User login
INTRODUCTION
In the normal person, the process of coagulation is finely controlled at many levels to ensure the appropriate amount of hemostasis at the appropriate location. Broadly defined, disseminated intravascular coagulation (DIC) is the name given to any process that disrupts this fine tuning, leading to unregulated coagulation. Defined this way, DIC may be found in a variety of patients with a variety of disease states, and can present with a spectrum of findings ranging from asymptomatic abnormal laboratory results to florid bleeding or thrombosis. It is important to remember that DIC is always a consequence of an underlying pathological process and not a disease in and of itself. This article first reviews concepts common to all forms of DIC, and then reviews the more common disease states that lead to DIC.
PATHOGENESIS
At the most basic level, DIC is the clinical manifestation of inappropriate thrombin activation.1–5 Inappropriate thrombin activation can be due to underlying conditions such as sepsis and obstetric disasters. The activation of thrombin leads to (1) conversion of fibrinogen to fibrin, (2) activation of platelets (and their consumption), (3) activation of factors V and VIII, (4) activation of protein C (and degradation of factors Va and VIIIa), (5) activation of endothelial cells, and (6) activation of fibrinolysis (Table 1). 
1. Conversion of fibrinogen to fibrin, which leads to the formation of fibrin monomers and excessive thrombus formation. These thrombi are rapidly dissolved by excessive fibrinolysis in most patients. In certain clinical situations, especially cancer, excessive thrombosis will occur. In patients with cancer, this is most often a deep venous thrombosis. Rare patients, especially those with pancreatic cancer, may have severe DIC with multiple arterial and venous thromboses. Nonbacterial thrombotic endocarditis can also be seen in these patients, leading to widespread embolic complications.
2. Activation of platelets and their consumption. Thrombin is the most potent physiologic activator of platelets, so in DIC there is increased activation of platelets. These activated platelets are consumed, resulting in thrombocytopenia. Platelet dysfunction is also present. Platelets that have been activated and have released their contents but still circulate are known as “exhausted” platelets; these cells can no longer function to support coagulation. The fibrin degradation products (FDP) in DIC can also bind to GP IIb/IIIa and inhibit further platelet aggregation.
3. Activation of factors V, VIII, XI, and XIII. Activation of these factors can promote thrombosis, but they are then rapidly cleared by antithrombin (XI) or activated protein C (V and VIII) or by binding to the fibrin clot (XIII). This can lead to depletion of all the prothrombotic clotting factors and antithrombin, which in turn can lead to both thrombosis and bleeding.
4. Activation of protein C further promotes degradation of factors Va and VIIIa, enhances fibrinolysis, and decreases protein C levels.
5. Activation of endothelial cells, especially in the skin, may lead to thrombosis, and in certain patients, especially those with meningococcemia, purpura fulminans. Endothelial damage will down-regulate thrombomodulin, preventing activation of protein C and leading to further reductions in levels of activated protein C.56. Activation of fibrinolysis leads to the breakdown of fibrin monomers, formation of fibrin thrombi, and increased circulating fibrinogen. In most patients with DIC, the fibrinolytic response is brisk.6 This is why most patients with DIC present with bleeding and prolonged clotting times.
PATTERNS OF DIC
The clinical manifestations of DIC in a given patient depend on the balance of thrombin activation and secondary fibrinolysis plus the patient’s ability to compensate for the DIC. Patients with DIC can present in 1 of 4 patterns:1–3
1. Asymptomatic. Patients can present with laboratory evidence of DIC but no bleeding or thrombosis. This is often seen in patients with sepsis or cancer. However, with further progression of the underlying disease, these patients can rapidly become symptomatic.
2. Bleeding. The bleeding is due to a combination of factor depletion, platelet dysfunction, thrombocytopenia, and excessive fibrinolysis.1 These patients may present with diffuse bleeding from multiple sites (eg, intravenous sites, areas of instrumentation).
3. Thrombosis. Despite the general activation of the coagulation process, thrombosis is unusual in most patients with acute DIC. The exceptions include patients with cancer, trauma patients, and certain obstetrical patients. Most often the thrombosis is venous, but arterial thrombosis and nonbacterial thrombotic endocarditis have been reported.7
4. Purpura fulminans. This form of DIC is discussed in more detail later (see Specific DIC Syndromes section).
DIAGNOSIS
There is no one test that will diagnose DIC; one must match the test to the clinical situation (Table 2).8 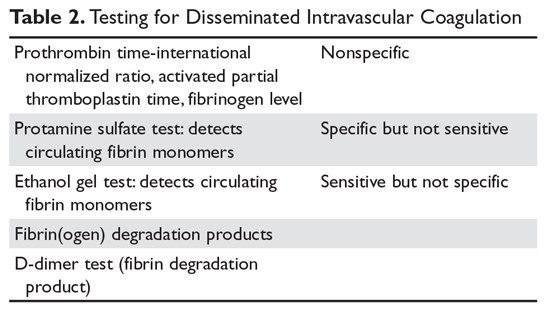
SCREENING TESTS
The prothrombin time-INR and activated thromboplastin time (aPPT) are usually elevated in severe DIC but may be normal or shortened in chronic forms.9 One may also see a shortened aPTT in severe acute DIC due to large amounts of activated thrombin and factor X “bypassing” the contact pathway. An aPTT as short as 10 seconds has been seen in acute DIC. The platelet count is usually reduced but may be normal in chronic DIC. Serum fibrinogen and platelets are decreased in acute DIC but again may be in the “normal” range in chronic DIC.10 The most sensitive screening test for DIC is a fall in the platelet count, with low counts seen in 98% of patients and counts under 50,000 cells/μL in 50%.9,11 The least specific test is fibrinogen, which tends to fall below normal only in severe acute DIC.9
SPECIFIC TESTS
This group of tests allows one to deduce that abnormally high concentrations of thrombin are present.
Ethanol Gel and Protamine Tests
Both of these older tests detected circulating fibrin monomers, whose appearance is an early sign of DIC. Circulating fibrin monomers are seen when thrombin acts on fibrinogen. Usually the monomer polymerizes with the fibrin clot, but when there is excess thrombin these monomers can circulate. Detection of circulating fibrin monomer means there is too much IIa and, ergo, DIC is present.
Fibrin(ogen) Degradation Products
Plasmin acts on the fibrin/fibrinogen molecule to cleave the molecule in specific places. The resulting degradation product levels will be elevated in situations of increased fibrin/fibrinogen destruction (DIC and fibrinolysis). The FDP are typically mildly elevated in renal and liver disease due to reduced clearance.
D-Dimers
When fibrin monomers bind to form a thrombus, factor XIII acts to bind their “D” domains together. This bond is resistant to plasmin and thus this degradation fragment is known as the “D-dimer.” High levels of D-dimer indicate that (1) IIa has acted on fibrinogen to form a fibrin monomer that bonded to another fibrin monomer, and (2) this thrombus was lysed by plasmin. Because D-dimers can be elevated (eg, with exercise, after surgery), an elevated D-dimer needs to be interpreted in the context of the clinical situation.11 Currently, this is the most common specific test for DIC performed.
Other Tests
Several other tests are sometimes helpful in diagnosing DIC.
Thrombin time. This test is performed by adding thrombin to plasma. Thrombin times are elevated in DIC (FDPs interfere with polymerization), in the presence of low fibrinogen levels, in dysfibrinogenemia, and in the presence of heparin (very sensitive).
Reptilase time is the same as thrombin time but is performed with a snake venom that is insensitive to heparin. Reptilase time is elevated in the same conditions as the thrombin time, with the exception of the presence of heparin. Thrombin time and reptilase time are most useful in evaluation of dysfibrinogenemia.
Prothrombin fragment 1.2 (F1.2). F1.2 is a small peptide cleaved off when prothrombin is activated to thrombin. Thus, high levels of F1.2 are found in DIC but can be seen in other thrombotic disorders. This test is still of limited clinical value.
DIC scoring system. A scoring system to both diagnose and quantify DIC has been proposed (Figure).11,12 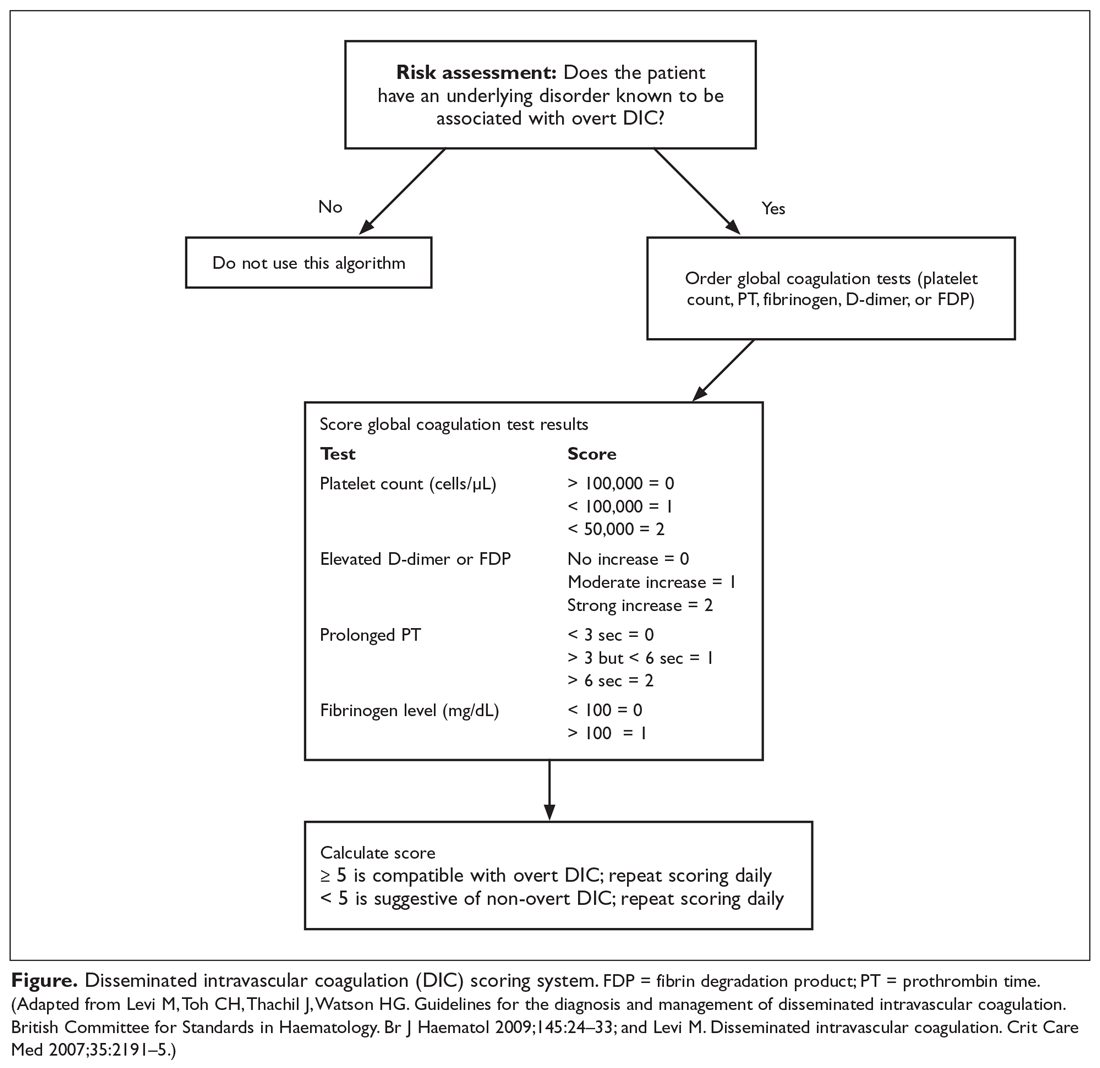
Thromboelastography (TEG). This is a point-of-care test that uses whole blood to determine specific coagulation parameters such as R time (time from start of test to clot formation), maximal amplitude (MA, maximum extent of thrombus), and LY30 (MA at 30 minutes, a measure of fibrinolysis).13 Studies have shown that TEG can identify DIC by demonstrating a shorter R time (excess thrombin generation) which prolongs as coagulation factors are consumed. The MA is decreased as fibrinogen is consumed and the LY30 shows excess fibrinolysis. TEG has been shown to be of particular value in the management of the complex coagulopathy of trauma.14
MIMICKERS OF DIC
It is important to recognize coagulation syndromes that are not DIC, especially those that have specific other therapies. The syndromes most frequently encountered are thrombotic thrombocytopenic purpura (TTP) and catastrophic antiphospholipid antibody syndrome (CAPS). One important clue to both of these syndromes is that, unlike DIC, there is no primary disorder (cancer, sepsis) that is driving the coagulation abnormalities.
TTP should be suspected when any patient presents with any combination of thrombocytopenia, microangiopathic hemolytic anemia (schistocytes and signs of hemolysis) plus end-organ damage.15–18 Patients with TTP most often present with intractable seizures, strokes, or sequelae of renal insufficiency. Many patients who present with TTP have been misdiagnosed as having sepsis, “lupus flare,” or vasculitis. The key diagnostic differentiator between TTP and DIC is the lack of activation of coagulation with TTP—fibrinogen is normal and D-dimers are minimally or not elevated. In TTP, lactate dehydrogenase is invariably elevated, often 2 to 3 times normal.19 The importance of identifying TTP is that untreated TTP is rapidly fatal. Mortality in the pre–plasma exchange era ranged from 95% to 100%. Today plasma exchange therapy is the foundation of TTP treatment and has reduced mortality to less than 20%.16,20–23Rarely patients with antiphospholipid antibody syndrome can present with fulminant multiorgan system failure.24–28 CAPS is caused by widespread microthrombi in multiple vascular fields. These patients will develop renal failure, encephalopathy, adult respiratory distress syndrome (often with pulmonary hemorrhage), cardiac failure, dramatic livedo reticularis, and worsening thrombocytopenia. Many of these patients have pre-existing autoimmune disorders and high-titer anticardiolipin antibodies. It appears that the best therapy for these patients is aggressive immunosuppression with steroids plus plasmapheresis, followed by rituximab or, if in the setting of lupus, intravenous cyclophosphamide monthly.27,29 Early recognition of CAPS can lead to quick therapy and resolution of the multiorgan system failure.
GENERAL THERAPY
The best way to treat DIC is to treat the underlying cause that is driving the thrombin generation.1,2,4,30,31 Fully addressing the underlying cause may not be possible or may take time, and in the meantime it is necessary to disrupt the cycle of thrombosis and/or hemorrhage. In the past, there was concern about using factor replacement due to fears of “feeding the fire,” or perpetuating the cycle of thrombosis. However, these concerns are not supported by evidence, and factors must be replaced if depletion occurs and bleeding ensues.32
Transfusion therapy of the patient with DIC is guided by the 5 laboratory tests that reflect the basic parameters essential for both hemostasis and blood volume status:33,34 hematocrit, platelet count, prothrombin time-INR, aPTT, and fibrinogen level. Decisions regarding replacement therapy are based on the results of these laboratory tests and the clinical situation of the patient (Table 3). 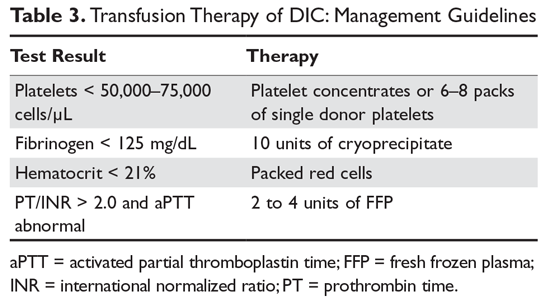
The basic 5 laboratory tests should be repeated after administering the blood products. This allows one to ensure that adequate replacement therapy was given for the coagulation defects. Frequent checks of the coagulation tests also allow rapid identification and treatment of new coagulation defects in a timely fashion. A flow chart of the test and the blood products administered should also be maintained. This is important in acute situations such as trauma or obstetrical bleeding.
In theory, since DIC is the manifestation of exuberant thrombin production, blocking thrombin with heparin should decrease or shut down DIC. However, studies have shown that in most patients heparin administration has led to excessive bleeding. Currently, heparin therapy is reserved for patients who have thrombosis as a component of their DIC.2,41,42 Given the coagulopathy that is often present, specific heparin levels instead of the aPTT should be used to monitor anticoagulation.43,44
SPECIFIC DIC SYNDROMES
SEPSIS/INFECTIOUS DISEASE
Any overwhelming infection can lead to DIC.45 Classically, it was believed that gram-negative bacteria can lead tissue factor exposure via production of endotoxin, but recent studies indicate that DIC can be seen with any overwhelming infection.46,47 There are several potential avenues by which infections can lead to DIC. As mentioned, gram-negative bacteria produce endotoxin that can directly lead to tissue factor exposure, resulting in excess thrombin generation. In addition, any infection can lead to expression of inflammatory cytokines that induce tissue-factor expression by endothelium and monocytes. Some viruses and Rickettsia species can directly infect the vascular endothelium, converting it from an antithrombotic to a prothrombotic phenotype.48 When fighting infections, neutrophils can extrude their contents, including DNA, to help trap organisms. These neutrophil extracellular traps (NETS) may play an important role in promoting coagulopathy.49,50 The hypotension produced by sepsis leads to tissue hypoxia, which results in more DIC. The coagulopathy in sepsis can range from subtle abnormalities of testing to purpura fulminans. Thrombocytopenia is worsened by cytokine-induced hemophagocytic syndrome.
As with all forms of DIC, empiric therapy targeting the most likely source of infection and maintaining hemodynamic stability is the key to therapy. As discussed below, heparin and other forms of coagulation replacement appear to be of no benefit in therapy.
PURPURA FULMINANS
DIC in association with necrosis of the skin is seen in primary and secondary purpura fulminans.51,52 Primary purpura fulminans is most often seen after a viral infection.53 In these patients, the purpura fulminans starts with a painful red area on an extremity that rapidly progresses to a black ischemic area. In many patients, acquired deficiency of protein S is found.51,54,55 Secondary purpura fulminans is most often associated with meningococcemia infections but can be seen in any patient with overwhelming infection.56–58 Post-splenectomy sepsis syndrome patients and those with functional hyposplenism due to chronic liver diseases are also at risk.59 Patients present with signs of sepsis, and the skin lesions often involve the extremities and may lead to amputations. As opposed to primary purpura fulminans, those with the secondary form will have symmetrical ischemia distally (toes and fingers) that ascends as the process progresses. Rarely, adrenal infarction (Waterhouse-Friderichsen syndrome) occurs, which leads to severe hypotension.45
Recently, Warkenten has reported on limb gangrene in critically ill patients complicating sepsis or cardiogenic shock.60,61 These patients have DIC that is complicated by shock liver. Deep venous thrombosis with ischemic gangrene then develops, which can result in tissue loss and even amputation. The pathogenesis is hypothesized to be hepatic dysfunction leading to sudden drops in protein C and S plasma levels, which then leads to thrombophilia with widespread microvascular thrombosis. Therapy for purpura fulminans is controversial. Primary purpura fulminans, especially in those with postvaricella autoimmune protein S deficiency, has responded to plasma infusion titrated to keep the protein S level above 25%.51 Intravenous immunoglobulin has also been reported to help decrease the anti-protein S antibodies. Heparin has been reported to control the DIC and extent of necrosis.62 The starting dose in these patients is 5 to 8 units/kg/hr.2
Sick patients with secondary purpura fulminans have been treated with plasma drips, plasmapheresis, and continuous plasma ultrafiltration.62–66 Heparin therapy alone has not been shown to improve survival.66 Much attention has been given to replacement of natural anticoagulants such as protein C and antithrombin as therapy for purpura fulminans, but unfortunately randomized trials using antithrombin have shown mostly negative results.51,55,67–69 Trials using protein C concentrates have shown more promise in controlling the coagulopathy of purpura fulminans, but this is not widely available.63,70–72 Unfortunately, many patients will need debridement and amputation for their necrotic limbs, with one review showing approximately 66% of patients needing amputations.52
TRAUMA
Currently, the most common cause of acute DIC is trauma. The coagulation defects that occur in trauma patients are complex in origin and still controversial (including if even calling it DIC is appropriate!).73–76 The most common etiologies are
- Generation of excess activated protein C leading to increased consumption of factor V and VIII and increased fibrinolysis;
- Tissue damage leading to generation of excess thrombin generation;
- Dilution of hemostatic factors by blood or fluid resuscitation; and
- Activation of endothelial cells leading to generation of a prothrombotic surface and shedding of glycocalyx with antithrombotic properties.
Trauma patients are prone to hypothermia, and this can be the major complicating factor in their bleeding.77,78 Patients may be out “in the field” for a prolonged period of time and be hypothermic on arrival.79 Packed red cells are stored at 4°C, and the infusion of 1 unit can lower the body temperature by 0.16°C.80 Hypothermia has profound effects on the coagulation system that are associated with clinical bleeding.77,81,82 Even modest hypothermia can greatly augment bleeding and needs to be treated or prevented.
The initial management of the bleeding trauma patient is administration of red cells and plasma (FFP) in a 1:1 ratio. This has been shown by clinical studies to lessen the risk of exsanguination in the first 24 hours and to be associated with improved clinical outcomes.83,84 The basic set of coagulation tests should also be obtained to guide product replacement, especially as the bleeding is brought under control. Hypothermia can be prevented by several measures, including transfusing the blood through blood warmers. Devices are available that can warm 1 unit of blood per minute. An increasingly used technique is to perform “damage control” surgery. Patients are initially stabilized with control of damaged vessels and packing of oozing sites.85 Then the patient is taken to the intensive care unit to be warmed and have coagulation defects corrected.
For trauma patients at risk of serious bleeding, the use of tranexamic acid reduced all- cause mortality (relative risk 0.91), with death due to bleeding also being reduced (relative risk 0.85).86 There was no increase in thrombosis, but benefit was restricted to patients treated within 3 hours of the trauma. The dose of tranexamic acid was a 1-g bolus followed by a 1-g continuous infusion over 8 hours.
PREGNANCY-RELATED DIC SYNDROMES
Acute DIC of Pregnancy
Pregnancy can be associated with the rapid onset of severe DIC in 2 situations, abruption and amniotic fluid embolism.87,88 The separation of the placenta from the uterine wall creates a space for blood to occupy. Given the richness of the placenta in tissue factor, this leads to activation of coagulation both locally and systemically. Release of blood when this space reaches the vaginal opening can lead to rapid hemorrhage, further augmenting the coagulation abnormalities. Placental insufficiency can lead to fetal demise, which can also worsen the DIC. Management depends on the size of the abruption and the clinical status of both mother and fetus.87 For severe bleeding and DIC, blood product support is crucial to allow safe delivery. In pregnancy, the fibrinogen goal needs to be higher—200 mg/dL.89 For smaller abruption, close observation with early delivery is indicated.
Amniotic fluid embolism is sudden, with the vascular collapse of the woman soon after delivery. Due to the presence of procoagulant rich fluid in the circulatory system, there is often overwhelming DIC. Therapy is directed at both supporting blood volume and correcting hemostatic defects.
HELLP
The acronym HELLP (hemolysis, elevated liver tests, low platelets) describes a variant of preeclampsia.90 Classically, HELLP syndrome occurs after 28 weeks of gestation in a patient with preeclampsia, but can occur as early as 22 weeks in patients with antiphospholipid antibody syndrome.91–93 The preeclampsia need not be severe. The first sign of HELLP is a decrease in the platelet count followed by abnormal liver function tests. Signs of hemolysis are present with abundant schistocytes on the smear and a high lactate dehydrogenase level. HELLP can progress to liver failure, and deaths are also reported due to hepatic rupture. Unlike TTP, fetal involvement is present in the HELLP syndrome, with fetal thrombocytopenia reported in 30% of cases. In severe cases, elevated D-dimers consistent with DIC are also found. Delivery of the child will most often result in cessation of the HELLP syndrome, but refractory cases will require dexamethasone and plasma exchange.94 Patients should be closely observed for 1 to 2 days after delivery as the hematologic picture can transiently worsen before improving.95
Acute Fatty Liver of Pregnancy
Fatty liver of pregnancy also occurs late in pregnancy and is only associated with preeclampsia in 50% of cases.96,97 Patients first present with nonspecific symptoms of nausea and vomiting but can progress to fulminant liver failure. Patients develop thrombocytopenia early in the course, but in the later stages can develop DIC and very low fibrinogen levels. Mortality rates without therapy can be as high as 90%. Low blood glucose and high ammonia levels can help distinguish fatty liver from other pregnancy complications.98 Treatment consists of prompt delivery of the child and aggressive blood product support.
Retained Dead Fetus Syndrome
Becoming rarer in modern practices, the presence of a dead fetus for many weeks (usually ≥ 5) can result in a chronic DIC state with fibrinogen depletion and coagulopathy. In some women, this is worsened at delivery. In a stable patient, a short trial of heparin prior to planning delivery can control the DIC to allow the coagulopathy to stabilize.
DRUG-INDUCED HEMOLYTIC-DIC SYNDROMES
A severe variant of the drug-induced immune complex hemolysis associated with DIC has been recognized. Rare patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone) have developed this syndrome.99–104 The clinical syndrome starts 7 to 10 days after the drug is administered. Often the patient has only received the antibiotic for surgical prophylaxis. The patient will develop severe Coombs’-positive hemolysis with hypotension and DIC. The patients are often believed to have sepsis and in the management of the supposed sepsis often are re-exposed to the cephalosporin, resulting in worsening of the clinical picture. The outcome is often fatal due to massive hemolysis and thrombosis.101,105–107
Quinine is associated with a unique syndrome of drug-induced DIC.108–111 Approximately 24 to 96 hours after quinine exposure, the patient becomes acutely ill with nausea and vomiting. The patient then develops a microangiopathic hemolytic anemia, DIC, and renal failure. Some patients, besides having antiplatelet antibodies, also have antibodies binding to red cells and neutrophils, which may lead to the more severe syndrome. Despite therapy, patients with quinine-induced TTP have a high incidence of chronic renal failure.
Treatment of the drug-induced hemolytic-DIC syndrome is anecdotal. Patients have responded to aggressive therapy, including plasma exchange, dialysis, and prednisone. Early recognition of the hemolytic anemia and the suspicion it is drug related is important for early diagnosis so that the incriminated drug can be discontinued.
CANCER
Cancers, primarily adenocarcinomas, can result in DIC. The classic Trousseau syndrome referred to the association of migratory superficial thrombophlebitis with cancer112 but now refers to cancer associated with thrombotic DIC.113,114 Highly vascular tumor cells are known to express tissue factor.114,115 In addition, some tumor cells can express a direct activator of factor X (“cancer procoagulant”). Unlike many DIC states, cancer presents with thrombosis instead of bleeding. This may be due to the inflammatory state which accompanies cancer, or it may be a unique part of the chronic nature of cancer DIC biology that allows time for the body to compensate for loss of coagulation factors. In some patients, thrombosis is the first sign of an underlying cancer, sometimes predating the cancer diagnosis by months.115 Rarely, the DIC can result in nonthrombotic endocarditis with micro-emboli leading to widespread small-vessel thrombosis.113
Since effective antineoplastic therapy is lacking for many tumors associated with Trousseau syndrome, DIC therapy is aimed at suppressing thrombosis. An exception is prostate cancer, where hormonal therapy can markedly decrease the DIC.116 Due to the tumor directly activating coagulation factors, inhibition of active enzymes via heparin has been shown to reduce rates of recurrence compared with warfarin.114,115 Clinical trials have demonstrated that heparin therapy is associated with a lower thrombosis recurrence rate than warfarin.117,118 In some patients, the thrombotic process is so vigorous that new thrombosis can be seen within hours of stopping heparin.112
ACUTE PROMYELOCYTIC LEUKEMIA
There are multiple hemostatic defects in patients with acute promyelocytic leukemia (APL).119 Most, if not all, patients with APL have evidence of DIC at the time of diagnosis. Patients with APL have a higher risk of death during induction therapy as compared with patients with other forms of leukemia, with death most often due to bleeding. Once in remission, APL patients have a higher cure rate than most patients with leukemia. APL is also unique among leukemias in that biologic therapy with retinoic acid or arsenic is effective in inducing remission and cure in most patients. Although effective therapy is available, early death rates due to bleeding have not changed.119
APL patients can present with pancytopenia due to leukemic marrow replacement or with diffuse bleeding due to DIC and thrombocytopenia. Life-threatening bleeding such as intracranial hemorrhage may occur at any time until the leukemia is put into remission. The etiology of the hemostatic defects in APL is complex and is thought to be the result of DIC, fibrinolysis, and the release of prothrombotic extracellular chromatin and other procoagulant enzymes.119,120 The diagnosis of APL can be straightforward when the leukemic cells are promyelocytes with abundant Auer rods, although some patients have the microgranular form without obvious Auer rods. The precise diagnosis requires molecular methods, including obtaining FISH for detecting the t(15;17) in PML/RARA fusion. Upon diagnosis of APL, one should obtain a complete coagulation profile, including INR, aPTT, fibrinogen, platelet count, and D-dimers. Change in fibrinogen levels tends to be a good marker of progress in treating the coagulation defects.
Therapy of APL involves treating both the leukemia and the coagulopathy. Currently, the standard treatment for APL is trans-retinoic acid (ATRA) in combination with chemotherapy or arsenic.121,122 This approach will induce remission in more than 90% of patients, and a sizable majority of these patients will be cured of their APL. ATRA therapy will also lead to early correction of the coagulation defects, often within the first week of therapy.123 This is in stark contrast to the chemotherapy era when the coagulation defects would become worse with therapy. Given the marked beneficial effect of ATRA on the coagulopathy of APL and its low toxicity profile, it should be empirically started for any patients suspected of having APL while genetic testing is being performed. Rare reports of massive thrombosis complicating therapy with ATRA exist, but the relationship to either the APL or ATRA is unknown.
Therapy for the coagulation defects consists of aggressive transfusion therapy support and possible use of other pharmacologic agents to control DIC.124,125 The fibrinogen level should be maintained at over 150 mg/dL and the platelet count at over 50,000 cells/µL.126 Controversy still exists over the role of heparin in therapy of APL.104 Although attractive for its ability to quench thrombin, heparin use can lead to profound bleeding and its use in treating APL has fallen out of favor.
SNAKEBITES
Snake envenomation can lead to direct activation of multiple coagulation enzymes, including factors V, X, thrombin, and protein C, and lead to cleavage of fibrinogen.127,128 Envenomation can also activate coagulation and damage vascular endothelium. The DIC can be enhanced by widespread tissue necrosis and hypotension. The key to management of snake bites is administration of specific antivenom. The role of prophylactic factor replacement is controversial, but this therapy is indicated if there is clinical bleeding.129 One confounder is that some snake venoms, especially rattlesnake, can induce reversible platelet aggregation, which corrects with antivenom.
LOCAL VASCULAR ABNORMALITIES
Abnormal vascular structures, such as vascular tumors, vascular malformations, and aneurysms, can lead to localized areas of thrombin generation that can “spill-over” into the general circulation, leading to DIC. The diagnosis Kasabach-Merritt phenomenon should be reserved for children with vascular tumors such as angioma or hemangioendothelioma.130 Therapy depends on the lesion. Embolization to reduce blood flow of vascular malformations can either be definitive therapy or stabilize the patient for surgery. Aneurysms can be repaired by surgery or stenting. Rare patients with aneurysms with significant coagulopathy may require heparin to raise the fibrinogen level before surgery. Kasabach-Merritt disease can respond to steroids or therapy such as vincristine or interferon.130 Increasing data shows that use of the mTOR inhibitor sirolimus can shrink these vascular abnormalities leading to lessening of the coagulopathy.131
CONCLUSION
At the most basic level, DIC is the excess activity of thrombin. However, the clinical presentation and therapy can differ greatly depending on the primary cause. Both diagnosis and therapy involve close coordination of laboratory data and clinical assessment.
1. Carey MJ, Rodgers GM. Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998;59:65–73.
2. De Jonge E, Levi M, Stoutenbeek CP, Van Deventer SJH. Current drug treatment strategies for disseminated intravascular coagulation. Drugs 1998;55:767–77.
3. Baker WF Jr. Clinical aspects of disseminated intravascular coagulation: a clinician’s point of view. Sem Thrombosis Hemostasis 1989;15:1–57.
4. Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med 1999;341:586–92.
5. Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers 2016;2:16037.
6. Kolev K, Longstaff C. Bleeding related to disturbed fibrinolysis. Br J Haematol 2016;175:12–23.
7. Sharma S, Mayberry JC, DeLoughery TG, Mullins RJ. Fatal cerebroembolism from nonbacterial thrombotic endocarditis in a trauma patient: case report and review. Mil Med 2000;165:83–5.
8. Toh CH, Alhamdi Y, Abrams ST. Current pathological and laboratory considerations in the diagnosis of disseminated intravascular coagulation. Ann Lab Med 2016;36:505–12.
9. Yu M, Nardella A, Pechet L. Screening tests of disseminated intravascular coagulation: guidelines for rapid and specific laboratory diagnosis. Crit Care Med 2000;28:1777–80.
10. Mant MJ, King EG. Severe, acute disseminated intravascular coagulation. A reappraisal of its pathophysiology, clinical significance, and therapy based on 47 patients. Am J Med 1979;67:557–63.
11. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol 2009;145:24–33.
12. Levi M. Disseminated intravascular coagulation. Crit Care Med 2007;35:2191–5.
13. Nogami K. The utility of thromboelastography in inherited and acquired bleeding disorders. Br J Haematol 2016;174:503–14.
14. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin 2017;33:119–34.
15. George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med 2006;354:1927–35.
16. George JN. How I treat patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Blood 2000;96:1223–9.
17. Murrin RJ, Murray JA. Thrombotic thrombocytopenic purpura: aetiology, pathophysiology and treatment. Blood Rev 2006;20:51–60.
18. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017;129:2836–46.
19. Patton JF, Manning KR, Case D, Owen J. Serum lactate dehydrogenase and platelet count predict survival in thrombotic thrombocytopenic purpura. Am J Hematol 1994;47:94–9.
20. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991;325:393–7.
21. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpurahemolytic uremic syndrome—clinical experience in 108 patients. N Engl J Med 1991;325:398–403.
22. Kaplan BS, Trachtman H. Improve survival with plasma exchange thrombotic thrombopenic purpura-hemolytic uremic syndrome. Am J Med 2001;110:156–7.
23. Kremer Hovinga JA, Coppo P, Lammle B, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers 2017;3:17020.
24. Asherson RA. The catastrophic antiphospholipid syndrome [editorial]. J Rheumatol 1992;19:508–12.
25. Asherson RA, Piette JC. The catastrophic antiphospholipid syndrome 1996: acute multi-organ failure associated with antiphospholipid antibodies: a review of 31 patients. Lupus 1996;5:414–7.
26. Asherson RA, Cervera R. Castastrophic antiphospholipid syndrome. Curr Opinion Hematol 2000;5:325–9.
27. Merrill JT, Asherson RA. Catastrophic antiphospholipid syndrome. Nat Clin Pract Rhuem 2006;2:81–9.
28. Rodriguez-Pinto I, Espinosa G, Cervera R. Catastrophic antiphospholipid syndrome: The current management approach. Best Pract Res Clin Rheumatol 2016;30:239–9.
29. Kazzaz NM, McCune WJ, Knight JS. Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol 2016;28:218–27.
30. Hoffman JN, Faist E. Coagulation inhibitor replacement during sepsis: useless? Crit Care Med 2000;28(9 Suppl):S74–6.
31. Wada H, Asakura H, Okamoto K, et al. Expert consensus for the treatment of disseminated intravascular coagulation in Japan. Japanese Society of Thrombosis Hemostasis/DIC subcommittee. Thromb Res 2010;125:6–11.
32. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
33. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
34. Stainsby D, MacLennan S, Hamilton PJ. Management of massive blood loss: a template guideline. Br J Anaesth 2000;85:487–91.
35. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999;340:409–17.
36. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
37. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
38. Miller RD, Robbins TO, Tong MJ, Barton SL. Coagulation defects associated with massive blood transfusions. Ann Surg 1971;174:794–801.
39. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
40. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
41. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
42. Callander N, Rapaport SI. Trousseau’s syndrome. West J Med 1993;158:364–71.
43. Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993;119:104–9.
44. Olson JD, Arkin CF, Brandt JT, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulant therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998;122:782–8.
45. Yoshikawa T, Tanaka KR, Guze LB. Infection and disseminated intravascular coagulation. Medicine (Baltimore) 1971;50:237–58.
46. Jagneaux T, Taylor DE, Kantrow SP. Coagulation in sepsis. Am J Med Sci 2004;328:196–204.
47. Lipinska-Gediga M. Coagulopathy in sepsis - a new look at an old problem. Anaesthesiol Intensive Ther 2016;48:352–9.
48. Van Gorp ECM, Suharti C, ten Cate H, et al. Review: Infections diseases and coagulation disorders. Journal of Infectious Diseases 1999;180:176–86.
49. McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017;129:1357–67.
50. Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011;118:1952–61.
51. Darmstadt GL. Acute infectious purpura fulminans: pathogenesis and medical management. Pediatr Dermatol 1998;15:169–83.
52. Davis MD, Dy KM, Nelson S. Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic. J Am Acad Dermatol 2007;57:944–56.
53. Spicer TE, Rau JM. Purpura fulminans. Am J Med 1976;61:566–71.
54. Josephson C, Nuss R, Jacobson L, et al. The varicellaautoantibody syndrome. Pediatr Res 2001;50:345–52.
55. Smith OP, White B. Infectious purpura fulminans: diagnosis and treatment. Br J Haematol 1999;104:202–7.
56. Gamper G, Oschatz E, Herkner H, et al. Sepsis-associated purpura fulminans in adults. Wien Klin Wochenschr 2001;113:107–12.
57. Ward KM, Celebi JT, Gmyrek R, Grossman ME. Acute infectious purpura fulminans associated with asplenism or hyposplenism. J Am Acad Dermatol 2002;47:493–6.
58. Childers BJ, Cobanov B. Acute infectious purpura fulminans: a 15-year retrospective review of 28 consecutive cases. Am Surg 2003;69:86–90.
59. Carpenter CT, Kaiser AB. Purpura fulminans in pneumococcal sepsis: case report and review. Scand J Infect Dis 1997;29:479–83.
60. Warkentin TE, Pai M. Shock, acute disseminated intravascular coagulation, and microvascular thrombosis: is ‘shock liver’ the unrecognized provocateur of ischemic limb necrosis: reply. J Thromb Haemost 2016;14:2317–9.
61. Warkentin TE. Ischemic limb gangrene with pulses. N Engl J Med 2015;373:642–55.
62. Duncan A. New therapies for severe meningococcal disease but better outcomes? Lancet 1997;350:1565–6.
63. Smith OP, White B, Vaughan D, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet1997;350:1590–3.
64. Branson HE, Katz J. A structured approach to the management of purpura fulminans. J Natl Med Assoc 1983;75:821–5.
65. Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth 2001;86:581–6.
66. Manios SG, Kanakoudi F, Maniati E. Fulminant meningococcemia. Heparin therapy and survival rate. Scand J Infect Dis 1971;3:127–33.
67. Giudici D, Baudo F, Palareti G, et al. Antithrombin replacement in patients with sepsis and septic shock. Haematologica 1999;84:452–60.
68. Fourrier F, Jourdain M, Tournoys A. Clinical trial results with antithrombin III in sepsis. Crit Care Med 2000;28(9 Suppl):S38–43.
69. Levi M, De Jonge E, van der PT, ten Cate H. Novel approaches to the management of disseminated intravascular coagulation. Crit Care Med 2000;28(9 Suppl):S20–4.
70. Rivard GE, David M, Farrell C, Schwarz HP. Treatment of purpura fulminans in meningococcemia with protein C concentrate. J Pediatr 1995;126:646–52.
71. White B, Livingstone W, Murphy C, et al. An open-label study of the role of adjuvant hemostatic support with protein C replacement therapy in purpura fulminans-associated meningococcemia. Blood 2000;96:3719–24.
72. Schellongowski P, Bauer E, Holzinger U, et al. Treatment of adult patients with sepsis-induced coagulopathy and purpura fulminans using a plasma-derived protein C concentrate (Ceprotin). Vox Sang 2006;90:294–301.
73. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
74. Cohen MJ, Christie SA. Coagulopathy of trauma. Crit Care Clin 2017;33:101–18.
75. Giordano S, Spiezia L, Campello E, Simioni P. The current understanding of trauma-induced coagulopathy (TIC): a focused review on pathophysiology. Intern Emerg Med 2017 May 5.
76. Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood 2016;128:1043–9.
77. Eddy VA, Morris JA Jr, Cullinane DC. Hypothermia, coagulopathy, and acidosis. Surg Clin North Am 2000;80:845–54.
78. Peng RY, Bongard FS. Hypothermia in trauma patients. J Am Coll Surg 1999;188:685–96.
79. Steinemann S, Shackford SR, Davis JW. Implications of admission hypothermia in trauma patients. J Trauma 1990;30:200–2.
80. Rajek A, Greif R, Sessler DI, et al. Core cooling by central venous infusion of ice-cold (4 degrees C and 20 degrees C) fluid: isolation of core and peripheral thermal compartments. Anesthesiol 2000;93:629–37.
81. Watts DD, Trask A, Soeken K, et al. Hypothermic coagulopathy in trauma: effect of varying levels of hypothermia on enzyme speed, platelet function, and fibrinolytic activity. J Trauma 1998;44:846–54.
82. Ferrara A, MacArthur JD, Wright HK, et al. Hypothermia and acidosis worsen coagulopathy in the patient requiring massive transfusion. Am J Surg 1990;160:515–8.
83. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
84. Johansson PI, Stensballe J, Oliveri R, Wade CE, Ostrowski SR, Holcomb JB. How I treat patients with massive hemorrhage. Blood 2014;124:3052–8.
85. Stone HH, Strom PR, Mullins RJ. Management of the major coagulopathy with onset during laparotomy. Ann Surg 1983;197:532–5.
86. WOMAN Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23–32.
87. Hall DR. Abruptio placentae and disseminated intravascular coagulopathy. Semin Perinatol 2009;33:189–95.
88. Thachil J, Toh CH. Disseminated intravascular coagulation in obstetric disorders and its acute haematological management. Blood Rev 2009;23:167–76.
89. Collins P, Abdul-Kadir R, Thachil J, Subcommittees on Women’ s Health Issues in T, Haemostasis, on Disseminated Intravascular C. Management of coagulopathy associated with postpartum hemorrhage: guidance from the SSC of the ISTH. J Thromb Haemost 2016;14:205–10.
90. Baxter JK, Weinstein L. HELLP syndrome: the state of the art. Obstet Gynecol Surv 2004;59:838–45.
91. Egerman RS, Sibai BM. HELLP syndrome. Clin Obstetr Gynecol 1999;42:381–9.
92. Saphier CJ, Repke JT. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome: a review of diagnosis and management. Sem Perinatol 1998;22:118–33.
93. Le Thi TD, Tieulie N, Costedoat N, et al. The HELLP syndrome in the antiphospholipid syndrome: retrospective study of 16 cases in 15 women. Ann Rheum Dis 2005;64:273–8.
94. Martin JN Jr, Perry KG Jr, Blake PG, et al. Better maternal outcomes are achieved with dexamethasone therapy for postpartum HELLP (hemolysis, elevated liver enzymes, and thrombocytopenia) syndrome. Am J Obstet Gynecol 1997;177:1011–7.
95. Magann EF, Martin JN Jr. Twelve steps to optimal management of HELLP syndrome. Clinical Obstet Gynecol 1999;42:532–50.
96. Jwayyed SM, Blanda M, Kubina M. Acute fatty liver of pregnancy. J Emerg Medi 1999;17:673–7.
97. Bacq Y. Acute fatty liver of pregnancy. Sem Perinatol 1998;22:134–40.
98. Egerman RS, Sibai BM. Imitators of preeclampsia and eclampsia. Clin Obstet Gynecol 1999;42:551–62.
99. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Medi Rev 1993;7:255–67.
100. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis 1992;15:863–5.
101. Garratty G, Nance S, Lloyd M, Domen R. Fatal immune hemolytic anemia due to cefotetan. Transfusion 1992;32:269–71.
102. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes:case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion 1999;39:306–9.
103. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion 1999;39:1239–46.
104. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol 2006;81:186–8.
105. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr 1995;126:813–5.
106. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1995;14:1116–7.
107. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone. J Pediatr 1995;126:816–7.
108. Gottschall JL, Elliot W, Lianos E, et al. Quinine-induced immune thrombocytopenia associated with hemolytic uremic syndrome: a new clinical entity. Blood 1991;77:306–10.
109. Gottschall JL, Neahring B, McFarland JG, et al. Quinine-induced immune thrombocytopenia with hemolytic uremic syndrome: clinical and serological findings in nine patients and review of literature. Am J Hematol 1994;47:283–9.
110. Crum NF, Gable P. Quinine-induced hemolytic-uremic syndrome. South Med J 2000;93:726–8.
111. Vesely T, Vesely JN, George JN. Quinine-Induced thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS): frequency, clinical features, and long-term outcomes. Blood 2000;96:629 [abstract].
112. Bell WR, Starksen NF, Tong S, Porterfield JK. Trousseau’s syndrome. Devastating coagulopathy in the absence of heparin. Am J Med 1985;79:423–30.
113. Sack GH, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinic, pathophysiologic, and therapeutic features. Medicine 1977;56:1–37.
114. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007;110:1723–9.
115. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol 2005;6:401–10.
116. de la Fouchardiere C, Flechon A, Droz JP. Coagulopathy in prostate cancer. Neth J Med 2003;61:347–54.
117. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. 8th ed. Chest 2008;133(6 Suppl):454S–545S.
118. Lee AY, Kamphuisen PW, Meyer G, et al. Tinzaparin vs warfarin for treatment of acute venous thromboembolism in patients with active cancer: a randomized clinical trial. JAMA 2015;314:677–86.
119. Choudhry A, DeLoughery TG. Bleeding and thrombosis in acute promyelocytic leukemia. Am J Hematol 2012;87:596–603.
120. Cao M, Li T, He Z, et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood 2017;129:1855–64.
121. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008;111:2505–15.
122. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013;369:111–21.
123. Dombret H, Scrobohaci ML, Ghorra P, et al. Coagulation disorders associated iwth acute promyelocytic leukemia: Corrective effect of all-trans retinoic acid treatment. Leukemia 1993;7:2–9.
124. Falanga A, Rickles FR. Management of thrombohemorrhagic syndromes (THS) in hematologic malignancies. Hematology Am Soc Hematol Educ Program 2007;2007:165–71
125. Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood 2009;114:5126–35.
126. Sanz MA, Grimwade D, Tallman MS, et al. Guidelines on the management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009;113:1875–91.
127. Lu Q, Clemetson JM, Clemetson KJ. Snake venoms and hemostasis. J Thromb Haemost 2005;3:1791–9.
128. Berling I, Isbister GK. Hematologic effects and complications of snake envenoming. Transfus Med Rev 2015;29:82–9.
129. Isbister GK, Jayamanne S, Mohamed F, et al. A randomized controlled trial of fresh frozen plasma for coagulopathy in Russell’s viper (Daboia russelii) envenoming. J Thromb Haemost 2017;15:645–54.
130. Rodriguez V, Lee A, Witman PM, Anderson PA. Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 2009;31:522–6.
131. Triana P, Dore M, Cerezo VN, et al. Sirolimus in the treatment of vascular anomalies. Eur J Pediatr Surg 2017;27:86–90.
INTRODUCTION
In the normal person, the process of coagulation is finely controlled at many levels to ensure the appropriate amount of hemostasis at the appropriate location. Broadly defined, disseminated intravascular coagulation (DIC) is the name given to any process that disrupts this fine tuning, leading to unregulated coagulation. Defined this way, DIC may be found in a variety of patients with a variety of disease states, and can present with a spectrum of findings ranging from asymptomatic abnormal laboratory results to florid bleeding or thrombosis. It is important to remember that DIC is always a consequence of an underlying pathological process and not a disease in and of itself. This article first reviews concepts common to all forms of DIC, and then reviews the more common disease states that lead to DIC.
PATHOGENESIS
At the most basic level, DIC is the clinical manifestation of inappropriate thrombin activation.1–5 Inappropriate thrombin activation can be due to underlying conditions such as sepsis and obstetric disasters. The activation of thrombin leads to (1) conversion of fibrinogen to fibrin, (2) activation of platelets (and their consumption), (3) activation of factors V and VIII, (4) activation of protein C (and degradation of factors Va and VIIIa), (5) activation of endothelial cells, and (6) activation of fibrinolysis (Table 1).
1. Conversion of fibrinogen to fibrin, which leads to the formation of fibrin monomers and excessive thrombus formation. These thrombi are rapidly dissolved by excessive fibrinolysis in most patients. In certain clinical situations, especially cancer, excessive thrombosis will occur. In patients with cancer, this is most often a deep venous thrombosis. Rare patients, especially those with pancreatic cancer, may have severe DIC with multiple arterial and venous thromboses. Nonbacterial thrombotic endocarditis can also be seen in these patients, leading to widespread embolic complications.
2. Activation of platelets and their consumption. Thrombin is the most potent physiologic activator of platelets, so in DIC there is increased activation of platelets. These activated platelets are consumed, resulting in thrombocytopenia. Platelet dysfunction is also present. Platelets that have been activated and have released their contents but still circulate are known as “exhausted” platelets; these cells can no longer function to support coagulation. The fibrin degradation products (FDP) in DIC can also bind to GP IIb/IIIa and inhibit further platelet aggregation.
3. Activation of factors V, VIII, XI, and XIII. Activation of these factors can promote thrombosis, but they are then rapidly cleared by antithrombin (XI) or activated protein C (V and VIII) or by binding to the fibrin clot (XIII). This can lead to depletion of all the prothrombotic clotting factors and antithrombin, which in turn can lead to both thrombosis and bleeding.
4. Activation of protein C further promotes degradation of factors Va and VIIIa, enhances fibrinolysis, and decreases protein C levels.
5. Activation of endothelial cells, especially in the skin, may lead to thrombosis, and in certain patients, especially those with meningococcemia, purpura fulminans. Endothelial damage will down-regulate thrombomodulin, preventing activation of protein C and leading to further reductions in levels of activated protein C.56. Activation of fibrinolysis leads to the breakdown of fibrin monomers, formation of fibrin thrombi, and increased circulating fibrinogen. In most patients with DIC, the fibrinolytic response is brisk.6 This is why most patients with DIC present with bleeding and prolonged clotting times.
PATTERNS OF DIC
The clinical manifestations of DIC in a given patient depend on the balance of thrombin activation and secondary fibrinolysis plus the patient’s ability to compensate for the DIC. Patients with DIC can present in 1 of 4 patterns:1–3
1. Asymptomatic. Patients can present with laboratory evidence of DIC but no bleeding or thrombosis. This is often seen in patients with sepsis or cancer. However, with further progression of the underlying disease, these patients can rapidly become symptomatic.
2. Bleeding. The bleeding is due to a combination of factor depletion, platelet dysfunction, thrombocytopenia, and excessive fibrinolysis.1 These patients may present with diffuse bleeding from multiple sites (eg, intravenous sites, areas of instrumentation).
3. Thrombosis. Despite the general activation of the coagulation process, thrombosis is unusual in most patients with acute DIC. The exceptions include patients with cancer, trauma patients, and certain obstetrical patients. Most often the thrombosis is venous, but arterial thrombosis and nonbacterial thrombotic endocarditis have been reported.7
4. Purpura fulminans. This form of DIC is discussed in more detail later (see Specific DIC Syndromes section).
DIAGNOSIS
There is no one test that will diagnose DIC; one must match the test to the clinical situation (Table 2).8
SCREENING TESTS
The prothrombin time-INR and activated thromboplastin time (aPPT) are usually elevated in severe DIC but may be normal or shortened in chronic forms.9 One may also see a shortened aPTT in severe acute DIC due to large amounts of activated thrombin and factor X “bypassing” the contact pathway. An aPTT as short as 10 seconds has been seen in acute DIC. The platelet count is usually reduced but may be normal in chronic DIC. Serum fibrinogen and platelets are decreased in acute DIC but again may be in the “normal” range in chronic DIC.10 The most sensitive screening test for DIC is a fall in the platelet count, with low counts seen in 98% of patients and counts under 50,000 cells/μL in 50%.9,11 The least specific test is fibrinogen, which tends to fall below normal only in severe acute DIC.9
SPECIFIC TESTS
This group of tests allows one to deduce that abnormally high concentrations of thrombin are present.
Ethanol Gel and Protamine Tests
Both of these older tests detected circulating fibrin monomers, whose appearance is an early sign of DIC. Circulating fibrin monomers are seen when thrombin acts on fibrinogen. Usually the monomer polymerizes with the fibrin clot, but when there is excess thrombin these monomers can circulate. Detection of circulating fibrin monomer means there is too much IIa and, ergo, DIC is present.
Fibrin(ogen) Degradation Products
Plasmin acts on the fibrin/fibrinogen molecule to cleave the molecule in specific places. The resulting degradation product levels will be elevated in situations of increased fibrin/fibrinogen destruction (DIC and fibrinolysis). The FDP are typically mildly elevated in renal and liver disease due to reduced clearance.
D-Dimers
When fibrin monomers bind to form a thrombus, factor XIII acts to bind their “D” domains together. This bond is resistant to plasmin and thus this degradation fragment is known as the “D-dimer.” High levels of D-dimer indicate that (1) IIa has acted on fibrinogen to form a fibrin monomer that bonded to another fibrin monomer, and (2) this thrombus was lysed by plasmin. Because D-dimers can be elevated (eg, with exercise, after surgery), an elevated D-dimer needs to be interpreted in the context of the clinical situation.11 Currently, this is the most common specific test for DIC performed.
Other Tests
Several other tests are sometimes helpful in diagnosing DIC.
Thrombin time. This test is performed by adding thrombin to plasma. Thrombin times are elevated in DIC (FDPs interfere with polymerization), in the presence of low fibrinogen levels, in dysfibrinogenemia, and in the presence of heparin (very sensitive).
Reptilase time is the same as thrombin time but is performed with a snake venom that is insensitive to heparin. Reptilase time is elevated in the same conditions as the thrombin time, with the exception of the presence of heparin. Thrombin time and reptilase time are most useful in evaluation of dysfibrinogenemia.
Prothrombin fragment 1.2 (F1.2). F1.2 is a small peptide cleaved off when prothrombin is activated to thrombin. Thus, high levels of F1.2 are found in DIC but can be seen in other thrombotic disorders. This test is still of limited clinical value.
DIC scoring system. A scoring system to both diagnose and quantify DIC has been proposed (Figure).11,12
Thromboelastography (TEG). This is a point-of-care test that uses whole blood to determine specific coagulation parameters such as R time (time from start of test to clot formation), maximal amplitude (MA, maximum extent of thrombus), and LY30 (MA at 30 minutes, a measure of fibrinolysis).13 Studies have shown that TEG can identify DIC by demonstrating a shorter R time (excess thrombin generation) which prolongs as coagulation factors are consumed. The MA is decreased as fibrinogen is consumed and the LY30 shows excess fibrinolysis. TEG has been shown to be of particular value in the management of the complex coagulopathy of trauma.14
MIMICKERS OF DIC
It is important to recognize coagulation syndromes that are not DIC, especially those that have specific other therapies. The syndromes most frequently encountered are thrombotic thrombocytopenic purpura (TTP) and catastrophic antiphospholipid antibody syndrome (CAPS). One important clue to both of these syndromes is that, unlike DIC, there is no primary disorder (cancer, sepsis) that is driving the coagulation abnormalities.
TTP should be suspected when any patient presents with any combination of thrombocytopenia, microangiopathic hemolytic anemia (schistocytes and signs of hemolysis) plus end-organ damage.15–18 Patients with TTP most often present with intractable seizures, strokes, or sequelae of renal insufficiency. Many patients who present with TTP have been misdiagnosed as having sepsis, “lupus flare,” or vasculitis. The key diagnostic differentiator between TTP and DIC is the lack of activation of coagulation with TTP—fibrinogen is normal and D-dimers are minimally or not elevated. In TTP, lactate dehydrogenase is invariably elevated, often 2 to 3 times normal.19 The importance of identifying TTP is that untreated TTP is rapidly fatal. Mortality in the pre–plasma exchange era ranged from 95% to 100%. Today plasma exchange therapy is the foundation of TTP treatment and has reduced mortality to less than 20%.16,20–23Rarely patients with antiphospholipid antibody syndrome can present with fulminant multiorgan system failure.24–28 CAPS is caused by widespread microthrombi in multiple vascular fields. These patients will develop renal failure, encephalopathy, adult respiratory distress syndrome (often with pulmonary hemorrhage), cardiac failure, dramatic livedo reticularis, and worsening thrombocytopenia. Many of these patients have pre-existing autoimmune disorders and high-titer anticardiolipin antibodies. It appears that the best therapy for these patients is aggressive immunosuppression with steroids plus plasmapheresis, followed by rituximab or, if in the setting of lupus, intravenous cyclophosphamide monthly.27,29 Early recognition of CAPS can lead to quick therapy and resolution of the multiorgan system failure.
GENERAL THERAPY
The best way to treat DIC is to treat the underlying cause that is driving the thrombin generation.1,2,4,30,31 Fully addressing the underlying cause may not be possible or may take time, and in the meantime it is necessary to disrupt the cycle of thrombosis and/or hemorrhage. In the past, there was concern about using factor replacement due to fears of “feeding the fire,” or perpetuating the cycle of thrombosis. However, these concerns are not supported by evidence, and factors must be replaced if depletion occurs and bleeding ensues.32
Transfusion therapy of the patient with DIC is guided by the 5 laboratory tests that reflect the basic parameters essential for both hemostasis and blood volume status:33,34 hematocrit, platelet count, prothrombin time-INR, aPTT, and fibrinogen level. Decisions regarding replacement therapy are based on the results of these laboratory tests and the clinical situation of the patient (Table 3).
The basic 5 laboratory tests should be repeated after administering the blood products. This allows one to ensure that adequate replacement therapy was given for the coagulation defects. Frequent checks of the coagulation tests also allow rapid identification and treatment of new coagulation defects in a timely fashion. A flow chart of the test and the blood products administered should also be maintained. This is important in acute situations such as trauma or obstetrical bleeding.
In theory, since DIC is the manifestation of exuberant thrombin production, blocking thrombin with heparin should decrease or shut down DIC. However, studies have shown that in most patients heparin administration has led to excessive bleeding. Currently, heparin therapy is reserved for patients who have thrombosis as a component of their DIC.2,41,42 Given the coagulopathy that is often present, specific heparin levels instead of the aPTT should be used to monitor anticoagulation.43,44
SPECIFIC DIC SYNDROMES
SEPSIS/INFECTIOUS DISEASE
Any overwhelming infection can lead to DIC.45 Classically, it was believed that gram-negative bacteria can lead tissue factor exposure via production of endotoxin, but recent studies indicate that DIC can be seen with any overwhelming infection.46,47 There are several potential avenues by which infections can lead to DIC. As mentioned, gram-negative bacteria produce endotoxin that can directly lead to tissue factor exposure, resulting in excess thrombin generation. In addition, any infection can lead to expression of inflammatory cytokines that induce tissue-factor expression by endothelium and monocytes. Some viruses and Rickettsia species can directly infect the vascular endothelium, converting it from an antithrombotic to a prothrombotic phenotype.48 When fighting infections, neutrophils can extrude their contents, including DNA, to help trap organisms. These neutrophil extracellular traps (NETS) may play an important role in promoting coagulopathy.49,50 The hypotension produced by sepsis leads to tissue hypoxia, which results in more DIC. The coagulopathy in sepsis can range from subtle abnormalities of testing to purpura fulminans. Thrombocytopenia is worsened by cytokine-induced hemophagocytic syndrome.
As with all forms of DIC, empiric therapy targeting the most likely source of infection and maintaining hemodynamic stability is the key to therapy. As discussed below, heparin and other forms of coagulation replacement appear to be of no benefit in therapy.
PURPURA FULMINANS
DIC in association with necrosis of the skin is seen in primary and secondary purpura fulminans.51,52 Primary purpura fulminans is most often seen after a viral infection.53 In these patients, the purpura fulminans starts with a painful red area on an extremity that rapidly progresses to a black ischemic area. In many patients, acquired deficiency of protein S is found.51,54,55 Secondary purpura fulminans is most often associated with meningococcemia infections but can be seen in any patient with overwhelming infection.56–58 Post-splenectomy sepsis syndrome patients and those with functional hyposplenism due to chronic liver diseases are also at risk.59 Patients present with signs of sepsis, and the skin lesions often involve the extremities and may lead to amputations. As opposed to primary purpura fulminans, those with the secondary form will have symmetrical ischemia distally (toes and fingers) that ascends as the process progresses. Rarely, adrenal infarction (Waterhouse-Friderichsen syndrome) occurs, which leads to severe hypotension.45
Recently, Warkenten has reported on limb gangrene in critically ill patients complicating sepsis or cardiogenic shock.60,61 These patients have DIC that is complicated by shock liver. Deep venous thrombosis with ischemic gangrene then develops, which can result in tissue loss and even amputation. The pathogenesis is hypothesized to be hepatic dysfunction leading to sudden drops in protein C and S plasma levels, which then leads to thrombophilia with widespread microvascular thrombosis. Therapy for purpura fulminans is controversial. Primary purpura fulminans, especially in those with postvaricella autoimmune protein S deficiency, has responded to plasma infusion titrated to keep the protein S level above 25%.51 Intravenous immunoglobulin has also been reported to help decrease the anti-protein S antibodies. Heparin has been reported to control the DIC and extent of necrosis.62 The starting dose in these patients is 5 to 8 units/kg/hr.2
Sick patients with secondary purpura fulminans have been treated with plasma drips, plasmapheresis, and continuous plasma ultrafiltration.62–66 Heparin therapy alone has not been shown to improve survival.66 Much attention has been given to replacement of natural anticoagulants such as protein C and antithrombin as therapy for purpura fulminans, but unfortunately randomized trials using antithrombin have shown mostly negative results.51,55,67–69 Trials using protein C concentrates have shown more promise in controlling the coagulopathy of purpura fulminans, but this is not widely available.63,70–72 Unfortunately, many patients will need debridement and amputation for their necrotic limbs, with one review showing approximately 66% of patients needing amputations.52
TRAUMA
Currently, the most common cause of acute DIC is trauma. The coagulation defects that occur in trauma patients are complex in origin and still controversial (including if even calling it DIC is appropriate!).73–76 The most common etiologies are
- Generation of excess activated protein C leading to increased consumption of factor V and VIII and increased fibrinolysis;
- Tissue damage leading to generation of excess thrombin generation;
- Dilution of hemostatic factors by blood or fluid resuscitation; and
- Activation of endothelial cells leading to generation of a prothrombotic surface and shedding of glycocalyx with antithrombotic properties.
Trauma patients are prone to hypothermia, and this can be the major complicating factor in their bleeding.77,78 Patients may be out “in the field” for a prolonged period of time and be hypothermic on arrival.79 Packed red cells are stored at 4°C, and the infusion of 1 unit can lower the body temperature by 0.16°C.80 Hypothermia has profound effects on the coagulation system that are associated with clinical bleeding.77,81,82 Even modest hypothermia can greatly augment bleeding and needs to be treated or prevented.
The initial management of the bleeding trauma patient is administration of red cells and plasma (FFP) in a 1:1 ratio. This has been shown by clinical studies to lessen the risk of exsanguination in the first 24 hours and to be associated with improved clinical outcomes.83,84 The basic set of coagulation tests should also be obtained to guide product replacement, especially as the bleeding is brought under control. Hypothermia can be prevented by several measures, including transfusing the blood through blood warmers. Devices are available that can warm 1 unit of blood per minute. An increasingly used technique is to perform “damage control” surgery. Patients are initially stabilized with control of damaged vessels and packing of oozing sites.85 Then the patient is taken to the intensive care unit to be warmed and have coagulation defects corrected.
For trauma patients at risk of serious bleeding, the use of tranexamic acid reduced all- cause mortality (relative risk 0.91), with death due to bleeding also being reduced (relative risk 0.85).86 There was no increase in thrombosis, but benefit was restricted to patients treated within 3 hours of the trauma. The dose of tranexamic acid was a 1-g bolus followed by a 1-g continuous infusion over 8 hours.
PREGNANCY-RELATED DIC SYNDROMES
Acute DIC of Pregnancy
Pregnancy can be associated with the rapid onset of severe DIC in 2 situations, abruption and amniotic fluid embolism.87,88 The separation of the placenta from the uterine wall creates a space for blood to occupy. Given the richness of the placenta in tissue factor, this leads to activation of coagulation both locally and systemically. Release of blood when this space reaches the vaginal opening can lead to rapid hemorrhage, further augmenting the coagulation abnormalities. Placental insufficiency can lead to fetal demise, which can also worsen the DIC. Management depends on the size of the abruption and the clinical status of both mother and fetus.87 For severe bleeding and DIC, blood product support is crucial to allow safe delivery. In pregnancy, the fibrinogen goal needs to be higher—200 mg/dL.89 For smaller abruption, close observation with early delivery is indicated.
Amniotic fluid embolism is sudden, with the vascular collapse of the woman soon after delivery. Due to the presence of procoagulant rich fluid in the circulatory system, there is often overwhelming DIC. Therapy is directed at both supporting blood volume and correcting hemostatic defects.
HELLP
The acronym HELLP (hemolysis, elevated liver tests, low platelets) describes a variant of preeclampsia.90 Classically, HELLP syndrome occurs after 28 weeks of gestation in a patient with preeclampsia, but can occur as early as 22 weeks in patients with antiphospholipid antibody syndrome.91–93 The preeclampsia need not be severe. The first sign of HELLP is a decrease in the platelet count followed by abnormal liver function tests. Signs of hemolysis are present with abundant schistocytes on the smear and a high lactate dehydrogenase level. HELLP can progress to liver failure, and deaths are also reported due to hepatic rupture. Unlike TTP, fetal involvement is present in the HELLP syndrome, with fetal thrombocytopenia reported in 30% of cases. In severe cases, elevated D-dimers consistent with DIC are also found. Delivery of the child will most often result in cessation of the HELLP syndrome, but refractory cases will require dexamethasone and plasma exchange.94 Patients should be closely observed for 1 to 2 days after delivery as the hematologic picture can transiently worsen before improving.95
Acute Fatty Liver of Pregnancy
Fatty liver of pregnancy also occurs late in pregnancy and is only associated with preeclampsia in 50% of cases.96,97 Patients first present with nonspecific symptoms of nausea and vomiting but can progress to fulminant liver failure. Patients develop thrombocytopenia early in the course, but in the later stages can develop DIC and very low fibrinogen levels. Mortality rates without therapy can be as high as 90%. Low blood glucose and high ammonia levels can help distinguish fatty liver from other pregnancy complications.98 Treatment consists of prompt delivery of the child and aggressive blood product support.
Retained Dead Fetus Syndrome
Becoming rarer in modern practices, the presence of a dead fetus for many weeks (usually ≥ 5) can result in a chronic DIC state with fibrinogen depletion and coagulopathy. In some women, this is worsened at delivery. In a stable patient, a short trial of heparin prior to planning delivery can control the DIC to allow the coagulopathy to stabilize.
DRUG-INDUCED HEMOLYTIC-DIC SYNDROMES
A severe variant of the drug-induced immune complex hemolysis associated with DIC has been recognized. Rare patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone) have developed this syndrome.99–104 The clinical syndrome starts 7 to 10 days after the drug is administered. Often the patient has only received the antibiotic for surgical prophylaxis. The patient will develop severe Coombs’-positive hemolysis with hypotension and DIC. The patients are often believed to have sepsis and in the management of the supposed sepsis often are re-exposed to the cephalosporin, resulting in worsening of the clinical picture. The outcome is often fatal due to massive hemolysis and thrombosis.101,105–107
Quinine is associated with a unique syndrome of drug-induced DIC.108–111 Approximately 24 to 96 hours after quinine exposure, the patient becomes acutely ill with nausea and vomiting. The patient then develops a microangiopathic hemolytic anemia, DIC, and renal failure. Some patients, besides having antiplatelet antibodies, also have antibodies binding to red cells and neutrophils, which may lead to the more severe syndrome. Despite therapy, patients with quinine-induced TTP have a high incidence of chronic renal failure.
Treatment of the drug-induced hemolytic-DIC syndrome is anecdotal. Patients have responded to aggressive therapy, including plasma exchange, dialysis, and prednisone. Early recognition of the hemolytic anemia and the suspicion it is drug related is important for early diagnosis so that the incriminated drug can be discontinued.
CANCER
Cancers, primarily adenocarcinomas, can result in DIC. The classic Trousseau syndrome referred to the association of migratory superficial thrombophlebitis with cancer112 but now refers to cancer associated with thrombotic DIC.113,114 Highly vascular tumor cells are known to express tissue factor.114,115 In addition, some tumor cells can express a direct activator of factor X (“cancer procoagulant”). Unlike many DIC states, cancer presents with thrombosis instead of bleeding. This may be due to the inflammatory state which accompanies cancer, or it may be a unique part of the chronic nature of cancer DIC biology that allows time for the body to compensate for loss of coagulation factors. In some patients, thrombosis is the first sign of an underlying cancer, sometimes predating the cancer diagnosis by months.115 Rarely, the DIC can result in nonthrombotic endocarditis with micro-emboli leading to widespread small-vessel thrombosis.113
Since effective antineoplastic therapy is lacking for many tumors associated with Trousseau syndrome, DIC therapy is aimed at suppressing thrombosis. An exception is prostate cancer, where hormonal therapy can markedly decrease the DIC.116 Due to the tumor directly activating coagulation factors, inhibition of active enzymes via heparin has been shown to reduce rates of recurrence compared with warfarin.114,115 Clinical trials have demonstrated that heparin therapy is associated with a lower thrombosis recurrence rate than warfarin.117,118 In some patients, the thrombotic process is so vigorous that new thrombosis can be seen within hours of stopping heparin.112
ACUTE PROMYELOCYTIC LEUKEMIA
There are multiple hemostatic defects in patients with acute promyelocytic leukemia (APL).119 Most, if not all, patients with APL have evidence of DIC at the time of diagnosis. Patients with APL have a higher risk of death during induction therapy as compared with patients with other forms of leukemia, with death most often due to bleeding. Once in remission, APL patients have a higher cure rate than most patients with leukemia. APL is also unique among leukemias in that biologic therapy with retinoic acid or arsenic is effective in inducing remission and cure in most patients. Although effective therapy is available, early death rates due to bleeding have not changed.119
APL patients can present with pancytopenia due to leukemic marrow replacement or with diffuse bleeding due to DIC and thrombocytopenia. Life-threatening bleeding such as intracranial hemorrhage may occur at any time until the leukemia is put into remission. The etiology of the hemostatic defects in APL is complex and is thought to be the result of DIC, fibrinolysis, and the release of prothrombotic extracellular chromatin and other procoagulant enzymes.119,120 The diagnosis of APL can be straightforward when the leukemic cells are promyelocytes with abundant Auer rods, although some patients have the microgranular form without obvious Auer rods. The precise diagnosis requires molecular methods, including obtaining FISH for detecting the t(15;17) in PML/RARA fusion. Upon diagnosis of APL, one should obtain a complete coagulation profile, including INR, aPTT, fibrinogen, platelet count, and D-dimers. Change in fibrinogen levels tends to be a good marker of progress in treating the coagulation defects.
Therapy of APL involves treating both the leukemia and the coagulopathy. Currently, the standard treatment for APL is trans-retinoic acid (ATRA) in combination with chemotherapy or arsenic.121,122 This approach will induce remission in more than 90% of patients, and a sizable majority of these patients will be cured of their APL. ATRA therapy will also lead to early correction of the coagulation defects, often within the first week of therapy.123 This is in stark contrast to the chemotherapy era when the coagulation defects would become worse with therapy. Given the marked beneficial effect of ATRA on the coagulopathy of APL and its low toxicity profile, it should be empirically started for any patients suspected of having APL while genetic testing is being performed. Rare reports of massive thrombosis complicating therapy with ATRA exist, but the relationship to either the APL or ATRA is unknown.
Therapy for the coagulation defects consists of aggressive transfusion therapy support and possible use of other pharmacologic agents to control DIC.124,125 The fibrinogen level should be maintained at over 150 mg/dL and the platelet count at over 50,000 cells/µL.126 Controversy still exists over the role of heparin in therapy of APL.104 Although attractive for its ability to quench thrombin, heparin use can lead to profound bleeding and its use in treating APL has fallen out of favor.
SNAKEBITES
Snake envenomation can lead to direct activation of multiple coagulation enzymes, including factors V, X, thrombin, and protein C, and lead to cleavage of fibrinogen.127,128 Envenomation can also activate coagulation and damage vascular endothelium. The DIC can be enhanced by widespread tissue necrosis and hypotension. The key to management of snake bites is administration of specific antivenom. The role of prophylactic factor replacement is controversial, but this therapy is indicated if there is clinical bleeding.129 One confounder is that some snake venoms, especially rattlesnake, can induce reversible platelet aggregation, which corrects with antivenom.
LOCAL VASCULAR ABNORMALITIES
Abnormal vascular structures, such as vascular tumors, vascular malformations, and aneurysms, can lead to localized areas of thrombin generation that can “spill-over” into the general circulation, leading to DIC. The diagnosis Kasabach-Merritt phenomenon should be reserved for children with vascular tumors such as angioma or hemangioendothelioma.130 Therapy depends on the lesion. Embolization to reduce blood flow of vascular malformations can either be definitive therapy or stabilize the patient for surgery. Aneurysms can be repaired by surgery or stenting. Rare patients with aneurysms with significant coagulopathy may require heparin to raise the fibrinogen level before surgery. Kasabach-Merritt disease can respond to steroids or therapy such as vincristine or interferon.130 Increasing data shows that use of the mTOR inhibitor sirolimus can shrink these vascular abnormalities leading to lessening of the coagulopathy.131
CONCLUSION
At the most basic level, DIC is the excess activity of thrombin. However, the clinical presentation and therapy can differ greatly depending on the primary cause. Both diagnosis and therapy involve close coordination of laboratory data and clinical assessment.
INTRODUCTION
In the normal person, the process of coagulation is finely controlled at many levels to ensure the appropriate amount of hemostasis at the appropriate location. Broadly defined, disseminated intravascular coagulation (DIC) is the name given to any process that disrupts this fine tuning, leading to unregulated coagulation. Defined this way, DIC may be found in a variety of patients with a variety of disease states, and can present with a spectrum of findings ranging from asymptomatic abnormal laboratory results to florid bleeding or thrombosis. It is important to remember that DIC is always a consequence of an underlying pathological process and not a disease in and of itself. This article first reviews concepts common to all forms of DIC, and then reviews the more common disease states that lead to DIC.
PATHOGENESIS
At the most basic level, DIC is the clinical manifestation of inappropriate thrombin activation.1–5 Inappropriate thrombin activation can be due to underlying conditions such as sepsis and obstetric disasters. The activation of thrombin leads to (1) conversion of fibrinogen to fibrin, (2) activation of platelets (and their consumption), (3) activation of factors V and VIII, (4) activation of protein C (and degradation of factors Va and VIIIa), (5) activation of endothelial cells, and (6) activation of fibrinolysis (Table 1).
1. Conversion of fibrinogen to fibrin, which leads to the formation of fibrin monomers and excessive thrombus formation. These thrombi are rapidly dissolved by excessive fibrinolysis in most patients. In certain clinical situations, especially cancer, excessive thrombosis will occur. In patients with cancer, this is most often a deep venous thrombosis. Rare patients, especially those with pancreatic cancer, may have severe DIC with multiple arterial and venous thromboses. Nonbacterial thrombotic endocarditis can also be seen in these patients, leading to widespread embolic complications.
2. Activation of platelets and their consumption. Thrombin is the most potent physiologic activator of platelets, so in DIC there is increased activation of platelets. These activated platelets are consumed, resulting in thrombocytopenia. Platelet dysfunction is also present. Platelets that have been activated and have released their contents but still circulate are known as “exhausted” platelets; these cells can no longer function to support coagulation. The fibrin degradation products (FDP) in DIC can also bind to GP IIb/IIIa and inhibit further platelet aggregation.
3. Activation of factors V, VIII, XI, and XIII. Activation of these factors can promote thrombosis, but they are then rapidly cleared by antithrombin (XI) or activated protein C (V and VIII) or by binding to the fibrin clot (XIII). This can lead to depletion of all the prothrombotic clotting factors and antithrombin, which in turn can lead to both thrombosis and bleeding.
4. Activation of protein C further promotes degradation of factors Va and VIIIa, enhances fibrinolysis, and decreases protein C levels.
5. Activation of endothelial cells, especially in the skin, may lead to thrombosis, and in certain patients, especially those with meningococcemia, purpura fulminans. Endothelial damage will down-regulate thrombomodulin, preventing activation of protein C and leading to further reductions in levels of activated protein C.56. Activation of fibrinolysis leads to the breakdown of fibrin monomers, formation of fibrin thrombi, and increased circulating fibrinogen. In most patients with DIC, the fibrinolytic response is brisk.6 This is why most patients with DIC present with bleeding and prolonged clotting times.
PATTERNS OF DIC
The clinical manifestations of DIC in a given patient depend on the balance of thrombin activation and secondary fibrinolysis plus the patient’s ability to compensate for the DIC. Patients with DIC can present in 1 of 4 patterns:1–3
1. Asymptomatic. Patients can present with laboratory evidence of DIC but no bleeding or thrombosis. This is often seen in patients with sepsis or cancer. However, with further progression of the underlying disease, these patients can rapidly become symptomatic.
2. Bleeding. The bleeding is due to a combination of factor depletion, platelet dysfunction, thrombocytopenia, and excessive fibrinolysis.1 These patients may present with diffuse bleeding from multiple sites (eg, intravenous sites, areas of instrumentation).
3. Thrombosis. Despite the general activation of the coagulation process, thrombosis is unusual in most patients with acute DIC. The exceptions include patients with cancer, trauma patients, and certain obstetrical patients. Most often the thrombosis is venous, but arterial thrombosis and nonbacterial thrombotic endocarditis have been reported.7
4. Purpura fulminans. This form of DIC is discussed in more detail later (see Specific DIC Syndromes section).
DIAGNOSIS
There is no one test that will diagnose DIC; one must match the test to the clinical situation (Table 2).8
SCREENING TESTS
The prothrombin time-INR and activated thromboplastin time (aPPT) are usually elevated in severe DIC but may be normal or shortened in chronic forms.9 One may also see a shortened aPTT in severe acute DIC due to large amounts of activated thrombin and factor X “bypassing” the contact pathway. An aPTT as short as 10 seconds has been seen in acute DIC. The platelet count is usually reduced but may be normal in chronic DIC. Serum fibrinogen and platelets are decreased in acute DIC but again may be in the “normal” range in chronic DIC.10 The most sensitive screening test for DIC is a fall in the platelet count, with low counts seen in 98% of patients and counts under 50,000 cells/μL in 50%.9,11 The least specific test is fibrinogen, which tends to fall below normal only in severe acute DIC.9
SPECIFIC TESTS
This group of tests allows one to deduce that abnormally high concentrations of thrombin are present.
Ethanol Gel and Protamine Tests
Both of these older tests detected circulating fibrin monomers, whose appearance is an early sign of DIC. Circulating fibrin monomers are seen when thrombin acts on fibrinogen. Usually the monomer polymerizes with the fibrin clot, but when there is excess thrombin these monomers can circulate. Detection of circulating fibrin monomer means there is too much IIa and, ergo, DIC is present.
Fibrin(ogen) Degradation Products
Plasmin acts on the fibrin/fibrinogen molecule to cleave the molecule in specific places. The resulting degradation product levels will be elevated in situations of increased fibrin/fibrinogen destruction (DIC and fibrinolysis). The FDP are typically mildly elevated in renal and liver disease due to reduced clearance.
D-Dimers
When fibrin monomers bind to form a thrombus, factor XIII acts to bind their “D” domains together. This bond is resistant to plasmin and thus this degradation fragment is known as the “D-dimer.” High levels of D-dimer indicate that (1) IIa has acted on fibrinogen to form a fibrin monomer that bonded to another fibrin monomer, and (2) this thrombus was lysed by plasmin. Because D-dimers can be elevated (eg, with exercise, after surgery), an elevated D-dimer needs to be interpreted in the context of the clinical situation.11 Currently, this is the most common specific test for DIC performed.
Other Tests
Several other tests are sometimes helpful in diagnosing DIC.
Thrombin time. This test is performed by adding thrombin to plasma. Thrombin times are elevated in DIC (FDPs interfere with polymerization), in the presence of low fibrinogen levels, in dysfibrinogenemia, and in the presence of heparin (very sensitive).
Reptilase time is the same as thrombin time but is performed with a snake venom that is insensitive to heparin. Reptilase time is elevated in the same conditions as the thrombin time, with the exception of the presence of heparin. Thrombin time and reptilase time are most useful in evaluation of dysfibrinogenemia.
Prothrombin fragment 1.2 (F1.2). F1.2 is a small peptide cleaved off when prothrombin is activated to thrombin. Thus, high levels of F1.2 are found in DIC but can be seen in other thrombotic disorders. This test is still of limited clinical value.
DIC scoring system. A scoring system to both diagnose and quantify DIC has been proposed (Figure).11,12
Thromboelastography (TEG). This is a point-of-care test that uses whole blood to determine specific coagulation parameters such as R time (time from start of test to clot formation), maximal amplitude (MA, maximum extent of thrombus), and LY30 (MA at 30 minutes, a measure of fibrinolysis).13 Studies have shown that TEG can identify DIC by demonstrating a shorter R time (excess thrombin generation) which prolongs as coagulation factors are consumed. The MA is decreased as fibrinogen is consumed and the LY30 shows excess fibrinolysis. TEG has been shown to be of particular value in the management of the complex coagulopathy of trauma.14
MIMICKERS OF DIC
It is important to recognize coagulation syndromes that are not DIC, especially those that have specific other therapies. The syndromes most frequently encountered are thrombotic thrombocytopenic purpura (TTP) and catastrophic antiphospholipid antibody syndrome (CAPS). One important clue to both of these syndromes is that, unlike DIC, there is no primary disorder (cancer, sepsis) that is driving the coagulation abnormalities.
TTP should be suspected when any patient presents with any combination of thrombocytopenia, microangiopathic hemolytic anemia (schistocytes and signs of hemolysis) plus end-organ damage.15–18 Patients with TTP most often present with intractable seizures, strokes, or sequelae of renal insufficiency. Many patients who present with TTP have been misdiagnosed as having sepsis, “lupus flare,” or vasculitis. The key diagnostic differentiator between TTP and DIC is the lack of activation of coagulation with TTP—fibrinogen is normal and D-dimers are minimally or not elevated. In TTP, lactate dehydrogenase is invariably elevated, often 2 to 3 times normal.19 The importance of identifying TTP is that untreated TTP is rapidly fatal. Mortality in the pre–plasma exchange era ranged from 95% to 100%. Today plasma exchange therapy is the foundation of TTP treatment and has reduced mortality to less than 20%.16,20–23Rarely patients with antiphospholipid antibody syndrome can present with fulminant multiorgan system failure.24–28 CAPS is caused by widespread microthrombi in multiple vascular fields. These patients will develop renal failure, encephalopathy, adult respiratory distress syndrome (often with pulmonary hemorrhage), cardiac failure, dramatic livedo reticularis, and worsening thrombocytopenia. Many of these patients have pre-existing autoimmune disorders and high-titer anticardiolipin antibodies. It appears that the best therapy for these patients is aggressive immunosuppression with steroids plus plasmapheresis, followed by rituximab or, if in the setting of lupus, intravenous cyclophosphamide monthly.27,29 Early recognition of CAPS can lead to quick therapy and resolution of the multiorgan system failure.
GENERAL THERAPY
The best way to treat DIC is to treat the underlying cause that is driving the thrombin generation.1,2,4,30,31 Fully addressing the underlying cause may not be possible or may take time, and in the meantime it is necessary to disrupt the cycle of thrombosis and/or hemorrhage. In the past, there was concern about using factor replacement due to fears of “feeding the fire,” or perpetuating the cycle of thrombosis. However, these concerns are not supported by evidence, and factors must be replaced if depletion occurs and bleeding ensues.32
Transfusion therapy of the patient with DIC is guided by the 5 laboratory tests that reflect the basic parameters essential for both hemostasis and blood volume status:33,34 hematocrit, platelet count, prothrombin time-INR, aPTT, and fibrinogen level. Decisions regarding replacement therapy are based on the results of these laboratory tests and the clinical situation of the patient (Table 3).
The basic 5 laboratory tests should be repeated after administering the blood products. This allows one to ensure that adequate replacement therapy was given for the coagulation defects. Frequent checks of the coagulation tests also allow rapid identification and treatment of new coagulation defects in a timely fashion. A flow chart of the test and the blood products administered should also be maintained. This is important in acute situations such as trauma or obstetrical bleeding.
In theory, since DIC is the manifestation of exuberant thrombin production, blocking thrombin with heparin should decrease or shut down DIC. However, studies have shown that in most patients heparin administration has led to excessive bleeding. Currently, heparin therapy is reserved for patients who have thrombosis as a component of their DIC.2,41,42 Given the coagulopathy that is often present, specific heparin levels instead of the aPTT should be used to monitor anticoagulation.43,44
SPECIFIC DIC SYNDROMES
SEPSIS/INFECTIOUS DISEASE
Any overwhelming infection can lead to DIC.45 Classically, it was believed that gram-negative bacteria can lead tissue factor exposure via production of endotoxin, but recent studies indicate that DIC can be seen with any overwhelming infection.46,47 There are several potential avenues by which infections can lead to DIC. As mentioned, gram-negative bacteria produce endotoxin that can directly lead to tissue factor exposure, resulting in excess thrombin generation. In addition, any infection can lead to expression of inflammatory cytokines that induce tissue-factor expression by endothelium and monocytes. Some viruses and Rickettsia species can directly infect the vascular endothelium, converting it from an antithrombotic to a prothrombotic phenotype.48 When fighting infections, neutrophils can extrude their contents, including DNA, to help trap organisms. These neutrophil extracellular traps (NETS) may play an important role in promoting coagulopathy.49,50 The hypotension produced by sepsis leads to tissue hypoxia, which results in more DIC. The coagulopathy in sepsis can range from subtle abnormalities of testing to purpura fulminans. Thrombocytopenia is worsened by cytokine-induced hemophagocytic syndrome.
As with all forms of DIC, empiric therapy targeting the most likely source of infection and maintaining hemodynamic stability is the key to therapy. As discussed below, heparin and other forms of coagulation replacement appear to be of no benefit in therapy.
PURPURA FULMINANS
DIC in association with necrosis of the skin is seen in primary and secondary purpura fulminans.51,52 Primary purpura fulminans is most often seen after a viral infection.53 In these patients, the purpura fulminans starts with a painful red area on an extremity that rapidly progresses to a black ischemic area. In many patients, acquired deficiency of protein S is found.51,54,55 Secondary purpura fulminans is most often associated with meningococcemia infections but can be seen in any patient with overwhelming infection.56–58 Post-splenectomy sepsis syndrome patients and those with functional hyposplenism due to chronic liver diseases are also at risk.59 Patients present with signs of sepsis, and the skin lesions often involve the extremities and may lead to amputations. As opposed to primary purpura fulminans, those with the secondary form will have symmetrical ischemia distally (toes and fingers) that ascends as the process progresses. Rarely, adrenal infarction (Waterhouse-Friderichsen syndrome) occurs, which leads to severe hypotension.45
Recently, Warkenten has reported on limb gangrene in critically ill patients complicating sepsis or cardiogenic shock.60,61 These patients have DIC that is complicated by shock liver. Deep venous thrombosis with ischemic gangrene then develops, which can result in tissue loss and even amputation. The pathogenesis is hypothesized to be hepatic dysfunction leading to sudden drops in protein C and S plasma levels, which then leads to thrombophilia with widespread microvascular thrombosis. Therapy for purpura fulminans is controversial. Primary purpura fulminans, especially in those with postvaricella autoimmune protein S deficiency, has responded to plasma infusion titrated to keep the protein S level above 25%.51 Intravenous immunoglobulin has also been reported to help decrease the anti-protein S antibodies. Heparin has been reported to control the DIC and extent of necrosis.62 The starting dose in these patients is 5 to 8 units/kg/hr.2
Sick patients with secondary purpura fulminans have been treated with plasma drips, plasmapheresis, and continuous plasma ultrafiltration.62–66 Heparin therapy alone has not been shown to improve survival.66 Much attention has been given to replacement of natural anticoagulants such as protein C and antithrombin as therapy for purpura fulminans, but unfortunately randomized trials using antithrombin have shown mostly negative results.51,55,67–69 Trials using protein C concentrates have shown more promise in controlling the coagulopathy of purpura fulminans, but this is not widely available.63,70–72 Unfortunately, many patients will need debridement and amputation for their necrotic limbs, with one review showing approximately 66% of patients needing amputations.52
TRAUMA
Currently, the most common cause of acute DIC is trauma. The coagulation defects that occur in trauma patients are complex in origin and still controversial (including if even calling it DIC is appropriate!).73–76 The most common etiologies are
- Generation of excess activated protein C leading to increased consumption of factor V and VIII and increased fibrinolysis;
- Tissue damage leading to generation of excess thrombin generation;
- Dilution of hemostatic factors by blood or fluid resuscitation; and
- Activation of endothelial cells leading to generation of a prothrombotic surface and shedding of glycocalyx with antithrombotic properties.
Trauma patients are prone to hypothermia, and this can be the major complicating factor in their bleeding.77,78 Patients may be out “in the field” for a prolonged period of time and be hypothermic on arrival.79 Packed red cells are stored at 4°C, and the infusion of 1 unit can lower the body temperature by 0.16°C.80 Hypothermia has profound effects on the coagulation system that are associated with clinical bleeding.77,81,82 Even modest hypothermia can greatly augment bleeding and needs to be treated or prevented.
The initial management of the bleeding trauma patient is administration of red cells and plasma (FFP) in a 1:1 ratio. This has been shown by clinical studies to lessen the risk of exsanguination in the first 24 hours and to be associated with improved clinical outcomes.83,84 The basic set of coagulation tests should also be obtained to guide product replacement, especially as the bleeding is brought under control. Hypothermia can be prevented by several measures, including transfusing the blood through blood warmers. Devices are available that can warm 1 unit of blood per minute. An increasingly used technique is to perform “damage control” surgery. Patients are initially stabilized with control of damaged vessels and packing of oozing sites.85 Then the patient is taken to the intensive care unit to be warmed and have coagulation defects corrected.
For trauma patients at risk of serious bleeding, the use of tranexamic acid reduced all- cause mortality (relative risk 0.91), with death due to bleeding also being reduced (relative risk 0.85).86 There was no increase in thrombosis, but benefit was restricted to patients treated within 3 hours of the trauma. The dose of tranexamic acid was a 1-g bolus followed by a 1-g continuous infusion over 8 hours.
PREGNANCY-RELATED DIC SYNDROMES
Acute DIC of Pregnancy
Pregnancy can be associated with the rapid onset of severe DIC in 2 situations, abruption and amniotic fluid embolism.87,88 The separation of the placenta from the uterine wall creates a space for blood to occupy. Given the richness of the placenta in tissue factor, this leads to activation of coagulation both locally and systemically. Release of blood when this space reaches the vaginal opening can lead to rapid hemorrhage, further augmenting the coagulation abnormalities. Placental insufficiency can lead to fetal demise, which can also worsen the DIC. Management depends on the size of the abruption and the clinical status of both mother and fetus.87 For severe bleeding and DIC, blood product support is crucial to allow safe delivery. In pregnancy, the fibrinogen goal needs to be higher—200 mg/dL.89 For smaller abruption, close observation with early delivery is indicated.
Amniotic fluid embolism is sudden, with the vascular collapse of the woman soon after delivery. Due to the presence of procoagulant rich fluid in the circulatory system, there is often overwhelming DIC. Therapy is directed at both supporting blood volume and correcting hemostatic defects.
HELLP
The acronym HELLP (hemolysis, elevated liver tests, low platelets) describes a variant of preeclampsia.90 Classically, HELLP syndrome occurs after 28 weeks of gestation in a patient with preeclampsia, but can occur as early as 22 weeks in patients with antiphospholipid antibody syndrome.91–93 The preeclampsia need not be severe. The first sign of HELLP is a decrease in the platelet count followed by abnormal liver function tests. Signs of hemolysis are present with abundant schistocytes on the smear and a high lactate dehydrogenase level. HELLP can progress to liver failure, and deaths are also reported due to hepatic rupture. Unlike TTP, fetal involvement is present in the HELLP syndrome, with fetal thrombocytopenia reported in 30% of cases. In severe cases, elevated D-dimers consistent with DIC are also found. Delivery of the child will most often result in cessation of the HELLP syndrome, but refractory cases will require dexamethasone and plasma exchange.94 Patients should be closely observed for 1 to 2 days after delivery as the hematologic picture can transiently worsen before improving.95
Acute Fatty Liver of Pregnancy
Fatty liver of pregnancy also occurs late in pregnancy and is only associated with preeclampsia in 50% of cases.96,97 Patients first present with nonspecific symptoms of nausea and vomiting but can progress to fulminant liver failure. Patients develop thrombocytopenia early in the course, but in the later stages can develop DIC and very low fibrinogen levels. Mortality rates without therapy can be as high as 90%. Low blood glucose and high ammonia levels can help distinguish fatty liver from other pregnancy complications.98 Treatment consists of prompt delivery of the child and aggressive blood product support.
Retained Dead Fetus Syndrome
Becoming rarer in modern practices, the presence of a dead fetus for many weeks (usually ≥ 5) can result in a chronic DIC state with fibrinogen depletion and coagulopathy. In some women, this is worsened at delivery. In a stable patient, a short trial of heparin prior to planning delivery can control the DIC to allow the coagulopathy to stabilize.
DRUG-INDUCED HEMOLYTIC-DIC SYNDROMES
A severe variant of the drug-induced immune complex hemolysis associated with DIC has been recognized. Rare patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone) have developed this syndrome.99–104 The clinical syndrome starts 7 to 10 days after the drug is administered. Often the patient has only received the antibiotic for surgical prophylaxis. The patient will develop severe Coombs’-positive hemolysis with hypotension and DIC. The patients are often believed to have sepsis and in the management of the supposed sepsis often are re-exposed to the cephalosporin, resulting in worsening of the clinical picture. The outcome is often fatal due to massive hemolysis and thrombosis.101,105–107
Quinine is associated with a unique syndrome of drug-induced DIC.108–111 Approximately 24 to 96 hours after quinine exposure, the patient becomes acutely ill with nausea and vomiting. The patient then develops a microangiopathic hemolytic anemia, DIC, and renal failure. Some patients, besides having antiplatelet antibodies, also have antibodies binding to red cells and neutrophils, which may lead to the more severe syndrome. Despite therapy, patients with quinine-induced TTP have a high incidence of chronic renal failure.
Treatment of the drug-induced hemolytic-DIC syndrome is anecdotal. Patients have responded to aggressive therapy, including plasma exchange, dialysis, and prednisone. Early recognition of the hemolytic anemia and the suspicion it is drug related is important for early diagnosis so that the incriminated drug can be discontinued.
CANCER
Cancers, primarily adenocarcinomas, can result in DIC. The classic Trousseau syndrome referred to the association of migratory superficial thrombophlebitis with cancer112 but now refers to cancer associated with thrombotic DIC.113,114 Highly vascular tumor cells are known to express tissue factor.114,115 In addition, some tumor cells can express a direct activator of factor X (“cancer procoagulant”). Unlike many DIC states, cancer presents with thrombosis instead of bleeding. This may be due to the inflammatory state which accompanies cancer, or it may be a unique part of the chronic nature of cancer DIC biology that allows time for the body to compensate for loss of coagulation factors. In some patients, thrombosis is the first sign of an underlying cancer, sometimes predating the cancer diagnosis by months.115 Rarely, the DIC can result in nonthrombotic endocarditis with micro-emboli leading to widespread small-vessel thrombosis.113
Since effective antineoplastic therapy is lacking for many tumors associated with Trousseau syndrome, DIC therapy is aimed at suppressing thrombosis. An exception is prostate cancer, where hormonal therapy can markedly decrease the DIC.116 Due to the tumor directly activating coagulation factors, inhibition of active enzymes via heparin has been shown to reduce rates of recurrence compared with warfarin.114,115 Clinical trials have demonstrated that heparin therapy is associated with a lower thrombosis recurrence rate than warfarin.117,118 In some patients, the thrombotic process is so vigorous that new thrombosis can be seen within hours of stopping heparin.112
ACUTE PROMYELOCYTIC LEUKEMIA
There are multiple hemostatic defects in patients with acute promyelocytic leukemia (APL).119 Most, if not all, patients with APL have evidence of DIC at the time of diagnosis. Patients with APL have a higher risk of death during induction therapy as compared with patients with other forms of leukemia, with death most often due to bleeding. Once in remission, APL patients have a higher cure rate than most patients with leukemia. APL is also unique among leukemias in that biologic therapy with retinoic acid or arsenic is effective in inducing remission and cure in most patients. Although effective therapy is available, early death rates due to bleeding have not changed.119
APL patients can present with pancytopenia due to leukemic marrow replacement or with diffuse bleeding due to DIC and thrombocytopenia. Life-threatening bleeding such as intracranial hemorrhage may occur at any time until the leukemia is put into remission. The etiology of the hemostatic defects in APL is complex and is thought to be the result of DIC, fibrinolysis, and the release of prothrombotic extracellular chromatin and other procoagulant enzymes.119,120 The diagnosis of APL can be straightforward when the leukemic cells are promyelocytes with abundant Auer rods, although some patients have the microgranular form without obvious Auer rods. The precise diagnosis requires molecular methods, including obtaining FISH for detecting the t(15;17) in PML/RARA fusion. Upon diagnosis of APL, one should obtain a complete coagulation profile, including INR, aPTT, fibrinogen, platelet count, and D-dimers. Change in fibrinogen levels tends to be a good marker of progress in treating the coagulation defects.
Therapy of APL involves treating both the leukemia and the coagulopathy. Currently, the standard treatment for APL is trans-retinoic acid (ATRA) in combination with chemotherapy or arsenic.121,122 This approach will induce remission in more than 90% of patients, and a sizable majority of these patients will be cured of their APL. ATRA therapy will also lead to early correction of the coagulation defects, often within the first week of therapy.123 This is in stark contrast to the chemotherapy era when the coagulation defects would become worse with therapy. Given the marked beneficial effect of ATRA on the coagulopathy of APL and its low toxicity profile, it should be empirically started for any patients suspected of having APL while genetic testing is being performed. Rare reports of massive thrombosis complicating therapy with ATRA exist, but the relationship to either the APL or ATRA is unknown.
Therapy for the coagulation defects consists of aggressive transfusion therapy support and possible use of other pharmacologic agents to control DIC.124,125 The fibrinogen level should be maintained at over 150 mg/dL and the platelet count at over 50,000 cells/µL.126 Controversy still exists over the role of heparin in therapy of APL.104 Although attractive for its ability to quench thrombin, heparin use can lead to profound bleeding and its use in treating APL has fallen out of favor.
SNAKEBITES
Snake envenomation can lead to direct activation of multiple coagulation enzymes, including factors V, X, thrombin, and protein C, and lead to cleavage of fibrinogen.127,128 Envenomation can also activate coagulation and damage vascular endothelium. The DIC can be enhanced by widespread tissue necrosis and hypotension. The key to management of snake bites is administration of specific antivenom. The role of prophylactic factor replacement is controversial, but this therapy is indicated if there is clinical bleeding.129 One confounder is that some snake venoms, especially rattlesnake, can induce reversible platelet aggregation, which corrects with antivenom.
LOCAL VASCULAR ABNORMALITIES
Abnormal vascular structures, such as vascular tumors, vascular malformations, and aneurysms, can lead to localized areas of thrombin generation that can “spill-over” into the general circulation, leading to DIC. The diagnosis Kasabach-Merritt phenomenon should be reserved for children with vascular tumors such as angioma or hemangioendothelioma.130 Therapy depends on the lesion. Embolization to reduce blood flow of vascular malformations can either be definitive therapy or stabilize the patient for surgery. Aneurysms can be repaired by surgery or stenting. Rare patients with aneurysms with significant coagulopathy may require heparin to raise the fibrinogen level before surgery. Kasabach-Merritt disease can respond to steroids or therapy such as vincristine or interferon.130 Increasing data shows that use of the mTOR inhibitor sirolimus can shrink these vascular abnormalities leading to lessening of the coagulopathy.131
CONCLUSION
At the most basic level, DIC is the excess activity of thrombin. However, the clinical presentation and therapy can differ greatly depending on the primary cause. Both diagnosis and therapy involve close coordination of laboratory data and clinical assessment.
1. Carey MJ, Rodgers GM. Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998;59:65–73.
2. De Jonge E, Levi M, Stoutenbeek CP, Van Deventer SJH. Current drug treatment strategies for disseminated intravascular coagulation. Drugs 1998;55:767–77.
3. Baker WF Jr. Clinical aspects of disseminated intravascular coagulation: a clinician’s point of view. Sem Thrombosis Hemostasis 1989;15:1–57.
4. Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med 1999;341:586–92.
5. Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers 2016;2:16037.
6. Kolev K, Longstaff C. Bleeding related to disturbed fibrinolysis. Br J Haematol 2016;175:12–23.
7. Sharma S, Mayberry JC, DeLoughery TG, Mullins RJ. Fatal cerebroembolism from nonbacterial thrombotic endocarditis in a trauma patient: case report and review. Mil Med 2000;165:83–5.
8. Toh CH, Alhamdi Y, Abrams ST. Current pathological and laboratory considerations in the diagnosis of disseminated intravascular coagulation. Ann Lab Med 2016;36:505–12.
9. Yu M, Nardella A, Pechet L. Screening tests of disseminated intravascular coagulation: guidelines for rapid and specific laboratory diagnosis. Crit Care Med 2000;28:1777–80.
10. Mant MJ, King EG. Severe, acute disseminated intravascular coagulation. A reappraisal of its pathophysiology, clinical significance, and therapy based on 47 patients. Am J Med 1979;67:557–63.
11. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol 2009;145:24–33.
12. Levi M. Disseminated intravascular coagulation. Crit Care Med 2007;35:2191–5.
13. Nogami K. The utility of thromboelastography in inherited and acquired bleeding disorders. Br J Haematol 2016;174:503–14.
14. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin 2017;33:119–34.
15. George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med 2006;354:1927–35.
16. George JN. How I treat patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Blood 2000;96:1223–9.
17. Murrin RJ, Murray JA. Thrombotic thrombocytopenic purpura: aetiology, pathophysiology and treatment. Blood Rev 2006;20:51–60.
18. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017;129:2836–46.
19. Patton JF, Manning KR, Case D, Owen J. Serum lactate dehydrogenase and platelet count predict survival in thrombotic thrombocytopenic purpura. Am J Hematol 1994;47:94–9.
20. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991;325:393–7.
21. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpurahemolytic uremic syndrome—clinical experience in 108 patients. N Engl J Med 1991;325:398–403.
22. Kaplan BS, Trachtman H. Improve survival with plasma exchange thrombotic thrombopenic purpura-hemolytic uremic syndrome. Am J Med 2001;110:156–7.
23. Kremer Hovinga JA, Coppo P, Lammle B, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers 2017;3:17020.
24. Asherson RA. The catastrophic antiphospholipid syndrome [editorial]. J Rheumatol 1992;19:508–12.
25. Asherson RA, Piette JC. The catastrophic antiphospholipid syndrome 1996: acute multi-organ failure associated with antiphospholipid antibodies: a review of 31 patients. Lupus 1996;5:414–7.
26. Asherson RA, Cervera R. Castastrophic antiphospholipid syndrome. Curr Opinion Hematol 2000;5:325–9.
27. Merrill JT, Asherson RA. Catastrophic antiphospholipid syndrome. Nat Clin Pract Rhuem 2006;2:81–9.
28. Rodriguez-Pinto I, Espinosa G, Cervera R. Catastrophic antiphospholipid syndrome: The current management approach. Best Pract Res Clin Rheumatol 2016;30:239–9.
29. Kazzaz NM, McCune WJ, Knight JS. Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol 2016;28:218–27.
30. Hoffman JN, Faist E. Coagulation inhibitor replacement during sepsis: useless? Crit Care Med 2000;28(9 Suppl):S74–6.
31. Wada H, Asakura H, Okamoto K, et al. Expert consensus for the treatment of disseminated intravascular coagulation in Japan. Japanese Society of Thrombosis Hemostasis/DIC subcommittee. Thromb Res 2010;125:6–11.
32. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
33. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
34. Stainsby D, MacLennan S, Hamilton PJ. Management of massive blood loss: a template guideline. Br J Anaesth 2000;85:487–91.
35. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999;340:409–17.
36. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
37. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
38. Miller RD, Robbins TO, Tong MJ, Barton SL. Coagulation defects associated with massive blood transfusions. Ann Surg 1971;174:794–801.
39. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
40. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
41. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
42. Callander N, Rapaport SI. Trousseau’s syndrome. West J Med 1993;158:364–71.
43. Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993;119:104–9.
44. Olson JD, Arkin CF, Brandt JT, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulant therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998;122:782–8.
45. Yoshikawa T, Tanaka KR, Guze LB. Infection and disseminated intravascular coagulation. Medicine (Baltimore) 1971;50:237–58.
46. Jagneaux T, Taylor DE, Kantrow SP. Coagulation in sepsis. Am J Med Sci 2004;328:196–204.
47. Lipinska-Gediga M. Coagulopathy in sepsis - a new look at an old problem. Anaesthesiol Intensive Ther 2016;48:352–9.
48. Van Gorp ECM, Suharti C, ten Cate H, et al. Review: Infections diseases and coagulation disorders. Journal of Infectious Diseases 1999;180:176–86.
49. McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017;129:1357–67.
50. Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011;118:1952–61.
51. Darmstadt GL. Acute infectious purpura fulminans: pathogenesis and medical management. Pediatr Dermatol 1998;15:169–83.
52. Davis MD, Dy KM, Nelson S. Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic. J Am Acad Dermatol 2007;57:944–56.
53. Spicer TE, Rau JM. Purpura fulminans. Am J Med 1976;61:566–71.
54. Josephson C, Nuss R, Jacobson L, et al. The varicellaautoantibody syndrome. Pediatr Res 2001;50:345–52.
55. Smith OP, White B. Infectious purpura fulminans: diagnosis and treatment. Br J Haematol 1999;104:202–7.
56. Gamper G, Oschatz E, Herkner H, et al. Sepsis-associated purpura fulminans in adults. Wien Klin Wochenschr 2001;113:107–12.
57. Ward KM, Celebi JT, Gmyrek R, Grossman ME. Acute infectious purpura fulminans associated with asplenism or hyposplenism. J Am Acad Dermatol 2002;47:493–6.
58. Childers BJ, Cobanov B. Acute infectious purpura fulminans: a 15-year retrospective review of 28 consecutive cases. Am Surg 2003;69:86–90.
59. Carpenter CT, Kaiser AB. Purpura fulminans in pneumococcal sepsis: case report and review. Scand J Infect Dis 1997;29:479–83.
60. Warkentin TE, Pai M. Shock, acute disseminated intravascular coagulation, and microvascular thrombosis: is ‘shock liver’ the unrecognized provocateur of ischemic limb necrosis: reply. J Thromb Haemost 2016;14:2317–9.
61. Warkentin TE. Ischemic limb gangrene with pulses. N Engl J Med 2015;373:642–55.
62. Duncan A. New therapies for severe meningococcal disease but better outcomes? Lancet 1997;350:1565–6.
63. Smith OP, White B, Vaughan D, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet1997;350:1590–3.
64. Branson HE, Katz J. A structured approach to the management of purpura fulminans. J Natl Med Assoc 1983;75:821–5.
65. Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth 2001;86:581–6.
66. Manios SG, Kanakoudi F, Maniati E. Fulminant meningococcemia. Heparin therapy and survival rate. Scand J Infect Dis 1971;3:127–33.
67. Giudici D, Baudo F, Palareti G, et al. Antithrombin replacement in patients with sepsis and septic shock. Haematologica 1999;84:452–60.
68. Fourrier F, Jourdain M, Tournoys A. Clinical trial results with antithrombin III in sepsis. Crit Care Med 2000;28(9 Suppl):S38–43.
69. Levi M, De Jonge E, van der PT, ten Cate H. Novel approaches to the management of disseminated intravascular coagulation. Crit Care Med 2000;28(9 Suppl):S20–4.
70. Rivard GE, David M, Farrell C, Schwarz HP. Treatment of purpura fulminans in meningococcemia with protein C concentrate. J Pediatr 1995;126:646–52.
71. White B, Livingstone W, Murphy C, et al. An open-label study of the role of adjuvant hemostatic support with protein C replacement therapy in purpura fulminans-associated meningococcemia. Blood 2000;96:3719–24.
72. Schellongowski P, Bauer E, Holzinger U, et al. Treatment of adult patients with sepsis-induced coagulopathy and purpura fulminans using a plasma-derived protein C concentrate (Ceprotin). Vox Sang 2006;90:294–301.
73. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
74. Cohen MJ, Christie SA. Coagulopathy of trauma. Crit Care Clin 2017;33:101–18.
75. Giordano S, Spiezia L, Campello E, Simioni P. The current understanding of trauma-induced coagulopathy (TIC): a focused review on pathophysiology. Intern Emerg Med 2017 May 5.
76. Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood 2016;128:1043–9.
77. Eddy VA, Morris JA Jr, Cullinane DC. Hypothermia, coagulopathy, and acidosis. Surg Clin North Am 2000;80:845–54.
78. Peng RY, Bongard FS. Hypothermia in trauma patients. J Am Coll Surg 1999;188:685–96.
79. Steinemann S, Shackford SR, Davis JW. Implications of admission hypothermia in trauma patients. J Trauma 1990;30:200–2.
80. Rajek A, Greif R, Sessler DI, et al. Core cooling by central venous infusion of ice-cold (4 degrees C and 20 degrees C) fluid: isolation of core and peripheral thermal compartments. Anesthesiol 2000;93:629–37.
81. Watts DD, Trask A, Soeken K, et al. Hypothermic coagulopathy in trauma: effect of varying levels of hypothermia on enzyme speed, platelet function, and fibrinolytic activity. J Trauma 1998;44:846–54.
82. Ferrara A, MacArthur JD, Wright HK, et al. Hypothermia and acidosis worsen coagulopathy in the patient requiring massive transfusion. Am J Surg 1990;160:515–8.
83. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
84. Johansson PI, Stensballe J, Oliveri R, Wade CE, Ostrowski SR, Holcomb JB. How I treat patients with massive hemorrhage. Blood 2014;124:3052–8.
85. Stone HH, Strom PR, Mullins RJ. Management of the major coagulopathy with onset during laparotomy. Ann Surg 1983;197:532–5.
86. WOMAN Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23–32.
87. Hall DR. Abruptio placentae and disseminated intravascular coagulopathy. Semin Perinatol 2009;33:189–95.
88. Thachil J, Toh CH. Disseminated intravascular coagulation in obstetric disorders and its acute haematological management. Blood Rev 2009;23:167–76.
89. Collins P, Abdul-Kadir R, Thachil J, Subcommittees on Women’ s Health Issues in T, Haemostasis, on Disseminated Intravascular C. Management of coagulopathy associated with postpartum hemorrhage: guidance from the SSC of the ISTH. J Thromb Haemost 2016;14:205–10.
90. Baxter JK, Weinstein L. HELLP syndrome: the state of the art. Obstet Gynecol Surv 2004;59:838–45.
91. Egerman RS, Sibai BM. HELLP syndrome. Clin Obstetr Gynecol 1999;42:381–9.
92. Saphier CJ, Repke JT. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome: a review of diagnosis and management. Sem Perinatol 1998;22:118–33.
93. Le Thi TD, Tieulie N, Costedoat N, et al. The HELLP syndrome in the antiphospholipid syndrome: retrospective study of 16 cases in 15 women. Ann Rheum Dis 2005;64:273–8.
94. Martin JN Jr, Perry KG Jr, Blake PG, et al. Better maternal outcomes are achieved with dexamethasone therapy for postpartum HELLP (hemolysis, elevated liver enzymes, and thrombocytopenia) syndrome. Am J Obstet Gynecol 1997;177:1011–7.
95. Magann EF, Martin JN Jr. Twelve steps to optimal management of HELLP syndrome. Clinical Obstet Gynecol 1999;42:532–50.
96. Jwayyed SM, Blanda M, Kubina M. Acute fatty liver of pregnancy. J Emerg Medi 1999;17:673–7.
97. Bacq Y. Acute fatty liver of pregnancy. Sem Perinatol 1998;22:134–40.
98. Egerman RS, Sibai BM. Imitators of preeclampsia and eclampsia. Clin Obstet Gynecol 1999;42:551–62.
99. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Medi Rev 1993;7:255–67.
100. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis 1992;15:863–5.
101. Garratty G, Nance S, Lloyd M, Domen R. Fatal immune hemolytic anemia due to cefotetan. Transfusion 1992;32:269–71.
102. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes:case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion 1999;39:306–9.
103. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion 1999;39:1239–46.
104. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol 2006;81:186–8.
105. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr 1995;126:813–5.
106. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1995;14:1116–7.
107. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone. J Pediatr 1995;126:816–7.
108. Gottschall JL, Elliot W, Lianos E, et al. Quinine-induced immune thrombocytopenia associated with hemolytic uremic syndrome: a new clinical entity. Blood 1991;77:306–10.
109. Gottschall JL, Neahring B, McFarland JG, et al. Quinine-induced immune thrombocytopenia with hemolytic uremic syndrome: clinical and serological findings in nine patients and review of literature. Am J Hematol 1994;47:283–9.
110. Crum NF, Gable P. Quinine-induced hemolytic-uremic syndrome. South Med J 2000;93:726–8.
111. Vesely T, Vesely JN, George JN. Quinine-Induced thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS): frequency, clinical features, and long-term outcomes. Blood 2000;96:629 [abstract].
112. Bell WR, Starksen NF, Tong S, Porterfield JK. Trousseau’s syndrome. Devastating coagulopathy in the absence of heparin. Am J Med 1985;79:423–30.
113. Sack GH, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinic, pathophysiologic, and therapeutic features. Medicine 1977;56:1–37.
114. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007;110:1723–9.
115. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol 2005;6:401–10.
116. de la Fouchardiere C, Flechon A, Droz JP. Coagulopathy in prostate cancer. Neth J Med 2003;61:347–54.
117. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. 8th ed. Chest 2008;133(6 Suppl):454S–545S.
118. Lee AY, Kamphuisen PW, Meyer G, et al. Tinzaparin vs warfarin for treatment of acute venous thromboembolism in patients with active cancer: a randomized clinical trial. JAMA 2015;314:677–86.
119. Choudhry A, DeLoughery TG. Bleeding and thrombosis in acute promyelocytic leukemia. Am J Hematol 2012;87:596–603.
120. Cao M, Li T, He Z, et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood 2017;129:1855–64.
121. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008;111:2505–15.
122. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013;369:111–21.
123. Dombret H, Scrobohaci ML, Ghorra P, et al. Coagulation disorders associated iwth acute promyelocytic leukemia: Corrective effect of all-trans retinoic acid treatment. Leukemia 1993;7:2–9.
124. Falanga A, Rickles FR. Management of thrombohemorrhagic syndromes (THS) in hematologic malignancies. Hematology Am Soc Hematol Educ Program 2007;2007:165–71
125. Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood 2009;114:5126–35.
126. Sanz MA, Grimwade D, Tallman MS, et al. Guidelines on the management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009;113:1875–91.
127. Lu Q, Clemetson JM, Clemetson KJ. Snake venoms and hemostasis. J Thromb Haemost 2005;3:1791–9.
128. Berling I, Isbister GK. Hematologic effects and complications of snake envenoming. Transfus Med Rev 2015;29:82–9.
129. Isbister GK, Jayamanne S, Mohamed F, et al. A randomized controlled trial of fresh frozen plasma for coagulopathy in Russell’s viper (Daboia russelii) envenoming. J Thromb Haemost 2017;15:645–54.
130. Rodriguez V, Lee A, Witman PM, Anderson PA. Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 2009;31:522–6.
131. Triana P, Dore M, Cerezo VN, et al. Sirolimus in the treatment of vascular anomalies. Eur J Pediatr Surg 2017;27:86–90.
1. Carey MJ, Rodgers GM. Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998;59:65–73.
2. De Jonge E, Levi M, Stoutenbeek CP, Van Deventer SJH. Current drug treatment strategies for disseminated intravascular coagulation. Drugs 1998;55:767–77.
3. Baker WF Jr. Clinical aspects of disseminated intravascular coagulation: a clinician’s point of view. Sem Thrombosis Hemostasis 1989;15:1–57.
4. Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med 1999;341:586–92.
5. Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers 2016;2:16037.
6. Kolev K, Longstaff C. Bleeding related to disturbed fibrinolysis. Br J Haematol 2016;175:12–23.
7. Sharma S, Mayberry JC, DeLoughery TG, Mullins RJ. Fatal cerebroembolism from nonbacterial thrombotic endocarditis in a trauma patient: case report and review. Mil Med 2000;165:83–5.
8. Toh CH, Alhamdi Y, Abrams ST. Current pathological and laboratory considerations in the diagnosis of disseminated intravascular coagulation. Ann Lab Med 2016;36:505–12.
9. Yu M, Nardella A, Pechet L. Screening tests of disseminated intravascular coagulation: guidelines for rapid and specific laboratory diagnosis. Crit Care Med 2000;28:1777–80.
10. Mant MJ, King EG. Severe, acute disseminated intravascular coagulation. A reappraisal of its pathophysiology, clinical significance, and therapy based on 47 patients. Am J Med 1979;67:557–63.
11. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol 2009;145:24–33.
12. Levi M. Disseminated intravascular coagulation. Crit Care Med 2007;35:2191–5.
13. Nogami K. The utility of thromboelastography in inherited and acquired bleeding disorders. Br J Haematol 2016;174:503–14.
14. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin 2017;33:119–34.
15. George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med 2006;354:1927–35.
16. George JN. How I treat patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Blood 2000;96:1223–9.
17. Murrin RJ, Murray JA. Thrombotic thrombocytopenic purpura: aetiology, pathophysiology and treatment. Blood Rev 2006;20:51–60.
18. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017;129:2836–46.
19. Patton JF, Manning KR, Case D, Owen J. Serum lactate dehydrogenase and platelet count predict survival in thrombotic thrombocytopenic purpura. Am J Hematol 1994;47:94–9.
20. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991;325:393–7.
21. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpurahemolytic uremic syndrome—clinical experience in 108 patients. N Engl J Med 1991;325:398–403.
22. Kaplan BS, Trachtman H. Improve survival with plasma exchange thrombotic thrombopenic purpura-hemolytic uremic syndrome. Am J Med 2001;110:156–7.
23. Kremer Hovinga JA, Coppo P, Lammle B, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers 2017;3:17020.
24. Asherson RA. The catastrophic antiphospholipid syndrome [editorial]. J Rheumatol 1992;19:508–12.
25. Asherson RA, Piette JC. The catastrophic antiphospholipid syndrome 1996: acute multi-organ failure associated with antiphospholipid antibodies: a review of 31 patients. Lupus 1996;5:414–7.
26. Asherson RA, Cervera R. Castastrophic antiphospholipid syndrome. Curr Opinion Hematol 2000;5:325–9.
27. Merrill JT, Asherson RA. Catastrophic antiphospholipid syndrome. Nat Clin Pract Rhuem 2006;2:81–9.
28. Rodriguez-Pinto I, Espinosa G, Cervera R. Catastrophic antiphospholipid syndrome: The current management approach. Best Pract Res Clin Rheumatol 2016;30:239–9.
29. Kazzaz NM, McCune WJ, Knight JS. Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol 2016;28:218–27.
30. Hoffman JN, Faist E. Coagulation inhibitor replacement during sepsis: useless? Crit Care Med 2000;28(9 Suppl):S74–6.
31. Wada H, Asakura H, Okamoto K, et al. Expert consensus for the treatment of disseminated intravascular coagulation in Japan. Japanese Society of Thrombosis Hemostasis/DIC subcommittee. Thromb Res 2010;125:6–11.
32. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
33. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
34. Stainsby D, MacLennan S, Hamilton PJ. Management of massive blood loss: a template guideline. Br J Anaesth 2000;85:487–91.
35. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999;340:409–17.
36. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
37. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
38. Miller RD, Robbins TO, Tong MJ, Barton SL. Coagulation defects associated with massive blood transfusions. Ann Surg 1971;174:794–801.
39. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
40. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
41. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
42. Callander N, Rapaport SI. Trousseau’s syndrome. West J Med 1993;158:364–71.
43. Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993;119:104–9.
44. Olson JD, Arkin CF, Brandt JT, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulant therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998;122:782–8.
45. Yoshikawa T, Tanaka KR, Guze LB. Infection and disseminated intravascular coagulation. Medicine (Baltimore) 1971;50:237–58.
46. Jagneaux T, Taylor DE, Kantrow SP. Coagulation in sepsis. Am J Med Sci 2004;328:196–204.
47. Lipinska-Gediga M. Coagulopathy in sepsis - a new look at an old problem. Anaesthesiol Intensive Ther 2016;48:352–9.
48. Van Gorp ECM, Suharti C, ten Cate H, et al. Review: Infections diseases and coagulation disorders. Journal of Infectious Diseases 1999;180:176–86.
49. McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017;129:1357–67.
50. Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011;118:1952–61.
51. Darmstadt GL. Acute infectious purpura fulminans: pathogenesis and medical management. Pediatr Dermatol 1998;15:169–83.
52. Davis MD, Dy KM, Nelson S. Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic. J Am Acad Dermatol 2007;57:944–56.
53. Spicer TE, Rau JM. Purpura fulminans. Am J Med 1976;61:566–71.
54. Josephson C, Nuss R, Jacobson L, et al. The varicellaautoantibody syndrome. Pediatr Res 2001;50:345–52.
55. Smith OP, White B. Infectious purpura fulminans: diagnosis and treatment. Br J Haematol 1999;104:202–7.
56. Gamper G, Oschatz E, Herkner H, et al. Sepsis-associated purpura fulminans in adults. Wien Klin Wochenschr 2001;113:107–12.
57. Ward KM, Celebi JT, Gmyrek R, Grossman ME. Acute infectious purpura fulminans associated with asplenism or hyposplenism. J Am Acad Dermatol 2002;47:493–6.
58. Childers BJ, Cobanov B. Acute infectious purpura fulminans: a 15-year retrospective review of 28 consecutive cases. Am Surg 2003;69:86–90.
59. Carpenter CT, Kaiser AB. Purpura fulminans in pneumococcal sepsis: case report and review. Scand J Infect Dis 1997;29:479–83.
60. Warkentin TE, Pai M. Shock, acute disseminated intravascular coagulation, and microvascular thrombosis: is ‘shock liver’ the unrecognized provocateur of ischemic limb necrosis: reply. J Thromb Haemost 2016;14:2317–9.
61. Warkentin TE. Ischemic limb gangrene with pulses. N Engl J Med 2015;373:642–55.
62. Duncan A. New therapies for severe meningococcal disease but better outcomes? Lancet 1997;350:1565–6.
63. Smith OP, White B, Vaughan D, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet1997;350:1590–3.
64. Branson HE, Katz J. A structured approach to the management of purpura fulminans. J Natl Med Assoc 1983;75:821–5.
65. Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth 2001;86:581–6.
66. Manios SG, Kanakoudi F, Maniati E. Fulminant meningococcemia. Heparin therapy and survival rate. Scand J Infect Dis 1971;3:127–33.
67. Giudici D, Baudo F, Palareti G, et al. Antithrombin replacement in patients with sepsis and septic shock. Haematologica 1999;84:452–60.
68. Fourrier F, Jourdain M, Tournoys A. Clinical trial results with antithrombin III in sepsis. Crit Care Med 2000;28(9 Suppl):S38–43.
69. Levi M, De Jonge E, van der PT, ten Cate H. Novel approaches to the management of disseminated intravascular coagulation. Crit Care Med 2000;28(9 Suppl):S20–4.
70. Rivard GE, David M, Farrell C, Schwarz HP. Treatment of purpura fulminans in meningococcemia with protein C concentrate. J Pediatr 1995;126:646–52.
71. White B, Livingstone W, Murphy C, et al. An open-label study of the role of adjuvant hemostatic support with protein C replacement therapy in purpura fulminans-associated meningococcemia. Blood 2000;96:3719–24.
72. Schellongowski P, Bauer E, Holzinger U, et al. Treatment of adult patients with sepsis-induced coagulopathy and purpura fulminans using a plasma-derived protein C concentrate (Ceprotin). Vox Sang 2006;90:294–301.
73. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
74. Cohen MJ, Christie SA. Coagulopathy of trauma. Crit Care Clin 2017;33:101–18.
75. Giordano S, Spiezia L, Campello E, Simioni P. The current understanding of trauma-induced coagulopathy (TIC): a focused review on pathophysiology. Intern Emerg Med 2017 May 5.
76. Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood 2016;128:1043–9.
77. Eddy VA, Morris JA Jr, Cullinane DC. Hypothermia, coagulopathy, and acidosis. Surg Clin North Am 2000;80:845–54.
78. Peng RY, Bongard FS. Hypothermia in trauma patients. J Am Coll Surg 1999;188:685–96.
79. Steinemann S, Shackford SR, Davis JW. Implications of admission hypothermia in trauma patients. J Trauma 1990;30:200–2.
80. Rajek A, Greif R, Sessler DI, et al. Core cooling by central venous infusion of ice-cold (4 degrees C and 20 degrees C) fluid: isolation of core and peripheral thermal compartments. Anesthesiol 2000;93:629–37.
81. Watts DD, Trask A, Soeken K, et al. Hypothermic coagulopathy in trauma: effect of varying levels of hypothermia on enzyme speed, platelet function, and fibrinolytic activity. J Trauma 1998;44:846–54.
82. Ferrara A, MacArthur JD, Wright HK, et al. Hypothermia and acidosis worsen coagulopathy in the patient requiring massive transfusion. Am J Surg 1990;160:515–8.
83. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
84. Johansson PI, Stensballe J, Oliveri R, Wade CE, Ostrowski SR, Holcomb JB. How I treat patients with massive hemorrhage. Blood 2014;124:3052–8.
85. Stone HH, Strom PR, Mullins RJ. Management of the major coagulopathy with onset during laparotomy. Ann Surg 1983;197:532–5.
86. WOMAN Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23–32.
87. Hall DR. Abruptio placentae and disseminated intravascular coagulopathy. Semin Perinatol 2009;33:189–95.
88. Thachil J, Toh CH. Disseminated intravascular coagulation in obstetric disorders and its acute haematological management. Blood Rev 2009;23:167–76.
89. Collins P, Abdul-Kadir R, Thachil J, Subcommittees on Women’ s Health Issues in T, Haemostasis, on Disseminated Intravascular C. Management of coagulopathy associated with postpartum hemorrhage: guidance from the SSC of the ISTH. J Thromb Haemost 2016;14:205–10.
90. Baxter JK, Weinstein L. HELLP syndrome: the state of the art. Obstet Gynecol Surv 2004;59:838–45.
91. Egerman RS, Sibai BM. HELLP syndrome. Clin Obstetr Gynecol 1999;42:381–9.
92. Saphier CJ, Repke JT. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome: a review of diagnosis and management. Sem Perinatol 1998;22:118–33.
93. Le Thi TD, Tieulie N, Costedoat N, et al. The HELLP syndrome in the antiphospholipid syndrome: retrospective study of 16 cases in 15 women. Ann Rheum Dis 2005;64:273–8.
94. Martin JN Jr, Perry KG Jr, Blake PG, et al. Better maternal outcomes are achieved with dexamethasone therapy for postpartum HELLP (hemolysis, elevated liver enzymes, and thrombocytopenia) syndrome. Am J Obstet Gynecol 1997;177:1011–7.
95. Magann EF, Martin JN Jr. Twelve steps to optimal management of HELLP syndrome. Clinical Obstet Gynecol 1999;42:532–50.
96. Jwayyed SM, Blanda M, Kubina M. Acute fatty liver of pregnancy. J Emerg Medi 1999;17:673–7.
97. Bacq Y. Acute fatty liver of pregnancy. Sem Perinatol 1998;22:134–40.
98. Egerman RS, Sibai BM. Imitators of preeclampsia and eclampsia. Clin Obstet Gynecol 1999;42:551–62.
99. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Medi Rev 1993;7:255–67.
100. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis 1992;15:863–5.
101. Garratty G, Nance S, Lloyd M, Domen R. Fatal immune hemolytic anemia due to cefotetan. Transfusion 1992;32:269–71.
102. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes:case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion 1999;39:306–9.
103. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion 1999;39:1239–46.
104. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol 2006;81:186–8.
105. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr 1995;126:813–5.
106. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1995;14:1116–7.
107. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone. J Pediatr 1995;126:816–7.
108. Gottschall JL, Elliot W, Lianos E, et al. Quinine-induced immune thrombocytopenia associated with hemolytic uremic syndrome: a new clinical entity. Blood 1991;77:306–10.
109. Gottschall JL, Neahring B, McFarland JG, et al. Quinine-induced immune thrombocytopenia with hemolytic uremic syndrome: clinical and serological findings in nine patients and review of literature. Am J Hematol 1994;47:283–9.
110. Crum NF, Gable P. Quinine-induced hemolytic-uremic syndrome. South Med J 2000;93:726–8.
111. Vesely T, Vesely JN, George JN. Quinine-Induced thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS): frequency, clinical features, and long-term outcomes. Blood 2000;96:629 [abstract].
112. Bell WR, Starksen NF, Tong S, Porterfield JK. Trousseau’s syndrome. Devastating coagulopathy in the absence of heparin. Am J Med 1985;79:423–30.
113. Sack GH, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinic, pathophysiologic, and therapeutic features. Medicine 1977;56:1–37.
114. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007;110:1723–9.
115. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol 2005;6:401–10.
116. de la Fouchardiere C, Flechon A, Droz JP. Coagulopathy in prostate cancer. Neth J Med 2003;61:347–54.
117. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. 8th ed. Chest 2008;133(6 Suppl):454S–545S.
118. Lee AY, Kamphuisen PW, Meyer G, et al. Tinzaparin vs warfarin for treatment of acute venous thromboembolism in patients with active cancer: a randomized clinical trial. JAMA 2015;314:677–86.
119. Choudhry A, DeLoughery TG. Bleeding and thrombosis in acute promyelocytic leukemia. Am J Hematol 2012;87:596–603.
120. Cao M, Li T, He Z, et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood 2017;129:1855–64.
121. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008;111:2505–15.
122. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013;369:111–21.
123. Dombret H, Scrobohaci ML, Ghorra P, et al. Coagulation disorders associated iwth acute promyelocytic leukemia: Corrective effect of all-trans retinoic acid treatment. Leukemia 1993;7:2–9.
124. Falanga A, Rickles FR. Management of thrombohemorrhagic syndromes (THS) in hematologic malignancies. Hematology Am Soc Hematol Educ Program 2007;2007:165–71
125. Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood 2009;114:5126–35.
126. Sanz MA, Grimwade D, Tallman MS, et al. Guidelines on the management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009;113:1875–91.
127. Lu Q, Clemetson JM, Clemetson KJ. Snake venoms and hemostasis. J Thromb Haemost 2005;3:1791–9.
128. Berling I, Isbister GK. Hematologic effects and complications of snake envenoming. Transfus Med Rev 2015;29:82–9.
129. Isbister GK, Jayamanne S, Mohamed F, et al. A randomized controlled trial of fresh frozen plasma for coagulopathy in Russell’s viper (Daboia russelii) envenoming. J Thromb Haemost 2017;15:645–54.
130. Rodriguez V, Lee A, Witman PM, Anderson PA. Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 2009;31:522–6.
131. Triana P, Dore M, Cerezo VN, et al. Sirolimus in the treatment of vascular anomalies. Eur J Pediatr Surg 2017;27:86–90.