User login
Findings may aid development of antithrombotic drugs
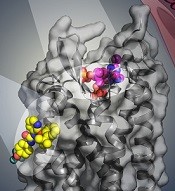
BPTU (yellow) bind to P2Y1R
Image courtesy of
Yekaterina Kadyshevskaya
Researchers say they have identified 2 disparate ligand-binding sites in the human P2Y1 receptor (P2Y1R), which plays a critical role in thrombosis.
Their research has provided a detailed molecular map of P2Y1R, a G protein-coupled receptor (GPCR), in complex with a nucleotide antagonist MRS2500 and a non-nucleotide antagonist BPTU.
The researchers believe their findings, published in Nature, could aid the development of new antithrombotic drugs.
Beili Wu, PhD, of the Shanghai Institute of Materia Medica in China, and her colleagues noted that the human purinergic receptors P2Y1R and P2Y12R play a major physiological role in adenosine 5′-diphosphate (ADP)-mediated platelet aggregation, an important component of thrombosis.
Although most of the available antithrombotic drugs act on P2Y12R, research has suggested that P2Y1R may be a promising antithrombotic drug target. In addition, P2Y1R inhibitors may offer a safety advantage over P2Y12R inhibitors by reducing the risk of bleeding. However, efforts to develop new drugs have been impeded by poor understanding of receptor-ligand interaction.
“The P2Y1R structures [we mapped] help us understand how this receptor and different types of experimental drugs interact at the molecular level and could enable further exploration to design new and safer antithrombotic drugs with reduced adverse effects,” Dr Wu said.
She and her colleagues found that the nucleotide ligand MRS2500 recognizes a binding site within the transmembrane bundle of P2Y1R. And it is different in shape and location from the nucleotide-binding site in P2Y12R.
“It is amazing to observe that 2 GPCRs recognize the same ligand in such different ways,” Dr Wu said. “The finding highlights the diversity of signal recognition mechanisms in GPCRs, and this is of great value to drug design for each receptor with high selectivity.”
The researchers also found that, instead of interacting within the transmembrane bundle, the non-nucleotide ligand BPTU binds to a pocket on the outer interface of P2Y1R embedded in the cell membrane.
This is the first structurally characterized, selective, and high-affinity GPCR ligand located entirely outside of the helical bundle, and it represents a new paradigm in ligand binding to alter signaling in GPCRs, according to the researchers.
The team believes this new understanding of the P2Y1R structure provides opportunities to broaden the scope of future GPCR drug discovery to target novel sites outside of the conventional GPCR ligand-binding pocket, which may improve drug selectivity and reduce side effects. ![]()
BPTU (yellow) bind to P2Y1R
Image courtesy of
Yekaterina Kadyshevskaya
Researchers say they have identified 2 disparate ligand-binding sites in the human P2Y1 receptor (P2Y1R), which plays a critical role in thrombosis.
Their research has provided a detailed molecular map of P2Y1R, a G protein-coupled receptor (GPCR), in complex with a nucleotide antagonist MRS2500 and a non-nucleotide antagonist BPTU.
The researchers believe their findings, published in Nature, could aid the development of new antithrombotic drugs.
Beili Wu, PhD, of the Shanghai Institute of Materia Medica in China, and her colleagues noted that the human purinergic receptors P2Y1R and P2Y12R play a major physiological role in adenosine 5′-diphosphate (ADP)-mediated platelet aggregation, an important component of thrombosis.
Although most of the available antithrombotic drugs act on P2Y12R, research has suggested that P2Y1R may be a promising antithrombotic drug target. In addition, P2Y1R inhibitors may offer a safety advantage over P2Y12R inhibitors by reducing the risk of bleeding. However, efforts to develop new drugs have been impeded by poor understanding of receptor-ligand interaction.
“The P2Y1R structures [we mapped] help us understand how this receptor and different types of experimental drugs interact at the molecular level and could enable further exploration to design new and safer antithrombotic drugs with reduced adverse effects,” Dr Wu said.
She and her colleagues found that the nucleotide ligand MRS2500 recognizes a binding site within the transmembrane bundle of P2Y1R. And it is different in shape and location from the nucleotide-binding site in P2Y12R.
“It is amazing to observe that 2 GPCRs recognize the same ligand in such different ways,” Dr Wu said. “The finding highlights the diversity of signal recognition mechanisms in GPCRs, and this is of great value to drug design for each receptor with high selectivity.”
The researchers also found that, instead of interacting within the transmembrane bundle, the non-nucleotide ligand BPTU binds to a pocket on the outer interface of P2Y1R embedded in the cell membrane.
This is the first structurally characterized, selective, and high-affinity GPCR ligand located entirely outside of the helical bundle, and it represents a new paradigm in ligand binding to alter signaling in GPCRs, according to the researchers.
The team believes this new understanding of the P2Y1R structure provides opportunities to broaden the scope of future GPCR drug discovery to target novel sites outside of the conventional GPCR ligand-binding pocket, which may improve drug selectivity and reduce side effects. ![]()
BPTU (yellow) bind to P2Y1R
Image courtesy of
Yekaterina Kadyshevskaya
Researchers say they have identified 2 disparate ligand-binding sites in the human P2Y1 receptor (P2Y1R), which plays a critical role in thrombosis.
Their research has provided a detailed molecular map of P2Y1R, a G protein-coupled receptor (GPCR), in complex with a nucleotide antagonist MRS2500 and a non-nucleotide antagonist BPTU.
The researchers believe their findings, published in Nature, could aid the development of new antithrombotic drugs.
Beili Wu, PhD, of the Shanghai Institute of Materia Medica in China, and her colleagues noted that the human purinergic receptors P2Y1R and P2Y12R play a major physiological role in adenosine 5′-diphosphate (ADP)-mediated platelet aggregation, an important component of thrombosis.
Although most of the available antithrombotic drugs act on P2Y12R, research has suggested that P2Y1R may be a promising antithrombotic drug target. In addition, P2Y1R inhibitors may offer a safety advantage over P2Y12R inhibitors by reducing the risk of bleeding. However, efforts to develop new drugs have been impeded by poor understanding of receptor-ligand interaction.
“The P2Y1R structures [we mapped] help us understand how this receptor and different types of experimental drugs interact at the molecular level and could enable further exploration to design new and safer antithrombotic drugs with reduced adverse effects,” Dr Wu said.
She and her colleagues found that the nucleotide ligand MRS2500 recognizes a binding site within the transmembrane bundle of P2Y1R. And it is different in shape and location from the nucleotide-binding site in P2Y12R.
“It is amazing to observe that 2 GPCRs recognize the same ligand in such different ways,” Dr Wu said. “The finding highlights the diversity of signal recognition mechanisms in GPCRs, and this is of great value to drug design for each receptor with high selectivity.”
The researchers also found that, instead of interacting within the transmembrane bundle, the non-nucleotide ligand BPTU binds to a pocket on the outer interface of P2Y1R embedded in the cell membrane.
This is the first structurally characterized, selective, and high-affinity GPCR ligand located entirely outside of the helical bundle, and it represents a new paradigm in ligand binding to alter signaling in GPCRs, according to the researchers.
The team believes this new understanding of the P2Y1R structure provides opportunities to broaden the scope of future GPCR drug discovery to target novel sites outside of the conventional GPCR ligand-binding pocket, which may improve drug selectivity and reduce side effects. ![]()
FDA strengthens warnings for anemia drug

Photo by Bill Branson
The US Food and Drug Administration (FDA) has strengthened an existing warning that serious, potentially fatal, allergic reactions can occur with the anemia drug Feraheme (ferumoxytol).
The FDA changed the drug’s prescribing information and approved a boxed warning detailing this risk.
The agency also added a new contraindication, which advises against the use of Feraheme in patients who have had an allergic reaction to any intravenous (IV) iron replacement product.
The FDA said it is continuing to monitor and evaluate the risk of serious allergic reactions with all IV iron products, and the agency will update the public as new information becomes available.
About Feraheme
Feraheme is an IV iron replacement product used to treat iron-deficiency anemia in patients with chronic kidney disease. Like other IV iron products, Feraheme may only be given where emergency personnel and equipment are immediately available to treat the potentially life-threatening allergic reactions that can occur with treatment.
All IV iron products carry a risk of potentially life-threatening allergic reactions. At the time of Feraheme’s approval in 2009, this risk was described in the “Warnings and Precautions” section of the drug label.
Since then, serious reactions, including deaths, have occurred, despite the proper use of therapies to treat these reactions and emergency resuscitation measures.
Serious reactions reported
In the initial clinical trials of Feraheme, conducted predominantly in patients with chronic kidney disease, serious hypersensitivity reactions were reported in 0.2% (3/1726) of patients receiving Feraheme.
Other adverse reactions potentially associated with hypersensitivity (eg, pruritus, rash, urticaria, or wheezing) were reported in 3.7% (63/1726) of these patients.
In other trials that did not include patients with chronic kidney disease, moderate to severe hypersensitivity reactions, including anaphylaxis, were reported in 2.6% (26/1014) of patients treated with Feraheme.
Since the approval of Feraheme on June 30, 2009, cases of serious hypersensitivity reactions, including death, have occurred.
A search of the FDA Adverse Event Reporting System database revealed 79 cases of anaphylactic reactions associated with Feraheme administration, reported from the time of approval to June 30, 2014. Of the 79 cases, 18 were fatal, despite immediate medical intervention and emergency resuscitation attempts.
The 79 patients ranged in age from 19 to 96 years. In nearly half of all cases, the anaphylactic reactions occurred with the first dose of Feraheme. For approximately 75% (60/79) of the cases, the reaction began during the infusion or within 5 minutes after administration was complete.
Frequently reported symptoms included cardiac arrest, hypotension, dyspnea, nausea, vomiting, and flushing. Of the 79 patients, 43% (34/79) had a medical history of drug allergy, and 24% had a history of multiple drug allergies.
Recommendations for administering Feraheme
Initial symptoms of fatal and serious hypersensitivity reactions associated with Feraheme may include hypotension, syncope, unresponsiveness, and cardiac/cardiorespiratory arrest, with or without signs of rash.
All IV iron products carry a risk of anaphylaxis, so these products should be administered only in patients who require IV iron therapy.
Feraheme is only approved for use in adults with iron-deficiency anemia in the setting of chronic kidney disease. The drug is contraindicated in patients with a history of hypersensitivity to Feraheme or any other IV iron product.
Only administer Feraheme and other IV iron products when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
Patients with a history of multiple drug allergies may have a greater risk of anaphylaxis with parenteral iron products. Carefully consider the potential risks and benefits before administering Feraheme to these patients.
Feraheme should only be administered as an IV infusion in 50-200 mL of 0.9% sodium chloride or 5% dextrose over a minimum period of 15 minutes following dilution. Do not administer Feraheme by undiluted IV injection.
Closely monitor patients for signs and symptoms of hypersensitivity reactions, including monitoring blood pressure and pulse during administration and for at least 30 minutes following each infusion of Feraheme.
Elderly patients 65 years of age and older with multiple or serious comorbidities who experience hypersensitivity reactions, hypotension, or both following administration of Feraheme may have more severe outcomes.
Advise patients to immediately report any signs and symptoms of hypersensitivity that may develop during and following Feraheme administration, such as respiratory distress, hypotension, dizziness or lightheadedness, edema, rash, or itching. Advise patients to seek immediate medical attention if these signs and symptoms occur.
Allow at least 30 minutes between administration of Feraheme and administration of other medications that could potentially cause serious hypersensitivity reactions, hypotension, or both, such as chemotherapeutic agents or monoclonal antibodies.
Report adverse events involving Feraheme to the FDA’s MedWatch Program. ![]()
Photo by Bill Branson
The US Food and Drug Administration (FDA) has strengthened an existing warning that serious, potentially fatal, allergic reactions can occur with the anemia drug Feraheme (ferumoxytol).
The FDA changed the drug’s prescribing information and approved a boxed warning detailing this risk.
The agency also added a new contraindication, which advises against the use of Feraheme in patients who have had an allergic reaction to any intravenous (IV) iron replacement product.
The FDA said it is continuing to monitor and evaluate the risk of serious allergic reactions with all IV iron products, and the agency will update the public as new information becomes available.
About Feraheme
Feraheme is an IV iron replacement product used to treat iron-deficiency anemia in patients with chronic kidney disease. Like other IV iron products, Feraheme may only be given where emergency personnel and equipment are immediately available to treat the potentially life-threatening allergic reactions that can occur with treatment.
All IV iron products carry a risk of potentially life-threatening allergic reactions. At the time of Feraheme’s approval in 2009, this risk was described in the “Warnings and Precautions” section of the drug label.
Since then, serious reactions, including deaths, have occurred, despite the proper use of therapies to treat these reactions and emergency resuscitation measures.
Serious reactions reported
In the initial clinical trials of Feraheme, conducted predominantly in patients with chronic kidney disease, serious hypersensitivity reactions were reported in 0.2% (3/1726) of patients receiving Feraheme.
Other adverse reactions potentially associated with hypersensitivity (eg, pruritus, rash, urticaria, or wheezing) were reported in 3.7% (63/1726) of these patients.
In other trials that did not include patients with chronic kidney disease, moderate to severe hypersensitivity reactions, including anaphylaxis, were reported in 2.6% (26/1014) of patients treated with Feraheme.
Since the approval of Feraheme on June 30, 2009, cases of serious hypersensitivity reactions, including death, have occurred.
A search of the FDA Adverse Event Reporting System database revealed 79 cases of anaphylactic reactions associated with Feraheme administration, reported from the time of approval to June 30, 2014. Of the 79 cases, 18 were fatal, despite immediate medical intervention and emergency resuscitation attempts.
The 79 patients ranged in age from 19 to 96 years. In nearly half of all cases, the anaphylactic reactions occurred with the first dose of Feraheme. For approximately 75% (60/79) of the cases, the reaction began during the infusion or within 5 minutes after administration was complete.
Frequently reported symptoms included cardiac arrest, hypotension, dyspnea, nausea, vomiting, and flushing. Of the 79 patients, 43% (34/79) had a medical history of drug allergy, and 24% had a history of multiple drug allergies.
Recommendations for administering Feraheme
Initial symptoms of fatal and serious hypersensitivity reactions associated with Feraheme may include hypotension, syncope, unresponsiveness, and cardiac/cardiorespiratory arrest, with or without signs of rash.
All IV iron products carry a risk of anaphylaxis, so these products should be administered only in patients who require IV iron therapy.
Feraheme is only approved for use in adults with iron-deficiency anemia in the setting of chronic kidney disease. The drug is contraindicated in patients with a history of hypersensitivity to Feraheme or any other IV iron product.
Only administer Feraheme and other IV iron products when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
Patients with a history of multiple drug allergies may have a greater risk of anaphylaxis with parenteral iron products. Carefully consider the potential risks and benefits before administering Feraheme to these patients.
Feraheme should only be administered as an IV infusion in 50-200 mL of 0.9% sodium chloride or 5% dextrose over a minimum period of 15 minutes following dilution. Do not administer Feraheme by undiluted IV injection.
Closely monitor patients for signs and symptoms of hypersensitivity reactions, including monitoring blood pressure and pulse during administration and for at least 30 minutes following each infusion of Feraheme.
Elderly patients 65 years of age and older with multiple or serious comorbidities who experience hypersensitivity reactions, hypotension, or both following administration of Feraheme may have more severe outcomes.
Advise patients to immediately report any signs and symptoms of hypersensitivity that may develop during and following Feraheme administration, such as respiratory distress, hypotension, dizziness or lightheadedness, edema, rash, or itching. Advise patients to seek immediate medical attention if these signs and symptoms occur.
Allow at least 30 minutes between administration of Feraheme and administration of other medications that could potentially cause serious hypersensitivity reactions, hypotension, or both, such as chemotherapeutic agents or monoclonal antibodies.
Report adverse events involving Feraheme to the FDA’s MedWatch Program. ![]()
Photo by Bill Branson
The US Food and Drug Administration (FDA) has strengthened an existing warning that serious, potentially fatal, allergic reactions can occur with the anemia drug Feraheme (ferumoxytol).
The FDA changed the drug’s prescribing information and approved a boxed warning detailing this risk.
The agency also added a new contraindication, which advises against the use of Feraheme in patients who have had an allergic reaction to any intravenous (IV) iron replacement product.
The FDA said it is continuing to monitor and evaluate the risk of serious allergic reactions with all IV iron products, and the agency will update the public as new information becomes available.
About Feraheme
Feraheme is an IV iron replacement product used to treat iron-deficiency anemia in patients with chronic kidney disease. Like other IV iron products, Feraheme may only be given where emergency personnel and equipment are immediately available to treat the potentially life-threatening allergic reactions that can occur with treatment.
All IV iron products carry a risk of potentially life-threatening allergic reactions. At the time of Feraheme’s approval in 2009, this risk was described in the “Warnings and Precautions” section of the drug label.
Since then, serious reactions, including deaths, have occurred, despite the proper use of therapies to treat these reactions and emergency resuscitation measures.
Serious reactions reported
In the initial clinical trials of Feraheme, conducted predominantly in patients with chronic kidney disease, serious hypersensitivity reactions were reported in 0.2% (3/1726) of patients receiving Feraheme.
Other adverse reactions potentially associated with hypersensitivity (eg, pruritus, rash, urticaria, or wheezing) were reported in 3.7% (63/1726) of these patients.
In other trials that did not include patients with chronic kidney disease, moderate to severe hypersensitivity reactions, including anaphylaxis, were reported in 2.6% (26/1014) of patients treated with Feraheme.
Since the approval of Feraheme on June 30, 2009, cases of serious hypersensitivity reactions, including death, have occurred.
A search of the FDA Adverse Event Reporting System database revealed 79 cases of anaphylactic reactions associated with Feraheme administration, reported from the time of approval to June 30, 2014. Of the 79 cases, 18 were fatal, despite immediate medical intervention and emergency resuscitation attempts.
The 79 patients ranged in age from 19 to 96 years. In nearly half of all cases, the anaphylactic reactions occurred with the first dose of Feraheme. For approximately 75% (60/79) of the cases, the reaction began during the infusion or within 5 minutes after administration was complete.
Frequently reported symptoms included cardiac arrest, hypotension, dyspnea, nausea, vomiting, and flushing. Of the 79 patients, 43% (34/79) had a medical history of drug allergy, and 24% had a history of multiple drug allergies.
Recommendations for administering Feraheme
Initial symptoms of fatal and serious hypersensitivity reactions associated with Feraheme may include hypotension, syncope, unresponsiveness, and cardiac/cardiorespiratory arrest, with or without signs of rash.
All IV iron products carry a risk of anaphylaxis, so these products should be administered only in patients who require IV iron therapy.
Feraheme is only approved for use in adults with iron-deficiency anemia in the setting of chronic kidney disease. The drug is contraindicated in patients with a history of hypersensitivity to Feraheme or any other IV iron product.
Only administer Feraheme and other IV iron products when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
Patients with a history of multiple drug allergies may have a greater risk of anaphylaxis with parenteral iron products. Carefully consider the potential risks and benefits before administering Feraheme to these patients.
Feraheme should only be administered as an IV infusion in 50-200 mL of 0.9% sodium chloride or 5% dextrose over a minimum period of 15 minutes following dilution. Do not administer Feraheme by undiluted IV injection.
Closely monitor patients for signs and symptoms of hypersensitivity reactions, including monitoring blood pressure and pulse during administration and for at least 30 minutes following each infusion of Feraheme.
Elderly patients 65 years of age and older with multiple or serious comorbidities who experience hypersensitivity reactions, hypotension, or both following administration of Feraheme may have more severe outcomes.
Advise patients to immediately report any signs and symptoms of hypersensitivity that may develop during and following Feraheme administration, such as respiratory distress, hypotension, dizziness or lightheadedness, edema, rash, or itching. Advise patients to seek immediate medical attention if these signs and symptoms occur.
Allow at least 30 minutes between administration of Feraheme and administration of other medications that could potentially cause serious hypersensitivity reactions, hypotension, or both, such as chemotherapeutic agents or monoclonal antibodies.
Report adverse events involving Feraheme to the FDA’s MedWatch Program. ![]()
Engineered protein overcomes radiation resistance in ALL

Photo courtesy of Dr Uckun
An engineered protein can enhance the effects of radiation and even overcome radiation resistance to treat B-precursor acute lymphoblastic leukemia (ALL), according to research published in EBioMedicine.
The protein, CD19L-sTRAIL, is a fusion of the CD19 ligand protein, which seeks out and binds to leukemia cells, with soluble TRAIL, a protein that can amplify the potency of radiation if it can be anchored on the membrane of leukemia cells.
Researchers found that CD19L-sTRAIL augmented the potency of radiation therapy even against the most aggressive and radiation-resistant forms of leukemia.
“Even very low doses of radiation were highly effective in mice challenged with aggressive human leukemia cells, when it was combined with [CD19L-sTRAIL],” said Fatih M. Uckun, MD, PhD, of The Saban Research Institute of Children’s Hospital Los Angeles in California.
“Due to its ability to selectively anchor to the surface of leukemia cells via its CD19L portion, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice.”
The researchers found that a combination of low-dose total body irradiation (TBI) and CD19L-sTRAIL greatly improved event-free survival (EFS) in mice that had received a typically fatal dose of cells from a patient with relapsed B-precursor ALL.
The median EFS for mice treated with CD19L-sTRAIL plus low-dose TBI was 72 days, which was significantly longer than the EFS for untreated control mice (17 days, P<0.0001), mice that received TBI alone (64 days, P=0.0014), mice that received CD19L-sTRAIL alone (20 days, P=0.0022), and mice that received a combination of vincristine, dexamethasone, and PEG-asparaginase (17 days, P=0.0033).
Dr Uckun and his colleagues noted that none of the mice that received CD19L-sTRAIL and TBI experienced a toxic death or signs of treatment-related toxicity.
The team also found that TBI plus CD19L-sTRAIL improved progression-free survival (PFS) in CD22ΔE12xBCR-ABL double transgenic mice with radiation-resistant, advanced stage, CD19+ murine B-precursor ALL with lymphomatous features.
The mean PFS was 24.0 ± 4.0 days for mice that received CD19L-sTRAIL plus TBI, which was significantly longer than the PFS for control mice (0 ± 0 days, P<0.0001), mice that received CD19L-sTRAIL alone (3.4 ± 0.9 days, P=0.0003), and mice that received TBI alone (9.0 ± 4.6 days, P=0.020).
Based on these results, Dr Uckun and his colleagues believe that incorporating CD19L-sTRAIL into the pre-transplant TBI regimens of patients with very high-risk B-precursor ALL could improve survival after hematopoietic stem cell transplant.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said. ![]()
Photo courtesy of Dr Uckun
An engineered protein can enhance the effects of radiation and even overcome radiation resistance to treat B-precursor acute lymphoblastic leukemia (ALL), according to research published in EBioMedicine.
The protein, CD19L-sTRAIL, is a fusion of the CD19 ligand protein, which seeks out and binds to leukemia cells, with soluble TRAIL, a protein that can amplify the potency of radiation if it can be anchored on the membrane of leukemia cells.
Researchers found that CD19L-sTRAIL augmented the potency of radiation therapy even against the most aggressive and radiation-resistant forms of leukemia.
“Even very low doses of radiation were highly effective in mice challenged with aggressive human leukemia cells, when it was combined with [CD19L-sTRAIL],” said Fatih M. Uckun, MD, PhD, of The Saban Research Institute of Children’s Hospital Los Angeles in California.
“Due to its ability to selectively anchor to the surface of leukemia cells via its CD19L portion, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice.”
The researchers found that a combination of low-dose total body irradiation (TBI) and CD19L-sTRAIL greatly improved event-free survival (EFS) in mice that had received a typically fatal dose of cells from a patient with relapsed B-precursor ALL.
The median EFS for mice treated with CD19L-sTRAIL plus low-dose TBI was 72 days, which was significantly longer than the EFS for untreated control mice (17 days, P<0.0001), mice that received TBI alone (64 days, P=0.0014), mice that received CD19L-sTRAIL alone (20 days, P=0.0022), and mice that received a combination of vincristine, dexamethasone, and PEG-asparaginase (17 days, P=0.0033).
Dr Uckun and his colleagues noted that none of the mice that received CD19L-sTRAIL and TBI experienced a toxic death or signs of treatment-related toxicity.
The team also found that TBI plus CD19L-sTRAIL improved progression-free survival (PFS) in CD22ΔE12xBCR-ABL double transgenic mice with radiation-resistant, advanced stage, CD19+ murine B-precursor ALL with lymphomatous features.
The mean PFS was 24.0 ± 4.0 days for mice that received CD19L-sTRAIL plus TBI, which was significantly longer than the PFS for control mice (0 ± 0 days, P<0.0001), mice that received CD19L-sTRAIL alone (3.4 ± 0.9 days, P=0.0003), and mice that received TBI alone (9.0 ± 4.6 days, P=0.020).
Based on these results, Dr Uckun and his colleagues believe that incorporating CD19L-sTRAIL into the pre-transplant TBI regimens of patients with very high-risk B-precursor ALL could improve survival after hematopoietic stem cell transplant.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said. ![]()
Photo courtesy of Dr Uckun
An engineered protein can enhance the effects of radiation and even overcome radiation resistance to treat B-precursor acute lymphoblastic leukemia (ALL), according to research published in EBioMedicine.
The protein, CD19L-sTRAIL, is a fusion of the CD19 ligand protein, which seeks out and binds to leukemia cells, with soluble TRAIL, a protein that can amplify the potency of radiation if it can be anchored on the membrane of leukemia cells.
Researchers found that CD19L-sTRAIL augmented the potency of radiation therapy even against the most aggressive and radiation-resistant forms of leukemia.
“Even very low doses of radiation were highly effective in mice challenged with aggressive human leukemia cells, when it was combined with [CD19L-sTRAIL],” said Fatih M. Uckun, MD, PhD, of The Saban Research Institute of Children’s Hospital Los Angeles in California.
“Due to its ability to selectively anchor to the surface of leukemia cells via its CD19L portion, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice.”
The researchers found that a combination of low-dose total body irradiation (TBI) and CD19L-sTRAIL greatly improved event-free survival (EFS) in mice that had received a typically fatal dose of cells from a patient with relapsed B-precursor ALL.
The median EFS for mice treated with CD19L-sTRAIL plus low-dose TBI was 72 days, which was significantly longer than the EFS for untreated control mice (17 days, P<0.0001), mice that received TBI alone (64 days, P=0.0014), mice that received CD19L-sTRAIL alone (20 days, P=0.0022), and mice that received a combination of vincristine, dexamethasone, and PEG-asparaginase (17 days, P=0.0033).
Dr Uckun and his colleagues noted that none of the mice that received CD19L-sTRAIL and TBI experienced a toxic death or signs of treatment-related toxicity.
The team also found that TBI plus CD19L-sTRAIL improved progression-free survival (PFS) in CD22ΔE12xBCR-ABL double transgenic mice with radiation-resistant, advanced stage, CD19+ murine B-precursor ALL with lymphomatous features.
The mean PFS was 24.0 ± 4.0 days for mice that received CD19L-sTRAIL plus TBI, which was significantly longer than the PFS for control mice (0 ± 0 days, P<0.0001), mice that received CD19L-sTRAIL alone (3.4 ± 0.9 days, P=0.0003), and mice that received TBI alone (9.0 ± 4.6 days, P=0.020).
Based on these results, Dr Uckun and his colleagues believe that incorporating CD19L-sTRAIL into the pre-transplant TBI regimens of patients with very high-risk B-precursor ALL could improve survival after hematopoietic stem cell transplant.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said. ![]()
Increasing cell signaling to eradicate Ph+ ALL

Photo by Aaron Logan
Increasing B-cell antigen receptor (BCR) signaling beyond “a point of no return” can lead to the selective elimination of leukemic cells, according to a group of researchers.
The team knew that proximal pre-BCR signaling is toxic to Philadelphia-chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) cells, and their experiments revealed that SYK tyrosine kinase activity mimicked constitutively active pre-BCR signaling.
So it was no surprise that pharmacologic hyperactivation of SYK prompted the removal of self-reactive B cells and selective killing of Ph+ ALL cells in vivo.
Markus Müschen, MD, PhD, of the University of California, San Francisco, and his colleagues described these findings in Nature.
When the researchers tested proximal pre-BCR signaling in mouse BCR-ABL1 cells, they found that an incremental increase of SYK activity could induce cell death.
Additional experiments showed that patient-derived Ph+ ALL cells have high levels of the inhibitory receptors PECAM1, CD300A, and LAIR1. And these receptors are needed to calibrate oncogenic signaling strength via recruitment of the inhibitory phosphatases PTPN6 and INPP5D.
So the researchers wondered if a small-molecule inhibitor of INPP5D, known as 3-a-aminocholestane (3AC), could induce SYK hyperactivation and target Ph+ ALL cells in mice.
They found that 3AC eliminated imatinib-resistant Ph+ ALL cells via rapid and massive cell death, and this significantly prolonged survival in the mice.
Dr Müschen and his colleagues noted that only Ph+ ALL cells were marked for destruction, which suggests a BCR-targeted drug could overcome imatinib resistance without affecting normal B cells.
The team also pointed out that a short exposure to 3AC was sufficient to commit the leukemia cells to death and to clear most of the disease burden from mice.
Dr Müschen said 3AC’s fast action was encouraging, because it remains unknown whether prolonged BCR hyperactivation is safe. That is why he and his colleagues are now focusing on formulating hyperactivating drugs that could be administered for only a few hours.
“These experiments show that we can engage signaling checkpoints in a very short period of time and that, once these checkpoints are engaged, the cell is irreversibly slated for death; it’s a point of no return,” Dr Müschen said.
“The next step is to work with medicinal chemists to make better ALL drugs that will overstimulate the B-cell receptor pathway and . . . could be used on a treatment schedule to elicit a very strong, but time-limited, spike in signaling to engage negative B-cell selection.” ![]()
Photo by Aaron Logan
Increasing B-cell antigen receptor (BCR) signaling beyond “a point of no return” can lead to the selective elimination of leukemic cells, according to a group of researchers.
The team knew that proximal pre-BCR signaling is toxic to Philadelphia-chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) cells, and their experiments revealed that SYK tyrosine kinase activity mimicked constitutively active pre-BCR signaling.
So it was no surprise that pharmacologic hyperactivation of SYK prompted the removal of self-reactive B cells and selective killing of Ph+ ALL cells in vivo.
Markus Müschen, MD, PhD, of the University of California, San Francisco, and his colleagues described these findings in Nature.
When the researchers tested proximal pre-BCR signaling in mouse BCR-ABL1 cells, they found that an incremental increase of SYK activity could induce cell death.
Additional experiments showed that patient-derived Ph+ ALL cells have high levels of the inhibitory receptors PECAM1, CD300A, and LAIR1. And these receptors are needed to calibrate oncogenic signaling strength via recruitment of the inhibitory phosphatases PTPN6 and INPP5D.
So the researchers wondered if a small-molecule inhibitor of INPP5D, known as 3-a-aminocholestane (3AC), could induce SYK hyperactivation and target Ph+ ALL cells in mice.
They found that 3AC eliminated imatinib-resistant Ph+ ALL cells via rapid and massive cell death, and this significantly prolonged survival in the mice.
Dr Müschen and his colleagues noted that only Ph+ ALL cells were marked for destruction, which suggests a BCR-targeted drug could overcome imatinib resistance without affecting normal B cells.
The team also pointed out that a short exposure to 3AC was sufficient to commit the leukemia cells to death and to clear most of the disease burden from mice.
Dr Müschen said 3AC’s fast action was encouraging, because it remains unknown whether prolonged BCR hyperactivation is safe. That is why he and his colleagues are now focusing on formulating hyperactivating drugs that could be administered for only a few hours.
“These experiments show that we can engage signaling checkpoints in a very short period of time and that, once these checkpoints are engaged, the cell is irreversibly slated for death; it’s a point of no return,” Dr Müschen said.
“The next step is to work with medicinal chemists to make better ALL drugs that will overstimulate the B-cell receptor pathway and . . . could be used on a treatment schedule to elicit a very strong, but time-limited, spike in signaling to engage negative B-cell selection.” ![]()
Photo by Aaron Logan
Increasing B-cell antigen receptor (BCR) signaling beyond “a point of no return” can lead to the selective elimination of leukemic cells, according to a group of researchers.
The team knew that proximal pre-BCR signaling is toxic to Philadelphia-chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) cells, and their experiments revealed that SYK tyrosine kinase activity mimicked constitutively active pre-BCR signaling.
So it was no surprise that pharmacologic hyperactivation of SYK prompted the removal of self-reactive B cells and selective killing of Ph+ ALL cells in vivo.
Markus Müschen, MD, PhD, of the University of California, San Francisco, and his colleagues described these findings in Nature.
When the researchers tested proximal pre-BCR signaling in mouse BCR-ABL1 cells, they found that an incremental increase of SYK activity could induce cell death.
Additional experiments showed that patient-derived Ph+ ALL cells have high levels of the inhibitory receptors PECAM1, CD300A, and LAIR1. And these receptors are needed to calibrate oncogenic signaling strength via recruitment of the inhibitory phosphatases PTPN6 and INPP5D.
So the researchers wondered if a small-molecule inhibitor of INPP5D, known as 3-a-aminocholestane (3AC), could induce SYK hyperactivation and target Ph+ ALL cells in mice.
They found that 3AC eliminated imatinib-resistant Ph+ ALL cells via rapid and massive cell death, and this significantly prolonged survival in the mice.
Dr Müschen and his colleagues noted that only Ph+ ALL cells were marked for destruction, which suggests a BCR-targeted drug could overcome imatinib resistance without affecting normal B cells.
The team also pointed out that a short exposure to 3AC was sufficient to commit the leukemia cells to death and to clear most of the disease burden from mice.
Dr Müschen said 3AC’s fast action was encouraging, because it remains unknown whether prolonged BCR hyperactivation is safe. That is why he and his colleagues are now focusing on formulating hyperactivating drugs that could be administered for only a few hours.
“These experiments show that we can engage signaling checkpoints in a very short period of time and that, once these checkpoints are engaged, the cell is irreversibly slated for death; it’s a point of no return,” Dr Müschen said.
“The next step is to work with medicinal chemists to make better ALL drugs that will overstimulate the B-cell receptor pathway and . . . could be used on a treatment schedule to elicit a very strong, but time-limited, spike in signaling to engage negative B-cell selection.” ![]()
Covert approach could treat TTP

Image by Andre E.X. Brown
A new approach for treating thrombotic thrombocytopenic purpura (TTP) has proven effective in mice, according to a paper published in Blood.
Researchers showed they could “hide” functional recombinant human ADAMTS13 (rADAMTS13) inside mouse platelets to prevent antibodies from impairing ADAMTS13 activity.
These rADAMTS13-expressing platelets were able to inhibit thrombosis and prevent the development of hereditary and acquired TTP in mice.
If a similar technique were to prove effective in humans, it could reduce the amount of plasma transfusions TTP patients require, the researchers said.
They also believe such a method could be used in emergency situations to treat strokes, heart attacks, malignant malaria, and pre-eclampsia, as all of these conditions are associated with relative deficiency of plasma ADAMTS13 activity.
This research began when X. Long Zheng, MD, PhD, of the University of Alabama at Birmingham, had the idea that he could treat TTP by hiding ADAMTS13 inside platelets where autoantibodies can’t see it.
He reasoned that, if he could fill platelets with ADAMTS13, the platelets would then carry the enzyme right to the place it was most needed to dissolve thrombi.
So he and his colleagues developed transgenic mice that expressed rADAMTS13 in their platelets.
The platelets had normal agglutination and aggregation. And they released rADAMTS13 upon stimulation with thrombin and collagen, although they released less when stimulated with 2MesADP.
Mice with rADAMTS13-expressing platelets were significantly protected in a vascular injury model of thrombus formation.
In mice that lacked plasma ADAMTS13 activity due to ADAMTS13 gene deletion and those that had antibody-mediated inhibition of plasma ADAMTS13 activity, rADAMTS13-expressing platelets protected the animals against TTP induced by bacterial toxin or recombinant von Willebrand factor.
These results, the researchers said, “suggest that platelets may be ideal carriers for antithrombic ADAMTS13, allowing its release at high concentrations at the site of thrombus formation without being inactivated by the potential circulating anti-ADAMTS13 inhibitors.”
To employ this treatment method in humans, Dr Zheng said researchers could investigate how to load ADAMTS13 inside donated platelets. They could then test these platelets to learn how many ADAMTS13-loaded platelets need to be transfused to produce antithrombotic and anti-TTP effects in patients with TTP. ![]()
Image by Andre E.X. Brown
A new approach for treating thrombotic thrombocytopenic purpura (TTP) has proven effective in mice, according to a paper published in Blood.
Researchers showed they could “hide” functional recombinant human ADAMTS13 (rADAMTS13) inside mouse platelets to prevent antibodies from impairing ADAMTS13 activity.
These rADAMTS13-expressing platelets were able to inhibit thrombosis and prevent the development of hereditary and acquired TTP in mice.
If a similar technique were to prove effective in humans, it could reduce the amount of plasma transfusions TTP patients require, the researchers said.
They also believe such a method could be used in emergency situations to treat strokes, heart attacks, malignant malaria, and pre-eclampsia, as all of these conditions are associated with relative deficiency of plasma ADAMTS13 activity.
This research began when X. Long Zheng, MD, PhD, of the University of Alabama at Birmingham, had the idea that he could treat TTP by hiding ADAMTS13 inside platelets where autoantibodies can’t see it.
He reasoned that, if he could fill platelets with ADAMTS13, the platelets would then carry the enzyme right to the place it was most needed to dissolve thrombi.
So he and his colleagues developed transgenic mice that expressed rADAMTS13 in their platelets.
The platelets had normal agglutination and aggregation. And they released rADAMTS13 upon stimulation with thrombin and collagen, although they released less when stimulated with 2MesADP.
Mice with rADAMTS13-expressing platelets were significantly protected in a vascular injury model of thrombus formation.
In mice that lacked plasma ADAMTS13 activity due to ADAMTS13 gene deletion and those that had antibody-mediated inhibition of plasma ADAMTS13 activity, rADAMTS13-expressing platelets protected the animals against TTP induced by bacterial toxin or recombinant von Willebrand factor.
These results, the researchers said, “suggest that platelets may be ideal carriers for antithrombic ADAMTS13, allowing its release at high concentrations at the site of thrombus formation without being inactivated by the potential circulating anti-ADAMTS13 inhibitors.”
To employ this treatment method in humans, Dr Zheng said researchers could investigate how to load ADAMTS13 inside donated platelets. They could then test these platelets to learn how many ADAMTS13-loaded platelets need to be transfused to produce antithrombotic and anti-TTP effects in patients with TTP. ![]()
Image by Andre E.X. Brown
A new approach for treating thrombotic thrombocytopenic purpura (TTP) has proven effective in mice, according to a paper published in Blood.
Researchers showed they could “hide” functional recombinant human ADAMTS13 (rADAMTS13) inside mouse platelets to prevent antibodies from impairing ADAMTS13 activity.
These rADAMTS13-expressing platelets were able to inhibit thrombosis and prevent the development of hereditary and acquired TTP in mice.
If a similar technique were to prove effective in humans, it could reduce the amount of plasma transfusions TTP patients require, the researchers said.
They also believe such a method could be used in emergency situations to treat strokes, heart attacks, malignant malaria, and pre-eclampsia, as all of these conditions are associated with relative deficiency of plasma ADAMTS13 activity.
This research began when X. Long Zheng, MD, PhD, of the University of Alabama at Birmingham, had the idea that he could treat TTP by hiding ADAMTS13 inside platelets where autoantibodies can’t see it.
He reasoned that, if he could fill platelets with ADAMTS13, the platelets would then carry the enzyme right to the place it was most needed to dissolve thrombi.
So he and his colleagues developed transgenic mice that expressed rADAMTS13 in their platelets.
The platelets had normal agglutination and aggregation. And they released rADAMTS13 upon stimulation with thrombin and collagen, although they released less when stimulated with 2MesADP.
Mice with rADAMTS13-expressing platelets were significantly protected in a vascular injury model of thrombus formation.
In mice that lacked plasma ADAMTS13 activity due to ADAMTS13 gene deletion and those that had antibody-mediated inhibition of plasma ADAMTS13 activity, rADAMTS13-expressing platelets protected the animals against TTP induced by bacterial toxin or recombinant von Willebrand factor.
These results, the researchers said, “suggest that platelets may be ideal carriers for antithrombic ADAMTS13, allowing its release at high concentrations at the site of thrombus formation without being inactivated by the potential circulating anti-ADAMTS13 inhibitors.”
To employ this treatment method in humans, Dr Zheng said researchers could investigate how to load ADAMTS13 inside donated platelets. They could then test these platelets to learn how many ADAMTS13-loaded platelets need to be transfused to produce antithrombotic and anti-TTP effects in patients with TTP. ![]()
FDA grants drug orphan status to treat PKD
Image courtesy of NHLBI
The US Food and Drug Administration (FDA) has granted orphan drug designation to AG-348 for the treatment of pyruvate kinase deficiency (PKD), a rare form of hemolytic anemia.
AG-348 is a small molecule allosteric activator of pyruvate kinase-R enzymes that directly targets the underlying metabolic defect in PKD.
The orphan designation will provide Agios Pharmaceuticals, the company developing AG-348, with certain benefits. These include market exclusivity upon regulatory approval and exemption from FDA application fees and tax credits for qualified clinical trials.
According to Agios, AG-348 exhibited favorable safety and pharmacokinetic profiles in a pair of phase 1 studies conducted in healthy volunteers.
Study investigators also observed dose-dependent changes in adenosine triphosphate (ATP) and 2,3-DPG blood levels, which are consistent with increased activity of the glycolytic pathway, the expected pharmacodynamic effect of AG-348.
Results from both studies were presented in a poster at the 2014 ASH Annual Meeting (abstract 4007*). One of the studies was a single ascending dose (SAD) study, and the other was a multiple ascending dose (MAD) study.
SAD study
In this study, healthy volunteers were randomized to receive AG-348 (n=36) or placebo (n=12). Patients were divided into 6 dosing cohorts: 30 mg, 120 mg, 360 mg, 700 mg, 1400 mg, and 2500 mg.
The maximum-tolerated dose of AG-348 was not reached, and there were no serious adverse events (AEs) or early withdrawals among AG-348-treated subjects. Overall, the rate of AEs was 33.3% in the placebo arm and 44.4% in the AG-348 arm.
The rate of AEs that were considered possibly treatment-related was 16.7% in the placebo arm and 30.6% in the AG-348 arm. The most common treatment-related AEs were headache (occurring in 16.7% and 11.1% of patients, respectively), nausea (0% and 13.9%, respectively), and vomiting (0% and 5.6%, respectively).
Exposure to AG-348, as measured by area under the concentration × time curve (AUC), increased in a dose-proportional manner after a single dose. And absorption was rapid (median Tmax ranged from 0.77 to 4.07 hours), although Tmax increased and there was a less-than-proportional increase in Cmax at higher doses.
When AG-348 was administered from 30 mg to 360 mg, there was a dose-dependent decrease in blood 2,3-DPG levels over 24 hours—up to a 49% mean decrease. Increasing the dose beyond 360 mg did not result in additional decreases in 2,3-DPG levels. And levels returned to placebo levels after about 72 hours.
There were minimal increases in blood ATP levels after AG-348 treatment at any dose.
MAD study
At the time of the presentation, 2 cohorts of 8 subjects each (6 receiving AG-348 and 2 receiving placebo) had completed treatment in the MAD study. One cohort received drug or placebo at 120 mg BID, and the other received 360 mg BID.
The pharmacokinetic results for day 1 of this study were consistent with those of the SAD study. However, the Cmax and AUC0-Ʈ were lower on day 14 than day 1. Investigators said this suggests that multiple doses of AG-348 increase the rate of its own metabolism.
They also said the decrease in exposure observed on day 14 is consistent with preclinical data that suggest AG-348 is a moderate inducer of CYP3A4, which is the major route of the oxidative metabolism of AG-348.
As in the SAD study, the investigators observed decreases in 2,3-DPG blood levels after the first dose in cohorts 1 and 2—up to a 48% mean decrease from baseline for both doses. Concentrations returned to placebo levels between 48 and 72 hours after the last dose.
Unlike in the SAD study, patients in this study had increases in ATP—up to a 52% mean increase from baseline in both dosing cohorts. ATP levels remained elevated through 72 hours after the last dose.
The investigators said the results of these 2 studies have informed dose selection for the planned phase 2 study of AG-348 in PKD patients, which is expected to begin soon. ![]()
*Information in the abstract differs from that presented at the meeting.
Image courtesy of NHLBI
The US Food and Drug Administration (FDA) has granted orphan drug designation to AG-348 for the treatment of pyruvate kinase deficiency (PKD), a rare form of hemolytic anemia.
AG-348 is a small molecule allosteric activator of pyruvate kinase-R enzymes that directly targets the underlying metabolic defect in PKD.
The orphan designation will provide Agios Pharmaceuticals, the company developing AG-348, with certain benefits. These include market exclusivity upon regulatory approval and exemption from FDA application fees and tax credits for qualified clinical trials.
According to Agios, AG-348 exhibited favorable safety and pharmacokinetic profiles in a pair of phase 1 studies conducted in healthy volunteers.
Study investigators also observed dose-dependent changes in adenosine triphosphate (ATP) and 2,3-DPG blood levels, which are consistent with increased activity of the glycolytic pathway, the expected pharmacodynamic effect of AG-348.
Results from both studies were presented in a poster at the 2014 ASH Annual Meeting (abstract 4007*). One of the studies was a single ascending dose (SAD) study, and the other was a multiple ascending dose (MAD) study.
SAD study
In this study, healthy volunteers were randomized to receive AG-348 (n=36) or placebo (n=12). Patients were divided into 6 dosing cohorts: 30 mg, 120 mg, 360 mg, 700 mg, 1400 mg, and 2500 mg.
The maximum-tolerated dose of AG-348 was not reached, and there were no serious adverse events (AEs) or early withdrawals among AG-348-treated subjects. Overall, the rate of AEs was 33.3% in the placebo arm and 44.4% in the AG-348 arm.
The rate of AEs that were considered possibly treatment-related was 16.7% in the placebo arm and 30.6% in the AG-348 arm. The most common treatment-related AEs were headache (occurring in 16.7% and 11.1% of patients, respectively), nausea (0% and 13.9%, respectively), and vomiting (0% and 5.6%, respectively).
Exposure to AG-348, as measured by area under the concentration × time curve (AUC), increased in a dose-proportional manner after a single dose. And absorption was rapid (median Tmax ranged from 0.77 to 4.07 hours), although Tmax increased and there was a less-than-proportional increase in Cmax at higher doses.
When AG-348 was administered from 30 mg to 360 mg, there was a dose-dependent decrease in blood 2,3-DPG levels over 24 hours—up to a 49% mean decrease. Increasing the dose beyond 360 mg did not result in additional decreases in 2,3-DPG levels. And levels returned to placebo levels after about 72 hours.
There were minimal increases in blood ATP levels after AG-348 treatment at any dose.
MAD study
At the time of the presentation, 2 cohorts of 8 subjects each (6 receiving AG-348 and 2 receiving placebo) had completed treatment in the MAD study. One cohort received drug or placebo at 120 mg BID, and the other received 360 mg BID.
The pharmacokinetic results for day 1 of this study were consistent with those of the SAD study. However, the Cmax and AUC0-Ʈ were lower on day 14 than day 1. Investigators said this suggests that multiple doses of AG-348 increase the rate of its own metabolism.
They also said the decrease in exposure observed on day 14 is consistent with preclinical data that suggest AG-348 is a moderate inducer of CYP3A4, which is the major route of the oxidative metabolism of AG-348.
As in the SAD study, the investigators observed decreases in 2,3-DPG blood levels after the first dose in cohorts 1 and 2—up to a 48% mean decrease from baseline for both doses. Concentrations returned to placebo levels between 48 and 72 hours after the last dose.
Unlike in the SAD study, patients in this study had increases in ATP—up to a 52% mean increase from baseline in both dosing cohorts. ATP levels remained elevated through 72 hours after the last dose.
The investigators said the results of these 2 studies have informed dose selection for the planned phase 2 study of AG-348 in PKD patients, which is expected to begin soon. ![]()
*Information in the abstract differs from that presented at the meeting.
Image courtesy of NHLBI
The US Food and Drug Administration (FDA) has granted orphan drug designation to AG-348 for the treatment of pyruvate kinase deficiency (PKD), a rare form of hemolytic anemia.
AG-348 is a small molecule allosteric activator of pyruvate kinase-R enzymes that directly targets the underlying metabolic defect in PKD.
The orphan designation will provide Agios Pharmaceuticals, the company developing AG-348, with certain benefits. These include market exclusivity upon regulatory approval and exemption from FDA application fees and tax credits for qualified clinical trials.
According to Agios, AG-348 exhibited favorable safety and pharmacokinetic profiles in a pair of phase 1 studies conducted in healthy volunteers.
Study investigators also observed dose-dependent changes in adenosine triphosphate (ATP) and 2,3-DPG blood levels, which are consistent with increased activity of the glycolytic pathway, the expected pharmacodynamic effect of AG-348.
Results from both studies were presented in a poster at the 2014 ASH Annual Meeting (abstract 4007*). One of the studies was a single ascending dose (SAD) study, and the other was a multiple ascending dose (MAD) study.
SAD study
In this study, healthy volunteers were randomized to receive AG-348 (n=36) or placebo (n=12). Patients were divided into 6 dosing cohorts: 30 mg, 120 mg, 360 mg, 700 mg, 1400 mg, and 2500 mg.
The maximum-tolerated dose of AG-348 was not reached, and there were no serious adverse events (AEs) or early withdrawals among AG-348-treated subjects. Overall, the rate of AEs was 33.3% in the placebo arm and 44.4% in the AG-348 arm.
The rate of AEs that were considered possibly treatment-related was 16.7% in the placebo arm and 30.6% in the AG-348 arm. The most common treatment-related AEs were headache (occurring in 16.7% and 11.1% of patients, respectively), nausea (0% and 13.9%, respectively), and vomiting (0% and 5.6%, respectively).
Exposure to AG-348, as measured by area under the concentration × time curve (AUC), increased in a dose-proportional manner after a single dose. And absorption was rapid (median Tmax ranged from 0.77 to 4.07 hours), although Tmax increased and there was a less-than-proportional increase in Cmax at higher doses.
When AG-348 was administered from 30 mg to 360 mg, there was a dose-dependent decrease in blood 2,3-DPG levels over 24 hours—up to a 49% mean decrease. Increasing the dose beyond 360 mg did not result in additional decreases in 2,3-DPG levels. And levels returned to placebo levels after about 72 hours.
There were minimal increases in blood ATP levels after AG-348 treatment at any dose.
MAD study
At the time of the presentation, 2 cohorts of 8 subjects each (6 receiving AG-348 and 2 receiving placebo) had completed treatment in the MAD study. One cohort received drug or placebo at 120 mg BID, and the other received 360 mg BID.
The pharmacokinetic results for day 1 of this study were consistent with those of the SAD study. However, the Cmax and AUC0-Ʈ were lower on day 14 than day 1. Investigators said this suggests that multiple doses of AG-348 increase the rate of its own metabolism.
They also said the decrease in exposure observed on day 14 is consistent with preclinical data that suggest AG-348 is a moderate inducer of CYP3A4, which is the major route of the oxidative metabolism of AG-348.
As in the SAD study, the investigators observed decreases in 2,3-DPG blood levels after the first dose in cohorts 1 and 2—up to a 48% mean decrease from baseline for both doses. Concentrations returned to placebo levels between 48 and 72 hours after the last dose.
Unlike in the SAD study, patients in this study had increases in ATP—up to a 52% mean increase from baseline in both dosing cohorts. ATP levels remained elevated through 72 hours after the last dose.
The investigators said the results of these 2 studies have informed dose selection for the planned phase 2 study of AG-348 in PKD patients, which is expected to begin soon. ![]()
*Information in the abstract differs from that presented at the meeting.
AML study shows transcription factors are ‘druggable’

In developing a compound that exhibits activity against a type of acute myeloid leukemia (AML), investigators have shown that transcription factors are, in fact, “druggable.”
The compound, AI-10-49, selectively binds to the mutated transcription factor CBFβ-SMMHC, and this prevents CBFβ-SMMHC from binding to another transcription factor, RUNX1.
In this way, AI-10-49 restores RUNX1 transcriptional activity, which leads to the death of inv(16) AML cells in vitro and in vivo.
“This drug that we’ve developed is . . . targeting a class of proteins that hasn’t been targeted for drug development very much in the past,” said study author John H. Bushweller, PhD, of the University of Virginia Health System in Charlottesville.
“It’s really a new paradigm, a new approach to try to treat these diseases. This class of proteins is very important for determining how much of many other proteins are made, so it’s a unique way of changing the way the cell behaves.”
Dr Bushweller and his colleagues described their work in Science.
The investigators noted that CBFβ-SMMHC is expressed in AML with the chromosome inversion inv(16)(p13q22). And CBFβ-SMMHC outcompetes wild-type CBFβ for binding to RUNX1, thereby deregulating RUNX1 activity in hematopoiesis and inducing AML.
“When you target a mutated protein in a cancer, you would ideally like to inhibit that mutated form of the protein but not affect the normal form of the protein that’s still there,” Dr Bushweller said. “In the case of [AI-10-49], we’ve achieved that.”
He and his colleagues tested AI-10-49 in 11 leukemia cells lines and found that only ME-1 cells were highly sensitive to the treatment. AI-10-49 effectively dissociated RUNX1 from CBFβ-SMMHC in ME-1 cells, with 90% dissociation after 6 hours of treatment.
But the drug had a modest effect on CBFβ-RUNX1 association. It also had negligible activity in normal human bone marrow cells.
The investigators then tested AI-10-49 in a mouse model of inv(16) AML. Control mice (vehicle-treated) succumbed to leukemia in a median of 33.5 days, compared to a median of 61 days for mice that received AI-10-49.
Dr Bushweller and his colleagues did not evaluate toxicity after long-term exposure to AI-10-49, but they found no evidence of toxicity after 7 days of AI-10-49 treatment.
Next, the investigators tested AI-10-49 in 4 primary inv(16) AML cell samples. They saw a reduction in leukemia cell viability when AI-10-49 was administered at 5 μM and 10 μM concentrations. Bivalent AI-10-49 was more potent than monovalent AI-10-47.
The team said these results suggest that direct inhibition of CBFβ-SMMHC may be an effective therapeutic approach for inv(16) AML, and the findings provide support for therapies targeting transcription factors. ![]()
In developing a compound that exhibits activity against a type of acute myeloid leukemia (AML), investigators have shown that transcription factors are, in fact, “druggable.”
The compound, AI-10-49, selectively binds to the mutated transcription factor CBFβ-SMMHC, and this prevents CBFβ-SMMHC from binding to another transcription factor, RUNX1.
In this way, AI-10-49 restores RUNX1 transcriptional activity, which leads to the death of inv(16) AML cells in vitro and in vivo.
“This drug that we’ve developed is . . . targeting a class of proteins that hasn’t been targeted for drug development very much in the past,” said study author John H. Bushweller, PhD, of the University of Virginia Health System in Charlottesville.
“It’s really a new paradigm, a new approach to try to treat these diseases. This class of proteins is very important for determining how much of many other proteins are made, so it’s a unique way of changing the way the cell behaves.”
Dr Bushweller and his colleagues described their work in Science.
The investigators noted that CBFβ-SMMHC is expressed in AML with the chromosome inversion inv(16)(p13q22). And CBFβ-SMMHC outcompetes wild-type CBFβ for binding to RUNX1, thereby deregulating RUNX1 activity in hematopoiesis and inducing AML.
“When you target a mutated protein in a cancer, you would ideally like to inhibit that mutated form of the protein but not affect the normal form of the protein that’s still there,” Dr Bushweller said. “In the case of [AI-10-49], we’ve achieved that.”
He and his colleagues tested AI-10-49 in 11 leukemia cells lines and found that only ME-1 cells were highly sensitive to the treatment. AI-10-49 effectively dissociated RUNX1 from CBFβ-SMMHC in ME-1 cells, with 90% dissociation after 6 hours of treatment.
But the drug had a modest effect on CBFβ-RUNX1 association. It also had negligible activity in normal human bone marrow cells.
The investigators then tested AI-10-49 in a mouse model of inv(16) AML. Control mice (vehicle-treated) succumbed to leukemia in a median of 33.5 days, compared to a median of 61 days for mice that received AI-10-49.
Dr Bushweller and his colleagues did not evaluate toxicity after long-term exposure to AI-10-49, but they found no evidence of toxicity after 7 days of AI-10-49 treatment.
Next, the investigators tested AI-10-49 in 4 primary inv(16) AML cell samples. They saw a reduction in leukemia cell viability when AI-10-49 was administered at 5 μM and 10 μM concentrations. Bivalent AI-10-49 was more potent than monovalent AI-10-47.
The team said these results suggest that direct inhibition of CBFβ-SMMHC may be an effective therapeutic approach for inv(16) AML, and the findings provide support for therapies targeting transcription factors. ![]()
In developing a compound that exhibits activity against a type of acute myeloid leukemia (AML), investigators have shown that transcription factors are, in fact, “druggable.”
The compound, AI-10-49, selectively binds to the mutated transcription factor CBFβ-SMMHC, and this prevents CBFβ-SMMHC from binding to another transcription factor, RUNX1.
In this way, AI-10-49 restores RUNX1 transcriptional activity, which leads to the death of inv(16) AML cells in vitro and in vivo.
“This drug that we’ve developed is . . . targeting a class of proteins that hasn’t been targeted for drug development very much in the past,” said study author John H. Bushweller, PhD, of the University of Virginia Health System in Charlottesville.
“It’s really a new paradigm, a new approach to try to treat these diseases. This class of proteins is very important for determining how much of many other proteins are made, so it’s a unique way of changing the way the cell behaves.”
Dr Bushweller and his colleagues described their work in Science.
The investigators noted that CBFβ-SMMHC is expressed in AML with the chromosome inversion inv(16)(p13q22). And CBFβ-SMMHC outcompetes wild-type CBFβ for binding to RUNX1, thereby deregulating RUNX1 activity in hematopoiesis and inducing AML.
“When you target a mutated protein in a cancer, you would ideally like to inhibit that mutated form of the protein but not affect the normal form of the protein that’s still there,” Dr Bushweller said. “In the case of [AI-10-49], we’ve achieved that.”
He and his colleagues tested AI-10-49 in 11 leukemia cells lines and found that only ME-1 cells were highly sensitive to the treatment. AI-10-49 effectively dissociated RUNX1 from CBFβ-SMMHC in ME-1 cells, with 90% dissociation after 6 hours of treatment.
But the drug had a modest effect on CBFβ-RUNX1 association. It also had negligible activity in normal human bone marrow cells.
The investigators then tested AI-10-49 in a mouse model of inv(16) AML. Control mice (vehicle-treated) succumbed to leukemia in a median of 33.5 days, compared to a median of 61 days for mice that received AI-10-49.
Dr Bushweller and his colleagues did not evaluate toxicity after long-term exposure to AI-10-49, but they found no evidence of toxicity after 7 days of AI-10-49 treatment.
Next, the investigators tested AI-10-49 in 4 primary inv(16) AML cell samples. They saw a reduction in leukemia cell viability when AI-10-49 was administered at 5 μM and 10 μM concentrations. Bivalent AI-10-49 was more potent than monovalent AI-10-47.
The team said these results suggest that direct inhibition of CBFβ-SMMHC may be an effective therapeutic approach for inv(16) AML, and the findings provide support for therapies targeting transcription factors.
Prevalence of SDB in SCD may be high
Image by Graham Beards
Results of a small study suggest there may be a high prevalence of sleep disordered breathing (SDB) in adults with sickle cell disease (SCD).
Of the 32 patients included in the study, 44% had a clinical diagnosis of SDB.
These patients had significantly increased REM latency, a significantly higher mean score on the Epworth Sleepiness Scale, and a significantly higher oxygen desaturation index (ODI) than patients who did not have SDB.
However, there was no significant difference between SDB and non-SDB patients with regard to insomnia, delayed sleep phase syndrome, nocturia, or SCD complications.
Sunil Sharma, MD, of Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia, Pennsylvania, and his colleagues recounted these results in the Journal of Clinical Sleep Medicine.
“Previous research identified pain and sleep disturbance as 2 common symptoms of adult sickle cell disorder,” Dr Sharma said. “We wanted to examine the reasons for the sleep disturbances, as it can have a strong impact on our patients’ quality of life and overall health. We discovered a high incidence of sleep disordered breathing in patients with sickle cell disease who also report trouble with sleep.”
Dr Sharma and his colleagues analyzed 32 consecutive adult SCD patients who had reported symptoms suggesting disordered sleep or had an Epworth Sleepiness Scale score of 10 or greater. The patients underwent a comprehensive sleep evaluation and overnight polysomnography in an accredited sleep center.
SDB was defined as having an apnea-hypopnea index (AHI, events/hour) of 5 or greater. SDB was considered mild if the AHI was 5 to < 15, moderate if the AHI was 15 to < 30, and severe if the AHI was ≥ 30. Once they determined which patients had SDB, the researchers compared these patients to those without the condition.
The team found that 44% of patients (n=14) had SDB. It was mild in 8 patients, moderate in 4, and severe in 2.
Compared to non-SDB patients, those with SDB had a significantly higher mean AHI (1.6 and 17, respectively; P=0.0001), a significantly increased REM latency (98 and 159 minutes, respectively; P=0.014), and a significantly higher mean score on the Epworth Sleepiness Scale (8.6 and 13, respectively; P=0.017).
Patients with SDB also had a significantly higher ODI than non-SDB patients (13 and 1.6, respectively; P=0.0009). Significant oxygen desaturation was defined as oxygen saturation < 89% for 5 cumulative minutes or more. The ODI was the number of recorded oxygen desaturations ≥ 4% per hour of sleep.
There was no significant difference between SDB and non-SDB patients in the incidence of nocturia (2.3 and 1.6, respectively; P=0.063), insomnia (57% and 72%, respectively; P=0.46), or delayed sleep phase syndrome (57% and 50%, respectively; P=0.73).
Delayed sleep phase syndrome was defined as a delay in sleep onset of 2 hours or greater from the desired sleep time and an inability to awaken at the desired time. Insomnia was defined as difficulty initiating sleep (sleep latency greater than 60 minutes) or difficulty maintaining sleep (more than 2 awakenings requiring more than 20 minutes to fall back asleep) on the majority of nights for more than 4 weeks.
There was no significant difference between SDB and non-SDB patients with regard to SCD complications, including crises during sleep (44% and 39%, respectively; P=0.67), average hospital admissions in the last 5 years (9.1 and 6.0, respectively; P=0.15), or average mini-crises per month (2.7 and 3.6, respectively; P=0.69).
Dr Sharma said the diagnosis of SDB could be missed in adults with SCD because they are not generally obese, a common risk factor for SDB, and daytime sleepiness is attributed to the pain medications used to treat the symptoms of SCD. He hopes this study will increase awareness among physicians who can screen patients for SDB.
“Our study suggests that patients with sickle cell disorder should be screened using a questionnaire to identify problems with sleep,” Dr Sharma said. “For further testing, an oxygen desaturation index is another low-cost screening tool that can identify sleep disordered breathing in this population.”
Image by Graham Beards
Results of a small study suggest there may be a high prevalence of sleep disordered breathing (SDB) in adults with sickle cell disease (SCD).
Of the 32 patients included in the study, 44% had a clinical diagnosis of SDB.
These patients had significantly increased REM latency, a significantly higher mean score on the Epworth Sleepiness Scale, and a significantly higher oxygen desaturation index (ODI) than patients who did not have SDB.
However, there was no significant difference between SDB and non-SDB patients with regard to insomnia, delayed sleep phase syndrome, nocturia, or SCD complications.
Sunil Sharma, MD, of Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia, Pennsylvania, and his colleagues recounted these results in the Journal of Clinical Sleep Medicine.
“Previous research identified pain and sleep disturbance as 2 common symptoms of adult sickle cell disorder,” Dr Sharma said. “We wanted to examine the reasons for the sleep disturbances, as it can have a strong impact on our patients’ quality of life and overall health. We discovered a high incidence of sleep disordered breathing in patients with sickle cell disease who also report trouble with sleep.”
Dr Sharma and his colleagues analyzed 32 consecutive adult SCD patients who had reported symptoms suggesting disordered sleep or had an Epworth Sleepiness Scale score of 10 or greater. The patients underwent a comprehensive sleep evaluation and overnight polysomnography in an accredited sleep center.
SDB was defined as having an apnea-hypopnea index (AHI, events/hour) of 5 or greater. SDB was considered mild if the AHI was 5 to < 15, moderate if the AHI was 15 to < 30, and severe if the AHI was ≥ 30. Once they determined which patients had SDB, the researchers compared these patients to those without the condition.
The team found that 44% of patients (n=14) had SDB. It was mild in 8 patients, moderate in 4, and severe in 2.
Compared to non-SDB patients, those with SDB had a significantly higher mean AHI (1.6 and 17, respectively; P=0.0001), a significantly increased REM latency (98 and 159 minutes, respectively; P=0.014), and a significantly higher mean score on the Epworth Sleepiness Scale (8.6 and 13, respectively; P=0.017).
Patients with SDB also had a significantly higher ODI than non-SDB patients (13 and 1.6, respectively; P=0.0009). Significant oxygen desaturation was defined as oxygen saturation < 89% for 5 cumulative minutes or more. The ODI was the number of recorded oxygen desaturations ≥ 4% per hour of sleep.
There was no significant difference between SDB and non-SDB patients in the incidence of nocturia (2.3 and 1.6, respectively; P=0.063), insomnia (57% and 72%, respectively; P=0.46), or delayed sleep phase syndrome (57% and 50%, respectively; P=0.73).
Delayed sleep phase syndrome was defined as a delay in sleep onset of 2 hours or greater from the desired sleep time and an inability to awaken at the desired time. Insomnia was defined as difficulty initiating sleep (sleep latency greater than 60 minutes) or difficulty maintaining sleep (more than 2 awakenings requiring more than 20 minutes to fall back asleep) on the majority of nights for more than 4 weeks.
There was no significant difference between SDB and non-SDB patients with regard to SCD complications, including crises during sleep (44% and 39%, respectively; P=0.67), average hospital admissions in the last 5 years (9.1 and 6.0, respectively; P=0.15), or average mini-crises per month (2.7 and 3.6, respectively; P=0.69).
Dr Sharma said the diagnosis of SDB could be missed in adults with SCD because they are not generally obese, a common risk factor for SDB, and daytime sleepiness is attributed to the pain medications used to treat the symptoms of SCD. He hopes this study will increase awareness among physicians who can screen patients for SDB.
“Our study suggests that patients with sickle cell disorder should be screened using a questionnaire to identify problems with sleep,” Dr Sharma said. “For further testing, an oxygen desaturation index is another low-cost screening tool that can identify sleep disordered breathing in this population.”
Image by Graham Beards
Results of a small study suggest there may be a high prevalence of sleep disordered breathing (SDB) in adults with sickle cell disease (SCD).
Of the 32 patients included in the study, 44% had a clinical diagnosis of SDB.
These patients had significantly increased REM latency, a significantly higher mean score on the Epworth Sleepiness Scale, and a significantly higher oxygen desaturation index (ODI) than patients who did not have SDB.
However, there was no significant difference between SDB and non-SDB patients with regard to insomnia, delayed sleep phase syndrome, nocturia, or SCD complications.
Sunil Sharma, MD, of Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia, Pennsylvania, and his colleagues recounted these results in the Journal of Clinical Sleep Medicine.
“Previous research identified pain and sleep disturbance as 2 common symptoms of adult sickle cell disorder,” Dr Sharma said. “We wanted to examine the reasons for the sleep disturbances, as it can have a strong impact on our patients’ quality of life and overall health. We discovered a high incidence of sleep disordered breathing in patients with sickle cell disease who also report trouble with sleep.”
Dr Sharma and his colleagues analyzed 32 consecutive adult SCD patients who had reported symptoms suggesting disordered sleep or had an Epworth Sleepiness Scale score of 10 or greater. The patients underwent a comprehensive sleep evaluation and overnight polysomnography in an accredited sleep center.
SDB was defined as having an apnea-hypopnea index (AHI, events/hour) of 5 or greater. SDB was considered mild if the AHI was 5 to < 15, moderate if the AHI was 15 to < 30, and severe if the AHI was ≥ 30. Once they determined which patients had SDB, the researchers compared these patients to those without the condition.
The team found that 44% of patients (n=14) had SDB. It was mild in 8 patients, moderate in 4, and severe in 2.
Compared to non-SDB patients, those with SDB had a significantly higher mean AHI (1.6 and 17, respectively; P=0.0001), a significantly increased REM latency (98 and 159 minutes, respectively; P=0.014), and a significantly higher mean score on the Epworth Sleepiness Scale (8.6 and 13, respectively; P=0.017).
Patients with SDB also had a significantly higher ODI than non-SDB patients (13 and 1.6, respectively; P=0.0009). Significant oxygen desaturation was defined as oxygen saturation < 89% for 5 cumulative minutes or more. The ODI was the number of recorded oxygen desaturations ≥ 4% per hour of sleep.
There was no significant difference between SDB and non-SDB patients in the incidence of nocturia (2.3 and 1.6, respectively; P=0.063), insomnia (57% and 72%, respectively; P=0.46), or delayed sleep phase syndrome (57% and 50%, respectively; P=0.73).
Delayed sleep phase syndrome was defined as a delay in sleep onset of 2 hours or greater from the desired sleep time and an inability to awaken at the desired time. Insomnia was defined as difficulty initiating sleep (sleep latency greater than 60 minutes) or difficulty maintaining sleep (more than 2 awakenings requiring more than 20 minutes to fall back asleep) on the majority of nights for more than 4 weeks.
There was no significant difference between SDB and non-SDB patients with regard to SCD complications, including crises during sleep (44% and 39%, respectively; P=0.67), average hospital admissions in the last 5 years (9.1 and 6.0, respectively; P=0.15), or average mini-crises per month (2.7 and 3.6, respectively; P=0.69).
Dr Sharma said the diagnosis of SDB could be missed in adults with SCD because they are not generally obese, a common risk factor for SDB, and daytime sleepiness is attributed to the pain medications used to treat the symptoms of SCD. He hopes this study will increase awareness among physicians who can screen patients for SDB.
“Our study suggests that patients with sickle cell disorder should be screened using a questionnaire to identify problems with sleep,” Dr Sharma said. “For further testing, an oxygen desaturation index is another low-cost screening tool that can identify sleep disordered breathing in this population.”
Israel approves ponatinib for CML, Ph+ ALL

Photo courtesy of the US FDA
The Israeli Ministry of Health has granted regulatory approval for the kinase inhibitor ponatinib (Iclusig) to treat certain adults with chronic myeloid leukemia (CML) or Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL).
The drug can now be used to treat adults with any phase of CML who have the T315I mutation or are resistant to/cannot tolerate dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ponatinib is also approved to treat patients with Ph+ ALL who have the T315I mutation or are resistant to/cannot tolerate dasatinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ariad Pharmaceuticals, Inc., the company developing ponatinib, said the drug should be available in Israel in the second quarter of 2015.
Trial results
The Ministry of Health’s decision to approve ponatinib was based on results from the phase 2 PACE trial, which included patients with CML or Ph+ ALL who were resistant to or intolerant of prior tyrosine kinase inhibitor therapy, or who had the T315I mutation.
The median follow-up times were 15.3 months in chronic-phase CML patients, 15.8 months in accelerated-phase CML patients, and 6.2 months in patients with blast-phase CML or Ph+ ALL.
In chronic-phase CML, the primary endpoint was major cytogenetic response, and it occurred in 56% of patients. Among chronic-phase patients with the T315I mutation, 70% achieved a major cytogenetic response. Among patients who had failed treatment with dasatinib or nilotinib, 51% achieved a major cytogenetic response.
In accelerated-phase CML, the primary endpoint was major hematologic response. This occurred in 57% of all patients in this group, 50% of patients with the T315I mutation, and 58% of patients who had failed treatment with dasatinib or nilotinib.
The primary endpoint was major hematologic response in blast-phase CML/Ph+ ALL as well. Thirty-four percent of all patients in this group met this endpoint, as did 33% of patients with the T315I mutation and 35% of patients who had failed treatment with dasatinib or nilotinib.
Common non-hematologic adverse events included rash (38%), abdominal pain (38%), headache (35%), dry skin (35%), constipation (34%), fatigue (27%), pyrexia (27%), nausea (26%), arthralgia (25%), hypertension (21%), increased lipase (19%), and increased amylase (7%).
Hematologic events of any grade included thrombocytopenia (42%), neutropenia (24%), and anemia (20%). Serious adverse events of arterial thromboembolism, including arterial stenosis, occurred in patients with cardiovascular risk factors.
Safety issues
Extended follow-up data from the PACE trial, collected in 2013, suggested ponatinib can increase the risk of thrombotic events. When these data came to light, officials in the European Union and the US, where ponatinib had already been approved, began to investigate the drug.
Ponatinib was pulled from the US market for a little over 2 months, and trials of the drug were placed on partial hold while the Food and Drug Administration evaluated the drug’s safety. Ponatinib went back on the market in January 2014, with new safety measures in place.
The drug was not pulled from the market in the European Union, but the European Medicine’s Agency released recommendations for safer use of ponatinib. The Committee for Medicinal Products for Human Use reviewed data on ponatinib and decided the drug’s benefits outweigh its risks.
Photo courtesy of the US FDA
The Israeli Ministry of Health has granted regulatory approval for the kinase inhibitor ponatinib (Iclusig) to treat certain adults with chronic myeloid leukemia (CML) or Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL).
The drug can now be used to treat adults with any phase of CML who have the T315I mutation or are resistant to/cannot tolerate dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ponatinib is also approved to treat patients with Ph+ ALL who have the T315I mutation or are resistant to/cannot tolerate dasatinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ariad Pharmaceuticals, Inc., the company developing ponatinib, said the drug should be available in Israel in the second quarter of 2015.
Trial results
The Ministry of Health’s decision to approve ponatinib was based on results from the phase 2 PACE trial, which included patients with CML or Ph+ ALL who were resistant to or intolerant of prior tyrosine kinase inhibitor therapy, or who had the T315I mutation.
The median follow-up times were 15.3 months in chronic-phase CML patients, 15.8 months in accelerated-phase CML patients, and 6.2 months in patients with blast-phase CML or Ph+ ALL.
In chronic-phase CML, the primary endpoint was major cytogenetic response, and it occurred in 56% of patients. Among chronic-phase patients with the T315I mutation, 70% achieved a major cytogenetic response. Among patients who had failed treatment with dasatinib or nilotinib, 51% achieved a major cytogenetic response.
In accelerated-phase CML, the primary endpoint was major hematologic response. This occurred in 57% of all patients in this group, 50% of patients with the T315I mutation, and 58% of patients who had failed treatment with dasatinib or nilotinib.
The primary endpoint was major hematologic response in blast-phase CML/Ph+ ALL as well. Thirty-four percent of all patients in this group met this endpoint, as did 33% of patients with the T315I mutation and 35% of patients who had failed treatment with dasatinib or nilotinib.
Common non-hematologic adverse events included rash (38%), abdominal pain (38%), headache (35%), dry skin (35%), constipation (34%), fatigue (27%), pyrexia (27%), nausea (26%), arthralgia (25%), hypertension (21%), increased lipase (19%), and increased amylase (7%).
Hematologic events of any grade included thrombocytopenia (42%), neutropenia (24%), and anemia (20%). Serious adverse events of arterial thromboembolism, including arterial stenosis, occurred in patients with cardiovascular risk factors.
Safety issues
Extended follow-up data from the PACE trial, collected in 2013, suggested ponatinib can increase the risk of thrombotic events. When these data came to light, officials in the European Union and the US, where ponatinib had already been approved, began to investigate the drug.
Ponatinib was pulled from the US market for a little over 2 months, and trials of the drug were placed on partial hold while the Food and Drug Administration evaluated the drug’s safety. Ponatinib went back on the market in January 2014, with new safety measures in place.
The drug was not pulled from the market in the European Union, but the European Medicine’s Agency released recommendations for safer use of ponatinib. The Committee for Medicinal Products for Human Use reviewed data on ponatinib and decided the drug’s benefits outweigh its risks.
Photo courtesy of the US FDA
The Israeli Ministry of Health has granted regulatory approval for the kinase inhibitor ponatinib (Iclusig) to treat certain adults with chronic myeloid leukemia (CML) or Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL).
The drug can now be used to treat adults with any phase of CML who have the T315I mutation or are resistant to/cannot tolerate dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ponatinib is also approved to treat patients with Ph+ ALL who have the T315I mutation or are resistant to/cannot tolerate dasatinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ariad Pharmaceuticals, Inc., the company developing ponatinib, said the drug should be available in Israel in the second quarter of 2015.
Trial results
The Ministry of Health’s decision to approve ponatinib was based on results from the phase 2 PACE trial, which included patients with CML or Ph+ ALL who were resistant to or intolerant of prior tyrosine kinase inhibitor therapy, or who had the T315I mutation.
The median follow-up times were 15.3 months in chronic-phase CML patients, 15.8 months in accelerated-phase CML patients, and 6.2 months in patients with blast-phase CML or Ph+ ALL.
In chronic-phase CML, the primary endpoint was major cytogenetic response, and it occurred in 56% of patients. Among chronic-phase patients with the T315I mutation, 70% achieved a major cytogenetic response. Among patients who had failed treatment with dasatinib or nilotinib, 51% achieved a major cytogenetic response.
In accelerated-phase CML, the primary endpoint was major hematologic response. This occurred in 57% of all patients in this group, 50% of patients with the T315I mutation, and 58% of patients who had failed treatment with dasatinib or nilotinib.
The primary endpoint was major hematologic response in blast-phase CML/Ph+ ALL as well. Thirty-four percent of all patients in this group met this endpoint, as did 33% of patients with the T315I mutation and 35% of patients who had failed treatment with dasatinib or nilotinib.
Common non-hematologic adverse events included rash (38%), abdominal pain (38%), headache (35%), dry skin (35%), constipation (34%), fatigue (27%), pyrexia (27%), nausea (26%), arthralgia (25%), hypertension (21%), increased lipase (19%), and increased amylase (7%).
Hematologic events of any grade included thrombocytopenia (42%), neutropenia (24%), and anemia (20%). Serious adverse events of arterial thromboembolism, including arterial stenosis, occurred in patients with cardiovascular risk factors.
Safety issues
Extended follow-up data from the PACE trial, collected in 2013, suggested ponatinib can increase the risk of thrombotic events. When these data came to light, officials in the European Union and the US, where ponatinib had already been approved, began to investigate the drug.
Ponatinib was pulled from the US market for a little over 2 months, and trials of the drug were placed on partial hold while the Food and Drug Administration evaluated the drug’s safety. Ponatinib went back on the market in January 2014, with new safety measures in place.
The drug was not pulled from the market in the European Union, but the European Medicine’s Agency released recommendations for safer use of ponatinib. The Committee for Medicinal Products for Human Use reviewed data on ponatinib and decided the drug’s benefits outweigh its risks.
Group identifies priorities for lymphoma research

Photo by Darren Baker
By agreeing upon—and addressing—the aspects of lymphoma research that need the most improvement, the research community could advance the treatment of these diseases, according to a report published in Blood.
The report’s authors said limitations in research infrastructure, funding, and collaborative approaches across research centers present potential challenges on the road to developing better treatments.
And they outlined several “priority areas” that, they believe, require particular attention.
“[Our report] draws focus to our most pressing needs, which, if unaddressed, will prevent transformative changes to how we study and treat these diseases,” said David M. Weinstock, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“Directing our collaborative efforts toward the most high-impact areas will enable us to more rapidly bring life-saving treatments to our patients.”
The report lists the following priority areas:
- Infrastructure
- Develop an adequate number of disease models for each lymphoma subtype
- Establish a central repository of biospecimens, cell lines, and in vivo models with open access
- Organize patient advocacy to support research.
- Research
- Catalogue how lymphoma cells differ across disease subtypes
- Better define and identify mutations and other abnormalities associated with the disease
- Develop strategies to identify high-risk patients who may benefit most from clinical trials
- Enhance efforts to use immune therapies to cure lymphoma
- Better understand how lymphoma cells communicate with normal cells.
“[W]e invite clinicians, scientists, advocates, and patients to weigh in on this strategic roadmap so that it reflects the input of everyone in the community,” Dr Weinstock said. “We will share these priorities with funding agencies, advocacy groups, and others who can help us address the challenges we have identified, and thereby accelerate the development of new approaches to understand and eradicate lymphoma.”
To weigh in, visit: http://www.hematology.org/lymphoma-roadmap.
This report was developed after a review of the state of the science in lymphoma that took place at a special ASH Meeting on Lymphoma Biology in August 2014. A second ASH Meeting on Lymphoma Biology is planned for the summer of 2016.
Photo by Darren Baker
By agreeing upon—and addressing—the aspects of lymphoma research that need the most improvement, the research community could advance the treatment of these diseases, according to a report published in Blood.
The report’s authors said limitations in research infrastructure, funding, and collaborative approaches across research centers present potential challenges on the road to developing better treatments.
And they outlined several “priority areas” that, they believe, require particular attention.
“[Our report] draws focus to our most pressing needs, which, if unaddressed, will prevent transformative changes to how we study and treat these diseases,” said David M. Weinstock, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“Directing our collaborative efforts toward the most high-impact areas will enable us to more rapidly bring life-saving treatments to our patients.”
The report lists the following priority areas:
- Infrastructure
- Develop an adequate number of disease models for each lymphoma subtype
- Establish a central repository of biospecimens, cell lines, and in vivo models with open access
- Organize patient advocacy to support research.
- Research
- Catalogue how lymphoma cells differ across disease subtypes
- Better define and identify mutations and other abnormalities associated with the disease
- Develop strategies to identify high-risk patients who may benefit most from clinical trials
- Enhance efforts to use immune therapies to cure lymphoma
- Better understand how lymphoma cells communicate with normal cells.
“[W]e invite clinicians, scientists, advocates, and patients to weigh in on this strategic roadmap so that it reflects the input of everyone in the community,” Dr Weinstock said. “We will share these priorities with funding agencies, advocacy groups, and others who can help us address the challenges we have identified, and thereby accelerate the development of new approaches to understand and eradicate lymphoma.”
To weigh in, visit: http://www.hematology.org/lymphoma-roadmap.
This report was developed after a review of the state of the science in lymphoma that took place at a special ASH Meeting on Lymphoma Biology in August 2014. A second ASH Meeting on Lymphoma Biology is planned for the summer of 2016.
Photo by Darren Baker
By agreeing upon—and addressing—the aspects of lymphoma research that need the most improvement, the research community could advance the treatment of these diseases, according to a report published in Blood.
The report’s authors said limitations in research infrastructure, funding, and collaborative approaches across research centers present potential challenges on the road to developing better treatments.
And they outlined several “priority areas” that, they believe, require particular attention.
“[Our report] draws focus to our most pressing needs, which, if unaddressed, will prevent transformative changes to how we study and treat these diseases,” said David M. Weinstock, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“Directing our collaborative efforts toward the most high-impact areas will enable us to more rapidly bring life-saving treatments to our patients.”
The report lists the following priority areas:
- Infrastructure
- Develop an adequate number of disease models for each lymphoma subtype
- Establish a central repository of biospecimens, cell lines, and in vivo models with open access
- Organize patient advocacy to support research.
- Research
- Catalogue how lymphoma cells differ across disease subtypes
- Better define and identify mutations and other abnormalities associated with the disease
- Develop strategies to identify high-risk patients who may benefit most from clinical trials
- Enhance efforts to use immune therapies to cure lymphoma
- Better understand how lymphoma cells communicate with normal cells.
“[W]e invite clinicians, scientists, advocates, and patients to weigh in on this strategic roadmap so that it reflects the input of everyone in the community,” Dr Weinstock said. “We will share these priorities with funding agencies, advocacy groups, and others who can help us address the challenges we have identified, and thereby accelerate the development of new approaches to understand and eradicate lymphoma.”
To weigh in, visit: http://www.hematology.org/lymphoma-roadmap.
This report was developed after a review of the state of the science in lymphoma that took place at a special ASH Meeting on Lymphoma Biology in August 2014. A second ASH Meeting on Lymphoma Biology is planned for the summer of 2016.