User login
Type 3 von Willebrand a rare but serious bleeding disorder
Type 3 von Willebrand disease (VWD) is rare, but this form of the disease is associated with severe bleeding, particularly in muscles and joints, a bleeding disorders expert said.
“There’s a virtually complete deficiency in von Willebrand factor [in type 3 disease], so usually it’s defined as below 5 or in some studies below 3 IU/dL, but also due to the very low levels of von Willebrand factor, there’s also a very low level of factor VIII,” said Jeroen Eikenboom, MD, PhD, from Leiden (the Netherlands) University Medical Center.
“The inheritance pattern is autosomal recessive, and the prevalence is about 1 in a million,” he said during the annual congress of the European Association for Haemophilia and Allied Disorders.
Erik Adolf von Willenbrand, MD, PhD, first described this form of VWD in a family from the Åland Islands, an autonomous region of Finland. The disease, later discovered to be caused in this family by a cytosine deletion in exon 18 of the von Willebrand factor (VWF) gene, was associated with fatal bleeding events in several family members.
“As VWF is the carrier protein of factor VIII, the very low VWF level leads to a strongly reduced factor VIII level, comparable to the levels seen in mild to moderate hemophilia A. As a consequence, VWD type 3 has the combined characteristics of a primary as well as a secondary hemostasis defect,” Dr. Eikenboom explained in an abstract accompanying his talk.
Compared with VWD type 1 or 2, type 3 VWD is associated with bleeding episodes more commonly seen in patients with hemophilia A, notably mucocutaneous bleeding, bleeding after trauma or during surgery, and bleeding into joints and/or muscles.
Treatment
The goals of treatment for patients with type 3 VWD are to correct the dual hemostasis defects of impaired platelet adhesion because of low VWF levels, and the intrinsic coagulation defect because of levels of factor VIII.
Desmopressin is not effective in type 3 VWD, Dr. Eikenboom said, so treatment requires the use of either plasma-derived VWF, with or without factor VIII, or recombinant VWF.
In the United States, the only standalone VWF concentrate approved by the Food and Drug Administration is a recombinant product (Vonvendi), Three other human plasma–derived concentrates containing both VWF and factor VIII are also licensed (Alpanate, Humate-P, Wilate).
Clinicians prescribing the combined factor concentrates need to be aware of differences in pharmacokinetics between the products.
For example, following infusion of Wilate, which has equal amounts of von Willebrand factor and factor VIII, there is an increase in circulation of both von Willebrand factor and factor VIII and a similar decline in each factor over time.
In contrast, following an infusion of Humate-P, which contains lower levels of factor VIII, “interestingly, you see a secondary rise of factor VIII in Humate-P–infused patients, whereas the secondary rise is not visible in the Wilate patients,” he said.
Approximately 22% of patients with type 3 VWD also receive prophylaxis with VWF concentrate, which has been shown to decrease the median annualized bleeding rate from 25% to 6.1%.
Dr. Eikenboom cautioned that 5%-10% of patients with type 3 VWD may develop allo-antibodies against VWF concentrates, which can complicate treatment and carries risk of anaphylactic shock.
“It’s also been mentioned in literature that there may be an association with partial or complete von Willebrand factor gene deletions or nonsense mutations and the development of allo-antibodies,” he said.
Prophylaxis burdensome but helpful
Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee, who specializes in the treatment of patients with von Willebrand disease, follows a number of both girls and boys with type 3 VWD.
“Those are the people who will have bleeding into their joints, and for the girls, worse periods than some of those with other types of von Willebrand disease, and it is true that if you want to stop their bleeding, you cannot use desmopressin like we use in most other von Willebrand patients. They will need factor, although for the heavy menstrual bleeding you can use hormones or tranexamic acid – there are some other options for that,” she said in an interview.
She also noted that type 3 von Willebrand disease can be highly variable. For patients with especially frequent joint bleeding, her center recommends prophylaxis.
“Prophylaxis can be very burdensome for patients. You’re talking about IV therapy several times a week, but it’s very helpful for the joint bleeds. Episodic prophylaxis can be very helpful for heavy menstrual bleeding, and we actually have type 2, type 3, and some type 1 patients with bad enough nose bleeds that they end up on prophylaxis,” she said.
Patients with gastrointestinal bleeding are the most challenging to care for, she noted.
“You can put them on factor prophylaxis, but even that isn’t always enough to help some adults with bad GI bleeding, and we’re investigating other options for that,” she said.
Dr. Eikenboom disclosed research support from CSL Behring and honoraria (directed to his institution) for educational activities sponsored by Roche and Celgene. Dr. Flood reported having no conflicts of interest to disclose.
Type 3 von Willebrand disease (VWD) is rare, but this form of the disease is associated with severe bleeding, particularly in muscles and joints, a bleeding disorders expert said.
“There’s a virtually complete deficiency in von Willebrand factor [in type 3 disease], so usually it’s defined as below 5 or in some studies below 3 IU/dL, but also due to the very low levels of von Willebrand factor, there’s also a very low level of factor VIII,” said Jeroen Eikenboom, MD, PhD, from Leiden (the Netherlands) University Medical Center.
“The inheritance pattern is autosomal recessive, and the prevalence is about 1 in a million,” he said during the annual congress of the European Association for Haemophilia and Allied Disorders.
Erik Adolf von Willenbrand, MD, PhD, first described this form of VWD in a family from the Åland Islands, an autonomous region of Finland. The disease, later discovered to be caused in this family by a cytosine deletion in exon 18 of the von Willebrand factor (VWF) gene, was associated with fatal bleeding events in several family members.
“As VWF is the carrier protein of factor VIII, the very low VWF level leads to a strongly reduced factor VIII level, comparable to the levels seen in mild to moderate hemophilia A. As a consequence, VWD type 3 has the combined characteristics of a primary as well as a secondary hemostasis defect,” Dr. Eikenboom explained in an abstract accompanying his talk.
Compared with VWD type 1 or 2, type 3 VWD is associated with bleeding episodes more commonly seen in patients with hemophilia A, notably mucocutaneous bleeding, bleeding after trauma or during surgery, and bleeding into joints and/or muscles.
Treatment
The goals of treatment for patients with type 3 VWD are to correct the dual hemostasis defects of impaired platelet adhesion because of low VWF levels, and the intrinsic coagulation defect because of levels of factor VIII.
Desmopressin is not effective in type 3 VWD, Dr. Eikenboom said, so treatment requires the use of either plasma-derived VWF, with or without factor VIII, or recombinant VWF.
In the United States, the only standalone VWF concentrate approved by the Food and Drug Administration is a recombinant product (Vonvendi), Three other human plasma–derived concentrates containing both VWF and factor VIII are also licensed (Alpanate, Humate-P, Wilate).
Clinicians prescribing the combined factor concentrates need to be aware of differences in pharmacokinetics between the products.
For example, following infusion of Wilate, which has equal amounts of von Willebrand factor and factor VIII, there is an increase in circulation of both von Willebrand factor and factor VIII and a similar decline in each factor over time.
In contrast, following an infusion of Humate-P, which contains lower levels of factor VIII, “interestingly, you see a secondary rise of factor VIII in Humate-P–infused patients, whereas the secondary rise is not visible in the Wilate patients,” he said.
Approximately 22% of patients with type 3 VWD also receive prophylaxis with VWF concentrate, which has been shown to decrease the median annualized bleeding rate from 25% to 6.1%.
Dr. Eikenboom cautioned that 5%-10% of patients with type 3 VWD may develop allo-antibodies against VWF concentrates, which can complicate treatment and carries risk of anaphylactic shock.
“It’s also been mentioned in literature that there may be an association with partial or complete von Willebrand factor gene deletions or nonsense mutations and the development of allo-antibodies,” he said.
Prophylaxis burdensome but helpful
Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee, who specializes in the treatment of patients with von Willebrand disease, follows a number of both girls and boys with type 3 VWD.
“Those are the people who will have bleeding into their joints, and for the girls, worse periods than some of those with other types of von Willebrand disease, and it is true that if you want to stop their bleeding, you cannot use desmopressin like we use in most other von Willebrand patients. They will need factor, although for the heavy menstrual bleeding you can use hormones or tranexamic acid – there are some other options for that,” she said in an interview.
She also noted that type 3 von Willebrand disease can be highly variable. For patients with especially frequent joint bleeding, her center recommends prophylaxis.
“Prophylaxis can be very burdensome for patients. You’re talking about IV therapy several times a week, but it’s very helpful for the joint bleeds. Episodic prophylaxis can be very helpful for heavy menstrual bleeding, and we actually have type 2, type 3, and some type 1 patients with bad enough nose bleeds that they end up on prophylaxis,” she said.
Patients with gastrointestinal bleeding are the most challenging to care for, she noted.
“You can put them on factor prophylaxis, but even that isn’t always enough to help some adults with bad GI bleeding, and we’re investigating other options for that,” she said.
Dr. Eikenboom disclosed research support from CSL Behring and honoraria (directed to his institution) for educational activities sponsored by Roche and Celgene. Dr. Flood reported having no conflicts of interest to disclose.
Type 3 von Willebrand disease (VWD) is rare, but this form of the disease is associated with severe bleeding, particularly in muscles and joints, a bleeding disorders expert said.
“There’s a virtually complete deficiency in von Willebrand factor [in type 3 disease], so usually it’s defined as below 5 or in some studies below 3 IU/dL, but also due to the very low levels of von Willebrand factor, there’s also a very low level of factor VIII,” said Jeroen Eikenboom, MD, PhD, from Leiden (the Netherlands) University Medical Center.
“The inheritance pattern is autosomal recessive, and the prevalence is about 1 in a million,” he said during the annual congress of the European Association for Haemophilia and Allied Disorders.
Erik Adolf von Willenbrand, MD, PhD, first described this form of VWD in a family from the Åland Islands, an autonomous region of Finland. The disease, later discovered to be caused in this family by a cytosine deletion in exon 18 of the von Willebrand factor (VWF) gene, was associated with fatal bleeding events in several family members.
“As VWF is the carrier protein of factor VIII, the very low VWF level leads to a strongly reduced factor VIII level, comparable to the levels seen in mild to moderate hemophilia A. As a consequence, VWD type 3 has the combined characteristics of a primary as well as a secondary hemostasis defect,” Dr. Eikenboom explained in an abstract accompanying his talk.
Compared with VWD type 1 or 2, type 3 VWD is associated with bleeding episodes more commonly seen in patients with hemophilia A, notably mucocutaneous bleeding, bleeding after trauma or during surgery, and bleeding into joints and/or muscles.
Treatment
The goals of treatment for patients with type 3 VWD are to correct the dual hemostasis defects of impaired platelet adhesion because of low VWF levels, and the intrinsic coagulation defect because of levels of factor VIII.
Desmopressin is not effective in type 3 VWD, Dr. Eikenboom said, so treatment requires the use of either plasma-derived VWF, with or without factor VIII, or recombinant VWF.
In the United States, the only standalone VWF concentrate approved by the Food and Drug Administration is a recombinant product (Vonvendi), Three other human plasma–derived concentrates containing both VWF and factor VIII are also licensed (Alpanate, Humate-P, Wilate).
Clinicians prescribing the combined factor concentrates need to be aware of differences in pharmacokinetics between the products.
For example, following infusion of Wilate, which has equal amounts of von Willebrand factor and factor VIII, there is an increase in circulation of both von Willebrand factor and factor VIII and a similar decline in each factor over time.
In contrast, following an infusion of Humate-P, which contains lower levels of factor VIII, “interestingly, you see a secondary rise of factor VIII in Humate-P–infused patients, whereas the secondary rise is not visible in the Wilate patients,” he said.
Approximately 22% of patients with type 3 VWD also receive prophylaxis with VWF concentrate, which has been shown to decrease the median annualized bleeding rate from 25% to 6.1%.
Dr. Eikenboom cautioned that 5%-10% of patients with type 3 VWD may develop allo-antibodies against VWF concentrates, which can complicate treatment and carries risk of anaphylactic shock.
“It’s also been mentioned in literature that there may be an association with partial or complete von Willebrand factor gene deletions or nonsense mutations and the development of allo-antibodies,” he said.
Prophylaxis burdensome but helpful
Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee, who specializes in the treatment of patients with von Willebrand disease, follows a number of both girls and boys with type 3 VWD.
“Those are the people who will have bleeding into their joints, and for the girls, worse periods than some of those with other types of von Willebrand disease, and it is true that if you want to stop their bleeding, you cannot use desmopressin like we use in most other von Willebrand patients. They will need factor, although for the heavy menstrual bleeding you can use hormones or tranexamic acid – there are some other options for that,” she said in an interview.
She also noted that type 3 von Willebrand disease can be highly variable. For patients with especially frequent joint bleeding, her center recommends prophylaxis.
“Prophylaxis can be very burdensome for patients. You’re talking about IV therapy several times a week, but it’s very helpful for the joint bleeds. Episodic prophylaxis can be very helpful for heavy menstrual bleeding, and we actually have type 2, type 3, and some type 1 patients with bad enough nose bleeds that they end up on prophylaxis,” she said.
Patients with gastrointestinal bleeding are the most challenging to care for, she noted.
“You can put them on factor prophylaxis, but even that isn’t always enough to help some adults with bad GI bleeding, and we’re investigating other options for that,” she said.
Dr. Eikenboom disclosed research support from CSL Behring and honoraria (directed to his institution) for educational activities sponsored by Roche and Celgene. Dr. Flood reported having no conflicts of interest to disclose.
FROM EAHAD 2021
Bleeding disorder diagnoses delayed by years in girls and women
Diagnosis of bleeding disorders in girls and women can lag behind diagnosis in boys and men by more than a decade, meaning needless delays in treatment and poor quality of life for many with hemophilia or related conditions.
“There is increasing awareness about issues faced by women and girls with inherited bleeding disorders, but disparities still exist both in both access to diagnosis and treatment,” said Roseline D’Oiron, MD, from Hôpital Bicêtre in Paris.
“Diagnosis, when it is made, is often made late, particularly in women. Indeed, a recent study from the European Hemophilia Consortium including more than 700 women with bleeding disorders showed that the median age at diagnosis was 16 years old,” she said during the annual congress of the European Association for Haemophilia and Allied Disorders.
She said that delayed diagnosis of bleeding disorders in women and girls may be caused by a lack of knowledge by patients, families, and general practitioners about family history of bleeding disorders, abnormal bleeding events, and heavy menstrual bleeding. In addition, despite the frequency and severity of heavy bleeding events, patients, their families, and caregivers may underestimate the effect on the patient’s quality of life.
Disparities documented
Dr. D’Oiron pointed to several studies showing clear sex-based disparities in time to diagnosis. For example, a study published in Haemophilia showed that in 22 girls with hemophilia A or hemophilia B, the diagnosis of severe hemophilia was delayed by a median of 6.5 months compared with the diagnosis in boys, and a diagnosis of moderate hemophilia in girls was delayed by a median of 39 months.
In a second, single-center study comparing 44 women and girls with mild hemophilia (factor VIII or factor XI levels from 5 to 50 IU/dL) with 77 men and boys with mild hemophilia, the mean age at diagnosis was 31.63 years versus 19.18 years, respectively – a delay of 12.45 years.
A third study comparing 442 girls/women and 442 boys/men with mild hemophilia in France showed a difference of 6.07 years in diagnosis: the median age for girls/women at diagnosis was 16.91 years versus10.84 years for boys/men.
Why it matters
Dr. D’Oiron described the case of a patient named Clare, who first experienced, at age 8, 12 hours of bleeding following a dental procedure. At age 12.5, she began having heavy menstrual bleeding, causing her to miss school for a few days each month, to be feel tired, and have poor-quality sleep.
Despite repeated bleeding episodes, severe anemia, and iron deficiency, her hemophilia was not suspected until after her 16th birthday, and a definitive diagnosis of hemophilia in both Clare and her mother was finally made when Clare was past 17, when a nonsense variant factor in F8, the gene encoding for factor VIII, was detected.
“For Clare, it took more the 8 years after the first bleeding symptoms, and nearly 4 years after presenting with heavy menstrual bleeding to recognize that she had a bleeding problem,” she said.
In total, Clare had about 450 days of heavy menstrual bleeding, causing her to miss an estimated 140 days of school because of the delayed diagnosis and treatment.
“In my view, this is the main argument why it is urgent for these patients to achieve diagnosis early: this is to reduce the duration [of] a very poor quality of life,” Dr. D’Oiron said.
Barriers to diagnosis
Patients and families have reported difficulty distinguishing normal bleeding from abnormal symptoms, and girls may be reluctant to discuss their symptoms with their family or peers. In addition, primary care practitioners may not recognize the severity of the symptoms and therefore may not refer patients to hematologists for further workup.
These findings emphasize the need for improved tools to help patients differentiate between normal and abnormal bleeding, using symptom recognition–based language tools that can lead to early testing and application of accurate diagnostic tools, she said.
Standardization of definitions can help to improve screening and diagnosis, Dr. D’Oiron said, pointing to a recent study in Blood Advances proposing definitions for future research in von Willebrand disease.
For example, the authors of that study proposed a definition of heavy menstrual bleeding to include any of the following:
- Bleeding lasting 8 or more days
- Bleeding that consistently soaks through one or more sanitary protections every 2 hours on multiple days
- Requires use of more than one sanitary protection item at a time
- Requires changing sanitary protection during the night
- Is associated with repeat passing of blood clots
- Has a Pictorial Blood Assessment Chart score greater than 100.
Problem and solutions
Answering the question posed in the title of her talk, Dr. D’Oiron said: “Yes, we do have a problem with the diagnosis of bleeding disorders in women and girls, but we also have solutions.”
The solutions include family and patient outreach efforts; communication to improve awareness; inclusion of general practitioners in the circle of care; and early screening, diagnosis, and treatment.
A bleeding disorders specialist who was not involved in the study said that Dr. D’Oiron’s report closely reflects what she sees in the clinic.
“I do pediatrics, and usually what happens is that I see a teenager with heavy menstrual bleeding and we take her history, and we find out that Mom and multiple female family members have had horrible menstrual bleeding, possibly many of whom have had hysterectomies for it, and then diagnosing the parents and other family members after diagnosing the girl that we’re seeing” said Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee.
“It is unfortunately a very real thing,” she added.
Reasons for the delay likely include lack of awareness of bleeding disorders.
“If you present to a hematologist, we think about bleeding disorders, but if you present to a primary care physician, they don’t always have that on their radar,” she said.
Additionally, a girl from a family with a history of heavy menstrual bleeding may just assume that what she is experiencing is “normal,” despite the serious affect it has on her quality of life, Dr. Flood said.
Dr. D’Oiron’s research is supported by her institution, the French Hemophilia Association, FranceCoag and Mhemon, the European Hemophilia Consortium, and the World Federation of Hemophilia. She reported advisory board or invited speaker activities for multiple companies. Dr. Flood reported having no conflicts of interest to disclose.
Diagnosis of bleeding disorders in girls and women can lag behind diagnosis in boys and men by more than a decade, meaning needless delays in treatment and poor quality of life for many with hemophilia or related conditions.
“There is increasing awareness about issues faced by women and girls with inherited bleeding disorders, but disparities still exist both in both access to diagnosis and treatment,” said Roseline D’Oiron, MD, from Hôpital Bicêtre in Paris.
“Diagnosis, when it is made, is often made late, particularly in women. Indeed, a recent study from the European Hemophilia Consortium including more than 700 women with bleeding disorders showed that the median age at diagnosis was 16 years old,” she said during the annual congress of the European Association for Haemophilia and Allied Disorders.
She said that delayed diagnosis of bleeding disorders in women and girls may be caused by a lack of knowledge by patients, families, and general practitioners about family history of bleeding disorders, abnormal bleeding events, and heavy menstrual bleeding. In addition, despite the frequency and severity of heavy bleeding events, patients, their families, and caregivers may underestimate the effect on the patient’s quality of life.
Disparities documented
Dr. D’Oiron pointed to several studies showing clear sex-based disparities in time to diagnosis. For example, a study published in Haemophilia showed that in 22 girls with hemophilia A or hemophilia B, the diagnosis of severe hemophilia was delayed by a median of 6.5 months compared with the diagnosis in boys, and a diagnosis of moderate hemophilia in girls was delayed by a median of 39 months.
In a second, single-center study comparing 44 women and girls with mild hemophilia (factor VIII or factor XI levels from 5 to 50 IU/dL) with 77 men and boys with mild hemophilia, the mean age at diagnosis was 31.63 years versus 19.18 years, respectively – a delay of 12.45 years.
A third study comparing 442 girls/women and 442 boys/men with mild hemophilia in France showed a difference of 6.07 years in diagnosis: the median age for girls/women at diagnosis was 16.91 years versus10.84 years for boys/men.
Why it matters
Dr. D’Oiron described the case of a patient named Clare, who first experienced, at age 8, 12 hours of bleeding following a dental procedure. At age 12.5, she began having heavy menstrual bleeding, causing her to miss school for a few days each month, to be feel tired, and have poor-quality sleep.
Despite repeated bleeding episodes, severe anemia, and iron deficiency, her hemophilia was not suspected until after her 16th birthday, and a definitive diagnosis of hemophilia in both Clare and her mother was finally made when Clare was past 17, when a nonsense variant factor in F8, the gene encoding for factor VIII, was detected.
“For Clare, it took more the 8 years after the first bleeding symptoms, and nearly 4 years after presenting with heavy menstrual bleeding to recognize that she had a bleeding problem,” she said.
In total, Clare had about 450 days of heavy menstrual bleeding, causing her to miss an estimated 140 days of school because of the delayed diagnosis and treatment.
“In my view, this is the main argument why it is urgent for these patients to achieve diagnosis early: this is to reduce the duration [of] a very poor quality of life,” Dr. D’Oiron said.
Barriers to diagnosis
Patients and families have reported difficulty distinguishing normal bleeding from abnormal symptoms, and girls may be reluctant to discuss their symptoms with their family or peers. In addition, primary care practitioners may not recognize the severity of the symptoms and therefore may not refer patients to hematologists for further workup.
These findings emphasize the need for improved tools to help patients differentiate between normal and abnormal bleeding, using symptom recognition–based language tools that can lead to early testing and application of accurate diagnostic tools, she said.
Standardization of definitions can help to improve screening and diagnosis, Dr. D’Oiron said, pointing to a recent study in Blood Advances proposing definitions for future research in von Willebrand disease.
For example, the authors of that study proposed a definition of heavy menstrual bleeding to include any of the following:
- Bleeding lasting 8 or more days
- Bleeding that consistently soaks through one or more sanitary protections every 2 hours on multiple days
- Requires use of more than one sanitary protection item at a time
- Requires changing sanitary protection during the night
- Is associated with repeat passing of blood clots
- Has a Pictorial Blood Assessment Chart score greater than 100.
Problem and solutions
Answering the question posed in the title of her talk, Dr. D’Oiron said: “Yes, we do have a problem with the diagnosis of bleeding disorders in women and girls, but we also have solutions.”
The solutions include family and patient outreach efforts; communication to improve awareness; inclusion of general practitioners in the circle of care; and early screening, diagnosis, and treatment.
A bleeding disorders specialist who was not involved in the study said that Dr. D’Oiron’s report closely reflects what she sees in the clinic.
“I do pediatrics, and usually what happens is that I see a teenager with heavy menstrual bleeding and we take her history, and we find out that Mom and multiple female family members have had horrible menstrual bleeding, possibly many of whom have had hysterectomies for it, and then diagnosing the parents and other family members after diagnosing the girl that we’re seeing” said Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee.
“It is unfortunately a very real thing,” she added.
Reasons for the delay likely include lack of awareness of bleeding disorders.
“If you present to a hematologist, we think about bleeding disorders, but if you present to a primary care physician, they don’t always have that on their radar,” she said.
Additionally, a girl from a family with a history of heavy menstrual bleeding may just assume that what she is experiencing is “normal,” despite the serious affect it has on her quality of life, Dr. Flood said.
Dr. D’Oiron’s research is supported by her institution, the French Hemophilia Association, FranceCoag and Mhemon, the European Hemophilia Consortium, and the World Federation of Hemophilia. She reported advisory board or invited speaker activities for multiple companies. Dr. Flood reported having no conflicts of interest to disclose.
Diagnosis of bleeding disorders in girls and women can lag behind diagnosis in boys and men by more than a decade, meaning needless delays in treatment and poor quality of life for many with hemophilia or related conditions.
“There is increasing awareness about issues faced by women and girls with inherited bleeding disorders, but disparities still exist both in both access to diagnosis and treatment,” said Roseline D’Oiron, MD, from Hôpital Bicêtre in Paris.
“Diagnosis, when it is made, is often made late, particularly in women. Indeed, a recent study from the European Hemophilia Consortium including more than 700 women with bleeding disorders showed that the median age at diagnosis was 16 years old,” she said during the annual congress of the European Association for Haemophilia and Allied Disorders.
She said that delayed diagnosis of bleeding disorders in women and girls may be caused by a lack of knowledge by patients, families, and general practitioners about family history of bleeding disorders, abnormal bleeding events, and heavy menstrual bleeding. In addition, despite the frequency and severity of heavy bleeding events, patients, their families, and caregivers may underestimate the effect on the patient’s quality of life.
Disparities documented
Dr. D’Oiron pointed to several studies showing clear sex-based disparities in time to diagnosis. For example, a study published in Haemophilia showed that in 22 girls with hemophilia A or hemophilia B, the diagnosis of severe hemophilia was delayed by a median of 6.5 months compared with the diagnosis in boys, and a diagnosis of moderate hemophilia in girls was delayed by a median of 39 months.
In a second, single-center study comparing 44 women and girls with mild hemophilia (factor VIII or factor XI levels from 5 to 50 IU/dL) with 77 men and boys with mild hemophilia, the mean age at diagnosis was 31.63 years versus 19.18 years, respectively – a delay of 12.45 years.
A third study comparing 442 girls/women and 442 boys/men with mild hemophilia in France showed a difference of 6.07 years in diagnosis: the median age for girls/women at diagnosis was 16.91 years versus10.84 years for boys/men.
Why it matters
Dr. D’Oiron described the case of a patient named Clare, who first experienced, at age 8, 12 hours of bleeding following a dental procedure. At age 12.5, she began having heavy menstrual bleeding, causing her to miss school for a few days each month, to be feel tired, and have poor-quality sleep.
Despite repeated bleeding episodes, severe anemia, and iron deficiency, her hemophilia was not suspected until after her 16th birthday, and a definitive diagnosis of hemophilia in both Clare and her mother was finally made when Clare was past 17, when a nonsense variant factor in F8, the gene encoding for factor VIII, was detected.
“For Clare, it took more the 8 years after the first bleeding symptoms, and nearly 4 years after presenting with heavy menstrual bleeding to recognize that she had a bleeding problem,” she said.
In total, Clare had about 450 days of heavy menstrual bleeding, causing her to miss an estimated 140 days of school because of the delayed diagnosis and treatment.
“In my view, this is the main argument why it is urgent for these patients to achieve diagnosis early: this is to reduce the duration [of] a very poor quality of life,” Dr. D’Oiron said.
Barriers to diagnosis
Patients and families have reported difficulty distinguishing normal bleeding from abnormal symptoms, and girls may be reluctant to discuss their symptoms with their family or peers. In addition, primary care practitioners may not recognize the severity of the symptoms and therefore may not refer patients to hematologists for further workup.
These findings emphasize the need for improved tools to help patients differentiate between normal and abnormal bleeding, using symptom recognition–based language tools that can lead to early testing and application of accurate diagnostic tools, she said.
Standardization of definitions can help to improve screening and diagnosis, Dr. D’Oiron said, pointing to a recent study in Blood Advances proposing definitions for future research in von Willebrand disease.
For example, the authors of that study proposed a definition of heavy menstrual bleeding to include any of the following:
- Bleeding lasting 8 or more days
- Bleeding that consistently soaks through one or more sanitary protections every 2 hours on multiple days
- Requires use of more than one sanitary protection item at a time
- Requires changing sanitary protection during the night
- Is associated with repeat passing of blood clots
- Has a Pictorial Blood Assessment Chart score greater than 100.
Problem and solutions
Answering the question posed in the title of her talk, Dr. D’Oiron said: “Yes, we do have a problem with the diagnosis of bleeding disorders in women and girls, but we also have solutions.”
The solutions include family and patient outreach efforts; communication to improve awareness; inclusion of general practitioners in the circle of care; and early screening, diagnosis, and treatment.
A bleeding disorders specialist who was not involved in the study said that Dr. D’Oiron’s report closely reflects what she sees in the clinic.
“I do pediatrics, and usually what happens is that I see a teenager with heavy menstrual bleeding and we take her history, and we find out that Mom and multiple female family members have had horrible menstrual bleeding, possibly many of whom have had hysterectomies for it, and then diagnosing the parents and other family members after diagnosing the girl that we’re seeing” said Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee.
“It is unfortunately a very real thing,” she added.
Reasons for the delay likely include lack of awareness of bleeding disorders.
“If you present to a hematologist, we think about bleeding disorders, but if you present to a primary care physician, they don’t always have that on their radar,” she said.
Additionally, a girl from a family with a history of heavy menstrual bleeding may just assume that what she is experiencing is “normal,” despite the serious affect it has on her quality of life, Dr. Flood said.
Dr. D’Oiron’s research is supported by her institution, the French Hemophilia Association, FranceCoag and Mhemon, the European Hemophilia Consortium, and the World Federation of Hemophilia. She reported advisory board or invited speaker activities for multiple companies. Dr. Flood reported having no conflicts of interest to disclose.
FROM EAHAD 2021
Maribavir seen as superior to other antivirals for CMV clearance post transplant
Maribavir, an investigational antiviral agent with a novel mechanism of action, was superior to other antiviral strategies at clearing cytomegalovirus (CMV) viremia and controlling symptoms in hematopoietic cell or solid-organ transplant recipients, results of a phase 3 clinical trial showed.
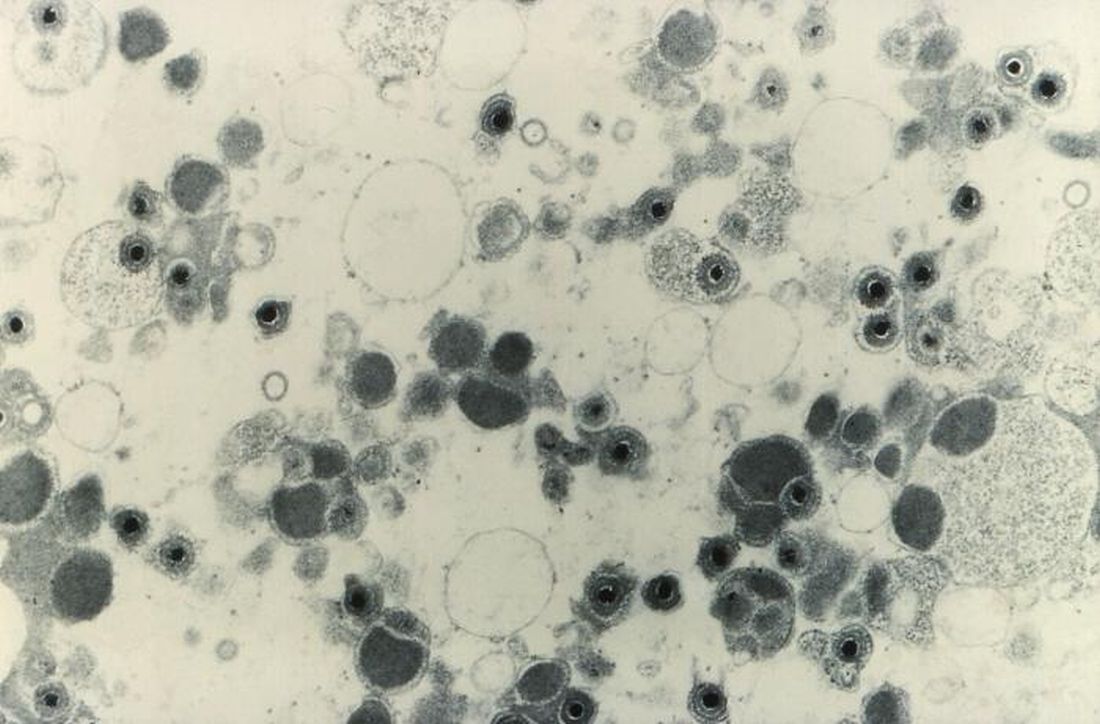
CMV viremia clearance at study week 8 was seen in 55.7% of all patients randomized to receive maribavir, compared with 23.9% for patients assigned to receive investigator-assigned therapy (IAT), Francisco Marty, MD, from the Dana-Farber Cancer Institute in Boston reported at the Transplant & Cellular Therapies Meetings.
“Maribavir’s benefit was driven by lower incidence of treatment-limiting toxicities, compared with IAT,” he said a late-breaking abstract session during the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
“Available anti-CMV antivirals are limited by development of resistance and toxicities, particularly myelosuppression with the use of valganciclovir and nephrotoxicity with the use of foscarnet and cidofovir. Alternative treatment options are required to address this unmet medical need,” he said.
Maribavir inhibits the CMV UL97 protein kinase and is thought to affect several critical processes in CMV replication, including viral DNA synthesis, viral gene expression, encapsidation, and egress of mature capsids from the nucleus.
Details of trial
In the phase 3 SHP620-30e trial (NCT02931539), Dr. Marty and colleagues enrolled patients with relapsed or refractory CMV infections after hematopoietic cell transplant (HCT) or solid-organ transplant (SOT) and after stratification by transplant type and screening CMV DNA level randomly assigned them on a 2:1 basis to receive either maribavir 400 mg twice daily (235 patients) or IAT (117 patients), consisting of either ganciclovir/valganciclovir, foscarnet, cidofovir, or combined foscarnet and val/ganciclovir.
The primary endpoint of viremia clearance at 8 weeks was defined as plasma CMV DNA less than 137 IU/mL in two consecutive tests at a central laboratory at least 5 days apart beginning at the end of week 8.
The trial met its primary endpoint, with a viremia clearance rate of 55.7% with maribavir versus 23.9% with IAT.
The viremia clearance rates were similar in each of the transplant groups: 55.9% versus 20.8%, respectively, in patients who underwent HCT, and 55.6% versus 26.1% in patients who underwent SOT (P < .001).
Clearance rates among patients with CMV DNA below 9,100 IU/mL at baseline were 62.1% with maribavir versus 24.7% with IAT. Among patients with baseline CMV DNA of 9100 IU/mL or above, the respective rates were 43.9% versus 21.9%.
CMV viremia clearance continued from week 8 to week 16 in 18.7% of patients assigned to maribavir and to 10.3% of patients randomized to IAT (P < .013).
The median time to first CMV viremia clearance as 22 days with maribavir versus 27 days with IAT (P = .039).
All-cause mortality was similar between the groups, at 11.5% versus 11.1%, respectively.
The incidences of serious and severe treatment-emergent adverse events (TEAE) were 38.5% and 32.1%, respectively, in the maribavir group, and 37.1% and 37.9% in the IAT group.
Any TEAE leading to study drug discontinuation was less common with maribavir, occurring in 13.2% of patients, compared with 31.9% of patients on IAT. Serious TEAEs leading to drug discontinuation occurred in 8.5% versus 14.7%, respectively.
Serious TEAEs leading to death occurred in 6.8% of patients on maribavir versus 5.2% of those on IAT.
Role of letermovir
In the question-and-answer session following the presentation, comoderator Monalisa Ghosh, MD, from the University of Michigan, Ann Arbor, asked whether any patients in the study were currently on letermovir (Prevymis) prophylaxis, and whether any patients had previously been treated with letermovir but had CMV reactivation and were then treated on study.
Dr. Marty noted that the trial was designed before letermovir was approved for CMV prophylaxis in adults who have undergone an allogeneic HCT.
“Nobody was on letermovir at the beginning of the trial,” he replied, but noted that some patients who were enrolled and had infections that were refractory or resistant to valganciclovir, foscarnet, or a combination of the two received letermovir as secondary prophylaxis.
“I haven’t got the data to tell you how often [letermovir] was used; I think part of the lack of mortality benefit [with maribavir] may be due to the fact that people jumped into secondary prophylaxis with letermovir to minimize the toxicities that we saw,” he said.
Although maribavir has not as of this writing received Food and Drug Administration approval, the drug may be available to some patients through a compassionate-use program from Takeda, Dr. Marty noted.
The study was funded by Shire ViroPharma. Dr. Marty disclosed research funding from Shire and from others. Dr. Ghosh had no relevant disclosures.
Maribavir, an investigational antiviral agent with a novel mechanism of action, was superior to other antiviral strategies at clearing cytomegalovirus (CMV) viremia and controlling symptoms in hematopoietic cell or solid-organ transplant recipients, results of a phase 3 clinical trial showed.
CMV viremia clearance at study week 8 was seen in 55.7% of all patients randomized to receive maribavir, compared with 23.9% for patients assigned to receive investigator-assigned therapy (IAT), Francisco Marty, MD, from the Dana-Farber Cancer Institute in Boston reported at the Transplant & Cellular Therapies Meetings.
“Maribavir’s benefit was driven by lower incidence of treatment-limiting toxicities, compared with IAT,” he said a late-breaking abstract session during the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
“Available anti-CMV antivirals are limited by development of resistance and toxicities, particularly myelosuppression with the use of valganciclovir and nephrotoxicity with the use of foscarnet and cidofovir. Alternative treatment options are required to address this unmet medical need,” he said.
Maribavir inhibits the CMV UL97 protein kinase and is thought to affect several critical processes in CMV replication, including viral DNA synthesis, viral gene expression, encapsidation, and egress of mature capsids from the nucleus.
Details of trial
In the phase 3 SHP620-30e trial (NCT02931539), Dr. Marty and colleagues enrolled patients with relapsed or refractory CMV infections after hematopoietic cell transplant (HCT) or solid-organ transplant (SOT) and after stratification by transplant type and screening CMV DNA level randomly assigned them on a 2:1 basis to receive either maribavir 400 mg twice daily (235 patients) or IAT (117 patients), consisting of either ganciclovir/valganciclovir, foscarnet, cidofovir, or combined foscarnet and val/ganciclovir.
The primary endpoint of viremia clearance at 8 weeks was defined as plasma CMV DNA less than 137 IU/mL in two consecutive tests at a central laboratory at least 5 days apart beginning at the end of week 8.
The trial met its primary endpoint, with a viremia clearance rate of 55.7% with maribavir versus 23.9% with IAT.
The viremia clearance rates were similar in each of the transplant groups: 55.9% versus 20.8%, respectively, in patients who underwent HCT, and 55.6% versus 26.1% in patients who underwent SOT (P < .001).
Clearance rates among patients with CMV DNA below 9,100 IU/mL at baseline were 62.1% with maribavir versus 24.7% with IAT. Among patients with baseline CMV DNA of 9100 IU/mL or above, the respective rates were 43.9% versus 21.9%.
CMV viremia clearance continued from week 8 to week 16 in 18.7% of patients assigned to maribavir and to 10.3% of patients randomized to IAT (P < .013).
The median time to first CMV viremia clearance as 22 days with maribavir versus 27 days with IAT (P = .039).
All-cause mortality was similar between the groups, at 11.5% versus 11.1%, respectively.
The incidences of serious and severe treatment-emergent adverse events (TEAE) were 38.5% and 32.1%, respectively, in the maribavir group, and 37.1% and 37.9% in the IAT group.
Any TEAE leading to study drug discontinuation was less common with maribavir, occurring in 13.2% of patients, compared with 31.9% of patients on IAT. Serious TEAEs leading to drug discontinuation occurred in 8.5% versus 14.7%, respectively.
Serious TEAEs leading to death occurred in 6.8% of patients on maribavir versus 5.2% of those on IAT.
Role of letermovir
In the question-and-answer session following the presentation, comoderator Monalisa Ghosh, MD, from the University of Michigan, Ann Arbor, asked whether any patients in the study were currently on letermovir (Prevymis) prophylaxis, and whether any patients had previously been treated with letermovir but had CMV reactivation and were then treated on study.
Dr. Marty noted that the trial was designed before letermovir was approved for CMV prophylaxis in adults who have undergone an allogeneic HCT.
“Nobody was on letermovir at the beginning of the trial,” he replied, but noted that some patients who were enrolled and had infections that were refractory or resistant to valganciclovir, foscarnet, or a combination of the two received letermovir as secondary prophylaxis.
“I haven’t got the data to tell you how often [letermovir] was used; I think part of the lack of mortality benefit [with maribavir] may be due to the fact that people jumped into secondary prophylaxis with letermovir to minimize the toxicities that we saw,” he said.
Although maribavir has not as of this writing received Food and Drug Administration approval, the drug may be available to some patients through a compassionate-use program from Takeda, Dr. Marty noted.
The study was funded by Shire ViroPharma. Dr. Marty disclosed research funding from Shire and from others. Dr. Ghosh had no relevant disclosures.
Maribavir, an investigational antiviral agent with a novel mechanism of action, was superior to other antiviral strategies at clearing cytomegalovirus (CMV) viremia and controlling symptoms in hematopoietic cell or solid-organ transplant recipients, results of a phase 3 clinical trial showed.
CMV viremia clearance at study week 8 was seen in 55.7% of all patients randomized to receive maribavir, compared with 23.9% for patients assigned to receive investigator-assigned therapy (IAT), Francisco Marty, MD, from the Dana-Farber Cancer Institute in Boston reported at the Transplant & Cellular Therapies Meetings.
“Maribavir’s benefit was driven by lower incidence of treatment-limiting toxicities, compared with IAT,” he said a late-breaking abstract session during the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
“Available anti-CMV antivirals are limited by development of resistance and toxicities, particularly myelosuppression with the use of valganciclovir and nephrotoxicity with the use of foscarnet and cidofovir. Alternative treatment options are required to address this unmet medical need,” he said.
Maribavir inhibits the CMV UL97 protein kinase and is thought to affect several critical processes in CMV replication, including viral DNA synthesis, viral gene expression, encapsidation, and egress of mature capsids from the nucleus.
Details of trial
In the phase 3 SHP620-30e trial (NCT02931539), Dr. Marty and colleagues enrolled patients with relapsed or refractory CMV infections after hematopoietic cell transplant (HCT) or solid-organ transplant (SOT) and after stratification by transplant type and screening CMV DNA level randomly assigned them on a 2:1 basis to receive either maribavir 400 mg twice daily (235 patients) or IAT (117 patients), consisting of either ganciclovir/valganciclovir, foscarnet, cidofovir, or combined foscarnet and val/ganciclovir.
The primary endpoint of viremia clearance at 8 weeks was defined as plasma CMV DNA less than 137 IU/mL in two consecutive tests at a central laboratory at least 5 days apart beginning at the end of week 8.
The trial met its primary endpoint, with a viremia clearance rate of 55.7% with maribavir versus 23.9% with IAT.
The viremia clearance rates were similar in each of the transplant groups: 55.9% versus 20.8%, respectively, in patients who underwent HCT, and 55.6% versus 26.1% in patients who underwent SOT (P < .001).
Clearance rates among patients with CMV DNA below 9,100 IU/mL at baseline were 62.1% with maribavir versus 24.7% with IAT. Among patients with baseline CMV DNA of 9100 IU/mL or above, the respective rates were 43.9% versus 21.9%.
CMV viremia clearance continued from week 8 to week 16 in 18.7% of patients assigned to maribavir and to 10.3% of patients randomized to IAT (P < .013).
The median time to first CMV viremia clearance as 22 days with maribavir versus 27 days with IAT (P = .039).
All-cause mortality was similar between the groups, at 11.5% versus 11.1%, respectively.
The incidences of serious and severe treatment-emergent adverse events (TEAE) were 38.5% and 32.1%, respectively, in the maribavir group, and 37.1% and 37.9% in the IAT group.
Any TEAE leading to study drug discontinuation was less common with maribavir, occurring in 13.2% of patients, compared with 31.9% of patients on IAT. Serious TEAEs leading to drug discontinuation occurred in 8.5% versus 14.7%, respectively.
Serious TEAEs leading to death occurred in 6.8% of patients on maribavir versus 5.2% of those on IAT.
Role of letermovir
In the question-and-answer session following the presentation, comoderator Monalisa Ghosh, MD, from the University of Michigan, Ann Arbor, asked whether any patients in the study were currently on letermovir (Prevymis) prophylaxis, and whether any patients had previously been treated with letermovir but had CMV reactivation and were then treated on study.
Dr. Marty noted that the trial was designed before letermovir was approved for CMV prophylaxis in adults who have undergone an allogeneic HCT.
“Nobody was on letermovir at the beginning of the trial,” he replied, but noted that some patients who were enrolled and had infections that were refractory or resistant to valganciclovir, foscarnet, or a combination of the two received letermovir as secondary prophylaxis.
“I haven’t got the data to tell you how often [letermovir] was used; I think part of the lack of mortality benefit [with maribavir] may be due to the fact that people jumped into secondary prophylaxis with letermovir to minimize the toxicities that we saw,” he said.
Although maribavir has not as of this writing received Food and Drug Administration approval, the drug may be available to some patients through a compassionate-use program from Takeda, Dr. Marty noted.
The study was funded by Shire ViroPharma. Dr. Marty disclosed research funding from Shire and from others. Dr. Ghosh had no relevant disclosures.
FROM TCT 2021
CAR-T in children branching out to solid tumors
Although the only pediatric indication for chimeric antigen receptor T-cell therapy currently approved by the Food and Drug Administration is B-lineage acute lymphoblastic leukemia (ALL) that is refractory to at least two frontline induction attempts or is in second or later relapse, clinical trials of CAR-T therapy for pediatric solid tumors are also currently in progress, said Gregory Yanik, MD, from the CS Mott Children’s Hospital at the University of Michigan, Ann Arbor, at the Transplant & Cellular Therapies Meetings.
In his presentation, Dr. Yanik discussed progress in solid tumor studies as well as some issues involving the current use of CAR-T therapy for ALL.
Solid tumor studies
Malignancies such as sarcomas, brain tumors, and neuroblastomas pose unique challenges, “In contrast to hematologic malignancies, the protein we’re targeting may not be present on the cell surface of all the tumor cells. There are lower-expression profiles, and this is a problem. In fact, many people have postulated that with CAR-T for pediatric solid tumors we’ll have to do repeated cycles, almost like we do with chemotherapy,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
There are currently 14 studies of CAR-T for central nervous system tumors in children, targeting either epidermal growth factor receptor (EGFR) in glioblastoma multiforme and high-grade gliomas, HER2 in a variety of CNS tumors, the GD2 antigen on pontine gliomas, and the checkpoint molecular B7H3 in medulloblastomas and pontine gliomas.
“In sarcomas in kids there are currently 12 trials in progress. Most of the targeting epitopes are targeting either HER2 or GD2. Repetitive CAR-T infusions are being used in several of these trials in sarcomas.
For neuroblastomas there are currently 13 studies in progress, nearly all of which target GD2. Some of the trials include combining CAR-T with immune checkpoint inhibitors or C7R, an engineered cytokine driver designed to prevent T-cell exhaustion.
In addition, several trials of tumor pulsed dendritic cell vaccines are underway for treatment of children with Wilms tumor, Dr. Yanik noted.
Unresolved procedural questions
It’s still early days in CAR-T therapy, and there are several still unanswered questions regarding optimal therapy for and management of patients undergoing CAR-T procedures, Dr. Yanik said.
For example, the optimal time to collect T cells during apheresis is still unclear, he said. Collecting prior to reinduction therapy raises the risk of transducing leukemic cells, while collecting after reinduction may result in inadequate quantity or quality of cells. Regardless of when cells are collected, apheresis should be performed only when the absolute lymphocyte count is above 500/mcL or the CD3 count is above 150/mcL at the time of apheresis.
In the case tisagenlecleucel (Kymriah), his center typically collects 1x109 CD3 cells regardless of age or weight.
The number of CAR T-cells infused also appears to matter, as responses are improved at CAR-T doses above 1.5x106/kg, while risk for higher-grade cytokine release syndrome (CRS) occurs at higher infusion doses.
Blinatumomab or inotuzumab?
Along with CAR-T, two other agents, the bispecific T-cell engager blinatumomab (Blincyto) and the antibody conjugate inotuzumab ozogamicin (Besponsa) are also approved for the treatment of patients with relapsed/refractory B-cell ALL.
Like CAR-T therapy, the primary toxicities associated with blinatumomab are CRS and neurologic adverse events, whereas at inotuzumab is largely associated with hematologic and hepatic toxicities.
The logistics of therapy differ widely, with a 28-day infusion required for blinatumomab, compared with weekly dosing of inotuzumab, and the multiple visits for apheresis and infusion required for CAR-T.
Blinatumomab is approved for both children and adults with relapsed/refractory ALL, but inotuzumab is approved only for adults, and CAR-T with tisagenlecleucel is approved only for children in this indication.
CD-19 expression
There is evidence to suggest that CD19 expression prior to CAR-T has an effect on outcomes, Dr. Yanik said.
“Does blinatumomab pre–CAR-T impact outcome? The answer is probably yes,” he said.
He referred to a study by investigators at the Children’s Hospital of Philadelphia showing that, “if you’re giving blinatumomab prior to CAR-T therapy, you’re potentially reducing the cell-surface expression of CD19 on your leukemic blasts, and now while you’re bringing these patients in for CAR-T therapy, you’re getting a much higher population of dim CD19 expressers, and this is associated with a higher relapse rate and lower remission rate.”
Predicting relapse
Dr. Yanik referred to a study, currently unpublished, which will show that next-generation sequencing (NGS) is more sensitive than flow cytometry for detection of minimal residual disease (MRD), and that MRD analysis of marrow was more sensitive than analysis of peripheral blood.
“Poor outcomes were seen post CAR-T for patients who were in morphologic remission on day 28 or day 100, but had positive MRD. This especially held true if it was next-gen sequencing MRD-positive at day 100, for which relapse rates were over 95%,” he said.
The absence of B-cells is a surrogate marker for the persistence of CAR-T, and conversely, the recovery of CD19-positive B cells may be a predictor for relapse, especially if the B-cell recovery occurs within the first 6 months following CAR-T infusion.
Transplant after CAR-T?
Bone marrow transplant after CAR-T is recommend for patients with high risk of relapse, including those with B-cell recovery within the first 6 months after CAR-T, patients with MRD positivity at days 28 or 100, and patients with mixed lineage leukemia.
“Should we transplant good-risk patients, meaning, if you have NGS-MRD negative patients, is there a role for transplant? You have to look at the risk versus benefit there. These patients may have a cure rate that’s in the 80%-plus range, could we potentially optimize that even more if we consolidate them with an allo[geneic] transplant,” Dr. Yank said.
Move CAR-T up front?
A Children’s Oncology Group study is currently examining whether giving CAR-T therapy to patients with MRD of 0.01% or greater following first consolidation could result in lower tumor burden, fewer relapse, and less CRS with CAR-T.
Dr. Yanik reported that he had no conflicts of interest to disclose.
Although the only pediatric indication for chimeric antigen receptor T-cell therapy currently approved by the Food and Drug Administration is B-lineage acute lymphoblastic leukemia (ALL) that is refractory to at least two frontline induction attempts or is in second or later relapse, clinical trials of CAR-T therapy for pediatric solid tumors are also currently in progress, said Gregory Yanik, MD, from the CS Mott Children’s Hospital at the University of Michigan, Ann Arbor, at the Transplant & Cellular Therapies Meetings.
In his presentation, Dr. Yanik discussed progress in solid tumor studies as well as some issues involving the current use of CAR-T therapy for ALL.
Solid tumor studies
Malignancies such as sarcomas, brain tumors, and neuroblastomas pose unique challenges, “In contrast to hematologic malignancies, the protein we’re targeting may not be present on the cell surface of all the tumor cells. There are lower-expression profiles, and this is a problem. In fact, many people have postulated that with CAR-T for pediatric solid tumors we’ll have to do repeated cycles, almost like we do with chemotherapy,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
There are currently 14 studies of CAR-T for central nervous system tumors in children, targeting either epidermal growth factor receptor (EGFR) in glioblastoma multiforme and high-grade gliomas, HER2 in a variety of CNS tumors, the GD2 antigen on pontine gliomas, and the checkpoint molecular B7H3 in medulloblastomas and pontine gliomas.
“In sarcomas in kids there are currently 12 trials in progress. Most of the targeting epitopes are targeting either HER2 or GD2. Repetitive CAR-T infusions are being used in several of these trials in sarcomas.
For neuroblastomas there are currently 13 studies in progress, nearly all of which target GD2. Some of the trials include combining CAR-T with immune checkpoint inhibitors or C7R, an engineered cytokine driver designed to prevent T-cell exhaustion.
In addition, several trials of tumor pulsed dendritic cell vaccines are underway for treatment of children with Wilms tumor, Dr. Yanik noted.
Unresolved procedural questions
It’s still early days in CAR-T therapy, and there are several still unanswered questions regarding optimal therapy for and management of patients undergoing CAR-T procedures, Dr. Yanik said.
For example, the optimal time to collect T cells during apheresis is still unclear, he said. Collecting prior to reinduction therapy raises the risk of transducing leukemic cells, while collecting after reinduction may result in inadequate quantity or quality of cells. Regardless of when cells are collected, apheresis should be performed only when the absolute lymphocyte count is above 500/mcL or the CD3 count is above 150/mcL at the time of apheresis.
In the case tisagenlecleucel (Kymriah), his center typically collects 1x109 CD3 cells regardless of age or weight.
The number of CAR T-cells infused also appears to matter, as responses are improved at CAR-T doses above 1.5x106/kg, while risk for higher-grade cytokine release syndrome (CRS) occurs at higher infusion doses.
Blinatumomab or inotuzumab?
Along with CAR-T, two other agents, the bispecific T-cell engager blinatumomab (Blincyto) and the antibody conjugate inotuzumab ozogamicin (Besponsa) are also approved for the treatment of patients with relapsed/refractory B-cell ALL.
Like CAR-T therapy, the primary toxicities associated with blinatumomab are CRS and neurologic adverse events, whereas at inotuzumab is largely associated with hematologic and hepatic toxicities.
The logistics of therapy differ widely, with a 28-day infusion required for blinatumomab, compared with weekly dosing of inotuzumab, and the multiple visits for apheresis and infusion required for CAR-T.
Blinatumomab is approved for both children and adults with relapsed/refractory ALL, but inotuzumab is approved only for adults, and CAR-T with tisagenlecleucel is approved only for children in this indication.
CD-19 expression
There is evidence to suggest that CD19 expression prior to CAR-T has an effect on outcomes, Dr. Yanik said.
“Does blinatumomab pre–CAR-T impact outcome? The answer is probably yes,” he said.
He referred to a study by investigators at the Children’s Hospital of Philadelphia showing that, “if you’re giving blinatumomab prior to CAR-T therapy, you’re potentially reducing the cell-surface expression of CD19 on your leukemic blasts, and now while you’re bringing these patients in for CAR-T therapy, you’re getting a much higher population of dim CD19 expressers, and this is associated with a higher relapse rate and lower remission rate.”
Predicting relapse
Dr. Yanik referred to a study, currently unpublished, which will show that next-generation sequencing (NGS) is more sensitive than flow cytometry for detection of minimal residual disease (MRD), and that MRD analysis of marrow was more sensitive than analysis of peripheral blood.
“Poor outcomes were seen post CAR-T for patients who were in morphologic remission on day 28 or day 100, but had positive MRD. This especially held true if it was next-gen sequencing MRD-positive at day 100, for which relapse rates were over 95%,” he said.
The absence of B-cells is a surrogate marker for the persistence of CAR-T, and conversely, the recovery of CD19-positive B cells may be a predictor for relapse, especially if the B-cell recovery occurs within the first 6 months following CAR-T infusion.
Transplant after CAR-T?
Bone marrow transplant after CAR-T is recommend for patients with high risk of relapse, including those with B-cell recovery within the first 6 months after CAR-T, patients with MRD positivity at days 28 or 100, and patients with mixed lineage leukemia.
“Should we transplant good-risk patients, meaning, if you have NGS-MRD negative patients, is there a role for transplant? You have to look at the risk versus benefit there. These patients may have a cure rate that’s in the 80%-plus range, could we potentially optimize that even more if we consolidate them with an allo[geneic] transplant,” Dr. Yank said.
Move CAR-T up front?
A Children’s Oncology Group study is currently examining whether giving CAR-T therapy to patients with MRD of 0.01% or greater following first consolidation could result in lower tumor burden, fewer relapse, and less CRS with CAR-T.
Dr. Yanik reported that he had no conflicts of interest to disclose.
Although the only pediatric indication for chimeric antigen receptor T-cell therapy currently approved by the Food and Drug Administration is B-lineage acute lymphoblastic leukemia (ALL) that is refractory to at least two frontline induction attempts or is in second or later relapse, clinical trials of CAR-T therapy for pediatric solid tumors are also currently in progress, said Gregory Yanik, MD, from the CS Mott Children’s Hospital at the University of Michigan, Ann Arbor, at the Transplant & Cellular Therapies Meetings.
In his presentation, Dr. Yanik discussed progress in solid tumor studies as well as some issues involving the current use of CAR-T therapy for ALL.
Solid tumor studies
Malignancies such as sarcomas, brain tumors, and neuroblastomas pose unique challenges, “In contrast to hematologic malignancies, the protein we’re targeting may not be present on the cell surface of all the tumor cells. There are lower-expression profiles, and this is a problem. In fact, many people have postulated that with CAR-T for pediatric solid tumors we’ll have to do repeated cycles, almost like we do with chemotherapy,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
There are currently 14 studies of CAR-T for central nervous system tumors in children, targeting either epidermal growth factor receptor (EGFR) in glioblastoma multiforme and high-grade gliomas, HER2 in a variety of CNS tumors, the GD2 antigen on pontine gliomas, and the checkpoint molecular B7H3 in medulloblastomas and pontine gliomas.
“In sarcomas in kids there are currently 12 trials in progress. Most of the targeting epitopes are targeting either HER2 or GD2. Repetitive CAR-T infusions are being used in several of these trials in sarcomas.
For neuroblastomas there are currently 13 studies in progress, nearly all of which target GD2. Some of the trials include combining CAR-T with immune checkpoint inhibitors or C7R, an engineered cytokine driver designed to prevent T-cell exhaustion.
In addition, several trials of tumor pulsed dendritic cell vaccines are underway for treatment of children with Wilms tumor, Dr. Yanik noted.
Unresolved procedural questions
It’s still early days in CAR-T therapy, and there are several still unanswered questions regarding optimal therapy for and management of patients undergoing CAR-T procedures, Dr. Yanik said.
For example, the optimal time to collect T cells during apheresis is still unclear, he said. Collecting prior to reinduction therapy raises the risk of transducing leukemic cells, while collecting after reinduction may result in inadequate quantity or quality of cells. Regardless of when cells are collected, apheresis should be performed only when the absolute lymphocyte count is above 500/mcL or the CD3 count is above 150/mcL at the time of apheresis.
In the case tisagenlecleucel (Kymriah), his center typically collects 1x109 CD3 cells regardless of age or weight.
The number of CAR T-cells infused also appears to matter, as responses are improved at CAR-T doses above 1.5x106/kg, while risk for higher-grade cytokine release syndrome (CRS) occurs at higher infusion doses.
Blinatumomab or inotuzumab?
Along with CAR-T, two other agents, the bispecific T-cell engager blinatumomab (Blincyto) and the antibody conjugate inotuzumab ozogamicin (Besponsa) are also approved for the treatment of patients with relapsed/refractory B-cell ALL.
Like CAR-T therapy, the primary toxicities associated with blinatumomab are CRS and neurologic adverse events, whereas at inotuzumab is largely associated with hematologic and hepatic toxicities.
The logistics of therapy differ widely, with a 28-day infusion required for blinatumomab, compared with weekly dosing of inotuzumab, and the multiple visits for apheresis and infusion required for CAR-T.
Blinatumomab is approved for both children and adults with relapsed/refractory ALL, but inotuzumab is approved only for adults, and CAR-T with tisagenlecleucel is approved only for children in this indication.
CD-19 expression
There is evidence to suggest that CD19 expression prior to CAR-T has an effect on outcomes, Dr. Yanik said.
“Does blinatumomab pre–CAR-T impact outcome? The answer is probably yes,” he said.
He referred to a study by investigators at the Children’s Hospital of Philadelphia showing that, “if you’re giving blinatumomab prior to CAR-T therapy, you’re potentially reducing the cell-surface expression of CD19 on your leukemic blasts, and now while you’re bringing these patients in for CAR-T therapy, you’re getting a much higher population of dim CD19 expressers, and this is associated with a higher relapse rate and lower remission rate.”
Predicting relapse
Dr. Yanik referred to a study, currently unpublished, which will show that next-generation sequencing (NGS) is more sensitive than flow cytometry for detection of minimal residual disease (MRD), and that MRD analysis of marrow was more sensitive than analysis of peripheral blood.
“Poor outcomes were seen post CAR-T for patients who were in morphologic remission on day 28 or day 100, but had positive MRD. This especially held true if it was next-gen sequencing MRD-positive at day 100, for which relapse rates were over 95%,” he said.
The absence of B-cells is a surrogate marker for the persistence of CAR-T, and conversely, the recovery of CD19-positive B cells may be a predictor for relapse, especially if the B-cell recovery occurs within the first 6 months following CAR-T infusion.
Transplant after CAR-T?
Bone marrow transplant after CAR-T is recommend for patients with high risk of relapse, including those with B-cell recovery within the first 6 months after CAR-T, patients with MRD positivity at days 28 or 100, and patients with mixed lineage leukemia.
“Should we transplant good-risk patients, meaning, if you have NGS-MRD negative patients, is there a role for transplant? You have to look at the risk versus benefit there. These patients may have a cure rate that’s in the 80%-plus range, could we potentially optimize that even more if we consolidate them with an allo[geneic] transplant,” Dr. Yank said.
Move CAR-T up front?
A Children’s Oncology Group study is currently examining whether giving CAR-T therapy to patients with MRD of 0.01% or greater following first consolidation could result in lower tumor burden, fewer relapse, and less CRS with CAR-T.
Dr. Yanik reported that he had no conflicts of interest to disclose.
FROM TCT 2021
Safety profiles of hemophilia agents vary widely
Despite their similar functions, each current and emerging therapy for treating hemophilia has a unique safety profile, and each needs to be weighed apart from agents both within and outside its pharmacologic class, a hemophilia specialist said.
“My view is that each new molecule coming to the hemophilia space, including variant factor molecules, needs to be scrutinized separately, without class assumptions or extrapolations, and it’s clear that thrombosis risk has become a priority safety consideration,” said Dan Hart, MBChB, MRCP, FRCPath, PhD, from Barts and the London School of Medicine and Dentistry.
He reviewed the comparative safety of standard and novel therapies for hemophilia at the annual congress of the European Association for Haemophilia and Allied Disorders.
Factor inhibitors
Inhibitors occur in both hemophilia A and hemophilia B, and are primarily seen in patients with childhood exposure to factor concentrates. Inhibitors, which include anti–factor VIII and factor IX alloantibodies, are more common among patients with severe hemophilia and those with more disruptive factor VIII and factor IX mutations.
“There can be transient vs. persistent inhibitors, and arguably the more you look, the more you find, but clinically we never miss high-titer inhibitors that have a big impact on individuals and the subsequent decisions about management,” he said.
Hamster vs. human
It’s currently unclear whether there is an immunologic advantage for previously untreated patients to be started on factor VIII concentrates derived from recombinant human cells lines, or from products derived from Chinese hamster ovary (CHO) or baby hamster kidney (BHK) cell lines, Dr. Hart said.
“We need to ensure that we’re not selective about comparator choice for new products in the absence of head-to-head studies,” he said.
Route of administration matters
Inhibitors appear to be a more common occurrence among patients who received factor concentrates subcutaneously, compared with intravenously, Dr. Hart noted, pointing to a 2011 study indicating a background annual risk of 5 cases of inhibitor development per 1,000 treatment years in previously treated patients who received intravenous therapy (Blood. 2011 Jun 9;117[23]:6367-70).
In contrast, in a phase 1 trial of subcutaneous turoctocog alfa pegol, 5 out of 26 patients had detectable N8-GP–binding antibodies after 42-91 exposure days. Of these patients, one developed an inhibitor to factor VIII, and anti–N8-GP antibody appearance was associated with a decline in factor VIII plasma activity in four of the five patients. In addition, five patients reported a total of nine bleeding episodes requiring treatment during prophylaxis. As a result of this trial, further clinical development of the subcutaneous version was suspended. (J Thromb Haemost. 2020 Feb;18[2]:341-51).
Other subcutaneously administered factors are currently in development, Dr. Hart noted.
Nonfactor inhibitors?
“The nonfactor agents do have the risk of generating antibodies: Monoclonal antibodies outside the hemophilia setting provoke antidrug antibodies,” he said.
Although there is no consensus regarding which assay can best monitor antidrug antibodies (ADA), enzyme-linked immunosorbent assay (ELISA) can detect neutralizing antibodies and other antibodies.
In the hemophilia setting, surrogate markers for loss of drug efficacy include longer activated partial thromboplastin time (ATTP) or a drop in serum drug levels. Worsening bleeding phenotype can also be a marker for loss of efficacy, albeit an imperfect one.
Emicizumab (Hemlibara), the first nonfactor monoclonal agent to make it to market, has the largest dataset available, and evidence suggests a rate of neutralizing antibodies with this agent of less than 1% in the HAVEN clinical trial series, but 5.2% in the single-arm STASEY trial.
“We shouldn’t assume that other biophenotypics will have a similar ADA rate, and this needs to be evaluated for each molecule, as it will need to be for other monoclonals” such as anti–tissue factor pathway (TFPI) antibodies, Dr. Hart emphasized.
Pegylation
Pegylated compounds include polyethylene glycol, an inert polymer, covalently bound to the therapeutic protein to extend its half-life, and theoretically, reduce immunogenicity.
Many patients may already have exposure to pegylated products in the form of peginterferon to treat hepatitis C, consumer products such as toothpaste, cough medicine, and cosmetics, and, more recently, in vaccines against COVID-19.
Safety considerations with pegylated agents in hemophilia include concerns about accumulation of polyethylene glycol (PEG), although “some of the preclinical models looking at excretion of PEG are difficult to interpret in my view, and people debate about whether studies are long enough, but it’s undoubtedly the case that toxicology dosing is order of magnitude higher than the routine dosing in hemophilia,” he said.
After more than 5 years of experience with pegylated products there is no clinical evidence of concern, although “it’s not clear, actually, what we’re looking for, whether it’s a clinical parameter, or imaging or histological parameter.”
Patients may also not have lifelong exposure to pegylated products, as it is unlikely that they will stay on the same product for decades, Dr. Hart said.
Thrombosis
As of June 30, 2020, more than 7,200 persons with hemophilia have received emicizumab, and there have been 23 reported thrombotic events, 19 of which occurred in the postmarketing period. Of the reported cases, six patients had a myocardial infarction, and all of these patients had at least one cardiovascular risk factor.
The antithrombin agent fitusiran was associated with one fatal thrombotic event in a phase 2, open-label extension trial, leading to a pause and resumption with mitigation protocols, but that trial has since been paused again because of additional, nonfatal thrombotic events.
Nonfatal thrombotic events have also occurred in clinical trials for the investigational anti-TFPI monoclonal antibodies BAY 1093884 and concizumab, but none have thus far been reported in phase 3 trial of marstacimab.
“We need renewed efforts for prospective reporting and independent review of all adverse events of all agents, old and new: This will need some guidance nationally and internationally, and I think the relevant trial [serious adverse events] need to be reported in peer review literature, and clinicaltrials.gov updated in a timely manner, regardless of whether that strategy was successful or unsuccessful,” Dr. Hart said.
Risk with longer-acting agents?
In the question and answer following his presentation, Christoph Königs, MD, PhD, from University Hospital Frankfurt, asked whether there was potential for increased thrombosis risk with second-generation extended half-life (EHL) molecules in clinical trials.
“As we edge towards normalization of hemostasis, clearly the other non–hemophilia dependent issues of thrombosis risk come into play,” Dr. Hart acknowledged. “I think it will be an inevitability that there will be events, and we need to understand what the denominators are – hence my pitch for there being a renewed effort to try and collate sufficient data that we can really define events happening with people treated with standard half-life [products] through into the novel agents,” he said.
Dr. Hart disclosed grant/research support and speaker bureau activities for Bayer, Octapharma, Takeda, and others. Dr. Königs has reported no relevant disclosures.
Despite their similar functions, each current and emerging therapy for treating hemophilia has a unique safety profile, and each needs to be weighed apart from agents both within and outside its pharmacologic class, a hemophilia specialist said.
“My view is that each new molecule coming to the hemophilia space, including variant factor molecules, needs to be scrutinized separately, without class assumptions or extrapolations, and it’s clear that thrombosis risk has become a priority safety consideration,” said Dan Hart, MBChB, MRCP, FRCPath, PhD, from Barts and the London School of Medicine and Dentistry.
He reviewed the comparative safety of standard and novel therapies for hemophilia at the annual congress of the European Association for Haemophilia and Allied Disorders.
Factor inhibitors
Inhibitors occur in both hemophilia A and hemophilia B, and are primarily seen in patients with childhood exposure to factor concentrates. Inhibitors, which include anti–factor VIII and factor IX alloantibodies, are more common among patients with severe hemophilia and those with more disruptive factor VIII and factor IX mutations.
“There can be transient vs. persistent inhibitors, and arguably the more you look, the more you find, but clinically we never miss high-titer inhibitors that have a big impact on individuals and the subsequent decisions about management,” he said.
Hamster vs. human
It’s currently unclear whether there is an immunologic advantage for previously untreated patients to be started on factor VIII concentrates derived from recombinant human cells lines, or from products derived from Chinese hamster ovary (CHO) or baby hamster kidney (BHK) cell lines, Dr. Hart said.
“We need to ensure that we’re not selective about comparator choice for new products in the absence of head-to-head studies,” he said.
Route of administration matters
Inhibitors appear to be a more common occurrence among patients who received factor concentrates subcutaneously, compared with intravenously, Dr. Hart noted, pointing to a 2011 study indicating a background annual risk of 5 cases of inhibitor development per 1,000 treatment years in previously treated patients who received intravenous therapy (Blood. 2011 Jun 9;117[23]:6367-70).
In contrast, in a phase 1 trial of subcutaneous turoctocog alfa pegol, 5 out of 26 patients had detectable N8-GP–binding antibodies after 42-91 exposure days. Of these patients, one developed an inhibitor to factor VIII, and anti–N8-GP antibody appearance was associated with a decline in factor VIII plasma activity in four of the five patients. In addition, five patients reported a total of nine bleeding episodes requiring treatment during prophylaxis. As a result of this trial, further clinical development of the subcutaneous version was suspended. (J Thromb Haemost. 2020 Feb;18[2]:341-51).
Other subcutaneously administered factors are currently in development, Dr. Hart noted.
Nonfactor inhibitors?
“The nonfactor agents do have the risk of generating antibodies: Monoclonal antibodies outside the hemophilia setting provoke antidrug antibodies,” he said.
Although there is no consensus regarding which assay can best monitor antidrug antibodies (ADA), enzyme-linked immunosorbent assay (ELISA) can detect neutralizing antibodies and other antibodies.
In the hemophilia setting, surrogate markers for loss of drug efficacy include longer activated partial thromboplastin time (ATTP) or a drop in serum drug levels. Worsening bleeding phenotype can also be a marker for loss of efficacy, albeit an imperfect one.
Emicizumab (Hemlibara), the first nonfactor monoclonal agent to make it to market, has the largest dataset available, and evidence suggests a rate of neutralizing antibodies with this agent of less than 1% in the HAVEN clinical trial series, but 5.2% in the single-arm STASEY trial.
“We shouldn’t assume that other biophenotypics will have a similar ADA rate, and this needs to be evaluated for each molecule, as it will need to be for other monoclonals” such as anti–tissue factor pathway (TFPI) antibodies, Dr. Hart emphasized.
Pegylation
Pegylated compounds include polyethylene glycol, an inert polymer, covalently bound to the therapeutic protein to extend its half-life, and theoretically, reduce immunogenicity.
Many patients may already have exposure to pegylated products in the form of peginterferon to treat hepatitis C, consumer products such as toothpaste, cough medicine, and cosmetics, and, more recently, in vaccines against COVID-19.
Safety considerations with pegylated agents in hemophilia include concerns about accumulation of polyethylene glycol (PEG), although “some of the preclinical models looking at excretion of PEG are difficult to interpret in my view, and people debate about whether studies are long enough, but it’s undoubtedly the case that toxicology dosing is order of magnitude higher than the routine dosing in hemophilia,” he said.
After more than 5 years of experience with pegylated products there is no clinical evidence of concern, although “it’s not clear, actually, what we’re looking for, whether it’s a clinical parameter, or imaging or histological parameter.”
Patients may also not have lifelong exposure to pegylated products, as it is unlikely that they will stay on the same product for decades, Dr. Hart said.
Thrombosis
As of June 30, 2020, more than 7,200 persons with hemophilia have received emicizumab, and there have been 23 reported thrombotic events, 19 of which occurred in the postmarketing period. Of the reported cases, six patients had a myocardial infarction, and all of these patients had at least one cardiovascular risk factor.
The antithrombin agent fitusiran was associated with one fatal thrombotic event in a phase 2, open-label extension trial, leading to a pause and resumption with mitigation protocols, but that trial has since been paused again because of additional, nonfatal thrombotic events.
Nonfatal thrombotic events have also occurred in clinical trials for the investigational anti-TFPI monoclonal antibodies BAY 1093884 and concizumab, but none have thus far been reported in phase 3 trial of marstacimab.
“We need renewed efforts for prospective reporting and independent review of all adverse events of all agents, old and new: This will need some guidance nationally and internationally, and I think the relevant trial [serious adverse events] need to be reported in peer review literature, and clinicaltrials.gov updated in a timely manner, regardless of whether that strategy was successful or unsuccessful,” Dr. Hart said.
Risk with longer-acting agents?
In the question and answer following his presentation, Christoph Königs, MD, PhD, from University Hospital Frankfurt, asked whether there was potential for increased thrombosis risk with second-generation extended half-life (EHL) molecules in clinical trials.
“As we edge towards normalization of hemostasis, clearly the other non–hemophilia dependent issues of thrombosis risk come into play,” Dr. Hart acknowledged. “I think it will be an inevitability that there will be events, and we need to understand what the denominators are – hence my pitch for there being a renewed effort to try and collate sufficient data that we can really define events happening with people treated with standard half-life [products] through into the novel agents,” he said.
Dr. Hart disclosed grant/research support and speaker bureau activities for Bayer, Octapharma, Takeda, and others. Dr. Königs has reported no relevant disclosures.
Despite their similar functions, each current and emerging therapy for treating hemophilia has a unique safety profile, and each needs to be weighed apart from agents both within and outside its pharmacologic class, a hemophilia specialist said.
“My view is that each new molecule coming to the hemophilia space, including variant factor molecules, needs to be scrutinized separately, without class assumptions or extrapolations, and it’s clear that thrombosis risk has become a priority safety consideration,” said Dan Hart, MBChB, MRCP, FRCPath, PhD, from Barts and the London School of Medicine and Dentistry.
He reviewed the comparative safety of standard and novel therapies for hemophilia at the annual congress of the European Association for Haemophilia and Allied Disorders.
Factor inhibitors
Inhibitors occur in both hemophilia A and hemophilia B, and are primarily seen in patients with childhood exposure to factor concentrates. Inhibitors, which include anti–factor VIII and factor IX alloantibodies, are more common among patients with severe hemophilia and those with more disruptive factor VIII and factor IX mutations.
“There can be transient vs. persistent inhibitors, and arguably the more you look, the more you find, but clinically we never miss high-titer inhibitors that have a big impact on individuals and the subsequent decisions about management,” he said.
Hamster vs. human
It’s currently unclear whether there is an immunologic advantage for previously untreated patients to be started on factor VIII concentrates derived from recombinant human cells lines, or from products derived from Chinese hamster ovary (CHO) or baby hamster kidney (BHK) cell lines, Dr. Hart said.
“We need to ensure that we’re not selective about comparator choice for new products in the absence of head-to-head studies,” he said.
Route of administration matters
Inhibitors appear to be a more common occurrence among patients who received factor concentrates subcutaneously, compared with intravenously, Dr. Hart noted, pointing to a 2011 study indicating a background annual risk of 5 cases of inhibitor development per 1,000 treatment years in previously treated patients who received intravenous therapy (Blood. 2011 Jun 9;117[23]:6367-70).
In contrast, in a phase 1 trial of subcutaneous turoctocog alfa pegol, 5 out of 26 patients had detectable N8-GP–binding antibodies after 42-91 exposure days. Of these patients, one developed an inhibitor to factor VIII, and anti–N8-GP antibody appearance was associated with a decline in factor VIII plasma activity in four of the five patients. In addition, five patients reported a total of nine bleeding episodes requiring treatment during prophylaxis. As a result of this trial, further clinical development of the subcutaneous version was suspended. (J Thromb Haemost. 2020 Feb;18[2]:341-51).
Other subcutaneously administered factors are currently in development, Dr. Hart noted.
Nonfactor inhibitors?
“The nonfactor agents do have the risk of generating antibodies: Monoclonal antibodies outside the hemophilia setting provoke antidrug antibodies,” he said.
Although there is no consensus regarding which assay can best monitor antidrug antibodies (ADA), enzyme-linked immunosorbent assay (ELISA) can detect neutralizing antibodies and other antibodies.
In the hemophilia setting, surrogate markers for loss of drug efficacy include longer activated partial thromboplastin time (ATTP) or a drop in serum drug levels. Worsening bleeding phenotype can also be a marker for loss of efficacy, albeit an imperfect one.
Emicizumab (Hemlibara), the first nonfactor monoclonal agent to make it to market, has the largest dataset available, and evidence suggests a rate of neutralizing antibodies with this agent of less than 1% in the HAVEN clinical trial series, but 5.2% in the single-arm STASEY trial.
“We shouldn’t assume that other biophenotypics will have a similar ADA rate, and this needs to be evaluated for each molecule, as it will need to be for other monoclonals” such as anti–tissue factor pathway (TFPI) antibodies, Dr. Hart emphasized.
Pegylation
Pegylated compounds include polyethylene glycol, an inert polymer, covalently bound to the therapeutic protein to extend its half-life, and theoretically, reduce immunogenicity.
Many patients may already have exposure to pegylated products in the form of peginterferon to treat hepatitis C, consumer products such as toothpaste, cough medicine, and cosmetics, and, more recently, in vaccines against COVID-19.
Safety considerations with pegylated agents in hemophilia include concerns about accumulation of polyethylene glycol (PEG), although “some of the preclinical models looking at excretion of PEG are difficult to interpret in my view, and people debate about whether studies are long enough, but it’s undoubtedly the case that toxicology dosing is order of magnitude higher than the routine dosing in hemophilia,” he said.
After more than 5 years of experience with pegylated products there is no clinical evidence of concern, although “it’s not clear, actually, what we’re looking for, whether it’s a clinical parameter, or imaging or histological parameter.”
Patients may also not have lifelong exposure to pegylated products, as it is unlikely that they will stay on the same product for decades, Dr. Hart said.
Thrombosis
As of June 30, 2020, more than 7,200 persons with hemophilia have received emicizumab, and there have been 23 reported thrombotic events, 19 of which occurred in the postmarketing period. Of the reported cases, six patients had a myocardial infarction, and all of these patients had at least one cardiovascular risk factor.
The antithrombin agent fitusiran was associated with one fatal thrombotic event in a phase 2, open-label extension trial, leading to a pause and resumption with mitigation protocols, but that trial has since been paused again because of additional, nonfatal thrombotic events.
Nonfatal thrombotic events have also occurred in clinical trials for the investigational anti-TFPI monoclonal antibodies BAY 1093884 and concizumab, but none have thus far been reported in phase 3 trial of marstacimab.
“We need renewed efforts for prospective reporting and independent review of all adverse events of all agents, old and new: This will need some guidance nationally and internationally, and I think the relevant trial [serious adverse events] need to be reported in peer review literature, and clinicaltrials.gov updated in a timely manner, regardless of whether that strategy was successful or unsuccessful,” Dr. Hart said.
Risk with longer-acting agents?
In the question and answer following his presentation, Christoph Königs, MD, PhD, from University Hospital Frankfurt, asked whether there was potential for increased thrombosis risk with second-generation extended half-life (EHL) molecules in clinical trials.
“As we edge towards normalization of hemostasis, clearly the other non–hemophilia dependent issues of thrombosis risk come into play,” Dr. Hart acknowledged. “I think it will be an inevitability that there will be events, and we need to understand what the denominators are – hence my pitch for there being a renewed effort to try and collate sufficient data that we can really define events happening with people treated with standard half-life [products] through into the novel agents,” he said.
Dr. Hart disclosed grant/research support and speaker bureau activities for Bayer, Octapharma, Takeda, and others. Dr. Königs has reported no relevant disclosures.
FROM EAHAD 2021
Using engineered T cells reduced acute, chronic GVHD
A novel T-cell engineered product, Orca-T (Orca Bio), was associated with lower incidence of both acute and chronic graft-versus-host disease (GVHD) and more than double the rate of GVHD-free and relapse-free survival, compared with the current standard of care for patients undergoing hematopoietic stem cell transplants (HSCT), investigators said.
In both a multicenter phase 1 trial (NCT04013685) and single-center phase 1/2 trial (NCT01660607) with a total of 50 patients, those who received Orca-T with single-agent GVHD prophylaxis had a 1-year GVHD-free and relapse-free survival rate of 75%, compared with 31% for patients who received standard of care with two-agent prophylaxis, reported Everett H. Meyer, MD, PhD, from the Stanford (Calif.) University.
“Orca-T has good evidence for reduced acute graft-versus-host disease, reduced chromic graft-versus-host disease, and a low nonrelapse mortality,” he said at the Transplant & Cellular Therapies Meetings.
The product can be quickly manufactured and delivered to treatment centers across the continental United States, with “vein-to-vein” time of less than 72 hours, he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
Orca-T consists of highly purified, donor-derived T-regulatory (Treg) cells that are sorted and delivered on day 0 with hematopoietic stem cells, without immunosuppressants, followed 2 days later with infusion of a matching dose of conventional T cells.
“The Treg cells are allowed to expand to create the right microenvironment for the [conventional T cells],” he explained.
In preclinical studies, donor-derived, high-purity Tregs delivered prior to adoptive transfer of conventional T cells prevented GVHD while maintaining graft-versus-tumor immunity, he said.
Two T-cell infusions
He reported updated results from current studies on a total of 50 adults, with a cohort of 144 patients treated concurrently with standard of care as controls.
The Orca-T–treated patients had a median age of 47 and 52% were male. Indications for transplant included acute myeloid and acute lymphoblastic leukemia, chronic myeloid leukemia, B-cell lymphoma, myelodysplastic syndrome/myelofibrosis, and other unspecified indications.
In both the Orca-T and control cohorts, patients underwent myeloablative conditioning from 10 to 2 days prior to stem cell infusion.
As noted patients in the experimental arm received infusion of hematopoietic stem/progenitor cells and Tregs, followed 2 days later by conventional T-cell infusion, and, on the day after that, tacrolimus at a target dose of 4.6 ng/mL. The conventional T cells were reserved from donor apheresis and were otherwise unmanipulated prior to infusion into the recipient, Dr. Meyer noted.
Patients in the standard-of-care arm received tacrolimus on the day before standard infusion of the apheresis product, followed by methotrexate prophylaxis on days 1, 3, 6 and 11.
Time to neutrophil engraftment, platelet engraftment, and from day 0 to hospital discharge were all significantly shorter in the Orca-T group, at 12 versus 14 days (P < .0001), 11 vs. 17 days (P < .0001), and 15 vs. 17 days (P = .01) respectively.
At 100 days of follow-up, the rate of grade 2 or greater acute GVHD was 30% among standard-of-care patients versus 10% among Orca-T–treated patients. At 1-year follow-up, respective rates of chronic GVHD were 46% vs. 3%.
Safety
“In general, the protocol is extremely well tolerated by our patients. We’ve seen no exceptional infectious disease complications, and we’ve seen no other major complications,” Dr. Meyer said.
Cytomegalovirus prophylaxis was used variably, depending on the center and on the attending physician. Epstein-Barr virus reactivation occurred in eight patients, with one requiring therapy, but there was no biopsy or radiographic evidence of posttransplant lymphoproliferative disorder.
In all, 18% of patients had serious adverse events during the reporting period, all of which resolved. There were no treatment-related deaths in the Orca-T arm, compared with 11% of controls.
Engraftment differences explored
In the question-and-answer session following the presentation, Christopher J. Gamper, MD, PhD, from the Johns Hopkins Hospital in Baltimore, told Dr. Meyer that “your outcomes from Orca-T look excellent,” and asked about the cost differential, compared with similar, unmanipulated transplants performed with standard GVHD prophylaxis.
“Is this recovered by lower costs for treatment of GVHD?” he asked.
“I have not done an economic cost analysis of course, and I think others may be looking into this,” Dr. Meyer replied. “Graft engineering can be expensive, although it’s an engineering proposition and one could imagine that the costs will go down substantially over time.”
Session moderator Alan Hanash, MD, PhD, from Memorial Sloan Kettering Cancer Center in New York, commented on the differences in engraftment between the experimental controls arms, and asked Dr. Meyer: “Do you think this is due to the difference in prophylaxis? Absence of methotrexate? Do you think that it could be a direct impact of regulatory T cells on hematopoietic engraftment?”
“Certainly not having methotrexate is beneficial for engraftment, and may account for the differences we see, Dr. Meyer said. “However, it is possible that Tregs could be playing a facilitative role. There certainly is good preclinical literature that Tregs, particularly in the bone marrow space, can facilitate bone marrow engraftment.”
The Orca-T trials are sponsored by Orca Bio and Stanford, with support from the National Institutes of Health. Dr. Meyer receives research support from Orca and is a scientific adviser to GigaGen, Triursus, Incyte, and Indee Labs. Dr. Hanash and Dr. Gamper had no relevant disclosures.
A novel T-cell engineered product, Orca-T (Orca Bio), was associated with lower incidence of both acute and chronic graft-versus-host disease (GVHD) and more than double the rate of GVHD-free and relapse-free survival, compared with the current standard of care for patients undergoing hematopoietic stem cell transplants (HSCT), investigators said.
In both a multicenter phase 1 trial (NCT04013685) and single-center phase 1/2 trial (NCT01660607) with a total of 50 patients, those who received Orca-T with single-agent GVHD prophylaxis had a 1-year GVHD-free and relapse-free survival rate of 75%, compared with 31% for patients who received standard of care with two-agent prophylaxis, reported Everett H. Meyer, MD, PhD, from the Stanford (Calif.) University.
“Orca-T has good evidence for reduced acute graft-versus-host disease, reduced chromic graft-versus-host disease, and a low nonrelapse mortality,” he said at the Transplant & Cellular Therapies Meetings.
The product can be quickly manufactured and delivered to treatment centers across the continental United States, with “vein-to-vein” time of less than 72 hours, he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
Orca-T consists of highly purified, donor-derived T-regulatory (Treg) cells that are sorted and delivered on day 0 with hematopoietic stem cells, without immunosuppressants, followed 2 days later with infusion of a matching dose of conventional T cells.
“The Treg cells are allowed to expand to create the right microenvironment for the [conventional T cells],” he explained.
In preclinical studies, donor-derived, high-purity Tregs delivered prior to adoptive transfer of conventional T cells prevented GVHD while maintaining graft-versus-tumor immunity, he said.
Two T-cell infusions
He reported updated results from current studies on a total of 50 adults, with a cohort of 144 patients treated concurrently with standard of care as controls.
The Orca-T–treated patients had a median age of 47 and 52% were male. Indications for transplant included acute myeloid and acute lymphoblastic leukemia, chronic myeloid leukemia, B-cell lymphoma, myelodysplastic syndrome/myelofibrosis, and other unspecified indications.
In both the Orca-T and control cohorts, patients underwent myeloablative conditioning from 10 to 2 days prior to stem cell infusion.
As noted patients in the experimental arm received infusion of hematopoietic stem/progenitor cells and Tregs, followed 2 days later by conventional T-cell infusion, and, on the day after that, tacrolimus at a target dose of 4.6 ng/mL. The conventional T cells were reserved from donor apheresis and were otherwise unmanipulated prior to infusion into the recipient, Dr. Meyer noted.
Patients in the standard-of-care arm received tacrolimus on the day before standard infusion of the apheresis product, followed by methotrexate prophylaxis on days 1, 3, 6 and 11.
Time to neutrophil engraftment, platelet engraftment, and from day 0 to hospital discharge were all significantly shorter in the Orca-T group, at 12 versus 14 days (P < .0001), 11 vs. 17 days (P < .0001), and 15 vs. 17 days (P = .01) respectively.
At 100 days of follow-up, the rate of grade 2 or greater acute GVHD was 30% among standard-of-care patients versus 10% among Orca-T–treated patients. At 1-year follow-up, respective rates of chronic GVHD were 46% vs. 3%.
Safety
“In general, the protocol is extremely well tolerated by our patients. We’ve seen no exceptional infectious disease complications, and we’ve seen no other major complications,” Dr. Meyer said.
Cytomegalovirus prophylaxis was used variably, depending on the center and on the attending physician. Epstein-Barr virus reactivation occurred in eight patients, with one requiring therapy, but there was no biopsy or radiographic evidence of posttransplant lymphoproliferative disorder.
In all, 18% of patients had serious adverse events during the reporting period, all of which resolved. There were no treatment-related deaths in the Orca-T arm, compared with 11% of controls.
Engraftment differences explored
In the question-and-answer session following the presentation, Christopher J. Gamper, MD, PhD, from the Johns Hopkins Hospital in Baltimore, told Dr. Meyer that “your outcomes from Orca-T look excellent,” and asked about the cost differential, compared with similar, unmanipulated transplants performed with standard GVHD prophylaxis.
“Is this recovered by lower costs for treatment of GVHD?” he asked.
“I have not done an economic cost analysis of course, and I think others may be looking into this,” Dr. Meyer replied. “Graft engineering can be expensive, although it’s an engineering proposition and one could imagine that the costs will go down substantially over time.”
Session moderator Alan Hanash, MD, PhD, from Memorial Sloan Kettering Cancer Center in New York, commented on the differences in engraftment between the experimental controls arms, and asked Dr. Meyer: “Do you think this is due to the difference in prophylaxis? Absence of methotrexate? Do you think that it could be a direct impact of regulatory T cells on hematopoietic engraftment?”
“Certainly not having methotrexate is beneficial for engraftment, and may account for the differences we see, Dr. Meyer said. “However, it is possible that Tregs could be playing a facilitative role. There certainly is good preclinical literature that Tregs, particularly in the bone marrow space, can facilitate bone marrow engraftment.”
The Orca-T trials are sponsored by Orca Bio and Stanford, with support from the National Institutes of Health. Dr. Meyer receives research support from Orca and is a scientific adviser to GigaGen, Triursus, Incyte, and Indee Labs. Dr. Hanash and Dr. Gamper had no relevant disclosures.
A novel T-cell engineered product, Orca-T (Orca Bio), was associated with lower incidence of both acute and chronic graft-versus-host disease (GVHD) and more than double the rate of GVHD-free and relapse-free survival, compared with the current standard of care for patients undergoing hematopoietic stem cell transplants (HSCT), investigators said.
In both a multicenter phase 1 trial (NCT04013685) and single-center phase 1/2 trial (NCT01660607) with a total of 50 patients, those who received Orca-T with single-agent GVHD prophylaxis had a 1-year GVHD-free and relapse-free survival rate of 75%, compared with 31% for patients who received standard of care with two-agent prophylaxis, reported Everett H. Meyer, MD, PhD, from the Stanford (Calif.) University.
“Orca-T has good evidence for reduced acute graft-versus-host disease, reduced chromic graft-versus-host disease, and a low nonrelapse mortality,” he said at the Transplant & Cellular Therapies Meetings.
The product can be quickly manufactured and delivered to treatment centers across the continental United States, with “vein-to-vein” time of less than 72 hours, he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
Orca-T consists of highly purified, donor-derived T-regulatory (Treg) cells that are sorted and delivered on day 0 with hematopoietic stem cells, without immunosuppressants, followed 2 days later with infusion of a matching dose of conventional T cells.
“The Treg cells are allowed to expand to create the right microenvironment for the [conventional T cells],” he explained.
In preclinical studies, donor-derived, high-purity Tregs delivered prior to adoptive transfer of conventional T cells prevented GVHD while maintaining graft-versus-tumor immunity, he said.
Two T-cell infusions
He reported updated results from current studies on a total of 50 adults, with a cohort of 144 patients treated concurrently with standard of care as controls.
The Orca-T–treated patients had a median age of 47 and 52% were male. Indications for transplant included acute myeloid and acute lymphoblastic leukemia, chronic myeloid leukemia, B-cell lymphoma, myelodysplastic syndrome/myelofibrosis, and other unspecified indications.
In both the Orca-T and control cohorts, patients underwent myeloablative conditioning from 10 to 2 days prior to stem cell infusion.
As noted patients in the experimental arm received infusion of hematopoietic stem/progenitor cells and Tregs, followed 2 days later by conventional T-cell infusion, and, on the day after that, tacrolimus at a target dose of 4.6 ng/mL. The conventional T cells were reserved from donor apheresis and were otherwise unmanipulated prior to infusion into the recipient, Dr. Meyer noted.
Patients in the standard-of-care arm received tacrolimus on the day before standard infusion of the apheresis product, followed by methotrexate prophylaxis on days 1, 3, 6 and 11.
Time to neutrophil engraftment, platelet engraftment, and from day 0 to hospital discharge were all significantly shorter in the Orca-T group, at 12 versus 14 days (P < .0001), 11 vs. 17 days (P < .0001), and 15 vs. 17 days (P = .01) respectively.
At 100 days of follow-up, the rate of grade 2 or greater acute GVHD was 30% among standard-of-care patients versus 10% among Orca-T–treated patients. At 1-year follow-up, respective rates of chronic GVHD were 46% vs. 3%.
Safety
“In general, the protocol is extremely well tolerated by our patients. We’ve seen no exceptional infectious disease complications, and we’ve seen no other major complications,” Dr. Meyer said.
Cytomegalovirus prophylaxis was used variably, depending on the center and on the attending physician. Epstein-Barr virus reactivation occurred in eight patients, with one requiring therapy, but there was no biopsy or radiographic evidence of posttransplant lymphoproliferative disorder.
In all, 18% of patients had serious adverse events during the reporting period, all of which resolved. There were no treatment-related deaths in the Orca-T arm, compared with 11% of controls.
Engraftment differences explored
In the question-and-answer session following the presentation, Christopher J. Gamper, MD, PhD, from the Johns Hopkins Hospital in Baltimore, told Dr. Meyer that “your outcomes from Orca-T look excellent,” and asked about the cost differential, compared with similar, unmanipulated transplants performed with standard GVHD prophylaxis.
“Is this recovered by lower costs for treatment of GVHD?” he asked.
“I have not done an economic cost analysis of course, and I think others may be looking into this,” Dr. Meyer replied. “Graft engineering can be expensive, although it’s an engineering proposition and one could imagine that the costs will go down substantially over time.”
Session moderator Alan Hanash, MD, PhD, from Memorial Sloan Kettering Cancer Center in New York, commented on the differences in engraftment between the experimental controls arms, and asked Dr. Meyer: “Do you think this is due to the difference in prophylaxis? Absence of methotrexate? Do you think that it could be a direct impact of regulatory T cells on hematopoietic engraftment?”
“Certainly not having methotrexate is beneficial for engraftment, and may account for the differences we see, Dr. Meyer said. “However, it is possible that Tregs could be playing a facilitative role. There certainly is good preclinical literature that Tregs, particularly in the bone marrow space, can facilitate bone marrow engraftment.”
The Orca-T trials are sponsored by Orca Bio and Stanford, with support from the National Institutes of Health. Dr. Meyer receives research support from Orca and is a scientific adviser to GigaGen, Triursus, Incyte, and Indee Labs. Dr. Hanash and Dr. Gamper had no relevant disclosures.
FROM TCT 2021
Transplant-related mortality higher with CD34 selection
In a clinical trial comparing three graft-versus-host disease (GVHD)–prevention regimens in patients undergoing hematopoietic stem cell transplants, a calcineurin inhibitor (CNI)–free strategy using CD34-selected peripheral blood stem cells (PBSCs) was associated with a nearly twofold increase in transplant-related mortality, compared with either a different CNI-free regimen or tacrolimus plus methotrexate, investigators reported.
In the phase 3 Progress II trial, patients who received CD34-selected PBSCs without post-transplant immune suppression had a hazard ratio for death of 1.74 compared with patients who received T-cell depletion with posttransplant cyclophosphamide, and a HR of 1.78, compared with patients who received tacrolimus and methotrexate after a bone marrow graft, Miguel-Angel Perales , MD, from Memorial Sloan Kettering Cancer Center, New York, reported at the Transplant & Cellular Therapies Meetings.
“CD34 selection was associated with worse overall survival, which offset any benefit from lower rates of moderate to severe chronic GVHD,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
Neither of the two CNI-free interventions were superior to tacrolimus/methotrexate with bone marrow–derived stem cells for preventing chronic GVHD, and there were no differences in the primary endpoint of chronic GVHD/relapse-free survival, Dr. Perales said.
T-cell depletion vs. CNI
The Progress II trial was designed to see whether either of two CNI-free, T-cell depletion approaches could improve chronic GVHD rates post transplant over a CNI-based regimen.
The investigators enrolled patients aged 65 years or younger with acute leukemia or myelodysplasia with fewer than 5% blasts and a HLA-matched related or unrelated donor.
The patients were randomly assigned to either bone marrow grafts with tacrolimus/methotrexate (118 patients), bone marrow with in vivo posttransplant cyclophosphamide (114), or PBSCs with ex vivo CD34-selected cells (114).
The primary endpoint of chronic GVHD/relapse-free survival (CRFS) was a time-to-event outcome defined as moderate to severe chronic GVHD according to National Institutes of Health consensus criteria, disease relapse or progression, or death from any cause.
As noted before, there were no between-arm differences in the primary CRFS endpoint, and in multivariate analysis controlling for donor type, patient characteristics, disease category and disease risk index, the only factor significantly predictive for CRFS was being aged 50 years or older.
The 2-year posttransplant survival rates were 61.6% in the CD34-selected arm, 76.7% in the posttransplant cyclophosphamide arm, and 74.2% in the tacrolimus/methotrexate arm.
As noted before, the HR for CRFS with CD34 versus tacrolimus/methotrexate was 1.74, and for CD34 versus cyclophosphamide was 1.78 (P = .02 for both comparisons). In contrast, there was no difference in CRFS between posttransplant cyclophosphamide and tacrolimus/methotrexate.
Both relapse-free survival and transplant-related mortality were worse with the CD34-selected group, compared with the other two groups, but there were no significant differences among the arms in disease relapse.
Hematologic recovery was faster in the CD34 arm, but there were no significant differences in graft failure.
In addition, the incidence of grade II-IV acute GVHD was increased in the posttransplant cyclophosphamide group, compared with the other two, while chronic GVHD and moderate to severe chronic GVHD were reduced in the CD34 group.
There were no differences in quality of life measures among the groups, Dr. Perales said.
Practice changing?
In the question-and answer-session following the presentation, comoderator Sarah Nikiforow , MD, PhD, from the Dana-Farber Cancer Institute in Boston, who was not involved in the study, asked whether the trial results could be considered as practice changing for any centers that historically have done CD34 selection, or whether CD34 selection is still a viable approach to GVHD prophylaxis.
“That’s obviously a key question from the study, and a question that we’re asking ourselves,” Dr. Perales said. “I think the lesson that we took from this study as it pertains to CD34 selection is obviously the increased mortality, likely related to regimen toxicity, and I think the use of high-dose radiation is something that we have to reexamine.”
He said that his center is also considering whether to reduce antithymocyte globulin dosing, move it earlier in the process, and to use pharmokinetic-directed ATG as a possible means of decreasing nonrelapse mortality.
“I think it remains a useful platform for adoptive cell therapy, potentially targeting relapsed disease,” he added.
The study was supported by the National Heart, Lung, and Blood Institute. Dr. Perales disclosed advisory board activities and consulting for multiple companies, and receiving research funding for clinical trials from several more. Dr. Nikiforow disclosed a consulting/advisory role for Kite Pharma, and travel accommodations and expense from Celyad Oncology.
In a clinical trial comparing three graft-versus-host disease (GVHD)–prevention regimens in patients undergoing hematopoietic stem cell transplants, a calcineurin inhibitor (CNI)–free strategy using CD34-selected peripheral blood stem cells (PBSCs) was associated with a nearly twofold increase in transplant-related mortality, compared with either a different CNI-free regimen or tacrolimus plus methotrexate, investigators reported.
In the phase 3 Progress II trial, patients who received CD34-selected PBSCs without post-transplant immune suppression had a hazard ratio for death of 1.74 compared with patients who received T-cell depletion with posttransplant cyclophosphamide, and a HR of 1.78, compared with patients who received tacrolimus and methotrexate after a bone marrow graft, Miguel-Angel Perales , MD, from Memorial Sloan Kettering Cancer Center, New York, reported at the Transplant & Cellular Therapies Meetings.
“CD34 selection was associated with worse overall survival, which offset any benefit from lower rates of moderate to severe chronic GVHD,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
Neither of the two CNI-free interventions were superior to tacrolimus/methotrexate with bone marrow–derived stem cells for preventing chronic GVHD, and there were no differences in the primary endpoint of chronic GVHD/relapse-free survival, Dr. Perales said.
T-cell depletion vs. CNI
The Progress II trial was designed to see whether either of two CNI-free, T-cell depletion approaches could improve chronic GVHD rates post transplant over a CNI-based regimen.
The investigators enrolled patients aged 65 years or younger with acute leukemia or myelodysplasia with fewer than 5% blasts and a HLA-matched related or unrelated donor.
The patients were randomly assigned to either bone marrow grafts with tacrolimus/methotrexate (118 patients), bone marrow with in vivo posttransplant cyclophosphamide (114), or PBSCs with ex vivo CD34-selected cells (114).
The primary endpoint of chronic GVHD/relapse-free survival (CRFS) was a time-to-event outcome defined as moderate to severe chronic GVHD according to National Institutes of Health consensus criteria, disease relapse or progression, or death from any cause.
As noted before, there were no between-arm differences in the primary CRFS endpoint, and in multivariate analysis controlling for donor type, patient characteristics, disease category and disease risk index, the only factor significantly predictive for CRFS was being aged 50 years or older.
The 2-year posttransplant survival rates were 61.6% in the CD34-selected arm, 76.7% in the posttransplant cyclophosphamide arm, and 74.2% in the tacrolimus/methotrexate arm.
As noted before, the HR for CRFS with CD34 versus tacrolimus/methotrexate was 1.74, and for CD34 versus cyclophosphamide was 1.78 (P = .02 for both comparisons). In contrast, there was no difference in CRFS between posttransplant cyclophosphamide and tacrolimus/methotrexate.
Both relapse-free survival and transplant-related mortality were worse with the CD34-selected group, compared with the other two groups, but there were no significant differences among the arms in disease relapse.
Hematologic recovery was faster in the CD34 arm, but there were no significant differences in graft failure.
In addition, the incidence of grade II-IV acute GVHD was increased in the posttransplant cyclophosphamide group, compared with the other two, while chronic GVHD and moderate to severe chronic GVHD were reduced in the CD34 group.
There were no differences in quality of life measures among the groups, Dr. Perales said.
Practice changing?
In the question-and answer-session following the presentation, comoderator Sarah Nikiforow , MD, PhD, from the Dana-Farber Cancer Institute in Boston, who was not involved in the study, asked whether the trial results could be considered as practice changing for any centers that historically have done CD34 selection, or whether CD34 selection is still a viable approach to GVHD prophylaxis.
“That’s obviously a key question from the study, and a question that we’re asking ourselves,” Dr. Perales said. “I think the lesson that we took from this study as it pertains to CD34 selection is obviously the increased mortality, likely related to regimen toxicity, and I think the use of high-dose radiation is something that we have to reexamine.”
He said that his center is also considering whether to reduce antithymocyte globulin dosing, move it earlier in the process, and to use pharmokinetic-directed ATG as a possible means of decreasing nonrelapse mortality.
“I think it remains a useful platform for adoptive cell therapy, potentially targeting relapsed disease,” he added.
The study was supported by the National Heart, Lung, and Blood Institute. Dr. Perales disclosed advisory board activities and consulting for multiple companies, and receiving research funding for clinical trials from several more. Dr. Nikiforow disclosed a consulting/advisory role for Kite Pharma, and travel accommodations and expense from Celyad Oncology.
In a clinical trial comparing three graft-versus-host disease (GVHD)–prevention regimens in patients undergoing hematopoietic stem cell transplants, a calcineurin inhibitor (CNI)–free strategy using CD34-selected peripheral blood stem cells (PBSCs) was associated with a nearly twofold increase in transplant-related mortality, compared with either a different CNI-free regimen or tacrolimus plus methotrexate, investigators reported.
In the phase 3 Progress II trial, patients who received CD34-selected PBSCs without post-transplant immune suppression had a hazard ratio for death of 1.74 compared with patients who received T-cell depletion with posttransplant cyclophosphamide, and a HR of 1.78, compared with patients who received tacrolimus and methotrexate after a bone marrow graft, Miguel-Angel Perales , MD, from Memorial Sloan Kettering Cancer Center, New York, reported at the Transplant & Cellular Therapies Meetings.
“CD34 selection was associated with worse overall survival, which offset any benefit from lower rates of moderate to severe chronic GVHD,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
Neither of the two CNI-free interventions were superior to tacrolimus/methotrexate with bone marrow–derived stem cells for preventing chronic GVHD, and there were no differences in the primary endpoint of chronic GVHD/relapse-free survival, Dr. Perales said.
T-cell depletion vs. CNI
The Progress II trial was designed to see whether either of two CNI-free, T-cell depletion approaches could improve chronic GVHD rates post transplant over a CNI-based regimen.
The investigators enrolled patients aged 65 years or younger with acute leukemia or myelodysplasia with fewer than 5% blasts and a HLA-matched related or unrelated donor.
The patients were randomly assigned to either bone marrow grafts with tacrolimus/methotrexate (118 patients), bone marrow with in vivo posttransplant cyclophosphamide (114), or PBSCs with ex vivo CD34-selected cells (114).
The primary endpoint of chronic GVHD/relapse-free survival (CRFS) was a time-to-event outcome defined as moderate to severe chronic GVHD according to National Institutes of Health consensus criteria, disease relapse or progression, or death from any cause.
As noted before, there were no between-arm differences in the primary CRFS endpoint, and in multivariate analysis controlling for donor type, patient characteristics, disease category and disease risk index, the only factor significantly predictive for CRFS was being aged 50 years or older.
The 2-year posttransplant survival rates were 61.6% in the CD34-selected arm, 76.7% in the posttransplant cyclophosphamide arm, and 74.2% in the tacrolimus/methotrexate arm.
As noted before, the HR for CRFS with CD34 versus tacrolimus/methotrexate was 1.74, and for CD34 versus cyclophosphamide was 1.78 (P = .02 for both comparisons). In contrast, there was no difference in CRFS between posttransplant cyclophosphamide and tacrolimus/methotrexate.
Both relapse-free survival and transplant-related mortality were worse with the CD34-selected group, compared with the other two groups, but there were no significant differences among the arms in disease relapse.
Hematologic recovery was faster in the CD34 arm, but there were no significant differences in graft failure.
In addition, the incidence of grade II-IV acute GVHD was increased in the posttransplant cyclophosphamide group, compared with the other two, while chronic GVHD and moderate to severe chronic GVHD were reduced in the CD34 group.
There were no differences in quality of life measures among the groups, Dr. Perales said.
Practice changing?
In the question-and answer-session following the presentation, comoderator Sarah Nikiforow , MD, PhD, from the Dana-Farber Cancer Institute in Boston, who was not involved in the study, asked whether the trial results could be considered as practice changing for any centers that historically have done CD34 selection, or whether CD34 selection is still a viable approach to GVHD prophylaxis.
“That’s obviously a key question from the study, and a question that we’re asking ourselves,” Dr. Perales said. “I think the lesson that we took from this study as it pertains to CD34 selection is obviously the increased mortality, likely related to regimen toxicity, and I think the use of high-dose radiation is something that we have to reexamine.”
He said that his center is also considering whether to reduce antithymocyte globulin dosing, move it earlier in the process, and to use pharmokinetic-directed ATG as a possible means of decreasing nonrelapse mortality.
“I think it remains a useful platform for adoptive cell therapy, potentially targeting relapsed disease,” he added.
The study was supported by the National Heart, Lung, and Blood Institute. Dr. Perales disclosed advisory board activities and consulting for multiple companies, and receiving research funding for clinical trials from several more. Dr. Nikiforow disclosed a consulting/advisory role for Kite Pharma, and travel accommodations and expense from Celyad Oncology.
FROM TCT 2021
Steroid complications in GVHD common, boost costs of care
Steroids are usually the first choice of therapy for the treatment of patients with graft-vs.-host disease (GVHD), but complications from steroid use may carry a high financial cost, investigators caution.
Among 689 patients with a diagnosis of GVHD following a hematopoietic stem cell transplant (HSCT) who received steroids, 685 (97%) had at least one steroid-related complication, resulting in nearly $165,000 in mean health-care costs over 24 months, said Elizabeth J. Bell, PhD, MPH, an epidemiologist at Optum Inc.
“For both acute and chronic GVHD, the standard of care for first-line treatment is systemic steroids. The complications associated with steroid treatment are well known. However, the health-care resources utilized and the costs incurred by these patients are not well-quantified,” she said at the Transplantation & Cellular Therapies Meetings (Abstract 12).
Dr. Bell reported the results of a retrospective database analysis on costs associated with steroid complications in HSCT recipients at the meeting, which was held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
She and colleagues from Optum, Incyte, and the University of Minnesota in Minneapolis looked at data on 689 patients with a diagnosis of GVHD after HSCT who received systemic steroids from July 1, 2010, through Aug. 31, 2019. The data were extracted from the Optum Research database, and included U.S. commercial and Medicare Advantage patients.
They looked at total complications and steroid-associated complications in each of four categories: infections; metabolic or endocrine complications (for example, diabetes, dyslipidemia); gastrointestinal (GI) complications (e.g., peptic ulcer disease); and bone or muscle complications (myopathy, etc).
They estimated costs based on International Classification of Diseases (ICD) codes for any steroid complications during the 24 months after steroid initiation, including those complications that may have been present at the time of GVHD diagnosis.
The median patient age was 55 years, and 60% of the sample were male. The mean Charlson Comorbidity Index score at baseline was 3.
Overall, 22% of patients had only acute GVHD, 21% had only chronic GVHD, and 39% had both acute and chronic disease. The GVHD type was unspecified in the remaining 18%.
The median time from GVHD diagnosis to initiating steroids was 30 days for patients with both acute and chronic disease, as well as those with both presentations. The median time to initiation was 36 days for patients with unspecified GVHD type.
The median cumulative duration of steroid use over 24 months was 62 days for patients with acute GVHD, 208 days for those with chronic GVHD, 166 days for those with both, and 74 days for patients with unspecified GVHD type.
As noted before, complications occurred in 97% of patients, with infections being the most common complications, occurring in 80% of patients, followed by metabolic/endocrine complications in 32%, gastrointestinal in 29%, and bone/muscle complications in 20%.
For the 665 patients who had any steroid-related complication, the mean costs of steroid-associated care in the 24 months after they were started on steroids was $164,787, and the median cost was $50,834.
Health care costs were highest among patients with infections, at a mean of $167,473, and a median of $57,680, followed by bone/muscle conditions ($75,289 and $2,057, respectively), GI conditions ($67,861 and $3,360), and metabolic or endocrine conditions ($47, 101 and $1,164).
In all categories, hospitalizations accounted for the large majority of costs.
Two-thirds (66%) of patients who experienced any steroid-related complication required hospitalization, primarily for infections.
Among all patients with complications, the median cumulative hospital stay over 24 months was 20 days, with bone/muscle complications and infections associated with a median of 19 and 18 days of hospitalization, respectively.
Dr. Bell acknowledged that the study was limited by use of ICD coding to identify steroid complication-related health-care utilization and costs, which can be imprecise, and by the fact that the analysis included only complications resulting in health care use as documented in medical claims. In addition, the investigators noted that they could not control for the possibility that steroids exacerbated conditions that existed at baseline.
“These findings emphasize the need to cautiously evaluate the treatment options for patients with GVHD. Future study with medical records is needed to provide insights on the clinical aspects of the complications (e.g., severity and suspected causality),” Dr. Bell and colleagues concluded in the study’s abstract.
Definitions questioned
An HSCT specialist approached for comment said that the findings of the study made sense, but she had questions regarding the study methodology.
“I would intuitively think that steroid-associated complications are a major cause of health care use in GVHD patients and it’s interesting to see that there is emerging data to support this hypothesis,” HSCT specialist Hélène Schoemans, MD of the University of Leuven, Belgium, said in an interview.
She noted, however, that “it is surprising that the period of steroid initiation was the same for acute and chronic GVHD,” and questioned whether that anomalous finding could be due to the study’s definition of acute and chronic GVHD or to how the period from baseline to steroid initiation was defined.
The questions about the definitions and timing of therapy make it uncertain as to whether the complications reported were caused by steroids or by some other factor, she suggested.
The study was supported by Optum Inc. Dr. Bell is an employee of the company, and a paid consultant of Incyte. Dr. Schoemans has received travel expenses from Celgene, Abbvie, and Incyte; is part of the advisory boards for Incyte; and has received speakers fees from Novartis, Incyte, Jazz Pharmaceuticals, and Takeda.
Steroids are usually the first choice of therapy for the treatment of patients with graft-vs.-host disease (GVHD), but complications from steroid use may carry a high financial cost, investigators caution.
Among 689 patients with a diagnosis of GVHD following a hematopoietic stem cell transplant (HSCT) who received steroids, 685 (97%) had at least one steroid-related complication, resulting in nearly $165,000 in mean health-care costs over 24 months, said Elizabeth J. Bell, PhD, MPH, an epidemiologist at Optum Inc.
“For both acute and chronic GVHD, the standard of care for first-line treatment is systemic steroids. The complications associated with steroid treatment are well known. However, the health-care resources utilized and the costs incurred by these patients are not well-quantified,” she said at the Transplantation & Cellular Therapies Meetings (Abstract 12).
Dr. Bell reported the results of a retrospective database analysis on costs associated with steroid complications in HSCT recipients at the meeting, which was held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
She and colleagues from Optum, Incyte, and the University of Minnesota in Minneapolis looked at data on 689 patients with a diagnosis of GVHD after HSCT who received systemic steroids from July 1, 2010, through Aug. 31, 2019. The data were extracted from the Optum Research database, and included U.S. commercial and Medicare Advantage patients.
They looked at total complications and steroid-associated complications in each of four categories: infections; metabolic or endocrine complications (for example, diabetes, dyslipidemia); gastrointestinal (GI) complications (e.g., peptic ulcer disease); and bone or muscle complications (myopathy, etc).
They estimated costs based on International Classification of Diseases (ICD) codes for any steroid complications during the 24 months after steroid initiation, including those complications that may have been present at the time of GVHD diagnosis.
The median patient age was 55 years, and 60% of the sample were male. The mean Charlson Comorbidity Index score at baseline was 3.
Overall, 22% of patients had only acute GVHD, 21% had only chronic GVHD, and 39% had both acute and chronic disease. The GVHD type was unspecified in the remaining 18%.
The median time from GVHD diagnosis to initiating steroids was 30 days for patients with both acute and chronic disease, as well as those with both presentations. The median time to initiation was 36 days for patients with unspecified GVHD type.
The median cumulative duration of steroid use over 24 months was 62 days for patients with acute GVHD, 208 days for those with chronic GVHD, 166 days for those with both, and 74 days for patients with unspecified GVHD type.
As noted before, complications occurred in 97% of patients, with infections being the most common complications, occurring in 80% of patients, followed by metabolic/endocrine complications in 32%, gastrointestinal in 29%, and bone/muscle complications in 20%.
For the 665 patients who had any steroid-related complication, the mean costs of steroid-associated care in the 24 months after they were started on steroids was $164,787, and the median cost was $50,834.
Health care costs were highest among patients with infections, at a mean of $167,473, and a median of $57,680, followed by bone/muscle conditions ($75,289 and $2,057, respectively), GI conditions ($67,861 and $3,360), and metabolic or endocrine conditions ($47, 101 and $1,164).
In all categories, hospitalizations accounted for the large majority of costs.
Two-thirds (66%) of patients who experienced any steroid-related complication required hospitalization, primarily for infections.
Among all patients with complications, the median cumulative hospital stay over 24 months was 20 days, with bone/muscle complications and infections associated with a median of 19 and 18 days of hospitalization, respectively.
Dr. Bell acknowledged that the study was limited by use of ICD coding to identify steroid complication-related health-care utilization and costs, which can be imprecise, and by the fact that the analysis included only complications resulting in health care use as documented in medical claims. In addition, the investigators noted that they could not control for the possibility that steroids exacerbated conditions that existed at baseline.
“These findings emphasize the need to cautiously evaluate the treatment options for patients with GVHD. Future study with medical records is needed to provide insights on the clinical aspects of the complications (e.g., severity and suspected causality),” Dr. Bell and colleagues concluded in the study’s abstract.
Definitions questioned
An HSCT specialist approached for comment said that the findings of the study made sense, but she had questions regarding the study methodology.
“I would intuitively think that steroid-associated complications are a major cause of health care use in GVHD patients and it’s interesting to see that there is emerging data to support this hypothesis,” HSCT specialist Hélène Schoemans, MD of the University of Leuven, Belgium, said in an interview.
She noted, however, that “it is surprising that the period of steroid initiation was the same for acute and chronic GVHD,” and questioned whether that anomalous finding could be due to the study’s definition of acute and chronic GVHD or to how the period from baseline to steroid initiation was defined.
The questions about the definitions and timing of therapy make it uncertain as to whether the complications reported were caused by steroids or by some other factor, she suggested.
The study was supported by Optum Inc. Dr. Bell is an employee of the company, and a paid consultant of Incyte. Dr. Schoemans has received travel expenses from Celgene, Abbvie, and Incyte; is part of the advisory boards for Incyte; and has received speakers fees from Novartis, Incyte, Jazz Pharmaceuticals, and Takeda.
Steroids are usually the first choice of therapy for the treatment of patients with graft-vs.-host disease (GVHD), but complications from steroid use may carry a high financial cost, investigators caution.
Among 689 patients with a diagnosis of GVHD following a hematopoietic stem cell transplant (HSCT) who received steroids, 685 (97%) had at least one steroid-related complication, resulting in nearly $165,000 in mean health-care costs over 24 months, said Elizabeth J. Bell, PhD, MPH, an epidemiologist at Optum Inc.
“For both acute and chronic GVHD, the standard of care for first-line treatment is systemic steroids. The complications associated with steroid treatment are well known. However, the health-care resources utilized and the costs incurred by these patients are not well-quantified,” she said at the Transplantation & Cellular Therapies Meetings (Abstract 12).
Dr. Bell reported the results of a retrospective database analysis on costs associated with steroid complications in HSCT recipients at the meeting, which was held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
She and colleagues from Optum, Incyte, and the University of Minnesota in Minneapolis looked at data on 689 patients with a diagnosis of GVHD after HSCT who received systemic steroids from July 1, 2010, through Aug. 31, 2019. The data were extracted from the Optum Research database, and included U.S. commercial and Medicare Advantage patients.
They looked at total complications and steroid-associated complications in each of four categories: infections; metabolic or endocrine complications (for example, diabetes, dyslipidemia); gastrointestinal (GI) complications (e.g., peptic ulcer disease); and bone or muscle complications (myopathy, etc).
They estimated costs based on International Classification of Diseases (ICD) codes for any steroid complications during the 24 months after steroid initiation, including those complications that may have been present at the time of GVHD diagnosis.
The median patient age was 55 years, and 60% of the sample were male. The mean Charlson Comorbidity Index score at baseline was 3.
Overall, 22% of patients had only acute GVHD, 21% had only chronic GVHD, and 39% had both acute and chronic disease. The GVHD type was unspecified in the remaining 18%.
The median time from GVHD diagnosis to initiating steroids was 30 days for patients with both acute and chronic disease, as well as those with both presentations. The median time to initiation was 36 days for patients with unspecified GVHD type.
The median cumulative duration of steroid use over 24 months was 62 days for patients with acute GVHD, 208 days for those with chronic GVHD, 166 days for those with both, and 74 days for patients with unspecified GVHD type.
As noted before, complications occurred in 97% of patients, with infections being the most common complications, occurring in 80% of patients, followed by metabolic/endocrine complications in 32%, gastrointestinal in 29%, and bone/muscle complications in 20%.
For the 665 patients who had any steroid-related complication, the mean costs of steroid-associated care in the 24 months after they were started on steroids was $164,787, and the median cost was $50,834.
Health care costs were highest among patients with infections, at a mean of $167,473, and a median of $57,680, followed by bone/muscle conditions ($75,289 and $2,057, respectively), GI conditions ($67,861 and $3,360), and metabolic or endocrine conditions ($47, 101 and $1,164).
In all categories, hospitalizations accounted for the large majority of costs.
Two-thirds (66%) of patients who experienced any steroid-related complication required hospitalization, primarily for infections.
Among all patients with complications, the median cumulative hospital stay over 24 months was 20 days, with bone/muscle complications and infections associated with a median of 19 and 18 days of hospitalization, respectively.
Dr. Bell acknowledged that the study was limited by use of ICD coding to identify steroid complication-related health-care utilization and costs, which can be imprecise, and by the fact that the analysis included only complications resulting in health care use as documented in medical claims. In addition, the investigators noted that they could not control for the possibility that steroids exacerbated conditions that existed at baseline.
“These findings emphasize the need to cautiously evaluate the treatment options for patients with GVHD. Future study with medical records is needed to provide insights on the clinical aspects of the complications (e.g., severity and suspected causality),” Dr. Bell and colleagues concluded in the study’s abstract.
Definitions questioned
An HSCT specialist approached for comment said that the findings of the study made sense, but she had questions regarding the study methodology.
“I would intuitively think that steroid-associated complications are a major cause of health care use in GVHD patients and it’s interesting to see that there is emerging data to support this hypothesis,” HSCT specialist Hélène Schoemans, MD of the University of Leuven, Belgium, said in an interview.
She noted, however, that “it is surprising that the period of steroid initiation was the same for acute and chronic GVHD,” and questioned whether that anomalous finding could be due to the study’s definition of acute and chronic GVHD or to how the period from baseline to steroid initiation was defined.
The questions about the definitions and timing of therapy make it uncertain as to whether the complications reported were caused by steroids or by some other factor, she suggested.
The study was supported by Optum Inc. Dr. Bell is an employee of the company, and a paid consultant of Incyte. Dr. Schoemans has received travel expenses from Celgene, Abbvie, and Incyte; is part of the advisory boards for Incyte; and has received speakers fees from Novartis, Incyte, Jazz Pharmaceuticals, and Takeda.
FROM TCT 2021
Chronic GVHD therapies offer hope for treating refractory disease
Despite improvements in prevention of graft-versus-host disease, chronic GVHD still occurs in 10%-50% of patients who undergo an allogeneic hematopoietic stem cell transplant, and these patients may require prolonged treatment with multiple lines of therapy, said a hematologist and transplant researcher.
“More effective, less toxic therapies for chronic GVHD are needed,” Stephanie Lee, MD, MPH, from the Fred Hutchinson Cancer Research Center in Seattle said at the Transplant & Cellular Therapies Meetings.
Dr. Lee reviewed clinical trials for chronic GVHD at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
Although the incidence of chronic GVHD has gradually declined over the last 40 years and both relapse-free and overall survival following a chronic GVHD diagnosis have improved, “for patients who are diagnosed with chronic GVHD, they still will see many lines of therapy and many years of therapy,” she said.
Among 148 patients with chronic GVHD treated at her center, for example, 66% went on to two lines of therapy, 50% went on to three lines, 37% required four lines of therapy, and 20% needed five lines or more.
Salvage therapies for patients with chronic GVHD have evolved away from immunomodulators and immunosuppressants in the early 1990s, toward monoclonal antibodies such as rituximab in the early 2000s, to interleukin-2 and to tyrosine kinase inhibitors such as ruxolitinib (Jakafi) and ibrutinib (Imbruvica).
There are currently 36 agents that are FDA approved for at least one indication and can also be prescribed for the treatment of chronic GVHD, Dr. Lee noted.
Treatment goals
Dr. Lee laid out six goals for treating patients with chronic GVHD. They include:
- Controlling current signs and symptoms, measured by response rates and patient-reported outcomes
- Preventing further tissue and organ damage
- Minimizing toxicity
- Maintaining graft-versus-tumor effect
- Achieving graft tolerance and stopping immunosuppression
- Decreasing nonrelapse mortality and improving survival
Active trials
Dr. Lee identified 33 trials with chronic GVHD as an indication that are currently recruiting, and an additional 13 trials that are active but closed to recruiting. The trials can be generally grouped by mechanism of action, and involve agents targeting T-regulatory cells, B cells and/or B-cell receptor (BCR) signaling, monocytes/macrophages, costimulatory blockage, a proteasome inhibition, Janus kinase (JAK) 1/2 inhibitors, ROCK2 inhibitors, hedgehog pathway inhibition, cellular therapy, and organ-targeted therapy.
Most of the trials have overall response rate as the primary endpoint, and all but five are currently in phase 1 or 2. The currently active phase 3 trials include two with ibrutinib, one with the investigational agent itacitinib, one with ruxolitinib, and one with mesenchymal stem cells.
“I’ll note that, when results are reported, the denominator really matters for the overall response rate, especially if you’re talking about small trials, because if you require the patient to be treated with an agent for a certain period of time, and you take out all the people who didn’t make it to that time point, then your overall response rate looks better,” she said.
BTK inhibitors
The first-in-class Bruton tyrosine kinase (BTK) inhibitor ibrutinib was the first and thus far only agent approved by the Food and Drug Administration for chronic GVHD. The approval was based on a single-arm, multicenter trial with 42 patients.
The ORR in this trial was 69%, consisting of 31% complete responses and 38% partial responses, with a duration of response longer than 10 months in slightly more than half of all patients. In all, 24% of patients had improvement of symptoms in two consecutive visits, and 29% continued on ibrutinib at the time of the primary analysis in 2017.
Based on these promising results, acalabrutinib, which is more potent and selective for BTK than ibrutinib, with no effect on either platelets or natural killer cells, is currently under investigation in a phase 2 trial in 50 patients at a dose of 100 mg orally twice daily.
JAK1/2 inhibition
The JAK1 inhibitor itacitinib failed to meet its primary ORR endpoint in the phase 3 GRAVITAS-301 study, according to a press release, but the manufacturer (Incyte) said that it is continuing its commitment to JAK inhibitors with ruxolitinib, which has shown activity against acute, steroid-refractory GVHD, and is being explored for prevention of chronic GVHD in the randomized, phase 3 REACH3 study.
The trial met its primary endpoint for a higher ORR at week 24 with ruxolitinib versus best available therapy, at 49.7% versus 25.6%, respectively, which translated into an odds ratio for response with the JAK inhibitor of 2.99 (P < .0001).
Selective T-cell expansion
Efavaleukin alfa is an IL-2-mutated protein (mutein), with a mutation in the IL-2RB-binding portion of IL-2 causing increased selectivity for regulatory T-cell expansion. It is bound to an IgG-Fc domain that is itself mutated, with reduced Fc receptor binding and IgG effector function to give it a longer half life. This agent is being studied in a phase 1/2 trial in a subcutaneous formulation delivered every 1 or 2 weeks to 68 patients.
Monocyte/macrophage depletion
Axatilimab is a high-affinity antibody targeting colony stimulating factor–1 receptor (CSF-1R) expressed on monocytes and macrophages. By blocking CSF-1R, it depletes circulation of nonclassical monocytes and prevents the differentiation and survival of M2 macrophages in tissue.
It is currently being investigated 30 patients in a phase 1/2 study in an intravenous formulation delivered over 30 minutes every 2-4 weeks.
Hedgehog pathway inhibition
There is evidence suggesting that hedgehog pathway inhibition can lessen fibrosis. Glasdegib (Daurismo) a potent selective oral inhibitor of the hedgehog signaling pathway, is approved for use with low-dose cytarabine for patients with newly diagnosed acute myeloid leukemia aged older than 75 years or have comorbidities precluding intensive chemotherapy.
This agent is associated with drug intolerance because of muscle spasms, dysgeusia, and alopecia, however.
The drug is currently in phase 1/2 at a dose of 50 mg orally per day in 20 patients.
ROCK2 inhibition
Belumosudil (formerly KD025) “appears to rebalance the immune system,” Dr. Lee said. Investigators think that the drug dampens an autoaggressive inflammatory response by selective inhibition of ROCK2.
This drug has been studied in a dose-escalation study and a phase 2 trial, in which 132 participants were randomized to receive belumosudil 200 mg either once or twice daily.
At a median follow-up of 8 months, the ORR with belumosudil 200 mg once and twice daily was 73% and 74%, respectively. Similar results were seen in patients who had previously received either ruxolitinib or ibrutinib. High response rates were seen in patients with severe chronic GVHD, involvement of four or more organs and a refractory response to their last line of therapy.
Hard-to-manage patients
“We’re very hopeful for many of these agents, but we have to acknowledge that there are still many management dilemmas, patients that we just don’t really know what to do with,” Dr. Lee said. “These include patients who have bad sclerosis and fasciitis, nonhealing skin ulcers, bronchiolitis obliterans, serositis that can be very difficult to manage, severe keratoconjunctivitis that can be eyesight threatening, nonhealing mouth ulcers, esophageal structures, and always patients who have frequent infections.
“We are hopeful that some these agents will be useful for our patients who have severe manifestations, but often the number of patients with these manifestations in the trials is too low to say something specific about them,” she added.
‘Exciting time’
“It’s an exciting time because there are a lot of different drugs that are being studied for chronic GVHD,” commented Betty Hamilton, MD, a hematologist/oncologist at the Cleveland Clinic.
“I think that where the field is going in terms of treatment is recognizing that chronic GVHD is a pretty heterogeneous disease, and we have to learn even more about the underlying biologic pathways to be able to determine which class of drugs to use and when,” she said in an interview.
She agreed with Dr. Lee that the goals of treating patients with chronic GVHD include improving symptoms and quality, preventing progression, ideally tapering patients off immunosuppression, and achieving a balance between preventing negative consequences of GVHD while maintain the benefits of a graft-versus-leukemia effect.
“In our center, drug choice is based on physician preference and comfort with how often they’ve used the drug, patients’ comorbidities, toxicities of the drug, and logistical considerations,” Dr. Hamilton said.
Dr. Lee disclosed consulting activities for Pfizer and Kadmon, travel and lodging from Amgen, and research funding from those companies and others. Dr. Hamilton disclosed consulting for Syndax and Incyte.
Despite improvements in prevention of graft-versus-host disease, chronic GVHD still occurs in 10%-50% of patients who undergo an allogeneic hematopoietic stem cell transplant, and these patients may require prolonged treatment with multiple lines of therapy, said a hematologist and transplant researcher.
“More effective, less toxic therapies for chronic GVHD are needed,” Stephanie Lee, MD, MPH, from the Fred Hutchinson Cancer Research Center in Seattle said at the Transplant & Cellular Therapies Meetings.
Dr. Lee reviewed clinical trials for chronic GVHD at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
Although the incidence of chronic GVHD has gradually declined over the last 40 years and both relapse-free and overall survival following a chronic GVHD diagnosis have improved, “for patients who are diagnosed with chronic GVHD, they still will see many lines of therapy and many years of therapy,” she said.
Among 148 patients with chronic GVHD treated at her center, for example, 66% went on to two lines of therapy, 50% went on to three lines, 37% required four lines of therapy, and 20% needed five lines or more.
Salvage therapies for patients with chronic GVHD have evolved away from immunomodulators and immunosuppressants in the early 1990s, toward monoclonal antibodies such as rituximab in the early 2000s, to interleukin-2 and to tyrosine kinase inhibitors such as ruxolitinib (Jakafi) and ibrutinib (Imbruvica).
There are currently 36 agents that are FDA approved for at least one indication and can also be prescribed for the treatment of chronic GVHD, Dr. Lee noted.
Treatment goals
Dr. Lee laid out six goals for treating patients with chronic GVHD. They include:
- Controlling current signs and symptoms, measured by response rates and patient-reported outcomes
- Preventing further tissue and organ damage
- Minimizing toxicity
- Maintaining graft-versus-tumor effect
- Achieving graft tolerance and stopping immunosuppression
- Decreasing nonrelapse mortality and improving survival
Active trials
Dr. Lee identified 33 trials with chronic GVHD as an indication that are currently recruiting, and an additional 13 trials that are active but closed to recruiting. The trials can be generally grouped by mechanism of action, and involve agents targeting T-regulatory cells, B cells and/or B-cell receptor (BCR) signaling, monocytes/macrophages, costimulatory blockage, a proteasome inhibition, Janus kinase (JAK) 1/2 inhibitors, ROCK2 inhibitors, hedgehog pathway inhibition, cellular therapy, and organ-targeted therapy.
Most of the trials have overall response rate as the primary endpoint, and all but five are currently in phase 1 or 2. The currently active phase 3 trials include two with ibrutinib, one with the investigational agent itacitinib, one with ruxolitinib, and one with mesenchymal stem cells.
“I’ll note that, when results are reported, the denominator really matters for the overall response rate, especially if you’re talking about small trials, because if you require the patient to be treated with an agent for a certain period of time, and you take out all the people who didn’t make it to that time point, then your overall response rate looks better,” she said.
BTK inhibitors
The first-in-class Bruton tyrosine kinase (BTK) inhibitor ibrutinib was the first and thus far only agent approved by the Food and Drug Administration for chronic GVHD. The approval was based on a single-arm, multicenter trial with 42 patients.
The ORR in this trial was 69%, consisting of 31% complete responses and 38% partial responses, with a duration of response longer than 10 months in slightly more than half of all patients. In all, 24% of patients had improvement of symptoms in two consecutive visits, and 29% continued on ibrutinib at the time of the primary analysis in 2017.
Based on these promising results, acalabrutinib, which is more potent and selective for BTK than ibrutinib, with no effect on either platelets or natural killer cells, is currently under investigation in a phase 2 trial in 50 patients at a dose of 100 mg orally twice daily.
JAK1/2 inhibition
The JAK1 inhibitor itacitinib failed to meet its primary ORR endpoint in the phase 3 GRAVITAS-301 study, according to a press release, but the manufacturer (Incyte) said that it is continuing its commitment to JAK inhibitors with ruxolitinib, which has shown activity against acute, steroid-refractory GVHD, and is being explored for prevention of chronic GVHD in the randomized, phase 3 REACH3 study.
The trial met its primary endpoint for a higher ORR at week 24 with ruxolitinib versus best available therapy, at 49.7% versus 25.6%, respectively, which translated into an odds ratio for response with the JAK inhibitor of 2.99 (P < .0001).
Selective T-cell expansion
Efavaleukin alfa is an IL-2-mutated protein (mutein), with a mutation in the IL-2RB-binding portion of IL-2 causing increased selectivity for regulatory T-cell expansion. It is bound to an IgG-Fc domain that is itself mutated, with reduced Fc receptor binding and IgG effector function to give it a longer half life. This agent is being studied in a phase 1/2 trial in a subcutaneous formulation delivered every 1 or 2 weeks to 68 patients.
Monocyte/macrophage depletion
Axatilimab is a high-affinity antibody targeting colony stimulating factor–1 receptor (CSF-1R) expressed on monocytes and macrophages. By blocking CSF-1R, it depletes circulation of nonclassical monocytes and prevents the differentiation and survival of M2 macrophages in tissue.
It is currently being investigated 30 patients in a phase 1/2 study in an intravenous formulation delivered over 30 minutes every 2-4 weeks.
Hedgehog pathway inhibition
There is evidence suggesting that hedgehog pathway inhibition can lessen fibrosis. Glasdegib (Daurismo) a potent selective oral inhibitor of the hedgehog signaling pathway, is approved for use with low-dose cytarabine for patients with newly diagnosed acute myeloid leukemia aged older than 75 years or have comorbidities precluding intensive chemotherapy.
This agent is associated with drug intolerance because of muscle spasms, dysgeusia, and alopecia, however.
The drug is currently in phase 1/2 at a dose of 50 mg orally per day in 20 patients.
ROCK2 inhibition
Belumosudil (formerly KD025) “appears to rebalance the immune system,” Dr. Lee said. Investigators think that the drug dampens an autoaggressive inflammatory response by selective inhibition of ROCK2.
This drug has been studied in a dose-escalation study and a phase 2 trial, in which 132 participants were randomized to receive belumosudil 200 mg either once or twice daily.
At a median follow-up of 8 months, the ORR with belumosudil 200 mg once and twice daily was 73% and 74%, respectively. Similar results were seen in patients who had previously received either ruxolitinib or ibrutinib. High response rates were seen in patients with severe chronic GVHD, involvement of four or more organs and a refractory response to their last line of therapy.
Hard-to-manage patients
“We’re very hopeful for many of these agents, but we have to acknowledge that there are still many management dilemmas, patients that we just don’t really know what to do with,” Dr. Lee said. “These include patients who have bad sclerosis and fasciitis, nonhealing skin ulcers, bronchiolitis obliterans, serositis that can be very difficult to manage, severe keratoconjunctivitis that can be eyesight threatening, nonhealing mouth ulcers, esophageal structures, and always patients who have frequent infections.
“We are hopeful that some these agents will be useful for our patients who have severe manifestations, but often the number of patients with these manifestations in the trials is too low to say something specific about them,” she added.
‘Exciting time’
“It’s an exciting time because there are a lot of different drugs that are being studied for chronic GVHD,” commented Betty Hamilton, MD, a hematologist/oncologist at the Cleveland Clinic.
“I think that where the field is going in terms of treatment is recognizing that chronic GVHD is a pretty heterogeneous disease, and we have to learn even more about the underlying biologic pathways to be able to determine which class of drugs to use and when,” she said in an interview.
She agreed with Dr. Lee that the goals of treating patients with chronic GVHD include improving symptoms and quality, preventing progression, ideally tapering patients off immunosuppression, and achieving a balance between preventing negative consequences of GVHD while maintain the benefits of a graft-versus-leukemia effect.
“In our center, drug choice is based on physician preference and comfort with how often they’ve used the drug, patients’ comorbidities, toxicities of the drug, and logistical considerations,” Dr. Hamilton said.
Dr. Lee disclosed consulting activities for Pfizer and Kadmon, travel and lodging from Amgen, and research funding from those companies and others. Dr. Hamilton disclosed consulting for Syndax and Incyte.
Despite improvements in prevention of graft-versus-host disease, chronic GVHD still occurs in 10%-50% of patients who undergo an allogeneic hematopoietic stem cell transplant, and these patients may require prolonged treatment with multiple lines of therapy, said a hematologist and transplant researcher.
“More effective, less toxic therapies for chronic GVHD are needed,” Stephanie Lee, MD, MPH, from the Fred Hutchinson Cancer Research Center in Seattle said at the Transplant & Cellular Therapies Meetings.
Dr. Lee reviewed clinical trials for chronic GVHD at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
Although the incidence of chronic GVHD has gradually declined over the last 40 years and both relapse-free and overall survival following a chronic GVHD diagnosis have improved, “for patients who are diagnosed with chronic GVHD, they still will see many lines of therapy and many years of therapy,” she said.
Among 148 patients with chronic GVHD treated at her center, for example, 66% went on to two lines of therapy, 50% went on to three lines, 37% required four lines of therapy, and 20% needed five lines or more.
Salvage therapies for patients with chronic GVHD have evolved away from immunomodulators and immunosuppressants in the early 1990s, toward monoclonal antibodies such as rituximab in the early 2000s, to interleukin-2 and to tyrosine kinase inhibitors such as ruxolitinib (Jakafi) and ibrutinib (Imbruvica).
There are currently 36 agents that are FDA approved for at least one indication and can also be prescribed for the treatment of chronic GVHD, Dr. Lee noted.
Treatment goals
Dr. Lee laid out six goals for treating patients with chronic GVHD. They include:
- Controlling current signs and symptoms, measured by response rates and patient-reported outcomes
- Preventing further tissue and organ damage
- Minimizing toxicity
- Maintaining graft-versus-tumor effect
- Achieving graft tolerance and stopping immunosuppression
- Decreasing nonrelapse mortality and improving survival
Active trials
Dr. Lee identified 33 trials with chronic GVHD as an indication that are currently recruiting, and an additional 13 trials that are active but closed to recruiting. The trials can be generally grouped by mechanism of action, and involve agents targeting T-regulatory cells, B cells and/or B-cell receptor (BCR) signaling, monocytes/macrophages, costimulatory blockage, a proteasome inhibition, Janus kinase (JAK) 1/2 inhibitors, ROCK2 inhibitors, hedgehog pathway inhibition, cellular therapy, and organ-targeted therapy.
Most of the trials have overall response rate as the primary endpoint, and all but five are currently in phase 1 or 2. The currently active phase 3 trials include two with ibrutinib, one with the investigational agent itacitinib, one with ruxolitinib, and one with mesenchymal stem cells.
“I’ll note that, when results are reported, the denominator really matters for the overall response rate, especially if you’re talking about small trials, because if you require the patient to be treated with an agent for a certain period of time, and you take out all the people who didn’t make it to that time point, then your overall response rate looks better,” she said.
BTK inhibitors
The first-in-class Bruton tyrosine kinase (BTK) inhibitor ibrutinib was the first and thus far only agent approved by the Food and Drug Administration for chronic GVHD. The approval was based on a single-arm, multicenter trial with 42 patients.
The ORR in this trial was 69%, consisting of 31% complete responses and 38% partial responses, with a duration of response longer than 10 months in slightly more than half of all patients. In all, 24% of patients had improvement of symptoms in two consecutive visits, and 29% continued on ibrutinib at the time of the primary analysis in 2017.
Based on these promising results, acalabrutinib, which is more potent and selective for BTK than ibrutinib, with no effect on either platelets or natural killer cells, is currently under investigation in a phase 2 trial in 50 patients at a dose of 100 mg orally twice daily.
JAK1/2 inhibition
The JAK1 inhibitor itacitinib failed to meet its primary ORR endpoint in the phase 3 GRAVITAS-301 study, according to a press release, but the manufacturer (Incyte) said that it is continuing its commitment to JAK inhibitors with ruxolitinib, which has shown activity against acute, steroid-refractory GVHD, and is being explored for prevention of chronic GVHD in the randomized, phase 3 REACH3 study.
The trial met its primary endpoint for a higher ORR at week 24 with ruxolitinib versus best available therapy, at 49.7% versus 25.6%, respectively, which translated into an odds ratio for response with the JAK inhibitor of 2.99 (P < .0001).
Selective T-cell expansion
Efavaleukin alfa is an IL-2-mutated protein (mutein), with a mutation in the IL-2RB-binding portion of IL-2 causing increased selectivity for regulatory T-cell expansion. It is bound to an IgG-Fc domain that is itself mutated, with reduced Fc receptor binding and IgG effector function to give it a longer half life. This agent is being studied in a phase 1/2 trial in a subcutaneous formulation delivered every 1 or 2 weeks to 68 patients.
Monocyte/macrophage depletion
Axatilimab is a high-affinity antibody targeting colony stimulating factor–1 receptor (CSF-1R) expressed on monocytes and macrophages. By blocking CSF-1R, it depletes circulation of nonclassical monocytes and prevents the differentiation and survival of M2 macrophages in tissue.
It is currently being investigated 30 patients in a phase 1/2 study in an intravenous formulation delivered over 30 minutes every 2-4 weeks.
Hedgehog pathway inhibition
There is evidence suggesting that hedgehog pathway inhibition can lessen fibrosis. Glasdegib (Daurismo) a potent selective oral inhibitor of the hedgehog signaling pathway, is approved for use with low-dose cytarabine for patients with newly diagnosed acute myeloid leukemia aged older than 75 years or have comorbidities precluding intensive chemotherapy.
This agent is associated with drug intolerance because of muscle spasms, dysgeusia, and alopecia, however.
The drug is currently in phase 1/2 at a dose of 50 mg orally per day in 20 patients.
ROCK2 inhibition
Belumosudil (formerly KD025) “appears to rebalance the immune system,” Dr. Lee said. Investigators think that the drug dampens an autoaggressive inflammatory response by selective inhibition of ROCK2.
This drug has been studied in a dose-escalation study and a phase 2 trial, in which 132 participants were randomized to receive belumosudil 200 mg either once or twice daily.
At a median follow-up of 8 months, the ORR with belumosudil 200 mg once and twice daily was 73% and 74%, respectively. Similar results were seen in patients who had previously received either ruxolitinib or ibrutinib. High response rates were seen in patients with severe chronic GVHD, involvement of four or more organs and a refractory response to their last line of therapy.
Hard-to-manage patients
“We’re very hopeful for many of these agents, but we have to acknowledge that there are still many management dilemmas, patients that we just don’t really know what to do with,” Dr. Lee said. “These include patients who have bad sclerosis and fasciitis, nonhealing skin ulcers, bronchiolitis obliterans, serositis that can be very difficult to manage, severe keratoconjunctivitis that can be eyesight threatening, nonhealing mouth ulcers, esophageal structures, and always patients who have frequent infections.
“We are hopeful that some these agents will be useful for our patients who have severe manifestations, but often the number of patients with these manifestations in the trials is too low to say something specific about them,” she added.
‘Exciting time’
“It’s an exciting time because there are a lot of different drugs that are being studied for chronic GVHD,” commented Betty Hamilton, MD, a hematologist/oncologist at the Cleveland Clinic.
“I think that where the field is going in terms of treatment is recognizing that chronic GVHD is a pretty heterogeneous disease, and we have to learn even more about the underlying biologic pathways to be able to determine which class of drugs to use and when,” she said in an interview.
She agreed with Dr. Lee that the goals of treating patients with chronic GVHD include improving symptoms and quality, preventing progression, ideally tapering patients off immunosuppression, and achieving a balance between preventing negative consequences of GVHD while maintain the benefits of a graft-versus-leukemia effect.
“In our center, drug choice is based on physician preference and comfort with how often they’ve used the drug, patients’ comorbidities, toxicities of the drug, and logistical considerations,” Dr. Hamilton said.
Dr. Lee disclosed consulting activities for Pfizer and Kadmon, travel and lodging from Amgen, and research funding from those companies and others. Dr. Hamilton disclosed consulting for Syndax and Incyte.
FROM TCT 2021
MRI-guided prostate biopsy prevails in PRECISE trial
Here’s welcome news for men of a certain age: New results support a less invasive approach to investigations for suspicion of prostate cancer.
An approach using MRI of the prostate followed by targeted biopsy (TB) in men with images suggesting a high risk beat the conventional approach of using transrectal ultrasound (TRUS)–guided 12-core systematic biopsy.
The results come from the randomized phase 3 PRECISION trial and were published online in JAMA Oncology.
“What the trial showed is that by taking an imaging-first strategy, you could reduce the number of patients needing a biopsy by about 40% and actually find more significant cancer (35% vs. 30%) and reduce the diagnosis of grade group 1 cancers that we don’t want to find by more than half,” lead author Laurence Klotz, CM, MD, of the Sunnybrook Health Sciences Centre in Toronto commented in an interview with this news organization.
These results from the PRECISE trial support and slightly improve upon findings from the European-based PRECISION trial.
The European trial had already provided “compelling evidence in favor of MRI and targeted biopsy,” noted Olivier Rouvière, MD, PhD, from the University of Lyon (France) in an accompanying editorial. But he argued that it was worth duplicating the trial, as “it must not be forgotten that, in science, testing the robustness of an effect and the factors influencing it is are as important as demonstrating this effect in the first place.”
The results from both trials suggest that, instead of replacing TRUS biopsy entirely, MRI results could be used to guide patients to the appropriate diagnostic pathway, Dr. Rouvière commented.
“Using only MRI findings to decide which patients should undergo biopsy is probably insufficient,” he added. “Most likely, MRI findings will be used in conjunction with other biomarkers such as PSA [prostate-specific antigen] density to select, among the patients with positive MRI findings, those who need targeted biopsy (and those who may safely avoid it), and among the patients with negative MRI findings, those who may still deserve systematic biopsy,” he wrote.
Details of PRECISE findings
The Canadian PRECISE study was developed as a noninferiority trial in coordination with the European PRECISION study, but the Canadian version had several additional features. It added risk-based eligibility, systematic follow-up of all patients for 2 years, a repeat MRI in all untreated patients, investigation of fluid- and tissue-based biomarkers in the cohort, and an economic analysis.
PRECISE was conducted in five Canadian academic health sciences centers from January 2017 through November 2019. A total of 453 biopsy-naive men with a clinical suspicion of prostate cancer who were referred for prostate biopsy were enrolled. Of this group, 421 were evaluable per protocol.
The investigators defined clinical suspicion as a 5% or greater chance of grade group 2 or greater prostate cancer. Patients were also required to have serum prostate-specific antigen PSA levels of 20 ng/mL or less and no contraindication to MRI.
The patients randomly assigned to MRI underwent an MRI-targeted biopsy (MRI-TB) only if a lesion with a Prostate Imaging Reporting and Data System (PI-RADS) score of 3 or greater was identified, whereas all men in the other arm of the trial underwent a systematic TRUS-guided 12-core biopsy.
The MRI approach identified more clinically significant cancers. Grade 2 or higher tumors were found in 79 (35%) of 227 men allocated to MRI-TB vs. 67 (30%) of 225 men who underwent TRUS biopsy.
MRI also reduced the need for a biopsy, allowing many men to avoid the associated pain, discomfort, and infection risks. Of 221 men who were randomly assigned to the MRI group, 83 (37%) had a negative MRI result and avoided biopsy. In contrast, all men in the TRUS group had a biopsy.
In addition, MRI was associated with a marked reduction in the diagnosis of clinically insignificant International Society of Urological Pathology (ISUP) grade group 1 cancers (10% with MRI-TB vs. 22% with TRUS). Detection of such early cancers, under conventional protocols, often leads to unnecessary therapies or invasive procedures with significant side effects.
These results led the researchers to conclude that the strategy of MRI followed by MRI-guided biopsy only in men at risk of prostate cancer “offers substantial advantages over an initial systematic biopsy.”
The MRI strategy “results in similar detection rates of clinically significant prostate cancer … while avoiding biopsy in more than one-third of men and reducing the diagnosis of clinically insignificant cancer,” the investigators point out.
The investigators acknowledged that the performance of the MRI-targeted biopsy varied between centers, with differences in both positive MRI rates and target biopsy yields.
“This difference occurred despite the fact that all MRIs were reviewed, and biopsies performed, by experienced radiologists or urologists. This underscores the need for quality control measures to enable the broad application of MRI,” they write.
What’s next?
Dr. Klotz noted that an important addition to PRECISE is the planned follow-up of patients who did not receive treatment on study.
“We know MRI is not perfect, so what happens to the guys who avoid the biopsy? How much at risk are they of having something missed? So the only way to know that really is with long-term follow-up on the patients,” he said in an interview.
In addition to repeating MRI at 2 years, the investigators plan to follow these patients for up to 8 years.
“But certainly, compared to the traditional strategy of systematic biopsy, this looks a lot better,” Dr. Klotz commented.
In his editorial, Dr. Rouvière commented that the intersite variability seen in the study “sounds like a warning to the MRI pathway.”
“Considering that all participating centers were experienced high-volume tertiary centers, the intersite variability is probably much higher among less experienced centers,” he wrote. “Thus, whatever the diagnostic pathway, continuing education and quality insurance programs will become major issues in the future, not only for prostate MRI interpretation, but also for targeted biopsy, which does have a learning curve, even with magnetic resonance and ultrasound fusion systems, and even for systematic biopsy.”
PRECISE was funded by the Ontario Institute of Cancer Research and Prostate Cancer Canada. Dr. Klotz and Dr. Rouvière have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Here’s welcome news for men of a certain age: New results support a less invasive approach to investigations for suspicion of prostate cancer.
An approach using MRI of the prostate followed by targeted biopsy (TB) in men with images suggesting a high risk beat the conventional approach of using transrectal ultrasound (TRUS)–guided 12-core systematic biopsy.
The results come from the randomized phase 3 PRECISION trial and were published online in JAMA Oncology.
“What the trial showed is that by taking an imaging-first strategy, you could reduce the number of patients needing a biopsy by about 40% and actually find more significant cancer (35% vs. 30%) and reduce the diagnosis of grade group 1 cancers that we don’t want to find by more than half,” lead author Laurence Klotz, CM, MD, of the Sunnybrook Health Sciences Centre in Toronto commented in an interview with this news organization.
These results from the PRECISE trial support and slightly improve upon findings from the European-based PRECISION trial.
The European trial had already provided “compelling evidence in favor of MRI and targeted biopsy,” noted Olivier Rouvière, MD, PhD, from the University of Lyon (France) in an accompanying editorial. But he argued that it was worth duplicating the trial, as “it must not be forgotten that, in science, testing the robustness of an effect and the factors influencing it is are as important as demonstrating this effect in the first place.”
The results from both trials suggest that, instead of replacing TRUS biopsy entirely, MRI results could be used to guide patients to the appropriate diagnostic pathway, Dr. Rouvière commented.
“Using only MRI findings to decide which patients should undergo biopsy is probably insufficient,” he added. “Most likely, MRI findings will be used in conjunction with other biomarkers such as PSA [prostate-specific antigen] density to select, among the patients with positive MRI findings, those who need targeted biopsy (and those who may safely avoid it), and among the patients with negative MRI findings, those who may still deserve systematic biopsy,” he wrote.
Details of PRECISE findings
The Canadian PRECISE study was developed as a noninferiority trial in coordination with the European PRECISION study, but the Canadian version had several additional features. It added risk-based eligibility, systematic follow-up of all patients for 2 years, a repeat MRI in all untreated patients, investigation of fluid- and tissue-based biomarkers in the cohort, and an economic analysis.
PRECISE was conducted in five Canadian academic health sciences centers from January 2017 through November 2019. A total of 453 biopsy-naive men with a clinical suspicion of prostate cancer who were referred for prostate biopsy were enrolled. Of this group, 421 were evaluable per protocol.
The investigators defined clinical suspicion as a 5% or greater chance of grade group 2 or greater prostate cancer. Patients were also required to have serum prostate-specific antigen PSA levels of 20 ng/mL or less and no contraindication to MRI.
The patients randomly assigned to MRI underwent an MRI-targeted biopsy (MRI-TB) only if a lesion with a Prostate Imaging Reporting and Data System (PI-RADS) score of 3 or greater was identified, whereas all men in the other arm of the trial underwent a systematic TRUS-guided 12-core biopsy.
The MRI approach identified more clinically significant cancers. Grade 2 or higher tumors were found in 79 (35%) of 227 men allocated to MRI-TB vs. 67 (30%) of 225 men who underwent TRUS biopsy.
MRI also reduced the need for a biopsy, allowing many men to avoid the associated pain, discomfort, and infection risks. Of 221 men who were randomly assigned to the MRI group, 83 (37%) had a negative MRI result and avoided biopsy. In contrast, all men in the TRUS group had a biopsy.
In addition, MRI was associated with a marked reduction in the diagnosis of clinically insignificant International Society of Urological Pathology (ISUP) grade group 1 cancers (10% with MRI-TB vs. 22% with TRUS). Detection of such early cancers, under conventional protocols, often leads to unnecessary therapies or invasive procedures with significant side effects.
These results led the researchers to conclude that the strategy of MRI followed by MRI-guided biopsy only in men at risk of prostate cancer “offers substantial advantages over an initial systematic biopsy.”
The MRI strategy “results in similar detection rates of clinically significant prostate cancer … while avoiding biopsy in more than one-third of men and reducing the diagnosis of clinically insignificant cancer,” the investigators point out.
The investigators acknowledged that the performance of the MRI-targeted biopsy varied between centers, with differences in both positive MRI rates and target biopsy yields.
“This difference occurred despite the fact that all MRIs were reviewed, and biopsies performed, by experienced radiologists or urologists. This underscores the need for quality control measures to enable the broad application of MRI,” they write.
What’s next?
Dr. Klotz noted that an important addition to PRECISE is the planned follow-up of patients who did not receive treatment on study.
“We know MRI is not perfect, so what happens to the guys who avoid the biopsy? How much at risk are they of having something missed? So the only way to know that really is with long-term follow-up on the patients,” he said in an interview.
In addition to repeating MRI at 2 years, the investigators plan to follow these patients for up to 8 years.
“But certainly, compared to the traditional strategy of systematic biopsy, this looks a lot better,” Dr. Klotz commented.
In his editorial, Dr. Rouvière commented that the intersite variability seen in the study “sounds like a warning to the MRI pathway.”
“Considering that all participating centers were experienced high-volume tertiary centers, the intersite variability is probably much higher among less experienced centers,” he wrote. “Thus, whatever the diagnostic pathway, continuing education and quality insurance programs will become major issues in the future, not only for prostate MRI interpretation, but also for targeted biopsy, which does have a learning curve, even with magnetic resonance and ultrasound fusion systems, and even for systematic biopsy.”
PRECISE was funded by the Ontario Institute of Cancer Research and Prostate Cancer Canada. Dr. Klotz and Dr. Rouvière have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Here’s welcome news for men of a certain age: New results support a less invasive approach to investigations for suspicion of prostate cancer.
An approach using MRI of the prostate followed by targeted biopsy (TB) in men with images suggesting a high risk beat the conventional approach of using transrectal ultrasound (TRUS)–guided 12-core systematic biopsy.
The results come from the randomized phase 3 PRECISION trial and were published online in JAMA Oncology.
“What the trial showed is that by taking an imaging-first strategy, you could reduce the number of patients needing a biopsy by about 40% and actually find more significant cancer (35% vs. 30%) and reduce the diagnosis of grade group 1 cancers that we don’t want to find by more than half,” lead author Laurence Klotz, CM, MD, of the Sunnybrook Health Sciences Centre in Toronto commented in an interview with this news organization.
These results from the PRECISE trial support and slightly improve upon findings from the European-based PRECISION trial.
The European trial had already provided “compelling evidence in favor of MRI and targeted biopsy,” noted Olivier Rouvière, MD, PhD, from the University of Lyon (France) in an accompanying editorial. But he argued that it was worth duplicating the trial, as “it must not be forgotten that, in science, testing the robustness of an effect and the factors influencing it is are as important as demonstrating this effect in the first place.”
The results from both trials suggest that, instead of replacing TRUS biopsy entirely, MRI results could be used to guide patients to the appropriate diagnostic pathway, Dr. Rouvière commented.
“Using only MRI findings to decide which patients should undergo biopsy is probably insufficient,” he added. “Most likely, MRI findings will be used in conjunction with other biomarkers such as PSA [prostate-specific antigen] density to select, among the patients with positive MRI findings, those who need targeted biopsy (and those who may safely avoid it), and among the patients with negative MRI findings, those who may still deserve systematic biopsy,” he wrote.
Details of PRECISE findings
The Canadian PRECISE study was developed as a noninferiority trial in coordination with the European PRECISION study, but the Canadian version had several additional features. It added risk-based eligibility, systematic follow-up of all patients for 2 years, a repeat MRI in all untreated patients, investigation of fluid- and tissue-based biomarkers in the cohort, and an economic analysis.
PRECISE was conducted in five Canadian academic health sciences centers from January 2017 through November 2019. A total of 453 biopsy-naive men with a clinical suspicion of prostate cancer who were referred for prostate biopsy were enrolled. Of this group, 421 were evaluable per protocol.
The investigators defined clinical suspicion as a 5% or greater chance of grade group 2 or greater prostate cancer. Patients were also required to have serum prostate-specific antigen PSA levels of 20 ng/mL or less and no contraindication to MRI.
The patients randomly assigned to MRI underwent an MRI-targeted biopsy (MRI-TB) only if a lesion with a Prostate Imaging Reporting and Data System (PI-RADS) score of 3 or greater was identified, whereas all men in the other arm of the trial underwent a systematic TRUS-guided 12-core biopsy.
The MRI approach identified more clinically significant cancers. Grade 2 or higher tumors were found in 79 (35%) of 227 men allocated to MRI-TB vs. 67 (30%) of 225 men who underwent TRUS biopsy.
MRI also reduced the need for a biopsy, allowing many men to avoid the associated pain, discomfort, and infection risks. Of 221 men who were randomly assigned to the MRI group, 83 (37%) had a negative MRI result and avoided biopsy. In contrast, all men in the TRUS group had a biopsy.
In addition, MRI was associated with a marked reduction in the diagnosis of clinically insignificant International Society of Urological Pathology (ISUP) grade group 1 cancers (10% with MRI-TB vs. 22% with TRUS). Detection of such early cancers, under conventional protocols, often leads to unnecessary therapies or invasive procedures with significant side effects.
These results led the researchers to conclude that the strategy of MRI followed by MRI-guided biopsy only in men at risk of prostate cancer “offers substantial advantages over an initial systematic biopsy.”
The MRI strategy “results in similar detection rates of clinically significant prostate cancer … while avoiding biopsy in more than one-third of men and reducing the diagnosis of clinically insignificant cancer,” the investigators point out.
The investigators acknowledged that the performance of the MRI-targeted biopsy varied between centers, with differences in both positive MRI rates and target biopsy yields.
“This difference occurred despite the fact that all MRIs were reviewed, and biopsies performed, by experienced radiologists or urologists. This underscores the need for quality control measures to enable the broad application of MRI,” they write.
What’s next?
Dr. Klotz noted that an important addition to PRECISE is the planned follow-up of patients who did not receive treatment on study.
“We know MRI is not perfect, so what happens to the guys who avoid the biopsy? How much at risk are they of having something missed? So the only way to know that really is with long-term follow-up on the patients,” he said in an interview.
In addition to repeating MRI at 2 years, the investigators plan to follow these patients for up to 8 years.
“But certainly, compared to the traditional strategy of systematic biopsy, this looks a lot better,” Dr. Klotz commented.
In his editorial, Dr. Rouvière commented that the intersite variability seen in the study “sounds like a warning to the MRI pathway.”
“Considering that all participating centers were experienced high-volume tertiary centers, the intersite variability is probably much higher among less experienced centers,” he wrote. “Thus, whatever the diagnostic pathway, continuing education and quality insurance programs will become major issues in the future, not only for prostate MRI interpretation, but also for targeted biopsy, which does have a learning curve, even with magnetic resonance and ultrasound fusion systems, and even for systematic biopsy.”
PRECISE was funded by the Ontario Institute of Cancer Research and Prostate Cancer Canada. Dr. Klotz and Dr. Rouvière have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.