User login
Breakthrough designation for pathogen-reduced cryoprecipitate

The U.S. Food and Drug Administration (FDA) has granted breakthrough device designation for pathogen-reduced cryoprecipitate manufactured from plasma treated with the INTERCEPT Blood System.
Cerus Corporation aims to seek FDA approval for this pathogen-reduced cryoprecipitate to treat massive hemorrhage.
“The proposed label indication for pathogen-reduced cryoprecipitate would be to control massive bleeding associated with fibrinogen deficiency,” said Laurence Corash, chief scientific officer of Cerus.
The proposed shelf life for pathogen-reduced cyroprecipitate is 5 days at room temperature.
The INTERCEPT Blood System is already FDA-approved for the inactivation of pathogens in platelets and plasma.
“Cryo[precipitate] is a natural extension to our current FDA-approved INTERCEPT Blood System for plasma,” said William “Obi” Greenman, president and chief executive officer of Cerus.
If the FDA granted approval, INTERCEPT-treated plasma could be manufactured into pathogen-reduced cryoprecipitate.
About breakthrough device designation
The FDA says it grants breakthrough designation to devices “that provide more effective treatment or diagnosis of life-threatening or irreversibly debilitating human diseases or conditions.”
Breakthrough designation is intended to expedite the development, assessment, and review of medical devices. The breakthrough devices program supersedes the FDA’s expedited access pathway and priority review programs.
The U.S. Food and Drug Administration (FDA) has granted breakthrough device designation for pathogen-reduced cryoprecipitate manufactured from plasma treated with the INTERCEPT Blood System.
Cerus Corporation aims to seek FDA approval for this pathogen-reduced cryoprecipitate to treat massive hemorrhage.
“The proposed label indication for pathogen-reduced cryoprecipitate would be to control massive bleeding associated with fibrinogen deficiency,” said Laurence Corash, chief scientific officer of Cerus.
The proposed shelf life for pathogen-reduced cyroprecipitate is 5 days at room temperature.
The INTERCEPT Blood System is already FDA-approved for the inactivation of pathogens in platelets and plasma.
“Cryo[precipitate] is a natural extension to our current FDA-approved INTERCEPT Blood System for plasma,” said William “Obi” Greenman, president and chief executive officer of Cerus.
If the FDA granted approval, INTERCEPT-treated plasma could be manufactured into pathogen-reduced cryoprecipitate.
About breakthrough device designation
The FDA says it grants breakthrough designation to devices “that provide more effective treatment or diagnosis of life-threatening or irreversibly debilitating human diseases or conditions.”
Breakthrough designation is intended to expedite the development, assessment, and review of medical devices. The breakthrough devices program supersedes the FDA’s expedited access pathway and priority review programs.
The U.S. Food and Drug Administration (FDA) has granted breakthrough device designation for pathogen-reduced cryoprecipitate manufactured from plasma treated with the INTERCEPT Blood System.
Cerus Corporation aims to seek FDA approval for this pathogen-reduced cryoprecipitate to treat massive hemorrhage.
“The proposed label indication for pathogen-reduced cryoprecipitate would be to control massive bleeding associated with fibrinogen deficiency,” said Laurence Corash, chief scientific officer of Cerus.
The proposed shelf life for pathogen-reduced cyroprecipitate is 5 days at room temperature.
The INTERCEPT Blood System is already FDA-approved for the inactivation of pathogens in platelets and plasma.
“Cryo[precipitate] is a natural extension to our current FDA-approved INTERCEPT Blood System for plasma,” said William “Obi” Greenman, president and chief executive officer of Cerus.
If the FDA granted approval, INTERCEPT-treated plasma could be manufactured into pathogen-reduced cryoprecipitate.
About breakthrough device designation
The FDA says it grants breakthrough designation to devices “that provide more effective treatment or diagnosis of life-threatening or irreversibly debilitating human diseases or conditions.”
Breakthrough designation is intended to expedite the development, assessment, and review of medical devices. The breakthrough devices program supersedes the FDA’s expedited access pathway and priority review programs.
Lower threshold for platelet transfusions appears safer
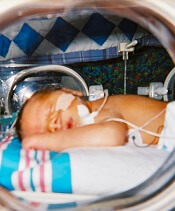
A lower threshold for platelet transfusions may be safer for preterm infants with severe thrombocytopenia, a new study suggests.
Researchers randomized preterm infants with severe thrombocytopenia to receive transfusions at platelet count thresholds of 50,000/mm3 or 25,000/mm3.
The team found that patients in the high-threshold group had a significantly higher risk of major bleeding or death.
Anna Curley, MD, of the National Maternity Hospital in Dublin, Ireland, and her colleagues reported this finding in The New England Journal of Medicine.
The researchers studied 660 infants with a mean gestational age of 26.6 weeks. They were randomized to receive platelet transfusions at a high platelet-count threshold of 50,000/mm3 or a low threshold of 25,000/mm3.
Within 28 days of randomization, a new major bleeding episode or death occurred in 26% of the high-threshold group and 19% of the low-threshold group.
When the researchers adjusted for gestational age, intrauterine growth restriction, and trial site, the odds ratio (OR) for major bleeding or death was 1.57 (95% confidence interval [CI], 1.06-2.32; P=0.02).
The OR for death alone was 1.56 (95% CI, 0.95-2.55), and the hazard ratio for at least one major bleeding episode was 1.32 (95% CI, 1.00-1.74).
The rates of serious adverse events, not including major bleeding, were similar between the high- and low-threshold groups—25% and 22%, respectively (OR=1.14; 95% CI, 0.78-1.67).
The researchers said the results of this trial suggest that reducing the transfusion threshold from 50,000/mm3 to 25,000/mm3 may prevent death or major bleeding in 7 of 100 preterm neonates with severe thrombocytopenia.
However, the team also acknowledged that it isn’t clear why reducing the threshold may reduce the risk of mortality or major bleeding in this patient group.
The researchers said a range of factors might play a role in adverse outcomes of platelet transfusion in preterm neonates, including inflammatory consequences, hemodynamic shifts, fragility of the germinal matrix, disturbances in organ and brain blood flow, preterm lungs with a large capillary bed and abundant immune cells, platelet-derived reactive oxygen species, proangiogenic factors, and vessel occlusion by platelet microthrombi.
This study was supported by the National Health Service Blood and Transplant Research and Development Committee, Sanquin Research, Addenbrooke’s Charitable Trust, the Neonatal Breath of Life Fund, and the National Institute for Health Research Clinical Research Network.
One study author reported consulting for Sanquin Research, and another declared travel funds from Cerus Corporation.
A lower threshold for platelet transfusions may be safer for preterm infants with severe thrombocytopenia, a new study suggests.
Researchers randomized preterm infants with severe thrombocytopenia to receive transfusions at platelet count thresholds of 50,000/mm3 or 25,000/mm3.
The team found that patients in the high-threshold group had a significantly higher risk of major bleeding or death.
Anna Curley, MD, of the National Maternity Hospital in Dublin, Ireland, and her colleagues reported this finding in The New England Journal of Medicine.
The researchers studied 660 infants with a mean gestational age of 26.6 weeks. They were randomized to receive platelet transfusions at a high platelet-count threshold of 50,000/mm3 or a low threshold of 25,000/mm3.
Within 28 days of randomization, a new major bleeding episode or death occurred in 26% of the high-threshold group and 19% of the low-threshold group.
When the researchers adjusted for gestational age, intrauterine growth restriction, and trial site, the odds ratio (OR) for major bleeding or death was 1.57 (95% confidence interval [CI], 1.06-2.32; P=0.02).
The OR for death alone was 1.56 (95% CI, 0.95-2.55), and the hazard ratio for at least one major bleeding episode was 1.32 (95% CI, 1.00-1.74).
The rates of serious adverse events, not including major bleeding, were similar between the high- and low-threshold groups—25% and 22%, respectively (OR=1.14; 95% CI, 0.78-1.67).
The researchers said the results of this trial suggest that reducing the transfusion threshold from 50,000/mm3 to 25,000/mm3 may prevent death or major bleeding in 7 of 100 preterm neonates with severe thrombocytopenia.
However, the team also acknowledged that it isn’t clear why reducing the threshold may reduce the risk of mortality or major bleeding in this patient group.
The researchers said a range of factors might play a role in adverse outcomes of platelet transfusion in preterm neonates, including inflammatory consequences, hemodynamic shifts, fragility of the germinal matrix, disturbances in organ and brain blood flow, preterm lungs with a large capillary bed and abundant immune cells, platelet-derived reactive oxygen species, proangiogenic factors, and vessel occlusion by platelet microthrombi.
This study was supported by the National Health Service Blood and Transplant Research and Development Committee, Sanquin Research, Addenbrooke’s Charitable Trust, the Neonatal Breath of Life Fund, and the National Institute for Health Research Clinical Research Network.
One study author reported consulting for Sanquin Research, and another declared travel funds from Cerus Corporation.
A lower threshold for platelet transfusions may be safer for preterm infants with severe thrombocytopenia, a new study suggests.
Researchers randomized preterm infants with severe thrombocytopenia to receive transfusions at platelet count thresholds of 50,000/mm3 or 25,000/mm3.
The team found that patients in the high-threshold group had a significantly higher risk of major bleeding or death.
Anna Curley, MD, of the National Maternity Hospital in Dublin, Ireland, and her colleagues reported this finding in The New England Journal of Medicine.
The researchers studied 660 infants with a mean gestational age of 26.6 weeks. They were randomized to receive platelet transfusions at a high platelet-count threshold of 50,000/mm3 or a low threshold of 25,000/mm3.
Within 28 days of randomization, a new major bleeding episode or death occurred in 26% of the high-threshold group and 19% of the low-threshold group.
When the researchers adjusted for gestational age, intrauterine growth restriction, and trial site, the odds ratio (OR) for major bleeding or death was 1.57 (95% confidence interval [CI], 1.06-2.32; P=0.02).
The OR for death alone was 1.56 (95% CI, 0.95-2.55), and the hazard ratio for at least one major bleeding episode was 1.32 (95% CI, 1.00-1.74).
The rates of serious adverse events, not including major bleeding, were similar between the high- and low-threshold groups—25% and 22%, respectively (OR=1.14; 95% CI, 0.78-1.67).
The researchers said the results of this trial suggest that reducing the transfusion threshold from 50,000/mm3 to 25,000/mm3 may prevent death or major bleeding in 7 of 100 preterm neonates with severe thrombocytopenia.
However, the team also acknowledged that it isn’t clear why reducing the threshold may reduce the risk of mortality or major bleeding in this patient group.
The researchers said a range of factors might play a role in adverse outcomes of platelet transfusion in preterm neonates, including inflammatory consequences, hemodynamic shifts, fragility of the germinal matrix, disturbances in organ and brain blood flow, preterm lungs with a large capillary bed and abundant immune cells, platelet-derived reactive oxygen species, proangiogenic factors, and vessel occlusion by platelet microthrombi.
This study was supported by the National Health Service Blood and Transplant Research and Development Committee, Sanquin Research, Addenbrooke’s Charitable Trust, the Neonatal Breath of Life Fund, and the National Institute for Health Research Clinical Research Network.
One study author reported consulting for Sanquin Research, and another declared travel funds from Cerus Corporation.
Health Canada approves pralatrexate for rel/ref PTCL

Health Canada has granted conditional approval for pralatrexate (Folotyn®) to treat relapsed or refractory peripheral T-cell lymphoma (PTCL).
Pralatrexate received a Notice of Compliance with Conditions (NOC/c), which is an approval granted on the basis of promising evidence of clinical effectiveness that must be verified in additional clinical trials.
To be approved under Health Canada’s NOC/c policy, a product must be intended for the treatment, prevention, or diagnosis of a serious, life-threatening, or severely debilitating illness.
In addition, the product must have demonstrated promising benefit, be of high quality, have an acceptable safety profile based on a benefit/risk assessment, and either respond to a serious unmet need or provide a significant improvement over existing therapies.
The NOC/c for pralatrexate is based on response rates in the single-arm, phase 2 PROPEL trial. Results from PROPEL were published in the Journal of Clinical Oncology in 2011.
The trial enrolled 115 patients with relapsed or refractory PTCL who had received a median of three (range, 1-12) prior systemic therapies.
The patients received pralatrexate once weekly at a dose of 30 mg/m2 for 6 weeks in 7-week cycles until disease progression or unacceptable toxicity.
In the 109 evaluable patients, the response rate was 29% (32/109). The complete response rate was 11% (n=12), and the partial response rate was 18% (n=20).
The median duration of response was 10.1 months, the median progression-free survival was 3.5 months, and the median overall survival was 14.5 months.
The most common adverse events (AEs) were mucositis (71%), thrombocytopenia (41%), nausea (41%), fatigue (36%), pyrexia (34%), anemia (34%), constipation (34%), edema (31%), cough (29%), epistaxis (26%), vomiting (25%), neutropenia (25%), and diarrhea (23%).
The most common grade 3/4 AEs were thrombocytopenia (32%), mucositis (22%), neutropenia (22%), and anemia (18%).
The product monograph for pralatrexate contains a boxed warning highlighting the risk of AEs associated with pralatrexate use, including dermatologic reactions, bone marrow suppression, infection, mucosal inflammation, tumor lysis syndrome, potential fetal harm, and pulmonary toxicity.
The product monograph is available for download from the website of Servier Canada, the company marketing pralatrexate in Canada.
Health Canada has granted conditional approval for pralatrexate (Folotyn®) to treat relapsed or refractory peripheral T-cell lymphoma (PTCL).
Pralatrexate received a Notice of Compliance with Conditions (NOC/c), which is an approval granted on the basis of promising evidence of clinical effectiveness that must be verified in additional clinical trials.
To be approved under Health Canada’s NOC/c policy, a product must be intended for the treatment, prevention, or diagnosis of a serious, life-threatening, or severely debilitating illness.
In addition, the product must have demonstrated promising benefit, be of high quality, have an acceptable safety profile based on a benefit/risk assessment, and either respond to a serious unmet need or provide a significant improvement over existing therapies.
The NOC/c for pralatrexate is based on response rates in the single-arm, phase 2 PROPEL trial. Results from PROPEL were published in the Journal of Clinical Oncology in 2011.
The trial enrolled 115 patients with relapsed or refractory PTCL who had received a median of three (range, 1-12) prior systemic therapies.
The patients received pralatrexate once weekly at a dose of 30 mg/m2 for 6 weeks in 7-week cycles until disease progression or unacceptable toxicity.
In the 109 evaluable patients, the response rate was 29% (32/109). The complete response rate was 11% (n=12), and the partial response rate was 18% (n=20).
The median duration of response was 10.1 months, the median progression-free survival was 3.5 months, and the median overall survival was 14.5 months.
The most common adverse events (AEs) were mucositis (71%), thrombocytopenia (41%), nausea (41%), fatigue (36%), pyrexia (34%), anemia (34%), constipation (34%), edema (31%), cough (29%), epistaxis (26%), vomiting (25%), neutropenia (25%), and diarrhea (23%).
The most common grade 3/4 AEs were thrombocytopenia (32%), mucositis (22%), neutropenia (22%), and anemia (18%).
The product monograph for pralatrexate contains a boxed warning highlighting the risk of AEs associated with pralatrexate use, including dermatologic reactions, bone marrow suppression, infection, mucosal inflammation, tumor lysis syndrome, potential fetal harm, and pulmonary toxicity.
The product monograph is available for download from the website of Servier Canada, the company marketing pralatrexate in Canada.
Health Canada has granted conditional approval for pralatrexate (Folotyn®) to treat relapsed or refractory peripheral T-cell lymphoma (PTCL).
Pralatrexate received a Notice of Compliance with Conditions (NOC/c), which is an approval granted on the basis of promising evidence of clinical effectiveness that must be verified in additional clinical trials.
To be approved under Health Canada’s NOC/c policy, a product must be intended for the treatment, prevention, or diagnosis of a serious, life-threatening, or severely debilitating illness.
In addition, the product must have demonstrated promising benefit, be of high quality, have an acceptable safety profile based on a benefit/risk assessment, and either respond to a serious unmet need or provide a significant improvement over existing therapies.
The NOC/c for pralatrexate is based on response rates in the single-arm, phase 2 PROPEL trial. Results from PROPEL were published in the Journal of Clinical Oncology in 2011.
The trial enrolled 115 patients with relapsed or refractory PTCL who had received a median of three (range, 1-12) prior systemic therapies.
The patients received pralatrexate once weekly at a dose of 30 mg/m2 for 6 weeks in 7-week cycles until disease progression or unacceptable toxicity.
In the 109 evaluable patients, the response rate was 29% (32/109). The complete response rate was 11% (n=12), and the partial response rate was 18% (n=20).
The median duration of response was 10.1 months, the median progression-free survival was 3.5 months, and the median overall survival was 14.5 months.
The most common adverse events (AEs) were mucositis (71%), thrombocytopenia (41%), nausea (41%), fatigue (36%), pyrexia (34%), anemia (34%), constipation (34%), edema (31%), cough (29%), epistaxis (26%), vomiting (25%), neutropenia (25%), and diarrhea (23%).
The most common grade 3/4 AEs were thrombocytopenia (32%), mucositis (22%), neutropenia (22%), and anemia (18%).
The product monograph for pralatrexate contains a boxed warning highlighting the risk of AEs associated with pralatrexate use, including dermatologic reactions, bone marrow suppression, infection, mucosal inflammation, tumor lysis syndrome, potential fetal harm, and pulmonary toxicity.
The product monograph is available for download from the website of Servier Canada, the company marketing pralatrexate in Canada.
EC approves venetoclax in combo with rituximab

The European Commission (EC) has approved a new indication for venetoclax (Venclyxto®).
The drug is now approved for use in combination with rituximab to treat patients with relapsed/refractory chronic lymphocytic leukemia (CLL) who have received at least one prior therapy.
The approval is valid in all member states of the European Union as well as Iceland, Liechtenstein, and Norway.
The EC’s approval is based on results from the phase 3 MURANO trial, which were published in The New England Journal of Medicine in March.
The trial included 389 CLL patients who were randomized to receive venetoclax plus rituximab (VEN+R) or bendamustine plus rituximab (B+R). The median follow-up was 23.8 months.
According to an independent review committee, the overall response rate was 92.3% in the VEN+R arm and 72.3% in the B+R arm. The investigator-assessed overall response rates were 93.3% and 67.7%, respectively.
According to investigators, the median progression-free survival (PFS) was not reached in the VEN+R arm and was 17.0 months in the B+R arm (hazard ratio [HR]=0.17; P<0.0001).
According to the independent review committee, the median PFS was not reached in the VEN+R arm and was 18.1 months in the B+R arm (HR=0.20; P<0.0001).
Investigators said the 2-year PFS rate was 84.9% in the VEN+R arm and 36.3% in the B+R arm.
They said the 2-year overall survival rates were 91.9% and 86.6%, respectively (HR=0.48; P<0.0001). The median overall survival was not reached in either arm.
Grade 3/4 adverse events (AEs) with at least a 2% difference in incidence between the treatment arms (in the VEN+R and B+R arms, respectively) included:
- Neutropenia (57.7% and 38.8%)
- Infections and infestations (17.5% and 21.8%)
- Anemia (10.8% and 13.8%)
- Thrombocytopenia (5.7% and 10.1%)
- Febrile neutropenia (3.6% and 9.6%)
- Pneumonia (5.2% and 8.0%)
- Infusion-related reactions (1.5% and 5.3%)
- Tumor lysis syndrome (3.1% and 1.1%)
- Hypotension (0% and 2.7%)
- Hyperglycemia (2.1% and 0%)
- Hypogammaglobulinemia (2.1% and 0%).
Serious AEs with at least a 2% difference in incidence between the arms (in the VEN+R and B+R arms, respectively) were:
- Pneumonia (8.2% and 8.0%)
- Febrile neutropenia (3.6% and 8.5%)
- Pyrexia (2.6% and 6.9%)
- Anemia (1.5% and 2.7%)
- Infusion-related reactions (0.5% and 3.2%)
- Sepsis (0.5% and 2.1%)
- Tumor lysis syndrome (2.1% and 0.5%)
- Hypotension (0% and 2.7%).
Fatal AEs occurred in 5.2% of patients in the VEN+R arm and 5.9% in the B+R arm.
Fatal AEs in the VEN+R arm included pneumonia (n=3), sepsis (n=1), thrombocytopenia (n=1), cardiac failure (n=1), myocardial infarction (n=1), sudden cardiac death (n=1), colorectal cancer (n=1), status epilepticus (n=1), and acute respiratory failure (n=1). Two cases of pneumonia occurred in the setting of progression/Richter’s transformation.
The European Commission (EC) has approved a new indication for venetoclax (Venclyxto®).
The drug is now approved for use in combination with rituximab to treat patients with relapsed/refractory chronic lymphocytic leukemia (CLL) who have received at least one prior therapy.
The approval is valid in all member states of the European Union as well as Iceland, Liechtenstein, and Norway.
The EC’s approval is based on results from the phase 3 MURANO trial, which were published in The New England Journal of Medicine in March.
The trial included 389 CLL patients who were randomized to receive venetoclax plus rituximab (VEN+R) or bendamustine plus rituximab (B+R). The median follow-up was 23.8 months.
According to an independent review committee, the overall response rate was 92.3% in the VEN+R arm and 72.3% in the B+R arm. The investigator-assessed overall response rates were 93.3% and 67.7%, respectively.
According to investigators, the median progression-free survival (PFS) was not reached in the VEN+R arm and was 17.0 months in the B+R arm (hazard ratio [HR]=0.17; P<0.0001).
According to the independent review committee, the median PFS was not reached in the VEN+R arm and was 18.1 months in the B+R arm (HR=0.20; P<0.0001).
Investigators said the 2-year PFS rate was 84.9% in the VEN+R arm and 36.3% in the B+R arm.
They said the 2-year overall survival rates were 91.9% and 86.6%, respectively (HR=0.48; P<0.0001). The median overall survival was not reached in either arm.
Grade 3/4 adverse events (AEs) with at least a 2% difference in incidence between the treatment arms (in the VEN+R and B+R arms, respectively) included:
- Neutropenia (57.7% and 38.8%)
- Infections and infestations (17.5% and 21.8%)
- Anemia (10.8% and 13.8%)
- Thrombocytopenia (5.7% and 10.1%)
- Febrile neutropenia (3.6% and 9.6%)
- Pneumonia (5.2% and 8.0%)
- Infusion-related reactions (1.5% and 5.3%)
- Tumor lysis syndrome (3.1% and 1.1%)
- Hypotension (0% and 2.7%)
- Hyperglycemia (2.1% and 0%)
- Hypogammaglobulinemia (2.1% and 0%).
Serious AEs with at least a 2% difference in incidence between the arms (in the VEN+R and B+R arms, respectively) were:
- Pneumonia (8.2% and 8.0%)
- Febrile neutropenia (3.6% and 8.5%)
- Pyrexia (2.6% and 6.9%)
- Anemia (1.5% and 2.7%)
- Infusion-related reactions (0.5% and 3.2%)
- Sepsis (0.5% and 2.1%)
- Tumor lysis syndrome (2.1% and 0.5%)
- Hypotension (0% and 2.7%).
Fatal AEs occurred in 5.2% of patients in the VEN+R arm and 5.9% in the B+R arm.
Fatal AEs in the VEN+R arm included pneumonia (n=3), sepsis (n=1), thrombocytopenia (n=1), cardiac failure (n=1), myocardial infarction (n=1), sudden cardiac death (n=1), colorectal cancer (n=1), status epilepticus (n=1), and acute respiratory failure (n=1). Two cases of pneumonia occurred in the setting of progression/Richter’s transformation.
The European Commission (EC) has approved a new indication for venetoclax (Venclyxto®).
The drug is now approved for use in combination with rituximab to treat patients with relapsed/refractory chronic lymphocytic leukemia (CLL) who have received at least one prior therapy.
The approval is valid in all member states of the European Union as well as Iceland, Liechtenstein, and Norway.
The EC’s approval is based on results from the phase 3 MURANO trial, which were published in The New England Journal of Medicine in March.
The trial included 389 CLL patients who were randomized to receive venetoclax plus rituximab (VEN+R) or bendamustine plus rituximab (B+R). The median follow-up was 23.8 months.
According to an independent review committee, the overall response rate was 92.3% in the VEN+R arm and 72.3% in the B+R arm. The investigator-assessed overall response rates were 93.3% and 67.7%, respectively.
According to investigators, the median progression-free survival (PFS) was not reached in the VEN+R arm and was 17.0 months in the B+R arm (hazard ratio [HR]=0.17; P<0.0001).
According to the independent review committee, the median PFS was not reached in the VEN+R arm and was 18.1 months in the B+R arm (HR=0.20; P<0.0001).
Investigators said the 2-year PFS rate was 84.9% in the VEN+R arm and 36.3% in the B+R arm.
They said the 2-year overall survival rates were 91.9% and 86.6%, respectively (HR=0.48; P<0.0001). The median overall survival was not reached in either arm.
Grade 3/4 adverse events (AEs) with at least a 2% difference in incidence between the treatment arms (in the VEN+R and B+R arms, respectively) included:
- Neutropenia (57.7% and 38.8%)
- Infections and infestations (17.5% and 21.8%)
- Anemia (10.8% and 13.8%)
- Thrombocytopenia (5.7% and 10.1%)
- Febrile neutropenia (3.6% and 9.6%)
- Pneumonia (5.2% and 8.0%)
- Infusion-related reactions (1.5% and 5.3%)
- Tumor lysis syndrome (3.1% and 1.1%)
- Hypotension (0% and 2.7%)
- Hyperglycemia (2.1% and 0%)
- Hypogammaglobulinemia (2.1% and 0%).
Serious AEs with at least a 2% difference in incidence between the arms (in the VEN+R and B+R arms, respectively) were:
- Pneumonia (8.2% and 8.0%)
- Febrile neutropenia (3.6% and 8.5%)
- Pyrexia (2.6% and 6.9%)
- Anemia (1.5% and 2.7%)
- Infusion-related reactions (0.5% and 3.2%)
- Sepsis (0.5% and 2.1%)
- Tumor lysis syndrome (2.1% and 0.5%)
- Hypotension (0% and 2.7%).
Fatal AEs occurred in 5.2% of patients in the VEN+R arm and 5.9% in the B+R arm.
Fatal AEs in the VEN+R arm included pneumonia (n=3), sepsis (n=1), thrombocytopenia (n=1), cardiac failure (n=1), myocardial infarction (n=1), sudden cardiac death (n=1), colorectal cancer (n=1), status epilepticus (n=1), and acute respiratory failure (n=1). Two cases of pneumonia occurred in the setting of progression/Richter’s transformation.
Test strips for warfarin monitoring recalled due to serious risks

The U.S. Food and Drug Administration (FDA) announced a Class I recall of test strips used with medical devices that monitor warfarin levels, which means use of these strips may cause serious injuries or death.
Roche Diagnostics has recalled certain test strip lots used with its CoaguChek test meter devices.
The recall involves more than 1.1 million packages of CoaguChek XS PT Test Strips that were distributed nationwide from January 12, 2018, to October 29, 2018.
The test strips are used with the CoaguChek XS plus, CoaguChek XS Pro, CoaguChek XS professional, CoaguChek XS PST, and CoaguChek Vantus test meter devices.
The FDA said patients and healthcare professionals should not rely on these test meter devices to monitor warfarin levels if they’re using test strips affected by the recall. Instead, patients should have blood drawn from a vein and have their levels measured by a laboratory test or use an alternative meter device.
“These strips are widely used, and we are working diligently to warn healthcare providers and the public about the dangers associated with this recall,” said Jeffrey Shuren, MD, director of the FDA’s Center for Devices and Radiological Health.
“Using faulty strips can lead to serious errors in medication dosage that could cause serious harm or death in some patients. We are also working with the company on the swift removal of the recalled strips and to ensure the new corrected strips are distributed to patients and healthcare providers as quickly as possible.”
The FDA’s warning concerning the CoaguChek XS PT Test Strips is based on medical device reports submitted by Roche Diagnostics indicating that the test strips may provide results that are higher than the actual international normalized ratio (INR).
As a result of incorrect INR results, some patients may be prescribed an insufficient warfarin dose or instructed to interrupt warfarin use, which may increase the risk of thrombosis.
Approximately 90 medical device reports and two serious patient injuries involving strokes were reported to the FDA.
Roche Diagnostics attributes the cause of the problem to a recent recalibration of the test strips to a different international standard.
The company plans to provide new batches of recalibrated test strips, based on the previous international standard, to their customers by the end of November.
The FDA reviewed validation data submitted by the company for these recalibrated strips.
Patients who are using CoaguChek meters should contact their healthcare provider to get information about alternative test methods and to address questions regarding their individual testing schedule. Patients should also contact their self-testing service providers to find out when they will be getting their corrected test strips.
For questions about the recall or to report adverse reactions or quality problems, contact Roche Diagnostics Point of Care Technical Service at 1-800-428-4674.
Adverse reactions can also be reported to the FDA through MedWatch, the agency’s voluntary reporting program.
The U.S. Food and Drug Administration (FDA) announced a Class I recall of test strips used with medical devices that monitor warfarin levels, which means use of these strips may cause serious injuries or death.
Roche Diagnostics has recalled certain test strip lots used with its CoaguChek test meter devices.
The recall involves more than 1.1 million packages of CoaguChek XS PT Test Strips that were distributed nationwide from January 12, 2018, to October 29, 2018.
The test strips are used with the CoaguChek XS plus, CoaguChek XS Pro, CoaguChek XS professional, CoaguChek XS PST, and CoaguChek Vantus test meter devices.
The FDA said patients and healthcare professionals should not rely on these test meter devices to monitor warfarin levels if they’re using test strips affected by the recall. Instead, patients should have blood drawn from a vein and have their levels measured by a laboratory test or use an alternative meter device.
“These strips are widely used, and we are working diligently to warn healthcare providers and the public about the dangers associated with this recall,” said Jeffrey Shuren, MD, director of the FDA’s Center for Devices and Radiological Health.
“Using faulty strips can lead to serious errors in medication dosage that could cause serious harm or death in some patients. We are also working with the company on the swift removal of the recalled strips and to ensure the new corrected strips are distributed to patients and healthcare providers as quickly as possible.”
The FDA’s warning concerning the CoaguChek XS PT Test Strips is based on medical device reports submitted by Roche Diagnostics indicating that the test strips may provide results that are higher than the actual international normalized ratio (INR).
As a result of incorrect INR results, some patients may be prescribed an insufficient warfarin dose or instructed to interrupt warfarin use, which may increase the risk of thrombosis.
Approximately 90 medical device reports and two serious patient injuries involving strokes were reported to the FDA.
Roche Diagnostics attributes the cause of the problem to a recent recalibration of the test strips to a different international standard.
The company plans to provide new batches of recalibrated test strips, based on the previous international standard, to their customers by the end of November.
The FDA reviewed validation data submitted by the company for these recalibrated strips.
Patients who are using CoaguChek meters should contact their healthcare provider to get information about alternative test methods and to address questions regarding their individual testing schedule. Patients should also contact their self-testing service providers to find out when they will be getting their corrected test strips.
For questions about the recall or to report adverse reactions or quality problems, contact Roche Diagnostics Point of Care Technical Service at 1-800-428-4674.
Adverse reactions can also be reported to the FDA through MedWatch, the agency’s voluntary reporting program.
The U.S. Food and Drug Administration (FDA) announced a Class I recall of test strips used with medical devices that monitor warfarin levels, which means use of these strips may cause serious injuries or death.
Roche Diagnostics has recalled certain test strip lots used with its CoaguChek test meter devices.
The recall involves more than 1.1 million packages of CoaguChek XS PT Test Strips that were distributed nationwide from January 12, 2018, to October 29, 2018.
The test strips are used with the CoaguChek XS plus, CoaguChek XS Pro, CoaguChek XS professional, CoaguChek XS PST, and CoaguChek Vantus test meter devices.
The FDA said patients and healthcare professionals should not rely on these test meter devices to monitor warfarin levels if they’re using test strips affected by the recall. Instead, patients should have blood drawn from a vein and have their levels measured by a laboratory test or use an alternative meter device.
“These strips are widely used, and we are working diligently to warn healthcare providers and the public about the dangers associated with this recall,” said Jeffrey Shuren, MD, director of the FDA’s Center for Devices and Radiological Health.
“Using faulty strips can lead to serious errors in medication dosage that could cause serious harm or death in some patients. We are also working with the company on the swift removal of the recalled strips and to ensure the new corrected strips are distributed to patients and healthcare providers as quickly as possible.”
The FDA’s warning concerning the CoaguChek XS PT Test Strips is based on medical device reports submitted by Roche Diagnostics indicating that the test strips may provide results that are higher than the actual international normalized ratio (INR).
As a result of incorrect INR results, some patients may be prescribed an insufficient warfarin dose or instructed to interrupt warfarin use, which may increase the risk of thrombosis.
Approximately 90 medical device reports and two serious patient injuries involving strokes were reported to the FDA.
Roche Diagnostics attributes the cause of the problem to a recent recalibration of the test strips to a different international standard.
The company plans to provide new batches of recalibrated test strips, based on the previous international standard, to their customers by the end of November.
The FDA reviewed validation data submitted by the company for these recalibrated strips.
Patients who are using CoaguChek meters should contact their healthcare provider to get information about alternative test methods and to address questions regarding their individual testing schedule. Patients should also contact their self-testing service providers to find out when they will be getting their corrected test strips.
For questions about the recall or to report adverse reactions or quality problems, contact Roche Diagnostics Point of Care Technical Service at 1-800-428-4674.
Adverse reactions can also be reported to the FDA through MedWatch, the agency’s voluntary reporting program.
Novel risk factors for febrile neutropenia in NHL, other cancers

A retrospective study has revealed new potential risk factors for chemotherapy-induced febrile neutropenia (FN) in patients with solid tumors and non-Hodgkin lymphoma (NHL).
Researchers found the timing and duration of corticosteroid use were both associated with FN.
The team also observed “marginal” associations between FN and certain dermatologic and mucosal conditions as well as the use of intravenous (IV) antibiotics before chemotherapy.
On the other hand, there was no association between oral antibiotic use and FN or between radiation therapy (RT) and FN.
Chun Rebecca Chao, PhD, of Kaiser Permanente Southern California in Pasadena, and her colleagues reported these findings in JNCCN.
“Febrile neutropenia is life-threatening and often requires hospitalization,” Dr. Chao noted. “Furthermore, FN can lead to chemotherapy dose delay and dose reduction, which, in turn, negatively impacts antitumor efficacy. However, it can be prevented if high-risk individuals are identified and treated prophylactically.”
With this in mind, Dr. Chao and her colleagues set out to identify novel risk factors for FN by analyzing 15,971 patients who were treated with myelosuppressive chemotherapy at Kaiser Permanente Southern California between 2000 and 2009.
Patients had been diagnosed with NHL (n=1,617) or breast (n=6,323), lung (n=3,584), colorectal (n=3,062), ovarian (n=924), or gastric (n=461) cancers.
In all, 4.3% of patients developed FN during their first cycle of chemotherapy.
Corticosteroid use
The researchers found corticosteroid use was associated with an increased risk of FN in a propensity score-adjusted (PSA) model (adjusted for age, sex, socioeconomic factors, comorbidities, etc.). The hazard ratio (HR) was 1.53 (95% CI, 1.17-1.98; P<0.01) for patients who received corticosteroids.
A longer duration of corticosteroid use was associated with a greater risk of FN. The adjusted HR (compared to no corticosteroid use) was:
- 1.78 for corticosteroid treatment lasting less than 15 days (P<0.01)
- 1.84 for treatment lasting 15 to 29 days (P<0.01)
- 2.27 for treatment lasting 30 to 44 days (P<0.01)
- 2.86 for treatment lasting 45 to 90 days (P<0.01).
More recent corticosteroid use was associated with a greater risk of FN as well. The adjusted HR was:
- 1.88 for corticosteroid treatment less than 15 days before chemotherapy (P<0.01)
- 1.13 for treatment 15 to 29 days before chemotherapy (P=0.72)
- 1.22 for treatment 30 to 44 days before chemotherapy (P=0.66)
- 1.41 for treatment 45 to 90 days before chemotherapy (P=0.32).
“One way to reduce the incidence rate for FN could be to schedule prior corticosteroid use and subsequent chemotherapy with at least 2 weeks between them, given the magnitude of the risk increase and prevalence of this risk factor,” Dr. Chao said.
Other potential risk factors
The researchers found a “marginally” increased risk of FN in patients with certain dermatologic conditions (dermatitis, psoriasis, pruritus, etc.) and mucosal conditions (gastritis, stomatitis, mucositis, etc.).
In the PSA model, the HR was 1.40 (95% CI, 0.98-1.93; P=0.05) for patients with these conditions.
IV antibiotic use was also found to be marginally associated with an increased risk of FN in a restricted analysis covering patients treated in 2008 and 2009. In the PSA model, the HR was 1.35 (95% CI, 0.97-1.87; P=0.08).
On the other hand, there was no association between FN and oral antibiotic use in the restricted analysis. In the PSA model, the HR was 1.07 (95% CI, 0.77-1.48; P=0.70) for patients who received oral antibiotics.
Dr. Chao and her colleagues said these results suggest IV antibiotics may have a more profound impact than oral antibiotics on the balance of bacterial flora and other immune functions. Another possible explanation is that patients who received IV antibiotics were generally sicker and more prone to severe infection than patients who received oral antibiotics.
As with oral antibiotics, the researchers found no association between FN and the following factors (with the PSA model):
- Prior surgery (HR=0.89; 95% CI, 0.72-1.11; P=0.30)
- Prior RT (HR=0.91; 95% CI, 0.64-1.27; P=0.61)
- Concurrent RT (HR=1.32; 95% CI, 0.69-2.37; P=0.37).
The researchers noted that they did not account for radiation field or dose in this study, so additional evaluation of RT as a risk factor is needed.
In closing, Dr. Chao and her colleagues said these results suggest corticosteroid use, IV antibiotics, and certain dermatologic and mucosal conditions should be taken into consideration when monitoring patients receiving myelosuppressive chemotherapy and when evaluating the need for prophylactic granulocyte colony-stimulating factor or chemotherapy dose reduction.
Dr. Chao and her colleagues received funding from Amgen, Inc., to perform this study.
A retrospective study has revealed new potential risk factors for chemotherapy-induced febrile neutropenia (FN) in patients with solid tumors and non-Hodgkin lymphoma (NHL).
Researchers found the timing and duration of corticosteroid use were both associated with FN.
The team also observed “marginal” associations between FN and certain dermatologic and mucosal conditions as well as the use of intravenous (IV) antibiotics before chemotherapy.
On the other hand, there was no association between oral antibiotic use and FN or between radiation therapy (RT) and FN.
Chun Rebecca Chao, PhD, of Kaiser Permanente Southern California in Pasadena, and her colleagues reported these findings in JNCCN.
“Febrile neutropenia is life-threatening and often requires hospitalization,” Dr. Chao noted. “Furthermore, FN can lead to chemotherapy dose delay and dose reduction, which, in turn, negatively impacts antitumor efficacy. However, it can be prevented if high-risk individuals are identified and treated prophylactically.”
With this in mind, Dr. Chao and her colleagues set out to identify novel risk factors for FN by analyzing 15,971 patients who were treated with myelosuppressive chemotherapy at Kaiser Permanente Southern California between 2000 and 2009.
Patients had been diagnosed with NHL (n=1,617) or breast (n=6,323), lung (n=3,584), colorectal (n=3,062), ovarian (n=924), or gastric (n=461) cancers.
In all, 4.3% of patients developed FN during their first cycle of chemotherapy.
Corticosteroid use
The researchers found corticosteroid use was associated with an increased risk of FN in a propensity score-adjusted (PSA) model (adjusted for age, sex, socioeconomic factors, comorbidities, etc.). The hazard ratio (HR) was 1.53 (95% CI, 1.17-1.98; P<0.01) for patients who received corticosteroids.
A longer duration of corticosteroid use was associated with a greater risk of FN. The adjusted HR (compared to no corticosteroid use) was:
- 1.78 for corticosteroid treatment lasting less than 15 days (P<0.01)
- 1.84 for treatment lasting 15 to 29 days (P<0.01)
- 2.27 for treatment lasting 30 to 44 days (P<0.01)
- 2.86 for treatment lasting 45 to 90 days (P<0.01).
More recent corticosteroid use was associated with a greater risk of FN as well. The adjusted HR was:
- 1.88 for corticosteroid treatment less than 15 days before chemotherapy (P<0.01)
- 1.13 for treatment 15 to 29 days before chemotherapy (P=0.72)
- 1.22 for treatment 30 to 44 days before chemotherapy (P=0.66)
- 1.41 for treatment 45 to 90 days before chemotherapy (P=0.32).
“One way to reduce the incidence rate for FN could be to schedule prior corticosteroid use and subsequent chemotherapy with at least 2 weeks between them, given the magnitude of the risk increase and prevalence of this risk factor,” Dr. Chao said.
Other potential risk factors
The researchers found a “marginally” increased risk of FN in patients with certain dermatologic conditions (dermatitis, psoriasis, pruritus, etc.) and mucosal conditions (gastritis, stomatitis, mucositis, etc.).
In the PSA model, the HR was 1.40 (95% CI, 0.98-1.93; P=0.05) for patients with these conditions.
IV antibiotic use was also found to be marginally associated with an increased risk of FN in a restricted analysis covering patients treated in 2008 and 2009. In the PSA model, the HR was 1.35 (95% CI, 0.97-1.87; P=0.08).
On the other hand, there was no association between FN and oral antibiotic use in the restricted analysis. In the PSA model, the HR was 1.07 (95% CI, 0.77-1.48; P=0.70) for patients who received oral antibiotics.
Dr. Chao and her colleagues said these results suggest IV antibiotics may have a more profound impact than oral antibiotics on the balance of bacterial flora and other immune functions. Another possible explanation is that patients who received IV antibiotics were generally sicker and more prone to severe infection than patients who received oral antibiotics.
As with oral antibiotics, the researchers found no association between FN and the following factors (with the PSA model):
- Prior surgery (HR=0.89; 95% CI, 0.72-1.11; P=0.30)
- Prior RT (HR=0.91; 95% CI, 0.64-1.27; P=0.61)
- Concurrent RT (HR=1.32; 95% CI, 0.69-2.37; P=0.37).
The researchers noted that they did not account for radiation field or dose in this study, so additional evaluation of RT as a risk factor is needed.
In closing, Dr. Chao and her colleagues said these results suggest corticosteroid use, IV antibiotics, and certain dermatologic and mucosal conditions should be taken into consideration when monitoring patients receiving myelosuppressive chemotherapy and when evaluating the need for prophylactic granulocyte colony-stimulating factor or chemotherapy dose reduction.
Dr. Chao and her colleagues received funding from Amgen, Inc., to perform this study.
A retrospective study has revealed new potential risk factors for chemotherapy-induced febrile neutropenia (FN) in patients with solid tumors and non-Hodgkin lymphoma (NHL).
Researchers found the timing and duration of corticosteroid use were both associated with FN.
The team also observed “marginal” associations between FN and certain dermatologic and mucosal conditions as well as the use of intravenous (IV) antibiotics before chemotherapy.
On the other hand, there was no association between oral antibiotic use and FN or between radiation therapy (RT) and FN.
Chun Rebecca Chao, PhD, of Kaiser Permanente Southern California in Pasadena, and her colleagues reported these findings in JNCCN.
“Febrile neutropenia is life-threatening and often requires hospitalization,” Dr. Chao noted. “Furthermore, FN can lead to chemotherapy dose delay and dose reduction, which, in turn, negatively impacts antitumor efficacy. However, it can be prevented if high-risk individuals are identified and treated prophylactically.”
With this in mind, Dr. Chao and her colleagues set out to identify novel risk factors for FN by analyzing 15,971 patients who were treated with myelosuppressive chemotherapy at Kaiser Permanente Southern California between 2000 and 2009.
Patients had been diagnosed with NHL (n=1,617) or breast (n=6,323), lung (n=3,584), colorectal (n=3,062), ovarian (n=924), or gastric (n=461) cancers.
In all, 4.3% of patients developed FN during their first cycle of chemotherapy.
Corticosteroid use
The researchers found corticosteroid use was associated with an increased risk of FN in a propensity score-adjusted (PSA) model (adjusted for age, sex, socioeconomic factors, comorbidities, etc.). The hazard ratio (HR) was 1.53 (95% CI, 1.17-1.98; P<0.01) for patients who received corticosteroids.
A longer duration of corticosteroid use was associated with a greater risk of FN. The adjusted HR (compared to no corticosteroid use) was:
- 1.78 for corticosteroid treatment lasting less than 15 days (P<0.01)
- 1.84 for treatment lasting 15 to 29 days (P<0.01)
- 2.27 for treatment lasting 30 to 44 days (P<0.01)
- 2.86 for treatment lasting 45 to 90 days (P<0.01).
More recent corticosteroid use was associated with a greater risk of FN as well. The adjusted HR was:
- 1.88 for corticosteroid treatment less than 15 days before chemotherapy (P<0.01)
- 1.13 for treatment 15 to 29 days before chemotherapy (P=0.72)
- 1.22 for treatment 30 to 44 days before chemotherapy (P=0.66)
- 1.41 for treatment 45 to 90 days before chemotherapy (P=0.32).
“One way to reduce the incidence rate for FN could be to schedule prior corticosteroid use and subsequent chemotherapy with at least 2 weeks between them, given the magnitude of the risk increase and prevalence of this risk factor,” Dr. Chao said.
Other potential risk factors
The researchers found a “marginally” increased risk of FN in patients with certain dermatologic conditions (dermatitis, psoriasis, pruritus, etc.) and mucosal conditions (gastritis, stomatitis, mucositis, etc.).
In the PSA model, the HR was 1.40 (95% CI, 0.98-1.93; P=0.05) for patients with these conditions.
IV antibiotic use was also found to be marginally associated with an increased risk of FN in a restricted analysis covering patients treated in 2008 and 2009. In the PSA model, the HR was 1.35 (95% CI, 0.97-1.87; P=0.08).
On the other hand, there was no association between FN and oral antibiotic use in the restricted analysis. In the PSA model, the HR was 1.07 (95% CI, 0.77-1.48; P=0.70) for patients who received oral antibiotics.
Dr. Chao and her colleagues said these results suggest IV antibiotics may have a more profound impact than oral antibiotics on the balance of bacterial flora and other immune functions. Another possible explanation is that patients who received IV antibiotics were generally sicker and more prone to severe infection than patients who received oral antibiotics.
As with oral antibiotics, the researchers found no association between FN and the following factors (with the PSA model):
- Prior surgery (HR=0.89; 95% CI, 0.72-1.11; P=0.30)
- Prior RT (HR=0.91; 95% CI, 0.64-1.27; P=0.61)
- Concurrent RT (HR=1.32; 95% CI, 0.69-2.37; P=0.37).
The researchers noted that they did not account for radiation field or dose in this study, so additional evaluation of RT as a risk factor is needed.
In closing, Dr. Chao and her colleagues said these results suggest corticosteroid use, IV antibiotics, and certain dermatologic and mucosal conditions should be taken into consideration when monitoring patients receiving myelosuppressive chemotherapy and when evaluating the need for prophylactic granulocyte colony-stimulating factor or chemotherapy dose reduction.
Dr. Chao and her colleagues received funding from Amgen, Inc., to perform this study.
EVI1 overexpression promotes leukemogenesis, study suggests

Preclinical research suggests the oncoprotein EVI1 can promote leukemogenesis by suppressing erythropoiesis and lymphopoiesis while shifting differentiation toward the expansion of myeloid cells.
Researchers developed a new mouse model that mimics chromosomal rearrangements at 3q26, which are associated with poor-prognosis acute myeloid leukemia (AML), myelodysplastic syndromes, and myeloproliferative neoplasms.
Using the mouse model, the team demonstrated that EVI1 overexpression distorts hematopoiesis and markedly expands premalignant myelopoiesis that eventually results in leukemic transformation.
Archibald Perkins, MD, PhD, of the University of Rochester Medical Center in New York, and his colleagues published these findings in Nature Communications.
The team demonstrated that the “myeloid-skewed phenotype” is dependent upon EVI1-binding DNA. This upregulates Spi1 and encodes the master myeloid regulator PU.1.
When the researchers knocked down Spi1, the myeloid skewing diminished.
“It’s not so pie-in-the-sky anymore,” Dr. Perkins said, “to think we can interrupt the process within the genome that leads to leukemia.”
The researchers first created a mouse model of 3q26 AML with a tetracycline-inducible allele of EVI1 by inserting tetracycline operons within the first exon. This allowed the induction of all three isoforms of EVI1.
These mice were viable and fertile but had no phenotype, which indicated that the allele functioned normally unless induced.
To assess the effect of EVI1 overexpression, the researchers transplanted oncogene-expressing bone marrow mixed 1:1 with wild-type bone marrow into recipient mice.
After confirming successful engraftment, the researchers fed the mice doxycycline-treated food to induce EVI1. The team analyzed cells in the peripheral blood and bone marrow at 10 weeks post-induction.
The researchers observed a more than two-fold expansion of the EVI1-overexpressing compartment in the mouse model.
Suppression of erythropoiesis
The researchers analyzed erythroid lineage in the transplanted mice at 2, 6, and 10 weeks post-induction and found the EVI1-overexpressing cells did not contribute effectively to erythropoiesis.
Using flow cytometry, the researchers quantitated apoptosis and proliferation in erythroid progenitors. They observed a six-fold increase in apoptosis within the erythroblasts compared to wild-type cells.
They also observed a drop in the proliferation of proerythroblasts and erythroblasts compared to wild-type.
Suppression of lymphopoiesis
The researchers observed significantly lower numbers of EVI1-overexpressing B-lineage cells within the bone marrow at 6 and 10 weeks.
And at 10 weeks post-induction, the team observed a decrease in peripheral T cells from approximately 1,800 cells/µL to approximately 750 cells/µL.
EVI1 nearly eliminated the peripheral B cells completely, they noted.
Expansion of myelopoiesis
The team reported that, at 2 weeks post-induction, the EVI1-overexpressing bone marrow and control bone marrow showed the same number of myeloid cells.
But at 6 and 10 weeks post-induction, the EVI1-overexpressing myeloid compartment expanded markedly.
The researchers aged a cohort of five mice transplanted with the 1:1 mix of wild-type and EVI1 bone marrow cells to determine if chronic overexpression of EVI1 results in leukemia.
All five mice died at 90 to 119 days of doxycycline treatment. Analysis revealed AML in all mice. Bone marrows were replete with blasts, and the peripheral blood revealed severe anemia.
The researchers then proceeded to establish the relationship between EVI1 and Spi1/PU.1 transcriptional regulation.
They documented binding of EVI1 to the regulatory element -14kbURE, which, together with EVI1., induced upregulation of PU.1.
When the team knocked down PU.1, myeloid skewing diminished. This, they say, indicates PU.1 is necessary for EVI1-induced myeloid expansion.
Funding for this research was provided by the National Institutes of Health, New York State Stem Cell Science, the Wilmot Cancer Institute, and the Clinical and Translational Science Institute at the University of Rochester.
The authors had no competing interests to disclose.
Preclinical research suggests the oncoprotein EVI1 can promote leukemogenesis by suppressing erythropoiesis and lymphopoiesis while shifting differentiation toward the expansion of myeloid cells.
Researchers developed a new mouse model that mimics chromosomal rearrangements at 3q26, which are associated with poor-prognosis acute myeloid leukemia (AML), myelodysplastic syndromes, and myeloproliferative neoplasms.
Using the mouse model, the team demonstrated that EVI1 overexpression distorts hematopoiesis and markedly expands premalignant myelopoiesis that eventually results in leukemic transformation.
Archibald Perkins, MD, PhD, of the University of Rochester Medical Center in New York, and his colleagues published these findings in Nature Communications.
The team demonstrated that the “myeloid-skewed phenotype” is dependent upon EVI1-binding DNA. This upregulates Spi1 and encodes the master myeloid regulator PU.1.
When the researchers knocked down Spi1, the myeloid skewing diminished.
“It’s not so pie-in-the-sky anymore,” Dr. Perkins said, “to think we can interrupt the process within the genome that leads to leukemia.”
The researchers first created a mouse model of 3q26 AML with a tetracycline-inducible allele of EVI1 by inserting tetracycline operons within the first exon. This allowed the induction of all three isoforms of EVI1.
These mice were viable and fertile but had no phenotype, which indicated that the allele functioned normally unless induced.
To assess the effect of EVI1 overexpression, the researchers transplanted oncogene-expressing bone marrow mixed 1:1 with wild-type bone marrow into recipient mice.
After confirming successful engraftment, the researchers fed the mice doxycycline-treated food to induce EVI1. The team analyzed cells in the peripheral blood and bone marrow at 10 weeks post-induction.
The researchers observed a more than two-fold expansion of the EVI1-overexpressing compartment in the mouse model.
Suppression of erythropoiesis
The researchers analyzed erythroid lineage in the transplanted mice at 2, 6, and 10 weeks post-induction and found the EVI1-overexpressing cells did not contribute effectively to erythropoiesis.
Using flow cytometry, the researchers quantitated apoptosis and proliferation in erythroid progenitors. They observed a six-fold increase in apoptosis within the erythroblasts compared to wild-type cells.
They also observed a drop in the proliferation of proerythroblasts and erythroblasts compared to wild-type.
Suppression of lymphopoiesis
The researchers observed significantly lower numbers of EVI1-overexpressing B-lineage cells within the bone marrow at 6 and 10 weeks.
And at 10 weeks post-induction, the team observed a decrease in peripheral T cells from approximately 1,800 cells/µL to approximately 750 cells/µL.
EVI1 nearly eliminated the peripheral B cells completely, they noted.
Expansion of myelopoiesis
The team reported that, at 2 weeks post-induction, the EVI1-overexpressing bone marrow and control bone marrow showed the same number of myeloid cells.
But at 6 and 10 weeks post-induction, the EVI1-overexpressing myeloid compartment expanded markedly.
The researchers aged a cohort of five mice transplanted with the 1:1 mix of wild-type and EVI1 bone marrow cells to determine if chronic overexpression of EVI1 results in leukemia.
All five mice died at 90 to 119 days of doxycycline treatment. Analysis revealed AML in all mice. Bone marrows were replete with blasts, and the peripheral blood revealed severe anemia.
The researchers then proceeded to establish the relationship between EVI1 and Spi1/PU.1 transcriptional regulation.
They documented binding of EVI1 to the regulatory element -14kbURE, which, together with EVI1., induced upregulation of PU.1.
When the team knocked down PU.1, myeloid skewing diminished. This, they say, indicates PU.1 is necessary for EVI1-induced myeloid expansion.
Funding for this research was provided by the National Institutes of Health, New York State Stem Cell Science, the Wilmot Cancer Institute, and the Clinical and Translational Science Institute at the University of Rochester.
The authors had no competing interests to disclose.
Preclinical research suggests the oncoprotein EVI1 can promote leukemogenesis by suppressing erythropoiesis and lymphopoiesis while shifting differentiation toward the expansion of myeloid cells.
Researchers developed a new mouse model that mimics chromosomal rearrangements at 3q26, which are associated with poor-prognosis acute myeloid leukemia (AML), myelodysplastic syndromes, and myeloproliferative neoplasms.
Using the mouse model, the team demonstrated that EVI1 overexpression distorts hematopoiesis and markedly expands premalignant myelopoiesis that eventually results in leukemic transformation.
Archibald Perkins, MD, PhD, of the University of Rochester Medical Center in New York, and his colleagues published these findings in Nature Communications.
The team demonstrated that the “myeloid-skewed phenotype” is dependent upon EVI1-binding DNA. This upregulates Spi1 and encodes the master myeloid regulator PU.1.
When the researchers knocked down Spi1, the myeloid skewing diminished.
“It’s not so pie-in-the-sky anymore,” Dr. Perkins said, “to think we can interrupt the process within the genome that leads to leukemia.”
The researchers first created a mouse model of 3q26 AML with a tetracycline-inducible allele of EVI1 by inserting tetracycline operons within the first exon. This allowed the induction of all three isoforms of EVI1.
These mice were viable and fertile but had no phenotype, which indicated that the allele functioned normally unless induced.
To assess the effect of EVI1 overexpression, the researchers transplanted oncogene-expressing bone marrow mixed 1:1 with wild-type bone marrow into recipient mice.
After confirming successful engraftment, the researchers fed the mice doxycycline-treated food to induce EVI1. The team analyzed cells in the peripheral blood and bone marrow at 10 weeks post-induction.
The researchers observed a more than two-fold expansion of the EVI1-overexpressing compartment in the mouse model.
Suppression of erythropoiesis
The researchers analyzed erythroid lineage in the transplanted mice at 2, 6, and 10 weeks post-induction and found the EVI1-overexpressing cells did not contribute effectively to erythropoiesis.
Using flow cytometry, the researchers quantitated apoptosis and proliferation in erythroid progenitors. They observed a six-fold increase in apoptosis within the erythroblasts compared to wild-type cells.
They also observed a drop in the proliferation of proerythroblasts and erythroblasts compared to wild-type.
Suppression of lymphopoiesis
The researchers observed significantly lower numbers of EVI1-overexpressing B-lineage cells within the bone marrow at 6 and 10 weeks.
And at 10 weeks post-induction, the team observed a decrease in peripheral T cells from approximately 1,800 cells/µL to approximately 750 cells/µL.
EVI1 nearly eliminated the peripheral B cells completely, they noted.
Expansion of myelopoiesis
The team reported that, at 2 weeks post-induction, the EVI1-overexpressing bone marrow and control bone marrow showed the same number of myeloid cells.
But at 6 and 10 weeks post-induction, the EVI1-overexpressing myeloid compartment expanded markedly.
The researchers aged a cohort of five mice transplanted with the 1:1 mix of wild-type and EVI1 bone marrow cells to determine if chronic overexpression of EVI1 results in leukemia.
All five mice died at 90 to 119 days of doxycycline treatment. Analysis revealed AML in all mice. Bone marrows were replete with blasts, and the peripheral blood revealed severe anemia.
The researchers then proceeded to establish the relationship between EVI1 and Spi1/PU.1 transcriptional regulation.
They documented binding of EVI1 to the regulatory element -14kbURE, which, together with EVI1., induced upregulation of PU.1.
When the team knocked down PU.1, myeloid skewing diminished. This, they say, indicates PU.1 is necessary for EVI1-induced myeloid expansion.
Funding for this research was provided by the National Institutes of Health, New York State Stem Cell Science, the Wilmot Cancer Institute, and the Clinical and Translational Science Institute at the University of Rochester.
The authors had no competing interests to disclose.
Palliative care guidelines relevant for hematologists, doc says

The latest edition of the national palliative care guidelines provides new clinical strategies relevant to hematology practice in the United States, according to a physician-researcher specializing in hematology.
The Clinical Practice Guidelines for Quality Palliative Care, 4th edition, represents a “blueprint for what it looks like to provide high-quality, comprehensive palliative care to people with serious illness,” said Thomas W. LeBlanc, MD, a physician-researcher at Duke University School of Medicine in Durham, North Carolina.
However, unlike previous editions, this update to the guidelines emphasizes the importance of palliative care provided by both primary care and specialty care clinicians.
“Part of this report is about trying to raise the game of everybody in medicine and provide a higher basic level of primary palliative care to all people with serious illness, but then also to figure out who has higher levels of needs where the specialists should be applied, since they are a scarce resource,” Dr. LeBlanc said.
The latest edition helps establish a foundation for gold standard palliative care for people living with serious illness, regardless of diagnosis, prognosis, setting, or age, according to The National Coalition for Hospice and Palliative Care, which published the clinical practice guidelines.
The update was developed by the National Consensus Project for Quality Palliative Care (NCP), which includes 16 national organizations with palliative care and hospice expertise, and is endorsed by more than 80 national organizations, including the American Society of Hematology.
One key reason for the update, according to NCP, was to acknowledge that today’s healthcare system may not be meeting patients’ palliative care needs.
Specifically, the guidelines call on clinicians who don’t practice palliative care to integrate palliative care principles into their routine assessment of seriously ill patients with conditions such as heart failure, lung disease, and cancer.
That differs from the way palliative care is traditionally practiced, in which specially trained doctors, nurses, and other specialists provide that support.
An issue with that traditional model is a shortage of specialized clinicians to meet palliative care needs, said Dr. LeBlanc, whose clinical practice and research focuses on palliative care needs of patients with hematologic malignancies.
“Palliative care has matured as a field such that we are now actually facing workforce shortage issues and really fundamental questions about who really needs us the most and how we increase our reach to improve the lives of more patients and families facing serious illness,” he said.
That’s a major driver behind the emphasis in the latest guidelines on providing palliative care in the community, coordinating care, and dealing with care transitions, Dr. LeBlanc added.
“I hope that this document will help to demonstrate the value and the need for palliative care specialists and for improvements in primary care in the care of patients with hematologic diseases in general,” he said. “To me, this adds increasing legitimacy to this whole field.”
The latest edition of the national palliative care guidelines provides new clinical strategies relevant to hematology practice in the United States, according to a physician-researcher specializing in hematology.
The Clinical Practice Guidelines for Quality Palliative Care, 4th edition, represents a “blueprint for what it looks like to provide high-quality, comprehensive palliative care to people with serious illness,” said Thomas W. LeBlanc, MD, a physician-researcher at Duke University School of Medicine in Durham, North Carolina.
However, unlike previous editions, this update to the guidelines emphasizes the importance of palliative care provided by both primary care and specialty care clinicians.
“Part of this report is about trying to raise the game of everybody in medicine and provide a higher basic level of primary palliative care to all people with serious illness, but then also to figure out who has higher levels of needs where the specialists should be applied, since they are a scarce resource,” Dr. LeBlanc said.
The latest edition helps establish a foundation for gold standard palliative care for people living with serious illness, regardless of diagnosis, prognosis, setting, or age, according to The National Coalition for Hospice and Palliative Care, which published the clinical practice guidelines.
The update was developed by the National Consensus Project for Quality Palliative Care (NCP), which includes 16 national organizations with palliative care and hospice expertise, and is endorsed by more than 80 national organizations, including the American Society of Hematology.
One key reason for the update, according to NCP, was to acknowledge that today’s healthcare system may not be meeting patients’ palliative care needs.
Specifically, the guidelines call on clinicians who don’t practice palliative care to integrate palliative care principles into their routine assessment of seriously ill patients with conditions such as heart failure, lung disease, and cancer.
That differs from the way palliative care is traditionally practiced, in which specially trained doctors, nurses, and other specialists provide that support.
An issue with that traditional model is a shortage of specialized clinicians to meet palliative care needs, said Dr. LeBlanc, whose clinical practice and research focuses on palliative care needs of patients with hematologic malignancies.
“Palliative care has matured as a field such that we are now actually facing workforce shortage issues and really fundamental questions about who really needs us the most and how we increase our reach to improve the lives of more patients and families facing serious illness,” he said.
That’s a major driver behind the emphasis in the latest guidelines on providing palliative care in the community, coordinating care, and dealing with care transitions, Dr. LeBlanc added.
“I hope that this document will help to demonstrate the value and the need for palliative care specialists and for improvements in primary care in the care of patients with hematologic diseases in general,” he said. “To me, this adds increasing legitimacy to this whole field.”
The latest edition of the national palliative care guidelines provides new clinical strategies relevant to hematology practice in the United States, according to a physician-researcher specializing in hematology.
The Clinical Practice Guidelines for Quality Palliative Care, 4th edition, represents a “blueprint for what it looks like to provide high-quality, comprehensive palliative care to people with serious illness,” said Thomas W. LeBlanc, MD, a physician-researcher at Duke University School of Medicine in Durham, North Carolina.
However, unlike previous editions, this update to the guidelines emphasizes the importance of palliative care provided by both primary care and specialty care clinicians.
“Part of this report is about trying to raise the game of everybody in medicine and provide a higher basic level of primary palliative care to all people with serious illness, but then also to figure out who has higher levels of needs where the specialists should be applied, since they are a scarce resource,” Dr. LeBlanc said.
The latest edition helps establish a foundation for gold standard palliative care for people living with serious illness, regardless of diagnosis, prognosis, setting, or age, according to The National Coalition for Hospice and Palliative Care, which published the clinical practice guidelines.
The update was developed by the National Consensus Project for Quality Palliative Care (NCP), which includes 16 national organizations with palliative care and hospice expertise, and is endorsed by more than 80 national organizations, including the American Society of Hematology.
One key reason for the update, according to NCP, was to acknowledge that today’s healthcare system may not be meeting patients’ palliative care needs.
Specifically, the guidelines call on clinicians who don’t practice palliative care to integrate palliative care principles into their routine assessment of seriously ill patients with conditions such as heart failure, lung disease, and cancer.
That differs from the way palliative care is traditionally practiced, in which specially trained doctors, nurses, and other specialists provide that support.
An issue with that traditional model is a shortage of specialized clinicians to meet palliative care needs, said Dr. LeBlanc, whose clinical practice and research focuses on palliative care needs of patients with hematologic malignancies.
“Palliative care has matured as a field such that we are now actually facing workforce shortage issues and really fundamental questions about who really needs us the most and how we increase our reach to improve the lives of more patients and families facing serious illness,” he said.
That’s a major driver behind the emphasis in the latest guidelines on providing palliative care in the community, coordinating care, and dealing with care transitions, Dr. LeBlanc added.
“I hope that this document will help to demonstrate the value and the need for palliative care specialists and for improvements in primary care in the care of patients with hematologic diseases in general,” he said. “To me, this adds increasing legitimacy to this whole field.”
Testing platelets reduces waste, cuts costs
BOSTON—Rapid bacterial testing of platelets in a hospital blood bank can result in both significant cost savings and reduced wastage of blood products, according to investigators.
In a single-center study, rapid bacterial testing of 6- or 7-day-old apheresis platelets resulted in projected annual cost savings of nearly $89,000 and cut the rate of platelet wastage from expiration by more than half.
Adam L. Booth, MD, of the University of Texas in Galveston, and his colleagues described this study in a poster presentation at AABB 2018 (abstract INV4).
Platelets typically have a shelf life of 5 days because longer storage increases the risk for bacterial growth and the potential for transfusion-transmitted infections, Dr. Booth and his colleagues noted.
In March 2016, the U.S. Food and Drug Administration (FDA) published a draft guidance proposing a change in regulations to allow for an extended shelf life if platelets are collected in an FDA-approved, 7-day storage container with labeling that requires testing every product with a bacterial detection device, or if the platelets are individually tested for bacterial detection using an approved device.
Dr. Booth and his colleagues wanted to see what effect the regulations, if implemented as expected, might have on acquisition costs and wastage of apheresis platelets.
The investigators reviewed their center’s platelet acquisition costs and wastage from expiration 12 months before and 6 months after implementation of a rapid bacterial testing protocol, with 6-month results projected out to 1 year for comparison purposes.
The team looked at data on bacterial testing of 6-day and 7-day-old apheresis platelets, and they excluded data on platelet units that were due to expire on day 5 because they were not stored in FDA-approved containers.
Prior to testing, the annual wastage rate was 24%, or 332 of 1,371 platelet units purchased. Using a mean per-unit cost of $516.96, the annual cost was more than $171,000.
After the start of testing, the annualized rate of wastage dropped to 10%, or 117 of 1,168 platelet units. So the annualized cost was more than $60,000.
The difference in cost—minus the cost of rapid bacterial testing (roughly $22,500)—resulted in an annual savings for the institution of nearly $89,000.
The number of units transfused and the associated costs of transfusions were similar between the time periods studied.
This study was internally funded. The authors reported having no conflicts of interest.
BOSTON—Rapid bacterial testing of platelets in a hospital blood bank can result in both significant cost savings and reduced wastage of blood products, according to investigators.
In a single-center study, rapid bacterial testing of 6- or 7-day-old apheresis platelets resulted in projected annual cost savings of nearly $89,000 and cut the rate of platelet wastage from expiration by more than half.
Adam L. Booth, MD, of the University of Texas in Galveston, and his colleagues described this study in a poster presentation at AABB 2018 (abstract INV4).
Platelets typically have a shelf life of 5 days because longer storage increases the risk for bacterial growth and the potential for transfusion-transmitted infections, Dr. Booth and his colleagues noted.
In March 2016, the U.S. Food and Drug Administration (FDA) published a draft guidance proposing a change in regulations to allow for an extended shelf life if platelets are collected in an FDA-approved, 7-day storage container with labeling that requires testing every product with a bacterial detection device, or if the platelets are individually tested for bacterial detection using an approved device.
Dr. Booth and his colleagues wanted to see what effect the regulations, if implemented as expected, might have on acquisition costs and wastage of apheresis platelets.
The investigators reviewed their center’s platelet acquisition costs and wastage from expiration 12 months before and 6 months after implementation of a rapid bacterial testing protocol, with 6-month results projected out to 1 year for comparison purposes.
The team looked at data on bacterial testing of 6-day and 7-day-old apheresis platelets, and they excluded data on platelet units that were due to expire on day 5 because they were not stored in FDA-approved containers.
Prior to testing, the annual wastage rate was 24%, or 332 of 1,371 platelet units purchased. Using a mean per-unit cost of $516.96, the annual cost was more than $171,000.
After the start of testing, the annualized rate of wastage dropped to 10%, or 117 of 1,168 platelet units. So the annualized cost was more than $60,000.
The difference in cost—minus the cost of rapid bacterial testing (roughly $22,500)—resulted in an annual savings for the institution of nearly $89,000.
The number of units transfused and the associated costs of transfusions were similar between the time periods studied.
This study was internally funded. The authors reported having no conflicts of interest.
BOSTON—Rapid bacterial testing of platelets in a hospital blood bank can result in both significant cost savings and reduced wastage of blood products, according to investigators.
In a single-center study, rapid bacterial testing of 6- or 7-day-old apheresis platelets resulted in projected annual cost savings of nearly $89,000 and cut the rate of platelet wastage from expiration by more than half.
Adam L. Booth, MD, of the University of Texas in Galveston, and his colleagues described this study in a poster presentation at AABB 2018 (abstract INV4).
Platelets typically have a shelf life of 5 days because longer storage increases the risk for bacterial growth and the potential for transfusion-transmitted infections, Dr. Booth and his colleagues noted.
In March 2016, the U.S. Food and Drug Administration (FDA) published a draft guidance proposing a change in regulations to allow for an extended shelf life if platelets are collected in an FDA-approved, 7-day storage container with labeling that requires testing every product with a bacterial detection device, or if the platelets are individually tested for bacterial detection using an approved device.
Dr. Booth and his colleagues wanted to see what effect the regulations, if implemented as expected, might have on acquisition costs and wastage of apheresis platelets.
The investigators reviewed their center’s platelet acquisition costs and wastage from expiration 12 months before and 6 months after implementation of a rapid bacterial testing protocol, with 6-month results projected out to 1 year for comparison purposes.
The team looked at data on bacterial testing of 6-day and 7-day-old apheresis platelets, and they excluded data on platelet units that were due to expire on day 5 because they were not stored in FDA-approved containers.
Prior to testing, the annual wastage rate was 24%, or 332 of 1,371 platelet units purchased. Using a mean per-unit cost of $516.96, the annual cost was more than $171,000.
After the start of testing, the annualized rate of wastage dropped to 10%, or 117 of 1,168 platelet units. So the annualized cost was more than $60,000.
The difference in cost—minus the cost of rapid bacterial testing (roughly $22,500)—resulted in an annual savings for the institution of nearly $89,000.
The number of units transfused and the associated costs of transfusions were similar between the time periods studied.
This study was internally funded. The authors reported having no conflicts of interest.
Team finds potential therapeutic target for AML
Researchers have found the cancer-associated pseudokinase Tribbles 2 (TRIB2) to be a potential therapeutic target in solid tumors and blood cancers, including acute myeloid leukemia (AML).
Previous research had described TRIB2 as a target of small-molecule protein kinase inhibitors originally designed to interfere with kinase domains of the epidermal growth factor receptor (EGFR) tyrosine kinase family.
Using a thermal shift assay, the team discovered TRIB2-binding compounds within the Published Kinase Inhibitor Set (PKIS). They then employed a biochemical drug repurposing approach to classify compounds that either stabilized or destabilized TRIB2 in vitro.
The researchers found that afatinib, which is already approved by the U.S. Food and Drug Administration to treat non-small cell lung cancer, led to rapid TRIB2 degradation in human AML cells.
Patrick A. Eyers, PhD, of the University of Liverpool in the U.K., and his colleagues published their findings in Science Signaling.
The team found afatinib to be relatively specific for EGFR and human epidermal growth factor receptor 2 (HER2) at nanomolar concentrations in cells.
The researchers confirmed that at least two TRIB2 Cys residues interact with afatinib in vitro.
The team also discovered TRIB2 could be destabilized by neratinib and osimertinib in vitro.
“Our data prove that the cellular mechanism by which TRIB2 stability is regulated by compounds is proteasome-based,” the researchers wrote, “and we speculate that an afatinib-induced conformational change might induce TRIB2 ubiquitination.”
The researchers plan to study further TRIB2 small-molecule interactions with dynamic changes in ubiquitination status.
Furthermore, they report their work demonstrates that covalent inhibitors such as afatinib have TRIB2-degrading activity in human cells at micromolar concentrations.
The researchers determined that afatinib has similar efficacy to the TRIB2-destabilizing quinazoline neratinib at similar ranges.
The team believes their data “raise the intriguing possibility that clinical inhibitors might be used as TRIB2-degrading agents in research, and possibly clinical, contexts.”
“A long-standing goal in cancer research is drug-induced degradation of oncogenic proteins,” Dr. Eyers commented. “Our study highlights how information obtained with ‘off-target’ effects of known drugs is potentially useful because it might be exploited in the future to help eliminate a protein that is involved in a completely different type of cancer.”
The TRIB proteins play many diverse roles in cell signaling, development, and cancer. According to a paper in Developmental Dynamics, they were named after the small, round, fictional organisms from the original Star Trek television series. Their major role was to eat and reproduce.
This work was funded by two U.K. Biotechnology and Biological Sciences Research Council Doctoral Training Partnership studentships, a Tools and Resources Development Fund award, Royal Society Research Grants, North West Cancer Research grants, and funding from the National Institutes of Health.
The authors disclosed no perceived conflicts of interest, although several authors are affiliated with the Structural Genomics Consortium at the University of North Carolina at Chapel Hill, which receives direct funds from various pharmaceutical companies but remains entirely independent.
Researchers have found the cancer-associated pseudokinase Tribbles 2 (TRIB2) to be a potential therapeutic target in solid tumors and blood cancers, including acute myeloid leukemia (AML).
Previous research had described TRIB2 as a target of small-molecule protein kinase inhibitors originally designed to interfere with kinase domains of the epidermal growth factor receptor (EGFR) tyrosine kinase family.
Using a thermal shift assay, the team discovered TRIB2-binding compounds within the Published Kinase Inhibitor Set (PKIS). They then employed a biochemical drug repurposing approach to classify compounds that either stabilized or destabilized TRIB2 in vitro.
The researchers found that afatinib, which is already approved by the U.S. Food and Drug Administration to treat non-small cell lung cancer, led to rapid TRIB2 degradation in human AML cells.
Patrick A. Eyers, PhD, of the University of Liverpool in the U.K., and his colleagues published their findings in Science Signaling.
The team found afatinib to be relatively specific for EGFR and human epidermal growth factor receptor 2 (HER2) at nanomolar concentrations in cells.
The researchers confirmed that at least two TRIB2 Cys residues interact with afatinib in vitro.
The team also discovered TRIB2 could be destabilized by neratinib and osimertinib in vitro.
“Our data prove that the cellular mechanism by which TRIB2 stability is regulated by compounds is proteasome-based,” the researchers wrote, “and we speculate that an afatinib-induced conformational change might induce TRIB2 ubiquitination.”
The researchers plan to study further TRIB2 small-molecule interactions with dynamic changes in ubiquitination status.
Furthermore, they report their work demonstrates that covalent inhibitors such as afatinib have TRIB2-degrading activity in human cells at micromolar concentrations.
The researchers determined that afatinib has similar efficacy to the TRIB2-destabilizing quinazoline neratinib at similar ranges.
The team believes their data “raise the intriguing possibility that clinical inhibitors might be used as TRIB2-degrading agents in research, and possibly clinical, contexts.”
“A long-standing goal in cancer research is drug-induced degradation of oncogenic proteins,” Dr. Eyers commented. “Our study highlights how information obtained with ‘off-target’ effects of known drugs is potentially useful because it might be exploited in the future to help eliminate a protein that is involved in a completely different type of cancer.”
The TRIB proteins play many diverse roles in cell signaling, development, and cancer. According to a paper in Developmental Dynamics, they were named after the small, round, fictional organisms from the original Star Trek television series. Their major role was to eat and reproduce.
This work was funded by two U.K. Biotechnology and Biological Sciences Research Council Doctoral Training Partnership studentships, a Tools and Resources Development Fund award, Royal Society Research Grants, North West Cancer Research grants, and funding from the National Institutes of Health.
The authors disclosed no perceived conflicts of interest, although several authors are affiliated with the Structural Genomics Consortium at the University of North Carolina at Chapel Hill, which receives direct funds from various pharmaceutical companies but remains entirely independent.
Researchers have found the cancer-associated pseudokinase Tribbles 2 (TRIB2) to be a potential therapeutic target in solid tumors and blood cancers, including acute myeloid leukemia (AML).
Previous research had described TRIB2 as a target of small-molecule protein kinase inhibitors originally designed to interfere with kinase domains of the epidermal growth factor receptor (EGFR) tyrosine kinase family.
Using a thermal shift assay, the team discovered TRIB2-binding compounds within the Published Kinase Inhibitor Set (PKIS). They then employed a biochemical drug repurposing approach to classify compounds that either stabilized or destabilized TRIB2 in vitro.
The researchers found that afatinib, which is already approved by the U.S. Food and Drug Administration to treat non-small cell lung cancer, led to rapid TRIB2 degradation in human AML cells.
Patrick A. Eyers, PhD, of the University of Liverpool in the U.K., and his colleagues published their findings in Science Signaling.
The team found afatinib to be relatively specific for EGFR and human epidermal growth factor receptor 2 (HER2) at nanomolar concentrations in cells.
The researchers confirmed that at least two TRIB2 Cys residues interact with afatinib in vitro.
The team also discovered TRIB2 could be destabilized by neratinib and osimertinib in vitro.
“Our data prove that the cellular mechanism by which TRIB2 stability is regulated by compounds is proteasome-based,” the researchers wrote, “and we speculate that an afatinib-induced conformational change might induce TRIB2 ubiquitination.”
The researchers plan to study further TRIB2 small-molecule interactions with dynamic changes in ubiquitination status.
Furthermore, they report their work demonstrates that covalent inhibitors such as afatinib have TRIB2-degrading activity in human cells at micromolar concentrations.
The researchers determined that afatinib has similar efficacy to the TRIB2-destabilizing quinazoline neratinib at similar ranges.
The team believes their data “raise the intriguing possibility that clinical inhibitors might be used as TRIB2-degrading agents in research, and possibly clinical, contexts.”
“A long-standing goal in cancer research is drug-induced degradation of oncogenic proteins,” Dr. Eyers commented. “Our study highlights how information obtained with ‘off-target’ effects of known drugs is potentially useful because it might be exploited in the future to help eliminate a protein that is involved in a completely different type of cancer.”
The TRIB proteins play many diverse roles in cell signaling, development, and cancer. According to a paper in Developmental Dynamics, they were named after the small, round, fictional organisms from the original Star Trek television series. Their major role was to eat and reproduce.
This work was funded by two U.K. Biotechnology and Biological Sciences Research Council Doctoral Training Partnership studentships, a Tools and Resources Development Fund award, Royal Society Research Grants, North West Cancer Research grants, and funding from the National Institutes of Health.
The authors disclosed no perceived conflicts of interest, although several authors are affiliated with the Structural Genomics Consortium at the University of North Carolina at Chapel Hill, which receives direct funds from various pharmaceutical companies but remains entirely independent.