User login
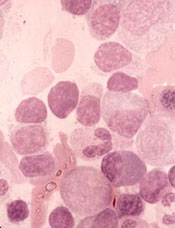
CRISPR bests TALEN in iPSCs

Credit: Salk Institute
The gene-editing technology CRISPR can precisely and efficiently alter human stem cells, according to research published in Molecular Therapy.
Using JAK2 and other genes as models, researchers showed that CRISPR offers advantages over TALEN, another gene-editing technique, for manipulating induced pluripotent stem cells (iPSCs).
And, unlike in a previous study, CRISPR did not produce any off-target effects.
The team believes their findings could streamline and speed up efforts to modify human iPSCs for use as treatments or in the development of model systems to study diseases and test drugs.
“Stem cell technology is quickly advancing, and we think that the days when we can use iPSCs for human therapy aren’t that far away,” said study author Zhaohui Ye, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“This is one of the first studies to detail the use of CRISPR in human iPSCs, showcasing its potential in these cells.”
CRISPR originated from a microbial immune system that contains DNA segments known as “clustered regularly interspaced short palindromic repeats.” The system makes use of an enzyme that nicks together DNA with a piece of small RNA that guides the tool to where researchers want to introduce cuts or other changes in the genome.
Previous research has shown that CRISPR can generate genomic changes or mutations through these interventions more efficiently than other gene-editing techniques, such as TALEN, which is short for “transcription activator-like effector nuclease.”
Despite CRISPR’s advantages, a recent study suggested it might also produce a large number of off-target effects in human cancer cell lines; specifically, modification of genes that researchers didn’t mean to change.
To see if this unwanted effect occurred in other human cell types, Dr Ye and his colleagues pitted CRISPR against TALEN in human iPSCs. The researchers compared the ability of both techniques to either cut out pieces of known genes in iPSCs or cut out a piece of these genes and replace it with another.
As model genes, the researchers used JAK2, a gene that, when mutated, causes myeloproliferative neoplasms; SERPINA1, a gene that, when mutated, causes alpha1-antitrypsin deficiency, an inherited disorder that may cause lung and liver disease; and AAVS1, a gene that’s been recently discovered to be a “safe harbor” in the human genome for inserting foreign genes.
The comparison showed that, when simply cutting out portions of genes, the CRISPR system was significantly more efficient than TALEN in all 3 gene systems, inducing up to 100 times more cuts.
However, when using these genome-editing tools for replacing portions of the genes, such as the disease-causing mutations in JAK2 and SERPINA1 genes, CRISPR and TALEN showed about the same efficiency in patient-derived iPSCs.
Contrary to results of the human cancer cell line study, both CRISPR and TALEN had the same targeting specificity in human iPSCs, hitting only the genes they were designed to affect.
The researchers also found that the CRISPR system has a second advantage over TALEN. It can be designed to target only the mutation-containing gene without affecting the healthy gene in patients where only one copy of a gene is affected.
These findings, according to the researchers, offer reassurance that CRISPR will be a useful tool for editing the genes of human iPSCs with little risk of off-target effects. ![]()
Credit: Salk Institute
The gene-editing technology CRISPR can precisely and efficiently alter human stem cells, according to research published in Molecular Therapy.
Using JAK2 and other genes as models, researchers showed that CRISPR offers advantages over TALEN, another gene-editing technique, for manipulating induced pluripotent stem cells (iPSCs).
And, unlike in a previous study, CRISPR did not produce any off-target effects.
The team believes their findings could streamline and speed up efforts to modify human iPSCs for use as treatments or in the development of model systems to study diseases and test drugs.
“Stem cell technology is quickly advancing, and we think that the days when we can use iPSCs for human therapy aren’t that far away,” said study author Zhaohui Ye, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“This is one of the first studies to detail the use of CRISPR in human iPSCs, showcasing its potential in these cells.”
CRISPR originated from a microbial immune system that contains DNA segments known as “clustered regularly interspaced short palindromic repeats.” The system makes use of an enzyme that nicks together DNA with a piece of small RNA that guides the tool to where researchers want to introduce cuts or other changes in the genome.
Previous research has shown that CRISPR can generate genomic changes or mutations through these interventions more efficiently than other gene-editing techniques, such as TALEN, which is short for “transcription activator-like effector nuclease.”
Despite CRISPR’s advantages, a recent study suggested it might also produce a large number of off-target effects in human cancer cell lines; specifically, modification of genes that researchers didn’t mean to change.
To see if this unwanted effect occurred in other human cell types, Dr Ye and his colleagues pitted CRISPR against TALEN in human iPSCs. The researchers compared the ability of both techniques to either cut out pieces of known genes in iPSCs or cut out a piece of these genes and replace it with another.
As model genes, the researchers used JAK2, a gene that, when mutated, causes myeloproliferative neoplasms; SERPINA1, a gene that, when mutated, causes alpha1-antitrypsin deficiency, an inherited disorder that may cause lung and liver disease; and AAVS1, a gene that’s been recently discovered to be a “safe harbor” in the human genome for inserting foreign genes.
The comparison showed that, when simply cutting out portions of genes, the CRISPR system was significantly more efficient than TALEN in all 3 gene systems, inducing up to 100 times more cuts.
However, when using these genome-editing tools for replacing portions of the genes, such as the disease-causing mutations in JAK2 and SERPINA1 genes, CRISPR and TALEN showed about the same efficiency in patient-derived iPSCs.
Contrary to results of the human cancer cell line study, both CRISPR and TALEN had the same targeting specificity in human iPSCs, hitting only the genes they were designed to affect.
The researchers also found that the CRISPR system has a second advantage over TALEN. It can be designed to target only the mutation-containing gene without affecting the healthy gene in patients where only one copy of a gene is affected.
These findings, according to the researchers, offer reassurance that CRISPR will be a useful tool for editing the genes of human iPSCs with little risk of off-target effects. ![]()
Credit: Salk Institute
The gene-editing technology CRISPR can precisely and efficiently alter human stem cells, according to research published in Molecular Therapy.
Using JAK2 and other genes as models, researchers showed that CRISPR offers advantages over TALEN, another gene-editing technique, for manipulating induced pluripotent stem cells (iPSCs).
And, unlike in a previous study, CRISPR did not produce any off-target effects.
The team believes their findings could streamline and speed up efforts to modify human iPSCs for use as treatments or in the development of model systems to study diseases and test drugs.
“Stem cell technology is quickly advancing, and we think that the days when we can use iPSCs for human therapy aren’t that far away,” said study author Zhaohui Ye, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“This is one of the first studies to detail the use of CRISPR in human iPSCs, showcasing its potential in these cells.”
CRISPR originated from a microbial immune system that contains DNA segments known as “clustered regularly interspaced short palindromic repeats.” The system makes use of an enzyme that nicks together DNA with a piece of small RNA that guides the tool to where researchers want to introduce cuts or other changes in the genome.
Previous research has shown that CRISPR can generate genomic changes or mutations through these interventions more efficiently than other gene-editing techniques, such as TALEN, which is short for “transcription activator-like effector nuclease.”
Despite CRISPR’s advantages, a recent study suggested it might also produce a large number of off-target effects in human cancer cell lines; specifically, modification of genes that researchers didn’t mean to change.
To see if this unwanted effect occurred in other human cell types, Dr Ye and his colleagues pitted CRISPR against TALEN in human iPSCs. The researchers compared the ability of both techniques to either cut out pieces of known genes in iPSCs or cut out a piece of these genes and replace it with another.
As model genes, the researchers used JAK2, a gene that, when mutated, causes myeloproliferative neoplasms; SERPINA1, a gene that, when mutated, causes alpha1-antitrypsin deficiency, an inherited disorder that may cause lung and liver disease; and AAVS1, a gene that’s been recently discovered to be a “safe harbor” in the human genome for inserting foreign genes.
The comparison showed that, when simply cutting out portions of genes, the CRISPR system was significantly more efficient than TALEN in all 3 gene systems, inducing up to 100 times more cuts.
However, when using these genome-editing tools for replacing portions of the genes, such as the disease-causing mutations in JAK2 and SERPINA1 genes, CRISPR and TALEN showed about the same efficiency in patient-derived iPSCs.
Contrary to results of the human cancer cell line study, both CRISPR and TALEN had the same targeting specificity in human iPSCs, hitting only the genes they were designed to affect.
The researchers also found that the CRISPR system has a second advantage over TALEN. It can be designed to target only the mutation-containing gene without affecting the healthy gene in patients where only one copy of a gene is affected.
These findings, according to the researchers, offer reassurance that CRISPR will be a useful tool for editing the genes of human iPSCs with little risk of off-target effects. ![]()
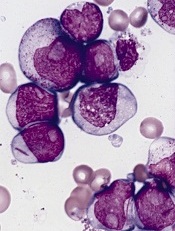
Ibrutinib proves active in high-risk CLL

Credit: Mary Ann Thompson
Single-agent ibrutinib can elicit a high response rate in patients with high-risk chronic lymphocytic leukemia (CLL), results of a phase 2 trial suggest.
The Bruton’s tyrosine kinase inhibitor prompted a 92% objective response rate in patients who had previously untreated or relapsed/refractory CLL with either 17p deletion (del 17p) or tumor protein 53 (TP53) aberrations.
Researchers reported this and other results of the trial in The Lancet Oncology.
“Ibrutinib treatment results observed in CLL patients with del 17p or TP53 aberrations are very encouraging given that these patients have a high relapse rate after chemotherapy and are in need of tolerable, effective, and durable treatment options,” said study author Mohammed Farooqui, DO, of the National Heart, Lung, and Blood Institute in Bethesda, Maryland.
He and his colleagues studied 51 patients in this trial, 35 with previously untreated CLL and 16 with relapsed or refractory CLL. Forty-seven of the patients (92%) had del 17p, and 4 patients carried the TP53 aberration but did not have del 17p.
The study’s primary endpoint was overall response rate after 24 weeks. Secondary endpoints included safety, overall survival, progression-free survival, best response, and nodal response.
The median follow-up for all patients was 24 months (15 months for the previously untreated cohort). At 24 weeks, 48 patients were evaluable for response, assessed according to the modified IWCLL 2008 criteria.
Response rates
At 24 weeks, 92% (n=44) of the 48 evaluable patients achieved an objective response. Fifty percent of all evaluable patients achieved a partial response (n=24)—55% of previously untreated patients (n=18) and 40% of relapsed/refractory patients (n=6).
As for best response, 10% of all patients achieved a complete response (n=5)—12% of previously untreated patients (n=4) and 7% of relapsed/refractory patients (n=1). And 67% of patients had a partial response (n=32)—70% of previously untreated patients (n=23) and 60% of relapsed/refractory patients (n=9).
After 8 weeks on therapy, ibrutinib was associated with a more than 50% mean reduction in tumor burden in the bone marrow (44%), lymph nodes (70%), and spleen (79%). After 24 weeks of therapy, the rates of tumor burden reduction (> 50%) increased to 83%, 93%, and 95%, respectively.
Survival and safety
The estimated progression-free survival at 24 months for all patients on an intention-to-treat basis was 82%. Forty-two of the 51 patients (82%) continued on ibrutinib treatment without disease progression.
The estimated overall survival at 24 months was 80% for all patients—84% for previously untreated patients and 74% for patients with relapsed or refractory disease.
At the final follow-up, 8 (16%) patients had died—5 (10%) from progressive disease, 2 (4%) from infection, and 1 (2%) patient with a sudden, unexplained death that may have been treatment-related.
The most common adverse events (occurring in more than 30% of all patients) potentially related to ibrutinib were arthralgia (59%), diarrhea (51%), rash (47%), nail ridging (43%), bruising (33%), and muscle spasms (31%).
The most frequent grade 3 or 4 hematologic adverse events were neutropenia (24%), anemia (14%), and thrombocytopenia (10%). The most common nonhematologic grade 3 adverse event was pneumonia, which occurred in 3 patients (6%).
Nine patients (18%) discontinued treatment. The reasons for discontinuation included disease progression in 5 patients (10%) and death for 3 patients (6%).
This research was sponsored by the Intramural Research Program of the National Heart, Lung, and Blood Institute and the National Cancer Institute; Danish Cancer Society; Novo Nordisk Foundation; National Institutes of Health Medical Research Scholars Program; and Pharmacyclics Inc.
Ibrutinib is jointly developed and commercialized by Pharmacyclics and Janssen Biotech, Inc. ![]()
Credit: Mary Ann Thompson
Single-agent ibrutinib can elicit a high response rate in patients with high-risk chronic lymphocytic leukemia (CLL), results of a phase 2 trial suggest.
The Bruton’s tyrosine kinase inhibitor prompted a 92% objective response rate in patients who had previously untreated or relapsed/refractory CLL with either 17p deletion (del 17p) or tumor protein 53 (TP53) aberrations.
Researchers reported this and other results of the trial in The Lancet Oncology.
“Ibrutinib treatment results observed in CLL patients with del 17p or TP53 aberrations are very encouraging given that these patients have a high relapse rate after chemotherapy and are in need of tolerable, effective, and durable treatment options,” said study author Mohammed Farooqui, DO, of the National Heart, Lung, and Blood Institute in Bethesda, Maryland.
He and his colleagues studied 51 patients in this trial, 35 with previously untreated CLL and 16 with relapsed or refractory CLL. Forty-seven of the patients (92%) had del 17p, and 4 patients carried the TP53 aberration but did not have del 17p.
The study’s primary endpoint was overall response rate after 24 weeks. Secondary endpoints included safety, overall survival, progression-free survival, best response, and nodal response.
The median follow-up for all patients was 24 months (15 months for the previously untreated cohort). At 24 weeks, 48 patients were evaluable for response, assessed according to the modified IWCLL 2008 criteria.
Response rates
At 24 weeks, 92% (n=44) of the 48 evaluable patients achieved an objective response. Fifty percent of all evaluable patients achieved a partial response (n=24)—55% of previously untreated patients (n=18) and 40% of relapsed/refractory patients (n=6).
As for best response, 10% of all patients achieved a complete response (n=5)—12% of previously untreated patients (n=4) and 7% of relapsed/refractory patients (n=1). And 67% of patients had a partial response (n=32)—70% of previously untreated patients (n=23) and 60% of relapsed/refractory patients (n=9).
After 8 weeks on therapy, ibrutinib was associated with a more than 50% mean reduction in tumor burden in the bone marrow (44%), lymph nodes (70%), and spleen (79%). After 24 weeks of therapy, the rates of tumor burden reduction (> 50%) increased to 83%, 93%, and 95%, respectively.
Survival and safety
The estimated progression-free survival at 24 months for all patients on an intention-to-treat basis was 82%. Forty-two of the 51 patients (82%) continued on ibrutinib treatment without disease progression.
The estimated overall survival at 24 months was 80% for all patients—84% for previously untreated patients and 74% for patients with relapsed or refractory disease.
At the final follow-up, 8 (16%) patients had died—5 (10%) from progressive disease, 2 (4%) from infection, and 1 (2%) patient with a sudden, unexplained death that may have been treatment-related.
The most common adverse events (occurring in more than 30% of all patients) potentially related to ibrutinib were arthralgia (59%), diarrhea (51%), rash (47%), nail ridging (43%), bruising (33%), and muscle spasms (31%).
The most frequent grade 3 or 4 hematologic adverse events were neutropenia (24%), anemia (14%), and thrombocytopenia (10%). The most common nonhematologic grade 3 adverse event was pneumonia, which occurred in 3 patients (6%).
Nine patients (18%) discontinued treatment. The reasons for discontinuation included disease progression in 5 patients (10%) and death for 3 patients (6%).
This research was sponsored by the Intramural Research Program of the National Heart, Lung, and Blood Institute and the National Cancer Institute; Danish Cancer Society; Novo Nordisk Foundation; National Institutes of Health Medical Research Scholars Program; and Pharmacyclics Inc.
Ibrutinib is jointly developed and commercialized by Pharmacyclics and Janssen Biotech, Inc. ![]()
Credit: Mary Ann Thompson
Single-agent ibrutinib can elicit a high response rate in patients with high-risk chronic lymphocytic leukemia (CLL), results of a phase 2 trial suggest.
The Bruton’s tyrosine kinase inhibitor prompted a 92% objective response rate in patients who had previously untreated or relapsed/refractory CLL with either 17p deletion (del 17p) or tumor protein 53 (TP53) aberrations.
Researchers reported this and other results of the trial in The Lancet Oncology.
“Ibrutinib treatment results observed in CLL patients with del 17p or TP53 aberrations are very encouraging given that these patients have a high relapse rate after chemotherapy and are in need of tolerable, effective, and durable treatment options,” said study author Mohammed Farooqui, DO, of the National Heart, Lung, and Blood Institute in Bethesda, Maryland.
He and his colleagues studied 51 patients in this trial, 35 with previously untreated CLL and 16 with relapsed or refractory CLL. Forty-seven of the patients (92%) had del 17p, and 4 patients carried the TP53 aberration but did not have del 17p.
The study’s primary endpoint was overall response rate after 24 weeks. Secondary endpoints included safety, overall survival, progression-free survival, best response, and nodal response.
The median follow-up for all patients was 24 months (15 months for the previously untreated cohort). At 24 weeks, 48 patients were evaluable for response, assessed according to the modified IWCLL 2008 criteria.
Response rates
At 24 weeks, 92% (n=44) of the 48 evaluable patients achieved an objective response. Fifty percent of all evaluable patients achieved a partial response (n=24)—55% of previously untreated patients (n=18) and 40% of relapsed/refractory patients (n=6).
As for best response, 10% of all patients achieved a complete response (n=5)—12% of previously untreated patients (n=4) and 7% of relapsed/refractory patients (n=1). And 67% of patients had a partial response (n=32)—70% of previously untreated patients (n=23) and 60% of relapsed/refractory patients (n=9).
After 8 weeks on therapy, ibrutinib was associated with a more than 50% mean reduction in tumor burden in the bone marrow (44%), lymph nodes (70%), and spleen (79%). After 24 weeks of therapy, the rates of tumor burden reduction (> 50%) increased to 83%, 93%, and 95%, respectively.
Survival and safety
The estimated progression-free survival at 24 months for all patients on an intention-to-treat basis was 82%. Forty-two of the 51 patients (82%) continued on ibrutinib treatment without disease progression.
The estimated overall survival at 24 months was 80% for all patients—84% for previously untreated patients and 74% for patients with relapsed or refractory disease.
At the final follow-up, 8 (16%) patients had died—5 (10%) from progressive disease, 2 (4%) from infection, and 1 (2%) patient with a sudden, unexplained death that may have been treatment-related.
The most common adverse events (occurring in more than 30% of all patients) potentially related to ibrutinib were arthralgia (59%), diarrhea (51%), rash (47%), nail ridging (43%), bruising (33%), and muscle spasms (31%).
The most frequent grade 3 or 4 hematologic adverse events were neutropenia (24%), anemia (14%), and thrombocytopenia (10%). The most common nonhematologic grade 3 adverse event was pneumonia, which occurred in 3 patients (6%).
Nine patients (18%) discontinued treatment. The reasons for discontinuation included disease progression in 5 patients (10%) and death for 3 patients (6%).
This research was sponsored by the Intramural Research Program of the National Heart, Lung, and Blood Institute and the National Cancer Institute; Danish Cancer Society; Novo Nordisk Foundation; National Institutes of Health Medical Research Scholars Program; and Pharmacyclics Inc.
Ibrutinib is jointly developed and commercialized by Pharmacyclics and Janssen Biotech, Inc. ![]()
Whole plant treats malaria better

artemisinin is derived
Credit: Jorge Ferreira
Preclinical research suggests that using the whole plant Artemesia annua, from which the drug artemisinin is extracted, may treat malaria more effectively than artemisinin itself.
Whole-plant treatment withstood the evolution of resistance and remained effective for up to 3 times longer than pure artemisinin.
Whole-plant therapy was also more effective in killing rodent parasites that have previously evolved resistance to pure artemisinin.
Stephen Rich, PhD, of the University of Massachusetts Amherst, and his colleagues reported these findings in PNAS.
The team previously showed that the whole-plant approach is more effective at killing rodent malaria than purified artemisinin.
In the present study, the investigators conducted a series of experiments to determine the rates at which parasites become resistant to whole-plant treatment compared to the rate with pure artemisinin, and if the whole-plant treatment can overcome resistance to pharmaceutical artemisinin.
The team chose 2 rodent malaria species for particular characteristics. They chose Plasmodium yoelii because an artemisinin-resistant strain exists and could be used to test whether the whole plant can overcome that resistance.
And they chose Plasmodium chabaudi because, among several species of rodent malaria, it most closely biologically resembles the deadliest of the 5 human malaria parasites, Plasmodium falciparum.
“Conducting these experiments in different rodent malaria species also provides a robust test of the therapy,” Dr Rich noted.
To determine the respective evolutionary rates of resistance to whole-plant therapy and artemisinin, Dr Rich and his colleagues conducted artificial evolution experiments. The goal was to compare the rates at which resistance to these two treatments arises in serial passage among wild-type parasite lines.
In this technique, parasite proliferation rates determine resistance. Resistant parasites are expected to reach a certain target level at the same time, whether treatment is present or absent. Sensitive parasite strains will grow more slowly in the presence of treatment and reach the target later than untreated strains.
The investigators found that artemisinin-treated parasites achieved stable resistance to low-dose (100 mg/kg) therapy on passage 16. Those parasites were then treated with a doubled artemisinin dose, and they became resistant to this after an additional 24 passages.
By comparison, parasites did not become resistant to even the low dose of whole-plant therapy (100 mg/kg) after 49 passages.
From this, the investigators concluded that the whole-plant therapy lasts at least 3 times longer than its artemisinin counterpart, and at least twice as long as the doubled dose of pure artemisinin.
“This is especially important given the recent reports of resistance to artemisinin in malaria-endemic regions of the world,” Dr Rich said.
He and his colleagues also tested whether dried, whole-plant therapy can overcome existing resistance to pharmaceutical artemisinin.
They fed groups of mice infected with artemisinin-resistant malaria either the whole-plant therapy or artemisinin mixed with water. Single treatments were given in low (40 mg) and high (200 mg) doses. Control groups received a mouse chow placebo.
The investigators then measured the parasite levels in the rodents’ bloodstream at 9 points after treatment began.
Mice given either the low or high dose of whole-plant therapy showed a significantly greater reduction in parasitemia than those in their respective artemisinin groups. As expected for these resistant parasites, parasitemia in mice in the low-dose artemisinin group did not differ from controls.
The investigators said consuming the whole plant may be more effective than the single purified drug because the whole plant “may constitute a naturally occurring combination therapy that augments artemisinin delivery and synergizes the drug’s activity.”
Dr Rich did note that the exact mechanisms of whole-plant therapy’s effectiveness still need to be identified. But he also said the antimalarial activity of whole-plant therapy against artemisinin-resistant parasites provides “compelling reasons to further explore the role of non-pharmaceutical forms of artemisinin to treat human malaria.” ![]()
artemisinin is derived
Credit: Jorge Ferreira
Preclinical research suggests that using the whole plant Artemesia annua, from which the drug artemisinin is extracted, may treat malaria more effectively than artemisinin itself.
Whole-plant treatment withstood the evolution of resistance and remained effective for up to 3 times longer than pure artemisinin.
Whole-plant therapy was also more effective in killing rodent parasites that have previously evolved resistance to pure artemisinin.
Stephen Rich, PhD, of the University of Massachusetts Amherst, and his colleagues reported these findings in PNAS.
The team previously showed that the whole-plant approach is more effective at killing rodent malaria than purified artemisinin.
In the present study, the investigators conducted a series of experiments to determine the rates at which parasites become resistant to whole-plant treatment compared to the rate with pure artemisinin, and if the whole-plant treatment can overcome resistance to pharmaceutical artemisinin.
The team chose 2 rodent malaria species for particular characteristics. They chose Plasmodium yoelii because an artemisinin-resistant strain exists and could be used to test whether the whole plant can overcome that resistance.
And they chose Plasmodium chabaudi because, among several species of rodent malaria, it most closely biologically resembles the deadliest of the 5 human malaria parasites, Plasmodium falciparum.
“Conducting these experiments in different rodent malaria species also provides a robust test of the therapy,” Dr Rich noted.
To determine the respective evolutionary rates of resistance to whole-plant therapy and artemisinin, Dr Rich and his colleagues conducted artificial evolution experiments. The goal was to compare the rates at which resistance to these two treatments arises in serial passage among wild-type parasite lines.
In this technique, parasite proliferation rates determine resistance. Resistant parasites are expected to reach a certain target level at the same time, whether treatment is present or absent. Sensitive parasite strains will grow more slowly in the presence of treatment and reach the target later than untreated strains.
The investigators found that artemisinin-treated parasites achieved stable resistance to low-dose (100 mg/kg) therapy on passage 16. Those parasites were then treated with a doubled artemisinin dose, and they became resistant to this after an additional 24 passages.
By comparison, parasites did not become resistant to even the low dose of whole-plant therapy (100 mg/kg) after 49 passages.
From this, the investigators concluded that the whole-plant therapy lasts at least 3 times longer than its artemisinin counterpart, and at least twice as long as the doubled dose of pure artemisinin.
“This is especially important given the recent reports of resistance to artemisinin in malaria-endemic regions of the world,” Dr Rich said.
He and his colleagues also tested whether dried, whole-plant therapy can overcome existing resistance to pharmaceutical artemisinin.
They fed groups of mice infected with artemisinin-resistant malaria either the whole-plant therapy or artemisinin mixed with water. Single treatments were given in low (40 mg) and high (200 mg) doses. Control groups received a mouse chow placebo.
The investigators then measured the parasite levels in the rodents’ bloodstream at 9 points after treatment began.
Mice given either the low or high dose of whole-plant therapy showed a significantly greater reduction in parasitemia than those in their respective artemisinin groups. As expected for these resistant parasites, parasitemia in mice in the low-dose artemisinin group did not differ from controls.
The investigators said consuming the whole plant may be more effective than the single purified drug because the whole plant “may constitute a naturally occurring combination therapy that augments artemisinin delivery and synergizes the drug’s activity.”
Dr Rich did note that the exact mechanisms of whole-plant therapy’s effectiveness still need to be identified. But he also said the antimalarial activity of whole-plant therapy against artemisinin-resistant parasites provides “compelling reasons to further explore the role of non-pharmaceutical forms of artemisinin to treat human malaria.” ![]()
artemisinin is derived
Credit: Jorge Ferreira
Preclinical research suggests that using the whole plant Artemesia annua, from which the drug artemisinin is extracted, may treat malaria more effectively than artemisinin itself.
Whole-plant treatment withstood the evolution of resistance and remained effective for up to 3 times longer than pure artemisinin.
Whole-plant therapy was also more effective in killing rodent parasites that have previously evolved resistance to pure artemisinin.
Stephen Rich, PhD, of the University of Massachusetts Amherst, and his colleagues reported these findings in PNAS.
The team previously showed that the whole-plant approach is more effective at killing rodent malaria than purified artemisinin.
In the present study, the investigators conducted a series of experiments to determine the rates at which parasites become resistant to whole-plant treatment compared to the rate with pure artemisinin, and if the whole-plant treatment can overcome resistance to pharmaceutical artemisinin.
The team chose 2 rodent malaria species for particular characteristics. They chose Plasmodium yoelii because an artemisinin-resistant strain exists and could be used to test whether the whole plant can overcome that resistance.
And they chose Plasmodium chabaudi because, among several species of rodent malaria, it most closely biologically resembles the deadliest of the 5 human malaria parasites, Plasmodium falciparum.
“Conducting these experiments in different rodent malaria species also provides a robust test of the therapy,” Dr Rich noted.
To determine the respective evolutionary rates of resistance to whole-plant therapy and artemisinin, Dr Rich and his colleagues conducted artificial evolution experiments. The goal was to compare the rates at which resistance to these two treatments arises in serial passage among wild-type parasite lines.
In this technique, parasite proliferation rates determine resistance. Resistant parasites are expected to reach a certain target level at the same time, whether treatment is present or absent. Sensitive parasite strains will grow more slowly in the presence of treatment and reach the target later than untreated strains.
The investigators found that artemisinin-treated parasites achieved stable resistance to low-dose (100 mg/kg) therapy on passage 16. Those parasites were then treated with a doubled artemisinin dose, and they became resistant to this after an additional 24 passages.
By comparison, parasites did not become resistant to even the low dose of whole-plant therapy (100 mg/kg) after 49 passages.
From this, the investigators concluded that the whole-plant therapy lasts at least 3 times longer than its artemisinin counterpart, and at least twice as long as the doubled dose of pure artemisinin.
“This is especially important given the recent reports of resistance to artemisinin in malaria-endemic regions of the world,” Dr Rich said.
He and his colleagues also tested whether dried, whole-plant therapy can overcome existing resistance to pharmaceutical artemisinin.
They fed groups of mice infected with artemisinin-resistant malaria either the whole-plant therapy or artemisinin mixed with water. Single treatments were given in low (40 mg) and high (200 mg) doses. Control groups received a mouse chow placebo.
The investigators then measured the parasite levels in the rodents’ bloodstream at 9 points after treatment began.
Mice given either the low or high dose of whole-plant therapy showed a significantly greater reduction in parasitemia than those in their respective artemisinin groups. As expected for these resistant parasites, parasitemia in mice in the low-dose artemisinin group did not differ from controls.
The investigators said consuming the whole plant may be more effective than the single purified drug because the whole plant “may constitute a naturally occurring combination therapy that augments artemisinin delivery and synergizes the drug’s activity.”
Dr Rich did note that the exact mechanisms of whole-plant therapy’s effectiveness still need to be identified. But he also said the antimalarial activity of whole-plant therapy against artemisinin-resistant parasites provides “compelling reasons to further explore the role of non-pharmaceutical forms of artemisinin to treat human malaria.” ![]()
Drug gets orphan designation for WM
The US Food and Drug Administration (FDA) has granted orphan drug designation for IMO-8400, an antagonist of the endosomal Toll-like receptors (TLRs) 7, 8 and 9, for the treatment of Waldenström’s macroglobulinemia (WM).
The designation provides the drug’s maker, Idera Pharmaceuticals, with certain incentives, including eligibility for federal grants, research and development tax credits, and 7 years of marketing exclusivity if the product is approved.
Preclinical studies have shown that, in WM and other B‐cell lymphomas characterized by the MYD88 L265P oncogenic mutation, TLR signaling is overactivated. And this enables tumor cell survival and proliferation.
About 90% of WM patients are reported to harbor the MYD88 L265P mutation.
In research presented at the 2014 AACR Annual Meeting, investigators showed that IMO-8400 decreased the viability of mutated WM cells and diffuse large B-cell lymphoma (DLBCL) cells in vitro. The drug also decreased tumor growth and prolonged survival in mice with MYD88 L265P-positive DLBCL.
Now, Idera is conducting a phase 1/2 trial (NCT02092909) of IMO-8400 in patients with WM who have a history of relapse or failure to respond to one or more prior therapies. The protocol includes 3 dose-escalation cohorts of IMO-8400 administered subcutaneously.
The trial’s independent data review committee has completed its review of 4-week safety data from the second dose cohort (1.2 mg/kg/week) and has determined that Idera may open enrollment in the third dose cohort (2.4 mg/kg/week).
Final 24-week safety and clinical activity data are anticipated in the second half of 2015.
Aside from WM, Idera is pursuing clinical development of IMO-8400 in DLBCL patients harboring the MYD88 L265P mutation and in rare autoimmune diseases, including dermatomyositis. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for IMO-8400, an antagonist of the endosomal Toll-like receptors (TLRs) 7, 8 and 9, for the treatment of Waldenström’s macroglobulinemia (WM).
The designation provides the drug’s maker, Idera Pharmaceuticals, with certain incentives, including eligibility for federal grants, research and development tax credits, and 7 years of marketing exclusivity if the product is approved.
Preclinical studies have shown that, in WM and other B‐cell lymphomas characterized by the MYD88 L265P oncogenic mutation, TLR signaling is overactivated. And this enables tumor cell survival and proliferation.
About 90% of WM patients are reported to harbor the MYD88 L265P mutation.
In research presented at the 2014 AACR Annual Meeting, investigators showed that IMO-8400 decreased the viability of mutated WM cells and diffuse large B-cell lymphoma (DLBCL) cells in vitro. The drug also decreased tumor growth and prolonged survival in mice with MYD88 L265P-positive DLBCL.
Now, Idera is conducting a phase 1/2 trial (NCT02092909) of IMO-8400 in patients with WM who have a history of relapse or failure to respond to one or more prior therapies. The protocol includes 3 dose-escalation cohorts of IMO-8400 administered subcutaneously.
The trial’s independent data review committee has completed its review of 4-week safety data from the second dose cohort (1.2 mg/kg/week) and has determined that Idera may open enrollment in the third dose cohort (2.4 mg/kg/week).
Final 24-week safety and clinical activity data are anticipated in the second half of 2015.
Aside from WM, Idera is pursuing clinical development of IMO-8400 in DLBCL patients harboring the MYD88 L265P mutation and in rare autoimmune diseases, including dermatomyositis. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for IMO-8400, an antagonist of the endosomal Toll-like receptors (TLRs) 7, 8 and 9, for the treatment of Waldenström’s macroglobulinemia (WM).
The designation provides the drug’s maker, Idera Pharmaceuticals, with certain incentives, including eligibility for federal grants, research and development tax credits, and 7 years of marketing exclusivity if the product is approved.
Preclinical studies have shown that, in WM and other B‐cell lymphomas characterized by the MYD88 L265P oncogenic mutation, TLR signaling is overactivated. And this enables tumor cell survival and proliferation.
About 90% of WM patients are reported to harbor the MYD88 L265P mutation.
In research presented at the 2014 AACR Annual Meeting, investigators showed that IMO-8400 decreased the viability of mutated WM cells and diffuse large B-cell lymphoma (DLBCL) cells in vitro. The drug also decreased tumor growth and prolonged survival in mice with MYD88 L265P-positive DLBCL.
Now, Idera is conducting a phase 1/2 trial (NCT02092909) of IMO-8400 in patients with WM who have a history of relapse or failure to respond to one or more prior therapies. The protocol includes 3 dose-escalation cohorts of IMO-8400 administered subcutaneously.
The trial’s independent data review committee has completed its review of 4-week safety data from the second dose cohort (1.2 mg/kg/week) and has determined that Idera may open enrollment in the third dose cohort (2.4 mg/kg/week).
Final 24-week safety and clinical activity data are anticipated in the second half of 2015.
Aside from WM, Idera is pursuing clinical development of IMO-8400 in DLBCL patients harboring the MYD88 L265P mutation and in rare autoimmune diseases, including dermatomyositis. ![]()
Certain cancers primarily result from ‘bad luck’
in the bone marrow
Scientists have created a statistical model that measures the proportion of cancer incidence, across many tissue types, caused mainly by random mutations that occur when stem cells divide.
By their measure, two-thirds of adult cancers—including certain leukemias—can be explained primarily by “bad luck,” when these random mutations occur in genes that can drive cancer growth.
The remaining third are due to environmental factors and inherited genes.
“All cancers are caused by a combination of bad luck, the environment, and heredity, and we’ve created a model that may help quantify how much of these three factors contribute to cancer development,” said Bert Vogelstein, MD, of the Johns Hopkins University School of Medicine.
Dr Vogelstein and Cristian Tomasetti, PhD, also of the Johns Hopkins University School of Medicine, detailed these findings in Science.
The pair came to their conclusions by searching the scientific literature for information on the cumulative number of stem cell divisions in 31 tissue types during an average individual’s lifetime.
The researchers knew that cancer arises when tissue-specific stem cells make random mistakes, or mutations. But the actual contribution of these random mistakes to cancer incidence, in comparison to the contribution of hereditary or environmental factors, was unclear.
To sort out the role of random mutations in cancer risk, the team charted the number of stem cell divisions in 31 tissues and compared these rates with the lifetime risks of cancer in the same tissues among Americans.
From this data scatterplot, Drs Tomasetti and Vogelstein determined the correlation between the total number of stem cell divisions and cancer risk to be 0.804. Mathematically, the closer this value is to 1, the more stem cell divisions and cancer risk are correlated.
“Our study shows, in general, that a change in the number of stem cell divisions in a tissue type is highly correlated with a change in the incidence of cancer in that same tissue,” Dr Vogelstein said.
One example is in colon tissue, which undergoes 4 times more stem cell divisions than small intestine tissue in humans. Likewise, colon cancer is much more prevalent than small intestinal cancer.
“You could argue that the colon is exposed to more environmental factors than the small intestine, which increases the potential rate of acquired mutations,” Dr Tomasetti said.
However, the scientists observed the opposite in mouse colons, which had a lower number of stem cell divisions than in their small intestines. In mice, cancer incidence is lower in the colon than in the small intestine. The researchers believe this supports the role of the total number of stem cell divisions in the development of cancer.
Using statistical theory, the pair calculated how much of the variation in cancer risk can be explained by the number of stem cell divisions, which is 0.804 squared, or, in percentage form, approximately 65%.
Finally, the scientists classified the types of cancers they studied into two groups. They calculated which cancer types had an incidence predicted by the number of stem cell divisions and which had higher incidence.
They found that 22 cancer types—including acute myeloid leukemia and chronic lymphocytic leukemia—could be largely explained by the “bad luck” factor of random DNA mutations during cell division.
The other 9 cancer types had incidences higher than predicted by “bad luck” and were presumably due to a combination of bad luck plus environmental or inherited factors.
“We found that the types of cancer that had higher risk than predicted by the number of stem cell divisions were precisely the ones you’d expect, including lung cancer, which is linked to smoking; skin cancer, linked to sun exposure; and forms of cancers associated with hereditary syndromes,” Dr Vogelstein said.
“This study shows that you can add to your risk of getting cancers by smoking or other poor lifestyle factors. However, many forms of cancer are due largely to the bad luck of acquiring a mutation in a cancer driver gene regardless of lifestyle and heredity factors. The best way to eradicate these cancers will be through early detection, when they are still curable by surgery.”
The researchers noted that some cancers, such as breast and prostate cancer, were not included in the report because the team was unable to find reliable stem cell division rates in the scientific literature.
They hope other scientists will help refine their statistical model by finding more precise stem cell division rates. ![]()
in the bone marrow
Scientists have created a statistical model that measures the proportion of cancer incidence, across many tissue types, caused mainly by random mutations that occur when stem cells divide.
By their measure, two-thirds of adult cancers—including certain leukemias—can be explained primarily by “bad luck,” when these random mutations occur in genes that can drive cancer growth.
The remaining third are due to environmental factors and inherited genes.
“All cancers are caused by a combination of bad luck, the environment, and heredity, and we’ve created a model that may help quantify how much of these three factors contribute to cancer development,” said Bert Vogelstein, MD, of the Johns Hopkins University School of Medicine.
Dr Vogelstein and Cristian Tomasetti, PhD, also of the Johns Hopkins University School of Medicine, detailed these findings in Science.
The pair came to their conclusions by searching the scientific literature for information on the cumulative number of stem cell divisions in 31 tissue types during an average individual’s lifetime.
The researchers knew that cancer arises when tissue-specific stem cells make random mistakes, or mutations. But the actual contribution of these random mistakes to cancer incidence, in comparison to the contribution of hereditary or environmental factors, was unclear.
To sort out the role of random mutations in cancer risk, the team charted the number of stem cell divisions in 31 tissues and compared these rates with the lifetime risks of cancer in the same tissues among Americans.
From this data scatterplot, Drs Tomasetti and Vogelstein determined the correlation between the total number of stem cell divisions and cancer risk to be 0.804. Mathematically, the closer this value is to 1, the more stem cell divisions and cancer risk are correlated.
“Our study shows, in general, that a change in the number of stem cell divisions in a tissue type is highly correlated with a change in the incidence of cancer in that same tissue,” Dr Vogelstein said.
One example is in colon tissue, which undergoes 4 times more stem cell divisions than small intestine tissue in humans. Likewise, colon cancer is much more prevalent than small intestinal cancer.
“You could argue that the colon is exposed to more environmental factors than the small intestine, which increases the potential rate of acquired mutations,” Dr Tomasetti said.
However, the scientists observed the opposite in mouse colons, which had a lower number of stem cell divisions than in their small intestines. In mice, cancer incidence is lower in the colon than in the small intestine. The researchers believe this supports the role of the total number of stem cell divisions in the development of cancer.
Using statistical theory, the pair calculated how much of the variation in cancer risk can be explained by the number of stem cell divisions, which is 0.804 squared, or, in percentage form, approximately 65%.
Finally, the scientists classified the types of cancers they studied into two groups. They calculated which cancer types had an incidence predicted by the number of stem cell divisions and which had higher incidence.
They found that 22 cancer types—including acute myeloid leukemia and chronic lymphocytic leukemia—could be largely explained by the “bad luck” factor of random DNA mutations during cell division.
The other 9 cancer types had incidences higher than predicted by “bad luck” and were presumably due to a combination of bad luck plus environmental or inherited factors.
“We found that the types of cancer that had higher risk than predicted by the number of stem cell divisions were precisely the ones you’d expect, including lung cancer, which is linked to smoking; skin cancer, linked to sun exposure; and forms of cancers associated with hereditary syndromes,” Dr Vogelstein said.
“This study shows that you can add to your risk of getting cancers by smoking or other poor lifestyle factors. However, many forms of cancer are due largely to the bad luck of acquiring a mutation in a cancer driver gene regardless of lifestyle and heredity factors. The best way to eradicate these cancers will be through early detection, when they are still curable by surgery.”
The researchers noted that some cancers, such as breast and prostate cancer, were not included in the report because the team was unable to find reliable stem cell division rates in the scientific literature.
They hope other scientists will help refine their statistical model by finding more precise stem cell division rates. ![]()
in the bone marrow
Scientists have created a statistical model that measures the proportion of cancer incidence, across many tissue types, caused mainly by random mutations that occur when stem cells divide.
By their measure, two-thirds of adult cancers—including certain leukemias—can be explained primarily by “bad luck,” when these random mutations occur in genes that can drive cancer growth.
The remaining third are due to environmental factors and inherited genes.
“All cancers are caused by a combination of bad luck, the environment, and heredity, and we’ve created a model that may help quantify how much of these three factors contribute to cancer development,” said Bert Vogelstein, MD, of the Johns Hopkins University School of Medicine.
Dr Vogelstein and Cristian Tomasetti, PhD, also of the Johns Hopkins University School of Medicine, detailed these findings in Science.
The pair came to their conclusions by searching the scientific literature for information on the cumulative number of stem cell divisions in 31 tissue types during an average individual’s lifetime.
The researchers knew that cancer arises when tissue-specific stem cells make random mistakes, or mutations. But the actual contribution of these random mistakes to cancer incidence, in comparison to the contribution of hereditary or environmental factors, was unclear.
To sort out the role of random mutations in cancer risk, the team charted the number of stem cell divisions in 31 tissues and compared these rates with the lifetime risks of cancer in the same tissues among Americans.
From this data scatterplot, Drs Tomasetti and Vogelstein determined the correlation between the total number of stem cell divisions and cancer risk to be 0.804. Mathematically, the closer this value is to 1, the more stem cell divisions and cancer risk are correlated.
“Our study shows, in general, that a change in the number of stem cell divisions in a tissue type is highly correlated with a change in the incidence of cancer in that same tissue,” Dr Vogelstein said.
One example is in colon tissue, which undergoes 4 times more stem cell divisions than small intestine tissue in humans. Likewise, colon cancer is much more prevalent than small intestinal cancer.
“You could argue that the colon is exposed to more environmental factors than the small intestine, which increases the potential rate of acquired mutations,” Dr Tomasetti said.
However, the scientists observed the opposite in mouse colons, which had a lower number of stem cell divisions than in their small intestines. In mice, cancer incidence is lower in the colon than in the small intestine. The researchers believe this supports the role of the total number of stem cell divisions in the development of cancer.
Using statistical theory, the pair calculated how much of the variation in cancer risk can be explained by the number of stem cell divisions, which is 0.804 squared, or, in percentage form, approximately 65%.
Finally, the scientists classified the types of cancers they studied into two groups. They calculated which cancer types had an incidence predicted by the number of stem cell divisions and which had higher incidence.
They found that 22 cancer types—including acute myeloid leukemia and chronic lymphocytic leukemia—could be largely explained by the “bad luck” factor of random DNA mutations during cell division.
The other 9 cancer types had incidences higher than predicted by “bad luck” and were presumably due to a combination of bad luck plus environmental or inherited factors.
“We found that the types of cancer that had higher risk than predicted by the number of stem cell divisions were precisely the ones you’d expect, including lung cancer, which is linked to smoking; skin cancer, linked to sun exposure; and forms of cancers associated with hereditary syndromes,” Dr Vogelstein said.
“This study shows that you can add to your risk of getting cancers by smoking or other poor lifestyle factors. However, many forms of cancer are due largely to the bad luck of acquiring a mutation in a cancer driver gene regardless of lifestyle and heredity factors. The best way to eradicate these cancers will be through early detection, when they are still curable by surgery.”
The researchers noted that some cancers, such as breast and prostate cancer, were not included in the report because the team was unable to find reliable stem cell division rates in the scientific literature.
They hope other scientists will help refine their statistical model by finding more precise stem cell division rates. ![]()
New data added to obinutuzumab label

Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved a supplemental biologics license application for obinutuzumab (Gazyva) in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia (CLL).
The approval adds to the drug’s label data from stage 2 of the CLL11 study, which showed that obinutuzumab plus chlorambucil offers significant clinical improvements when compared head-to-head with rituximab plus chlorambucil.
This includes progression-free survival (PFS), complete response (CR), and minimal residual disease (MRD) data from stage 2 of the study. In addition, overall survival data was added from stage 1, in which researchers compared obinutuzumab plus chlorambucil to chlorambucil alone.
The label now reflects that obinutuzumab plus chlorambucil improved PFS compared to rituximab plus chlorambucil. The median PFS was 26.7 months and 14.9 months, respectively (hazard ratio=0.42, P<0.0001).
Additionally, obinutuzumab plus chlorambucil nearly tripled the CR rate when compared to rituximab plus chlorambucil. The CR rates were 26.1% and 8.8%, respectively.
Of the patients who achieved a CR with or without complete recovery from abnormal blood cell counts, 19% (18/94) of patients in the obinutuzumab arm and 6% (2/34) in the rituximab arm were MRD negative in the bone marrow.
Forty-one percent (39/94) of patients in the obinutuzumab arm and 12% (4/34) in the rituximab arm were MRD-negative in the peripheral blood.
At nearly 2 years, the rate of death was 9% (22/238) for patients who received obinutuzumab plus chlorambucil and 20% (24/118) for those who received chlorambucil alone (hazard ratio=0.41). The median overall survival has not yet been reached.
About obinutuzumab
Obinutuzumab is an engineered monoclonal antibody designed to attach to CD20 on B cells. The drug attacks targeted cells both directly and together with the body’s immune system.
The prescribing information for obinutuzumab includes warnings that the drug can cause serious or life-threatening side effects. These include hepatitis B reactivation, progressive multifocal leukoencephalopathy, infusion reactions, tumor lysis syndrome, infections, and neutropenia.
The most common side effects of the drug are infusion reactions, neutropenia, thrombocytopenia, anemia, fever, cough, nausea, and diarrhea.
Obinutuzumab was FDA-approved for use in combination with chlorambucil to treat previously untreated CLL in November 2013. The drug (which is known as Gazyvaro in Europe) was approved by the European Commission for the same indication in July 2014.
Obinutuzumab was discovered by Roche Glycart AG, an independent research unit of Roche. In the US, the drug is part of a collaboration between Genentech and Biogen Idec.![]()
Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved a supplemental biologics license application for obinutuzumab (Gazyva) in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia (CLL).
The approval adds to the drug’s label data from stage 2 of the CLL11 study, which showed that obinutuzumab plus chlorambucil offers significant clinical improvements when compared head-to-head with rituximab plus chlorambucil.
This includes progression-free survival (PFS), complete response (CR), and minimal residual disease (MRD) data from stage 2 of the study. In addition, overall survival data was added from stage 1, in which researchers compared obinutuzumab plus chlorambucil to chlorambucil alone.
The label now reflects that obinutuzumab plus chlorambucil improved PFS compared to rituximab plus chlorambucil. The median PFS was 26.7 months and 14.9 months, respectively (hazard ratio=0.42, P<0.0001).
Additionally, obinutuzumab plus chlorambucil nearly tripled the CR rate when compared to rituximab plus chlorambucil. The CR rates were 26.1% and 8.8%, respectively.
Of the patients who achieved a CR with or without complete recovery from abnormal blood cell counts, 19% (18/94) of patients in the obinutuzumab arm and 6% (2/34) in the rituximab arm were MRD negative in the bone marrow.
Forty-one percent (39/94) of patients in the obinutuzumab arm and 12% (4/34) in the rituximab arm were MRD-negative in the peripheral blood.
At nearly 2 years, the rate of death was 9% (22/238) for patients who received obinutuzumab plus chlorambucil and 20% (24/118) for those who received chlorambucil alone (hazard ratio=0.41). The median overall survival has not yet been reached.
About obinutuzumab
Obinutuzumab is an engineered monoclonal antibody designed to attach to CD20 on B cells. The drug attacks targeted cells both directly and together with the body’s immune system.
The prescribing information for obinutuzumab includes warnings that the drug can cause serious or life-threatening side effects. These include hepatitis B reactivation, progressive multifocal leukoencephalopathy, infusion reactions, tumor lysis syndrome, infections, and neutropenia.
The most common side effects of the drug are infusion reactions, neutropenia, thrombocytopenia, anemia, fever, cough, nausea, and diarrhea.
Obinutuzumab was FDA-approved for use in combination with chlorambucil to treat previously untreated CLL in November 2013. The drug (which is known as Gazyvaro in Europe) was approved by the European Commission for the same indication in July 2014.
Obinutuzumab was discovered by Roche Glycart AG, an independent research unit of Roche. In the US, the drug is part of a collaboration between Genentech and Biogen Idec.![]()
Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved a supplemental biologics license application for obinutuzumab (Gazyva) in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia (CLL).
The approval adds to the drug’s label data from stage 2 of the CLL11 study, which showed that obinutuzumab plus chlorambucil offers significant clinical improvements when compared head-to-head with rituximab plus chlorambucil.
This includes progression-free survival (PFS), complete response (CR), and minimal residual disease (MRD) data from stage 2 of the study. In addition, overall survival data was added from stage 1, in which researchers compared obinutuzumab plus chlorambucil to chlorambucil alone.
The label now reflects that obinutuzumab plus chlorambucil improved PFS compared to rituximab plus chlorambucil. The median PFS was 26.7 months and 14.9 months, respectively (hazard ratio=0.42, P<0.0001).
Additionally, obinutuzumab plus chlorambucil nearly tripled the CR rate when compared to rituximab plus chlorambucil. The CR rates were 26.1% and 8.8%, respectively.
Of the patients who achieved a CR with or without complete recovery from abnormal blood cell counts, 19% (18/94) of patients in the obinutuzumab arm and 6% (2/34) in the rituximab arm were MRD negative in the bone marrow.
Forty-one percent (39/94) of patients in the obinutuzumab arm and 12% (4/34) in the rituximab arm were MRD-negative in the peripheral blood.
At nearly 2 years, the rate of death was 9% (22/238) for patients who received obinutuzumab plus chlorambucil and 20% (24/118) for those who received chlorambucil alone (hazard ratio=0.41). The median overall survival has not yet been reached.
About obinutuzumab
Obinutuzumab is an engineered monoclonal antibody designed to attach to CD20 on B cells. The drug attacks targeted cells both directly and together with the body’s immune system.
The prescribing information for obinutuzumab includes warnings that the drug can cause serious or life-threatening side effects. These include hepatitis B reactivation, progressive multifocal leukoencephalopathy, infusion reactions, tumor lysis syndrome, infections, and neutropenia.
The most common side effects of the drug are infusion reactions, neutropenia, thrombocytopenia, anemia, fever, cough, nausea, and diarrhea.
Obinutuzumab was FDA-approved for use in combination with chlorambucil to treat previously untreated CLL in November 2013. The drug (which is known as Gazyvaro in Europe) was approved by the European Commission for the same indication in July 2014.
Obinutuzumab was discovered by Roche Glycart AG, an independent research unit of Roche. In the US, the drug is part of a collaboration between Genentech and Biogen Idec.![]()
FDA releases warning about IV solutions

The US Food and Drug Administration (FDA) is alerting healthcare professionals not to use Wallcur, LLC, simulated intravenous (IV) products in human or animal patients, as the products are for training purposes only.
The FDA has become aware that some Wallcur training IV products have been distributed to healthcare facilities and administered to patients.
There have been reports of serious adverse events associated with some of these products, such as Practi IV Solution Bags.
Before administering IV solutions to patients, healthcare providers should carefully check the labels to ensure that products are not training products.
Wallcur’s training products, which may bear the words “for clinical simulation,” are not to be administered to patients.
If you suspect that any Wallcur training IV products may have been administered to a patient, whether or not the incident has resulted in an adverse event, please report the incident to the FDA’s MedWatch Adverse Event Reporting Program.
The FDA said it will continue to investigate and monitor this issue. The agency is also working with the Centers for Disease Control and Prevention to inform healthcare professionals and state health departments. ![]()
The US Food and Drug Administration (FDA) is alerting healthcare professionals not to use Wallcur, LLC, simulated intravenous (IV) products in human or animal patients, as the products are for training purposes only.
The FDA has become aware that some Wallcur training IV products have been distributed to healthcare facilities and administered to patients.
There have been reports of serious adverse events associated with some of these products, such as Practi IV Solution Bags.
Before administering IV solutions to patients, healthcare providers should carefully check the labels to ensure that products are not training products.
Wallcur’s training products, which may bear the words “for clinical simulation,” are not to be administered to patients.
If you suspect that any Wallcur training IV products may have been administered to a patient, whether or not the incident has resulted in an adverse event, please report the incident to the FDA’s MedWatch Adverse Event Reporting Program.
The FDA said it will continue to investigate and monitor this issue. The agency is also working with the Centers for Disease Control and Prevention to inform healthcare professionals and state health departments. ![]()
The US Food and Drug Administration (FDA) is alerting healthcare professionals not to use Wallcur, LLC, simulated intravenous (IV) products in human or animal patients, as the products are for training purposes only.
The FDA has become aware that some Wallcur training IV products have been distributed to healthcare facilities and administered to patients.
There have been reports of serious adverse events associated with some of these products, such as Practi IV Solution Bags.
Before administering IV solutions to patients, healthcare providers should carefully check the labels to ensure that products are not training products.
Wallcur’s training products, which may bear the words “for clinical simulation,” are not to be administered to patients.
If you suspect that any Wallcur training IV products may have been administered to a patient, whether or not the incident has resulted in an adverse event, please report the incident to the FDA’s MedWatch Adverse Event Reporting Program.
The FDA said it will continue to investigate and monitor this issue. The agency is also working with the Centers for Disease Control and Prevention to inform healthcare professionals and state health departments.
Teams map paths to drug resistance in cancers, MPNs

Researchers say they’ve discovered key events that prompt drug resistance in myeloproliferative neoplasms (MPNs) and certain solid tumor malignancies.
By mapping the steps that myelofibrosis, melanoma, and breast cancer cells use to become resistant to drugs, the investigators believe they have identified better targets for blocking those pathways to ensure current therapies are effective.
The groups reported these findings in two papers published in Science Signaling.
“In our studies, we developed a screening technology that allows us to quickly identify the routes cells can use to become resistant, and, using that information, we were able to show that these mechanisms seen in the laboratory are actually also occurring in patients’ tumors,” said Kris Wood, PhD, an assistant professor at Duke University in Durham, North Carolina, and senior author of both studies.
In the first study, Dr Wood and his colleagues conducted a broad survey of the known cell-signaling pathways that, when activated, have the potential to trigger drug resistance.
Using this screening technology, the researchers were able to corroborate the results of earlier drug-resistance studies, while also finding pathways that had not previously been described.
The team identified the MAPK and PI3K pathways, which are known to mediate drug resistance. But they also found that activation of the Notch1 pathway caused drug resistance, and inhibiting Notch1 signaling restored drug efficacy.
“Interestingly, the mechanisms are quite similar among all 3 of the cancer types,” Dr Wood noted. “In breast cancer and melanoma, our findings suggest the same Notch1 pathway as a potential driver of resistance to a wide array of targeted therapies—a role that has not been widely acknowledged previously.”
The investigators found that Notch signaling mediated drug resistance to an estrogen receptor-targeted therapy used in breast cancer and to a kinase-targeted therapy used in melanoma. And tumors from some patients with relapsed breast cancer or melanoma had increased markers of Notch1 signaling.
In the second study, the researchers used the aforementioned pathway-centric screen to track a pair of separate signaling pathways downstream of RAS, a protein frequently activated in MPNs.
The team found the pathway mediated by RAS promoted drug resistance in hematopoietic cell lines containing an activating mutation in JAK2. And RAS signaling led to the phosphorylation-mediated inactivation of the pro-apoptotic protein BAD, which enabled cell survival.
Combining JAK inhibitors with inhibitors of kinases downstream of RAS signaling prompted apoptosis in cell lines that were resistant to JAK inhibitors. This suggests such combinations may be effective for treating drug-resistant MPNs.
“Together, these findings improve our ability to stratify patients into groups more and less likely to respond to therapy and design drug combinations that work together to block or delay resistance,” Dr Wood concluded.
Researchers say they’ve discovered key events that prompt drug resistance in myeloproliferative neoplasms (MPNs) and certain solid tumor malignancies.
By mapping the steps that myelofibrosis, melanoma, and breast cancer cells use to become resistant to drugs, the investigators believe they have identified better targets for blocking those pathways to ensure current therapies are effective.
The groups reported these findings in two papers published in Science Signaling.
“In our studies, we developed a screening technology that allows us to quickly identify the routes cells can use to become resistant, and, using that information, we were able to show that these mechanisms seen in the laboratory are actually also occurring in patients’ tumors,” said Kris Wood, PhD, an assistant professor at Duke University in Durham, North Carolina, and senior author of both studies.
In the first study, Dr Wood and his colleagues conducted a broad survey of the known cell-signaling pathways that, when activated, have the potential to trigger drug resistance.
Using this screening technology, the researchers were able to corroborate the results of earlier drug-resistance studies, while also finding pathways that had not previously been described.
The team identified the MAPK and PI3K pathways, which are known to mediate drug resistance. But they also found that activation of the Notch1 pathway caused drug resistance, and inhibiting Notch1 signaling restored drug efficacy.
“Interestingly, the mechanisms are quite similar among all 3 of the cancer types,” Dr Wood noted. “In breast cancer and melanoma, our findings suggest the same Notch1 pathway as a potential driver of resistance to a wide array of targeted therapies—a role that has not been widely acknowledged previously.”
The investigators found that Notch signaling mediated drug resistance to an estrogen receptor-targeted therapy used in breast cancer and to a kinase-targeted therapy used in melanoma. And tumors from some patients with relapsed breast cancer or melanoma had increased markers of Notch1 signaling.
In the second study, the researchers used the aforementioned pathway-centric screen to track a pair of separate signaling pathways downstream of RAS, a protein frequently activated in MPNs.
The team found the pathway mediated by RAS promoted drug resistance in hematopoietic cell lines containing an activating mutation in JAK2. And RAS signaling led to the phosphorylation-mediated inactivation of the pro-apoptotic protein BAD, which enabled cell survival.
Combining JAK inhibitors with inhibitors of kinases downstream of RAS signaling prompted apoptosis in cell lines that were resistant to JAK inhibitors. This suggests such combinations may be effective for treating drug-resistant MPNs.
“Together, these findings improve our ability to stratify patients into groups more and less likely to respond to therapy and design drug combinations that work together to block or delay resistance,” Dr Wood concluded.
Researchers say they’ve discovered key events that prompt drug resistance in myeloproliferative neoplasms (MPNs) and certain solid tumor malignancies.
By mapping the steps that myelofibrosis, melanoma, and breast cancer cells use to become resistant to drugs, the investigators believe they have identified better targets for blocking those pathways to ensure current therapies are effective.
The groups reported these findings in two papers published in Science Signaling.
“In our studies, we developed a screening technology that allows us to quickly identify the routes cells can use to become resistant, and, using that information, we were able to show that these mechanisms seen in the laboratory are actually also occurring in patients’ tumors,” said Kris Wood, PhD, an assistant professor at Duke University in Durham, North Carolina, and senior author of both studies.
In the first study, Dr Wood and his colleagues conducted a broad survey of the known cell-signaling pathways that, when activated, have the potential to trigger drug resistance.
Using this screening technology, the researchers were able to corroborate the results of earlier drug-resistance studies, while also finding pathways that had not previously been described.
The team identified the MAPK and PI3K pathways, which are known to mediate drug resistance. But they also found that activation of the Notch1 pathway caused drug resistance, and inhibiting Notch1 signaling restored drug efficacy.
“Interestingly, the mechanisms are quite similar among all 3 of the cancer types,” Dr Wood noted. “In breast cancer and melanoma, our findings suggest the same Notch1 pathway as a potential driver of resistance to a wide array of targeted therapies—a role that has not been widely acknowledged previously.”
The investigators found that Notch signaling mediated drug resistance to an estrogen receptor-targeted therapy used in breast cancer and to a kinase-targeted therapy used in melanoma. And tumors from some patients with relapsed breast cancer or melanoma had increased markers of Notch1 signaling.
In the second study, the researchers used the aforementioned pathway-centric screen to track a pair of separate signaling pathways downstream of RAS, a protein frequently activated in MPNs.
The team found the pathway mediated by RAS promoted drug resistance in hematopoietic cell lines containing an activating mutation in JAK2. And RAS signaling led to the phosphorylation-mediated inactivation of the pro-apoptotic protein BAD, which enabled cell survival.
Combining JAK inhibitors with inhibitors of kinases downstream of RAS signaling prompted apoptosis in cell lines that were resistant to JAK inhibitors. This suggests such combinations may be effective for treating drug-resistant MPNs.
“Together, these findings improve our ability to stratify patients into groups more and less likely to respond to therapy and design drug combinations that work together to block or delay resistance,” Dr Wood concluded.
Test predicts response to GVHD treatment
A newly developed algorithm could enable risk-adapted therapy for graft-vs-host disease (GVHD), researchers have reported in Lancet Haematology.
The problem with treating GVHD, the team said, is that the severity of symptoms at disease onset does not accurately define risk.
Patients with low-risk GVHD are often overtreated and experience significant side effects, while patients with high-risk GVHD can be undertreated and see their GVHD progress.
“Our goal is to provide the right treatment for each patient,” said study author James L. M. Ferrara, MD, DSc, of the Icahn School of Medicine at Mount Sinai.
“We hope to identify those patients at higher risk and design an aggressive intervention while tailoring a less-aggressive approach for those with low-risk.”
With that goal in mind, Dr Ferrara and his colleagues developed and tested a scoring system for GVHD. They collected plasma from 492 patients newly diagnosed with varying grades of GVHD and randomly assigned them to training (n=328) and test (n=164) sets.
The team used the concentrations of 3 validated biomarkers—TNFR1, ST2, and Reg3α—to create an algorithm that calculated the probability of non-relapse mortality (NRM) 6 months after GVHD onset for patients in the training set.
The researchers ranked the probabilities and identified thresholds that created 3 NRM scores. They then tested the algorithm in the test set of patients and a validation cohort of 300 additional patients who were enrolled on trials of GVHD treatment.
Results showed the algorithm works. The cumulative incidence of 6-month NRM significantly increased as the Ann Arbor GVHD score increased, and the response to primary GVHD treatment within 28 days decreased as the GVHD score increased.
In the validation set, the incidence of NRM was 8% for score 1, 27% for score 2, and 46% for score 3 (P<0.0001). Treatment response measured 86% for score 1, 67% for score 2, and 46% for score 3 (P<0.0001).
“This new scoring system will help identify patient who may not respond to standard treatments and may require an experimental and more aggressive approach,” Dr Ferrara said. “And it will also help guide treatment for patients with lower-risk GVHD who may be overtreated. This will allow us to personalize treatment at the onset of the disease. Future algorithms will prove increasingly useful to develop precision medicine for all [stem cell transplant] patients.”
To capitalize on this discovery, Dr Ferrara created the Mount Sinai Acute GVHD International Consortium (MAGIC), which consists of a group of 10 stem cell transplant centers in the US and Europe that will collaborate to use this new scoring system to test new treatments for acute GVHD.
Dr Ferrara and his colleagues have also written a protocol to treat high-risk GVHD that has been approved by the US Food and Drug Administration.
A newly developed algorithm could enable risk-adapted therapy for graft-vs-host disease (GVHD), researchers have reported in Lancet Haematology.
The problem with treating GVHD, the team said, is that the severity of symptoms at disease onset does not accurately define risk.
Patients with low-risk GVHD are often overtreated and experience significant side effects, while patients with high-risk GVHD can be undertreated and see their GVHD progress.
“Our goal is to provide the right treatment for each patient,” said study author James L. M. Ferrara, MD, DSc, of the Icahn School of Medicine at Mount Sinai.
“We hope to identify those patients at higher risk and design an aggressive intervention while tailoring a less-aggressive approach for those with low-risk.”
With that goal in mind, Dr Ferrara and his colleagues developed and tested a scoring system for GVHD. They collected plasma from 492 patients newly diagnosed with varying grades of GVHD and randomly assigned them to training (n=328) and test (n=164) sets.
The team used the concentrations of 3 validated biomarkers—TNFR1, ST2, and Reg3α—to create an algorithm that calculated the probability of non-relapse mortality (NRM) 6 months after GVHD onset for patients in the training set.
The researchers ranked the probabilities and identified thresholds that created 3 NRM scores. They then tested the algorithm in the test set of patients and a validation cohort of 300 additional patients who were enrolled on trials of GVHD treatment.
Results showed the algorithm works. The cumulative incidence of 6-month NRM significantly increased as the Ann Arbor GVHD score increased, and the response to primary GVHD treatment within 28 days decreased as the GVHD score increased.
In the validation set, the incidence of NRM was 8% for score 1, 27% for score 2, and 46% for score 3 (P<0.0001). Treatment response measured 86% for score 1, 67% for score 2, and 46% for score 3 (P<0.0001).
“This new scoring system will help identify patient who may not respond to standard treatments and may require an experimental and more aggressive approach,” Dr Ferrara said. “And it will also help guide treatment for patients with lower-risk GVHD who may be overtreated. This will allow us to personalize treatment at the onset of the disease. Future algorithms will prove increasingly useful to develop precision medicine for all [stem cell transplant] patients.”
To capitalize on this discovery, Dr Ferrara created the Mount Sinai Acute GVHD International Consortium (MAGIC), which consists of a group of 10 stem cell transplant centers in the US and Europe that will collaborate to use this new scoring system to test new treatments for acute GVHD.
Dr Ferrara and his colleagues have also written a protocol to treat high-risk GVHD that has been approved by the US Food and Drug Administration.
A newly developed algorithm could enable risk-adapted therapy for graft-vs-host disease (GVHD), researchers have reported in Lancet Haematology.
The problem with treating GVHD, the team said, is that the severity of symptoms at disease onset does not accurately define risk.
Patients with low-risk GVHD are often overtreated and experience significant side effects, while patients with high-risk GVHD can be undertreated and see their GVHD progress.
“Our goal is to provide the right treatment for each patient,” said study author James L. M. Ferrara, MD, DSc, of the Icahn School of Medicine at Mount Sinai.
“We hope to identify those patients at higher risk and design an aggressive intervention while tailoring a less-aggressive approach for those with low-risk.”
With that goal in mind, Dr Ferrara and his colleagues developed and tested a scoring system for GVHD. They collected plasma from 492 patients newly diagnosed with varying grades of GVHD and randomly assigned them to training (n=328) and test (n=164) sets.
The team used the concentrations of 3 validated biomarkers—TNFR1, ST2, and Reg3α—to create an algorithm that calculated the probability of non-relapse mortality (NRM) 6 months after GVHD onset for patients in the training set.
The researchers ranked the probabilities and identified thresholds that created 3 NRM scores. They then tested the algorithm in the test set of patients and a validation cohort of 300 additional patients who were enrolled on trials of GVHD treatment.
Results showed the algorithm works. The cumulative incidence of 6-month NRM significantly increased as the Ann Arbor GVHD score increased, and the response to primary GVHD treatment within 28 days decreased as the GVHD score increased.
In the validation set, the incidence of NRM was 8% for score 1, 27% for score 2, and 46% for score 3 (P<0.0001). Treatment response measured 86% for score 1, 67% for score 2, and 46% for score 3 (P<0.0001).
“This new scoring system will help identify patient who may not respond to standard treatments and may require an experimental and more aggressive approach,” Dr Ferrara said. “And it will also help guide treatment for patients with lower-risk GVHD who may be overtreated. This will allow us to personalize treatment at the onset of the disease. Future algorithms will prove increasingly useful to develop precision medicine for all [stem cell transplant] patients.”
To capitalize on this discovery, Dr Ferrara created the Mount Sinai Acute GVHD International Consortium (MAGIC), which consists of a group of 10 stem cell transplant centers in the US and Europe that will collaborate to use this new scoring system to test new treatments for acute GVHD.
Dr Ferrara and his colleagues have also written a protocol to treat high-risk GVHD that has been approved by the US Food and Drug Administration.
FDA extends approved use of neutropenia drug

The US Food and Drug Administration (FDA) has approved tbo-filgrastim (Granix) injection for administration by patients and caregivers, allowing physicians to prescribe the drug for in-office or at-home use.
Tbo-filgrastim is a leukocyte growth factor used to reduce the duration of severe neutropenia in patients with non-myeloid malignancies who are receiving myelosuppressive anticancer drugs associated with a clinically significant incidence of febrile neutropenia.
The drug has been commercially available in the US since November 2013, but, at present, it can only be administered by a healthcare professional.
Teva Pharmaceutical Industries, Ltd., the company developing tbo-filgrastim, plans to launch the new syringe for administration by patients and caregivers in early 2015.
Clinical trials
Researchers evaluated tbo-filgrastim in 3 phase 3 trials of patients receiving myelosuppressive chemotherapy for breast cancer, lung cancer, and non-Hodgkin lymphoma (NHL).
In the NHL study, investigators compared tbo-filgrastim to filgrastim for the prevention of chemotherapy-induced neutropenia in 92 patients.
For cycle 1, patients were randomized to daily injections (subcutaneous 5 µg/kg/day) of tbo-filgrastim (n=63) or filgrastim (n=29) for at least 5 days and a maximum of 14 days. In subsequent cycles, all patients received tbo-filgrastim.
In cycle 1, the mean duration of severe neutropenia was 0.5 days in the tbo-filgrastim arm and 0.9 days in the filgrastim arm (P=0.1055). The incidence of febrile neutropenia was 11.1% and 20.7%, respectively (P=0.1232).
In the lung cancer trial, researchers compared tbo-filgrastim to filgrastim in 240 patients receiving platinum-based chemotherapy. In cycle 1, patients were randomized to daily injections (subcutaneous 5 µg/kg/d) of tbo-filgrastim (n=160) or filgrastim (n=80) for at least 5 days and a maximum of 14 days. In subsequent cycles, all patients received tbo-filgrastim.
In cycle 1, the mean duration of severe neutropenia was 0.5 days in the tbo-filgrastim arm and 0.3 days in the filgrastim arm. There was no significant difference in the incidence of febrile neutropenia in cycle 1 between the two arms (P=0.2347).
In the breast cancer trial, investigators compared tbo-filgrastim to filgrastim or placebo in 348 patients receiving chemotherapy. Patients were randomized to daily injections (subcutaneous 5 µg/kg/day) for at least 5 days and a maximum of 14 days in each cycle of tbo-filgrastim (n=140), filgrastim (n=136), or placebo (n=72).
The mean duration of severe neutropenia in cycle 1 was 1.1 days in the tbo-filgrastim arm, 1.1 days in the filgrastim arm, and 3.9 days in the placebo arm.
In all the trials, bone pain was the most frequent treatment-emergent adverse event, occurring in at least 1% of patients treated with tbo-filgrastim at the recommended dose. The overall incidence of bone pain in cycle 1 was 3.4% for tbo-filgrastim, 1.4% for placebo, and 7.5% for filgrastim.
According to the drug’s prescribing information, tbo-filgrastim may pose a risk of splenic rupture, acute respiratory distress syndrome, serious allergic reactions, severe and sometimes fatal sickle cell crises, and capillary leak syndrome. The possibility that the drug acts as a growth factor for tumors cannot be excluded.
The US Food and Drug Administration (FDA) has approved tbo-filgrastim (Granix) injection for administration by patients and caregivers, allowing physicians to prescribe the drug for in-office or at-home use.
Tbo-filgrastim is a leukocyte growth factor used to reduce the duration of severe neutropenia in patients with non-myeloid malignancies who are receiving myelosuppressive anticancer drugs associated with a clinically significant incidence of febrile neutropenia.
The drug has been commercially available in the US since November 2013, but, at present, it can only be administered by a healthcare professional.
Teva Pharmaceutical Industries, Ltd., the company developing tbo-filgrastim, plans to launch the new syringe for administration by patients and caregivers in early 2015.
Clinical trials
Researchers evaluated tbo-filgrastim in 3 phase 3 trials of patients receiving myelosuppressive chemotherapy for breast cancer, lung cancer, and non-Hodgkin lymphoma (NHL).
In the NHL study, investigators compared tbo-filgrastim to filgrastim for the prevention of chemotherapy-induced neutropenia in 92 patients.
For cycle 1, patients were randomized to daily injections (subcutaneous 5 µg/kg/day) of tbo-filgrastim (n=63) or filgrastim (n=29) for at least 5 days and a maximum of 14 days. In subsequent cycles, all patients received tbo-filgrastim.
In cycle 1, the mean duration of severe neutropenia was 0.5 days in the tbo-filgrastim arm and 0.9 days in the filgrastim arm (P=0.1055). The incidence of febrile neutropenia was 11.1% and 20.7%, respectively (P=0.1232).
In the lung cancer trial, researchers compared tbo-filgrastim to filgrastim in 240 patients receiving platinum-based chemotherapy. In cycle 1, patients were randomized to daily injections (subcutaneous 5 µg/kg/d) of tbo-filgrastim (n=160) or filgrastim (n=80) for at least 5 days and a maximum of 14 days. In subsequent cycles, all patients received tbo-filgrastim.
In cycle 1, the mean duration of severe neutropenia was 0.5 days in the tbo-filgrastim arm and 0.3 days in the filgrastim arm. There was no significant difference in the incidence of febrile neutropenia in cycle 1 between the two arms (P=0.2347).
In the breast cancer trial, investigators compared tbo-filgrastim to filgrastim or placebo in 348 patients receiving chemotherapy. Patients were randomized to daily injections (subcutaneous 5 µg/kg/day) for at least 5 days and a maximum of 14 days in each cycle of tbo-filgrastim (n=140), filgrastim (n=136), or placebo (n=72).
The mean duration of severe neutropenia in cycle 1 was 1.1 days in the tbo-filgrastim arm, 1.1 days in the filgrastim arm, and 3.9 days in the placebo arm.
In all the trials, bone pain was the most frequent treatment-emergent adverse event, occurring in at least 1% of patients treated with tbo-filgrastim at the recommended dose. The overall incidence of bone pain in cycle 1 was 3.4% for tbo-filgrastim, 1.4% for placebo, and 7.5% for filgrastim.
According to the drug’s prescribing information, tbo-filgrastim may pose a risk of splenic rupture, acute respiratory distress syndrome, serious allergic reactions, severe and sometimes fatal sickle cell crises, and capillary leak syndrome. The possibility that the drug acts as a growth factor for tumors cannot be excluded.
The US Food and Drug Administration (FDA) has approved tbo-filgrastim (Granix) injection for administration by patients and caregivers, allowing physicians to prescribe the drug for in-office or at-home use.
Tbo-filgrastim is a leukocyte growth factor used to reduce the duration of severe neutropenia in patients with non-myeloid malignancies who are receiving myelosuppressive anticancer drugs associated with a clinically significant incidence of febrile neutropenia.
The drug has been commercially available in the US since November 2013, but, at present, it can only be administered by a healthcare professional.
Teva Pharmaceutical Industries, Ltd., the company developing tbo-filgrastim, plans to launch the new syringe for administration by patients and caregivers in early 2015.
Clinical trials
Researchers evaluated tbo-filgrastim in 3 phase 3 trials of patients receiving myelosuppressive chemotherapy for breast cancer, lung cancer, and non-Hodgkin lymphoma (NHL).
In the NHL study, investigators compared tbo-filgrastim to filgrastim for the prevention of chemotherapy-induced neutropenia in 92 patients.
For cycle 1, patients were randomized to daily injections (subcutaneous 5 µg/kg/day) of tbo-filgrastim (n=63) or filgrastim (n=29) for at least 5 days and a maximum of 14 days. In subsequent cycles, all patients received tbo-filgrastim.
In cycle 1, the mean duration of severe neutropenia was 0.5 days in the tbo-filgrastim arm and 0.9 days in the filgrastim arm (P=0.1055). The incidence of febrile neutropenia was 11.1% and 20.7%, respectively (P=0.1232).
In the lung cancer trial, researchers compared tbo-filgrastim to filgrastim in 240 patients receiving platinum-based chemotherapy. In cycle 1, patients were randomized to daily injections (subcutaneous 5 µg/kg/d) of tbo-filgrastim (n=160) or filgrastim (n=80) for at least 5 days and a maximum of 14 days. In subsequent cycles, all patients received tbo-filgrastim.
In cycle 1, the mean duration of severe neutropenia was 0.5 days in the tbo-filgrastim arm and 0.3 days in the filgrastim arm. There was no significant difference in the incidence of febrile neutropenia in cycle 1 between the two arms (P=0.2347).
In the breast cancer trial, investigators compared tbo-filgrastim to filgrastim or placebo in 348 patients receiving chemotherapy. Patients were randomized to daily injections (subcutaneous 5 µg/kg/day) for at least 5 days and a maximum of 14 days in each cycle of tbo-filgrastim (n=140), filgrastim (n=136), or placebo (n=72).
The mean duration of severe neutropenia in cycle 1 was 1.1 days in the tbo-filgrastim arm, 1.1 days in the filgrastim arm, and 3.9 days in the placebo arm.
In all the trials, bone pain was the most frequent treatment-emergent adverse event, occurring in at least 1% of patients treated with tbo-filgrastim at the recommended dose. The overall incidence of bone pain in cycle 1 was 3.4% for tbo-filgrastim, 1.4% for placebo, and 7.5% for filgrastim.
According to the drug’s prescribing information, tbo-filgrastim may pose a risk of splenic rupture, acute respiratory distress syndrome, serious allergic reactions, severe and sometimes fatal sickle cell crises, and capillary leak syndrome. The possibility that the drug acts as a growth factor for tumors cannot be excluded.