Complication rates after antireflux surgery depend on the surgeon’s experience and technique. Death is exceedingly rare. In most high-volume centers, the need to convert from laparoscopic to open fundoplication occurs in fewer than 2.4% of patients.2
Potential perioperative complications include perforation (4%), wound infection (3%), and pneumothorax (2%).2
Antireflux surgery is also associated with a significant risk of dysphagia, bloating, an inability to burp, and excessive flatulence, all of which can markedly impair the quality of life.
A major consideration is that fundoplication is generally irreversible. Reoperation rates have been reported to range from 0% to 15%.2 Furthermore, up to 50% of patients still need medical therapy after surgery.60,61 Of note, only about 25% of patients on medical therapy after surgery will actually have an abnormal pH study.61
MORE STUDY NEEDED
Future studies directly comparing medical and surgical therapy for carefully selected patients with extraesophageal manifestations of GERD and refractory symptoms should help further delineate outcome in this difficult group of patients.
Under development are new drugs that may inhibit transient relaxation of the lower esophageal sphincter, as well as minimally invasive procedures, which may alter the indications for surgery in coming years.36
Acknowledgment: The research for this article was supported in part by a grant from the National Institutes of Health (T32 DK07634).
For most patients with gastroesophageal reflux disease (GERD), a proton pump inhibitor (PPI) is the first choice for treatment.1 But some patients have symptoms that persist despite PPI therapy, some desire surgery despite successful PPI therapy, and some have persistent extraesophageal symptoms or other complications of reflux. For these patients, surgery is an option.2
In this article, we review the management of GERD and clarify the indications for antireflux surgery based on evidence of safety and efficacy.
GERD DEFINED: SYMPTOMS OR COMPLICATIONS
Defining the role of antireflux surgery is difficult, given the variety of presentations and the absence of a gold standard for diagnosing GERD. Most adults experience several episodes of physiologic reflux daily without symptoms.3 But a broad array of symptoms have been attributed to GERD, including chest pain, cough, and sore throat, and some conditions caused by acid reflux (eg, Barrett esophagus) can be asymptomatic.4,5
HEARTBURN ISN’T ALWAYS GERD
Typical GERD presents with the classic symptoms of pyrosis (heartburn) or acid regurgitation, or both.
Although these symptoms are often thought to be specific for GERD, other causes of esophageal injury— eg, eosinophilic esophagitis, infection (Candida, cytomegalovirus, herpes simplex virus), pill-induced esophagitis, or radiation therapy—can produce similar symptoms. Other causes, including coronary artery disease, biliary colic, foregut malignancy, or peptic ulcer disease, should also be considered in patients with supposedly typical GERD. Life-threatening mimics of GERD, such as unstable angina, should be excluded if they are likely, before proceeding with evaluating for possible GERD. Therefore, the initial history and examination should focus on appropriate diagnosis, with careful delineation of symptom quality.
Alarm features for advanced pathology6–8 include involuntary weight loss, dysphagia, vomiting, evidence of gastrointestinal blood loss, anemia, chest pain, and an epigastric mass.7 Admittedly, these features are only mediocre for detecting or excluding gastric or esophageal cancer, with a sensitivity of 67% and a specificity 66%.9 Nevertheless, they should prompt an endoscopic examination. In patients who have alarm features but have not yet been treated for GERD, upper endoscopy can identify an abnormality in about 60% of patients.10–12
PPIs HAVE REPLACED ANTACIDS AND HISTAMINE-2 RECEPTOR ANTAGONISTS
When the symptoms suggest GERD and no alarm features are present, an initial trial of the following lifestyle changes is reasonable:
- Avoiding acidic or refluxogenic foods (coffee, alcohol, chocolate, peppermint, fatty foods, citrus foods)
- Avoiding certain medications (anticholinergics, estrogens, calcium-channel blockers, nitroglycerine, benzodiazepines)
- Losing weight
- Quitting smoking
- Raising the head of the bed
- Staying upright for 2 to 3 hours after meals.
For someone with mild symptoms, these changes pose minimal risk. Unfortunately, they are unlikely to provide adequate symptom control for most patients.13–17
Before PPIs were invented, drug therapy for GERD symptoms that did not resolve with lifestyle changes consisted of antacids and, later, histamine-2 receptor antagonists. When maximal therapy failed to control symptoms, fundoplication surgery was considered an appropriate next step.
PPIs substantially changed the management of GERD, suppressing acid secretion much better than histamine-2 receptor antagonists. Taken 30 minutes before breakfast, a single daily dose of a PPI normalizes esophageal acid exposure in 67% of patients.18 Adding a second dose 30 minutes before dinner raises the number to more than 90%.19
PPIs have consistently outperformed histamine-2 blockers in the healing of esophagitis and in improving heartburn symptoms and are now the first-line medical therapy for uncomplicated GERD.6,8,20–25
WHEN PPIs WORK, SURGERY OFFERS NO ADVANTAGE
The LOTUS trial (Long-Term Usage of Esomeprazole vs Surgery for Treatment of Chronic GERD) compared long-term drug therapy with surgery to maintain remission of symptoms in GERD.27 In this trial, 554 patients whose symptoms initially responded to the PPI esomeprazole (Nexium) were randomized to continue to receive esomeprazole (n = 266) or to undergo laparoscopic antireflux surgery (288 were randomly assigned, and 248 had the operation). Dose adjustment of the esomeprazole was allowed (20–40 mg/day). A total of 372 patients completed 5 years of follow-up (192 esomeprazole, 180 surgery).
Symptoms stayed in remission in 92% of the esomeprazole group and 85% of the surgery group (P = .048). However, the difference was no longer statistically significant after modeling the effects of study dropout. The rate of severe adverse events was similar in both groups: 24.1% with esomeprazole and 28.6% with surgery.
These findings indicate that if symptoms fully abate with medical therapy, surgery offers no advantage. In addition, patients who desire surgery in the hope of avoiding lifelong drug therapy should be made aware that drug therapy and reoperation are often necessary after surgery.28 In most cases, antireflux surgery is unnecessary for patients whose GERD fully responds to PPI therapy.
IF PPIs FAIL, FURTHER TESTING NEEDED
But many patients who take PPIs still have symptoms, even though these drugs suppress acid secretion and heal esophagitis. In fact, symptoms completely resolve in only about one-half of patients with erosive disease and one-third of those without erosive disease.21
Reasons for an incomplete symptomatic response to PPIs are various. Acid reflux can persist, but this accounts for only 10% of cases.29 About one-third of patients have persistent reflux that is weakly acidic, with a pH higher than 4.29. However, most patients with persistent typical GERD symptoms do not have significant, persistent reflux, or their symptoms are not related to reflux events. In these cases, an alternative cause of the refractory symptoms should be sought.
Further diagnostic testing is indicated when symptoms persist despite PPI therapy. Upper endoscopy will reveal an abnormality such as persistent erosive esophagitis, eosinophilic esophagitis, esophageal stricture, Barrett esophagus, or esophageal cancer in roughly 10% of patients in whom empiric therapy fails.10
Although patients with persistent symptoms have not been enrolled in many randomized controlled trials, a multivariate analysis showed that failure of medical therapy heralds a poor response to surgery.30 Data such as these have led most experts to discourage fundoplication for such patients now, unlike in the pre-PPI era.
pH and intraluminal impedance testing
However, this recommendation against surgery is not a hard-and-fast rule.
In patients with esophageal regurgitation, most will not achieve adequate relief of symptoms with PPI therapy alone.34 The therapeutic gain of PPI therapy vs placebo averaged just 17% in seven randomized, controlled trials, more than 20% less than the response rate for heartburn.34 This is likely because of structural abnormalities such as reduced lower esophageal sphincter pressure, hiatal hernia, or delayed gastric emptying. Antireflux surgery can correct these structural abnormalities or prevent them from causing so much trouble; however, the presence of true regurgitation should first be confirmed by MII testing. If regurgitation is confirmed, antireflux surgery is warranted, particularly in patients with nocturnal symptoms who may be at high risk of aspiration. With careful patient selection, regurgitation symptoms improve in about 90% after surgery.2
In patients with heartburn, if esophageal acid exposure continues to be abnormal on MII-pH testing, then an escalation of therapy may improve symptoms, particularly if symptoms occur during reflux or if they partially responded to PPI therapy. Options in this scenario include alteration or intensification of acid-suppressive therapy, treatment with baclofen (Lioresal), and antireflux surgery.18,35,36 In randomized controlled trials of patients whose symptoms partially responded to PPIs, antireflux surgery has performed similarly to PPIs in terms of improving typical GERD symptoms, particularly regurgitation.27,37–41 Although this scenario is a reasonable indication for antireflux surgery, recommendations should be made with appropriate restraint since it is not easily reversible, some patients experience complications, and up to one-third will have no therapeutic benefit.30
Nonacid reflux. In some cases, MII-pH testing during PPI therapy will reveal reflux of weakly acidic (pH > 4) or alkaline stomach contents, often called “nonacid reflux.”29 Nonacid reflux is often present in patients with esophagitis that persists despite PPI therapy, indicating that even weakly acidic stomach contents can injure the mucosa.42 Since intensifying the acid-suppressive therapy is unlikely to improve these symptoms, antireflux surgery may have a role.
In one study,43 nonacid reflux was well controlled by laparoscopic Nissen fundoplication, although 15 (48%) of 31 patients had persistent symptoms of GERD after surgery. No patient had a strong symptom correlation with postoperative reflux events, suggesting an alternative cause of the persistent symptoms. Therefore, antireflux surgery for nonacid reflux should be limited exclusively to patients with strong symptom correlation, and even then it should be considered with restraint, given the limited evidence for benefit and the potential for harm.
If testing is negative. In studies investigating the diagnostic yield of MII-pH testing, more than 50% of patients who had refractory symptoms had a negative MII-pH test.29 In such situations, when the symptoms are strongly correlated with reflux events, the patient is said to have “esophageal hypersensitivity.” A few small studies have suggested that such patients may benefit from surgery, but these data have not been replicated in randomized controlled trials.32
When the testing is negative and there is no correlation between the patient’s symptoms and reflux events, the patient is unlikely to benefit from antireflux surgery. Care of these patients is beyond the scope of this review.
SURGERY RARELY IMPROVES COUGH, ASTHMA, OR LARYNGITIS
GERD has been implicated as a cause of chronic cough, asthma, and laryngitis, although each of these has many potential causes.44–46 Despite these associations, the evidence for therapeutic benefit from antireflux therapy is weak.
PPI therapy shows no benefit over placebo for chronic cough of uncertain etiology, but some benefit if GERD is objectively demonstrated.47 Laryngitis resolved in just 15% of patients on esomeprazole vs 16% of patients on placebo after excluding patients with moderate to severe heartburn.48
In a large randomized controlled trial in patients with asthma, there was no overall improvement in peak flow for the PPI group vs the placebo group, although significant improvement occurred in patients with heartburn and nocturnal respiratory symptoms.46
Potent antisecretory therapy seems to improve extraesophageal symptoms when typical GERD symptoms are also present, but it otherwise has shown little evidence of benefit.
The evidence for a benefit from antireflux surgery in patients with extraesophageal GERD syndromes is even more limited. Although one systematic review49 found that cough and other laryngeal symptoms improved in 60% to 100% of patients with objective evidence of GERD who underwent fundoplication, virtually all of the studies were uncontrolled case series.49
The lone randomized controlled trial in the systematic review compared Nissen fundoplication with ranitidine (Zantac) or antacids only for patients with asthma and GERD, and found no significant difference in peak expiratory flow among the three groups after 2 years. However, asthma symptom scores improved in 75% of the surgical group, 9% of the medical group, and 4% of the control group.50
In a study that was not included in the prior systematic review, patients with laryngopharyngeal reflux unresponsive to aggressive acid suppression who subsequently underwent fundoplication fared no better than those who did not.51
Thus, based on the available data, antireflux surgery is only rarely indicated for extraesophageal symptoms, especially in patients who have no typical GERD symptoms or in patients whose symptoms are refractory to medical therapy.
SURGERY FOR EROSIVE ESOPHAGITIS OR BARRETT ESOPHAGUS IF PPI FAILS
Lifelong antireflux therapy is indicated for patients with severe erosive esophagitis or Barrett esophagus. Erosive esophagitis recurs in more than 80% within 12 months of discontinuing antisecretory therapy.52 Both Barrett esophagus and esophageal adenocarcinoma are strongly associated with GERD, and nearly 10% of patients with chronic reflux have Barrett esophagus.53,54 It is suspected that suppressing reflux reduces the rate of progression of Barrett esophagus to esophageal adenocarcinoma, but this remains to be proven.
Perhaps the strongest indication for surgery in the PPI era is for patients who have persistent symptoms and severe erosive esophagitis (Los Angeles grade C or D) despite high-dose PPI therapy. If other causes of persistent esophagitis have been ruled out, fundoplication can induce healing and improve symptoms.55,56 In these cases, surgery is done to induce remission of the disease when maximal medical therapy has been truly unsuccessful.
Randomized controlled trials suggest that medical and surgical therapies are equally effective for preventing the recurrence of erosive esophagitis or the progression of Barrett esophagus. In a study of 225 patients, at 7 years of follow-up, esophagitis had recurred in 10.4% of patients on omeprazole vs 11.8% of those who had undergone antireflux surgery.40 Similarly, open Nissen fundoplication was no different from drug therapy (histamine-2 receptor antagonist or PPI) for progression of Barrett esophagus over a median of 5 years.57 A meta-analysis with nearly 5,000 person-years each in the medical and surgical groups also found no significant difference in rates of cancer progression.58
Notably, symptoms such as dysphagia, flatulence, and the inability to burp occurred significantly more often in the surgical groups in these studies.
In view of these data, antireflux surgery has no significant advantage over medical therapy for maintaining healing of erosive esophagitis or preventing progression of Barrett esophagus. Thus, it should be reserved for patients who do not desire lifelong drug therapy, provided they understand that there is no therapeutic advantage for their esophagitis or for Barrett esophagus.
SPECIFIC INDICATIONS FOR ANTIREFLUX SURGERY
Now that we have PPIs, several situations remain in which surgery for GERD is either indicated or worth considering.
Antireflux surgery is clearly indicated for:
- Patients with erosive esophagitis that does not heal with maximal drug therapy
- Patients with volume regurgitation, particularly if it occurs at night or if there is evidence of aspiration
- Patients who require lifelong treatment for reflux but who have had a serious adverse event related to PPI therapy, such as refractory Clostridium difficile infection.
Antireflux surgery is also worth considering in patients who for personal reasons wish to avoid long-term or lifelong drug therapy.
Patients should be informed, however, that antireflux surgery has not been shown to be better than medical therapy for maintaining remission of symptoms, for preventing progression of Barrett esophagus, or for maintaining healing of erosive esophagitis. Medical therapy is still the first option for these patients.
Surgery may also be considered in patients with persistent symptoms who have a partial response to medical therapy, who show persistent acidic or weakly acidic reflux on MII-pH testing, and whose symptoms have been correlated with reflux events. Although surgery is not sure to improve their symptoms, benefit is more likely in this patient population compared with those without these characteristics.
Extraesophageal GERD
In patients suspected of having extraesophageal GERD, surgery should be considered if typical GERD symptoms are present and improve with PPI therapy, if the extraesophageal syndrome partially responds to PPI therapy, and if MII-pH testing demonstrates a correlation between symptoms and reflux. Surgery may have a stronger indication in this setting if the patient has nocturnal reflux or extraesophageal symptoms.
When is surgery not an option?
In general, surgery should not be considered in patients who do not have a partial response to PPI therapy or who do not have a strong symptom-reflux correlation on MII-pH testing. In all cases of failed medical therapy without persistent severe erosive disease, the threshold for opting for surgery should be high, given the uncertain response of these patients to surgery.
Peristaltic dysfunction is a relative but not an absolute contraindication to surgery.59
RISKS, BENEFITS OF SURGERY FOR GERD
The patient’s preference for surgery over drug therapy should always be balanced against the risks of surgery, including both short-term and long-term adverse events, to allow the patient to make an adequately informed decision (Table 2).2,26
Adverse events associated with PPI therapy are rare and in many cases the association is debatable.26 Nonetheless, long-term PPI therapy has been most strongly associated with an increased risk of C difficile infection and other enteric infections, although the absolute risk of these events remains low.
Complication rates after antireflux surgery depend on the surgeon’s experience and technique. Death is exceedingly rare. In most high-volume centers, the need to convert from laparoscopic to open fundoplication occurs in fewer than 2.4% of patients.2
Potential perioperative complications include perforation (4%), wound infection (3%), and pneumothorax (2%).2
Antireflux surgery is also associated with a significant risk of dysphagia, bloating, an inability to burp, and excessive flatulence, all of which can markedly impair the quality of life.
A major consideration is that fundoplication is generally irreversible. Reoperation rates have been reported to range from 0% to 15%.2 Furthermore, up to 50% of patients still need medical therapy after surgery.60,61 Of note, only about 25% of patients on medical therapy after surgery will actually have an abnormal pH study.61
MORE STUDY NEEDED
Future studies directly comparing medical and surgical therapy for carefully selected patients with extraesophageal manifestations of GERD and refractory symptoms should help further delineate outcome in this difficult group of patients.
Under development are new drugs that may inhibit transient relaxation of the lower esophageal sphincter, as well as minimally invasive procedures, which may alter the indications for surgery in coming years.36
Acknowledgment: The research for this article was supported in part by a grant from the National Institutes of Health (T32 DK07634).
- Finks JF, Wei Y, Birkmeyer JD. The rise and fall of antireflux surgery in the United States. Surg Endosc 2006; 20:1698–1701.
- Stefanidis D, Hope WW, Kohn GP, Reardon PR, Richardson WS, Fanelli RD; SAGES Guidelines Committee. Guidelines for surgical treatment of gastroesophageal reflux disease. Surg Endosc 2010; 24:2647–2669.
- Richter JE. Typical and atypical presentations of gastroesophageal reflux disease. The role of esophageal testing in diagnosis and management. Gastroenterol Clin North Am 1996; 25:75–102.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Dickman R, Kim JL, Camargo L, et al. Correlation of gastroesophageal reflux disease symptoms characteristics with long-segment Barrett’s esophagus. Dis Esophagus 2006; 19:360–365.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Armstrong D, Marshall JK, Chiba N, et al; Canadian Association of Gastroenterology GERD Consensus Group. Canadian Consensus Conference on the management of gastroesophageal reflux disease in adults - update 2004. Can J Gastroenterol 2005; 19:15–35.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.
- Vakil N, Moayyedi P, Fennerty MB, Talley NJ. Limited value of alarm features in the diagnosis of upper gastrointestinal malignancy: systematic review and meta-analysis. Gastroenterology 2006; 131:390–401.
- Poh CH, Gasiorowska A, Navarro-Rodriguez T, et al. Upper GI tract findings in patients with heartburn in whom proton pump inhibitor treatment failed versus those not receiving antireflux treatment. Gastrointest Endosc 2010; 71:28–34.
- Dickman R, Mattek N, Holub J, Peters D, Fass R. Prevalence of upper gastrointestinal tract findings in patients with noncardiac chest pain versus those with gastroesophageal reflux disease (GERD)-related symptoms: results from a national endoscopic database. Am J Gastroenterol 2007; 102:1173–1179.
- Voutilainen M, Sipponen P, Mecklin JP, Juhola M, Färkkilä M. Gastroesophageal reflux disease: prevalence, clinical, endoscopic and histopathological findings in 1,128 consecutive patients referred for endoscopy due to dyspeptic and reflux symptoms. Digestion 2000; 61:6–13.
- Fraser-Moodie CA, Norton B, Gornall C, Magnago S, Weale AR, Holmes GK. Weight loss has an independent beneficial effect on symptoms of gastro-oesophageal reflux in patients who are overweight. Scand J Gastroenterol 1999; 34:337–340.
- Jacobson BC, Somers SC, Fuchs CS, Kelly CP, Camargo CA. Bodymass index and symptoms of gastroesophageal reflux in women. N Engl J Med 2006; 354:2340–2348.
- Kjellin A, Ramel S, Rössner S, Thor K. Gastroesophageal reflux in obese patients is not reduced by weight reduction. Scand J Gastroenterol 1996; 31:1047–1051.
- Waring JP, Eastwood TF, Austin JM, Sanowski RA. The immediate effects of cessation of cigarette smoking on gastroesophageal reflux. Am J Gastroenterol 1989; 84:1076–1078.
- Pehl C, Waizenhoefer A, Wendl B, Schmidt T, Schepp W, Pfeiffer A. Effect of low and high fat meals on lower esophageal sphincter motility and gastroesophageal reflux in healthy subjects. Am J Gastroenterol 1999; 94:1192–1196.
- Bajbouj M, Becker V, Phillip V, Wilhelm D, Schmid RM, Meining A. High-dose esomeprazole for treatment of symptomatic refractory gastroesophageal reflux disease—a prospective pH-metry/impedance-controlled study. Digestion 2009; 80:112–118.
- Charbel S, Khandwala F, Vaezi MF. The role of esophageal pH monitoring in symptomatic patients on PPI therapy. Am J Gastroenterol 2005; 100:283–289.
- Khan M, Santana J, Donnellan C, Preston C, Moayyedi P. Medical treatments in the short term management of reflux oesophagitis. Cochrane Database Syst Rev 2007;CD003244.
- Dean BB, Gano AD, Knight K, Ofman JJ, Fass R. Effectiveness of proton pump inhibitors in nonerosive reflux disease. Clin Gastroenterol Hepatol 2004; 2:656–664.
- Sabesin SM, Berlin RG, Humphries TJ, Bradstreet DC, Walton-Bowen KL, Zaidi S. Famotidine relieves symptoms of gastroesophageal reflux disease and heals erosions and ulcerations. Results of a multicenter, placebo-controlled, dose-ranging study. USA Merck Gastroesophageal Reflux Disease Study Group. Arch Intern Med 1991; 151:2394–2400.
- van Pinxteren B, Numans ME, Bonis PA, Lau J. Short-term treatment with proton pump inhibitors, H2-receptor antagonists and prokinetics for gastro-oesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Database Syst Rev 2004;CD002095.
- Chiba N, De Gara CJ, Wilkinson JM, Hunt RH. Speed of healing and symptom relief in grade II to IV gastroesophageal reflux disease: a meta-analysis. Gastroenterology 1997; 112:1798–1810.
- Venables TL, Newland RD, Patel AC, Hole J, Wilcock C, Turbitt ML. Omeprazole 10 milligrams once daily, omeprazole 20 milligrams once daily, or ranitidine 150 milligrams twice daily, evaluated as initial therapy for the relief of symptoms of gastro-oesophageal reflux disease in general practice. Scand J Gastroenterol 1997; 32:965–973.
- Madanick RD. Proton pump inhibitor side effects and drug interactions: much ado about nothing? Cleve Clin J Med 2011; 78:39–49.
- Galmiche JP, Hatlebakk J, Attwood S, et al; LOTUS Trial Collaborators. Laparoscopic antireflux surgery vs esomeprazole treatment for chronic GERD: the LOTUS randomized clinical trial. JAMA 2011; 305:1969–1977.
- Spechler SJ, Lee E, Ahnen D, et al. Long-term outcome of medical and surgical therapies for gastroesophageal reflux disease: followup of a randomized controlled trial. JAMA 2001; 285:2331–2338.
- Mainie I, Tutuian R, Shay S, et al. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: a multicentre study using combined ambulatory impedance-pH monitoring. Gut 2006; 55:1398–1402.
- Campos GM, Peters JH, DeMeester TR, et al. Multivariate analysis of factors predicting outcome after laparoscopic Nissen fundoplication. J Gastrointest Surg 1999; 3:292–300.
- Becker V, Bajbouj M, Waller K, Schmid RM, Meining A. Clinical trial: persistent gastro-oesophageal reflux symptoms despite standard therapy with proton pump inhibitors - a follow-up study of intraluminal-impedance guided therapy. Aliment Pharmacol Ther 2007; 26:1355–1360.
- Mainie I, Tutuian R, Agrawal A, Adams D, Castell DO. Combined multichannel intraluminal impedance-pH monitoring to select patients with persistent gastro-oesophageal reflux for laparoscopic Nissen fundoplication. Br J Surg 2006; 93:1483–1487.
- del Genio G, Tolone S, del Genio F, et al. Prospective assessment of patient selection for antireflux surgery by combined multichannel intraluminal impedance pH monitoring. J Gastrointest Surg 2008; 12:1491–1496.
- Kahrilas PJ, Howden CW, Hughes N. Response of regurgitation to proton pump inhibitor therapy in clinical trials of gastroesophageal reflux disease. Am J Gastroenterol 2011; 106:1419–1425.
- Koek GH, Sifrim D, Lerut T, Janssens J, Tack J. Effect of the GABA(B) agonist baclofen in patients with symptoms and duodeno-gastro-oesophageal reflux refractory to proton pump inhibitors. Gut 2003; 52:1397–1402.
- Boeckxstaens GE. Reflux inhibitors: a new approach for GERD? Curr Opin Pharmacol 2008; 8:685–689.
- Anvari M, Allen C, Marshall J, et al. A randomized controlled trial of laparoscopic nissen fundoplication versus proton pump inhibitors for treatment of patients with chronic gastroesophageal reflux disease: One-year follow-up. Surg Innov 2006; 13:238–249.
- Mahon D, Rhodes M, Decadt B, et al. Randomized clinical trial of laparoscopic Nissen fundoplication compared with proton-pump inhibitors for treatment of chronic gastro-oesophageal reflux. Br J Surg 2005; 92:695–699.
- Mehta S, Bennett J, Mahon D, Rhodes M. Prospective trial of laparoscopic nissen fundoplication versus proton pump inhibitor therapy for gastroesophageal reflux disease: Seven-year follow-up. J Gastrointest Surg 2006; 10:1312–1316.
- Lundell L, Miettinen P, Myrvold HE, et al; Nordic GORD Study Group. Seven-year follow-up of a randomized clinical trial comparing proton-pump inhibition with surgical therapy for reflux oesophagitis. Br J Surg 2007; 94:198–203.
- Lundell L, Attwood S, Ell C, et al; LOTUS trial collaborators. Comparing laparoscopic antireflux surgery with esomeprazole in the management of patients with chronic gastro-oesophageal reflux disease: a 3-year interim analysis of the LOTUS trial. Gut 2008; 57:1207–1213.
- Frazzoni M, Conigliaro R, Melotti G. Weakly acidic refluxes have a major role in the pathogenesis of proton pump inhibitor-resistant reflux oesophagitis. Aliment Pharmacol Ther 2011; 33:601–606.
- Broeders JA, Bredenoord AJ, Hazebroek EJ, Broeders IA, Gooszen HG, Smout AJ. Effects of anti-reflux surgery on weakly acidic reflux and belching. Gut 2011; 60:435–441.
- American Gastroenterological Association medical position statement: guidelines on the use of esophageal pH recording. Gastroenterology 1996; 110:1981.
- el-Serag HB, Sonnenberg A. Comorbid occurrence of laryngeal or pulmonary disease with esophagitis in United States military veterans. Gastroenterology 1997; 113:755–760.
- Kiljander TO, Laitinen JO. The prevalence of gastroesophageal reflux disease in adult asthmatics. Chest 2004; 126:1490–1494.
- Chang AB, Lasserson TJ, Kiljander TO, Connor FL, Gaffney JT, Garske LA. Systematic review and meta-analysis of randomised controlled trials of gastro-oesophageal reflux interventions for chronic cough associated with gastro-oesophageal reflux. BMJ 2006; 332:11–17.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116:254–260.
- Iqbal M, Batch AJ, Spychal RT, Cooper BT. Outcome of surgical fundoplication for extraesophageal (atypical) manifestations of gastroesophageal reflux disease in adults: a systematic review. J Laparoendosc Adv Surg Tech A 2008; 18:789–796.
- Sontag SJ, O’Connell S, Khandelwal S, et al. Asthmatics with gastroesophageal reflux: long term results of a randomized trial of medical and surgical antireflux therapies. Am J Gastroenterol 2003; 98:987–999.
- Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngopharyngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol 2006; 4:433–441.
- Johnson DA, Benjamin SB, Vakil NB, et al. Esomeprazole once daily for 6 months is effective therapy for maintaining healed erosive esophagitis and for controlling gastroesophageal reflux disease symptoms: a randomized, double-blind, placebo-controlled study of efficacy and safety. Am J Gastroenterol 2001; 96:27–34.
- Winters C, Spurling TJ, Chobanian SJ, et al. Barrett’s esophagus. A prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology 1987; 92:118–124.
- Westhoff B, Brotze S, Weston A, et al. The frequency of Barrett’s esophagus in high-risk patients with chronic GERD. Gastrointest Endosc 2005; 61:226–231.
- Rosenthal R, Peterli R, Guenin MO, von Flüe M, Ackermann C. Laparoscopic antireflux surgery: long-term outcomes and quality of life. J Laparoendosc Adv Surg Tech A 2006; 16:557–561.
- Broeders JA, Draaisma WA, Bredenoord AJ, Smout AJ, Broeders IA, Gooszen HG. Long-term outcome of Nissen fundoplication in non-erosive and erosive gastro-oesophageal reflux disease. Br J Surg 2010; 97:845–352.
- Parrilla P, Martínez de Haro LF, Ortiz A, et al. Long-term results of a randomized prospective study comparing medical and surgical treatment of Barrett’s esophagus. Ann Surg 2003; 237:291–298.
- Corey KE, Schmitz SM, Shaheen NJ. Does a surgical antireflux procedure decrease the incidence of esophageal adenocarcinoma in Barrett’s esophagus? A meta-analysis. Am J Gastroenterol 2003; 98:2390–2394.
- Pandolfino JE, Kahrilas PJ; American Gastroenterological Association. AGA technical review on the clinical use of esophageal manometry. Gastroenterology 2005; 128:209–224.
- Dominitz JA, Dire CA, Billingsley KG, Todd-Stenberg JA. Complications and antireflux medication use after antireflux surgery. Clin Gastroenterol Hepatol 2006; 4:299–305.
- Lord RV, Kaminski A, Oberg S, et al. Absence of gastroesophageal reflux disease in a majority of patients taking acid suppression medications after Nissen fundoplication. J Gastrointest Surg 2002; 6:3–9.
- Finks JF, Wei Y, Birkmeyer JD. The rise and fall of antireflux surgery in the United States. Surg Endosc 2006; 20:1698–1701.
- Stefanidis D, Hope WW, Kohn GP, Reardon PR, Richardson WS, Fanelli RD; SAGES Guidelines Committee. Guidelines for surgical treatment of gastroesophageal reflux disease. Surg Endosc 2010; 24:2647–2669.
- Richter JE. Typical and atypical presentations of gastroesophageal reflux disease. The role of esophageal testing in diagnosis and management. Gastroenterol Clin North Am 1996; 25:75–102.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Dickman R, Kim JL, Camargo L, et al. Correlation of gastroesophageal reflux disease symptoms characteristics with long-segment Barrett’s esophagus. Dis Esophagus 2006; 19:360–365.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Armstrong D, Marshall JK, Chiba N, et al; Canadian Association of Gastroenterology GERD Consensus Group. Canadian Consensus Conference on the management of gastroesophageal reflux disease in adults - update 2004. Can J Gastroenterol 2005; 19:15–35.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.
- Vakil N, Moayyedi P, Fennerty MB, Talley NJ. Limited value of alarm features in the diagnosis of upper gastrointestinal malignancy: systematic review and meta-analysis. Gastroenterology 2006; 131:390–401.
- Poh CH, Gasiorowska A, Navarro-Rodriguez T, et al. Upper GI tract findings in patients with heartburn in whom proton pump inhibitor treatment failed versus those not receiving antireflux treatment. Gastrointest Endosc 2010; 71:28–34.
- Dickman R, Mattek N, Holub J, Peters D, Fass R. Prevalence of upper gastrointestinal tract findings in patients with noncardiac chest pain versus those with gastroesophageal reflux disease (GERD)-related symptoms: results from a national endoscopic database. Am J Gastroenterol 2007; 102:1173–1179.
- Voutilainen M, Sipponen P, Mecklin JP, Juhola M, Färkkilä M. Gastroesophageal reflux disease: prevalence, clinical, endoscopic and histopathological findings in 1,128 consecutive patients referred for endoscopy due to dyspeptic and reflux symptoms. Digestion 2000; 61:6–13.
- Fraser-Moodie CA, Norton B, Gornall C, Magnago S, Weale AR, Holmes GK. Weight loss has an independent beneficial effect on symptoms of gastro-oesophageal reflux in patients who are overweight. Scand J Gastroenterol 1999; 34:337–340.
- Jacobson BC, Somers SC, Fuchs CS, Kelly CP, Camargo CA. Bodymass index and symptoms of gastroesophageal reflux in women. N Engl J Med 2006; 354:2340–2348.
- Kjellin A, Ramel S, Rössner S, Thor K. Gastroesophageal reflux in obese patients is not reduced by weight reduction. Scand J Gastroenterol 1996; 31:1047–1051.
- Waring JP, Eastwood TF, Austin JM, Sanowski RA. The immediate effects of cessation of cigarette smoking on gastroesophageal reflux. Am J Gastroenterol 1989; 84:1076–1078.
- Pehl C, Waizenhoefer A, Wendl B, Schmidt T, Schepp W, Pfeiffer A. Effect of low and high fat meals on lower esophageal sphincter motility and gastroesophageal reflux in healthy subjects. Am J Gastroenterol 1999; 94:1192–1196.
- Bajbouj M, Becker V, Phillip V, Wilhelm D, Schmid RM, Meining A. High-dose esomeprazole for treatment of symptomatic refractory gastroesophageal reflux disease—a prospective pH-metry/impedance-controlled study. Digestion 2009; 80:112–118.
- Charbel S, Khandwala F, Vaezi MF. The role of esophageal pH monitoring in symptomatic patients on PPI therapy. Am J Gastroenterol 2005; 100:283–289.
- Khan M, Santana J, Donnellan C, Preston C, Moayyedi P. Medical treatments in the short term management of reflux oesophagitis. Cochrane Database Syst Rev 2007;CD003244.
- Dean BB, Gano AD, Knight K, Ofman JJ, Fass R. Effectiveness of proton pump inhibitors in nonerosive reflux disease. Clin Gastroenterol Hepatol 2004; 2:656–664.
- Sabesin SM, Berlin RG, Humphries TJ, Bradstreet DC, Walton-Bowen KL, Zaidi S. Famotidine relieves symptoms of gastroesophageal reflux disease and heals erosions and ulcerations. Results of a multicenter, placebo-controlled, dose-ranging study. USA Merck Gastroesophageal Reflux Disease Study Group. Arch Intern Med 1991; 151:2394–2400.
- van Pinxteren B, Numans ME, Bonis PA, Lau J. Short-term treatment with proton pump inhibitors, H2-receptor antagonists and prokinetics for gastro-oesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Database Syst Rev 2004;CD002095.
- Chiba N, De Gara CJ, Wilkinson JM, Hunt RH. Speed of healing and symptom relief in grade II to IV gastroesophageal reflux disease: a meta-analysis. Gastroenterology 1997; 112:1798–1810.
- Venables TL, Newland RD, Patel AC, Hole J, Wilcock C, Turbitt ML. Omeprazole 10 milligrams once daily, omeprazole 20 milligrams once daily, or ranitidine 150 milligrams twice daily, evaluated as initial therapy for the relief of symptoms of gastro-oesophageal reflux disease in general practice. Scand J Gastroenterol 1997; 32:965–973.
- Madanick RD. Proton pump inhibitor side effects and drug interactions: much ado about nothing? Cleve Clin J Med 2011; 78:39–49.
- Galmiche JP, Hatlebakk J, Attwood S, et al; LOTUS Trial Collaborators. Laparoscopic antireflux surgery vs esomeprazole treatment for chronic GERD: the LOTUS randomized clinical trial. JAMA 2011; 305:1969–1977.
- Spechler SJ, Lee E, Ahnen D, et al. Long-term outcome of medical and surgical therapies for gastroesophageal reflux disease: followup of a randomized controlled trial. JAMA 2001; 285:2331–2338.
- Mainie I, Tutuian R, Shay S, et al. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: a multicentre study using combined ambulatory impedance-pH monitoring. Gut 2006; 55:1398–1402.
- Campos GM, Peters JH, DeMeester TR, et al. Multivariate analysis of factors predicting outcome after laparoscopic Nissen fundoplication. J Gastrointest Surg 1999; 3:292–300.
- Becker V, Bajbouj M, Waller K, Schmid RM, Meining A. Clinical trial: persistent gastro-oesophageal reflux symptoms despite standard therapy with proton pump inhibitors - a follow-up study of intraluminal-impedance guided therapy. Aliment Pharmacol Ther 2007; 26:1355–1360.
- Mainie I, Tutuian R, Agrawal A, Adams D, Castell DO. Combined multichannel intraluminal impedance-pH monitoring to select patients with persistent gastro-oesophageal reflux for laparoscopic Nissen fundoplication. Br J Surg 2006; 93:1483–1487.
- del Genio G, Tolone S, del Genio F, et al. Prospective assessment of patient selection for antireflux surgery by combined multichannel intraluminal impedance pH monitoring. J Gastrointest Surg 2008; 12:1491–1496.
- Kahrilas PJ, Howden CW, Hughes N. Response of regurgitation to proton pump inhibitor therapy in clinical trials of gastroesophageal reflux disease. Am J Gastroenterol 2011; 106:1419–1425.
- Koek GH, Sifrim D, Lerut T, Janssens J, Tack J. Effect of the GABA(B) agonist baclofen in patients with symptoms and duodeno-gastro-oesophageal reflux refractory to proton pump inhibitors. Gut 2003; 52:1397–1402.
- Boeckxstaens GE. Reflux inhibitors: a new approach for GERD? Curr Opin Pharmacol 2008; 8:685–689.
- Anvari M, Allen C, Marshall J, et al. A randomized controlled trial of laparoscopic nissen fundoplication versus proton pump inhibitors for treatment of patients with chronic gastroesophageal reflux disease: One-year follow-up. Surg Innov 2006; 13:238–249.
- Mahon D, Rhodes M, Decadt B, et al. Randomized clinical trial of laparoscopic Nissen fundoplication compared with proton-pump inhibitors for treatment of chronic gastro-oesophageal reflux. Br J Surg 2005; 92:695–699.
- Mehta S, Bennett J, Mahon D, Rhodes M. Prospective trial of laparoscopic nissen fundoplication versus proton pump inhibitor therapy for gastroesophageal reflux disease: Seven-year follow-up. J Gastrointest Surg 2006; 10:1312–1316.
- Lundell L, Miettinen P, Myrvold HE, et al; Nordic GORD Study Group. Seven-year follow-up of a randomized clinical trial comparing proton-pump inhibition with surgical therapy for reflux oesophagitis. Br J Surg 2007; 94:198–203.
- Lundell L, Attwood S, Ell C, et al; LOTUS trial collaborators. Comparing laparoscopic antireflux surgery with esomeprazole in the management of patients with chronic gastro-oesophageal reflux disease: a 3-year interim analysis of the LOTUS trial. Gut 2008; 57:1207–1213.
- Frazzoni M, Conigliaro R, Melotti G. Weakly acidic refluxes have a major role in the pathogenesis of proton pump inhibitor-resistant reflux oesophagitis. Aliment Pharmacol Ther 2011; 33:601–606.
- Broeders JA, Bredenoord AJ, Hazebroek EJ, Broeders IA, Gooszen HG, Smout AJ. Effects of anti-reflux surgery on weakly acidic reflux and belching. Gut 2011; 60:435–441.
- American Gastroenterological Association medical position statement: guidelines on the use of esophageal pH recording. Gastroenterology 1996; 110:1981.
- el-Serag HB, Sonnenberg A. Comorbid occurrence of laryngeal or pulmonary disease with esophagitis in United States military veterans. Gastroenterology 1997; 113:755–760.
- Kiljander TO, Laitinen JO. The prevalence of gastroesophageal reflux disease in adult asthmatics. Chest 2004; 126:1490–1494.
- Chang AB, Lasserson TJ, Kiljander TO, Connor FL, Gaffney JT, Garske LA. Systematic review and meta-analysis of randomised controlled trials of gastro-oesophageal reflux interventions for chronic cough associated with gastro-oesophageal reflux. BMJ 2006; 332:11–17.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116:254–260.
- Iqbal M, Batch AJ, Spychal RT, Cooper BT. Outcome of surgical fundoplication for extraesophageal (atypical) manifestations of gastroesophageal reflux disease in adults: a systematic review. J Laparoendosc Adv Surg Tech A 2008; 18:789–796.
- Sontag SJ, O’Connell S, Khandelwal S, et al. Asthmatics with gastroesophageal reflux: long term results of a randomized trial of medical and surgical antireflux therapies. Am J Gastroenterol 2003; 98:987–999.
- Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngopharyngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol 2006; 4:433–441.
- Johnson DA, Benjamin SB, Vakil NB, et al. Esomeprazole once daily for 6 months is effective therapy for maintaining healed erosive esophagitis and for controlling gastroesophageal reflux disease symptoms: a randomized, double-blind, placebo-controlled study of efficacy and safety. Am J Gastroenterol 2001; 96:27–34.
- Winters C, Spurling TJ, Chobanian SJ, et al. Barrett’s esophagus. A prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology 1987; 92:118–124.
- Westhoff B, Brotze S, Weston A, et al. The frequency of Barrett’s esophagus in high-risk patients with chronic GERD. Gastrointest Endosc 2005; 61:226–231.
- Rosenthal R, Peterli R, Guenin MO, von Flüe M, Ackermann C. Laparoscopic antireflux surgery: long-term outcomes and quality of life. J Laparoendosc Adv Surg Tech A 2006; 16:557–561.
- Broeders JA, Draaisma WA, Bredenoord AJ, Smout AJ, Broeders IA, Gooszen HG. Long-term outcome of Nissen fundoplication in non-erosive and erosive gastro-oesophageal reflux disease. Br J Surg 2010; 97:845–352.
- Parrilla P, Martínez de Haro LF, Ortiz A, et al. Long-term results of a randomized prospective study comparing medical and surgical treatment of Barrett’s esophagus. Ann Surg 2003; 237:291–298.
- Corey KE, Schmitz SM, Shaheen NJ. Does a surgical antireflux procedure decrease the incidence of esophageal adenocarcinoma in Barrett’s esophagus? A meta-analysis. Am J Gastroenterol 2003; 98:2390–2394.
- Pandolfino JE, Kahrilas PJ; American Gastroenterological Association. AGA technical review on the clinical use of esophageal manometry. Gastroenterology 2005; 128:209–224.
- Dominitz JA, Dire CA, Billingsley KG, Todd-Stenberg JA. Complications and antireflux medication use after antireflux surgery. Clin Gastroenterol Hepatol 2006; 4:299–305.
- Lord RV, Kaminski A, Oberg S, et al. Absence of gastroesophageal reflux disease in a majority of patients taking acid suppression medications after Nissen fundoplication. J Gastrointest Surg 2002; 6:3–9.
KEY POINTS
- If a PPI in twice-daily doses fails to relieve GERD symptoms, a pH study combined with multichannel intraluminal impedance testing can help in deciding whether to try surgery.
- Antireflux surgery can be considered for erosive esophagitis that does not resolve with drug therapy, for volume regurgitation (particularly if it occurs at night or if there is a risk of aspiration), and for patients who need lifelong treatment for reflux but have had a serious adverse event related to PPI therapy.
- Studies are needed to directly compare medical and surgical therapy in patients with extraesophageal manifestations of GERD and refractory symptoms, a difficult group of patients.
- Drugs that inhibit transient relaxation of the lower esophageal sphincter are under investigation, as are minimally invasive procedures to manipulate the physical barrier to reflux.