User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
The Post-PASI Era: Considering Comorbidities to Select Appropriate Systemic Psoriasis Treatments
The Post-PASI Era: Considering Comorbidities to Select Appropriate Systemic Psoriasis Treatments
Psoriasis treatments have come a long way in the past 20 years. We now have more than a dozen systemic targeted treatments for psoriatic disease, with more on the way; however, with each successive class of medications introduced, the gap has narrowed in terms of increasing efficacy. In an era of medications reporting complete clearance rates in the 70% range, the average improvement in Psoriasis Area and Severity Index (PASI) for most biologics has remained at 90% to 95% in the past half-decade. While this is a far cry from the mean PASI improvements of 70% seen with the first biologics,1 it is becoming more challenging to base our treatment decisions solely on PASI outcome measures.
How, then, do we approach rational selection of a systemic psoriasis treatment? We could try to delineate based on mechanism of action, but it may be disingenuous to dissect minor differences in pathways (eg, IL-17 vs IL-23) that are fundamentally related and on the same continuum in psoriasis pathophysiology. Therefore, the most meaningful way to select an appropriate therapeutic may be to adopt a patient-centered approach that accounts for both individual preferences and specific medical needs by evaluating for other comorbidities2 to exclude or select certain medicines or types of treatments. We have long known to avoid tumor necrosis factor (TNF) α inhibitors in patients with congestive heart failure or a history of demyelinating disorders while regularly considering the presence of psoriatic arthritis and family planning when making treatment decisions. Now, we can be more nuanced in our approaches to psoriasis biologics. Specifically, the most important comorbidities to consider broadly encompass cardiometabolic disorders, gastrointestinal conditions, and psychiatric conditions.
Cardiometabolic Disorders
Possibly the hottest topic in psoriasis for some years now, the relationship between cardiometabolic disorders and psoriasis is of great interest to clinicians, scientists, and patients alike. There is a clear link between development of atherosclerosis and Th17-related immune mechanisms that also are implicated in the pathogenesis of psoriasis.3 Furthermore, the incidence of cardiovascular disease is markedly increased in patients with psoriasis, which is an independent risk factor for myocardial infarction, particularly among younger patients.4,5 Although several retrospective studies6-8 have shown that TNF-α inhibitors are associated with a reduction in cardiovascular outcomes, it is yet to be seen whether biologic treatment actually has a direct impact on cardiovascular outcomes, multiple studies investigating the effect of biologics on arterial inflammation markers notwithstanding.9
There are some direct factors to keep in mind when considering cardiometabolic comorbidities in patients with psoriasis. Obesity is common in the psoriasis population and can have a direct negative effect on cardiovascular health.10 However, the data on obesity and psoriasis are somewhat mixed with regard to treatment outcomes. In general, with increased volumes of distribution for biologics in patients with obesity, it has been shown that treatment success is more difficult to achieve in those with a body mass index greater than 30.11 Rather surprisingly, a separate nationwide study in South Korea found that patients on biologics for psoriasis were more likely to experience weight gain, even after controlling for factors such as exercise, smoking, and drinking,12 but it is unclear whether this is driven mostly by a known connection between weight gain and TNF-α inhibitors.13 These contrasting results point to the need for further studies in this area, as our intuitive approach would involve promoting weight loss while starting on a systemic treatment for psoriasis—but perhaps it is important not to assume that one will come with the other in tow, reinforcing the need to discuss a healthy diet with our patients with psoriasis regardless of treatment decisions.
The data that we have do not directly answer the big questions about biologic treatment and cardiovascular health, but we are starting to see interesting signals. For example, in a report of tildrakizumab treatment in patients with and without metabolic syndrome, the rates of major adverse cardiovascular events as well as cardiac disorders were essentially the same in both groups after receiving treatment for up to 244 weeks.14 This is interesting, more because of the lack of an increase in cardiovascular adverse events in the metabolic syndrome group, who entered the trial on average 25 kg to 30 kg heavier than those without metabolic syndrome. There is an increased risk for adverse cardiovascular events among patients with metabolic syndrome, a roughly 2-fold relative risk in as few as 5 to 6 years of follow-up.15 While the cohorts in the tildrakizumab study14 were too small to draw firm conclusions, the data are interesting and a step in the right direction; we need much larger data sets for analysis. Among other agents, similar efficacy and safety have been reported for guselkumab in a long-term psoriasis study; as a class, IL-23 inhibitors also tend to perform well from an efficacy standpoint in patients with obesity.16
Overall, when assessing the evidence for cardiometabolic disorders, it is reasonable to consider starting a biologic from the IL-17 or IL-23 inhibitor classes— thus avoiding both the potential downside of weight gain and contraindication in patients with congestive heart failure associated with TNF-α inhibitors. It is important to counsel patients about weight loss in conjunction with these treatments, both to improve efficacy and reduce cardiovascular risk factors. There may be a preference for IL-23 inhibitors in patients with obesity, as this class of medications maintains efficacy particularly well in these patients. Patients with psoriasis should be counseled to follow up with a primary care physician given their higher risk for metabolic syndrome and adverse cardiovascular outcomes.
Gastrointestinal Conditions
Psoriasis and inflammatory bowel disease (IBD) have a bidirectional association, and patients with psoriasis are about 1.7 times more likely to have either Crohn disease or ulcerative colitis.17,18 This association may be related to a shared pathogenesis with regard to immune dysregulation and overactivated inflammatory pathways, but there are some important differences to consider from a therapeutic standpoint. Given the increased expression of IL-17 in patients with IBD,19 a phase II trial of secukinumab yielded surprising results—not only was secukinumab ineffective in treating Crohn disease, but there also were higher rates of adverse events20 (as noted on the product label for all IL-17 inhibitors). We have come to understand that there are regulatory subsets of IL-17 cells that are important in mucosal homeostasis and also regulate IL-10, which generally is considered an anti-inflammatory cytokine.21 Thus, while IL-17 inhibition can reduce some component of inflammatory signaling, it also can increase inflammatory signaling through indirect pathways while increasing intestinal permeability to microbes. Importantly, this process seems to occur via IL-23–independent pathways; as such, while direct inhibition of IL-17 can be deleterious, IL-23 inhibitors have become important therapeutics for IBD.22
IL-17 family, IL-17A clearly is the culprit for worsening colitis as evidenced by both human and animal models. On the contrary, IL-17F blockade has been shown to ameliorate colitis in a murine model, whereas IL-17A inhibition worsens it.23 Furthermore, dual blockade of IL-17A and IL-17F has a protective effect against colitis, suggesting that the IL-17F inhibition is dominant. This interesting finding has some mechanistic backing, since blockade of IL-17F induces Treg cells that serve to maintain gut epithelium homeostasis and integrity.24
Overall, IL-17A inhibitors should be avoided in patients with a history of IBD—namely, secukinumab and ixekizumab. While there may be theoretical reasons that brodalumab or bimekizumab may confer a somewhat different risk for IBD exacerbation, there may be better choices that would be expected to effectively treat both the psoriasis and IBD manifestations. Given the US Food and Drug Administration approval of IL-23 inhibitors for IBD and their high levels of efficacy in treating psoriasis, these likely will be the best choice for most patients. Another mainstay of IBD treatment is TNF-α inhibitors, but they come with other risks such as increased immunosuppression and increased risk for nonmelanoma skin cancer.
An important question remains: What about patients who do not have known IBD? Do we proactively change our treatment choice due to fear of IBD development given the higher incidence of both Crohn disease and ulcerative colitis in patients with psoriasis? What about patients with a family history of IBD? First-degree relatives of patients with Crohn disease and ulcerative colitis have an 8- and 4-fold higher risk for those same conditions, respectively.25 Postmarketing surveillance and database findings of low rates of IBD development with IL-17 inhibitors gives only modest reassurance, as dermatologists generally know to avoid these medications for patients with even questionable IBD symptoms. It is important to emphasize to our patients that in no case do we believe that a psoriasis medication actually will cause IBD—rather, someone with subclinical IBD could experience a flare and a first manifestation of colitis. The drug is not the culprit in inducing IBD but rather may serve to unmask existing disease.
One study suggested that for patients who move on to the IL-17 inhibitor secukinumab after being treated with TNF-α inhibitors for psoriasis, the rates of IBD development are higher (4.8%) than in those who start IL-17A inhibition without prior treatment (1%)(OR, 8.38; P=.018).26 This begs the question of whether subclinical IBD in many patients with psoriasis who are treated with TNF-α inhibitors can be unmasked later when they are transitioned to a treatment that either does not treat the IBD or could worsen it. There may be a mechanistic drive behind this sequencing of treatments that predisposes patients to colitis, which would suggest selecting an IL-23 inhibitor after failing/trying a TNF-α inhibitor. However, the data are very preliminary, and in real practice, other concerns such as severe psoriatic arthritis may outweigh these considerations, as the IL-17 inhibitor class still is considered to be more effective than IL-23 inhibition at treating psoriatic arthritis overall. For most patients with no personal history of IBD and no strong family history of IBD (ie, first-degree relatives), the choice of biologic should not be affected by concern over gastrointestinal issues.
Psychiatric Conditions
It has been well established that psoriasis is linked to higher rates of depression, anxiety, and suicidality.27 How do we take this into account when treating patients with psoriasis, especially when we have biologics with a warning label for suicidality and a Risk Evaluation and Mitigation Strategies program (brodalumab) and language around suicidal ideation in the label (bimekizumab)? While it is challenging to discuss mental health, it is not a conversation that we as dermatologists should shy away from. Appropriate treatment of psoriasis is an important tool to get our patients on the path to better mental health. A recent database study of more than 4000 patients showed that patients with psoriasis treated with biologics had a 17% lower risk for depression than those treated with conventional disease-modifying drugs such as methotrexate.28 The comparator of the conventional disease-modifying drug class is important as it serves as a control for disease severity. Too often, a higher rate of depression, anxiety, or suicidality can be attributed to a medication when we in fact may just be capturing the background of higher incidence of all 3 in patients with severe psoriasis.
Indeed, even with the medication that many worry about on this front (brodalumab), multiple studies have confirmed that the effect on mental health generally is a positive one, with decreases in depressive symptoms.29 In a cohort switched from TNF-α inhibitors to brodalumab, symptoms of depression actually improved,30 so attributing a direct treatment effect to negative mental health outcomes does not seem to be justified, especially in light of the low number of suicide events in global postmarketing surveillance for brodalumab, comparable to or lower than other biologics for psoriasis.31 Similarly, bimekizumab has language in the label about discussing suicidality with patients, although the rates of suicidal ideation and behavior are no different from other biologics and rates of depression improved with its use.32
Heightened awareness of our patients’ mental health is something that we as providers should embrace, even when it seems that we do not have much time to see each patient. The priority when a patient comes in with mental health symptoms should be to treat what is within our scope (ie, psoriasis) as quickly and effectively as possible— with a newer-generation biologic such as an IL-17 or IL-23 inhibitor—while encouraging the patient to seek care from a mental health professional. In these cases, one might even argue that the rapidity of action of IL-17 inhibitors may be of additional benefit.
Final Thoughts
We as dermatologists generally are tasked with seeing high volumes of patients, and an initial psoriasis consultation can be a lengthy visit; however, it is rewarding to establish this relationship with patients and a reminder of why we practice medicine to begin with. Psoriasis can be satisfying to treat, and we have so many highly effective medicines that can completely transform our patients’ lives. Applying an understanding of the interplay between psoriasis, its related comorbidities, and treatment choices can be a fulfilling exercise that captures the essence of shared decision-making, which can lead to better outcomes and satisfaction for both providers and patients.
- Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349:2014-2022. doi:10.1056/NEJMoa030409
- Thatiparthi A, Martin A, Liu J, et al. Biologic treatment algorithms for moderate-to-severe psoriasis with comorbid conditions and special populations: a review. Am J Clin Dermatol. 2021;22:425-442. doi:10.1007/s40257-021-00603-w
- Packard RR, Lichtman AH, Libby P. Innate and adaptive immunity in atherosclerosis. Semin Immunopathol. 2009;31:5-22. doi:10.1007 /s00281-009-0153-8
- Gelfand JM, Neimann AL, Shin DB, et al. Risk of myocardial infarction in patients with psoriasis. JAMA. 2006;296:1735-1741. doi:10.1001/jama.296.14.1735
- Miller IM, Ellervik C, Yazdanyar S, et al. Meta-analysis of psoriasis, cardiovascular disease, and associated risk factors. J Am Acad Dermatol. 2013;69:1014-1024. doi:10.1016/j.jaad.2013.06.053
- Wu JJ, Guerin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-alpha inhibitors versus methotrexate. J Am Acad Dermatol. 2017;76:81-90. doi:10.1016/j.jaad.2016.07.042
- Wu JJ, Poon KY, Channual JC, et al. Association between tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. Arch Dermatol. 2012;148:1244-1250. doi:10.1001 /archdermatol.2012.2502
- Wu JJ, Sundaram M, Cloutier M, et al. The risk of cardiovascular events in psoriasis patients treated with tumor necrosis factor-alpha inhibitors versus phototherapy: an observational cohort study. J Am Acad Dermatol. 2018;79:60-68. doi:10.1016/j.jaad.2018.02.050
- Cai J, Cui L, Wang Y, et al. Cardiometabolic comorbidities in patients with psoriasis: focusing on risk, biological therapy, and pathogenesis. Front Pharmacol. 2021;12:774808. doi:10.3389/fphar.2021.774808
- Powell-Wiley TM, Poirier P, Burke LE, et al. Obesity and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2021;143:E984-E1010. doi:10.1161/CIR.0000000000000973
- Pirro F, Caldarola G, Chiricozzi A, et al. Impact of body mass index on the efficacy of biological therapies in patients with psoriasis: a real-world study. Clin Drug Investig. 2021;41:917-925. doi:10.1007 /s40261-021-01080-z
- Kim H, Hong JY, Cheong S, et al. Impact of biologic agents on body weight and obesity-related disorders in patients with psoriasis: a nationwide population-based cohort study. Obes Res Clin Pract. 2023;17:210-217. doi:10.1016/j.orcp.2023.05.004
- Saraceno R, Schipani C, Mazzotta A, et al. Effect of anti-tumor necrosis factor-alpha therapies on body mass index in patients with psoriasis. Pharmacol Res. 2008;57:290-295. doi:10.1016/j.phrs.2008.02.006
- Fernandez AP, Dauden E, Gerdes S, et al. Tildrakizumab efficacy and safety in patients with psoriasis and concomitant metabolic syndrome: post hoc analysis of 5-year data from reSURFACE 1 and reSURFACE 2. J Eur Acad Dermatol Venereol. 2022;36:1774-1783. doi:10.1111/jdv.18167
- Mottillo S, Filion KB, Genest J, et al. The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J Am Coll Cardiol. 2010;56:1113-1132. doi:10.1016/j.jacc.2010.05.034
- Ricceri F, Chiricozzi A, Peris K, et al. Successful use of anti-IL-23 molecules in overweight-to-obese psoriatic patients: a multicentric retrospective study. Dermatol Ther. 2022;35:E15793. doi:10.1111/dth.15793
- Alinaghi F, Tekin HG, Burisch J, et al. Global prevalence and bidirectional association between psoriasis and inflammatory bowel disease— a systematic review and meta-analysis. J Crohns Colitis. 2020;14:351-360. doi:10.1093/ecco-jcc/jjz152
- Fu Y, Lee CH, Chi CC. Association of psoriasis with inflammatory bowel disease: a systematic review and meta-analysis. JAMA Dermatol. 2018;154:1417-1423. doi:10.1001/jamadermatol.2018.3631
- Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65-70. doi:10.1136/gut.52.1.65
- Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebocontrolled trial. Gut. 2012;61:1693-1700. doi:10.1136 /gutjnl-2011-301668
- Brockmann L, Tran A, Huang Y, et al. Intestinal microbiotaspecific Th17 cells possess regulatory properties and suppress effector T cells via c-MAF and IL-10. Immunity. 2023;56:2719-2735 e7. doi:10.1016/j.immuni.2023.11.003
- Lee JS, Tato CM, Joyce-Shaikh B, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity. 2015;43:727-738. doi:10.1016/j.immuni.2015.09.003
- Wedebye Schmidt EG, Larsen HL, Kristensen NN, et al. TH17 cell induction and effects of IL-17A and IL-17F blockade in experimental colitis. Inflamm Bowel Dis. 2013;19:1567-1576. doi:10.1097 /MIB.0b013e318286fa1c
- Tang C, Kakuta S, Shimizu K, et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing T(reg) cells through modification of the intestinal microbiota. Nat Immunol. 2018;19:755-765. doi:10.1038/s41590-018-0134-y
- El Hadad J, Schreiner P, Vavricka SR, Greuter T. The genetics of inflammatory bowel disease. Mol Diagn Ther. 2024;28:27-35. doi:10.1007 /s40291-023-00678-7
- Albayrak F, Gür M, Karatas¸ A, et al. Is the use of secukinumab after anti-TNF therapy greater than expected for the risk of developing inflammatory bowel disease? Reumatol Clin (Engl Ed). 2024;20:123-127. doi:10.1016/j.reumae.2023.11.002
- Kurd SK, Troxel AB, Crits-Christoph P, et al. The risk of depression, anxiety, and suicidality in patients with psoriasis: a populationbased cohort study. Arch Dermatol. 2010;146:891-895. doi:10.1001 /archdermatol.2010.186
- Strober B, Soliman AM, Truong B, et al. Association between biologic exposure and the risk of depression in patients with psoriasis: a retrospective analysis of large US administrative claims data. Am J Clin Dermatol. 2024;25:853-856. doi:10.1007/s40257 -024-00877-w
- Koo J, Ho RS, Thibodeaux Q. Depression and suicidality in psoriasis and clinical studies of brodalumab: a narrative review. Cutis. 2019;104:361-365.
- Andersch-Bjorkman Y, Micu E, Seifert O, et al. Effects of brodalumab on psoriasis and depressive symptoms in patients with insufficient response to TNF-alpha inhibitors. J Dermatol. 2023;50:1401-1414. doi:10.1111/1346-8138.16917
- Yeroushalmi S, Chung M, Bartholomew E, et al. Examining worldwide postmarketing suicides from biologics used for psoriasis with a focus on brodalumab: a cross-sectional analysis using the Food and Drug Administration Adverse Event Reporting System (FAERS). JAAD Int. 2022;9:119-121. doi:10.1016/j.jdin.2022.08.010
- Blauvelt A, Armstrong A, Merola JF, et al. Mental health outcomes in patients with moderate to severe psoriasis treated with bimekizumab: analysis of phase 2/3 randomized trials. J Am Acad Dermatol. 2024;91:72-81. doi:10.1016/j.jaad.2024.02.039
Psoriasis treatments have come a long way in the past 20 years. We now have more than a dozen systemic targeted treatments for psoriatic disease, with more on the way; however, with each successive class of medications introduced, the gap has narrowed in terms of increasing efficacy. In an era of medications reporting complete clearance rates in the 70% range, the average improvement in Psoriasis Area and Severity Index (PASI) for most biologics has remained at 90% to 95% in the past half-decade. While this is a far cry from the mean PASI improvements of 70% seen with the first biologics,1 it is becoming more challenging to base our treatment decisions solely on PASI outcome measures.
How, then, do we approach rational selection of a systemic psoriasis treatment? We could try to delineate based on mechanism of action, but it may be disingenuous to dissect minor differences in pathways (eg, IL-17 vs IL-23) that are fundamentally related and on the same continuum in psoriasis pathophysiology. Therefore, the most meaningful way to select an appropriate therapeutic may be to adopt a patient-centered approach that accounts for both individual preferences and specific medical needs by evaluating for other comorbidities2 to exclude or select certain medicines or types of treatments. We have long known to avoid tumor necrosis factor (TNF) α inhibitors in patients with congestive heart failure or a history of demyelinating disorders while regularly considering the presence of psoriatic arthritis and family planning when making treatment decisions. Now, we can be more nuanced in our approaches to psoriasis biologics. Specifically, the most important comorbidities to consider broadly encompass cardiometabolic disorders, gastrointestinal conditions, and psychiatric conditions.
Cardiometabolic Disorders
Possibly the hottest topic in psoriasis for some years now, the relationship between cardiometabolic disorders and psoriasis is of great interest to clinicians, scientists, and patients alike. There is a clear link between development of atherosclerosis and Th17-related immune mechanisms that also are implicated in the pathogenesis of psoriasis.3 Furthermore, the incidence of cardiovascular disease is markedly increased in patients with psoriasis, which is an independent risk factor for myocardial infarction, particularly among younger patients.4,5 Although several retrospective studies6-8 have shown that TNF-α inhibitors are associated with a reduction in cardiovascular outcomes, it is yet to be seen whether biologic treatment actually has a direct impact on cardiovascular outcomes, multiple studies investigating the effect of biologics on arterial inflammation markers notwithstanding.9
There are some direct factors to keep in mind when considering cardiometabolic comorbidities in patients with psoriasis. Obesity is common in the psoriasis population and can have a direct negative effect on cardiovascular health.10 However, the data on obesity and psoriasis are somewhat mixed with regard to treatment outcomes. In general, with increased volumes of distribution for biologics in patients with obesity, it has been shown that treatment success is more difficult to achieve in those with a body mass index greater than 30.11 Rather surprisingly, a separate nationwide study in South Korea found that patients on biologics for psoriasis were more likely to experience weight gain, even after controlling for factors such as exercise, smoking, and drinking,12 but it is unclear whether this is driven mostly by a known connection between weight gain and TNF-α inhibitors.13 These contrasting results point to the need for further studies in this area, as our intuitive approach would involve promoting weight loss while starting on a systemic treatment for psoriasis—but perhaps it is important not to assume that one will come with the other in tow, reinforcing the need to discuss a healthy diet with our patients with psoriasis regardless of treatment decisions.
The data that we have do not directly answer the big questions about biologic treatment and cardiovascular health, but we are starting to see interesting signals. For example, in a report of tildrakizumab treatment in patients with and without metabolic syndrome, the rates of major adverse cardiovascular events as well as cardiac disorders were essentially the same in both groups after receiving treatment for up to 244 weeks.14 This is interesting, more because of the lack of an increase in cardiovascular adverse events in the metabolic syndrome group, who entered the trial on average 25 kg to 30 kg heavier than those without metabolic syndrome. There is an increased risk for adverse cardiovascular events among patients with metabolic syndrome, a roughly 2-fold relative risk in as few as 5 to 6 years of follow-up.15 While the cohorts in the tildrakizumab study14 were too small to draw firm conclusions, the data are interesting and a step in the right direction; we need much larger data sets for analysis. Among other agents, similar efficacy and safety have been reported for guselkumab in a long-term psoriasis study; as a class, IL-23 inhibitors also tend to perform well from an efficacy standpoint in patients with obesity.16
Overall, when assessing the evidence for cardiometabolic disorders, it is reasonable to consider starting a biologic from the IL-17 or IL-23 inhibitor classes— thus avoiding both the potential downside of weight gain and contraindication in patients with congestive heart failure associated with TNF-α inhibitors. It is important to counsel patients about weight loss in conjunction with these treatments, both to improve efficacy and reduce cardiovascular risk factors. There may be a preference for IL-23 inhibitors in patients with obesity, as this class of medications maintains efficacy particularly well in these patients. Patients with psoriasis should be counseled to follow up with a primary care physician given their higher risk for metabolic syndrome and adverse cardiovascular outcomes.
Gastrointestinal Conditions
Psoriasis and inflammatory bowel disease (IBD) have a bidirectional association, and patients with psoriasis are about 1.7 times more likely to have either Crohn disease or ulcerative colitis.17,18 This association may be related to a shared pathogenesis with regard to immune dysregulation and overactivated inflammatory pathways, but there are some important differences to consider from a therapeutic standpoint. Given the increased expression of IL-17 in patients with IBD,19 a phase II trial of secukinumab yielded surprising results—not only was secukinumab ineffective in treating Crohn disease, but there also were higher rates of adverse events20 (as noted on the product label for all IL-17 inhibitors). We have come to understand that there are regulatory subsets of IL-17 cells that are important in mucosal homeostasis and also regulate IL-10, which generally is considered an anti-inflammatory cytokine.21 Thus, while IL-17 inhibition can reduce some component of inflammatory signaling, it also can increase inflammatory signaling through indirect pathways while increasing intestinal permeability to microbes. Importantly, this process seems to occur via IL-23–independent pathways; as such, while direct inhibition of IL-17 can be deleterious, IL-23 inhibitors have become important therapeutics for IBD.22
IL-17 family, IL-17A clearly is the culprit for worsening colitis as evidenced by both human and animal models. On the contrary, IL-17F blockade has been shown to ameliorate colitis in a murine model, whereas IL-17A inhibition worsens it.23 Furthermore, dual blockade of IL-17A and IL-17F has a protective effect against colitis, suggesting that the IL-17F inhibition is dominant. This interesting finding has some mechanistic backing, since blockade of IL-17F induces Treg cells that serve to maintain gut epithelium homeostasis and integrity.24
Overall, IL-17A inhibitors should be avoided in patients with a history of IBD—namely, secukinumab and ixekizumab. While there may be theoretical reasons that brodalumab or bimekizumab may confer a somewhat different risk for IBD exacerbation, there may be better choices that would be expected to effectively treat both the psoriasis and IBD manifestations. Given the US Food and Drug Administration approval of IL-23 inhibitors for IBD and their high levels of efficacy in treating psoriasis, these likely will be the best choice for most patients. Another mainstay of IBD treatment is TNF-α inhibitors, but they come with other risks such as increased immunosuppression and increased risk for nonmelanoma skin cancer.
An important question remains: What about patients who do not have known IBD? Do we proactively change our treatment choice due to fear of IBD development given the higher incidence of both Crohn disease and ulcerative colitis in patients with psoriasis? What about patients with a family history of IBD? First-degree relatives of patients with Crohn disease and ulcerative colitis have an 8- and 4-fold higher risk for those same conditions, respectively.25 Postmarketing surveillance and database findings of low rates of IBD development with IL-17 inhibitors gives only modest reassurance, as dermatologists generally know to avoid these medications for patients with even questionable IBD symptoms. It is important to emphasize to our patients that in no case do we believe that a psoriasis medication actually will cause IBD—rather, someone with subclinical IBD could experience a flare and a first manifestation of colitis. The drug is not the culprit in inducing IBD but rather may serve to unmask existing disease.
One study suggested that for patients who move on to the IL-17 inhibitor secukinumab after being treated with TNF-α inhibitors for psoriasis, the rates of IBD development are higher (4.8%) than in those who start IL-17A inhibition without prior treatment (1%)(OR, 8.38; P=.018).26 This begs the question of whether subclinical IBD in many patients with psoriasis who are treated with TNF-α inhibitors can be unmasked later when they are transitioned to a treatment that either does not treat the IBD or could worsen it. There may be a mechanistic drive behind this sequencing of treatments that predisposes patients to colitis, which would suggest selecting an IL-23 inhibitor after failing/trying a TNF-α inhibitor. However, the data are very preliminary, and in real practice, other concerns such as severe psoriatic arthritis may outweigh these considerations, as the IL-17 inhibitor class still is considered to be more effective than IL-23 inhibition at treating psoriatic arthritis overall. For most patients with no personal history of IBD and no strong family history of IBD (ie, first-degree relatives), the choice of biologic should not be affected by concern over gastrointestinal issues.
Psychiatric Conditions
It has been well established that psoriasis is linked to higher rates of depression, anxiety, and suicidality.27 How do we take this into account when treating patients with psoriasis, especially when we have biologics with a warning label for suicidality and a Risk Evaluation and Mitigation Strategies program (brodalumab) and language around suicidal ideation in the label (bimekizumab)? While it is challenging to discuss mental health, it is not a conversation that we as dermatologists should shy away from. Appropriate treatment of psoriasis is an important tool to get our patients on the path to better mental health. A recent database study of more than 4000 patients showed that patients with psoriasis treated with biologics had a 17% lower risk for depression than those treated with conventional disease-modifying drugs such as methotrexate.28 The comparator of the conventional disease-modifying drug class is important as it serves as a control for disease severity. Too often, a higher rate of depression, anxiety, or suicidality can be attributed to a medication when we in fact may just be capturing the background of higher incidence of all 3 in patients with severe psoriasis.
Indeed, even with the medication that many worry about on this front (brodalumab), multiple studies have confirmed that the effect on mental health generally is a positive one, with decreases in depressive symptoms.29 In a cohort switched from TNF-α inhibitors to brodalumab, symptoms of depression actually improved,30 so attributing a direct treatment effect to negative mental health outcomes does not seem to be justified, especially in light of the low number of suicide events in global postmarketing surveillance for brodalumab, comparable to or lower than other biologics for psoriasis.31 Similarly, bimekizumab has language in the label about discussing suicidality with patients, although the rates of suicidal ideation and behavior are no different from other biologics and rates of depression improved with its use.32
Heightened awareness of our patients’ mental health is something that we as providers should embrace, even when it seems that we do not have much time to see each patient. The priority when a patient comes in with mental health symptoms should be to treat what is within our scope (ie, psoriasis) as quickly and effectively as possible— with a newer-generation biologic such as an IL-17 or IL-23 inhibitor—while encouraging the patient to seek care from a mental health professional. In these cases, one might even argue that the rapidity of action of IL-17 inhibitors may be of additional benefit.
Final Thoughts
We as dermatologists generally are tasked with seeing high volumes of patients, and an initial psoriasis consultation can be a lengthy visit; however, it is rewarding to establish this relationship with patients and a reminder of why we practice medicine to begin with. Psoriasis can be satisfying to treat, and we have so many highly effective medicines that can completely transform our patients’ lives. Applying an understanding of the interplay between psoriasis, its related comorbidities, and treatment choices can be a fulfilling exercise that captures the essence of shared decision-making, which can lead to better outcomes and satisfaction for both providers and patients.
Psoriasis treatments have come a long way in the past 20 years. We now have more than a dozen systemic targeted treatments for psoriatic disease, with more on the way; however, with each successive class of medications introduced, the gap has narrowed in terms of increasing efficacy. In an era of medications reporting complete clearance rates in the 70% range, the average improvement in Psoriasis Area and Severity Index (PASI) for most biologics has remained at 90% to 95% in the past half-decade. While this is a far cry from the mean PASI improvements of 70% seen with the first biologics,1 it is becoming more challenging to base our treatment decisions solely on PASI outcome measures.
How, then, do we approach rational selection of a systemic psoriasis treatment? We could try to delineate based on mechanism of action, but it may be disingenuous to dissect minor differences in pathways (eg, IL-17 vs IL-23) that are fundamentally related and on the same continuum in psoriasis pathophysiology. Therefore, the most meaningful way to select an appropriate therapeutic may be to adopt a patient-centered approach that accounts for both individual preferences and specific medical needs by evaluating for other comorbidities2 to exclude or select certain medicines or types of treatments. We have long known to avoid tumor necrosis factor (TNF) α inhibitors in patients with congestive heart failure or a history of demyelinating disorders while regularly considering the presence of psoriatic arthritis and family planning when making treatment decisions. Now, we can be more nuanced in our approaches to psoriasis biologics. Specifically, the most important comorbidities to consider broadly encompass cardiometabolic disorders, gastrointestinal conditions, and psychiatric conditions.
Cardiometabolic Disorders
Possibly the hottest topic in psoriasis for some years now, the relationship between cardiometabolic disorders and psoriasis is of great interest to clinicians, scientists, and patients alike. There is a clear link between development of atherosclerosis and Th17-related immune mechanisms that also are implicated in the pathogenesis of psoriasis.3 Furthermore, the incidence of cardiovascular disease is markedly increased in patients with psoriasis, which is an independent risk factor for myocardial infarction, particularly among younger patients.4,5 Although several retrospective studies6-8 have shown that TNF-α inhibitors are associated with a reduction in cardiovascular outcomes, it is yet to be seen whether biologic treatment actually has a direct impact on cardiovascular outcomes, multiple studies investigating the effect of biologics on arterial inflammation markers notwithstanding.9
There are some direct factors to keep in mind when considering cardiometabolic comorbidities in patients with psoriasis. Obesity is common in the psoriasis population and can have a direct negative effect on cardiovascular health.10 However, the data on obesity and psoriasis are somewhat mixed with regard to treatment outcomes. In general, with increased volumes of distribution for biologics in patients with obesity, it has been shown that treatment success is more difficult to achieve in those with a body mass index greater than 30.11 Rather surprisingly, a separate nationwide study in South Korea found that patients on biologics for psoriasis were more likely to experience weight gain, even after controlling for factors such as exercise, smoking, and drinking,12 but it is unclear whether this is driven mostly by a known connection between weight gain and TNF-α inhibitors.13 These contrasting results point to the need for further studies in this area, as our intuitive approach would involve promoting weight loss while starting on a systemic treatment for psoriasis—but perhaps it is important not to assume that one will come with the other in tow, reinforcing the need to discuss a healthy diet with our patients with psoriasis regardless of treatment decisions.
The data that we have do not directly answer the big questions about biologic treatment and cardiovascular health, but we are starting to see interesting signals. For example, in a report of tildrakizumab treatment in patients with and without metabolic syndrome, the rates of major adverse cardiovascular events as well as cardiac disorders were essentially the same in both groups after receiving treatment for up to 244 weeks.14 This is interesting, more because of the lack of an increase in cardiovascular adverse events in the metabolic syndrome group, who entered the trial on average 25 kg to 30 kg heavier than those without metabolic syndrome. There is an increased risk for adverse cardiovascular events among patients with metabolic syndrome, a roughly 2-fold relative risk in as few as 5 to 6 years of follow-up.15 While the cohorts in the tildrakizumab study14 were too small to draw firm conclusions, the data are interesting and a step in the right direction; we need much larger data sets for analysis. Among other agents, similar efficacy and safety have been reported for guselkumab in a long-term psoriasis study; as a class, IL-23 inhibitors also tend to perform well from an efficacy standpoint in patients with obesity.16
Overall, when assessing the evidence for cardiometabolic disorders, it is reasonable to consider starting a biologic from the IL-17 or IL-23 inhibitor classes— thus avoiding both the potential downside of weight gain and contraindication in patients with congestive heart failure associated with TNF-α inhibitors. It is important to counsel patients about weight loss in conjunction with these treatments, both to improve efficacy and reduce cardiovascular risk factors. There may be a preference for IL-23 inhibitors in patients with obesity, as this class of medications maintains efficacy particularly well in these patients. Patients with psoriasis should be counseled to follow up with a primary care physician given their higher risk for metabolic syndrome and adverse cardiovascular outcomes.
Gastrointestinal Conditions
Psoriasis and inflammatory bowel disease (IBD) have a bidirectional association, and patients with psoriasis are about 1.7 times more likely to have either Crohn disease or ulcerative colitis.17,18 This association may be related to a shared pathogenesis with regard to immune dysregulation and overactivated inflammatory pathways, but there are some important differences to consider from a therapeutic standpoint. Given the increased expression of IL-17 in patients with IBD,19 a phase II trial of secukinumab yielded surprising results—not only was secukinumab ineffective in treating Crohn disease, but there also were higher rates of adverse events20 (as noted on the product label for all IL-17 inhibitors). We have come to understand that there are regulatory subsets of IL-17 cells that are important in mucosal homeostasis and also regulate IL-10, which generally is considered an anti-inflammatory cytokine.21 Thus, while IL-17 inhibition can reduce some component of inflammatory signaling, it also can increase inflammatory signaling through indirect pathways while increasing intestinal permeability to microbes. Importantly, this process seems to occur via IL-23–independent pathways; as such, while direct inhibition of IL-17 can be deleterious, IL-23 inhibitors have become important therapeutics for IBD.22
IL-17 family, IL-17A clearly is the culprit for worsening colitis as evidenced by both human and animal models. On the contrary, IL-17F blockade has been shown to ameliorate colitis in a murine model, whereas IL-17A inhibition worsens it.23 Furthermore, dual blockade of IL-17A and IL-17F has a protective effect against colitis, suggesting that the IL-17F inhibition is dominant. This interesting finding has some mechanistic backing, since blockade of IL-17F induces Treg cells that serve to maintain gut epithelium homeostasis and integrity.24
Overall, IL-17A inhibitors should be avoided in patients with a history of IBD—namely, secukinumab and ixekizumab. While there may be theoretical reasons that brodalumab or bimekizumab may confer a somewhat different risk for IBD exacerbation, there may be better choices that would be expected to effectively treat both the psoriasis and IBD manifestations. Given the US Food and Drug Administration approval of IL-23 inhibitors for IBD and their high levels of efficacy in treating psoriasis, these likely will be the best choice for most patients. Another mainstay of IBD treatment is TNF-α inhibitors, but they come with other risks such as increased immunosuppression and increased risk for nonmelanoma skin cancer.
An important question remains: What about patients who do not have known IBD? Do we proactively change our treatment choice due to fear of IBD development given the higher incidence of both Crohn disease and ulcerative colitis in patients with psoriasis? What about patients with a family history of IBD? First-degree relatives of patients with Crohn disease and ulcerative colitis have an 8- and 4-fold higher risk for those same conditions, respectively.25 Postmarketing surveillance and database findings of low rates of IBD development with IL-17 inhibitors gives only modest reassurance, as dermatologists generally know to avoid these medications for patients with even questionable IBD symptoms. It is important to emphasize to our patients that in no case do we believe that a psoriasis medication actually will cause IBD—rather, someone with subclinical IBD could experience a flare and a first manifestation of colitis. The drug is not the culprit in inducing IBD but rather may serve to unmask existing disease.
One study suggested that for patients who move on to the IL-17 inhibitor secukinumab after being treated with TNF-α inhibitors for psoriasis, the rates of IBD development are higher (4.8%) than in those who start IL-17A inhibition without prior treatment (1%)(OR, 8.38; P=.018).26 This begs the question of whether subclinical IBD in many patients with psoriasis who are treated with TNF-α inhibitors can be unmasked later when they are transitioned to a treatment that either does not treat the IBD or could worsen it. There may be a mechanistic drive behind this sequencing of treatments that predisposes patients to colitis, which would suggest selecting an IL-23 inhibitor after failing/trying a TNF-α inhibitor. However, the data are very preliminary, and in real practice, other concerns such as severe psoriatic arthritis may outweigh these considerations, as the IL-17 inhibitor class still is considered to be more effective than IL-23 inhibition at treating psoriatic arthritis overall. For most patients with no personal history of IBD and no strong family history of IBD (ie, first-degree relatives), the choice of biologic should not be affected by concern over gastrointestinal issues.
Psychiatric Conditions
It has been well established that psoriasis is linked to higher rates of depression, anxiety, and suicidality.27 How do we take this into account when treating patients with psoriasis, especially when we have biologics with a warning label for suicidality and a Risk Evaluation and Mitigation Strategies program (brodalumab) and language around suicidal ideation in the label (bimekizumab)? While it is challenging to discuss mental health, it is not a conversation that we as dermatologists should shy away from. Appropriate treatment of psoriasis is an important tool to get our patients on the path to better mental health. A recent database study of more than 4000 patients showed that patients with psoriasis treated with biologics had a 17% lower risk for depression than those treated with conventional disease-modifying drugs such as methotrexate.28 The comparator of the conventional disease-modifying drug class is important as it serves as a control for disease severity. Too often, a higher rate of depression, anxiety, or suicidality can be attributed to a medication when we in fact may just be capturing the background of higher incidence of all 3 in patients with severe psoriasis.
Indeed, even with the medication that many worry about on this front (brodalumab), multiple studies have confirmed that the effect on mental health generally is a positive one, with decreases in depressive symptoms.29 In a cohort switched from TNF-α inhibitors to brodalumab, symptoms of depression actually improved,30 so attributing a direct treatment effect to negative mental health outcomes does not seem to be justified, especially in light of the low number of suicide events in global postmarketing surveillance for brodalumab, comparable to or lower than other biologics for psoriasis.31 Similarly, bimekizumab has language in the label about discussing suicidality with patients, although the rates of suicidal ideation and behavior are no different from other biologics and rates of depression improved with its use.32
Heightened awareness of our patients’ mental health is something that we as providers should embrace, even when it seems that we do not have much time to see each patient. The priority when a patient comes in with mental health symptoms should be to treat what is within our scope (ie, psoriasis) as quickly and effectively as possible— with a newer-generation biologic such as an IL-17 or IL-23 inhibitor—while encouraging the patient to seek care from a mental health professional. In these cases, one might even argue that the rapidity of action of IL-17 inhibitors may be of additional benefit.
Final Thoughts
We as dermatologists generally are tasked with seeing high volumes of patients, and an initial psoriasis consultation can be a lengthy visit; however, it is rewarding to establish this relationship with patients and a reminder of why we practice medicine to begin with. Psoriasis can be satisfying to treat, and we have so many highly effective medicines that can completely transform our patients’ lives. Applying an understanding of the interplay between psoriasis, its related comorbidities, and treatment choices can be a fulfilling exercise that captures the essence of shared decision-making, which can lead to better outcomes and satisfaction for both providers and patients.
- Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349:2014-2022. doi:10.1056/NEJMoa030409
- Thatiparthi A, Martin A, Liu J, et al. Biologic treatment algorithms for moderate-to-severe psoriasis with comorbid conditions and special populations: a review. Am J Clin Dermatol. 2021;22:425-442. doi:10.1007/s40257-021-00603-w
- Packard RR, Lichtman AH, Libby P. Innate and adaptive immunity in atherosclerosis. Semin Immunopathol. 2009;31:5-22. doi:10.1007 /s00281-009-0153-8
- Gelfand JM, Neimann AL, Shin DB, et al. Risk of myocardial infarction in patients with psoriasis. JAMA. 2006;296:1735-1741. doi:10.1001/jama.296.14.1735
- Miller IM, Ellervik C, Yazdanyar S, et al. Meta-analysis of psoriasis, cardiovascular disease, and associated risk factors. J Am Acad Dermatol. 2013;69:1014-1024. doi:10.1016/j.jaad.2013.06.053
- Wu JJ, Guerin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-alpha inhibitors versus methotrexate. J Am Acad Dermatol. 2017;76:81-90. doi:10.1016/j.jaad.2016.07.042
- Wu JJ, Poon KY, Channual JC, et al. Association between tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. Arch Dermatol. 2012;148:1244-1250. doi:10.1001 /archdermatol.2012.2502
- Wu JJ, Sundaram M, Cloutier M, et al. The risk of cardiovascular events in psoriasis patients treated with tumor necrosis factor-alpha inhibitors versus phototherapy: an observational cohort study. J Am Acad Dermatol. 2018;79:60-68. doi:10.1016/j.jaad.2018.02.050
- Cai J, Cui L, Wang Y, et al. Cardiometabolic comorbidities in patients with psoriasis: focusing on risk, biological therapy, and pathogenesis. Front Pharmacol. 2021;12:774808. doi:10.3389/fphar.2021.774808
- Powell-Wiley TM, Poirier P, Burke LE, et al. Obesity and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2021;143:E984-E1010. doi:10.1161/CIR.0000000000000973
- Pirro F, Caldarola G, Chiricozzi A, et al. Impact of body mass index on the efficacy of biological therapies in patients with psoriasis: a real-world study. Clin Drug Investig. 2021;41:917-925. doi:10.1007 /s40261-021-01080-z
- Kim H, Hong JY, Cheong S, et al. Impact of biologic agents on body weight and obesity-related disorders in patients with psoriasis: a nationwide population-based cohort study. Obes Res Clin Pract. 2023;17:210-217. doi:10.1016/j.orcp.2023.05.004
- Saraceno R, Schipani C, Mazzotta A, et al. Effect of anti-tumor necrosis factor-alpha therapies on body mass index in patients with psoriasis. Pharmacol Res. 2008;57:290-295. doi:10.1016/j.phrs.2008.02.006
- Fernandez AP, Dauden E, Gerdes S, et al. Tildrakizumab efficacy and safety in patients with psoriasis and concomitant metabolic syndrome: post hoc analysis of 5-year data from reSURFACE 1 and reSURFACE 2. J Eur Acad Dermatol Venereol. 2022;36:1774-1783. doi:10.1111/jdv.18167
- Mottillo S, Filion KB, Genest J, et al. The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J Am Coll Cardiol. 2010;56:1113-1132. doi:10.1016/j.jacc.2010.05.034
- Ricceri F, Chiricozzi A, Peris K, et al. Successful use of anti-IL-23 molecules in overweight-to-obese psoriatic patients: a multicentric retrospective study. Dermatol Ther. 2022;35:E15793. doi:10.1111/dth.15793
- Alinaghi F, Tekin HG, Burisch J, et al. Global prevalence and bidirectional association between psoriasis and inflammatory bowel disease— a systematic review and meta-analysis. J Crohns Colitis. 2020;14:351-360. doi:10.1093/ecco-jcc/jjz152
- Fu Y, Lee CH, Chi CC. Association of psoriasis with inflammatory bowel disease: a systematic review and meta-analysis. JAMA Dermatol. 2018;154:1417-1423. doi:10.1001/jamadermatol.2018.3631
- Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65-70. doi:10.1136/gut.52.1.65
- Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebocontrolled trial. Gut. 2012;61:1693-1700. doi:10.1136 /gutjnl-2011-301668
- Brockmann L, Tran A, Huang Y, et al. Intestinal microbiotaspecific Th17 cells possess regulatory properties and suppress effector T cells via c-MAF and IL-10. Immunity. 2023;56:2719-2735 e7. doi:10.1016/j.immuni.2023.11.003
- Lee JS, Tato CM, Joyce-Shaikh B, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity. 2015;43:727-738. doi:10.1016/j.immuni.2015.09.003
- Wedebye Schmidt EG, Larsen HL, Kristensen NN, et al. TH17 cell induction and effects of IL-17A and IL-17F blockade in experimental colitis. Inflamm Bowel Dis. 2013;19:1567-1576. doi:10.1097 /MIB.0b013e318286fa1c
- Tang C, Kakuta S, Shimizu K, et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing T(reg) cells through modification of the intestinal microbiota. Nat Immunol. 2018;19:755-765. doi:10.1038/s41590-018-0134-y
- El Hadad J, Schreiner P, Vavricka SR, Greuter T. The genetics of inflammatory bowel disease. Mol Diagn Ther. 2024;28:27-35. doi:10.1007 /s40291-023-00678-7
- Albayrak F, Gür M, Karatas¸ A, et al. Is the use of secukinumab after anti-TNF therapy greater than expected for the risk of developing inflammatory bowel disease? Reumatol Clin (Engl Ed). 2024;20:123-127. doi:10.1016/j.reumae.2023.11.002
- Kurd SK, Troxel AB, Crits-Christoph P, et al. The risk of depression, anxiety, and suicidality in patients with psoriasis: a populationbased cohort study. Arch Dermatol. 2010;146:891-895. doi:10.1001 /archdermatol.2010.186
- Strober B, Soliman AM, Truong B, et al. Association between biologic exposure and the risk of depression in patients with psoriasis: a retrospective analysis of large US administrative claims data. Am J Clin Dermatol. 2024;25:853-856. doi:10.1007/s40257 -024-00877-w
- Koo J, Ho RS, Thibodeaux Q. Depression and suicidality in psoriasis and clinical studies of brodalumab: a narrative review. Cutis. 2019;104:361-365.
- Andersch-Bjorkman Y, Micu E, Seifert O, et al. Effects of brodalumab on psoriasis and depressive symptoms in patients with insufficient response to TNF-alpha inhibitors. J Dermatol. 2023;50:1401-1414. doi:10.1111/1346-8138.16917
- Yeroushalmi S, Chung M, Bartholomew E, et al. Examining worldwide postmarketing suicides from biologics used for psoriasis with a focus on brodalumab: a cross-sectional analysis using the Food and Drug Administration Adverse Event Reporting System (FAERS). JAAD Int. 2022;9:119-121. doi:10.1016/j.jdin.2022.08.010
- Blauvelt A, Armstrong A, Merola JF, et al. Mental health outcomes in patients with moderate to severe psoriasis treated with bimekizumab: analysis of phase 2/3 randomized trials. J Am Acad Dermatol. 2024;91:72-81. doi:10.1016/j.jaad.2024.02.039
- Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349:2014-2022. doi:10.1056/NEJMoa030409
- Thatiparthi A, Martin A, Liu J, et al. Biologic treatment algorithms for moderate-to-severe psoriasis with comorbid conditions and special populations: a review. Am J Clin Dermatol. 2021;22:425-442. doi:10.1007/s40257-021-00603-w
- Packard RR, Lichtman AH, Libby P. Innate and adaptive immunity in atherosclerosis. Semin Immunopathol. 2009;31:5-22. doi:10.1007 /s00281-009-0153-8
- Gelfand JM, Neimann AL, Shin DB, et al. Risk of myocardial infarction in patients with psoriasis. JAMA. 2006;296:1735-1741. doi:10.1001/jama.296.14.1735
- Miller IM, Ellervik C, Yazdanyar S, et al. Meta-analysis of psoriasis, cardiovascular disease, and associated risk factors. J Am Acad Dermatol. 2013;69:1014-1024. doi:10.1016/j.jaad.2013.06.053
- Wu JJ, Guerin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-alpha inhibitors versus methotrexate. J Am Acad Dermatol. 2017;76:81-90. doi:10.1016/j.jaad.2016.07.042
- Wu JJ, Poon KY, Channual JC, et al. Association between tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. Arch Dermatol. 2012;148:1244-1250. doi:10.1001 /archdermatol.2012.2502
- Wu JJ, Sundaram M, Cloutier M, et al. The risk of cardiovascular events in psoriasis patients treated with tumor necrosis factor-alpha inhibitors versus phototherapy: an observational cohort study. J Am Acad Dermatol. 2018;79:60-68. doi:10.1016/j.jaad.2018.02.050
- Cai J, Cui L, Wang Y, et al. Cardiometabolic comorbidities in patients with psoriasis: focusing on risk, biological therapy, and pathogenesis. Front Pharmacol. 2021;12:774808. doi:10.3389/fphar.2021.774808
- Powell-Wiley TM, Poirier P, Burke LE, et al. Obesity and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2021;143:E984-E1010. doi:10.1161/CIR.0000000000000973
- Pirro F, Caldarola G, Chiricozzi A, et al. Impact of body mass index on the efficacy of biological therapies in patients with psoriasis: a real-world study. Clin Drug Investig. 2021;41:917-925. doi:10.1007 /s40261-021-01080-z
- Kim H, Hong JY, Cheong S, et al. Impact of biologic agents on body weight and obesity-related disorders in patients with psoriasis: a nationwide population-based cohort study. Obes Res Clin Pract. 2023;17:210-217. doi:10.1016/j.orcp.2023.05.004
- Saraceno R, Schipani C, Mazzotta A, et al. Effect of anti-tumor necrosis factor-alpha therapies on body mass index in patients with psoriasis. Pharmacol Res. 2008;57:290-295. doi:10.1016/j.phrs.2008.02.006
- Fernandez AP, Dauden E, Gerdes S, et al. Tildrakizumab efficacy and safety in patients with psoriasis and concomitant metabolic syndrome: post hoc analysis of 5-year data from reSURFACE 1 and reSURFACE 2. J Eur Acad Dermatol Venereol. 2022;36:1774-1783. doi:10.1111/jdv.18167
- Mottillo S, Filion KB, Genest J, et al. The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J Am Coll Cardiol. 2010;56:1113-1132. doi:10.1016/j.jacc.2010.05.034
- Ricceri F, Chiricozzi A, Peris K, et al. Successful use of anti-IL-23 molecules in overweight-to-obese psoriatic patients: a multicentric retrospective study. Dermatol Ther. 2022;35:E15793. doi:10.1111/dth.15793
- Alinaghi F, Tekin HG, Burisch J, et al. Global prevalence and bidirectional association between psoriasis and inflammatory bowel disease— a systematic review and meta-analysis. J Crohns Colitis. 2020;14:351-360. doi:10.1093/ecco-jcc/jjz152
- Fu Y, Lee CH, Chi CC. Association of psoriasis with inflammatory bowel disease: a systematic review and meta-analysis. JAMA Dermatol. 2018;154:1417-1423. doi:10.1001/jamadermatol.2018.3631
- Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65-70. doi:10.1136/gut.52.1.65
- Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebocontrolled trial. Gut. 2012;61:1693-1700. doi:10.1136 /gutjnl-2011-301668
- Brockmann L, Tran A, Huang Y, et al. Intestinal microbiotaspecific Th17 cells possess regulatory properties and suppress effector T cells via c-MAF and IL-10. Immunity. 2023;56:2719-2735 e7. doi:10.1016/j.immuni.2023.11.003
- Lee JS, Tato CM, Joyce-Shaikh B, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity. 2015;43:727-738. doi:10.1016/j.immuni.2015.09.003
- Wedebye Schmidt EG, Larsen HL, Kristensen NN, et al. TH17 cell induction and effects of IL-17A and IL-17F blockade in experimental colitis. Inflamm Bowel Dis. 2013;19:1567-1576. doi:10.1097 /MIB.0b013e318286fa1c
- Tang C, Kakuta S, Shimizu K, et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing T(reg) cells through modification of the intestinal microbiota. Nat Immunol. 2018;19:755-765. doi:10.1038/s41590-018-0134-y
- El Hadad J, Schreiner P, Vavricka SR, Greuter T. The genetics of inflammatory bowel disease. Mol Diagn Ther. 2024;28:27-35. doi:10.1007 /s40291-023-00678-7
- Albayrak F, Gür M, Karatas¸ A, et al. Is the use of secukinumab after anti-TNF therapy greater than expected for the risk of developing inflammatory bowel disease? Reumatol Clin (Engl Ed). 2024;20:123-127. doi:10.1016/j.reumae.2023.11.002
- Kurd SK, Troxel AB, Crits-Christoph P, et al. The risk of depression, anxiety, and suicidality in patients with psoriasis: a populationbased cohort study. Arch Dermatol. 2010;146:891-895. doi:10.1001 /archdermatol.2010.186
- Strober B, Soliman AM, Truong B, et al. Association between biologic exposure and the risk of depression in patients with psoriasis: a retrospective analysis of large US administrative claims data. Am J Clin Dermatol. 2024;25:853-856. doi:10.1007/s40257 -024-00877-w
- Koo J, Ho RS, Thibodeaux Q. Depression and suicidality in psoriasis and clinical studies of brodalumab: a narrative review. Cutis. 2019;104:361-365.
- Andersch-Bjorkman Y, Micu E, Seifert O, et al. Effects of brodalumab on psoriasis and depressive symptoms in patients with insufficient response to TNF-alpha inhibitors. J Dermatol. 2023;50:1401-1414. doi:10.1111/1346-8138.16917
- Yeroushalmi S, Chung M, Bartholomew E, et al. Examining worldwide postmarketing suicides from biologics used for psoriasis with a focus on brodalumab: a cross-sectional analysis using the Food and Drug Administration Adverse Event Reporting System (FAERS). JAAD Int. 2022;9:119-121. doi:10.1016/j.jdin.2022.08.010
- Blauvelt A, Armstrong A, Merola JF, et al. Mental health outcomes in patients with moderate to severe psoriasis treated with bimekizumab: analysis of phase 2/3 randomized trials. J Am Acad Dermatol. 2024;91:72-81. doi:10.1016/j.jaad.2024.02.039
The Post-PASI Era: Considering Comorbidities to Select Appropriate Systemic Psoriasis Treatments
The Post-PASI Era: Considering Comorbidities to Select Appropriate Systemic Psoriasis Treatments
Cryotherapy for Treatment of Idiopathic Gingival Papillokeratosis With Crypt Formation
Cryotherapy for Treatment of Idiopathic Gingival Papillokeratosis With Crypt Formation
To the Editor:
Idiopathic gingival papillokeratosis with crypt formation (IGPC) is an uncommon benign condition that first was reported in 1967.1 The condition manifests as white plaques with a papillary appearance on the gingival tissue. While data on the prevalence of IGPC are limited, it is known to occur more frequently in younger patients (ie, 9-24 years1-3) and has been linked to use of orthodontic appliances.3,4 The lesions typically are asymptomatic with a bilateral appearance along the mucogingival junction. Research on IGPC has not identified the underlying mechanisms that trigger the hyperkeratinization and papillary alterations within the gingival tissue.
Management of IGPC can be challenging due to the rarity of the condition and its uncertain pathogenesis. Wiping or brushing the affected area offers only temporary improvement of symptoms and the appearance of the lesions. Surgical excision is another option; however, it can result in aesthetic and/or functional periodontal defects.2 Alternately, employing methods such as wiping or brushing the affected area offers only transient and temporary results in managing the condition. Additional investigative approaches and clinical studies are needed to identify more effective therapeutic modalities for the management of IGPC, particularly in pediatric patients, in whom aesthetic results may take on a heightened importance.1-3 We report a case of IGPC in which cryotherapy yielded satisfactory results with no recurrence of the lesions.
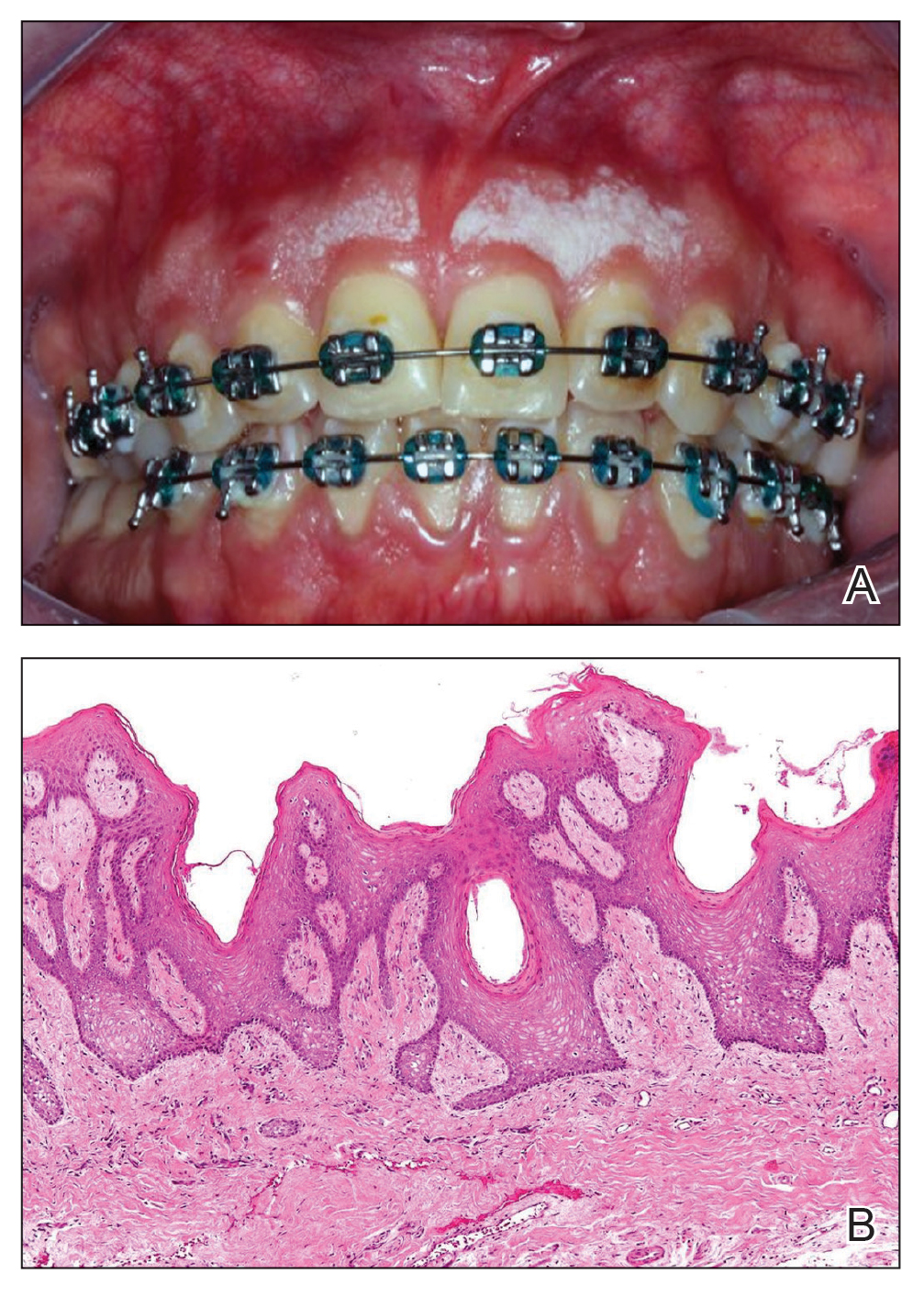
A 32-year-old woman presented to the dental clinic with white spots on the gingiva of 5 months’ duration. The patient reported a history of smoking cigarettes (3 packs per year) and drinking alcohol in social situations; her medical history was otherwise unremarkable. Clinical examination of the oral cavity revealed a bilateral, irregular, verrucouslike plaque throughout the vestibular upper attached gingiva. An incisional biopsy from the attached gingiva between teeth 13 and 23 was performed. Histopathologic analysis revealed parakeratosis and papillary acanthosis of the gingival mucosa associated with multifocal epithelial invaginations resembling crypts as well as long tapered epithelial ridges with no inflammation in the lamina propria. Based on the histopathologic findings, a diagnosis of IGPC was made (Figure 1).
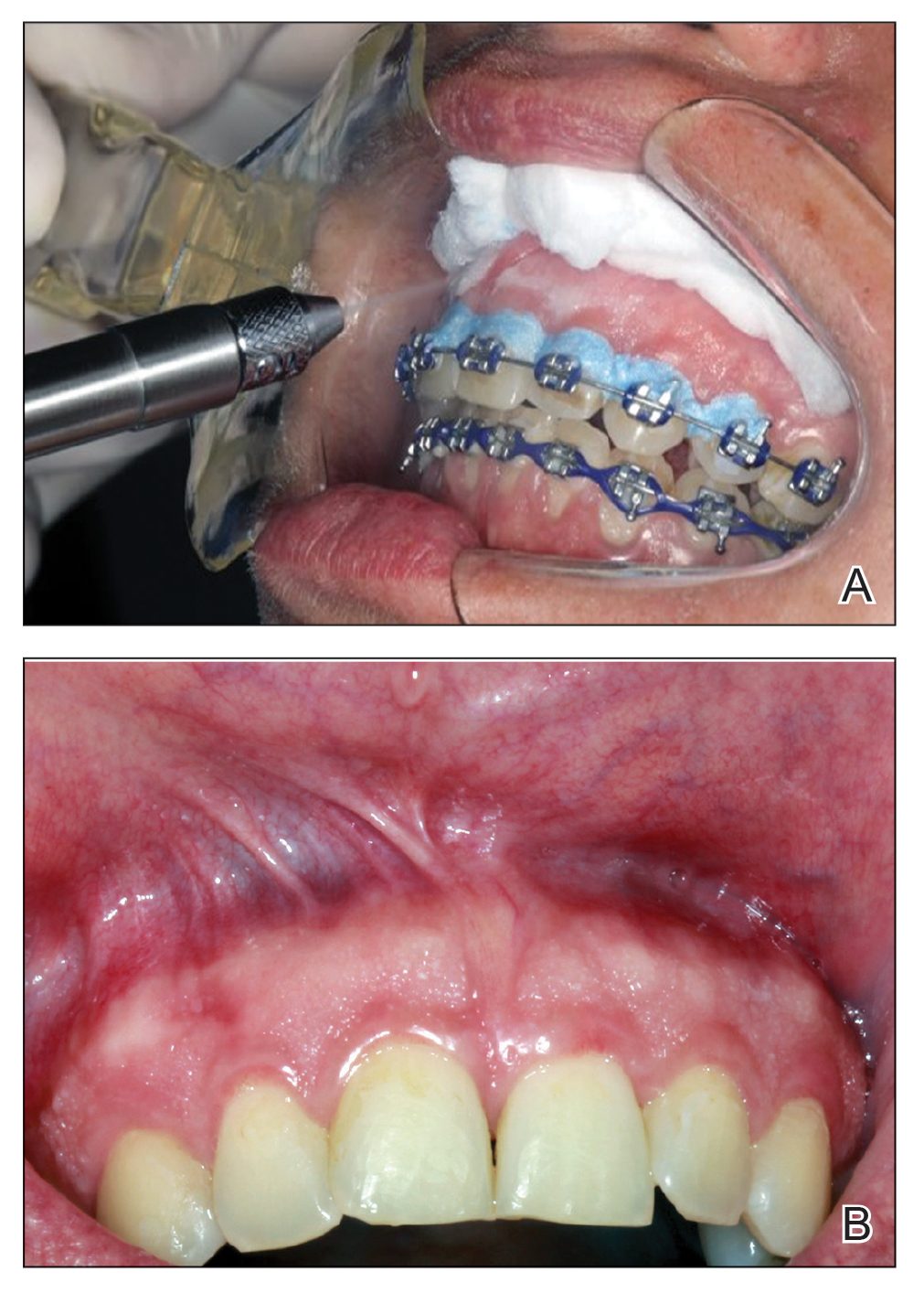
Given the patient’s clinical presentation, we suggested treatment with cryotherapy as a minimally invasive option that would preserve the gingival architecture and aesthetics while avoiding the potential complications of surgical excision. The patient consented to the procedure, and liquid nitrogen was administered through a handheld device using a 0.6-mm aperture spray tip. During application, the spray tip was positioned at a distance of 0.5 to 1.0 cm from the labial marginal gingiva at about a 45° angle. The freeze/thaw cycle involved a continuous one-way spray application of liquid nitrogen onto the lesion until solid ice formed over the entire area, followed by a waiting period until gradual thawing occurred.
A total of 5 cryotherapy sessions were conducted over an 8-week period; no recurrence of the lesions was observed during a 2-year follow-up period (Figure 2).
We present our case to add to the body of knowledge regarding management options for IGPC, specifically cryotherapy. Historically, brushing with a toothbrush and surgical excision have been the most commonly used interventions.2 Gently brushing the affected areas can help stimulate local blood circulation, which can improve the health of the gingival tissue, promote oxygenation and delivery of nutrients to the cells, and aid in the removal of metabolic waste. Surgical excision is the most commonly used treatment method for IGPC to ensure that the lesions are safely and completely removed; however, this option can result in aesthetic and/or functional periodontal defects. There also is a risk for recurrence, although Noonan et al2 reported no recurrence 4 years after performing a surgical excision for IGPC.
Cryotherapy reduces tissue sensitivity, provides local anesthesia, and reduces inflammation in the oral mucosa. Moreover, cryotherapy accelerates healing by stimulating vasoconstriction and reactive vasodilation, thus enhancing blood flow, oxygenation, and nutrient delivery for faster cell regeneration of the oral mucosa.4,5 Cryotherapy generally is regarded as a simple noninvasive procedure that is relatively safe when performed by qualified professionals.4,5 It can provide benefits such as minimal patient discomfort, rapid recovery, and potential reduction of complications associated with more invasive procedures.5
The efficacy of cryotherapy for IGPC may vary based on lesion severity, individual patient response, and the need for repeated treatment sessions. Robust scientific evidence concerning the long-term efficacy of cryotherapy as a treatment for IGPC is limited due to the rarity of this condition.
The etiopathogenesis of IGPC has been hypothesized to involve both genetic and environmental factors with equal significance. This suggestion is based on reports of IGPC occurring in multiple members of the same family and animal model studies indicating that gingival tissue is sensitive to environmental influences, such as nutritional factors.1,6 However, it is important to emphasize that these hypotheses remain speculative, and the true etiopathogenesis of IGPC remains uncertain.6 Microscopically, biopsy fragments from suspected cases of IGPC reveal gingival mucosa characterized by parakeratosis and papillary acanthosis accompanied by multifocal epithelial invaginations resembling crypts.2 Additionally, elongated and tapered epithelial ridges without inflammation in the lamina propria may be observed (as in our case), favoring the diagnosis of IGPC.3 The absence of inflammation is noteworthy because it suggests that the observed alterations are not attributed to typical inflammatory processes seen in some gingival conditions.
The limited number of studies reporting successful treatment outcomes with long-term follow-up for IGPC cases underscores the need for further exploration of effective treatment options. Cryotherapy emerges as a promising minimally invasive therapeutic approach, with our case offering support for its potential application. Additional research and clinical trials are essential to validate its efficacy and improve our understanding of cryotherapy as a treatment modality for IGPC lesions.
- Bennett JS, Grupe HE. Epithelial adnexal formations in human gingiva. Oral Surg Oral Med Oral Pathol. 1967;23:789-795. doi:10.1016/0030-4220(67)90371-4
- Noonan VL, Woo SB, Sundararajan D, et al. Idiopathic gingival papillokeratosis with crypt formation, a report of 7 cases of a previously undescribed entity: possible unusual oral epithelial nevus? Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:358-364. doi:10.1016/j.oooo.2016.10.018
- Romo SA, de Arruda JAA, Nava FJT, et al. Idiopathic gingival papillokeratosis with crypt formation: a clinicopathological entity in the young population? Int J Dermatol. 2023;62:E291-E293. doi: 10.1111/ijd.16579
- Farah CS, Savage NW. Cryotherapy for treatment of oral lesions. Aust Dent J. 2006;51:2-5. doi:10.1111/j.1834-7819.2006.tb00392.x
- Nogueira VKC, Fernandes D, Navarro CM, et al. Cryotherapy for localized juvenile spongiotic gingival hyperplasia: preliminary findings on two cases. Int J Paediatr Dent. 2017;27:231-235. doi:10.1111/ipd.12278
- Bernick S, Bavetta LA. The development of gingival sebaceous-like glands and cysts in rats of the Holtzman strain. Oral Surg Oral Med Oral Pathol Oral Radiol. 1962;15:351-354. doi:10.1016/0030-4220(62)90116-0
To the Editor:
Idiopathic gingival papillokeratosis with crypt formation (IGPC) is an uncommon benign condition that first was reported in 1967.1 The condition manifests as white plaques with a papillary appearance on the gingival tissue. While data on the prevalence of IGPC are limited, it is known to occur more frequently in younger patients (ie, 9-24 years1-3) and has been linked to use of orthodontic appliances.3,4 The lesions typically are asymptomatic with a bilateral appearance along the mucogingival junction. Research on IGPC has not identified the underlying mechanisms that trigger the hyperkeratinization and papillary alterations within the gingival tissue.
Management of IGPC can be challenging due to the rarity of the condition and its uncertain pathogenesis. Wiping or brushing the affected area offers only temporary improvement of symptoms and the appearance of the lesions. Surgical excision is another option; however, it can result in aesthetic and/or functional periodontal defects.2 Alternately, employing methods such as wiping or brushing the affected area offers only transient and temporary results in managing the condition. Additional investigative approaches and clinical studies are needed to identify more effective therapeutic modalities for the management of IGPC, particularly in pediatric patients, in whom aesthetic results may take on a heightened importance.1-3 We report a case of IGPC in which cryotherapy yielded satisfactory results with no recurrence of the lesions.
A 32-year-old woman presented to the dental clinic with white spots on the gingiva of 5 months’ duration. The patient reported a history of smoking cigarettes (3 packs per year) and drinking alcohol in social situations; her medical history was otherwise unremarkable. Clinical examination of the oral cavity revealed a bilateral, irregular, verrucouslike plaque throughout the vestibular upper attached gingiva. An incisional biopsy from the attached gingiva between teeth 13 and 23 was performed. Histopathologic analysis revealed parakeratosis and papillary acanthosis of the gingival mucosa associated with multifocal epithelial invaginations resembling crypts as well as long tapered epithelial ridges with no inflammation in the lamina propria. Based on the histopathologic findings, a diagnosis of IGPC was made (Figure 1).
Given the patient’s clinical presentation, we suggested treatment with cryotherapy as a minimally invasive option that would preserve the gingival architecture and aesthetics while avoiding the potential complications of surgical excision. The patient consented to the procedure, and liquid nitrogen was administered through a handheld device using a 0.6-mm aperture spray tip. During application, the spray tip was positioned at a distance of 0.5 to 1.0 cm from the labial marginal gingiva at about a 45° angle. The freeze/thaw cycle involved a continuous one-way spray application of liquid nitrogen onto the lesion until solid ice formed over the entire area, followed by a waiting period until gradual thawing occurred.
A total of 5 cryotherapy sessions were conducted over an 8-week period; no recurrence of the lesions was observed during a 2-year follow-up period (Figure 2).
We present our case to add to the body of knowledge regarding management options for IGPC, specifically cryotherapy. Historically, brushing with a toothbrush and surgical excision have been the most commonly used interventions.2 Gently brushing the affected areas can help stimulate local blood circulation, which can improve the health of the gingival tissue, promote oxygenation and delivery of nutrients to the cells, and aid in the removal of metabolic waste. Surgical excision is the most commonly used treatment method for IGPC to ensure that the lesions are safely and completely removed; however, this option can result in aesthetic and/or functional periodontal defects. There also is a risk for recurrence, although Noonan et al2 reported no recurrence 4 years after performing a surgical excision for IGPC.
Cryotherapy reduces tissue sensitivity, provides local anesthesia, and reduces inflammation in the oral mucosa. Moreover, cryotherapy accelerates healing by stimulating vasoconstriction and reactive vasodilation, thus enhancing blood flow, oxygenation, and nutrient delivery for faster cell regeneration of the oral mucosa.4,5 Cryotherapy generally is regarded as a simple noninvasive procedure that is relatively safe when performed by qualified professionals.4,5 It can provide benefits such as minimal patient discomfort, rapid recovery, and potential reduction of complications associated with more invasive procedures.5
The efficacy of cryotherapy for IGPC may vary based on lesion severity, individual patient response, and the need for repeated treatment sessions. Robust scientific evidence concerning the long-term efficacy of cryotherapy as a treatment for IGPC is limited due to the rarity of this condition.
The etiopathogenesis of IGPC has been hypothesized to involve both genetic and environmental factors with equal significance. This suggestion is based on reports of IGPC occurring in multiple members of the same family and animal model studies indicating that gingival tissue is sensitive to environmental influences, such as nutritional factors.1,6 However, it is important to emphasize that these hypotheses remain speculative, and the true etiopathogenesis of IGPC remains uncertain.6 Microscopically, biopsy fragments from suspected cases of IGPC reveal gingival mucosa characterized by parakeratosis and papillary acanthosis accompanied by multifocal epithelial invaginations resembling crypts.2 Additionally, elongated and tapered epithelial ridges without inflammation in the lamina propria may be observed (as in our case), favoring the diagnosis of IGPC.3 The absence of inflammation is noteworthy because it suggests that the observed alterations are not attributed to typical inflammatory processes seen in some gingival conditions.
The limited number of studies reporting successful treatment outcomes with long-term follow-up for IGPC cases underscores the need for further exploration of effective treatment options. Cryotherapy emerges as a promising minimally invasive therapeutic approach, with our case offering support for its potential application. Additional research and clinical trials are essential to validate its efficacy and improve our understanding of cryotherapy as a treatment modality for IGPC lesions.
To the Editor:
Idiopathic gingival papillokeratosis with crypt formation (IGPC) is an uncommon benign condition that first was reported in 1967.1 The condition manifests as white plaques with a papillary appearance on the gingival tissue. While data on the prevalence of IGPC are limited, it is known to occur more frequently in younger patients (ie, 9-24 years1-3) and has been linked to use of orthodontic appliances.3,4 The lesions typically are asymptomatic with a bilateral appearance along the mucogingival junction. Research on IGPC has not identified the underlying mechanisms that trigger the hyperkeratinization and papillary alterations within the gingival tissue.
Management of IGPC can be challenging due to the rarity of the condition and its uncertain pathogenesis. Wiping or brushing the affected area offers only temporary improvement of symptoms and the appearance of the lesions. Surgical excision is another option; however, it can result in aesthetic and/or functional periodontal defects.2 Alternately, employing methods such as wiping or brushing the affected area offers only transient and temporary results in managing the condition. Additional investigative approaches and clinical studies are needed to identify more effective therapeutic modalities for the management of IGPC, particularly in pediatric patients, in whom aesthetic results may take on a heightened importance.1-3 We report a case of IGPC in which cryotherapy yielded satisfactory results with no recurrence of the lesions.
A 32-year-old woman presented to the dental clinic with white spots on the gingiva of 5 months’ duration. The patient reported a history of smoking cigarettes (3 packs per year) and drinking alcohol in social situations; her medical history was otherwise unremarkable. Clinical examination of the oral cavity revealed a bilateral, irregular, verrucouslike plaque throughout the vestibular upper attached gingiva. An incisional biopsy from the attached gingiva between teeth 13 and 23 was performed. Histopathologic analysis revealed parakeratosis and papillary acanthosis of the gingival mucosa associated with multifocal epithelial invaginations resembling crypts as well as long tapered epithelial ridges with no inflammation in the lamina propria. Based on the histopathologic findings, a diagnosis of IGPC was made (Figure 1).
Given the patient’s clinical presentation, we suggested treatment with cryotherapy as a minimally invasive option that would preserve the gingival architecture and aesthetics while avoiding the potential complications of surgical excision. The patient consented to the procedure, and liquid nitrogen was administered through a handheld device using a 0.6-mm aperture spray tip. During application, the spray tip was positioned at a distance of 0.5 to 1.0 cm from the labial marginal gingiva at about a 45° angle. The freeze/thaw cycle involved a continuous one-way spray application of liquid nitrogen onto the lesion until solid ice formed over the entire area, followed by a waiting period until gradual thawing occurred.
A total of 5 cryotherapy sessions were conducted over an 8-week period; no recurrence of the lesions was observed during a 2-year follow-up period (Figure 2).
We present our case to add to the body of knowledge regarding management options for IGPC, specifically cryotherapy. Historically, brushing with a toothbrush and surgical excision have been the most commonly used interventions.2 Gently brushing the affected areas can help stimulate local blood circulation, which can improve the health of the gingival tissue, promote oxygenation and delivery of nutrients to the cells, and aid in the removal of metabolic waste. Surgical excision is the most commonly used treatment method for IGPC to ensure that the lesions are safely and completely removed; however, this option can result in aesthetic and/or functional periodontal defects. There also is a risk for recurrence, although Noonan et al2 reported no recurrence 4 years after performing a surgical excision for IGPC.
Cryotherapy reduces tissue sensitivity, provides local anesthesia, and reduces inflammation in the oral mucosa. Moreover, cryotherapy accelerates healing by stimulating vasoconstriction and reactive vasodilation, thus enhancing blood flow, oxygenation, and nutrient delivery for faster cell regeneration of the oral mucosa.4,5 Cryotherapy generally is regarded as a simple noninvasive procedure that is relatively safe when performed by qualified professionals.4,5 It can provide benefits such as minimal patient discomfort, rapid recovery, and potential reduction of complications associated with more invasive procedures.5
The efficacy of cryotherapy for IGPC may vary based on lesion severity, individual patient response, and the need for repeated treatment sessions. Robust scientific evidence concerning the long-term efficacy of cryotherapy as a treatment for IGPC is limited due to the rarity of this condition.
The etiopathogenesis of IGPC has been hypothesized to involve both genetic and environmental factors with equal significance. This suggestion is based on reports of IGPC occurring in multiple members of the same family and animal model studies indicating that gingival tissue is sensitive to environmental influences, such as nutritional factors.1,6 However, it is important to emphasize that these hypotheses remain speculative, and the true etiopathogenesis of IGPC remains uncertain.6 Microscopically, biopsy fragments from suspected cases of IGPC reveal gingival mucosa characterized by parakeratosis and papillary acanthosis accompanied by multifocal epithelial invaginations resembling crypts.2 Additionally, elongated and tapered epithelial ridges without inflammation in the lamina propria may be observed (as in our case), favoring the diagnosis of IGPC.3 The absence of inflammation is noteworthy because it suggests that the observed alterations are not attributed to typical inflammatory processes seen in some gingival conditions.
The limited number of studies reporting successful treatment outcomes with long-term follow-up for IGPC cases underscores the need for further exploration of effective treatment options. Cryotherapy emerges as a promising minimally invasive therapeutic approach, with our case offering support for its potential application. Additional research and clinical trials are essential to validate its efficacy and improve our understanding of cryotherapy as a treatment modality for IGPC lesions.
- Bennett JS, Grupe HE. Epithelial adnexal formations in human gingiva. Oral Surg Oral Med Oral Pathol. 1967;23:789-795. doi:10.1016/0030-4220(67)90371-4
- Noonan VL, Woo SB, Sundararajan D, et al. Idiopathic gingival papillokeratosis with crypt formation, a report of 7 cases of a previously undescribed entity: possible unusual oral epithelial nevus? Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:358-364. doi:10.1016/j.oooo.2016.10.018
- Romo SA, de Arruda JAA, Nava FJT, et al. Idiopathic gingival papillokeratosis with crypt formation: a clinicopathological entity in the young population? Int J Dermatol. 2023;62:E291-E293. doi: 10.1111/ijd.16579
- Farah CS, Savage NW. Cryotherapy for treatment of oral lesions. Aust Dent J. 2006;51:2-5. doi:10.1111/j.1834-7819.2006.tb00392.x
- Nogueira VKC, Fernandes D, Navarro CM, et al. Cryotherapy for localized juvenile spongiotic gingival hyperplasia: preliminary findings on two cases. Int J Paediatr Dent. 2017;27:231-235. doi:10.1111/ipd.12278
- Bernick S, Bavetta LA. The development of gingival sebaceous-like glands and cysts in rats of the Holtzman strain. Oral Surg Oral Med Oral Pathol Oral Radiol. 1962;15:351-354. doi:10.1016/0030-4220(62)90116-0
- Bennett JS, Grupe HE. Epithelial adnexal formations in human gingiva. Oral Surg Oral Med Oral Pathol. 1967;23:789-795. doi:10.1016/0030-4220(67)90371-4
- Noonan VL, Woo SB, Sundararajan D, et al. Idiopathic gingival papillokeratosis with crypt formation, a report of 7 cases of a previously undescribed entity: possible unusual oral epithelial nevus? Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:358-364. doi:10.1016/j.oooo.2016.10.018
- Romo SA, de Arruda JAA, Nava FJT, et al. Idiopathic gingival papillokeratosis with crypt formation: a clinicopathological entity in the young population? Int J Dermatol. 2023;62:E291-E293. doi: 10.1111/ijd.16579
- Farah CS, Savage NW. Cryotherapy for treatment of oral lesions. Aust Dent J. 2006;51:2-5. doi:10.1111/j.1834-7819.2006.tb00392.x
- Nogueira VKC, Fernandes D, Navarro CM, et al. Cryotherapy for localized juvenile spongiotic gingival hyperplasia: preliminary findings on two cases. Int J Paediatr Dent. 2017;27:231-235. doi:10.1111/ipd.12278
- Bernick S, Bavetta LA. The development of gingival sebaceous-like glands and cysts in rats of the Holtzman strain. Oral Surg Oral Med Oral Pathol Oral Radiol. 1962;15:351-354. doi:10.1016/0030-4220(62)90116-0
Cryotherapy for Treatment of Idiopathic Gingival Papillokeratosis With Crypt Formation
Cryotherapy for Treatment of Idiopathic Gingival Papillokeratosis With Crypt Formation
PRACTICE POINTS
- Surgical excision is an effective treatment for idiopathic gingival papillokeratosis with crypt formation (IGPC) but may result in periodontal defects that impact the aesthetic outcome.
- Cryotherapy is a novel therapeutic intervention for IGPC.

Exophytic Scaly Nodule on the Wrist
Exophytic Scaly Nodule on the Wrist
THE DIAGNOSIS: Atypical Spitz Tumor
The shave biopsy revealed extensive dermal proliferation with spitzoid cytomorphology containing large, spindled nuclei; prominent nucleoli; and abundant homogenous cytoplasm arranged in haphazard fascicles. The proliferation was associated with prominent pseudoepitheliomatous hyperplasia of the overlying epidermis, and anaplastic lymphoma kinase immunohistochemistry showed diffuse strong positivity. Fluorescence in situ hybridization confirmed fusion of the tropomyosin 3 (TPM3) and anaplastic lymphoma kinase (ALK) genes, which finalized the diagnosis of an ALK-mutated atypical spitz tumor. Due to the location and size of the lesion, Mohs micrographic surgery was performed to excise the tumor and clear the margins.
Spitz nevi are uncommon benign melanocytic neoplasms that typically occur in pediatric populations.1 Atypical spitz nevi comprised fewer than 17% of all childhood melanocytic nevi in the United States and can be considered in the broader category of spitzoid tumors. Spitz nevi are divided into 3 classes: Spitz nevus, atypical Spitz nevus, and spitzoid melanoma. Atypical Spitz nevi have typical Spitz nevus and spitzoid melanoma features and often can be difficult to distinguish on dermoscopy. Malignant Spitz tumors typically occur in the fifth decade of life, though the age distribution can vary widely.1
Black patients are less likely to be diagnosed with Spitz nevi, potentially due to a lower prevalence in this population, thus limiting the clinician’s clinical exposure and leading to increased rates of misdiagnoses.2 Spitz nevi usually manifest as well-circumscribed, dome-shaped papules and frequently are described as pink to red due to increased vascularity and limited melanin content1; however, these lesions may appear more violaceous, dusky, or dark brown in darker skin types. Additionally, approximately 71% of patients in a clinical review of Spitz nevi had a pigmented lesion, ranging from light brown to black.3 It is important for dermatologists to understand that the contrast in color between the nevus and the surrounding skin may not be as striking, prominent, or clinically concerning, particularly in darker skin types, such as in our patient.
Spitz nevi frequently manifest as rapidly growing solitary lesions most frequently developing in the lower legs (shown in 41% of lesions in one report).4 However, a recent retrospective review indicated that Spitz nevi in Black patients most commonly were found on the upper extremities, as was seen in our patient.2 Compared to typical and common Spitz nevi, atypical Spitz nevi often are greater than 10 mm in diameter and have features of ulceration.
Diagnosing atypical spitzoid melanocytic lesions requires adequate clinical suspicion and confirmation via biopsy. Under dermoscopy, typical Spitz nevi often display a starburst or globular pattern with pinpoint vessels, though it can have variable manifestations of both patterns. Atypical Spitz nevi can be challenging to distinguish from melanoma on dermoscopy since both conditions can have atypical pigment networks or structureless homogenous areas.1 Consequently, there often is a lower threshold for biopsy and possible follow-up excision for atypical Spitz nevi. Histopathology of atypical Spitz nevi includes epithelioid and spindle melanocytes but can share features of melanomas, including areas of prominent pagetoid spread, asymmetry, and poor circumscription.5 Furthermore, atypical Spitz nevi with ALK gene fusion, as seen in our patient, have been shown in the literature to demonstrate distinct histopathologic features, such as wedge-shaped extension into the dermis or a bulbous lower border that can resemble pseudoepitheliomatous hyperplasia.6
The differential diagnosis for this rapidly growing scaly nodule also should include pyogenic granuloma, bacillary angiomatosis, Kaposi sarcoma, and amelanotic melanoma. Pyogenic granuloma is a rapidly growing, benign, vascular tumor that often becomes ulcerated and can occur in any age group.7 Pyogenic granuloma frequently appears at sites of trauma as a solitary, bright pink to red, friable, pedunculated papule and often manifests on the arms, hands, and face, similar to atypical Spitz nevi, though they can appear anywhere on the body. Histology shows a lobular capillary network with a central feeder vessel.7
Bacillary angiomatosis is an uncommon cutaneous infection associated with vascular proliferation and neovascularization due to the gram-negative organism Bartonella henselae.8 Bacillary nodules typically are reddish to purple and appear on the arms, sometimes with central ulceration and bleeding. Patients may present with multiple papules and nodules of varying sizes, as the lesions can arise in crops and follow a sporotrichoid pattern. Most patients with bacillary angiomatosis are immunosuppressed, though it rarely can affect immunocompetent patients. Histologically, bacillary angiomatosis is similar to pyogenic granuloma, though Gram or Warthin-Starry stains can help differentiate B henselae.8
Kaposi sarcoma is a malignant vascular neoplasm that often manifests in immunocompromised patients as violaceous, purple, or red patches, plaques, and nodules on the skin or oral mucosa. Histopathology shows spindle cell proliferation of irregular complex vascular channels dissecting through the dermis. Human herpesvirus 8 immunohistochemistry can be used to confirm diagnosis on histopathology.9 In contrast, amelanotic melanoma consists of lack of pigmentation, asymmetry with polymorphous vascular pattern, and high mitotic rate and is commonly found in sun-exposed areas. Dermoscopic features include irregular globules with blue-whitish veil.10
Treatment of atypical Spitz nevi depends mainly on the age of the patient and the histologic features of the nevus. Adults with atypical Spitz nevi frequently require excision, while the preferred choice for treatment in children with common Spitz nevi is regular clinical monitoring when there are no concerning clinical, dermoscopic, or histologic features.8 Compared to common Spitz nevi, atypical Spitz nevi have more melanoma-like features, resulting in a stronger recommendation for excision. Excision allows for a more thorough histologic evaluation and minimizes the likelihood of a recurrent atypical lesion.11 In all cases, close clinical follow-up is recommended to monitor for reoccurrence.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084. doi:10.1016/j.jaad.2011.04.040
- Farid YI, Honda KS. Spitz nevi in African Americans: a retrospective chart review of 11 patients. J Cutan Pathol. 2021;48:511-518. doi:10.1111 /cup.13903
- Dal Pozzo V, Benelli C, Restano L, et al. Clinical review of 247 case records of Spitz nevus (epithelioid cell and/or spindle cell nevus). Dermatology 1997;194:20-25. doi: 10.1159/000246051
- Berlingeri-Ramos AC, Morales-Burgos A, Sanchez JL, et al. Spitz nevus in a Hispanic population: a clinicopathological study of 130 cases. Am J Dermatopathol 2010;32:267-275. doi: 10.1097 /DAD.0b013e3181c52b99
- Brown A, Sawyer JD, Neumeister MW. Spitz nevus: review and update. Clin Plast Surg 2021;48:677-686. doi: 10.1016/j.cps.2021.06.002 [published Online First: 20210818]
- Yeh I, de la Fouchardiere A, Pissaloux D, et al. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol 2015;39:581-91. doi: 10.1097/PAS.0000000000000387
- Sarwal P, Lapumnuaypol K. Pyogenic granuloma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556077/
- Akram SM, Anwar MY, Thandra KC, et al. Bacillary angiomatosis. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 4, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448092/
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Pizzichetta MA, Talamini R, Stanganelli I, et al. Amelanotic/ hypomelanotic melanoma: clinical and dermoscopic features. Br J Dermatol 2004;150(6):1117-1124. doi: 10.1111/j.1365-2133.2004.05928.x
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part II. natural history and management. J Am Acad Dermatol 2011;65:1087-1092. doi:10.1016/j.jaad.2011.06.045
THE DIAGNOSIS: Atypical Spitz Tumor
The shave biopsy revealed extensive dermal proliferation with spitzoid cytomorphology containing large, spindled nuclei; prominent nucleoli; and abundant homogenous cytoplasm arranged in haphazard fascicles. The proliferation was associated with prominent pseudoepitheliomatous hyperplasia of the overlying epidermis, and anaplastic lymphoma kinase immunohistochemistry showed diffuse strong positivity. Fluorescence in situ hybridization confirmed fusion of the tropomyosin 3 (TPM3) and anaplastic lymphoma kinase (ALK) genes, which finalized the diagnosis of an ALK-mutated atypical spitz tumor. Due to the location and size of the lesion, Mohs micrographic surgery was performed to excise the tumor and clear the margins.
Spitz nevi are uncommon benign melanocytic neoplasms that typically occur in pediatric populations.1 Atypical spitz nevi comprised fewer than 17% of all childhood melanocytic nevi in the United States and can be considered in the broader category of spitzoid tumors. Spitz nevi are divided into 3 classes: Spitz nevus, atypical Spitz nevus, and spitzoid melanoma. Atypical Spitz nevi have typical Spitz nevus and spitzoid melanoma features and often can be difficult to distinguish on dermoscopy. Malignant Spitz tumors typically occur in the fifth decade of life, though the age distribution can vary widely.1
Black patients are less likely to be diagnosed with Spitz nevi, potentially due to a lower prevalence in this population, thus limiting the clinician’s clinical exposure and leading to increased rates of misdiagnoses.2 Spitz nevi usually manifest as well-circumscribed, dome-shaped papules and frequently are described as pink to red due to increased vascularity and limited melanin content1; however, these lesions may appear more violaceous, dusky, or dark brown in darker skin types. Additionally, approximately 71% of patients in a clinical review of Spitz nevi had a pigmented lesion, ranging from light brown to black.3 It is important for dermatologists to understand that the contrast in color between the nevus and the surrounding skin may not be as striking, prominent, or clinically concerning, particularly in darker skin types, such as in our patient.
Spitz nevi frequently manifest as rapidly growing solitary lesions most frequently developing in the lower legs (shown in 41% of lesions in one report).4 However, a recent retrospective review indicated that Spitz nevi in Black patients most commonly were found on the upper extremities, as was seen in our patient.2 Compared to typical and common Spitz nevi, atypical Spitz nevi often are greater than 10 mm in diameter and have features of ulceration.
Diagnosing atypical spitzoid melanocytic lesions requires adequate clinical suspicion and confirmation via biopsy. Under dermoscopy, typical Spitz nevi often display a starburst or globular pattern with pinpoint vessels, though it can have variable manifestations of both patterns. Atypical Spitz nevi can be challenging to distinguish from melanoma on dermoscopy since both conditions can have atypical pigment networks or structureless homogenous areas.1 Consequently, there often is a lower threshold for biopsy and possible follow-up excision for atypical Spitz nevi. Histopathology of atypical Spitz nevi includes epithelioid and spindle melanocytes but can share features of melanomas, including areas of prominent pagetoid spread, asymmetry, and poor circumscription.5 Furthermore, atypical Spitz nevi with ALK gene fusion, as seen in our patient, have been shown in the literature to demonstrate distinct histopathologic features, such as wedge-shaped extension into the dermis or a bulbous lower border that can resemble pseudoepitheliomatous hyperplasia.6
The differential diagnosis for this rapidly growing scaly nodule also should include pyogenic granuloma, bacillary angiomatosis, Kaposi sarcoma, and amelanotic melanoma. Pyogenic granuloma is a rapidly growing, benign, vascular tumor that often becomes ulcerated and can occur in any age group.7 Pyogenic granuloma frequently appears at sites of trauma as a solitary, bright pink to red, friable, pedunculated papule and often manifests on the arms, hands, and face, similar to atypical Spitz nevi, though they can appear anywhere on the body. Histology shows a lobular capillary network with a central feeder vessel.7
Bacillary angiomatosis is an uncommon cutaneous infection associated with vascular proliferation and neovascularization due to the gram-negative organism Bartonella henselae.8 Bacillary nodules typically are reddish to purple and appear on the arms, sometimes with central ulceration and bleeding. Patients may present with multiple papules and nodules of varying sizes, as the lesions can arise in crops and follow a sporotrichoid pattern. Most patients with bacillary angiomatosis are immunosuppressed, though it rarely can affect immunocompetent patients. Histologically, bacillary angiomatosis is similar to pyogenic granuloma, though Gram or Warthin-Starry stains can help differentiate B henselae.8
Kaposi sarcoma is a malignant vascular neoplasm that often manifests in immunocompromised patients as violaceous, purple, or red patches, plaques, and nodules on the skin or oral mucosa. Histopathology shows spindle cell proliferation of irregular complex vascular channels dissecting through the dermis. Human herpesvirus 8 immunohistochemistry can be used to confirm diagnosis on histopathology.9 In contrast, amelanotic melanoma consists of lack of pigmentation, asymmetry with polymorphous vascular pattern, and high mitotic rate and is commonly found in sun-exposed areas. Dermoscopic features include irregular globules with blue-whitish veil.10
Treatment of atypical Spitz nevi depends mainly on the age of the patient and the histologic features of the nevus. Adults with atypical Spitz nevi frequently require excision, while the preferred choice for treatment in children with common Spitz nevi is regular clinical monitoring when there are no concerning clinical, dermoscopic, or histologic features.8 Compared to common Spitz nevi, atypical Spitz nevi have more melanoma-like features, resulting in a stronger recommendation for excision. Excision allows for a more thorough histologic evaluation and minimizes the likelihood of a recurrent atypical lesion.11 In all cases, close clinical follow-up is recommended to monitor for reoccurrence.
THE DIAGNOSIS: Atypical Spitz Tumor
The shave biopsy revealed extensive dermal proliferation with spitzoid cytomorphology containing large, spindled nuclei; prominent nucleoli; and abundant homogenous cytoplasm arranged in haphazard fascicles. The proliferation was associated with prominent pseudoepitheliomatous hyperplasia of the overlying epidermis, and anaplastic lymphoma kinase immunohistochemistry showed diffuse strong positivity. Fluorescence in situ hybridization confirmed fusion of the tropomyosin 3 (TPM3) and anaplastic lymphoma kinase (ALK) genes, which finalized the diagnosis of an ALK-mutated atypical spitz tumor. Due to the location and size of the lesion, Mohs micrographic surgery was performed to excise the tumor and clear the margins.
Spitz nevi are uncommon benign melanocytic neoplasms that typically occur in pediatric populations.1 Atypical spitz nevi comprised fewer than 17% of all childhood melanocytic nevi in the United States and can be considered in the broader category of spitzoid tumors. Spitz nevi are divided into 3 classes: Spitz nevus, atypical Spitz nevus, and spitzoid melanoma. Atypical Spitz nevi have typical Spitz nevus and spitzoid melanoma features and often can be difficult to distinguish on dermoscopy. Malignant Spitz tumors typically occur in the fifth decade of life, though the age distribution can vary widely.1
Black patients are less likely to be diagnosed with Spitz nevi, potentially due to a lower prevalence in this population, thus limiting the clinician’s clinical exposure and leading to increased rates of misdiagnoses.2 Spitz nevi usually manifest as well-circumscribed, dome-shaped papules and frequently are described as pink to red due to increased vascularity and limited melanin content1; however, these lesions may appear more violaceous, dusky, or dark brown in darker skin types. Additionally, approximately 71% of patients in a clinical review of Spitz nevi had a pigmented lesion, ranging from light brown to black.3 It is important for dermatologists to understand that the contrast in color between the nevus and the surrounding skin may not be as striking, prominent, or clinically concerning, particularly in darker skin types, such as in our patient.
Spitz nevi frequently manifest as rapidly growing solitary lesions most frequently developing in the lower legs (shown in 41% of lesions in one report).4 However, a recent retrospective review indicated that Spitz nevi in Black patients most commonly were found on the upper extremities, as was seen in our patient.2 Compared to typical and common Spitz nevi, atypical Spitz nevi often are greater than 10 mm in diameter and have features of ulceration.
Diagnosing atypical spitzoid melanocytic lesions requires adequate clinical suspicion and confirmation via biopsy. Under dermoscopy, typical Spitz nevi often display a starburst or globular pattern with pinpoint vessels, though it can have variable manifestations of both patterns. Atypical Spitz nevi can be challenging to distinguish from melanoma on dermoscopy since both conditions can have atypical pigment networks or structureless homogenous areas.1 Consequently, there often is a lower threshold for biopsy and possible follow-up excision for atypical Spitz nevi. Histopathology of atypical Spitz nevi includes epithelioid and spindle melanocytes but can share features of melanomas, including areas of prominent pagetoid spread, asymmetry, and poor circumscription.5 Furthermore, atypical Spitz nevi with ALK gene fusion, as seen in our patient, have been shown in the literature to demonstrate distinct histopathologic features, such as wedge-shaped extension into the dermis or a bulbous lower border that can resemble pseudoepitheliomatous hyperplasia.6
The differential diagnosis for this rapidly growing scaly nodule also should include pyogenic granuloma, bacillary angiomatosis, Kaposi sarcoma, and amelanotic melanoma. Pyogenic granuloma is a rapidly growing, benign, vascular tumor that often becomes ulcerated and can occur in any age group.7 Pyogenic granuloma frequently appears at sites of trauma as a solitary, bright pink to red, friable, pedunculated papule and often manifests on the arms, hands, and face, similar to atypical Spitz nevi, though they can appear anywhere on the body. Histology shows a lobular capillary network with a central feeder vessel.7
Bacillary angiomatosis is an uncommon cutaneous infection associated with vascular proliferation and neovascularization due to the gram-negative organism Bartonella henselae.8 Bacillary nodules typically are reddish to purple and appear on the arms, sometimes with central ulceration and bleeding. Patients may present with multiple papules and nodules of varying sizes, as the lesions can arise in crops and follow a sporotrichoid pattern. Most patients with bacillary angiomatosis are immunosuppressed, though it rarely can affect immunocompetent patients. Histologically, bacillary angiomatosis is similar to pyogenic granuloma, though Gram or Warthin-Starry stains can help differentiate B henselae.8
Kaposi sarcoma is a malignant vascular neoplasm that often manifests in immunocompromised patients as violaceous, purple, or red patches, plaques, and nodules on the skin or oral mucosa. Histopathology shows spindle cell proliferation of irregular complex vascular channels dissecting through the dermis. Human herpesvirus 8 immunohistochemistry can be used to confirm diagnosis on histopathology.9 In contrast, amelanotic melanoma consists of lack of pigmentation, asymmetry with polymorphous vascular pattern, and high mitotic rate and is commonly found in sun-exposed areas. Dermoscopic features include irregular globules with blue-whitish veil.10
Treatment of atypical Spitz nevi depends mainly on the age of the patient and the histologic features of the nevus. Adults with atypical Spitz nevi frequently require excision, while the preferred choice for treatment in children with common Spitz nevi is regular clinical monitoring when there are no concerning clinical, dermoscopic, or histologic features.8 Compared to common Spitz nevi, atypical Spitz nevi have more melanoma-like features, resulting in a stronger recommendation for excision. Excision allows for a more thorough histologic evaluation and minimizes the likelihood of a recurrent atypical lesion.11 In all cases, close clinical follow-up is recommended to monitor for reoccurrence.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084. doi:10.1016/j.jaad.2011.04.040
- Farid YI, Honda KS. Spitz nevi in African Americans: a retrospective chart review of 11 patients. J Cutan Pathol. 2021;48:511-518. doi:10.1111 /cup.13903
- Dal Pozzo V, Benelli C, Restano L, et al. Clinical review of 247 case records of Spitz nevus (epithelioid cell and/or spindle cell nevus). Dermatology 1997;194:20-25. doi: 10.1159/000246051
- Berlingeri-Ramos AC, Morales-Burgos A, Sanchez JL, et al. Spitz nevus in a Hispanic population: a clinicopathological study of 130 cases. Am J Dermatopathol 2010;32:267-275. doi: 10.1097 /DAD.0b013e3181c52b99
- Brown A, Sawyer JD, Neumeister MW. Spitz nevus: review and update. Clin Plast Surg 2021;48:677-686. doi: 10.1016/j.cps.2021.06.002 [published Online First: 20210818]
- Yeh I, de la Fouchardiere A, Pissaloux D, et al. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol 2015;39:581-91. doi: 10.1097/PAS.0000000000000387
- Sarwal P, Lapumnuaypol K. Pyogenic granuloma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556077/
- Akram SM, Anwar MY, Thandra KC, et al. Bacillary angiomatosis. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 4, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448092/
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Pizzichetta MA, Talamini R, Stanganelli I, et al. Amelanotic/ hypomelanotic melanoma: clinical and dermoscopic features. Br J Dermatol 2004;150(6):1117-1124. doi: 10.1111/j.1365-2133.2004.05928.x
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part II. natural history and management. J Am Acad Dermatol 2011;65:1087-1092. doi:10.1016/j.jaad.2011.06.045
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084. doi:10.1016/j.jaad.2011.04.040
- Farid YI, Honda KS. Spitz nevi in African Americans: a retrospective chart review of 11 patients. J Cutan Pathol. 2021;48:511-518. doi:10.1111 /cup.13903
- Dal Pozzo V, Benelli C, Restano L, et al. Clinical review of 247 case records of Spitz nevus (epithelioid cell and/or spindle cell nevus). Dermatology 1997;194:20-25. doi: 10.1159/000246051
- Berlingeri-Ramos AC, Morales-Burgos A, Sanchez JL, et al. Spitz nevus in a Hispanic population: a clinicopathological study of 130 cases. Am J Dermatopathol 2010;32:267-275. doi: 10.1097 /DAD.0b013e3181c52b99
- Brown A, Sawyer JD, Neumeister MW. Spitz nevus: review and update. Clin Plast Surg 2021;48:677-686. doi: 10.1016/j.cps.2021.06.002 [published Online First: 20210818]
- Yeh I, de la Fouchardiere A, Pissaloux D, et al. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol 2015;39:581-91. doi: 10.1097/PAS.0000000000000387
- Sarwal P, Lapumnuaypol K. Pyogenic granuloma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556077/
- Akram SM, Anwar MY, Thandra KC, et al. Bacillary angiomatosis. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 4, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448092/
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Pizzichetta MA, Talamini R, Stanganelli I, et al. Amelanotic/ hypomelanotic melanoma: clinical and dermoscopic features. Br J Dermatol 2004;150(6):1117-1124. doi: 10.1111/j.1365-2133.2004.05928.x
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part II. natural history and management. J Am Acad Dermatol 2011;65:1087-1092. doi:10.1016/j.jaad.2011.06.045
Exophytic Scaly Nodule on the Wrist
Exophytic Scaly Nodule on the Wrist
A 30-year-old Black man presented to the dermatology clinic with a rapidly growing, exophytic, scaly nodule on the right volar wrist of 2 months’ duration. The patient’s medical history was otherwise unremarkable. Physical examination revealed an irregularly bordered, red to violaceous, scaly, eroded, exophytic nodule on the wrist that was 2 cm in diameter with a surrounding adherent white-yellow crust. The patient had presumed the nodule was a wart and had been self-treating with over-the-counter salicylic acid and cryotherapy with no relief. He denied any bleeding or pruritus. The rest of the skin examination was unremarkable. A shave biopsy was performed for further evaluation.

Verrucous Plaques on Sun-Exposed Areas
Verrucous Plaques on Sun-Exposed Areas
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
Verrucous Plaques on Sun-Exposed Areas
Verrucous Plaques on Sun-Exposed Areas
A 54-year-old man with no notable medical history presented to an outpatient dermatology clinic with multiple skin lesions on sun-exposed areas including the face, chest, scalp, and bilateral upper extremities. The patient reported that he had not seen a doctor for 26 years. He noted that the lesions had been present for many years but was unsure of the exact timeframe. Physical examination revealed verrucous plaques with a violaceous rim and central hypopigmentation on the chest, scalp, face, and arms. Scarring alopecia also was noted on the scalp with no associated pain or pruritus. Antinuclear antibody and extractable nuclear antigen tests were negative, and urine analysis was normal. A shave biopsy of the chest was performed for histopathologic evaluation. Rapid plasma reagin tests and HIV antibody tests also were performed.
Bilateral Ankle Ulcerations and Gangrene of the Toes
Bilateral Ankle Ulcerations and Gangrene of the Toes
THE DIAGNOSIS: Rheumatoid Vasculitis
A diagnosis of rheumatoid vasculitis (RV) was made based on the clinical features, histopathology, and laboratory results in the setting of rheumatoid arthritis (RA). The distal gangrene was surgically managed with bilateral transmetatarsal amputation followed by ankle collagen graft placement. The patient was started on a prednisone taper for 1 month (40 mg/d for 3 days, then 30 mg/d for 3 days, then 20 mg/d for 24 days) before transitioning to rituximab (375 mg/m2 once weekly for 4 weeks), which improved the size and depth of the ulcers.
Rheumatoid vasculitis is an inflammatory disease that affects small- to medium-sized blood vessels in patients with RA. The pathogenesis involves immune complex deposition and complement system activation, leading to vessel wall destruction.1 Rheumatoid vasculitis is an extra-articular complication of RA that primarily is observed in seropositive patients with long-standing severe disease.1,2 The mean duration between RA diagnosis and RV onset is 10 to 14 years.2 Rheumatoid vasculitis manifests heterogeneously and can affect many organs; however, it most frequently affects the skin. Cutaneous manifestations vary in severity. Palpable purpura, pyoderma gangrenosum, and distal ulcers can be seen in addition to extensive digital ischemia with necrosis, as was present in our patient.1
When RA patients present with skin changes that are concerning for vasculitis, RV should be suspected. Currently, there are no validated diagnostic criteria for RV. Diagnosis is made based on clinical presentation and tissue biopsy. Histopathology shows small- and medium-sized vessel wall destruction with neutrophilic, granulomatous, or lymphocytic infiltration, which may be observed only in the lower dermis sparing superficial vessels.3 Direct immunofluorescence shows IgM, IgA, and C3 deposition within and around vessels.3,4 Laboratory findings including elevated inflammatory markers, positive rheumatoid factor, positive anti–cyclic citrullinated peptide, and hypocomplementemia support a diagnosis of RV.1,2
Mortality rates for RV remain high, necessitating aggressive treatment. High-dose corticosteroids typically are combined with immunosuppressant or biologic agents, frequently cyclophosphamide or rituximab.1 Consistent with other reported cases, our patient’s ulcers improved with rituximab and oral steroids.
The differential diagnosis for our patient included type I cryoglobulinemia, cutaneous polyarteritis nodosa (CPAN), peripheral vascular disease (PVD), and nonuremic calciphylaxis. Type I cryoglobulinemia manifests due to direct occlusion of vessels by precipitation of monoclonal immunoglobulin.5 It commonly is associated with lymphoproliferative diseases such as Waldenström macroglobulinemia and multiple myeloma. While our patient’s history of RA was a risk factor for mixed cryoglobulinemia as opposed to type I cryoglobulinemia, the clinical presentation aligned more closely with type I cryoglobulinemia. The clinical manifestations of type I cryoglobulinemia are related to intravascular obstruction, including Raynaud phenomenon, retiform purpura, ischemic ulcers, distal gangrene, and cold-induced urticaria.6-8 Type I cryoglobulinemia also frequently has neurologic and renal manifestations. Histopathology, along with the detection of serum cryoglobulins, is the gold standard for diagnosing cryoglobulinemia.6 On histopathology, type I cryoglobulinemia typically shows a thrombotic vasculopathy with amorphous eosinophilic periodic acid–Schiff–positive thrombi.7 False-negative results are particularly common with serum cryoglobulins, so repeat testing often is needed. While many clinical features overlap, RV is the most likely diagnosis in a patient with long-standing RA who is negative for cryoglobulins and has no history of lymphoproliferative disorders.
Cutaneous polyarteritis nodosa is a necrotizing vasculitis that similarly affects small- and medium-sized vessels. The exact etiology is unknown, but the high prevalence of anti–phosphatidylserine/prothrombin complex antibodies among patients with CPAN suggests that prothrombin bound to apoptotic endothelial cells may initiate the immune response.9 Underlying infection and inflammatory and autoimmune diseases (including group A beta-hemolytic streptococcus, hepatitis B, inflammatory bowel disease, myasthenia gravis, and RA) also may trigger CPAN.9,10,11 The most common clinical manifestations of CPAN are tender subcutaneous nodules, livedo reticularis, leg ulcers, and cutaneous necrosis. Extracutaneous symptoms such as myalgias and arthralgias also can be associated with CPAN. There is no specific serologic test to diagnose CPAN; the diagnosis is made based on clinicopathologic correlation, with characteristic histopathology showing leukocytoclastic vasculitis in the small- and medium-sized arteries of the deep dermis or hypodermis.9
Peripheral vascular disease is a manifestation of atherosclerosis that affects the legs. Risk factors for atherosclerosis, especially smoking and diabetes mellitus, similarly increase the risk for PVD.12 The most common clinical manifestation of PVD is intermittent claudication, but rarely PVD can progress to critical limb ischemia, which is characterized by pain at rest, nonhealing ulcers, or gangrene of the legs.12 Common findings on physical examination include diminished or absent pedal pulses, abnormal skin color, and skin that is cool to the touch.12 The standard diagnostic test for PVD affecting the legs is evaluation via the ankle-brachial index, with a score of 0.90 or lower being diagnostic of PVD, a score of 0.91 to 1.00 being borderline, and a score of 1.01 to 1.40 being normal.13
Calciphylaxis most frequently is seen in patients with end-stage kidney disease; however, it also has been less commonly reported in patients with normal kidney function, known as nonuremic calciphylaxis. It is characterized by calcification of arteries, arterioles, and soft tissues, which can lead to thrombosis and eventually ischemia and necrosis of the skin.14 Calciphylaxis initially causes tender, indurated, erythematous to purpuric plaques that quickly progress to retiform and stellate ulcers with overlying necrotic eschars.15 Disease typically occurs on the legs and areas that are rich in adipose tissue, such as the abdomen and thighs.16 Skin biopsy is needed for diagnosis of calciphylaxis. Characteristic histopathologic findings include calcification, microvascular thrombosis, and fibrointimal hyperplasia of small dermal and subcutaneous arteries and arterioles.16
We present a rare case of RV in a patient with well-controlled RA. While the incidence of RV is decreasing in the United States and United Kingdom due to the initiation of earlier and more aggressive RA therapies, mortality remains high.1 Thus, it is important to include RV in the differential diagnosis when there are skin changes concerning vasculitis in patients with seropositive, longstanding RA, even if the RA is well controlled.
- Kishore S, Maher L, Majithia V. Rheumatoid vasculitis: a diminishing yet devastating menace. Curr Rheumatol Rep. 2017;19:39. doi:10.1007/s11926-017-0667-3
- Makol A, Matteson EL, Warrington KJ. Rheumatoid vasculitis: an update. Curr Opin Rheumatol. 2015;27:63-70. doi:10.1097 /BOR.0000000000000126
- Patterson J. The vasculopathic reaction pattern. In: Patterson J, ed. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:241-301.
- Lora V, Cerroni L, Cota C. Skin manifestations of rheumatoid arthritis. G Ital Dermatol Venereol. 2018;153:243-255. doi:10.23736 /S0392-0488.18.05872-8
- Kolopp-Sarda MN, Miossec P. Cryoglobulinemic vasculitis: pathophysiological mechanisms and diagnosis. Curr Opin Rheumatol. 2021;33:1-7. doi:10.1097/BOR.0000000000000757
- Silva F, Pinto C, Barbosa A, et al. New insights in cryoglobulinemic vasculitis. J Autoimmun. 2019;105:102313. doi:10.1016 /j.jaut.2019.102313
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j.jbspin.2019.01.016
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756. doi:10.1111/j.1365-4632.2010.04522.
- Criado PR, Marques GF, Morita TC, et al. Epidemiological, clinical and laboratory profiles of cutaneous polyarteritis nodosa patients: report of 22 cases and literature review. Autoimmun Rev. 2016;15:558-563. doi:10.1016/j.autrev.2016.02.010
- Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol. 1997;136:706-713.
- Campia U, Gerhard-Herman M, Piazza G, et al. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133-1141. doi:10.1016/j.amjmed.2019.04.043
- Aboyans V, Criqui MH, Abraham P, et al. Measurement and interpretation of the ankle-brachial index: a scientific statement from the American Heart Association [published correction appears in Circulation. 2013 Jan 1;127:e264]. Circulation. 2012;126:2890-2909. doi:10.1161/CIR.0b013e318276fbcb
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Gomes F, La Feria P, Costa C, et al. Non-uremic calciphylaxis: a rare diagnosis with limited therapeutic strategies. Eur J Case Rep Intern Med.
THE DIAGNOSIS: Rheumatoid Vasculitis
A diagnosis of rheumatoid vasculitis (RV) was made based on the clinical features, histopathology, and laboratory results in the setting of rheumatoid arthritis (RA). The distal gangrene was surgically managed with bilateral transmetatarsal amputation followed by ankle collagen graft placement. The patient was started on a prednisone taper for 1 month (40 mg/d for 3 days, then 30 mg/d for 3 days, then 20 mg/d for 24 days) before transitioning to rituximab (375 mg/m2 once weekly for 4 weeks), which improved the size and depth of the ulcers.
Rheumatoid vasculitis is an inflammatory disease that affects small- to medium-sized blood vessels in patients with RA. The pathogenesis involves immune complex deposition and complement system activation, leading to vessel wall destruction.1 Rheumatoid vasculitis is an extra-articular complication of RA that primarily is observed in seropositive patients with long-standing severe disease.1,2 The mean duration between RA diagnosis and RV onset is 10 to 14 years.2 Rheumatoid vasculitis manifests heterogeneously and can affect many organs; however, it most frequently affects the skin. Cutaneous manifestations vary in severity. Palpable purpura, pyoderma gangrenosum, and distal ulcers can be seen in addition to extensive digital ischemia with necrosis, as was present in our patient.1
When RA patients present with skin changes that are concerning for vasculitis, RV should be suspected. Currently, there are no validated diagnostic criteria for RV. Diagnosis is made based on clinical presentation and tissue biopsy. Histopathology shows small- and medium-sized vessel wall destruction with neutrophilic, granulomatous, or lymphocytic infiltration, which may be observed only in the lower dermis sparing superficial vessels.3 Direct immunofluorescence shows IgM, IgA, and C3 deposition within and around vessels.3,4 Laboratory findings including elevated inflammatory markers, positive rheumatoid factor, positive anti–cyclic citrullinated peptide, and hypocomplementemia support a diagnosis of RV.1,2
Mortality rates for RV remain high, necessitating aggressive treatment. High-dose corticosteroids typically are combined with immunosuppressant or biologic agents, frequently cyclophosphamide or rituximab.1 Consistent with other reported cases, our patient’s ulcers improved with rituximab and oral steroids.
The differential diagnosis for our patient included type I cryoglobulinemia, cutaneous polyarteritis nodosa (CPAN), peripheral vascular disease (PVD), and nonuremic calciphylaxis. Type I cryoglobulinemia manifests due to direct occlusion of vessels by precipitation of monoclonal immunoglobulin.5 It commonly is associated with lymphoproliferative diseases such as Waldenström macroglobulinemia and multiple myeloma. While our patient’s history of RA was a risk factor for mixed cryoglobulinemia as opposed to type I cryoglobulinemia, the clinical presentation aligned more closely with type I cryoglobulinemia. The clinical manifestations of type I cryoglobulinemia are related to intravascular obstruction, including Raynaud phenomenon, retiform purpura, ischemic ulcers, distal gangrene, and cold-induced urticaria.6-8 Type I cryoglobulinemia also frequently has neurologic and renal manifestations. Histopathology, along with the detection of serum cryoglobulins, is the gold standard for diagnosing cryoglobulinemia.6 On histopathology, type I cryoglobulinemia typically shows a thrombotic vasculopathy with amorphous eosinophilic periodic acid–Schiff–positive thrombi.7 False-negative results are particularly common with serum cryoglobulins, so repeat testing often is needed. While many clinical features overlap, RV is the most likely diagnosis in a patient with long-standing RA who is negative for cryoglobulins and has no history of lymphoproliferative disorders.
Cutaneous polyarteritis nodosa is a necrotizing vasculitis that similarly affects small- and medium-sized vessels. The exact etiology is unknown, but the high prevalence of anti–phosphatidylserine/prothrombin complex antibodies among patients with CPAN suggests that prothrombin bound to apoptotic endothelial cells may initiate the immune response.9 Underlying infection and inflammatory and autoimmune diseases (including group A beta-hemolytic streptococcus, hepatitis B, inflammatory bowel disease, myasthenia gravis, and RA) also may trigger CPAN.9,10,11 The most common clinical manifestations of CPAN are tender subcutaneous nodules, livedo reticularis, leg ulcers, and cutaneous necrosis. Extracutaneous symptoms such as myalgias and arthralgias also can be associated with CPAN. There is no specific serologic test to diagnose CPAN; the diagnosis is made based on clinicopathologic correlation, with characteristic histopathology showing leukocytoclastic vasculitis in the small- and medium-sized arteries of the deep dermis or hypodermis.9
Peripheral vascular disease is a manifestation of atherosclerosis that affects the legs. Risk factors for atherosclerosis, especially smoking and diabetes mellitus, similarly increase the risk for PVD.12 The most common clinical manifestation of PVD is intermittent claudication, but rarely PVD can progress to critical limb ischemia, which is characterized by pain at rest, nonhealing ulcers, or gangrene of the legs.12 Common findings on physical examination include diminished or absent pedal pulses, abnormal skin color, and skin that is cool to the touch.12 The standard diagnostic test for PVD affecting the legs is evaluation via the ankle-brachial index, with a score of 0.90 or lower being diagnostic of PVD, a score of 0.91 to 1.00 being borderline, and a score of 1.01 to 1.40 being normal.13
Calciphylaxis most frequently is seen in patients with end-stage kidney disease; however, it also has been less commonly reported in patients with normal kidney function, known as nonuremic calciphylaxis. It is characterized by calcification of arteries, arterioles, and soft tissues, which can lead to thrombosis and eventually ischemia and necrosis of the skin.14 Calciphylaxis initially causes tender, indurated, erythematous to purpuric plaques that quickly progress to retiform and stellate ulcers with overlying necrotic eschars.15 Disease typically occurs on the legs and areas that are rich in adipose tissue, such as the abdomen and thighs.16 Skin biopsy is needed for diagnosis of calciphylaxis. Characteristic histopathologic findings include calcification, microvascular thrombosis, and fibrointimal hyperplasia of small dermal and subcutaneous arteries and arterioles.16
We present a rare case of RV in a patient with well-controlled RA. While the incidence of RV is decreasing in the United States and United Kingdom due to the initiation of earlier and more aggressive RA therapies, mortality remains high.1 Thus, it is important to include RV in the differential diagnosis when there are skin changes concerning vasculitis in patients with seropositive, longstanding RA, even if the RA is well controlled.
THE DIAGNOSIS: Rheumatoid Vasculitis
A diagnosis of rheumatoid vasculitis (RV) was made based on the clinical features, histopathology, and laboratory results in the setting of rheumatoid arthritis (RA). The distal gangrene was surgically managed with bilateral transmetatarsal amputation followed by ankle collagen graft placement. The patient was started on a prednisone taper for 1 month (40 mg/d for 3 days, then 30 mg/d for 3 days, then 20 mg/d for 24 days) before transitioning to rituximab (375 mg/m2 once weekly for 4 weeks), which improved the size and depth of the ulcers.
Rheumatoid vasculitis is an inflammatory disease that affects small- to medium-sized blood vessels in patients with RA. The pathogenesis involves immune complex deposition and complement system activation, leading to vessel wall destruction.1 Rheumatoid vasculitis is an extra-articular complication of RA that primarily is observed in seropositive patients with long-standing severe disease.1,2 The mean duration between RA diagnosis and RV onset is 10 to 14 years.2 Rheumatoid vasculitis manifests heterogeneously and can affect many organs; however, it most frequently affects the skin. Cutaneous manifestations vary in severity. Palpable purpura, pyoderma gangrenosum, and distal ulcers can be seen in addition to extensive digital ischemia with necrosis, as was present in our patient.1
When RA patients present with skin changes that are concerning for vasculitis, RV should be suspected. Currently, there are no validated diagnostic criteria for RV. Diagnosis is made based on clinical presentation and tissue biopsy. Histopathology shows small- and medium-sized vessel wall destruction with neutrophilic, granulomatous, or lymphocytic infiltration, which may be observed only in the lower dermis sparing superficial vessels.3 Direct immunofluorescence shows IgM, IgA, and C3 deposition within and around vessels.3,4 Laboratory findings including elevated inflammatory markers, positive rheumatoid factor, positive anti–cyclic citrullinated peptide, and hypocomplementemia support a diagnosis of RV.1,2
Mortality rates for RV remain high, necessitating aggressive treatment. High-dose corticosteroids typically are combined with immunosuppressant or biologic agents, frequently cyclophosphamide or rituximab.1 Consistent with other reported cases, our patient’s ulcers improved with rituximab and oral steroids.
The differential diagnosis for our patient included type I cryoglobulinemia, cutaneous polyarteritis nodosa (CPAN), peripheral vascular disease (PVD), and nonuremic calciphylaxis. Type I cryoglobulinemia manifests due to direct occlusion of vessels by precipitation of monoclonal immunoglobulin.5 It commonly is associated with lymphoproliferative diseases such as Waldenström macroglobulinemia and multiple myeloma. While our patient’s history of RA was a risk factor for mixed cryoglobulinemia as opposed to type I cryoglobulinemia, the clinical presentation aligned more closely with type I cryoglobulinemia. The clinical manifestations of type I cryoglobulinemia are related to intravascular obstruction, including Raynaud phenomenon, retiform purpura, ischemic ulcers, distal gangrene, and cold-induced urticaria.6-8 Type I cryoglobulinemia also frequently has neurologic and renal manifestations. Histopathology, along with the detection of serum cryoglobulins, is the gold standard for diagnosing cryoglobulinemia.6 On histopathology, type I cryoglobulinemia typically shows a thrombotic vasculopathy with amorphous eosinophilic periodic acid–Schiff–positive thrombi.7 False-negative results are particularly common with serum cryoglobulins, so repeat testing often is needed. While many clinical features overlap, RV is the most likely diagnosis in a patient with long-standing RA who is negative for cryoglobulins and has no history of lymphoproliferative disorders.
Cutaneous polyarteritis nodosa is a necrotizing vasculitis that similarly affects small- and medium-sized vessels. The exact etiology is unknown, but the high prevalence of anti–phosphatidylserine/prothrombin complex antibodies among patients with CPAN suggests that prothrombin bound to apoptotic endothelial cells may initiate the immune response.9 Underlying infection and inflammatory and autoimmune diseases (including group A beta-hemolytic streptococcus, hepatitis B, inflammatory bowel disease, myasthenia gravis, and RA) also may trigger CPAN.9,10,11 The most common clinical manifestations of CPAN are tender subcutaneous nodules, livedo reticularis, leg ulcers, and cutaneous necrosis. Extracutaneous symptoms such as myalgias and arthralgias also can be associated with CPAN. There is no specific serologic test to diagnose CPAN; the diagnosis is made based on clinicopathologic correlation, with characteristic histopathology showing leukocytoclastic vasculitis in the small- and medium-sized arteries of the deep dermis or hypodermis.9
Peripheral vascular disease is a manifestation of atherosclerosis that affects the legs. Risk factors for atherosclerosis, especially smoking and diabetes mellitus, similarly increase the risk for PVD.12 The most common clinical manifestation of PVD is intermittent claudication, but rarely PVD can progress to critical limb ischemia, which is characterized by pain at rest, nonhealing ulcers, or gangrene of the legs.12 Common findings on physical examination include diminished or absent pedal pulses, abnormal skin color, and skin that is cool to the touch.12 The standard diagnostic test for PVD affecting the legs is evaluation via the ankle-brachial index, with a score of 0.90 or lower being diagnostic of PVD, a score of 0.91 to 1.00 being borderline, and a score of 1.01 to 1.40 being normal.13
Calciphylaxis most frequently is seen in patients with end-stage kidney disease; however, it also has been less commonly reported in patients with normal kidney function, known as nonuremic calciphylaxis. It is characterized by calcification of arteries, arterioles, and soft tissues, which can lead to thrombosis and eventually ischemia and necrosis of the skin.14 Calciphylaxis initially causes tender, indurated, erythematous to purpuric plaques that quickly progress to retiform and stellate ulcers with overlying necrotic eschars.15 Disease typically occurs on the legs and areas that are rich in adipose tissue, such as the abdomen and thighs.16 Skin biopsy is needed for diagnosis of calciphylaxis. Characteristic histopathologic findings include calcification, microvascular thrombosis, and fibrointimal hyperplasia of small dermal and subcutaneous arteries and arterioles.16
We present a rare case of RV in a patient with well-controlled RA. While the incidence of RV is decreasing in the United States and United Kingdom due to the initiation of earlier and more aggressive RA therapies, mortality remains high.1 Thus, it is important to include RV in the differential diagnosis when there are skin changes concerning vasculitis in patients with seropositive, longstanding RA, even if the RA is well controlled.
- Kishore S, Maher L, Majithia V. Rheumatoid vasculitis: a diminishing yet devastating menace. Curr Rheumatol Rep. 2017;19:39. doi:10.1007/s11926-017-0667-3
- Makol A, Matteson EL, Warrington KJ. Rheumatoid vasculitis: an update. Curr Opin Rheumatol. 2015;27:63-70. doi:10.1097 /BOR.0000000000000126
- Patterson J. The vasculopathic reaction pattern. In: Patterson J, ed. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:241-301.
- Lora V, Cerroni L, Cota C. Skin manifestations of rheumatoid arthritis. G Ital Dermatol Venereol. 2018;153:243-255. doi:10.23736 /S0392-0488.18.05872-8
- Kolopp-Sarda MN, Miossec P. Cryoglobulinemic vasculitis: pathophysiological mechanisms and diagnosis. Curr Opin Rheumatol. 2021;33:1-7. doi:10.1097/BOR.0000000000000757
- Silva F, Pinto C, Barbosa A, et al. New insights in cryoglobulinemic vasculitis. J Autoimmun. 2019;105:102313. doi:10.1016 /j.jaut.2019.102313
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j.jbspin.2019.01.016
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756. doi:10.1111/j.1365-4632.2010.04522.
- Criado PR, Marques GF, Morita TC, et al. Epidemiological, clinical and laboratory profiles of cutaneous polyarteritis nodosa patients: report of 22 cases and literature review. Autoimmun Rev. 2016;15:558-563. doi:10.1016/j.autrev.2016.02.010
- Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol. 1997;136:706-713.
- Campia U, Gerhard-Herman M, Piazza G, et al. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133-1141. doi:10.1016/j.amjmed.2019.04.043
- Aboyans V, Criqui MH, Abraham P, et al. Measurement and interpretation of the ankle-brachial index: a scientific statement from the American Heart Association [published correction appears in Circulation. 2013 Jan 1;127:e264]. Circulation. 2012;126:2890-2909. doi:10.1161/CIR.0b013e318276fbcb
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Gomes F, La Feria P, Costa C, et al. Non-uremic calciphylaxis: a rare diagnosis with limited therapeutic strategies. Eur J Case Rep Intern Med.
- Kishore S, Maher L, Majithia V. Rheumatoid vasculitis: a diminishing yet devastating menace. Curr Rheumatol Rep. 2017;19:39. doi:10.1007/s11926-017-0667-3
- Makol A, Matteson EL, Warrington KJ. Rheumatoid vasculitis: an update. Curr Opin Rheumatol. 2015;27:63-70. doi:10.1097 /BOR.0000000000000126
- Patterson J. The vasculopathic reaction pattern. In: Patterson J, ed. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:241-301.
- Lora V, Cerroni L, Cota C. Skin manifestations of rheumatoid arthritis. G Ital Dermatol Venereol. 2018;153:243-255. doi:10.23736 /S0392-0488.18.05872-8
- Kolopp-Sarda MN, Miossec P. Cryoglobulinemic vasculitis: pathophysiological mechanisms and diagnosis. Curr Opin Rheumatol. 2021;33:1-7. doi:10.1097/BOR.0000000000000757
- Silva F, Pinto C, Barbosa A, et al. New insights in cryoglobulinemic vasculitis. J Autoimmun. 2019;105:102313. doi:10.1016 /j.jaut.2019.102313
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j.jbspin.2019.01.016
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756. doi:10.1111/j.1365-4632.2010.04522.
- Criado PR, Marques GF, Morita TC, et al. Epidemiological, clinical and laboratory profiles of cutaneous polyarteritis nodosa patients: report of 22 cases and literature review. Autoimmun Rev. 2016;15:558-563. doi:10.1016/j.autrev.2016.02.010
- Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol. 1997;136:706-713.
- Campia U, Gerhard-Herman M, Piazza G, et al. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133-1141. doi:10.1016/j.amjmed.2019.04.043
- Aboyans V, Criqui MH, Abraham P, et al. Measurement and interpretation of the ankle-brachial index: a scientific statement from the American Heart Association [published correction appears in Circulation. 2013 Jan 1;127:e264]. Circulation. 2012;126:2890-2909. doi:10.1161/CIR.0b013e318276fbcb
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Gomes F, La Feria P, Costa C, et al. Non-uremic calciphylaxis: a rare diagnosis with limited therapeutic strategies. Eur J Case Rep Intern Med.
Bilateral Ankle Ulcerations and Gangrene of the Toes
Bilateral Ankle Ulcerations and Gangrene of the Toes
A 74-year-old woman presented to the hospital with large tender ulcerations on both ankles as well as gangrene of the toes of 6 to 8 weeks’ duration. The patient had a history of hypertension as well as seropositive nonerosive rheumatoid arthritis that had been diagnosed 8 years prior and was well controlled with leflunomide and prednisone as needed for flares. She denied any history of similar ulcers as well as any recent illnesses, medication changes, or joint pain or swelling. She was evaluated by vascular surgery 1 week prior to the current presentation, at which time her ankle-brachial index score was normal. Skin examination revealed noninflammatory retiform purpura surrounding ulcerations on both ankles (top) and necrosis of all toes (bottom) with peripheral retiform purpura. Joint examination revealed swan neck deformities of multiple fingers with normal range of motion, and there was no effusion or tenderness of the joints of the fingers on palpation. No rheumatoid nodules were present. Laboratory testing revealed elevated rheumatoid factor, anti–cyclic citrullinated peptide, C-reactive protein, and anti–Sjögren syndrome–related antigen A levels and low C4 levels. Cryoglobulins, antineutrophil cytoplasmic antibodies, and serum protein electrophoresis were negative. Biopsy of an ulcer on the right ankle showed medium-sized vessel vasculitis with fibrinoid necrosis, including endothelium necrosis and a perivascular lymphocytic infiltrate. Direct immunofluorescence demonstrated dense, granular, intraperivascular deposition of IgM and IgG with slightly weaker deposition of IgA, C3, and C5b-9 in the dermis and subcutis with a greater effect on medium-sized vessels.
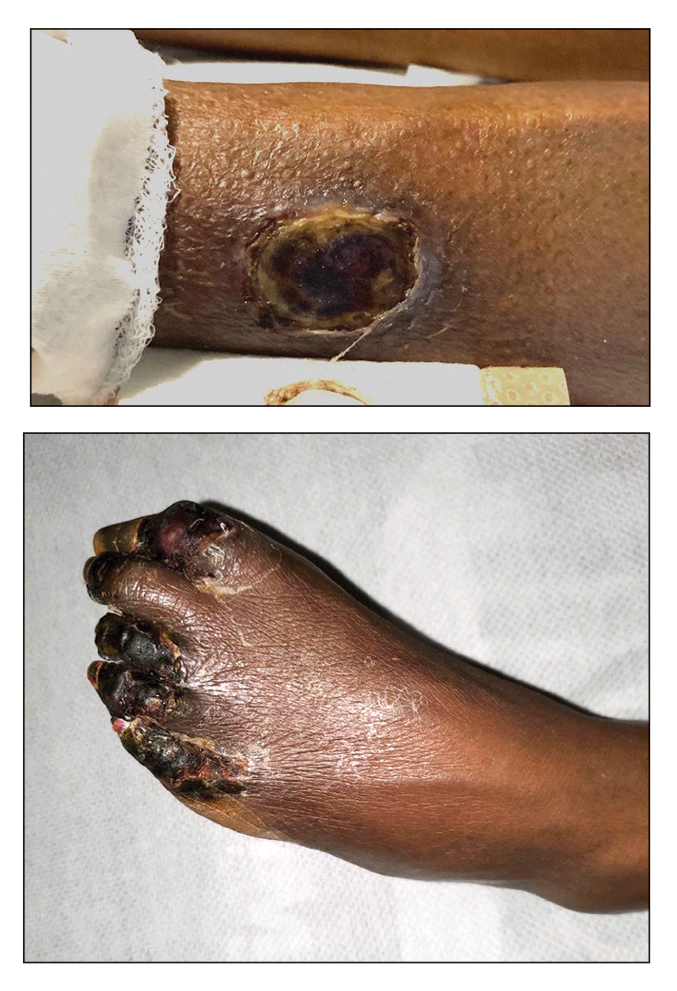
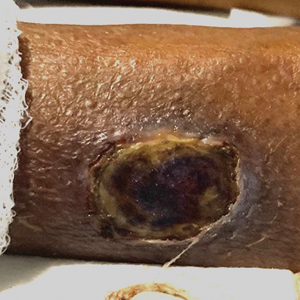
Cutaneous Metastasis of an Undiagnosed Prostatic Adenocarcinoma
Cutaneous Metastasis of an Undiagnosed Prostatic Adenocarcinoma
To the Editor:
Cutaneous metastasis of prostate cancer is rare and portends a bleak prognosis. Diagnosis of the primary cancer can be challenging, as skin metastasis can mimic a variety of conditions. We report a case of metastatic prostatic adenocarcinoma confirmed via biopsy of a new skin lesion.
A 97-year-old man presented to the dermatology clinic for routine follow-up of psoriasis. During the visit, a family member mentioned a new bleeding lesion on the left shoulder. It was not known how long the lesion had been present. Four months prior, the patient had a prostate-specific antigen (PSA) level of 582 ng/mL (reference range, 0-6.5 ng/mL), and computed tomography of the chest had shown innumerable pulmonary nodules in addition to lymphadenopathy of the left axilla, clavicle, and mediastinum. The imaging was ordered by the patient’s urologist as part of routine workup, as he had a history of obstructive renal failure and was being monitored for an indwelling catheter. Two months later, a bone scan ordered by the urologist due to high PSA levels showed extensive osteoblastic metastatic disease throughout the axial and proximal appendicular skeleton. The elevated PSA levels and findings of pulmonary and osteoblastic metastasis suggested a diagnosis of metastatic prostatic adenocarcinoma, but no confirmatory biopsy was performed following the imaging because the patient’s family declined additional workup or intervention.
Physical examination at the current presentation revealed an 8-mm brown papule with an overlying blue-white veil (Figure 1). There were no other skin findings. Primary differential diagnoses included metastatic prostate cancer, nodular melanoma, and traumatized seborrheic keratosis. A shave biopsy of the lesion showed multiple glandular structures infiltrating the dermis lined by monomorphic epithelial cells with prominent eosinophilic nucleoli (Figures 2 and 3). Focal cribriform architecture of the glands was present as well as dermal hemorrhage and a lymphohistiocytic infiltrate (Figure 2A). Interestingly, in-transit vascular metastases were confirmed with the support of ERG, CD34, and CD31 immunohistochemical staining of the vessels.
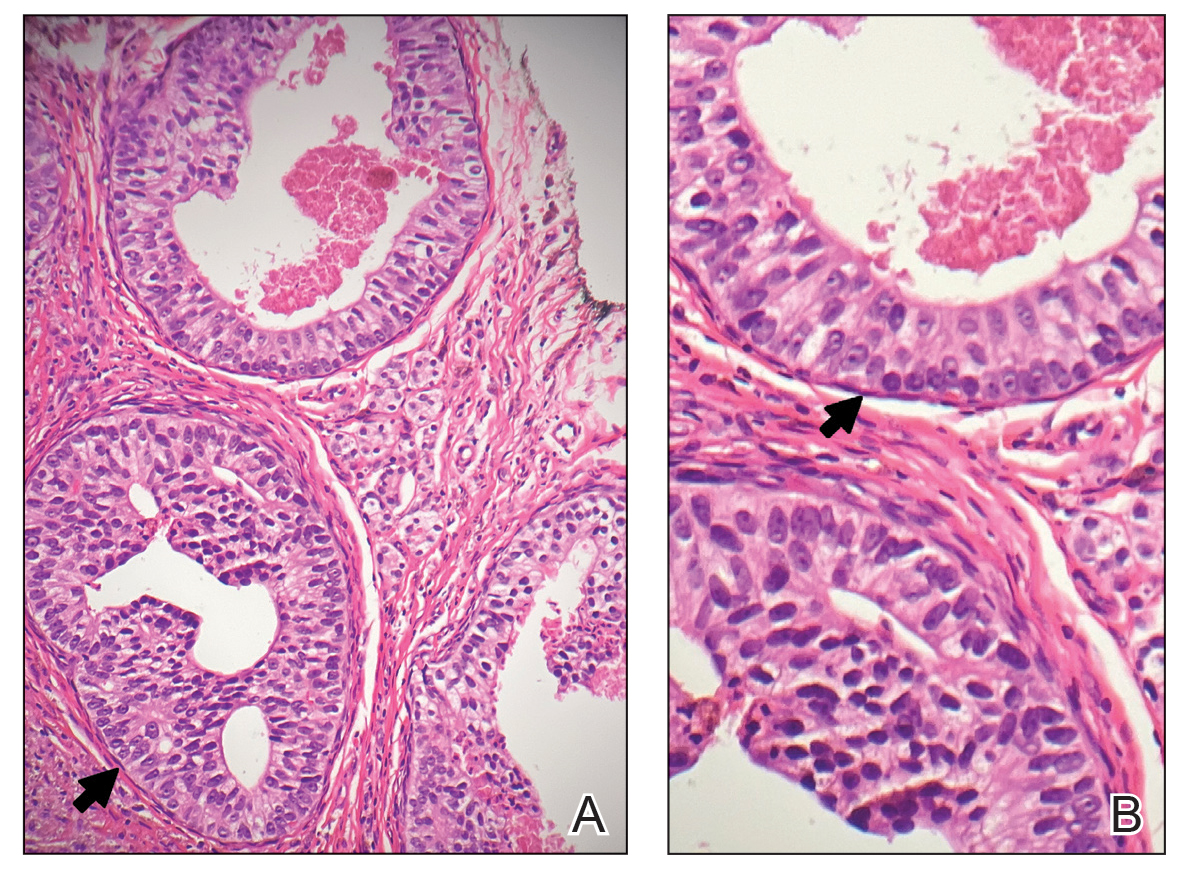
Immunohistochemical staining was positive for PSA (Figure 2B), NKX 3.1, and ERG in the invasive glandular structures, which also displayed patchy weak staining with AMACR. Staining was negative for prostein, cytokeratin (CK) 7, CK20, CK5/6, p63, p40, CDX2, and thyroid transcription factor 1. These findings were consistent with a diagnosis of cutaneous metastatic prostatic adenocarcinoma. Next-generation sequencing showed trans-membrane protease serine 2:v-ets erythroblastosis virus E26 oncogene homolog (TMPRSS2-ERG) fusion compatible with the positive ERG immunohistochemical staining. The patient and family declined any treatment due to his age, comorbidities, and rapid decline. He died 2 months after diagnosis of the skin metastasis.
Aside from nonmelanoma skin cancer, prostate cancer is the most common cancer and the second leading cause of cancer-related deaths among men in the United States.1 It most commonly metastasizes to the bones, nonregional lymph nodes, liver, and thorax.2 Metastasis to the skin is very rare, with only a 0.36% incidence.3 When prostate cancer does metastasize to the skin, the prognosis is poor, with an estimated mean survival of 7 months after diagnosis of cutaneous metastasis.4 Our patient’s survival time was even shorter—only 2 months after diagnosis of cutaneous metastasis, likely the result of his late diagnosis.
Clinically, cutaneous metastasis of prostate cancer can manifest as a wide variety of lesions; in one report of 78 cases, 56 (72%) were hard nodules, 11 (14%) were single nodules, 5 (7%) were edema or lymphedema, and 5 (7%) were an unspecific rash.4 Diagnosis of cutaneous metastasis of prostate cancer can be challenging, as it often is mistaken for other skin conditions including herpes zoster, basal cell carcinoma, angiosarcoma, cellulitis, mammary Paget disease, telangiectasia, pyoderma, morphea, and trichoepithelioma.5 In our patient, the clinical appearance of the lesion resembled a nodular melanoma. Thus, in patients with a history of prostate cancer, it is important to keep cutaneous metastasis in the differential when examining the skin because of the prognostic implications. Cutaneous metastasis of prostate cancer often indicates a poor prognosis.
In a report of 78 patients, the most common sites of skin metastasis for prostate cancer were the inguinal area and penis (28% [22/78]), abdomen (23% [18/78]), head and neck (16% [12/78]), and chest (14% [11/78]); the extremities and back were less frequently involved (10% [8/78] and 9% [7/78], respectively).4 Generally, cutaneous metastasis of internal malignancies involves the deep dermis and the subcutaneous tissue. It is common for cutaneous metastases to show histologic features of the primary tumor, as we saw in our patient. In a case series with 45 histologic diagnoses of cutaneous metastases from internal malignancies, 75.5% (34/45) of cases showed morphologic features of the primary tumor.6 However, this is not always the case, and the histologic appearance may vary. Metastatic prostate cancer may manifest as sheets, nests, or cords and often may have nuclear pleomorphism with prominent nucleoli.7
Immunohistochemical staining can help make a definitive diagnosis and differentiate the source of the tumor. Prostate cancer metastases often will stain positive for NKX3.1, PSA, AMACR, ERG, PSMA, and prosaposin, with PSA being the most specific marker.7,8 In our patient, no prostate biopsy had been performed, thus the skin biopsy was the diagnostic tissue for the prostatic adenocarcinoma.
Next-generation sequencing showed a TMPRSS2- ERG fusion, which commonly is seen in prostate cancer.9 A search of Google Scholar using the terms next-generation sequencing, cutaneous metastasis, and prostate adenocarcinoma yielded 3 additional cases of cutaneous metastasis of prostate cancer in which next-generation sequencing was performed.10-12 One case showed mutations of the tumor protein 53 (TP53) and phosphatase and tensin homolog (PTEN) genes; one showed just a TP53 mutation; and one showed inactivation of the breast cancer predisposition gene 2 (BRCA2) and amplification of MYC proto-oncogene, BHLH transcription factor (MYC) and fibroblast growth factor receptor 1 (FGFR1).10,11,12 While limited by a small number of reported cases, there does not appear to be a repeating mutation to suggest a genetic mechanism of skin metastasis.
The route of cutaneous metastasis of prostate cancer still is unclear, but hypothesized mechanisms include hematogenous or lymphatic spread, direct infiltration, or implantation from a surgical scar.11 When cutaneous involvement occurs in an area far from the primary tumor, it is thought to be the result of hematogenous spread, which would be consistent with our patient’s findings.13 Given the role of Batson venous plexus as a conduit from the prostate to the vertebral column for metastatic spread and considering the location of the lesion on our patient’s back, we hypothesized that the mechanism of metastasis to the skin was from vascular extension of the metastatic foci involving the vertebrae.
Our case highlights the importance of considering cutaneous involvement of prostatic adenocarcinoma in patients with new skin lesions, particularly in the setting of a known or suspected prostate malignancy. Skin metastasis can have a range of manifestations and provides prognostic information that can help determine the course of treatment.
- US Cancer Statistics Working Group. US cancer statistics data visualizations tool, based on 2022 submission data (1999-2020). US Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute. November 2023. Accessed November 11, 2024. https://www.cdc.gov/cancer/dataviz
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74:210-216. doi:10.1002/pros.22742
- Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026. doi:10.1016/j.urology.2004.01.014
- Wang SQ, Mecca PS, Myskowski PL, et al. Scrotal and penile papules and plaques as the initial manifestation of a cutaneous metastasis of adenocarcinoma of the prostate: case report and review of the literature. J Cutan Pathol. 2008;35:681-684. doi:10.1111/j.1600-0560.2007.00873.x
- Reddy S, Bang RH, Contreras ME. Telangiectatic cutaneous metastasis from carcinoma of the prostate. Br J Dermatol. 2007;156:598-600. doi:10.1111/j.1365-2133.2006.07696.x
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614. doi:10.1684/ejd.2017.3142
- Onalaja-Underwood AA, Sokumbi O. Eruptive papules as a cutaneous manifestation of metastatic prostate adenocarcinoma. Am J Dermatopathol. 2023;45:828-830. doi:10.1097/DAD.0000000000002559
- Oesterling JE. Prostate specific antigen: a critical assessment of the most useful tumor marker for adenocarcinoma of the prostate. J Urol. 1991;145:907-923. doi:10.1016/s0022-5347(17)38491-4
- Wang Z, Wang Y, Zhang J, et al. Significance of the TMPRSS2:ERG gene fusion in prostate cancer. Mol Med Rep. 2017;16:5450-5458. doi:10.3892/mmr.2017.7281
- Sharma H, Franklin M, Braunberger R, et al. Cutaneous metastasis from prostate cancer: a case report with literature review. Curr Probl Cancer Case Rep. 2022;7:100175. doi:10.1016/j.cpccr.2022.100175
- Dills A, Obi O, Bustos K, et al. Cutaneous manifestation of prostate adenocarcinoma: a rare presentation of a common disease. J Investig Med High Impact Case Rep. 2021;9:2324709621990769. doi:10.1177/2324709621990769
- Fadel CA, Kallab AM. Cutaneous scrotal metastasis secondary to primary prostate adenocarcinoma responding to immunotherapy. Ann Intern Med: Clinical Cases. 2022;1. doi:10.7326/aimcc.2022.0682
- Powell FC, Venencie PY, Winkelmann RK. Metastatic prostate carcinoma manifesting as penile nodules. Arch Dermatol. 1984;120:1604- 1606. doi:10.1001/archderm.1984.01650480066022
To the Editor:
Cutaneous metastasis of prostate cancer is rare and portends a bleak prognosis. Diagnosis of the primary cancer can be challenging, as skin metastasis can mimic a variety of conditions. We report a case of metastatic prostatic adenocarcinoma confirmed via biopsy of a new skin lesion.
A 97-year-old man presented to the dermatology clinic for routine follow-up of psoriasis. During the visit, a family member mentioned a new bleeding lesion on the left shoulder. It was not known how long the lesion had been present. Four months prior, the patient had a prostate-specific antigen (PSA) level of 582 ng/mL (reference range, 0-6.5 ng/mL), and computed tomography of the chest had shown innumerable pulmonary nodules in addition to lymphadenopathy of the left axilla, clavicle, and mediastinum. The imaging was ordered by the patient’s urologist as part of routine workup, as he had a history of obstructive renal failure and was being monitored for an indwelling catheter. Two months later, a bone scan ordered by the urologist due to high PSA levels showed extensive osteoblastic metastatic disease throughout the axial and proximal appendicular skeleton. The elevated PSA levels and findings of pulmonary and osteoblastic metastasis suggested a diagnosis of metastatic prostatic adenocarcinoma, but no confirmatory biopsy was performed following the imaging because the patient’s family declined additional workup or intervention.
Physical examination at the current presentation revealed an 8-mm brown papule with an overlying blue-white veil (Figure 1). There were no other skin findings. Primary differential diagnoses included metastatic prostate cancer, nodular melanoma, and traumatized seborrheic keratosis. A shave biopsy of the lesion showed multiple glandular structures infiltrating the dermis lined by monomorphic epithelial cells with prominent eosinophilic nucleoli (Figures 2 and 3). Focal cribriform architecture of the glands was present as well as dermal hemorrhage and a lymphohistiocytic infiltrate (Figure 2A). Interestingly, in-transit vascular metastases were confirmed with the support of ERG, CD34, and CD31 immunohistochemical staining of the vessels.
Immunohistochemical staining was positive for PSA (Figure 2B), NKX 3.1, and ERG in the invasive glandular structures, which also displayed patchy weak staining with AMACR. Staining was negative for prostein, cytokeratin (CK) 7, CK20, CK5/6, p63, p40, CDX2, and thyroid transcription factor 1. These findings were consistent with a diagnosis of cutaneous metastatic prostatic adenocarcinoma. Next-generation sequencing showed trans-membrane protease serine 2:v-ets erythroblastosis virus E26 oncogene homolog (TMPRSS2-ERG) fusion compatible with the positive ERG immunohistochemical staining. The patient and family declined any treatment due to his age, comorbidities, and rapid decline. He died 2 months after diagnosis of the skin metastasis.
Aside from nonmelanoma skin cancer, prostate cancer is the most common cancer and the second leading cause of cancer-related deaths among men in the United States.1 It most commonly metastasizes to the bones, nonregional lymph nodes, liver, and thorax.2 Metastasis to the skin is very rare, with only a 0.36% incidence.3 When prostate cancer does metastasize to the skin, the prognosis is poor, with an estimated mean survival of 7 months after diagnosis of cutaneous metastasis.4 Our patient’s survival time was even shorter—only 2 months after diagnosis of cutaneous metastasis, likely the result of his late diagnosis.
Clinically, cutaneous metastasis of prostate cancer can manifest as a wide variety of lesions; in one report of 78 cases, 56 (72%) were hard nodules, 11 (14%) were single nodules, 5 (7%) were edema or lymphedema, and 5 (7%) were an unspecific rash.4 Diagnosis of cutaneous metastasis of prostate cancer can be challenging, as it often is mistaken for other skin conditions including herpes zoster, basal cell carcinoma, angiosarcoma, cellulitis, mammary Paget disease, telangiectasia, pyoderma, morphea, and trichoepithelioma.5 In our patient, the clinical appearance of the lesion resembled a nodular melanoma. Thus, in patients with a history of prostate cancer, it is important to keep cutaneous metastasis in the differential when examining the skin because of the prognostic implications. Cutaneous metastasis of prostate cancer often indicates a poor prognosis.
In a report of 78 patients, the most common sites of skin metastasis for prostate cancer were the inguinal area and penis (28% [22/78]), abdomen (23% [18/78]), head and neck (16% [12/78]), and chest (14% [11/78]); the extremities and back were less frequently involved (10% [8/78] and 9% [7/78], respectively).4 Generally, cutaneous metastasis of internal malignancies involves the deep dermis and the subcutaneous tissue. It is common for cutaneous metastases to show histologic features of the primary tumor, as we saw in our patient. In a case series with 45 histologic diagnoses of cutaneous metastases from internal malignancies, 75.5% (34/45) of cases showed morphologic features of the primary tumor.6 However, this is not always the case, and the histologic appearance may vary. Metastatic prostate cancer may manifest as sheets, nests, or cords and often may have nuclear pleomorphism with prominent nucleoli.7
Immunohistochemical staining can help make a definitive diagnosis and differentiate the source of the tumor. Prostate cancer metastases often will stain positive for NKX3.1, PSA, AMACR, ERG, PSMA, and prosaposin, with PSA being the most specific marker.7,8 In our patient, no prostate biopsy had been performed, thus the skin biopsy was the diagnostic tissue for the prostatic adenocarcinoma.
Next-generation sequencing showed a TMPRSS2- ERG fusion, which commonly is seen in prostate cancer.9 A search of Google Scholar using the terms next-generation sequencing, cutaneous metastasis, and prostate adenocarcinoma yielded 3 additional cases of cutaneous metastasis of prostate cancer in which next-generation sequencing was performed.10-12 One case showed mutations of the tumor protein 53 (TP53) and phosphatase and tensin homolog (PTEN) genes; one showed just a TP53 mutation; and one showed inactivation of the breast cancer predisposition gene 2 (BRCA2) and amplification of MYC proto-oncogene, BHLH transcription factor (MYC) and fibroblast growth factor receptor 1 (FGFR1).10,11,12 While limited by a small number of reported cases, there does not appear to be a repeating mutation to suggest a genetic mechanism of skin metastasis.
The route of cutaneous metastasis of prostate cancer still is unclear, but hypothesized mechanisms include hematogenous or lymphatic spread, direct infiltration, or implantation from a surgical scar.11 When cutaneous involvement occurs in an area far from the primary tumor, it is thought to be the result of hematogenous spread, which would be consistent with our patient’s findings.13 Given the role of Batson venous plexus as a conduit from the prostate to the vertebral column for metastatic spread and considering the location of the lesion on our patient’s back, we hypothesized that the mechanism of metastasis to the skin was from vascular extension of the metastatic foci involving the vertebrae.
Our case highlights the importance of considering cutaneous involvement of prostatic adenocarcinoma in patients with new skin lesions, particularly in the setting of a known or suspected prostate malignancy. Skin metastasis can have a range of manifestations and provides prognostic information that can help determine the course of treatment.
To the Editor:
Cutaneous metastasis of prostate cancer is rare and portends a bleak prognosis. Diagnosis of the primary cancer can be challenging, as skin metastasis can mimic a variety of conditions. We report a case of metastatic prostatic adenocarcinoma confirmed via biopsy of a new skin lesion.
A 97-year-old man presented to the dermatology clinic for routine follow-up of psoriasis. During the visit, a family member mentioned a new bleeding lesion on the left shoulder. It was not known how long the lesion had been present. Four months prior, the patient had a prostate-specific antigen (PSA) level of 582 ng/mL (reference range, 0-6.5 ng/mL), and computed tomography of the chest had shown innumerable pulmonary nodules in addition to lymphadenopathy of the left axilla, clavicle, and mediastinum. The imaging was ordered by the patient’s urologist as part of routine workup, as he had a history of obstructive renal failure and was being monitored for an indwelling catheter. Two months later, a bone scan ordered by the urologist due to high PSA levels showed extensive osteoblastic metastatic disease throughout the axial and proximal appendicular skeleton. The elevated PSA levels and findings of pulmonary and osteoblastic metastasis suggested a diagnosis of metastatic prostatic adenocarcinoma, but no confirmatory biopsy was performed following the imaging because the patient’s family declined additional workup or intervention.
Physical examination at the current presentation revealed an 8-mm brown papule with an overlying blue-white veil (Figure 1). There were no other skin findings. Primary differential diagnoses included metastatic prostate cancer, nodular melanoma, and traumatized seborrheic keratosis. A shave biopsy of the lesion showed multiple glandular structures infiltrating the dermis lined by monomorphic epithelial cells with prominent eosinophilic nucleoli (Figures 2 and 3). Focal cribriform architecture of the glands was present as well as dermal hemorrhage and a lymphohistiocytic infiltrate (Figure 2A). Interestingly, in-transit vascular metastases were confirmed with the support of ERG, CD34, and CD31 immunohistochemical staining of the vessels.
Immunohistochemical staining was positive for PSA (Figure 2B), NKX 3.1, and ERG in the invasive glandular structures, which also displayed patchy weak staining with AMACR. Staining was negative for prostein, cytokeratin (CK) 7, CK20, CK5/6, p63, p40, CDX2, and thyroid transcription factor 1. These findings were consistent with a diagnosis of cutaneous metastatic prostatic adenocarcinoma. Next-generation sequencing showed trans-membrane protease serine 2:v-ets erythroblastosis virus E26 oncogene homolog (TMPRSS2-ERG) fusion compatible with the positive ERG immunohistochemical staining. The patient and family declined any treatment due to his age, comorbidities, and rapid decline. He died 2 months after diagnosis of the skin metastasis.
Aside from nonmelanoma skin cancer, prostate cancer is the most common cancer and the second leading cause of cancer-related deaths among men in the United States.1 It most commonly metastasizes to the bones, nonregional lymph nodes, liver, and thorax.2 Metastasis to the skin is very rare, with only a 0.36% incidence.3 When prostate cancer does metastasize to the skin, the prognosis is poor, with an estimated mean survival of 7 months after diagnosis of cutaneous metastasis.4 Our patient’s survival time was even shorter—only 2 months after diagnosis of cutaneous metastasis, likely the result of his late diagnosis.
Clinically, cutaneous metastasis of prostate cancer can manifest as a wide variety of lesions; in one report of 78 cases, 56 (72%) were hard nodules, 11 (14%) were single nodules, 5 (7%) were edema or lymphedema, and 5 (7%) were an unspecific rash.4 Diagnosis of cutaneous metastasis of prostate cancer can be challenging, as it often is mistaken for other skin conditions including herpes zoster, basal cell carcinoma, angiosarcoma, cellulitis, mammary Paget disease, telangiectasia, pyoderma, morphea, and trichoepithelioma.5 In our patient, the clinical appearance of the lesion resembled a nodular melanoma. Thus, in patients with a history of prostate cancer, it is important to keep cutaneous metastasis in the differential when examining the skin because of the prognostic implications. Cutaneous metastasis of prostate cancer often indicates a poor prognosis.
In a report of 78 patients, the most common sites of skin metastasis for prostate cancer were the inguinal area and penis (28% [22/78]), abdomen (23% [18/78]), head and neck (16% [12/78]), and chest (14% [11/78]); the extremities and back were less frequently involved (10% [8/78] and 9% [7/78], respectively).4 Generally, cutaneous metastasis of internal malignancies involves the deep dermis and the subcutaneous tissue. It is common for cutaneous metastases to show histologic features of the primary tumor, as we saw in our patient. In a case series with 45 histologic diagnoses of cutaneous metastases from internal malignancies, 75.5% (34/45) of cases showed morphologic features of the primary tumor.6 However, this is not always the case, and the histologic appearance may vary. Metastatic prostate cancer may manifest as sheets, nests, or cords and often may have nuclear pleomorphism with prominent nucleoli.7
Immunohistochemical staining can help make a definitive diagnosis and differentiate the source of the tumor. Prostate cancer metastases often will stain positive for NKX3.1, PSA, AMACR, ERG, PSMA, and prosaposin, with PSA being the most specific marker.7,8 In our patient, no prostate biopsy had been performed, thus the skin biopsy was the diagnostic tissue for the prostatic adenocarcinoma.
Next-generation sequencing showed a TMPRSS2- ERG fusion, which commonly is seen in prostate cancer.9 A search of Google Scholar using the terms next-generation sequencing, cutaneous metastasis, and prostate adenocarcinoma yielded 3 additional cases of cutaneous metastasis of prostate cancer in which next-generation sequencing was performed.10-12 One case showed mutations of the tumor protein 53 (TP53) and phosphatase and tensin homolog (PTEN) genes; one showed just a TP53 mutation; and one showed inactivation of the breast cancer predisposition gene 2 (BRCA2) and amplification of MYC proto-oncogene, BHLH transcription factor (MYC) and fibroblast growth factor receptor 1 (FGFR1).10,11,12 While limited by a small number of reported cases, there does not appear to be a repeating mutation to suggest a genetic mechanism of skin metastasis.
The route of cutaneous metastasis of prostate cancer still is unclear, but hypothesized mechanisms include hematogenous or lymphatic spread, direct infiltration, or implantation from a surgical scar.11 When cutaneous involvement occurs in an area far from the primary tumor, it is thought to be the result of hematogenous spread, which would be consistent with our patient’s findings.13 Given the role of Batson venous plexus as a conduit from the prostate to the vertebral column for metastatic spread and considering the location of the lesion on our patient’s back, we hypothesized that the mechanism of metastasis to the skin was from vascular extension of the metastatic foci involving the vertebrae.
Our case highlights the importance of considering cutaneous involvement of prostatic adenocarcinoma in patients with new skin lesions, particularly in the setting of a known or suspected prostate malignancy. Skin metastasis can have a range of manifestations and provides prognostic information that can help determine the course of treatment.
- US Cancer Statistics Working Group. US cancer statistics data visualizations tool, based on 2022 submission data (1999-2020). US Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute. November 2023. Accessed November 11, 2024. https://www.cdc.gov/cancer/dataviz
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74:210-216. doi:10.1002/pros.22742
- Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026. doi:10.1016/j.urology.2004.01.014
- Wang SQ, Mecca PS, Myskowski PL, et al. Scrotal and penile papules and plaques as the initial manifestation of a cutaneous metastasis of adenocarcinoma of the prostate: case report and review of the literature. J Cutan Pathol. 2008;35:681-684. doi:10.1111/j.1600-0560.2007.00873.x
- Reddy S, Bang RH, Contreras ME. Telangiectatic cutaneous metastasis from carcinoma of the prostate. Br J Dermatol. 2007;156:598-600. doi:10.1111/j.1365-2133.2006.07696.x
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614. doi:10.1684/ejd.2017.3142
- Onalaja-Underwood AA, Sokumbi O. Eruptive papules as a cutaneous manifestation of metastatic prostate adenocarcinoma. Am J Dermatopathol. 2023;45:828-830. doi:10.1097/DAD.0000000000002559
- Oesterling JE. Prostate specific antigen: a critical assessment of the most useful tumor marker for adenocarcinoma of the prostate. J Urol. 1991;145:907-923. doi:10.1016/s0022-5347(17)38491-4
- Wang Z, Wang Y, Zhang J, et al. Significance of the TMPRSS2:ERG gene fusion in prostate cancer. Mol Med Rep. 2017;16:5450-5458. doi:10.3892/mmr.2017.7281
- Sharma H, Franklin M, Braunberger R, et al. Cutaneous metastasis from prostate cancer: a case report with literature review. Curr Probl Cancer Case Rep. 2022;7:100175. doi:10.1016/j.cpccr.2022.100175
- Dills A, Obi O, Bustos K, et al. Cutaneous manifestation of prostate adenocarcinoma: a rare presentation of a common disease. J Investig Med High Impact Case Rep. 2021;9:2324709621990769. doi:10.1177/2324709621990769
- Fadel CA, Kallab AM. Cutaneous scrotal metastasis secondary to primary prostate adenocarcinoma responding to immunotherapy. Ann Intern Med: Clinical Cases. 2022;1. doi:10.7326/aimcc.2022.0682
- Powell FC, Venencie PY, Winkelmann RK. Metastatic prostate carcinoma manifesting as penile nodules. Arch Dermatol. 1984;120:1604- 1606. doi:10.1001/archderm.1984.01650480066022
- US Cancer Statistics Working Group. US cancer statistics data visualizations tool, based on 2022 submission data (1999-2020). US Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute. November 2023. Accessed November 11, 2024. https://www.cdc.gov/cancer/dataviz
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74:210-216. doi:10.1002/pros.22742
- Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026. doi:10.1016/j.urology.2004.01.014
- Wang SQ, Mecca PS, Myskowski PL, et al. Scrotal and penile papules and plaques as the initial manifestation of a cutaneous metastasis of adenocarcinoma of the prostate: case report and review of the literature. J Cutan Pathol. 2008;35:681-684. doi:10.1111/j.1600-0560.2007.00873.x
- Reddy S, Bang RH, Contreras ME. Telangiectatic cutaneous metastasis from carcinoma of the prostate. Br J Dermatol. 2007;156:598-600. doi:10.1111/j.1365-2133.2006.07696.x
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614. doi:10.1684/ejd.2017.3142
- Onalaja-Underwood AA, Sokumbi O. Eruptive papules as a cutaneous manifestation of metastatic prostate adenocarcinoma. Am J Dermatopathol. 2023;45:828-830. doi:10.1097/DAD.0000000000002559
- Oesterling JE. Prostate specific antigen: a critical assessment of the most useful tumor marker for adenocarcinoma of the prostate. J Urol. 1991;145:907-923. doi:10.1016/s0022-5347(17)38491-4
- Wang Z, Wang Y, Zhang J, et al. Significance of the TMPRSS2:ERG gene fusion in prostate cancer. Mol Med Rep. 2017;16:5450-5458. doi:10.3892/mmr.2017.7281
- Sharma H, Franklin M, Braunberger R, et al. Cutaneous metastasis from prostate cancer: a case report with literature review. Curr Probl Cancer Case Rep. 2022;7:100175. doi:10.1016/j.cpccr.2022.100175
- Dills A, Obi O, Bustos K, et al. Cutaneous manifestation of prostate adenocarcinoma: a rare presentation of a common disease. J Investig Med High Impact Case Rep. 2021;9:2324709621990769. doi:10.1177/2324709621990769
- Fadel CA, Kallab AM. Cutaneous scrotal metastasis secondary to primary prostate adenocarcinoma responding to immunotherapy. Ann Intern Med: Clinical Cases. 2022;1. doi:10.7326/aimcc.2022.0682
- Powell FC, Venencie PY, Winkelmann RK. Metastatic prostate carcinoma manifesting as penile nodules. Arch Dermatol. 1984;120:1604- 1606. doi:10.1001/archderm.1984.01650480066022
Cutaneous Metastasis of an Undiagnosed Prostatic Adenocarcinoma
Cutaneous Metastasis of an Undiagnosed Prostatic Adenocarcinoma
PRACTICE POINTS
- Cutaneous metastasis of prostate cancer can have various manifestations and portends a poor prognosis.
- New skin lesions that develop in patients with a high clinical suspicion for prostate cancer warrant consideration of cutaneous metastasis.

Indeterminate Cell Histiocytosis and a Review of Current Treatment
Indeterminate Cell Histiocytosis and a Review of Current Treatment
To the Editor:
Indeterminate cell histiocytosis (ICH) is a rare neoplastic dendritic cell disorder with a poorly understood histogenesis and pathogenesis.1 The clinical manifestation of ICH is broad and can include isolated or multiple papules or nodules on the face, neck, trunk, arms, or legs. Our case demonstrates a rare occurrence of ICH that initially was misdiagnosed and highlights the use of cobimetinib, a MEK inhibitor, as a potential new therapeutic option for ICH.
A 74-year-old man with a history of type 2 diabetes mellitus presented for evaluation of a progressive pruritic rash of approximately 5 years’ duration. The eruption previously had been diagnosed as Langerhans cell histiocytosis. It started on the chest and spread to the face, neck, trunk, and arms. The patient denied systemic symptoms and had no known history of malignancy.
Physical examination revealed pink to orange smooth papules, nodules, and small plaques on the ears, cheeks, trunk, neck, and arms (Figure 1). Baseline laboratory results showed a normal complete blood count and comprehensive metabolic panel, elevated lactate dehydrogenase and erythrocyte sedimentation rate, and hyperlipidemia. Serology for hepatitis B and C was negative. Bone marrow biopsy was normal, and positron emission tomography/ computed tomography demonstrated no evidence of extracutaneous disease. A punch biopsy of a lesion on the left forearm revealed epithelioid histiocytic proliferation in the dermis extending into the subcutis with a background infiltrate of small lymphocytes. Immunohistochemistry was positive for CD1a and CD56 and was variably positive for CD4 but negative for CD163, CD68, S100, Langerin, cyclin D1, myeloperoxidase, CD21, and CD23. No mutation was detected in BRAF codon 600. Given the negative Langerin stain, these findings were compatible with a diagnosis of ICH. After considering the lack of standard treatment options as well as the recent approval of cobimetinib for histiocytic disorders, we initiated treatment with cobimetinib at the standard dose of 60 mg daily for 21 days followed by a 7-day break.

One month into treatment, the patient’s lesions were less erythematous, and he reported improvement in pruritus. Two months into treatment, there was continued improvement in cutaneous symptoms with flattening of the lesions on the chest and back. At this time, the patient developed edema of the face and ears (Figure 2) and reported weakness, blurred vision, and decreased appetite. He was advised to take an additional 7-day treatment break before resuming cobimetinib at a decreased dose of 40 mg daily. The patient returned to the clinic 1 month later with improved systemic symptoms and continued flattening of the lesions. Five months into treatment, the lesions had continued to improve with complete resolution of the facial plaques (Figure 3).


Indeterminate cell histiocytosis is a rarely diagnosed condition characterized by the proliferation of indeterminate histiocytes that morphologically and immunophenotypically resemble Langerhans cells but lack their characteristic Birbeck granules.2 There is no standard treatment for ICH, but previous reports have described improvement with a variety of treatment options including methotrexate,3,4 UVB phototherapy,5 and topical delgocitinib 0.5%.6
Because histiocytic disorders are characterized by mutations in the mitogen-activated protein kinase pathway, it is possible that they would be responsive to MEK inhibition. Cobimetinib, a MEK inhibitor initially approved to treat metastatic melanoma, was approved by the US Food and Drug Administration to treat histiocytic disorders in October 2022.7 The approval followed the release of data from a phase 2 trial of cobimetinib in 18 adults with various histiocytic disorders, which demonstrated an 89% (16/18) overall response rate with 94% (17/18) of patients remaining progression free at 1 year.8 While cobimetinib has not specifically been studied in ICH, given the high response rate in histiocytic disorders and the lack of standard treatment options for ICH, the decision was made to initiate treatment with cobimetinib in our patient. Based on the observed improvement in our patient, we propose cobimetinib as a treatment option for patients with cutaneous ICH and recommend additional studies to confirm its safety and efficacy in patients with this disorder.
- Bakry OA, Samaka RM, Kandil MA, et al. Indeterminate cell histiocytosis with naïve cells. Rare Tumors. 2013;5:e13. doi:10.4081 /rt.2013.e13
- Manente L, Cotellessa C, Schmitt I, et al. Indeterminate cell histiocytosis: a rare histiocytic disorder. Am J Dermatopathol. 1997; 19:276-283. doi:10.1097/00000372-199706000-00014
- Lie E, Jedrych J, Sweren R, et al. Generalized indeterminate cell histiocytosis successfully treated with methotrexate. JAAD Case Rep. 2022;25:93-96. doi:10.1016/j.jdcr.2022.05.027
- Fournier J, Ingraffea A, Pedvis-Leftick A. Successful treatment of indeterminate cell histiocytosis with low-dose methotrexate. J Dermatol. 2011;38:937-939. doi:10.1111/j.1346-8138.2010.01148.x
- Logemann N, Thomas B, Yetto T. Indeterminate cell histiocytosis successfully treated with narrowband UVB. Dermatol Online J. 2013;19:20031. doi:10.5070/D31910020031
- Fujimoto RFT, Miura H, Takata M, et al. Indeterminate cell histiocytosis treated with 0.5% delgocitinib ointment. Br J Dermatol. 2023;188:E39. doi:10.1093/bjd/ljad029
- Diamond EL, Durham B, Dogan A, et al. Phase 2 trial of single-agent cobimetinib for adults with histiocytic neoplasms. Blood. 2023;142:1812. doi:10.1182/blood-2023-187508
- Diamond EL, Durham BH, Ulaner GA, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature. 2019;567:521-524. doi:10.1038/s41586-019-1012-y
To the Editor:
Indeterminate cell histiocytosis (ICH) is a rare neoplastic dendritic cell disorder with a poorly understood histogenesis and pathogenesis.1 The clinical manifestation of ICH is broad and can include isolated or multiple papules or nodules on the face, neck, trunk, arms, or legs. Our case demonstrates a rare occurrence of ICH that initially was misdiagnosed and highlights the use of cobimetinib, a MEK inhibitor, as a potential new therapeutic option for ICH.
A 74-year-old man with a history of type 2 diabetes mellitus presented for evaluation of a progressive pruritic rash of approximately 5 years’ duration. The eruption previously had been diagnosed as Langerhans cell histiocytosis. It started on the chest and spread to the face, neck, trunk, and arms. The patient denied systemic symptoms and had no known history of malignancy.
Physical examination revealed pink to orange smooth papules, nodules, and small plaques on the ears, cheeks, trunk, neck, and arms (Figure 1). Baseline laboratory results showed a normal complete blood count and comprehensive metabolic panel, elevated lactate dehydrogenase and erythrocyte sedimentation rate, and hyperlipidemia. Serology for hepatitis B and C was negative. Bone marrow biopsy was normal, and positron emission tomography/ computed tomography demonstrated no evidence of extracutaneous disease. A punch biopsy of a lesion on the left forearm revealed epithelioid histiocytic proliferation in the dermis extending into the subcutis with a background infiltrate of small lymphocytes. Immunohistochemistry was positive for CD1a and CD56 and was variably positive for CD4 but negative for CD163, CD68, S100, Langerin, cyclin D1, myeloperoxidase, CD21, and CD23. No mutation was detected in BRAF codon 600. Given the negative Langerin stain, these findings were compatible with a diagnosis of ICH. After considering the lack of standard treatment options as well as the recent approval of cobimetinib for histiocytic disorders, we initiated treatment with cobimetinib at the standard dose of 60 mg daily for 21 days followed by a 7-day break.
One month into treatment, the patient’s lesions were less erythematous, and he reported improvement in pruritus. Two months into treatment, there was continued improvement in cutaneous symptoms with flattening of the lesions on the chest and back. At this time, the patient developed edema of the face and ears (Figure 2) and reported weakness, blurred vision, and decreased appetite. He was advised to take an additional 7-day treatment break before resuming cobimetinib at a decreased dose of 40 mg daily. The patient returned to the clinic 1 month later with improved systemic symptoms and continued flattening of the lesions. Five months into treatment, the lesions had continued to improve with complete resolution of the facial plaques (Figure 3).
Indeterminate cell histiocytosis is a rarely diagnosed condition characterized by the proliferation of indeterminate histiocytes that morphologically and immunophenotypically resemble Langerhans cells but lack their characteristic Birbeck granules.2 There is no standard treatment for ICH, but previous reports have described improvement with a variety of treatment options including methotrexate,3,4 UVB phototherapy,5 and topical delgocitinib 0.5%.6
Because histiocytic disorders are characterized by mutations in the mitogen-activated protein kinase pathway, it is possible that they would be responsive to MEK inhibition. Cobimetinib, a MEK inhibitor initially approved to treat metastatic melanoma, was approved by the US Food and Drug Administration to treat histiocytic disorders in October 2022.7 The approval followed the release of data from a phase 2 trial of cobimetinib in 18 adults with various histiocytic disorders, which demonstrated an 89% (16/18) overall response rate with 94% (17/18) of patients remaining progression free at 1 year.8 While cobimetinib has not specifically been studied in ICH, given the high response rate in histiocytic disorders and the lack of standard treatment options for ICH, the decision was made to initiate treatment with cobimetinib in our patient. Based on the observed improvement in our patient, we propose cobimetinib as a treatment option for patients with cutaneous ICH and recommend additional studies to confirm its safety and efficacy in patients with this disorder.
To the Editor:
Indeterminate cell histiocytosis (ICH) is a rare neoplastic dendritic cell disorder with a poorly understood histogenesis and pathogenesis.1 The clinical manifestation of ICH is broad and can include isolated or multiple papules or nodules on the face, neck, trunk, arms, or legs. Our case demonstrates a rare occurrence of ICH that initially was misdiagnosed and highlights the use of cobimetinib, a MEK inhibitor, as a potential new therapeutic option for ICH.
A 74-year-old man with a history of type 2 diabetes mellitus presented for evaluation of a progressive pruritic rash of approximately 5 years’ duration. The eruption previously had been diagnosed as Langerhans cell histiocytosis. It started on the chest and spread to the face, neck, trunk, and arms. The patient denied systemic symptoms and had no known history of malignancy.
Physical examination revealed pink to orange smooth papules, nodules, and small plaques on the ears, cheeks, trunk, neck, and arms (Figure 1). Baseline laboratory results showed a normal complete blood count and comprehensive metabolic panel, elevated lactate dehydrogenase and erythrocyte sedimentation rate, and hyperlipidemia. Serology for hepatitis B and C was negative. Bone marrow biopsy was normal, and positron emission tomography/ computed tomography demonstrated no evidence of extracutaneous disease. A punch biopsy of a lesion on the left forearm revealed epithelioid histiocytic proliferation in the dermis extending into the subcutis with a background infiltrate of small lymphocytes. Immunohistochemistry was positive for CD1a and CD56 and was variably positive for CD4 but negative for CD163, CD68, S100, Langerin, cyclin D1, myeloperoxidase, CD21, and CD23. No mutation was detected in BRAF codon 600. Given the negative Langerin stain, these findings were compatible with a diagnosis of ICH. After considering the lack of standard treatment options as well as the recent approval of cobimetinib for histiocytic disorders, we initiated treatment with cobimetinib at the standard dose of 60 mg daily for 21 days followed by a 7-day break.
One month into treatment, the patient’s lesions were less erythematous, and he reported improvement in pruritus. Two months into treatment, there was continued improvement in cutaneous symptoms with flattening of the lesions on the chest and back. At this time, the patient developed edema of the face and ears (Figure 2) and reported weakness, blurred vision, and decreased appetite. He was advised to take an additional 7-day treatment break before resuming cobimetinib at a decreased dose of 40 mg daily. The patient returned to the clinic 1 month later with improved systemic symptoms and continued flattening of the lesions. Five months into treatment, the lesions had continued to improve with complete resolution of the facial plaques (Figure 3).
Indeterminate cell histiocytosis is a rarely diagnosed condition characterized by the proliferation of indeterminate histiocytes that morphologically and immunophenotypically resemble Langerhans cells but lack their characteristic Birbeck granules.2 There is no standard treatment for ICH, but previous reports have described improvement with a variety of treatment options including methotrexate,3,4 UVB phototherapy,5 and topical delgocitinib 0.5%.6
Because histiocytic disorders are characterized by mutations in the mitogen-activated protein kinase pathway, it is possible that they would be responsive to MEK inhibition. Cobimetinib, a MEK inhibitor initially approved to treat metastatic melanoma, was approved by the US Food and Drug Administration to treat histiocytic disorders in October 2022.7 The approval followed the release of data from a phase 2 trial of cobimetinib in 18 adults with various histiocytic disorders, which demonstrated an 89% (16/18) overall response rate with 94% (17/18) of patients remaining progression free at 1 year.8 While cobimetinib has not specifically been studied in ICH, given the high response rate in histiocytic disorders and the lack of standard treatment options for ICH, the decision was made to initiate treatment with cobimetinib in our patient. Based on the observed improvement in our patient, we propose cobimetinib as a treatment option for patients with cutaneous ICH and recommend additional studies to confirm its safety and efficacy in patients with this disorder.
- Bakry OA, Samaka RM, Kandil MA, et al. Indeterminate cell histiocytosis with naïve cells. Rare Tumors. 2013;5:e13. doi:10.4081 /rt.2013.e13
- Manente L, Cotellessa C, Schmitt I, et al. Indeterminate cell histiocytosis: a rare histiocytic disorder. Am J Dermatopathol. 1997; 19:276-283. doi:10.1097/00000372-199706000-00014
- Lie E, Jedrych J, Sweren R, et al. Generalized indeterminate cell histiocytosis successfully treated with methotrexate. JAAD Case Rep. 2022;25:93-96. doi:10.1016/j.jdcr.2022.05.027
- Fournier J, Ingraffea A, Pedvis-Leftick A. Successful treatment of indeterminate cell histiocytosis with low-dose methotrexate. J Dermatol. 2011;38:937-939. doi:10.1111/j.1346-8138.2010.01148.x
- Logemann N, Thomas B, Yetto T. Indeterminate cell histiocytosis successfully treated with narrowband UVB. Dermatol Online J. 2013;19:20031. doi:10.5070/D31910020031
- Fujimoto RFT, Miura H, Takata M, et al. Indeterminate cell histiocytosis treated with 0.5% delgocitinib ointment. Br J Dermatol. 2023;188:E39. doi:10.1093/bjd/ljad029
- Diamond EL, Durham B, Dogan A, et al. Phase 2 trial of single-agent cobimetinib for adults with histiocytic neoplasms. Blood. 2023;142:1812. doi:10.1182/blood-2023-187508
- Diamond EL, Durham BH, Ulaner GA, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature. 2019;567:521-524. doi:10.1038/s41586-019-1012-y
- Bakry OA, Samaka RM, Kandil MA, et al. Indeterminate cell histiocytosis with naïve cells. Rare Tumors. 2013;5:e13. doi:10.4081 /rt.2013.e13
- Manente L, Cotellessa C, Schmitt I, et al. Indeterminate cell histiocytosis: a rare histiocytic disorder. Am J Dermatopathol. 1997; 19:276-283. doi:10.1097/00000372-199706000-00014
- Lie E, Jedrych J, Sweren R, et al. Generalized indeterminate cell histiocytosis successfully treated with methotrexate. JAAD Case Rep. 2022;25:93-96. doi:10.1016/j.jdcr.2022.05.027
- Fournier J, Ingraffea A, Pedvis-Leftick A. Successful treatment of indeterminate cell histiocytosis with low-dose methotrexate. J Dermatol. 2011;38:937-939. doi:10.1111/j.1346-8138.2010.01148.x
- Logemann N, Thomas B, Yetto T. Indeterminate cell histiocytosis successfully treated with narrowband UVB. Dermatol Online J. 2013;19:20031. doi:10.5070/D31910020031
- Fujimoto RFT, Miura H, Takata M, et al. Indeterminate cell histiocytosis treated with 0.5% delgocitinib ointment. Br J Dermatol. 2023;188:E39. doi:10.1093/bjd/ljad029
- Diamond EL, Durham B, Dogan A, et al. Phase 2 trial of single-agent cobimetinib for adults with histiocytic neoplasms. Blood. 2023;142:1812. doi:10.1182/blood-2023-187508
- Diamond EL, Durham BH, Ulaner GA, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature. 2019;567:521-524. doi:10.1038/s41586-019-1012-y
Indeterminate Cell Histiocytosis and a Review of Current Treatment
Indeterminate Cell Histiocytosis and a Review of Current Treatment
PRACTICE POINTS
- Indeterminate cell histiocytosis (ICH) is a rare neoplastic dendritic cell disorder that can manifest as isolated or multiple papules or nodules on the face, neck, trunk, arms, or legs.
- Although there is no standard treatment for ICH, histiocytic disorders are characterized by mutations in the mitogen-activated protein kinase pathway and may be responsive to MEK inhibition.
- Cobimetinib, a MEK inhibitor initially approved to treat metastatic melanoma, was approved by the US Food and Drug Administration to treat histiocytic disorders in October 2022.

Bimekizumab for Hidradenitis Suppurativa: Pathophysiology and Promising Interventions
Bimekizumab for Hidradenitis Suppurativa: Pathophysiology and Promising Interventions
Hidradenitis suppurativa (HS) is a debilitating dermatologic condition characterized by recurrent episodes of neutrophilic inflammation affecting the apocrine and pilosebaceous units that most commonly affects individuals aged 20 to 40 years. Originating from the hair follicles, inflammation initiates the formation of painful nodules and abscesses that can progress to sinus tracts or fistulas accompanied by the development of extensive scarring, exquisite pain, and malodorous drainage.1 The lesions most commonly occur in intertriginous zones as well as areas rich in apocrine glands. The distinctive and sometimes irreversible clinical features of HS profoundly influence patients’ well-being and have lasting social, personal, and emotional impacts on their lives.2
Bimekizumab is a monoclonal antibody that specifically targets IL-17A and IL-17F, aiming to inhibit the downstream effects responsible for the chronic inflammation and tissue damage characteristic of HS.3 In HS lesions, IL-17 cytokines produced by T helper 17 (Th17) cells stimulate the production of chemokines (such as CC motif chemokine ligand 20) and neutrophil-attracting chemokines (including C-X-C motif chemokine ligands 1 and 8), cytokines (such as granulocyte colony-stimulating factor and IL-19), and epidermal antimicrobial proteins.1,2 This cascade results in the chemotaxis of monocytes and neutrophils in the skin, recruiting additional Th17 and myeloid cells and further amplifying IL-17 production.1
Bimekizumab’s mechanism of action strategically disrupts this feed-forward inflammatory loop, decreasing the transcription of neutrophil-attracting chemokines, IL-19, and epidermal antimicrobial proteins (Figure).1,2 This leads to diminished recruitment of Th17 cells and inhibits the chemotaxis of monocytes and neutrophils in the skin, effectively addressing the chronic inflammation and tissue damage characteristic of HS.
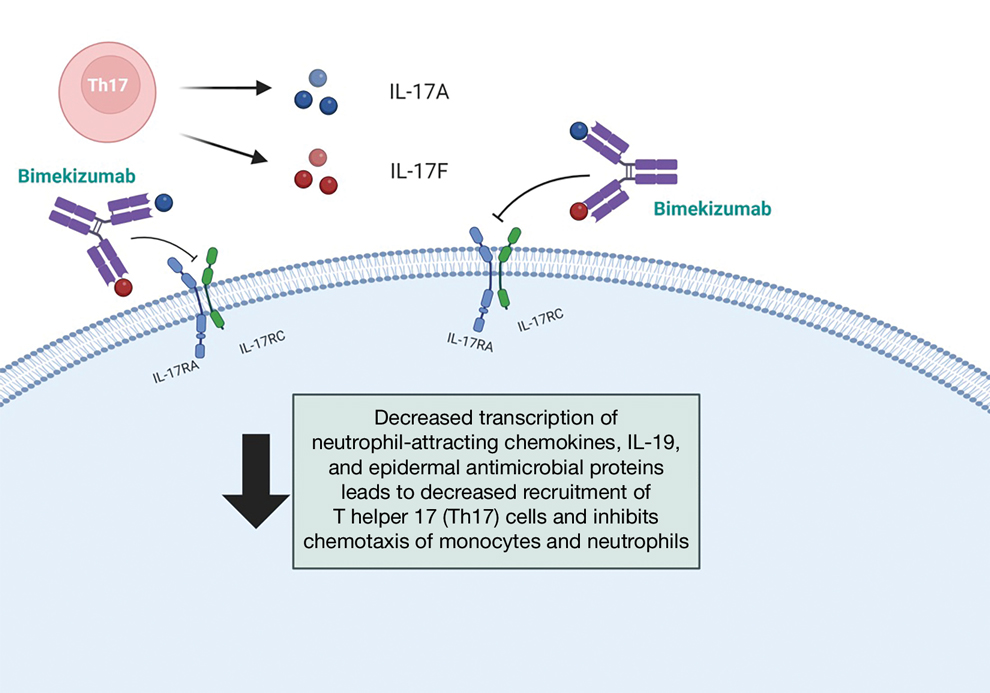
We present a comprehensive review of the current standards of care, the underlying molecular pathophysiology of HS, and evaluation of the efficacy and safety of bimekizumab.
Evaluating HS Severity
The Hurley staging system provides a valuable framework for evaluating the severity of HS based on lesion characteristics. Stage I is characterized by abscess formation without tracts or scars. Stage II is characterized by recurrent abscesses with sinus tracts and scarring. Stage III is characterized by diffuse involvement, multiple interconnected sinus tracts, and abscesses across an entire area, leaving little to no uninvolved skin.4
Treatment strategies for HS vary based on Hurley staging (eTable).5-11 For mild cases (stage I), topical and intralesional therapies are common, while moderate to severe cases (stages II and III) may require extensive surgical approaches or systemic drugs such as antibiotics, hormonal therapies, retinoids, or immunosuppressive/biologic agents.2
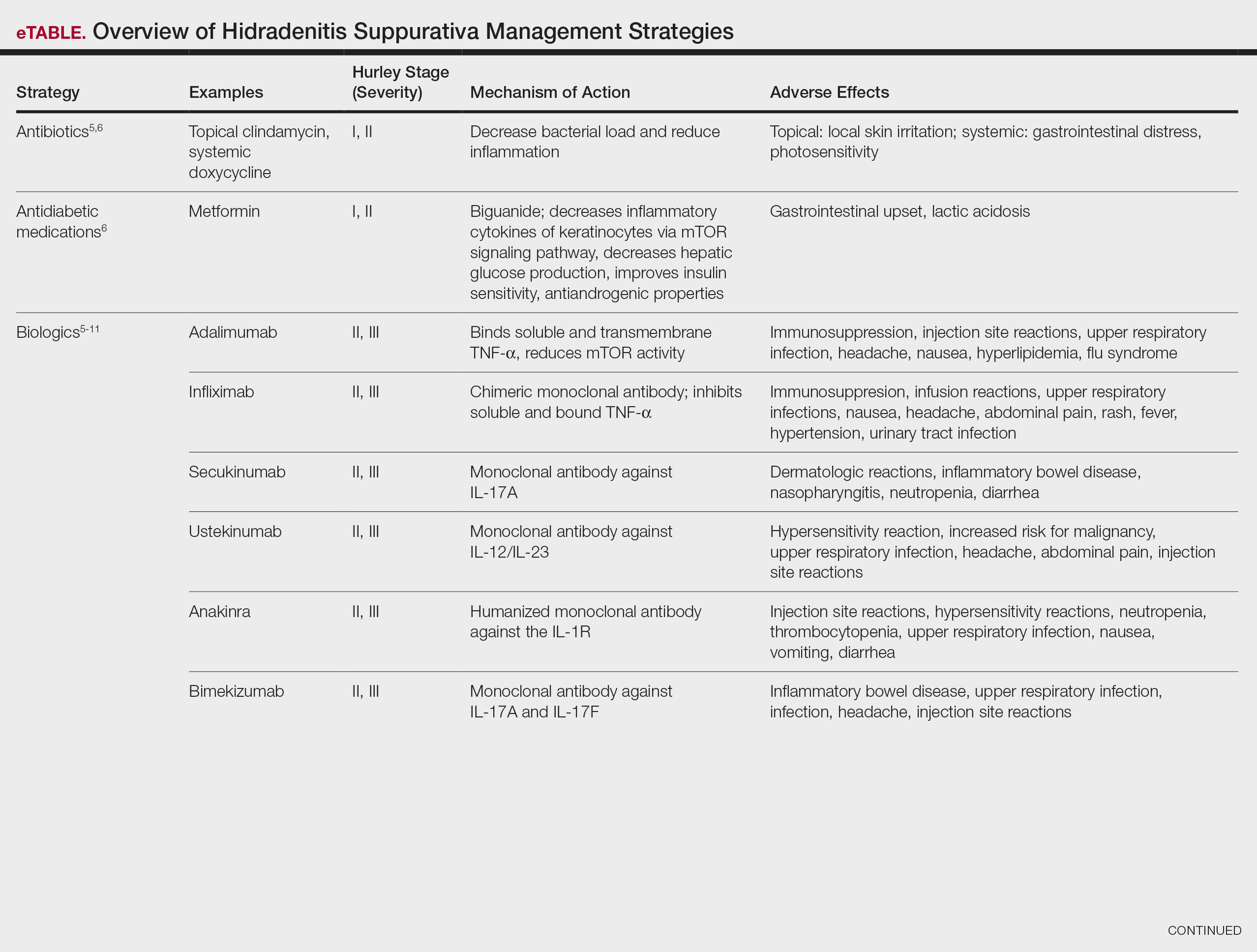
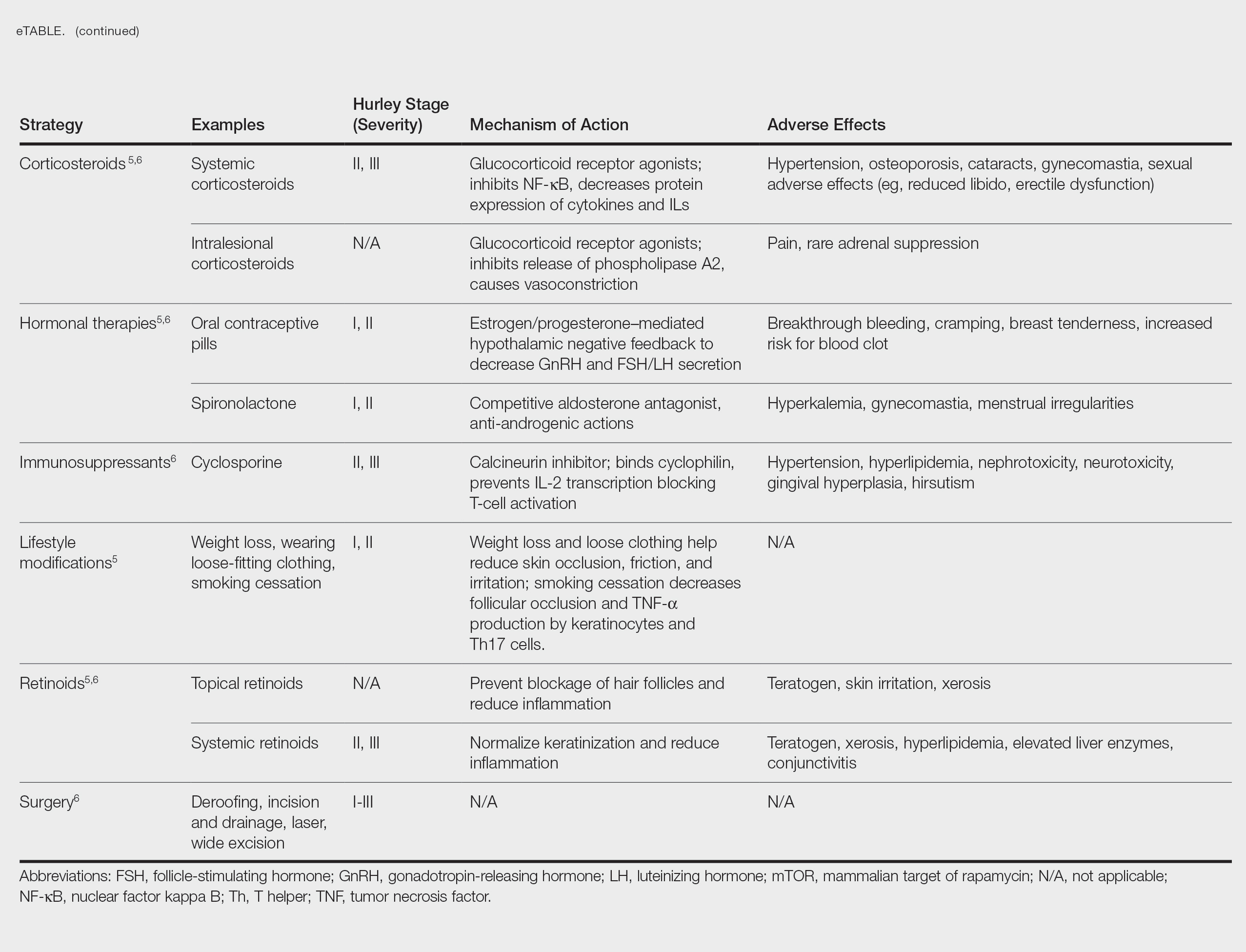
Adalimumab, an anti–tumor necrosis factor (TNF) α monoclonal antibody, was the first US Food and Drug Administration (FDA)–approved biologic for HS. Secukinumab, a monoclonal antibody against IL-17A, subsequently was approved by the FDA for moderate to severe HS.12 Off-label use of biologics including infliximab and ustekinumab expands the available treatment options for HS. In one Phase II randomized clinical trial (RCT), infliximab showed efficacy in reducing Hidradenitis Suppurativa Severity Index scores, with 26.7% (4/15) of patients achieving a 50% or greater reduction compared to placebo, although this was not statistically significant. Similarly, ustekinumab demonstrated promising results, with 47.1% (8/17) of patients achieving Hidradenitis Suppurativa Clinical Response (HiSCR) at week 40.2 This multifaceted approach aims to address the varying degrees of severity and optimize outcomes for individuals with HS.
Molecular Pathophysiology of HS
The pathogenesis of HS is multifactorial, involving a complex interplay of genetic, environmental, and behavioral factors.2 Approximately 33% to 40% of patients with HS worldwide report a first-degree relative with the condition, indicating a hereditary element with an autosomal-dominant transmission pattern and highlighting the global relevance of genetic factors in HS.4 Hidradenitis suppurativa is highly prevalent in individuals with obesity, likely due to increased intertriginous surface area, skin friction, sweat production, and hormonal changes in these patients. Smoking also commonly is associated with HS, with nicotine potentially contributing to increased follicular plugging.1 Hormonal influences also play a role, as evidenced by a greater prevalence of HS in females, disease onset typically occurring between puberty and menopause, and symptomatic fluctuations correlating with menstrual cycles and exogenous hormones.4
Altered infundibular keratinization with subsequent hyperkeratosis/occlusion and innate immune pathway activation are key events leading to development of HS.1 These events are mediated by release of pathogen- and danger-associated molecular patterns, leading to inflammasome-mediated IL-1α release, followed by downstream cytokine release.2 Elevated levels of TNF-α, IL-1Β, IL-10, IL-17, and particularly IL-17A have been detected in HS lesional skin. The IL-17 family comprises multiple members, namely IL-17A, IL-17C, IL-17E, and IL17F. IL-17A and IL-17F often are co-expressed and secreted predominantly by a subset of CD4+ T helper cells, namely Th17 cells.2 IL-17 cytokines exert pro-inflammatory effects, influencing immune cell activity and contributing to skin inflammation, particularly in HS.
Given the pivotal role of IL-17 in the pathogenesis of HS, the exploration of IL-17–targeted agents has become a focal point in clinical research. Bimekizumab, a novel IL-17 inhibitor, has emerged as a promising candidate, offering a potential breakthrough in the treatment landscape for individuals affected by HS.
Bimekizumab for HS Management
A phase II, double-blind, placebo-controlled RCT included 90 patients with moderate to severe HS (age range, 18-70 years) who were randomly assigned in a 2:1:1 ratio to receive either bimekizumab 320 mg every 2 weeks (with a 640-mg loading dose at baseline)(n=46), placebo (n=21), or adalimumab 40 mg once weekly from week 4 onward (following an initial 160-mg loading dose at baseline and 80-mg dose at week 2)(n=21). The study included a 12-week treatment period followed by a 20-week safety follow-up period. The primary endpoint was the achievement of HiSCR50—defined as a reduction of at least 50% nodules, coupled with no increase in the number of abscesses or draining fistulas relative to baseline—at week 12. Additionally, the study assessed the number of patients who achieved a modified HiSCR with 75% reduction (HiSCR75) of combined abscess and inflammatory nodule count or a modified HiSCR with 90% reduction (HiSCR90). At week 12, the modeled response rates were estimated using a Bayesian logistic regression model. For HiSCR50, the modeled rate for bimekizumab was 57.3%, with an observed rate of 62.5% (25/40), compared to a modeled rate of 26.1% for placebo (observed rate, 27.8% [5/18]). The posterior probability of superiority for bimekizumab over placebo was 0.998. By week 12, bimekizumab-treated patients achieved modeled HiSCR75 and HiSCR90 rates of 46.0% and 32.0%, respectively, with observed rates of 50.0% (20/40) for HiSCR75 and 35.0% (14/40) for HiSCR90. In comparison, placebo-treated patients achieved modeled HiSCR75 and HiSCR90 rates of 10.0% and 0%, respectively, with observed rates of 11.1% (2/18) for HiSCR75 and 0% (0/18) for HiSCR90. Adalimumab-treated participants demonstrated intermediate results, achieving modeled HiSCR75 and HiSCR90 rates of 35.0% and 15.0%, respectively, with observed rates of 38.88% (7/18) for HiSCR75 and 16.66% (3/18) for HiSCR90.7
Bimekizumab was effective in the treatment of moderate to severe HS with comparable results to adalimumab.7 The incidence of treatment-emergent adverse events was similar across treatment arms (bimekizumab, 69.6% [32/46]; placebo, 61.9% [13/21]; adalimumab, 71.4% [15/21]). The most common treatment-emergent adverse events in the biologic treatment arms were infections (43.5% [20/46] in the bimekizumab group and 42.9% [9/21] in the adalimumab group), skin and subcutaneous tissue disorders (28.3% [13/46] in the bimekizumab group and 42.9% [9/21] in the adalimumab group), and general disorders/administration site conditions (21.7% [10/46] in the bimekizumab group and 23.8% [5/21] in the adalimumab group). Serious adverse events occurred in 4.3% (2/46) of patients in the bimekizumab group, 9.5% (2/21) of patients in the placebo group, and 4.8% (1/21) of patients in the adalimumab group. Serious adverse events that required hospitalization were due to anemia and empyema in the bimekizumab group; worsening HS in the adalimumab group; and myocardial infarction, hypoesthesia, headache, and dizziness in the placebo group. No deaths occurred in this study. Overall, bimekizumab was well tolerated, and discontinuation rates were low across all arms. The primary reason for discontinuation was withdrawal of consent (not due to an adverse event) or loss to follow-up.7
Two completed 48-week phase III RCTs, BE HEARD I and BE HEARD II, evaluated the efficacy and safety of bimekizumab in patients with moderate to severe HS.13 In both trials, 2 bimekizumab dosing regimens (320 mg every 2 weeks and 320 mg every 4 weeks) were compared with placebo during the 16-week initial and 32-week maintenance treatment periods. The primary endpoint of week 16 was achieved by 47.8% (138/289) and 51.9% (151/291) of patients receiving bimekizumab every 2 weeks in BE HEARD I (n=505) and BE HEARD II (n=509), respectively, compared with 29.2% (21/72) and 32.4% (24/74) of the placebo group. The bimekizumab 320 mg every 4 weeks dosing regimen met the primary endpoint only in BE HEARD II, with 53.5% (77/144) of patients achieving HiSCR50 compared to 32.4% (24/74) with placebo (P=0.0038).13 Both trials met the key secondary endpoint of HiSCR75 at week 16 for bimekizumab 320 mg every 2 weeks vs placebo. In BE HEARD I, 33.6% (97/289) of patients receiving bimekizumab achieved HiSCR75 versus 18.1% (13/72) taking placebo. In BE HEARD II, 35.7% (104/291) of patients receiving bimekizumab achieved HiSCR75 vs 16.2% (12/74) taking placebo. Responses were maintained or increased through week 48 in both trials. The most common treatment-emergent adverse events through week 48 were worsening HS, COVID-19 infection, diarrhea, oral candidiasis, and headache.13
A smaller scale case series investigated the use of bimekizumab in 4 female patients aged 20 to 62 years with moderate to severe HS and concomitant plaque or inverse psoriasis.8 A monthly loading dose of 320 mg was given during weeks 0 to 12 followed by a maintenance dose of 320 mg administered every 8 weeks. The International Hidradenitis Suppurativa Score System, visual analogue scale, and Dermatology Life Quality Index were used to assess the effectiveness of therapy by comparing scores before and after 4 and 16 weeks of treatment. A reduction of pain and improvement of HS lesions was observed in 3 (75.0%) patients after the first dosage of bimekizumab, with completed remission of HS by week 16. The fourth patient (25.0%) experienced substantial improvement in all measures, although not complete remission. All 4 patients remained on bimekizumab, and no adverse effects were reported.8
A meta-analysis evaluated 16 RCTs of 9 biologics and 3 small-molecule inhibitors in 2076 patients with HS.10 Secukinumab was not included in this meta-analysis. Only adalimumab (risk ratio, 1.77; 95% CI, 1.44-2.17) and bimekizumab (risk ratio, 2.25; 95% CI, 1.03-4.92) were superior to placebo in achieving HiSCR response at weeks 12 to 16 in 5 RCTs and 1 RCT, respectively; however, no statistically significant differences were noted between adalimumab and bimekizumab (P=.56). This analysis concluded that adalimumab and bimekizumab are the only 2 biologics efficacious in reaching HiSCR and consistently improved both disease severity and quality of life in patients with HS with an acceptable safety profile.10 Furthermore, these biologics had no increase in serious adverse events when compared to placebo.10
A network meta-analysis of 10 clinical trials involving more than 900 total participants evaluated nonsurgical therapies for HS. The analysis used Surface Under the Cumulative Ranking curve (SUCRA) values to estimate the efficacy of treatments in achieving clinical response according to HiSCR criteria. These values range from 0% to 100%, with 100% representing the best possible ranking for efficacy. Bimekizumab showed the highest estimated efficacy with a SUCRA value of 67%, followed by adalimumab (64%), anakinra (49%), and placebo (19%). These SUCRA values indicate the relative ranking of treatments, with higher values suggesting greater likelihood of achieving clinical response, rather than representing the actual percentage of patients achieving HiSCR. Bimekizumab was found to be more efficacious than placebo (P<.05).14
Building on the initial evidence of bimekizumab’s efficacy, BE HEARD I and BE HEARD II addressed some limitations of prior studies, including small sample sizes and insufficient stratification.13 Notably, stratification by baseline Hurley stage severity (ie, the most severe stage of disease assigned at baseline) and baseline systemic antibiotic use helped mitigate bias and ensured a more robust assessment of treatment efficacy; however, certain limitations persist. While the trials demonstrated rapid and clinically meaningful responses maintained up to 48 weeks, longer-term data beyond this period are limited, leaving gaps in understanding the durability of treatment effects over years. Additionally, despite appropriate stratification, the generalizability of the findings to broader patient populations remains unclear, as trial participants may not fully represent the diversity of patients seen in clinical practice.13
Future research is needed to address these limitations. The use of validated HS biomarkers as endpoints could enhance the ability to evaluate biologic efficacy and identify predictors of response. Comparative studies with other biologics also are warranted to establish the relative efficacy of bimekizumab within the growing therapeutic landscape for HS. Finally, real-world evidence from larger and more diverse populations will be critical to confirm the trial findings and assess long-term safety and effectiveness in routine clinical practice.13
Conclusion
The existing literature and recent phase III RCTs, BE HEARD I and BE HEARD II, demonstrate that bimekizumab is an effective treatment for moderate to severe HS, with robust efficacy according to HiSCR scores and sustained responses through 48 weeks. These trials addressed some prior limitations, including small sample sizes and insufficient stratification, providing a more comprehensive evaluation of bimekizumab’s clinical impact. The safety profile of bimekizumab remains favorable, with low discontinuation rates and manageable adverse events, such as infection, gastrointestinal upset, headache, and injection-site reactions. Long-term efficacy and safety data beyond 48 weeks still are needed to fully establish its durability and impact in diverse populations. The recent FDA approval of bimekizumab for moderate to severe HS provides patients with a new treatment option, offering a more positive clinical outlook.
- Malvaso D, Calabrese L, Chiricozzi A, et al. IL-17 inhibition: a valid therapeutic strategy in the management of hidradenitis suppurativa. Pharmaceutics. 2023;15:2450. doi:10.3390 /pharmaceutics15102450
- Markota C¡agalj A, Marinovic´ B, Bukvic´ Mokos Z. New and emerging targeted therapies for hidradenitis suppurativa. Int J Mol Sci. 2022;23:3753. doi:10.3390/ijms23073753
- Zouboulis CC, Frew JW, Giamarellos-Bourboulis EJ, et al. Target molecules for future hidradenitis suppurativa treatment. Exp Dermatol. 2021;30 suppl 1:8-17. doi:10.1111/exd.14338
- Ballard K, Shuman VL. Hidradenitis suppurativa. StatPearls [Internet]. Updated May 6, 2024. Accessed December 5, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534867/
- Rathod U, Prasad PN, Patel BM, et al. Hidradenitis suppurativa: a literature review comparing current therapeutic modalities. Cureus. 2023;15:E43695. doi:10.7759/cureus.43695
- Goldburg SR, Strober BE, Payette MJ. Hidradenitis suppurativa: current and emerging treatments. J Am Acad Dermatol. 2020;82:1061-1082. doi:10.1016/j.jaad.2019.08.089
- Glatt S, Jemec GBE, Forman S, et al. Efficacy and safety of bimekizumab in moderate to severe hidradenitis suppurativa: a phase 2, doubleblind, placebo-controlled randomized clinical trial. JAMA Dermatol. 2021;157:1279-1288. doi:10.1001/jamadermatol.2021.2905
- Molinelli E, Gambini D, Maurizi A, et al. Bimekizumab in hidradenitis suppurativa: a valid and effective emerging treatment. Clin Exp Dermatol. 2023;48:1272-1274. doi:10.1093/ced/llad229
- Martora F, Megna M, Battista T, et al. Adalimumab, ustekinumab, and secukinumab in the management of hidradenitis suppurativa: a review of the real-life experience. Clin Cosmet Investig Dermatol. 2023;16:135-148. doi:10.2147/CCID.S391356
- Huang CH, Huang IH, Tai CC, et al. Biologics and small molecule inhibitors for treating hidradenitis suppurativa: a systematic review and meta-analysis. Biomedicines. 2022;10:1303. doi:10.3390 /biomedicines10061303
- Ojeda Gómez A, Madero Velázquez L, Buendía Sanchez L, et al. Inflammatory bowel disease new-onset during secukinumab therapy: real-world data from a tertiary center. Rev Esp Enferm Dig. 2021;113: 858-859. doi:10.17235/reed.2021.8397/2021
- Martora F, Marasca C, Cacciapuoti S, et al. Secukinumab in hidradenitis suppurativa patients who failed adalimumab: a 52-week real-life study. Clin Cosmet Investig Dermatol. 2024;17:159-166. doi:10.2147 /CCID.S449367
- Kimball AB, Jemec GBE, Sayed CJ, et al. Efficacy and safety of bimekizumab in patients with moderate-to-severe hidradenitis suppurativa (BE HEARD I and BE HEARD II): two 48-week, randomised, double-blind, placebo-controlled, multicentre phase 3 trials. Lancet. 2024;403:2504-2519. doi:10.1016 /S0140-6736(24)00101-6
- Gupta AK, Shear NH, Piguet V, et al. Efficacy of non-surgical monotherapies for hidradenitis suppurativa: a systematic review and network meta-analyses of randomized trials. J Dermatolog Treat. 2022;33:2149-2160. doi:10.1080/09546634.2021.1927949
Hidradenitis suppurativa (HS) is a debilitating dermatologic condition characterized by recurrent episodes of neutrophilic inflammation affecting the apocrine and pilosebaceous units that most commonly affects individuals aged 20 to 40 years. Originating from the hair follicles, inflammation initiates the formation of painful nodules and abscesses that can progress to sinus tracts or fistulas accompanied by the development of extensive scarring, exquisite pain, and malodorous drainage.1 The lesions most commonly occur in intertriginous zones as well as areas rich in apocrine glands. The distinctive and sometimes irreversible clinical features of HS profoundly influence patients’ well-being and have lasting social, personal, and emotional impacts on their lives.2
Bimekizumab is a monoclonal antibody that specifically targets IL-17A and IL-17F, aiming to inhibit the downstream effects responsible for the chronic inflammation and tissue damage characteristic of HS.3 In HS lesions, IL-17 cytokines produced by T helper 17 (Th17) cells stimulate the production of chemokines (such as CC motif chemokine ligand 20) and neutrophil-attracting chemokines (including C-X-C motif chemokine ligands 1 and 8), cytokines (such as granulocyte colony-stimulating factor and IL-19), and epidermal antimicrobial proteins.1,2 This cascade results in the chemotaxis of monocytes and neutrophils in the skin, recruiting additional Th17 and myeloid cells and further amplifying IL-17 production.1
Bimekizumab’s mechanism of action strategically disrupts this feed-forward inflammatory loop, decreasing the transcription of neutrophil-attracting chemokines, IL-19, and epidermal antimicrobial proteins (Figure).1,2 This leads to diminished recruitment of Th17 cells and inhibits the chemotaxis of monocytes and neutrophils in the skin, effectively addressing the chronic inflammation and tissue damage characteristic of HS.
We present a comprehensive review of the current standards of care, the underlying molecular pathophysiology of HS, and evaluation of the efficacy and safety of bimekizumab.
Evaluating HS Severity
The Hurley staging system provides a valuable framework for evaluating the severity of HS based on lesion characteristics. Stage I is characterized by abscess formation without tracts or scars. Stage II is characterized by recurrent abscesses with sinus tracts and scarring. Stage III is characterized by diffuse involvement, multiple interconnected sinus tracts, and abscesses across an entire area, leaving little to no uninvolved skin.4
Treatment strategies for HS vary based on Hurley staging (eTable).5-11 For mild cases (stage I), topical and intralesional therapies are common, while moderate to severe cases (stages II and III) may require extensive surgical approaches or systemic drugs such as antibiotics, hormonal therapies, retinoids, or immunosuppressive/biologic agents.2
Adalimumab, an anti–tumor necrosis factor (TNF) α monoclonal antibody, was the first US Food and Drug Administration (FDA)–approved biologic for HS. Secukinumab, a monoclonal antibody against IL-17A, subsequently was approved by the FDA for moderate to severe HS.12 Off-label use of biologics including infliximab and ustekinumab expands the available treatment options for HS. In one Phase II randomized clinical trial (RCT), infliximab showed efficacy in reducing Hidradenitis Suppurativa Severity Index scores, with 26.7% (4/15) of patients achieving a 50% or greater reduction compared to placebo, although this was not statistically significant. Similarly, ustekinumab demonstrated promising results, with 47.1% (8/17) of patients achieving Hidradenitis Suppurativa Clinical Response (HiSCR) at week 40.2 This multifaceted approach aims to address the varying degrees of severity and optimize outcomes for individuals with HS.
Molecular Pathophysiology of HS
The pathogenesis of HS is multifactorial, involving a complex interplay of genetic, environmental, and behavioral factors.2 Approximately 33% to 40% of patients with HS worldwide report a first-degree relative with the condition, indicating a hereditary element with an autosomal-dominant transmission pattern and highlighting the global relevance of genetic factors in HS.4 Hidradenitis suppurativa is highly prevalent in individuals with obesity, likely due to increased intertriginous surface area, skin friction, sweat production, and hormonal changes in these patients. Smoking also commonly is associated with HS, with nicotine potentially contributing to increased follicular plugging.1 Hormonal influences also play a role, as evidenced by a greater prevalence of HS in females, disease onset typically occurring between puberty and menopause, and symptomatic fluctuations correlating with menstrual cycles and exogenous hormones.4
Altered infundibular keratinization with subsequent hyperkeratosis/occlusion and innate immune pathway activation are key events leading to development of HS.1 These events are mediated by release of pathogen- and danger-associated molecular patterns, leading to inflammasome-mediated IL-1α release, followed by downstream cytokine release.2 Elevated levels of TNF-α, IL-1Β, IL-10, IL-17, and particularly IL-17A have been detected in HS lesional skin. The IL-17 family comprises multiple members, namely IL-17A, IL-17C, IL-17E, and IL17F. IL-17A and IL-17F often are co-expressed and secreted predominantly by a subset of CD4+ T helper cells, namely Th17 cells.2 IL-17 cytokines exert pro-inflammatory effects, influencing immune cell activity and contributing to skin inflammation, particularly in HS.
Given the pivotal role of IL-17 in the pathogenesis of HS, the exploration of IL-17–targeted agents has become a focal point in clinical research. Bimekizumab, a novel IL-17 inhibitor, has emerged as a promising candidate, offering a potential breakthrough in the treatment landscape for individuals affected by HS.
Bimekizumab for HS Management
A phase II, double-blind, placebo-controlled RCT included 90 patients with moderate to severe HS (age range, 18-70 years) who were randomly assigned in a 2:1:1 ratio to receive either bimekizumab 320 mg every 2 weeks (with a 640-mg loading dose at baseline)(n=46), placebo (n=21), or adalimumab 40 mg once weekly from week 4 onward (following an initial 160-mg loading dose at baseline and 80-mg dose at week 2)(n=21). The study included a 12-week treatment period followed by a 20-week safety follow-up period. The primary endpoint was the achievement of HiSCR50—defined as a reduction of at least 50% nodules, coupled with no increase in the number of abscesses or draining fistulas relative to baseline—at week 12. Additionally, the study assessed the number of patients who achieved a modified HiSCR with 75% reduction (HiSCR75) of combined abscess and inflammatory nodule count or a modified HiSCR with 90% reduction (HiSCR90). At week 12, the modeled response rates were estimated using a Bayesian logistic regression model. For HiSCR50, the modeled rate for bimekizumab was 57.3%, with an observed rate of 62.5% (25/40), compared to a modeled rate of 26.1% for placebo (observed rate, 27.8% [5/18]). The posterior probability of superiority for bimekizumab over placebo was 0.998. By week 12, bimekizumab-treated patients achieved modeled HiSCR75 and HiSCR90 rates of 46.0% and 32.0%, respectively, with observed rates of 50.0% (20/40) for HiSCR75 and 35.0% (14/40) for HiSCR90. In comparison, placebo-treated patients achieved modeled HiSCR75 and HiSCR90 rates of 10.0% and 0%, respectively, with observed rates of 11.1% (2/18) for HiSCR75 and 0% (0/18) for HiSCR90. Adalimumab-treated participants demonstrated intermediate results, achieving modeled HiSCR75 and HiSCR90 rates of 35.0% and 15.0%, respectively, with observed rates of 38.88% (7/18) for HiSCR75 and 16.66% (3/18) for HiSCR90.7
Bimekizumab was effective in the treatment of moderate to severe HS with comparable results to adalimumab.7 The incidence of treatment-emergent adverse events was similar across treatment arms (bimekizumab, 69.6% [32/46]; placebo, 61.9% [13/21]; adalimumab, 71.4% [15/21]). The most common treatment-emergent adverse events in the biologic treatment arms were infections (43.5% [20/46] in the bimekizumab group and 42.9% [9/21] in the adalimumab group), skin and subcutaneous tissue disorders (28.3% [13/46] in the bimekizumab group and 42.9% [9/21] in the adalimumab group), and general disorders/administration site conditions (21.7% [10/46] in the bimekizumab group and 23.8% [5/21] in the adalimumab group). Serious adverse events occurred in 4.3% (2/46) of patients in the bimekizumab group, 9.5% (2/21) of patients in the placebo group, and 4.8% (1/21) of patients in the adalimumab group. Serious adverse events that required hospitalization were due to anemia and empyema in the bimekizumab group; worsening HS in the adalimumab group; and myocardial infarction, hypoesthesia, headache, and dizziness in the placebo group. No deaths occurred in this study. Overall, bimekizumab was well tolerated, and discontinuation rates were low across all arms. The primary reason for discontinuation was withdrawal of consent (not due to an adverse event) or loss to follow-up.7
Two completed 48-week phase III RCTs, BE HEARD I and BE HEARD II, evaluated the efficacy and safety of bimekizumab in patients with moderate to severe HS.13 In both trials, 2 bimekizumab dosing regimens (320 mg every 2 weeks and 320 mg every 4 weeks) were compared with placebo during the 16-week initial and 32-week maintenance treatment periods. The primary endpoint of week 16 was achieved by 47.8% (138/289) and 51.9% (151/291) of patients receiving bimekizumab every 2 weeks in BE HEARD I (n=505) and BE HEARD II (n=509), respectively, compared with 29.2% (21/72) and 32.4% (24/74) of the placebo group. The bimekizumab 320 mg every 4 weeks dosing regimen met the primary endpoint only in BE HEARD II, with 53.5% (77/144) of patients achieving HiSCR50 compared to 32.4% (24/74) with placebo (P=0.0038).13 Both trials met the key secondary endpoint of HiSCR75 at week 16 for bimekizumab 320 mg every 2 weeks vs placebo. In BE HEARD I, 33.6% (97/289) of patients receiving bimekizumab achieved HiSCR75 versus 18.1% (13/72) taking placebo. In BE HEARD II, 35.7% (104/291) of patients receiving bimekizumab achieved HiSCR75 vs 16.2% (12/74) taking placebo. Responses were maintained or increased through week 48 in both trials. The most common treatment-emergent adverse events through week 48 were worsening HS, COVID-19 infection, diarrhea, oral candidiasis, and headache.13
A smaller scale case series investigated the use of bimekizumab in 4 female patients aged 20 to 62 years with moderate to severe HS and concomitant plaque or inverse psoriasis.8 A monthly loading dose of 320 mg was given during weeks 0 to 12 followed by a maintenance dose of 320 mg administered every 8 weeks. The International Hidradenitis Suppurativa Score System, visual analogue scale, and Dermatology Life Quality Index were used to assess the effectiveness of therapy by comparing scores before and after 4 and 16 weeks of treatment. A reduction of pain and improvement of HS lesions was observed in 3 (75.0%) patients after the first dosage of bimekizumab, with completed remission of HS by week 16. The fourth patient (25.0%) experienced substantial improvement in all measures, although not complete remission. All 4 patients remained on bimekizumab, and no adverse effects were reported.8
A meta-analysis evaluated 16 RCTs of 9 biologics and 3 small-molecule inhibitors in 2076 patients with HS.10 Secukinumab was not included in this meta-analysis. Only adalimumab (risk ratio, 1.77; 95% CI, 1.44-2.17) and bimekizumab (risk ratio, 2.25; 95% CI, 1.03-4.92) were superior to placebo in achieving HiSCR response at weeks 12 to 16 in 5 RCTs and 1 RCT, respectively; however, no statistically significant differences were noted between adalimumab and bimekizumab (P=.56). This analysis concluded that adalimumab and bimekizumab are the only 2 biologics efficacious in reaching HiSCR and consistently improved both disease severity and quality of life in patients with HS with an acceptable safety profile.10 Furthermore, these biologics had no increase in serious adverse events when compared to placebo.10
A network meta-analysis of 10 clinical trials involving more than 900 total participants evaluated nonsurgical therapies for HS. The analysis used Surface Under the Cumulative Ranking curve (SUCRA) values to estimate the efficacy of treatments in achieving clinical response according to HiSCR criteria. These values range from 0% to 100%, with 100% representing the best possible ranking for efficacy. Bimekizumab showed the highest estimated efficacy with a SUCRA value of 67%, followed by adalimumab (64%), anakinra (49%), and placebo (19%). These SUCRA values indicate the relative ranking of treatments, with higher values suggesting greater likelihood of achieving clinical response, rather than representing the actual percentage of patients achieving HiSCR. Bimekizumab was found to be more efficacious than placebo (P<.05).14
Building on the initial evidence of bimekizumab’s efficacy, BE HEARD I and BE HEARD II addressed some limitations of prior studies, including small sample sizes and insufficient stratification.13 Notably, stratification by baseline Hurley stage severity (ie, the most severe stage of disease assigned at baseline) and baseline systemic antibiotic use helped mitigate bias and ensured a more robust assessment of treatment efficacy; however, certain limitations persist. While the trials demonstrated rapid and clinically meaningful responses maintained up to 48 weeks, longer-term data beyond this period are limited, leaving gaps in understanding the durability of treatment effects over years. Additionally, despite appropriate stratification, the generalizability of the findings to broader patient populations remains unclear, as trial participants may not fully represent the diversity of patients seen in clinical practice.13
Future research is needed to address these limitations. The use of validated HS biomarkers as endpoints could enhance the ability to evaluate biologic efficacy and identify predictors of response. Comparative studies with other biologics also are warranted to establish the relative efficacy of bimekizumab within the growing therapeutic landscape for HS. Finally, real-world evidence from larger and more diverse populations will be critical to confirm the trial findings and assess long-term safety and effectiveness in routine clinical practice.13
Conclusion
The existing literature and recent phase III RCTs, BE HEARD I and BE HEARD II, demonstrate that bimekizumab is an effective treatment for moderate to severe HS, with robust efficacy according to HiSCR scores and sustained responses through 48 weeks. These trials addressed some prior limitations, including small sample sizes and insufficient stratification, providing a more comprehensive evaluation of bimekizumab’s clinical impact. The safety profile of bimekizumab remains favorable, with low discontinuation rates and manageable adverse events, such as infection, gastrointestinal upset, headache, and injection-site reactions. Long-term efficacy and safety data beyond 48 weeks still are needed to fully establish its durability and impact in diverse populations. The recent FDA approval of bimekizumab for moderate to severe HS provides patients with a new treatment option, offering a more positive clinical outlook.
Hidradenitis suppurativa (HS) is a debilitating dermatologic condition characterized by recurrent episodes of neutrophilic inflammation affecting the apocrine and pilosebaceous units that most commonly affects individuals aged 20 to 40 years. Originating from the hair follicles, inflammation initiates the formation of painful nodules and abscesses that can progress to sinus tracts or fistulas accompanied by the development of extensive scarring, exquisite pain, and malodorous drainage.1 The lesions most commonly occur in intertriginous zones as well as areas rich in apocrine glands. The distinctive and sometimes irreversible clinical features of HS profoundly influence patients’ well-being and have lasting social, personal, and emotional impacts on their lives.2
Bimekizumab is a monoclonal antibody that specifically targets IL-17A and IL-17F, aiming to inhibit the downstream effects responsible for the chronic inflammation and tissue damage characteristic of HS.3 In HS lesions, IL-17 cytokines produced by T helper 17 (Th17) cells stimulate the production of chemokines (such as CC motif chemokine ligand 20) and neutrophil-attracting chemokines (including C-X-C motif chemokine ligands 1 and 8), cytokines (such as granulocyte colony-stimulating factor and IL-19), and epidermal antimicrobial proteins.1,2 This cascade results in the chemotaxis of monocytes and neutrophils in the skin, recruiting additional Th17 and myeloid cells and further amplifying IL-17 production.1
Bimekizumab’s mechanism of action strategically disrupts this feed-forward inflammatory loop, decreasing the transcription of neutrophil-attracting chemokines, IL-19, and epidermal antimicrobial proteins (Figure).1,2 This leads to diminished recruitment of Th17 cells and inhibits the chemotaxis of monocytes and neutrophils in the skin, effectively addressing the chronic inflammation and tissue damage characteristic of HS.
We present a comprehensive review of the current standards of care, the underlying molecular pathophysiology of HS, and evaluation of the efficacy and safety of bimekizumab.
Evaluating HS Severity
The Hurley staging system provides a valuable framework for evaluating the severity of HS based on lesion characteristics. Stage I is characterized by abscess formation without tracts or scars. Stage II is characterized by recurrent abscesses with sinus tracts and scarring. Stage III is characterized by diffuse involvement, multiple interconnected sinus tracts, and abscesses across an entire area, leaving little to no uninvolved skin.4
Treatment strategies for HS vary based on Hurley staging (eTable).5-11 For mild cases (stage I), topical and intralesional therapies are common, while moderate to severe cases (stages II and III) may require extensive surgical approaches or systemic drugs such as antibiotics, hormonal therapies, retinoids, or immunosuppressive/biologic agents.2
Adalimumab, an anti–tumor necrosis factor (TNF) α monoclonal antibody, was the first US Food and Drug Administration (FDA)–approved biologic for HS. Secukinumab, a monoclonal antibody against IL-17A, subsequently was approved by the FDA for moderate to severe HS.12 Off-label use of biologics including infliximab and ustekinumab expands the available treatment options for HS. In one Phase II randomized clinical trial (RCT), infliximab showed efficacy in reducing Hidradenitis Suppurativa Severity Index scores, with 26.7% (4/15) of patients achieving a 50% or greater reduction compared to placebo, although this was not statistically significant. Similarly, ustekinumab demonstrated promising results, with 47.1% (8/17) of patients achieving Hidradenitis Suppurativa Clinical Response (HiSCR) at week 40.2 This multifaceted approach aims to address the varying degrees of severity and optimize outcomes for individuals with HS.
Molecular Pathophysiology of HS
The pathogenesis of HS is multifactorial, involving a complex interplay of genetic, environmental, and behavioral factors.2 Approximately 33% to 40% of patients with HS worldwide report a first-degree relative with the condition, indicating a hereditary element with an autosomal-dominant transmission pattern and highlighting the global relevance of genetic factors in HS.4 Hidradenitis suppurativa is highly prevalent in individuals with obesity, likely due to increased intertriginous surface area, skin friction, sweat production, and hormonal changes in these patients. Smoking also commonly is associated with HS, with nicotine potentially contributing to increased follicular plugging.1 Hormonal influences also play a role, as evidenced by a greater prevalence of HS in females, disease onset typically occurring between puberty and menopause, and symptomatic fluctuations correlating with menstrual cycles and exogenous hormones.4
Altered infundibular keratinization with subsequent hyperkeratosis/occlusion and innate immune pathway activation are key events leading to development of HS.1 These events are mediated by release of pathogen- and danger-associated molecular patterns, leading to inflammasome-mediated IL-1α release, followed by downstream cytokine release.2 Elevated levels of TNF-α, IL-1Β, IL-10, IL-17, and particularly IL-17A have been detected in HS lesional skin. The IL-17 family comprises multiple members, namely IL-17A, IL-17C, IL-17E, and IL17F. IL-17A and IL-17F often are co-expressed and secreted predominantly by a subset of CD4+ T helper cells, namely Th17 cells.2 IL-17 cytokines exert pro-inflammatory effects, influencing immune cell activity and contributing to skin inflammation, particularly in HS.
Given the pivotal role of IL-17 in the pathogenesis of HS, the exploration of IL-17–targeted agents has become a focal point in clinical research. Bimekizumab, a novel IL-17 inhibitor, has emerged as a promising candidate, offering a potential breakthrough in the treatment landscape for individuals affected by HS.
Bimekizumab for HS Management
A phase II, double-blind, placebo-controlled RCT included 90 patients with moderate to severe HS (age range, 18-70 years) who were randomly assigned in a 2:1:1 ratio to receive either bimekizumab 320 mg every 2 weeks (with a 640-mg loading dose at baseline)(n=46), placebo (n=21), or adalimumab 40 mg once weekly from week 4 onward (following an initial 160-mg loading dose at baseline and 80-mg dose at week 2)(n=21). The study included a 12-week treatment period followed by a 20-week safety follow-up period. The primary endpoint was the achievement of HiSCR50—defined as a reduction of at least 50% nodules, coupled with no increase in the number of abscesses or draining fistulas relative to baseline—at week 12. Additionally, the study assessed the number of patients who achieved a modified HiSCR with 75% reduction (HiSCR75) of combined abscess and inflammatory nodule count or a modified HiSCR with 90% reduction (HiSCR90). At week 12, the modeled response rates were estimated using a Bayesian logistic regression model. For HiSCR50, the modeled rate for bimekizumab was 57.3%, with an observed rate of 62.5% (25/40), compared to a modeled rate of 26.1% for placebo (observed rate, 27.8% [5/18]). The posterior probability of superiority for bimekizumab over placebo was 0.998. By week 12, bimekizumab-treated patients achieved modeled HiSCR75 and HiSCR90 rates of 46.0% and 32.0%, respectively, with observed rates of 50.0% (20/40) for HiSCR75 and 35.0% (14/40) for HiSCR90. In comparison, placebo-treated patients achieved modeled HiSCR75 and HiSCR90 rates of 10.0% and 0%, respectively, with observed rates of 11.1% (2/18) for HiSCR75 and 0% (0/18) for HiSCR90. Adalimumab-treated participants demonstrated intermediate results, achieving modeled HiSCR75 and HiSCR90 rates of 35.0% and 15.0%, respectively, with observed rates of 38.88% (7/18) for HiSCR75 and 16.66% (3/18) for HiSCR90.7
Bimekizumab was effective in the treatment of moderate to severe HS with comparable results to adalimumab.7 The incidence of treatment-emergent adverse events was similar across treatment arms (bimekizumab, 69.6% [32/46]; placebo, 61.9% [13/21]; adalimumab, 71.4% [15/21]). The most common treatment-emergent adverse events in the biologic treatment arms were infections (43.5% [20/46] in the bimekizumab group and 42.9% [9/21] in the adalimumab group), skin and subcutaneous tissue disorders (28.3% [13/46] in the bimekizumab group and 42.9% [9/21] in the adalimumab group), and general disorders/administration site conditions (21.7% [10/46] in the bimekizumab group and 23.8% [5/21] in the adalimumab group). Serious adverse events occurred in 4.3% (2/46) of patients in the bimekizumab group, 9.5% (2/21) of patients in the placebo group, and 4.8% (1/21) of patients in the adalimumab group. Serious adverse events that required hospitalization were due to anemia and empyema in the bimekizumab group; worsening HS in the adalimumab group; and myocardial infarction, hypoesthesia, headache, and dizziness in the placebo group. No deaths occurred in this study. Overall, bimekizumab was well tolerated, and discontinuation rates were low across all arms. The primary reason for discontinuation was withdrawal of consent (not due to an adverse event) or loss to follow-up.7
Two completed 48-week phase III RCTs, BE HEARD I and BE HEARD II, evaluated the efficacy and safety of bimekizumab in patients with moderate to severe HS.13 In both trials, 2 bimekizumab dosing regimens (320 mg every 2 weeks and 320 mg every 4 weeks) were compared with placebo during the 16-week initial and 32-week maintenance treatment periods. The primary endpoint of week 16 was achieved by 47.8% (138/289) and 51.9% (151/291) of patients receiving bimekizumab every 2 weeks in BE HEARD I (n=505) and BE HEARD II (n=509), respectively, compared with 29.2% (21/72) and 32.4% (24/74) of the placebo group. The bimekizumab 320 mg every 4 weeks dosing regimen met the primary endpoint only in BE HEARD II, with 53.5% (77/144) of patients achieving HiSCR50 compared to 32.4% (24/74) with placebo (P=0.0038).13 Both trials met the key secondary endpoint of HiSCR75 at week 16 for bimekizumab 320 mg every 2 weeks vs placebo. In BE HEARD I, 33.6% (97/289) of patients receiving bimekizumab achieved HiSCR75 versus 18.1% (13/72) taking placebo. In BE HEARD II, 35.7% (104/291) of patients receiving bimekizumab achieved HiSCR75 vs 16.2% (12/74) taking placebo. Responses were maintained or increased through week 48 in both trials. The most common treatment-emergent adverse events through week 48 were worsening HS, COVID-19 infection, diarrhea, oral candidiasis, and headache.13
A smaller scale case series investigated the use of bimekizumab in 4 female patients aged 20 to 62 years with moderate to severe HS and concomitant plaque or inverse psoriasis.8 A monthly loading dose of 320 mg was given during weeks 0 to 12 followed by a maintenance dose of 320 mg administered every 8 weeks. The International Hidradenitis Suppurativa Score System, visual analogue scale, and Dermatology Life Quality Index were used to assess the effectiveness of therapy by comparing scores before and after 4 and 16 weeks of treatment. A reduction of pain and improvement of HS lesions was observed in 3 (75.0%) patients after the first dosage of bimekizumab, with completed remission of HS by week 16. The fourth patient (25.0%) experienced substantial improvement in all measures, although not complete remission. All 4 patients remained on bimekizumab, and no adverse effects were reported.8
A meta-analysis evaluated 16 RCTs of 9 biologics and 3 small-molecule inhibitors in 2076 patients with HS.10 Secukinumab was not included in this meta-analysis. Only adalimumab (risk ratio, 1.77; 95% CI, 1.44-2.17) and bimekizumab (risk ratio, 2.25; 95% CI, 1.03-4.92) were superior to placebo in achieving HiSCR response at weeks 12 to 16 in 5 RCTs and 1 RCT, respectively; however, no statistically significant differences were noted between adalimumab and bimekizumab (P=.56). This analysis concluded that adalimumab and bimekizumab are the only 2 biologics efficacious in reaching HiSCR and consistently improved both disease severity and quality of life in patients with HS with an acceptable safety profile.10 Furthermore, these biologics had no increase in serious adverse events when compared to placebo.10
A network meta-analysis of 10 clinical trials involving more than 900 total participants evaluated nonsurgical therapies for HS. The analysis used Surface Under the Cumulative Ranking curve (SUCRA) values to estimate the efficacy of treatments in achieving clinical response according to HiSCR criteria. These values range from 0% to 100%, with 100% representing the best possible ranking for efficacy. Bimekizumab showed the highest estimated efficacy with a SUCRA value of 67%, followed by adalimumab (64%), anakinra (49%), and placebo (19%). These SUCRA values indicate the relative ranking of treatments, with higher values suggesting greater likelihood of achieving clinical response, rather than representing the actual percentage of patients achieving HiSCR. Bimekizumab was found to be more efficacious than placebo (P<.05).14
Building on the initial evidence of bimekizumab’s efficacy, BE HEARD I and BE HEARD II addressed some limitations of prior studies, including small sample sizes and insufficient stratification.13 Notably, stratification by baseline Hurley stage severity (ie, the most severe stage of disease assigned at baseline) and baseline systemic antibiotic use helped mitigate bias and ensured a more robust assessment of treatment efficacy; however, certain limitations persist. While the trials demonstrated rapid and clinically meaningful responses maintained up to 48 weeks, longer-term data beyond this period are limited, leaving gaps in understanding the durability of treatment effects over years. Additionally, despite appropriate stratification, the generalizability of the findings to broader patient populations remains unclear, as trial participants may not fully represent the diversity of patients seen in clinical practice.13
Future research is needed to address these limitations. The use of validated HS biomarkers as endpoints could enhance the ability to evaluate biologic efficacy and identify predictors of response. Comparative studies with other biologics also are warranted to establish the relative efficacy of bimekizumab within the growing therapeutic landscape for HS. Finally, real-world evidence from larger and more diverse populations will be critical to confirm the trial findings and assess long-term safety and effectiveness in routine clinical practice.13
Conclusion
The existing literature and recent phase III RCTs, BE HEARD I and BE HEARD II, demonstrate that bimekizumab is an effective treatment for moderate to severe HS, with robust efficacy according to HiSCR scores and sustained responses through 48 weeks. These trials addressed some prior limitations, including small sample sizes and insufficient stratification, providing a more comprehensive evaluation of bimekizumab’s clinical impact. The safety profile of bimekizumab remains favorable, with low discontinuation rates and manageable adverse events, such as infection, gastrointestinal upset, headache, and injection-site reactions. Long-term efficacy and safety data beyond 48 weeks still are needed to fully establish its durability and impact in diverse populations. The recent FDA approval of bimekizumab for moderate to severe HS provides patients with a new treatment option, offering a more positive clinical outlook.
- Malvaso D, Calabrese L, Chiricozzi A, et al. IL-17 inhibition: a valid therapeutic strategy in the management of hidradenitis suppurativa. Pharmaceutics. 2023;15:2450. doi:10.3390 /pharmaceutics15102450
- Markota C¡agalj A, Marinovic´ B, Bukvic´ Mokos Z. New and emerging targeted therapies for hidradenitis suppurativa. Int J Mol Sci. 2022;23:3753. doi:10.3390/ijms23073753
- Zouboulis CC, Frew JW, Giamarellos-Bourboulis EJ, et al. Target molecules for future hidradenitis suppurativa treatment. Exp Dermatol. 2021;30 suppl 1:8-17. doi:10.1111/exd.14338
- Ballard K, Shuman VL. Hidradenitis suppurativa. StatPearls [Internet]. Updated May 6, 2024. Accessed December 5, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534867/
- Rathod U, Prasad PN, Patel BM, et al. Hidradenitis suppurativa: a literature review comparing current therapeutic modalities. Cureus. 2023;15:E43695. doi:10.7759/cureus.43695
- Goldburg SR, Strober BE, Payette MJ. Hidradenitis suppurativa: current and emerging treatments. J Am Acad Dermatol. 2020;82:1061-1082. doi:10.1016/j.jaad.2019.08.089
- Glatt S, Jemec GBE, Forman S, et al. Efficacy and safety of bimekizumab in moderate to severe hidradenitis suppurativa: a phase 2, doubleblind, placebo-controlled randomized clinical trial. JAMA Dermatol. 2021;157:1279-1288. doi:10.1001/jamadermatol.2021.2905
- Molinelli E, Gambini D, Maurizi A, et al. Bimekizumab in hidradenitis suppurativa: a valid and effective emerging treatment. Clin Exp Dermatol. 2023;48:1272-1274. doi:10.1093/ced/llad229
- Martora F, Megna M, Battista T, et al. Adalimumab, ustekinumab, and secukinumab in the management of hidradenitis suppurativa: a review of the real-life experience. Clin Cosmet Investig Dermatol. 2023;16:135-148. doi:10.2147/CCID.S391356
- Huang CH, Huang IH, Tai CC, et al. Biologics and small molecule inhibitors for treating hidradenitis suppurativa: a systematic review and meta-analysis. Biomedicines. 2022;10:1303. doi:10.3390 /biomedicines10061303
- Ojeda Gómez A, Madero Velázquez L, Buendía Sanchez L, et al. Inflammatory bowel disease new-onset during secukinumab therapy: real-world data from a tertiary center. Rev Esp Enferm Dig. 2021;113: 858-859. doi:10.17235/reed.2021.8397/2021
- Martora F, Marasca C, Cacciapuoti S, et al. Secukinumab in hidradenitis suppurativa patients who failed adalimumab: a 52-week real-life study. Clin Cosmet Investig Dermatol. 2024;17:159-166. doi:10.2147 /CCID.S449367
- Kimball AB, Jemec GBE, Sayed CJ, et al. Efficacy and safety of bimekizumab in patients with moderate-to-severe hidradenitis suppurativa (BE HEARD I and BE HEARD II): two 48-week, randomised, double-blind, placebo-controlled, multicentre phase 3 trials. Lancet. 2024;403:2504-2519. doi:10.1016 /S0140-6736(24)00101-6
- Gupta AK, Shear NH, Piguet V, et al. Efficacy of non-surgical monotherapies for hidradenitis suppurativa: a systematic review and network meta-analyses of randomized trials. J Dermatolog Treat. 2022;33:2149-2160. doi:10.1080/09546634.2021.1927949
- Malvaso D, Calabrese L, Chiricozzi A, et al. IL-17 inhibition: a valid therapeutic strategy in the management of hidradenitis suppurativa. Pharmaceutics. 2023;15:2450. doi:10.3390 /pharmaceutics15102450
- Markota C¡agalj A, Marinovic´ B, Bukvic´ Mokos Z. New and emerging targeted therapies for hidradenitis suppurativa. Int J Mol Sci. 2022;23:3753. doi:10.3390/ijms23073753
- Zouboulis CC, Frew JW, Giamarellos-Bourboulis EJ, et al. Target molecules for future hidradenitis suppurativa treatment. Exp Dermatol. 2021;30 suppl 1:8-17. doi:10.1111/exd.14338
- Ballard K, Shuman VL. Hidradenitis suppurativa. StatPearls [Internet]. Updated May 6, 2024. Accessed December 5, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534867/
- Rathod U, Prasad PN, Patel BM, et al. Hidradenitis suppurativa: a literature review comparing current therapeutic modalities. Cureus. 2023;15:E43695. doi:10.7759/cureus.43695
- Goldburg SR, Strober BE, Payette MJ. Hidradenitis suppurativa: current and emerging treatments. J Am Acad Dermatol. 2020;82:1061-1082. doi:10.1016/j.jaad.2019.08.089
- Glatt S, Jemec GBE, Forman S, et al. Efficacy and safety of bimekizumab in moderate to severe hidradenitis suppurativa: a phase 2, doubleblind, placebo-controlled randomized clinical trial. JAMA Dermatol. 2021;157:1279-1288. doi:10.1001/jamadermatol.2021.2905
- Molinelli E, Gambini D, Maurizi A, et al. Bimekizumab in hidradenitis suppurativa: a valid and effective emerging treatment. Clin Exp Dermatol. 2023;48:1272-1274. doi:10.1093/ced/llad229
- Martora F, Megna M, Battista T, et al. Adalimumab, ustekinumab, and secukinumab in the management of hidradenitis suppurativa: a review of the real-life experience. Clin Cosmet Investig Dermatol. 2023;16:135-148. doi:10.2147/CCID.S391356
- Huang CH, Huang IH, Tai CC, et al. Biologics and small molecule inhibitors for treating hidradenitis suppurativa: a systematic review and meta-analysis. Biomedicines. 2022;10:1303. doi:10.3390 /biomedicines10061303
- Ojeda Gómez A, Madero Velázquez L, Buendía Sanchez L, et al. Inflammatory bowel disease new-onset during secukinumab therapy: real-world data from a tertiary center. Rev Esp Enferm Dig. 2021;113: 858-859. doi:10.17235/reed.2021.8397/2021
- Martora F, Marasca C, Cacciapuoti S, et al. Secukinumab in hidradenitis suppurativa patients who failed adalimumab: a 52-week real-life study. Clin Cosmet Investig Dermatol. 2024;17:159-166. doi:10.2147 /CCID.S449367
- Kimball AB, Jemec GBE, Sayed CJ, et al. Efficacy and safety of bimekizumab in patients with moderate-to-severe hidradenitis suppurativa (BE HEARD I and BE HEARD II): two 48-week, randomised, double-blind, placebo-controlled, multicentre phase 3 trials. Lancet. 2024;403:2504-2519. doi:10.1016 /S0140-6736(24)00101-6
- Gupta AK, Shear NH, Piguet V, et al. Efficacy of non-surgical monotherapies for hidradenitis suppurativa: a systematic review and network meta-analyses of randomized trials. J Dermatolog Treat. 2022;33:2149-2160. doi:10.1080/09546634.2021.1927949
Bimekizumab for Hidradenitis Suppurativa: Pathophysiology and Promising Interventions
Bimekizumab for Hidradenitis Suppurativa: Pathophysiology and Promising Interventions
PRACTICE POINTS
- Management of hidradenitis suppurativa (HS) includes lifestyle modifications as well as topical and systemic antibiotics, intralesional and systemic corticosteroids, retinoids, hormonal therapies, immunosuppressants, biologic agents, and minor to invasive surgical procedures.
- Adalimumab, secukinumab, and more recently bimekizumab are biologics that are approved by the US Food and Drug Administration for the treatment of moderate to severe HS.
- Bimekizumab is a monoclonal antibody targeting IL-17A and IL-17F that has demonstrated strong clinical efficacy in generating a sustained clinical response in moderate to severe HS-related clinical features.
Solitary Lesion on the Umbilicus
Solitary Lesion on the Umbilicus
THE DIAGNOSIS: Cutaneous Endometriosis
Endometriosis is the ectopic presence of endometrial tissue and occurs in approximately 13% of women of childbearing age.1 This non-neoplastic lesion can manifest on the skin in less than 5.5% of endometriosis cases worldwide. Historically, secondary cutaneous endometriosis (CE) most frequently has been associated with prior gynecologic surgery (often cesarean section)2; however, an increased incidence of primary CE in patients without prior surgical history recently has been documented in the literature.3 While secondary CE usually manifests at the site of a surgical scar, primary CE has a predilection for the umbilicus (Villar nodule). In both primary and secondary CE, patients present clinically with a solitary nodule and abdominal pain that may be exacerbated during menstruation. Bleeding without associated pain may be more common in primary CE, while bleeding with pain may be more common in secondary CE. Cutaneous endometriosis often is overlooked given its low incidence, leading to delayed diagnosis. Primary CE often is misdiagnosed clinically as a pyogenic granuloma, Sister Mary Joseph nodule, or keloid, while secondary CE may be mistaken for a fibroma, incisional hernia, or granuloma.2
Primary and secondary CE have identical histopathologic features. Glands of variable size consisting of a single epithelial layer of columnar cells are present in the reticular dermis or subcutis (quiz image).4 The accompanying periglandular stroma often is uniform, consisting of spindle-shaped basophilic cells with abundant vascular structures. The stroma may contain moderate numbers of mitotic figures, a chronic inflammatory infiltrate, and extravasated red blood cells. The ectopic tissue may be inactive or display morphologic changes resembling those of the endometrium in the normal menstrual cycle.4 As the ectopic tissue progresses through the stages of menstruation, the glandular morphology also transforms. The proliferative stage demonstrates increased epithelial mitotic figures, the secretory stage exhibits intraluminal secretion, and during menstruation there are degenerative epithelial cells and evidence of vascular congestion. A mixture of glandular stages may be seen in biopsy results. Robust immunohistochemical expression of CD10 in the endometrial stroma can aid in diagnosis (Figure 1). Estrogen and progesterone receptor immunostaining also shows strong nuclear positivity, except in decidualized tissue.4 Unlike intestinal glands, endometrial glands do not express CDX2 or CK20.5 Complete surgical excision of CE usually is curative; however, recurrence has been documented in 10% (3/30) of cases.2
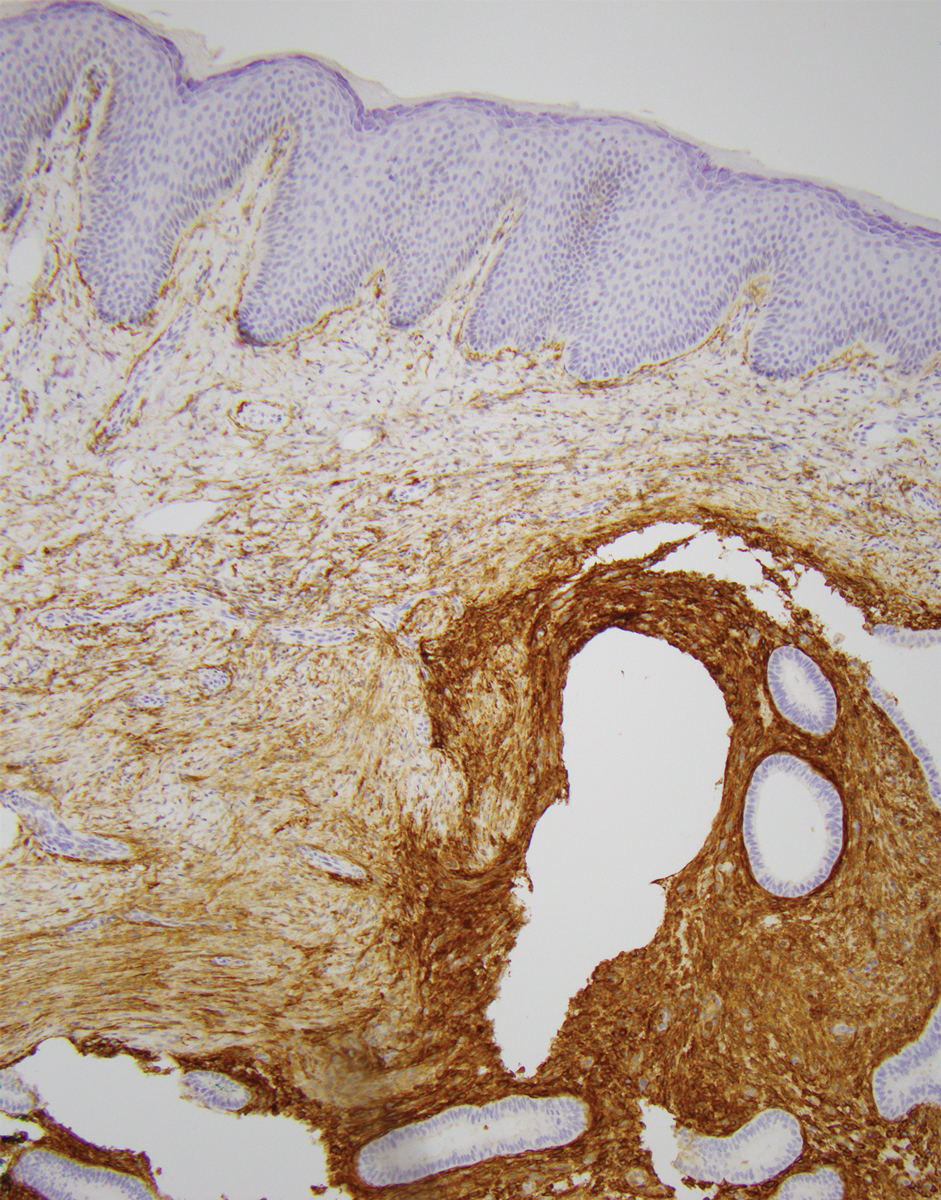
Breast carcinoma is the most common internal malignancy associated with cutaneous metastasis and may develop prior to visceral diagnosis. It is possible that tumor cells travel through the communicating networks of the cutaneous lymphatic ducts and the mammary lymphatic plexus; however, cutaneous manifestation often is located on the ipsilateral breast, and therefore tumor expansion rather than true metastasis cannot always be ruled out. On histopathology, findings of breast adenocarcinoma include tumor cells that tend to show either interstitial, nodular, mixed, or intravascular growth patterns (Figure 2). Tumor cells may invade the stroma in clusters or as individual cells. Sites of distant metastasis may show an increased likelihood of vascular and lymphatic invasion.6
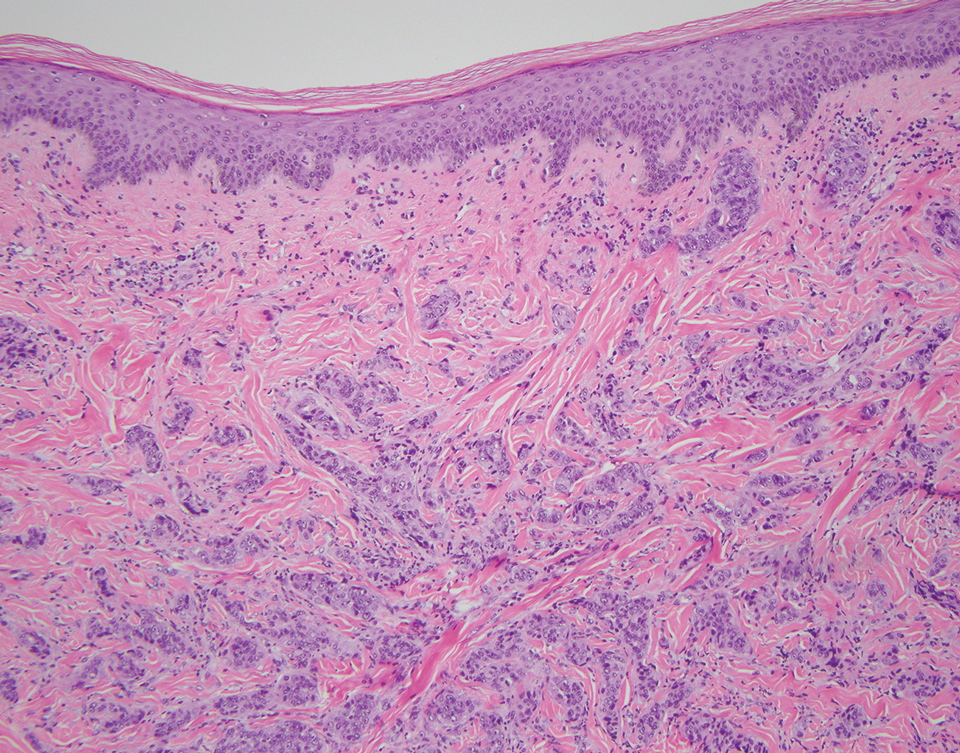
Nodular hidradenoma often manifests as a solitary nodule in the head or neck region, predominantly in women.7 Pathology shows well-demarcated intradermal aggregates of tumor cells within a hyalinized stroma; connection to the epidermis is not a feature of nodular hidradenoma. The epithelial component consists of polygonal cells with eosinophilic to amphophilic cytoplasm as well as large glycogenated cells with pale to clear cytoplasm (leading to the alternative term clear cell hidradenoma)(Figure 3). The cystic portion represents deterioration of tumor cells. Surgical excision usually is curative, although lesions may recur. Malignant transformation is rare.7
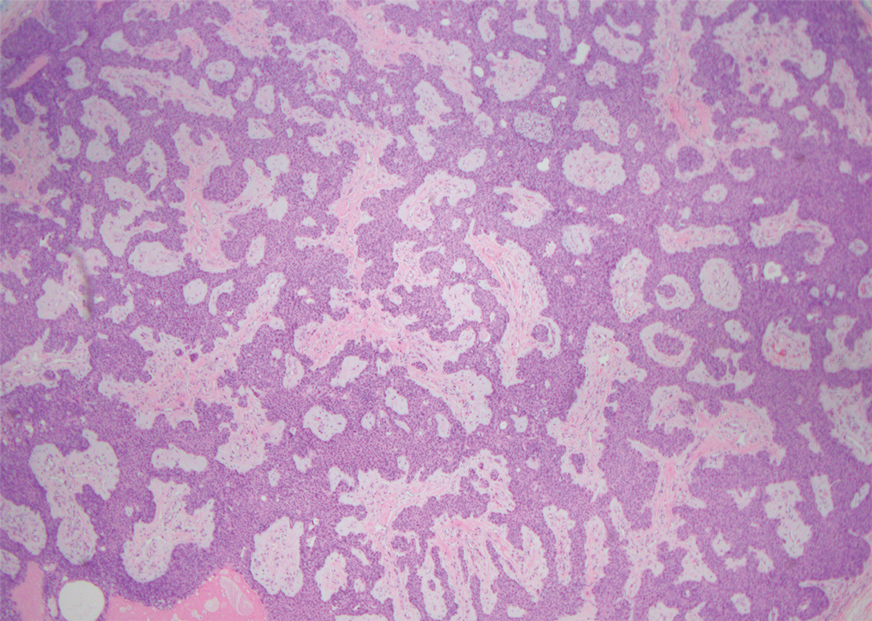
Sister Mary Joseph nodule is a cutaneous involvement of the umbilicus by a metastatic malignancy, often from an intra-abdominal primary malignancy (most commonly ovarian carcinoma in women and colonic carcinoma in men). Clinically, patients present with a solitary firm nodule or plaque within the umbilicus.8,9 Histopathology recapitulates the primary tumor (Figure 4).9 Sister Mary Joseph nodule portends a poor prognosis, with a survival rate of less than 8 months from the time of diagnosis.10
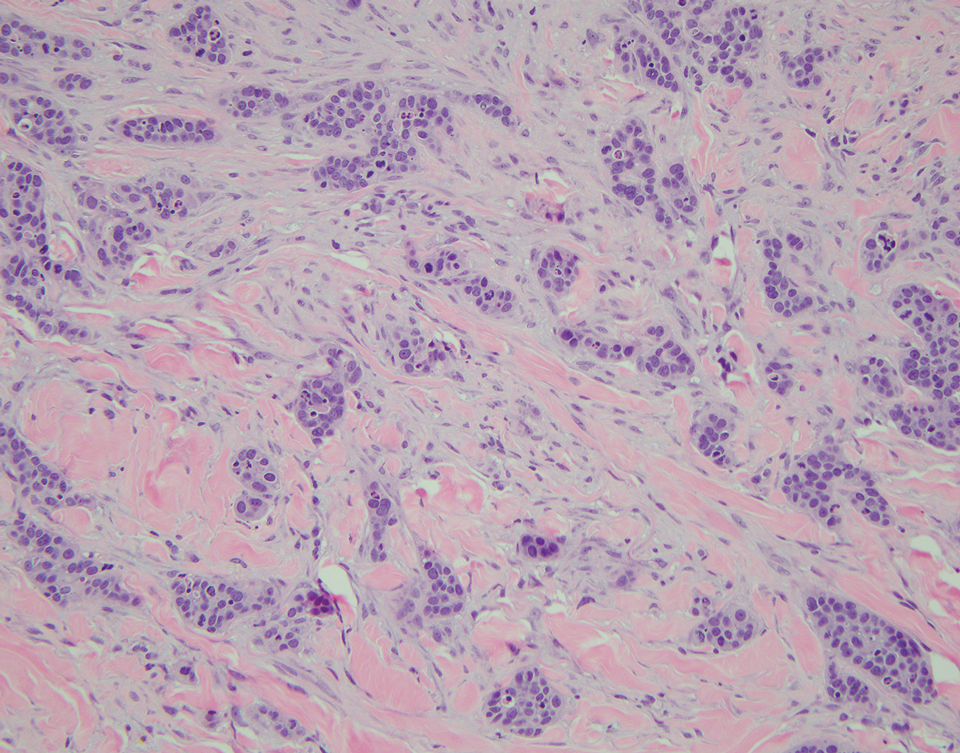
Urachal duct cyst develops from a remnant of the urachus that closed appropriately at the umbilicus and bladder but did not completely regress. It may manifest as an extraperitoneal mass at the umbilicus. Clinically, urachal duct cysts may be asymptomatic until an inciting event (eg, inflammation, deposition of calculus, or malignancy) occurs.11 Histopathology shows cystically dilated structures lined with a transitional epithelium (Figure 5).12 Urachal duct cysts usually are diagnosed in children or young adults and subsequently are excised.11
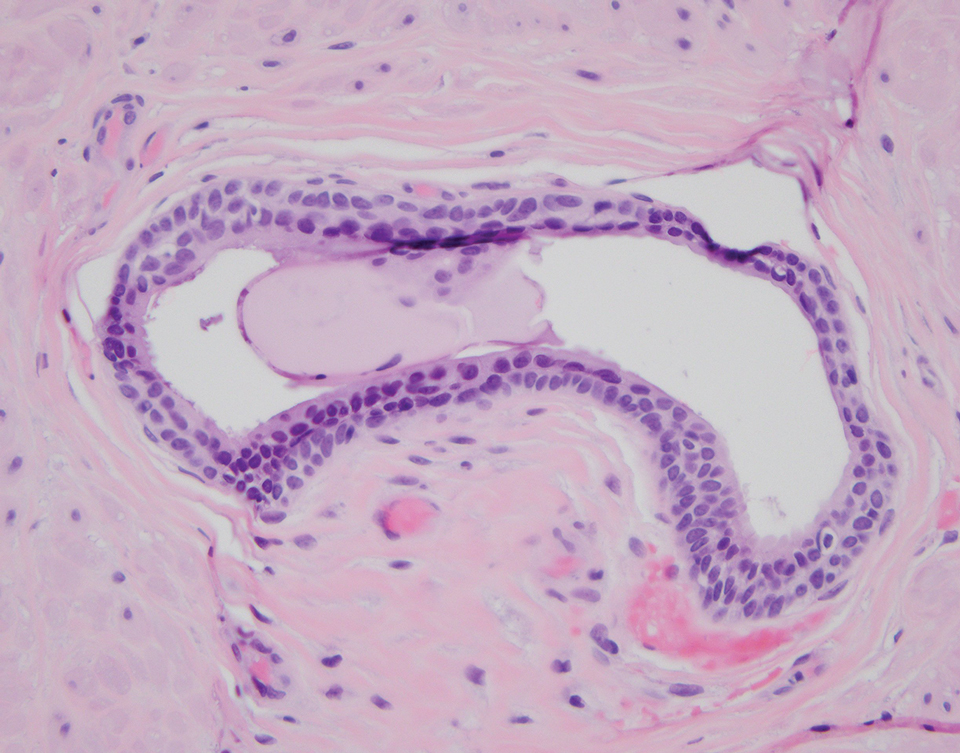
- Harder C, Velho RV, Brandes I, et al. Assessing the true prevalence of endometriosis: a narrative review of literature data. Int J Gynaecol Obstet. 2024;167:883-900. doi:10.1002/ijgo.15756
- Lopez-Soto A, Sanchez-Zapata MI, Martinez-Cendan JP, et al. Cutaneous endometriosis: presentation of 33 cases and literature review. Eur J Obstet Gynecol Reprod Biol. Feb 2018;221:58-63. doi:10.1016 /j.ejogrb.2017.11.024
- Dridi D, Chiaffarino F, Parazzini F, et al. Umbilical endometriosis: a systematic literature review and pathogenic theory proposal. J Clin Med. 2022;11:995. doi:10.3390/jcm11040995
- Farooq U, Laureano AC, Miteva M, Elgart GW. Cutaneous endometriosis: diagnostic immunohistochemistry and clinicopathologic correlation. J Cutan Pathol. 2011;38:525-528. doi:10.1111/j.1600-0560.2011.01681.x
- Gadducci A, Zannoni GF. Endometriosis-associated extraovarian malignancies: a challenging question for the clinician and the pathologist. Anticancer Res. 2020;40:2429-2438. doi:10.21873/anticanres.14212
- Ronen S, Suster D, Chen WS, et al. Histologic patterns of cutaneous metastases of breast carcinoma: a clinicopathologic study of 232 cases. Am J Dermatopathol. 2021;43:401-411. doi:10.1097 /DAD.0000000000001841
- Nandeesh BN, Rajalakshmi T. A study of histopathologic spectrum of nodular hidradenoma. Am J Dermatopathol. 2012;34:461-470. doi:10.1097/DAD.0b013e31821a4d33
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273. doi:10.1097/MAJ.0b013e3181954187
- Powell FC, Cooper AJ, Massa MC, et al. Sister Mary Joseph’s nodule: a clinical and histologic study. J Am Acad Dermatol. 1984;10:610-615. doi:10.1016/s0190-9622(84)80265-0
- Hugen N, Kanne H, Simmer F, et al. Umbilical metastases: real-world data shows abysmal outcome. Int J Cancer. 2021;149: 1266-1273. doi:10.1002/ijc.33684
- Al-Salem A. An Illustrated Guide to Pediatric Urology. 1st ed. Springer Cham; 2016.
- Schubert GE, Pavkovic MB, Bethke-Bedürftig BA. Tubular urachal remnants in adult bladders. J Urol. 1982;127:40-42. doi:10.1016/s0022- 5347(17)53595-8
THE DIAGNOSIS: Cutaneous Endometriosis
Endometriosis is the ectopic presence of endometrial tissue and occurs in approximately 13% of women of childbearing age.1 This non-neoplastic lesion can manifest on the skin in less than 5.5% of endometriosis cases worldwide. Historically, secondary cutaneous endometriosis (CE) most frequently has been associated with prior gynecologic surgery (often cesarean section)2; however, an increased incidence of primary CE in patients without prior surgical history recently has been documented in the literature.3 While secondary CE usually manifests at the site of a surgical scar, primary CE has a predilection for the umbilicus (Villar nodule). In both primary and secondary CE, patients present clinically with a solitary nodule and abdominal pain that may be exacerbated during menstruation. Bleeding without associated pain may be more common in primary CE, while bleeding with pain may be more common in secondary CE. Cutaneous endometriosis often is overlooked given its low incidence, leading to delayed diagnosis. Primary CE often is misdiagnosed clinically as a pyogenic granuloma, Sister Mary Joseph nodule, or keloid, while secondary CE may be mistaken for a fibroma, incisional hernia, or granuloma.2
Primary and secondary CE have identical histopathologic features. Glands of variable size consisting of a single epithelial layer of columnar cells are present in the reticular dermis or subcutis (quiz image).4 The accompanying periglandular stroma often is uniform, consisting of spindle-shaped basophilic cells with abundant vascular structures. The stroma may contain moderate numbers of mitotic figures, a chronic inflammatory infiltrate, and extravasated red blood cells. The ectopic tissue may be inactive or display morphologic changes resembling those of the endometrium in the normal menstrual cycle.4 As the ectopic tissue progresses through the stages of menstruation, the glandular morphology also transforms. The proliferative stage demonstrates increased epithelial mitotic figures, the secretory stage exhibits intraluminal secretion, and during menstruation there are degenerative epithelial cells and evidence of vascular congestion. A mixture of glandular stages may be seen in biopsy results. Robust immunohistochemical expression of CD10 in the endometrial stroma can aid in diagnosis (Figure 1). Estrogen and progesterone receptor immunostaining also shows strong nuclear positivity, except in decidualized tissue.4 Unlike intestinal glands, endometrial glands do not express CDX2 or CK20.5 Complete surgical excision of CE usually is curative; however, recurrence has been documented in 10% (3/30) of cases.2
Breast carcinoma is the most common internal malignancy associated with cutaneous metastasis and may develop prior to visceral diagnosis. It is possible that tumor cells travel through the communicating networks of the cutaneous lymphatic ducts and the mammary lymphatic plexus; however, cutaneous manifestation often is located on the ipsilateral breast, and therefore tumor expansion rather than true metastasis cannot always be ruled out. On histopathology, findings of breast adenocarcinoma include tumor cells that tend to show either interstitial, nodular, mixed, or intravascular growth patterns (Figure 2). Tumor cells may invade the stroma in clusters or as individual cells. Sites of distant metastasis may show an increased likelihood of vascular and lymphatic invasion.6
Nodular hidradenoma often manifests as a solitary nodule in the head or neck region, predominantly in women.7 Pathology shows well-demarcated intradermal aggregates of tumor cells within a hyalinized stroma; connection to the epidermis is not a feature of nodular hidradenoma. The epithelial component consists of polygonal cells with eosinophilic to amphophilic cytoplasm as well as large glycogenated cells with pale to clear cytoplasm (leading to the alternative term clear cell hidradenoma)(Figure 3). The cystic portion represents deterioration of tumor cells. Surgical excision usually is curative, although lesions may recur. Malignant transformation is rare.7
Sister Mary Joseph nodule is a cutaneous involvement of the umbilicus by a metastatic malignancy, often from an intra-abdominal primary malignancy (most commonly ovarian carcinoma in women and colonic carcinoma in men). Clinically, patients present with a solitary firm nodule or plaque within the umbilicus.8,9 Histopathology recapitulates the primary tumor (Figure 4).9 Sister Mary Joseph nodule portends a poor prognosis, with a survival rate of less than 8 months from the time of diagnosis.10
Urachal duct cyst develops from a remnant of the urachus that closed appropriately at the umbilicus and bladder but did not completely regress. It may manifest as an extraperitoneal mass at the umbilicus. Clinically, urachal duct cysts may be asymptomatic until an inciting event (eg, inflammation, deposition of calculus, or malignancy) occurs.11 Histopathology shows cystically dilated structures lined with a transitional epithelium (Figure 5).12 Urachal duct cysts usually are diagnosed in children or young adults and subsequently are excised.11
THE DIAGNOSIS: Cutaneous Endometriosis
Endometriosis is the ectopic presence of endometrial tissue and occurs in approximately 13% of women of childbearing age.1 This non-neoplastic lesion can manifest on the skin in less than 5.5% of endometriosis cases worldwide. Historically, secondary cutaneous endometriosis (CE) most frequently has been associated with prior gynecologic surgery (often cesarean section)2; however, an increased incidence of primary CE in patients without prior surgical history recently has been documented in the literature.3 While secondary CE usually manifests at the site of a surgical scar, primary CE has a predilection for the umbilicus (Villar nodule). In both primary and secondary CE, patients present clinically with a solitary nodule and abdominal pain that may be exacerbated during menstruation. Bleeding without associated pain may be more common in primary CE, while bleeding with pain may be more common in secondary CE. Cutaneous endometriosis often is overlooked given its low incidence, leading to delayed diagnosis. Primary CE often is misdiagnosed clinically as a pyogenic granuloma, Sister Mary Joseph nodule, or keloid, while secondary CE may be mistaken for a fibroma, incisional hernia, or granuloma.2
Primary and secondary CE have identical histopathologic features. Glands of variable size consisting of a single epithelial layer of columnar cells are present in the reticular dermis or subcutis (quiz image).4 The accompanying periglandular stroma often is uniform, consisting of spindle-shaped basophilic cells with abundant vascular structures. The stroma may contain moderate numbers of mitotic figures, a chronic inflammatory infiltrate, and extravasated red blood cells. The ectopic tissue may be inactive or display morphologic changes resembling those of the endometrium in the normal menstrual cycle.4 As the ectopic tissue progresses through the stages of menstruation, the glandular morphology also transforms. The proliferative stage demonstrates increased epithelial mitotic figures, the secretory stage exhibits intraluminal secretion, and during menstruation there are degenerative epithelial cells and evidence of vascular congestion. A mixture of glandular stages may be seen in biopsy results. Robust immunohistochemical expression of CD10 in the endometrial stroma can aid in diagnosis (Figure 1). Estrogen and progesterone receptor immunostaining also shows strong nuclear positivity, except in decidualized tissue.4 Unlike intestinal glands, endometrial glands do not express CDX2 or CK20.5 Complete surgical excision of CE usually is curative; however, recurrence has been documented in 10% (3/30) of cases.2
Breast carcinoma is the most common internal malignancy associated with cutaneous metastasis and may develop prior to visceral diagnosis. It is possible that tumor cells travel through the communicating networks of the cutaneous lymphatic ducts and the mammary lymphatic plexus; however, cutaneous manifestation often is located on the ipsilateral breast, and therefore tumor expansion rather than true metastasis cannot always be ruled out. On histopathology, findings of breast adenocarcinoma include tumor cells that tend to show either interstitial, nodular, mixed, or intravascular growth patterns (Figure 2). Tumor cells may invade the stroma in clusters or as individual cells. Sites of distant metastasis may show an increased likelihood of vascular and lymphatic invasion.6
Nodular hidradenoma often manifests as a solitary nodule in the head or neck region, predominantly in women.7 Pathology shows well-demarcated intradermal aggregates of tumor cells within a hyalinized stroma; connection to the epidermis is not a feature of nodular hidradenoma. The epithelial component consists of polygonal cells with eosinophilic to amphophilic cytoplasm as well as large glycogenated cells with pale to clear cytoplasm (leading to the alternative term clear cell hidradenoma)(Figure 3). The cystic portion represents deterioration of tumor cells. Surgical excision usually is curative, although lesions may recur. Malignant transformation is rare.7
Sister Mary Joseph nodule is a cutaneous involvement of the umbilicus by a metastatic malignancy, often from an intra-abdominal primary malignancy (most commonly ovarian carcinoma in women and colonic carcinoma in men). Clinically, patients present with a solitary firm nodule or plaque within the umbilicus.8,9 Histopathology recapitulates the primary tumor (Figure 4).9 Sister Mary Joseph nodule portends a poor prognosis, with a survival rate of less than 8 months from the time of diagnosis.10
Urachal duct cyst develops from a remnant of the urachus that closed appropriately at the umbilicus and bladder but did not completely regress. It may manifest as an extraperitoneal mass at the umbilicus. Clinically, urachal duct cysts may be asymptomatic until an inciting event (eg, inflammation, deposition of calculus, or malignancy) occurs.11 Histopathology shows cystically dilated structures lined with a transitional epithelium (Figure 5).12 Urachal duct cysts usually are diagnosed in children or young adults and subsequently are excised.11
- Harder C, Velho RV, Brandes I, et al. Assessing the true prevalence of endometriosis: a narrative review of literature data. Int J Gynaecol Obstet. 2024;167:883-900. doi:10.1002/ijgo.15756
- Lopez-Soto A, Sanchez-Zapata MI, Martinez-Cendan JP, et al. Cutaneous endometriosis: presentation of 33 cases and literature review. Eur J Obstet Gynecol Reprod Biol. Feb 2018;221:58-63. doi:10.1016 /j.ejogrb.2017.11.024
- Dridi D, Chiaffarino F, Parazzini F, et al. Umbilical endometriosis: a systematic literature review and pathogenic theory proposal. J Clin Med. 2022;11:995. doi:10.3390/jcm11040995
- Farooq U, Laureano AC, Miteva M, Elgart GW. Cutaneous endometriosis: diagnostic immunohistochemistry and clinicopathologic correlation. J Cutan Pathol. 2011;38:525-528. doi:10.1111/j.1600-0560.2011.01681.x
- Gadducci A, Zannoni GF. Endometriosis-associated extraovarian malignancies: a challenging question for the clinician and the pathologist. Anticancer Res. 2020;40:2429-2438. doi:10.21873/anticanres.14212
- Ronen S, Suster D, Chen WS, et al. Histologic patterns of cutaneous metastases of breast carcinoma: a clinicopathologic study of 232 cases. Am J Dermatopathol. 2021;43:401-411. doi:10.1097 /DAD.0000000000001841
- Nandeesh BN, Rajalakshmi T. A study of histopathologic spectrum of nodular hidradenoma. Am J Dermatopathol. 2012;34:461-470. doi:10.1097/DAD.0b013e31821a4d33
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273. doi:10.1097/MAJ.0b013e3181954187
- Powell FC, Cooper AJ, Massa MC, et al. Sister Mary Joseph’s nodule: a clinical and histologic study. J Am Acad Dermatol. 1984;10:610-615. doi:10.1016/s0190-9622(84)80265-0
- Hugen N, Kanne H, Simmer F, et al. Umbilical metastases: real-world data shows abysmal outcome. Int J Cancer. 2021;149: 1266-1273. doi:10.1002/ijc.33684
- Al-Salem A. An Illustrated Guide to Pediatric Urology. 1st ed. Springer Cham; 2016.
- Schubert GE, Pavkovic MB, Bethke-Bedürftig BA. Tubular urachal remnants in adult bladders. J Urol. 1982;127:40-42. doi:10.1016/s0022- 5347(17)53595-8
- Harder C, Velho RV, Brandes I, et al. Assessing the true prevalence of endometriosis: a narrative review of literature data. Int J Gynaecol Obstet. 2024;167:883-900. doi:10.1002/ijgo.15756
- Lopez-Soto A, Sanchez-Zapata MI, Martinez-Cendan JP, et al. Cutaneous endometriosis: presentation of 33 cases and literature review. Eur J Obstet Gynecol Reprod Biol. Feb 2018;221:58-63. doi:10.1016 /j.ejogrb.2017.11.024
- Dridi D, Chiaffarino F, Parazzini F, et al. Umbilical endometriosis: a systematic literature review and pathogenic theory proposal. J Clin Med. 2022;11:995. doi:10.3390/jcm11040995
- Farooq U, Laureano AC, Miteva M, Elgart GW. Cutaneous endometriosis: diagnostic immunohistochemistry and clinicopathologic correlation. J Cutan Pathol. 2011;38:525-528. doi:10.1111/j.1600-0560.2011.01681.x
- Gadducci A, Zannoni GF. Endometriosis-associated extraovarian malignancies: a challenging question for the clinician and the pathologist. Anticancer Res. 2020;40:2429-2438. doi:10.21873/anticanres.14212
- Ronen S, Suster D, Chen WS, et al. Histologic patterns of cutaneous metastases of breast carcinoma: a clinicopathologic study of 232 cases. Am J Dermatopathol. 2021;43:401-411. doi:10.1097 /DAD.0000000000001841
- Nandeesh BN, Rajalakshmi T. A study of histopathologic spectrum of nodular hidradenoma. Am J Dermatopathol. 2012;34:461-470. doi:10.1097/DAD.0b013e31821a4d33
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273. doi:10.1097/MAJ.0b013e3181954187
- Powell FC, Cooper AJ, Massa MC, et al. Sister Mary Joseph’s nodule: a clinical and histologic study. J Am Acad Dermatol. 1984;10:610-615. doi:10.1016/s0190-9622(84)80265-0
- Hugen N, Kanne H, Simmer F, et al. Umbilical metastases: real-world data shows abysmal outcome. Int J Cancer. 2021;149: 1266-1273. doi:10.1002/ijc.33684
- Al-Salem A. An Illustrated Guide to Pediatric Urology. 1st ed. Springer Cham; 2016.
- Schubert GE, Pavkovic MB, Bethke-Bedürftig BA. Tubular urachal remnants in adult bladders. J Urol. 1982;127:40-42. doi:10.1016/s0022- 5347(17)53595-8
Solitary Lesion on the Umbilicus
Solitary Lesion on the Umbilicus
A 33-year-old woman with no notable medical or surgical history presented to our clinic with a solitary indurated nodule on the umbilicus that had been progressively enlarging for 1 year. The patient reported that she had undergone piercing of the umbilicus more than 5 years prior. She noted that the lesion was uncomfortable and pruritic and occasionally bled spontaneously. Physical examination revealed no other mucosal or cutaneous findings. A shave biopsy of the nodule was performed.
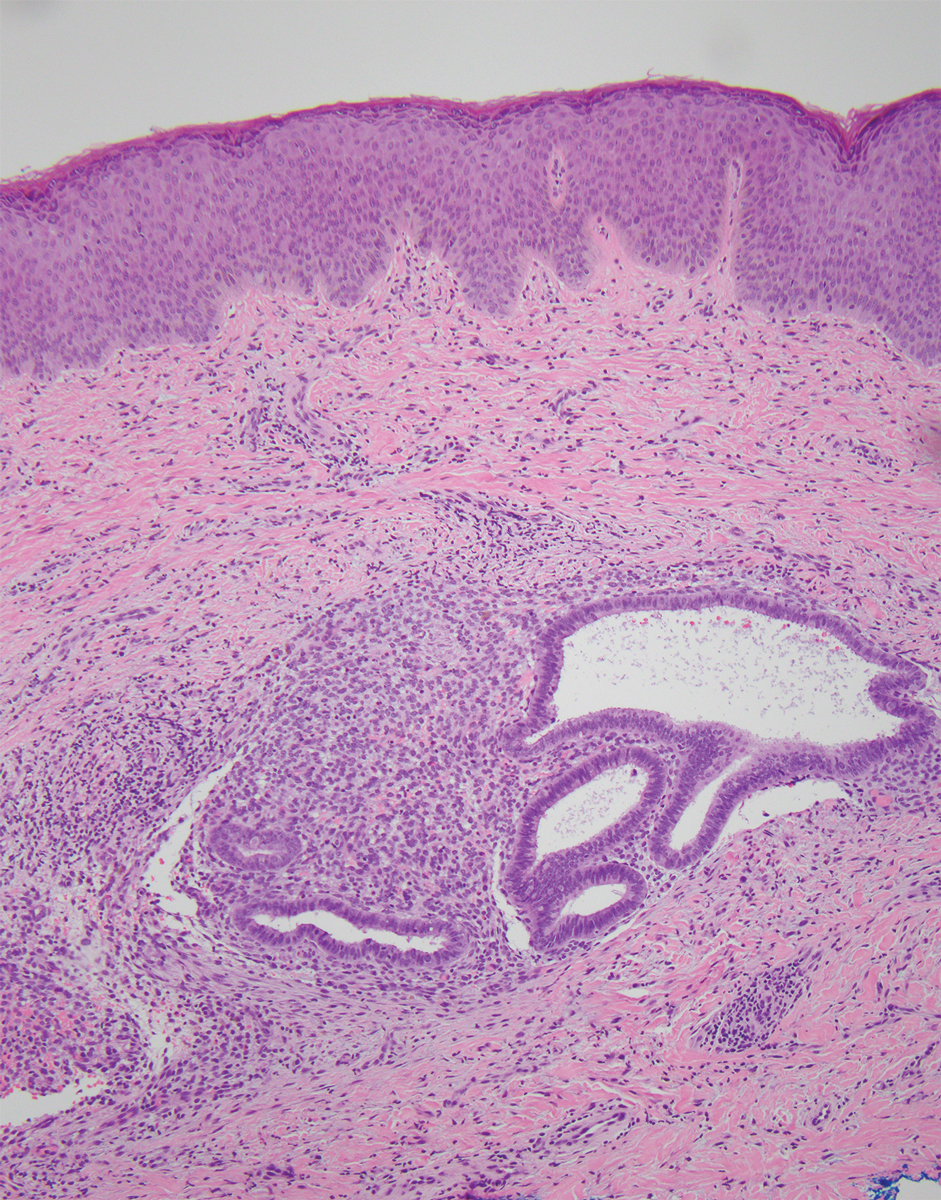

Treatment of Seborrheic Dermatitis in Black Patients
Treatment of Seborrheic Dermatitis in Black Patients
Seborrheic dermatitis (SD) is a common chronic inflammatory skin condition that predominantly affects areas with high concentrations of sebaceous glands such as the scalp and face. Up to 5% of the worldwide population is affected by SD each year, causing a major burden of disease for patients and the health care system.1 In 2023, the cost of medical treatment for SD in the United States was $300 million, with outpatient office visits alone costing $58 million and prescription drugs costing $109 million. Indirect costs of disease (eg, lost workdays) account for another $51 million.1 Since SD frequently manifests on the face, it tends to have negative effects on the patient’s quality of life, resulting in psychological distress and low self-esteem.2
Patients with SD may describe symptoms of excessive dandruff and itching along with hyperpigmentation or hypopigmentation of the skin; Black patients tend to present with the classic manifestations: a combination of scaling, flaking, and erythematous patches on the scalp, ears, and face, particularly around the eyebrows, eyelids, and nose. With SD being the second most common diagnosis in Black patients who seek care from a dermatologist, it is important to have effective treatment approaches for SD in this patient population.3
In this study, we aimed to evaluate medical and nonmedical treatment options for SD in Black patients by identifying common practices and products mentioned on consumer websites and in the medical literature.
Methods
A Google search was conducted during 2 time periods (September 2022—October 2022 and March 2023—April 2023) using the terms products for itchy scalp in Black patients, products for dandruff in Black patients, itchy scalp in Black women, itchy scalp in Black men, treatment for scalp itch in Black patients, and dry scalp in Black hair. Products that were recommended by at least 1 website on the first page of search results were included in our list of products, and the ingredients were reviewed by the authors. We excluded individual retailer websites as well as those that did not provide specific recommendations on products or ingredients to use when treating SD. To ensure reliability and standardization, we did not review products that were suggested by ads in the shopping section on the first page of search results.
We also evaluated medical treatments used for SD in dermatology literature. A PubMed search of articles indexed for MEDLINE using the terms seborrheic dermatitis treatment for Black patients, treatment for dandruff for Black patients, and seborrheic dermatitis and skin of color was conducted. We excluded articles that did not address treatment options for SD, were specific to treating SD in patient populations with specific comorbidities being studied, discussed SD in animals, or were published prior to 1990.
Results
We identified 16 unique consumer websites with product or ingredient recommendations for SD in Black patients, none of which were provided by authors with a medical or scientific background; however, 4 (25%) websites included insights from board-certified dermatologists. A total of 16 ingredients were recommended, 15 (94%) of which were mentioned at least twice in our search results (eTable 1).
Overall, we noticed that ingredients labeled as natural or organic were common in over-the-counter (OTC) products, and ingredients such as sulfates and parabens were avoided. Common OTC ingredients for antidandruff and anti-itch shampoos and conditioners include zinc pyrithione, selenium sulfide, coal tar, salicylic acid, and citric acid. Additionally, coconut oil, tea tree oil, apple cider vinegar, and charcoal are common natural alternatives used to address SD symptoms.
Our review of the literature yielded limited recommendations tailored specifically to Black patients with SD. Of 108 abstracts, articles, or textbook chapters providing treatment recommendations for SD, 6 (6%) specifically discussed treatments for Black patients. All articles were written by authors with medical or scientific backgrounds. Of the treatment options discussed, topical antifungals generally were considered first-line for SD in all patients, with ketoconazole shampoo being a common first choice.4,5
Comment
Our study indicated that many consumer websites recommend unstudied nonmedical treatments for SD. Zinc pyrithione was one of the most commonly mentioned ingredients in OTC products to treat SD targeted toward Black patients, as its properties have contributed to ease of hair combing and less frizz.6 Zinc pyrithione has antifungal properties that reduce the proliferation of Malassezia furfur as well as anti-inflammatory properties that reduce irritation, pruritus, and erythema in areas affected by SD.7 Tea tree and peppermint oils also were commonly mentioned; the theory is that these oils mitigate SD by reducing yeast growth and soothing inflammation through antioxidant activity.8,9 Coal tar also is used due to its keratoplastic properties, which slow the growth of skin cells and ultimately reduce scaling and dryness.10 Yeast thrives in basic pH conditions; apple cider vinegar is used as an ingredient in OTC products for SD because its acidic pH creates a less favorable environment for yeast to grow.11 Although many of the ingredients found in OTC products we identified have not yet been studied, they have properties that theoretically would be helpful in treating SD.
Our review of the medical literature revealed that while there are treatments that are effective for SD, the recommended use may not consider the cultural differences that exist for Black patients. For instance, reports in the literature regarding ketoconazole shampoo revealed that ketoconazole increases the risk for hair shaft dryness, damage, and subsequent breakage, especially in Black women who also may be using heat styling or chemical relaxers.5 As a result, ketoconazole should be used with caution in Black women, with an emphasis on direct application to the scalp rather than the hair shafts.12 Additional options reported for Black patients include ciclopirox olamine and zinc pyrithione, which may have fewer risks.13
When prescribing medicated shampoos, traditional instructions regarding frequency of use to control symptoms of SD range from 2 to 3 times weekly to daily for a specified period of time determined by the dermatologist.14 However, frequency of hair washing varies greatly among Black patients, sometimes occurring only once monthly. The frequency also may change based on styling techniques (eg, braids, weaves, and wigs).15 Based on previous research underscoring the tendency for Black patients to use medicated shampoos less frequently than White patients, it is important for clinicians to understand that these cultural practices can undermine the effectiveness when medicated shampoos are prescribed for SD.16
Additionally, topical corticosteroids often are used in conjunction with antifungals to help decrease inflammation of the scalp.17 An option reported for Black patients is topical fluocinolone 0.01%; however, package instructions state to apply topically to the scalp nightly and wash the hair thoroughly each morning, which may not be feasible for Black patients based on previously mentioned differences in hair-washing techniques. An alternative option may be to apply the medication 3 to 4 times per week, washing the hair weekly rather than daily.18 Fluocinolone can be used as an ointment, solution, oil, or cream.19,20 When comparing treatment vehicles for SD, a study conducted by Chappell et al21 found that Black patients preferred using ointment or oil vehicles; White patients preferred foams and sprays, which may not be suitable for Afro hair patterns. As such, using less-drying modalities may increase compliance and treatment success in Black patients. For patients who may have involvement on the hairline, face, or ears along with hypopigmentation (which is a common skin concern associated with SD), calcineurin inhibitors can be used until resolution occurs.5,22 High et al15 found that twice-daily use of pimecrolimus rapidly normalized skin pigmentation during the first 2 weeks of use. Overall, personalization of treatment may not only avoid adverse effects but also ensure patient compliance, with the overall goal of treating to reduce yeast activity, pruritus, and dyschromia.22
Interestingly, after the website searches were completed for this study, the US Food and Drug Administration approved topical roflumilast foam for SD. In a phase III trial of 457 total patients, 36 Black patients were included.23 It was determined that 79.5% of patients overall throughout the trial achieved Investigator Global Assessment success (score of 0 [clear] or 1 [almost clear]) plus ≥2-point improvement from baseline (on a scale of 0 [clear] to 4 [severe]) at weeks 2, 4, and 8. Although there currently are no long-term studies, roflumilast may be a promising option for Black patients with SD.23
Aside from developing an individualized treatment approach for Black patients with SD, it is important to ask targeted questions during the clinical encounter to identify factors that may be exacerbating symptoms, especially due to the wide range of hair care practices used by the Black community (eTable 2). Asking targeted questions is especially important, as prior studies have shown that extensions, hair relaxers, and particular hair products can irritate the scalp and increase the likelihood of developing SD.21,24 Rucker Wright et al25 evaluated different hair care practices among young Black females and their association with the development of SD. The authors found that using hair extensions (either braided, cornrowed, or ponytails), chemical relaxers, and hair oils every 2 weeks was associated with SD. The study also found that SD rates were roughly 20% higher among Black girls with extensions compared to Black girls without extensions, regardless of how frequently hair was washed.25

Many Black patients grease the scalp with oils that are beneficial for lubrication and reduction of abrasive damage caused by grooming; however, they also may increase incidence of SD.26 Tight curls worn by Black patients also can impede sebum from traveling down the hair shaft, leading to oil buildup on the scalp. This is the ideal environment for increased Malassezia density and higher risk for SD development.27 To balance the beneficial effects of hair oils with the increased susceptibility for SD, providers should emphasize applying these oils only to distal hair shafts, which are more likely to be damaged, and avoiding application to the scalp.19
Conclusion
Given its long-term relapsing and remitting nature, SD can be distressing for Black patients, many of whom may seek additional treatment options aside from those recommended by health care professionals. In order to better educate patients, it is important for dermatologists to know not only the common ingredients that may be present in OTC products but also the thought process behind why patients use them. Additionally, prescription treatments for Black patients with SD may require nuanced alterations to the product instructions that may prevent health disparities and provide culturally sensitive care. Overall, the literature regarding treatment for Black patients with SD is limited, and more high-quality studies are needed.
- Tucker D, Masood S. Seborrheic dermatitis. StatPearls [Internet]. Updated March 1, 2024. Accessed December 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK551707/
- Borda LJ, Wikramanayake TC. Seborrheic dermatitis and dandruff: a comprehensive review. J Clin Investig Dermatol. 2015;3:10.13188 /2373-1044.1000019.
- American Academy of Dermatology. Seborrheic dermatitis by the numbers. American Academy of Dermatology Skin Disease Briefs. Updated May 5, 2018. Accessed November 22, 2024. https://www.aad.org/asset/49w949DPcF8RSJYIRHfDon
- Davis SA, Naarahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Borda LJ, Perper M, Keri JE. Treatment of seborrheic dermatitis: a comprehensive review. J Dermatolog Treat. 2019;30:158-169.
- Draelos ZD, Kenneally DC, Hodges LT, et al. A comparison of hair quality and cosmetic acceptance following the use of two anti-dandruff shampoos. J Investig Dermatol Symp Proc. 2005;10:201-214.
- Barak-Shinar D, Green LJ. Scalp seborrheic dermatitis and dandruff therapy using a herbal and zinc pyrithione-based therapy of shampoo and scalp lotion. J Clin Aesthet Dermatol. 2018;11:26-31.
- Satchell AC, Saurajen A, Bell C, et al. Treatment of dandruff with 5% tea tree oil shampoo. J Am Acad Dermatol. 2002;47:852-855.
- Herro E, Jacob SE. Mentha piperita (peppermint). Dermatitis. 2010;21:327-329.
- Sanfilippo A, English JC. An overview of medicated shampoos used in dandruff treatment. Pharm Ther. 2006;31:396-400.
- Arun PVPS, Vineetha Y, Waheed M, et al. Quantification of the minimum amount of lemon juice and apple cider vinegar required for the growth inhibition of dandruff causing fungi Malassezia furfur. Int J Sci Res in Biological Sciences. 2019;6:144-147.
- Gao HY, Li Wan Po A. Topical formulations of fluocinolone acetonide. Are creams, gels and ointments bioequivalent and does dilution affect activity? Eur J Clin Pharmacol. 1994;46:71-75.
- Pauporte M, Maibach H, Lowe N, et al. Fluocinolone acetonide topical oil for scalp psoriasis. J Dermatolog Treat. 2004;15:360-364.
- Elgash M, Dlova N, Ogunleye T, et al. Seborrheic dermatitis in skin of color: clinical considerations. J Drugs Dermatol. 2019;18:24-27.
- High WA, Pandya AG. Pilot trial of 1% pimecrolimus cream in the treatment of seborrheic dermatitis in African American adults with associated hypopigmentation. J Am Acad Dermatol. 2006;54:1083-1088.
- Hollins LC, Butt M, Hong J, et al. Research in brief: survey of hair care practices in various ethnic and racial pediatric populations. Pediatr Dermatol. 2022;39:494-496.
- Halder RM, Roberts CI, Nootheti PK. Cutaneous diseases in the black races. Dermatol Clin. 2003;21:679-687, ix.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Friedmann DP, Mishra V, Batty T. Progressive facial papules in an African- American patient: an atypical presentation of seborrheic dermatitis. J Clin Aesthet Dermatol. 2018;11:44-45.
- Clark GW, Pope SM, Jaboori KA. Diagnosis and treatment of seborrheic dermatitis. Am Fam Physician. 2015;91:185-190.
- Chappell J, Mattox A, Simonetta C, et al. Seborrheic dermatitis of the scalp in populations practicing less frequent hair washing: ketoconazole 2% foam versus ketoconazole 2% shampoo. three-year data. J Am Acad Dermatol. 2014;70:AB54.
- Dadzie OE, Salam A. The hair grooming practices of women of African descent in London, United Kingdom: findings of a cross-sectional study. J Eur Acad Dermatol Venereol. 2016;30:1021-1024.
- Blauvelt A, Draelos ZD, Stein Gold L, et al. Roflumilast foam 0.3% for adolescent and adult patients with seborrheic dermatitis: a randomized, double-blinded, vehicle-controlled, phase 3 trial. J Am Acad Dermatol. 2024;90:986-993.
- Taylor SC, Barbosa V, Burgess C, et al. Hair and scalp disorders in adult and pediatric patients with skin of color. Cutis. 2017;100:31-35.
- Rucker Wright D, Gathers R, Kapke A, et al. Hair care practices and their association with scalp and hair disorders in African American girls. J Am Acad Dermatol. 2011;64:253-262.
- Raffi J, Suresh R, Agbai O. Clinical recognition and management of alopecia in women of color. Int J Womens Dermatol. 2019;5:314-319.
- Mayo T, Dinkins J, Elewski B. Hair oils may worsen seborrheic dermatitis in Black patients. Skin Appendage Disord. 2023;9:151-152.
Seborrheic dermatitis (SD) is a common chronic inflammatory skin condition that predominantly affects areas with high concentrations of sebaceous glands such as the scalp and face. Up to 5% of the worldwide population is affected by SD each year, causing a major burden of disease for patients and the health care system.1 In 2023, the cost of medical treatment for SD in the United States was $300 million, with outpatient office visits alone costing $58 million and prescription drugs costing $109 million. Indirect costs of disease (eg, lost workdays) account for another $51 million.1 Since SD frequently manifests on the face, it tends to have negative effects on the patient’s quality of life, resulting in psychological distress and low self-esteem.2
Patients with SD may describe symptoms of excessive dandruff and itching along with hyperpigmentation or hypopigmentation of the skin; Black patients tend to present with the classic manifestations: a combination of scaling, flaking, and erythematous patches on the scalp, ears, and face, particularly around the eyebrows, eyelids, and nose. With SD being the second most common diagnosis in Black patients who seek care from a dermatologist, it is important to have effective treatment approaches for SD in this patient population.3
In this study, we aimed to evaluate medical and nonmedical treatment options for SD in Black patients by identifying common practices and products mentioned on consumer websites and in the medical literature.
Methods
A Google search was conducted during 2 time periods (September 2022—October 2022 and March 2023—April 2023) using the terms products for itchy scalp in Black patients, products for dandruff in Black patients, itchy scalp in Black women, itchy scalp in Black men, treatment for scalp itch in Black patients, and dry scalp in Black hair. Products that were recommended by at least 1 website on the first page of search results were included in our list of products, and the ingredients were reviewed by the authors. We excluded individual retailer websites as well as those that did not provide specific recommendations on products or ingredients to use when treating SD. To ensure reliability and standardization, we did not review products that were suggested by ads in the shopping section on the first page of search results.
We also evaluated medical treatments used for SD in dermatology literature. A PubMed search of articles indexed for MEDLINE using the terms seborrheic dermatitis treatment for Black patients, treatment for dandruff for Black patients, and seborrheic dermatitis and skin of color was conducted. We excluded articles that did not address treatment options for SD, were specific to treating SD in patient populations with specific comorbidities being studied, discussed SD in animals, or were published prior to 1990.
Results
We identified 16 unique consumer websites with product or ingredient recommendations for SD in Black patients, none of which were provided by authors with a medical or scientific background; however, 4 (25%) websites included insights from board-certified dermatologists. A total of 16 ingredients were recommended, 15 (94%) of which were mentioned at least twice in our search results (eTable 1).
Overall, we noticed that ingredients labeled as natural or organic were common in over-the-counter (OTC) products, and ingredients such as sulfates and parabens were avoided. Common OTC ingredients for antidandruff and anti-itch shampoos and conditioners include zinc pyrithione, selenium sulfide, coal tar, salicylic acid, and citric acid. Additionally, coconut oil, tea tree oil, apple cider vinegar, and charcoal are common natural alternatives used to address SD symptoms.
Our review of the literature yielded limited recommendations tailored specifically to Black patients with SD. Of 108 abstracts, articles, or textbook chapters providing treatment recommendations for SD, 6 (6%) specifically discussed treatments for Black patients. All articles were written by authors with medical or scientific backgrounds. Of the treatment options discussed, topical antifungals generally were considered first-line for SD in all patients, with ketoconazole shampoo being a common first choice.4,5
Comment
Our study indicated that many consumer websites recommend unstudied nonmedical treatments for SD. Zinc pyrithione was one of the most commonly mentioned ingredients in OTC products to treat SD targeted toward Black patients, as its properties have contributed to ease of hair combing and less frizz.6 Zinc pyrithione has antifungal properties that reduce the proliferation of Malassezia furfur as well as anti-inflammatory properties that reduce irritation, pruritus, and erythema in areas affected by SD.7 Tea tree and peppermint oils also were commonly mentioned; the theory is that these oils mitigate SD by reducing yeast growth and soothing inflammation through antioxidant activity.8,9 Coal tar also is used due to its keratoplastic properties, which slow the growth of skin cells and ultimately reduce scaling and dryness.10 Yeast thrives in basic pH conditions; apple cider vinegar is used as an ingredient in OTC products for SD because its acidic pH creates a less favorable environment for yeast to grow.11 Although many of the ingredients found in OTC products we identified have not yet been studied, they have properties that theoretically would be helpful in treating SD.
Our review of the medical literature revealed that while there are treatments that are effective for SD, the recommended use may not consider the cultural differences that exist for Black patients. For instance, reports in the literature regarding ketoconazole shampoo revealed that ketoconazole increases the risk for hair shaft dryness, damage, and subsequent breakage, especially in Black women who also may be using heat styling or chemical relaxers.5 As a result, ketoconazole should be used with caution in Black women, with an emphasis on direct application to the scalp rather than the hair shafts.12 Additional options reported for Black patients include ciclopirox olamine and zinc pyrithione, which may have fewer risks.13
When prescribing medicated shampoos, traditional instructions regarding frequency of use to control symptoms of SD range from 2 to 3 times weekly to daily for a specified period of time determined by the dermatologist.14 However, frequency of hair washing varies greatly among Black patients, sometimes occurring only once monthly. The frequency also may change based on styling techniques (eg, braids, weaves, and wigs).15 Based on previous research underscoring the tendency for Black patients to use medicated shampoos less frequently than White patients, it is important for clinicians to understand that these cultural practices can undermine the effectiveness when medicated shampoos are prescribed for SD.16
Additionally, topical corticosteroids often are used in conjunction with antifungals to help decrease inflammation of the scalp.17 An option reported for Black patients is topical fluocinolone 0.01%; however, package instructions state to apply topically to the scalp nightly and wash the hair thoroughly each morning, which may not be feasible for Black patients based on previously mentioned differences in hair-washing techniques. An alternative option may be to apply the medication 3 to 4 times per week, washing the hair weekly rather than daily.18 Fluocinolone can be used as an ointment, solution, oil, or cream.19,20 When comparing treatment vehicles for SD, a study conducted by Chappell et al21 found that Black patients preferred using ointment or oil vehicles; White patients preferred foams and sprays, which may not be suitable for Afro hair patterns. As such, using less-drying modalities may increase compliance and treatment success in Black patients. For patients who may have involvement on the hairline, face, or ears along with hypopigmentation (which is a common skin concern associated with SD), calcineurin inhibitors can be used until resolution occurs.5,22 High et al15 found that twice-daily use of pimecrolimus rapidly normalized skin pigmentation during the first 2 weeks of use. Overall, personalization of treatment may not only avoid adverse effects but also ensure patient compliance, with the overall goal of treating to reduce yeast activity, pruritus, and dyschromia.22
Interestingly, after the website searches were completed for this study, the US Food and Drug Administration approved topical roflumilast foam for SD. In a phase III trial of 457 total patients, 36 Black patients were included.23 It was determined that 79.5% of patients overall throughout the trial achieved Investigator Global Assessment success (score of 0 [clear] or 1 [almost clear]) plus ≥2-point improvement from baseline (on a scale of 0 [clear] to 4 [severe]) at weeks 2, 4, and 8. Although there currently are no long-term studies, roflumilast may be a promising option for Black patients with SD.23
Aside from developing an individualized treatment approach for Black patients with SD, it is important to ask targeted questions during the clinical encounter to identify factors that may be exacerbating symptoms, especially due to the wide range of hair care practices used by the Black community (eTable 2). Asking targeted questions is especially important, as prior studies have shown that extensions, hair relaxers, and particular hair products can irritate the scalp and increase the likelihood of developing SD.21,24 Rucker Wright et al25 evaluated different hair care practices among young Black females and their association with the development of SD. The authors found that using hair extensions (either braided, cornrowed, or ponytails), chemical relaxers, and hair oils every 2 weeks was associated with SD. The study also found that SD rates were roughly 20% higher among Black girls with extensions compared to Black girls without extensions, regardless of how frequently hair was washed.25
Many Black patients grease the scalp with oils that are beneficial for lubrication and reduction of abrasive damage caused by grooming; however, they also may increase incidence of SD.26 Tight curls worn by Black patients also can impede sebum from traveling down the hair shaft, leading to oil buildup on the scalp. This is the ideal environment for increased Malassezia density and higher risk for SD development.27 To balance the beneficial effects of hair oils with the increased susceptibility for SD, providers should emphasize applying these oils only to distal hair shafts, which are more likely to be damaged, and avoiding application to the scalp.19
Conclusion
Given its long-term relapsing and remitting nature, SD can be distressing for Black patients, many of whom may seek additional treatment options aside from those recommended by health care professionals. In order to better educate patients, it is important for dermatologists to know not only the common ingredients that may be present in OTC products but also the thought process behind why patients use them. Additionally, prescription treatments for Black patients with SD may require nuanced alterations to the product instructions that may prevent health disparities and provide culturally sensitive care. Overall, the literature regarding treatment for Black patients with SD is limited, and more high-quality studies are needed.
Seborrheic dermatitis (SD) is a common chronic inflammatory skin condition that predominantly affects areas with high concentrations of sebaceous glands such as the scalp and face. Up to 5% of the worldwide population is affected by SD each year, causing a major burden of disease for patients and the health care system.1 In 2023, the cost of medical treatment for SD in the United States was $300 million, with outpatient office visits alone costing $58 million and prescription drugs costing $109 million. Indirect costs of disease (eg, lost workdays) account for another $51 million.1 Since SD frequently manifests on the face, it tends to have negative effects on the patient’s quality of life, resulting in psychological distress and low self-esteem.2
Patients with SD may describe symptoms of excessive dandruff and itching along with hyperpigmentation or hypopigmentation of the skin; Black patients tend to present with the classic manifestations: a combination of scaling, flaking, and erythematous patches on the scalp, ears, and face, particularly around the eyebrows, eyelids, and nose. With SD being the second most common diagnosis in Black patients who seek care from a dermatologist, it is important to have effective treatment approaches for SD in this patient population.3
In this study, we aimed to evaluate medical and nonmedical treatment options for SD in Black patients by identifying common practices and products mentioned on consumer websites and in the medical literature.
Methods
A Google search was conducted during 2 time periods (September 2022—October 2022 and March 2023—April 2023) using the terms products for itchy scalp in Black patients, products for dandruff in Black patients, itchy scalp in Black women, itchy scalp in Black men, treatment for scalp itch in Black patients, and dry scalp in Black hair. Products that were recommended by at least 1 website on the first page of search results were included in our list of products, and the ingredients were reviewed by the authors. We excluded individual retailer websites as well as those that did not provide specific recommendations on products or ingredients to use when treating SD. To ensure reliability and standardization, we did not review products that were suggested by ads in the shopping section on the first page of search results.
We also evaluated medical treatments used for SD in dermatology literature. A PubMed search of articles indexed for MEDLINE using the terms seborrheic dermatitis treatment for Black patients, treatment for dandruff for Black patients, and seborrheic dermatitis and skin of color was conducted. We excluded articles that did not address treatment options for SD, were specific to treating SD in patient populations with specific comorbidities being studied, discussed SD in animals, or were published prior to 1990.
Results
We identified 16 unique consumer websites with product or ingredient recommendations for SD in Black patients, none of which were provided by authors with a medical or scientific background; however, 4 (25%) websites included insights from board-certified dermatologists. A total of 16 ingredients were recommended, 15 (94%) of which were mentioned at least twice in our search results (eTable 1).
Overall, we noticed that ingredients labeled as natural or organic were common in over-the-counter (OTC) products, and ingredients such as sulfates and parabens were avoided. Common OTC ingredients for antidandruff and anti-itch shampoos and conditioners include zinc pyrithione, selenium sulfide, coal tar, salicylic acid, and citric acid. Additionally, coconut oil, tea tree oil, apple cider vinegar, and charcoal are common natural alternatives used to address SD symptoms.
Our review of the literature yielded limited recommendations tailored specifically to Black patients with SD. Of 108 abstracts, articles, or textbook chapters providing treatment recommendations for SD, 6 (6%) specifically discussed treatments for Black patients. All articles were written by authors with medical or scientific backgrounds. Of the treatment options discussed, topical antifungals generally were considered first-line for SD in all patients, with ketoconazole shampoo being a common first choice.4,5
Comment
Our study indicated that many consumer websites recommend unstudied nonmedical treatments for SD. Zinc pyrithione was one of the most commonly mentioned ingredients in OTC products to treat SD targeted toward Black patients, as its properties have contributed to ease of hair combing and less frizz.6 Zinc pyrithione has antifungal properties that reduce the proliferation of Malassezia furfur as well as anti-inflammatory properties that reduce irritation, pruritus, and erythema in areas affected by SD.7 Tea tree and peppermint oils also were commonly mentioned; the theory is that these oils mitigate SD by reducing yeast growth and soothing inflammation through antioxidant activity.8,9 Coal tar also is used due to its keratoplastic properties, which slow the growth of skin cells and ultimately reduce scaling and dryness.10 Yeast thrives in basic pH conditions; apple cider vinegar is used as an ingredient in OTC products for SD because its acidic pH creates a less favorable environment for yeast to grow.11 Although many of the ingredients found in OTC products we identified have not yet been studied, they have properties that theoretically would be helpful in treating SD.
Our review of the medical literature revealed that while there are treatments that are effective for SD, the recommended use may not consider the cultural differences that exist for Black patients. For instance, reports in the literature regarding ketoconazole shampoo revealed that ketoconazole increases the risk for hair shaft dryness, damage, and subsequent breakage, especially in Black women who also may be using heat styling or chemical relaxers.5 As a result, ketoconazole should be used with caution in Black women, with an emphasis on direct application to the scalp rather than the hair shafts.12 Additional options reported for Black patients include ciclopirox olamine and zinc pyrithione, which may have fewer risks.13
When prescribing medicated shampoos, traditional instructions regarding frequency of use to control symptoms of SD range from 2 to 3 times weekly to daily for a specified period of time determined by the dermatologist.14 However, frequency of hair washing varies greatly among Black patients, sometimes occurring only once monthly. The frequency also may change based on styling techniques (eg, braids, weaves, and wigs).15 Based on previous research underscoring the tendency for Black patients to use medicated shampoos less frequently than White patients, it is important for clinicians to understand that these cultural practices can undermine the effectiveness when medicated shampoos are prescribed for SD.16
Additionally, topical corticosteroids often are used in conjunction with antifungals to help decrease inflammation of the scalp.17 An option reported for Black patients is topical fluocinolone 0.01%; however, package instructions state to apply topically to the scalp nightly and wash the hair thoroughly each morning, which may not be feasible for Black patients based on previously mentioned differences in hair-washing techniques. An alternative option may be to apply the medication 3 to 4 times per week, washing the hair weekly rather than daily.18 Fluocinolone can be used as an ointment, solution, oil, or cream.19,20 When comparing treatment vehicles for SD, a study conducted by Chappell et al21 found that Black patients preferred using ointment or oil vehicles; White patients preferred foams and sprays, which may not be suitable for Afro hair patterns. As such, using less-drying modalities may increase compliance and treatment success in Black patients. For patients who may have involvement on the hairline, face, or ears along with hypopigmentation (which is a common skin concern associated with SD), calcineurin inhibitors can be used until resolution occurs.5,22 High et al15 found that twice-daily use of pimecrolimus rapidly normalized skin pigmentation during the first 2 weeks of use. Overall, personalization of treatment may not only avoid adverse effects but also ensure patient compliance, with the overall goal of treating to reduce yeast activity, pruritus, and dyschromia.22
Interestingly, after the website searches were completed for this study, the US Food and Drug Administration approved topical roflumilast foam for SD. In a phase III trial of 457 total patients, 36 Black patients were included.23 It was determined that 79.5% of patients overall throughout the trial achieved Investigator Global Assessment success (score of 0 [clear] or 1 [almost clear]) plus ≥2-point improvement from baseline (on a scale of 0 [clear] to 4 [severe]) at weeks 2, 4, and 8. Although there currently are no long-term studies, roflumilast may be a promising option for Black patients with SD.23
Aside from developing an individualized treatment approach for Black patients with SD, it is important to ask targeted questions during the clinical encounter to identify factors that may be exacerbating symptoms, especially due to the wide range of hair care practices used by the Black community (eTable 2). Asking targeted questions is especially important, as prior studies have shown that extensions, hair relaxers, and particular hair products can irritate the scalp and increase the likelihood of developing SD.21,24 Rucker Wright et al25 evaluated different hair care practices among young Black females and their association with the development of SD. The authors found that using hair extensions (either braided, cornrowed, or ponytails), chemical relaxers, and hair oils every 2 weeks was associated with SD. The study also found that SD rates were roughly 20% higher among Black girls with extensions compared to Black girls without extensions, regardless of how frequently hair was washed.25
Many Black patients grease the scalp with oils that are beneficial for lubrication and reduction of abrasive damage caused by grooming; however, they also may increase incidence of SD.26 Tight curls worn by Black patients also can impede sebum from traveling down the hair shaft, leading to oil buildup on the scalp. This is the ideal environment for increased Malassezia density and higher risk for SD development.27 To balance the beneficial effects of hair oils with the increased susceptibility for SD, providers should emphasize applying these oils only to distal hair shafts, which are more likely to be damaged, and avoiding application to the scalp.19
Conclusion
Given its long-term relapsing and remitting nature, SD can be distressing for Black patients, many of whom may seek additional treatment options aside from those recommended by health care professionals. In order to better educate patients, it is important for dermatologists to know not only the common ingredients that may be present in OTC products but also the thought process behind why patients use them. Additionally, prescription treatments for Black patients with SD may require nuanced alterations to the product instructions that may prevent health disparities and provide culturally sensitive care. Overall, the literature regarding treatment for Black patients with SD is limited, and more high-quality studies are needed.
- Tucker D, Masood S. Seborrheic dermatitis. StatPearls [Internet]. Updated March 1, 2024. Accessed December 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK551707/
- Borda LJ, Wikramanayake TC. Seborrheic dermatitis and dandruff: a comprehensive review. J Clin Investig Dermatol. 2015;3:10.13188 /2373-1044.1000019.
- American Academy of Dermatology. Seborrheic dermatitis by the numbers. American Academy of Dermatology Skin Disease Briefs. Updated May 5, 2018. Accessed November 22, 2024. https://www.aad.org/asset/49w949DPcF8RSJYIRHfDon
- Davis SA, Naarahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Borda LJ, Perper M, Keri JE. Treatment of seborrheic dermatitis: a comprehensive review. J Dermatolog Treat. 2019;30:158-169.
- Draelos ZD, Kenneally DC, Hodges LT, et al. A comparison of hair quality and cosmetic acceptance following the use of two anti-dandruff shampoos. J Investig Dermatol Symp Proc. 2005;10:201-214.
- Barak-Shinar D, Green LJ. Scalp seborrheic dermatitis and dandruff therapy using a herbal and zinc pyrithione-based therapy of shampoo and scalp lotion. J Clin Aesthet Dermatol. 2018;11:26-31.
- Satchell AC, Saurajen A, Bell C, et al. Treatment of dandruff with 5% tea tree oil shampoo. J Am Acad Dermatol. 2002;47:852-855.
- Herro E, Jacob SE. Mentha piperita (peppermint). Dermatitis. 2010;21:327-329.
- Sanfilippo A, English JC. An overview of medicated shampoos used in dandruff treatment. Pharm Ther. 2006;31:396-400.
- Arun PVPS, Vineetha Y, Waheed M, et al. Quantification of the minimum amount of lemon juice and apple cider vinegar required for the growth inhibition of dandruff causing fungi Malassezia furfur. Int J Sci Res in Biological Sciences. 2019;6:144-147.
- Gao HY, Li Wan Po A. Topical formulations of fluocinolone acetonide. Are creams, gels and ointments bioequivalent and does dilution affect activity? Eur J Clin Pharmacol. 1994;46:71-75.
- Pauporte M, Maibach H, Lowe N, et al. Fluocinolone acetonide topical oil for scalp psoriasis. J Dermatolog Treat. 2004;15:360-364.
- Elgash M, Dlova N, Ogunleye T, et al. Seborrheic dermatitis in skin of color: clinical considerations. J Drugs Dermatol. 2019;18:24-27.
- High WA, Pandya AG. Pilot trial of 1% pimecrolimus cream in the treatment of seborrheic dermatitis in African American adults with associated hypopigmentation. J Am Acad Dermatol. 2006;54:1083-1088.
- Hollins LC, Butt M, Hong J, et al. Research in brief: survey of hair care practices in various ethnic and racial pediatric populations. Pediatr Dermatol. 2022;39:494-496.
- Halder RM, Roberts CI, Nootheti PK. Cutaneous diseases in the black races. Dermatol Clin. 2003;21:679-687, ix.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Friedmann DP, Mishra V, Batty T. Progressive facial papules in an African- American patient: an atypical presentation of seborrheic dermatitis. J Clin Aesthet Dermatol. 2018;11:44-45.
- Clark GW, Pope SM, Jaboori KA. Diagnosis and treatment of seborrheic dermatitis. Am Fam Physician. 2015;91:185-190.
- Chappell J, Mattox A, Simonetta C, et al. Seborrheic dermatitis of the scalp in populations practicing less frequent hair washing: ketoconazole 2% foam versus ketoconazole 2% shampoo. three-year data. J Am Acad Dermatol. 2014;70:AB54.
- Dadzie OE, Salam A. The hair grooming practices of women of African descent in London, United Kingdom: findings of a cross-sectional study. J Eur Acad Dermatol Venereol. 2016;30:1021-1024.
- Blauvelt A, Draelos ZD, Stein Gold L, et al. Roflumilast foam 0.3% for adolescent and adult patients with seborrheic dermatitis: a randomized, double-blinded, vehicle-controlled, phase 3 trial. J Am Acad Dermatol. 2024;90:986-993.
- Taylor SC, Barbosa V, Burgess C, et al. Hair and scalp disorders in adult and pediatric patients with skin of color. Cutis. 2017;100:31-35.
- Rucker Wright D, Gathers R, Kapke A, et al. Hair care practices and their association with scalp and hair disorders in African American girls. J Am Acad Dermatol. 2011;64:253-262.
- Raffi J, Suresh R, Agbai O. Clinical recognition and management of alopecia in women of color. Int J Womens Dermatol. 2019;5:314-319.
- Mayo T, Dinkins J, Elewski B. Hair oils may worsen seborrheic dermatitis in Black patients. Skin Appendage Disord. 2023;9:151-152.
- Tucker D, Masood S. Seborrheic dermatitis. StatPearls [Internet]. Updated March 1, 2024. Accessed December 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK551707/
- Borda LJ, Wikramanayake TC. Seborrheic dermatitis and dandruff: a comprehensive review. J Clin Investig Dermatol. 2015;3:10.13188 /2373-1044.1000019.
- American Academy of Dermatology. Seborrheic dermatitis by the numbers. American Academy of Dermatology Skin Disease Briefs. Updated May 5, 2018. Accessed November 22, 2024. https://www.aad.org/asset/49w949DPcF8RSJYIRHfDon
- Davis SA, Naarahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Borda LJ, Perper M, Keri JE. Treatment of seborrheic dermatitis: a comprehensive review. J Dermatolog Treat. 2019;30:158-169.
- Draelos ZD, Kenneally DC, Hodges LT, et al. A comparison of hair quality and cosmetic acceptance following the use of two anti-dandruff shampoos. J Investig Dermatol Symp Proc. 2005;10:201-214.
- Barak-Shinar D, Green LJ. Scalp seborrheic dermatitis and dandruff therapy using a herbal and zinc pyrithione-based therapy of shampoo and scalp lotion. J Clin Aesthet Dermatol. 2018;11:26-31.
- Satchell AC, Saurajen A, Bell C, et al. Treatment of dandruff with 5% tea tree oil shampoo. J Am Acad Dermatol. 2002;47:852-855.
- Herro E, Jacob SE. Mentha piperita (peppermint). Dermatitis. 2010;21:327-329.
- Sanfilippo A, English JC. An overview of medicated shampoos used in dandruff treatment. Pharm Ther. 2006;31:396-400.
- Arun PVPS, Vineetha Y, Waheed M, et al. Quantification of the minimum amount of lemon juice and apple cider vinegar required for the growth inhibition of dandruff causing fungi Malassezia furfur. Int J Sci Res in Biological Sciences. 2019;6:144-147.
- Gao HY, Li Wan Po A. Topical formulations of fluocinolone acetonide. Are creams, gels and ointments bioequivalent and does dilution affect activity? Eur J Clin Pharmacol. 1994;46:71-75.
- Pauporte M, Maibach H, Lowe N, et al. Fluocinolone acetonide topical oil for scalp psoriasis. J Dermatolog Treat. 2004;15:360-364.
- Elgash M, Dlova N, Ogunleye T, et al. Seborrheic dermatitis in skin of color: clinical considerations. J Drugs Dermatol. 2019;18:24-27.
- High WA, Pandya AG. Pilot trial of 1% pimecrolimus cream in the treatment of seborrheic dermatitis in African American adults with associated hypopigmentation. J Am Acad Dermatol. 2006;54:1083-1088.
- Hollins LC, Butt M, Hong J, et al. Research in brief: survey of hair care practices in various ethnic and racial pediatric populations. Pediatr Dermatol. 2022;39:494-496.
- Halder RM, Roberts CI, Nootheti PK. Cutaneous diseases in the black races. Dermatol Clin. 2003;21:679-687, ix.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Friedmann DP, Mishra V, Batty T. Progressive facial papules in an African- American patient: an atypical presentation of seborrheic dermatitis. J Clin Aesthet Dermatol. 2018;11:44-45.
- Clark GW, Pope SM, Jaboori KA. Diagnosis and treatment of seborrheic dermatitis. Am Fam Physician. 2015;91:185-190.
- Chappell J, Mattox A, Simonetta C, et al. Seborrheic dermatitis of the scalp in populations practicing less frequent hair washing: ketoconazole 2% foam versus ketoconazole 2% shampoo. three-year data. J Am Acad Dermatol. 2014;70:AB54.
- Dadzie OE, Salam A. The hair grooming practices of women of African descent in London, United Kingdom: findings of a cross-sectional study. J Eur Acad Dermatol Venereol. 2016;30:1021-1024.
- Blauvelt A, Draelos ZD, Stein Gold L, et al. Roflumilast foam 0.3% for adolescent and adult patients with seborrheic dermatitis: a randomized, double-blinded, vehicle-controlled, phase 3 trial. J Am Acad Dermatol. 2024;90:986-993.
- Taylor SC, Barbosa V, Burgess C, et al. Hair and scalp disorders in adult and pediatric patients with skin of color. Cutis. 2017;100:31-35.
- Rucker Wright D, Gathers R, Kapke A, et al. Hair care practices and their association with scalp and hair disorders in African American girls. J Am Acad Dermatol. 2011;64:253-262.
- Raffi J, Suresh R, Agbai O. Clinical recognition and management of alopecia in women of color. Int J Womens Dermatol. 2019;5:314-319.
- Mayo T, Dinkins J, Elewski B. Hair oils may worsen seborrheic dermatitis in Black patients. Skin Appendage Disord. 2023;9:151-152.
Treatment of Seborrheic Dermatitis in Black Patients
Treatment of Seborrheic Dermatitis in Black Patients
PRACTICE POINTS
- Cultural awareness when treating Black patients with seborrheic dermatitis is vital to providing appropriate care, as hair care practices may impact treatment options and regimen.
- Knowledge about over-the-counter products that are targeted toward Black patients and the ingredients they contain can assist in providing better counseling to patients and improve shared decision-making.