User login
Management of Blood Pressure after Stroke
Hospitalists are on the front lines of care for patients with cerebrovascular accidents. After the first steps in acute stroke management occur in the emergency room, care is frequently transferred to the hospitalist. This review focuses on the evidence‐based management of blood pressure following acute ischemic stroke. Management of blood pressure after stroke is still controversial, and current consensus statement guidelines acknowledge that optimal treatment has not yet been established.1, 2 As such, it is essential to understand the changes in normal homeostatic physiologic processes that occur after stroke and their subsequent effects on neurologic function. Only then can the appropriate blood pressure target and antihypertensive regimen be chosen.
Physiology of Cerebral Perfusion
Once a stroke has occurred and perfusion to a section of brain tissue has been acutely compromised, systemic pressure tends to rise. This rise is presumably due in part to increased adrenergic tone and activation of the renin‐aldosterone system and potentially is part of Cushing's reflex in cases in which intracranial pressure is elevated.3 The increase in mean arterial pressure may represent a protective response. In the first major study on the topic, in 1981, on admission for stroke mean blood pressure was 163/90 mm Hg for patients without a history of hypertension and 214/118 mm Hg for patients who had been treated for hypertension previously.4 This rise in blood pressure in response to endogenous mechanisms is attenuated over the first 24 hours, and even without intervention, blood pressure tends to fall spontaneously over the next 10 days.47
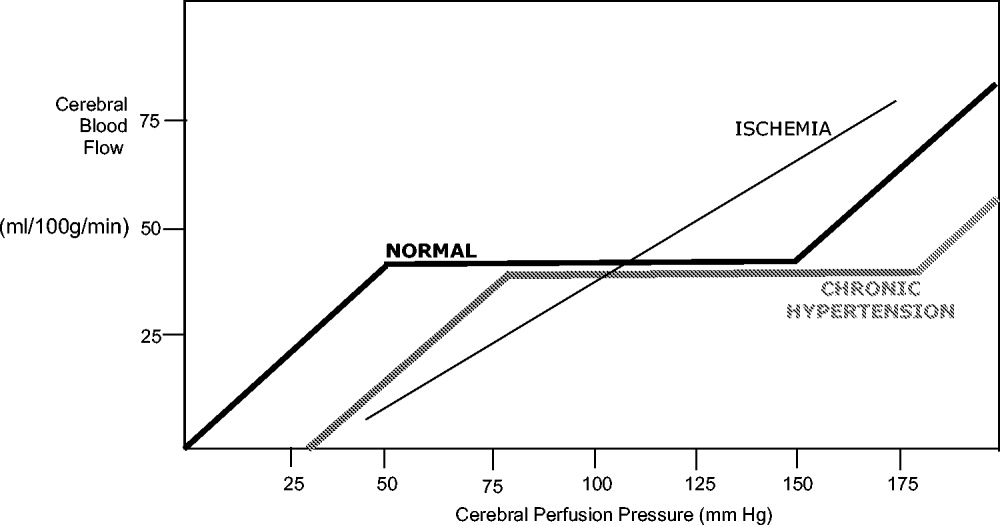
Under normal circumstances, cerebral blood flow (CBF) is tightly autoregulated across a wide range of cerebral perfusion pressures by alteration in cerebrovascular resistance via arteriolar constriction.89 This allows CBF to remain constant even if cerebral perfusion pressure (CPP) fluctuates from 60 to 150 mm Hg.9 In patients with chronic hypertension, autoregulation works best with blood pressure in a higher range because of vascular smooth muscle hypertrophy and structural changes in the cerebral vessels (see Table 1).
| Cerebral blood flow (CBF) of 50‐70 mL/100 g/minnormal |
| CBF of 20‐50 mL/100 g/minreduced flow compensated for by increased oxygen extraction |
| CBF of 15‐20 mL/100 g/minneuronal quiescence |
| CBF 15 mL/100 g/minneuronal death |
After an acute ischemic stroke, autoregulation is lost, and local CBF becomes linearly associated with cerebral perfusion pressure. Loss of autoregulation occurs because of several local factors as a result of the infarct. Acidosis and hypoxia in the region of the stroke lead to vasodilatation in the perfusing vessels.9 This may improve local circulation in collateral vessels as a response to the obstruction in the primary supplying artery, but the consequence of maximal vasodilatation is loss of the ability to autoregulate. Normal CPP is driven by the mean arterial pressure minus the intracranial pressure. However, if intracranial vessels have lost the ability to accommodate changes in perfusion pressure, then blood flow to the area of injury becomes linearly correlated with mean arterial pressure.
Surrounding a central core of irreversible necrosis may be a zone of at‐risk tissue that is susceptible to reduction below the threshold of viability in response to any decrement in systemic mean blood pressure.11 This critical concept in stroke management is referred to as the peri‐infarct penumbra (see Fig. 1 and Table 2).
| Mean arterial pressure (MAP) = ⅔ diastolic blood pressure (DBP) + ⅓ systolic blood pressure (SBP) |
| Cerebral perfusion pressure (CPP) = mean arterial pressure (MAP) intracranial pressure (ICP) |
| Cerebral blood flow (CBF) = cerebral perfusion pressure (CPP)/cerebrovascular resistance (CVR) |
Blood Pressure Management after Stroke
The presence of a systemic blood pressure‐dependent peri‐infarct penumbra that might be compromised by blood pressure reduction and thus extend the infarct is the principal argument for allowing permissive hypertension. This is bolstered by the observation that decreases in blood pressure in the first 24 hours are associated with a significant risk of poor neurologic outcome.6 In their an observational study Vlcek et al. found that a greater than 25% drop in diastolic blood pressure (DBP) was associated with a 4‐fold risk of severe disability.5 In a 2003 observational study Oliveria et al. found that reduction in systolic blood pressure (SBP) in the first 24 hours was independently associated with poor outcomes, with a doubling of the risk for poor outcomes for every 10% drop in systolic blood pressure.6 In both these studies, the worsened outcome did not depend on whether the drop was spontaneous or induced by medications. Case reports suggest that large drops in blood pressure can be catastrophic and that even moderate lowering of blood pressure after acute stroke can be associated with clinical deterioration.8, 12
The primary argument for lowering blood pressure after an acute stroke hinges on secondary prevention of new ischemic events, minimization of cerebral edema, and prevention of hemorrhagic conversion. It has long been recognized that hypertension itself is a risk factor for stroke13 and that reduction in chronic hypertension is part of secondary prevention for cerebrovascular accidents.14 Further, hypertension is the most common risk factor for intracerebral hemorrhage, and it would stand to reason that damage to the brain parenchyma from ischemic stroke would increase the risk of pressure‐induced bleeding in an acute setting.15
Because there are compelling theoretical arguments both for and against lowering blood pressure after an acute stroke, it is necessary to look at the results of randomized clinical trials for guidance in weighing the risks and benefits. The overall goal of blood pressure management is to maximize perfusion to the ischemic penumbra while minimizing the hypertensive risk of hemorrhagic transformation.
Lisk et al. conducted a small randomized trial in 1993 of antihypertensive therapy versus placebo after ischemic stroke looking at SPECT perfusion and found lower CBF if mean arterial pressure (MAP) dropped more than 16%.16 This is in contrast with the results of a 1997 randomized trial of perindopril after cerebrovascular accident that found no decrease in Doppler CBF despite a 10% decrease in blood pressure in the active treatment group.17 Neither study was powered to detect significant differences in clinical outcome.
Early randomized controlled trials of antihypertensive agents after acute stroke investigated neuroprotection via mechanisms other than the antihypertensive effect. Nimodipine, a dihydropyridine derivative thought to prevent neuronal death via blockade of calcium channels, has been studied extensively.1820 A meta‐analysis of the 9 early trials of nimodipine, from 1988 to 1992, suggested that nimodipine was potentially beneficial in neurologic score and functional outcome only if used within the first 12 hours and that it could be harmful if started after 24 hours.21 Two additional studies were done in 1994 using both intravenous and oral nimodipine formulations. They demonstrated worsened neurologic function and higher mortality, respectively, which was hypothesized to be a result of the detrimental hemodynamic effects of nimodipine.18, 22
The BEST trial used beta‐blockers in the early period after acute stroke and failed to find benefit, whereas the FIST trial had similar results with the calcium channel blocker flunarizine.23, 24 The ACCESS trial in 2003 was a prospective, randomized, controlled trial using oral candesartan in the first 24 hours after stroke.25 It did find improved mortality in the candesartan arm after 1 year, yet there were no significant differences in blood pressure between candesartan and placebo. Thus, the improved outcome was presumed to be a result of mechanisms other than antihypertensive effect.
Some significant caveats should be kept in mind when interpreting the results of these studies. None of the trials was designed to titrate blood pressure to a prespecified goal in a prospective randomized fashion. It is also difficult to tease out the effect of the active medical intervention from the effect of spontaneous drops in blood pressure, and many trials did not find a significant difference in blood pressure between the medication and placebo groups. A large randomized trial to evaluate interventions to a predefined blood pressure target is needed.
The Cochrane Stroke Group reviewed 32 trials involving 5368 patients and concluded there was not enough evidence to reliably evaluate the effect of altering blood pressure on outcomes.3
Recommendations for Treatment of Hypertension in Acute Stroke
The decision of when to initiate antihypertensive therapy has not been clearly delineated. However, most experts agree that blood pressure targets need to take into account whether thrombolytics are used. The available data provide little evidence that lowering blood pressure decreases adverse events; however, there is some evidence that lowering blood pressure can worsen outcomes by expanding the area of ischemia. Thus, in treating most acute ischemic strokes, antihypertensive therapy can and should be withheld. The exception to this is stroke in a patient with comorbid hypertensive organ damage such as myocardial infarction, aortic dissection, acute hypertensive renal failure, pulmonary edema, or encephalopathy. The consensus statement of the American Stroke Association is to withhold treatment until blood pressure exceeds 220/120 mm Hg,1 roughly corresponding to a MAP of 150 mm Hg, the normal upper limit of cerebral autoregulation. Treatment optimally should be titratable in order to avoid overcorrection, preferably with minimal cerebral venodilatation effect. The parenteral agents labetolol, fenoldopam, nicardipine, and nitroprusside are most commonly used. A disadvantage of nitrates or nitroprusside is that their venodilating effect may raise intracranial pressure. Clonidine may induce central nervous system depression, which can complicate interpretation of the mental status of a patient with an acute neurologic event. Sublingual nifedipine should be avoided because of its well‐documented tendency to cause overcorrection of blood pressure.26 No large direct comparison trial has evaluated which of these antihypertensive agents is superior in clinical outcomes. For the hospitalist, choice of antihypertensive agent should be driven by a need for rapid control of blood pressure to target without overcorrection (see Table 3). Issues such as intracranial pressure, heart rate, and comorbidities may also play a role in choice of medication.
| Not eligible for thrombolytic therapy | |
|---|---|
| |
| Systolic 220 or diastolic 120 | Observe unless other end‐organ involvement (aortic dissection, acute myocardial infarction, pulmonary edema, hypertensive encephalopathy) |
| Systolic > 220 or diastolic 121‐140 | Aim for 10%‐15% reduction in blood pressure |
| Labetalol 10‐20 mg IV over 1‐2 minutes; repeat or double every 10 minutes; maximum 300 mg | |
| or | |
| Nicardipine 5 mg/hour IV; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| or | |
| Nitroprusside 0.5 g/kg/minute IV; titrate to goal | |
| Diastolic > 140 | Aim for 10%‐15% reduction in blood pressure |
| Nitroprusside 0.5 g/kg/minute IV; titrate to desired blood pressure | |
| Eligible for thrombolytic therapy | |
| Pretreatment | |
| Systolic > 185 or diastolic > 110 | Labetalol 10‐20mg IV |
| May repeat 1 or Nitropaste 1‐2 inches | |
| During and after tPA | |
| Systolic 180‐230 or Diastolic 105‐120 | Labetalol 10 mg |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute | |
| Systolic > 230 or diastolic 121‐140 | Labetalol 10mg IV |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute or | |
| Nicardipine 5 mg/hour; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| If not at goal with labetalol or nicardipine, consider nitroprusside | |
| Diastolic>140 | Nitroprusside 0.5 g/kg/minute IV infusion; titrate to desired blood pressure |
The risk of hemorrhagic conversion is greater when thrombolysis is used for the treatment of cerebrovascular accident, in the NINDS trial rising from 0.6% in patients receiving a placebo to 6.4% in patients receiving tPA.27 Given that the goal of thrombolysis is to restore perfusion through the previously blocked vessel and that the risk of hemorrhage rises after thrombolysis, the balance between maintaining CPP and decreasing the risk of bleeding shifts toward maintaining a lower blood pressure postthrombolysis. The NINDS thrombolytic trial required patients to have a blood pressure less than 185/110 mm Hg to be included.27 Once a thrombolytic had been administered, the permitted maximum blood pressure dropped to 180/105 mm Hg. Blood pressure needs to be monitored closely, initially every 15 minutes for 2 hours, then every 30 minutes for the next 6 hours, and then hourly until the end of the first 24 hours. After this, blood pressure monitoring intervals can be more spaced out depending on the need for active antihypertensive therapy.28 However, arterial punctures for invasive monitoring are not recommended if thrombolytics are administered. Review of the experience of the NINDS trial suggests that approximately one third of patients receiving thrombolytics will require pharmacologic therapy to reach the recommended blood pressure goals in the first 24 hours.28
Although guidelines for the management of blood pressure after cerebrovascular accident from major organizations such as the American Stroke Association and the European Stroke Initiative are published and widely quoted in reviews of stroke management, physician adherence to these guidelines is poor.1, 2933 A 2002 review of prescribing practices in a Canadian hospital found excessive reduction of blood pressure in 60% of patients where nitroglycerin was used, and sublingual nifedipine was still the second most commonly prescribed medication.34 A similar 2004 study by Lindenauer et al. found that only 26% of patients who had antihypertensive medication initiated in the hospital met consensus guidelines for therapy.35
Induced Hypertension
Hypotension should be avoided after acute stroke. Observational studies have demonstrated an increased mortality rate of patients who present with hypotension.42 Patients with acute ischemic stroke and loss of autoregulation may have impaired tolerance to even mild levels of hypotension.8 First‐line therapy for hypotension after stroke is volume resuscitation and optimization of cardiac function by correcting arrhythmias.1 If this should fail, vasopressor support is advisable to avoid ongoing hypotension and cerebral hypoperfusion.
The same physiologic arguments that favor permissively managing high blood pressure and avoiding hypotension raise the question of whether inducing hypertension through the use of vasopressors might improve outcomes of stroke. This hypothesis has been bolstered by animal studies and imaging data that suggest improved perfusion to the area of injury with vasopressor‐induced hypertension.3638 However, there are concerns that this strategy may increase edema and hemorrhage and create the potential for vasopressor‐induced myocardial ischemia or arrhythmias. Although the results of small trials have appeared promising, particularly for patients with large vessel stenosis, at this point induced hypertension is still considered experimental until the results of larger randomized, controlled trials provide greater support for this procedure.1, 3941
Chronic Blood Pressure Control for Secondary Prevention of Stroke
Multiple trials have demonstrated that interventions to treat chronic hypertension can reduce the rate of future strokes.14, 43, 44 The PROGRESS trial demonstrated that blood pressure reduction with combination therapy decreased stroke recurrence by 43%.43 Clearly long‐term blood pressure control is a vital component of secondary prevention of stroke. Based on the results of the PROGRESS and ACCESS trials, it is suggested that angiotensin‐converting enzyme (ACE) inhibitors or angiotensin receptor blockers, potentially with additional diuretic therapy, are beneficial in the treatment of chronic hypertension following cerebrovascular accident.25, 42 The issue of optimal medication class is not settled. Recommendations are to treat chronically elevated blood pressure to lower and maintain it at less than 140/80 mm Hg. Because there is no single threshold level of blood pressure beyond which the risk of cerebrovascular disease increases, reduction below this cut point may still offer incremental benefit.13, 45, 46 The timing of initiation of therapy to reach secondary‐prevention goals is not addressed in current guidelines, although some authors recommend waiting days to weeks after an acute stroke before starting therapy.1, 37 For a patient who remains hypertensive at discharge, it is incumbent on the hospitalist to communicate the plan for initiation of antihypertensive agents to the patient and the primary care physician in order to ensure that this critical secondary prevention measure is addressed.
Areas of Ambiguity and Need for Future Research
Within an evidence‐guided practice, there are still many areas for which the evidence to date does not answer important clinical questions about patient management. Whether a patient's prestroke baseline blood pressure should be considered in individualizing blood pressure goals is unclear, as is the question of whether to continue or hold previous chronic antihypertensive therapy. Given the many stroke subtypes, from tiny lacunar infarcts with little collateralization to massive malignant middle‐cerebral infarctions, should blood pressure management be individualized according to the likely size and duration of the peri‐infarct penumbra? Research is needed to determine whether reducing blood pressure to a specific target will improve outcome and the optimal timing of therapy to achieve secondary prevention goals. Finally, using the concept of the penumbra as the theoretical physiologic basis for current blood pressure recommendations, there exists a potential for rapidly evolving neuroimaging techniques to help define the presence, size, and duration of the penumbra to guide these decisions.
CONCLUSIONS
Management of blood pressure after acute ischemic stroke differs from that of many other hypertensive conditions because of the need to preserve perfusion of the peri‐infarct penumbra. Evidence to date suggests little benefit and potential harm from acutely lowering blood pressure after stroke, although there have not been large randomized trials examining outcomes when blood pressure is lowered to a prespecified goal. Current consensus guidelines suggest that blood pressure may be managed permissively up to 220/120 mm Hg when thrombolysis is not administered and to 180/105 mm Hg when thrombolysis is performed. Data suggest that low‐dose antihypertensive therapy such as ACE inhibitors or angiotensin receptor blockers may be safe in the acute setting if pressure is reduced by less than 10%‐15%.17, 25 However, the evidence is not sufficiently strong for this practice to be routinely recommended. At this point, it is recommended that hypotension be aggressively treated; however, induced hypertension remains a promising but unproven therapy. Evidence is strong that long‐term blood pressure control is key to secondary prevention of stroke, but the timing of its initiation remains poorly determined.
- ,,, et al.Guidelines for the early management of patients with ischemic stroke.Stroke.2003;34:1056–1083.
- ,,,.Guidelines for the early management of patients with ischemic stroke: 2005 guidelines update.Stroke.2005;36:916–921.
- Blood Pressure in Acute Stroke Collaboration (BASC).Vasoactive drugs for acute strokeCochrane Database Syst Rev.2004;1:CD002839.
- ,.Blood pressure after stroke.JAMA.1981;246:2177–2180.
- ,,,,,.Association between course of blood pressure within the first 24 hours and functional recovery after acute ischemic stroke.Ann Emerg Med.2003;42:619–626.
- ,,,,,.Detrimental effect of blood pressure reduction in the first 24 hours of acute stroke onset.Neurology.2003;61:1047–1050.
- ,,,, et al.Blood pressure during the first minutes of focal cerebral ischemia.Ann Emerg Med.1993;22:1438–1443.
- ,,,,.Stroke precipitated by moderate blood pressure reduction.J Emerg Med.2000;19:339–346.
- .Hemodynamics and metabolism in ischemic cerebrovascular disease.Neurologic Clinics.1992;10(1):31–48.
- ,.The 5 Ps of acute ischemic stroke treatment: parenchyma, pipes, perfusion, penumbra, and prevention of complications.South Med J.2003;96:336–341.
- .Viability Thresholds and the Penumbra of Focal Ischemia.Annals of Neurology.1994;36:557–565.
- ,,.Hazards of therapy for excessive hypertension in acute stroke.Acta Med Scand.1980;207:253–257.
- Prospective Studies Collaboration.Age‐specific relevance of usual blood pressure to vascular mortality: a meta‐analysis of individual data for one million adults in 61 prospective studies.Lancet.2002;360:1903–1913.
- ,,,.Secondary prevention of ischemic cerebrovascular disease. What is the evidence?Angiology.2005;56:539–552.
- ,,.Intracerebral hemorrhage.Emerg Med Clin North Am.2002;20:631–655.
- ,,, et al.Should hypertension be treated after acute stroke?Arch Neurol.1993;50:855–862.
- ,,.Perindopril reduces blood pressure but not cerebral blood flow in patients with recent cerebral ischemic stroke.Stroke.1997;28:580–583.
- ,,,,.Intravenous Nimodipine West European Stroke Trial (INWEST) of nimodipine in the treatment of acute ischaemic stroke.Cerebrovasc Dis.1994;4:204–210.
- The American Nimodipine Study Group.Clinical trial of nimodipine in acute ischemic stroke.Stroke.1992;23(1):3–8.
- ,,, et al.Placebo‐controlled trial of nimodipine in the treatment of acute ischemic cerebral infarction.Stroke.1990;21:1023–1028.
- ,,, et al.Meta‐analysis of oral nimodipine trials in acute ischemic stroke.Cerebrovasc Dis.1994;4:197–203.
- ,,, et al.A randomized, double‐blind, placebo‐controlled trial of nimodipine in acute ischemic hemispheric stroke.Stroke.1994;25:1348–1353.
- ,,,.Low dose B blockade in acute stroke (“BEST” trial): an evaluationBMJ.1988;296:737–741.
- ,,, et al.Flunarizine in stroke treatment (FIST): a double‐blind, placebo‐controlled trial in Scandinavia and the Netherlands.Acta Neurol Scand.1996;93:56–60.
- ,,, et al.The ACCESS study: Evaluation of acute candesartan cilexetil therapy in stroke survivors.Stroke.2003;34:1699–1703.
- .FDA gives Calcium channel blockers clean bill of health but warns of short‐acting nifedipine hazards.JAMA.1996;275:1638.
- The National Institute of Neurological Disorders and Stroke rt‐PA Stroke Study Group.Tissue plasminogen activator for acute ischemic stroke.N Engl J Med.1995;333:1581–1587.
- ,,, et al.Hypertension and its treatment in the NINDS rt‐PA Stroke Trial.Stroke.1998;29:1504–1509.
- ,.Optimizing blood pressure in neurologic emergencies.Neurocrit Care.2004;1:287–299.
- .Management of acute stroke.South M J.2003;96:380–385.
- .Management of acute stroke.Lancet Neurol.2002;1(1):41–50.
- ,.Acute ischemic stroke: emergent evaluation and management.Emerg Med Clin North Am.2002;20:609–630.
- ,.Post‐emergency department management of stroke.Emerg Med Clin North Am.2002;20:687–702.
- ,,.Blood pressure management in acute stroke: comparison of current guidelines with prescribing patterns.Can J Neurol Sci.2002;29(2):125–131.
- ,,,,,.Use of antihypertensive agents in the management of patients with acute ischemic stroke.Neurology.2004;63:318–323.
- ,,,.Induced hypertension during ischemia reduces infarct area after temporary middle cerebral artery occlusion in rats.Surg Neurol.1996;46:229–234.
- ,,,.Effects of induced hypertension on intracranial pressure and flow velocities of the middle cerebral arteries in patients with large hemispheric stroke.Stroke.2002;33:998–1004.
- ,,,.Induced hypertension improves cerebral blood flow in acute ischemic stroke.Neurology.2005;64:1979.
- ,,,,.A pilot study of drug induced hypertension for treatment of acute stroke.Neurology.2001;56:1210–1213.
- ,,,,,.Pharmacologic elevation of blood pressure in acute stroke: clinical effects and safety.Stroke.1997;28:2133–2138.
- ,,,,.Feasibility and safety of norephinephrine‐induced arterial hypertension in acute ischemic stroke.Neurology.2004;62:1193–1195.
- ,,,,.Initial emergency department blood pressure as predictor of survival after acute ischemic stroke.Neurology.2005;65:1179–1183.
- The PROGRESS Collaborative Group.Randomized trial of a perindopril‐based blood‐pressure‐lowering regimen among 6105 individuals with previous stroke of transient ischaemic attack.Lancet.2001;358:1033–1041.
- ,,, et al.Effect of treating isolated systolic hypertension on the risk of developing various types and subtypes of stroke.JAMA.2000;284:465–471.
- ,.Treatment of chronic hypertension for the prevention of stroke.South Med J.2003;96:359–362.
- ,,,.Epidemiologic assessment of the role of blood pressure in stroke: the Framingham Study.JAMA.1996;276:1269–1278.
Hospitalists are on the front lines of care for patients with cerebrovascular accidents. After the first steps in acute stroke management occur in the emergency room, care is frequently transferred to the hospitalist. This review focuses on the evidence‐based management of blood pressure following acute ischemic stroke. Management of blood pressure after stroke is still controversial, and current consensus statement guidelines acknowledge that optimal treatment has not yet been established.1, 2 As such, it is essential to understand the changes in normal homeostatic physiologic processes that occur after stroke and their subsequent effects on neurologic function. Only then can the appropriate blood pressure target and antihypertensive regimen be chosen.
Physiology of Cerebral Perfusion
Once a stroke has occurred and perfusion to a section of brain tissue has been acutely compromised, systemic pressure tends to rise. This rise is presumably due in part to increased adrenergic tone and activation of the renin‐aldosterone system and potentially is part of Cushing's reflex in cases in which intracranial pressure is elevated.3 The increase in mean arterial pressure may represent a protective response. In the first major study on the topic, in 1981, on admission for stroke mean blood pressure was 163/90 mm Hg for patients without a history of hypertension and 214/118 mm Hg for patients who had been treated for hypertension previously.4 This rise in blood pressure in response to endogenous mechanisms is attenuated over the first 24 hours, and even without intervention, blood pressure tends to fall spontaneously over the next 10 days.47
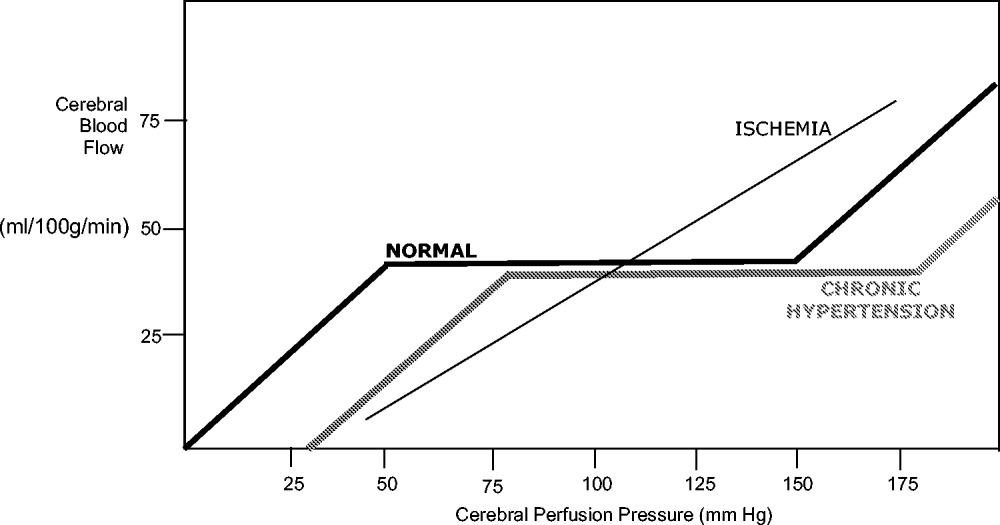
Under normal circumstances, cerebral blood flow (CBF) is tightly autoregulated across a wide range of cerebral perfusion pressures by alteration in cerebrovascular resistance via arteriolar constriction.89 This allows CBF to remain constant even if cerebral perfusion pressure (CPP) fluctuates from 60 to 150 mm Hg.9 In patients with chronic hypertension, autoregulation works best with blood pressure in a higher range because of vascular smooth muscle hypertrophy and structural changes in the cerebral vessels (see Table 1).
| Cerebral blood flow (CBF) of 50‐70 mL/100 g/minnormal |
| CBF of 20‐50 mL/100 g/minreduced flow compensated for by increased oxygen extraction |
| CBF of 15‐20 mL/100 g/minneuronal quiescence |
| CBF 15 mL/100 g/minneuronal death |
After an acute ischemic stroke, autoregulation is lost, and local CBF becomes linearly associated with cerebral perfusion pressure. Loss of autoregulation occurs because of several local factors as a result of the infarct. Acidosis and hypoxia in the region of the stroke lead to vasodilatation in the perfusing vessels.9 This may improve local circulation in collateral vessels as a response to the obstruction in the primary supplying artery, but the consequence of maximal vasodilatation is loss of the ability to autoregulate. Normal CPP is driven by the mean arterial pressure minus the intracranial pressure. However, if intracranial vessels have lost the ability to accommodate changes in perfusion pressure, then blood flow to the area of injury becomes linearly correlated with mean arterial pressure.
Surrounding a central core of irreversible necrosis may be a zone of at‐risk tissue that is susceptible to reduction below the threshold of viability in response to any decrement in systemic mean blood pressure.11 This critical concept in stroke management is referred to as the peri‐infarct penumbra (see Fig. 1 and Table 2).
| Mean arterial pressure (MAP) = ⅔ diastolic blood pressure (DBP) + ⅓ systolic blood pressure (SBP) |
| Cerebral perfusion pressure (CPP) = mean arterial pressure (MAP) intracranial pressure (ICP) |
| Cerebral blood flow (CBF) = cerebral perfusion pressure (CPP)/cerebrovascular resistance (CVR) |
Blood Pressure Management after Stroke
The presence of a systemic blood pressure‐dependent peri‐infarct penumbra that might be compromised by blood pressure reduction and thus extend the infarct is the principal argument for allowing permissive hypertension. This is bolstered by the observation that decreases in blood pressure in the first 24 hours are associated with a significant risk of poor neurologic outcome.6 In their an observational study Vlcek et al. found that a greater than 25% drop in diastolic blood pressure (DBP) was associated with a 4‐fold risk of severe disability.5 In a 2003 observational study Oliveria et al. found that reduction in systolic blood pressure (SBP) in the first 24 hours was independently associated with poor outcomes, with a doubling of the risk for poor outcomes for every 10% drop in systolic blood pressure.6 In both these studies, the worsened outcome did not depend on whether the drop was spontaneous or induced by medications. Case reports suggest that large drops in blood pressure can be catastrophic and that even moderate lowering of blood pressure after acute stroke can be associated with clinical deterioration.8, 12
The primary argument for lowering blood pressure after an acute stroke hinges on secondary prevention of new ischemic events, minimization of cerebral edema, and prevention of hemorrhagic conversion. It has long been recognized that hypertension itself is a risk factor for stroke13 and that reduction in chronic hypertension is part of secondary prevention for cerebrovascular accidents.14 Further, hypertension is the most common risk factor for intracerebral hemorrhage, and it would stand to reason that damage to the brain parenchyma from ischemic stroke would increase the risk of pressure‐induced bleeding in an acute setting.15
Because there are compelling theoretical arguments both for and against lowering blood pressure after an acute stroke, it is necessary to look at the results of randomized clinical trials for guidance in weighing the risks and benefits. The overall goal of blood pressure management is to maximize perfusion to the ischemic penumbra while minimizing the hypertensive risk of hemorrhagic transformation.
Lisk et al. conducted a small randomized trial in 1993 of antihypertensive therapy versus placebo after ischemic stroke looking at SPECT perfusion and found lower CBF if mean arterial pressure (MAP) dropped more than 16%.16 This is in contrast with the results of a 1997 randomized trial of perindopril after cerebrovascular accident that found no decrease in Doppler CBF despite a 10% decrease in blood pressure in the active treatment group.17 Neither study was powered to detect significant differences in clinical outcome.
Early randomized controlled trials of antihypertensive agents after acute stroke investigated neuroprotection via mechanisms other than the antihypertensive effect. Nimodipine, a dihydropyridine derivative thought to prevent neuronal death via blockade of calcium channels, has been studied extensively.1820 A meta‐analysis of the 9 early trials of nimodipine, from 1988 to 1992, suggested that nimodipine was potentially beneficial in neurologic score and functional outcome only if used within the first 12 hours and that it could be harmful if started after 24 hours.21 Two additional studies were done in 1994 using both intravenous and oral nimodipine formulations. They demonstrated worsened neurologic function and higher mortality, respectively, which was hypothesized to be a result of the detrimental hemodynamic effects of nimodipine.18, 22
The BEST trial used beta‐blockers in the early period after acute stroke and failed to find benefit, whereas the FIST trial had similar results with the calcium channel blocker flunarizine.23, 24 The ACCESS trial in 2003 was a prospective, randomized, controlled trial using oral candesartan in the first 24 hours after stroke.25 It did find improved mortality in the candesartan arm after 1 year, yet there were no significant differences in blood pressure between candesartan and placebo. Thus, the improved outcome was presumed to be a result of mechanisms other than antihypertensive effect.
Some significant caveats should be kept in mind when interpreting the results of these studies. None of the trials was designed to titrate blood pressure to a prespecified goal in a prospective randomized fashion. It is also difficult to tease out the effect of the active medical intervention from the effect of spontaneous drops in blood pressure, and many trials did not find a significant difference in blood pressure between the medication and placebo groups. A large randomized trial to evaluate interventions to a predefined blood pressure target is needed.
The Cochrane Stroke Group reviewed 32 trials involving 5368 patients and concluded there was not enough evidence to reliably evaluate the effect of altering blood pressure on outcomes.3
Recommendations for Treatment of Hypertension in Acute Stroke
The decision of when to initiate antihypertensive therapy has not been clearly delineated. However, most experts agree that blood pressure targets need to take into account whether thrombolytics are used. The available data provide little evidence that lowering blood pressure decreases adverse events; however, there is some evidence that lowering blood pressure can worsen outcomes by expanding the area of ischemia. Thus, in treating most acute ischemic strokes, antihypertensive therapy can and should be withheld. The exception to this is stroke in a patient with comorbid hypertensive organ damage such as myocardial infarction, aortic dissection, acute hypertensive renal failure, pulmonary edema, or encephalopathy. The consensus statement of the American Stroke Association is to withhold treatment until blood pressure exceeds 220/120 mm Hg,1 roughly corresponding to a MAP of 150 mm Hg, the normal upper limit of cerebral autoregulation. Treatment optimally should be titratable in order to avoid overcorrection, preferably with minimal cerebral venodilatation effect. The parenteral agents labetolol, fenoldopam, nicardipine, and nitroprusside are most commonly used. A disadvantage of nitrates or nitroprusside is that their venodilating effect may raise intracranial pressure. Clonidine may induce central nervous system depression, which can complicate interpretation of the mental status of a patient with an acute neurologic event. Sublingual nifedipine should be avoided because of its well‐documented tendency to cause overcorrection of blood pressure.26 No large direct comparison trial has evaluated which of these antihypertensive agents is superior in clinical outcomes. For the hospitalist, choice of antihypertensive agent should be driven by a need for rapid control of blood pressure to target without overcorrection (see Table 3). Issues such as intracranial pressure, heart rate, and comorbidities may also play a role in choice of medication.
| Not eligible for thrombolytic therapy | |
|---|---|
| |
| Systolic 220 or diastolic 120 | Observe unless other end‐organ involvement (aortic dissection, acute myocardial infarction, pulmonary edema, hypertensive encephalopathy) |
| Systolic > 220 or diastolic 121‐140 | Aim for 10%‐15% reduction in blood pressure |
| Labetalol 10‐20 mg IV over 1‐2 minutes; repeat or double every 10 minutes; maximum 300 mg | |
| or | |
| Nicardipine 5 mg/hour IV; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| or | |
| Nitroprusside 0.5 g/kg/minute IV; titrate to goal | |
| Diastolic > 140 | Aim for 10%‐15% reduction in blood pressure |
| Nitroprusside 0.5 g/kg/minute IV; titrate to desired blood pressure | |
| Eligible for thrombolytic therapy | |
| Pretreatment | |
| Systolic > 185 or diastolic > 110 | Labetalol 10‐20mg IV |
| May repeat 1 or Nitropaste 1‐2 inches | |
| During and after tPA | |
| Systolic 180‐230 or Diastolic 105‐120 | Labetalol 10 mg |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute | |
| Systolic > 230 or diastolic 121‐140 | Labetalol 10mg IV |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute or | |
| Nicardipine 5 mg/hour; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| If not at goal with labetalol or nicardipine, consider nitroprusside | |
| Diastolic>140 | Nitroprusside 0.5 g/kg/minute IV infusion; titrate to desired blood pressure |
The risk of hemorrhagic conversion is greater when thrombolysis is used for the treatment of cerebrovascular accident, in the NINDS trial rising from 0.6% in patients receiving a placebo to 6.4% in patients receiving tPA.27 Given that the goal of thrombolysis is to restore perfusion through the previously blocked vessel and that the risk of hemorrhage rises after thrombolysis, the balance between maintaining CPP and decreasing the risk of bleeding shifts toward maintaining a lower blood pressure postthrombolysis. The NINDS thrombolytic trial required patients to have a blood pressure less than 185/110 mm Hg to be included.27 Once a thrombolytic had been administered, the permitted maximum blood pressure dropped to 180/105 mm Hg. Blood pressure needs to be monitored closely, initially every 15 minutes for 2 hours, then every 30 minutes for the next 6 hours, and then hourly until the end of the first 24 hours. After this, blood pressure monitoring intervals can be more spaced out depending on the need for active antihypertensive therapy.28 However, arterial punctures for invasive monitoring are not recommended if thrombolytics are administered. Review of the experience of the NINDS trial suggests that approximately one third of patients receiving thrombolytics will require pharmacologic therapy to reach the recommended blood pressure goals in the first 24 hours.28
Although guidelines for the management of blood pressure after cerebrovascular accident from major organizations such as the American Stroke Association and the European Stroke Initiative are published and widely quoted in reviews of stroke management, physician adherence to these guidelines is poor.1, 2933 A 2002 review of prescribing practices in a Canadian hospital found excessive reduction of blood pressure in 60% of patients where nitroglycerin was used, and sublingual nifedipine was still the second most commonly prescribed medication.34 A similar 2004 study by Lindenauer et al. found that only 26% of patients who had antihypertensive medication initiated in the hospital met consensus guidelines for therapy.35
Induced Hypertension
Hypotension should be avoided after acute stroke. Observational studies have demonstrated an increased mortality rate of patients who present with hypotension.42 Patients with acute ischemic stroke and loss of autoregulation may have impaired tolerance to even mild levels of hypotension.8 First‐line therapy for hypotension after stroke is volume resuscitation and optimization of cardiac function by correcting arrhythmias.1 If this should fail, vasopressor support is advisable to avoid ongoing hypotension and cerebral hypoperfusion.
The same physiologic arguments that favor permissively managing high blood pressure and avoiding hypotension raise the question of whether inducing hypertension through the use of vasopressors might improve outcomes of stroke. This hypothesis has been bolstered by animal studies and imaging data that suggest improved perfusion to the area of injury with vasopressor‐induced hypertension.3638 However, there are concerns that this strategy may increase edema and hemorrhage and create the potential for vasopressor‐induced myocardial ischemia or arrhythmias. Although the results of small trials have appeared promising, particularly for patients with large vessel stenosis, at this point induced hypertension is still considered experimental until the results of larger randomized, controlled trials provide greater support for this procedure.1, 3941
Chronic Blood Pressure Control for Secondary Prevention of Stroke
Multiple trials have demonstrated that interventions to treat chronic hypertension can reduce the rate of future strokes.14, 43, 44 The PROGRESS trial demonstrated that blood pressure reduction with combination therapy decreased stroke recurrence by 43%.43 Clearly long‐term blood pressure control is a vital component of secondary prevention of stroke. Based on the results of the PROGRESS and ACCESS trials, it is suggested that angiotensin‐converting enzyme (ACE) inhibitors or angiotensin receptor blockers, potentially with additional diuretic therapy, are beneficial in the treatment of chronic hypertension following cerebrovascular accident.25, 42 The issue of optimal medication class is not settled. Recommendations are to treat chronically elevated blood pressure to lower and maintain it at less than 140/80 mm Hg. Because there is no single threshold level of blood pressure beyond which the risk of cerebrovascular disease increases, reduction below this cut point may still offer incremental benefit.13, 45, 46 The timing of initiation of therapy to reach secondary‐prevention goals is not addressed in current guidelines, although some authors recommend waiting days to weeks after an acute stroke before starting therapy.1, 37 For a patient who remains hypertensive at discharge, it is incumbent on the hospitalist to communicate the plan for initiation of antihypertensive agents to the patient and the primary care physician in order to ensure that this critical secondary prevention measure is addressed.
Areas of Ambiguity and Need for Future Research
Within an evidence‐guided practice, there are still many areas for which the evidence to date does not answer important clinical questions about patient management. Whether a patient's prestroke baseline blood pressure should be considered in individualizing blood pressure goals is unclear, as is the question of whether to continue or hold previous chronic antihypertensive therapy. Given the many stroke subtypes, from tiny lacunar infarcts with little collateralization to massive malignant middle‐cerebral infarctions, should blood pressure management be individualized according to the likely size and duration of the peri‐infarct penumbra? Research is needed to determine whether reducing blood pressure to a specific target will improve outcome and the optimal timing of therapy to achieve secondary prevention goals. Finally, using the concept of the penumbra as the theoretical physiologic basis for current blood pressure recommendations, there exists a potential for rapidly evolving neuroimaging techniques to help define the presence, size, and duration of the penumbra to guide these decisions.
CONCLUSIONS
Management of blood pressure after acute ischemic stroke differs from that of many other hypertensive conditions because of the need to preserve perfusion of the peri‐infarct penumbra. Evidence to date suggests little benefit and potential harm from acutely lowering blood pressure after stroke, although there have not been large randomized trials examining outcomes when blood pressure is lowered to a prespecified goal. Current consensus guidelines suggest that blood pressure may be managed permissively up to 220/120 mm Hg when thrombolysis is not administered and to 180/105 mm Hg when thrombolysis is performed. Data suggest that low‐dose antihypertensive therapy such as ACE inhibitors or angiotensin receptor blockers may be safe in the acute setting if pressure is reduced by less than 10%‐15%.17, 25 However, the evidence is not sufficiently strong for this practice to be routinely recommended. At this point, it is recommended that hypotension be aggressively treated; however, induced hypertension remains a promising but unproven therapy. Evidence is strong that long‐term blood pressure control is key to secondary prevention of stroke, but the timing of its initiation remains poorly determined.
Hospitalists are on the front lines of care for patients with cerebrovascular accidents. After the first steps in acute stroke management occur in the emergency room, care is frequently transferred to the hospitalist. This review focuses on the evidence‐based management of blood pressure following acute ischemic stroke. Management of blood pressure after stroke is still controversial, and current consensus statement guidelines acknowledge that optimal treatment has not yet been established.1, 2 As such, it is essential to understand the changes in normal homeostatic physiologic processes that occur after stroke and their subsequent effects on neurologic function. Only then can the appropriate blood pressure target and antihypertensive regimen be chosen.
Physiology of Cerebral Perfusion
Once a stroke has occurred and perfusion to a section of brain tissue has been acutely compromised, systemic pressure tends to rise. This rise is presumably due in part to increased adrenergic tone and activation of the renin‐aldosterone system and potentially is part of Cushing's reflex in cases in which intracranial pressure is elevated.3 The increase in mean arterial pressure may represent a protective response. In the first major study on the topic, in 1981, on admission for stroke mean blood pressure was 163/90 mm Hg for patients without a history of hypertension and 214/118 mm Hg for patients who had been treated for hypertension previously.4 This rise in blood pressure in response to endogenous mechanisms is attenuated over the first 24 hours, and even without intervention, blood pressure tends to fall spontaneously over the next 10 days.47
Under normal circumstances, cerebral blood flow (CBF) is tightly autoregulated across a wide range of cerebral perfusion pressures by alteration in cerebrovascular resistance via arteriolar constriction.89 This allows CBF to remain constant even if cerebral perfusion pressure (CPP) fluctuates from 60 to 150 mm Hg.9 In patients with chronic hypertension, autoregulation works best with blood pressure in a higher range because of vascular smooth muscle hypertrophy and structural changes in the cerebral vessels (see Table 1).
| Cerebral blood flow (CBF) of 50‐70 mL/100 g/minnormal |
| CBF of 20‐50 mL/100 g/minreduced flow compensated for by increased oxygen extraction |
| CBF of 15‐20 mL/100 g/minneuronal quiescence |
| CBF 15 mL/100 g/minneuronal death |
After an acute ischemic stroke, autoregulation is lost, and local CBF becomes linearly associated with cerebral perfusion pressure. Loss of autoregulation occurs because of several local factors as a result of the infarct. Acidosis and hypoxia in the region of the stroke lead to vasodilatation in the perfusing vessels.9 This may improve local circulation in collateral vessels as a response to the obstruction in the primary supplying artery, but the consequence of maximal vasodilatation is loss of the ability to autoregulate. Normal CPP is driven by the mean arterial pressure minus the intracranial pressure. However, if intracranial vessels have lost the ability to accommodate changes in perfusion pressure, then blood flow to the area of injury becomes linearly correlated with mean arterial pressure.
Surrounding a central core of irreversible necrosis may be a zone of at‐risk tissue that is susceptible to reduction below the threshold of viability in response to any decrement in systemic mean blood pressure.11 This critical concept in stroke management is referred to as the peri‐infarct penumbra (see Fig. 1 and Table 2).
| Mean arterial pressure (MAP) = ⅔ diastolic blood pressure (DBP) + ⅓ systolic blood pressure (SBP) |
| Cerebral perfusion pressure (CPP) = mean arterial pressure (MAP) intracranial pressure (ICP) |
| Cerebral blood flow (CBF) = cerebral perfusion pressure (CPP)/cerebrovascular resistance (CVR) |
Blood Pressure Management after Stroke
The presence of a systemic blood pressure‐dependent peri‐infarct penumbra that might be compromised by blood pressure reduction and thus extend the infarct is the principal argument for allowing permissive hypertension. This is bolstered by the observation that decreases in blood pressure in the first 24 hours are associated with a significant risk of poor neurologic outcome.6 In their an observational study Vlcek et al. found that a greater than 25% drop in diastolic blood pressure (DBP) was associated with a 4‐fold risk of severe disability.5 In a 2003 observational study Oliveria et al. found that reduction in systolic blood pressure (SBP) in the first 24 hours was independently associated with poor outcomes, with a doubling of the risk for poor outcomes for every 10% drop in systolic blood pressure.6 In both these studies, the worsened outcome did not depend on whether the drop was spontaneous or induced by medications. Case reports suggest that large drops in blood pressure can be catastrophic and that even moderate lowering of blood pressure after acute stroke can be associated with clinical deterioration.8, 12
The primary argument for lowering blood pressure after an acute stroke hinges on secondary prevention of new ischemic events, minimization of cerebral edema, and prevention of hemorrhagic conversion. It has long been recognized that hypertension itself is a risk factor for stroke13 and that reduction in chronic hypertension is part of secondary prevention for cerebrovascular accidents.14 Further, hypertension is the most common risk factor for intracerebral hemorrhage, and it would stand to reason that damage to the brain parenchyma from ischemic stroke would increase the risk of pressure‐induced bleeding in an acute setting.15
Because there are compelling theoretical arguments both for and against lowering blood pressure after an acute stroke, it is necessary to look at the results of randomized clinical trials for guidance in weighing the risks and benefits. The overall goal of blood pressure management is to maximize perfusion to the ischemic penumbra while minimizing the hypertensive risk of hemorrhagic transformation.
Lisk et al. conducted a small randomized trial in 1993 of antihypertensive therapy versus placebo after ischemic stroke looking at SPECT perfusion and found lower CBF if mean arterial pressure (MAP) dropped more than 16%.16 This is in contrast with the results of a 1997 randomized trial of perindopril after cerebrovascular accident that found no decrease in Doppler CBF despite a 10% decrease in blood pressure in the active treatment group.17 Neither study was powered to detect significant differences in clinical outcome.
Early randomized controlled trials of antihypertensive agents after acute stroke investigated neuroprotection via mechanisms other than the antihypertensive effect. Nimodipine, a dihydropyridine derivative thought to prevent neuronal death via blockade of calcium channels, has been studied extensively.1820 A meta‐analysis of the 9 early trials of nimodipine, from 1988 to 1992, suggested that nimodipine was potentially beneficial in neurologic score and functional outcome only if used within the first 12 hours and that it could be harmful if started after 24 hours.21 Two additional studies were done in 1994 using both intravenous and oral nimodipine formulations. They demonstrated worsened neurologic function and higher mortality, respectively, which was hypothesized to be a result of the detrimental hemodynamic effects of nimodipine.18, 22
The BEST trial used beta‐blockers in the early period after acute stroke and failed to find benefit, whereas the FIST trial had similar results with the calcium channel blocker flunarizine.23, 24 The ACCESS trial in 2003 was a prospective, randomized, controlled trial using oral candesartan in the first 24 hours after stroke.25 It did find improved mortality in the candesartan arm after 1 year, yet there were no significant differences in blood pressure between candesartan and placebo. Thus, the improved outcome was presumed to be a result of mechanisms other than antihypertensive effect.
Some significant caveats should be kept in mind when interpreting the results of these studies. None of the trials was designed to titrate blood pressure to a prespecified goal in a prospective randomized fashion. It is also difficult to tease out the effect of the active medical intervention from the effect of spontaneous drops in blood pressure, and many trials did not find a significant difference in blood pressure between the medication and placebo groups. A large randomized trial to evaluate interventions to a predefined blood pressure target is needed.
The Cochrane Stroke Group reviewed 32 trials involving 5368 patients and concluded there was not enough evidence to reliably evaluate the effect of altering blood pressure on outcomes.3
Recommendations for Treatment of Hypertension in Acute Stroke
The decision of when to initiate antihypertensive therapy has not been clearly delineated. However, most experts agree that blood pressure targets need to take into account whether thrombolytics are used. The available data provide little evidence that lowering blood pressure decreases adverse events; however, there is some evidence that lowering blood pressure can worsen outcomes by expanding the area of ischemia. Thus, in treating most acute ischemic strokes, antihypertensive therapy can and should be withheld. The exception to this is stroke in a patient with comorbid hypertensive organ damage such as myocardial infarction, aortic dissection, acute hypertensive renal failure, pulmonary edema, or encephalopathy. The consensus statement of the American Stroke Association is to withhold treatment until blood pressure exceeds 220/120 mm Hg,1 roughly corresponding to a MAP of 150 mm Hg, the normal upper limit of cerebral autoregulation. Treatment optimally should be titratable in order to avoid overcorrection, preferably with minimal cerebral venodilatation effect. The parenteral agents labetolol, fenoldopam, nicardipine, and nitroprusside are most commonly used. A disadvantage of nitrates or nitroprusside is that their venodilating effect may raise intracranial pressure. Clonidine may induce central nervous system depression, which can complicate interpretation of the mental status of a patient with an acute neurologic event. Sublingual nifedipine should be avoided because of its well‐documented tendency to cause overcorrection of blood pressure.26 No large direct comparison trial has evaluated which of these antihypertensive agents is superior in clinical outcomes. For the hospitalist, choice of antihypertensive agent should be driven by a need for rapid control of blood pressure to target without overcorrection (see Table 3). Issues such as intracranial pressure, heart rate, and comorbidities may also play a role in choice of medication.
| Not eligible for thrombolytic therapy | |
|---|---|
| |
| Systolic 220 or diastolic 120 | Observe unless other end‐organ involvement (aortic dissection, acute myocardial infarction, pulmonary edema, hypertensive encephalopathy) |
| Systolic > 220 or diastolic 121‐140 | Aim for 10%‐15% reduction in blood pressure |
| Labetalol 10‐20 mg IV over 1‐2 minutes; repeat or double every 10 minutes; maximum 300 mg | |
| or | |
| Nicardipine 5 mg/hour IV; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| or | |
| Nitroprusside 0.5 g/kg/minute IV; titrate to goal | |
| Diastolic > 140 | Aim for 10%‐15% reduction in blood pressure |
| Nitroprusside 0.5 g/kg/minute IV; titrate to desired blood pressure | |
| Eligible for thrombolytic therapy | |
| Pretreatment | |
| Systolic > 185 or diastolic > 110 | Labetalol 10‐20mg IV |
| May repeat 1 or Nitropaste 1‐2 inches | |
| During and after tPA | |
| Systolic 180‐230 or Diastolic 105‐120 | Labetalol 10 mg |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute | |
| Systolic > 230 or diastolic 121‐140 | Labetalol 10mg IV |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute or | |
| Nicardipine 5 mg/hour; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| If not at goal with labetalol or nicardipine, consider nitroprusside | |
| Diastolic>140 | Nitroprusside 0.5 g/kg/minute IV infusion; titrate to desired blood pressure |
The risk of hemorrhagic conversion is greater when thrombolysis is used for the treatment of cerebrovascular accident, in the NINDS trial rising from 0.6% in patients receiving a placebo to 6.4% in patients receiving tPA.27 Given that the goal of thrombolysis is to restore perfusion through the previously blocked vessel and that the risk of hemorrhage rises after thrombolysis, the balance between maintaining CPP and decreasing the risk of bleeding shifts toward maintaining a lower blood pressure postthrombolysis. The NINDS thrombolytic trial required patients to have a blood pressure less than 185/110 mm Hg to be included.27 Once a thrombolytic had been administered, the permitted maximum blood pressure dropped to 180/105 mm Hg. Blood pressure needs to be monitored closely, initially every 15 minutes for 2 hours, then every 30 minutes for the next 6 hours, and then hourly until the end of the first 24 hours. After this, blood pressure monitoring intervals can be more spaced out depending on the need for active antihypertensive therapy.28 However, arterial punctures for invasive monitoring are not recommended if thrombolytics are administered. Review of the experience of the NINDS trial suggests that approximately one third of patients receiving thrombolytics will require pharmacologic therapy to reach the recommended blood pressure goals in the first 24 hours.28
Although guidelines for the management of blood pressure after cerebrovascular accident from major organizations such as the American Stroke Association and the European Stroke Initiative are published and widely quoted in reviews of stroke management, physician adherence to these guidelines is poor.1, 2933 A 2002 review of prescribing practices in a Canadian hospital found excessive reduction of blood pressure in 60% of patients where nitroglycerin was used, and sublingual nifedipine was still the second most commonly prescribed medication.34 A similar 2004 study by Lindenauer et al. found that only 26% of patients who had antihypertensive medication initiated in the hospital met consensus guidelines for therapy.35
Induced Hypertension
Hypotension should be avoided after acute stroke. Observational studies have demonstrated an increased mortality rate of patients who present with hypotension.42 Patients with acute ischemic stroke and loss of autoregulation may have impaired tolerance to even mild levels of hypotension.8 First‐line therapy for hypotension after stroke is volume resuscitation and optimization of cardiac function by correcting arrhythmias.1 If this should fail, vasopressor support is advisable to avoid ongoing hypotension and cerebral hypoperfusion.
The same physiologic arguments that favor permissively managing high blood pressure and avoiding hypotension raise the question of whether inducing hypertension through the use of vasopressors might improve outcomes of stroke. This hypothesis has been bolstered by animal studies and imaging data that suggest improved perfusion to the area of injury with vasopressor‐induced hypertension.3638 However, there are concerns that this strategy may increase edema and hemorrhage and create the potential for vasopressor‐induced myocardial ischemia or arrhythmias. Although the results of small trials have appeared promising, particularly for patients with large vessel stenosis, at this point induced hypertension is still considered experimental until the results of larger randomized, controlled trials provide greater support for this procedure.1, 3941
Chronic Blood Pressure Control for Secondary Prevention of Stroke
Multiple trials have demonstrated that interventions to treat chronic hypertension can reduce the rate of future strokes.14, 43, 44 The PROGRESS trial demonstrated that blood pressure reduction with combination therapy decreased stroke recurrence by 43%.43 Clearly long‐term blood pressure control is a vital component of secondary prevention of stroke. Based on the results of the PROGRESS and ACCESS trials, it is suggested that angiotensin‐converting enzyme (ACE) inhibitors or angiotensin receptor blockers, potentially with additional diuretic therapy, are beneficial in the treatment of chronic hypertension following cerebrovascular accident.25, 42 The issue of optimal medication class is not settled. Recommendations are to treat chronically elevated blood pressure to lower and maintain it at less than 140/80 mm Hg. Because there is no single threshold level of blood pressure beyond which the risk of cerebrovascular disease increases, reduction below this cut point may still offer incremental benefit.13, 45, 46 The timing of initiation of therapy to reach secondary‐prevention goals is not addressed in current guidelines, although some authors recommend waiting days to weeks after an acute stroke before starting therapy.1, 37 For a patient who remains hypertensive at discharge, it is incumbent on the hospitalist to communicate the plan for initiation of antihypertensive agents to the patient and the primary care physician in order to ensure that this critical secondary prevention measure is addressed.
Areas of Ambiguity and Need for Future Research
Within an evidence‐guided practice, there are still many areas for which the evidence to date does not answer important clinical questions about patient management. Whether a patient's prestroke baseline blood pressure should be considered in individualizing blood pressure goals is unclear, as is the question of whether to continue or hold previous chronic antihypertensive therapy. Given the many stroke subtypes, from tiny lacunar infarcts with little collateralization to massive malignant middle‐cerebral infarctions, should blood pressure management be individualized according to the likely size and duration of the peri‐infarct penumbra? Research is needed to determine whether reducing blood pressure to a specific target will improve outcome and the optimal timing of therapy to achieve secondary prevention goals. Finally, using the concept of the penumbra as the theoretical physiologic basis for current blood pressure recommendations, there exists a potential for rapidly evolving neuroimaging techniques to help define the presence, size, and duration of the penumbra to guide these decisions.
CONCLUSIONS
Management of blood pressure after acute ischemic stroke differs from that of many other hypertensive conditions because of the need to preserve perfusion of the peri‐infarct penumbra. Evidence to date suggests little benefit and potential harm from acutely lowering blood pressure after stroke, although there have not been large randomized trials examining outcomes when blood pressure is lowered to a prespecified goal. Current consensus guidelines suggest that blood pressure may be managed permissively up to 220/120 mm Hg when thrombolysis is not administered and to 180/105 mm Hg when thrombolysis is performed. Data suggest that low‐dose antihypertensive therapy such as ACE inhibitors or angiotensin receptor blockers may be safe in the acute setting if pressure is reduced by less than 10%‐15%.17, 25 However, the evidence is not sufficiently strong for this practice to be routinely recommended. At this point, it is recommended that hypotension be aggressively treated; however, induced hypertension remains a promising but unproven therapy. Evidence is strong that long‐term blood pressure control is key to secondary prevention of stroke, but the timing of its initiation remains poorly determined.
- ,,, et al.Guidelines for the early management of patients with ischemic stroke.Stroke.2003;34:1056–1083.
- ,,,.Guidelines for the early management of patients with ischemic stroke: 2005 guidelines update.Stroke.2005;36:916–921.
- Blood Pressure in Acute Stroke Collaboration (BASC).Vasoactive drugs for acute strokeCochrane Database Syst Rev.2004;1:CD002839.
- ,.Blood pressure after stroke.JAMA.1981;246:2177–2180.
- ,,,,,.Association between course of blood pressure within the first 24 hours and functional recovery after acute ischemic stroke.Ann Emerg Med.2003;42:619–626.
- ,,,,,.Detrimental effect of blood pressure reduction in the first 24 hours of acute stroke onset.Neurology.2003;61:1047–1050.
- ,,,, et al.Blood pressure during the first minutes of focal cerebral ischemia.Ann Emerg Med.1993;22:1438–1443.
- ,,,,.Stroke precipitated by moderate blood pressure reduction.J Emerg Med.2000;19:339–346.
- .Hemodynamics and metabolism in ischemic cerebrovascular disease.Neurologic Clinics.1992;10(1):31–48.
- ,.The 5 Ps of acute ischemic stroke treatment: parenchyma, pipes, perfusion, penumbra, and prevention of complications.South Med J.2003;96:336–341.
- .Viability Thresholds and the Penumbra of Focal Ischemia.Annals of Neurology.1994;36:557–565.
- ,,.Hazards of therapy for excessive hypertension in acute stroke.Acta Med Scand.1980;207:253–257.
- Prospective Studies Collaboration.Age‐specific relevance of usual blood pressure to vascular mortality: a meta‐analysis of individual data for one million adults in 61 prospective studies.Lancet.2002;360:1903–1913.
- ,,,.Secondary prevention of ischemic cerebrovascular disease. What is the evidence?Angiology.2005;56:539–552.
- ,,.Intracerebral hemorrhage.Emerg Med Clin North Am.2002;20:631–655.
- ,,, et al.Should hypertension be treated after acute stroke?Arch Neurol.1993;50:855–862.
- ,,.Perindopril reduces blood pressure but not cerebral blood flow in patients with recent cerebral ischemic stroke.Stroke.1997;28:580–583.
- ,,,,.Intravenous Nimodipine West European Stroke Trial (INWEST) of nimodipine in the treatment of acute ischaemic stroke.Cerebrovasc Dis.1994;4:204–210.
- The American Nimodipine Study Group.Clinical trial of nimodipine in acute ischemic stroke.Stroke.1992;23(1):3–8.
- ,,, et al.Placebo‐controlled trial of nimodipine in the treatment of acute ischemic cerebral infarction.Stroke.1990;21:1023–1028.
- ,,, et al.Meta‐analysis of oral nimodipine trials in acute ischemic stroke.Cerebrovasc Dis.1994;4:197–203.
- ,,, et al.A randomized, double‐blind, placebo‐controlled trial of nimodipine in acute ischemic hemispheric stroke.Stroke.1994;25:1348–1353.
- ,,,.Low dose B blockade in acute stroke (“BEST” trial): an evaluationBMJ.1988;296:737–741.
- ,,, et al.Flunarizine in stroke treatment (FIST): a double‐blind, placebo‐controlled trial in Scandinavia and the Netherlands.Acta Neurol Scand.1996;93:56–60.
- ,,, et al.The ACCESS study: Evaluation of acute candesartan cilexetil therapy in stroke survivors.Stroke.2003;34:1699–1703.
- .FDA gives Calcium channel blockers clean bill of health but warns of short‐acting nifedipine hazards.JAMA.1996;275:1638.
- The National Institute of Neurological Disorders and Stroke rt‐PA Stroke Study Group.Tissue plasminogen activator for acute ischemic stroke.N Engl J Med.1995;333:1581–1587.
- ,,, et al.Hypertension and its treatment in the NINDS rt‐PA Stroke Trial.Stroke.1998;29:1504–1509.
- ,.Optimizing blood pressure in neurologic emergencies.Neurocrit Care.2004;1:287–299.
- .Management of acute stroke.South M J.2003;96:380–385.
- .Management of acute stroke.Lancet Neurol.2002;1(1):41–50.
- ,.Acute ischemic stroke: emergent evaluation and management.Emerg Med Clin North Am.2002;20:609–630.
- ,.Post‐emergency department management of stroke.Emerg Med Clin North Am.2002;20:687–702.
- ,,.Blood pressure management in acute stroke: comparison of current guidelines with prescribing patterns.Can J Neurol Sci.2002;29(2):125–131.
- ,,,,,.Use of antihypertensive agents in the management of patients with acute ischemic stroke.Neurology.2004;63:318–323.
- ,,,.Induced hypertension during ischemia reduces infarct area after temporary middle cerebral artery occlusion in rats.Surg Neurol.1996;46:229–234.
- ,,,.Effects of induced hypertension on intracranial pressure and flow velocities of the middle cerebral arteries in patients with large hemispheric stroke.Stroke.2002;33:998–1004.
- ,,,.Induced hypertension improves cerebral blood flow in acute ischemic stroke.Neurology.2005;64:1979.
- ,,,,.A pilot study of drug induced hypertension for treatment of acute stroke.Neurology.2001;56:1210–1213.
- ,,,,,.Pharmacologic elevation of blood pressure in acute stroke: clinical effects and safety.Stroke.1997;28:2133–2138.
- ,,,,.Feasibility and safety of norephinephrine‐induced arterial hypertension in acute ischemic stroke.Neurology.2004;62:1193–1195.
- ,,,,.Initial emergency department blood pressure as predictor of survival after acute ischemic stroke.Neurology.2005;65:1179–1183.
- The PROGRESS Collaborative Group.Randomized trial of a perindopril‐based blood‐pressure‐lowering regimen among 6105 individuals with previous stroke of transient ischaemic attack.Lancet.2001;358:1033–1041.
- ,,, et al.Effect of treating isolated systolic hypertension on the risk of developing various types and subtypes of stroke.JAMA.2000;284:465–471.
- ,.Treatment of chronic hypertension for the prevention of stroke.South Med J.2003;96:359–362.
- ,,,.Epidemiologic assessment of the role of blood pressure in stroke: the Framingham Study.JAMA.1996;276:1269–1278.
- ,,, et al.Guidelines for the early management of patients with ischemic stroke.Stroke.2003;34:1056–1083.
- ,,,.Guidelines for the early management of patients with ischemic stroke: 2005 guidelines update.Stroke.2005;36:916–921.
- Blood Pressure in Acute Stroke Collaboration (BASC).Vasoactive drugs for acute strokeCochrane Database Syst Rev.2004;1:CD002839.
- ,.Blood pressure after stroke.JAMA.1981;246:2177–2180.
- ,,,,,.Association between course of blood pressure within the first 24 hours and functional recovery after acute ischemic stroke.Ann Emerg Med.2003;42:619–626.
- ,,,,,.Detrimental effect of blood pressure reduction in the first 24 hours of acute stroke onset.Neurology.2003;61:1047–1050.
- ,,,, et al.Blood pressure during the first minutes of focal cerebral ischemia.Ann Emerg Med.1993;22:1438–1443.
- ,,,,.Stroke precipitated by moderate blood pressure reduction.J Emerg Med.2000;19:339–346.
- .Hemodynamics and metabolism in ischemic cerebrovascular disease.Neurologic Clinics.1992;10(1):31–48.
- ,.The 5 Ps of acute ischemic stroke treatment: parenchyma, pipes, perfusion, penumbra, and prevention of complications.South Med J.2003;96:336–341.
- .Viability Thresholds and the Penumbra of Focal Ischemia.Annals of Neurology.1994;36:557–565.
- ,,.Hazards of therapy for excessive hypertension in acute stroke.Acta Med Scand.1980;207:253–257.
- Prospective Studies Collaboration.Age‐specific relevance of usual blood pressure to vascular mortality: a meta‐analysis of individual data for one million adults in 61 prospective studies.Lancet.2002;360:1903–1913.
- ,,,.Secondary prevention of ischemic cerebrovascular disease. What is the evidence?Angiology.2005;56:539–552.
- ,,.Intracerebral hemorrhage.Emerg Med Clin North Am.2002;20:631–655.
- ,,, et al.Should hypertension be treated after acute stroke?Arch Neurol.1993;50:855–862.
- ,,.Perindopril reduces blood pressure but not cerebral blood flow in patients with recent cerebral ischemic stroke.Stroke.1997;28:580–583.
- ,,,,.Intravenous Nimodipine West European Stroke Trial (INWEST) of nimodipine in the treatment of acute ischaemic stroke.Cerebrovasc Dis.1994;4:204–210.
- The American Nimodipine Study Group.Clinical trial of nimodipine in acute ischemic stroke.Stroke.1992;23(1):3–8.
- ,,, et al.Placebo‐controlled trial of nimodipine in the treatment of acute ischemic cerebral infarction.Stroke.1990;21:1023–1028.
- ,,, et al.Meta‐analysis of oral nimodipine trials in acute ischemic stroke.Cerebrovasc Dis.1994;4:197–203.
- ,,, et al.A randomized, double‐blind, placebo‐controlled trial of nimodipine in acute ischemic hemispheric stroke.Stroke.1994;25:1348–1353.
- ,,,.Low dose B blockade in acute stroke (“BEST” trial): an evaluationBMJ.1988;296:737–741.
- ,,, et al.Flunarizine in stroke treatment (FIST): a double‐blind, placebo‐controlled trial in Scandinavia and the Netherlands.Acta Neurol Scand.1996;93:56–60.
- ,,, et al.The ACCESS study: Evaluation of acute candesartan cilexetil therapy in stroke survivors.Stroke.2003;34:1699–1703.
- .FDA gives Calcium channel blockers clean bill of health but warns of short‐acting nifedipine hazards.JAMA.1996;275:1638.
- The National Institute of Neurological Disorders and Stroke rt‐PA Stroke Study Group.Tissue plasminogen activator for acute ischemic stroke.N Engl J Med.1995;333:1581–1587.
- ,,, et al.Hypertension and its treatment in the NINDS rt‐PA Stroke Trial.Stroke.1998;29:1504–1509.
- ,.Optimizing blood pressure in neurologic emergencies.Neurocrit Care.2004;1:287–299.
- .Management of acute stroke.South M J.2003;96:380–385.
- .Management of acute stroke.Lancet Neurol.2002;1(1):41–50.
- ,.Acute ischemic stroke: emergent evaluation and management.Emerg Med Clin North Am.2002;20:609–630.
- ,.Post‐emergency department management of stroke.Emerg Med Clin North Am.2002;20:687–702.
- ,,.Blood pressure management in acute stroke: comparison of current guidelines with prescribing patterns.Can J Neurol Sci.2002;29(2):125–131.
- ,,,,,.Use of antihypertensive agents in the management of patients with acute ischemic stroke.Neurology.2004;63:318–323.
- ,,,.Induced hypertension during ischemia reduces infarct area after temporary middle cerebral artery occlusion in rats.Surg Neurol.1996;46:229–234.
- ,,,.Effects of induced hypertension on intracranial pressure and flow velocities of the middle cerebral arteries in patients with large hemispheric stroke.Stroke.2002;33:998–1004.
- ,,,.Induced hypertension improves cerebral blood flow in acute ischemic stroke.Neurology.2005;64:1979.
- ,,,,.A pilot study of drug induced hypertension for treatment of acute stroke.Neurology.2001;56:1210–1213.
- ,,,,,.Pharmacologic elevation of blood pressure in acute stroke: clinical effects and safety.Stroke.1997;28:2133–2138.
- ,,,,.Feasibility and safety of norephinephrine‐induced arterial hypertension in acute ischemic stroke.Neurology.2004;62:1193–1195.
- ,,,,.Initial emergency department blood pressure as predictor of survival after acute ischemic stroke.Neurology.2005;65:1179–1183.
- The PROGRESS Collaborative Group.Randomized trial of a perindopril‐based blood‐pressure‐lowering regimen among 6105 individuals with previous stroke of transient ischaemic attack.Lancet.2001;358:1033–1041.
- ,,, et al.Effect of treating isolated systolic hypertension on the risk of developing various types and subtypes of stroke.JAMA.2000;284:465–471.
- ,.Treatment of chronic hypertension for the prevention of stroke.South Med J.2003;96:359–362.
- ,,,.Epidemiologic assessment of the role of blood pressure in stroke: the Framingham Study.JAMA.1996;276:1269–1278.
In the Literature
Repeat Testing for C. Diff?
By Jeff Glasheen, MD
Mohan SS, McDermott BP, Parchuri S, et al. Lack of value of repeat stool testing for Clostridium difficile toxin. Am J Med. 2006 Apr;119(4):356.e7-356.e8
Clostridium difficile is a common complication of antibiotic and chemotherapeutic use, especially in hospitalized patients. Yet most nosocomial diarrhea is not caused by C. difficile. Most antibiotics can cause loose stools through changes in the gastrointestinal flora that result in inadequate digestion and absorption of carbohydrates and a resultant osmotic diarrhea. Further, antibiotics such as erythromycin and amoxicillin/clavulanate may result in diarrhea via increases in GI tract motility. While osmotic and motility causes of diarrhea tend to improve with antibiotic discontinuation, C. difficile-associated diarrhea is associated with significant morbidity that often continues until adequately treated.
Thus having a test that differentiates between C. difficile and non-clostridial diarrhea is essential. The most commonly used test is the enzyme immunoassay (EIA) that detects toxins A and B. The sensitivity and specificity of this test has been reported to range between 50%-90% and 70%-95%, respectively. The authors of this paper evaluated the utility of repeat EIA testing in patients with a one negative test in the setting of nosocomial diarrhea associated with antibiotic use.
The authors reviewed 474 sequential EIA tests for C. difficile in 396 patients over a 10-month period at a large university-affiliated community hospital with an EIA sensitivity and specificity of 80%-90% and 80%-95%, respectively. Tests were considered to be “repeat” if they occurred within seven days of the original negative test. Of the 78 repeat tests (16.5% of all tests), only one was positive, resulting in a 0.8% conversion rate. At an institutional cost of $128 per test the total cost of EIA testing over the 10-month period was $60,672. The cost of repeat testing alone was $9,984. The authors conclude that there is limited value—and high cost—in repeat EIA testing and that alternative sources of diarrhea should be sought or we should repeat EIA testing in patients with continued nosocomial diarrhea and a negative EIA test.
While prior studies have shown incremental benefit of retesting for C. difficile with the EIA assay, this study’s authors conclude that repeat “C. difficile testing is not clinically justified and is economically wasteful.” Unfortunately, the authors did not utilize a strong enough research design to defend this statement. From the data presented, all we can conclude is that repeat testing with an EIA assay did not add significant value to the diagnostic workup. The lack of a negative gold standard test, such as the cytotoxin assay or follow up outcomes, such as resolution of diarrhea, leaves the reader pondering if the repeat EIA assays were negative because the patients did not have C. difficile or because the test was not sensitive enough to detect the toxin. The reported sensitivity for the EIA assay used was 80-90%, meaning that 10%-20% of patients with C. difficile disease would have had a false negative test. While a second negative test would lower the likelihood of true disease, it would not lower it enough in a patient with a high pre-test probability of disease to sufficiently rule out the disease such that further testing is indicated.
Additionally, while we can extrapolate that the EIA test was of little utility to the patients studied here, no patient-specific data is presented. Thus it is difficult to determine if our patient population is represented in the study. More knowledge about the patients would allow the reader to use published prediction rules to better delineate how likely it was that this cohort was at high risk for having toxin-mediated diarrhea. Perhaps a more reasonable approach to this clinical conundrum would be to send the repeat stool test for a cytotoxin assay or to treat the patient empirically in situations where either the likelihood of disease or the disease burden is high.
Stroke after MI
By Ethan Cumbler, MD
Witt BJ, Ballman KV, Brown RD Jr, et al. The incidence of stroke after myocardial infarction: a meta-analysis. Am J Med. 2006 Apr;119(4):354.e1-9.
Stroke and myocardial infarction (MI) share many of the risk factors leading to atherosclerosis, including hypertension, hyperlipidemia, diabetes, tobacco abuse, and age. Logically, patients at risk for one event would also be at risk for the other, yet this interaction appears to be more complex. The study by Witt, et al., aims to elucidate the rate of in-hospital stroke in patients initially admitted with an MI.
The authors analyzed 22 observational studies of myocardial infarction that recorded the incidence of cerebrovascular accidents after acute MI. Clinical trials were excluded from the analysis in order to provide representation of an unselected population. Of the trials reviewed, 11 were included in the analysis of in-hospital strokes, three for the 30-day time point, and two for the one-year time point. The other trials used different time points and were not included in the analysis. The patients had a mean age ranging between 59 and 72.7 years, and all had a predominance of males. The rate of in-hospital stroke was 11.1 events per 1,000 hospitalizations. This incidence rose to 12.2/1,000 at 30 days and 21.4/1,000 at one year.
Plausible hypotheses for why the rate of cerebrovascular accident would be particularly high in the post-MI period include the potential for localized wall motion dysmotility or low flow leading to intracardiac thrombosis, event-related arrhythmia, or procedure-related embolic events. The studies from which this meta-analysis was derived were not designed in such a way for a causal relationship to be identified. However, age, diabetes, hypertension, prior stroke, anterior location of MI, atrial fibrillation, heart failure, and nonwhite race were all found to have an association with increased risk for stroke. Interestingly, angina on presentation was associated with an apparent decreased risk, theorized to potentially be due to ischemic preconditioning.
While this study shares the usual limitations of meta-analyses of observational studies, the authors have done an excellent summation of the data available including both English and non-English language articles in the analysis. Notably, the review included studies spanning more than 25 years and, thus, included data from studies done in the era prior to modern therapy for cardiac ischemia including potent antiplatelet and statin therapy. The three studies with in-hospital time points started in the 1990s had a lower average rate of stroke, which may reflect the effect of more potent anti-platelet agents used in today’s therapy for acute coronary syndromes.
The implication for the hospitalist is to recognize that patients admitted for MI are at high risk for stroke during the index hospitalization. A low threshold for suspicion of a cerebrovascular event needs to be maintained for post-MI patients with new neurologic symptoms. Future studies will be needed to address the risk/benefit of anticoagulation in high-risk patients for stroke following a myocardial infarction.
Predicting PE in the ED Using Revised Geneva Score
By Jeffrey Carter, MD
Le Gal G, Righini M, Roy PM, et al. Prediction of pulmonary embolism in the emergency department: the revised Geneva score. Ann Intern Med. 2006 Feb 7;144(3):165-171.
Introduction: Pulmonary embolism remains a common life-threatening disorder with imperfect diagnostic modalities and strategies. Much of the current literature focuses on the development and validation of clinical probability assessments that identify low-risk patients who can be safely managed without invasive testing or lung scanning.
Two common scoring systems include the Geneva and Wells criteria, which employ a combination of historical information, clinical and laboratory data to stratify patients into three risk groups.1,2 It has recently been shown that a single negative D-dimer test safely excludes patients with suspected VTE in a large cohort, with a three-month follow-up rate of VTE of 0%.3 Criticism of current clinical probability assessment strategies include the inclusion of subjective criteria or the need for blood gas values.1,2 In this paper, the authors sought to derive and validate a scoring system based on readily available objective clinical data.
Methods: The cohort evaluated in this study consists of the same 965 patients used to prospectively evaluate an emergency department diagnostic strategy of VTE.3 Patients presenting to the emergency ward with complaints of chest pain or shortness of breath were evaluated with a standard diagnostic workup that included a checklist of demographic and historical information, signs and symptoms possibly consistent with VTE, ABG, chest X-ray, and EKG results, as well as the likelihood of another diagnosis. However, the goal was to create a scoring system based on readily available objective data, so ABG, chest X-ray and electrocardiogram results were not evaluated as possible components of the score. Clinical variables associated with pulmonary embolism were evaluated for statistical significance in both a univariate and multivariate model. These variables were then validated both internally and with an external cohort.
Results: The incidence of VTE was 23% (222/965). Ten clinical variables were found to be statistically significantly associated with VTE; these comprise the Revised Geneva Score.
Clinical probability is based on points: 0-3 is low probability; 4-10 is intermediate probability, and >10 is high probability. Low, intermediate, and high clinical probabilities had respective rates of VTE of 8%, 28%, and 74%. These percentages were found in the validation cohort and were similar to those in the derivation cohort.
Discussion: This study focuses on the derivation and validation of a clinical scoring system that can provide a numerical estimate of likelihood of VTE. The diagnostic strategy by which VTE is confirmed or excluded is from derivation cohort and is published elsewhere.3 In that study patients classified as no VTE and not treated had a 90-day risk of VTE of 1%. The nine-point revised Geneva score does indeed provide an accurate assessment of risk of VTE, and can thus help guide clinical decision-making. However it is not clear that the revised Geneva score will help decrease invasive diagnostic tests. In the low-risk group, 8% is too great a risk of a life-threatening illness to forego further diagnostic evaluation. Coupled with a negative ELISA D-dimer, exclusion of these patients is safe, but these patients cannot be safely excluded without risk stratification.3
References
- Wells PS, Anderson DR, Rodger M, et al. Derivation of a simple clinical model to categorize patients’ probability of pulmonary embolism: increasing the model’s utility with the SimpliRED D-dimer. Thromb Haemost. 2000 Mar;83:416-420.
- Wicki J, Perneger TV, Junof AF, et al. Assessing clinical probability of pulmonary embolism in the emergency ward: a simple score. Arch Intern Med. 2001 Jan 8;161(1):92-97.
- Perrier A, Roy PM, Aujesky D, et al. Diagnosing pulmonary embolism in outpatients with clinical assessment, D-dimer measurement, venous ultrasound and helical computed tomography: a multicenter management study. Am J Med. 2004 Mar 1;116(5):291-299.
LMWH for Inpatient Palliative Care
By Jeanie Youngwerth, MD
Noble SI, Nelson A, Turner C, et al. Acceptability of low molecular weight heparin thromboprophylaxis for patients receiving palliative care: qualitative study. BMJ. 2006 Mar 11:332(7541);577-580.ePub 2006 Feb 3.
Venous thromboembolism is a major risk factor for patients with malignancy. VTE may reduce survival time in patients receiving palliative care, with one in seven inpatients with cancer dying from pulmonary embolism. The American College of Chest Physicians recommend low molecular weight heparin (LMWH) in hospitalized patients with cancer as level 1A evidence in their 2004 consensus statement on VTE. There are no thromboprophylaxis guidelines in the United Kingdom. Many physicians view daily injections of LMWH as unnecessary distress for palliative care patients. This study focused on what inpatients with advanced cancer receiving palliative care thought about the effect of thromboprophylaxis on overall quality of life.
This was a qualitative study of 28 inpatients receiving palliative care who had advanced metastatic cancer and who were in a regional cancer center in Wales. The patients had received LMWH for at least five consecutive days. The patients were audiotaped and then had semi-structured interviews transcribed regarding cancer treatments they had received. These interviews covered the patients’ insight into prognosis, their understanding of thromboprophylaxis, their beliefs concerning the effects of thromboprophylaxis on overall quality of life, and the negative aspects of heparin treatment.
The main outcome measures were recurring themes of the effect of thromboprophylaxis on overall quality of life. Major themes identified were insight into prognosis, knowledge and understanding of thromboprophylaxis, acceptability of heparin thromboprophylaxis, reassurance, and optimism. Minor themes identified were bruising, negative impact of antiembolic stockings, and anger at paternalistic views toward terminally ill patients.
Most patients showed clear insight into the nature of their condition and understood heparin prophylaxis for VTE. Many patients identified immobility and surgery as VTE risk factors, with little understanding of cancer as a risk factor. All knew that VTE could cause death, but none were aware of the common symptoms. All patients found LMWH thromboprophylaxis acceptable. Patients believed taking measures to prevent symptoms might improve their quality of life and felt that the medical team had not given up on them.
The only negative experiences of LMWH thromboprophylaxis was bruising. All of the patients who wore antiembolic stockings during previous admissions found them uncomfortable. Patients expressed their need to be involved in decision-making, particularly with respect to withdrawal or non-administration of treatment.
This small, qualitative study showed that, contrary to many physicians’ beliefs, patients in palliative care units believe that LMWH injections are acceptable as thromboprophylaxis, but antiembolic stockings are not. The effect of daily injections had little or no effect on the quality of life, with many patients having positive feelings that things were being done to prevent new problems from occurring.
This study was limited by the small sample size, the qualitative nature that could introduce interpretation bias, and the fact that only patients using LMWH were included. Additionally, the United Kingdom has aggressively educated the public on the risks of VTE associated with long flights such that baseline knowledge may differ in other parts of the world. Resource utilization, including drug costs and length of stay, as well as effect on mortality were not studied.
The implications for hospitalists are that many inpatients with advanced cancer receiving palliative care may find LMWH thromboprophylaxis acceptable therapy, and that discussions with the patient regarding this option should be explored together.
Cost Analysis: Intensive Glycemic Control in Critically Ill Adults
By Whitney Woodmansee, MD
Krinsley JS, Jones, RL. Cost analysis of intensive glycemic control in critically ill adult patients. Chest. 2006;129:644-650.
Several studies have demonstrated improved outcomes and decreased mortality in ICU patients treated with intensive control of blood glucose levels. This study sought to identify the costs associated with intensive glycemic control in the ICU.
An ICU patient database was analyzed for cost data related to intensive glycemic control. A baseline group of 800 consecutive ICU patients admitted prior to initiation (baseline) of an intensive glucose management (blood glucose levels between 80-140 mg/dl protocol were compared with a treatment group of 800 consecutive patients admitted after initiation of the protocol). Previously reported outcomes of these patients demonstrated significant improvement in mortality with tight glycemic control. Costs related to ICU and non-ICU length of stay; duration of mechanical ventilation; and all laboratory, pharmacy, and radiology services were analyzed between groups. Resource utilization was determined by assessing charges from the database and adjusting for inflation and applying Medicare cost, charge ratios for each category, and fiscal year. Costs associated with the intensive glycemic control protocol were determined. Unfortunately, only cost estimates for insulin and associated disposable supplies for each group were available for analysis.
Baseline and treatment populations did not differ significantly regarding demographics such as age, gender, race, admitting diagnosis, diabetes prevalence, or APACHE II scores. There were fewer patients in the treatment group that required mechanical ventilation during their ICU stay (40.6% versus 33.6%). Intensive glucose management was associated with a 13.9% reduction in total ICU days and duration of mechanical ventilation (median of two days decreased to 1.7 days p=0.045). There was a $1,580 adjusted cost savings per patient in the intensive treatment group compared with the baseline group (p<0.001). This reduction in cost was primarily driven by a decrease in laboratory and radiology costs in the ventilated patients. There were nonsignificant cost reductions in the unventilated patients.
Intensive control of hyperglycemia in the hospitalized ICU patient appears to be associated with reduction of morbidity and mortality. This suggests that tight glycemic control also leads to reductions in overall patient care costs—particularly in the ventilated ICU patient. Although not a randomized control trial, database analysis of costs and resource utilization demonstrated an overall cost savings in the treatment group (after initiation of an intensive glycemic control protocol) compared with the baseline group (before protocol initiation). One caveat is that the authors used estimates when determining the costs associated with the implementation of the intensive glucose management protocol. Nevertheless, intensive glycemic control was associated with an overall reduction in patient costs related to decreased ICU days and mechanical ventilation as well as resource utilization in a patient population already shown to have improved mortality. These results, if confirmed, suggest that tight glycemic control in the ICU is cost effective and should become standard medical practice. TH
Repeat Testing for C. Diff?
By Jeff Glasheen, MD
Mohan SS, McDermott BP, Parchuri S, et al. Lack of value of repeat stool testing for Clostridium difficile toxin. Am J Med. 2006 Apr;119(4):356.e7-356.e8
Clostridium difficile is a common complication of antibiotic and chemotherapeutic use, especially in hospitalized patients. Yet most nosocomial diarrhea is not caused by C. difficile. Most antibiotics can cause loose stools through changes in the gastrointestinal flora that result in inadequate digestion and absorption of carbohydrates and a resultant osmotic diarrhea. Further, antibiotics such as erythromycin and amoxicillin/clavulanate may result in diarrhea via increases in GI tract motility. While osmotic and motility causes of diarrhea tend to improve with antibiotic discontinuation, C. difficile-associated diarrhea is associated with significant morbidity that often continues until adequately treated.
Thus having a test that differentiates between C. difficile and non-clostridial diarrhea is essential. The most commonly used test is the enzyme immunoassay (EIA) that detects toxins A and B. The sensitivity and specificity of this test has been reported to range between 50%-90% and 70%-95%, respectively. The authors of this paper evaluated the utility of repeat EIA testing in patients with a one negative test in the setting of nosocomial diarrhea associated with antibiotic use.
The authors reviewed 474 sequential EIA tests for C. difficile in 396 patients over a 10-month period at a large university-affiliated community hospital with an EIA sensitivity and specificity of 80%-90% and 80%-95%, respectively. Tests were considered to be “repeat” if they occurred within seven days of the original negative test. Of the 78 repeat tests (16.5% of all tests), only one was positive, resulting in a 0.8% conversion rate. At an institutional cost of $128 per test the total cost of EIA testing over the 10-month period was $60,672. The cost of repeat testing alone was $9,984. The authors conclude that there is limited value—and high cost—in repeat EIA testing and that alternative sources of diarrhea should be sought or we should repeat EIA testing in patients with continued nosocomial diarrhea and a negative EIA test.
While prior studies have shown incremental benefit of retesting for C. difficile with the EIA assay, this study’s authors conclude that repeat “C. difficile testing is not clinically justified and is economically wasteful.” Unfortunately, the authors did not utilize a strong enough research design to defend this statement. From the data presented, all we can conclude is that repeat testing with an EIA assay did not add significant value to the diagnostic workup. The lack of a negative gold standard test, such as the cytotoxin assay or follow up outcomes, such as resolution of diarrhea, leaves the reader pondering if the repeat EIA assays were negative because the patients did not have C. difficile or because the test was not sensitive enough to detect the toxin. The reported sensitivity for the EIA assay used was 80-90%, meaning that 10%-20% of patients with C. difficile disease would have had a false negative test. While a second negative test would lower the likelihood of true disease, it would not lower it enough in a patient with a high pre-test probability of disease to sufficiently rule out the disease such that further testing is indicated.
Additionally, while we can extrapolate that the EIA test was of little utility to the patients studied here, no patient-specific data is presented. Thus it is difficult to determine if our patient population is represented in the study. More knowledge about the patients would allow the reader to use published prediction rules to better delineate how likely it was that this cohort was at high risk for having toxin-mediated diarrhea. Perhaps a more reasonable approach to this clinical conundrum would be to send the repeat stool test for a cytotoxin assay or to treat the patient empirically in situations where either the likelihood of disease or the disease burden is high.
Stroke after MI
By Ethan Cumbler, MD
Witt BJ, Ballman KV, Brown RD Jr, et al. The incidence of stroke after myocardial infarction: a meta-analysis. Am J Med. 2006 Apr;119(4):354.e1-9.
Stroke and myocardial infarction (MI) share many of the risk factors leading to atherosclerosis, including hypertension, hyperlipidemia, diabetes, tobacco abuse, and age. Logically, patients at risk for one event would also be at risk for the other, yet this interaction appears to be more complex. The study by Witt, et al., aims to elucidate the rate of in-hospital stroke in patients initially admitted with an MI.
The authors analyzed 22 observational studies of myocardial infarction that recorded the incidence of cerebrovascular accidents after acute MI. Clinical trials were excluded from the analysis in order to provide representation of an unselected population. Of the trials reviewed, 11 were included in the analysis of in-hospital strokes, three for the 30-day time point, and two for the one-year time point. The other trials used different time points and were not included in the analysis. The patients had a mean age ranging between 59 and 72.7 years, and all had a predominance of males. The rate of in-hospital stroke was 11.1 events per 1,000 hospitalizations. This incidence rose to 12.2/1,000 at 30 days and 21.4/1,000 at one year.
Plausible hypotheses for why the rate of cerebrovascular accident would be particularly high in the post-MI period include the potential for localized wall motion dysmotility or low flow leading to intracardiac thrombosis, event-related arrhythmia, or procedure-related embolic events. The studies from which this meta-analysis was derived were not designed in such a way for a causal relationship to be identified. However, age, diabetes, hypertension, prior stroke, anterior location of MI, atrial fibrillation, heart failure, and nonwhite race were all found to have an association with increased risk for stroke. Interestingly, angina on presentation was associated with an apparent decreased risk, theorized to potentially be due to ischemic preconditioning.
While this study shares the usual limitations of meta-analyses of observational studies, the authors have done an excellent summation of the data available including both English and non-English language articles in the analysis. Notably, the review included studies spanning more than 25 years and, thus, included data from studies done in the era prior to modern therapy for cardiac ischemia including potent antiplatelet and statin therapy. The three studies with in-hospital time points started in the 1990s had a lower average rate of stroke, which may reflect the effect of more potent anti-platelet agents used in today’s therapy for acute coronary syndromes.
The implication for the hospitalist is to recognize that patients admitted for MI are at high risk for stroke during the index hospitalization. A low threshold for suspicion of a cerebrovascular event needs to be maintained for post-MI patients with new neurologic symptoms. Future studies will be needed to address the risk/benefit of anticoagulation in high-risk patients for stroke following a myocardial infarction.
Predicting PE in the ED Using Revised Geneva Score
By Jeffrey Carter, MD
Le Gal G, Righini M, Roy PM, et al. Prediction of pulmonary embolism in the emergency department: the revised Geneva score. Ann Intern Med. 2006 Feb 7;144(3):165-171.
Introduction: Pulmonary embolism remains a common life-threatening disorder with imperfect diagnostic modalities and strategies. Much of the current literature focuses on the development and validation of clinical probability assessments that identify low-risk patients who can be safely managed without invasive testing or lung scanning.
Two common scoring systems include the Geneva and Wells criteria, which employ a combination of historical information, clinical and laboratory data to stratify patients into three risk groups.1,2 It has recently been shown that a single negative D-dimer test safely excludes patients with suspected VTE in a large cohort, with a three-month follow-up rate of VTE of 0%.3 Criticism of current clinical probability assessment strategies include the inclusion of subjective criteria or the need for blood gas values.1,2 In this paper, the authors sought to derive and validate a scoring system based on readily available objective clinical data.
Methods: The cohort evaluated in this study consists of the same 965 patients used to prospectively evaluate an emergency department diagnostic strategy of VTE.3 Patients presenting to the emergency ward with complaints of chest pain or shortness of breath were evaluated with a standard diagnostic workup that included a checklist of demographic and historical information, signs and symptoms possibly consistent with VTE, ABG, chest X-ray, and EKG results, as well as the likelihood of another diagnosis. However, the goal was to create a scoring system based on readily available objective data, so ABG, chest X-ray and electrocardiogram results were not evaluated as possible components of the score. Clinical variables associated with pulmonary embolism were evaluated for statistical significance in both a univariate and multivariate model. These variables were then validated both internally and with an external cohort.
Results: The incidence of VTE was 23% (222/965). Ten clinical variables were found to be statistically significantly associated with VTE; these comprise the Revised Geneva Score.
Clinical probability is based on points: 0-3 is low probability; 4-10 is intermediate probability, and >10 is high probability. Low, intermediate, and high clinical probabilities had respective rates of VTE of 8%, 28%, and 74%. These percentages were found in the validation cohort and were similar to those in the derivation cohort.
Discussion: This study focuses on the derivation and validation of a clinical scoring system that can provide a numerical estimate of likelihood of VTE. The diagnostic strategy by which VTE is confirmed or excluded is from derivation cohort and is published elsewhere.3 In that study patients classified as no VTE and not treated had a 90-day risk of VTE of 1%. The nine-point revised Geneva score does indeed provide an accurate assessment of risk of VTE, and can thus help guide clinical decision-making. However it is not clear that the revised Geneva score will help decrease invasive diagnostic tests. In the low-risk group, 8% is too great a risk of a life-threatening illness to forego further diagnostic evaluation. Coupled with a negative ELISA D-dimer, exclusion of these patients is safe, but these patients cannot be safely excluded without risk stratification.3
References
- Wells PS, Anderson DR, Rodger M, et al. Derivation of a simple clinical model to categorize patients’ probability of pulmonary embolism: increasing the model’s utility with the SimpliRED D-dimer. Thromb Haemost. 2000 Mar;83:416-420.
- Wicki J, Perneger TV, Junof AF, et al. Assessing clinical probability of pulmonary embolism in the emergency ward: a simple score. Arch Intern Med. 2001 Jan 8;161(1):92-97.
- Perrier A, Roy PM, Aujesky D, et al. Diagnosing pulmonary embolism in outpatients with clinical assessment, D-dimer measurement, venous ultrasound and helical computed tomography: a multicenter management study. Am J Med. 2004 Mar 1;116(5):291-299.
LMWH for Inpatient Palliative Care
By Jeanie Youngwerth, MD
Noble SI, Nelson A, Turner C, et al. Acceptability of low molecular weight heparin thromboprophylaxis for patients receiving palliative care: qualitative study. BMJ. 2006 Mar 11:332(7541);577-580.ePub 2006 Feb 3.
Venous thromboembolism is a major risk factor for patients with malignancy. VTE may reduce survival time in patients receiving palliative care, with one in seven inpatients with cancer dying from pulmonary embolism. The American College of Chest Physicians recommend low molecular weight heparin (LMWH) in hospitalized patients with cancer as level 1A evidence in their 2004 consensus statement on VTE. There are no thromboprophylaxis guidelines in the United Kingdom. Many physicians view daily injections of LMWH as unnecessary distress for palliative care patients. This study focused on what inpatients with advanced cancer receiving palliative care thought about the effect of thromboprophylaxis on overall quality of life.
This was a qualitative study of 28 inpatients receiving palliative care who had advanced metastatic cancer and who were in a regional cancer center in Wales. The patients had received LMWH for at least five consecutive days. The patients were audiotaped and then had semi-structured interviews transcribed regarding cancer treatments they had received. These interviews covered the patients’ insight into prognosis, their understanding of thromboprophylaxis, their beliefs concerning the effects of thromboprophylaxis on overall quality of life, and the negative aspects of heparin treatment.
The main outcome measures were recurring themes of the effect of thromboprophylaxis on overall quality of life. Major themes identified were insight into prognosis, knowledge and understanding of thromboprophylaxis, acceptability of heparin thromboprophylaxis, reassurance, and optimism. Minor themes identified were bruising, negative impact of antiembolic stockings, and anger at paternalistic views toward terminally ill patients.
Most patients showed clear insight into the nature of their condition and understood heparin prophylaxis for VTE. Many patients identified immobility and surgery as VTE risk factors, with little understanding of cancer as a risk factor. All knew that VTE could cause death, but none were aware of the common symptoms. All patients found LMWH thromboprophylaxis acceptable. Patients believed taking measures to prevent symptoms might improve their quality of life and felt that the medical team had not given up on them.
The only negative experiences of LMWH thromboprophylaxis was bruising. All of the patients who wore antiembolic stockings during previous admissions found them uncomfortable. Patients expressed their need to be involved in decision-making, particularly with respect to withdrawal or non-administration of treatment.
This small, qualitative study showed that, contrary to many physicians’ beliefs, patients in palliative care units believe that LMWH injections are acceptable as thromboprophylaxis, but antiembolic stockings are not. The effect of daily injections had little or no effect on the quality of life, with many patients having positive feelings that things were being done to prevent new problems from occurring.
This study was limited by the small sample size, the qualitative nature that could introduce interpretation bias, and the fact that only patients using LMWH were included. Additionally, the United Kingdom has aggressively educated the public on the risks of VTE associated with long flights such that baseline knowledge may differ in other parts of the world. Resource utilization, including drug costs and length of stay, as well as effect on mortality were not studied.
The implications for hospitalists are that many inpatients with advanced cancer receiving palliative care may find LMWH thromboprophylaxis acceptable therapy, and that discussions with the patient regarding this option should be explored together.
Cost Analysis: Intensive Glycemic Control in Critically Ill Adults
By Whitney Woodmansee, MD
Krinsley JS, Jones, RL. Cost analysis of intensive glycemic control in critically ill adult patients. Chest. 2006;129:644-650.
Several studies have demonstrated improved outcomes and decreased mortality in ICU patients treated with intensive control of blood glucose levels. This study sought to identify the costs associated with intensive glycemic control in the ICU.
An ICU patient database was analyzed for cost data related to intensive glycemic control. A baseline group of 800 consecutive ICU patients admitted prior to initiation (baseline) of an intensive glucose management (blood glucose levels between 80-140 mg/dl protocol were compared with a treatment group of 800 consecutive patients admitted after initiation of the protocol). Previously reported outcomes of these patients demonstrated significant improvement in mortality with tight glycemic control. Costs related to ICU and non-ICU length of stay; duration of mechanical ventilation; and all laboratory, pharmacy, and radiology services were analyzed between groups. Resource utilization was determined by assessing charges from the database and adjusting for inflation and applying Medicare cost, charge ratios for each category, and fiscal year. Costs associated with the intensive glycemic control protocol were determined. Unfortunately, only cost estimates for insulin and associated disposable supplies for each group were available for analysis.
Baseline and treatment populations did not differ significantly regarding demographics such as age, gender, race, admitting diagnosis, diabetes prevalence, or APACHE II scores. There were fewer patients in the treatment group that required mechanical ventilation during their ICU stay (40.6% versus 33.6%). Intensive glucose management was associated with a 13.9% reduction in total ICU days and duration of mechanical ventilation (median of two days decreased to 1.7 days p=0.045). There was a $1,580 adjusted cost savings per patient in the intensive treatment group compared with the baseline group (p<0.001). This reduction in cost was primarily driven by a decrease in laboratory and radiology costs in the ventilated patients. There were nonsignificant cost reductions in the unventilated patients.
Intensive control of hyperglycemia in the hospitalized ICU patient appears to be associated with reduction of morbidity and mortality. This suggests that tight glycemic control also leads to reductions in overall patient care costs—particularly in the ventilated ICU patient. Although not a randomized control trial, database analysis of costs and resource utilization demonstrated an overall cost savings in the treatment group (after initiation of an intensive glycemic control protocol) compared with the baseline group (before protocol initiation). One caveat is that the authors used estimates when determining the costs associated with the implementation of the intensive glucose management protocol. Nevertheless, intensive glycemic control was associated with an overall reduction in patient costs related to decreased ICU days and mechanical ventilation as well as resource utilization in a patient population already shown to have improved mortality. These results, if confirmed, suggest that tight glycemic control in the ICU is cost effective and should become standard medical practice. TH
Repeat Testing for C. Diff?
By Jeff Glasheen, MD
Mohan SS, McDermott BP, Parchuri S, et al. Lack of value of repeat stool testing for Clostridium difficile toxin. Am J Med. 2006 Apr;119(4):356.e7-356.e8
Clostridium difficile is a common complication of antibiotic and chemotherapeutic use, especially in hospitalized patients. Yet most nosocomial diarrhea is not caused by C. difficile. Most antibiotics can cause loose stools through changes in the gastrointestinal flora that result in inadequate digestion and absorption of carbohydrates and a resultant osmotic diarrhea. Further, antibiotics such as erythromycin and amoxicillin/clavulanate may result in diarrhea via increases in GI tract motility. While osmotic and motility causes of diarrhea tend to improve with antibiotic discontinuation, C. difficile-associated diarrhea is associated with significant morbidity that often continues until adequately treated.
Thus having a test that differentiates between C. difficile and non-clostridial diarrhea is essential. The most commonly used test is the enzyme immunoassay (EIA) that detects toxins A and B. The sensitivity and specificity of this test has been reported to range between 50%-90% and 70%-95%, respectively. The authors of this paper evaluated the utility of repeat EIA testing in patients with a one negative test in the setting of nosocomial diarrhea associated with antibiotic use.
The authors reviewed 474 sequential EIA tests for C. difficile in 396 patients over a 10-month period at a large university-affiliated community hospital with an EIA sensitivity and specificity of 80%-90% and 80%-95%, respectively. Tests were considered to be “repeat” if they occurred within seven days of the original negative test. Of the 78 repeat tests (16.5% of all tests), only one was positive, resulting in a 0.8% conversion rate. At an institutional cost of $128 per test the total cost of EIA testing over the 10-month period was $60,672. The cost of repeat testing alone was $9,984. The authors conclude that there is limited value—and high cost—in repeat EIA testing and that alternative sources of diarrhea should be sought or we should repeat EIA testing in patients with continued nosocomial diarrhea and a negative EIA test.
While prior studies have shown incremental benefit of retesting for C. difficile with the EIA assay, this study’s authors conclude that repeat “C. difficile testing is not clinically justified and is economically wasteful.” Unfortunately, the authors did not utilize a strong enough research design to defend this statement. From the data presented, all we can conclude is that repeat testing with an EIA assay did not add significant value to the diagnostic workup. The lack of a negative gold standard test, such as the cytotoxin assay or follow up outcomes, such as resolution of diarrhea, leaves the reader pondering if the repeat EIA assays were negative because the patients did not have C. difficile or because the test was not sensitive enough to detect the toxin. The reported sensitivity for the EIA assay used was 80-90%, meaning that 10%-20% of patients with C. difficile disease would have had a false negative test. While a second negative test would lower the likelihood of true disease, it would not lower it enough in a patient with a high pre-test probability of disease to sufficiently rule out the disease such that further testing is indicated.
Additionally, while we can extrapolate that the EIA test was of little utility to the patients studied here, no patient-specific data is presented. Thus it is difficult to determine if our patient population is represented in the study. More knowledge about the patients would allow the reader to use published prediction rules to better delineate how likely it was that this cohort was at high risk for having toxin-mediated diarrhea. Perhaps a more reasonable approach to this clinical conundrum would be to send the repeat stool test for a cytotoxin assay or to treat the patient empirically in situations where either the likelihood of disease or the disease burden is high.
Stroke after MI
By Ethan Cumbler, MD
Witt BJ, Ballman KV, Brown RD Jr, et al. The incidence of stroke after myocardial infarction: a meta-analysis. Am J Med. 2006 Apr;119(4):354.e1-9.
Stroke and myocardial infarction (MI) share many of the risk factors leading to atherosclerosis, including hypertension, hyperlipidemia, diabetes, tobacco abuse, and age. Logically, patients at risk for one event would also be at risk for the other, yet this interaction appears to be more complex. The study by Witt, et al., aims to elucidate the rate of in-hospital stroke in patients initially admitted with an MI.
The authors analyzed 22 observational studies of myocardial infarction that recorded the incidence of cerebrovascular accidents after acute MI. Clinical trials were excluded from the analysis in order to provide representation of an unselected population. Of the trials reviewed, 11 were included in the analysis of in-hospital strokes, three for the 30-day time point, and two for the one-year time point. The other trials used different time points and were not included in the analysis. The patients had a mean age ranging between 59 and 72.7 years, and all had a predominance of males. The rate of in-hospital stroke was 11.1 events per 1,000 hospitalizations. This incidence rose to 12.2/1,000 at 30 days and 21.4/1,000 at one year.
Plausible hypotheses for why the rate of cerebrovascular accident would be particularly high in the post-MI period include the potential for localized wall motion dysmotility or low flow leading to intracardiac thrombosis, event-related arrhythmia, or procedure-related embolic events. The studies from which this meta-analysis was derived were not designed in such a way for a causal relationship to be identified. However, age, diabetes, hypertension, prior stroke, anterior location of MI, atrial fibrillation, heart failure, and nonwhite race were all found to have an association with increased risk for stroke. Interestingly, angina on presentation was associated with an apparent decreased risk, theorized to potentially be due to ischemic preconditioning.
While this study shares the usual limitations of meta-analyses of observational studies, the authors have done an excellent summation of the data available including both English and non-English language articles in the analysis. Notably, the review included studies spanning more than 25 years and, thus, included data from studies done in the era prior to modern therapy for cardiac ischemia including potent antiplatelet and statin therapy. The three studies with in-hospital time points started in the 1990s had a lower average rate of stroke, which may reflect the effect of more potent anti-platelet agents used in today’s therapy for acute coronary syndromes.
The implication for the hospitalist is to recognize that patients admitted for MI are at high risk for stroke during the index hospitalization. A low threshold for suspicion of a cerebrovascular event needs to be maintained for post-MI patients with new neurologic symptoms. Future studies will be needed to address the risk/benefit of anticoagulation in high-risk patients for stroke following a myocardial infarction.
Predicting PE in the ED Using Revised Geneva Score
By Jeffrey Carter, MD
Le Gal G, Righini M, Roy PM, et al. Prediction of pulmonary embolism in the emergency department: the revised Geneva score. Ann Intern Med. 2006 Feb 7;144(3):165-171.
Introduction: Pulmonary embolism remains a common life-threatening disorder with imperfect diagnostic modalities and strategies. Much of the current literature focuses on the development and validation of clinical probability assessments that identify low-risk patients who can be safely managed without invasive testing or lung scanning.
Two common scoring systems include the Geneva and Wells criteria, which employ a combination of historical information, clinical and laboratory data to stratify patients into three risk groups.1,2 It has recently been shown that a single negative D-dimer test safely excludes patients with suspected VTE in a large cohort, with a three-month follow-up rate of VTE of 0%.3 Criticism of current clinical probability assessment strategies include the inclusion of subjective criteria or the need for blood gas values.1,2 In this paper, the authors sought to derive and validate a scoring system based on readily available objective clinical data.
Methods: The cohort evaluated in this study consists of the same 965 patients used to prospectively evaluate an emergency department diagnostic strategy of VTE.3 Patients presenting to the emergency ward with complaints of chest pain or shortness of breath were evaluated with a standard diagnostic workup that included a checklist of demographic and historical information, signs and symptoms possibly consistent with VTE, ABG, chest X-ray, and EKG results, as well as the likelihood of another diagnosis. However, the goal was to create a scoring system based on readily available objective data, so ABG, chest X-ray and electrocardiogram results were not evaluated as possible components of the score. Clinical variables associated with pulmonary embolism were evaluated for statistical significance in both a univariate and multivariate model. These variables were then validated both internally and with an external cohort.
Results: The incidence of VTE was 23% (222/965). Ten clinical variables were found to be statistically significantly associated with VTE; these comprise the Revised Geneva Score.
Clinical probability is based on points: 0-3 is low probability; 4-10 is intermediate probability, and >10 is high probability. Low, intermediate, and high clinical probabilities had respective rates of VTE of 8%, 28%, and 74%. These percentages were found in the validation cohort and were similar to those in the derivation cohort.
Discussion: This study focuses on the derivation and validation of a clinical scoring system that can provide a numerical estimate of likelihood of VTE. The diagnostic strategy by which VTE is confirmed or excluded is from derivation cohort and is published elsewhere.3 In that study patients classified as no VTE and not treated had a 90-day risk of VTE of 1%. The nine-point revised Geneva score does indeed provide an accurate assessment of risk of VTE, and can thus help guide clinical decision-making. However it is not clear that the revised Geneva score will help decrease invasive diagnostic tests. In the low-risk group, 8% is too great a risk of a life-threatening illness to forego further diagnostic evaluation. Coupled with a negative ELISA D-dimer, exclusion of these patients is safe, but these patients cannot be safely excluded without risk stratification.3
References
- Wells PS, Anderson DR, Rodger M, et al. Derivation of a simple clinical model to categorize patients’ probability of pulmonary embolism: increasing the model’s utility with the SimpliRED D-dimer. Thromb Haemost. 2000 Mar;83:416-420.
- Wicki J, Perneger TV, Junof AF, et al. Assessing clinical probability of pulmonary embolism in the emergency ward: a simple score. Arch Intern Med. 2001 Jan 8;161(1):92-97.
- Perrier A, Roy PM, Aujesky D, et al. Diagnosing pulmonary embolism in outpatients with clinical assessment, D-dimer measurement, venous ultrasound and helical computed tomography: a multicenter management study. Am J Med. 2004 Mar 1;116(5):291-299.
LMWH for Inpatient Palliative Care
By Jeanie Youngwerth, MD
Noble SI, Nelson A, Turner C, et al. Acceptability of low molecular weight heparin thromboprophylaxis for patients receiving palliative care: qualitative study. BMJ. 2006 Mar 11:332(7541);577-580.ePub 2006 Feb 3.
Venous thromboembolism is a major risk factor for patients with malignancy. VTE may reduce survival time in patients receiving palliative care, with one in seven inpatients with cancer dying from pulmonary embolism. The American College of Chest Physicians recommend low molecular weight heparin (LMWH) in hospitalized patients with cancer as level 1A evidence in their 2004 consensus statement on VTE. There are no thromboprophylaxis guidelines in the United Kingdom. Many physicians view daily injections of LMWH as unnecessary distress for palliative care patients. This study focused on what inpatients with advanced cancer receiving palliative care thought about the effect of thromboprophylaxis on overall quality of life.
This was a qualitative study of 28 inpatients receiving palliative care who had advanced metastatic cancer and who were in a regional cancer center in Wales. The patients had received LMWH for at least five consecutive days. The patients were audiotaped and then had semi-structured interviews transcribed regarding cancer treatments they had received. These interviews covered the patients’ insight into prognosis, their understanding of thromboprophylaxis, their beliefs concerning the effects of thromboprophylaxis on overall quality of life, and the negative aspects of heparin treatment.
The main outcome measures were recurring themes of the effect of thromboprophylaxis on overall quality of life. Major themes identified were insight into prognosis, knowledge and understanding of thromboprophylaxis, acceptability of heparin thromboprophylaxis, reassurance, and optimism. Minor themes identified were bruising, negative impact of antiembolic stockings, and anger at paternalistic views toward terminally ill patients.
Most patients showed clear insight into the nature of their condition and understood heparin prophylaxis for VTE. Many patients identified immobility and surgery as VTE risk factors, with little understanding of cancer as a risk factor. All knew that VTE could cause death, but none were aware of the common symptoms. All patients found LMWH thromboprophylaxis acceptable. Patients believed taking measures to prevent symptoms might improve their quality of life and felt that the medical team had not given up on them.
The only negative experiences of LMWH thromboprophylaxis was bruising. All of the patients who wore antiembolic stockings during previous admissions found them uncomfortable. Patients expressed their need to be involved in decision-making, particularly with respect to withdrawal or non-administration of treatment.
This small, qualitative study showed that, contrary to many physicians’ beliefs, patients in palliative care units believe that LMWH injections are acceptable as thromboprophylaxis, but antiembolic stockings are not. The effect of daily injections had little or no effect on the quality of life, with many patients having positive feelings that things were being done to prevent new problems from occurring.
This study was limited by the small sample size, the qualitative nature that could introduce interpretation bias, and the fact that only patients using LMWH were included. Additionally, the United Kingdom has aggressively educated the public on the risks of VTE associated with long flights such that baseline knowledge may differ in other parts of the world. Resource utilization, including drug costs and length of stay, as well as effect on mortality were not studied.
The implications for hospitalists are that many inpatients with advanced cancer receiving palliative care may find LMWH thromboprophylaxis acceptable therapy, and that discussions with the patient regarding this option should be explored together.
Cost Analysis: Intensive Glycemic Control in Critically Ill Adults
By Whitney Woodmansee, MD
Krinsley JS, Jones, RL. Cost analysis of intensive glycemic control in critically ill adult patients. Chest. 2006;129:644-650.
Several studies have demonstrated improved outcomes and decreased mortality in ICU patients treated with intensive control of blood glucose levels. This study sought to identify the costs associated with intensive glycemic control in the ICU.
An ICU patient database was analyzed for cost data related to intensive glycemic control. A baseline group of 800 consecutive ICU patients admitted prior to initiation (baseline) of an intensive glucose management (blood glucose levels between 80-140 mg/dl protocol were compared with a treatment group of 800 consecutive patients admitted after initiation of the protocol). Previously reported outcomes of these patients demonstrated significant improvement in mortality with tight glycemic control. Costs related to ICU and non-ICU length of stay; duration of mechanical ventilation; and all laboratory, pharmacy, and radiology services were analyzed between groups. Resource utilization was determined by assessing charges from the database and adjusting for inflation and applying Medicare cost, charge ratios for each category, and fiscal year. Costs associated with the intensive glycemic control protocol were determined. Unfortunately, only cost estimates for insulin and associated disposable supplies for each group were available for analysis.
Baseline and treatment populations did not differ significantly regarding demographics such as age, gender, race, admitting diagnosis, diabetes prevalence, or APACHE II scores. There were fewer patients in the treatment group that required mechanical ventilation during their ICU stay (40.6% versus 33.6%). Intensive glucose management was associated with a 13.9% reduction in total ICU days and duration of mechanical ventilation (median of two days decreased to 1.7 days p=0.045). There was a $1,580 adjusted cost savings per patient in the intensive treatment group compared with the baseline group (p<0.001). This reduction in cost was primarily driven by a decrease in laboratory and radiology costs in the ventilated patients. There were nonsignificant cost reductions in the unventilated patients.
Intensive control of hyperglycemia in the hospitalized ICU patient appears to be associated with reduction of morbidity and mortality. This suggests that tight glycemic control also leads to reductions in overall patient care costs—particularly in the ventilated ICU patient. Although not a randomized control trial, database analysis of costs and resource utilization demonstrated an overall cost savings in the treatment group (after initiation of an intensive glycemic control protocol) compared with the baseline group (before protocol initiation). One caveat is that the authors used estimates when determining the costs associated with the implementation of the intensive glucose management protocol. Nevertheless, intensive glycemic control was associated with an overall reduction in patient costs related to decreased ICU days and mechanical ventilation as well as resource utilization in a patient population already shown to have improved mortality. These results, if confirmed, suggest that tight glycemic control in the ICU is cost effective and should become standard medical practice. TH