User login
Our mission: To meet your needs
Many of you know me from my published research and books, national presentations, or CME broadcasts. What you might not know is how much I enjoy the clinical practice of psychiatry and helping seriously ill patients regain their wellness and return to social and vocational functioning.
Over the years, my patients have been my best teachers about disease burden, effective treatment strategies, coping with illness, and recovery. I never cease to be amazed at how vital a strong physician-patient alliance is to achieving successful clinical outcomes. My patients also have been my best source of research questions about the causes and treatment of psychiatric brain disorders. The more I learn, the more I am humbled by how much remains to be discovered in our field.
The mission of clinically focused journals such as Current Psychiatry is to inform readers about practical applications of the latest medical advances. That’s why many of you read Current Psychiatry from cover to cover. Psychiatry is a vibrant, rapidly growing medical specialty with a solid neuroscience foundation and a promising future, but we face many unmet needs in daily practice. Most relate to the diagnosis and treatment of common psychiatric disorders.
Needed: Diagnostic clarity
The schema of psychiatric diagnosis needs to become anchored in scientific evidence, for example. DSMIV-TR’s clusters of inclusion and exclusion symptom-focused criteria are unsatisfactory and not validated by empiric biomedical findings. Many patients have multiple comorbidities, which raises questions about:
- which illness is primary or secondary
- what is the shared neurobiology
- why does a class of drugs approved for one diagnosis—such as selective serotonin reuptake inhibitors, atypical antipsychotics, or anticonvulsants—help many other symptoms or diagnoses?
When it comes to personality disorders, why do axis II symptoms emerge during an acute axis I onset and disappear when the axis I episode remits? Why, on the other hand, do the features of “real” axis II disorders endure for a lifetime? Why the distinction between axis I and II anyway?
Why do addictive disorders such as substance abuse plague so many of our patients? Why do symptoms such as insomnia, anxiety, dysphoria, or aggression occur in disparate psychiatric disorders such as anxiety, mood, psychotic, or personality disorders?
Every day these nosologic questions challenge us and influence our management decisions and prognostic formulations. Much research is needed because specific, effective treatment requires diagnostic clarity and validity.
Needed: More effective treatments
This brings us to a perennial unmet need: more effective treatments. Psychiatry is experiencing a relative drought of innovative biological and psychological treatments. Pharmacologic agents are approved for a limited number of DSM-IV-TR diagnoses, which has created an extensive “black market psychopharmacology.” Approved psychotropics are used for unapproved indications, in unapproved doses, and in unapproved combinations—particularly in child and adolescent psychiatry with its serious dearth of controlled studies. We urgently need:
- nondopaminergic approaches to schizophrenia (where so many clues point to glutamate pathways) that can effectively treat cognitive deficits and negative symptoms
- antidepressants with rapid onset of action, especially for patients with suicidal intent
- an unambiguously effective mood stabilizer which, as monotherapy, restores balance to all phases of bipolar disorder
- effective treatment for alcoholism and drug dependencies.
We also await breakthrough medications for brain disorders with no known treatments, such as antisocial behavior, hypochondriasis, autism, pedophilia, and dissociative disorders, to name a few. Finally, we need drugs designed to avoid metabolic complications and other serious illnesses that can disrupt quality of life and lead to premature death.
Needed: Medical care for the mentally ill
Speaking of early mortality—and its association with chronic psychosis, mood, and anxiety disorders—another unmet need is the integration of primary care and psychiatric care for public-sector patients whose nonpsychiatric medical needs are woefully neglected. The mental health system is widely described as “broken,” and to fix it we need to advocate tirelessly for a more rational system of comprehensive medical care for our seriously mentally ill patients.
We also need to advocate for parity in reimbursement for treating psychiatric brain disorders, to eliminate the stigma of having an emotional or behavioral ailment, and to end the shameful incarceration of the criminally mentally ill in lieu of proper and humane treatment in a medical setting.
What do you need?
Current Psychiatry will continue to bring you the latest advances regarding the causes and treatments of psychiatric illness. Please contact me by:
- e-mail ([email protected])
- or fax (201-391-2778) about your major unmet clinical needs. Besides publishing your letters, I pledge that we will listen to you and meet your needs. That, ultimately, is Current Psychiatry’s mission.
I look forward to hearing from you.

Many of you know me from my published research and books, national presentations, or CME broadcasts. What you might not know is how much I enjoy the clinical practice of psychiatry and helping seriously ill patients regain their wellness and return to social and vocational functioning.
Over the years, my patients have been my best teachers about disease burden, effective treatment strategies, coping with illness, and recovery. I never cease to be amazed at how vital a strong physician-patient alliance is to achieving successful clinical outcomes. My patients also have been my best source of research questions about the causes and treatment of psychiatric brain disorders. The more I learn, the more I am humbled by how much remains to be discovered in our field.
The mission of clinically focused journals such as Current Psychiatry is to inform readers about practical applications of the latest medical advances. That’s why many of you read Current Psychiatry from cover to cover. Psychiatry is a vibrant, rapidly growing medical specialty with a solid neuroscience foundation and a promising future, but we face many unmet needs in daily practice. Most relate to the diagnosis and treatment of common psychiatric disorders.
Needed: Diagnostic clarity
The schema of psychiatric diagnosis needs to become anchored in scientific evidence, for example. DSMIV-TR’s clusters of inclusion and exclusion symptom-focused criteria are unsatisfactory and not validated by empiric biomedical findings. Many patients have multiple comorbidities, which raises questions about:
- which illness is primary or secondary
- what is the shared neurobiology
- why does a class of drugs approved for one diagnosis—such as selective serotonin reuptake inhibitors, atypical antipsychotics, or anticonvulsants—help many other symptoms or diagnoses?
When it comes to personality disorders, why do axis II symptoms emerge during an acute axis I onset and disappear when the axis I episode remits? Why, on the other hand, do the features of “real” axis II disorders endure for a lifetime? Why the distinction between axis I and II anyway?
Why do addictive disorders such as substance abuse plague so many of our patients? Why do symptoms such as insomnia, anxiety, dysphoria, or aggression occur in disparate psychiatric disorders such as anxiety, mood, psychotic, or personality disorders?
Every day these nosologic questions challenge us and influence our management decisions and prognostic formulations. Much research is needed because specific, effective treatment requires diagnostic clarity and validity.
Needed: More effective treatments
This brings us to a perennial unmet need: more effective treatments. Psychiatry is experiencing a relative drought of innovative biological and psychological treatments. Pharmacologic agents are approved for a limited number of DSM-IV-TR diagnoses, which has created an extensive “black market psychopharmacology.” Approved psychotropics are used for unapproved indications, in unapproved doses, and in unapproved combinations—particularly in child and adolescent psychiatry with its serious dearth of controlled studies. We urgently need:
- nondopaminergic approaches to schizophrenia (where so many clues point to glutamate pathways) that can effectively treat cognitive deficits and negative symptoms
- antidepressants with rapid onset of action, especially for patients with suicidal intent
- an unambiguously effective mood stabilizer which, as monotherapy, restores balance to all phases of bipolar disorder
- effective treatment for alcoholism and drug dependencies.
We also await breakthrough medications for brain disorders with no known treatments, such as antisocial behavior, hypochondriasis, autism, pedophilia, and dissociative disorders, to name a few. Finally, we need drugs designed to avoid metabolic complications and other serious illnesses that can disrupt quality of life and lead to premature death.
Needed: Medical care for the mentally ill
Speaking of early mortality—and its association with chronic psychosis, mood, and anxiety disorders—another unmet need is the integration of primary care and psychiatric care for public-sector patients whose nonpsychiatric medical needs are woefully neglected. The mental health system is widely described as “broken,” and to fix it we need to advocate tirelessly for a more rational system of comprehensive medical care for our seriously mentally ill patients.
We also need to advocate for parity in reimbursement for treating psychiatric brain disorders, to eliminate the stigma of having an emotional or behavioral ailment, and to end the shameful incarceration of the criminally mentally ill in lieu of proper and humane treatment in a medical setting.
What do you need?
Current Psychiatry will continue to bring you the latest advances regarding the causes and treatments of psychiatric illness. Please contact me by:
- e-mail ([email protected])
- or fax (201-391-2778) about your major unmet clinical needs. Besides publishing your letters, I pledge that we will listen to you and meet your needs. That, ultimately, is Current Psychiatry’s mission.
I look forward to hearing from you.
Many of you know me from my published research and books, national presentations, or CME broadcasts. What you might not know is how much I enjoy the clinical practice of psychiatry and helping seriously ill patients regain their wellness and return to social and vocational functioning.
Over the years, my patients have been my best teachers about disease burden, effective treatment strategies, coping with illness, and recovery. I never cease to be amazed at how vital a strong physician-patient alliance is to achieving successful clinical outcomes. My patients also have been my best source of research questions about the causes and treatment of psychiatric brain disorders. The more I learn, the more I am humbled by how much remains to be discovered in our field.
The mission of clinically focused journals such as Current Psychiatry is to inform readers about practical applications of the latest medical advances. That’s why many of you read Current Psychiatry from cover to cover. Psychiatry is a vibrant, rapidly growing medical specialty with a solid neuroscience foundation and a promising future, but we face many unmet needs in daily practice. Most relate to the diagnosis and treatment of common psychiatric disorders.
Needed: Diagnostic clarity
The schema of psychiatric diagnosis needs to become anchored in scientific evidence, for example. DSMIV-TR’s clusters of inclusion and exclusion symptom-focused criteria are unsatisfactory and not validated by empiric biomedical findings. Many patients have multiple comorbidities, which raises questions about:
- which illness is primary or secondary
- what is the shared neurobiology
- why does a class of drugs approved for one diagnosis—such as selective serotonin reuptake inhibitors, atypical antipsychotics, or anticonvulsants—help many other symptoms or diagnoses?
When it comes to personality disorders, why do axis II symptoms emerge during an acute axis I onset and disappear when the axis I episode remits? Why, on the other hand, do the features of “real” axis II disorders endure for a lifetime? Why the distinction between axis I and II anyway?
Why do addictive disorders such as substance abuse plague so many of our patients? Why do symptoms such as insomnia, anxiety, dysphoria, or aggression occur in disparate psychiatric disorders such as anxiety, mood, psychotic, or personality disorders?
Every day these nosologic questions challenge us and influence our management decisions and prognostic formulations. Much research is needed because specific, effective treatment requires diagnostic clarity and validity.
Needed: More effective treatments
This brings us to a perennial unmet need: more effective treatments. Psychiatry is experiencing a relative drought of innovative biological and psychological treatments. Pharmacologic agents are approved for a limited number of DSM-IV-TR diagnoses, which has created an extensive “black market psychopharmacology.” Approved psychotropics are used for unapproved indications, in unapproved doses, and in unapproved combinations—particularly in child and adolescent psychiatry with its serious dearth of controlled studies. We urgently need:
- nondopaminergic approaches to schizophrenia (where so many clues point to glutamate pathways) that can effectively treat cognitive deficits and negative symptoms
- antidepressants with rapid onset of action, especially for patients with suicidal intent
- an unambiguously effective mood stabilizer which, as monotherapy, restores balance to all phases of bipolar disorder
- effective treatment for alcoholism and drug dependencies.
We also await breakthrough medications for brain disorders with no known treatments, such as antisocial behavior, hypochondriasis, autism, pedophilia, and dissociative disorders, to name a few. Finally, we need drugs designed to avoid metabolic complications and other serious illnesses that can disrupt quality of life and lead to premature death.
Needed: Medical care for the mentally ill
Speaking of early mortality—and its association with chronic psychosis, mood, and anxiety disorders—another unmet need is the integration of primary care and psychiatric care for public-sector patients whose nonpsychiatric medical needs are woefully neglected. The mental health system is widely described as “broken,” and to fix it we need to advocate tirelessly for a more rational system of comprehensive medical care for our seriously mentally ill patients.
We also need to advocate for parity in reimbursement for treating psychiatric brain disorders, to eliminate the stigma of having an emotional or behavioral ailment, and to end the shameful incarceration of the criminally mentally ill in lieu of proper and humane treatment in a medical setting.
What do you need?
Current Psychiatry will continue to bring you the latest advances regarding the causes and treatments of psychiatric illness. Please contact me by:
- e-mail ([email protected])
- or fax (201-391-2778) about your major unmet clinical needs. Besides publishing your letters, I pledge that we will listen to you and meet your needs. That, ultimately, is Current Psychiatry’s mission.
I look forward to hearing from you.
Schizophrenia is psychotic bipolar disorder? What a polarizing idea!
A hundred years ago, KraepelinDrs. Lake and Hurwitz for highlighting the diagnostic and treatment errors in a bipolar patient with severe psychotic features who was misdiagnosed as having schizophrenia. Errors such as this were common with DSM I and II but declined with the more reliable diagnostic schemas of DSM III and IV. I am puzzled, however, by their leap to the radical conclusion that schizophrenia does not exist and that all patients diagnosed with schizophrenia have psychotic bipolar disorder. This is not as egregious as Szasz’ absurd proclamation 4 decades ago that schizophrenia is a “myth,” but it is a significant scientific “transgression,” given the evidence that distinguishes schizophrenia from bipolar disorder.
Symptoms. Beyond a doubt, these two brain diseases have overlapping clinical features, pharmacotherapies, and even outcomes in a subgroup of patients. However, these diseases have major differences, as outlined in the accompanying table.
Brain anomalies. Neuroimaging studies indicate that schizophrenia is associated with more-severe and pervasive morphologic brain anomalies (dysplasia and hypoplasia) than bipolar disorder, although some bipolar patients have reduced cerebral and frontal volumes and marked cognitive deficits.3 Progressive neuro tissue loss has been observed early in schizophrenia but not in bipolar disorder.
Recent genetic studies indicate that several genes are found exclusively in schizophrenia or in bipolar disorder cohorts,4 but some are shared by both disorders and may be related to delusional symptoms.5 Familial transmission appears to differ: transgenerational studies find an abundance of mood disorders in family members of bipolar probands but relatively sparse occurrence of psychosis in families of probands with schizophrenia.
Table
Symptom differences between schizophrenia and bipolar disorder
| Symptom | Schizophrenia | Bipolar disorder |
|---|---|---|
| Psychosis | Auditory hallucinations and bizarre delusions are more common | Grandiosity is more common |
| Paranoia | Occurs in both, but more systematic in schizophrenia | |
| Core psychopathology | Far more negative symptoms and cognition dysfunction | Far more mood lability and affective cyclicity |
| Thought disorder | Far more disorganized and derailed thoughts | More likely to have racing thoughts and flight of ideas |
| Between-episode interpersonal skills | Withdrawn, alogic, seclusive | Much more interactive and verbal |
Treatment. There is no doubt that monotherapy with antipsychotics (old and new) has similar efficacy6 in schizophrenia and bipolar mania (and even in bipolar depression, with some atypicals7). However, there is minimal, if any, evidence that monotherapy mood stabilizers (lithium or anti-convulsants) have any tangible efficacy in schizophrenia. Electroconvulsive therapy is remarkably efficious in all phases of bipolar disorder but of dubious, if any, lasting benefit in schizophrenia.
Course. Both the premorbid history and post-treatment functional outcome tend to be more favorable in patients with bipolar disorder than schizophrenia. Most patients with schizophrenia experience significant clinical, social, and vocational deterioration, compared with a relative minority of bipolar patients.
In summary, schizophrenia and bipolar disorder are clearly distinct in their pure forms, although many patients have varying mixtures of both. Schizoaffective disorder is one of the most extensively investigated. Although many scholars have studied schizoaffective disorder, the evidence defies lumping it with either end of the continuum.
I agree with Drs. Lake and Hurwitz that most cases of schizoaffective disorder, especially the “schizomanic” type, are probably bipolar disorder with severe psychotic features. To assert, however, that schizophrenia does not exist at all and should be reclassified as bipolar disorder with psychotic features would contradict a massive body of clinical and biological evidence. It would cause Kraepelin to squirm in his grave.
1. Kraepelin E, Lange J. Psychiatrie. In: Klinische Psychiatrie, vol 3 (8th ed). Leipzig, Germany: Barth; 1923.
2. Nasrallah HA. The continuum of psychoses between schizophrenia and bipolar disorder. Neurol Psychiatry Brain Res 1994;2:206-9.
3. Coffman JA, Bornstein RA, Olson SC, et al. Cognitive impairment and cerebral structure by MRI in bipolar disorder. Biol Psychiatry 1990;27(11):1188-96.
4. Weinberger DR. Genetic mechanisms of psychosis: in vivo and postmortem genomics. Clin Ther 2005;27(suppl):8-15.
5. Schulze TG, Ohlfaun S, Czerski PM, et al. Genotype-phenotype studies in bipolar disorder showing association between the DAOA/G30 locus and persecutory delusions: a first step toward a molecular genetic classification of psychiatric phenotypes. Arch Gen Psychiatry 2005;162:2101-8.
6. Tandon R, Fleischhacker WW. Comparative efficacy of antipsy chotics in the treatment of schizophrenia: a critical assessment. Schizophr Res 2005;79:145-55.
7. Calabrese JR, Keck P, Jr, McFadden W, et al:. A randomized double-blind, placebo-controlled trial of quetiapine in the treatment of bipolar I or II depression. Am J Psychiatry 2005;162:1351-60.
A hundred years ago, KraepelinDrs. Lake and Hurwitz for highlighting the diagnostic and treatment errors in a bipolar patient with severe psychotic features who was misdiagnosed as having schizophrenia. Errors such as this were common with DSM I and II but declined with the more reliable diagnostic schemas of DSM III and IV. I am puzzled, however, by their leap to the radical conclusion that schizophrenia does not exist and that all patients diagnosed with schizophrenia have psychotic bipolar disorder. This is not as egregious as Szasz’ absurd proclamation 4 decades ago that schizophrenia is a “myth,” but it is a significant scientific “transgression,” given the evidence that distinguishes schizophrenia from bipolar disorder.
Symptoms. Beyond a doubt, these two brain diseases have overlapping clinical features, pharmacotherapies, and even outcomes in a subgroup of patients. However, these diseases have major differences, as outlined in the accompanying table.
Brain anomalies. Neuroimaging studies indicate that schizophrenia is associated with more-severe and pervasive morphologic brain anomalies (dysplasia and hypoplasia) than bipolar disorder, although some bipolar patients have reduced cerebral and frontal volumes and marked cognitive deficits.3 Progressive neuro tissue loss has been observed early in schizophrenia but not in bipolar disorder.
Recent genetic studies indicate that several genes are found exclusively in schizophrenia or in bipolar disorder cohorts,4 but some are shared by both disorders and may be related to delusional symptoms.5 Familial transmission appears to differ: transgenerational studies find an abundance of mood disorders in family members of bipolar probands but relatively sparse occurrence of psychosis in families of probands with schizophrenia.
Table
Symptom differences between schizophrenia and bipolar disorder
| Symptom | Schizophrenia | Bipolar disorder |
|---|---|---|
| Psychosis | Auditory hallucinations and bizarre delusions are more common | Grandiosity is more common |
| Paranoia | Occurs in both, but more systematic in schizophrenia | |
| Core psychopathology | Far more negative symptoms and cognition dysfunction | Far more mood lability and affective cyclicity |
| Thought disorder | Far more disorganized and derailed thoughts | More likely to have racing thoughts and flight of ideas |
| Between-episode interpersonal skills | Withdrawn, alogic, seclusive | Much more interactive and verbal |
Treatment. There is no doubt that monotherapy with antipsychotics (old and new) has similar efficacy6 in schizophrenia and bipolar mania (and even in bipolar depression, with some atypicals7). However, there is minimal, if any, evidence that monotherapy mood stabilizers (lithium or anti-convulsants) have any tangible efficacy in schizophrenia. Electroconvulsive therapy is remarkably efficious in all phases of bipolar disorder but of dubious, if any, lasting benefit in schizophrenia.
Course. Both the premorbid history and post-treatment functional outcome tend to be more favorable in patients with bipolar disorder than schizophrenia. Most patients with schizophrenia experience significant clinical, social, and vocational deterioration, compared with a relative minority of bipolar patients.
In summary, schizophrenia and bipolar disorder are clearly distinct in their pure forms, although many patients have varying mixtures of both. Schizoaffective disorder is one of the most extensively investigated. Although many scholars have studied schizoaffective disorder, the evidence defies lumping it with either end of the continuum.
I agree with Drs. Lake and Hurwitz that most cases of schizoaffective disorder, especially the “schizomanic” type, are probably bipolar disorder with severe psychotic features. To assert, however, that schizophrenia does not exist at all and should be reclassified as bipolar disorder with psychotic features would contradict a massive body of clinical and biological evidence. It would cause Kraepelin to squirm in his grave.
A hundred years ago, KraepelinDrs. Lake and Hurwitz for highlighting the diagnostic and treatment errors in a bipolar patient with severe psychotic features who was misdiagnosed as having schizophrenia. Errors such as this were common with DSM I and II but declined with the more reliable diagnostic schemas of DSM III and IV. I am puzzled, however, by their leap to the radical conclusion that schizophrenia does not exist and that all patients diagnosed with schizophrenia have psychotic bipolar disorder. This is not as egregious as Szasz’ absurd proclamation 4 decades ago that schizophrenia is a “myth,” but it is a significant scientific “transgression,” given the evidence that distinguishes schizophrenia from bipolar disorder.
Symptoms. Beyond a doubt, these two brain diseases have overlapping clinical features, pharmacotherapies, and even outcomes in a subgroup of patients. However, these diseases have major differences, as outlined in the accompanying table.
Brain anomalies. Neuroimaging studies indicate that schizophrenia is associated with more-severe and pervasive morphologic brain anomalies (dysplasia and hypoplasia) than bipolar disorder, although some bipolar patients have reduced cerebral and frontal volumes and marked cognitive deficits.3 Progressive neuro tissue loss has been observed early in schizophrenia but not in bipolar disorder.
Recent genetic studies indicate that several genes are found exclusively in schizophrenia or in bipolar disorder cohorts,4 but some are shared by both disorders and may be related to delusional symptoms.5 Familial transmission appears to differ: transgenerational studies find an abundance of mood disorders in family members of bipolar probands but relatively sparse occurrence of psychosis in families of probands with schizophrenia.
Table
Symptom differences between schizophrenia and bipolar disorder
| Symptom | Schizophrenia | Bipolar disorder |
|---|---|---|
| Psychosis | Auditory hallucinations and bizarre delusions are more common | Grandiosity is more common |
| Paranoia | Occurs in both, but more systematic in schizophrenia | |
| Core psychopathology | Far more negative symptoms and cognition dysfunction | Far more mood lability and affective cyclicity |
| Thought disorder | Far more disorganized and derailed thoughts | More likely to have racing thoughts and flight of ideas |
| Between-episode interpersonal skills | Withdrawn, alogic, seclusive | Much more interactive and verbal |
Treatment. There is no doubt that monotherapy with antipsychotics (old and new) has similar efficacy6 in schizophrenia and bipolar mania (and even in bipolar depression, with some atypicals7). However, there is minimal, if any, evidence that monotherapy mood stabilizers (lithium or anti-convulsants) have any tangible efficacy in schizophrenia. Electroconvulsive therapy is remarkably efficious in all phases of bipolar disorder but of dubious, if any, lasting benefit in schizophrenia.
Course. Both the premorbid history and post-treatment functional outcome tend to be more favorable in patients with bipolar disorder than schizophrenia. Most patients with schizophrenia experience significant clinical, social, and vocational deterioration, compared with a relative minority of bipolar patients.
In summary, schizophrenia and bipolar disorder are clearly distinct in their pure forms, although many patients have varying mixtures of both. Schizoaffective disorder is one of the most extensively investigated. Although many scholars have studied schizoaffective disorder, the evidence defies lumping it with either end of the continuum.
I agree with Drs. Lake and Hurwitz that most cases of schizoaffective disorder, especially the “schizomanic” type, are probably bipolar disorder with severe psychotic features. To assert, however, that schizophrenia does not exist at all and should be reclassified as bipolar disorder with psychotic features would contradict a massive body of clinical and biological evidence. It would cause Kraepelin to squirm in his grave.
1. Kraepelin E, Lange J. Psychiatrie. In: Klinische Psychiatrie, vol 3 (8th ed). Leipzig, Germany: Barth; 1923.
2. Nasrallah HA. The continuum of psychoses between schizophrenia and bipolar disorder. Neurol Psychiatry Brain Res 1994;2:206-9.
3. Coffman JA, Bornstein RA, Olson SC, et al. Cognitive impairment and cerebral structure by MRI in bipolar disorder. Biol Psychiatry 1990;27(11):1188-96.
4. Weinberger DR. Genetic mechanisms of psychosis: in vivo and postmortem genomics. Clin Ther 2005;27(suppl):8-15.
5. Schulze TG, Ohlfaun S, Czerski PM, et al. Genotype-phenotype studies in bipolar disorder showing association between the DAOA/G30 locus and persecutory delusions: a first step toward a molecular genetic classification of psychiatric phenotypes. Arch Gen Psychiatry 2005;162:2101-8.
6. Tandon R, Fleischhacker WW. Comparative efficacy of antipsy chotics in the treatment of schizophrenia: a critical assessment. Schizophr Res 2005;79:145-55.
7. Calabrese JR, Keck P, Jr, McFadden W, et al:. A randomized double-blind, placebo-controlled trial of quetiapine in the treatment of bipolar I or II depression. Am J Psychiatry 2005;162:1351-60.
1. Kraepelin E, Lange J. Psychiatrie. In: Klinische Psychiatrie, vol 3 (8th ed). Leipzig, Germany: Barth; 1923.
2. Nasrallah HA. The continuum of psychoses between schizophrenia and bipolar disorder. Neurol Psychiatry Brain Res 1994;2:206-9.
3. Coffman JA, Bornstein RA, Olson SC, et al. Cognitive impairment and cerebral structure by MRI in bipolar disorder. Biol Psychiatry 1990;27(11):1188-96.
4. Weinberger DR. Genetic mechanisms of psychosis: in vivo and postmortem genomics. Clin Ther 2005;27(suppl):8-15.
5. Schulze TG, Ohlfaun S, Czerski PM, et al. Genotype-phenotype studies in bipolar disorder showing association between the DAOA/G30 locus and persecutory delusions: a first step toward a molecular genetic classification of psychiatric phenotypes. Arch Gen Psychiatry 2005;162:2101-8.
6. Tandon R, Fleischhacker WW. Comparative efficacy of antipsy chotics in the treatment of schizophrenia: a critical assessment. Schizophr Res 2005;79:145-55.
7. Calabrese JR, Keck P, Jr, McFadden W, et al:. A randomized double-blind, placebo-controlled trial of quetiapine in the treatment of bipolar I or II depression. Am J Psychiatry 2005;162:1351-60.
CATIE’s surprises: In antipsychotics’ square-off, were there winners or losers?
Investigators faced a dilemma while designing the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). More than 200 enrollees with chronic schizophrenia had pre-existing tardive dyskinesia (TD). Would it be ethical to give them the antipsychotic most likely to worsen their TD? Would exempting them from taking that drug influence the trial’s outcome?
This issue and others had to be resolved before the largest controlled study of “real world” schizophrenia could begin. Now that data are unfolding, groups with diverse agendas are debating CATIE’s methods and surprising results. This article describes how the trial’s design and findings could transform public policy and clinical practice.
poll here
Efficacy vs Effectiveness
The National Institute of Mental Health funded the prospective CATIE schizophrenia study to compare the effectiveness of atypical antipsychotics versus each other and versus a first-generation (typical) antipsychotic.
All approved atypicals have shown similar efficacy compared with placebo in short-term trials (usually 6 weeks).1 The CATIE trial’s rationale is that short-term efficacy studies required for FDA approval may not necessarily reflect the drugs’ effectiveness in long-term schizophrenia management. Effectiveness measures take into account efficacy as well as safety, tolerability, and unpredictable patient behaviors in the real world.
CATIE’s ‘Real World’ Patients
CATIE investigators enrolled a community sample of chronic schizophrenia patients similar to those many psychiatrists see. Very liberal inclusion and exclusion criteria (Table 1) allowed enrollees to have a history of substance abuse, comorbid psychiatric or medical disorders, be receiving other medications, or show evidence of TD. Their schizophrenia ranged from minimal to severe.2,3
The 1,493 patients who completed the study (Table 2) were enrolled at 57 outpatient treatment settings. One site’s 33 patients were eliminated from analysis because of doubts about the integrity of the data, leaving a total of 1,460 subjects.4
Table 1
Criteria for enrolling patients in the CATIE schizophrenia trial
| Inclusion criteria | Ages 18 to 65 yrs |
| DSM-IV diagnosis of schizophrenia | |
| Able to take oral medication | |
| Able to give informed consent | |
| Exclusion criteria | Diagnosis of schizoaffective disorder, mental retardation, or other cognitive disorders |
| History of serious adverse reactions to one of the study medications | |
| Had only one schizophrenic episode | |
| History of treatment resistance, defined as persistence of severe symptoms despite adequate trials of one of the study antipsychotics or prior treatment with clozapine | |
| Pregnant or breast feeding | |
| Serious and unstable medical conditions |
Table 2
CATIE’s 1,460 ‘real world’ schizophrenia patients at trial entry
| Mean age | 40.6±11.1 yrs |
| Mean age of first treatment | 24.0±8.9 yrs |
| Mean duration of treatment | 14.4±10.7 yrs |
| Gender | 74% male |
| Race | 60% white, 35% black, 5% other |
| Mean education | 12.1±2.3 years |
| Marital status | 59% never married |
| 29% previously married | |
| 11% married | |
| Employment status | 85% unemployed |
| Mean PANSS total score | 75.7±17.6 |
| Mean CGI | 4.0±0.9 |
| Psychiatric comorbidities | 29% drug dependence/abuse |
| 28% depression | |
| 25% alcohol dependence/abuse | |
| 14% anxiety disorder | |
| 5% obsessive-compulsive disorder | |
| Illness severity | 4% severe |
| 20% marked | |
| 47% moderate | |
| 23% mild | |
| 6% minimal | |
| PANSS: Positive and Negative Syndrome Scale | |
| CGI: Clinician-rated Clinical Global Impressions severity score | |
| Source: Reference 5. | |
Medications. Before randomization, 28% of enrollees were not receiving antipsychotics. The remainder were receiving:
- olanzapine (22%)
- risperidone (19%)
- quetiapine (7%)
- ziprasidone (0%; approved after the trial began)
- any combination of olanzapine, risperidone, and quetiapine (7%)
- typical antipsychotics (16%).
Metabolic profile. These outpatients had a high rate of metabolic disorders: 42%—twice the rate in the general population—met criteria for metabolic syndrome,5 putting them at high risk to die of cardiovascular causes within 10 years.6 They had relatively poor physical health self-ratings and increased somatic preoccupation.7 Most worrisome, many were receiving no medications for their metabolic disorders, including 45% of those with diabetes, 89% with hyperlipidemia, and 62% with hypertension.8
Substance abuse. At enrollment, 40% of patients were abstinent from substance use, 22% were using substances without abuse or dependence, and 37% had substance abuse or dependence. Compared with nonusers, substance abusers tended to be male with more childhood problems, higher positive symptoms on the Positive and Negative Syndrome Scale (PANSS), and more likely to have had a recent illness exacerbation.9
Tardive dyskinesia. The 231 subjects who met criteria for probable TD10 were older than the overall sample with more years of antipsychotic treatment, especially with conventional neuroleptics and anticholinergics. Substance abuse was associated with TD, as were severity of psychopathology, extrapyramidal symptoms (EPS), and akathisia.11
Violent behavior. A history of serious violent behavior was reported in:
- 5.4% of patients with high positive and low negative PANSS symptom scores
- 1.7% of patients with low positive and high negative PANSS symptom scores.
Consent. Patients’ capacity to give consent to participate in the study was assessed with the MacArthur Competence Assessment Tool for Clinical Research (MacCAT-CR). Psychosis severity (PANSS positive symptom scale) was not found to affect decision-making capacity, but negative symptoms and diminished working memory did.12
CATIE’s Unique Design
Defining effectiveness. CATIE was designed in three phases (Figure). Phase 1—discussed here—was a blinded, controlled comparison of four atypical antipsychotics and perphenazine. Results of phases 2 and 3 have yet to be published. The primary effectiveness endpoint, “all-cause discontinuation,” was defined as:
- lack of efficacy (patient was switched to another drug assigned at random)
- lack of tolerability (patient requested a drug change)
- safety problem (investigator initiated a switch)
- patient’s decision for any reason (often dropping out of the study).
The longer subjects stayed on the first antipsychotic they received, the more effective that drug was considered to be.
Figure CATIE schizophrenia trial design
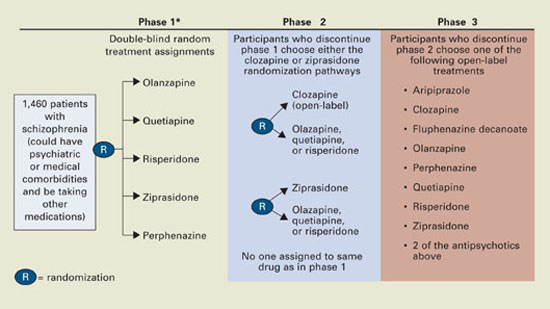
* Phase 1A: participants with tardive dyskinesia (N=231) do not get randomized to perphenazine; phase 1B: participants who fail perphenazine will be randomized to an atypical (olanzapine, quetiapine, or risperidone) before eligibility for phase 2.
Source: Reference 2.Medications. Three atypicals—risperidone, olanzapine, and quetiapine—were approved for schizophrenia when the trial began in 1999. Recruitment ended in June 2003, the last subject completed the 18-month trial in December 2004, and data analysis began in January 2005. Ziprasidone was added to phase 1 after 40% of the sample had been enrolled, and aripiprazole was included as an option in the unblinded phase 3.
Perphenazine was chosen to represent typical antipsychotics because it has medium potency and less risk of EPS than high-potency drugs such as haloperidol and is associated with less weight gain than low-potency drugs such as thioridazine.
Dosing. Pharmaceutical manufacturers donated the antipsychotics and were invited to recommend their respective drugs’ starting dosages, dose increments, and maximum dosages. Olanzapine’s maker requested a higher starting dosage (7.5 mg/d instead of 5.0 mg/d) and a maximum dosage 50% higher than the FDA-approved range (30 mg/d instead of 20 mg/d). The others recommended the FDA-approved dosage ranges or less:
- quetiapine, 200 to 800 mg/d
- risperidone, 1.5 to 6 mg/d
- ziprasidone, 40 to 160 mg/d
- perphenazine, 8 to 32 mg/d.
The study team accepted their recommendations.
The medications were packaged in identical capsules. Quetiapine and ziprasidone were given twice daily because of product labeling; risperidone, olanzapine, and perphenazine were given once daily to one-half the patients assigned to them and twice daily to the others to prevent raters from guessing which drug a patient was receiving.
Tardive dyskinesia. For ethical reasons, the 231 patients with TD at enrollment were randomly assigned in phase 1 to atypicals but not to perphenazine because of the well-established link between typical antipsychotics and TD. This exception could have contributed to the closer-than-expected differences in EPS and perhaps in efficacy, given reports that TD patients have more negative symptoms and cognitive dysfunction.13 However, a statistical analysis took that into account.
CATIE’s Key Findings
Discontinuation. A disappointingly high discontinuation rate (74% overall) within a few months was the most important finding (Table 3). A recent effectiveness study with a design similar to the CATIE trial found a similarly high rate of all-cause discontinuation (70%) in patients with first-episode psychosis.14 Thus, patient-initiated drug discontinuation appears to be a core illness behavior from schizophrenia onset to chronic illness.
The high discontinuation rate shows that we need to modify our approach to schizophrenia, emphasizing full adherence to antipsychotic therapy from the onset of the illness.
Table 3
All-cause discontinuation rates in the CATIE trial
| Antipsychotic | Percent discontinued | Duration on antipsychotic (months)* | Dosage (mg/d)* |
| Olanzapine | 64% | 9.2 | 20.1 |
| Perphenazine | 75% | 4.6 | 20.8 |
| Quetiapine | 82% | 4.8 | 543.4 |
| Risperidone | 74% | 5.6 | 3.9 |
| Ziprasidone | 79% | 3.5 | 112.8 |
| Overall | 74% | Median 6.0; mean 8.3 | |
| Notes | |||
| *Mean modal | |||
| Olanzapine’s discontinuation rate was significantly lower than those of perphenazine, quetiapine, and risperidone but not of ziprasidone. | |||
| Olanzapine’s maximum dosage was 30 mg/d (50% higher than FDA-approved 20 mg/d); other agents were dosed within approved ranges. | |||
| Patients reached maximum daily antipsychotic dosages at these rates: 40% with olanzapine, 40% with perphenazine, 44% with quetiapine, 40% with risperidone, and 48% with ziprasidone. | |||
Effectiveness—measured as all-cause discontinuation or switching—was the primary outcome of phase 1. The unexpected finding that perphenazine and the atypicals had similar effectiveness could influence clinical practice. Insurers, for example, might consider promoting cheaper typical antipsychotics for first-line use. CATIE’s cost-effectiveness arm (Rosenheck et al, submitted for publication) will provide additional data on this issue.
Before rushing to use older antipsychotics as first-line treatments for schizophrenia, however, policymakers should consider three factors in the study design that could have enhanced perphenazine’s efficacy and safety profiles.
First, perphenazine was given at lower dosages (up to 32 mg/d) than “real world” clinicians used a decade ago (up to 64 mg/d). Thus, lower rates of serious side effects, especially TD, might have occurred in the study than in past clinical practice. Since atypical antipsychotics were approved, clinicians see far fewer psychiatric patients with pill-rolling tremors, rigid posture, or a shuffling gait, compared with 10 to 15 years ago when typical antipsychotics were widely used.
Second, perphenazine was associated with the highest EPS rate (17%), though its mean modal dosage (20.8 mg/d) is considered moderate. Discontinuation because of EPS was highest with perphenazine and lowest with quetiapine.
Third, excluding enrollees with TD from perphenazine may have increased perphenazine’s effectiveness, whereas including them in the atypicals groups may have reduced the atypicals’ effectiveness. TD patients are at increased risk to develop EPS; they had more-severe illness and a higher substance abuse rate among CATIE patients.11 Even so, investigators did control for TD in the data analysis and found no significant difference between typical and atypical antipsychotics.
No ‘Winners’ or ‘Losers’
Effectiveness, tolerability, and safety findings for each antipsychotic are compared in Tables 4A and 4B. Careful review shows no clear “winners” or “losers;” each agent has weaknesses but also strengths that may benefit individual patients.
Efficacy. Olanzapine showed a relatively higher efficacy and lower discontinuation rate but also had the highest risk of adverse metabolic effects. Some have attributed its greater efficacy to its higher dosing compared with the other antipsychotics. Some also have argued that the antipsychotics that showed lower efficacy, such as quetiapine and ziprasidone, were underdosed in this chronic schizophrenia population with a mean duration of illness of 14 years. Perphenazine, too, was dosed at the lower end of its range (mean modal dose 20.8 mg/d) compared with the old community standard of 36 to 64 mg/d.
Generally, a mean modal dosage of 20.1 mg/d for olanzapine is considered equivalent to ziprasidone, 160 mg; quetiapine, 800 mg; and risperidone, 6 mg. In CATIE phase 1, mean modal dosages were:
- ziprasidone, 112.8 mg/d (30% below 160 mg)
- quetiapine, 543.4 mg/d (32% below 800)
- risperidone, 3.9 mg/d (35% below 6 mg).
Olanzapine’s starting dosage of 7.5 mg/d was relatively higher than those of the other atypicals, which may have produced more-rapid onset of efficacy.
Switching. Another potential “advantage” for olanzapine was that 22% of subjects were taking it when they enrolled. By random assignment, 23% of patients who were taking olanzapine stayed on olanzapine and did not switch. By comparison:
- No patients assigned to ziprasidone were taking it before entering the trial.
- Only 5% of those taking quetiapine stayed on that drug after randomization.
- Few were receiving perphenazine before enrollment.
Switching antipsychotics may increase side effect risk or efficacy problems. For example, a patient switched from olanzapine or quetiapine to ziprasidone or perphenazine may experience insomnia during the transition, which may lead to tolerability complaints.
Metabolic side effects seen in this trial support past observations and reports that olanzapine is associated with higher risk for weight gain, hyperglycemia, and hyperlipidemia than other antipsychotics.15 Data on metabolic changes in CATIE patients taking olanzapine are being analyzed.
Hyperprolactinemia was most common with risperidone and practically nonexistent with other antipsychotics—even perphenazine. On the other hand, risperidone had the most favorable tolerability profile. This implies that elevated prolactin does not necessarily lead to antipsychotic discontinuation because of tolerability among patients with schizophrenia.
QTC interval and cataract data were benign across all antipsychotics. These findings appear to exonerate ziprasidone and quetiapine, respectively, which have been perceived as associated with these side effects.
When data become available, the next article in this series will discuss CATIE phase 2 findings. This phase includes patients who did not improve with the phase 1 regimens because of efficacy or tolerability problems and were switched to other antipsychotic therapies.
Related resources
- Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia study. www.catie.unc.edu/schizophrenia
- Schizophrenia Research Forum. NARSAD, The Mental Health Research Association.www.schizophreniaforum.org
Drug brand names
- Aripiprazole • Abilify
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr Nasrallah receives grants/research support from AstraZeneca, Janssen Pharmaceutica, Eli Lilly & Co., and Pfizer. He is a consultant, advisory board member, and speaker for Abbott Laboratories, AstraZeneca, Janssen Pharmaceutica, Pfizer, and Shire Pharmaceuticals Group.
1. Tandon R, Jibson MD. Efficacy of newer generation antipsychotics in the treatment of schizophrenia. Psychoneuroendocrinol 2003;28(suppl 1):9-26.
2. Stroup TS, McEvoy JP, Swartz MS, et al. The National Institute of Mental Health Clinical Antipsychotic Trial of Intervention Effectiveness (CATIE). Project: schizophrenia trial design and protocol development. Schizophr Bull 2003;29:15-31.
3. Swartz MS, Perkins DO, Stroup TS, et al. Assessing clinical and functional outcomes in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia trial. Schizophr Bull 2003;29:33-43.
4. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353:1209-23.
5. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the CATIE schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res 2005;80:19-32.
6. Goff D, Sullivan LM, McEvoy JP, et al. A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr Res 2005;80:45-53.
7. Meyer JM, Nasrallah HA, McEvoy JP, et al. The Clinical Antipsychotic Trial of Intervention Effectiveness (CATIE) schizophrenia trial: clinical comparison of subgroups with and without the metabolic syndrome. Schizophr Res 2005;80:9-18.
8. Nasrallah HA, McEvoy JP, Meyer JM, et al. Low rates of treatment for metabolic disorders in the CATIE schizophrenia trial. Neuropsychopharmacol 2005;(suppl 1):204.-
9. Swartz MS, et al. (unpublished data).
10. Schooler NR, Kane JM. Research diagnosis for tardive dyskinesia. Arch Gen Psychiatry 1982;39:486-7.
11. Miller DD, McEvoy JP, Davis SM, et al. Clinical correlates of tardive dyskinesia in schizophrenia: baseline data from the CATIE schizophrenia trial. Schizophr Res 2005;80:33-43.
12. Stroup TS, Applebaum P, Swartz M, et al. Decision-making capacity for research participation among individuals in the CATIE schizophrenia trial. Schizophr Res 2005;80:1-8.
13. Waddington JL, Youssef HA, Dolphin C, et al. Cognitive function, negative symptoms and tardive dyskinesia in schizophrenia. Their association in relation to topography of involuntary movements and criterion of their abnormality. Arch Gen Psychiatry 1987;44:907-12.
14. Keefe R. The CAFÉ effectiveness study. Amsterdam: European College of Neuropsychopharmacology annual meeting, 2005;
15. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, and North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs, obesity, and diabetes. Diabetes Care 2004;27:596-601.
Investigators faced a dilemma while designing the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). More than 200 enrollees with chronic schizophrenia had pre-existing tardive dyskinesia (TD). Would it be ethical to give them the antipsychotic most likely to worsen their TD? Would exempting them from taking that drug influence the trial’s outcome?
This issue and others had to be resolved before the largest controlled study of “real world” schizophrenia could begin. Now that data are unfolding, groups with diverse agendas are debating CATIE’s methods and surprising results. This article describes how the trial’s design and findings could transform public policy and clinical practice.
poll here
Efficacy vs Effectiveness
The National Institute of Mental Health funded the prospective CATIE schizophrenia study to compare the effectiveness of atypical antipsychotics versus each other and versus a first-generation (typical) antipsychotic.
All approved atypicals have shown similar efficacy compared with placebo in short-term trials (usually 6 weeks).1 The CATIE trial’s rationale is that short-term efficacy studies required for FDA approval may not necessarily reflect the drugs’ effectiveness in long-term schizophrenia management. Effectiveness measures take into account efficacy as well as safety, tolerability, and unpredictable patient behaviors in the real world.
CATIE’s ‘Real World’ Patients
CATIE investigators enrolled a community sample of chronic schizophrenia patients similar to those many psychiatrists see. Very liberal inclusion and exclusion criteria (Table 1) allowed enrollees to have a history of substance abuse, comorbid psychiatric or medical disorders, be receiving other medications, or show evidence of TD. Their schizophrenia ranged from minimal to severe.2,3
The 1,493 patients who completed the study (Table 2) were enrolled at 57 outpatient treatment settings. One site’s 33 patients were eliminated from analysis because of doubts about the integrity of the data, leaving a total of 1,460 subjects.4
Table 1
Criteria for enrolling patients in the CATIE schizophrenia trial
| Inclusion criteria | Ages 18 to 65 yrs |
| DSM-IV diagnosis of schizophrenia | |
| Able to take oral medication | |
| Able to give informed consent | |
| Exclusion criteria | Diagnosis of schizoaffective disorder, mental retardation, or other cognitive disorders |
| History of serious adverse reactions to one of the study medications | |
| Had only one schizophrenic episode | |
| History of treatment resistance, defined as persistence of severe symptoms despite adequate trials of one of the study antipsychotics or prior treatment with clozapine | |
| Pregnant or breast feeding | |
| Serious and unstable medical conditions |
Table 2
CATIE’s 1,460 ‘real world’ schizophrenia patients at trial entry
| Mean age | 40.6±11.1 yrs |
| Mean age of first treatment | 24.0±8.9 yrs |
| Mean duration of treatment | 14.4±10.7 yrs |
| Gender | 74% male |
| Race | 60% white, 35% black, 5% other |
| Mean education | 12.1±2.3 years |
| Marital status | 59% never married |
| 29% previously married | |
| 11% married | |
| Employment status | 85% unemployed |
| Mean PANSS total score | 75.7±17.6 |
| Mean CGI | 4.0±0.9 |
| Psychiatric comorbidities | 29% drug dependence/abuse |
| 28% depression | |
| 25% alcohol dependence/abuse | |
| 14% anxiety disorder | |
| 5% obsessive-compulsive disorder | |
| Illness severity | 4% severe |
| 20% marked | |
| 47% moderate | |
| 23% mild | |
| 6% minimal | |
| PANSS: Positive and Negative Syndrome Scale | |
| CGI: Clinician-rated Clinical Global Impressions severity score | |
| Source: Reference 5. | |
Medications. Before randomization, 28% of enrollees were not receiving antipsychotics. The remainder were receiving:
- olanzapine (22%)
- risperidone (19%)
- quetiapine (7%)
- ziprasidone (0%; approved after the trial began)
- any combination of olanzapine, risperidone, and quetiapine (7%)
- typical antipsychotics (16%).
Metabolic profile. These outpatients had a high rate of metabolic disorders: 42%—twice the rate in the general population—met criteria for metabolic syndrome,5 putting them at high risk to die of cardiovascular causes within 10 years.6 They had relatively poor physical health self-ratings and increased somatic preoccupation.7 Most worrisome, many were receiving no medications for their metabolic disorders, including 45% of those with diabetes, 89% with hyperlipidemia, and 62% with hypertension.8
Substance abuse. At enrollment, 40% of patients were abstinent from substance use, 22% were using substances without abuse or dependence, and 37% had substance abuse or dependence. Compared with nonusers, substance abusers tended to be male with more childhood problems, higher positive symptoms on the Positive and Negative Syndrome Scale (PANSS), and more likely to have had a recent illness exacerbation.9
Tardive dyskinesia. The 231 subjects who met criteria for probable TD10 were older than the overall sample with more years of antipsychotic treatment, especially with conventional neuroleptics and anticholinergics. Substance abuse was associated with TD, as were severity of psychopathology, extrapyramidal symptoms (EPS), and akathisia.11
Violent behavior. A history of serious violent behavior was reported in:
- 5.4% of patients with high positive and low negative PANSS symptom scores
- 1.7% of patients with low positive and high negative PANSS symptom scores.
Consent. Patients’ capacity to give consent to participate in the study was assessed with the MacArthur Competence Assessment Tool for Clinical Research (MacCAT-CR). Psychosis severity (PANSS positive symptom scale) was not found to affect decision-making capacity, but negative symptoms and diminished working memory did.12
CATIE’s Unique Design
Defining effectiveness. CATIE was designed in three phases (Figure). Phase 1—discussed here—was a blinded, controlled comparison of four atypical antipsychotics and perphenazine. Results of phases 2 and 3 have yet to be published. The primary effectiveness endpoint, “all-cause discontinuation,” was defined as:
- lack of efficacy (patient was switched to another drug assigned at random)
- lack of tolerability (patient requested a drug change)
- safety problem (investigator initiated a switch)
- patient’s decision for any reason (often dropping out of the study).
The longer subjects stayed on the first antipsychotic they received, the more effective that drug was considered to be.
Figure CATIE schizophrenia trial design
* Phase 1A: participants with tardive dyskinesia (N=231) do not get randomized to perphenazine; phase 1B: participants who fail perphenazine will be randomized to an atypical (olanzapine, quetiapine, or risperidone) before eligibility for phase 2.
Source: Reference 2.Medications. Three atypicals—risperidone, olanzapine, and quetiapine—were approved for schizophrenia when the trial began in 1999. Recruitment ended in June 2003, the last subject completed the 18-month trial in December 2004, and data analysis began in January 2005. Ziprasidone was added to phase 1 after 40% of the sample had been enrolled, and aripiprazole was included as an option in the unblinded phase 3.
Perphenazine was chosen to represent typical antipsychotics because it has medium potency and less risk of EPS than high-potency drugs such as haloperidol and is associated with less weight gain than low-potency drugs such as thioridazine.
Dosing. Pharmaceutical manufacturers donated the antipsychotics and were invited to recommend their respective drugs’ starting dosages, dose increments, and maximum dosages. Olanzapine’s maker requested a higher starting dosage (7.5 mg/d instead of 5.0 mg/d) and a maximum dosage 50% higher than the FDA-approved range (30 mg/d instead of 20 mg/d). The others recommended the FDA-approved dosage ranges or less:
- quetiapine, 200 to 800 mg/d
- risperidone, 1.5 to 6 mg/d
- ziprasidone, 40 to 160 mg/d
- perphenazine, 8 to 32 mg/d.
The study team accepted their recommendations.
The medications were packaged in identical capsules. Quetiapine and ziprasidone were given twice daily because of product labeling; risperidone, olanzapine, and perphenazine were given once daily to one-half the patients assigned to them and twice daily to the others to prevent raters from guessing which drug a patient was receiving.
Tardive dyskinesia. For ethical reasons, the 231 patients with TD at enrollment were randomly assigned in phase 1 to atypicals but not to perphenazine because of the well-established link between typical antipsychotics and TD. This exception could have contributed to the closer-than-expected differences in EPS and perhaps in efficacy, given reports that TD patients have more negative symptoms and cognitive dysfunction.13 However, a statistical analysis took that into account.
CATIE’s Key Findings
Discontinuation. A disappointingly high discontinuation rate (74% overall) within a few months was the most important finding (Table 3). A recent effectiveness study with a design similar to the CATIE trial found a similarly high rate of all-cause discontinuation (70%) in patients with first-episode psychosis.14 Thus, patient-initiated drug discontinuation appears to be a core illness behavior from schizophrenia onset to chronic illness.
The high discontinuation rate shows that we need to modify our approach to schizophrenia, emphasizing full adherence to antipsychotic therapy from the onset of the illness.
Table 3
All-cause discontinuation rates in the CATIE trial
| Antipsychotic | Percent discontinued | Duration on antipsychotic (months)* | Dosage (mg/d)* |
| Olanzapine | 64% | 9.2 | 20.1 |
| Perphenazine | 75% | 4.6 | 20.8 |
| Quetiapine | 82% | 4.8 | 543.4 |
| Risperidone | 74% | 5.6 | 3.9 |
| Ziprasidone | 79% | 3.5 | 112.8 |
| Overall | 74% | Median 6.0; mean 8.3 | |
| Notes | |||
| *Mean modal | |||
| Olanzapine’s discontinuation rate was significantly lower than those of perphenazine, quetiapine, and risperidone but not of ziprasidone. | |||
| Olanzapine’s maximum dosage was 30 mg/d (50% higher than FDA-approved 20 mg/d); other agents were dosed within approved ranges. | |||
| Patients reached maximum daily antipsychotic dosages at these rates: 40% with olanzapine, 40% with perphenazine, 44% with quetiapine, 40% with risperidone, and 48% with ziprasidone. | |||
Effectiveness—measured as all-cause discontinuation or switching—was the primary outcome of phase 1. The unexpected finding that perphenazine and the atypicals had similar effectiveness could influence clinical practice. Insurers, for example, might consider promoting cheaper typical antipsychotics for first-line use. CATIE’s cost-effectiveness arm (Rosenheck et al, submitted for publication) will provide additional data on this issue.
Before rushing to use older antipsychotics as first-line treatments for schizophrenia, however, policymakers should consider three factors in the study design that could have enhanced perphenazine’s efficacy and safety profiles.
First, perphenazine was given at lower dosages (up to 32 mg/d) than “real world” clinicians used a decade ago (up to 64 mg/d). Thus, lower rates of serious side effects, especially TD, might have occurred in the study than in past clinical practice. Since atypical antipsychotics were approved, clinicians see far fewer psychiatric patients with pill-rolling tremors, rigid posture, or a shuffling gait, compared with 10 to 15 years ago when typical antipsychotics were widely used.
Second, perphenazine was associated with the highest EPS rate (17%), though its mean modal dosage (20.8 mg/d) is considered moderate. Discontinuation because of EPS was highest with perphenazine and lowest with quetiapine.
Third, excluding enrollees with TD from perphenazine may have increased perphenazine’s effectiveness, whereas including them in the atypicals groups may have reduced the atypicals’ effectiveness. TD patients are at increased risk to develop EPS; they had more-severe illness and a higher substance abuse rate among CATIE patients.11 Even so, investigators did control for TD in the data analysis and found no significant difference between typical and atypical antipsychotics.
No ‘Winners’ or ‘Losers’
Effectiveness, tolerability, and safety findings for each antipsychotic are compared in Tables 4A and 4B. Careful review shows no clear “winners” or “losers;” each agent has weaknesses but also strengths that may benefit individual patients.
Efficacy. Olanzapine showed a relatively higher efficacy and lower discontinuation rate but also had the highest risk of adverse metabolic effects. Some have attributed its greater efficacy to its higher dosing compared with the other antipsychotics. Some also have argued that the antipsychotics that showed lower efficacy, such as quetiapine and ziprasidone, were underdosed in this chronic schizophrenia population with a mean duration of illness of 14 years. Perphenazine, too, was dosed at the lower end of its range (mean modal dose 20.8 mg/d) compared with the old community standard of 36 to 64 mg/d.
Generally, a mean modal dosage of 20.1 mg/d for olanzapine is considered equivalent to ziprasidone, 160 mg; quetiapine, 800 mg; and risperidone, 6 mg. In CATIE phase 1, mean modal dosages were:
- ziprasidone, 112.8 mg/d (30% below 160 mg)
- quetiapine, 543.4 mg/d (32% below 800)
- risperidone, 3.9 mg/d (35% below 6 mg).
Olanzapine’s starting dosage of 7.5 mg/d was relatively higher than those of the other atypicals, which may have produced more-rapid onset of efficacy.
Switching. Another potential “advantage” for olanzapine was that 22% of subjects were taking it when they enrolled. By random assignment, 23% of patients who were taking olanzapine stayed on olanzapine and did not switch. By comparison:
- No patients assigned to ziprasidone were taking it before entering the trial.
- Only 5% of those taking quetiapine stayed on that drug after randomization.
- Few were receiving perphenazine before enrollment.
Switching antipsychotics may increase side effect risk or efficacy problems. For example, a patient switched from olanzapine or quetiapine to ziprasidone or perphenazine may experience insomnia during the transition, which may lead to tolerability complaints.
Metabolic side effects seen in this trial support past observations and reports that olanzapine is associated with higher risk for weight gain, hyperglycemia, and hyperlipidemia than other antipsychotics.15 Data on metabolic changes in CATIE patients taking olanzapine are being analyzed.
Hyperprolactinemia was most common with risperidone and practically nonexistent with other antipsychotics—even perphenazine. On the other hand, risperidone had the most favorable tolerability profile. This implies that elevated prolactin does not necessarily lead to antipsychotic discontinuation because of tolerability among patients with schizophrenia.
QTC interval and cataract data were benign across all antipsychotics. These findings appear to exonerate ziprasidone and quetiapine, respectively, which have been perceived as associated with these side effects.
When data become available, the next article in this series will discuss CATIE phase 2 findings. This phase includes patients who did not improve with the phase 1 regimens because of efficacy or tolerability problems and were switched to other antipsychotic therapies.
Related resources
- Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia study. www.catie.unc.edu/schizophrenia
- Schizophrenia Research Forum. NARSAD, The Mental Health Research Association.www.schizophreniaforum.org
Drug brand names
- Aripiprazole • Abilify
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr Nasrallah receives grants/research support from AstraZeneca, Janssen Pharmaceutica, Eli Lilly & Co., and Pfizer. He is a consultant, advisory board member, and speaker for Abbott Laboratories, AstraZeneca, Janssen Pharmaceutica, Pfizer, and Shire Pharmaceuticals Group.
Investigators faced a dilemma while designing the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). More than 200 enrollees with chronic schizophrenia had pre-existing tardive dyskinesia (TD). Would it be ethical to give them the antipsychotic most likely to worsen their TD? Would exempting them from taking that drug influence the trial’s outcome?
This issue and others had to be resolved before the largest controlled study of “real world” schizophrenia could begin. Now that data are unfolding, groups with diverse agendas are debating CATIE’s methods and surprising results. This article describes how the trial’s design and findings could transform public policy and clinical practice.
poll here
Efficacy vs Effectiveness
The National Institute of Mental Health funded the prospective CATIE schizophrenia study to compare the effectiveness of atypical antipsychotics versus each other and versus a first-generation (typical) antipsychotic.
All approved atypicals have shown similar efficacy compared with placebo in short-term trials (usually 6 weeks).1 The CATIE trial’s rationale is that short-term efficacy studies required for FDA approval may not necessarily reflect the drugs’ effectiveness in long-term schizophrenia management. Effectiveness measures take into account efficacy as well as safety, tolerability, and unpredictable patient behaviors in the real world.
CATIE’s ‘Real World’ Patients
CATIE investigators enrolled a community sample of chronic schizophrenia patients similar to those many psychiatrists see. Very liberal inclusion and exclusion criteria (Table 1) allowed enrollees to have a history of substance abuse, comorbid psychiatric or medical disorders, be receiving other medications, or show evidence of TD. Their schizophrenia ranged from minimal to severe.2,3
The 1,493 patients who completed the study (Table 2) were enrolled at 57 outpatient treatment settings. One site’s 33 patients were eliminated from analysis because of doubts about the integrity of the data, leaving a total of 1,460 subjects.4
Table 1
Criteria for enrolling patients in the CATIE schizophrenia trial
| Inclusion criteria | Ages 18 to 65 yrs |
| DSM-IV diagnosis of schizophrenia | |
| Able to take oral medication | |
| Able to give informed consent | |
| Exclusion criteria | Diagnosis of schizoaffective disorder, mental retardation, or other cognitive disorders |
| History of serious adverse reactions to one of the study medications | |
| Had only one schizophrenic episode | |
| History of treatment resistance, defined as persistence of severe symptoms despite adequate trials of one of the study antipsychotics or prior treatment with clozapine | |
| Pregnant or breast feeding | |
| Serious and unstable medical conditions |
Table 2
CATIE’s 1,460 ‘real world’ schizophrenia patients at trial entry
| Mean age | 40.6±11.1 yrs |
| Mean age of first treatment | 24.0±8.9 yrs |
| Mean duration of treatment | 14.4±10.7 yrs |
| Gender | 74% male |
| Race | 60% white, 35% black, 5% other |
| Mean education | 12.1±2.3 years |
| Marital status | 59% never married |
| 29% previously married | |
| 11% married | |
| Employment status | 85% unemployed |
| Mean PANSS total score | 75.7±17.6 |
| Mean CGI | 4.0±0.9 |
| Psychiatric comorbidities | 29% drug dependence/abuse |
| 28% depression | |
| 25% alcohol dependence/abuse | |
| 14% anxiety disorder | |
| 5% obsessive-compulsive disorder | |
| Illness severity | 4% severe |
| 20% marked | |
| 47% moderate | |
| 23% mild | |
| 6% minimal | |
| PANSS: Positive and Negative Syndrome Scale | |
| CGI: Clinician-rated Clinical Global Impressions severity score | |
| Source: Reference 5. | |
Medications. Before randomization, 28% of enrollees were not receiving antipsychotics. The remainder were receiving:
- olanzapine (22%)
- risperidone (19%)
- quetiapine (7%)
- ziprasidone (0%; approved after the trial began)
- any combination of olanzapine, risperidone, and quetiapine (7%)
- typical antipsychotics (16%).
Metabolic profile. These outpatients had a high rate of metabolic disorders: 42%—twice the rate in the general population—met criteria for metabolic syndrome,5 putting them at high risk to die of cardiovascular causes within 10 years.6 They had relatively poor physical health self-ratings and increased somatic preoccupation.7 Most worrisome, many were receiving no medications for their metabolic disorders, including 45% of those with diabetes, 89% with hyperlipidemia, and 62% with hypertension.8
Substance abuse. At enrollment, 40% of patients were abstinent from substance use, 22% were using substances without abuse or dependence, and 37% had substance abuse or dependence. Compared with nonusers, substance abusers tended to be male with more childhood problems, higher positive symptoms on the Positive and Negative Syndrome Scale (PANSS), and more likely to have had a recent illness exacerbation.9
Tardive dyskinesia. The 231 subjects who met criteria for probable TD10 were older than the overall sample with more years of antipsychotic treatment, especially with conventional neuroleptics and anticholinergics. Substance abuse was associated with TD, as were severity of psychopathology, extrapyramidal symptoms (EPS), and akathisia.11
Violent behavior. A history of serious violent behavior was reported in:
- 5.4% of patients with high positive and low negative PANSS symptom scores
- 1.7% of patients with low positive and high negative PANSS symptom scores.
Consent. Patients’ capacity to give consent to participate in the study was assessed with the MacArthur Competence Assessment Tool for Clinical Research (MacCAT-CR). Psychosis severity (PANSS positive symptom scale) was not found to affect decision-making capacity, but negative symptoms and diminished working memory did.12
CATIE’s Unique Design
Defining effectiveness. CATIE was designed in three phases (Figure). Phase 1—discussed here—was a blinded, controlled comparison of four atypical antipsychotics and perphenazine. Results of phases 2 and 3 have yet to be published. The primary effectiveness endpoint, “all-cause discontinuation,” was defined as:
- lack of efficacy (patient was switched to another drug assigned at random)
- lack of tolerability (patient requested a drug change)
- safety problem (investigator initiated a switch)
- patient’s decision for any reason (often dropping out of the study).
The longer subjects stayed on the first antipsychotic they received, the more effective that drug was considered to be.
Figure CATIE schizophrenia trial design
* Phase 1A: participants with tardive dyskinesia (N=231) do not get randomized to perphenazine; phase 1B: participants who fail perphenazine will be randomized to an atypical (olanzapine, quetiapine, or risperidone) before eligibility for phase 2.
Source: Reference 2.Medications. Three atypicals—risperidone, olanzapine, and quetiapine—were approved for schizophrenia when the trial began in 1999. Recruitment ended in June 2003, the last subject completed the 18-month trial in December 2004, and data analysis began in January 2005. Ziprasidone was added to phase 1 after 40% of the sample had been enrolled, and aripiprazole was included as an option in the unblinded phase 3.
Perphenazine was chosen to represent typical antipsychotics because it has medium potency and less risk of EPS than high-potency drugs such as haloperidol and is associated with less weight gain than low-potency drugs such as thioridazine.
Dosing. Pharmaceutical manufacturers donated the antipsychotics and were invited to recommend their respective drugs’ starting dosages, dose increments, and maximum dosages. Olanzapine’s maker requested a higher starting dosage (7.5 mg/d instead of 5.0 mg/d) and a maximum dosage 50% higher than the FDA-approved range (30 mg/d instead of 20 mg/d). The others recommended the FDA-approved dosage ranges or less:
- quetiapine, 200 to 800 mg/d
- risperidone, 1.5 to 6 mg/d
- ziprasidone, 40 to 160 mg/d
- perphenazine, 8 to 32 mg/d.
The study team accepted their recommendations.
The medications were packaged in identical capsules. Quetiapine and ziprasidone were given twice daily because of product labeling; risperidone, olanzapine, and perphenazine were given once daily to one-half the patients assigned to them and twice daily to the others to prevent raters from guessing which drug a patient was receiving.
Tardive dyskinesia. For ethical reasons, the 231 patients with TD at enrollment were randomly assigned in phase 1 to atypicals but not to perphenazine because of the well-established link between typical antipsychotics and TD. This exception could have contributed to the closer-than-expected differences in EPS and perhaps in efficacy, given reports that TD patients have more negative symptoms and cognitive dysfunction.13 However, a statistical analysis took that into account.
CATIE’s Key Findings
Discontinuation. A disappointingly high discontinuation rate (74% overall) within a few months was the most important finding (Table 3). A recent effectiveness study with a design similar to the CATIE trial found a similarly high rate of all-cause discontinuation (70%) in patients with first-episode psychosis.14 Thus, patient-initiated drug discontinuation appears to be a core illness behavior from schizophrenia onset to chronic illness.
The high discontinuation rate shows that we need to modify our approach to schizophrenia, emphasizing full adherence to antipsychotic therapy from the onset of the illness.
Table 3
All-cause discontinuation rates in the CATIE trial
| Antipsychotic | Percent discontinued | Duration on antipsychotic (months)* | Dosage (mg/d)* |
| Olanzapine | 64% | 9.2 | 20.1 |
| Perphenazine | 75% | 4.6 | 20.8 |
| Quetiapine | 82% | 4.8 | 543.4 |
| Risperidone | 74% | 5.6 | 3.9 |
| Ziprasidone | 79% | 3.5 | 112.8 |
| Overall | 74% | Median 6.0; mean 8.3 | |
| Notes | |||
| *Mean modal | |||
| Olanzapine’s discontinuation rate was significantly lower than those of perphenazine, quetiapine, and risperidone but not of ziprasidone. | |||
| Olanzapine’s maximum dosage was 30 mg/d (50% higher than FDA-approved 20 mg/d); other agents were dosed within approved ranges. | |||
| Patients reached maximum daily antipsychotic dosages at these rates: 40% with olanzapine, 40% with perphenazine, 44% with quetiapine, 40% with risperidone, and 48% with ziprasidone. | |||
Effectiveness—measured as all-cause discontinuation or switching—was the primary outcome of phase 1. The unexpected finding that perphenazine and the atypicals had similar effectiveness could influence clinical practice. Insurers, for example, might consider promoting cheaper typical antipsychotics for first-line use. CATIE’s cost-effectiveness arm (Rosenheck et al, submitted for publication) will provide additional data on this issue.
Before rushing to use older antipsychotics as first-line treatments for schizophrenia, however, policymakers should consider three factors in the study design that could have enhanced perphenazine’s efficacy and safety profiles.
First, perphenazine was given at lower dosages (up to 32 mg/d) than “real world” clinicians used a decade ago (up to 64 mg/d). Thus, lower rates of serious side effects, especially TD, might have occurred in the study than in past clinical practice. Since atypical antipsychotics were approved, clinicians see far fewer psychiatric patients with pill-rolling tremors, rigid posture, or a shuffling gait, compared with 10 to 15 years ago when typical antipsychotics were widely used.
Second, perphenazine was associated with the highest EPS rate (17%), though its mean modal dosage (20.8 mg/d) is considered moderate. Discontinuation because of EPS was highest with perphenazine and lowest with quetiapine.
Third, excluding enrollees with TD from perphenazine may have increased perphenazine’s effectiveness, whereas including them in the atypicals groups may have reduced the atypicals’ effectiveness. TD patients are at increased risk to develop EPS; they had more-severe illness and a higher substance abuse rate among CATIE patients.11 Even so, investigators did control for TD in the data analysis and found no significant difference between typical and atypical antipsychotics.
No ‘Winners’ or ‘Losers’
Effectiveness, tolerability, and safety findings for each antipsychotic are compared in Tables 4A and 4B. Careful review shows no clear “winners” or “losers;” each agent has weaknesses but also strengths that may benefit individual patients.
Efficacy. Olanzapine showed a relatively higher efficacy and lower discontinuation rate but also had the highest risk of adverse metabolic effects. Some have attributed its greater efficacy to its higher dosing compared with the other antipsychotics. Some also have argued that the antipsychotics that showed lower efficacy, such as quetiapine and ziprasidone, were underdosed in this chronic schizophrenia population with a mean duration of illness of 14 years. Perphenazine, too, was dosed at the lower end of its range (mean modal dose 20.8 mg/d) compared with the old community standard of 36 to 64 mg/d.
Generally, a mean modal dosage of 20.1 mg/d for olanzapine is considered equivalent to ziprasidone, 160 mg; quetiapine, 800 mg; and risperidone, 6 mg. In CATIE phase 1, mean modal dosages were:
- ziprasidone, 112.8 mg/d (30% below 160 mg)
- quetiapine, 543.4 mg/d (32% below 800)
- risperidone, 3.9 mg/d (35% below 6 mg).
Olanzapine’s starting dosage of 7.5 mg/d was relatively higher than those of the other atypicals, which may have produced more-rapid onset of efficacy.
Switching. Another potential “advantage” for olanzapine was that 22% of subjects were taking it when they enrolled. By random assignment, 23% of patients who were taking olanzapine stayed on olanzapine and did not switch. By comparison:
- No patients assigned to ziprasidone were taking it before entering the trial.
- Only 5% of those taking quetiapine stayed on that drug after randomization.
- Few were receiving perphenazine before enrollment.
Switching antipsychotics may increase side effect risk or efficacy problems. For example, a patient switched from olanzapine or quetiapine to ziprasidone or perphenazine may experience insomnia during the transition, which may lead to tolerability complaints.
Metabolic side effects seen in this trial support past observations and reports that olanzapine is associated with higher risk for weight gain, hyperglycemia, and hyperlipidemia than other antipsychotics.15 Data on metabolic changes in CATIE patients taking olanzapine are being analyzed.
Hyperprolactinemia was most common with risperidone and practically nonexistent with other antipsychotics—even perphenazine. On the other hand, risperidone had the most favorable tolerability profile. This implies that elevated prolactin does not necessarily lead to antipsychotic discontinuation because of tolerability among patients with schizophrenia.
QTC interval and cataract data were benign across all antipsychotics. These findings appear to exonerate ziprasidone and quetiapine, respectively, which have been perceived as associated with these side effects.
When data become available, the next article in this series will discuss CATIE phase 2 findings. This phase includes patients who did not improve with the phase 1 regimens because of efficacy or tolerability problems and were switched to other antipsychotic therapies.
Related resources
- Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia study. www.catie.unc.edu/schizophrenia
- Schizophrenia Research Forum. NARSAD, The Mental Health Research Association.www.schizophreniaforum.org
Drug brand names
- Aripiprazole • Abilify
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr Nasrallah receives grants/research support from AstraZeneca, Janssen Pharmaceutica, Eli Lilly & Co., and Pfizer. He is a consultant, advisory board member, and speaker for Abbott Laboratories, AstraZeneca, Janssen Pharmaceutica, Pfizer, and Shire Pharmaceuticals Group.
1. Tandon R, Jibson MD. Efficacy of newer generation antipsychotics in the treatment of schizophrenia. Psychoneuroendocrinol 2003;28(suppl 1):9-26.
2. Stroup TS, McEvoy JP, Swartz MS, et al. The National Institute of Mental Health Clinical Antipsychotic Trial of Intervention Effectiveness (CATIE). Project: schizophrenia trial design and protocol development. Schizophr Bull 2003;29:15-31.
3. Swartz MS, Perkins DO, Stroup TS, et al. Assessing clinical and functional outcomes in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia trial. Schizophr Bull 2003;29:33-43.
4. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353:1209-23.
5. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the CATIE schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res 2005;80:19-32.
6. Goff D, Sullivan LM, McEvoy JP, et al. A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr Res 2005;80:45-53.
7. Meyer JM, Nasrallah HA, McEvoy JP, et al. The Clinical Antipsychotic Trial of Intervention Effectiveness (CATIE) schizophrenia trial: clinical comparison of subgroups with and without the metabolic syndrome. Schizophr Res 2005;80:9-18.
8. Nasrallah HA, McEvoy JP, Meyer JM, et al. Low rates of treatment for metabolic disorders in the CATIE schizophrenia trial. Neuropsychopharmacol 2005;(suppl 1):204.-
9. Swartz MS, et al. (unpublished data).
10. Schooler NR, Kane JM. Research diagnosis for tardive dyskinesia. Arch Gen Psychiatry 1982;39:486-7.
11. Miller DD, McEvoy JP, Davis SM, et al. Clinical correlates of tardive dyskinesia in schizophrenia: baseline data from the CATIE schizophrenia trial. Schizophr Res 2005;80:33-43.
12. Stroup TS, Applebaum P, Swartz M, et al. Decision-making capacity for research participation among individuals in the CATIE schizophrenia trial. Schizophr Res 2005;80:1-8.
13. Waddington JL, Youssef HA, Dolphin C, et al. Cognitive function, negative symptoms and tardive dyskinesia in schizophrenia. Their association in relation to topography of involuntary movements and criterion of their abnormality. Arch Gen Psychiatry 1987;44:907-12.
14. Keefe R. The CAFÉ effectiveness study. Amsterdam: European College of Neuropsychopharmacology annual meeting, 2005;
15. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, and North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs, obesity, and diabetes. Diabetes Care 2004;27:596-601.
1. Tandon R, Jibson MD. Efficacy of newer generation antipsychotics in the treatment of schizophrenia. Psychoneuroendocrinol 2003;28(suppl 1):9-26.
2. Stroup TS, McEvoy JP, Swartz MS, et al. The National Institute of Mental Health Clinical Antipsychotic Trial of Intervention Effectiveness (CATIE). Project: schizophrenia trial design and protocol development. Schizophr Bull 2003;29:15-31.
3. Swartz MS, Perkins DO, Stroup TS, et al. Assessing clinical and functional outcomes in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia trial. Schizophr Bull 2003;29:33-43.
4. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353:1209-23.
5. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the CATIE schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res 2005;80:19-32.
6. Goff D, Sullivan LM, McEvoy JP, et al. A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr Res 2005;80:45-53.
7. Meyer JM, Nasrallah HA, McEvoy JP, et al. The Clinical Antipsychotic Trial of Intervention Effectiveness (CATIE) schizophrenia trial: clinical comparison of subgroups with and without the metabolic syndrome. Schizophr Res 2005;80:9-18.
8. Nasrallah HA, McEvoy JP, Meyer JM, et al. Low rates of treatment for metabolic disorders in the CATIE schizophrenia trial. Neuropsychopharmacol 2005;(suppl 1):204.-
9. Swartz MS, et al. (unpublished data).
10. Schooler NR, Kane JM. Research diagnosis for tardive dyskinesia. Arch Gen Psychiatry 1982;39:486-7.
11. Miller DD, McEvoy JP, Davis SM, et al. Clinical correlates of tardive dyskinesia in schizophrenia: baseline data from the CATIE schizophrenia trial. Schizophr Res 2005;80:33-43.
12. Stroup TS, Applebaum P, Swartz M, et al. Decision-making capacity for research participation among individuals in the CATIE schizophrenia trial. Schizophr Res 2005;80:1-8.
13. Waddington JL, Youssef HA, Dolphin C, et al. Cognitive function, negative symptoms and tardive dyskinesia in schizophrenia. Their association in relation to topography of involuntary movements and criterion of their abnormality. Arch Gen Psychiatry 1987;44:907-12.
14. Keefe R. The CAFÉ effectiveness study. Amsterdam: European College of Neuropsychopharmacology annual meeting, 2005;
15. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, and North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs, obesity, and diabetes. Diabetes Care 2004;27:596-601.
Commentary: When medical illness complicates schizophrenia and bipolar disorder
Patients with schizophrenia or bipolar disorder face a higher risk of premature death, compared with the general population. Their overall mortality rate is elevated not only by higher suicide rates but also by higher rates of medical comorbidities, including obesity, diabetes mellitus, cardiovascular and pulmonary diseases, HIV infection, and cancer.1,2
Schizophrenia and bipolar disorder are also frequently complicated by psychiatric comorbidities including alcohol and substance use disorders (such as nicotine dependence), anxiety disorders, and eating disorders (such as binge eating).3,4 Even so, the impact of psychiatric comorbidity on medical morbidity and mortality has not been adequately investigated and has—until rather recently—received little attention from clinical researchers.
Within the past 5 years or so, epidemiologic and population studies have shown increased morbidity and mortality from medical illnesses in patients with serious and persistent mental illness.5,6 These findings have sparked renewed interest in behavioral, biological, and psychosocial factors associated with schizophrenia and bipolar disorder that may contribute to comorbid medical illnesses. These factors include:
- negative and depressive symptoms
- physical inactivity and poor diet
- hypothalamic-pituitary-adrenal axis dysregulation associated with acute psychotic and affective episodes
- amotivation
- social isolation
- limited access to primary and preventive health care.7
The degree to which these factors may elevate medical comorbidity rates in patients with Commentary these serious psychiatric illnesses has not been well-studied or described.
TREATMENT IMPLICATIONS
These observations raise concerns about safe and effective use of medications by patients with schizophrenia or bipolar disorder as well as medical illness. Issues for clinicians to consider include:
- possible pharmacokinetic and pharmacodynamic interactions in patients taking concomitant medications for psychiatric and medical illnesses
- potential beneficial and adverse effects of psychotropics on medical illnesses
- and—conversely—potential beneficial and adverse effects on mood and psychotic disorders of medications used to treat medical illnesses.
Being aware of metabolic effects when prescribing psychotropics is not a new idea. Lithium, for example, has long been known to cause weight gain, suppress thyroid hormone, and interact with thiazide diuretics when used to treat bipolar disorder. Thus, therapeutic blood monitoring is recommended for patients receiving acute and maintenance lithium therapy.
More recently, atypical antipsychotics such as olanzapine and clozapine (and risperidone and quetiapine to a lesser extent) have been shown to produce substantial weight gain and other metabolic effects that increase the risk of diabetes and cardiovascular disease.7 As a result, the American Diabetes Association—along with the American Psychiatric Association and other groups—now recommends that psychiatrists and other physicians monitor patients’ body weight and body mass index, vital signs, serum glucose, and lipids when prescribing these agents.7-11 Careful monitoring should improve the medical care of patients with schizophrenia and bipolar disorder and help protect them from the medical risks associated with overweight and obesity.
Similar precautions are needed when schizophrenia and bipolar disorder co-occur with other medical illnesses treated by complex pharmacologic regimens, such as pulmonary disease, cancer, and HIV. Three points to keep in mind are:
- careful selection of treatments for co-occurring medical illnesses, considering potential effects on the primary psychiatric disorder
- the impact of psychotropics on patients’ medical illnesses
- and the potential for drug interactions.
1. Kilbourne AM, Cornelius JR, Han X, et al. Burden of general medical conditions among individuals with bipolar disorder. Bipolar Disord 2004;6:368-73.
2. Meyer JM, Nasrallah HA. Medical illness and schizophrenia Washington, DC: American Psychiatric Press, Inc, 2003.
3. McElroy SL, Altshuler LL, Suppes T, et al. Axis I psychiatric comorbidity and its relationship with historical illness variables in 288 patients with bipolar disorder. Am J Psychiatry 2001;158:420-6.
4. Green AI, Canuso CM, Brenner MJ, et al. Detection and management of comorbidity in patients with schizophrenia. Psychiatr Clin North Am 2003;26:115-39.
5. Buda M, Tsuang MT, Fleming JA. Causes of death in DSM-III schizophrenics and other psychotics (atypical group). Comparison with the general population. Arch Gen Psychiatry 1988;45:283-5.
6. Osby U, Brandt L, Correia N, et al. Excess mortality in bipolar and unipolar disorder in Sweden. Arch Gen Psychiatry 2001;58:844-50.
7. Keck PE, Jr, Buse JB, Dagago-Jack S, et al. Managing metabolic concerns in patients with severe mental illness: a special report (Postgrad Med). Minneapolis, MN: McGraw-Hill, 2003.
8. American Diabetes Association. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care 2004;27:596-601.
9. Marder SR, Essock SM, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry 2004;161:1334-9.
10. Chue P, Kovacs CS. Safety and tolerability of atypical antipsychotics in patients with bipolar disorder: prevalence, monitoring and management. Bipolar Disord 2003;5(suppl 2):62-79.
11. Nasrallah HA, Newcomer JW. Atypical antipsychotics and metabolic dysregulation. Evaluating the risk/benefit equation and improving standard of care. J Clin Psychopharmacol 2004;24(suppl 2):S7-S14.
Patients with schizophrenia or bipolar disorder face a higher risk of premature death, compared with the general population. Their overall mortality rate is elevated not only by higher suicide rates but also by higher rates of medical comorbidities, including obesity, diabetes mellitus, cardiovascular and pulmonary diseases, HIV infection, and cancer.1,2
Schizophrenia and bipolar disorder are also frequently complicated by psychiatric comorbidities including alcohol and substance use disorders (such as nicotine dependence), anxiety disorders, and eating disorders (such as binge eating).3,4 Even so, the impact of psychiatric comorbidity on medical morbidity and mortality has not been adequately investigated and has—until rather recently—received little attention from clinical researchers.
Within the past 5 years or so, epidemiologic and population studies have shown increased morbidity and mortality from medical illnesses in patients with serious and persistent mental illness.5,6 These findings have sparked renewed interest in behavioral, biological, and psychosocial factors associated with schizophrenia and bipolar disorder that may contribute to comorbid medical illnesses. These factors include:
- negative and depressive symptoms
- physical inactivity and poor diet
- hypothalamic-pituitary-adrenal axis dysregulation associated with acute psychotic and affective episodes
- amotivation
- social isolation
- limited access to primary and preventive health care.7
The degree to which these factors may elevate medical comorbidity rates in patients with Commentary these serious psychiatric illnesses has not been well-studied or described.
TREATMENT IMPLICATIONS
These observations raise concerns about safe and effective use of medications by patients with schizophrenia or bipolar disorder as well as medical illness. Issues for clinicians to consider include:
- possible pharmacokinetic and pharmacodynamic interactions in patients taking concomitant medications for psychiatric and medical illnesses
- potential beneficial and adverse effects of psychotropics on medical illnesses
- and—conversely—potential beneficial and adverse effects on mood and psychotic disorders of medications used to treat medical illnesses.
Being aware of metabolic effects when prescribing psychotropics is not a new idea. Lithium, for example, has long been known to cause weight gain, suppress thyroid hormone, and interact with thiazide diuretics when used to treat bipolar disorder. Thus, therapeutic blood monitoring is recommended for patients receiving acute and maintenance lithium therapy.
More recently, atypical antipsychotics such as olanzapine and clozapine (and risperidone and quetiapine to a lesser extent) have been shown to produce substantial weight gain and other metabolic effects that increase the risk of diabetes and cardiovascular disease.7 As a result, the American Diabetes Association—along with the American Psychiatric Association and other groups—now recommends that psychiatrists and other physicians monitor patients’ body weight and body mass index, vital signs, serum glucose, and lipids when prescribing these agents.7-11 Careful monitoring should improve the medical care of patients with schizophrenia and bipolar disorder and help protect them from the medical risks associated with overweight and obesity.
Similar precautions are needed when schizophrenia and bipolar disorder co-occur with other medical illnesses treated by complex pharmacologic regimens, such as pulmonary disease, cancer, and HIV. Three points to keep in mind are:
- careful selection of treatments for co-occurring medical illnesses, considering potential effects on the primary psychiatric disorder
- the impact of psychotropics on patients’ medical illnesses
- and the potential for drug interactions.
Patients with schizophrenia or bipolar disorder face a higher risk of premature death, compared with the general population. Their overall mortality rate is elevated not only by higher suicide rates but also by higher rates of medical comorbidities, including obesity, diabetes mellitus, cardiovascular and pulmonary diseases, HIV infection, and cancer.1,2
Schizophrenia and bipolar disorder are also frequently complicated by psychiatric comorbidities including alcohol and substance use disorders (such as nicotine dependence), anxiety disorders, and eating disorders (such as binge eating).3,4 Even so, the impact of psychiatric comorbidity on medical morbidity and mortality has not been adequately investigated and has—until rather recently—received little attention from clinical researchers.
Within the past 5 years or so, epidemiologic and population studies have shown increased morbidity and mortality from medical illnesses in patients with serious and persistent mental illness.5,6 These findings have sparked renewed interest in behavioral, biological, and psychosocial factors associated with schizophrenia and bipolar disorder that may contribute to comorbid medical illnesses. These factors include:
- negative and depressive symptoms
- physical inactivity and poor diet
- hypothalamic-pituitary-adrenal axis dysregulation associated with acute psychotic and affective episodes
- amotivation
- social isolation
- limited access to primary and preventive health care.7
The degree to which these factors may elevate medical comorbidity rates in patients with Commentary these serious psychiatric illnesses has not been well-studied or described.
TREATMENT IMPLICATIONS
These observations raise concerns about safe and effective use of medications by patients with schizophrenia or bipolar disorder as well as medical illness. Issues for clinicians to consider include:
- possible pharmacokinetic and pharmacodynamic interactions in patients taking concomitant medications for psychiatric and medical illnesses
- potential beneficial and adverse effects of psychotropics on medical illnesses
- and—conversely—potential beneficial and adverse effects on mood and psychotic disorders of medications used to treat medical illnesses.
Being aware of metabolic effects when prescribing psychotropics is not a new idea. Lithium, for example, has long been known to cause weight gain, suppress thyroid hormone, and interact with thiazide diuretics when used to treat bipolar disorder. Thus, therapeutic blood monitoring is recommended for patients receiving acute and maintenance lithium therapy.
More recently, atypical antipsychotics such as olanzapine and clozapine (and risperidone and quetiapine to a lesser extent) have been shown to produce substantial weight gain and other metabolic effects that increase the risk of diabetes and cardiovascular disease.7 As a result, the American Diabetes Association—along with the American Psychiatric Association and other groups—now recommends that psychiatrists and other physicians monitor patients’ body weight and body mass index, vital signs, serum glucose, and lipids when prescribing these agents.7-11 Careful monitoring should improve the medical care of patients with schizophrenia and bipolar disorder and help protect them from the medical risks associated with overweight and obesity.
Similar precautions are needed when schizophrenia and bipolar disorder co-occur with other medical illnesses treated by complex pharmacologic regimens, such as pulmonary disease, cancer, and HIV. Three points to keep in mind are:
- careful selection of treatments for co-occurring medical illnesses, considering potential effects on the primary psychiatric disorder
- the impact of psychotropics on patients’ medical illnesses
- and the potential for drug interactions.
1. Kilbourne AM, Cornelius JR, Han X, et al. Burden of general medical conditions among individuals with bipolar disorder. Bipolar Disord 2004;6:368-73.
2. Meyer JM, Nasrallah HA. Medical illness and schizophrenia Washington, DC: American Psychiatric Press, Inc, 2003.
3. McElroy SL, Altshuler LL, Suppes T, et al. Axis I psychiatric comorbidity and its relationship with historical illness variables in 288 patients with bipolar disorder. Am J Psychiatry 2001;158:420-6.
4. Green AI, Canuso CM, Brenner MJ, et al. Detection and management of comorbidity in patients with schizophrenia. Psychiatr Clin North Am 2003;26:115-39.
5. Buda M, Tsuang MT, Fleming JA. Causes of death in DSM-III schizophrenics and other psychotics (atypical group). Comparison with the general population. Arch Gen Psychiatry 1988;45:283-5.
6. Osby U, Brandt L, Correia N, et al. Excess mortality in bipolar and unipolar disorder in Sweden. Arch Gen Psychiatry 2001;58:844-50.
7. Keck PE, Jr, Buse JB, Dagago-Jack S, et al. Managing metabolic concerns in patients with severe mental illness: a special report (Postgrad Med). Minneapolis, MN: McGraw-Hill, 2003.
8. American Diabetes Association. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care 2004;27:596-601.
9. Marder SR, Essock SM, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry 2004;161:1334-9.
10. Chue P, Kovacs CS. Safety and tolerability of atypical antipsychotics in patients with bipolar disorder: prevalence, monitoring and management. Bipolar Disord 2003;5(suppl 2):62-79.
11. Nasrallah HA, Newcomer JW. Atypical antipsychotics and metabolic dysregulation. Evaluating the risk/benefit equation and improving standard of care. J Clin Psychopharmacol 2004;24(suppl 2):S7-S14.
1. Kilbourne AM, Cornelius JR, Han X, et al. Burden of general medical conditions among individuals with bipolar disorder. Bipolar Disord 2004;6:368-73.
2. Meyer JM, Nasrallah HA. Medical illness and schizophrenia Washington, DC: American Psychiatric Press, Inc, 2003.
3. McElroy SL, Altshuler LL, Suppes T, et al. Axis I psychiatric comorbidity and its relationship with historical illness variables in 288 patients with bipolar disorder. Am J Psychiatry 2001;158:420-6.
4. Green AI, Canuso CM, Brenner MJ, et al. Detection and management of comorbidity in patients with schizophrenia. Psychiatr Clin North Am 2003;26:115-39.
5. Buda M, Tsuang MT, Fleming JA. Causes of death in DSM-III schizophrenics and other psychotics (atypical group). Comparison with the general population. Arch Gen Psychiatry 1988;45:283-5.
6. Osby U, Brandt L, Correia N, et al. Excess mortality in bipolar and unipolar disorder in Sweden. Arch Gen Psychiatry 2001;58:844-50.
7. Keck PE, Jr, Buse JB, Dagago-Jack S, et al. Managing metabolic concerns in patients with severe mental illness: a special report (Postgrad Med). Minneapolis, MN: McGraw-Hill, 2003.
8. American Diabetes Association. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care 2004;27:596-601.
9. Marder SR, Essock SM, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry 2004;161:1334-9.
10. Chue P, Kovacs CS. Safety and tolerability of atypical antipsychotics in patients with bipolar disorder: prevalence, monitoring and management. Bipolar Disord 2003;5(suppl 2):62-79.
11. Nasrallah HA, Newcomer JW. Atypical antipsychotics and metabolic dysregulation. Evaluating the risk/benefit equation and improving standard of care. J Clin Psychopharmacol 2004;24(suppl 2):S7-S14.
IM risperidone: Long-acting atypical antipsychotic
Patients with chronic schizophrenia are notoriously inconsistent in adhering to medications.1 Partial compliance is a serious problem because the resulting psychotic relapses often lead to progressive neurologic and clinical deterioration as well as social and vocational impairment.
Long-acting, “depot” formulations of first-generation antipsychotics (haloperidol decanoate and fluphenazine decanoate) have been used over the past 25 years to ensure adherence in the least-compliant patients. These formulations, however, are not widely used because of the risks of tardive dyskinesia (TD) and other movement disorders.
Atypical antipsychotics—with their reduced risk of TD—have become the standard of care in managing schizophrenia and related psychosis over the long term.2 All are administered orally, however, and—until now—none has been available in a long-acting formulation.
The FDA recently approved a long-acting, injectable form of risperidone (Table 1), based on results of a 12-week, placebo-controlled trial and a 1-year open-label trial. The clinical and functional improvements seen in chronic schizophrenia patients who received long-acting risperidone for 1 to 3 years may change prevailing notions of the potential to stabilize and restore function in this severe brain disorder.
Table 1
Injectable, long-acting risperidone: Fast facts
| Drug brand name: Risperdal Consta |
| Class: Long-acting injectable atypical antipsychotic |
| FDA-approved indications: Schizophrenia |
| Approval date: Oct. 29, 2003 |
| Manufacturer: Janssen Pharmaceutica |
| Dosing forms: 25 mg, 37.5 mg, 50 mg |
| Recommended dosage: Most adult patients will be started at 25 mg every 2 weeks, with dosages titrated upward as needed |
| Estimated date of availability: By January 2004 |
| Dosing equivalencies (approximate): 25 mg IM biweekly = 3 mg/d oral 37.5 mg IM biweekly = 4.5 mg/d oral 50 mg IM biweekly = 5 to 6 mg/d oral |
HOW LONG-ACTING RISPERIDONE WORKS
Long-acting injectable risperidone has the same mechanism of action as oral risperidone. The injectable form is delivered into muscle tissue by microspheres that encapsulate the drug into a biodegradable polymer. The microspheres undergo gradual hydrolysis, resulting in a gradual release of risperidone into the blood stream. The drug then crosses the blood-brain barrier to block dopamine D2 and serotonin 5HT2A receptors in brain tissue, which is accepted as the pharmacodynamic basis for the efficacy of atypical antipsychotics. Risperidone’s receptor-binding profile is shown in Table 2.
CLINICAL PHARMACOKINETICS
Full release of long-acting risperidone from the gradually hydrolyzing microspheres starts about 3 weeks after an intramuscular (IM) injection. Thus, supplemental oral risperidone is recommended during the first 3 weeks of IM injections. Release is then maintained for 4 to 6 weeks. Steady-state plasma levels are reached after four biweekly injections. Risperidone is absorbed completely from the microspheres, which are biodegradable to carbon dioxide and water.
In plasma, risperidone is oxidized by the cytochrome P-450 isoenzyme CYP 2D6 to 9-hydroxy risperidone, an active metabolite similar to risperidone in its pharmacologic characteristics and efficacy. Risperidone is also metabolized via N-dealkylation. The plasma protein binding of risperidone is 90% and that of 9-hydroxy risperidone is 77%. After several biweekly IM injections of 25 or 50 mg during clinical trials, median trough and peak plasma concentrations of active moiety fluctuated between 9.9 and 19.2 ng/ml and 17.9 and 45.5 ng/ml, respectively.
Table 2
Receptor-binding profile of risperidone long-acting formulation
| Receptor type | Effects |
|---|---|
| Dopamine D2 | Antagonism (< haloperidol) |
| Serotonin 5HT2A | Antagonism (170 times > haloperidol) |
| Alpha 1 | Low affinity |
| Alpha 2 | Low affinity |
| Histaminic | Low affinity |
| Muscarinic | No affinity |
Risperidone plasma concentrations may be affected by interactions with other psychotropics that inhibit or induce the oxidative enzyme CYP 2D6 (Table 3).
Clearance of risperidone and 9-hydroxy risperidone is decreased by 60% in patients with severe kidney disease, as compared with healthy subjects. Plasma levels and maximum drug concentrations are 25 to 32% lower with long-acting risperidone than with oral risperidone. This difference may account for the injectable formulation’s more favorable side-effect profile because lower peaks means a lower likelihood of side effects.
Table 3
Potential drug-drug interactions with risperidone long-acting microspheres
| Drug | CYP enzyme affected | Effect on plasma concentration of risperidone |
|---|---|---|
| Fluphenazine | 2D6 | Increase |
| Paroxetine | 2D6 | Increase |
| Carbamazepine | 2D6 | Decrease |
RESULTS FROM CLINICAL TRIALS
Long-acting risperidone was tested at doses of 25, 50, and 75 mg in a 12-week, double-blind trial of 400 patients with acute relapse of schizophrenia.4 During the 3-week initial titration, patients also received the usual dosage of oral risperidone (3 to 5 mg/d) for schizophrenia. Oral risperidone can be discontinued 3 weeks after the first injection (ie, 1 week after the second injection). Measurement of serum concentrations is not needed because the microspheres encapsulating risperidone have been shown in bioavailability studies to begin disintegrating and releasing risperidone 3 weeks after being deposited into muscle tissue.
All three doses were more effective than placebo in reducing total, positive, and negative symptom scores, as measured by the Positive and Negative Syndrome Scale (PANSS). The 75-mg dose showed no greater efficacy than the 50-mg dose.
In a second, open-label study, 775 stable outpatients with schizophrenia or schizoaffective disorder received biweekly injections of 25, 50, or 75 mg of long-acting risperidone for 1 year. All three doses improved the baseline PANSS scores significantly, above and beyond the patients’ stable clinical status. These results indicate that injectable long-acting risperidone can further stabilize schizophrenia beyond the usual response to oral antipsychotics.5
Notably, patients’ quality-of-life scores—as measured by the 36-item Short-Form Health Survey (SF-36)—were significantly lower than U.S. norms at baseline. At the end of the study, patients’ scores had increased to within the norm range.6 The completion rate in this 1-year study was 65%; patients dropped out because of insufficient response (7%) or adverse events (5%), or they withdrew consent (15%) or were lost to follow-up (3%).
SAFETY AND TOLERABILITY
Few side effects were seen in the 12-week and 1-year trials. Extrapyramidal symptoms as measured by the Extrapyramidal Symptom Rating Scale declined from baseline by 67% with the 25-mg dose, by 50% with the 50-mg dose, and by 33% with the 75-mg dose in the 12-week study. Patients who had TD at baseline also improved by the end of the 1-year study, suggesting that long-acting risperidone has a low risk of TD.7 Also:
- Although prolactin levels were elevated compared with baseline, they were 18% lower with long-acting risperidone than with oral risperidone, possibly because of lower plasma peaks of the drug in the long-acting formulation.
- Injection site pain or redness was minimal, as measured by patient ratings.
- Mean weight gain after 12 weeks was 0.5 kg with the 25-mg dose, 1.2 kg with the 50-mg dose, and 1.9 kg with the 75-mg dose. After 52 weeks, weight gain was 1.8 kg, 2.1 kg, and 2.7 kg, respectively.
- QTc prolongation—as measured with random ECGs—was negligible with all doses.
REPARATIVE EFFECTS?
Based on clinical trial results, long-acting risperidone appears to be highly effective in treating and preventing relapse of acute psychotic episodes in schizophrenia. Its injectable formulation ensures that compliance is far more consistent than with oral atypical antipsychotics.
Patients who had been disabled with chronic schizophrenia improved dramatically after about 1 year of biweekly injections of long-acting risperidone. Many were able to return to school to finish a degree, go back to holding full-time jobs, or develop close personal relationships such as dating. Total PANSS scores after 1 year of treatment approached the low 40s in some patients, which is similar to what a healthy person might score on the PANSS on certain days. This pattern, which justifies the term “recovery,” suggests that uninterrupted, long-term atypical antipsychotic treatment may have reparative and/or neuroprotective effects on the brain in schizophrenia.8
Candidates for long-acting injectable risperidone include:
- first-episode patients
- patients with a history of partial or complete noncompliance
- patients who become violent or assaultive when they relapse
- and those receiving depot injections of haloperidol decanoate or fluphenazine decanoate.
Long-acting injectable atypical antipsychotics may become the standard of care for treating newonset schizophrenia.9 The goal would be to return patients to baseline functioning as soon as possible, rather than resorting to a long-acting antipsychotic only after repetitive relapses, adverse neuroplastic changes, and psychosocial decline.
Related resources
- Lasser R, Bossie C, Zhu Y, Gharahawi G. Does constant therapy infer optimal efficacy in schizophrenia? Moving to an advanced pharmacotherapeutic option. Schizophr Res 2003;60:291.
- Risperdal Consta Web site. www.risperdalconsta.com
Drug brand names
- Carbamazepine • Tegretol
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Paroxetine • Paxil
Disclosure
The author participated in the 12-week controlled clinical trial of long-acting, injectable risperidone and in an open-label extension for more than 3 additional years.
Dr. Nasrallah reports that he also receives research/grant support from and is a consultant to and speaker for AstraZeneca Pharmaceuticals, Janssen Pharmaceutica, and Pfizer Inc., and is a consultant to and speaker for Bristol-Myers Squibb Co. and Abbott Laboratories.
Acknowledgment
The author thanks Peggy Grause for her assistance in preparing this manuscript for publication.
1. Barnes TR, Curson DA. Long-acting depot antipsychotics: a riskbenefit assessment. Drug Safety 1994;10:464-79.
2. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. Philadelphia: Handbooks in Health Care, 2002.
3. Kane JM, Eerdekens M, Lindenmayer JP, et al. Long-acting injectable risperidone: efficacy and safety of the first long-acting atypical antipsychotic. Am J Psychiatry 2003;160:1125-32.
4. Remington G, Light M, Lasser R, et al. Can stable patients with schizophrenia improve? The impact of partial compliance versus constant therapy. Schizophr Res 2003;60:300.-
5. Nasrallah HA, Duchesne J, Mehnert A, Janagap C. Improved quality of life in schizophrenia with long-acting intramuscular risperidone. J Neuropsychopharmacol 2002;5(suppl):390.-
6. Chouinard G, Lasser R, Bossie C, et al. Does a long-acting atypical antipsychotic offer a low risk of tardive dyskinesia in patients with schizophrenia? Schizophr Res 2003;60:277.-
7. Nasrallah HA, Mahadik S, Evans D, Keshavan M. Are atypical antipsychotics neuroprotective? Evidence from animal and human studies. Biol Psychiatry 2002;51(suppl 8S):25.-
8. Thompson PM, Vidal C, Giedd JN, et al. Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. PNAS 2001;98:11,650-5.
Patients with chronic schizophrenia are notoriously inconsistent in adhering to medications.1 Partial compliance is a serious problem because the resulting psychotic relapses often lead to progressive neurologic and clinical deterioration as well as social and vocational impairment.
Long-acting, “depot” formulations of first-generation antipsychotics (haloperidol decanoate and fluphenazine decanoate) have been used over the past 25 years to ensure adherence in the least-compliant patients. These formulations, however, are not widely used because of the risks of tardive dyskinesia (TD) and other movement disorders.
Atypical antipsychotics—with their reduced risk of TD—have become the standard of care in managing schizophrenia and related psychosis over the long term.2 All are administered orally, however, and—until now—none has been available in a long-acting formulation.
The FDA recently approved a long-acting, injectable form of risperidone (Table 1), based on results of a 12-week, placebo-controlled trial and a 1-year open-label trial. The clinical and functional improvements seen in chronic schizophrenia patients who received long-acting risperidone for 1 to 3 years may change prevailing notions of the potential to stabilize and restore function in this severe brain disorder.
Table 1
Injectable, long-acting risperidone: Fast facts
| Drug brand name: Risperdal Consta |
| Class: Long-acting injectable atypical antipsychotic |
| FDA-approved indications: Schizophrenia |
| Approval date: Oct. 29, 2003 |
| Manufacturer: Janssen Pharmaceutica |
| Dosing forms: 25 mg, 37.5 mg, 50 mg |
| Recommended dosage: Most adult patients will be started at 25 mg every 2 weeks, with dosages titrated upward as needed |
| Estimated date of availability: By January 2004 |
| Dosing equivalencies (approximate): 25 mg IM biweekly = 3 mg/d oral 37.5 mg IM biweekly = 4.5 mg/d oral 50 mg IM biweekly = 5 to 6 mg/d oral |
HOW LONG-ACTING RISPERIDONE WORKS
Long-acting injectable risperidone has the same mechanism of action as oral risperidone. The injectable form is delivered into muscle tissue by microspheres that encapsulate the drug into a biodegradable polymer. The microspheres undergo gradual hydrolysis, resulting in a gradual release of risperidone into the blood stream. The drug then crosses the blood-brain barrier to block dopamine D2 and serotonin 5HT2A receptors in brain tissue, which is accepted as the pharmacodynamic basis for the efficacy of atypical antipsychotics. Risperidone’s receptor-binding profile is shown in Table 2.
CLINICAL PHARMACOKINETICS
Full release of long-acting risperidone from the gradually hydrolyzing microspheres starts about 3 weeks after an intramuscular (IM) injection. Thus, supplemental oral risperidone is recommended during the first 3 weeks of IM injections. Release is then maintained for 4 to 6 weeks. Steady-state plasma levels are reached after four biweekly injections. Risperidone is absorbed completely from the microspheres, which are biodegradable to carbon dioxide and water.
In plasma, risperidone is oxidized by the cytochrome P-450 isoenzyme CYP 2D6 to 9-hydroxy risperidone, an active metabolite similar to risperidone in its pharmacologic characteristics and efficacy. Risperidone is also metabolized via N-dealkylation. The plasma protein binding of risperidone is 90% and that of 9-hydroxy risperidone is 77%. After several biweekly IM injections of 25 or 50 mg during clinical trials, median trough and peak plasma concentrations of active moiety fluctuated between 9.9 and 19.2 ng/ml and 17.9 and 45.5 ng/ml, respectively.
Table 2
Receptor-binding profile of risperidone long-acting formulation
| Receptor type | Effects |
|---|---|
| Dopamine D2 | Antagonism (< haloperidol) |
| Serotonin 5HT2A | Antagonism (170 times > haloperidol) |
| Alpha 1 | Low affinity |
| Alpha 2 | Low affinity |
| Histaminic | Low affinity |
| Muscarinic | No affinity |
Risperidone plasma concentrations may be affected by interactions with other psychotropics that inhibit or induce the oxidative enzyme CYP 2D6 (Table 3).
Clearance of risperidone and 9-hydroxy risperidone is decreased by 60% in patients with severe kidney disease, as compared with healthy subjects. Plasma levels and maximum drug concentrations are 25 to 32% lower with long-acting risperidone than with oral risperidone. This difference may account for the injectable formulation’s more favorable side-effect profile because lower peaks means a lower likelihood of side effects.
Table 3
Potential drug-drug interactions with risperidone long-acting microspheres
| Drug | CYP enzyme affected | Effect on plasma concentration of risperidone |
|---|---|---|
| Fluphenazine | 2D6 | Increase |
| Paroxetine | 2D6 | Increase |
| Carbamazepine | 2D6 | Decrease |
RESULTS FROM CLINICAL TRIALS
Long-acting risperidone was tested at doses of 25, 50, and 75 mg in a 12-week, double-blind trial of 400 patients with acute relapse of schizophrenia.4 During the 3-week initial titration, patients also received the usual dosage of oral risperidone (3 to 5 mg/d) for schizophrenia. Oral risperidone can be discontinued 3 weeks after the first injection (ie, 1 week after the second injection). Measurement of serum concentrations is not needed because the microspheres encapsulating risperidone have been shown in bioavailability studies to begin disintegrating and releasing risperidone 3 weeks after being deposited into muscle tissue.
All three doses were more effective than placebo in reducing total, positive, and negative symptom scores, as measured by the Positive and Negative Syndrome Scale (PANSS). The 75-mg dose showed no greater efficacy than the 50-mg dose.
In a second, open-label study, 775 stable outpatients with schizophrenia or schizoaffective disorder received biweekly injections of 25, 50, or 75 mg of long-acting risperidone for 1 year. All three doses improved the baseline PANSS scores significantly, above and beyond the patients’ stable clinical status. These results indicate that injectable long-acting risperidone can further stabilize schizophrenia beyond the usual response to oral antipsychotics.5
Notably, patients’ quality-of-life scores—as measured by the 36-item Short-Form Health Survey (SF-36)—were significantly lower than U.S. norms at baseline. At the end of the study, patients’ scores had increased to within the norm range.6 The completion rate in this 1-year study was 65%; patients dropped out because of insufficient response (7%) or adverse events (5%), or they withdrew consent (15%) or were lost to follow-up (3%).
SAFETY AND TOLERABILITY
Few side effects were seen in the 12-week and 1-year trials. Extrapyramidal symptoms as measured by the Extrapyramidal Symptom Rating Scale declined from baseline by 67% with the 25-mg dose, by 50% with the 50-mg dose, and by 33% with the 75-mg dose in the 12-week study. Patients who had TD at baseline also improved by the end of the 1-year study, suggesting that long-acting risperidone has a low risk of TD.7 Also:
- Although prolactin levels were elevated compared with baseline, they were 18% lower with long-acting risperidone than with oral risperidone, possibly because of lower plasma peaks of the drug in the long-acting formulation.
- Injection site pain or redness was minimal, as measured by patient ratings.
- Mean weight gain after 12 weeks was 0.5 kg with the 25-mg dose, 1.2 kg with the 50-mg dose, and 1.9 kg with the 75-mg dose. After 52 weeks, weight gain was 1.8 kg, 2.1 kg, and 2.7 kg, respectively.
- QTc prolongation—as measured with random ECGs—was negligible with all doses.
REPARATIVE EFFECTS?
Based on clinical trial results, long-acting risperidone appears to be highly effective in treating and preventing relapse of acute psychotic episodes in schizophrenia. Its injectable formulation ensures that compliance is far more consistent than with oral atypical antipsychotics.
Patients who had been disabled with chronic schizophrenia improved dramatically after about 1 year of biweekly injections of long-acting risperidone. Many were able to return to school to finish a degree, go back to holding full-time jobs, or develop close personal relationships such as dating. Total PANSS scores after 1 year of treatment approached the low 40s in some patients, which is similar to what a healthy person might score on the PANSS on certain days. This pattern, which justifies the term “recovery,” suggests that uninterrupted, long-term atypical antipsychotic treatment may have reparative and/or neuroprotective effects on the brain in schizophrenia.8
Candidates for long-acting injectable risperidone include:
- first-episode patients
- patients with a history of partial or complete noncompliance
- patients who become violent or assaultive when they relapse
- and those receiving depot injections of haloperidol decanoate or fluphenazine decanoate.
Long-acting injectable atypical antipsychotics may become the standard of care for treating newonset schizophrenia.9 The goal would be to return patients to baseline functioning as soon as possible, rather than resorting to a long-acting antipsychotic only after repetitive relapses, adverse neuroplastic changes, and psychosocial decline.
Related resources
- Lasser R, Bossie C, Zhu Y, Gharahawi G. Does constant therapy infer optimal efficacy in schizophrenia? Moving to an advanced pharmacotherapeutic option. Schizophr Res 2003;60:291.
- Risperdal Consta Web site. www.risperdalconsta.com
Drug brand names
- Carbamazepine • Tegretol
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Paroxetine • Paxil
Disclosure
The author participated in the 12-week controlled clinical trial of long-acting, injectable risperidone and in an open-label extension for more than 3 additional years.
Dr. Nasrallah reports that he also receives research/grant support from and is a consultant to and speaker for AstraZeneca Pharmaceuticals, Janssen Pharmaceutica, and Pfizer Inc., and is a consultant to and speaker for Bristol-Myers Squibb Co. and Abbott Laboratories.
Acknowledgment
The author thanks Peggy Grause for her assistance in preparing this manuscript for publication.
Patients with chronic schizophrenia are notoriously inconsistent in adhering to medications.1 Partial compliance is a serious problem because the resulting psychotic relapses often lead to progressive neurologic and clinical deterioration as well as social and vocational impairment.
Long-acting, “depot” formulations of first-generation antipsychotics (haloperidol decanoate and fluphenazine decanoate) have been used over the past 25 years to ensure adherence in the least-compliant patients. These formulations, however, are not widely used because of the risks of tardive dyskinesia (TD) and other movement disorders.
Atypical antipsychotics—with their reduced risk of TD—have become the standard of care in managing schizophrenia and related psychosis over the long term.2 All are administered orally, however, and—until now—none has been available in a long-acting formulation.
The FDA recently approved a long-acting, injectable form of risperidone (Table 1), based on results of a 12-week, placebo-controlled trial and a 1-year open-label trial. The clinical and functional improvements seen in chronic schizophrenia patients who received long-acting risperidone for 1 to 3 years may change prevailing notions of the potential to stabilize and restore function in this severe brain disorder.
Table 1
Injectable, long-acting risperidone: Fast facts
| Drug brand name: Risperdal Consta |
| Class: Long-acting injectable atypical antipsychotic |
| FDA-approved indications: Schizophrenia |
| Approval date: Oct. 29, 2003 |
| Manufacturer: Janssen Pharmaceutica |
| Dosing forms: 25 mg, 37.5 mg, 50 mg |
| Recommended dosage: Most adult patients will be started at 25 mg every 2 weeks, with dosages titrated upward as needed |
| Estimated date of availability: By January 2004 |
| Dosing equivalencies (approximate): 25 mg IM biweekly = 3 mg/d oral 37.5 mg IM biweekly = 4.5 mg/d oral 50 mg IM biweekly = 5 to 6 mg/d oral |
HOW LONG-ACTING RISPERIDONE WORKS
Long-acting injectable risperidone has the same mechanism of action as oral risperidone. The injectable form is delivered into muscle tissue by microspheres that encapsulate the drug into a biodegradable polymer. The microspheres undergo gradual hydrolysis, resulting in a gradual release of risperidone into the blood stream. The drug then crosses the blood-brain barrier to block dopamine D2 and serotonin 5HT2A receptors in brain tissue, which is accepted as the pharmacodynamic basis for the efficacy of atypical antipsychotics. Risperidone’s receptor-binding profile is shown in Table 2.
CLINICAL PHARMACOKINETICS
Full release of long-acting risperidone from the gradually hydrolyzing microspheres starts about 3 weeks after an intramuscular (IM) injection. Thus, supplemental oral risperidone is recommended during the first 3 weeks of IM injections. Release is then maintained for 4 to 6 weeks. Steady-state plasma levels are reached after four biweekly injections. Risperidone is absorbed completely from the microspheres, which are biodegradable to carbon dioxide and water.
In plasma, risperidone is oxidized by the cytochrome P-450 isoenzyme CYP 2D6 to 9-hydroxy risperidone, an active metabolite similar to risperidone in its pharmacologic characteristics and efficacy. Risperidone is also metabolized via N-dealkylation. The plasma protein binding of risperidone is 90% and that of 9-hydroxy risperidone is 77%. After several biweekly IM injections of 25 or 50 mg during clinical trials, median trough and peak plasma concentrations of active moiety fluctuated between 9.9 and 19.2 ng/ml and 17.9 and 45.5 ng/ml, respectively.
Table 2
Receptor-binding profile of risperidone long-acting formulation
| Receptor type | Effects |
|---|---|
| Dopamine D2 | Antagonism (< haloperidol) |
| Serotonin 5HT2A | Antagonism (170 times > haloperidol) |
| Alpha 1 | Low affinity |
| Alpha 2 | Low affinity |
| Histaminic | Low affinity |
| Muscarinic | No affinity |
Risperidone plasma concentrations may be affected by interactions with other psychotropics that inhibit or induce the oxidative enzyme CYP 2D6 (Table 3).
Clearance of risperidone and 9-hydroxy risperidone is decreased by 60% in patients with severe kidney disease, as compared with healthy subjects. Plasma levels and maximum drug concentrations are 25 to 32% lower with long-acting risperidone than with oral risperidone. This difference may account for the injectable formulation’s more favorable side-effect profile because lower peaks means a lower likelihood of side effects.
Table 3
Potential drug-drug interactions with risperidone long-acting microspheres
| Drug | CYP enzyme affected | Effect on plasma concentration of risperidone |
|---|---|---|
| Fluphenazine | 2D6 | Increase |
| Paroxetine | 2D6 | Increase |
| Carbamazepine | 2D6 | Decrease |
RESULTS FROM CLINICAL TRIALS
Long-acting risperidone was tested at doses of 25, 50, and 75 mg in a 12-week, double-blind trial of 400 patients with acute relapse of schizophrenia.4 During the 3-week initial titration, patients also received the usual dosage of oral risperidone (3 to 5 mg/d) for schizophrenia. Oral risperidone can be discontinued 3 weeks after the first injection (ie, 1 week after the second injection). Measurement of serum concentrations is not needed because the microspheres encapsulating risperidone have been shown in bioavailability studies to begin disintegrating and releasing risperidone 3 weeks after being deposited into muscle tissue.
All three doses were more effective than placebo in reducing total, positive, and negative symptom scores, as measured by the Positive and Negative Syndrome Scale (PANSS). The 75-mg dose showed no greater efficacy than the 50-mg dose.
In a second, open-label study, 775 stable outpatients with schizophrenia or schizoaffective disorder received biweekly injections of 25, 50, or 75 mg of long-acting risperidone for 1 year. All three doses improved the baseline PANSS scores significantly, above and beyond the patients’ stable clinical status. These results indicate that injectable long-acting risperidone can further stabilize schizophrenia beyond the usual response to oral antipsychotics.5
Notably, patients’ quality-of-life scores—as measured by the 36-item Short-Form Health Survey (SF-36)—were significantly lower than U.S. norms at baseline. At the end of the study, patients’ scores had increased to within the norm range.6 The completion rate in this 1-year study was 65%; patients dropped out because of insufficient response (7%) or adverse events (5%), or they withdrew consent (15%) or were lost to follow-up (3%).
SAFETY AND TOLERABILITY
Few side effects were seen in the 12-week and 1-year trials. Extrapyramidal symptoms as measured by the Extrapyramidal Symptom Rating Scale declined from baseline by 67% with the 25-mg dose, by 50% with the 50-mg dose, and by 33% with the 75-mg dose in the 12-week study. Patients who had TD at baseline also improved by the end of the 1-year study, suggesting that long-acting risperidone has a low risk of TD.7 Also:
- Although prolactin levels were elevated compared with baseline, they were 18% lower with long-acting risperidone than with oral risperidone, possibly because of lower plasma peaks of the drug in the long-acting formulation.
- Injection site pain or redness was minimal, as measured by patient ratings.
- Mean weight gain after 12 weeks was 0.5 kg with the 25-mg dose, 1.2 kg with the 50-mg dose, and 1.9 kg with the 75-mg dose. After 52 weeks, weight gain was 1.8 kg, 2.1 kg, and 2.7 kg, respectively.
- QTc prolongation—as measured with random ECGs—was negligible with all doses.
REPARATIVE EFFECTS?
Based on clinical trial results, long-acting risperidone appears to be highly effective in treating and preventing relapse of acute psychotic episodes in schizophrenia. Its injectable formulation ensures that compliance is far more consistent than with oral atypical antipsychotics.
Patients who had been disabled with chronic schizophrenia improved dramatically after about 1 year of biweekly injections of long-acting risperidone. Many were able to return to school to finish a degree, go back to holding full-time jobs, or develop close personal relationships such as dating. Total PANSS scores after 1 year of treatment approached the low 40s in some patients, which is similar to what a healthy person might score on the PANSS on certain days. This pattern, which justifies the term “recovery,” suggests that uninterrupted, long-term atypical antipsychotic treatment may have reparative and/or neuroprotective effects on the brain in schizophrenia.8
Candidates for long-acting injectable risperidone include:
- first-episode patients
- patients with a history of partial or complete noncompliance
- patients who become violent or assaultive when they relapse
- and those receiving depot injections of haloperidol decanoate or fluphenazine decanoate.
Long-acting injectable atypical antipsychotics may become the standard of care for treating newonset schizophrenia.9 The goal would be to return patients to baseline functioning as soon as possible, rather than resorting to a long-acting antipsychotic only after repetitive relapses, adverse neuroplastic changes, and psychosocial decline.
Related resources
- Lasser R, Bossie C, Zhu Y, Gharahawi G. Does constant therapy infer optimal efficacy in schizophrenia? Moving to an advanced pharmacotherapeutic option. Schizophr Res 2003;60:291.
- Risperdal Consta Web site. www.risperdalconsta.com
Drug brand names
- Carbamazepine • Tegretol
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Paroxetine • Paxil
Disclosure
The author participated in the 12-week controlled clinical trial of long-acting, injectable risperidone and in an open-label extension for more than 3 additional years.
Dr. Nasrallah reports that he also receives research/grant support from and is a consultant to and speaker for AstraZeneca Pharmaceuticals, Janssen Pharmaceutica, and Pfizer Inc., and is a consultant to and speaker for Bristol-Myers Squibb Co. and Abbott Laboratories.
Acknowledgment
The author thanks Peggy Grause for her assistance in preparing this manuscript for publication.
1. Barnes TR, Curson DA. Long-acting depot antipsychotics: a riskbenefit assessment. Drug Safety 1994;10:464-79.
2. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. Philadelphia: Handbooks in Health Care, 2002.
3. Kane JM, Eerdekens M, Lindenmayer JP, et al. Long-acting injectable risperidone: efficacy and safety of the first long-acting atypical antipsychotic. Am J Psychiatry 2003;160:1125-32.
4. Remington G, Light M, Lasser R, et al. Can stable patients with schizophrenia improve? The impact of partial compliance versus constant therapy. Schizophr Res 2003;60:300.-
5. Nasrallah HA, Duchesne J, Mehnert A, Janagap C. Improved quality of life in schizophrenia with long-acting intramuscular risperidone. J Neuropsychopharmacol 2002;5(suppl):390.-
6. Chouinard G, Lasser R, Bossie C, et al. Does a long-acting atypical antipsychotic offer a low risk of tardive dyskinesia in patients with schizophrenia? Schizophr Res 2003;60:277.-
7. Nasrallah HA, Mahadik S, Evans D, Keshavan M. Are atypical antipsychotics neuroprotective? Evidence from animal and human studies. Biol Psychiatry 2002;51(suppl 8S):25.-
8. Thompson PM, Vidal C, Giedd JN, et al. Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. PNAS 2001;98:11,650-5.
1. Barnes TR, Curson DA. Long-acting depot antipsychotics: a riskbenefit assessment. Drug Safety 1994;10:464-79.
2. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. Philadelphia: Handbooks in Health Care, 2002.
3. Kane JM, Eerdekens M, Lindenmayer JP, et al. Long-acting injectable risperidone: efficacy and safety of the first long-acting atypical antipsychotic. Am J Psychiatry 2003;160:1125-32.
4. Remington G, Light M, Lasser R, et al. Can stable patients with schizophrenia improve? The impact of partial compliance versus constant therapy. Schizophr Res 2003;60:300.-
5. Nasrallah HA, Duchesne J, Mehnert A, Janagap C. Improved quality of life in schizophrenia with long-acting intramuscular risperidone. J Neuropsychopharmacol 2002;5(suppl):390.-
6. Chouinard G, Lasser R, Bossie C, et al. Does a long-acting atypical antipsychotic offer a low risk of tardive dyskinesia in patients with schizophrenia? Schizophr Res 2003;60:277.-
7. Nasrallah HA, Mahadik S, Evans D, Keshavan M. Are atypical antipsychotics neuroprotective? Evidence from animal and human studies. Biol Psychiatry 2002;51(suppl 8S):25.-
8. Thompson PM, Vidal C, Giedd JN, et al. Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. PNAS 2001;98:11,650-5.